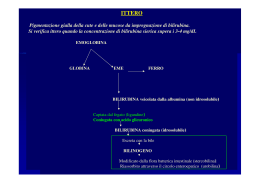
Journal Club del 31/01/2014 Scuola di Specializzazione in Pediatria Direttore Prof. G. Buonocore ITTERO NEONATALE Ed un caso particolare … Dott.ssa Cerrone Cosimina Dott. Coriolani Gianni Illustrazione del caso clinico Anamnesi R.B. è nato a 33 settimane di EG da parto spontaneo cefalico, da gravidanza normodecorsa. Segnalato polidramnios. Madre di 32 anni. Sierologia materna: immunità per Rosolia, negatività per Toxoplasmosi, HbsAg, HCV, HIV, Lue. Tampone vaginale negativo per GBS. In Isola Neonatale… Alla nascita: peso 2060 gr (50° CT), lunghezza 42 cm (25-50° CT), circonferenza cranica 33 cm, (50° CT). Apgar 8 (1')-9 (5'). Alla nascita è stata praticata SLI (sustained lung inflation) , successivamente R.B. è stato trasferito in TIN per le cure e gli accertamenti del caso. In Terapia Intensiva Neonatale… All’arrivo in TIN il piccolo è stato posto in isolette con FiO2 0,23 (SpO2: 100%, FC 146 bpm) e reperito accesso venoso centrale. All’esame obiettivo si evidenziava addome “marcatamente disteso”. Venivano eseguiti Rx torace - Diretta addome Rx torace “Non alterazioni pleuroparenchimali in atto, regolare la ventilazione polmonare” MA … ? Cosa si legge in letteratura ? “Double bubble sign” “ … The double bubble sign is a classic radiographic manifestation of duodenal obstruction, the cause of which could be intrinsic (such as duodenal atresia, duodenal stenosis or duodenal web) or extrinsic (such as annular pancreas or rotational anomalies). Identification of the typical double bubble sign requires immediate investigation, as any cause of duodenal obstruction may require surgical treatment. …” Emerg Med J 2011;28:1084 Haw-Chiao Yang et al. Tornando a R.B: Rx addome LL “distensione gastrica e del bulbo duodenale con livelli idroaerei senza apprezzabile meteorismo a valle, verosimilmente riferibile a stenosi duodenale” Nelle ore seguenti … “Incremento ulteriore della gastrectasia nota” E allora .. : L’ intervento !! In seconda giornata di vita, R.B. è stato sottoposto ad intervento chirurgico: “confezionamento di una anastomosi a diamante duodeno-duodenale e … scollamento dei monconi duodenali del pancreas anulare !!”. Immagine tratta da BMJ Case Reports 2012; Pansini et al. “Intraoperatively: dilation of the stomach (black arrowheads) and the duodenum due to a complete annular band of pancreatic tissue that surrounded and constricted the second part of the duodenum (white arrows); distal to this site the jejunum (black arrows) was collapsed and atrophic.” Che cosa è il PANCREAS ANULARE ? • “… Annular pancreas first described by Tiedemann in 1818, is a rare congenital anomaly which most commonly manifests itself in children …” [Tieddmann F (1818) Dtsch Arch Physiol 4:403]. • Normally pancreas develops from one dorsal and two ventral buds that first appear as evaginations of the primitive foregut at around 5th week of gestation. The two ventral buds fuse early. Due to selective expansion of the duodenum by about seventh week, the ventral bud rotate with gut, passing behind the duodenum from right to left and eventually fusing with dorsal bud. [Lee PC, Lebnethal E (1993) Prenatal and postnatal development of the human exocrine pancreas. In: Go VL, Dimagno EP, Gardner JD et al (eds) Pancreas: Pathology, pathobiology and disease 2nd ed. Raven Press, NY, p 57]. Drawings illustrate the normal embryologic development of the pancreas and biliary tree. The ventral pancreatic bud (arrow in a and b) and biliary system arise from the hepatic diverticulum, and the dorsal pancreatic bud (arrowhead in a and b) arises from the dorsal mesogastrium. After clockwise rotation of the ventral bud around the caudal part of the foregut, there