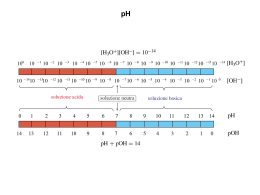
Chimica (a.a. 2011/2012, 6CFU) Equilibri ionici in soluzione acquosa (I parte): Acidi e Basi: la teoria di Brönsted-Lowry Forza relativa di acidi e basi Autoionizzazione (autodissociazione) dell’acqua Il pH e il pOH Calcolo del pH di soluzioni di acidi forti e basi forti Acidi deboli La costante di acidità Ka Basi deboli: La costante di basicità Kb Calcolo del pH per un acido debole monovalente (monoprotico) Calcolo del pH per un base debole monovalente Che cosa è un acido? E una base? ACIDI E BASI NELLA VITA QUOTIDIANA Acidi Basi Molti dei prodotti, normalmente usati in casa, presentano caratteristiche acide o basiche. Ne sono esempi: saponi, detersivi, medicine e alimenti Etimologia: Acidi e Basi Acido è una parola che deriva dal latino “acetum” (aceto). Col tempo la parola si è estesa a tutte le sostanze che hanno un analogo sapore “acidulo” (aspro). Basi o alcali (dall’arabo al kali = la cenere delle piante) sono le sostanze in grado di neutralizzare gli acidi. Qual è la definizione di acido e di base dal punto di vista della chimica? La storia ……………….… Antoine Lavoisier 1743-1794 Humpry Davy 1778-1829 Lavoisier definì gli acidi come combinazioni di radicali ed ossigeno (oxys genes = generatore d’acido), e le basi come una combinazione di metalli ed ossigeno. La sua idea fu in genere accettata, nonostante che acidi come l’acido cloridrico fossero noti da secoli. Si credeva allora che il cloro fosse un ossido piuttosto che un elemento. Nel 1809 Gay-Lussac (1778-1850), in collaborazione con Trénard (1777-1857) trovò che i cloruri, allora chiamati muriati, non contenevano ossigeno. Comunque essi erano talmente persuasi della validità delle idee di Lavoisier che preferirono dubitare dei propri risultati piuttosto che proporre il nuovo concetto. Fu il chimico inglese Humpry Davy (1778-1829) a cambiare la teoria acido-base provando che il cloro era un elemento e che l’acido cloridrico non conteneva ossigeno. Egli considerava l’idrogeno come il principio degli acidi (1810) e la sua teoria fu accettata in breve tempo. Un approccio moderno al problema è stato possibile solo dopo che Arrhenius propose la sua teoria della dissociazione elettrolitica, che includeva il concetto di elettroliti forti, cioè, sali, acidi e basi. Egli proclamò le sue convinzioni all’Accademia Svedese delle scienze nel 1883 e pubblicò la sua teoria nel 1887. Secondo Arrhenius, un acido si dissocia in ioni idrogeno ed alcuni anioni, mentre una base si dissocia in ioni idrossido e nei corrispondenti cationi. Il concetto di Arrhenius forniva anche una spiegazione delle diverse forze dei diversi acidi, basate sui loro differenti gradi di dissociazione. Svante August Arrhenius Wijk (Svezia), 1859 – Stoccolma, 1927 Premio Nobel per la Chimica 1903 ……….. la storia ……………….… Johannes Nicolaus BrØnsted 1879-1947 Thomas Martin Lowry 1874-1936 Sorsero, tuttavia, ulteriori difficoltà nel tentativo di spiegare l’acidità delle soluzioni organiche. Una soluzione di ammoniaca in etere esibisce delle proprietà basiche, ma si può dimostrare che in una tale soluzione non esiste lo ione idrossido. Arthur Lapworth (1872-1941), professore dell’Università di Manchester, insegnante di chimica organica, fisica ed inorganica, descrisse gli acidi come donatori di ioni idrogeno. T. M. Lowry (1874-1936) era uno dei suoi allievi. Lui ed il professore danese Johann N. Brønsted (1879-1947) svilupparono indipendentemente le idee di Lapworth di acidi e basi in quella che è attualmente chiamata teoria di Brønsted- Lowry (1923). Acidi e Basi Definizione di Arrhenius (1887): acidi e basi sono composti che in acqua danno luogo a dissociazione elettrolitica: Acido → H+(aq) + ... Base → OH-(aq) + ... Gli acidi e le basi secondo la teoria di Arrhenius: Le sostanze che dissociandosi in acqua dando ioni idrogeno (protoni) sono acide: HCl H2O → H+ + Cl- (H+ raggio ionico di 10-15 m!, esso in soluzione acquosa si trova come ione idrossonio: H3O+ ovvero H(H2O)+n con 1≤n≤7, n numero intero) Gli acidi e le basi secondo la teoria di Arrhenius: HCl H2O → H+ + ClH2O H3O+ Gli acidi e le basi secondo la teoria di Arrhenius: Le sostanze che dissociandosi in acqua danno ioni idrossido (ossidrile) sono basiche: H2O NaOH → Na+ + OH- Arrhenius distinse poi gli acidi e le basi a seconda della loro forza. Un acido forte è una sostanza che in acqua si ionizza completamente per dare ioni H+ (es. HCl): HCl(aq) → H2O H+(aq) + Cl-(aq) Una base forte è una sostanza che in acqua si ionizza completamente per dare ioni OH- (es. NaOH): NaOH(aq) → H2O Na+(aq) + OH-(aq) Acidi e basi deboli non sono completamente soluzione ma danno luogo ad un equilibrio CH3COOH(aq) Acido acetico H2O ionizzati H+(aq) + CH3COO-(aq) in Nonostante i suoi primi successi e la sua utilità, la teoria di Arrhenius ha dei limiti. Il primo problema è di natura formale ed emerse dopo i primi studi sull’atomo. Lo ione H+ è un protone semplice e non può esistere in acqua come tale, ma solo legato ad una molecola di acqua sottoforma di ione H3O+ in cui lo ione H+ è legato all’ossigeno (grazie al doppietto solitario: si forma un vero è proprio legame covalente) dell’acqua (talvolta questo legame è detto dativo, tuttavia questa dizione è in disuso, oltre ad essere formalmente errata, nella chimica moderna). Questo può essere facilmente preso in considerazione: HCl(aq) +H2O(l) → CH3COOH(aq)+H2O(l) H3O+(aq) + Cl-(aq) H3O+(aq) + CH3COO-(aq) Inoltre la teoria di Arrhenius è limitata a soluzioni acquose e non considera basiche sostanze che non contengono ioni OH- (ad esempio NH3). Lo Ione Idrossonio (detto anche idronio o ossonio ) • Il catione idrogeno, un protone nudo, è una particella caratterizzata da un’elevata densità di carica (è, infatti, molto piccolo) e non esiste come tale in soluzione. Esso tende ad interagire con l’atomo di ossigeno di una molecola di acqua per formare lo ione H3O+ o idrossonio. •Lo ione idrossonio, a sua volta, interagisce con le molecole d’acqua che lo circondano formando altre specie: H5O2+, H7O3+, H9O4+ H(H2O)+4 • Questo fenomeno, che può essere dedotto da misure di conducibilità, è DETTO IDRATAZIONE • Il protone idratato si indica come H+(aq) Insufficienza della definizione di Arrhenius: Molte basi, come per esempio NH3, non possono formare OH-(aq) per semplice dissociazione elettrolitica. La reazione acido-base deve essere di tipo diverso !!!! Secondo la teoria di Br Brø ønsted nsted-Lowry (1923 1923): ): Un ACIDO è una qualunque sostanza, che è capace di donare uno ione idrogeno (protone) ad un altra sostanza in una reazione chimica. Una BASE è una sostanza che accetta lo ione idrogeno (protone) dall'acido. N.B.: questa definizione non è vincolata alla presenza del solvente; una reazione acido-base può avvenire quindi in un solvente qualunque, in assenza di solvente ed in qualunque stato di aggregazione delle sostanze. Noi, tuttavia , tratteremo solo soluzioni acquose! EQUILIBRI ACIDO-BASE Secondo Brønsted e Lowry un acido è una sostanza capace di cedere uno o più protoni una base è una sostanza capace di acquistarli Si introduce così il concetto di sistema acido-base nel quale l’acido manifesta le sue proprietà qualora sia disponibile una sostanza (la base) che renda possibile il trasferimento di protoni sistema coniugato acido base (1) Acido1 + Base2 Base1 + sistema coniugato acido base (2) Acido2 EQUILIBRI ACIDO-BASE Forza di acidi e basi Secondo la teoria di Brønsted e Lowry una delle due sostanze che partecipano all’equilibrio acido-base può essere il solvente stesso, ad esempio l’acqua La forza degli acidi e delle basi è determinata sulla base del solvente nel quale si instaura l’equilibrio sistema coniugato acido base (1) Acido1 + H2O Base1 + H3O+ sistema coniugato acido base (2) Tanto maggiore la capacità dell’acido di trasferire il proprio protone all’acqua, tanto più forte sarà l’acido (riassumendo riassumendo)) Definizione di Br Brø ønsted e Lowry Acido = donatore di protoni (H+) Base = accettore di protoni (H+) Poiché il protone non può esistere isolato, in soluzione, un acido si manifesta tale solo se è in presenza di una base che possa accettare protoni, così come una base si manifesta tale solo in presenza di un acido che possa fornire protoni: è perciò più corretto parlare di sistemi acido- base che di acidi e basi singolarmente. “coppie acidoacido-base coniugati” coniugati” ione nitrito acido nitroso ione idrossonio HNO2(aq aq)) + H2O( O(ll) ⇄ NO2-(aq aq)) + H3O+(aq aq)) acido 1 base 2 base 1 NO2- base coniugata dell’acido HNO2 Posso H2O base coniugata dell’acido H3O+ anche dire acido 2 HNO2 acido coniugato della base NO2H3O+ acido coniugato della base H2O HNO2 acido coniugato della base NO2oppure NO2- base coniugata dell’ acido HNO2 In generale coppia coniugata acido acido-base: HNO2/NO2- H2O base coniugata dell’acido H3O+ oppure H3O+ acido coniugato della base H2O In generale coppia coniugata acido acido-base: H3O+/H2O Il caso dell’ammoniaca (NH3) : ione ammonio ammoniaca ione ossidrile NH3(aq aq)) + H2O( O(ll) ⇄ NH4+(aq aq)) + OH-(aq aq)) base 1 acido 2 acido 1 base 2 NH4+ acido coniugato della base NH3 oppure NH3 base coniugata dell’ acido NH4+ In generale coppia coniugata acido acido-base: NH4+/NH3 OH- base coniugata dell’acido H2O oppure H2O acido coniugato della base OHIn generale coppia coniugata acido acido-base: H2O/OH- Una specie può comportarsi da acido o da base a seconda della specie con cui viene fatta reagire. Ad esempio H2O si comporta da base con HCl (accetta un protone formando H3O+), ma si comporta da acido con NH3 (cede il protone formando OH-). Specie di questo tipo, che possono agire sia come acido che come base in dipendenza dell’altro reagente, vengono dette anfiprotiche (o anfotere). La teoria di Brønsted e Lowry è più generale della teoria di Arrhenius. In particolare: - Una base è una sostanza che accetta protoni (lo ione OH-, che secondo Arrhenius una sostanza deve necessariamente possedere per essere una base, è solo un esempio di base); - Acidi e basi possono essere sia sostanze molecolari sia ioni; - Possiamo avere solventi diversi dall’acqua; - Alcune sostanze possono agire da acido o da base a seconda della specie con cui reagiscono; Forza relativa di acidi e basi La forza relativa di un acido (o di una base) può essere considerata in funzione della loro tendenza a perdere (accettare) un protone. Gli acidi più forti sono quelli che perdono più facilmente i loro protoni. Analogamente le basi più forti sono quelle che accettano un protone più facilmente. Un acido forte è una sostanza che in acqua è completamente ionizzato: HCl(aq) + H2O(l) → H3O+(aq) + Cl-(aq) acido base acido base La reazione è spostata completamente verso destra, per cui l’acido cloridrico è un acido forte. Se si considera la reazione inversa, questa avviene in piccolissima parte (praticamente non avviene). In essa lo ione Cl- agisce come base accettando un protone dall’acido H3O+, ma è una base estremamente debole. HCl(aq) + H2O(l) → H3O+(aq) + Cl-(aq) acido base acido base più forte più debole Questa reazione si può considerare anche in funzione della forza relativa tra HCl e H3O+. HCl perde il protone più facilmente di H3O+ ed è quindi un acido più forte e la reazione è spostata verso destra. Se consideriamo la ionizzazione dell’acido acetico: CH3COOH(aq) +H2O(l) H3O+(aq) + CH3COO-(aq) acido acido più forte più debole Sperimentalmente si vede che solo l’1% delle molecole di CH3COOH sono ionizzate. Questo vuol dire che H3O+ è un acido più forte di CH3COOH. L’equilibrio è spostato verso sinistra. CH3COO- è la base coniugata di CH3COOH. Poiché l’equilibrio è spostato verso sinistra questo vuol dire CH3COO- è sì una base debole ma sicuramente più forte di H2O. In generale in una reazione acido-base la reazione è spostata nella direzione dal più forte al più debole membro della coppia coniugata acido-base. Se consideriamo la ionizzazione dell’acido fluoridrico: HF(aq) +H2O(l) acido più debole H3O+(aq) + F-(aq) acido più forte Qui solo il 3% delle molecole di HF sono ionizzate. Questo vuol dire che HF è un acido più debole di H3O+, ma è più forte di CH3COOH. F-, la base coniugata di HF, è una base più forte di H2O, ma più debole di CH3COO-. Gli acidi più forti hanno le basi coniugate più deboli, e le basi più forti hanno gli acidi coniugati più deboli. In base ai dati precedenti possiamo stabilire la seguente scala di acidità: HCl>H3O+>HF>CH3COOH ACIDI FORTI ed ACIDI DEBOLI Si può quindi scrivere la seguente tabella ACIDI FORTI ed ACIDI DEBOLI In realtà nella tabella HNO3, H2SO4, HCl, HBr, HI e HClO4 sono tutti acidi forti (=completamente dissociati in acqua) ed è quindi evidente che per stabilire la forza relativa di questi acidi occorre procedere diversamente da quanto fatto in precedenza. Gli acidi precedenti, sciolti in acqua, sono tutti ionizzati al 100%, ma se usiamo un solvente meno basico dell’acqua (come, ad esempio, l’etere etilico CH3CH2OCH2CH3) si può notare una differenza. Per esempio né HCl, né HI sono completamente ionizzati, ma si trova che HI è ionizzato in percentuale maggiore rispetto ad HCl ed è quindi un acido più forte. Poiché per gli acidi forti la forza in acqua sembra essere la stessa si parla di EFFETTO LIVELLANTE DELL’ACQUA sugli acidi forti. Riassumendo … • In una reazione acido-base la direzione favorita della reazione è dal più forte al più debole membro della coppia coniugata acido-base: HCl + OHCl+ H2O Acido (1) forte Base (2) forte H2O + Acido (1) debole I- Base (2) debole Base (1) debole OHBase (1) forte Acido (2) debole + HI Acido (2) forte • IL “FORTE” SPOSTA SEMPRE IL “DEBOLE” • PIÙ FORTE È UN ACIDO E PIÙ DEBOLE È LA SUA BASE CONIUGATA E VICEVERSA • In acqua HCl e HClO4 sono entrambi completamente dissociati: l’acqua ha un EFFETTO LIVELLANTE su questi acidi. Per determinare quale sia più forte bisogna usare un solvente che sia una base più debole dell’acqua: in etere dietilico HClO4 è dissociato completamente mentre HCl no (quindi, HClO4 è più forte) L’ACQUA:: ACIDO o BASE L’ACQUA L’acqua è una sostanza ANFIPROTICA = può comportarsi sia da acido che da base (di Brønsted-Lowry): ANFOTERA = sostanza che può comportarsi sia da acido che da base L’autoprotolisi L’ autoprotolisi dell’acqua Il prodotto della concentrazione di OH- per quella dello ione H3O+ in una qualunque soluzione acquosa è costante a temperatura costante. Esso corrisponde alla costante dell'equilibrio di autoprotolisi (o autodissociazione o autoionizzazione) dell'acqua che a 25°C è uguale a 1,0 x 10-14. H2O(l) + H2O(l) ⇄ H3O+(aq aq)) + OH-(aq) Per l'equilibrio vale la seguente costante: + − K W = [ H 3O ] ⋅ [OH ] = 1,0 ⋅ 10 −14 detto prodotto ionico dell’acqua Il pH e la sua scala: il grado di acidità di una soluzione acquosa non viene valutato direttamente con il valore della concentrazione molare (attività) dello ione H+ (cioè H3O+) ma , per agilità di calcolo, come il suo cologaritmo (in base 10), indicato con il simbolo pH: pH = -log10 a H3O+ = -log10 [H3O+] pOH = -log10 [OH-] pH + pOH = pKw = 14 Soluzioni basiche pH basicità crescente 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 14 13 12 11 10 9 8 7 6 5 acidità crescente Soluzioni acide 4 3 2 1 0 pOH Il concetto di pH nacque all’inizio (1909) del secolo scorso in una fabbrica di birra. Storia e mistero di un simbolo Ph ⇓ pH + ⇓ pH ⇓ pH Originariamente Sørensen utilizzò come simbolo "Ph" versione originale in un articolo in francese tipografi tedeschi tipografi americani (anni ‘20) attuale scrittura in linea S.V.L. Sørensen 1868-1939 (chimico danese) Direttore del prestigioso Laboratorio Carlsberg, fondato dall'industriale danese che creò l'omonima birreria Sørensen studiando l’azione degli acidi sulla catalisi enzimatica, dovendo esprimere con numeri semplici il grado di acidità propose l’uso del pH = - log [H+] N.B. il simbolo p non è un reale operatore matematico ma dobbiamo solo ricordare che corrispondete a –log10. …il pH e la sua scala… Una soluzione è definita: Neutra: quando il pH assume esattamente il valore 7 Acida: quando il pH assume valori minori di 7 Basica: quando il pH assume valori maggiori di 7 Nella seguente figura vengono riportati i valori di pH di alcune soluzioni comuni: …il pH e la sua scala… La scala qui mostrata va da pH 0 a 14. Sono possibili valori leggermente negativi di pH così come valori di pH più alti di 14: • pH<0: soluzioni estremamente acide. • pH>14 : soluzioni estremamente basiche. [OH-] [H3O+] pOH pH Autoprotonazione dell’acqua 2 H2O(l) H3O+(aq) + OH-(aq) Kw = [H3O+]· [OH-] A 25°C si ha Kw = 10-14 Soluzione neutra: [H3O+] = [OH-] Soluzione acida: [H3O+] > [OH-] Soluzione basica: [H3O+] < [OH-] Kw = + [H3O ]·[OH ] Soluzione neutra neutra:: [H 3O + ] = [OH − ] = K w Soluzione acida acida:: [H 3O + ] > K w ; [OH − ] < K w + − Soluzione basica basica:: [H 3O ] < K w ; [OH ] > K w a 25° 25°C [H3O+] > [OH-] [H3O+] 100 10-1 10-2 10-3 10-4 10-5 Kw 10-6 soluzione acida [H3O+] > 10-7 [OH-] < 10-7 [H3O+] < [OH-] 10-7 10-8 10-9 10-10 10-11 10-12 10-13 10-14 soluzione basica soluzione neutra [H3O+] = [OH-] = 10-7 [H3O+] < 10-7 [OH-] > 10-7 L’autoprotonazione dell’acqua è una reazione endotermica (assorbe calore) H3O+(aq) + OH-(aq) 2 H2O(l) + − Kw = [H3O ] [OH ] ∆H° > 0 Perché?:: Perché? Nella lezione #12 abbiamo dimostrato che la reazione neutralizzazione)) è esotermica (∆H< H<0 0) inversa (neutralizzazione T > 25°C 10-13 lnKw ∆H° > 0 25°C 10-14 10-15 10-16 T < 25°C 1/T Soluzione neutra: 1/T [H3O+ ] = [OH− ] = Kw ; pH = −log10Kw A 25° 25°C: pH = 7; a T < 25° 25°C: pH > 7; a T > 25° 25°C: pH < 7 T( °C) 0 10 20 25 30 40 50 60 Kw 1,14 · 10-15 2,92 · 10-15 6,81 · 10-15 1,01 · 10-14 1,47 · 10-14 2,92 · 10-14 5,47 · 10-14 9,61 · 10-14 pH 7,47 7,27 7,08 7,00 6,92 6,77 6,63 6,51 Calcolo del pH di soluzioni di acidi forti e basi forti Comuni acidi forti e basi forti Acidi Basi HCl LiOH HBr NaOH HI KOH HClO4 RbOH HNO3 CsOH H2SO4 * Mg(OH)2 Ca(OH)2 Sr(OH)2 Ba(OH)2 *H2SO4 ionizza in due stadi distinti ed è un acido forte solo nella prima ionizzazione Calcolo del pH di soluzioni di acidi forti Si disciolga un acido forte, per esempio HCl, in acqua, questo si dissocia completamente secondo l’equazione: HCl(aq) + H2O(l) → H3O+(aq) + Cl-(aq) nella soluzione è presente autodissociazione dell’acqua: anche l’equilibrio di 2 H2O(l) ⇄ H3O+(aq) +OH- (aq) che pure fornisce ioni H3O+. Nella maggior parete dei casi, tuttavia, questi ultimi sono trascurabili nei confronti di quelli che derivano dalla dissociazione (completa) dell’acido. Calcolo del pH di soluzioni di acidi forti Si calcoli il pH di una soluzione 0,10 M di HNO3 HNO3 è un acido forte, quindi in H2O si dissocia completamente: HNO3(aq) + H2O(l) → NO3-(aq) + H3O+(aq) [H3O+] derivante dall’acido = [HNO3]= 0,100 M pH = -log10[H3O+] = -log10 [0,10] = 1 Il pH risultante è acido Si calcolino il pH ed il pOH di una soluzione acquosa 1,0·10-4 M di HClO4 HClO4 è un acido forte, quindi in H2O si dissocia completamente: HClO4(aq) + H2O(l) → ClO4-(aq) + H3O+(aq) [H3O+] derivante dall’acido = [HClO4]= 1,0 · 10-4 M pH = -log [1,0 · 10-4] = 4 poiché [H3O+]·[OH-] =KW= 1,0 ·10-14 M risulta che: [OH-] = 1,0 · 10-14/1,0 · 10-4 = 1,0 ·10-10 M pOH = 10,0 Si noti che pH + pOH = pKw= 14 Nel problema non si è tenuto conto del contributo degli ioni H3O+ derivanti dalla dissociazione dell’H2O Si verifica a posteriori che l’approssimazione fatta sia lecita: dato che la [OH-] = 10-10 M deriva dalla dissociazione delle molecole di H2O, la [H3O+] derivante dalla medesima dissociazione sarà uguale, cioè pari a 10-10 M. Questa concentrazione è trascurabile rispetto alla concentrazione di [H3O+] derivante dall’acido (10-4 M) Si calcoli il pH di una soluzione 5,0 · 10-8 M di HClO4 (acido forte: totalmente ionizzato) HClO4 è un acido forte con Ka molto grande (possiamo considerarla infinita) quindi in H2O si dissocia completamente: HClO4 (aq) + H2O (l) → ClO4-(aq) + H3O+(aq) [H3O+] derivante dall’acido = [HClO4]= 5,0·10-8 M + −8 pH = −log10 [H 3O ] = −log10 [5,0 ⋅10 ] = +7,30 Impossibile: la soluzione è basica?! Tale concentrazione è paragonabile all’autodissociazione delle molecole di acqua che, quindi, contribuirà al pH della soluzione. 