Cromatografia
Il termine cromatografia indica un insieme di tecniche che
hanno lo scopo di separare una miscela nei suoi componenti,
per permetterne il riconoscimento qualitativo e quantitativo
Queste tecniche sono basate sulla distribuzione differenziale
dei vari componenti fra due fasi, una chiamata fase fissa o
fase stazionaria e l’altra chiamata fase mobile o eluente, che
fluisce in continuo attraverso la fase fissa
Le tecniche sono molto utilizzate in campo ambientale,
biologico, farmaceutico, ecc., essendo particolarmente utili
nell’analisi di miscele complesse come sono la maggior parte
dei campioni di natura organica
1
La separazione dei componenti di una miscela si ottiene in seguito alle
interazioni chimiche o fisiche che avvengono tra le molecole disciolte nella fase
mobile e la fase stazionaria: queste interazioni possono avvenire con la
superficie adsorbente del supporto solido, o con un liquido che ricopre il
supporto in modo omogeneo o ancora con molecole ad esso legate in maniera
covalente.
Dal momento che i diversi componenti della miscela da separare interagiscono
in maniera differente con la fase stazionaria, si muovono lungo la colonna
cromatografica con velocità diverse: i composti che sono più “affini” alla fase
stazionaria si muovono in media più lentamente di quelli che sono più “affini”
alla fase mobile.
2
La Nascita della Cromatografia
La nascita della cromatografia si deve al botanico russo Mikhail Semenovich Tswett,
che per primo la utilizzò nel 1903 per separare i pigmenti naturali contenuti in
estratti vegetali.
Mikhail S. Tsvett
(1872–1919)
Scrive Tswett nel 1906:
“Se una soluzione di clorofilla in etere di petrolio è filtrata attraverso una colonna di adsorbente – io
utilizzo soprattutto carbonato di calcio, accuratamente pressato, all'interno di uno stretto tubo di vetro
– allora i pigmenti, si separano, secondo la sequenza di adsorbimento, dall'alto in basso, in molte zone
colorate […] Proprio come i raggi di luce nello spettro, i diversi componenti della miscela appaiono
separati, sulla colonna di carbonato di calcio, secondo una legge e possono essere misurati sia in modo
qualitativo che quantitativo. Io chiamo questa preparazione cromatogramma ed il metodo
corrispondente lo chiamo metodo cromatografico. Ovviamente i fenomeni di adsorbimento fin qui
descritti non sono ristretti ai soli pigmenti della clorofilla ed è lecito presumere che tutti i composti
chimici, sia colorati che incolori, siano soggetti alle stesse leggi.”
3
Nell’esperimento di Tswett, la fase stazionaria era carbonato di calcio mentre la fase
mobile o eluente era etere di petrolio, un solvente organico composto da idrocarburi a
basso punto di ebollizione. I composti da separare sono introdotti nella fase mobile e si
ripartiscono lungo la colonna cromatografica in funzione della loro affinità relativa per la
fase mobile e per la fase stazionaria.
4
La cromatografia è nata dunque all’inizio del XX secolo
come tecnica per la separazione di pigmenti fogliari, ma
già il suo inventore era consapevole delle sue potenzialità
applicative
Tswett chiamò questa tecnica cromatografia dal
greco scrittura del colore o, visto il significato del suo
cognome in russo, scrittura di Tswett
5
Twsett era uno spirito inquieto e mobile, in quanto
rimangono tracce di suoi soggiorni - spesso con
collaborazioni scientifiche - in università tedesche,
olandesi, belghe e francesi. Nel 1910 era apparsa una
sua importante opera di, intitolata "Cromofille dei
regni vegetale e animale", in cui erano descritte
dettagliatamente sia le tecniche sperimentali , scritto
però in russo. Il chimico tedesco Willstätter si era
fatto tradurre per uso privato il libro di Twsett e
questa traduzione, passata nelle mani del suo allievo
Kuhn. Nel1930, ad appena 22 anni, Edgard Lederer
era giunto per il suo lavoro di postdottorato sui
pigmenti della carota ad Heidelberg, nel laboratorio
La scienza è spesso
diretto da Kuhn , messo sulla strada giusta dalla
lettura di un libro sui carotenoidi dell'americano L.S.
influenzata da
Palmer, edito nel 1922, dove si faceva riferimento al
avvenimenti casuali:
Sorprendentemente, nel 1930 volume di Twsett.
Lederer venne in possesso
della 'traduzione privata‘ del
libro di Tswett e così riprese il
cammino della cromatografia.
