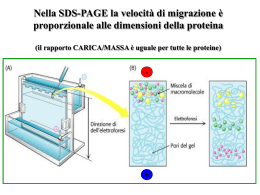
Proteine: dove, quando e perchè Paolo Plevani Milano, CusMiBio 21-23 Settembre 2011 Il dogma centrale della Biologia Molecolare DNA Trascrizione RNA Traduzione Proteina Proteoma Proteome = Proteins encoded by the genome Il proteoma comprende tu9e le proteine espresse in una data cellula in un certo momento, incluse tu9e le isoforme della stessa proteina (splicing alterna=vo) e le loro modificazioni post-‐traduzionali. Mentre il genoma è costante per una data cellula ed iden=co per tu9e le cellule di un organismo (~ 25.000 geni nell’uomo), il numero di proteine prodo9e è molto piu’ elevato (un fa9ore 10-‐100) in virtù del meccanismo , di splicing alterna=vo e delle modificazioni post-‐traduzionali. Inoltre, il proteoma è dinamico nel tempo e in risposta a fa9ori esterni, e differisce in maniera sostanziale tra i diversi =pi cellulari. Proteine: dove, quando e perchè Analisi di proteine: Tanti metodi: Elettroforesi e Western Blot Elettroforesi su gel Metodo per separare macro-‐molecole (Proteine, DNA, RNA, etc.) sulla base delle loro proprieta’ chimico-‐fisiche quali: (1) dimensioni (2) forma (3) carica ele9rica Elettroforesi di Proteine • Piu’ complessa dell’ele9roforesi di DNA – Proteine diverse hanno cariche diverse – Le proteine variano molto per forma (stru9ura) • Generalmente il gel e’ fa9o di poliacrilammide Perche’ usare Gel di Poliacrilammide per separare le proteine? • I gel di poliacrilammide hanno una trama piu’ compa9a • I pori hanno dimensioni minori che nei gel di agarosio • Le proteine sono molto piu’ piccole del DNA – Amminoacido medio = 110 Da – Paio di nucleo=di medio = 649 Da – 1 kilobase di DNA = 650 kDa – 1 kilobase di DNA codifica 333 amminoacidi = 36 kDa Ele9roforesi di proteine Condizioni Non Denaturan= • Non-‐denaturante (na=vo): nessun pre-‐tra9amento delle proteine prima dell’ele9roforesi – Le proteine conservano la loro stru9ura 2° e 3°; pon= disolfuro etc. – Le proteine conservano la loro carica normale – Le proteine sono separate sulla base di carica, dimensioni e forma Nome Carica Massa Proteina Q +3 30kD Proteina R -4 42kD Forma Ele9roforesi di proteine in condizioni denaturan= • Le proteine sono tra9ate con SDS (detergente anionico) prima dell’ ele9roforesi (SDS-‐PAGE) – Le molecole di SDS si legano alle proteine – Le proteine perdono la loro normale forma – Le proteine hanno tu9e lo stesso rapporto carica/massa – Le proteine vengono separate esclusivamente sulla base delle loro dimensioni Carica Massa +3 30kD -4 42kD Carica Massa SDS -300 30kD -420 42kD SDS-‐Polyacrylamide Gel Electrophoresis (SDS-‐ PAGE) CH3 CH2 CH2 " SDS (Sodio Dodecil CH2 Solfato) CH2 e denatura le CH2 proteine Aggiunge cariche negative alle proteine CH2 CH2 CH2 CH2 CH2 O S - O O Solubilizza CH2 SDS O Proteina Nativa Carica netta: -4 Proteina trattata con SDS Carica netta: Molto (-) Come funziona un Gel SDS-‐PAGE ? • Le proteine cariche nega=vamente si muovono verso l’ele9rodo posi=vo • Proteine piu’ piccole si muovono piu’ velocemente s-s SDS, calore Proteine con SDS – • Le proteine si separano per dimensione + Estrazione delle proteine Lisi delle cellule con un tampone di lisi che dissocia le proteine e inibisce le proteasi cellulari Concentrazione dei sali Detergenti (Triton X-100, NP40, SDS) pH Inibitori di proteasi Bassa temperatura (ghiaccio) Proteasi Inibitori proteasi: Leupeptina, Pepstatina, Aprotinina, PMSF Cosa c’e’ nel