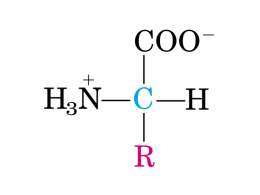
orletto a spazzola Digestione delle proteine: 6 fasi 1. Idrolisi gastrica del legame peptidico 2. Digestione a peptidi più piccoli da parte delle proteasi pancreatiche nel lume dell’intestino tenue 3. Idrolisi degli oligopeptidi operata da peptidasi dell’orletto a spazzola degli enterociti 4. Ulteriore digestione dei di- e tri-peptidi da peptidasicitoplasmatiche nell’enterocita 5. Metabolismo degli AA negli enterociti 6. Trasporto degli AA attraverso la membrana basolaterale e invio al sangue portale e quindi al fegato Origine Zimogeno/ Enzima Attivazione Substrato Prodotto finale Stomaco Pepsinogeno/pepsina pH 1-2, autoattivazione Proteine Peptidi Pancreas Tripsinogeno/tripsina Enteropeptidasi Proteine, peptidi Peptidi, dipeptidi Tripsina Proteine, peptidi Peptidi, dipeptidi Tripsina Proteine, peptidi Peptidi, dipeptidi Estremità Cterminale polipeptidi Peptidi, aminoacidi Estremità Nterminale di oligopeptidi Peptidi, aminoacidi (membrana enterociti duodenali) Chimotripsinogeno/ chimotripsina Pro-elastasi/elastasi Pro-carbossipeptidasi Intestino tenue (membrana e citoplasma) Aminopeptidasi Tripsina Assente Specificità di taglio delle proteasi dell’apparato gastrointestinale tripsina – amminoacidi basici (arginina, lisina), versante -COOchimotripsina - amminoacidi aromatici (Phe, Tyr) ,versante -COO- elastasi – glicina, versante -COOcarbossipeptidasi a - amminoacidi aromatici carbossipeptidasi b - amminoacidi basici ASSORBIMENTO degli AMINOACIDI presenza di proteine trasportatrici sulle membrane apicali e basolaterali degli enterociti Ogni sistema trasporta aminoacidi o dipeptidi con proprietà strutturali diverse Alcuni sfruttano un sistema di trasporto attivo, mediante co-trasporto con Na+ o H+ Destino delle proteine (amminoacidi) della dieta durante la fase di assorbimento (circa 2 ore) Sintesi proteica Glucosio (7-9 g) 10 g 4g Intestino (Glutammina) 12 g Amminoacidi 30g Fegato Urea CO2 4g (Amminoacidi ramificati) Muscolo e rene ATP Funzioni degli amminoacidi •Substrati per la sintesi proteica (20 +1), suscettibili di modificazioni post-traduzionali (es. idrossiprolina, idrossilisina, acido γ-carbossiglutammico) •Intermedi metabolici (es. ornitina) •Fonte energetica (glucogenici, chetogenici) •Trasporto di gruppi amminici (glutammina, alanina) ALTRI DESTINI METABOLICI DEGLI AMMINOACIDI Tirosina Triptofano Glicina Arginina Aspartato Glutammato Istidina Glutammina Lisina Metionina Cisteina → → → → → → → → → → → melanina , catecolammine, ormoni tiroidei niacina (NAD+, NADP+), serotonina purine, eme, acido glicocolico, creatina, creatinina creatina, creatinina, ossido nitrico (NO) purine gamma-aminobutirrato (GABA) istamina purine carnitina carnitina, creatina taurina Biosintesi di alcuni neurotrasmettitori da amminoacidi Notare il ruolo delle decarbossilasi Fosfocreatina composto fosforilato di riserva del muscolo (concentrazione muscolare circa 30 mM) Sintesi a partire da glicina, arginina e metionina La CREATININA è il prodotto di degradazione della fosfocreatina muscolare Sintesi delle purine: gli atomi di azoto derivano dagli amminoacidi aspartato, glicina e glutammina Sintesi del glutatione: glutammato, cisteina, glicina Sintesi dell’ossido nitrico (NO) a partire dal gruppo guanidinico dell’arginina (ossido nitrico sintasi) L’ossido nitrico è un importante mediatore di processi fisiologici (controllo della pressione sanguigna, trasmissione nervosa, coagulazione del sangue) Sulla base delle vie cataboliche energetiche dello scheletro carbonioso gli amminoacidi possono essere suddivisi in due gruppi Glucogenici degradati a piruvato, alfa-chetoglutarato, succinil-CoA, fumarato, o ossalacetato (produrranno glucosio tramite gluconeogenesi) Chetogenici degradati ad acetil-CoA o acetoacetato (produrranno acidi grassi o corpi chetonici) Alcuni amminoacidi appartengono ad entrambi i gruppi Leucina e lisina (producono solo acetoacetato o acetilCoA) sono esclusivamente