7
Richard Kuhn (1900-1967)
Edgar Lederer (1908-1988)
8
Archer Martin (1910-2002)
Richard Synge (1914-1994
The Nobel Prize in Chemistry 1952 was awarded jointly to Archer John Porter
Martin and Richard Laurence Millington Synge
"for their invention of partition chromatography"
LONGITUDINAL DIFFUSION AND RESISTANCE TO MASS
TRANSFER AS CAUSES OF NONIDEALITY IN
CHROMATOGRAPHY
J. J. VAN DEEMTER, F. J. ZUIDERWEG
Koninklijke/Shell-Laboratorium, Amsterdam (N.V. De Bataafsche Petroleum
Maatschappij)
and
A. KLINKENBERG
N. V. De Bataafsche Petroleum Maatschappij, The Hague
(Received 1 February 1956)
Chem. Engng Sci. 5, 271-289, 1956.
10
1898-1903 D. Trebot Day, geologo americano, separò alcuni idrocarburi utilizzando colonne di farina
fossile.
1903-1906 M. Tswett, botanico russo, separò una serie di pigmenti colorati presenti in un estratto di
foglie verdi utilizzando CaCO3 ed etere di petrolio; coniò anche il nome cromatografia che significa
"scrittura mediante il colore".
1930-1931 R. Kuhn e E. Lederer utilizzarono la cromatografia per la separazione di carotenoidi e
xantofille.
1938 N.A. Izmailov e M.S.Shaiber descrissero per la prima volta la cromatografia su strato sottile
1941 A.J.P. Martin e R.L.M. Synge presentarono il primo lavoro sulla cromatografia di ripartizione. Essi
introdussero la teoria dei piatti basata su di un'analogia con la teoria della distillazione e dell'estrazione
controcorrente.
1952 A.J.P. Martin e R.L.M. Synge ebbero il premio Nobel per la chimica per lo sviluppo della
cromatografia di ripartizione. Nello stesso anno, in collaborazione con A.T.James realizza la cromatografia
gas-liquido.
1956 J.J.van Deemter sviluppò la teoria della separazione cromatografica.
1958 E.Stahl mise a punto la tecnica della cromatografia su strato sottile.
1966 Inizia lo sviluppo della moderna tecnica della cromatografia in fase liquida ad alta prestazione.
J. Calvin Giddings (1930-1996)
12
R.A. Dewar (1908-1981) I.G. McWilliam (1933)
V. Pretorius (1928-1989)
Il rivelatore a ionizzazione di fiamma fu inventato
indipendentemente da due gruppi di ricercatori in Australia e
Sud Africa
Nel 1957 M:J.E. Golay realizza le colonne capillari di
vetro e ne sviluppa la teoria
14
-James Lovelock in 2005
15
Basi del procedimento cromatografico
Il campione è introdotto nella fase mobile, che può essere un gas, un liquido
o un fluido supercritico.
La fase mobile viene fatta eluire in continuo attraverso la fase stazionaria,
che deve essere immiscibile nell’eluente.
La fase stazionaria (liquida o solida) si trova all’interno di una colonna
oppure è supportata su una superficie piana.
La fase mobile e la fase stazionaria sono scelte in modo che i componenti
della miscela da separare si distribuiscano tra le due fasi
i componenti più affini alla fase stazionaria passeranno più tempo in
questa fase, quindi si sposteranno più lentamente attraverso il sistema
i componenti più affini alla fase mobile si sposteranno invece più
velocemente
La separazione dei componenti avviene in quanto ogni sostanza ha una
distribuzione caratteristica tra le due fasi (costante di ripartizione Kd=Cs/Cm)
Visualizzazione della separazione
Ponendo all’uscita della colonna un
rivelatore che misuri la concentrazione
del soluto nell’eluato (cioè la fase
mobile che esce dalla colonna) e
riportando il segnale in funzione del
tempo
si
può
ottenere
un
cromatogramma
La posizione dei picchi sull’asse dei
tempi, o tempo di ritenzione, serve per
identificare i componenti del campione
L’area sottesa dai picchi è proporzionale
alla
quantità
di
ogni
singolo
componente e può essere utilizzata a
scopo quantitativo
17
Forma generale delle isoterme di
adsorbimento
ISOTERME DI RIPARTIZIONE
Picco gaussiano
19
ISOTERME NON LINEARI
PRINCIPI TEORICI DELLA CROMATOGRAFIA
Esempio: separazione di un campione a tre componenti in una colonna chiusa.