is fusion of the dorsal pancreas (located anterior) and ventral pancreas (located posterior). Finally, the ventral and dorsal pancreatic ducts fuse, and the pancreas is predominantly drained through the ventral duct, which joins the common bile duct (CBD) at the level of the major papilla. The dorsal duct empties at the level of the minor papilla. [Mortelé KJ et al. Multimodality imaging of pancreatic and biliary congenital anomalies. Radiographics 2006;26:715–31.] “ .. Annular pancreas is a rare congenital anomaly in which incomplete rotation of the ventral anlage leads to a segment of the pancreas encircling the second part of the duodenum” [Choi BI et al. Radiology 1990; 174:161–163]. Annular pancreas has a prevalence of one in 2,000 persons (> Males) and occurs either as an isolated finding or with other congenital abnormalities including down’s syndrome, tracheoesophageal fistula, esophageal atresia, imperforate anus and hirschprung disease [Nijs E, et al. Pediatr Radiol 2005;35:358–373.;Jimenez Jcet al. J Pediatr Surg 2004;39:1654–1657.] In approximately one-half of symptomatic cases, annular pancreas will manifest in the neonate with gastrointestinal obstruction or bile duct obstruction, possibly associated with pancreatitis. In general, annular pancreas obstructs the duodenum in 10% of cases. [Mortelé KJ, et al. Radiographics 2006;26:715–31.] There are two types of annular pancreas: the extramural type and the intramural type [Choi BI et al. Radiology 1990; 174:161–163]. Annular pancreas can be diagnosed on the basis of CT and MR imaging findings that reveal pancreatic tissue and an annular duct encircling the descending duodenum [Leyendecker JR, AJR Am J Roentgenol 2002;179:1465– 1471.] [Mortelé KJ, et al. Radiographics 2006;26:715–31.] Annular pancreas. (a) Axial T2-weighted MR image shows the pancreas (arrow) encircling the descending portion o the duodenum. (b) Coronal MR cholangiopancreatogram shows the duct of Wirsung (arrowhead) encircling the duodenum. Surgical bypass of the duodenum is indicated in severe stenosis. Local resection of the annular segment is avoided because of the fear of development of pancreatic fistula and is often difficult because of dense adhesions due to local fibrosis. Duodeneo-duodenostomy or duodeno-jejunostomy are the procedures of choice [Thomford NR, (1972). Ann Surg 176:159]. Gastrojejunostomy is an alternative option in case of grossly fibrotic duodenal C-loop [De Ugarte DA, (2006) Am Surg 72:71]. Riassumendo … Dall’analisi di studi (reviews e case reports) pubblicati in letteratura emerge che: Il riscontro ecografico prenatale di polidramnios è il primo indizio di una atresia intestinale ▸ Nel periodo neonatale, il vomito è il sintomo precoce più comune ▸ Il segno della doppia bolla alla diretta addome eseguita dopo la nascita, senza evidenza di meteorismo a valle, è essenzialmente patognomonico per atresia duodenale. [BMJ Case Reports 2012; Pansini et al.] I neonati con pancreas anulare associato con ostruzione duodenale sono frequentemente prematuri o piccoli per l'età gestazionale. [ed Sci Monit. 2002 Jun;8(6):CR434-7. Sencan A et al.] I pazienti con pancreas anulare hanno ostruzione duodenale pre-ampullare, che è più comunemente parziale. Il trattamento più appropriato è la duodeno-duodenostomia. La prognosi è eccellente, nonostante la frequente associazione con anomalie cromosomiche e malformazioni congenite maggiori. [J Pediatr. Surg. 2004Nov;39(11):1654-7. Jimenez et al] Tuttavia studi di follow-up a lungo termine hanno evidenziato: Elevata incidenza (70-80% !!) di complicanze post-operatorie quali: megaduodeno con sindrome dell’ansa cieca, gastrite da reflusso biliare, ittero colestatico, reflusso