chetogenici In ROSSO solo glucogenici; in AZZURRO solo chetogenici; in VERDE entrambi Alanina, glicina, cisteina, serina, treonina piruvato (glucogenici) Treonina anche acetilCoA (anche chetogenico) Asparagina, aspartato ossalacetato (glucogenici) Arginina, glutammato, glutammina, istidina, prolina alfachetoglutarato (glucogenici) Isoleucina, metionina, treonina, valina succinil-CoA, tramite propionilCoA (glucogenici) Isoleucina anche acetil-CoA (anche chetogenico) Leucina e lisina acetilCoA e/o acetoacetato (SOLO chetogenici) Triptofano alanina piruvato) acetoacetilCoA (sia glucogenico che chetogenico) Fenilalanina e tirosina fumarato acetoacetato ( sia glucogenici che chetogenici) Schema del catabolismo degli amminoacidi Esclusivamente chetogenici ALCUNE RIFLESSIONI •Il catabolismo dello scheletro carbonioso degli amminoacidi produce energia attraverso l’interazione con la via glucogenetica ed il ciclo di Krebs •Il catabolismo dei carboidrati e quello delle proteine sono in stretta correlazione •L’efficienza del catabolismo delle proteine (molecole di ATP prodotte) è inferiore rispetto a quello dei carboidrati e lipidi - non tutto lo scheletro carbonioso degli amminoacidi è soggetto ad ossidazione -la formazione di urea richiede il consumo di 3 molecole di ATP •Molte malattie genetiche dell’uomo sono inerenti al catabolismo degli amminoacidi Malattia Processo difettoso Enzima difettoso Sintomi ed effetti Albinismo Sintesi melanina da Tyr Tirosinasi Mancanza di pigmentazione Alcaptonuria Degradazione Tyr Omogentisato diossigenasi Pigmentazione scura dell’urina, artriti Argininemia Ciclo dell’urea Arginasi Ritardo mentale Omocistinuria Degradazione Met Cistationina beta sintasi Alterazione viluppo osseo, ritardo mentale Malattie urine a sciroppo d’acero Degradazione aa ramificati Complesso alfachetoacido deidrogenasi a catena ramificata Vomito, ritardo mentale, morte precoce Acidemia metilmalonica Conversione propionilCoA a succinilCoA MetilmalonilCoA mutasi Vomito, ritardo mentale, morte precoce Fenilalanina idrossilasi Vomito neonatale, ritardo mentale Fenilchetonuria Conversione Phe a (più frequente) Tyr Classificazione in base alla struttura presentano una diversa percentuale di azoto Classificazione nutrizionale Essenziali Non essenziali Essenziali in certe condizioni (condizionatamente) Valina Alanina Arginina Leucina Glutammato Glicina Isoleucina Aspartato Prolina Lisina Asparagina Glutammina Istidina Serina Triptofano Cisteina Fenilalanina Tirosina Metionina Treonina •Tirosina e Cisteina diventano essenziali se mancano fenilalanina e metionina, da cui derivano (semi-indispensabili) •La Tirosina è essenziale nel cervello, dove la sua sintesi dalla fenilalanina non avviene •L’Arginina è essenziale nell’infanzia e nello sviluppo •La sintesi dei non essenziali può essere compromessa per scarso apporto dei carboidrati •L’essenzialità dell’istidina è controversa Biosintesi degli amminoacidi non essenziali o dei condizionatamente essenziali nell’uomo piruvato → alanina ossalacetato → aspartato (+ glutammina) → asparagina α-chetoglutarato → glutammato + NH3→ glutammina glutammato → prolina, arginina 3-fosfoglicerato → serina → glicina fenilalanina → tirosina metionina → cisteina La velocità di sintesi può non essere sufficiente ai bisogni in alcune condizioni (malati, stress, neonati prematuri o sottopeso, ustionati) Biosintesi della serina dal 3- fosfo glicerato e conversione della serina in glicina Metionina Biosintesi della Cys dalla Hcys +Ser Contenuto in proteine di un UOMO ADULTO: circa 12 kg/70 kg peso actina, miosina, collagene ed emoglobina costituiscono circa la metà di tutte le proteine 40% nel muscolo: possono diventare fonte di amminoacidi in condizioni di stress, a discapito però di proteine funzionali 10% tessuti viscerali (fegato, intestino): scarsamente mobilizzate in condizioni di stress 30% nella pelle e nel sangue: diventano fonte di amminoacidi in deficit di proteine alimentari
Scaricare