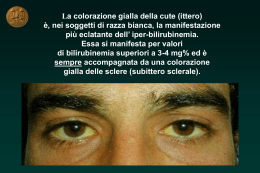
Il bambino con transaminasi elevate Massimo Resti Unità Operativa Pediatria Medica, Epatologia Pediatrica, Azienda Ospedaliero-Universitaria Meyer, Firenze Di fronte a un bambino che presenta un aumento delle transaminasi, indipendentemente dai valori il primo passo da compiere per procedere in maniera sistematica, è fare un’accurata anamnesi. All’anamnesi familiare devono essere indagati consanguineità dei genitori, precedenti aborti, decessi infantili o neonatali sine causa. Questi reperti possono suggerire che alla base dell’ipertransaminasemia ci possa essere una patologia di natura metabolico-ereditaria. A questo proposito è opportuno identificare la presenza di familiari con epatopatie. Ad esempio, la presenza di familiarità per colelitiasi precoce è frequente nei bambini con litiasi biliare. Il contatto stretto con persone affette da epatite B o C, una storia di trasfusioni ematiche in era prescreening o la familiarità per malattie autoimmuni potrebbero orientare in modo semplice ma deciso la diagnosi. Se scrupolosa, l’anamnesi è utile anche per porre il sospetto di malattie epatiche rare. Una storia di colestasi gravidica o successiva all’assunzione di estrogeni da parte della madre è suggestiva di una colestasi familiare intraepatica di tipo 3, patologia rara che fa parte del gruppo delle colestasi familiari progressive intraepatiche. La presenza di familiari con enfisema polmonare in età giovanile, invece, deve far prendere in considerazione un possibile deficit di α-1-antitripsina. L’anamnesi fisiologica e patologica remota sono allo stesso modo importanti. Devono essere indagati l’uso e l’abuso di farmaci in gravidanza (ipertransaminasemie neonatali da assunzione di paracetamolo materno) e valutati gli esami di screening effettuati nel corso del periodo gestazionale per confermare o escludere eventuali malattie infettive a trasmissione verticale che possono interessare il fegato. Le informazioni ottenute devono poi essere integrate con i dati che fanno riferimento all’epoca neonatale: peso alla nascita, crescita postnatale, giornata di comparsa e durata dell’ittero e tipo di allattamento effettuato. Le domande rivolte al padre e alla madre del bambino, inoltre, devono essere mirate a ottenere informazioni sul suo sviluppo psicologico e motorio, sui suoi comportamenti e le sue abitudini. Ad esempio, una ritardata acquisizione delle tappe motorie potrebbe essere la manifestazione di una patologia muscolare non ancora evidente clinicamente, ma testimoniata dall'aumento delle transaminasi che in questo caso sarebbero di origine muscolare. Inoltre, una storia caratterizzata da ridotto accrescimento staturo-ponderale a seguito del divezzamento e dell’introduzione del glutine rappresenta spesso il primo segno di una malattia celiaca in cui ALT e AST possono essere alterate. 1 È importante indagare eventuali viaggi in paesi ad alta endemia di epatiti infettive e la possibile ingestione di cibi contaminati che costituiscono oggi i fattori di rischio più importanti per l’epatite A. L’insieme di questi elementi può condizionare l’iter diagnostico, evitando sprechi di tempo ed esecuzione di esami inutili. Esame Obiettivo L'esame obiettivo del paziente con ipertransaminasemia inizia con la valutazione dello stato generale e delle caratteristiche fisiche del paziente. Come già detto, uno scarso accrescimento staturo-ponderale può essere causato dalla malattia celiaca; l'obesità può essere accompagnata da steatosi e steatoepatite. Una facies particolare con fronte bombata, mento appuntito, occhi infossati e ipertelorismo può essere notata da un occhio attento inducendo il sospetto di sindrome di Alagille (colestasi cronica da scarsità dei dotti biliari associata a difetti cardiaci, manifestazioni oculari, anomalie scheletriche e facies tipica). Alterazioni particolari del volto possono essere presenti anche in alcune malattie metaboliche, come