gastroesofageo, transito intestinale ritardato, ostruzione intestinale, ritardo di crescita, diarrea cronica. [J Pediatr Surg 1990 Nov;25(11):1127-30. Spigland et al.; Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi.1998 Mar-Apr;39(2):89-93. Lyn YT et al. ] Il ristagno e l’ostruzione funzionale nel duodeno prossimale sono i principali fattori che influenzano la morbilità tra questi pazienti (per cui si rendono necessarie tecniche chirurgiche che favoriscano il più precoce ripristino del transito intestinale). In conclusione, uno stretto follow-up a lungo termine è essenziale per i bambini trattati per pancreas anulare poiché molti di questi svilupperanno complicanze, anche se l’immediato periodo postoperatorio non è complicato e la sopravvivenza è eccellente. [J Pediatr Surg. 1990 Nov;25(11):1127-30. Complications associated with surgical treatment of congenital intrinsic duodenal obstruction. Spigland N,] Torniamo al nostro R.B. Dopo l’intervento R.B. è rimasto in ventilazione assistita per circa 48 ore (FiO2 21%). In seguito in respirazione spontanea. In sesta giornata di vita (5°post-operatoria) il quadro clinico del bambino è peggiorato: dal sondaggio gastrico oltre a bile è stata rilevata la presenza di materiale denso l’addome del piccolo ha iniziato ad essere meteorico l’Rx dell'addome mostrava segni di perforazione … “E’ apprezzabile aria libera in addome di discreta entità in esiti di intervento di atresia duodenale” Perforazione intestinale, cosa si fa? R.B. è stato quindi sottoposto a nuovo intervento chirurgico con sutura della precedente anastomosi, posizionamento di drenaggio peritoneale e applicazione di gastrostomia. Rx di controllo post-intervento: “Non segni di perforazione di visceri cavi addominali. E’ presente minima quantità di aria libera in prossimità della regione gastrica compatibile con esiti di recente intervento e dalla presenza di drenaggio intraaddominale” Ed ora cosa succede .. ? In decima giornata di vita dal drenaggio peritoneale ha iniziato ad essere apprezzabile fuoriuscita di liquido biliare (probabile peritonite biliare), tuttavia le radiografie dell’addome non mostravano segni di pneumoperitoneo e lo studio con gastrografin non mostrava spandimento di liquido in addome. 1 2 1) 2) 3) 3 Opacizzazione del fondo gastrico ed iniziale del corpo; Progressione del gastrografin verosimilmente nel colon ascendente Distensione con ristagno a livello dello stomaco; non segni indiretti di alterata canalizzazione con presenza di gastrografin a livello del sigma E’ stato pertanto concordato con i colleghi chirurghi di rinforzare la terapia antibiotica con Metronidazolo e Vancocina. Dal giorno 17/10 dal drenaggio peritoneale non è più comparso liquido biliare ed il giorno 21/10 il drenaggio è stato pertanto rimosso. Le condizioni di R.B. sono progressivamente migliorate ed in 20° giornata di vita è stato effettuato il primo tentativo di alimentazione per via enterale, ben tollerato. Dopo un’ulteriore settimana anche la gastrostomia è stata definitivamente rimossa. Altra problematica: l’ ITTERO Per la comparsa di ittero a bilirubina indiretta, con positività del test di Coombs diretto, R.B. ha effettuato fototerapia radiante e somministrazione di Immunoglobuline ev (valore max di BT indiretta 12,2 mg/dl in terza giornata di vita). Definizione di ittero L’iperbilirubinemia neonatale è evento fisiologico che interessa circa il 60% di tutti i nati a termine. [Task force per l’iperbilirubinemia neonatale. SIN 2013] In particolari condizioni patologiche o parafisiologiche (come il nato pretermine !) può costituire un rischio di danno neurologico acuto e cronico. L’ ittero neonatale viene classicamente distinto in : Fisiologico Patologico ITTERO FISIOLOGICO Presenta le seguenti caratteristiche: Compare nel 60% dei neonati tra il 2°- 4° giorno di vita Ittero a bilirubina indiretta; Raggiunge la massima intensità tra il 3°-5° giorno di vita nel neonato a termine e in 7° giornata nel neonato pretermine; Raramente la bilirubinemia supera i 12-13 mg/dl; L’aumento della bilirubinemia è <0,5 mg/dl/ora È causato da aumentata sintesi della bilirubina (emolisi dei GR fetali), ridotta capacita del fegato di captare la bilirubina dal sangue, deficit di coniugazione epatica, deficit di escrezione e aumentato circolo entero-epatico. [Task force per l’iperbilirubinemia neonatale. SIN 2013] ITTERO PATOLOGICO Si distingue dall’ittero fisiologico perché generalmente: Compare nelle prime 24 ore di vita; La bilirubinemia spesso supera i 15 mg/dl; L’aumento della bilirubinemia è >5 mg/die; L’ittero si può prolungare oltre i primi 14 giorni di vita del neonato a termine e oltre i primi 21 giorni nel pretermine; Può essere a bilirubina indiretta o diretta Richiede sempre il trattamento [Task force per l’iperbilirubinemia neonatale. SIN 2013] AUMENTO BILIRUBINA INDIRETTA Aumentata produzione di bilirubina Ittero fisiologico MEN Anemia emolitica Sferocitosi G6PD Policitemia [Hct > 65%] Riassorbimento di ematoma Diminuita eliminazione per scarsa coniugazione epatica Latte materno Sd Gilbert Sd Crigler-Najjar Ipotiroidismo Farmaci [Task force per l’iperbilirubinemia neonatale. SIN 2013] A) CAUSE PIU' COMUNI DI IPERBILIRUBINEMIA INDIRETTA IPERBILIRUBINEMIA NEONATALE TRANSITORIA INCOMPATIBILITA' ABO: si verifica quando la madre è di gruppo O e il feto di gruppo A o B. IMMUNIZZAZIONE MATERNO-FETALE DEL SISTEMA Rh: causata da anticorpi anti-D prodottida madri Rh negative contro i GR Rh positivi del feto. EMATOMA SUBDURALE, CEFALOEMATOMA SOFFUSIONI EMORRAGICHE ESTESE NEONATO DA MADRE DIABETICA POLICITEMIA/IPERVISCOSITA' INFEZIONI (l'ittero come unico segno di sepsi è raro) ITTERO DA LATTE MATERNO: comparsa tardiva (fine 1a sett.-2a sett. di vita) picco più alto di bilirubinemia declino più lento della bilirubinemia (settimane o mesi) l'interruzione del latte per 24-48 ore provoca un rapido declino B) CAUSE MENO COMUNI DI IPERBILIRUBINEMIA INDIRETTA: DEFICIT DI G6PD: malattia legata al cromosoma X, frequente nei maschi. DEFICIT DI PIRUVATO KINASI SFEROCITOSI CONGENITA SINDROME DI CRIGLER-NAJJAR IPOTIROIDISMO EMOGLOBINOPATIE ( e TALASSEMIA) Perché trattare l’iperbilirubinemia? Complicanza temibile dell’iperbilirubinemia è l’encefalopatia. Il rischio di danno neurologico dipende da: concentrazione sierica della bilirubina indiretta, età gestazionale, condizioni genetiche (es.deficit G6PD, deficit piruvato chinasi, sferocitosi), condizioni intercorrenti (sepsi, disidratazione, acidosi, ipoalbuminemia) Sintomi: inizialmente il neonato è letargico, ipotonico, presenta difficoltà di alimentazione e vomito, successivamente diventa irritabile con pianto stridulo, tremori intensi, opistotono e convulsioni. [Journal Clinical Neonatology vol.2 Issue 2. 2013 Bhutani and Wong] [Journal Clinical Neonatology vol.2 Issue 2. 2013 Bhutani and Wong] Le prime 24-48 h di vita sono critiche !! [Journal Clinical Neonatology vol.2 Issue 2. 2013 Bhutani and Wong] Terapia Fototerapia • La fototerapia intensiva è il trattamento di scelta per ridurre (in 2-4 ore) la iperbilirubinemia non coniugata indipendentemente dalla sua eziologia. • L’impiego ottimale della fototerapia è stato definito da specifici range di bilirubina plasmatica totale che sono stati correlati all’età gestazionale, all’età post-natale (in ore), e al loro potenziale rischio per neurotossicità. [Journal Clinical Neonatology vol.2 Issue 2. 