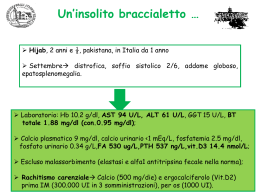
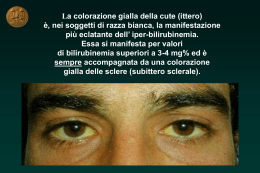
La Colestasi Neonatale Marco Sciveres MD Gastroenterologia, Epatologia e Trapianto di fegato pediatrico ISMETT, Palermo La bile Sospensione micellare in mezzo acquoso • Funzione digestiva (acidi biliari emulsione dei lipidi) • Funzione escretrice (bilirubina, colesterolo, esotossici, rame, zinco ed altri oligoelementi) 2 1 La bile Principali componenti: Sali biliari Colesterolo Fosfolipidi Bilirubina Elettroliti Oligoelementi Acqua 3 Formazione e secrezione della bile 620 ml/24 ore •Epatociti 70% Acidi biliari dipendente +++ •Colangiociti 30% 4 2 Colestasi genetiche Malattia Cromosoma PFIC1, GFC, BRIC1 PFIC2, BRIC2 PFIC3, ICP 18 2 7 Inborn errors in primary bile acid synthesis 16 7 8 20 14 7 3 2/9 15 9 16 15 2 Alagille Syndrome α1 antitrypsin deficiency Cystic fibrosis Sclerosing cholangitis (ichthyosis) Biliary atresia (polysplenia sd) Aagenaes syndrome Amish hypercholanemia NAICC ARC syndrome Progressive cholestasis childhood Gene FIC1 alias ATP8B1 BSEP alias ABCB11 MDR3 alias ABCB4 3β -HSD(HSD3B7) ∆4-3oxo-5β R (AKR1D1) oxystérol 7α OHase (CYP7B1) Jagged 1 α1 antitrypsin CFTR Claudin CFC1 / INV ? ? ZO-2, BAAT Cirhin VPS33B Villin? 5 Definizione di colestasi neonatale Qualsiasi ittero che compare entro 24 ore, dura più di 15 giorni o supera i 11 mg/dl è da considerarsi patologico e merita l’esecuzione del dosaggio delle frazioni della bilirubina. Una quota di bilirubina diretta > 10% è patologica Segni clinici della colestasi neonatale: ittero, decolorazione delle feci, urine ipercromiche, epatomegalia (+/-) Frequenza 1/2500 nati Colore delle feci !!! 6 3 Oltralpe… 7 Cause di colestasi neonatale Malattie dell’epatocita 33% • PFIC 10% • Deficit α1−antitripsina 10% • Colestasi benigna transitoria 10% • Infezioni (E. Coli, virus) 2% • Malattie metaboliche 1% Vie biliari intraepatiche • Sindrome d’Alagille • Fibrosi cistica 15% 14% 1% O. Bernard. Comunicazione personale. 8 4 Cause di colestasi neonatale Via biliari intra ed extraepatiche • Atresia delle vie biliari 47% 45% • Colangite sclerosante neonatale 2% Vie biliari extraepatiche 5% • Litiasi biliare • Perforazione/cisti/stenosi del coledoco O. Bernard. Comunicazione personale. 9 Cause di colestasi neonatale In ordine di probabilità… 1. Atresia vie biliari 45% 2. S.me di Alagille 14% 3. PFIC 10% 4. Deficit α-1-AT 10% 5. Col. transitoria 10% 6. Litiasi, malattie VBE 5% 7. Infezioni 2% 8. Colangite sclerosante 2% 9. Mal. Metaboliche 1% 10. Fibrosi cistica 1% ALTRE VBE CBT AVB A1-AT PFIC AGS 10 5 Take home message 1 • Neonato itterico = guardare le feci prima di ogni esame !!! • Sospetto di colestasi = somministrazione di Vit K parenterale (rischio emorragia!!) • L’atresia delle vie biliari è l’unica condizione che si beneficia di una terapia precoce (Intervento secondo Kasai) • Colestasi neonatale = AVB fino a prova del contrario • Colestasi neonatale = Diagnosi urgente • Colestasi neonatale = inviare presso il centro di riferimento • Ruolo del pediatra e della neonatologia +++ 11 Karim • • • • • Primogenito, genitori non consanguinei Anamnesi familiare muta Nato a termine, termine, parto spontaneo, spontaneo, PN 4100 g Ittero “fisiologico” fisiologico”, trattato con fototerapia. fototerapia. Colore del meconio? meconio? MA.. • Ad un mese di vita.. ..ancora ..ancora itterico, itterico, feci acoliche ed urine ipercromiche. ipercromiche. 