2 H2O(l) ⇄ H3O+(aq) + OH-(aq) 5,0·10-8 Ciniziali) Variazione di C(Δ) +x 5,0·10-8+x Cequilibrio) ---- +x +x Kw = [H3O+]·[OH-] allora: Kw = (5,0·10-8 + x)· x = 1,0 ·10-14 risolvendo l’equazione di secondo grado: x2+5,0·10-8x - 1,0 ·10-14=0 si ottiene: x = 7,81· 10−8 M la concentrazione totale di [H3O+] = 5,0·10-8+x = 1,281 · 10-7 M pH = −log10[H3O+] = -log10[1,281 ·10−7] = 6,89 Si noti che il pH è acido come atteso. In generale, solo quando gli ioni H3O+ derivanti da un acido forte sono in concentrazione << 10-6 M occorre tenere conto del contributo della dissociazione dell’acqua per il calcolo del pH. Calcolo del pH di soluzioni molto diluite di acidi forti Trattazione generale (trattamento sistematico) sistematico):: Indichiamo con HX il generico acido forte di concentrazione 10-8<Ca<10-6 mol/L Essendo la concentrazione dell’acido estremamente bassa, non è più possibile trascurare il contributo degli ioni H3O+ provenienti dalla dissociazione dell’acqua. Il problema deve essere risolto impostando i bilanci di carica e di massa relativi agli equilibri di dissociazione delle varie specie presenti in soluzione. Teniamo presente che l’acido HX è un acido forte, quindi completamente dissociato: HX (aq) + H2O (l) → H3O+(aq) + X-(aq) ed è presente l’equilibrio di dissociazione dell’acqua: 2 H2O(l) ⇄ H3O+(aq) + OH-(aq) Le relazioni che legano le varie specie presenti in soluzione sono: [X − ] = C a a) Bilancio di massa sul X− b) Bilancio di carica (la soluzione deve essere elettroneutra): [H 3O + ] = [OH − ] + [ X − ] c) Equilibrio dell’acqua + − K w = [H 3O ] ⋅ [OH ] Sostituendo [X-] = Ca nella (b) e ricavando [OH-] dalla (c), l’equazione (b) diventa: Kw [H 3O ] = Ca + + [H 3O ] + da cui risolvendo rispetto ad [H3O+] si ottiene: + 2 + [H 3O ] − C a ⋅ [H 3O ] − K w = 0 Calcolo del pH: basi forti Esercizio 1: Si calcoli il pH di una soluzione acquosa 5,0 · 10-4 M di NaOH. Risultato: 10,7 Esercizio 2: Si calcoli il pH di una soluzione acquosa 5,0 · 10-4 M di Ba(OH)2. Risultato: 11,0 Perchè se la concentrazione è la stessa ? Entrambe sono totalmente dissociate in acqua: 1) NaOH (aq) → Na 2) Ba(OH)2 (aq) + (aq) → Ba + OH ++ (aq) − (aq) + 2 OH − (aq) Tuttavia nel secondo caso la concentrazione dell’OH− è doppia: 1) [OH − ] = [NaOH] = Na + = 5 ⋅10 −4 M [ ] [ ] 2) [OH − ] = 2 ⋅ [Ba(OH)2 ] = 2 ⋅ Ba + + = 2 ⋅ 5 ⋅10 − 4 M = 10 −3 M 1) pOH = −log[OH − ] = − log [5 ⋅10 −4 ] = 3,3 2) pOH = −log[OH − ] = − log [10 −3 ] = 3,0 1) pH = 14 − pOH = 14 − 3,3 = 10,7 2) pOH = 14 − pOH = 14 − 3,0 = 11,0 Calcolo del pH di soluzioni molto diluite di basi forti trattamento sistematico Indichiamo con MOH la generica base forte di concentrazione 10-8<Cb<10-6 mol/L Essendo la concentrazione della base estremamente bassa, non è più possibile trascurare il contributo degli ioni H3O+ provenienti dalla dissociazione dell’acqua. Il problema deve essere risolto impostando i bilanci di carica e di massa relativi agli equilibri di dissociazione delle varie specie presenti in soluzione. Teniamo presente che la base MOH è un base forte, quindi completamente dissociata ne catione M+ e anione OH−: MOH H2O (aq) → M+(aq) + OH-(aq) ed è presente l’equilibrio di dissociazione dell’acqua: 2 H2O(l) ⇄ H3O+(aq) + OH-(aq) Le relazioni che legano le varie specie presenti in soluzione sono: a) Bilancio di massa sul M+ [M + ] = C b + + − [H 3O ] + [M ] = [OH ] b) Bilancio di carica c) Equilibrio dell’acqua + − K w = [H 3O ] ⋅ [OH ] Sostituendo [M+] = Cb nella (b) e ricavando [H3O+] dalla (c), l’equazione (b) diventa: Kw − Cb + = [OH ] − [OH ] da cui risolvendo rispetto ad [H3O+] si ottiene: − 2 − [OH ] − C b ⋅ [OH ] − K w = 0 Calcolo del pH di soluzioni di acidi forti e basi forti trattamento sistematico Concentrazioni (10-6-10-8M), qui è necessario il trattamento sistematico Concentrazioni alte (≥ 10-6M), il pH è calcolato da H3O+ e OHderivante dall’acido o dalla base Bassissime concentrazioni (≤10-8M), il pH=7 H3O+,OHnon sono abbastanza per cambiare il pH Applicazione della legge di azione delle masse agli equilibri acido-base Per calcolare il pH di Acidi e basi deboli abbiamo bisogno di introdurre rispettivamente le costanti di ionizzazione acida (Ka) e la costante di ionizzazione basica (Kb) La forza di un acido (o di una base) si “misura” con il valore della relativa costante (termodinamica) del seguente equilibrio omogeneo in fase liquida (acquosa): HA + H2O ⇄ H3O+ + APer gli acidi forti la costante è >>1 : in altre parole sono completamente ionizzati e la concentrazione di H3O+ e di Asono uguali alla concentrazione iniziale dell’acido In questo caso non si parla più di equilibrio!! Ma di reazione completamente spostata a destra. Acidi e basi deboli Per acidi deboli la costante è <1 : in altre parole sono poco ionizzati e in soluzione sono presenti contemporaneamente i prodotti (H3O+ e A-) e il reagente (cioè l’acido indissociato sottoforma di molecola discreta) tanto minore è il valore di Ka, tanto minore è la frazione di molecole dissociate nei loro ioni La costante Ka (è detta costante di dissociazione (o ionizzazione) acida) (è una costante di un equilibrio omogeneo in fase liquida, una KC): Consideriamo l’acqua un liquido puro # come HClO2(aq) aq) + H2O(l) ⇄ ClO2-(aq) aq) + H3O+(aq) aq) Ka = a ClO − ⋅ a H O + 2 3 a H 2O ⋅ a HClO2 − 2 + [ClO ] ⋅ [H 3O ] = [HClO 2 ] # la sua attività è unitaria e stiamo, inoltre, assumendo che la costante di acidità (secondo Brönsted-Lowry) numericamente coincida con quella di dissociazione… #oppure si può dire che essendo la concentrazione dell’acqua pressoché costante anche il rapporto KC/[H2O] sarà costante: [ClO 2− ] ⋅ [H 3O + ] KC [ClO −2 ] ⋅ [H 3O + ] KC = ⇒ Ka = = [H 2 O] ⋅ [HClO 2 ] [H 2 O] [HClO 2 ] Spesso sono deboli gli acidi organici con il gruppo funzionale –COOH pKa = −log10(Ka) Conviene fare come il pH più piccolo è il valore del pK più forte è l’acido (ovvero è meno debole) La costante (di basicità) Kb (è una costante di un equilibrio omogeneo in fase liquida, una KC): Consideriamo l’acqua come un liquido puro # NH3(aq aq)) + H2O( O(ll) ⇄ NH4+(aq aq)) + OH-(aq aq)) Kb = a NH + ⋅ a OH − 4 a H 2O ⋅ a NH3 + 4 − [NH ] ⋅ [OH ] = [NH 3 ] oppure + 4 − + 4 − KC [NH ] ⋅ [OH ] [NH ] ⋅ [OH ] KC = ⇒ Kb = = [H 2 O] ⋅ [NH 3 ] [H 2 O] [NH 3 ] pKb = −log10(Kb) Spesso sono deboli le basi che derivano dall’ammoniaca per sostituzione di –H con gruppi organici Calcolo del pH per un acido debole (Ka < 1) monovalente (monoprotico) di concentrazione (iniziale) Ca (ignoriamo la dissociazione dell’acqua ed assumiamo che gli ioni H3O+ derivino solamente dalla dissociazione dell’acido debole; tale assunzione è quasi sempre verificata, ovvero è verificata quando il valore del pH viene inferiore o uguale a 6) Indichiamo con HA il generico acido debole: (i) (eq) HA(aq)) + H2O( HA(aq O(ll) ⇄ A-(aq aq)) + H3O+(aq aq)) ~0 Ca --(Ca – x) x x [A − ] [H 3O + ] Ka = [HA] Ka = x = [A − ] = [H 3O + ] x2 (c a − x) Generalmente dovremmo risolvere un’equazione di secondo grado! Se dovessimo tener conto dell’autodissociazione dell’acqua si può dimostrare che dovremmo addirittura risolvere un’equazione di terzo grado rispetto ad H3O+ Vediamo se si possono fare delle approssimazioni…….. ….riscrivo la costante Ka così: + 2 [H 3O ] Ka = [HA] da cui + [H 3O ] = K a ⋅ [HA] Se α < 0,05, cioè se si dissociano (ionizzano) meno di 5 mol di HA su 100 mol, posso effettuare questa approssimazione: [HA] ≅ C a + [ H 3O ] = K a ⋅ C a Concentrazione iniziale Perchè posso effettuare questa approssimazione? Basta ricordare che per un elettrolita debole uno-unovalente vale la legge di diluizione di Ostwald (vedi lezione 17): α2 K= ⋅ C0 1− α Nel nostro caso se ⇒ C0 1 − α = 2 K α Ca > 380 ⇒ α < 0,05 Ka Invece quando Ca ≤ 380 Ka ⇒ α ≥ 0,05 + [HA] = C a − [H 3O ] + + [H 3O ] = K a ⋅ (C a − [H 3O ]) Dobbiamo risolvere l’equazione di secondo grado: + 2 + [ H 3O ] + K a ⋅ [ H 3O ] − K a ⋅ C a = 0 …. ricordando che … ax + bx + c = 0 2 − b ± b − 4ac x= 2a 2 − K a + K a + 4 Ca K a [ H 3O ] = 2 2 + Calcolo del pH per un Base debole monovalente (Kb < 1) di concentrazione (iniziale) Cb (ignoriamo la dissociazione dell’acqua ed assumiamo che gli ioni OH− derivino solamente dalla dissociazione della base debole );vale un ragionamento analogo a quello degli acidi deboli. Indichiamo con B la generica base debole: B(aq B( aq)) + H2O( O(ll) ⇄ BH+(aq aq)) + OH-(aq aq)) (i) Cb --~0 (eq) (Cb – x) x x 2 x [BH ] [OH ] x = [BH + ] = [OH − ] K = Kb = b (C b − x) [B] Cb Se > 380 ⇒ α < 0,05 Cb − x ≅ Cb Kb x2 [OH − ]2 Kb = Kb ≅ Cb Cb + − − [OH ] = K b ⋅ C b Se Cb ≤ 380 Kb ⇒ α ≥ 0,05 − [B] = C b − [OH ] [BH + ] [OH − ] [OH − ]2 Kb = = − C b − [OH ] C b − [OH − ] Dobbiamo risolvere l’equazione di secondo grado: − 2 − [OH ] + K b ⋅ [OH ] − K b ⋅ C b = 0 Chimica (a.a. 2011/2012, 6CFU) Equilibri ionici in soluzione acquosa (II parte): Idrolisi delle soluzioni saline Calcolo del pH nelle idrolisi Acidi e basi polifunzionali Le soluzioni tampone IDROLISI DEI SALI Molti sali contengono un anione o un catione che possono reagire con acqua rendendo le loro soluzioni ACIDE o BASICHE Molti sali reagiscono con l’acqua alterando il suo rapporto molare tra [H3O+] e [OH-] La soluzione assume un pH maggiore oppure minore di 7 (generalmente è un effetto causato da uno degli ioni del sale) Come prevedere? Basicità e acidità di cationi ed anioni − Perché? 2 Vediamo di razionalizzare questa tabella? * * Cationi di metalli piccoli e con alta carica legano fortemente molecole d’acqua, indeboliscono il legame O-H con effetti acidi Soluzioni di Sali in acqua Un sale deriva dalla reazione di neutralizzazione di un acido e di una base in soluzione acquosa. In soluzione acquosa i sali sono dissociati negli ioni costituenti: H2O → Na+ (aq) + Cl- (aq) H2O NH4Cl(s) → NH4+ (aq) + Cl- (aq) H2O CH3COONa(s) → CH3COO- (aq) + Na+ (aq) 2O CH COO- (aq) + NH + (aq) CH3COONH4(s) H→ 3 4 NaCl(s) Abbiamo visto che nella teoria di Brønsted e Lowry anche gli ioni possono comportarsi da acidi o da basi, in particolare se sono gli acidi o le basi coniugati di basi o acidi deboli, rispettivamente. Ioni che invece sono i coniugati di acidi o basi forti invece non hanno proprietà basiche o acide. Se abbiamo una soluzione di NaCl, poiché gli ioni costituenti sono i coniugati di un acido forte (HCl) e di una base forte (NaOH), Na+ e Cl- non hanno alcuna tendenza ad accettare o donare protoni all’acqua Na+ (aq) + H2O Cl- (aq) + H2O / / → → nessuna reazione nessuna reazione La soluzione risulterà quindi neutra. Consideriamo ora una soluzione ottenuta sciogliendo NH4Cl in acqua H O 2 NH4Cl (s) → NH4+ (aq) + Cl-(aq) Mentre lo ione Cl- è stabile in acqua, lo ione NH4+ è l’acido coniugato della base debole NH3 è quindi in grado di cedere un protone all’acqua secondo la reazione: NH4+ (aq) + H2O(l) NH3 (aq) + H3O+ (aq) NH4+ (aq) + H2O(l) NH3 (aq) + H3O+ (aq) La soluzione risulterà dunque acida. La reazione tra NH4+ e H2O fondamentalmente non è diversa dalle altre reazioni acido-base. Tuttavia una reazione tra uno ione e l’acqua viene detta REAZIONE DI IDROLISI. Si dice allora che lo ione ammonio si idrolizza (a differenza dello ione cloruro). Quando sciogliamo in acqua del cianuro di sodio, NaCN: NaCN (s) H2O Na+ (aq) + CN- (aq) in questo caso lo ione Na+ è stabile, mentre lo ione CN- è la base coniugata dell’acido cianidrico HCN, è quindi in grado di accettare un protone dall’acqua secondo la reazione: CN- (aq) + H2O (l) HCN (aq) + OH- (aq) la soluzione risulta dunque basica. Lo ione cianuro si idrolizza. Costante di IDROLISI (BASICA BASICA)) Prendiamo ad esempio il nitrito di sodio NaNO2 che si dissocia completamente il acqua a dare il catione Na+ (acido debolissimo coniugato della base fortissima NaOH) e l’anione NO2- (base debole coniugata dell’acido debole HNO2): NaNO2(s) → Na+(aq aq)) + NO2-(aq aq)) NO2- è la base coniugata di HNO2, e quest’ultimo è un acido debole governato dall’equilibrio: − + + HNO2(aq) aq) + H2O( O(ll) ⇄ NO2 (aq aq)) + H3O (aq aq)) 2 3 a IDROLISI BASICA: 2 [NO ] [H O ] K = [HNO ] NO2-(aq aq)) + H2O( O(ll) ⇄ HNO2(aq aq)) + OH-(aq aq)) [HNO 2 ] [OH − ] costante d' idrolisi = K i = K b = − [NO 2 ] Si dimostra che la costante di idrolisi è una costante basica Kb che è uguale: KW Kb = Ka Costante di IDROLISI (ACICA ACICA)) Prendiamo ad esempio il cloruro di ammonio NH4Cl che si dissocia completamente in acqua a dare l’anione Cl- (base debolissima coniugato della acido fortissimo HCl) e il catione NH4+ (acido debole coniugato della base debole NH3): NH4Cl( Cl(ss) → NH4+(aq aq)) + Cl-(aq aq)) NH4+ è l’acido coniugato di NH3, e quest’ultimo è una base debole governata dall’equilibrio: + − 4 NH3(aq aq)) + H2O( O(ll) ⇄ NH4+(aq aq)) + OH-(aq aq)) b 3 IDROLISI ACIDA ACIDA:: [NH ] [OH ] K = [NH ] NH4+(aq aq)) + H2O( O(ll) ⇄ NH3(aq aq)) + H3O+(aq aq)) + [NH 3 ] [H 3O ] costante d' idrolisi = K i = K a = + [NH 4 ] Si dimostra che la costante di idrolisi è una costante acida Ka che è uguale: KW Ka = Kb KW Dimostrazione di K b = Ka ovvero K a ⋅ K b = K W In tutti i casi, casi, per una coppia acidoacido-base coniugati si ha: Ka ⋅ Kb = KW Per esempio, per la coppia NH4+/NH3: Kb = + 4 − [NH ] [OH ] [NH 3 ] [NH 3 ] [H 3O + ] costante d' idrolisi = K i = K a = [NH +4 ] Se moltiplico e divido Kb per la concentrazione di [H3O+] [NH 4+ ] [OH − ] [H 3O + ] K w Kb = ⋅ = + [NH 3 ] [H 3O ] K a KaKb = Kw Oppure se moltiplico e divido Ka per la concentrazione di [OH-] [NH 3 ] [H 3O + ] [OH − ] K w Ka = ⋅ = + − [NH 4 ] [OH ] K b KaKb = Kw Inoltre KW Kb = Ka ovvero Ka ⋅ Kb = KW tale relazione mette in evidenza che la base coniugata di un acido debole (o, viceversa, l’acido coniugato di una base debole) è debole e che il coniugato di un forte è debolissimo. Calcolo del pH nelle idrolisi di sali basici (IDROLISI BASICA): SALE BASICO derivante da ACIDO DEBOLE e BASE FORTE Esempio l’acetato di sodio (CH3COONa) di concentrazione iniziale Cs CH3COONa (acetato di sodio) → CH3COO- + Na+ (l’acetato di sodio è come se provenisse dalla reazione di NaOH e CH3COOH) (i) (eq) CH3COO- (aq) + H2O(l) CH3COOH(aq) + OH- (aq) Cs Cs-x --x ~0 x x = [OH-] = [CH3COOH] − K [CH 3COOH] [OH ] Ki = Kb = W = Ka [CH 3COO − ] quando Cs > 380 ⇒ α < 0,05 Kb CS − x ≅ CS [OH − ]2 Kb = CS 2 x Kb = (CS − x) Cioè meno del 5% di CH3COO- si idrolizza − [OH ] = K b ⋅ Cs CS ≤ 380 se Kb ⇒ α ≥ 0,05 Cioè più del 5% di CH3COO- si idrolizza − − 2 [CH COOH] [OH ] [OH ] 3 − − = [CH 3COO ] = C s − [OH ] K b = − CS − [OH ] CS − [OH − ] Dobbiamo risolvere l’equazione di secondo grado: − 2 − [OH ] + K b ⋅ [OH ] − K b ⋅ CS = 0 −Kb + K + 4CS Kb [OH ] = 2 − Ricorda SEMPRE che 2 b KW Kb = Ka Calcolo del pH nelle idrolisi di sali acidi (IDROLISI ACIDA): SALE ACIDO derivante da ACIDO FORTE e BASE DEBOLE Esempio il cloruro di ammonio (NH4Cl) di concentrazione iniziale Cs NH4Cl (cloruro di ammonio) → NH4+ + Cl(il cloruro di ammonio è come se provenisse dalla reazione di HCl e NH3) (i) (eq) + NH4+(aq) aq) + H2O(l) ⇄ NH3 (aq) + H3O (aq) aq) CS --~0 x x CS-x x = [NH3] = [H3O+] + K W [NH 3 ] [H 3O ] Ki = Ka = = + Kb [NH 4 ] quando Cs > 380 ⇒ α < 0,05 Ka CS − x ≅ CS [H 3O + ]2 Ka = CS 2 x Ka = (CS − x) Cioè meno del 5% di NH4+ si idrolizza + [H 3O ] = K a ⋅ CS se CS ≤ 380 Ka ⇒ α ≥ 0,05 + + [NH 4 ] = Cs − [H 3O ] Cioè più del 5% di NH4+ si idrolizza [NH 3 ] [H 3O + ] [H 3O + ]2 [H 3O + ]2 Ka = = = + + + [NH 4 ] [NH 4 ] Cs − [H 3O ] Dobbiamo risolvere l’equazione di secondo grado: + + [H 3O ] + K a ⋅ [H 3O ] − K a ⋅ CS = 0 2 − Ka + Ka + 4 CS Ka [H3O ] = 2 KW Ka = SEMPRE che Kb 2 + ricorda Sali di acidi deboli e basi deboli Ad esempio NH4CN → NH4+ + CN(è come se derivasse da NH3 e HCN) CH3COONH4 → CH3COO- + NH4+ (è come se derivasse da NH3 e CH3COOH) NH4F → NH4+ + F(è come se derivasse da NH3 e HF) Sia il catione che l’anione hanno la tendenza ad idrolizzarsi, ma l’effetto che il sale ha sul pH dipende dalla capacità del catione di generare H+ (ovvero H3O+) rispetto a quella dell’anione di generare OH- (e viceversa ) Considerare caso per caso 1° caso NH4CN → NH4+ + CN- Cianuro di ammonio (è come se derivasse da NH3 e HCN) [1] NH4+ + H2O → K b,NH3 = 1,8 ⋅10−5 [2] KW 10 −14 −10 K idrolisi,1 = K a = = = 5,55 ⋅ 10 K b,NH3 1,8 ⋅10 −5 CN- + H2O → K a,HCN = 6,16 ⋅10 −10 NH3 + H3O+ HCN + OH- KW 1,0 ⋅10 −14 −5 K idrolisi,2 = K b = = = 1,62 ⋅ 10 K a, HCN 6,16 ⋅10 −10 Poiché Kidrolisi idrolisi,2 ,2 >>> Kidrolisi idrolisi,1 ,1 SOLUZIONE BASICA pH>7 2° caso Acetato di ammonio CH3COONH4 → NH4+ + CH3COO(è come se derivasse da NH3 e CH3COOH) [1] NH4+ + H2O → K b,NH3 = 1,8 ⋅10−5 [2] NH3 + H3O+ KW 10 −14 −10 K idrolisi,1 = K a = = = 5,55 ⋅ 10 K b,NH3 1,8 ⋅10 −5 CH3COO- + H2O → K a,CH 3COOH = 1,8 ⋅10−5 CH3COOH + OH- K idrolisi,2 = K b = Poiché KW K a,CH 3COOH 10 −14 −10 = = 5,55 ⋅ 10 1,8 ⋅10 −5 Kidrolisi idrolisi,2 ,2 = Kidrolisi idrolisi,1 ,1 SOLUZIONE NEUTRA pH=7 3° caso Fluoruro di ammonio NH4F → NH4+ + F(è come se derivasse da NH3 e HF) [1] NH4+ + H2O → K b,NH3 = 1,8 ⋅10−5 [2] KW 10 −14 −10 K idrolisi,1 = K a = = = 5,55 ⋅ 10 K b,NH3 1,8 ⋅10 −5 F- + H2O → K a,HF = 6,16 ⋅10 −10 NH3 + H3O+ HF + OH- KW 1,0 ⋅10 −14 −11 K idrolisi,2 = K b = = = 1,58 ⋅ 10 K a, HF 6,31 ⋅10 − 4 Poiché Kidrolisi idrolisi,2 ,2 <<< Kidrolisi idrolisi,1 ,1 SOLUZIONE ACIDA pH<7 pH di Soluzioni di Sali In termini generali: Sali derivanti di ACIDI FORTI E BASI FORTI (per esempio: NaCl, KCl, KNO3, BaI2, NaClO4, etc.) NON SI IDROLIZZANO IDROLIZZANO. La soluzione ha pH= pH=7 7 (soluzioni soluzioni neutre neutre) Sali derivanti di ACIDI DEBOLI E BASI FORTI (per esempio: CH3COONa, KNO2, NaClO2, etc.) SI IDROLIZZANO IDROLIZZANO. Il pH> pH>7 7 (L’anione si comporta da base, soluzioni basiche basiche) Sali di derivanti ACIDI FORTI E BASI DEBOLI (NH4Cl, NH4NO3, NH4Br, NH4ClO4, etc.) SI IDROLIZZANO IDROLIZZANO. Il pH< pH<7 7 (Il catione si comporta da acido, soluzioni acide acide) Sali di derivanti ACIDI DEBOLI E BASI DEBOLI (NH4CN, NH4NO2, CH3COONH4, etc.) SI IDROLIZZANO IDROLIZZANO. Il pH dipende dalla forza relativa dell’acido e della base (I cationi si comportano da acidi e gli anioni da basi). pH di Soluzioni di Sali Riassumendo * * * * Per il livello del nostro corso solo per questi casi bisogna essere in grado di calcolare il pH. Acidi e basi poliprotici Diciamo acido poliprotico un composto atto a cedere più di un protone. Molti acidi comuni hanno struttura poliprotica: tra di essi l'acido solforico, H2S04, l' acido carbonico, H2C03, capaci entrambi di cedere due protoni, e l'acido fosforico, H3P04, che può cederne tre. Nei sistemi biologici gli acidi poliprotici assolvono un ruolo decisivo; molti enzimi si possono considerare acidi poliprotici che espletano le funzioni vitali cedendo un protone dopo l'altro. Una base poliprotica è una specie che è in grado di accettare più di un protone; tra gli esempi ricordiamo gli anioni CO32- e SO32-, capaci di accettare due protoni, e lo ione PO43-, capace di accettarne tre. La differenza principale tra un acido poliprotico e uno monoprotico è che il primo cede i suoi protoni in successione. Ad esempio, l'acido carbonico può cedere un protone formando HCO3-, e successivamente questo ione può cedere il protone rimanente dando CO32-. Nel valutare il pH della soluzione di un acido poliprotico o di un suo sale occorre sapere quando e come tener conto di queste serie di processi di deprotonazione. Acidi e basi poliprotici Specie che cedono o accettano più di un protone sono dette poliprotiche Un acido è detto monoprotico se può liberare un solo protone e poliprotico se può liberarne più di uno (diprotico, triprotico, ecc.) Una base é detta monoprotica se può fissare (accettare) un solo protone e poliprotica se può fissarne (accettarne) più di uno (diprotica, triprotica, ecc.) Esempi di Acidi Poliprotici Gli acidi poliprotici danno luogo a tanti equilibri diversi con l'acqua quanti sono i protoni che possono cedere. Ad esempio, H3PO4 dà luogo agli equilibri seguenti: H3PO4(aq) + H2O(l) ⇄ H2PO4-(aq) + H3O+(aq) Ka1 = 7,1×10-3 H2PO4-(aq) + H2O(l) ⇄ HPO42-(aq) + H3O+(aq) Ka2 = 6,2×10-8 HPO4-(aq) + H2O(l) ⇄ PO43-(aq) + H3O+(aq) Ka3 = 4,4×10-13 H2CO3(aq) + H2O(l) ⇄ HCO3-(aq) + H3O+(aq) Ka1 = 4,4×10-7 HCO3-(aq) + H2O(l) ⇄ CO32-(aq) + H3O+(aq) Ka2 = 4,7×10-11 H2SO4(aq) + H2O(l) ⇄ HSO4-(aq) + H3O+(aq) Ka1 = ~102 (forte) HSO4-(aq) + H2O(l) ⇄ SO42-(aq) + H3O+(aq) Ka2 = 1,05×10-2 Regola generale: i successivi stadi sono caratterizzati da costanti di equilibrio via via decrescenti. La forza di un acido poliprotico diminuisce di un fattore 104÷105, o superiore, in ogni dissociazione successiva. Calcolo del pH in Soluzioni di Acidi Poliprotici • Per esempio, nel caso di un acido diprotico, in soluzione acquosa si instaurano i seguenti equilibri: [ H O ]⋅ [HA ] = + H2A + H2O HA− + H2O H3O+ + HA- H3O+ + A2− • Si verifica sempre che: Ka1 > Ka2 • Spesso: Ka1 >> Ka2 K a1 3 - [H 2 A] [H 3O + ] ⋅ [A 22−− ] K a2 = [HA - ] Calcolo del pH in Soluzioni di Acidi Poliprotici • Se Ka1/Ka2 > 103 – Si può assumere con buona approssimazione che H3O+ sia dovuto esclusivamente al primo equilibrio di dissociazione • Se Ca/Ka1 > 380 – È possibile utilizzare l’espressione approssimata per il calcolo di [H3O+] di un acido debole [H 3O + ] = K a1 ⋅ C H 2 A Altrimenti si utilizza la soluzione dell’equazione quadratica: [H O ] = + 3 − K a1 + K a1 + 4 ⋅ K a1 ⋅ C H 2 A 2 2 Che è comunque un’approssimazione, perché si trascura il contributo dell’autodissociazione dell’acqua In generale: il pH di un acido poliprotico, le cui deprotonazioni sono tutte deboli, si calcola considerando esclusivamente il primo equilibrio di deprotonazione assumendo che le deprotonazioni successive siano prive di importanza. Determinazione delle Concentrazioni Ioniche di un Acido diprotico. Un caso particolare : H2SO4 (l’acido solforico) L’acido solforico differisce dalla maggior parte degli acidi poliprotici perché nella sua prima ionizzazione è un acido forte e nella seconda è debole. Nel primo stadio la ionizzazione è completa!!! Infatti, l’acido solforico è l'unico acido poliprotico comune che subisca in modo completo la prima deprotonazione; la seconda aggiunge poco alla molarità di H30+, il pH complessivo sarà appena un po’ minore di quello dovuto alla sola prima deprotonazione. Ad esempio, in H2S04(aq) 0,010 M la prima deprotonazione H2SO4(aq) + H2O(l) →HSO4-(aq) + H3O+(aq) Ne discende una molarità di H30+ uguale a quella iniziale dell'acido, 0,010 mol/L, valore che corrisponde a pH = 2,0. Tuttavia anche la base coniugata, HSO4-, contribuisce con una parte dei propri protoni, per cui è necessario tener conto del secondo equilibrio di trasferimenuo protonico: HSO4-(aq) + H2O(l) ⇄ SO42-(aq) + H3O+(aq) Ka2 = 1,05×10-2 In definitiva, per calcolare il pH della soluzione di acido solforico costruiremo una tabella di equilibrio nella quale le concentrazioni iniziali siano quelle dovute alla prima deprotonazione: [HSO4-] = 0,010 mol·L-1, [H30+] = 0,010 mol·L-1 e [SO42-]=0 mol·L-1. H3O+ + SO42- HSO4- + H2O Concentrazione iniziale Variazioni Concentrazione all’equilibrio 0,010 -x 0,010-x + 0,010 +x 0,010+x ~0 +x +x ( [H 3O + ] ⋅ [SO 24− ] 0,010 + x ) ⋅ x −2 K a2 = = 1,05 ⋅10 = − [HSO 4 ] 0,010 − x si ottiene l’equazione di secondo grado: x 2 + 2,05 ⋅10 −2 x − 1,05 ⋅10 −4 = 0 x = 0,00424 [H 3O + ] = 0,010 + x = 0,010 + 0,00424 = 0,01424 la cui radice positiva: pH = −log 10 [H 3O + ] = −log 10[0,01424] ≅ +1,85 In generale per l’acido solforico: Quando la sua concentrazione è compresa fra 6·10–4 M e 0,15 M, per il calcolo del pH si procede come visto poco anzi. Invece l’acido solforico può essere considerato completamente dissociato in entrambi gli stadi se la sua concentrazione è minore di 6·10–4 M: H2SO4 (aq) + 2 H2O(l) → SO42-(aq) + 2 H3O+(aq) [H 3O + ] ≡ 2 ⋅ C a = 2 ⋅ [H 2SO 4 ] pH = −log 10[2 ⋅ C a ] Se l’acido solforico è in concentrazione maggiore di 0,15 M, il secondo stadio di dissociazione è completamente spostato a sinistra e l’acido si comporta da monoprotico secondo la reazione: H2SO4(aq) + H2O(l) → HSO4−(aq) + H3O+(aq) [H 3O + ] ≡ C a = [H 2SO 4 ] pH = −log 10[C a ] Il pH delle Sostanze anfiprotiche La base coniugata di un acido poliprotico è necessariamente anfiprotica, potendo agire sia da acido sia da base; essa può infatti cedere l'atomo di idrogeno acido rimanente oppure accettare un atomo di idrogeno ricostituendo l'acido originale. Per fare un esempio, lo ione idrogenosolfuro, HS−, agisce in acqua sia da acido sia da base: HS− + H2O H3O+ + S2- [H 3O + ] ⋅ [S2− ] −19 K a2 = = 1 , 0 ⋅ 10 [HS- ] HS− + H2O H2S + OH− K w [H 2S] ⋅ [OH − ] 1,0 ⋅10 −14 −7 K idrolisi = K b = = = ≅ 1,124 ⋅ 10 K a1 [HS − ] 8,9 ⋅10 −8 Sorge l'interrogativo se la soluzione di un sale di HS-, poniamo NaHS, sia acida grazie al carattere acido di HS-, oppure basica, a causa del carattere basico dello stesso ione. Si dimostra che adittando alcuni criteri logici e svolgendo un po' di algebra, risulta che il pH della soluzione è + 3 a1 a2 10 a1 a2 indipendentemente dalla concentrazione del sale.