2013 Bhutani and Wong] [Task force per l’iperbilirubinemia neonatale. SIN 2013] Exanguinotrasfusione Procedura invasiva, da considerare quando compaiono segni di neurotossicità, o nei bambini asintomatici che presentano: valori di BT>25 mg/dl, o quando la fototerapia intensiva non ha determinato significative riduzioni di BT in b con severa iperbilirubinemia, o in bambini che hanno ABR alterati con precedenti OEA normali. [Journal Clinical Neonatology vol.2 Issue 2. 2013 Bhutani and Wong] I valori indicativi per il trattamento con EXT dipendono dall’età gestazione e dall’età post-natale. [Task force per l’iperbilirubinemia neonatale. SIN 2013] Opzioni farmacologiche Gli interventi farmacologici hanno un ruolo limitato durante le emergenze, includono: Infusione di albumina: quando si riscontrano bassi livelli di albumina plasmatica (non ci sono però evidenze scientifiche che supportano questa pratica); Gammaglobuline : da utilizzare in caso di iperbilirubinemia da isoimmunizzazione Fenobarbitale: aumenta l’escrezione di bilirubina a livello epatico. Non raccomandato in E.G <32 wk, inefficace <12h di vita. Tin Mesoporfirina(SnMP) Inibitore dell’enzima eme-ossigenasi, blocca la trasformazione dell’eme a biliverdina e bilirubina. Sembra causare fotosensibilizzazione (specialmente con fototerapia a luce bianca intensa). È attualmente in fase di studi farmacologici e tossicologici ed è efficace nel ridurre i livelli di BT senza significativi effetti collaterali. [NeoReviews 2007; 8:e77-84. Wong et al.] … torniamo al nostro caso: Dalla fine del mese di Ottobre R.B. ha iniziato a presentare: feci acoliche rapido incremento della transaminasemia (valori massimi GOT: 202 UI/l, GPT: UI/l 128), degli acidi biliari sierici (v. max. 82,9 um/l), della gamma-GT (v. max 120 UI/l); FA moderatamente aumentata (743 UI/L) nuovo incremento della bilirubina totale (v.max. pari a 6,8 mg/dl) con prevalenza di bilirubina diretta (v.max. 5,4 mg/dl) Albumina, PT, PTT, Fibrinogeno, INR: nella norma Aspetto delle feci di un neonato sano (a) e di un neonato con colestasi incompleta (b) e completa (c). Nel sospetto di un ittero colestatico Il… piccolo è stato pertanto sottoposto a numerosi accertamenti di tipo medico: dosaggio di alfa 1 antitripsina (178 mg/dl, da ricontrollare), valutazione complesso TORCH: negativa markers epatitici maggiori e minori: negativi funzionalità tiroidea: TSH: 0,407 uUI/ml; FT4: 11.8 pg/ml. FT3 3.3 pg/ml controlli seriati ecografici dell'addome: risultati nella norma. ITTERO A BILIRUBINA DIRETTA- COLESTASI Gli itteri colestatici in periedo neonatale si caratterizzano: Frequenza relativamente elevata (1 su 2500 nati) Molteplicità di cause; Talora prognosi grave per evoluzione in insuffienza epatica e necessità di trapianto di fegato Manifestazioni cliniche Esordio abitualmente nel 1° mese di vita post-natale, o comunque nel primo trimestre Ittero prolungato (> 2 settimane) associato a decolarazione parziale o totale delle feci (feci acoliche), urine ipercromiche, deficit di crescita e a riscontro di una epatomegalia di grado variabile; talora può esordire con un quadro di sindrome emorragica per deficit dei fattori vitamina kdipendenti. [Medico e Bambino 3/1999. Maggiore e Caprai] È un ittero a bilirubina diretta, che si diagnostica: con valori di BT <5 mg/dl: BD>1 mg/dl con valori di BT >5 mg/dl: BD >20% BT [Neoreviews 2013 14(2) Feldman and Sokol] Medico e Bambino 3/1999 Maggiore e Caprai Diagnosi: Sintomi (valutare anche eventuali dismorfismi e sintomi neurologici associati) Esami di laboratorio (I livello): incremento livelli di BT e BD; incremento di transaminasi (GOT-GPT), incremento gamma-GT (può essere normale nella colestasi familiare progressiva), aumento sali