12 6 Karim • • • • • • 45 giorni di vita Itterico, Itterico, fegato a 2 cm dal’ dal’arco, arco, lievemente indurito GB 9130, Hb 9.6 g/dl, Piastrine 391000, Bilirubina tot/dir 13.3/10.45 mg/dl, AST 8.8 x N, ALT 5.6 x N, GGT 688 U/l, albumina 3.2 g/dl, PT 99% α1-antitripsina 1.03 g/l Rx rachide, rachide, ecocardiografia normali ECOGRAFIA: Fegato ad ecostruttura omogenea. omogenea. Colecisti visualizzata (dt 1.4 x 0.6 cm) non particolarmente sovradistesa. sovradistesa. 13 Karim •ColangioRM 14 7 Karim • Esplorazione chirurgica • Colecistografia intraoperatoria: dotto cistico e coledoco normali e colecisti ipoplasica, non visualizzate le vie biliari intra-epatiche nè il carrèfour biliare. Portoenteroanastomosi secondo Kasai 15 Protocollo diagnostico Colestasi + decolorazione franca e persistente delle feci Ecografia: Vie biliari dilatate? SI NO Litiasi S.me Alagille? Perforazione Deficit a1-antitripsina? Cisti del coledoco ATRESIA BILIARE escluse (Fibrosi Cistica?) Colangiografia +/- esplorazione chirurgica Colangite sclerosante 16 8 Matilde • Primogenita, genitori non consanguinei. Parto a termine, spontaneo. PN 3.450 g. Feci riferite normocromiche. • A 24 gg di vita: macroematuria, IVU, ospedalizzazione • Durante la degenza si evidenzia un ittero a bilirubina diretta e feci ipocoliche. • Feci 17 Matilde • 34 gg di vita • Itterica, fegato palpabile a 2 cm dall’arco, di consistenza quasi normale. • Feci ipocoliche che si “schiariscono” di giorno in giorno. • GB 16.400, Hb 13.4 g/dl, piastrine 744.000 • Bilirubina tot/dir 7,05/5,4 mg/dl, AST 4 x N, ALT N, GGT 246 U/l, albumina 3,3 g/dl, PT 127% • Ecocardiografia e RX rachide normali • ECOGRAFIA: Fegato a margini regolari, colecisti non visualizzata. 18 9 Matilde Esplorazione chirurgica + colangiografia Assenza del coledoco, colecisti abbozzata e priva di lume, trasformazione fibrosa delle vie biliari extraepatiche fino alla confluenza col cistico. Portoenteroanastomosi secondo Kasai 19 Matilde. Esplorazione chirurgica 20 10 Protocollo diagnostico 2 Colestasi + decolorazione incompleta o intermittente delle feci + GGT elevate Ecografia (RM) Litiasi Perforazione SI NO Escludere: Vie biliari dilatate? AGS, DeficitA1AT, FC Cisti del coledoco Valutare tutte le cause di Ecografia (RM) NO Segni suggestivi di AVB? Colestasi neonatale SI Colangiografia +/- esplorazione chirurgica Biopsia 21 Protocollo diagnostico 2 Colestasi + decolorazione incompleta o intermittente delle feci + GGT elevate Ecografia (RM) Litiasi Perforazione SI Vie biliari dilatate? NO Escludere: AGS, DeficitA1AT, FC Cisti del coledoco Valutare tutte le cause di Colestasi neonatale NO Ecografia (RM) Segni suggestivi di AVB? SI Biopsia Colangiografia +/- esplorazione chirurgica 22 11 Take home message 2 “TRAPPOLE” DA EVITARE!! •In neonatologia: feci (o meconio!!) decolorate: normale •Dal pediatra a 15 gg: ittero prolungato: fisiologico •Dal pediatra a 30 gg: ittero banale