* [H O ] = K ⋅ K *A patto che Csale >> K W /K a2 1 ⇒ pH = − log 2 e Csale >> K a1 Comunque non vedremo esercizi sulle sostanze anfiprotiche …… Ma è bene che sappiate …. (K ⋅K ) .. che quando il bicarbonato di sodio (NaHCO3) è sciolto in acqua, produce una soluzione lievemente basica. Per esempio una soluzione di 50 g in un litro di acqua a 25 °C ha il pH di circa 8,34: NaHCO3 → Na++HCO3- g 50 g 50 g = = ≅ 0,595 M g L PM NaHCO3 L 84,0066 L mol Per l’acido carbonico (H2CO3) da cui deriva il bicarbonato: Csale = [HCO 3− ] = 50 Csale >> K W /K a2 e Csale >> K a1 K a1 = 4,4 ⋅10−7 K a2 = 4,7 ⋅10−11 [H 3O + ] = K a1 ⋅ K a2 = 2,068 ⋅10 −17 = 4,55 ⋅10 −9 M ( ) pH = −log 10 4,55 ⋅10 −9 = 8,34 Quando in una soluzione sono presenti sia l’acido debole (HA HA)) che la sua base coniugata (A-) … che succede? Come si calcola il pH? HA(aq) + H2O(l) ⇄ A-(aq) + H3O+(aq) Ca CS ? Quando in una soluzione sono presenti sia l’acido debole che la sua base coniugata … che succede? Come si calcola il pH? Si ha una Soluzione tampone Un soluzione tampone è una soluzione che varia in maniera trascurabile il proprio pH in seguito all’aggiunta di quantità moderate di un acido o di una base forte. Se ad un litro di acqua pura vengono aggiunte 0,01 moli di HCl il pH varia da 7 a 2 (pH=-log(0,01) =2,0), cioè di 5 unità. L’aggiunta della stessa quantità di HCl ad un litro di soluzione tampone può far variare il pH di circa 0,1 unità. Per questo motivo le soluzioni tampone anche a livello biologico. Per esempio il tampone ed ha un pH di circa 7,4 trasportatore di ossigeno risulterebbe valore dovesse variare. sono molto importanti, sangue è una soluzione e la sua funzione di compromessa se tale I tamponi possono essere costituiti da: - un acido debole e la sua base coniugata (es. HCN/CN-) - una base debole e il sua acido coniugato (es. NH3/NH4+) Una soluzione tampone contiene quindi una coppia acidobase coniugata cioè un acido e la sua base coniugata in equilibrio fra di loro. All’aggiunta di un acido o di una base forte l’equilibrio si sposta dalla parte della base o dell’acido deboli che costituiscono il tampone “assorbendo” così l’eccesso di ioni H+ (ovvero H3O+) o di OH-. Consideriamo ad esempio il tampone costituito da HClO e ClO- Quando in una soluzione sono presenti sia l’acido debole che la sua base coniugata … Per esempio: HClO(aq)) + H2O( HClO(aq O(ll) ⇄ ClO-(aq aq)) + H3O+(aq aq)) (i) Ca CS --(eq) Ca-x CS+x x [ClO − ] [H 3O + ] (Cs + x)x Ka = = [HClO] (Ca − x) [H 3O + ] = x quando Ca e Cs sono paragonabili (e non molto piccole), cioè 0,1 < (Ca /Cs) < 10 Se si calcola x si vede che x << Ca e x << Cs na (Cs + x)x Cs [H 3O + ] C n Ka = ≅ ⇒ [H 3O + ] ≅ K a a = K a V = K a a nS (Ca − x) Ca CS nS V na [H 3O ] = K a ns + Le soluzioni tampone Qualora, in una soluzione acquosa, siano presenti un acido debole ed la sua base coniugata (ad esempio: CH3COOH e CH3COO- oppure NH4+ e NH3, etc.) si ha una SOLUZIONE TAMPONE quando il rapporto fra le concentrazioni stechiometriche dell’acido e della base è compreso tra 0,1 e 10 Le soluzioni tampone hanno proprietà chimiche peculiari: • Il pH non varia al variare della comunque non deve essere eccessiva); diluizione (che • Il pH tende a rimanere costante per moderate (non eccessive) aggiunte di acidi e basi forti. Tampone: l’acido acetico (CH3COOH) e il sale acetato (CH3COO-) Cacido=Ca Csale=Cs CH3COOH(aq) + H2O(l) ⇄ CH3COO- (aq) + H3O+ (aq) [CH 3COO − ] [H 3O + ] Ka = [CH 3COOH] Esplicitando [H3O+]: [CH 3COOH] [H 3O ] = K a − [CH 3COO ] + − e quindi: quando [CH 3COO ] pH = pK a + log10 [CH 3COOH] Ca 0,1 < < 10 Cs CS pH = pK a + log 10 Ca Oppure Nota come equazione di Henderson e Hasselbach nS pH = pK a + log 10 na Se le concentrazioni delle due specie CH3COOH e CH3COO- sono uguali: pH = pKa il tampone si dice perfetto Tampone: ad esempio la coppia coniugata NH3/NH4+ NH3(aq) aq) + H2O( O(ll) ⇄ NH4+(aq) aq) + OH-(aq) aq) (i) Cb (eq) Cb-x Cs Cs+x x Se 0,1 < Cs/Cb < 10, con un ragionamento analogo al precedente si nb ottiene: nb Cb −x Cb V [OH ] = K b ⋅ ≅ Kb ⋅ = Kb ⋅ = Kb ⋅ ns ns Cs + x Cs nb V − [OH ] = K b ns nb ns − pOH = −log10 OH = −log10 K b ⋅ = pK b + log10 ns nb pH = 14 − pOH − [ ] Sei metodi di preparazione di un tampone Un tampone è sempre costituito da un acido debole HA e la sua base coniugata A- oppure da una base debole B e il suo acido coniugato B+. In pratica può essere preparato in vari modi: Come opera un tampone Esempio di Soluzioni Tampone: il tampone acido acetico-acetato Lo ione acetato, la base coniugata dell’acido acetico, assorbe i protoni quando viene aggiunto un acido forte. In tal modo il rapporto [base coniugata]/[acido] è approssimativamente costante, per cui la variazione di pH è minima. Analogamente, l’acido acetico agisce da donatore di protoni quando viene aggiunta una base forte, mantenendo il rapporto [base coniugata]/[acido] approssimativamente costante e minimizzando la variazione di pH Capacità Tampone e Intervallo di Tamponamento CAPACITA’ TAMPONE = Quantità di acido o base che il tampone è in grado di neutralizzare senza che il suo pH vari apprezzabilmente E’ MASSIMA QUANTO LE CONCENTRAZIONI DI ACIDO DEBOLE E BASE CONIUGATA SONO ALTE E APPROSSIMATIVAMENTE UGUALI TRA LORO INTERVALLO DI TAMPONAMENTO = Intervallo di pH entro il quale un tampone neutralizza efficacemente gli acidi e le basi aggiunte mantenendo il pH circa costante È PARI A CIRCA 2 UNITA’ DI pH CENTRATE INTORNO AL VALORE DEL pKa DELL’ACIDO DEBOLE O DEL pKb DELLA BASE DEBOLE Calcolo della variazione di pH di Soluzioni Tampone dopo aggiunta di piccole quantità di acido o base forti 1. Si assume che la reazione di neutralizzazione proceda a completezza e si determinano le nuove concentrazioni stechiometriche 2. Si usano le concentrazioni determinate per ricavare [H3O+] ed il pH Lo schema può essere applicato anche alla coppia acido-base BH+/B, dove B è la base e BH+ è l’acido. Chimica (a.a. 2011/2012, 6CFU) Equilibri ionici in soluzione acquosa (III parte): Esercitazione sul pH: acidi e basi forti, acidi deboli monoprotici e basi deboli monovalenti, acidi deboli poliprotici, idrolisi e soluzioni tampone. Esercizi riassuntivi e integrativi Esercitazione Dal libro Nobile-Mastrorilli (Capitolo 15), oltre a studiare gli esercizi svolti, dovete essere in grado di svolgere i seguenti problemi: 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 alle pag.538-541 Inoltre studiare le dispense tratte dal libro Stechiometria per la chimica generale di Paola Michelin Lausarot e Gian Angelo Vaglio: Cap 13,14 e 16 e svolgere i relativi esercizi. Leggere soltanto il cap. 15 Infine svolgere gli esercizi integrativi riportati alla fine di questa lezione. Acidi e basi forti Un’industria scarica 30 litri/min di una soluzione acquosa di acido cloridrico (HCl) a pH=2. Quanto ossido di magnesio (MgO) serve per neutralizzare la soluzione scaricata in un’ora di lavorazione? Svolgimento: Tipologia di esercizio: In un’ora di lavorazione: 1h=60 min L ⋅ 60 min = 180 L Vsoluzione = 30 min acido cloridrico (HCl) elettrolita forte: HCl(aq) + H2O(l) → H3O+(aq) + Cl-(aq) [HCl] = [H 3O + ] = 10− pH = 10−2 M Reazione di neutralizzare (reazione spostata a destra ha un K=1/Kw=10+14) H3O+(aq aq)) + OH (aq aq)) → 2 H2O(l) L’ossido di magnesio (MgO) fornisce l’OH− MgO(s) + H2O(l)→ Mg(OH)2 → Mg2+(aq) + 2 OH−(aq) Ogni mol di MgO dà 2 mol di OH− n OH − = 2 ⋅ n MgO Vsoluzione = 180 L n H O + = 10 [H 3O + ] = 10 −2 M −2 3 mol ⋅180 L = 1,8 mol L n H O + = n OH − H3O+(aq aq)) + OH (aq aq)) → 2 H2O(l) 3 Neutralizzazione pH=7 (neutro) n OH − = 1,8 mol n MgO ma n OH − = 2 ⋅ n MgO 1 1 = ⋅ n OH − = ⋅1,8 mol = 0,9 mol 2 2 m MgO = n MgO ⋅ PM MgO g = 0,9 mol ⋅ 40,3044 ≅ 36,27 g mol Tipologia di esercizio: Acidi e basi forti In una fossa d’irrigazione, una cisterna scarica abusivamente 1 m3 di una soluzione di acido nitrico a pH=2. Con quanta acqua pura questo scarto si deve miscelare perché il pH diventi 7? Quanta calce (CaO) sarebbe bastata per neutralizzare preventivamente la soluzione? Svolgimento: acido nitrico (HNO3) elettrolita forte: HNO3(aq) + H2O(l) → H3O+(aq) + NO3−(aq) [HNO3 ]iniziale = [H 3O + ] = 10 − pH = 10 −2 M Se il pH diventa 7 [H 3O + ] = 10 − pH = 10 −7 M = [HNO 3 ]finale [HNO3 ]iniziale ⋅ Viniziale = [HNO3 ]finale ⋅ Vfinale Viniziale 3 1000 dm L 3 3 =1m =1m ⋅ ⋅ = 1000 L 3 3 1m dm Considerando i volumi additivi Vfinale = Viniziale + VH 2O mol mol ⋅1000 L = 10 −7 ⋅ (1000 L + VH 2O ) L L −4 − 7 mol 10 mol = 10 mol + 10 ⋅ VH 2O L 10 mol − 10 −4 mol VH 2O = 99999000 L VH 2O = − 7 mol 10 Circa 100 milioni di litri L Ovvero 10000 m3 10 − 2 Reazione di neutralizzare (reazione spostata a destra ha un K=1/Kw=10+14) H3O+(aq aq)) + OH (aq aq)) → 2 H2O(l) L’ossido di magnesio (CaO) fornisce l’OH− CaO(s) + H2O(l)→ Ca(OH)2 → Ca2+(aq) + 2 OH−(aq) Ogni mol di CaO dà 2 mol di OH− n OH − = 2 ⋅ n CaO ! Viniziale = 1000 L n H O + = 10 [H 3O + ] = 10 −2 M 3 −2 mol ⋅1000 L = 10 mol L n H O + = n OH − H3O+(aq aq)) + OH (aq aq)) → 2 H2O(l) 3 Neutralizzazione pH=7 (neutro) n OH − = 10 mol n CaO ma n OH − = 2 ⋅ n CaO 1 1 = ⋅ n OH − = ⋅10 mol = 5 mol 2 2 m CaO = n CaO ⋅ PM CaO g = 5 mol ⋅ 56,077 ≅ 280,4 g mol Sarebbero bastati circa 280,4 g di calce (CaO) per neutralizzare preventivamente la soluzione !!! Tipologia di esercizio: Acidi e basi forti In 100 litri di una soluzione di acido cloridrico a pH=2 si pongono 8,1 grammi di alluminio metallico. Determinare il pH della soluzione al termine del processo che avviene (il volume rimane invariato). Svolgimento: acido cloridrico (HCl) elettrolita forte: HCl(aq) + H2O(l) → H3O+(aq) + Cl-(aq) [HCl] = [H + ] ≡ [H 3O + ] = 10 − pH = 1⋅10 −2 M Vsoluzione = 100 L n H + = n H O + = n HCl = [HCl] ⋅ Vsoluzione = 1⋅10 − 2 3 n Al = mol ⋅100 L = 1 ,0 mol L m Al 8,1 g = ≅ 0,30 mol PA Al 26,981538 g Tra l’alluminio metallico e l’HCl avviene la reazione redox: 6 HCl (aq) + 2 Al(s) → 3 H 2 (aq) + 2 AlCl3(aq) O in forma ionica 6 H + (aq) + 2 Al(s) → 3 H 2 (aq) + 2 Al3+ (aq) che potremmo anche scrivere così: 6 H 3O + (aq) + 2 Al(s) → 3 H 2 (aq) + 2Al3+ (aq) + 6 H 2 O (l) Qualunque modo di scrivere la reazione si scelga si giungerà al medesimo risultato: 6H mol iniziali) Variazione di mol) mol finali) + (aq) + 2 Al(s) → 3 H 2 (aq) + 2 Al 1,0 0,3 6 − 0,3 ⋅ 2 0,1 [H 3O + ]finale ≡ [H + ]finale = n H+ − 0,3 -finale Vsoluzione = 3+ -- -- 3 + 0,3 ⋅ 2 0,45 + 0,3 0,1 mol mol = 1⋅10 −3 L 100 L pH = −log10 [H 3 O + ] = −log10 [1 ⋅10 −3 ] = 3 0,3 (aq) Tipologia di esercizio: Acidi e basi forti Esercizi simili. Prova tu!!! ☺In una cisterna che contiene 12,50 m3 di acqua vengono aggiunti 3,650 kg di acido cloridrico. Calcolare il pH della soluzione. Determinare la massa di idrossido di calcio necessaria per neutralizzare la soluzione. (R. pH = 2,096; ~4,509 kg) ☺ A 50 mL di una soluzione acquosa di acido nitrico di concentrazione incognita, si aggiungono 125 mL di una soluzione di idrossido di bario 10−2 M. La soluzione risultante è neutra: 2 HNO3 (aq) + Ba(OH)2 (aq) → Ba(NO3)2 (aq) + 2 H2O (aq) determinare la concentrazione iniziale dell'acido. (R. [HNO3]=5·10-2 M) ☺ In 300 L di acqua vengono sciolti 67,327 g di KOH. Calcolare il pH della soluzione. Quanti litri di una soluzione 0,5 M di acido cloridrico occorre aggiungere alla soluzione per ridurre a 7 il valore di pH? (R. pH = 11,60; 2,4 L ) Tipologia di esercizio: acidi deboli o basi deboli Calcolare il pH di una soluzione 0,1 M di HIO4 (Ka=2,3·10-2 mol/L) L'acido periodico è un acido monoprotico non troppo debole; ovvero il valore della costante di trasferimento protonico non è troppo piccolo. Si può scrivere: Svolgimento: i) eq) − HIO4(aq) + H2O(l) ⇄ IO4-(aq) + H3O+(aq) Ca Ca – x x x [IO4 ] ⋅ [H 3O + ] Ka = = 2,3 ⋅ 10− 2 [HIO4 ] x2 Ka = Ca − x Ca 0,1 = ≈ 4,35 < 380 ⇒ α > 0,05 −2 K a 2,3 ⋅10 Cioè più del 5% di HIO4 si ionizza, vale a dire non posso trascura la x al denominatore nell'equazione della costante di equilibrio: 2 x Ka = Ca − x Dobbiamo risolvere l’equazione di secondo grado: x2 + Ka x - KaCa = 0 x 2 + 2,3 ⋅10 −2 x − 2,3 ⋅10 −2 ⋅ 0,1 = 0 x 2 + 2,3 ⋅10 −2 x − 2,3 ⋅10 −3 = 0 − 2,3 ⋅10 −2 + (−2,3 ⋅10 − 2 ) 2 + 4 ⋅ 2,3 ⋅10 −3 x = [H 3 O ] = ≅ 3,782 ⋅10 − 2 [mol/L] 2 + pH = −log10 [H 3 O + ] = −log10 [3,782 ⋅10 −2 ] ≅ +1,42 Se il valore di x fosse stato trascurato rispetto a 0,1 M (Ca) nell'equazione della costante di equilibrio si sarebbe ottenuto: [H 3 O + ] = 2,3 ⋅ 10−2 ⋅ 0,1 = 2,3 ⋅ 10−2 ≅ 4,796 ⋅ 10−2 pH = −log10 [4,796 ⋅10 −2 ] ≅ +1,32 Avremmo commesso un errore dell’ordine del 7% sul pH e di oltre il 20% sulla concentrazione di H3O+ !!! Tipologia di esercizio: acidi deboli o basi deboli Calcolare le concentrazioni di HA, H+ e A- e il pH di una soluzione 0,10 mol/L di un acido monoprotico debole, HA, con Ka=1,4·10-5 Svolgimento: HA(aq) + H2O(l) Conc. iniziali 0,10 Variazioni -x Concentrazioni all’equilibrio H3O+ (aq) + A-(aq) - 0,10-x - +x +x x x Dall’equazione della costante di equilibrio si ha: [H + ] [A - ] Ka = [HA] Ca > 380 Ka 2 x 1,4 ×10-5 = 0,10 - x ⇒ α > 0,05 ⇒ C a − x = 0,10 − x ≅ 0,10 2 x 1,4 ×10-5 = 0,10 - x 2 x 1,4 ×10-5 = 0,10 x2 = 1,4·10-5· 0,10 = 1,4×10-6 x = 1,4 × 10 -6 = 1,2 × 10 -3 M Verifichiamo innanzitutto l’approssimazione fatta: x = 0,0012 << 0,10 0,10-x = 0,10 - 0,0012 = 0,0998 ≈ 0,10 Le concentrazioni all’equilibrio sono quindi: [H3O+] = x = 1,2×10-3 M [A-] = x = 1,2·10-3 [HA] = 0,10 - x = 0,0998 ≈ 0,10 pH = -log(1,2×10-3 )=2,92 Talvolta per gli acidi deboli ionizzazione o le basi deboli si richiede il grado di Grado di ionizzazione Il grado di ionizzazione di un acido (o base) è definito come il rapporto fra la concentrazione di acido (base) ionizzato all’equilibrio e la concentrazione totale presente inizialmente. Moltiplicato per 100 corrisponde alla percentuale di acido (base) che è ionizzato all’equilibrio. Nel caso precedente si ha: [A − ] [A − ] 1,2 × 10-3 = = = 1,2 ×10-2 grado di ionizzazione = [HA] 0 Ca 0,10 % ionizzazione = 1,2×10-2 × 100 = 1,2% Tipologia di esercizio: acidi deboli o basi deboli Calcolare le concentrazioni di tutte le specie e il pH di una soluzione 0,10 M di metilammina (Kb=4,3×10-4). Svolgimento: CH3NH2(aq) + H20(l) CH3NH3+ (aq) + OH-(aq) Conc. iniziali 0,10 Variazioni -x +x +x 0,10-x x x Concentrazioni all’equilibrio 0 ~0 + [CH NH ] [OH ] 3 3 Dall’equazione della costante di equilibrio si ha: Kb = 2 [CH 3 NH 2 ] C 0,10 x b = ≤ 380 4,3 ×10 −5 = -4 K b 4,3 ⋅10 0,10 - x ⇒ 4,3 ×10-4 ⋅ (0,10 - x ) = x 2 ⇒ = x 2 + 4,3 ×10 −4 x − 4,3 ×10 −5 = 0 x = [OH − ] = − 4,3 × 10 − 4 + (4,3 ×10 ) −4 2 2 + 4 ⋅ 4,3 × 10 −5 mol = 6,346 × 10 −3 L Le concentrazioni all’equilibrio sono quindi: [OH-] = x = 6,346×10-3 M [CH3NH3+] = x = 6,346×10-3 M [CH3NH2] = 0,10 - x = 0,10 - 6,6×10-3 = 0,09354 Il calcolo non dà direttamente [H3O+] e per calcolare il pH conviene prima calcolare il pOH e poi sottrarre a 14 pOH = -log(6,6× ×10-3 ) ≈ 2,2 pH = 14-pOH=14-2,2 = 11,8 Il grado di ionizzazione di tale base vale: grado di ionizzazione = [BH + ] [BH + ] 6,6 × 10-3 M = = = 6,346 × 10- 2 [B]0 Cb 0,10 M % ionizzazione = 6,346× ×10-2 × 100 = 6,346 % Tipologia di esercizio: acidi deboli o basi deboli Svolgimento: TABELLA 15.1 Costanti di dissociazione di acidi deboli. (i) (eq) − + HF(aq) + H2O(l) ⇄ F-(aq) + H3O+(aq) Ca Ca – x x x [F ] ⋅ [H 3O ] Ka = = 6,31⋅10 − 4 M [HF] x2 Ka = Ca − x Punto c) Ca 0,4 = ≈ 634 > 380 ⇒ α < 0,05 ⇒ x << C a −4 K a 6,31 ⋅10 Possiamo effettuare questa approssimazione: + [ H 3O ] = K a ⋅ C a + −4 [H 3 O ] = 6,31⋅10 ⋅ 0,40 = 2,524 ⋅10 −4 pH = −log10 [H 3O ] = −log10 [ 2,524 ⋅ 10 ] ≅ + 1,80 + −4 Punto d) Ca 0,01 = ≈ 15,85 < 380 ⇒ α > 0,05 −4 K a 6,31 ⋅10 Dobbiamo risolvere l’equazione di secondo grado: x 2 + K a x - K aC a = 0 x 2 + 6,31 ⋅10 −4 x − 6,31 ⋅10 −4 ⋅ 0,01 = 0 x 2 + 6,31 ⋅10 − 4 x − 6,31 ⋅10 −6 = 0 −4 −4 2 −6 6,31 ⋅ 10 + ( − 6,31 ⋅ 10 ) + 4 ⋅ 6,31 ⋅ 10 x = [H 3 O + ] = ≅ 2,22 ⋅10 −3 2 pH = −log10[H 3O ] = −log10[2,22 ⋅ 10 ] = +2,65 + −3 Se avessi usato l’equazione approssimata: [H 3 O + ] = 6,31⋅10 −4 ⋅ 0,01 = 6,31⋅10 −6 ≅ 2,51⋅10 −3 pH = −log10 [2,51⋅10 −3 ] = +2,60 Errore di ~ 2% Terminare l’esercizio da soli (punti a,b,e) prendendo le Ka dalla Tab.15.1 Tipologia di esercizio: n NH = 3 VNH V 3 molare a c.n. = 2,8 l l 22,414 mol acidi deboli o basi deboli Svolgimento: ≅ 0,125 mol ⇒ [NH ] = C 3 b = n NH V 3 soluzione = 0,125 mol = 0,1 M 1,25 l NH3(aq) + H2O(l) ⇄ NH4+(aq) + OH-(aq) eq) Cb – x x x [NH 4+ ] ⋅ [OH − ] Kb = = 1,8 ⋅10 −5 M [NH 3 ] Cb 0,1 +3 = ≈ 5,56 ⋅ 10 > 380 −5 K b 1,8 ⋅10 x = [OH-] = [NH4+] x2 Kb = (C b − x) ⇒ [OH − ] = K b ⋅ C b pOH = -log10 [OH − ] = -log10 [ K b ⋅ C b ] = -log10 [ 1,8 ⋅10 −5 ⋅ 0,1 ] pH = 14 - pOH = 14 + log 10 [ 1,8 ⋅10 −5 ⋅ 0,1 ] ≅ 11,13 Terminare l’esercizio da soli (punti b,c) prendendo la Kb di CH3NH2 dalla Tab.15.2 Tipologia di esercizio: acidi deboli e basi deboli Svolgimento: L’acido benzoico è un acido organico monoprotico debole: HA (i) (eq) − HA(aq) + H2O(l) ⇄ A-(aq) + H3O+(aq) Ca ----x x Ca – x + 2 [A ] ⋅ [H 3O ] Ka = [HA] x Ka = Ca − x x = [A-] = [H3O+] = 10-pH = 10-2,95 = 1,112·10-3 M ( ) −3 2 x 1,112 ⋅10 −5 Ka = = ≅ 6,67 ⋅ 10 M −3 C a − x (0,02 − 1,112 ⋅10 ) 2 Tipologia di esercizio: Acidi poliprotici Svolgimento: Punto c) Come se fosse H2SO3 H2SO3(aq) + H2O(l) ⇄ HSO3-(aq) + H3O+(aq) Ka1 = 1,41·10-2 HSO3-(aq) + H2O(l) ⇄ SO32-(aq) + H3O+(aq) Ka2 = 6,31·10-8 Oppure per la prima dissociazione si scrive: SO2(aq) + 2H2O(l) ⇄ HSO3-(aq) + H3O+(aq) Ka1 = 1,41×10-2 Bisogna calcolare la concentrazione di SO2(aq) che coincide con quella di H2SO3 Vmolare a c.n L V = 22,414 = mol n [SO 2 (aq)] ≡ [H 2SO 3 ] = ⇒ n SO 2 = n SO 2 VSoluzione = VSO 2 L 22,414 mol K a,1 = 4,5 L L 22,414 mol ≅ 0,200 mol 0,200 mol mol = 0,1 2,0 L L SO2(aq) + 2 H2O(l) ⇄ HSO3−(aq) + H3O+(aq) Ca Ca – x x x − 3 = + [HSO ] ⋅ [H 3O ] = 1,41⋅10 − 2 [SO 2 ] Ca 0,1 = ≈ 7,09 < 380 ⇒ −2 K a 1,41 ⋅10 x = [HSO 3− ] = [H 3O + ] x2 K a,1 = (Ca − x) Non possiamo trascurare la x x 2 + K a,1 ⋅ x − K a,1 ⋅ C a = 0 x 2 + 1,41 ⋅10 −2 ⋅ x − 1,41 ⋅10 −3 = 0 x = [H 3O + ] = −2 − 1,41⋅10 + (1,41⋅10 ) −2 2 + 4 ⋅1,41⋅10 −3 2 = 3,116 ⋅10 − 2 pH = −log 10 [H 3O + ] = −log 10 [3,116 ⋅10 −2 ] ≅ +1,51 Punto d) una soluzione che contiene 210 mg di H3AsO4 in 500 mL d’acqua H3AsO4(aq) + H2O(l) ⇄ H2AsO4−(aq) + H3O+(aq) Ka1 = 5,49·10−3 H2AsO4-(aq) + H2O(l) ⇄ HAsO42−(aq) + H3O+(aq) Ka2 = 1,74·10−7 HAsO42−(aq) + H2O(l) ⇄ AsO43−(aq) + H3O+(aq) Ka3 = 5,13·10−12 m H 3AsO4 [H 3AsO 4 ] = n H 3AsO4 Vsoluzione = PM H 3AsO4 Vsoluzione 0,210 g 141,9430 g/mol = ≅ 2,96 ⋅10 −3 M = C a 0,5 L K a,1 K a,2 5,49·10−3 +4 +3 = = 3,15 ⋅ 10 > 10 1,74·10−7 H3AsO4(aq) + H2O(l) i) Ca Eq) Ca – x − 4 + [H 2 AsO ] ⋅ [H 3O ] K a,1 = = 5,49 ⋅10 −3 [H 2 AsO4 ] H2AsO4−(aq) + H3O+(aq) x x x2 K a,1 = (Ca − x) Ca 2,96·10−3 = ≤ 380 ⇒Non possiamo trascurare la x −3 K a,1 5,49·10 x 2 + 5,49 ⋅10 −3 ⋅ x − 1,62504 ⋅10 −5 = 0 x 2 + K a,1 ⋅ x − K a,1 ⋅ C a = 0 + x = [H 3O ] = − 5,49 ⋅10 −3 + (5,49 ⋅10 ) −3 2 + 4 ⋅1,60728 ⋅10 −5 2 ≅ 2,132 ⋅10 −3 pH = −log 10 [H 3O + ] = −log 10 [2,132 ⋅10 −3 ] ≅ +2,67 Terminare l’esercizio da soli (punti a,b,e) prendendo le costanti dalla Tab.15.1 Tipologia di esercizio: Svolgimento: Punto a) CH3COOK(aq) → CH3COO-(aq) + K+(aq) (i) (eq) Idrolisi CH3COO- (aq) + H2O(l) ⇄ CS CS-x [CH3COOK] = [CH3COO-] = CS CH3COOH(aq) + OH- (aq) --x --x 2 − K W 1 ⋅10 −14 [CH COOH] ⋅ [OH ] x −10 3 Ki = Kb = = = 5,55 ⋅ 10 = = −5 − K a 1,8 ⋅10 [CH 3COO ] Cs − x CS KW 0,15 +8 − = ≅ 2,7 ⋅ 10 > 380 ⇒ x = [OH ] = K ⋅ C = ⋅ CS b S -10 K b 5,55 ⋅10 Ka KW ⋅ CS pOH = −log10 [OH ] = −log10 [ K b ⋅ CS ] = −log10 Ka − KW ⋅ CS pH = 14 − pOH = 14 + log10 Ka 1⋅10 -14 KW pH = 14 + log10 ⋅ CS = 14 + log10 ⋅ 0,15 ≅ 8,96 5 1,8 ⋅10 Ka Il pH è basico (come atteso) Punto c) una soluzione di Na2HPO3 al 10%w di densità d = 1,08 g/ml Na2HPO3 in acqua è completamente dissociato: Na2HPO3 (aq) 2 Na+ (aq) + HPO32- (aq) HPO32- (anione fosfito) è la base coniugata di H2PO3- (anione idrogenofosfito) L’ HPO32- (anione fosfito) è soggetto ad idrolisi (basica) secondo l’equilibrio: HPO32- + H20 ⇄ H2PO3- + 0H- KW K idrolisi,1 = K b = Ka Quale Ka bisogna usare? La Ka2 dell’acido fosforoso (H3PO3) H3PO3(aq) + H2O(l) ⇄ H2PO3-(aq) + H3O+(aq) ‾(aq) H2PO3 i) eq) + H2O(l) ⇄ H2PO3 2-(aq) O+(aq) + H3 HPO32- + H20 ⇄ H2PO3- + 0HCs ----Cs-x x x Ka1 = 5,0·10-2 Ka2 = 2,0·10-7 [H 2 PO 3− ]eq = [OH − ]eq = x [HPO 32− ]eq = CS − x − − [H PO ][OH ] K W 1,0 ⋅10 −14 2 3 −8 K idrolisi = K b = = = 5 ⋅10 = −7 K a,2 2,0 ⋅10 [HPO 32− ] x2 5,0 ⋅10 = CS − x −8 Dalla tabella 15.1 CS = ? Ricordiamo che dalla percentuale in peso alla molarità di una soluzione ci si può arrivare direttamente usando la formula: ml g C% 1000 ⋅ d ⋅ L mL 100 [C] = g PM mol purché la densità sia espressa in g/mL (si veda la lezione #10) ml g 10 1000 ⋅1,08 ⋅ l ml 100 ≅ 0,857 M CS = [Na 2 HPO 3 ] = g 125,9549 mol 2 CS CS 0,857 x +8 = = = 1,714 ⋅10 > 380 ⇒ 2,128 ⋅10 − 4 = −8 K idrolisi K b 5 ⋅10 CS x = [OH − ] = K b ⋅ CS = 5 ⋅10 −8 ⋅ 0,857 pOH = -log10 [OH − ] = −log10 [ 5 ⋅10 −8 ⋅ 0,857 ] pH = 14 − pOH = 14 + log10 [ 5 ⋅10 −8 ⋅ 0,857 ] pH ≅ 10,32 Il pH è basico (come atteso) Punto e) una soluzione di acetato di sodio avente frazione molare χ= 1,8 · 10–4 e densità d = 1,0 g/ml CH3COONa(aq) → CH3COO-(aq) + Na+(aq) (i) (eq) CH3COO- (aq) + H2O(l) [CH3COONa] = [CH3COO-] = Cs CH3COOH(aq) + OH- (aq) Cs Cs-x --x --x − 2 K W 1 ⋅10 −14 [CH COOH] [OH ] x ⋅ −10 3 Ki = Kb = = = 5,55 ⋅ 10 = = −5 − K a 1,8 ⋅10 [CH 3COO ] Cs − x 1,8•10-4 mol di CH3COONa 1 mol di soluzione 0,99982 mol di H2O m CH COONa = n CH COONa ⋅ PM CH COONa = 1,8 ⋅ 10 -4 mol ⋅ 82,03427 3 3 3 m H O = n H O ⋅ PM H O = 0,99982 mol ⋅ 18,0152 2 2 2 g ≅ 0,001477 g mol g ≅ 18,0120 g mol Vsoluzione m CH 3COONa + m H 2O 0,001477 g + 18,0120 g m soluzione = = = ≅ 18,0135 ml = 0,0180135 [L] g d d 1,0 mL [CH 3 COONa ] ≡ [CH 3 COO - ]= n CH 3COONa Vsoluzione CS 0,01 +7 = = 1,8 ⋅ 10 > 380 -10 K b 5,55 ⋅10 − pOH = -log10 [OH ] = -log10 [ pH = 14 - pOH = 14 + log10 1,8 ⋅ 10 -4 [mol] = ≅ 0,010 M 0,0180135 [L ] ⇒ [OH − ] = K b ⋅ CS = KW ⋅ CS Ka KW K b ⋅ C S ] = -log10 ⋅ CS K a KW ⋅ CS Ka 1 ⋅ 10 -14 KW ≅ 8,37 pH = 14 + log10 ⋅ C S = 14 + log10 ⋅ 0,01 K 1,8 ⋅ 10 -5 a Il pH è basico (come atteso) Terminare l’esercizio da soli (punti b,d) prendendo le costanti dalla Tab.15.1 Tipologia di esercizio: Idrolisi Calcolare il pH di una soluzione 5·10-2M di cloruro di ammonio (Kb,NH3 = 1,8·10-5 mol/L). Il cloruro di ammonio (NH4Cl) è un sale che in acqua è