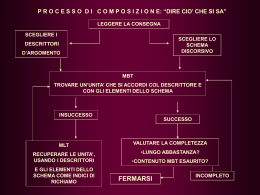
Analisi QSAR di Chiara Cappelletti, Davide Cittaro, Morena Spreafico, Marco Stefani INTRODUZIONE Dato un insieme di molecole congeneri vogliamo quantificare le differenze dell’attività biologica in termini di differenze strutturali. A tale scopo abbiamo utilizzato il metodo di regressione lineare dei Minimi Quadrati Ordinari formulando dei modelli QSAR. Le differenze strutturali vengono rappresentate da un insieme di descrittori molecolari appositamente scelti in modo da rendere il più completa possibile la rappresentazione delle molecole. Con un numero di descrittori grande, probabilmente riusciremo ad ottenere una buona descrizione della realtà in esame, ma il modello quasi sicuramente incorrerà nel fenomeno dell’over-fitting, riducendo anche drasticamente la capacità predittiva per le nuove molecole. Un buon modello mantiene generalmente il rapporto variabili-oggetti inferiore a 5, perciò diventa indispensabile selezionare adeguatamente i descrittori in modo da ottenere un modello con una buona capacità sia descrittiva che predittiva. Un ulteriore vincolo è dato dall’uso del metodo dei minimi quadrati, che impone l’eliminazione delle variabili troppo correlate. L’esplorazione esaustiva di tutte le possibili combinazioni di un insieme realistico (varie decine o centinaia) di descrittori richiederebbe un tempo di calcolo troppo elevato; di fatto, ciò impedisce di utilizzare degli algoritmi deterministici in grado di garantire la soluzione ottimale, e quindi s’impone l’utilizzo di metodi approssimati. La nostra scelta ricade sugli algoritmi genetici che permettono di esplorare in tempi ragionevoli lo spazio delle soluzioni. Ottenuto il modello, un passo molto importante consiste nella validazione del modello stesso, cioè la verifica della sua capacità predittiva nei confronti di molecole che non sono state utilizzate per la costruzione del modello. Se il modello è robusto, il passo successivo consiste nell’interpretazione del ruolo dei descrittori scelti. ALGORITMI GENETICI PER LA SELEZIONE DELLE VARIABILI Gli algoritmi genetici, proposti da Holland nel 1975, sono una strategia di ottimizzazione che si ispira alla genetica delle popolazioni. Il processo di ricerca di una soluzione avviene tramite un particolare bilanciamento tra l’esigenza di esplorare nuove regioni nello spazio delle soluzioni, e quella di sfruttare con una ricerca locale le informazioni a disposizione. La descrizione dei valori che ogni variabile può assumere avviene mediante un codice binario, ove ciascun termine binario costituisce un bit, cioè un numero uguale a 0 o 1. Il linguaggio degli algoritmi genetici ha un lessico appropriato in analogia con la genetica delle popolazioni: o Gene: ciascuna codifica binaria di un numero (una variabile, un parametro numerico) o Individuo o Cromosoma: l’insieme di più geni, cioè la concatenazione di più bits, ed è associato ad una particolare soluzione o Popolazione: l’insieme di individui o cromosomi o Riproduzione, Cross-over, Mutazione: rappresentano gli operatori genetici che regolano il processo di ricerca della soluzione ottima Gli algoritmi genetici sono organizzati in tre fasi: 1. Fase iniziale: viene effettuata una codifica delle variabili mediante il codice binario. Ciascuna condizione viene rappresentata da una stringa di 0 o di 1. Nella selezione delle variabili che ottimizzano un modello di regressione, lo 0 indica l’assenza della variabile nel modello mentre l’1 indica la presenza. Viene estratto un certo numero di cromosomi a caso e per ciascuno di essi viene calcolata la risposta corrispondente; i migliori entrano a far parte della popolazione iniziale. 2. Fase evolutiva: è costituita da una fase di generazione di cromosomi che derivano dall’accoppiamento dei cromosomi presenti nella popolazione e calcolo delle risposte corrispondenti, e da una fase di mutazione dove ciascun cromosoma può casualmente subire delle mutazioni. In questo caso, viene calcolata la corrispondente risposta. Se le risposte calcolate sono migliori della risposta corrispondente all’ultimo cromosoma della popolazione (il peggiore della popolazione esistente), i cromosomi corrispondenti entrano a far parte della popolazione, nella posizione che loro compete in base alla qualità della risposta. In caso contrario, i cromosomi vengono scartati. Esaminando più nel dettaglio, durante la riproduzione sono estratti dei cromosomi destinati all’accoppiamento, con probabilità proporzionale alla qualità del cromosoma. Durante il cross-over vengono generati i cromosomi derivanti dall’accoppiamento, scambiando i geni dei cromosomi genitori con una probabilità prefissata (cross-over probability). Durante la mutazione alcuni geni cambiano il loro valore da 0 a 1 e viceversa, secondo una determinata probabilità (mutation probability). La fase evolutiva procede fino a che non si raggiunge una condizione che pone termine alla procedura. 