da latte materno •Ecografia a 40 gg: via biliare principale normale (!!) •In ospedale: feci falsamente colorate da latti sintetici •A tutte le età: epatite neonatale banale (CMV, infezioni) •Buona crescita ponderale 23 Atresia delle vie biliari 24 12 Atresia delle vie biliari Colangiopatia infiammatoria fibrosante con esito in stenosi/obliterazione segmentaria /diffusa delle vie biliari intra ed extraepatiche Causa sconosciuta Esordio neonatale Rara. Incidenza variabile da 1: 20.000 (Francia, UK) a 1:10.000 (Giappone) fino a 1: 3.000 (Polinesia francese). Attesi circa 40 casi/anno in Italia Evoluzione spontanea: letale nel 90% dei casi entro i 3 anni di vita 25 AVB Varianti cliniche Forma embrionaria 10% dei casi Meconio decolorato o feci precocemente acoliche Anomalia di sviluppo delle vie biliari? Sindrome malformativa associata (sindrome da polisplenia) Origine genetica?? Geni implicati: INV, CFC1, ZIC3 (geni di lateralizzazione) 26 13 AVB Varianti cliniche Forma perinatale 90% dei casi Feci normali alla nascita che si decolorano rapidamente reazione infiammatoria/immune. Alterazione secondaria delle vie biliari Virus? (reovirus, rotavirus A e C, betaretrovirus, etc..) Ischemia? GVH materna? 27 AVB ipotesi eziologiche 28 14 AVB Varianti anatomiche 29 AVB Trattamento Portoenterostomia secondo Kasai 30 15 AVB Sopravvivenza con fegato nativo Totale 263 pazienti 23% (63) dei pazienti > 20 anni 23% Bilirubina < 1 mg/dl: 21 pazienti ALT e GGT N: 12 pazienti 7 gravidanze (9 figli) 31 Salvatore • Primogenito di genitori non consanguinei. Gravidanza normale conclusa con TC. PN 3520 g. • Dalla nascita feci color mastice. Colore del meconio sconosciuto. • Ittero a bilirubina prevalentemente diretta • Un cugino di primo grado, ormai adolescente, con una “vecchia” diagnosi di “paucità delle vie biliari” • Un papà con un viso particolare.. 32 16 Salvatore All’ingresso.. Neonato, normoreattivo, cute e sclere itteriche. • Soffio sistolico 1-2/6, appena udibile anche posteriormente. • Fegato a 2-3 cm dall’arco costale, di consistenza leggermente aumentata. Milza non apprezzabile. • Feci color mastice. • Bilirubina totale/diretta 9.67/ 7.43 mg/dl, AST 4 x N ALT 5 x N gGT 2 x N Colesterolo totale 167 mg/dl, Attività protrombinica 100 % 33 Salvatore Ecocardio: Stenosi all’origine del ramo sinistro dell’arteria polmonare con gradiente sistolico massimo di circa 45 mmHg. Ramo destro dell’arteria polmonare di buon calibro 34 17 Salvatore Studio del rachide: L5 a “farfalla” 35 Salvatore: Sindrome di Alagille Ricapitolando.. • Ittero colestatico a bilirubina diretta. • Facies tipica • Ecografia epatobiliare: Colecisti presente, alitiasica. • RX colonna: L5 a farfalla • Consulenza oculistica assenza di embriotoxon, cristallino trasparente. • Ecocardio: Stenosi all’origine del ramo sinistro dell’arteria polmonare • Mutazioni JAGGED 1: mutazione Arg184Leu in eterozigosi di origine paterna 36 18 Salvatore 37 Sindrome di Alagille 38 19 Sindrome di Alagille: facies 39 AGS: stenosi periferica dell’arteria polmonare 40 20 AGS: embriotoxon 41 AGS: vertebre a farfalla 42 21 AGS: paucità duttulare AH VP VP VB AE Fegato AGS Fegato Normale 43 AGS: I 5 criteri