completamente dissociato in catione ammonio (NH4+) e anione cloruro (Cl−). Lo ione ammonio (NH4+), che è presente nella soluzione, ha proprietà acide (è l'acido debole coniugato della base debole ammoniaca, NH3) ed è soggetto a reazione di idrolisi (acida): NH4Cl(aq) →NH4+(aq)+ Cl−(aq) (i) (eq) NH4+(aq) + H2O(l) CS CS – x NH3(aq) + H3O+(aq) ----x x K W [NH 3 ] [H 3O + ] costante d' idrolisi = K i = K a = = Kb [NH 4+ ] K W 1,0 ⋅10 −14 x2 −10 = = 5,55 ⋅10 = −5 K b 1,8 ⋅10 (CS − x) Cs 5 ⋅10 −2 = > 380 ⇒ −10 K a 5,55 ⋅10 Cioè meno del 5% di NH4+ si idrolizza CS − x ≅ CS + [H 3O + ]2 Ka = CS [H 3O ] = K a ⋅ CS = 5,55 ⋅10 −10 ⋅ 5,0 ⋅10 −2 pH = −log 10 [H 3O + ] = −log 10 [ 5,55 ⋅10 −10 ⋅ 5,0 ⋅10 −2 ] ≅ +5,28 Tipologia di esercizio: Ca(CH3COO)2(aq) → 2 CH3COO-(aq) Svolgimento: + Ca++(aq) Cs = [CH3COO-] = 2•[Ca(CH3COO)2]=2• [Ca++] CH3COOH(aq) + H2O(l) (i) Ca m Ca(CH3COO) 2 Vsoluzione CH3COO-(aq) + H3O+(aq) Cs+x Innanzitutto, calcoliamo Cs e Ca: CS = 2 ⋅ Ca = [CH3COOH] Cs (eq) Ca−x n Ca(CH3COO) 2 = 2⋅ Tampone PM Ca(CH3COO) 2 Vsoluzione --x 15,8 [g ] 158,169 g 0,1 [mol] mol = 2⋅ ≅ 2⋅ = 0,4 M 0,5 [L] 0,5 [L] m CH COOH 3 Ca = n CH COOH 3 Vsoluzione m CH COOH 3 = PM CH COOH 3 Vsoluzione % w CH 3 COOH % w CH 3 COOH = ⋅ m soluzione = ⋅ Vsoluzione ⋅ d = 100 100 1,2 g = ⋅ 500 [ml] ⋅ 1,0 = 6,0 [g ] 100 ml m CH COOH 3 Ca = n CH COOH 3 Vsoluzione = PM CH COOH 3 Vsoluzione C a 0,2 0,1 < = = 0,5 < 10 Cs 0,4 6,0 [g ] 60,0524 g mol 0,1 [mol] = = = 0,2 M 0,5 [l] 0,5 [l] Abbiamo un tampone e possiamo trascurare la x [CH 3COO − ] [H 3O + ] (Cs + x)x Ka = = [CH 3COOH] (Ca − x) na n C [H O ] C Ka ≅ s 3 ⇒ [H 3O + ] ≅ K a a = K a V = K a a ns Ca ns Cs V + Ca pH = −log 10[H 3O ] = −log 10[K a ] Cs + 0,2 = −log 10[1,8 ⋅10 ⋅ ] = + 5,05 0,4 -5 Oppure na pH = −log 10[H 3O ] = −log 10[K a ] ns + 0,1 = −log 10[1,8 ⋅10 ⋅ ] = + 5,05 0,2 -5 Tipologia di esercizio: Tampone (esempio di preparazione) Svolgimento: acido debole + base forte → sale + acqua CH3COO COOH H(aq) + NaOH NaOH(aq) → CH3COONa(aq) + H2O(aq) meglio in forma ionica: CH3COO COOH H(aq) + OH-(aq) → CH3COO-(aq) + H2O(aq) La reazione avviene completamente (spostata a destra: irreversibile) perché ha una costante molto grande (è l’inverso della costante di idrolisi basica 1/Ki=Ka/KW≈1,8·10+9). Ricorda il forte (NaOH: base forte) sposta completamente il debole (CH3COOH: acido debole) Calcoliamo le moli di acido debole (CH3COOH) e di base forte (NaOH). Il numero di moli di acido acetico contenuto in 400 ml di soluzione 0,0010 M è n CH 3COOH = [CH 3COOH]⋅ Vsoluzione,CH 3COOH mol = 0,001 ⋅ 0,400 l = 4 ⋅10 − 4 [mol ] L Il numero di moli di idrossido di sodio contenuto in 500 ml di soluzione 0,00040 M è n NaOH = [NaOH]⋅ Vsoluzione,NaOH = 0,0004 mol ⋅ 0,500 l = 2 ⋅10 − 4 [mol ] ≡ n OH − L Moli i) CH3COO COOH H(aq) + OH-(aq) → CH3COO-(aq) + H2O(aq) 4•10-4 2•10-4 --- ∆) -2•10-4 Moli f) 2•10-4 -2•10-4 +2•10-4 +2•10-4 0 2•10-4 2•10-4 Abbiamo un tampone: 0,1< na/ns<10 CH3COOH(aq) + H2O(l) ⇄ CH3COO−(aq) + H3O+(aq) (i) Ca CS --x CS+x (eq) Ca-x na -4 CS [H 3O ] Ca n 2 ⋅ 10 + Ka ≅ ⇒ [H 3O ] ≅ K a = Ka V = Ka a = Ka nS Ca CS nS 2 ⋅10 -4 V + pH = −log10[H 3O ] = −log10[K a ] = −log10[1,8 ⋅ 10 ] = + 4,74 + -5 Tipologia di esercizio con Acido debole libero; Tampone; idrolisi (basica) Ovvero preparazione di un tampone e sua successiva distruzione (“morte”) Esercizio A 160 ml di una soluzione 0,01 M di acido formico (HCOOH) si fanno due aggiunte successive di 40 mL di idrossido di sodio (NaOH) 0,02 M. Calcolare il pH della soluzione iniziale di HCOOH e il pH delle soluzioni ottenute in seguito a ciascuna aggiunta di NaOH. (La costante di dissociazione dell’acido formico è Ka = 1,8·10–4 mol/L e si ritengano, inoltre, i volumi additivi). Svolgimento: pH della soluzione iniziale di HCOOH HCOOH (aq) + H2O(l) ⇄ HCOO-(aq) + H3O+(aq) (i) Ca (eq) Ca−x --- ---- x x x = [HCOO − ] = [H 3O + ] − + [HCOO ] ⋅ [H 3O ] Ka = [HA] x2 Ka = Ca − x Ca 0,01 = < 380 –4 K a 1,8·10 ⇒ α > 0,05 Cioè più del 5% di HCOOH si ionizza, vale a dire non posso trascura la x al denominatore nell'equazione della costante di equilibrio K a ⋅ (C a − x ) = x 2 x2 Ka = Ca − x x 2 + K a x − K a Ca = 0 x = [H 3O + ] = − Ka + (K a )2 + 4K a Ca 2 = − 1,8 ⋅10 −4 + 3,24 ⋅10 −8 + 4 ⋅1,8 ⋅10 −4 ⋅1⋅10 − 2 = ≅ 1,255 ⋅10 −3 M 2 pH = −log 10 [H 3O + ] = −log 10[1,255 ⋅10 −3 ] = + 2,90 Prima aggiunta di 40 mL di idrossido di sodio (NaOH) 0,02 M. HCOOH HCOO H(aq) + NaOH NaOH(aq) → HCOONa(aq) + H2O(aq) meglio in forma ionica: HCOOH HCOO H(aq) + OH-(aq) → HCOO-(aq) + H2O(aq) La reazione avviene completamente (spostata a destra: irreversibile) perché ha una costante molto grande (è l’inverso della costante di idrolisi basica 1/Ki=Ka/KW≈1,8·10+10). Ricorda il forte (NaOH: base forte) sposta completamente il debole (HCOOH: acido debole) n OH − = n NaOH = C NaOH ⋅ VNaOH = 0,02 (mol/L ) ⋅ 0,040 L = 0,0008 mol n HCOOH = C HCOOH ⋅ VHCOOH = 0,01 (mol/L ) ⋅ 0,160 L = 0,0016 mol Moli OH− + HCOOH Iniziali 0,0008 0,0016 0 variazione -0,0008 -0,0008 +0,0008 Totale fine reazione 0 0,0008 0,0008 Abbiamo un tampone n 0,1 < a < 10 ns → Quindi HCOO− [H 3O + ] = K a + H2O na ns na - 4 0,0008 pH = −log10 [H 3O ] = −log10 [K a ] = −log10 [1,8 ⋅10 ⋅ ] ns 0,0008 + pH = −log 10 [1,8 ⋅10 -4 ] = pKa ≅ +3,745 Seconda aggiunta di 40 mL di idrossido di sodio (NaOH) 0,02 M. n OH − = n NaOH = C NaOH ⋅ VNaOH = 0,02 (mol/L ) ⋅ 0,040 L = 0,0008 mol n HCOO − = 0,0008 mol n HCOOH residuo = 0,0008 mol Moli OH− + HCOOH Iniziali 0,0008 0,0008 0,0008 variazione -0,0008 -0,0008 +0,0008 Totale fine reazione 0 0 0,0016 → HCOO− + H2O Non abbiamo più il tampone Abbiamo una soluzione di anione formiato (HCOO-) Considerando i volumi additivi: [HCOO ] = − n HCOO− Vtotale = n HCOO− V1 + V2 + V3 = 0,0016 [mol ] = 8 ⋅10 −3 M = CS (0,160 + 0,040 + 0,040) [L] dà idrolisi basica perché esso è la base coniugata di una acido debole (HCOOH) (i) (eq) HCOO- (aq) + H2O(l) ⇄ CS CS-x HCOOH(aq) + OH- (aq) --x --x x = [HCOOH] = [OH − ] K W 1⋅10 −14 [HCOOH] ⋅ [OH − ] x2 −11 Ki = K b = = = 5,55 ⋅10 = = −4 − K a 1,8 ⋅10 [HCOO ] Cs − x KW CS 7,754 ⋅10 −3 − = > 380 ⇒ x = [OH ] = K ⋅ C = ⋅ CS b S -11 K b 5,55 ⋅10 Ka − pOH = -log10 [OH ] = -log10 [ pH = 14 - pOH = 14 + log10 KW K b ⋅ CS ] = -log10 ⋅ CS Ka KW ⋅ CS Ka 1⋅10-14 KW − 3 pH = 14 + log 10 ⋅ CS = 14 + log 10 ⋅ 8 ⋅10 ≅ 7,82 4 1,8 ⋅10 Ka Esercizio simile da studiare per bene a pag. 519-521 del libro di testo (Nobile-Mastrorilli) Tipologia di esercizio: Tampone Calcolare quanti millilitri di una soluzione 0,12 M di cloruro di ammonio si devono aggiungere a 100 mL di ammoniaca 0,2 M per ottenere una soluzione tampone avente pH=9,72. Per l’ammoniaca, Kb = 1,8·10−5 M. Svolgimento: NH3(aq 3(aq)) + H2O(l) ⇄ (i) Cb (eq) Cb−x [NH +4 ] ⋅ [OH − ] Kb = = 1,8·10−5 M [NH 3 ] NH4+(aq + OH aq)) (aq aq)) Cs Cs+x x Per un tampone base debole/acido coniugato: nb n NH3 Cb −x Cb nb − V [OH ] = K b ⋅ ≅ Kb ⋅ = Kb ⋅ = Kb ⋅ = Kb ⋅ ns Cs + x Cs ns n NH + 4 V pH = 9,72 ⇒ pOH = 14 − 9,72 = 4,28 ⇒ [OH − ] = 1⋅10 −4,28 M − [OH ] = K b ⋅ n NH3 n NH + 1⋅10 4 − 4,28 −5 = 1,8 ⋅10 ⋅ n NH3 n NH + 4 n NH 3 = [NH 3 ] ⋅ Vsoluzione, NH 3 = 0,2 (mol/L) ⋅ 0,1 (L) = 0,02 mol n NH + = [NH 4 Cl] ⋅ Vsoluzione, NH 4Cl = 0,12 (mol/L) ⋅ Vx (L) 4 1⋅10 − 4,28 = 1,8 ⋅10 −5 ⋅ 0,02 0,12 ⋅ Vx 1,8 ⋅10 −5 0,02 Vx = ⋅ ≅ 0,05716 L = 57,16 mL − 4,28 1⋅10 0,12 Vx (L) = ? Tipologia di esercizio con idrolisi (acida); Tampone Svolgimento: acido forte + base debole (tipo ammoniaca) → HX(aq) Nel caso in esame: + HCl(aq) + oppure in forma ionica: Ovvero: :B(aq) CH3NH2(aq) → → sale BHX(aq) CH3NH3Cl(aq) H+(aq) + CH3NH2(aq) → CH3NH3+(aq) H3O+(aq) + CH3NH2(aq) → CH3NH3+(aq) + H2O(aq) Calcoliamo le moli di base debole (CH3NH2) e di acido forte (HCl). Il numero di moli di CH3NH2 contenuto in 50 mL di soluzione 2,0M è n CH NH = [CH3 NH 2 ] ⋅ Vsoluzione,CH NH 3 2 3 2 mol = 2,0 ⋅ 0,050 l = 0,1 [mol ] l Il numero di moli di HCl contenuto in 50 mL di soluzione al 7,0% in peso (d= 1,03 g/ml) è n HCl = [HCl] ⋅ Vsoluzione,HCl Ma, bisogna calcolarla? m HCl n HCl % w HCl = ⋅ Vsoluzione,HCl ⋅ d 100 n HCl m HCl = PM HCl 7,0 g % w HCl [ ] ⋅ 50 ml ⋅ 1,03 ⋅ Vsoluzione,HCl ⋅ d ml 100 100 = = ≅ 0,1 [mol] PMHCl g 36,4609 mol n HCl = n H O = 0,1 [mol ] 3 + 1° aggiunta: H3O+(aq) + CH3NH2(aq) → Moli i) 0,1 ∆) -0,1 f) 0 H2O(aq) + CH3NH3+(aq) 0,1 -- -- -0,1 +0,1 +0,1 0 0,1 0,1 Abbiamo una soluzione di ione metilammonio Considerando i volumi additivi: [CH NH ] = 3 + 3 n CH NH+ 3 Vtotale 3 0,1 [mol ] = = = 1,0 M V1 + V2 (0,050 + 0,050) [L] n CH NH+ 3 3 dà idrolisi acida perché esso è l’acido coniugato di una base debole: CH3NH2 Idrolisi acida: (i) (eq) CH3NH3+(aq aq)) + H2O( O(ll) CS CS-x Ki = Ka = x x KW x2 = K b Cs − x + K W [CH 3 NH 2 ] ⋅ [H 3O ] = Kb [CH 3 NH 3+ ] Cs C 1 = s = > 380 −14 K 1,0 ⋅10 Ki W Kb 4,26 · 10 – 4 CH3NH2(aq aq)) + H3O+(aq aq)) ------ x << Cs KW x = [H 3O ] = ⋅ Cs Kb + KW ⋅ Cs = pH = −log10[H 3O ] = −log10 Kb + 1,0 ⋅ 10 −14 = −log10 ⋅ 1,0 = 5,31 –4 4,26 · 10 2° aggiunta: H3O+(aq) + CH3NH2(aq) Moli i) 0 0,1 H2O(l) + CH3NH3+(aq) 0 0,1 Abbiamo un tampone: 0,1< nb/ns<10 + CH3NH2( + H O ⇄ CH NH + OH 2(aq aq)) 2 (l) 3 3 (aq aq)) (aq aq)) (i) Cb (eq) Cb-x [CH 3 NH 3+ ] ⋅ [OH − ] Kb = [CH 3 NH 2 ] Cs Cs+x nb nb Cb − x Cb − V [OH ] = K b ⋅ ≅ Kb ⋅ = Kb ⋅ = Kb ⋅ ns ns Cs + x Cs V x nb 0,1 [OH ] = K b ⋅ = K b ⋅ = Kb ns 0,1 − pOH = −log10 [OH ] = −log10 K b = −log10 (4,26 ⋅ 10 − -4 ) pH = 14 − pOH = 14 + log10 (4,26 ⋅ 10 ) ≅ +10,63 -4 Tipologia di esercizio: Calcolo della variazione di pH di Soluzioni Tampone dopo aggiunta di piccole o moderate quantità di acido o base forte Qual è la variazione di pH quando 1,00 mL di una soluzione 1,00 M di HCl è aggiunto a 1) 1,00 L di acqua pura ; 2) 1,00 L di tampone che ha [CH3COOH] = 0,700 M e [CH3COO−] = 0,600 M Soluzione della parte 1) Svolgimento: Prima dell’aggiunta di HCl il pH di 1,00 L di acqua pura è 2 H 2O → ← H 3O + + OH − K W = [H 3O + ] ⋅ [OH − ] = 1⋅10 −14 [H 3O + ] = 1 ⋅10 −14 = 1 ⋅10 −7 M pH iniziale = −log10 [1⋅10 −7 ] = +7 dopo l'aggiunta di 1,00 mL di HCl 1,00 M a 1,00 L di acqua? n H O + = n HCl = C HCl ⋅ VHCl = 1 (mol/L ) ⋅ 0,001 L = 0,001 mol 3 0,001 mol 0,001 mol 0,001 mol mol [H 3O ] = = = ≅ 0,001 Vfinale VHCl + VH 2O 0,001 L + 1 L L + pH finale = −log10 [1 ⋅10 −3 ] = +3 ∆pH = pH iniziale − pH finale = 7 − 3 = 4 Soluzione della parte 2) Prima dell’aggiunta di HCl il pH di tampone con [CH3COOH] = 0,700 M e [CH3COO−]=0,600 M è: CH3COOH(aq) + H2O(l) ⇄ CH3COO-(aq) + H3O+(aq) (i) Ca Cs Cs+x (eq) Ca−x [CH 3COO − ] [H 3O + ] (Cs + x)x Ka = = [CH 3COOH] (Ca − x) --x C a 0,7 0,1 < = ≅ 1,17 < 10 Cs 0,6 na Cs [H 3O + ] C n ⇒ Ka ≅ ⇒ [H 3O + ] ≅ K a a = K a V = K a a ns Ca Cs ns V Ca -5 0,7 pH = −log10 [H 3O ] = −log10 [K a ] = −log10 [1,8 ⋅10 ⋅ ] ≅ + 4,678 Cs 0,6 + Soluzione della parte 2) dopo l'aggiunta di 1,00 mL di HCl 1,00 M ? Ricorda la slide (44) di lezione 19 Parte stechiometria: H3O+ (da HCl) + CH3COO− (dal tampone) → CH3COOH (dal tampone) + H2O La reazione avviene completamente (spostata a destra: irreversibile) perché ha una costante molto grande (è l’inverso della costante acida 1/Ka ≈5,55·10+4). Ricorda il forte (HCl: acido forte) sposta completamente il debole (CH3COO−: base debole) Parte stechiometria (più facile ragionare con le moli): n H O + = n HCl = C HCl ⋅ VHCl = 1 (mol/L ) ⋅ 0,001 L = 0,001 mol 3 n CH COO − = C CH COO − ⋅ Vtampone = 0,600 (mol/L ) ⋅1,0 L = 0,600 mol 3 3 n CH 3COOH = C CH 3COOH ⋅ Vtampone = 0,700 (mol/L ) ⋅1,0 L = 0,700 mol Moli H3O+ + CH3COO− Iniziali 0,001 0,600 0,700 variazione -0,001 -0,001 +0,001 Totale fine reazione 0 0,599 0,701 → CH3COOH n Abbiamo ancora un tampone 0,1 < a < 10 ns na + [H 3O ] = K a Quindi ns + H2O na -5 0,701 pH = −log10 [H 3O ] = −log10 [K a ] = −log10 [1,8 ⋅10 ⋅ ] ≅ + 4,676 ns 0,599 ∆pH = pH iniziale − pH finale = 4,678 − 4,676 = 0,002 + La variazione di pH è pressoché NULLA c.v.d. Un paragone scherzoso Come un tampone (per esempio: CH3COOH/CH3COO−) resiste alle aggiunte di basi forti (OH−) e acidi forti (H3O+) È come se avessimo due tipi di squali uno ghiotto di OHe l’altro di H3O+ Purché la popolazione dei due tipi di squalo rimanga confrontabile, il pH tende a rimanere pressoché costante per moderate (non eccessive) aggiunte di acidi e basi forti. Tipologia di esercizio: Calcolo della variazione di pH di Soluzioni Tampone dopo aggiunta di piccole o moderate quantità di acido o base forte Un tampone viene preparato sciogliendo 2,0 g di acetato di sodio (CH3COONa) e 1,2 g di acido acetico (CH3COOH, Ka=1,8·10-5 M) in tanta acqua da avere 200 mL di soluzione. A questa soluzione vengono aggiunti 5,0 mL di una soluzione di NaOH 0,2 M. Si calcoli il pH del sistema prima e dopo l’aggiunta della base. Svolgimento: Prima dell’aggiunta di 5,0 mL NaOH 0,2 M (i) (eq) CH3COOH(aq) + H2O(l) ⇄ CH3COO-(aq) + H3O+(aq) Ca Cs --Ca−x Cs+x x [CH 3COO − ] [H 3O + ] (Cs + x)x Ka = = [CH 3COOH] (Ca − x) 1,2 g C a n a n CH 3COOH 60,0520 g/mol 0,01998 mol 0,1 < = = = = ≅ 0,82 < 10 2,0 g Cs n s n CH COO − 0,02438 mol 3 82,0338 g/mol n a + C [H O ] C n ⇒ Ka ≅ s 3 ⇒ [H 3O + ] ≅ K a a = K a V = K a a ns Ca Cs ns V Prima : pH = −log10 [H 3O + ] = −log10 [K a na 0,01998 ] = −log10 [1,8 ⋅10-5 ⋅ ] ≅ + 4,83 ns 0,02438 Dopo l’aggiunta di 5,0 mL NaOH 0,2 M n OH − = n NaOH = C NaOH ⋅ VNaOH = 0,2 (mol/L ) ⋅ 0,005 L = 0,0010 mol Parte stechiometria: OH- (da NaOH) + CH3COOH (dal tampone) → CH3COO− (dal tampone) + H2O La reazione avviene completamente (spostata a destra: irreversibile) perché ha una costante molto grande (è l’inverso della costante di idrolisi basica 1/Ki=Ka/KW≈1,8·10+9). Ricorda il forte (NaOH: base forte) sposta completamente il debole (CH3COOH: acido debole) Moli OH− Iniziali 0,00100 0,01998 0,02438 variazione -0,00100 -0,00100 +0,00100 Totale fine reazione 0 0,01898 0,02538 + CH3COOH → Abbiamo ancora un tampone 0,1 < na < 10 ns + H2O na 0,01898 ] = −log10 [1,8 ⋅10-5 ⋅ ] ≅ + 4,87 ns 0,02538 = 4,83 − 4,87 = 0,04 Dopo : pH = −log10 [H 3O + ] = −log10 [K a ∆pH = pH iniziale − pH finale CH3COO− Tipologia di esercizio: Calcolo della variazione di pH di Soluzioni Tampone dopo aggiunta di piccole o moderate quantità di acido o base forte Dopo aver calcolato il pH di una soluzione 0,30 M in NH3 (Kb=1,8·10-5 M) e 0,36 M in NH4Cl, calcolare il pH dopo l'aggiunta di 20,0 mL di una soluzione 0,050 M NaOH a 80,0 mL di tale soluzione tampone? Svolgimento: Prima dell’aggiunta di NaOH: (i) NH4+(aq) + OH aq) (aq) aq) + − [NH ] ⋅ [OH ] Cs 4 Kb = [NH 3 ] x Cs+x NH3( 3(aq aq)) + H2O(l) ⇄ Cb (eq eq)) Cb-x C 0,30 M 0,1 < b = = 0,83 < 10 Cs 0,36 M [ Cb −x Cb [OH ] = K b ⋅ ≅ Kb ⋅ Cs + x Cs − ] 0,30 pOH = −log10 OH − = −log10 1,8 ⋅10 −5 ⋅ 0,36 − 5 0,30 Prima : pH = 14 − pOH = 14 + log 10 1,8 ⋅10 ⋅ ≅ +9,176 0,36 il pH dopo l'aggiunta di 20,0 mL di una soluzione 0,050 M NaOH a 80,0 mL di tale soluzione tampone? n OH − = n NaOH = C NaOH ⋅ VNaOH = 0,050 (mol/L ) ⋅ 0,020 L = 0,0010 mol n NH + = C NH + ⋅ Vtampone = 0,36 (mol/L ) ⋅ 0,080 L = 0,0288 mol 4 4 n NH3 = C NH 3 ⋅ Vtampone = 0,30 (mol/L ) ⋅ 0,080 L = 0,0240 mol Parte stechiometria: OH- (da NaOH) + NH4+ (dal tampone) → NH3 (dal tampone) + H2O La reazione avviene completamente (spostata a destra: irreversibile) perché ha una costante molto grande (è l’inverso della costante basica 1/Kb ≈5,55·10+4). Ricorda il forte (NaOH: base forte) sposta completamente il debole (NH4+: acido debole) Moli OH− + NH4+ Iniziali 0,0010 0,0288 0,0240 variazione -0,0010 -0,0010 +0,0010 0,0278 0,0250 Totale fine 0 reazione Abbiamo ancora un tampone NH3 → + H2O nb C n [OH − ] ≅ K b ⋅ b = K b ⋅ V = K b ⋅ b ns Cs ns V nb < 10 ns 0,025 Dopo : pH = 14 − pOH = 14 + log 10 1,8 ⋅10 −5 ⋅ ≅ +9,209 0,0278 0,1 < ∆pH = pH iniziale − pH finale = 9,176 − 9,209 = 0,033 Tipologia di esercizio: Calcolo della variazione di pH di Soluzioni Tampone dopo aggiunta di piccole o moderate quantità di acido o base forte Calcolare la variazione di pH di 1,0 L di una soluzione 0,25 M in NH3 e 0,15 M in NH4Br dopo l'aggiunta di 0,15 mol di HCl. (si consideri nulla la variazione di volume della soluzione in seguito all’aggiunta di HCl) Svolgimento: Prima dell’aggiunta di HCl (i) NH4+(aq) + OH aq) (aq) aq) + − [NH ] [OH ] ⋅ Cs 4 Kb = [NH 3 ] x Cs+x NH3( 3(aq aq)) + H2O(l) ⇄ Cb (eq eq)) Cb−x 0,1 < C b 0,25 M = = 1,667 < 10 Cs 0,15 M pOH = −log10 [OH − ] = K b ⋅ Cb −x C ≅ Kb ⋅ b Cs + x Cs [OH ] = −log − −5 0,25 ⋅ 10 1,8 ⋅10 0,15 − 5 0,25 Prima : pH = 14 − pOH = 14 + log 10 1,8 ⋅10 ⋅ ≅ +9,477 0,15 il pH dopo l'aggiunta di 0,15 mol di HCl? n HCl ≡ n H O + = 0,15 mol 3 n NH + = C NH + ⋅ Vtampone = 0,15 (mol/L ) ⋅1,0 L = 0,15 mol 4 4 n NH 3 = C NH 3 ⋅ Vtampone = 0,25 (mol/L ) ⋅1,0 L = 0,25 mol Parte stechiometria: H3O+ (da HCl) + NH3 (dal tampone) → NH4+ (dal tampone) + H2O La reazione avviene completamente (spostata a destra: irreversibile) perché ha una costante molto grande (è l’inverso della costante di idrolisi acida 1/Ki=Kb/KW≈1,8·10+9). Ricorda il forte (HCl: acido forte) sposta completamente il debole (NH3: base debole) Moli H3O+ + NH3 Iniziali 0,15 0,25 0,15 variazione -0,15 -0,15 +0,15 Totale fine reazione 0 0,10 0,30 Abbiamo ancora un tampone → NH4+ + H2O nb n C n 0,1 < b < 10 [OH − ] ≅ K b ⋅ b = K b ⋅ V = K b ⋅ b ns Cs ns ns V 0,10 Dopo : pH = 14 − pOH = 14 + log 10 1,8 ⋅10 −5 ⋅ ≅ +8,778 0,30 ∆pH = pH iniziale − pH finale = + 9,477 − 8,778 ≅ 0,70 Esercizio di ricapitolazione Una soluzione 0,240 M di un acido debole (HA) monorprotico ha pH=1,824. Calcolare: (a) la costante di dissociazione di HA; (b) il pH di una soluzione 0,875 M del suo sale NaA; (c) il pH di una soluzione preparata miscelando 200 mL di una soluzione 0,6 M di HA con 100 mL di soluzione di NaA 0,3 M. Svolgimento (a) HA(aq) + H2O(l) ⇄ H3O+(aq) + A-(aq) Conc. iniziali) CHA - - Conc. all’eq) CHA −x x x [H 3O + ] ⋅ [A − ] x2 Ka = = [HA] C HA − x e per definizione pH = - log10 [H 3 O + ] ⇔ [H 3 O + ] = x = 10-pH = 10−1,824 = 0,015 M x2 (0,015)2 Ka = = = 1,0 ⋅10−3 [mol/L] C HA − x 0,240 − 0,015 (b) Il sale in oggetto NaA, essendo formato dall’anione A di un acido debole (HA) e dal catione Na+ di una base forte (NaOH), sarà completamente dissociato in acqua. [NaA] = [Na + ] = [A − ] = Cs = 0,875 M L’anione A- dà reazione di idrolisi: A-(aq) + H2O(l) ⇄ HA(aq) +OH-(aq) Conc. iniziali) Cs - - Conc. all’eq) Cs -x x x [HA] ⋅ [OH - ] Ki = [A - ] K w 1,0 ⋅10-14 -11 Ki = K b = = = 1,0 ⋅ 10 (mol/L) ⇒ -3 K a 1,0 ⋅10 CS 0,875 +10 = = 8,75 ⋅ 10 >> 380 ⇒ -11 K i 1,0 ⋅10 [HA] ⋅ [OH - ] x2 x2 Kb = = ≅ [A ] Cs − x Cs [ x2 ⇔ Kb = ⇒ x = K b ⋅ Cs Cs ] [ ] ⇒ pOH = −log10 OH − = −log10 K b ⋅ Cs = −log10 1,0 ⋅10-11 ⋅ 0,875 = −log10 2,958 ⋅10-6 ≅ 5,53 ⇒ pH = 14 − pOH = 14 − 5,53 = 8,47 (c) La soluzione preparata è un tampone: 100 mL di [NaA] = 0,3 M ⇒ n NaA = n A - 200 mL di [HA ] = 0,6 M ⇒ n HA mol = 0,3 ⋅ 0,100 L = 0,03 mol L mol = 0,6 ⋅ 0,200 L = 0,12 mol L HA(aq) + H2O(l) ⇄ H3O+(aq) + A-(aq) moli iniziali) 0,120 - 0,030 moli all’eq) 0,120-x x 0,030+x x (0,030 + x ) ⋅ [H 3O ] ⋅ [A ] VT VT = Ka = (0,120 − x ) ⇔ equazione di 2° grado (VT : volume totale della soluzione) [HA] VT + − Tuttavia n An HA n A0,03 = = 0,25 ⇒ 0,1 < < 10 ⇔ x << 0,12 e 0,03 ⇒ soluzione tampone 0,12 n HA ⇒ [ ] (0,120 - x ) x n 0,12 VT mol = H 3O + = K a ⋅ ≈ K a ⋅ acido = 1,0 ⋅10 −3 ⋅ = 4,0 ⋅10 −3 (0,030 + x ) VT n sale 0,30 L VT ⇒ pH = −log 10[ H 3O + ] = −log 10[4,0 ⋅10 −3 ] ≅ 2,398 Esercizio di ricapitolazione A quale volume è necessario diluire 30 mL di una soluzione 0,09 M di acido acetico (CH3COOH, Ka=1.8.10-5) per ottenere una soluzione a pH 3,5. Quale sarà il nuovo valore di pH se alla soluzione si aggiungono 0,3 g di CH3COONa (Si consideri nulla la variazione di volume della soluzione in seguito all’aggiunta del sale). Svolgimento: 30 mL C2=? CH3COOH(aq) + H2O(l) ⇄ CH3COO−(aq) + H3O+(aq) C1=0,09 M (i) C2 ----(eq) C2 – x x x − + [CH 3COO ] ⋅ [H 3O ] Ka = [CH 3COOH] x2 Ka = (C 2 − x) x = [CH3COO−] = [H3O+] = 10−pH = 10−3, 5 x2 1,8 ⋅10 = ⇒ (C 2 − x) −5 (C 2 − 10 −3,5 ) ⋅1,8 ⋅10 −5 = 10 −7 ⇒ (10 −3,5 ) 2 1,8 ⋅10 = (C 2 − 10 −3 ,5 ) −5 1,8 ⋅10 −5 C 2 − 1,8 ⋅10 −8,5 = 10 −7 10 −7 + 1,8 ⋅10 −8,5 ⇒ C2 = ≅ 0,005872 M −5 1,8 ⋅10 C1 ⋅ V1 = C 2 ⋅ V2 C1 ⋅ V1 n CH 3COOH 0,09 [mol/L] ⋅ 0,030 [L] 0,0027 [mol] V2 = = = = = 0,4598 [L] C2 C2 0,005872 [mol/L] 0,005872 [mol/L] Quale sarà il nuovo valore di pH se alla soluzione si aggiungono 0,3 g di CH3COONa? n CH 3COONa = m CH 3COONa PM CH 3COONa [CH COO ] = C − 3 S = = 0,3 g = 3,657 ⋅10 −3 mol = n CH COO − 3 82,0338 g/mol n CH COO − 3 V 3,657 ⋅10 −3 mol = 0,4598 L CH3COOH(aq) + H2O(l) ⇄ CH3COO−(aq) + H3O+(aq) (i) C2 CS --(eq) C2 – x CS +x x Abbiamo un tampone, inoltre : 0,1 < C a = C 2 < 10 CS CS (CS + x) ⋅ [H 3O + ] CS [H 3O + ] ≅ Ka = (Ca − x) Ca na n CH3COOH Ca na 2,7 ⋅10 −3 + −5 V ⇒ [H 3O ] ≅ K a = Ka = Ka = Ka = 1,8 ⋅10 ⋅ ≅ 1,329 ⋅10 −5 −3 nS CS nS n CH COO − 3,657 ⋅10 3 V ⇒ pH = −log 10[H 3O + ] = −log 10 [1,329 ⋅10 −5 ] ≅ 4,876 Esercizio di ricapitolazione Il pH di una soluzione 0,1 M di un acido debole monoprotico (HA) vale 2,75. Calcolare il numero di moli del sale KA (sale di potassio di A-) che occorre aggiungere ad un litro di tale soluzione per portare il valore del pH a 5,00. (Si consideri nulla la variazione di volume della soluzione in seguito all’aggiunta del sale). Svolgimento: I primi dati del problema servono per calcolare la Ka dell’acido debole HA(aq) ⇄ H3O+(aq) + A-(aq) Conc. iniziali) CHA - - Conc. all’eq) CHA −x x x x = [H 3O + ] = [A − ] e per definizione + H2O(l) [H 3O + ] ⋅ [A − ] x2 Ka = = [HA] C HA − x pH = − log10 [H 3 O + ] ⇔ [H 3 O + ] = x = 10− pH = 10−2,75 = 1,778 ⋅10−3 M x2 (1,778 ⋅10−3 ) 2 −5 Ka = = ≅ 3,22 ⋅ 10 [mol/L] −3 C HA − x 0,1 − 1,778 ⋅10 Seconda parte: Il numero di moli del sale KA (sale di potassio di A-) che occorre aggiungere ad un litro di tale soluzione per portare il valore del pH a 5,00 ? pH = − log10 [H 3 O + ] ⇔ [H 3 O + ] = x = 1⋅10− pH = 1⋅10−5 M È chiaro che si forma un tampone per aggiunta di KA (comunque lo verifichiamo a posteriori) HA(aq) Se nHA 0,1< <10 nA− + H2O(l) ⇄ H3O+(aq) + A-(aq) Moli iniziali) nHA - nA- Moli all’eq) nHA−x x nA+x abbiamo un tampone ⇒ [H 3O + ] ≅ K a n HA n A− n HA = C HA ⋅ Vsoluzione = 0,1(mol/L ) ⋅1,0 L = 0,1 mol 0,1 1 ⋅10 = 3,22 ⋅10 ⋅ n A− −5 −5 Si verifica che n A− 0,1 = 3,22 ⋅10 ⋅ = 0,322 [mol] −5 1⋅10 nHA 0,1 < <10 nA− −5 Esercizio di ricapitolazione In una soluzione di 100 mL di acido cloridrico (HCl) ha pH 2,31, si fanno assorbire 25 mL misurati a c.n. di ammoniaca gassosa, calcolare il pH finale della soluzione sapendo che Kb=1,8.10-5 M. (Assumere che il volume della soluzione resti invariato in seguito all’assorbimento del