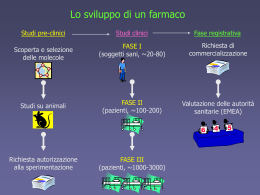
UNIVERSITA’ DEGLI STUDI “G. d’ANNUNZIO” CHIETI SCUOLA DI SPECIALIZZAZIONE IN IGIENE E MEDICINA PREVENTIVA Direttore: Prof. Ferdinando Romano La sperimentazione clinica: normativa e protocolli Specializzando Dott. Antonello TEDESCHI Relatore Prof. Francesco SCHIOPPA Anno Accademico 2002/2003 Evoluzione della normativa controllo della qualità: corrispondenza tra il contenuto della confezione e la formula dichiarata; controllo della sicurezza: (primum non nocere); hanno costituito i primi criteri di accettabilità dei farmaci controllo dell’efficacia: non più basato su criteri di soggettività ma sulla dimostrazione oggettiva dell’attività terapeutica per mezzo dello studio clinico controllato fondato su modelli statistici; Evoluzione della normativa (II) dimostrazione dell’attendibilità e credibilità dei dati garantita dalle buone pratiche farmaceutiche: GMP (Good Manifacturing Practice) GLP (Good Laboratory Practice) GCP (Good Clinical Practice) con l’intento di garantire l’integrità dei dati e la riproducibilità delle procedure con cui i dati stessi vengono prodotti. Evoluzione della normativa (III) armonizzazione internazionale attraverso: creazione dell’Agenzia Europea per la valutazione dei farmaci (EMEA); istituzione di procedure di registrazione comunitarie che coinvolgono strutture sopranazionali sulla base di un identico dossier di registrazione; creazione del Comitato delle Specialità Farmaceutiche (CPMP) che costituisce l’organo tecnico di consultazione dell’EMEA; standardizzazione del formato del dossier di registrazione tramite l’NTA (Notice to Applicants) linea guida per la stesura del dossier. L’osservanza delle GCP, in Europa, è richiesta dalla direttiva 91/507; recepita, in Italia, con il D.M. 15 luglio 1997. I concetti fondamentali delle GCP sono: ETICA: proteggere i soggetti partecipanti allo studio; RESPONSABILITA’: obblighi dello sponsor, del ricercatore, del monitor; GESTIONE DEI DATI: come i dati devono essere raccolti ed elaborati; STATISTICA: modelli per giustificare il disegno sperimentale e l’analisi dei dati; ASSICURAZIONE DI QUALITA’: come i dati devono essere controllati. Le GMP assicurano la riproducibilità delle caratteristiche dei lotti attraverso le procedure di convalida del metodo di fabbricazione. GLP: gli studi che devono essere condotti in GLP sono quelli di: mutagenesi, tossico-cinetica, farmacodinamica (variabilità individuale, gruppi a rischio). Lo studio tossicologico va deciso caso per caso in relazione alle caratteristiche del farmaco, all’impiego terapeutico previsto, ed alla popolazione cui è rivolto. Va giustificata con evidenze sperimentali la forma farmaceutica selezionata e la sua composizione in relazione all’impiego terapeutico. La scelta degli eccipienti va giustificata ed ottimizzata. Impurezze: possono richiedere approfondimenti tossicofarmacologici. Controllo dei livelli plasmatici utilizzando modelli statistici (valutazione efficacia e tollerabilità). Metodi di registrazione Formato unico del dossier di registrazione. In Europa sono vigenti tre procedure di registrazione: PROCEDURA NAZIONALE: per prodotti di interesse nazionale; e strutture sopranazionali non vengono coinvolte. PROCEDURA CENTRALIZZATA: è direttamente gestita dall’EMEA e da luogo a decisioni vincolanti per l’intera comunità. PROCEDURA DECENTRALIZZATA: basata sul mutuo riconoscimento da parte di un paese membro della registrazione già rilasciata da un altro stato membro. In condizioni particolari la registrazione può essere concessa prima del termine dell’iter sperimentale subordinandola alla presentazione di dati successivamente. Sviluppo preclinico e clinico di un farmaco FASI PRECLINICHE: tappe precedenti l’impiego sull’uomo. tossicità acuta: in due specie animali seguendo due vie di somministrazione, una delle quali è quella che si prevede verrà utilizzata nell’uomo; tossicità subacuta: per tre mesi ed in due o più specie; alla fine di tale periodo organi e tessuti degli animali impiegati vengono studiati sotto il profilo anatomopatologico; tossicità a lungo termine: da sei mesi a due anni; carcinogenesi: topi e ratti per la durata media della loro vita; studi tossicologici sulla fertilità e teratogenesi LE FASI CLINICHE (I, II, III, IV) valutano efficacia e tollerabilità sull’uomo. FASE I: condotta su un numero ristretto di volontari, sani, maschi e adulti, con somministrazione singola e ripetuta; per valutare il range posologico, la tollerabilità, l’assorbimento, la distribuzione, l’eliminazione, ecc. Possono, inoltre, essere valutate solo reazioni avverse al farmaco (RAF) assai comuni, di lieve entità e correlate all’attività farmacologica. FASE II: studi inizialmente in aperto e successivamente controllati su alcune centinaia di persone, con esclusione di sottogruppi in trattamento con altri farmaci o affetti da altra patologia, donne in età fertile, bambini ed anziani. FASE III: condotta su alcune migliaia di pazienti, comprendenti anche sottogruppi precedentemente esclusi: nefropatici, epatopatici, fasce estreme di età. Permette di valutare anche RAF di una certa rarità (1:500-1:1000). Gli studi condotti sull’uomo, impiegando un farmaco prima della sua commercializzazione, non possono fornire un’esatta riproduzione delle condizioni reali a causa di: ristretto numero di pazienti; tempi di terapia relativamente brevi; artificialità delle indagini condotte in maniera controllata in ospedale ed in popolazioni selezionate. FASE IV (FARMACOVIGILANZA): condotta dopo l’immissione