maggiori N° Fegato Cuore Occhio Vertebre Facies Lykavieris 172 161/172 162/172 119/142 123/155 165/170 Depreterre 27 25/27 26/27 9/16 6/18 19/27 Emerick 92 88/92 90/92 65/83 37/71 86/92 QuinosQuinos-Tejeira 43 43/43 42/43 16/22 12/32 42/43 334 317/334 320/334 209/263 95 % 96 % 79 % 178/276 312/332 64 % 94 % 44 22 AGS: anomalie renali Presenti nel 60% dei pazienti •Mesangiolipidosi •Tubulopatia •Nefropatia interstiziale •Agenesia renale •Ipoplasia renale •Cisti renali isolate •Rene multicistico •Rene a ferro di cavallo •Insufficienza renale 45 AGS: sindrome plurimalformativa Anomalie cardiache T. di Fallot, DIV, DIA Anomalie vascolari Anomalie scheletriche Coartazione/stenosi aortica, stenosi arterie renali, ipoplasia arterie iliache, stenosi arterie mesenteriche, malformazioni circolo cerebrale, Moya Moya. Spina bifida, clinodattilia, sinostosi radio-ulnare, Anomalie dell’orecchio Agenesia dei canali semicircolari, otite cronica, sordità Anomalie oculari Retinite pigmentosa, cataratta, miopia, strabismo, glaucoma, « drusen » del NO, ectopia pupillare cheratocono, distrofie maculare Anomalie pancreatiche Insufficienza pancreatica esocrina, diabete mellito Anomalie polmonari Malformazioni digestive Anomalie del laringe Stenosi tracheali e bronchiali Atresie/stenosi intestinali Voce « acuta » 46 23 AGS: gene JAG 1 La proteina Jag-1 è ligando della famiglia di recettori Notch. Ruolo nella differenziazione dei tessuti ectodermici e nell’angiogenesi. Localizzazione: 20p12.1 47 AGS: gene JAG-1 Trasmissione autosomica dominante ad espressività variabile Assenza di correlazione genotipo-fenotipo Cuore Colestasi Vertebre a farfalla 48 24 AGS: prognosi Sopravvivenza complessiva A 10 anni: 68% 75% Esordio tardivo A 20 anni: 62% 30% Cause del decesso Esordio neonatale Cause epatiche: 33% Cause extraepatiche: 67% Esordio neonatale = prognosi peggiore 49 Take home message 3 SINDROME DI ALAGILLE: • Causa frequente di colestasi neonatale (14%) : meglio escluderla prima dell’esplorazione chirurgica • Ampio spettro fenotipico • Forme neonatali: epatopatia più severa • Arteriopatia multiorgano !! • Nessuna terapia specifica • Forme epatiche severe: trapianto di fegato • Mortalità da cause extraepatiche prevalente 50 25 Tutta colpa di Jacob Byler?.. e Nancy?? Jacob Byler Nancy Kauffman 51 Colestasi a GGT normali 52 26 Colestasi familiari progressive (PFIC 1-2) a GGT normali P-type ATPase: FIC1 canalicular bile-salt–export pump: BSEP 53 PFIC 1: ruolo della proteina FIC1 Ipotesi: • FIC1 trasporta la fosfatidilserina e la fosfatidiletanolamina dal lato esterno a quello interno della membrana cellulare. • Questa attività può regolare la funzione di trasportatori (es. BSEP e trasporto canalicolare degli acidi biliari ) attraverso il mantenimento dell’asimmetria della membrana. 