3. Fase finale: la procedura viene fermata in base a criteri prestabiliti, quali, ad esempio, il raggiungimento di un numero massimo di iterazioni o quando la popolazione non si rinnova per un certo numero di iterazioni. Al termine della procedura si ottiene la selezione delle variabili migliori tra quelle che l’algoritmo ha considerato; se tutto lo spazio delle soluzioni possibili è stato indagato, quella proposta è la soluzione ottima. DESCRIZIONE DEL PROBLEMA Le molecole utilizzate per lo studio sono 14 isomeri delle PCDD (Poli-Cloro-dibenzo-diossina); è importante sottolineare che le molecole analizzate sono strettamente congeneri e differiscono solamente nel numero e nella posizione degli atomi di Cloro, pertanto rappresentano un set molto omogeneo di composti per il quale ci si può aspettare una buona correlazione tra le strutture e la attività biologica. La risposta biologica per la quale viene costruito un modello di regressione è l’affinità di binding. Al fine di calcolare i descrittori molecolari le 14 diossine sono state disegnate con il programma MOE (Molecular Operating Environment) e successivamente sono state minimizzate attraverso i metodi della meccanica molecolare. In particolare è stato utilizzato il campo di forza MMFF94 adatto a descrivere le proprietà di piccole molecole organiche. I DESCRITTORI MOLECOLARI La conformazione spaziale delle 14 molecole minimizzate è il punto di partenza per il calcolo dell’insieme dei descrittori molecolari, effettuato con DRAGON. Il linguaggio con cui descriviamo la realtà non coincide con la realtà stessa: per questo motivo le parole che utilizziamo per esprimerci svolgono un ruolo importantissimo nella rappresentazione della realtà. Nella costruzione di modelli QSAR i descrittori molecolari svolgono lo stesso compito delle parole in un discorso: la qualità del modello finale, che ha lo scopo di rappresentare una parte di realtà, nella fattispecie l’attività biologica dei composti analizzati, dipende strettamente dai descrittori utilizzati. Essi sono il modo attraverso il quale l’informazione chimica è trasformata e codificata. Ognuno di essi è un numero che descrive un aspetto strutturale della molecola: più un descrittore sarà in grado di spiegare bene quell’aspetto, ovvero più il linguaggio è accurato e chiaro, più il modello sarà buono.Per questo motivo la scelta dei descrittori riveste un ruolo fondamentale nella formulazione di un modello. Le classi di descrittori utilizzate in questo lavoro sono: COSTITUZIONALI Sono descrittori molto semplici, indipendenti dalla connettività e dalla conformazione. Indicano il numero di alcuni atomi, o di legami, o la somma di alcune proprietà (volumi, elettronegatività, polarizzabilità, etc.). Sono facili da ricavare ma la descrizione che danno della molecola è piuttosto generica. Tuttavia, ciò non vuol dire che apportino un’informazione poco importante, sono infatti descrittori molto semplici che si ritrovano spesso nei modelli calcolati. TOPOLOGICI Largamente utilizzati negli studi QSAR, sono descrittori 2D che derivano dal grafo molecolare (solitamente tolti gli atomi di idrogeno) e dalle matrici di connettività. Sono prevalentemente fondati sui concetti di entropia e di contenuto di informazione ed hanno la proprietà di essere invarianti al grafo molecolare, cioè il loro valore è indipendente da come vengono numerati i vertici del grafo. Sono semplici da calcolare ma i loro limiti più evidenti sono legati alla totale mancanza di informazione riguardo alla geometria 3D e agli aspetti conformazionali, e alla non sempre chiara interpretabilità chimica. A dispetto di ciò, si dimostrano spesso estremamente efficaci per il loro alto potere modellante. WHIM Il calcolo di questi descrittori prevede la centratura della molecola nel centro delle coordinate e, mediante l’analisi delle componenti principali, la determinazione degli assi principali. La rappresentazione della molecola avviene mediante una proiezione degli atomi sui tre assi principali e sul calcolo di alcuni parametri statistici (gli indici WHIM) per ciascuno di questi assi, senza applicare schemi di pesatura oppure pesando attraverso le masse atomiche, i volumi di Van der Waals, l’elettronegatività atomica di Mulliken, la polarizzabilità atomica o gli indici elettrotopologici