gas). Risoluzione: 100 mL di acido cloridrico (HCl) con pH 2,31: ⇒ [H 3O + ] = 10 − pH = C HCl = 10 −2,31 ≅ 4,898 ⋅10 −3 M ⇒ n H O + = [H 3O + ] ⋅ VHCl = 4,898 ⋅10 −3 M ⋅ 0,100 L = 4,898 ⋅10 − 4 mol 3 + 25 mL misurati a c.n. di ammoniaca gassosa V mL ⇒ Vm = = 22414 n mol ⇒ n NH 3 = VNH 3 Vm = 25 mL ≅ 11,15 ⋅10 − 4 mol mL 22414 mol H3O+(aq) + NH3(aq) → NH4+(aq) + H2O(aq) MOLI I) 4,898 ⋅10 −4 −4 Δ) − 4,898 ⋅10 TOTALE) −−− −−− −−− − 4,898 ⋅10 −4 + 4,898 ⋅10 −4 + 4,898 ⋅10 −4 6,252 ⋅10 −4 4,898 ⋅10 −4 11,15 ⋅10 −4 Ho la formazione di un tampone ammoniaca/ione ammonio NH4+(aq + OH aq)) (aq aq)) NH3(aq 3(aq)) + H2O(l) (i) Cb (eq) Cb−x Cs Cs+x x [NH +4 ] ⋅ [OH − ] Kb = [NH 4 ] nb Cb −x Cb nb − V [OH ] = K b ⋅ ≅ Kb ⋅ = Kb ⋅ = Kb ⋅ ns Cs + x Cs ns V −4 n NH3 n NH3 nb 6,252 ⋅10 −5 6,252 [OH ] = K b ⋅ = Kb ⋅ = Kb ⋅ = 1,8 ⋅10 −5 ⋅ = 1 , 8 ⋅ 10 ⋅ −4 nS n NH + n NH + 4,898 ⋅10 4,898 − 4 4 [ ] 6,252 pOH = −log10 OH − = −log10 1,8 ⋅10 −5 ⋅ 4,898 −5 6,252 pH = 14 − pOH = 14 + log 10 1,8 ⋅10 ⋅ ≅ +9,36 4,898 Degli ottimi Esercizi riassuntivi e integrativi sono i seguenti: Ricorda che hai bisogno di una calcolatrice scientifica per calcolare il pH + pH = −log 10 [H 3O ] Esercizi riassuntivi e integrativi 1) Si prepara un sistema tampone mescolando 184,0 mL di una soluzione acquosa 0,442 M di acido cloridrico (HCl) con 0,50 L di una soluzione acquosa 0,400 M di acetato di sodio (CH3COONa). Si calcoli la massa in grammi di idrossido di potassio (KOH) che deve essere aggiunta a 100,0 mL di tale tampone per farne variare il pH di 0,15 unità. (Per l’acido acetico (CH3COOH) pKa=4,75) [0,130 g] 2) Si abbiano, alla temperatura di 25,0°C, 250,0 mL di una soluzione acquosa contenente, contemporaneamente, 2,35 g di HNO2 e 1,72 g di NaNO2. A questa soluzione si aggiungano 10,0 mL di una soluzione 0,10 M di HCl. Si calcoli il pH del sistema prima e dopo l'aggiunta dell'acido forte (per HNO2, Ka = 4,0 ·10−4 M). [pH iniziale: 3,10, pH finale: 3,07] 3) Una soluzione acquosa 0,10 M del sale sodico, NaA, dell'acido debole HA presenta, a 25,0°C, un valore di pH pari a 11,30. Si calcoli, sempre a 25,0°, il valore del pH assunto dal sistema ottenuto aggiungendo a 100,0 mL della soluzione del sale 40,0 mL di una soluzione 0,25 M di HCl. Si considerino i volumi additivi. [pH=5,38] 4) Una soluzione acquosa 0,10 M di un acido debole monoprotico presenta, a 298,0 K, pH = 3,15. A 2,50 L di tale soluzione vengono aggiunti 24,0 mL di una soluzione acquosa di NaOH (7,50% in peso; d = 1,015g/mL). Considerando i volumi additivi, calcolare il pH della soluzione. [pH =4,65] Esercizi riassuntivi e integrativi 5) A 100,0 mL di una soluzione acquosa 0,10 M del sale di sodio, NaA, dell'acido debole monoprotico HA (per HA: Ka = 1,0·10−5 M) vengono aggiunte due porzioni successive, da 25,0 mL l'una, di una soluzione acquosa 0,20 M di HCl. Considerando i volumi additivi, si calcoli il pH del sistema prima dell'aggiunta dell'acido cloridrico e dopo ciascuna delle due aggiunte; si tenga presente che tutte le operazioni vengono condotte alla temperatura T= 298,0 K. [All’inizio: pH = 9,00; dopo la prima aggiunta: pH = 5,00 ; dopo la seconda aggiunta: pH = 3,09] 6) Alla temperatura di 298,0 K, a 50,0 mL di una soluzione acquosa 0,1 M di acido nitroso (Ka = 4,0·10−4M) vengono aggiunte due porzioni, ciascuna da 12,5 mL, di una soluzione acquosa 0,20 M di idrossido di sodio. Considerando i volumi additivi, calcolare il pH del sistema prima dell'aggiunta della base e dopo ciascuna delle due aggiunte. [pH iniziale: 2,21; pH dopo la prima aggiunta: 3,40; pH dopo la seconda aggiunta: 8,11] 7) Una soluzione acquosa 0,10 M di cianuro di potassio, KCN, presenta, a 298,0 K, pH = 10,95. Si calcoli, alla stessa temperatura, il pH presentato da una soluzione acquosa 0,20 M di acido cianidrico, HCN. [pH = 4,80] 8) Una soluzione acquosa 0,10 M di cloruro di ammonio, NH4Cl, presenta, a 298,0 K, pH = 5,13. Si calcoli, alla stessa temperatura, il pH presentato da una soluzione acquosa 0,20 M di ammoniaca, NH3. [pH =11,28] Esercizi riassuntivi e integrativi 9) Alla temperatura di 298,0 K, si è in possesso delle seguenti due soluzioni: (a) 100 mL di un acido debole HA (Ka = 1·10−4 M) di concentrazione 0,1 M; (b) 100 mL di idrossido di potassio di concentrazione 0,20 M; Considerando i volumi additivi, indicare quale volume della soluzione b) deve essere aggiunto a 50 mL della soluzione a) per ottenere un sistema finale il cui pH valga 4,00 [V = 12,5 mL] 10) Si abbiano, alla temperatura di 25,0°C, 300,0 mL di una soluzione acquosa contenente, contemporaneamente, 1,18g dell'acido debole monoprotico HA (PMHA=47,0 dalton) e 3,45g del suo sale di sodio. A questa soluzione si aggiungano 10,0 mL di una soluzione 1,0 M di HCl. Si calcoli il pH del sistema prima e dopo l'aggiunta dell'acido forte (si considerino i volumi additivi; per HA, Ka = 4,5·10−4 M). [pH prima dell’aggiunta: 3,65; pH dopo l’aggiunta: 3,40] 11) Una soluzione acquosa di KOH presenta un pH pari a 13,18. 230,0 mL di questa soluzione vengono aggiunti a 402,0 mL di una soluzione acquosa 0, 13M di cloruro di ammonio. Si calcoli il valore assunto dal pH del sistema dopo il mescolamento delle due soluzioni (considerare i volumi additivi; T = 298,0 K; per NH3: Kb=1,8·10−5) [pH = 9,56] Esercizi riassuntivi e integrativi 12) A 25,0°C , il pH di una soluzione acquosa 0,100M di acido fluoridrico (HF) è pari a 2,24. Si calcoli, alla stessa temperatura, il valore del pH di una soluzione acquosa 0,400 M di fluoruro di sodio NaF. [pH = 8,53] 13) Calcolare la variazione di pH che si ha in 2,00L di una soluzione contenente contemporaneamente NaBrO2 (0,40M) e HBrO2 (0,50 M) per l’aggiunta di 10,00 g di NaOH. (a T = 298 K; l’aggiunta dell’idrossido di sodio non causa variazioni nel volume della soluzione; per l’acido bromoso, HBrO2, Ka= 2,04·10−6 M). [pH iniziale: 5,59; pH finale: 5,84; ∆pH = 0,25] 14) Si abbiano, alla temperatura di 25,0°C, 250,0 mL di una soluzione acquosa contenente, contemporaneamente, 1,72g di HClO2 e 5,33g di KClO2. A questa soluzione si aggiungano 10,0 mL di una soluzione 1,0 M di NaOH. Si calcoli il pH del sistema prima e dopo I'aggiunta della base forte (si considerino i volumi additivi; per HClO2, Ka = 6,5 ·1 0−4 M) [pH iniziale: 3,49; pH finale: 3,78] 15) A 25,0°C, il pH di una soluzione acquosa 0,100 M di nitrito di potassio (KNO2) è pari a 8,17. Si calcoli, alla stessa temperatura, il valore del pH di una soluzione acquosa 0,250 M di acido nitroso (HNO2). [pH = 1,98] Esercizi riassuntivi e integrativi 16) Una soluzione acquosa di HCl presenta un pH pari a 1,20. 115,0 mL di questa soluzione vengono aggiunti a 90,0 mL di una soluzione acquosa 0,13 M di acetato di sodio (CH3COONa). Si calcoli il valore assunto dal pH del sistema dopo il mescolamento delle due soluzioni (considerare i volumi additivi; T = 298,0 K; per CH3COOH, Ka=1,8·10-5 M). [pH = 4,53] 17) Alla temperatura di 25,0°C , a 100,0 mL di una soluzione acquosa 0,100 M di un acido debole monoprotico vengono aggiunti 40,0 mL di una soluzione 0, 150 M di idrossido di sodio. Calcolare il valore del pH della soluzione risultante, sapendo che, per l’acido debole, Ka = 4,60·10−4 M. Si considerino i volumi additivi. [pH = 3,51] 18) La soluzione 0,10 M dell’acido debole monoprotico HA presenta, alla temperatura di 298,0 K, un valore del pH pari a 2,65. Considerando i volumi additivi, calcolare il volume di acqua che occorre aggiungere a 10,0 mL di tale soluzione perché il pH assuma il valore di 3,50. [V = 431,2 mL di acqua] 19) 100,0 mL di una soluzione 6,25·10-2 M dell'acido monoprotico debole HA (Ka=1,541·10−5 M) vengono aggiunte, a 25,0°C, due porzioni, ciascuna da 25,0 mL, di una soluzione 0, 125M di idrossido di sodio. Considerando i volumi additivi, si calcoli il pH del sistema prima e dopo l’aggiunta di ciascuna porzione della soluzione basica. [pH1 = 3,02; pH2 = 4,84; pH3=8,73] Esercizi riassuntivi e integrativi 20) A 100,0 mL di una soluzione acquosa di concentrazione 5,011·10−2 M dell'acido monoprotico debole HA vengono aggiunti 100,2 mg di idrossido di sodio solido, senza che si verifichino variazioni di volume. In tali condizioni, il sistema presenta, a 25,0°C, un valore del pH pari a 3,67. Si calcoli il valore del pH della soluzione iniziale dell'acido. [pH = 2,50] 21) Una soluzione acquosa di concentrazione 6,227·10−2 M dell'acido monoprotico debole HA presenta, a 25,0°C, un valore del pH pari a 2,94. A 50,0 mL di tale soluzione vengono aggiunti 70,3 mg di idrossido di potassio solido, senza che si verifichino variazioni di volume. Si calcoli il valore del pH del sistema in queste ultime condizioni. [pH = 4,49] 22) A 100,0 mL di una soluzione acquosa 0,10 M del sale di potassio, KA, dell'acido debole monoprotico HA (per HA: Ka = 5,0·10-6 M) vengono aggiunte due porzioni successive, da 20,0 mL l’una, di una soluzione acquosa 0,25 M di HCl. Si calcoli il pH del sistema prima dell'aggiunta dell'acido cloridrico e dopo ciascuna delle due aggiunte, tenendo presente che tutte le operazioni vengono condotte alla temperatura T = 298,0 K e considerando i volumi additivi. [pH1=9,15; pH3=5,30; pH3=3,22] Esercizi riassuntivi e integrativi 23) Una soluzione acquosa di idrossido di sodio (NaOH) presenta, alla temperatura di 25,0°C, pH=10,50. Si calcoli la quantità in peso di acido acetico (CH3COOH) che è necessario aggiungere a 125,0 mL di tale soluzione affinché il pH assuma il valore di 5,00. Si consideri invariato il volume del sistema dopo l'aggiunta dell'acido; per l’acido acetico Ka=1,8·10−5 M. [3,68 mg] 24) A 1,00 L di una soluzione acquosa contenente contemporaneamente 0,10 mol di NH3 e 0,100 mol di NH4Cl vengono aggiunte 10,0 mmol di HCl (il volume della soluzione resta invariato). Calcolare il pH del sistema tampone prima e dopo l’aggiunta dell’acido (Per NH3, Kb = 1,8·10−5 M; T = 298,0 K) [Prima pH=9,26; dopo pH=9,17] 25) Alla temperatura di 298,0 K, una soluzione 0,20 M dell’acido HA presenta un valore del pH pari a 2,40. A 15,0 mL di tale soluzione si aggiunge acqua fino ad avere un volume finale pari a 350,0 mL. Si calcoli il valore del pH della soluzione diluita dell'acido HA. [pH = 3,10] Esercizi riassuntivi e integrativi 26) Calcolare il pH di una soluzione 0,001 M di idrogenosolfato di potassio (KHSO4). La costante della seconda reazione di trasferimento protonico di H2SO4 vale Ka=1,2·10−2 M. [pH=3,03] 27) 200 mL di una soluzione di HCl 5,0 M vengono addizionati a 130 mL di una soluzione di NH3 (Kb=1,8·10−5 M) al 32% in peso, d=0,89 g/mL. Si calcoli il pH del sistema dopo il mescolamento. [pH=9,33] 28) Calcolare il volume di una soluzione 0,2M di NaOH che bisogna aggiungere a 100 mL di una soluzione 0,1 M di un acido debole monoprotico (Ka =2,6·10−5 M) per ottenere una soluzione a pH =5,8. [V=47,1 mL] 29) Il pH di una soluzione 0,2 M di cianuro di potassio (KCN) è pari a 11,30. Calcolare il volume di una soluzione concentrata di acido perclorico (HClO4, d=1,67 g/mL; 69,5% in peso) che bisogna aggiungere a 100 mL della soluzione di partenza per portarne il pH al valore di 9,60. [V=0,58 mL] Esercizi riassuntivi e integrativi 30) A 25,0 mL di una soluzione 0,2 M di un acido debole HA (pH = 2,78) vengono aggiunte due porzioni successive, ciascuna da 25,0 mL, di una soluzione 0,1 M di KOH. Si determini il valore del pH dopo ogni aggiunta della soluzione di base. [pH=4,86; pH=8,84] 31) Si dispone di una soluzione di NH3 (Kb =1,8·10-5 M) di pH pari a 11,28 e di una soluzione di HCl di pH=1,00. Si determini il rapporto dei volumi delle due soluzioni, perché, una volta mescolatele, il sistema assuma pH =9,5. [VNH3 /VHCl=1,378] 32) 500 mL di una soluzione 0,2 M di un acido debole monoprotico HA vengono addizionati di 800 mL di una soluzione 0,03 M di NaOH; il pH della soluzione risultante è pari a 2,80. Si calcoli il valore della costante di dissociazione, Ka, dell'acido debole. [Ka=5,00·10-4 M] 33) Mescolando campioni di stesso volume di soluzioni di pari concentrazione (0,001 M) di un acido debole monoprotico HA e di idrossido di potassio si ottiene un sistema il cui pH è pari a 8,70. Si calcolino: (a) la costante di dissociazione, Ka, di HA; (b) il pH della soluzione iniziale di HA. [Ka = 1,97·10-7 M, pH=4,85] 34) Calcolare a quale volume occorre diluire 100 mL di una soluzione 0,1 M di CH3COOH per ottenere una soluzione avente pH = 3,5. Calcolare inoltre la % in peso di una soluzione di KOH (densità = 1,15 g/mL), sapendo che se ne devono usare 20 mL per portare il pH della soluzione di acido acetico, dopo la diluizione, a 6,3. [Vdiluzione=1,8 L; %W KOH=2,37] Esercizi riassuntivi e integrativi 35) I vari tipi di candeggina altro non sono che soluzioni diluite (all’incirca al 5% in peso) di ipoclorito di sodio (NaClO). Qual è il pH di una soluzione di tale candeggiante che contenga ipoclorito di sodio in quantità pari a 50,0 g/L? La costante di dissociazione (Ka) dell’acido ipocloroso (HClO) è 3,1·10-8 M. [10,67] 36) Una soluzione di acetato di sodio (CH3COONa) ha pH=9,00. Calcolare quanta acqua bisogna aggiungere a 0,5 litri di questa soluzione affinché il pH diventi uguale ad 8,00. Per l’acido acetico (CH3COOH), Ka = 1,8·10−5 M. [V H2O=49,5 L] Chimica (a.a. 2010/2011, 6CFU) Equilibri ionici in soluzione acquosa (IV parte): Equilibri di solubilità Esercitazione Esercizi integrativi EQUILIBRI DI SOLUBILIT SOLUBILITÀ À Alcuni elettroliti forti (cioè quelli che si dissociano completamente in ioni) sono molto solubili: NaCl(s) → Na+(aq) + Cl-(aq) KOH(s) → K+(aq + OH (aq aq)) aq)) Alcuni elettroliti forti sono poco solubili: AgCl(s) ⇄ Ag+(aq) + Cl-(aq aq)) CaCO3(s) ⇄ Ca2+(aq) + CO32-(aq aq)) Ni(OH)2(s) ⇄ Ni2+(aq) + 2 OH-(aq aq)) 3Ca3(PO4)2(s) ⇄ 3 Ca2+(aq aq)) + 2 PO4 (aq aq)) dando origine a soluzioni sature o sovrasature ove si stabiliscono gli equilibri sopra riportati. Esempio di soluzione satura Si stabilisce l’equilibrio eterogeneo [solidoliquido (soluzione)]: Ca(OH)2(s) ⇄ Ca2+(aq + 2 OH aq)) (aq aq)) Ca2+(aq) + 2 OH-(aq) Ca(OH)2(s) All’equilibrio corrisponde una costante di equilibrio: K= 2 a Ca 2+ ⋅ a OH − a Ca(OH)2 che assumendo comportamento ideale della soluzione e attività unitaria per il solido diventa: 2+ − 2 K = [Ca ] ⋅ [OH ] –6 3 K = 4,68 · 10 M Visibile il corpo di fondo (il solido: idrossido di calcio) della soluzione Molti composti ionici sono, quindi, poco solubili o praticamente insolubili e instaurano un equilibrio con gli ioni costituenti nella soluzione satura. Ad esempio per il cloruro di argento si ha: AgCl(s) Ag+(aq) + Cl-(aq) cui corrisponde (ad ad un data temperatura) temperatura una costante di equilibrio Ks, detta prodotto di solubilità: + È una costante di un equilibrio eterogeneo dove assegniamo attività unitaria al solido − K s = [Ag ] ⋅ [Cl ] (KS è anche indicata come KPS o come KSP (testi americani) e raramente come PS) Altri esempi di sali poco solubili sono: PbI2(s) Ca3(PO4)2(s) Pb2+(aq) + 2I-(aq) K s = [Pb 2+ ] ⋅ [I − ]2 = 8,49 · 10 –9 M 3 3 Ca2+(aq) + 2 PO43-(aq) 2+ 3 3− 2 4 K s = [Ca ] ⋅ [PO ] = 2,07 · 10 –33 M 5 REGOLE QUALITATIVE DI SOLUBILITA’ NITRATI ED ACETATI di tutti i metalli sono SOLUBILI in acqua. L’Ag acetato è poco solubile. Tutti i Sali d’AMMONIO, POTASSIO e SODIO sono SOLUBILI in acqua. CLORURI, BROMURI e IODURI di tutti i metalli, ESCLUSO PIOMBO, ARGENTO E MERCURIO(I), sono SOLUBILI in acqua. Lo ioduro di mercurio(II), HgI2, è INSOLUBILE in acqua, mentre PbCl2, PbBr2 e PbI2 diventano solubili in acqua molto calda. I cloruri, bromuri e ioduri insolubili in acqua sono insolubili anche in ACIDI diluiti. i SOLFATI di tutti i metalli, ESCLUSO PIOMBO, BARIO, CALCIO E MERCURIO(I), sono SOLUBILI in acqua. Il solfato di Ag è poco solubile. I solfati insolubili in acqua sono insolubili anche in ACIDI diluiti. REGOLE QUALITATIVE DI SOLUBILITA’ CARBONATI, FOSFATI, BORATI, CROMATI ed ARSENIATI di tutti i metalli, escluso sodio, potassio ed ammonio, sono INSOLUBILI in acqua ma solubili in ACIDI diluiti. i SOLFURI di tutti i metalli, escluso magnesio, bario, calcio, sodio, ammonio e potassio, sono INSOLUBILI in acqua. BaS, CaS e MgS sono moderatamente solubili. IDROSSIDI di sodio, potassio e ammonio sono MOLTO SOLUBILI in acqua. Gli IDROSSIDI di calcio e bario sono moderatamente solubili. Ossidi e idrossidi di tutti gli altri metalli sono insolubili. Per calcoli quantitativi dobbiamo usare valori KS (prodotti di solubilità) che generalmente sono tabulati ad una certa temperatura (di solito quella standard 25° 25°C) Per esempio nel libro Nobile-Mastrorilli trovate:. TABELLA 15.3 Prodotti di solubilità dei principali sali insolubili. Altri esempi di sali poco solubili sono: 2CaCO3(s) ⇄ Ca2+(aq + CO aq)) 3 (aq aq)) 2+ 2− 3 K s = [Ca ] [CO ] a 25°C KS = 4,96 · 10–9 M2 2Ag2CO3(s) ⇄ 2 Ag+(aq + CO aq)) 3 (aq aq)) + 2 2− 3 K s = [Ag ] [CO ] a 25°C KS = 8,45×10-12 M2 Fe(OH)3(s) ⇄ Fe3+(aq aq)) + 3 OH (aq aq)) 3+ − 3 K s = [Fe ] [OH ] a 25°C KS = 2,64 · 10–39 M4 Si intende con solubilità il numero di moli di sale che si sciolgono per litro di soluzione. Essa è indicata con s ed è legata alle concentrazione dei vari ioni in cui si dissocia il sale dai coefficienti stechiometrici. Solubilità = concentrazione (molare: Solubilità (molare: mol· mol· L-1) del soluto che si scioglie nella soluzione, all’equilibrio, ad una certa temperatura. temperatura. Solubilità Solubilit à (S, in mol mol·· L-1) in acqua pura: Ag2CO3(s) ⇄ 2 Ag+(aq) + CO32-(aq) 2S S K S = 8,45 ⋅10 −12 M 3 = [Ag + ]2 ⋅ [CO 32− ] = (2S) 2 ⋅ S = 4S3 S=3 −12 Ks 8,45 ⋅ 10 =3 = 1,283 ⋅10 − 4 M 4 4 a 25°C Un altro esempio Solubilità Solubilit à ((S S, in mol· mol· L-1) in acqua pura del carbonato di calcio: CaCO3(s) ⇄ Ca2+(aq) + CO32-(aq) S S 2+ 2− 3 −9 K S = [Ca ] ⋅ [CO ] = 4,96 ⋅10 [M ] = S S = K S = 4,96 ⋅10 −9 2 −5 = 7,04 ⋅10 M a 25°C 2 Un altro esempio Solubilità Solubilit à (s, in mol mol·· L-1) in acqua pura del carbonato di calcio: Fe(OH)3(s) ⇄ Fe3+(aq) + 3 OH-(aq) S 3S KS=[Fe3+]·[OH-]3=2,64·10−39 M4 Solubilità a 25°C in acqua pura: K S = S ⋅ (3S) 3 = 27S4 −39 K 2,64 ·10 S=4 S =4 = 9,94 ⋅10 −11 M 27 27 Solubilità a 25°C in soluzione acida tamponata a pH=4, cioè pOH=10 e quindi [OH-]=10-10 M: ( K S = S ⋅ (10 −10 ) 3 = S ⋅ 1 ⋅10 −30 ) KS 2,64 ⋅10 −39 −9 S= = = 2 , 64 ⋅ 1 0 M −30 −30 1 ⋅10 1 ⋅10 Generalizzando Per il generico elettrolita (sale) AaBb poco solubile, per ogni sua mole che si dissocia in a mol di cationi Ab+ e in b mol di anioni Ba- abbiamo che : A a Bb (s) aA b+ (aq) +bB a− (aq) Concentraz. iniziali (Δ) Variaz. concentraz. --- -+aS -+bS Concentraz. Equilibrio -- aS bS S = solubilità molare (mol/L) di AaBb b+ a a− b K s = [A ] ⋅ [B ] = (aS) ⋅ (bS) a Dato tabulato (false dimensioni: Ma+b) a una certa temperatura (generalmente quella standard: 298,15 k) b mol a + b K s S = b a b ⋅a L Esercizio: Calcolo del Ks dalla solubilità – Calcolare il KS del PbI2 sapendo che la sua solubilità è di 1,285·10−3 mol/L. − PbI2(s) ⇄ Pb2+(aq aq)) + 2 I (aq aq)) Concentraz. iniziali Variaz. concentraz. Concentraz. Equilibrio - 0 +S S 0 +2 S 2 S KS=[Pb2+]·[I-]2 [Pb2+]=S [I-]=2S KS=[Pb2+]·[I-]2=S·(2S)2=4S3 KS=4S3 =4(1,285·10-3)3= 8,49·10-9 M3 Esercizio: Calcolo della solubilità dal KS – Calcolare la solubilità dell’AgCl sapendo che il suo Ks vale 1,77·10-10M − AgCl(s) ⇄ Ag+(aq + Cl aq)) (aq aq)) Concentraz. iniziali Variaz. concentraz. Concentraz. Equilibrio - KS=[Ag+]·[Cl-]= 1,77·10-10 M2=S⋅⋅S=S2 S = K S = 1,77 ⋅10 -10 = 1,33 ⋅10 -5 M 0 +S S 0 +S S S = KS solubilità = 1,33 ⋅10 −5 M Calcolo della solubilità dal Ks (un altro esempio) Calcolare la solubilità del CaF2 sapendo che il suo Ks vale 1,46·10-10 M3. CaF2(s) ⇄ Ca2+(aq) +2 F-(aq) Concentraz. iniziali Variaz. concentraz. Concentraz. Equilibrio - 0 +S S K S = [Ca 2+ ] ⋅ [F − ]2 = S·(2S)2 = 4S3 = 1,46 ⋅10 −10 M 3 Ks S= 4 3 −10 K 1 , 46 ⋅ 10 S=3 s =3 ≅ 3,32 ⋅10 −4 M 4 4 0 +2S 2S EFFETTO DELLO IONE COMUNE Consideriamo ora la solubilità di un sale poco solubile in una soluzione contenente uno dei due ioni implicati nell’equilibrio di solubilità, ad esempio, di PbI2(s) in una soluzione di NaI: In base al principio di Le Chatelier, l’equilibrio viene spostato verso sinistra dalla presenza di I- per cui possiamo prevedere che la solubilità in NaI sia inferiore a quella dell’acqua pura. Calcoliamo ad esempio la solubilità di AgBr, per il quale KS=5,56×10-13 M2 in una soluzione di NaBr 0,10 M. In soluzione l’NaBr (elettrolita forte) è dissociato completamente NaBr(s) → Na+(aq) + Br-(aq) per cui la soluzione è 0,10 M in Br- che uno ione in comune con l’ AgBr: Le concentrazioni all’equilibrio vanno calcolate da: AgBr(s) Concentraz. iniziali Variaz. concentraz. Concentraz. Equilibrio - Ag+(aq) + Br-(aq) 0 +S S 0,10 +S 0,10+S K S = 5,56 ⋅10 −13 = [Ag + ]·[Br − ] = S·(0,10 + S) Si noti che S è molto piccola per cui 0,10+S≈0,10 K S = S ⋅ 0,10 KS 5,56 ⋅10 −13 S= = = 5,8 ⋅10 −12 M 0,10 0,10 Si noti come la solubilità è inferiore a quella in acqua pura: S = K S = 5,56 ⋅10 −13 = 7,46 ⋅10−7 M EFFETTO DELLO IONE COMUNE MgF2(s) ⇄ Mg2+(aq) +2 F-(aq) K S = [Mg 2+ ] ⋅ [F − ]2 = S·(2S)2 = 4S3 = 7,42 ⋅10 −11 M 3 −11 K 7 , 42 ⋅ 10 S=3 s =3 ≅ 2,65 ⋅10 − 4 M 4 4 Se aggiungo NaF la solubilità a mano a mano diminuisce …… Per esempio [F-]=0,040 M K S = [Mg 2+ ] ⋅ [F − ]2 = S·(2S + 0,040) 2 ≅ S·(0,040)2 = = 7,42 ⋅10 −11 M 3 KS 7,42 ⋅10 −11 S= = = 2 0,10 (0,040) = 4,6375 ⋅10 −8 M in acqua pura EFFETTO DELLO IONE COMUNE Soluzione sovrassatura di AgCl Soluzione sovrassatura di AgCl dove è stata aggiunta una aliquota di una soluzione di nitrato di argento (AgNO3 sale solubile di argento: AgNO3 → Ag+ +NO3- ) La quantità di precipitato aumenta Precipitazione Consideriamo due soluzioni di sali solubili contenenti ciascuna uno dei due ioni che partecipano ad un equilibrio di solubilità di un sale poco solubile. Prendiamo ad esempio una soluzione di AgNO3 (che è solubile e dissocia negli ioni Ag+ e NO3-) ed una di NaCl (che è solubile e dissocia negli ioni Na+ e Cl- ): se le mescoliamo abbiamo nella soluzione risultante la presenza degli ioni Ag+ e Cl – che partecipano all’equilibrio di solubilità: AgCl(s) ⇄ Ag+(aq) + Cl−(aq) Se le concentrazioni [Ag+] e [Cl –] subito dopo il mescolamento sono sufficientemente elevate si ha formazione immediata nel corpo della soluzione di AgCl solido: la soluzione diventa improvvisamente torbida per la presenza di particelle di AgCl in sospensione e solo dopo un certo tempo il solido si deposita sul fondo lasciando in soluzione concentrazioni [Ag+] e [Cl –] compatibili con l’equilibrio. Tale fenomeno è noto come