54 27 PFIC 2: ruolo della proteina BSEP 55 PFIC a GGT normali: caratteristiche comuni • Clinica: colestasi criptogenetica (dai primi mesi o anni di vita), prurito severo • Biochimica: Attività sierica GGT e colesterolo persistentemente normali • Istologia: non vera proliferazione duttulare (metaplasia biliare degli epatociti), fibrosi lobulare, colestasi canalicolare, cirrosi • Radiologia: albero biliare normale • Bile: bassa concentrazione di sali biliari • Evoluzione: litiasi biliare, cirrosi, insufficienza epatica • Trattamento: Acido Ursodessossicolico, (diversione biliare), trapianto di fegato 56 28 PFIC a GGT normali: differenze Colestasi Insufficienza epatica Segni extraepatici ALT AFP colelitiasi Istologia epatica PFIC1 PFIC2 Infanzia. Ricorrente o persistente Neonatale. Persistente Ritardata o assente Precoce Possibili (diarrea, scarsa crescita) assenti 2xN normale 10 X N elevata no ? si Colestasi canalicolare Fibrosi lieve Colestasi canalicolare infiammazione fibrosi severa epatociti giganti 57 Colestasi familiari progressive (PFIC 3) a GGT elevata phospholipid transporter multidrugresistance-3 Pglycoprotein 58 29 PFIC 3: ruolo della proteina MDR3 MDR3 or ABCB4 (7q21): Codifica la proteina MDR3 che trasporta I fosfolipidi attraverso la membrana canaliculare dell’epatocita. La secrezione biliare dei fosfolipidi è cruciale nel proteggere la membrana dei colangiociti dagli effetti tossici di alte concentrazioni di acidi biliari ad effetto detergente. 59 Fenotipo PFIC 3: difetto di MDR3* • • • • • • • Clinica: colestasi cronica criptogenica (dai primi mesi all’età di giovani adulti), prurito lieve. Biochimica: attività GGT persistentemente elevata Istologia: proliferazione duttulare, fibrosi portale, cirrosi biliare Radiologia: albero biliare normale. Bile: fosfolipidi bassi, rapporto fosfolipidi/acidi biliari e fosfolipidi/colesterolo aumentato. Alto indice di saturazione del colesterolo Evoluzione: litiasi biliare, cirrosi, ipertensione portale, insufficienza epatica Trattamento: acido ursodesossicolico, trapianto di fegato *Jacquemin E et al. Gastroenterology 2001; 120:1448 60 30 Take home message 4 COLESTASI FAMILIARI: • Alto livello di sospetto (PFIC 1-2) in presenza di colestasi a GGT normali. • Ampio spettro fenotipico • Esordio neonatale: + PFIC2 • Prognosi eventualmente severa • Possono giovarsi dell’acido ursodesossicolico • Caratterizzazione fondamentale per la comprensione della fisiopatologia della secrezione biliare nell’uomo 61 Protocollo diagnostico 3 Colestasi + decolorazione incompleta o intermittente delle feci GGT normali Ecografia (RM) Litiasi Perforazione SI Vie biliari dilatate? NO Sospetto PFIC 1-2, deficit di sintesi AB Escludere: AGS, DeficitA1AT, FC Cisti del coledoco Valutare tutte le cause di Colestasi neonatale NO Ecografia (RM) Segni suggestivi di AVB? SI Biopsia Colangiografia +/- esplorazione chirurgica 62 31 Take home message 5 PRESA IN CARICO NEONATO COLESTATICO • Sorveglianza complicazioni ipertensione portale (varici esofagee ++) • Carenza in vitamine liposolubili: ADEK parenterali • Malassorbimento: incremento dell’apporto calorico, acidi grassi a media catena, nutrizione enterale continua, nutrizione parenterale. • Miglioramento del flusso biliare: UDCA • Prurito: Rifampicina 63 Indicazioni al trapianto di fegato Patients who underwent Transplantation (n=141) (n=206) Other 22% Cholestatic Disease (n=161) Cholestatic Disease 78% Biliary Atresia (n=123) Byler (n=12) Sclerosing Cholangitis (n=2) Alagille (n=17) Other (n=7) 32 Grazie 65 33
Scaricare