di Kier. Si ottengono così dei descrittori WHIM direzionali, che, per ogni schema di pesatura, possono poi dar luogo anche ad indici globali, cioè non direzionali. GETAWAY Sono dei descrittori basati su una matrice d’influenza, chiamata Molecular Influence Matrix, simile alla matrice dei Leverages solitamente usata nella regressione. Un primo set di descrittori, chiamato H-GETAWAY, viene ottenuto utilizzando solo le informazioni provenienti dalla Molecular Influence Matrix, mentre un secondo set, chiamato R-GETAWAY, combina questa informazione con le distanze interatomiche nella molecola. Le tre tipologie di descrittori sono state trattate separatamente considerando per ciascun insieme i descrittori con una correlazione pairwise inferiore al 95%. Sono state ottenute rispettivamente 39, 11 e 22 variabili. I PARAMETRI DI PERFORMANCE Per ciascuno dei tre insiemi di descrittori abbiamo utilizzato gli algoritmi genetici per selezionare la migliore combinazione di descrittori. Ogni modello è identificato da un cromosoma di lunghezza pari al numero di descrittori per ogni insieme; il numero di descrittori selezionati in ciascun cromosoma può variare tra 3 e 5. La popolazione è costituita da 100 individui e sono state effettuate 10000 iterazioni per ogni insieme di descrittori. La bontà dei modelli è valutata mediante un parametro di fitting, il coefficiente di determinazione R2, e due parametri di validazione, R2cv e R2LOO. Il coefficiente di determinazione R² viene definito come: ( yi − yˆ i ) 2 RSS ∑ R = 1− = 1− TSS ∑ ( yi − y i ) 2 2 Questo valore, moltiplicato per 100, rappresenta la varianza percentuale spiegata dal modello in fitting. La radice quadrata del parametro precedente è una quantità nota come coefficiente di correlazione multipla, R, e rappresenta la correlazione tra la risposta sperimentale e i predittori. Oltre a questi parametri, che misurano il fitting, ve ne sono altri che misurano la capacità predittiva del modello. Il parametro definito PRESS (Predictive Error Sum of Squares) è formalmente analogo a RSS, con la differenza che al posto delle risposte calcolate vengono utilizzate le risposte predette. PRESS = ∑ (y i i − yˆ i / i ) 2 Analogamente al parametro R², sostituendo RSS con PRESS, viene definita la varianza spiegata dal modello in predizione, ovvero: Q2 = R2cv = 1- PRESS TSS Diversamente da R2, questo parametro presenta un massimo per la complessità ottimale del modello e ridiscende ogni volta che aggiungiamo al modello variabili non predittive. Inoltre Q2 viene esplicitamente valutato rispetto al potere predittivo e non al livello di fitting del modello. E’ perciò un parametro molto importante, soprattutto nel caso in cui il modello presuppone un suo utilizzo per applicazioni successive su dati la cui risposta non è ancora nota. La validazione è stata fatta secondo due diverse tecniche: attraverso il metodo Leave One Out, sintetizzata dal parametro R2LOO, e attraverso il metodo Leave Many Out, descritta da R2cv. Entrambe sono tecniche di cross validation; con questo termine si indica una validazione incrociata, in cui un oggetto o un gruppo di oggetti viene utilizzato per la predizione una sola volta. METODO LEAVE ONE OUT Secondo questo metodo, dati n oggetti, si calcolano n modelli in ciascuno dei quali viene escluso un oggetto alla volta: ciascun modello viene calcolato con gli n-1 oggetti restanti e viene utilizzato per predire la risposta, che può essere di tipo qualitativo in un modello di classificazione o quantitativo in un modello di regressione. Lo scarto tra la risposta predetta e quella sperimentale viene accumulato per tutti gli oggetti che a turno vengono esclusi dal modello. Solitamente il modello finale viene costruito considerando comunque tutti gli oggetti, in modo da sfruttare tutta l’informazione disponibile. Questo metodo è l’unico che consente un confronto univoco tra modelli ottenuti con metodi diversi, ed è quello che comporta la minima perturbazione in quanto viene escluso un solo oggetto per volta, e per questo motivo talvolta fornisce risultati troppo ottimistici. METODO LEAVE MANY OUT E’ una generalizzazione del metodo precedente. Infatti i dati vengono suddivisi in G gruppi di cancellazione in modo tale che k oggetti pari a n/G vengono assegnati all’insieme di valutazione e quindi sono esclusi dal training set: il modello, stimato da un training set costituito da n-k oggetti, viene utilizzato per predire la risposta dei k esclusi. I k oggetti vengono esclusi una volta soltanto; una volta esclusi, vengono successivamente reinseriti nel modello e ne vengono esclusi altri k. Rispetto