precipitazione precipitazione. Formazione di precipitati (Precipitazione Precipitazione)) NaCl(s) → Na+(aq) + Cl-(aq) Na+(aq) Cl-(aq) AgNO3(s) → Ag+(aq) + NO3-(aq) AgCl(s) ⇄ Ag+(aq) + Cl-(aq) Ks =1,77·10-10 M Ag+(aq) NO3-(aq) Na+(aq) Cl-(aq) Ag+(aq) NO3-(aq) Na+(aq) Cl-(aq) Ag+(aq) NO3-(aq) AgCl((s) ⇄ Ag+(aq) + Cl-(aq) AgCl Esempio: aggiunta di una soluzione di NaCl ad una di AgNO3 NaCl AgNO3 AgCl NaCl(aq) + AgNO3(aq) → AgCl(s) + NaNO3(aq) In forma ionica: Ag+(aq + Cl aq)) (aq aq)) → AgCl(s) Altro esempio: aggiunta di una soluzione di K2CrO4 ad una di Pb(NO3)2 Pb(NO3)2 K2CrO4 PbCrO4 Altro esempio: aggiunta di una soluzione di K2CrO4 ad una di Pb(NO3)2 K2CrO4 (aq) + Pb(NO3)2(aq) → PbCrO4 (s) + 2 KNO3 (aq) In forma ionica: 2Pb2+(aq + CrO aq)) 4 (aq aq)) → PbCrO4(s) (s) Altro esempio: aggiunta di una soluzione di Pb(NO3)2 ad una di NaI Pb(NO3)2 NaI PbI2 Altro esempio: aggiunta di una soluzione di Pb(NO3)2 ad una di NaI 2 NaI (aq) + Pb(NO3)2(aq) → PbI2 (s) + 2 NaNO3 (aq) In forma ionica: Pb2+(aq + 2 I aq)) (aq aq)) → PbI2(s) (s) Per determinare se si ha o no precipitazione in seguito al mescolamento delle due soluzioni, si fa uso del criterio generale per determinare in che direzione procede una reazione per raggiungere l’equilibrio a partire da concentrazioni iniziali note di reagenti e prodotti: Se QS > KS la reazione procede verso sinistra Se QS < KS la reazione procede verso destra Se QS = KS la reazione è all’equilibrio QS corrisponde ad un prodotto ionico che ha la stessa espressione del KS, tuttavia le concentrazioni dei due ioni non sono quelle all’equilibrio ma quelle della soluzione subito dopo il dal mescolamento. Ad esempio per la solubilità di AgCl: KS= [Ag+]eq[Cl-]eq QS = [Ag+]0[Cl-]0 Questa situazione corrisponde a mescolare i due prodotti della reazione di equilibrio di solubilità e determinare in che direzione si sposta la reazione per raggiungere l’equilibrio. Ad esempio per l’equilibrio di solubilità: AgCl(s) ⇄ Ag+(aq) + Cl-(aq) poiché solo uno spostamento a sinistra di tale reazione corrisponde alla formazione di AgCl(s) cioè alla precipitazione, si deriva immediatamente il seguente criterio: Se QS > KS si ha precipitazione Se QS ≤ KS non si ha precipitazione Le concentrazioni da inserire in QS sono quelle dopo il mescolamento e si ottengono a partire da quelle delle due soluzioni mescolate tenendo conto della diluizione. diluizione. Esempio – Se mescoliamo 1,0 litri di una soluzione 0,10 M di Pb(NO3)2 e 1,0 litri di una soluzione 0,20 M di NaCl, si avrà precipitazione di PbCl2? (Ks=1,6·10-5 M) Poiché il PbCl2 dissocia secondo l’equazione: PbCl2(s) ⇄ Pb2+(aq) + 2 Cl-(aq) i due ioni da considerare nelle due soluzioni sono Pb2+ e ClIl Pb(NO3)2 è solubile e dissocia completamente negli ioni Pb2+ e NO3per cui nella prima soluzione [Pb2+]=0,10 M. Analogamente nella seconda soluzione [Cl-]=0,20 M. Per prima cosa calcoliamo le concentrazioni di questi due ioni nella soluzione risultante dopo il mescolamento. I volumi delle due soluzioni mescolate sono V1=V2=1,0 litri e quindi dopo il mescolamento il volume finale è: Vfin= V1+V2= (1,0+1,0) L= 2,0 L Nella soluzione iniziale di Pb(NO3)2 la concentrazione degli ioni Pb2+ è 0,10 M e quindi ci sono 0,10 mol di tale ione. Dopo il mescolamento, lo stesso numero di moli è diluito nel volume finale di 2,0 litri e quindi la sua concentrazione sarà: [Pb2+]=0,1 mol/2,0 L=0,05 M Analogamente la concentrazione degli ioni Cl- dopo il mescolamento sarà: [Cl-]=0,2 mol/2,0 L=0,1 M Si calcola QS per la reazione di equilibrio di solubilità: QS=[Pb2+][Cl-]2=(0,05)·(0,10)2=5,0·10-4 M2 e lo si confronta con il valore dato KS=1,6×10-5 M2. Poichè QS>KS, la reazione è spostata verso sinistra e quindi si ha la precipitazione di PbCl2. Man mano che PbCl2 precipita le concentrazioni di Pb2+ e Cldiminuiscono fino a che il loro prodotto ionico diventa uguale a KS e la precipitazione si arresta. Può essere interessante stabilire quanto dello ione Pb2+ inizialmente presente rimane in soluzione dopo la precipitazione e quanto invece è precipitato come PbCl2. Siccome Pb2+ e Cl- sono stati mescolati in quantità stechiometriche nessuno dei due è in eccesso e si ha [Pb2+]=½[Cl-]=x=ovvero S e quindi all’equilibrio: KS=[Pb2+]·[Cl-]2=(S)·(2S)2=4S3 -5 K 1,6 ⋅ 10 [Pb 2+ ] = S = 3 S = 3 = 1,6 ⋅10 -2 M 4 4 che corrisponde ad un numero di moli pari a: [Pb2+]× ×Vfin= 1,6·10-2 M·2 L=0,032 mol Le moli di Pb2+ inizialmente presenti erano 0,1 per cui ne sono precipitate: 0,068 × 100 = 68% 0,1-0,032=0,068 mol cioè il 0,10 La massa effettiva di PbCl2 precipitata è m PbCl2 = n PbCl2 ⋅ PM PbCl2 = 0,068 mol ⋅ 278,1 (g/mol ) = 18,9108 g Da un punto di vista pratico a che serve conoscere i prodotti di solubilità (KS), ad esempio: intonaci e pietre Risale ad oltre due millenni fa la scoperta delle capacità “adesive” delle malte: La reazione si fa avvenire a T=800-900˚C (è una reazione endotermica) e poiché è una reazione di equilibrio bisogna allontanare la CO2 gassosa per ottenere una conversione completa CaCO3 calore (T=800-900°C) CaO + CO2 La calce viva (ossido di calcio, CaO) lentamente! anche 9-12 mesi +CO2 la calce spenta reagisce con la CO2 dell’aria e si ritrasforma in carbonato di calcio +H2O reazione molto esotermica Ca(OH2) calce spenta (idrossido di calcio Ca(OH)2) che può succedere? • malte, intonaci, pietre calcaree, sono quindi costituite di (o contengono) carbonato di calcio: SO2 da combustione + ossidazione (aria: O2+N2) CaCO3 SO3 in acqua CaSO4 + CO2 +H2O CaSO4·2H2O gesso che può succedere? Il calcare (CaCO3) e il gesso (CaSO4·2H2O; cioè solfato di calcio più due molecole di acqua di idratazione) hanno solubilità diversa (il gesso è circa 100 volte più solubile del calcare): CaCO3(s) ⇄ Ca2+ (aq) + CO32-(aq) K S = [Ca 2+ ] ⋅ [CO 32− ] = 4,96 ⋅10 −9 [M 2 ] = S2 S = K S = 4,96 ⋅10 −9 = 7,04 ⋅10 −5 M CaSO4(s) ⇄ Ca2+(aq) + SO42-(aq) K S = [Ca 2+ ] ⋅ [SO 24− ] = 7,1 ⋅10 −5 [M 2 ] = S2 S = K S = 7,1⋅10 −5 = 8,43 ⋅10 −3 M perciò: il gesso viene portato via dall’acqua piovana molto più facilmente che può succedere? statua danneggiata gesso formatosi in una matrice di marmo (solfatazione del marmo) il solfato di calcio (gesso) è meccanicamente fragile; inoltre assorbe acqua causando una espansione dei volumi con seri danni alla struttura di marmo Esercitazione Dal libro Nobile-Mastrorilli (Cap 15: Equilibri in soluzione: pH e prodotto di solubilità), oltre a studiare gli esercizi svolti, dovete essere in grado di svolgere i seguenti problemi: Ks e solubilità (pag. 541-543) 1, 2, 3, 4, 5, 6, 7, 8. Inoltre studiare le dispense tratte dal libro Stechiometria per la chimica generale di Paola Michelin Lausarot e Gian Angelo Vaglio Cap 17 e svolgere i relativi esercizi. Infine svolgere gli esercizi integrativi riportati alla fine di questa lezione. Si dissocia in Cu+ e SCN− (anione isotiocianato) Periodato di torio (lV) Svolgimento (punto d): Ag 2SO 4 (s) Eq) 2 Ag + (aq) + SO 24- (aq) 2S K S = [Ag + ]2 ⋅ [SO 24− ] = 1,20 ⋅10 −5 S –5 K 1,2 · 10 K s = [2S]2 ⋅ [S] ⇒ S = 3 S = 3 ≅ 1,442 ⋅10 − 2 [mol ] L 4 4 mol S' mg = S ⋅ PM Ag 2SO 4 L L mg g ⋅ 1000 mol g mg mol g mg 1000 4497 S' mg = 1,442 ⋅10 − 2 ⋅ 311,7936 ⋅ ≅ mol L L g L Terminare l’esercizio da soli (punti a,b,c,e,f,g) prendendo le KS dalla Tab.15.3 Calcolare la solubilità espressa in mol/L del diioduro di piombo (PbI2): (a) in acqua pura; (b) in una soluzione 0,06 M di ioduro di potassio (sale solubile) -9 3 Sapendo che K S = 8,49 ⋅ 10 (mol/L) . Svolgimento: PbI 2 ( s ) (punto a): i) → ← Pb 2+ ( aq ) + 2I − ( aq ) -S Eq) -2S K s = 8,49 ⋅ 10 (mol/L) = S ⋅ (2S) -9 (punto b): i) Eq) PbI 2(s) 3 → ← K S = 8,49 ⋅ 10 -9 (mol/L)3 2 -9 K 8,49 ⋅ 10 ⇒ S=3 s =3 = 1,285 ⋅10 −3 (mol/L) 4 4 Pb 2+ (aq) + 2I − (aq) -S 0,06 0,06 + 2S 2S <<< 0,06 ⇔ 0,06 + 2S ≈ 0,06 K s = [Pb 2+ ] ⋅ [I − ]2 = 8,49 ⋅ 10 −9 [S] ⋅ [0,06]2 = 8,49 ⋅ 10 −9 8,49 ⋅ 10 −9 −6 ⇔ S= = 2,36 ⋅ 10 (mol/L) 2 [0,06] L’approssimazione 2S << 0,06 (mol/L) è verificata. Calcolare la solubilità del PbCl2 (espressa in g/L) in una soluzione 0,10 M di AlCl3 (sale solubile) sapendo che K S, PbCl 2 = 1,17 ⋅ 10 -5 (mol/L)3 Svolgimento: PbCl 2 ( s ) i) Eq) → ← Pb 2+ ( aq ) + 2 Cl - ( aq ) N.B. : -- ma S 0,3 0,3 + 2S [AlCl3 ] = 0,1 M = [Al3+ ] [Cl ] = 3 ⋅ [Al ] = 0,3 M − 2S <<< 0,3 ⇔ 3+ 0,3 + 2S ≈ 0,3 K s = [Pb 2+ ] ⋅ [Cl − ]2 = 1,17 ⋅ 10 −5 = [S] ⋅ [0,3]2 = 1,17 ⋅ 10 −5 1,17 ⋅ 10 −5 −4 ⇔ S= = 1,3 ⋅ 10 (mol/L) 2 [0,3] L’approssimazione 2S << 0,3 (mol/L) è verificata. mol mol g g g S = 5,41 ⋅10 −3 ⋅ PM PbCl 2 = 1,3 ⋅10 − 4 ⋅ 278,106 ≅ 3,6154 ⋅10 − 2 L L mol L L Svolgimento: a) Pb(IO 3 ) 2 (s) 2+ Pb (aq) + 2 IO 3− S K s = [S] ⋅ [2S]2 (aq) K s = [Pb 2+ ] ⋅ [IO3− ]2 2S –13 K 3,68 · 10 ⇒ S=3 s =3 = 4,51 ⋅10 −5 [mol ] L 4 4 mg mol g mg S' = S ⋅ PM Pb(IO3 ) 2 ⋅1000 ⋅ mL g L mol 1 ml 1000 L = mg 1 mol g mg = 4,51⋅10 ⋅ 557,0054 ⋅ 1000 ⋅ ≅ 0 , 0251 mol mL g 1000 ml L L −5 b) HIO3 a pH=2 significa che [H3O+]=[H+]=10-pH HIO3 è un acido forte: [H + ] = [IO3- ] = 10 − pH = 10 −2 = 0,01 M 2+ Pb (aq) + 2IO 3- (aq) Pb(IO 3 ) 2 (s) K s = [Pb + + ] ⋅ [IO3- ]2 i) - 0,01 eq) s 0,01+2s S << 0,01 ⇒ 0,01 + 2S ≈ 0,01 K s = [S] ⋅ [0,01] 2 Ks 3,68 · 10 –13 – 9 mol ⇒ S= = = 3,68 · 10 [ ] 2 –4 L 1 · 10 [0,01] mg mol S' mg = S ⋅ PM ⋅ 1000 ⋅ Pb(IO3 ) 2 ml g L 1 mL 1000 L = mg 1 mol g −6 = 3,68 ⋅10 −9 ⋅ 557,0054 ⋅ 1000 ⋅ ≅ 2 , 05 ⋅ 10 mol g 1000 ml L L L’approssimazione S << 0,01 (mol/L) è verificata mg mL Svolgimento: a) Ag 2 Cr2 O 7 (s) + 2 Ag (aq) + Cr2 O 72- (aq) K s = [Ag + ]2 ⋅ [Cr2 O 72- ] = ? 2S S K s = [2S] ⋅ [S] 2 mg S' 100 mL mol S = = mg 1 g L PM 1000 ⋅ ⋅ Ag 2 Cr2 O 7 g 1000 ml mol L mg 12,96 12,96 100 ml mol = = = 3,002 ⋅10 − 4 mg 1 431,7238 ⋅100 L g 431,7238 ⋅1000 ⋅ g 1000 ml mol L K s = [2S]2 ⋅ [S] −4 2 −4 K s = [2 ⋅ 3,002 ⋅10 ] ⋅ [3,002 ⋅10 ] = 1,082 ⋅10 b) Ag 2 Cr2 O 7 (s) 2 Ag + (aq) + Cr2 O i) 0,1 eq) 0,1+2S −10 mol L 3 27 (aq) -S S << 0,1 ⇒ 0,1 + 2S ≈ 0,1 Ks 1,082 ⋅10 −10 −8 K s = [0,1] ⋅ [S] ⇒ S = = = 1 , 082 ⋅ 10 [mol/L] 2 –2 [0,1] 1 · 10 2 mol S' g = S ⋅ PM Ag 2Cr2O7 L L = 1,082 ⋅10 −8 g mol = L’approssimazione S << 0,1 mol/L è verificata mol g −6 4 31 , 7238 4 , 671 10 ⋅ = ⋅ L mol g L Calcolare la solubilità, in moli/litro, di Cr(OH)3 in acqua pura e in una soluzione tamponata a pH = 4,0, sapendo che KS dell’idrossido di cromo(III) è uguale a 1,0·10−30 M4. Svolgimento: K s = [Cr + ] ⋅ [OH − ]3 = 1,0 ⋅10 −30 M 4 In acqua pura Cr(OH) 3(s) ← → 3+ (aq) Cr + 3 OH S 3S K s = [S] ⋅ [3S]3 = 27S4 − (aq) −30 4 ⋅ K 1,0 10 M s =4 ≅ 1,387 ⋅10 −8 M S=4 27 27 In una soluzione tamponata a pH = 4,0 pH = 4 ⇒ pOH = 10 ⇒ [OH − ] = 1,0 ⋅10 − pOH = 1,0 ⋅10 −10 M i) eq) Cr(OH) 3(s) ← → 3+ − Cr(aq) + 3 OH (aq) -1,0·10−10 S 1,0·10−10 [S] ⋅ [1,0 ⋅10 −10 ]3 = 1,0 ⋅10 −30 M 4 La soluzione tampone fa sì che rimanga costante S = 1,0 M Svolgimento (punto d): Il problema esplicitamente ci dice che avremo un precipitato, tuttavia dimostriamolo [ZnCl2 ] = 1,466 mol 400 mL di cloruro di zinco 1,466 M [Zn ] 2+ ZnCl2 (aq) → Zn 2+ (aq) + 2Cl − (aq) L mol = 1,466 L [Cl ] = 2 ⋅1,466 mol L − mol [KF] = 0,75 L 600 mL di fluoruro di potassio 0,75 M [K ] = 0,75 mol L + KF(aq) → K + (aq) +F − (aq) [F ] = 0,75 mol L − Formalmente è come se avvenisse la reazione: ZnCl2 + 2 KF → ZnF2 + 2 KCl Ma l’unico sale insolubile è il fluoruro di zinco (vedi tabella 15.3) La reazione vera è quella in forma ionica : Zn 2+ (aq) +2F − (aq) → ZnF2 ( s ) Che è l’inverso dell’equilibrio di solubilità: ZnF2 ( s ) → ← Zn 2+ (aq) + 2 F− (aq) Si ha precipitato se K S = [ Zn 2+ ] ⋅ [F− ]2 = 3,04 · 10 –2 M 3 QS = [ Zn 2+ ]0 ⋅ [F− ]02 > K S = 3,04 · 10 –2 M 3 N.B.: Concentrazioni dopo la diluizione Dobbiamo calcolarle con la formula delle diluizioni (Mi·Vi= Mf·Vf): [Zn 2+ ]0 = n Zn 2+ Vfinale,soluzione = VZnCl2 ⋅ [ ZnCl 2 ] VZnCl2 + VKF = 0,40 (L) ⋅1,466 (mol/L) 0,5864 mol = = 0,5864 (mol/L) 0,40 (L) + 0,60 (L) 1,00 L [F− ]0 = n F− Vfinale,soluzione = VKF ⋅ [KF] 0,600 (L) ⋅ 0,75 (mol/L) 0,45 mol/L = = = 0,45 (mol/L) VKF + VZnCl2 0,60 (L) + 0,40 (L) 1,00 L QS = [Zn 2+ ]0 ⋅ [F− ]02 = 0,5864 (mol/L) ⋅ [0,45 (mol/L)]2 = 1,18746 · 10 –1 M 3 > K S = 3,04 · 10 –2 M 3 Si ha formazione di precipitato …… ma quanto ne precipita? Possiamo ragionare in moli o con le concentrazioni dopo la diluizione (visto che il volume è lo stesso adesso: 1 litro) Moli I) ∆) Tot) Conc I) Eq) Zn 2+ (aq) + 2 F− (aq) → ZnF2 ( s ) 0,45 − 0,45 − + 0,225 0,3614 0 0,225 [Zn 2+ ] = 0,3614 mol = 0,3614 M L 0,5864 − 0,225 ZnF2 ( s ) → ← Zn 2+ (aq) + 2 F − (aq) 0,3614 − 0,3614 + S 2S Moli precipitate a cui tuttavia dobbiamo sottrarre quelle che si risolubilizzano per l’equilibrio K S = [ Zn 2+ ] ⋅ [F− ]2 = 3,04 · 10 –2 M 3 = (0,3614 + S) ⋅ ( 2S) 2 Pongo in prima approssimazione: Purtroppo S è confrontabile con 0,3614 0,3614 + S ≅ 0,3614 3,04 · 10 –2 mol S= = 0,145 0,3614 ⋅ 4 L 3,04 · 10 –2 = (0,3614) ⋅ ( 2S) 2 Piuttosto che risolvere l’equazione di 3° grado in S scelgo di usare un metodo iterativo –2 3,04 · 10 = (0,3614 + 0,145) ⋅ ( 2S') 3,04 · 10 –2 mol S' = = 0,1225 (0,3614 + 0,145)⋅ 4 L 3,04 · 10 –2 = (0,3614 + 0,1225) ⋅ ( 2S' ') 3,04 · 10 –2 mol S' ' = = 0,1253 (0,3614 + 0,1225) ⋅ 4 L 2 2 3,04 · 10 = (0,3614 + 0,1253) ⋅ ( 2S' ' ') –2 2 3,04 · 10 –2 mol S' ' ' = = 0,125 (0,3614 + 0,1253) ⋅ 4 L n ZnF2 ridisciolt e = S' ' '⋅Vsoluzione = 0,125 (mol/L ) ⋅1[L] = 0,125 mol converge n ZnF2 precipitat e effettive = 0,225 mol − 0,125 mol = 0,100 mol m ZnF2 precipitat o = 0,100 mol ⋅ PM ZnF2 = 0,100 mol ⋅103,39 g = 10,339 g mol Terminare l’esercizio da soli (punti a,b,c) prendendo KS dalla Tab.15.3 Esercizi riassuntivi e integrativi I) Il carbonato di calcio, CaC03, utilizzato dai sistemi viventi per costruire le conchiglie, si deposita in strati calcarei che, dopo compressione e riscaldamento, portano alla formazione del marmo. Determinare la solubilità in mg/L del carbonato di calcio sapendo che il suo KS è pari a 8,7·10-9 M2. [R. 9,34 mg/L] II) Calcolare la solubilità dell'idrossido rameico (in g/L) in soluzione acquosa tamponata a pH 7,0. Il prodotto di solubilità dell'idrossido rameico è di 2,2·10-20. [2,1·10-4 g/L] III) Calcolare la solubilità (in g/L) dell’idrossido ferroso Fe(OH)2 in una soluzione 0,1 M di NaOH con KS=1,52·10−14 M3. [1,36·10-10 g/L ] IV) Il solfato rameico è un efficace alghicida, e talora viene aggiunto all’acqua delle piscine per evitare la crescita delle alghe. Quale deve essere il pH massimo dell’acqua di una piscina per evitare la precipitazione dell'idrossido rameico (KS,Cu(OH)2 = 1,6·10-19 M3) quando si aggiungono 4,0 g di solfato rameico pentaidrato (CuSO4·5 H2O) ogni 10 litri d'acqua? [pH=6,0] V) Il fosfato tricalcico (KS, Ca3(PO4)2= 1,3·10-32 M5) è uno dei componenti più comuni dei calcoli renali (altri sali comuni in queste affezioni sono: carbonato di calcio, ossalato di calcio, fosfato ammonico magnesiaco). Qual è la concentrazione, in grammi/litro, di fosfato tricalcico nelle urine di un paziente che abbia calcoli renali di questo tipo? [5,1·10-5 g/L] Esercizi riassuntivi e integrativi VI) Molte acque naturali contengono sali di ferro provenienti dai vari minerali ferrosi attraverso cui le acque sono filtrate. Nella maggioranza dei casi le acque naturali sono leggermente alcaline. L'idrossido ferrico (di colore giallastro) è talmente poco solubile che lo ione ferrico è presente nelle acque naturali come idrossido ferrico solido finemente disperso (stato colloidale). In zone dove il contenuto di ferro nell'acqua è moderatamente elevato, non è raro trovare acque di colore giallognolo a causa della torbidità dovuta all'idrossido ferrico. Quale valore massimo di pH dovrebbe avere un’acqua naturale che contenga 33 mg/L di ione ferrico affinché non si formi precipitato di idrossido ferrico? Il prodotto di solubilità dell'idrossido ferrico è di 6·10-38 M4. [2,67] VII) L'applicazione dei raggi X in medicina è estremamente diffusa. L'osservazione delle cavità interne del corpo richiede il riempimento di tali cavità con materiali di contrasto più opachi o più trasparenti ai raggi X rispetto ai tessuti circostanti. Il solfato di bario (BaSO4), che è un sale insolubile opaco ai raggi X (KS = 1,1·10-10 M2), viene impiegato negli esami del tratto gastrointestinale. Lo ione bario però è tossico, per cui è necessario, onde diminuirne la solubilità per effetto dello ione comune, aggiungere alla sospensione di solfato di bario un eccesso di ioni solfato. Nella preparazione di una sospensione acquosa di solfato di bario viene aggiunta una quantità di epsomite (solfato di magnesio eptaidrato: MgSO4·7H2O, molto solubile), tale da fornire una concentrazione 0,015 M in ioni magnesio. Qual è la concentrazione molare di ioni bario liberi in questa sospensione? Quale sarebbe stata se non si fosse aggiunta epsomite? [7,3·10-9 M; 1,0·10-5 g/L] Esercizi riassuntivi e integrativi 1) Una soluzione satura di cloruro di piombo(II) contiene, a 20°C, 4,5 g di sale per litro. Si calcoli il valore del prodotto di solubilità del cloruro di piombo a 20°C. [KS=1,69·1O−5 M3] 2) A 25°C, il prodotto di solubilità del cromato di argento (Ag2CrO4) vale KS=1,9·1O−12 M3. Si calcoli la massa in grammi di cromato di argento che si scioglie in 2,8 L di a) acqua, b) una soluzione 0,250 M di nitrato di argento (AgNO3) e c) una soluzione 0,250 M di cromato di sodio (Na2CrO4). [a) 7,25·1O-2 g, b) 2,824·1O-8 g, c) 1,281·1O-3 g] 3) Si calcolino le concentrazioni degli ioni stronzio e solfato presenti nella soluzione ottenuta mescolando 2,0 L di una soluzione 0,10 M di cloruro di stronzio con 0,50 L di una soluzione 0,40 M di solfato di sodio. Alla temperatura dell'esperienza, il prodotto di solubilità del solfato di stronzio vale KS=7,6·1O−7M2. ([Sr2+]= [SO42-] =8,72·10-4 M) 4) Una soluzione viene preparata sciogliendo 1,00 g di nitrato di argento (AgNO3) e 1,50 g di cromato di potassio (K2CrO4) in tanta acqua da avere un volume finale di soluzione pari a 25,0 mL. Sapendo che, alla temperatura dell'esperienza, il prodotto di solubilità del cromato di argento vale KS=1,9·1O-12 M3, si calcoli la concentrazione di tutte le specie ioniche presenti nella soluzione. ([NO3-]=0,235 M, [K+]=0,618 M, [CrO42-]= 0,191 M, [Ag+]=3,16·1O-6 M] 5) Ad una certa temperatura, il prodotto di solubilità del cloruro di piombo(II) vale KS=1,70·10-5 M3. Si calcoli la solubilità del sale a) in acqua pura e b) in una soluzione 3,0 M di acido cloridrico, esprimendola in g/L. [a) 4,51 g/L; b) 5,25·10−4 g/L] 6) La solubilità del carbonato di calcio in acqua vale S=7,04 mg/L, a 25°C. Si calcoli la perdita in peso di un precipitato di carbonato di calcio se questa viene lavato a) con 1,0 L di acqua; b) con 1,0 L di una soluzione 0,02 M di carbonato di sodio. [a) 7,04 mg, b) 2,48·10−2 mg] Esercizi riassuntivi e integrativi 7) Il prodotto di solubilità del bromato di argento vale, a 25°C, Ks = 5,8 . 1O−5M2. Si calcoli la concentrazione degli ioni presenti in una soluzione ottenuta mescolando 40,0 mL di una soluzione 0,02 M di nitrato di argento (AgNO3) con 60,0 mL di una soluzione 0,04 M di bromato di potassio (KBrO3). ([K+]=2,4·1O-2 M; [NO3-]=8,0·10-3M; [Ag+]=3,03 ·10-3M; [BrO3-]=1,87·10-3M] 8) Quando 50,O mL di una soluzione 4,0·10-2M di LaCl3 sono mescolati con 50,0 mL di una soluzione 9,6 ·10-2 M di NaIO3 si ottiene un precipitato di La(IO3)3 del peso di 663,6 mg. Si calcoli il valore del prodotto di solubilità dello iodato di lantanio e la solubilità della sua soluzione satura. [Ks = 5,8·10-8 M4; sol. sat.: S= 6,81·10-3 M] 9) 500,0 mL di una soluzione 0,10 M di nitrato di argento vengono mescolati con 1500,0 mL di una soluzione 0,10 M di cromato di potassio. Si ha la precipitazione di cromato di argento, il cui prodotto di solubilità , alla temperatura dell'esperienza, vale Ks = 1,0·10-12M3. Si calcoli la concentrazione dello ione argento rimasto nella soluzione. ([Ag+]=4·10-6 M) 10) Si mescolano 150 ml di una soluzione 0,4 M di MnCl2 con 230 ml di una soluzione 0,2 M di K2S. Calcolare i grammi di MnS che precipitano e le concentrazioni degli ioni Mn2+ e S2− dopo il mescolamento. Per MnS, KS = 3,2·10−11 M2. [mMnS : 4,002 g; [ Mn2+]=0,037 M; [S2−]= 8,64 ·10−10 M] 11) Calcolare quanti grammi di Ca3(PO4)2 che ha KS=1,4 ·10−30 M5 si sciolgono in 100 mL di una soluzione 0,1 M di CaCl2. [5,8·10−13g] 12) Si mescolano 120 mL di una soluzione 0,15 M di Pb(NO3)2 con 250 mL di una soluzione 0.,03M di K2CrO4. Sapendo che per PbCrO4 KS=1,77·10−14 M2, trovare i grammi di cromato di piombo che precipitano e le concentrazioni residue di Pb2+ e di CrO42− dopo la precipitazione. [mPbCrO4 =2,42 g;[ Pb2+] = 2,842 M; [CrO42−]=6,23·10−13 M] Chimica (a.a. 2011/2012, 6CFU) Elettrochimica: • • • • • • • • • • • • • • • trasformazione di energia chimica in elettrica e viceversa (Elettrochimica) brevi cenni sulla conducibilità degli elettroliti (conduttori di II specie) il potere ossidante la pila Daniell ed il ponte salino schematizzazione di una pila misura del potenziale di elettrodo e forza elettromotrice potenziale standard di riduzione E° la serie elettrochimica l’equazione di Nernst le pile da un punto di vista termodinamico la f.e.m. e la dimostrazione della legge di Nernst tipi di elettrodi :1a specie e 2a specie le pile di concentrazione Esercitazione sulle pile. Esercizi riassuntivi e integrativi ELETTROCHIMICA TRASFORMAZIONE DELL’ENERGIA CHIMICA (PROVENIENTE DA REAZIONI SPONTANEE) IN ENERGIA ELETTRICA (“ ELETTRICITÀ”). TRASFORMAZIONE DELL’ENERGIA ELETTRICA IN ENERGIA CHIMICA (REAZIONI NON SPONTANEE) PILE ELETTROLISI (CELLE GALVANICHE o VOLTAICHE ) (CELLE ELETTROLITICHE) Brevi cenni sulla conducibilità degli elettroliti Classificazione di conduttore Conduttori di I specie (metallici) Meccanismo di conduzione Spostamento di elettroni All’aumentare della temperatura la conducibilità diminuisce Conduttori di II specie (elettrolitici) Spostamento di ioni positivi e negativi All’aumentare della temperatura la conducibilità aumenta I sistemi elettrochimici Sono costituiti da (almeno) due elettrodi (conduttori di 1a specie) e da un’elettrolita (conduttore 2a specie). Quando il sistema elettrochimico è percorso da corrente elettrica, questa è corrente elettronica negli elettrodi e nel circuito esterno, ed è corrente ionica nell’elettrolita. • Nel primo caso i portatori di carica elettrica sono gli elettroni, nel secondo sono ioni. • Contemporaneamente, agli elettrodi avvengono reazioni elettrochimiche (redox = ossidoriduzioni). Due esperimenti: (1) Nessuna reazione Cu Cu(ss) + Zn2+(aq) Cu( aq) → Cu2+(aq) aq) + Zn( Zn(s) Zn2+ (2) Cu2+ Zn D’altro canto, avviene la seguente reazione : Cu2+(aq