La fase stazionaria consiste di particelle solide porose contenute all’interno di
un tubo lungo e sottile (colonna). Nel passaggio attraverso la colonna ogni
componente X si distribuisce fra la fase stazionaria (s) e la mobile (m):
Xm Xs
A: il campione viene iniettato
all’entrata della colonna
B D: la fase mobile fa spostare il
campione attraverso la fase stazionaria
Flusso del solvente
Il coefficiente di ripartizione (o di distribuzione) del componente X è
definito come:
[X]s
KX
[X]m
A
B
C
D
21
22
Tempo di ritenzione
Il tempo di ritenzione tR è il tempo che impiega un componente della miscela iniettata ad
uscire dalla colonna o, tecnicamente, ad essere rivelato come picco dal detector. Un tipico
cromatogramma per una miscela a due componenti ha due situazioni diverse:
• il picco a sinistra rappresenta un soluto che non ha alcuna interazione con la fase
stazionaria ed esce al cosiddetto tempo morto, tM
• il picco a destra rappresenta un soluto che ha, invece, interazione con la fase
stazionaria ed esce al tempo tR > tM
23
Un parametro importante che viene usato molto spesso per descrivere la velocità di
migrazione dell’analita lungo la colonna è il fattore di capacità, k’.
k' A
n totale di moli di A nella fase stazionari a K A VS
n totale di moli di A nella fase mobile
VM
poiché CS / CM = KA .
VS / VM = ψ; k’A = KA × ψ.
Ma k’ è anche uguale al rapporto dei
tempi, per cui
k' A
tR tM
tM
Due sostanze saranno separabili se presentano valori diversi di k’.
La selettività quantifica l’entità della separazione fra due specie: riguarda la
capacità di un sistema cromatografico di distinguere fra due componenti ed è
dipendente dalla distribuzione relativa delle specie fra la fase mobile e quella
stazionaria, con (tR)B> (tR)A.
k'B ( tR )B tM
k' A ( tR )A tM
24
Se con Q indichiamo la portata della fase mobile (in inglese “flow rate”), Q
= V/t; t =V/Q, ed essendo Q=costante, se indichiamo il volume di ritensione
con VR ed il volume di fase mobile con VM, abbiamo
tR – tM / tM = VR - VM / VM = KA × VS / VM
VR - VM = KA × VS; V’R = KA × VS
25
RELAZIONI TERMODINAMICHE
La costante di equilibrio tra le fasi può essere messa
in relazione con la energia libera del processo
(funzione di Gibbs)
log K = - ΔG0/2,3RT
E ricordando la relazione tra K e k’
log k’ = log VS/VM - ΔG0/2,3RT
Log k’ = logψ - ΔH0/2,3RT + ΔS0/2,3R
26
Ma anche
log K = - ΔH0/2,3RT + ΔS0/2,3R
log VR’ = - ΔH0/2,3RT + ΔS0/2,3R – log Vs
27
28
EQUILIBRI CHIMICI SECONDARI
Quando più di un processo contribuisce alla ritenzione,
trascurando gli effetti di correlazione, possiamo scrivere:
k’ = Σi ψi Ki = Σi k’ i
Se fi e fj sono, rispettivamente, la frazione di soluto in
forma i e j si ha:
k’ = fi k’i +fj k’j
Es.
AH ↔ A– + H+ ; Ka = [A–] [H+] / [AH] ; f– = [A–] / [AH] + [A–]
f– = 1/{1 + ([H+] / Ka)} ;
k’ = 1/{1 + ([H+] / Ka)} k’ A–+ (1 - f–) k’ AH
29
Dispersion Forces
• London's dispersion forces arise from charge fluctuations throughout a
molecule resulting from electron/nuclei vibrations.
Ud = 3hν0α2 /4r6
•
•
•
•
where (α) is the polarizability of the molecule,
(ν0) is a characteristic frequency of the molecule,
(h) is Plank's constant,
and (r) is the distance between the molecules
30
Dipole-Dipole Interactions
• The interaction energy (UP) between two dipolar molecules is
given, to a first approximation, by
• Up =2α2µ2 /r6
• where (α) is the polarizability of the molecule,
• (µ) is the dipole moment of the molecule,
• and (r) is the distance between the molecules.
31
Dipole-Induced-Dipole Interactions
32
Ionic Forces
33
Interazione soluto-fasi
Le interazioni che si verificano tra le sostanze da separare e le due fasi
(mobile e stazionaria) sono deboli: se così non fosse non ci sarebbe
trattenimento sulla fase stazionaria oppure, al contrario, eluizione. Sono
sfruttate a scopo separativo le seguenti interazioni:
• legami a idrogeno
• interazioni dipolo-dipolo
• interazioni dipolo-dipolo indotto
• forze di Van der Waals
• formazione di composti di interazione
• attrazione coulombiana
• interazioni steriche
In tutte queste interazioni svolge un ruolo solitamente decisivo la polarità
delle due fasi. Spesso possono essere presenti più tipi di interazione nello
stesso processo cromatografico
34
Hydrophobic and Hydrophilic
Interactions
• n-heptane and water are immiscible, not
because water molecules repel n-heptane
molecules, they are immiscible because the
forces between two n-heptane molecules and
the forces between two water molecules are
much greater than the forces between a nheptane molecule and a water molecule.