al metodo precedente, questo è più perturbativo, e valuta in modo più severo la predittività, ma i risultati ottenuti dipendono dalla ripartizione degli oggetti all’interno dei gruppi di cancellazione. La funzione obiettivo che guida la selezione delle variabili negli Algoritmi Genetici è il parametro R2LOO e utilizza il metodo di regressione lineare dei Minimi Quadrati Ordinari. RISULTATI Riportiamo il miglior modello a 3, 4 e 5 variabili per ciascun insieme di descrittori. I modelli scelti sono quelli che si comportano meglio per i tre parametri. Costituzionali e Topologici Getaway WHIM R2 0.994 0.943 0.978 0.921 0.952 0.924 0.638 0.635 R2LOO 0.938 0.894 0.93 0.858 0.912 0.125 0.173 0.335 R2cv 0.984 0.88 0.891 0.845 0.372 -0.921 0.122 0.288 N 5 4 5 4 3 5 4 3 Tabella 1 – Performance dei migliori modelli calcolati Il fatto che nel gruppo Costituzionali e Topologici non vengano selezionati modelli a tre variabili ci fa supporre che tre sole variabili non siano sufficienti a raggiungere la qualità dei modelli a 4 e 5 variabili. I descrittori WHIM hanno una performance decisamente peggiore rispetto agli altri due insiemi. Il valore negativo di R2cv per il modello a 5 variabili significa che i residui del modello sono tanto superiori ai residui calcolati sul valor medio da far preferire il modello Zero, che seglie sempre il valore medio. Il suo alto valore di fitting non è sufficiente a renderlo un buon modello per la pessima capacità predittiva. I modelli con basso numero di variabili sono preferibili perchè più semplici e più facilmente interpretabili, e per la consuetudine di mantenere il rapporto oggetti/variabili superiore a 5, i modelli a 3 variabili sarebbero stati preferibili. Nel nostro caso però il modello a 3 variabili ha un R2cv così basso da escluderne la scelta. I descrittori Costituzionali e Topologici si comportano complessivamente meglio e quindi saranno analizzati più nel dettaglio. ID 1 2 3 4 5 6 7 8 R2 0.994 0.983 0.986 0.943 0.943 0.952 0.928 0.92 R2LOO 0.938 0.844 0.905 0.893 0.894 0.823 0.861 0.812 R2cv N 0.984 5 0.95 5 0.933 5 0.888 5 0.88 4 0.821 4 0.794 4 0.089 4 1 1 1 3 3 4 3 1 3 3 3 11 11 17 11 20 Variabili 20 20 15 20 25 25 20 25 23 21 20 25 36 29 25 29 37 37 37 36 Tabella 2 – Modelli per i descrittori Costituzionali e Topologici Osserviamo che negli otto modelli selezionati solo 1/3 dei descrittori totali è rappresentato. Il fatto che pochi descrittori ricorrano frequentemente è indice di robustezza dei modelli. Secondo noi il miglior modello è il numero 5, perché ha 4 variabili e perché tra i modelli a 4 variabili ha la migliore performance, sia in fitting che in predizione. L’equazione del modello è la seguente: y = 36.324 - 0.009 * MSD -328.954 * TIE -10.725 * Xindex + 2.351 * PCR Riportiamo anche l’equazione in forma standardizzata: y = -2.692 * MSD -1.053 * TIE -1.377 * Xindex + 0.422 * PCR Risposta predetta - sperimentale 9 Valore predetto 8 7 6 5 4 3 3 4 5 6 7 8 9 Valore sperimentale Figura 1 – Risposta predetta – sperimentale per il modello scelto Nel grafico si osserva il confronto tra risposta sperimentale e risposta predetta dal modello. I punti più vicini alla bisettrice sono quelli meglio predetti. Si osserva un buon accordo del modello con i dati sperimentali per tutte le molecole: Molecola 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Valore sperimentale Valore predetto 8.00 7.73 7.15 7.39 7.10 7.05 6.79 6.96 6.92 6.79 6.55 5.94 6.10 6.03 5.96 6.13 5.88 5.91 5.49 5.27 5.19 5.69 5.00 5.10 4.89 4.86 4.00 3.83 Tabella 3 – Valori predetti dal modello I descrittori coinvolti sono: • • • • MSD: mean square distance index (Balaban) TIE: E-state topological parameter Xindex: Balaban X index PCR: ratio of multiple path counts to path counts I descrittori selezionati appartengono tutti alla categoria dei Topologici e con le nostre conoscenze non è possibile interpretare il loro significato riguardo all’attività biologica. L’omogeneità dei descrittori all’interno di ciascuno dei tre gruppi studiati ci offre una visione parziale della realtà analizzata, limitando la comprensione del fenomeno biologico. Un approccio differente potrebbe essere quello di creare dei gruppi “misti”, che tengano conto di informazioni di diverso tipo, per avere una migliore visione d’insieme e un modello predittivo più completo. Nonostante l’utilizzo di descrittori relativamente omogenei, i risultati ottenuti sono decisamente buoni. Questo comportamento potrebbe essere spiegato da un lato con la congenericità dei composti analizzati, dall’altro con l’efficacia degli Algoritmi Genetici nella selezione delle variabili.
Scaricare