biliari, fosfatasi alcalina, severa coagulopatia (deficit fattori vitamino K-dipendenti; esame urine, urinocoltura (escludere una colestasi intraepatica secondaria ad infezione delle vie urinarie), emocoltura; elettroforesi proteine sieriche Indagini di II livello : dosaggio α1 anti-tripsina; Screening per galattosemia, fibrosi cistica, ipotiroidismo; Indagini sierologiche (TORCH): solo una sierologia IgM precoce nelle prime sett di vita concordante con quella materna è un indice a favore di fetopatia da CMV; Indagini metaboliche se sospetto errori congeniti del metabolismo (utile EGA, dosaggio lattato/piruvato arteriosi, ammoniemia, amminoacidogramma plasmatico-urinario e acidi organici urinari) Indagini strumentali: Ecografia addominale: esame da eseguire a digiuno. Evidenzia anomalie anatomiche, cisti del coledoco, colecisti non visualizzabile, segno “triangular cord” (struttura triangolare iperecogena, posta al di sopra della vena porta, indice di tessuto fibroso che ha sostituito i dotti epatici, tipico nella atresia delle vie biliari) [Neoreviews 2013 14(2) Feldman and Sokol] Colangio-RMN: insufficienti dati raccomandano tale esame per la diagnosi routinaria di colestasi neonatale, ma dipende da caso a caso… [Eur J Pediatr (2011) 170:279-284 De Bruyne et al] Ulteriori indagini: Scintigrafia epato-biliare: aiuta a distinguere cause ostruttive Vs non ostruttive di colestasi. Presenta alta SE ma bassa SP per diagnosi di atresia delle vie biliari. La scarsa captazione del radioisotopo a livello epatico o la mancata visualizzazione nel fegato è indice di disfunzione epatica e la mancata comparsa del radionucleotide a livello del piccolo intestino entro 4-24 ore suggerisce ostruzione o atresia delle vie biliari. Biopsia epatica percutanea: permette la diagnosi eziologica della colestasi. Nel nostro caso…. In data 13/11/13 è stata eseguita Colangio-RMN: “Minima dilatazione delle vie biliari, soprattutto dell'epatocoledoco e del dotto biliare sinistro. Colecisti a morfologia tubulare. Non visualizzabile il dotto di Wirsung. Aspetto edematoso dell'ansa duodenale ricostruita. Non liquido libero in addome. Nella norma per morfologia e segnale i parenchimi addominali” Il successivo RX digerente con contrasto (gastrografin) non ha aggiunto elementi significativi a questo quadro. Al controllo ecotomografico addominale alcuni giorni dopo: “Fegato di volume ed ecostruttura normali. Colecisti contratta; vie biliari non dilatate. Pancreas malvalutabile per meteorismo. Milza e reni nella norma.” Veniva dimesso con diagnosi di Ittero colestatico da verosimile ostruzione meccanica, in terapia con Deursil 30 mg per 2 vv/die (alcune forme di colestasi intraepatica rispondono favorevolmente a dosi elevate di ac.ursodesossicolico) Trattamento: Anche la terapia nutrizionale è di fondamentale importanza nel neonato con colestasi: il deficit di crescita è spesso secondario a malassorbimento di grassi, alterato metabolismo proteico e dei carboidrati, con aumentato fabbisogno calorico (circa 125% dell’intake raccomandato basandosi sul peso corporeo ideale). Intake proteico 2-3 g/kg/die , Integrare vitamine liposolubili, Privilegiare nutrizione ENTERALE il prima possibile, Formule con trigliceridi a media catena che possano essere direttamente assorbiti nel piccolo intestino Trattamento: Dipende dall’eziologia della colestasi. Atresia delle vie biliari: • • • Colpisce 1/6000-18000 nati vivi, caratterizzata da colangiopatia fibrosante idiopatica che porta ad ostruzione completa delle vie biliari extra-epatiche, cirrosi epatica progressiva e morte nei casi non trattati. Nel 20% dei casi associata a malformazioni congenite. Sintomi: ittero e feci acoliche persistenti a 3-6 settimane