Thus, water molecules and n-heptane
molecules interact very much more strongly
with themselves than with each other.
35
Molecular Forces and Chromatographic
Selectivity
• To choose a suitable stationary phase for a
particular separation it is necessary to select a
substance with which the solutes will interact
relatively strongly. If the solutes to be
separated are predominantly dispersive, then
a hydrocarbon-like stationary phase would be
appropriate.
36
Chromatogram of the Hydrocarbons Contained
in Unleaded Gasoline Using a Dispersive (Nonpolar) Stationary Phase
37
Two separations by GC illustrate the different selectivity
that can be obtained by using dispersive or polar stationary
phases
Polyethylene Glycol (polar)
Carbopack (dispersive)
38
IONIC INTERACTION
39
Meccanismi della separazione
In base ai tipi di interazione prima descritti possiamo suddividere i
meccanismi di separazione impiegati in cromatografia in:
• adsorbimento
• ripartizione
• scambio ionico
• affinità
• esclusione
Il meccanismo di esclusione dimensionale è dovuto solo all’ingombro sterico
è non prevede interazioni con la fase stazionaria
.
40
Adsorbimento
La fase stazionaria è un solido di granulometria piccola, impaccato
in colonna o steso su un supporto; sulla superficie dei granuli si
trovano siti attivi che possono stabilire legami deboli (reversibili!)
con le molecole della miscela da separare. Si parla quindi di
cromatografia di adsorbimento, che può essere gas-solido o
liquido-solido a seconda della natura della
fase mobile
La cromatografia di adsorbimento è
utilizzata per separare sostanze
neutre polari o non polari, di natura
organica o inorganica
Ripartizione
La fase stazionaria è un liquido che impregna un solido
granulare inerte o è ad esso chimicamente legato; in
questo liquido le molecole da separare sono solubili; la
fase stazionaria e la fase mobile devono invece essere
immiscibili. Durante l’eluizione le molecole si ripartiscono
dinamicamente tra le due fasi secondo la diversa solubilità
di ognuna.
Si parla quindi di
cromatografia di ripartizione,
che può essere
gas-liquido o liquido-liquido
a seconda della
natura della fase mobile
Scambio ionico
La fase stazionaria è costituita da un polimero o da silice
contenente siti attivi ionizzati o ionizzabili, i cui controioni possono
essere scambiati con altri ioni aventi carica dello stesso segno. Il
meccanismo di separazione è basato sulla competizione per i siti di
scambio tra gli ioni presenti nella fase mobile e quelli presenti nel
campione. Si parla di cromatografia di scambio ionico (IEC)
La cromatografia a
scambio ionico è
impiegata per la
separazione di sostanze
ioniche o ionizzabili
43
Affinità
In questo caso si utilizzano reazioni di tipo chimico o
biochimico, reversibili e molto specifiche, in modo che le
molecole da separare interagiscano con la fase stazionaria e
si ottenga così l’eluizione selettiva di alcuni componenti della
miscela. Si parla di cromatografia di affinità (AFC)
La cromatografia di affinità è
impiegata nella separazione
di molecole di interesse
prevalentemente biochimico
44
Esclusione dimensionale
La fase stazionaria è un solido poroso o un gel. Le molecole
dell’analita, disciolte nella fase mobile, penetrano nei pori se
le loro dimensioni sono compatibili e vi rimangono per un
certo tempo; le molecole più grandi sono invece escluse dai
pori ed escono dalla colonna in tempi brevi
Si parla di cromatografia di esclusione
dimensionale (SEC) con le varianti Gel
permeazione per la separazione di
sostanze insolubili in acqua e Gel
filtrazione per la separazione di
sostanze solubili in acqua
La tecnica è impiegata per la
separazione di molecole di grandi
dimensioni
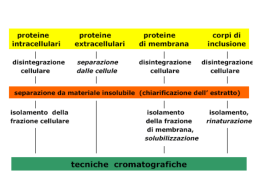
CLASSIFICAZIONE DEI METODI CROMATOGRAFICI
Non è più usata
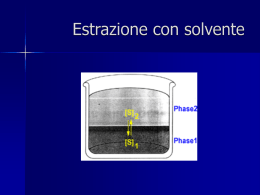
Equilibrio tra le fasi
Dc = C1/C2, dove C indica la somma della concentrazione
delle varie forme in cui può essere presente la sostanza che si
ripartisce tra le fasi.
Equilibrio
FASE 1
prima
dopo
0
V1
C1
V1
C0
V2
C2
V2
Concentrazione
Volume
FASE 2
Concentrazione
Volume
47
Scaricare