aq)) + Zn Zn((s) → Cu( Cu(ss) + Zn2+(aq aq)) potere ossidante di Cu2+/Cu > potere ossidante di Zn2+/Zn Cu Zn H2 H+ H+ 2 H+(aq aq)) + Cu(s Cu(s) → H2(g) + Cu2+(aq aq)) 2 H+(aq aq)) + Zn(s Zn(s) → H2(g) + Zn2+(aq aq)) pot. oss. Cu2+/Cu > pot. oss. H+/H2 > pot. oss. Zn2+/Zn Cu Ag+ Zn Ag+ 2 Ag+(aq aq)) + Cu(s Cu(s) → 2 Ag(s Ag(s) + Cu2+(aq aq)) Ag+/Ag Cu2+/Cu H+/H2 Zn2+/Zn Scala qualitativa del potere ossidante Cu2+(aq) + Zn(s) → Cu(s) + Zn2+(aq) Zn(s) → Zn2+(aq) + 2 e- ossidazione (zona anodica) Cu2+(aq) + 2 e- → Cu(s) riduzione (zona catodica) Zn e- Zn2+ Cu Cu2+ Se provo a collegare le due barrette metalliche si verifica un flusso di elettroni e quindi una corrente elettrica (PILA). Tale corrente, tuttavia, cesserà immediatamente di esistere. Perché ? Innanzitutto, dobbiamo tener conto che (1) la che è (2) la che è soluzione di ioni Zn2+ è una soluzione di ZnSO4, elettrolita forte (α α=1) totalmente dissociata in Zn2+ e SO42soluzione di ioni Cu2+ è una soluzione di CuSO4, elettrolita forte (α α=1) totalmente dissociata in Cu2+ e SO42- Quando gli elettroni passando dall’elettrodo di Zn a quello di Cu, neutralizzano gli ioni Cu2+ della soluzione che istantaneamente si depositano come atomi neutri sull’elettrodo stesso. D’altra parte all’elettrodo di Zn, al quale sono stati sottratti elettroni, manda in soluzione altri ioni Zn2+ per reintegrare gli elettroni perduti, ma anche questi nuovi elettroni passano nell’elettrodo di Cu ove neutralizzano altri ioni Cu2+ , che si depositano; e così via ……….. Il descritto meccanismo va però completato: infatti quanto detto comporta che, appena chiuso il circuito esterno, la soluzione anodica sia carica positivamente (formazione di ioni Zn2+ ) e quella catodica sia carica negativamente (scomparsa di ioni Cu2+): ciò deve arrestare il funzionamento della pila, perché risultano impedite, per azione elettrostatica, sia l’ulteriore formazione di ioni Zn2+ che l’ulteriore scarica di ioni Cu2+. Zn + + + ++ Eccesso di cariche positive dovute a ioni Zn2+ Cu -- -Eccesso di cariche negative dovute a ioni SO42- In qualche modo, perché continui a fluire corrente (a funzionare la pila), dobbiamo far in modo che gli ioni di Zn2+ migrino a destra e gli ioni SO42- a sinistra. Ciò significa che dobbiamo mettere in contatto le due soluzioni: ma così non avremmo più una pila perché si verificherebbe la reazione di ossidoriduzione !!!!! Questo fenomeno è aggirato tramite l’utilizzo di un setto poroso (membrana) oppure un ponte salino. Quest’ultimo è costituito da una soluzione concentrata di un sale, avente lo scopo di mantenere l’elettroneutralità delle due soluzioni durante il funzionamento della cella galvanica. Riassumendo: in assenza di un ponte salino una cella galvanica (detta indifferentemente: cella voltaica o pila voltaica o cella galvanica o semplicemente pila) non può funzionare perché genererebbe due soluzioni non elettroneutre: difetto di ioni SO42Zn(s) + Cu2+(aq) eccesso di ioni SO42Zn2+(aq) + Cu(s) Una importante convenzione da ricordare L’elettrodo dove avviene la riduzione si dice catodo ed in una pila è il polo positivo (+) L’elettrodo dove avviene la ossidazione si dice anodo ed in una pila è il polo negativo (-) Voltmetro Zn - + Cu ∆E = 0 Cu2+ Zn2+ Cl- ← ponte salino → K+ Il sistema (elettrochimico) costituito da due elettrodi immersi i due opportune soluzioni separate fra loro da un setto poroso (o un ponte salino), prende il nome di pila elettrica (o cella galvanica o cella voltaica) ed ha una straordinaria importanza teorica e pratica. UNA CELLA GALVANICA: la pila Daniell Zn voltmetro semicella semicella Zn2+/Zn Cu2+/Cu 2e- + Cu2+ Zn2+ + 2e- catodo anodo Zn + Cu2+ Zn2+ + Cu Cu La pila Daniell: ponte salino Reazione all’anodo = ossidazione: Reazione al catodo = riduzione: Zn(s) → Zn2+(aq) + 2e- Cu2+(aq) + 2e- → Cu(s) La pila Daniell: Setto poroso (membrana) Riepilogando:le due versioni della Pila Daniell e- e- V V (-) Zn Zn2+(aq) + SO42-(aq) (+) Setto poroso (-) Cu Zn Cu2+(aq) + SO42-(aq) Cu2+(aq) + 2 e- → Cu(s) Riduzione Zn(s) → Zn2+(aq) + 2 eZn(s) + Cu2+(aq) → Zn2+(aq) + Cu(s) Zn2+(aq) + SO42-(aq) Catodo (polo +) Ossidazione Ponte salino (+) Cu Cu2+(aq) + SO42-(aq) Zn Anodo (polo -) Reazione totale della pila Cu2+(aq) Schematizzazione di una pila: Indicare SEMPRE la polarità (+; -) Metallo M() Composti allo stato solido e/o gassoso (indicare P) M() Composti in soluzione (Cmolare) ponte salino o membrana Ad esempio, la Pila già incontrata (Daniell): Zn(−) Cu(+) ++ Zn + + (C Zn ++ ) Cu + + (CCu ) oppure ( −) Zn Composti allo stato solido e/o gassoso (indicare P) Composti in soluzione (Cmolare) Zn(−) oppure (+) [Zn + + ] = x M [Cu ++ ] = x M Cu Cu(+ ) [Zn + + ] = x M [Cu + + ] = x M oppure: (−) Zn [Zn + + ] = x M [Cu + + ] = x M Cu (+) Misura del potenziale di elettrodo e Forza elettromotrice Per misurare il potenziale di un elettrodo bisognerebbe disporre di uno strumento da inserire all’interfaccia tra la fase solida (metallo) e quella liquida (soluzione): ciò è, ovviamente, impossibile. Poiché il valore assoluto del potenziale dell’elettrodo (e della relativa coppia redox) non può essere conosciuto, bisogna assegnargliene uno relativo, riferito a una coppia alla quale viene attribuito arbitrariamente il valore zero: a tale scopo si deve costruire una pila con l’elettrodo in esame e con quello della coppia di riferimento e misurarne la differenza di potenziale, con uno strumento, il potenziometro, che consente di ottenerne il valore senza che la pila eroghi corrente. Questa differenza di potenziale è la massima che si possa ottenere tra i due elettrodi e prende il nome di forza elettromotrice (f.e.m.): se durante la misura la pila erogasse corrente, come avviene quando si usa un comune voltmetro, si avrebbe una caduta di tensione e il valore misurato sarebbe inferiore alla fem. Indicando con ∆E la forza elettromotrice e con EC ed EA i potenziali del catodo (+) e dell’anodo (–), si può scrivere ∆E = EC – EA o, con la notazione che indica il segno dell’elettrodo: ∆E = E(+) – E(–) Più tardi, torneremo sul significato termodinamico di forza elettromotrice. Necessità di poter confrontare quantitativamente il potere ossidante o riducente di una qualsivoglia coppia redox! Elettrodo standard a idrogeno Elettrodo di riferimento: è stato deciso di misurare la tendenza riducente o ossidante di ciascuna coppia redox nei confronti di questo elettrodo standard. Potenziale standard di riduzione E°: Ox + ne - → Red Potenziale relativo alla semireazione di riduzione. Si costruisce una scala di potenziali di riduzione rispetto al potenziale dell’elettrodo standard a idrogeno. Condizioni standard: la temperatura è di 25°C (298,15 K); tutte le specie in soluzione hanno concentrazione 1 M; tutte le specie gassose hanno pressione di 1 atm; tutti i solidi ed i liquidi puri hanno attività unitaria. Elettrodo standard di riferimento: H+(aq) + e- → ½ H2(g) E°H /H = 0 V + H2(1 atm) 2 Pt 25 25°°C = 298 K [H+]= 1 M; pH=0 Potenziale standard di riduzione: riduzione: Cu2+(aq) + 2 e- → Cu(s) E°= + 0,342 V H2(1 atm) Cu Pt 25 25°°C [Cu2+]=1,0 M pH=0 H2(g) + Cu2+(aq) → 2 H+(aq) + Cu(s) ∆E = E C - E A = E°Cu 2+ /Cu − E°H + /H = 0,342V ⇒ E°Cu 2+ /Cu = ∆E + E°H + /H = 0,342V + 0V ⇒ E°Cu 2+ /Cu = +0,342V 2 2 Potenziale standard di riduzione: Zn2+(aq) + 2 e- → Zn(s) H2(1 atm) pH=0 E°= - 0,762 V Zn Pt 25° 25°C [Zn2+]=1,0 M 2 H+(aq) + Zn(s) → H2(g) + Zn2+(aq) ∆E = E C - E A = E° H + /H − E° Zn 2+ /Zn = 0,762V ⇒ E° Zn 2+ /Zn = E° H + /H − ∆E = 0V - 0,762V ⇒ E° Zn 2+ /Zn = −0,762V 2 2 Potenziale standard di riduzione: NO3-(aq) + 3 e- + 4 H+(aq) → NO(g) + 2 H2O(l) H2 (1 atm) Pt Pt 25°°C 25 pH=0 E°°= + 0,957 V NO (1 atm) [NO3-] = 1,0 M pH = 0 2NO3-(aq) + 3H2 + 2 H+(aq) → 2NO(g) + 4 H2O(l) ∆E = E C - E A = E° NO - /NO − E°H + /H = 0,957V ⇒ E° NO - /NO = ∆E + E°H + /H = 0,957V + 0V ⇒ E° NO - /NO = +0,957V 3 2 3 2 3 La serie elettrochimica dei potenziali standard di riduzione (25°C) Semireazione E° (V) Semireazione E° (V) F2(g) + 2e- ⇔ 2F- +2,866 2H+(aq) + 2e- ⇔ H2(g) +0,000 PbO2(s) + SO42-(aq) + 4H+ + 2e- ⇔ PbSO4(s) + H2O +1,691 Sn2+(aq) + 2e- ⇔ Sn(s) -0,130 HClO2(aq) + 3H+(aq) + 3e- ⇔ Cl-(aq) + 2H20 +1,560 Ni2+(aq) + 2e- ⇔ Ni(s) -0,257 +1,507 Co2+(aq) + 2e- ⇔ Co(s) -0,280 +1,455 PbSO4(s) + 2e- ⇔ Pb(s) + SO42-(aq) -0,360 +1,498 Cd2+(aq) + 2e- ⇔ Cd(s) -0,403 Fe2+(aq) + 2e- ⇔ Fe(s) -0,447 Cr3+(aq) + 3e- ⇔ Cr(s) -0,744 Zn2+(aq) + 2e- ⇔ Zn(s) -0,762 - MnO4 (aq) + 8H+ 5e- (aq) + + 2e- PbO2(s) + 4H+ (aq) Au3+ 3e- ⇔ Au(s) (aq) - + BrO3 (aq) + Cl2 (g) + O2(g) + 6H+ 2e- 4H+ ⇔ (aq) + Cl- (aq) + ⇔ ⇔ 5e- Mn2+ Pb2+ (aq) (aq) + 4H20 + 2H2O ⇔ Br2(l) + 3H2O (aq) 4e- ⇔ 2H2O +1,482 +1,358 +1,229 Br2 (aq) + 2e- ⇔ 2Br-(aq) +1,066 NO3-(aq) + 4H+(aq) + 3e- ⇔ NO(g) + 2H2O +0,962 2H2O(aq) + 2e- ⇔ H2(g) + 2OH-(aq) -0,828 Ag+(aq) + e- ⇔ Ag(s) +0,800 Mg2+(aq) + 2e- ⇔ Mg(s) -2,372 Fe3+(aq) + e- ⇔ Fe2+(aq) +0,771 Na+(aq) + e- ⇔ Na(s) -2,710 I2(s) + 2e- ⇔ 2I-(aq) +0,536 Ca2+(aq) + 2e- ⇔ Ca(s) -2,868 Cu+(aq) + e- ⇔ Cu(s) +0,521 K+(aq) + e- ⇔ K(s) -2,931 Cu2+(aq) + 2e- ⇔ Cu(s) +0,342 Li+(aq) + e- ⇔ Li(s) -3,040 Sul Nobile-Mastrorilli la trovate alla tabella 16.1 di pag. 558 0,268 Per la pila Daniell abbiamo,ad esempio, in condizioni standard: ∆E = E − E = 0,342 − (−0,762) = 1,104 V o + o − - + Zn [Zn2+]=1,0 M Ossidazione (anodo) Cu 25 25°°C [Cu2+]=1,0 M Riduzione (catodo) Curiosità: la pila Daniell fu la prima cella galvanica ad essere prodotta su scala industriale per essere utilizzata come generatore di energia elettrica necessaria al funzionamento delle prime linee telegrafiche e telefoniche. Per calcolare il potenziale di un elettrodo non allo stato standard (allo stato attuale) si usa l’equazione di Nernst (legge di Nernst): ν0xOx + ne– → νRedRed Potenziale attuale Concentrazione molare della specie ossidata Potenziale standard RT [Ox ] E = E° + ln ν Red nF [Red ] ν Ox Walter Hermann Nernst Briesen 1864 – Berlino 1940 Premio Nobel per la Chimica 1920 A 25°C: Numero degli elettroni scambiati nella semireazione Costante di Faraday [ 0,0591 Ox] E = E° + log 10 ν Red n [Red ] ν Ox Concentrazione molare della specie ridotta νOx coefficiente stechiometrico di Ox; νRed coefficiente stechiometrico di Red Vediamo le pile da un punto di vista termodinamico (interpretazione termodinamica delle pile) Zn H2 [H+] =1 M Zn ( s ) + 2 H + ( aq ) → Zn + + ( aq ) + H 2 ( g ) + Q Zn( s ) → Zn + + (aq ) + 2e − 2 H + (aq ) + 2e − → H 2 ( g ) Se R → ∞ Q ≠ Q1 Zn ( s ) + 2 H + ( aq ) → Zn + + ( aq ) + H 2 ( g ) + Q1 Perché? Che significa da un punto di vista termodinamico? Per la reazione di ossidoriduzione Zn ( s ) + 2 H + ( aq ) → Zn + + ( aq ) + H 2 ( g ) + Q ∆U = Q − L vale Nella Pila dove L é solo lavoro di tipo PV : L = P∆V Zn ( s ) + 2 H + ( aq ) → Zn + + ( aq ) + H 2 ( g ) + Q1 ∆U = Q1 − L − Lutile dove Lutile = Lelettrico Se R→∞ significa che I≈0, vale a dire circuito aperto: condizioni di reversibilità, Q1 diventa un differenziale esatto dQ1 = TdS ⇔ Q1 = T ∆ S ∆ U = T ∆ S − P ∆ V − Lelettrico − Lelettrico = ∆ U + P ∆ V − T ∆ S − Lelettrico = ∆ H − T ∆ S − Lelettrico = ∆ G = − Lutile Lelettrico = carica × differenza di potenziale = q ⋅ V [Coulomb × Volt = Joule ] La costante di Faraday F è la carica di una mole di elettroni, F=96485 C/mol di e-; (F/NA)=(96485 C/mol/6,022·10+23 elettrone/mol)=1,602177·10-19C: carica di un elettrone. L’energia chimica massima della pila che può essere convertita in energia (lavoro) elettrica è la variazione di energia libera di Gibbs (∆ ∆G): ∆G = – n·F·∆ ∆E dove n·F = q è la carica e ∆E è la differenza di potenziale (V) a circuito aperto che è la cosiddetta forza elettromotrice (acronimo: f.e.m) e rappresenta la massima d.d.p. ottenibile da una pila. f.e.m. (acronimo di forza elettromotrice) Si definisce forza elettromotrice (f.e.m.) di una pila la differenza di potenziale (d.d.p.) MASSIMA che può esistere tra i due elettrodi di una pila (a resistenza infinita, cioè a circuito aperto: corrente zero). Essa misura la tendenza della reazione ad avvenire (SPONTANEITÀ). Si può misurare con un opportuno strumento che si chiama POTENZIOMETRO. Dimostrazione della legge di Nernst: Si considerino le due coppie redox Ox1/Red1 e Ox2/Red2; se il potenziale E1 della coppia 1 è maggiore di quello E2 della coppia 2, la reazione spontanea tra la forma ossidata Ox1 della coppia ossidante e quella ridotta Red2 della coppia riducente è Ox1 + Red2 → Red1 + Ox2 Se n1 e n2 sono gli elettroni in gioco nelle due semireazioni, queste possono essere scritte nella forma: Ox1 + n1 e– → Red1 Red2 → Ox2 + n2 e– e, indicando con n il minimo comune multiplo tra n1 e n2 e con a e b due numeri primi tra loro tali che: a n1 = b n2 = n la reazione bilanciata è a Ox1 + b Red2 → a Red1 + b Ox2 Dimostrazione della legge di Nernst: a Ox1 + b Red 2 → a Red1 + b Ox2 ∆G = ∆G ° + RT ln Q < 0 ∆G = −nF∆E Red 1 ] ⋅ [Ox 2 ] [ Q= [Ox1 ]a ⋅ [Red 2 ]b a b ∆G° = −nF∆E ° RT [Red 1 ] ⋅ [Ox 2 ] ∆E = ∆E ° − ln a b nF [Ox1 ] ⋅ [Red 2 ] a b [ 2,3 ⋅ RT Red1 ] ⋅ [Ox 2 ] ∆E = ∆E° − log 10 a b nF [Ox1 ] ⋅ [Red 2 ] a b [ 0,0591 Red 1 ] ⋅ [Ox 2 ] ∆E = ∆E° − log n [Ox1 ]a ⋅ [Red 2 ]b a b 0,0591 [ Red 1 ] 0,0591 [ Ox 2 ] E1 − E 2 = (E°1 − E°2 ) − log − log a b n n [Ox1 ] [Red 2 ] a a 0,0591 [ Ox1 ] E1 − E 2 = E°1 + log a n [ ] Red 1 0,0591 [ Ox1 ] E1 = E°1 + log n [Red 1 ]a b 0,0591 [ Ox 2 ] − E° 2 + log b n [ ] Red 2 [ 0,0591 Ox 2 ] E 2 = E° 2 + log n [Red 2 ]b a 0,0591 [Ox1 ] E1 = E°1 + log n [Red 1 ] b b a 0,0591 [Ox 2 ] E 2 = E° 2 + log n [Red 2 ] b [Ox1 ] 0,0591 E1 = E°1 + log [ Red 1 ] n a [ Ox 2 ] 0,0591 log E 2 = E°2 + [ n Red 2 ] b a n1 = b n2 = n [ 0,0591 Ox1 ] E1 = E°1 + log [Red1 ] n1 [ 0,0591 Ox 2 ] E 2 = E°2 + log [Red 2 ] n2 Quindi, in generale, per la generica coppia RedOx: il potenziale di riduzione attuale è: Ox + ne– → Red 0,0591 [ Ox] E = E° + log [Red ] n e− o ancor più in generale: ν0xOx + ne– → νRedRed dove • n= numero di elettroni scambiati nella semireazione • νOx coefficiente stechiometrico di Ox • νRed coefficiente stechiometrico di Red E = E°Ox/Red a 0,0591 + log 10 n a ν Ox Ox ν Red Red Come di solito, ai liquidi ed ai solidi puri assegniamo attività unitaria; per i gas ammettiamo un comportamento ideale e, quindi, all’attività sostituiamo la pressione; anche per le soluzioni ammettiamo un comportamento ideale e, quindi, sostituiamo all’attività la concentrazione molare. ν Ox 0,0591 [Ox] E = E°Ox/Red + log 10 ν Red n [Red] Esempio di applicazione della legge di Nernst: MnO4-(aq) + 8 H+(aq) + 5 e- → Mn2+(aq) + 4 H2O(l) E MnO − /Mn 2+ = E°MnO − /Mn 2+ 4 4 E MnO − /Mn 2+ = E°MnO − /Mn 2+ 4 4 E MnO − /Mn 2+ 4 E°°=1,491 V a MnO − ⋅ a 8H + 0,0591 4 + log10 5 a Mn 2+ ⋅ a 4H 2 O = 1 0,0591 [MnO −4 ] ⋅ [ H + ]8 + log 10 2+ 5 [Mn ] 0,0591 [MnO 4− ] ⋅ [ H + ]8 = 1,491 + log 10 2+ 5 [Mn ] Altri esempi di applicazione della legge di Nernst: H+(aq) + e- → ½ H2(g) E°= 0 V [H + ] [H + ] E = 0 + 0,0591 ⋅ log 10 1/ 2 = 0,0591 ⋅ log 10 1/ 2 PH 2 PH 2 Cl2(g) + 2 e- → 2 Cl-(aq) E°= + 1,358 V PCl 2 0,0591 E = 1,358 + log 10 2 [Cl − ]2 PbO2(s) + 4 H+(aq) + 2 e- → Pb2+(aq) + 2 H2O(l) E°=1,460 V 0,0591 [H + ]4 E = 1,460 + log 10 2 [Pb 2+ ] NO3-(aq) + 3 e- + 4 H+(aq) → NO(g) + 2 H2O(l) [NO 3− ][H + ]4 0,0591 E = 0,960 + log10 3 PNO E°= + 0,960 V Un’altra utilità della legge di Nernst: ∆G° == −nF ∆E° Nota la differenza di potenziale tra due coppie coniugate redox è possibile prevedere la variazione di energia libera –∆ ∆G°= RT ln K ∆G°/RT K = e –∆ K = e nF∆∆E°/RT sostituendo i valori delle costanti e passando dalla base naturale a quella decimale: K = 10 n∆E ° 0,0591 ove n = m.c.m. della reazione (globale) redox Nota la differenza di potenziale standard tra due coppie coniugate redox è possibile prevedere la condizione di equilibrio, cioè è possibile calcolare la costante (K) termodinamica di equilibrio. Tipi di elettrodi (semielementi) Elettrodi di 1a SPECIE: se entrambe le forme (ossidata e ridotta) della coppia redox, o almeno una di essa sono in soluzione. Il potenziale attuale (E) di un tale elettrodo dipende dalla concentrazione di entrambe le forme (o almeno da una delle due). Elettrodi di 2a SPECIE: se entrambe le forme (ossidata e ridotta) della coppia redox sono un solido oppure un liquido insolubile; quasi tutte le coppie che vengono usate per questi elettrodi sono costituite da un metallo (M), un suo sale poco solubile (MmAn) e l’anione (Am-) di quest’ultimo introdotto in soluzione da un elettrolita forte solubile. Il potenziale attuale (E) di un tale elettrodo dipende dalla concentrazione dell’anione Anpoiché le concentrazioni (attività) di solidi puri o liquidi puri sono poste per convenzione unitarie. Elettrodi di 1a specie: Un elettrodo è di prima specie se entrambe le forme (ossidata e ridotta) della coppia redox, o almeno una di esse, sono in soluzione. Esempio, coppia redox costituita da un metallo e dai suoi ioni: M n+ (aq) EM n+ − + ne → M (s) /M = E° M n+ /M 0,0591 n+ + ⋅ log [M ] n Elettrodi di 1a specie: Un elettrodo è di prima specie se entrambe le forme (ossidata e ridotta) della coppia redox, o almeno una di esse, sono in soluzione. Esempio, coppia redox che presenta sia la forma ossidata che quella ridotta in soluzione: Fe 3+ (aq) − + e → Fe 2+ (aq) E Fe3+ / Fe 2+ = E°Fe3+ / Fe 2+ Pt 2+ 3+ Fe (C Fe 2+ ), Fe (C Fe3+ ) 3+ 0,0591 [Fe ] + ⋅ log 1 [Fe2+ ] Altri esempi di elettrodi di 1a specie: gli elettrodi a gas possiamo considerarli come elettrodi di prima specie Pt H +(aq) + e - → 1 H 2 (g) 2 E° = 0 V (P in atm) H 2 (g) H + (C H + ) [H + ] [H + ] E = E° + 0,0591 ⋅ log 10 1 / 2 = 0,0591 ⋅ log 10 1 / 2 PH 2 PH 2 1 2 Cl2(g) + e - → Cl-(aq) E = E° + 0,0591 ⋅ log 10 1 2 Pt E° = 1,358 V (P in atm) Cl2 (g) Cl− (CCl − ) PCl1 22 − [Cl ] O 2 + 2 H + + 2 e – → H 2O = 1,358 + 0,0591 ⋅ log 10 E° = 1,229 PCl1 22 [Cl − ] Pt (P in atm) O2 (g) H + (C H + ) 0,0591 0,0591 12 + 2 ⋅ log PO 2 ⋅ [H ] = 1,229 + ⋅ log PO1 22 ⋅ [H + ]2 E = E° + 2 2 Elettrodi di 2a specie Un elettrodo è di seconda specie se sia la forma ossidata che quella ridotta è un solido insolubile; quasi tutte le coppie che vengono usate per questi elettrodi sono costituite da un metallo (M), un suo sale poco solubile (MmAn) e l’anione (Am-) di quest’ultimo introdotto in soluzione da un elettrolita forte solubile. Esempio 1: l’elettrodo ad argentoargento-argento cloruro − AgCl (s) + e → Ag (s) + Cl E AgCl/Ag,Cl − = E°AgCl/Ag,Cl − Ag − (aq) 0,0591 1 + ⋅ log 1 [Cl− ] AgCl (s) Cl− (C Cl − ) − E AgCl/Ag,Cl − = 0,222 − 0,0591 ⋅ log [Cl ] In una soluzione satura di KCl: E AgCl/Ag,Cl − = 0,198 V Esempio 1: l’elettrodo ad argentoargento-argento cloruro Ag AgCl (s) Cl− (C Cl − ) Dato che il potenziale varia al variare della concentrazione di KCl (cioè Cl-), questa deve essere nota (solitamente soluzione satura circa 4,6M) Elettrodi di 2a specie Esempio 2: l’elettrodo a calomelano 1 2 − Hg 2Cl2 (s) + e → Hg (s) + Cl E Hg Cl 2 2 /Hg,Cl E Hg Cl 2 - = E°Hg Cl 2 − (aq) 2 /Hg,Cl - Hg − Hg 2Cl2 (s) Cl (C Cl − ) 0,0591 1 + ⋅ log 1 [Cl− ] − 2 /Hg,Cl = 0,268 − 0,0591 ⋅ log [Cl ] In una soluzione satura di KCl: E Hg Cl 2 2 /Hg,Cl - = 0,244 V Esempio 2: l’elettrodo a calomelano Nelle misure potenziometriche raramente è utilizzato un elettrodo a idrogeno (difficoltà operative). Un elettrodo di riferimento ideale deve avere un potenziale noto, costante e indipendente dalla soluzione da analizzare. Un elettrodo di riferimento ideale deve inoltre essere robusto e facile da assemblare. Hg Hg 2Cl2 (s) Cl− (C Cl − ) Ponte salino nelle pile con un elettrodo di 2a specie In una pila con entrambi gli elettrodi di seconda specie, la forma ossidata della coppia ossidante e quella ridotta della coppia riducente, entrambe solide e confinate ai due elettrodi, non possono venire a contatto diretto tra loro: in questi casi non è, quindi, necessario il ponte salino per separare fisicamente le due semicelle. Se dei due elettrodi uno è di seconda specie e l’altro di prima, invece, il ponte salino può essere necessario o inutile: ciò dipende dalla possibilità, legata al loro stato fisico, che la forma ossidata della coppia ossidante e quella ridotta della coppia riducente vengano o meno a contatto tra loro e si scambino direttamente gli elettroni mettendo in corto circuito la pila. Per verificare quale delle due circostanze si verifica e, quindi, per determinare se occorre o meno usare il ponte salino, bisogna scrivere la reazione di ossidoriduzione nella direzione in cui avviene spontaneamente, partendo dai potenziali delle due coppie. Ponte salino nelle pile con un elettrodo di 2a specie Esempio 1. Pila costruita con le coppie H+/H2 (E° = 0,000 V) AgCl / Ag, Cl- (E° = 0,222 V) − (+) AgCl (s) + e − → Ag (s) + Cl(aq) 1 (− −) H 2 (g) → H + (aq) + e − 2 1 − + + H (aq) AgCl (s) + H 2 (g) → Ag (s) + Cl(aq) 2 Pt (−) H 2 (g) (PH 2 = 1 atm ) H + (pH = 0) Ponte salino NON NECESSARIO Ag (+) − Cl(aq) (1,0 M) AgCl (s) Ponte salino nelle pile con un elettrodo di 2a specie Esempio 2. Pila costruita con le coppie H+/H2 (E° = 0,00 V) AgI / Ag, I- (E° = -0,15 V) (− −) − Ag (s) + I (aq) → AgI (s) + e − (+ +) 1 H (aq) + e → H 2 (g) 2 + − Ag (s) + I − (aq) + H + (aq) I − (aq) 1 → AgI (s) + H 2 (g) 2 Pt (+) Ag (−) AgI (s) Ponte salino NECESSARIO (1,0 M) H + (pH = 0) H 2 (g) (PH 2 = 1 atm) Noi,tuttavia, considereremo sempre l’uso del ponte salino nei nostri esercizi esercizi.. Un caso particolare: Pile di concentrazione (+) M n + (s) + n e − → M (s) E C = E° M n+ /M + ( −) [ ] 0,0591 0,0591 ⋅ log M n + = E° M n+ /M + ⋅ log C1 n n M (s) → M n + (s) + n e − E A = E° M n + /M + M (−) M n + (C 2 ) [ ] 0,0591 0,0591 ⋅ log M n + = E° M n + /M + ⋅ log C 2 n n M (+) M n+ (C1 ) ∆E = E C − E A = E°M n+ /M + 0,0591 0,0591 C 0,0591 ⋅ log C1 − E°M n+ /M + ⋅ log C 2 = ⋅ log 1 n n n C2 0,0591 C1 ∆E = E C − E A = ⋅ log n C2 Le pile di concentrazione, ad esempio: 0,059 E = Eo + log10 [ Ag + ] 1 [ Ag + ]1 C1 ∆E = 0,0591 ⋅ log10 = 0,0591 ⋅ log10 + C2 [ Ag ] 2 C1 = C2 pila scarica C1 > C2 catodo a destra C1 < C2 catodo a sinistra Un particolare esempio: pila di concentrazione con elettrodi ad idrogeno in cui [H+]1> [H+]2 (vale a dire pH1 < pH2) Il pH-metro • Il pH-metro misura il pH di una soluzione usando un elettrodo che risponde alla concentrazione di H+. • L’elettrodo produce un voltaggio proporzionale alla conc. di H+. • Il voltaggio viene convertito in misura di Deve essere calibrato prima pH su un display. dell’uso utilizzando soluzioni standard a pH noto (vale a dire: soluzioni tampone). Il pH-metro Esercitazione Dal libro Nobile-Mastrorili, oltre agli esercizi svolti e