le glicogenosi. Durante la valutazione clinica, la ricerca di subittero e ittero costituisce una tappa preliminare. Questi segni, infatti, possono essere indicativi sia di una sindrome colestatica sia di un’epatite acuta o cronica. Tipiche della colestasi, a livello cutaneo, sono le lesioni da grattamento e la presenza di xantomi e xantelasmi. Inoltre, di fronte ad un paziente itterico, è necessario ricercare la presenza di eritema palmare e spider naevi, spesso non chiaramente evidenti ma indicativi di epatopatia cronica, ma anche reperti macroscopici come l’alterazione dello stato di coscienza, suggestivo di un’encefalopatia epatica e le manifestazioni emorragiche riconducibili a un deficit di sintesi dei fattori della coagulazione. È importante valutare le dimensioni epatiche e saggiare anche la consistenza del fegato ingrandito. L’epatomegalia è comune a molte malattie infettive del bambino ed è aspecifica, tuttavia la consistenza dura, invece che parenchimatosa, può orientare la diagnosi verso una patologia epatica cronica. La splenomegalia è allo stesso modo aspecifica, può essere dovuta a un'infezione virale come quella da Epstein-Barr Virus (EBV), ma può essere espressione di un’ipertensione portale da cirrosi soprattutto se associata a circoli venosi superficiali, eritema palmare e spider naevi . Infine, occorre porre attenzione anche segni e sintomi di patologia muscolare come l'astenia, i dolori muscolari, la difficoltà nell'assumere la posizione eretta, la presenza di pseudoipertrofia dei polpacci. Le più frequenti eziologie sono riportate nella tabella 1 e 2. 2 Bibliografia 1. Bugeac N, Pacht A, Mandel H, et al. The significance of isolated elevation of serum aminotransferases in infants and young children. Arch Dis Child. 2007;92(12):1109-12; 2. El-Koofy NM, El-Mahdy R, Fahmy ME, et al. Alagille syndrome: clinical and ocular pathognomonic features. Eur J Ophthalmol. 2011;21(2):199-206; 3. Farre C, Esteve M, Curcoy A, Cabré E, et al. Hypertransaminasemia in pediatric celiac disease patients and its prevalence as a diagnostic clue. Am J Gastroenterol. 2002;97(12):3176-81; 4. Giboney PT. Mildly elevated liver transaminase levels in the asymptomatic patient. Am Fam Physician. 2005 15;71(6):1105-10; 5. Gregorio GV, Portmann B, Reid F, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology 1997; 25: 541-547; 6. Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol. 2007;5(7):776-82; 7. Kelly DA. Managing liver failure.Postgrad Med J. 2002;78(925):660-7; 8. Loudianos G, Gitlin JD. Wilson's disease. Semin Liver Dis. 2000;20(3):353-64; 9. Mele A, Tosti ME, Spada E, et al. SEIEVA Collaborative Group. Epidemiology of acute viral hepatitis: twenty years of surveillance through SEIEVA in Italy and a review of the literature. Rapporti ISTISAN. 2006;12; 10. Mieli-Vergani G, Vergani D. Autoimmune hepatitis. Nat Rev Gastroenterol Hepatol. 2011;8(6):320-9; 11. Russmann S, Kullak-Ublick GA, Grattagliano I. Current concepts of mechanisms in druginduced hepatotoxicity. Curr Med Chem. 2009;16(23):3041-53; 3 Tabelle e Figure Tabella I. Principali cause di ipertransaminasemia in età neonatale e nel primo anno di vita Nome della patologia Infezioni virali Emocromatosi neonatale Deficit congeniti di sintesi degli acidi biliari primari Malattie metaboliche galattosemia tirosinemia malattie mitocondriali deficit α-1antitripsina malattie da accumulo di glicogeno Epatopatie ischemiche (cardiopatie congenite, miocarditi, asfissia neonatale) Epatopatie cellulari e biliari colestasi intraepatiche familiari progressive deficit FIC1 (ATP8B1) deficit BSEP (ABCB11) deficit MDR3 (ABCB4) epatite gigantocellulare con anemia emolitica atresia delle vie biliari sindrome di Alagille epatite idiopatica neonatale Caratteristiche fenotipico/diagnostiche e commenti Herpes virus (HSV 1 e 2 tipici dell’età neonatale, spesso responsabili di grave insufficienza epatica; CMV, HHV6, HHV7), enterovirus, adenovirus, parvovirus