in commercio del farmaco identifica RAF estremamente rare, nonché associazioni con altri farmaci, o in presenza di patologie associate. Fin dagli anni 60 la normativa sancisce l’obbligo, da parte dei medici, di segnalare le possibili reazioni avverse nonché: il monitoraggio delle aziende farmaceutiche degli effetti collaterali dei farmaci; l’uso di apposite schede da parte degli informatori scientifici per le segnalazioni; l’obbligo della sorveglianza per tutta la vita commerciale del farmaco; l’istituzione del Dipartimento per la valutazione dei medicinali e la farmacovigilanza presso il ministero della Salute; l’istituzione di un responsabile della farmacovigilanza presso le ASL e le aziende farmaceutiche. Bioetica AUTONOMIA: in passato, i soggetti non venivano informati non solo dei rischi, dei benefici e delle procedure che la sperimentazione comportava, ma anche del fatto stesso di essere coinvolti in una sperimentazione. Ciò ha portato alla nascita del consenso informato: completa informazione degli obiettivi, disagi, rischi e libertà di decidere se aderire o meno. Per i minori, l’incompleta comprensione rende necessario il consenso del tutore a meno che si tratti di interventi salva-vita o se gli stessi risultati non possono essere ottenuti con adulti normali. Bioetica (II) NON MALEFICENZA (NON NOCERE): il divieto di arrecare danno al paziente appariva messo in pericolo dai possibili benefici derivanti dal progresso della scienza medica. GIUSTIZIA: per es. la sperimentazione di un trattamento che sarà comunque assai costoso e non sarà mai disponibile ai gruppi più disagiati non può essere effettuata su una popolazione proveniente da tali gruppi. Farmaci per l’AIDS: le associazioni di sieropositivi hanno chiesto la presenza di loro rappresentanti non solo nei comitati etici ma anche nelle stesse commissioni incaricate di elaborare il disegno dello studio Bioetica (III) COMITATI ETICI: struttura indipendente formata da medici e non medici per la tutela dei diritti, della sicurezza e del benessere dei soggetti coinvolti in uno studio clinico. Da parere favorevole al protocollo, all’idoneità degli sperimentatori e delle strutture, dei metodi e del materiale. DICHIARAZIONE DI HELSINKI: nella ricerca sull’uomo il benessere della singola persona deve sempre prevalere sui possibili vantaggi per la scienza e la società. Bioetica (IV) ART. 32 COSTITUZIONE: nessuno può essere obbligato ad un trattamento sanitario se non per disposizioni di legge; la legge non può in nessun caso violare i limiti imposti dal rispetto della persona umana. ART. 5 CODICE CIVILE: assunzione di rischi non suscettibili di produrre minorazioni gravi e permanenti. La normativa recente ISPEZIONI: il Ministero della Salute diffida dal condurre sperimentazioni senza osservare le procedure autorizzative e la normativa vigente: non sono un fatto meramente burocratico ma finalizzate alla tutela della salute e dei diritti dei volontari nonché dei futuri pazienti. Mette in atto attività di informazione, formazione, richiamo scritto, ed ispezione degli stabilimenti e controllo dei medicinali da parte di ispettori. La normativa recente (II) PLACEBO: il D.M. del 18 marzo 1998 dice che i pazienti del gruppo di controllo non possono essere privati di una terapia efficace se questa è disponibile e non possono essere trattati con un placebo se ciò comporta sofferenza, prolungamento di malattia o rischio. L’uso del placebo appare incompatibile con la Dichiarazione di Helsinki: ad ogni paziente, inclusi quelli del gruppo di controllo dovrebbe essere assicurata la miglior prova diagnostica ed il miglior mezzo terapeutico. Le sperimentazioni di ogni fase possono essere effettuate negli ospedali pubblici o in quelli ad essi equiparati (Legge 833), negli enti di ricerca (Legge 833), nelle Università e negli istituti di ricovero e cura a carattere scientifico (D.M. 13 maggio 1999). Nelle unità dove non sono previsti posti letto, il medico deve essere presente almeno durante il tempo di permanenza dei pazienti, e deve essere comunque reperibile durante le ore in cui i pazienti non sono presenti presso l’unità stessa. FARMACI DI DERIVAZIONE BOVINA ED EMODERIVATI: il Ministero della Salute ha imposto il divieto di commercializzare e sottoporre a sperimentazione clinica questo tipo di farmaci se non accompagnati da certificati che assicurano la loro sicurezza. I REGISTRI: sono istituiti alcuni registri presso il Dipartimento per la valutazione dei medicinali e la Farmacovigilanza: il registro dei comitati etici; il registro dei giudizi di notorietà; il registro delle sperimentazioni; il registro dei centri privati; i registri, compilati e consultati per via telematica costituiscono la banca dati della Sperimentazione clinica (Osservatorio Nazionale). MMG e PLS: i medici di medicina generale ed i pediatri di libera scelta possono condurre sperimentazioni che si riferiscono alle affezioni non richiedenti ricovero ospedaliero, largamente diffuse sul territorio. Conclusioni I vantaggi ottenuti con la globalizzazione delle procedure della Sperimentazione e della Registrazione hanno comunque comportato un aumento dei costi, tale da mettere in pericolo nel prossimo futuro aziende o stati che non dispongono di risorse economiche e di tecnologie proprie delle multinazionali. Inoltre c’è il problema della sperimentazione rivolta a farmaci per le malattie rare: la maggior parte delle aziende incentivano ovviamente le ricerche su quei farmaci dalla cui vendita potranno ricavare dei profitti.
Scaricare