commentati, potete risolvere gli esercizi: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 alle pagine 617-621 N.B. Per la risoluzione degli esercizi del libro Nobile-Mastrorili utilizzare anche i dati riportati nella tabella 16.1. Si considerino i volumi additivi e le pressioni dei gas che gorgogliano sugli elettrodi pari a 1 atm. Infine svolgere gli esercizi riassuntivi e integrativi riportati alla fine di questa lezione. Considerare i volumi additivi invece le pressioni dei gas che gorgogliano sugli elettrodi sono indicate volta per volta. I potenziali standard sono indicati esercizio per esercizio Esercizio N.2 di pagina 617 Anodo: E A = E° Fe 2 + Catodo: E°Fe 2+ Fe → Fe 2 + + 2e − + Fe [ = −0,447 V Fe ] 0,0591 0,0591 log 10 Fe 2+ = −0,447 + log 10 [0,20] = −0,468 V 2 2 − IO3 + 4e − + 2H 2O → IO− + 4OH − E° IO − ,OH− = 0,15 V IO3- − [IO 3 ] 0,0591 0,0591 0,8 E C = E° + log 10 = 0,15 + log = 0,168 V 10 − − 4 4 4 4 [IO ] ⋅ [OH ] (0,8) ⋅ (0,5) fem = ∆E = E C − E A = 0,168 − (−0,468) = +0,636 V Fe → Fe 2 + + 2e − ×2 IO 3 + 4e - + 2H 2 O → IO - + 4OH − - 2Fe + IO 3 + 2H 2 O → 2Fe 2+ + IO - + 4OH − - Esercizio N.3 di pagina 618 Prima di disegnare lo schema della pila vediamo dall’equazione di Nernst quale semielemento si comporta da anodo o da catodo: 2H + + 2e − → H 2 E° H + =0V H2 [ ] 0,0591 [H + ]2 0,0591 10−3 E =0+ log 10 = log 10 2 PH 2 2 1 Hg 2Cl 2 + 2 e – → 2 Hg + 2 Cl – Quindi: anodo (-) : catodo (+ ) : E° Hg Cl 2 2 2 0,0591 = ⋅ (−6) ≅ −0,177 V 2 = +0,268 V ≡ E attuale Hg,Cl - H 2 → 2H + + 2e − Hg 2 Cl 2 + 2 e – → 2 Hg + 2 Cl – E A = −0,177 V E C = +0,268 V reazione complessiva : H 2 + Hg 2 Cl 2 → 2H + + 2 Hg + 2 Cl – fem = ∆E = E C − E A = 0,268 − ( −0,177) = +0,445 V ….prosieguo dell’esercizio N.3 di pagina 618 Lo schema della pila: Non è necessario il ponte salino (||) perché la specie ridotta (H2) della coppia riducente non viene a contatto con la forma ossidata della coppia ossidante (Hg2Cl2): + H 2 + Hg 2Cl 2 → 2H + 2 Hg + 2 Cl Esercizio simile il N.7 di pagina 619 Noi,tuttavia, considereremo sempre l’uso del ponte salino nei nostri esercizi esercizi.. – Esercizio N.10 di pagina 619 H+ − 3 − IO + H 2 O 2 → I + O 2 E° IO − /I- = 1,085 V E°O 2 /H2O 2 = 0,695 V 3 (-) H 2 O 2 → O 2 + 2H + + 2e − ×3 mcm = 6 = n (+) IO3− + 6e − + 6H + → I − + 3H 2 O − 3 − IO + 3H 2 O 2 → I + 3O 2 + 3H 2 O K = 10 K = 10 n∆E ° 0,0591 = 10 6⋅(1, 085− 0 , 695) 0 , 0591 = 10 6⋅0 , 39 0 , 0591 n∆E ° 0,0591 = 10 +39,594 ≅ 3,92 ⋅10 +39 ….prosieguo dell’esercizio N.10 di pagina 575 Lo schema della pila: 0,0591 PO2 ⋅ [H + ]2 0,0591 1 ⋅ [0,10] + log 10 = 0,695 + log 10 = 0,665 V 2 [H 2 O 2 ] 2 [0,10] 2 E A = E°O 2 /H2O 2 E C = E°IO − /I3 − [IO 3 ] ⋅ [H + ]6 0,0591 0,0591 [0,10] ⋅ [0,10]6 + log 10 = 1,085 + log 10 = 1,026 V − 6 [I ] 6 [0,10] fem = ∆E = E C − E A = 1,026 − 0,665 = +0,361 V Esercizi sulle applicazioni dell’equazione di Nernst Usando l’equazione di Nernst è possibile da misure di f.e.m. delle pile risalire alle concentrazioni di specie in soluzione. Questo approccio è particolarmente utile quando si tratta di determinare concentrazioni particolarmente basse come nel caso della misura del pH o della solubilità di sali poco solubili. Misure di pH Supponiamo di avere una soluzione acquosa di cui si voglia misurare il pH: a tale scopo si costruisce una cella voltaica costituita da un elettrodo ad idrogeno in cui una barra di platino è immersa nella soluzione di cui si vuole misurare il pH e in cui gorgoglia H2 gassoso ad 1 atmosfera ed un altro elettrodo quale ad esempio l’elettrodo standard di zinco Zn(-) Pt(+) |Zn2+(aq)(1M)||H+(aq)([H+] incognita)|H2(1atm) La f.e.m. di questa pila dipende dalle concentrazioni degli ioni H+ tramite l’equazione di Nernst ∆E = E + − E − = EH0 2 0,0591 [H + ]2 0,0591 0 + ⋅ log 10 − EZn − ⋅ log 10 [Zn + + ] 2 PH 2 2 in cui: E 0H 2 = 0 V E 0Zn = − 0,76 V PH 2 = 1 atm [Zn + + ] = 1,0 M si ha, quindi, 0,0591 [H + ]2 0,0591 ∆E = 0 + ⋅ log 10 + 0,76 − ⋅ log 10 1 2 1 2 da cui si ricava 0,76 − ∆E pH = 0,0591 Determinazione del Ks Lo stesso principio permette di determinare la concentrazione di uno ione implicato in un equilibrio di solubilità in una opportuna soluzione satura del sale poco solubile (in genere in acqua pura o in una soluzione a concentrazione nota dell’altro ione) e da questa risalire poi al Ks. Generalmente si fa uso di pile di concentrazione che semplificano l’equazione di Nernst. Esempio – Determinare il Ks di AgCl sapendo che la f.e.m. della seguente pila Ag(-) Ag(+) AgCl | Cl-(1M) || Ag+(aq)(1M) | , è di 0,58V a 25°C. La “reazione” di cella è Ag+(aq)(1M) → Ag+(aq)(incogn.) , in cui Ag+(incogn.) è la concentrazione di ioni Ag+ nella soluzione satura di AgCl(s) contenente Cl- 1M. [Ag + ]+ 1 ∆E = 0,0591 ⋅ log 10 = 0,0591 ⋅ log 10 + + [Ag ]− [Ag ]− + 0,58 = −0.0591 ⋅ log 10[Ag ]− + [Ag ] = 10 − 9,80 = 1,58 ×10 + log 10 [Ag ]− = −9,80 −10 Dall’espressione per Ks AgCl(s) Ag+(aq) + Cl-(aq) K s = [Ag + ] ⋅ [Cl − ] , segue K s = (1,58 ⋅10 −10 ) ⋅ (1) = 1,58 ⋅10 −10 mol L 2 Esercizio riassuntivo Determinare la costante di dissociazione Ka dell’ acido formico HCOOH sapendo che la f.e.m. della pila schematizzata di seguito è 0,358V e che il potenziale standard dell’elettrodo a cloruro di argento è 0,222 V: Ag( + ) Pt(-) H 2(g)(1atm) [HCOOH] = 0,14 M [Cl - ] = 1,0 M AgCl (s) + 2 1 0,0591 [H ] 0 0 ∆E = E + − E − = E AgCl,Cl − /Ag + 0,0591⋅ log − − E H + /H − ⋅ log 2 [Cl ] 2 PH 2 0,358V 0,222 V 1 0,358 − 0,222 log x = − 0,0591 0,0591 0,358 = 0,222 − ⋅ 2 ⋅ log x 2 x = [H + ] ≡ [H 3O + ] = 10 − 0,358−0,222 0,0591 = 10 −2,30 = 5,0 ⋅10 −3 M x HCOOH(aq) + H2O(l) ⇄ HCOO- (aq) + H3O+ (aq) (i) (eq) 0,14 0,14-x --x ~0 x [HCOO − ] ⋅ [H 3O + ] x2 (5 ⋅10 −3 ) 2 Ka = = = = −3 [HCOOH] 0,14 − x 0,14 − 5 ⋅10 (5 ⋅10 −3 ) 2 2,5 ⋅10 −5 −4 Ka = 1,85 10 = ≅ ⋅ −3 0,14 − 5 ⋅10 0,135 mol L Esercizio riassuntivo Calcolare KS a 25°C del AgBrO3, essendo noto che a questa temperatura, la f.e.m. della pila: Ag(-) Ag(+) | soluzione satura di AgBrO3 || Ag+(0,0734M) | è uguale a 0,066 V. [Ag + ]+ 0,0734 ∆E = 0,066 = 0,0591 ⋅ log = 0,0591 ⋅ log [Ag + ]− [Ag + ]− log 0,0734 0,066 0,0734 = ⇔ = 10 + + [Ag ]− 0,0591 [Ag ]− 0,066 0,0591 ⇒ [Ag + ]− = 0,0734 10 0,066 0,0591 = 5,61⋅10 −3 M + − AgBrO 3(s) → ← Ag ( aq ) + BrO 3 ( aq ) S + S − ( K S = [Ag ] ⋅ [BrO 3 ] = S = 5,61⋅10 2 ) −3 2 = 3,14 ⋅10 −5 M 2 Esercizio (riassuntivo) N.12 di pagina 620 Questa non è altro che una pila di concentrazione al [Ag+]: Ag + (aq ) + e − → Ag(s) [Ag + ]1 [Ag + ] + ∆E = 0,0591 ⋅ log = 0,0591 ⋅ log = E+ − E+ + [Ag ] 2 [Ag ]Adesso dobbiamo capire quale semielemento è il catodo (C+: ovvero concentrazione maggiore per Ag+) La traccia dell’esercizio ammette implicitamente che vi sarà precipitazione del Ag2CrO4 2AgNO 3 ( aq) + Na 2 CrO 4 (aq ) → Ag 2 CrO 4 ( s ) + 2NaNO 3 (aq ) i) c) f) tot) Volume finale = 2,0 L ⇔ [CrO 24− ] = Ag 2 CrO 4 ( s ) → ← 2Ag + ( aq ) + CrO -24 ( aq ) 2S 0,3 + S 0,2 0,2 0 0,7 0,1 0,6 0,1 0,1 0,2 0,2 0,6 mol = 0,3 M 2,0 L ⇒ K S = [Ag + ]2 ⋅ [CrO -24 ] = ? S << 0,3 [Ag + ] = 2S K S = [Ag + ]2 ⋅ [CrO -4 ] = [2S]2 ⋅ 0,3 ….prosieguo dell’esercizio N.12 di pagina 620 A questo punto è chiaro che il catodo è quello dove è presente la soluzione 0,1 M di AgNO3 [Ag + ]1 [Ag + ] + 0,1 ∆E = 0,0591 ⋅ log = 0,0591 ⋅ log = 0,0591 ⋅ log 2S [Ag + ] 2 [Ag + ]- 0,1 0,278 = 0,0591 ⋅ log 2S 0,278 0,1 0,1 = log ⇔ = 10 0,0591 2s 2S + 2 0,278 0,0591 ⇒ 2S = 10 [ K S = [Ag ] ⋅ [CrO ] = [2S] ⋅ 0,3 = 1,977 ⋅ 10 4 2 0,1 0,278 0,0591 = 1,977 ⋅ 10 −6 ] ⋅ 0,3 ≅ 1,17 ⋅10 −6 2 −12 M3 Esercizio riassuntivo Per la seguente pila a 25°C: Pt Cl2(g) (1 atm) Pt ClO3- (1·10-3 M) MnO4- (1·10-3 M) H+ (1·10-1 M) H+ (? M) Mn2+ (1·10-1 M) Calcolare a quale pH dell’elettrodo di destra la pila risulta scarica (f.e.m.=0) sapendo che, per le coppie ClO3-,H+/Cl2 e MnO4−,H+/Mn2+, i potenziali standard di riduzione valgono, rispettivamente, 1,470 V e 1,507 V. Svolgimento: f.e.m. = 0 ⇔ f.e.m. = E sinistra − E destra = E destra − E sinistra = 0 ⇒ E sinistra = E destra È indifferente scegliere il polo di destra o sinistra come anodo o catodo. La semireazione a sinistra scritta nel senso della riduzione è ( +5) − 3 − (0) + 2 Cl O + 10 e + 12 H → Cl 2 + 6 H 2 O da cui il potenziale è ricavabile mediante l’equazione di Nernst: E sinistra [ ] [ ] 2 0,0591 ClO3− ⋅ H + log = E° ClO− ,H + /Cl + 3 2 10 PCl2 = 1,470 + [ ] 12 [ ] 0,0591 1⋅10 −3 ⋅ [0,1] log = 1,470 + 10 1 0,0591 0,0591 log 1⋅10 −18 = 1,470 + ⋅ (− 18) 10 10 2 12 = La semireazione a destra scritta anch’essa nel senso della riduzione è ( +7) − 4 − + ( +2) Mn O + 5 e + 8 H → M n + + + 4 H 2 O e permette di impostare l’equazione di Nernst così: E destra [ ][ ] [ ] 0,0591 MnO 4− ⋅ H + = E° MnO− ,H + /Mn ++ + log 4 5 Mn + + = 1,507 + E sinistra ([ ][ ] ) 0,0591 log 1 ⋅10 − 2 ⋅ H + 5 = E destra 1,470 + 8 [ ][ ] 1 ⋅10 −3 ⋅ H + 0,0591 = 1,507 + log 5 [0,1] 0,0591 8 = 1,507 + ⋅ (− 2 ) + 0,0591⋅ log H + 5 5 8 [ ] [ ] 0,0591 0,0591 8 ⋅ (− 18) = 1,507 + ⋅ (− 2 ) + 0,0591 ⋅ log H + 10 5 5 [ ] 14,70 − 1,0638 = 15,07 − 0,2364 + 0,9456 ⋅ log H + [ ] log H + = − 1,1974 ≅ −1,267 0,9456 pH ≅ 1,27 ⇔ [H ] = 10 + −1,267 ≅ 0,0542 M 8 = Esercizio riassuntivo (simile) Sapendo che, per le coppie Co3+/Co2+ e MnO4−,H+/Mn2+, i potenziali standard di riduzione valgono, rispettivamente 1,30 V e 1,51 V, stabilire per quale valore del pH è nulla la forza elettromotrice (f.e.m.) della seguente pila: Pt Pt 3+ -3 - -3 Co (1·10 M) MnO4 (1·10 M) Co2+ (1·10-1 M) H (? M) + 2+ -1 Mn (1·10 M) Prova tu!! Risultato: pH=3,22 Esercizio riassuntivo Sapendo che la costante di dissociazione dell'ammoniaca vale Kb=1,810-5M e che il potenziale standard di riduzione della coppia O2/OH− vale E0= 0,40 V, calcolare la fem della pila: Pt Pt H2(g) P = 1 atm NH4Cl -3 (1·10 M) -4 NH4Cl (5·10 M) NH3 (1,5·10-3 M) O2(g) P = 1 atm e il pH delle due soluzioni elettrodiche. Svolgimento: Semielemento di sinistra: NH4+(aq) + H2O(l) ⇄ NH3(aq) + H3O+(aq) CS --~0 NH4Cl(aq) →NH4+(aq)+ Cl−(aq) CS – x x x K W [NH 3 ] [H 3O + ] costante d' idrolisi = K i = K a = = Kb [NH 4+ ] −14 K W 1,0 ⋅10 x −10 = = 5,56 ⋅10 = −5 K b 1,8 ⋅10 (CS − x) 2 Cs 1 ⋅10 −3 = > 380 ⇒ −10 K a 5,56 ⋅10 C S − x ≅ CS [H 3O + ]2 Ka = CS [H 3O + ] = K a ⋅ CS = 5,56 ⋅10 −10 ⋅1 ⋅10 −3 = 7,45 ⋅10 −7 M ≡ [H + ] pH = −log 10[H 3O + ] = −log 10[7,45 ⋅10 −7 ] ≅ +6,13 2 H + (aq) + 2e − → H 2 (g) E sinistra = E 0H + /H2 0,0591 [H + ]2 0,0591 [7,45 ⋅10 −7 ]2 + log = 0+ log = −0,362 V 2 PH 2 2 1 Semielemento di destra: [OH − ] = K b ⋅ C b 1,5 ⋅10 −3 M 0,1 < = = 3 < 10 ⇒ tampone −4 Cs 5 ⋅10 M Cb −x C ≅ Kb ⋅ b Cs + x Cs ⇒ [H 3O + ] = KW C Kb ⋅ b Cs K W Cs 1 ⋅10 −14 5 ⋅10 − 4 −10 [H 3O ] = ⋅ = ⋅ = 1,852 ⋅ 10 M −5 −3 K b C b 1,8 ⋅10 1,5 ⋅10 + ( ) pH = −log 1,852 ⋅10 −10 ≅ +9,73 O 2 (g) + 4e − + 2 H 2 O → 4 OH − E destra PO 2 0,0591 0,0591 [H + ]4 = E° + ⋅ log ⋅ = 0,40 + ⋅ log = − 4 4 4 [OH ] 4 [KW ] 0,0591 [1,852 ⋅10 −10 ]4 = 0,40 + ⋅ log = 0,652 V −14 4 4 [1 ⋅10 ] E destra = 0,652 V E sinistra = −0,362 V Catodo (+) Anodo (-) O 2 (g) + 4e − + 2 H 2 O → 4 OH − H 2 (g) → 2 H + (aq) + 2e − 2 H 2 (g) + O 2 (g) → 2 H 2O fem = E destra − E sinistra = E + − E − = 0,652 V − (− 0,362 V ) = +1,014 V x 2 Esercizio riassuntivo Assegnata la seguente pila a 25°C: Pt (+) [MnO4-]=0,1 M [Mn++] = 0,1 M [H+] = 0,001 M Ag (-) Ag3PO4 soluzione satura calcolarne la f.e.m. sapendo che i potenziali standard di riduzione del catodo ( E° MnO −4 ,H + /Mn ++ ) e delll’anodo ( E° Ag+ /Ag ) valgono rispettivamente 1,507 V e 0,800 V e che il prodotto di solubilità (KS) del Ag3PO4 a 25°C è uguale a 8,88·10–17 (mol/L)4. Svolgimento: Semielemento di sinistra (catodo +): E + = E°MnO− ,H + /Mn 2+ 4 MnO 4− + 5e − + 8H + → Mn 2+ + 4H 2O 0,0591 [MnO 4− ] ⋅ [H + ]8 0,0591 0,1⋅ [0,001]8 + log = 1,507 + log ≅ 1,223 V 2+ 5 [Mn ] 5 [0,1] Semielemento di destra (anodo -): Ag → Ag + + e - E − = E° Ag + /Ag + 0,0591 ⋅ log[Ag + ] = 0,800 + 0,0591 ⋅ log[Ag + ] [Ag + ] = ? Ag 3 PO 4 (s) + 3 Ag (aq) + PO34− (aq) 3S S K s = [ Ag + ]3 ⋅ [PO 34- ] = 8,88·10–17 (mol/L) 4 K S = [3S] ⋅ [S] = 27S4 3 Nota bene: –17 K 8,88·10 ⇒S=4 S =4 = 4,258·10– 5 M 27 27 [Ag + ] = 3 S = 3 ⋅ 4,258·10–4 M E − = 0,800 + 0,0591⋅ log[3 ⋅ 4,258·10–5 ] ≅ 0,570 V fem = ∆E = E + − E − = 1,223 V − 0,570 V = 0,653 V Esercizio riassuntivo (effetto delle pressioni sugli elettrodi) La pila: Pt (-) H2(g) P= 2 atm Pt (+) [HA] = 1,0 M [HCl]=1·10-4 M H2 P= 1 atm ha una fem pari a 0,068V a 25°C. Determinare il pH della soluzione di HA e la costante di equilibrio dell’acido. Svolgimento: ∆E = E + − E − = E 0H 2 + 0,0591⋅ log [H + ]+ PHcatodo 2 + [H ] - − E 0H 2 + 0,0591⋅ log anodo P H 2 anodo + P [H ]+ [H ][H ]+ H2 ∆E = 0,0591 ⋅ log − 0,0591 ⋅ log = 0,0591 ⋅ log ⋅ + catodo anodo catodo [H ] PH 2 PH 2 P H2 anodo [H + ] −4 P ⋅ 1 10 2 H 2 + fem = 0,068 = ∆E = 0,0591⋅ log ⋅ = 0,0591⋅ log ⋅ + + catodo [H ]- [H ] PH 1 2 + + calcolare 1 ⋅10 −4 ⋅ 2 0,068 = log 0,0591 x 1 ⋅10 −4 2 ⋅ 0,068 = 0,0591 ⋅ log x 1 2 ⋅10 −4 1,1506 = log x ( 1,1506 = log ( ) 2 ⋅10 −4 − log x ) log x = log 2 ⋅10 −4 − 1,1506 = −3,8495 − 1,1506 = 5,0001 x = [H + ] = [H 3O + ] = 10 −5, 0001 ≅ 1 ⋅10 −5 M pH = 5 HA(aq) + H2O(l) ⇄ A− (aq) + H3O+ (aq) (i) (eq) 1,0 1,0-x x x [A − ] ⋅ [H 3O + ] x2 (1 ⋅10 −5 ) 2 Ka = = = = −5 [HA] 1 − x 1 − 1 ⋅10 (1 ⋅10 −5 ) 2 −10 Ka = ≅ 1 ⋅ 10 [mol/l] −5 1 − 1⋅10 Esercizio riassuntivo (effetto delle pressioni sugli elettrodi) La forza elettromotrice (f.e.m.) della seguente pila: Pt (+) Ag (-) Cl2(g)+Ne(g) PTOT= 1atm [HCl] = 0,01 M [AgNO ]=0,10 M 3 vale ∆E= 0,735V a 25,0°C. Sapendo che E0Ag+/Ag= 0,800V e E0Cl2/Cl=1,360V, si calcoli la frazione molare di Ne nella miscela gassosa che gorgoglia sull'elettrodo di platino. [χ χNe=0,164] Svolgimento: Semielemento di sinistra (catodo +): Cl2 + 2e − → 2Cl− [Cl− ] ≡ [HCl] = 0,01 M E + = E°Cl /Cl− + 2 PCl 2 PCl 2 0,0591 0,0591 log 1 , 360 log = + 2 [Cl − ]2 2 [0,01]2 Semielemento di destra (anodo -): Ag → Ag + + e - [Ag + ] ≡ [AgNO 3 ] = 0,1 M E − = E° Ag + /Ag + 0,0591 ⋅ log[Ag + ] = 0,800 + 0,0591 ⋅ log[ 0,1] fem = 0,735V = ∆E = E + − E PCl 2 0,0591 0,735 = 1,360 + log − (0,800 + 0,0591⋅ log[ 0,1]) 2 2 [0,01] ( ) 0,0591 0,0591 0,735 = 1,360 + log PCl 2 − log[ 0,01]2 − 0,800 − 0,0591 ⋅ log[ 0,1] 2 2 ( ) 0,0591 0,735 = 1,360 + log PCl 2 + 0,1182 − 0,800 + 0,0591 2 0,0591 log PCl 2 = 0,735 − 1,360 − 0,1182 + 0,800 − 0,0591 = −0,0023 2 0,0023⋅2 − 0,0023 ⋅ 2 log PCl 2 = − ⇒ PCl 2 = 10 0, 0591 ≅ 0,836 atm 0,0591 ( ) ( ) χ Ne PNe PTOT - PCl 2 1 atm - 0,836 atm = = = = 0,164 PTOT PTOT 1 atm Esercizio riassuntivo Determinare di quanto varia la f.e.m. della pila: Fe Ag | [Fe+2]=5,0·10-2 M | | Ag2CrO4 sol. satura | se nel semielemento di destra, il cui volume è 0,5 litri, si aggiungono 81 mg di Na2CrO4 solido (elettrolita forte e completamente solubile). Sono noti i valori dei potenziali standard di riduzione delle coppie Fe+2/Fe e Ag+/Ag (-0,44 V e + 0,80 V) e quello del prodotto di solubilità di Ag2CrO4 (9·10-12 M3 ). [R. ∆fem=∆ ∆E2-∆ ∆E1=-0,025 V] Svolgimento: Semielemento di sinistra: E sinistra = E° Fe 2+ /Fe + Fe 2 + + 2e − → Fe 0,0591 0,0591 log[Fe 2 + ] = −0,44 + log[ 5 ⋅10 − 2 ] ≅ -0,478 V 2 2 È molto probabile (quasi certo) che sia l’anodo (ossidazione): Fe → Fe 2+ + 2e − Ag + + e − → Ag Semielemento di destra ? E destra = E° Ag + /Ag + 0,0591 ⋅ log[Ag + ] = 0,800 + 0,0591 ⋅ log[Ag + ] Prima dell’aggiunta Ag 2CrO 4 (s) 2 Ag + (aq) 2S + CrO S 2− 4 (aq) K S = [Ag + ]2 ⋅ [CrO 24− ] = 9 ⋅10−12 M 3 [Ag + ] = 2S −12 9 ⋅ 10 K S = [2S]2 ⋅ [S] = 9 ⋅10 −12 ⇒ S = 3 = 1,31⋅10 − 4 M 4 [Ag + ] = 2 ⋅1,31⋅10−4 M = 2,62 ⋅10−4 M E destra = 0,800 + 0,0591 ⋅ log[2,62 ⋅10 −4 ] ≅ 0,588V È il catodo (riduzione): Ag + + e − → Ag fem = E destra − E sinistra = ∆E1 = 0,588 − (− 0,478 ) = 1,066 V 81 mg di Na2CrO4 solido aggiunto in 0,5 litri n Na 2CrO 4 = [CrO ] = 24 n CrO 24 V Ag 2CrO 4 (s) i) m Na 2CrO 4 PM Na 2CrO 4 0,081 g ≅ 5 ⋅10 −4 mol ≡ n CrO 24 161,9732(g/mol ) 5 ⋅10 −4 mol = = 1 ⋅10 −3 M 0,5 L + 2 Ag (aq) + CrO 24− (aq) -- eq) = 2S 1·10-3 1·10-3+S K S = [2S]2 ⋅ [0,001] = 9 ⋅10 −12 K S = [Ag + ]2 ⋅ [CrO24− ] = 9 ⋅10−12 M 3 [Ag + ] = 2S S < 0,001 ⇒ 0,001 + S ≈ 0,001 9 ⋅10 −12 ⇒S= = 4,743 ⋅10 −5 M 4 ⋅ 0,001 [Ag + ] = 2 ⋅ 4,743 ⋅10−5 M = 9,486 ⋅10−5 M E destra = 0,800 + 0,0591 ⋅ log[9,486 ⋅10 −5 ] ≅ 0,563 V Ag + + e − → Ag fem = E destra − E sinistra = ∆E 2 = 0,563 − (− 0,478 ) = 1,041 V È ancora ancora il catodo (riduzione): ∆fem = ∆E 2 − ∆E1 = 1,041 V − 1,066 V = −0,025 V Esercizi riassuntivi e integrativi 1. Calcolare la f.e.m. di una pila formata da un elettrodo standard ad idrogeno e da un elettrodo di cromo immerso i una soluzione 1,0·10−3M dello ione Cr3+ (E°Cr+3/Cr =-0,742 V). Scrivere, inoltre, la reazione complessiva e calcolarne la costante di equilibrio. [R. f.e.m.=0,801 V] 2. Una pila di concentrazione è formata da due semicelle nelle quali gli elettrodi di platino sono immersi in due soluzioni, una 0,100 M di HCl e l’altra 0,100 M di CH3COOH (Ka=1,8 10-5). H2 alla pressione di 1 atm è fatto gorgogliare sui due elettrodi e la temperatura è di 298 K. Calcolare la forza elettromotrice della pila. [R. f.e.m.=0,111 V] 3. Una pila è formata da un elettrodo a idrogeno (PH2= 1 atm) immerso in una soluzione di acida e da un elettrodo di rame immerso in una soluzione di CuSO4 0,100 M. La f.e.m. della pila è 0,400V a 298 K. Calcolare [H+] nella semicella a idrogeno. [R. [H+]= 3,31·10-2M] 4. Un elettrodo standard a idrogeno è collegato a un elettrodo a idrogeno (PH2= 1 atm; T=298K) immerso in una soluzione 0,105 M di un acido debole monoprotico (HA). La f.e.m. della pila è 0,170V. Calcolare Ka dell’acido debole. [R. Ka= 1,75·10-5 M] 5. Calcolare la f.e.m. della seguente pila a concentrazione: Ag Ag | Ag2SO4 (soluzione satura) | | Ag+(0,100 V) | sapendo che il prodotto di solubilità di Ag2SO4 è 1,6 10-5 M3. [R=0,029 V] Esercizi riassuntivi e integrativi 6. A 25°C, la f.e.m. della pila Pt(+) Pt (-) (1 atm) H2 | [HCl]= ? M | | [HA]=0,10 M | (1 atm) H2 vale 0,0677 V. Si calcoli la concentrazione di HCl al catodo. Per l’acido debole monoprotico (HA) la costante di acidità Ka è pari a 5,10·10-5 (mol/L). [R. [HCl] =0,096 M] 7. Determinare la costante di equilibrio della reazione Zn + 2Ag+ = Zn2+ + 2Ag sapendo che la f.e.m. della pila Zn(-) Ag(+) | [ZnSO4]=5·10-3 M | | [AgNO3]=0,01 M | è pari a 1,51 V a 25°C. [R. K=6,3·10+52] 8. Si calcoli la f.e.m., a 25°C, della pila Cu(-) Pt(+) | [Cu2+]=0,1 M | | [Fe3+]=0,1M; [Fe2+]=0,1M | sapendo che la costante di equilibrio della reazione: 2Fe3+ + Cu = 2Fe2+ + Cu2+ alla temperatura di 25° C, vale 4,86·10+14. [R. f.e.m.=0,464 V] Esercizi riassuntivi e integrativi 9. Sapendo che la costante di equilibrio dell'acido debole HA vale KA= 5,0 ·1 0-4M, si calcoli la forza elettromotrice della pila: Pt Pt H2(0,5 atm)| [KCl]=0,10 M | | [HA]=0,10 M |H2(1 atm) Si calcoli anche la forza elettromotrice della pila quando, nella soluzione del semielemento di destra (di volume pari a 1 litro), si aggiungono 5,0 ·1 0-2mol di KOH solido. [Prima dell'aggiunta: ∆E=0,277V; dopo dell'aggiunta: ∆E=0,209V] 10. La costante di equilibrio della reazione: Hg2+ + 2Fe+2 = Hg + 2Fe+3 vale K=5,66 M-1 a 25°C. Si determini la f.e.m della pila: Pt Hg | [Fe+2]=1·10-2 M; [Fe+3]=5·10-3 M | | [Hg+2]=0,1 M | [R. f.e.m.=0,01049 V] Esercizi riassuntivi e integrativi 11. A 25,0°C, la forza elettromotrice della seguente pila : Pt(+) Pt(-) H2 (1 atm) | [HCl]= x M | | [HA] =0,01 M | H2 (1 atm) vale ΔE=67,7 mV. Si calcoli la concentrazione di HCl nella soluzione del semielemento positivo della pila. Per HA Ka = 5,10·10-4M. [ R.: [HCl]=0,096M] 12. Sapendo che i potenziali standard delle coppie: Fe3+/Fe2+ e Cr2072-,H+/Cr3+ valgono, rispettivamente, +0,77 V e + 1,33V, si stabilisca per quale valore del pH è nulla la forza elettromotrice della pila: Pt Pt | [Fe3+]=0,05 M, [Fe2+]=0,10 M | | [Cr2O72-] =0,02 M,[Cr3+]=0,10M, [H+]=?M | [ R.: pH=4,21] 13. Sapendo che i potenziali standard delle coppie Au3+/Au+ e Mn04-,H+/Mn2+ valgono, rispettivamente, +1,29V e +1,51V, si stabilisca per quale valore del pH è nulla, alla temperatura di 25,0°C, la forza elettromotrice della pila: Pt Pt | [Au3+] = 0,05M, [Au+] = 0,10 M | | [Mn04-] = 0,02M, [Mn2+] = 0,1 M, [H+]=?M | [R.: pH = 2,33] Esercizi riassuntivi e integrativi 14. La forza elettromotrice della seguente pila: Pt(-) Cu(+) (Ptot=1,0 atm) (H2 + Ar) | [HCl]=0,001 M | | [CuSO4]=0,01 M | vale M = 0,454 V a 25,0°C. Sapendo che: E°Cu2+/Cu=0,340V, si calcoli la frazione molare di Ar nella miscela gassosa che gorgoglia sull'elettrodo di platino. [χ χAr=0,279] 15. Il potenziale di un elettrodo di idrogeno (PH2=1 atm) immerso in una soluzione 0,74 M di cianuro di potassio (KCN) vale E=-0,68 V, a 25°C. Determinare il valore della costante di dissociazione dell'acido cianidrico (HCN), alla stessa temperatura. [Ka=7,20·10-10M] 16. La fem della pila, a 25°C, Pt(-) Ag(+) (1,0 atm) H2 | [NaCl]=0,10 M | | [AgNO3]=0,30 M | vale 1,183V. Se si aggiungono, nel semielemento di destra (V=1000 mL), 0,10 mol di K3PO4, si ha la precipitazione di Ag3PO4 e la fem nella nuova situazione (polarità degli elettrodi invariate) diventa pari a 0,958V. Si calcoli il valore del prodotto di solubilità di Ag3PO4. [KS=1,646·10-18 M4] Esercizi riassuntivi e integrativi 17. Sia data la pila, a 25°C: Ag Ag | Ag2CrO4 soluzione satura | | [AgNO3]=0,10 M | Nella soluzione del semielemento di destra (V = 1000 mL) si aggiungono 0,1 mol di NaCl solido. Sapendo che i prodotti di solubilità di Ag2CrO4 e di AgCl valgono, rispettivamente, 1,9·10-12M3 e 1,032·10-10 M2,calcolare la fem della pila prima e dopo l’aggiunta di NaCl. [∆Eprima=0,166 V; ∆Edopo=0,070 V ] 18. A 25°C, la fem della pila: Pt Pt (1,0 atm) H2 | [NaNO3]=0,10 M | | [NaNO2]=0,01 M | H2 (1,0 atm) vale 0,040 V. Si determini il valore della costante di dissociazione acida (Ka) di HNO2. [Ka=4,43·10-4 M] Da gennaio 2012 a settembre 2012 potrete trovarvi ogni venerdì mattina (salvo esami o impegni istituzionali) presso la stanza 125 (primo piano). Per comunicazioni telematiche la mia e-mail è [email protected] (specificare sempre l’oggetto: STUDENTE TARANTO). Solo per comunicazioni urgenti telefonare al numero 080-5963601 oppure 080-5963600 chiedendo del Dott. Giuseppe Romanazzi. Gli appelli degli esami di profitto per l’anno solare 2012 relativi all’insegnamento di Chimica (6 cfu) per il Corso di Laurea (interfacoltà) in Ingegneria per l’Ambiente e il Territorio (DM 270/2004) seguiranno il seguente calendario: Le date potranno subire variazioni (ovviamente piccole posticipazioni) “In bocca al lupo per la prova di esame e per il prosieguo dei vostri studi” THE END
Scaricare