Eziopatogenesi alloimmune. Transaminasi solo ai limiti superiori della norma, insufficienza epatica già presente alla nascita, ittero, ipoglicemia, depositi di ferro nelle ghiandole salivari iperbilirubinemia coniugata, aumento delle transaminasi con normale γ-GT, malassorbimento di vitamine liposolubili, colesterolo plasmatico spesso basso o ai limiti inferiori della norma deficit di crescita, vomito, diarrea, ittero che di solito insorge dopo la prima settimana di vita, sonnolenza, epatomegalia, edemi, ascite, sepsi da Gram negativi e in particolar modo Escherichia coli, ipoglicemia. La comparsa della cataratta è solitamente tardiva ma può essere già presente alla nascita esordio clinico spesso postneonatale, grave coagulopatia, normali transaminasi, alti livelli di α-fetoproteina, ittero assente o moderato, rachitismo, nefropatia compromissione spesso multiorgano (muscolare, neurologica): ipotonia, difficoltà ad alimentarsi, convulsioni, alterata funzionalità epatica, acidosi lattica e ipoglicemia manifestazione neonatale della malattia presente in circa 1/4 dei pazienti con fenotipo PiZ: epatite neonatale idiopatica colestatica con ittero, feci acoliche e epatomegalia a insorgenza precoce. Bassi livelli sierici di α-1antitripsina le glicogenosi I, II, III, IV e VI sono le forme con prevalente interessamento epatico: i criteri di sospetto diagnostico sono dati da ipoglicemia, iperlattacidemia, iperuricemia, ipertrigliceridemia e ipercolesterolemia caratterizzate da una marcata ipertransaminasemia con funzionalità epatica inizialmente normale esordio clinico tipicamente nei primi sei mesi di vita (possibile esordio più tardivo); andamento rapidamente progressivo (cirrosi spesso entro il compimento del decimo anno di vita). Sintomi classici: prurito, deficit di crescita, steatorrea, ittero, epatomegalia, stanchezza, deficit di vitamine liposolubili, litiasi delle vie biliari, cirrosi. Possibile la trasformazione epatica maligna. Si caratterizzano per la colestasi a basse γ-GT (FIC1 e BSEP) e alte γ-GT (MDR3) patologia rara e con elevata mortalità: grave citolisi epatica, talvolta accompagnata da insufficienza epatocellulare, associata ad anemia emolitica Coombs positiva diagnosi clinica auspicabile entro i primi 45 giorni di vita: ittero, feci acoliche, urine ipercromiche, aumento delle γ-GT, coagulopatia colestasi cronica ad esordio neonatale con paucità dei dotti biliari interlobulari; anomalie cardiache, facies caratteristica, anomalie scheletriche, embriotoxon posteriore diagnosi di esclusione 4 Tabella II. Principali cause di ipertransaminasemia dopo il primo anno di vita Nome della patologia Infezioni virali Infezioni batteriche Xenobiotici Epatopatie autoimmuni Malattia di Wilson Steatosi e steatoepatite non alcolica Epatiti idiopatiche Caratteristiche fenotipico/diagnostiche e commenti virus epatotropi maggiori (HAV, HBV, HCV, HEV) EBV, CMV, rotavirus, adenovirus, enterovirus, echovirus salmonellosi, tubercolosi, bartonella, sepsi paracetamolo, isoniazide e propiltiouracile carbamazepina, sulfalazina, antibiotici (ampicillina/amoxicillina-clavulanico), antitubercolari (isoniazide), chemioterapici (ciclofosfamide, dacarbazina), anestetici (alotano), antiaritmici (amiodarone), antidepressivi (trazodone), farmaci per la tiroide e droghe ricreazionali (cocaina ed estasi). Tra gli alimenti è importante ricordare l’intossicazione da funghi (Amanita phalloides e la specie Lepiotae). ipertransaminasemia, aumento delle IgG, VES, ipergammaglobulinemia, altre malattie autoimmuni associate, autoanticorpi (ANA e anti-ML nel tipo 1, LKM nel tipo 2), colestasi (colangite sclerosante autoimmune) bassi livelli di ceruloplasmina circolante, aumentata escrezione urinaria di rame, anello di Kayser-Fleischer assume le caratteristiche dell’epidemia, patologie potenzialmente cirrogene, associate alla sindrome metabolica diagnosi di esclusione, epatiti non-A non-G 5 6
Scaricare