di vita. Il neonato dovrà essere sottoposto a intervento chirurgico (epatoportoenterostomia secondo Kasai) entro 6 settimane di vita. Il tasso di successo (nel ristabilire flusso biliare) è strettamente correlato all’epoca di vita: 80% se entro 30-45 gg di vita. [Serinet et al. Pediatrics 2009; 123(5): 280-86] • • • • Deficit α1-antitripsina: Causa ereditaria più frequente di colestasi neonatale. Condizione autosomica recessiva, colpisce 1/2000 nati vivi. Negli individui affetti questa proteina rimane negli epatociti e non viene secreta nel sangue, con conseguente ridotta attività di tale enzima (con incapacità a neutralizzare l’elastasi neutrofila nei polmoni, da cui enfisema precoce). L’accumulo di tale proteina può causare cirrosi progressiva ed insufficienza epatica; Può richiedere trapianto di fegato. [Neoreviews 2013 14(2) Feldman and Sokol] • • • Sindrome Di Alagille: Embrio-fetopatia multisistemica, a marcata variabilità fenotipica, associata a mutazione del gene JAGGED1, che determina alterazione nei sistemi di programmazione del destino cellulare di numerosi tessuti ed in particolare del sistema vascolare. Incidenza:1/70.000 nati vivi. Si manifesta con epatopatia colestatica, cardiopatia congenita, dismorfismi, alterazioni renali e scheletriche. Il trattamento della malattia biliare si basa su acido ursodesossicolico e interventi chirurgici di derivazione biliare. [Medico e Bambino 10/2013 Nastasio, Maggiore et al] Colestasi associata a NP 18-67% dei neonati che ricevono NP prolungata (oltre 14 gg) sviluppano danno epatico e coestasi [Javid et al. J Pediatrs Surg 2011; 46(10):1913-17] L’incidenza correla inversamente con il peso alla nascita e direttamente con la durata della NP [Christensen et al. J Perinatol 2007; 27(5):284-290] La patogenesi è multifattoriale Per tali soggetti sono attualmente raccomandati la restrizione lipidica (1-1.5 g/kg/die) e il precoce passaggio a nutrizione enterale [Rangel et al J Pediatr Surg 2012;47(1):225-240] Galottosemia: • • Condizione autosomica recessiva, colpisce 1/50.000 nati vivi, associato a deficit dell’enzima galattosio-1-fosfato uridil transferasi. Sintomi: ittero colestatico, vomito, ipoglicemia, diarrea, epatomegalia, ascite. Trattamento: dieta priva di galattosio e lattosio. Colestasi intraepatica familiare progressiva : • Condizione autosomica recessiva, associata a mutazione dei geni implicati nella formazione dei canalicoli biliari con precoce comparsa di colestasi, grave prurito entro l’anno di vita!, diarrea, pancreatite, perdita di udito. Peculiarità di questa patologia è che i valori di gammaGT possono essere normali/bassi, con basso colesterolo tot. Trattamento chirurgico, eventuale trapianto di fegato • • • Se trovi ………...Sospetti ………… Tornando al nostro caso, durante il follow-up... Al controllo in Dimissione Protetta (19/11) il piccolo si presentava in buone condizioni generali. A domicilio ha presentato alvo regolare con feci ipocoliche verde chiaro ma mai acoliche, non episodi di vomito. Si è ben alimentato al seno. Peso 3,200 kg (in crescita). Ha eseguito ecografia addome che è risultato nella norma ed esami ematochimici: Emocromo: nella norma Bilirubina totale: 6 mg/dl; Bilirubina diretta 4,9 mg/dl. GOT: 219 UI/l GPT: 133 UI/l Fosfatasi alcalina: 1019 UI/l Gamma-GT: 172 UI/l Acidi bialiari: 68,9 uMol/l Amilasi e lipasi: nella norma. E ancora, dopo circa 2 mesi: Al controllo ambulatoriale del 03/01/14: Bilirubina totale: 0.7 mg/dl; Bilirubina diretta <0.15 mg/dl. GOT: 62UI/l GPT: 64 UI/l Fosfatasi alcalina: 815 UI/l Gamma-GT: 70 UI/l Acidi biliari: 61.8 uMol/l Medico e Bambino 3/1999 Maggiore e Caprai Se siamo sconvolti NOI… Figuriamoci VOI !! GRAZIE PER L’ATTENZIONE!!
Scaricare