tampone di caricamento? • Un tampone (Tris) per fornire il giusto pH (6,8) • SDS (Sodio Dodecil Solfato) per solubilizzare le proteine e fornire loro una carica nega=va complessiva • Glicerolo per rendere pesan= i campioni e farli scendere nei pozzee • Un agente riducente (DTT o β-‐Mercaptoetanolo) per rompere i legami disolfuro • Un colorante (Blu di Bromofenolo) per visualizzare i campioni SDS-PAGE Colorazione con Blu di Coomassie SDS-‐PAGE: separazione di proteine in base al loro peso molecolare In presenza di SDS e di un riducente dei legami disolfuro, le diverse proteine vengono separate in elettroforesi su gel esclusivamente sulla base di differenze di peso molecolare delle loro catene polipeptidiche. Il Gel • Come funziona: – Stacking gel: 4-‐5% gel superiore, pH 6.8 8-‐14% gel separatore, pH 8.8 – Le molecole di acrilammide sono tenute assieme dalla bis-‐acrilammide, formando una stru9ura a laece – Polimerizzazione piu’ rapida aggiungendo catalizzatori: TEMED e APS Il Gel Componen= del gel SDS-‐PAGE • Soluzione di Acrilammide/Bis-‐Acrilammide • Tris-‐HCl pH 6.8 oppure Tris-‐HCl pH 8.8 • SDS • ddH2O • TEMED • APS (ammonio persolfato) • Buffer di corsa (Tris, Glicina, SDS) • Come funziona: – Proteine vengono caricate, viene applicata una corrente ele9rica Il Gel • Includere un marker di peso molecolare colorato • 10-‐50ug di estra9o proteico totale • 0.1-‐1ug di proteina purificata – – Ioni Cloruro (-‐) / Proteina / Glicina (+) Stacking pH 6,8 – Si muovono verso il gel separartore (“resolving) Resolving pH 8,8 • Differenze di pH causano la ionizzazione della glicina, perme9endo alle proteine di migrare nel gel separatore + Il Gel • % di acrilammide consigliata • Dimensioni delle proteine 40-‐200 kDa 21-‐100 kDa 10-‐40 kDa 8% 10% 12% Visualizzazione delle proteine nel gel I due metodi piu’ usati sono: Colorazione con il Coomassie Brilliant Colorazione con l’argento “Silver staining” (puo’ essere visualizzato ~1ng di proteina per banda) Coomassie staining Silver staining SDS-‐PAGE: determinazione del peso molecolare di una proteina Stima dei pesi molecolari Western Bloeng Southern Blot: Per l’analisi del DNA. (sonda = DNA o RNA). Northern Blot: Per l’analisi dell’ RNA. (sonda = DNA o RNA). Western Blot: Per l’analisi delle proteine. (sonda = anticorpo). Cosa e’ un Western Blot? • Una tecnica in cui le proteine sono separate mediante ele9roforesi su gel e successivamente trasferite su un supporto (membrana o filtro). Successivamente una specifica proteina viene iden=ficata mediante la sua reazione specifica con un an=corpo. A cosa serve il Western Blot? Qual e’ la proteina che mi interessa? – SDS-‐PAGE (non certo) • Si basa sul confronto di peso molecolare – Western blot (certo) • Si basa su una reazione specifica an=gene-‐an=corpo – ? + Fasi di un Western Blot • Prima fase: ele,roforesi su gel. (Le proteine del campione vengono separate su un gel in base alle loro dimensioni) • Seconda fase: trasferimento su membrana. (Le proteine nel gel sono poi trasferite su una membrana di nitrocellulosa mediante un campo ele9rico) • Terza fase: saturazione o “blocking”. (La saturazione e’ usata per prevenire le interazioni non specifiche tra l’an=corpo e la membrana) Fasi di un Western Blot • Quarta fase: legame dell’an>corpo primario. (L’an=corpo riconosce la proteina specifica immobilizzata sulla membrana) • Quinta fase: legame dell’an>corpo secondario. (L’an=corpo secondario, coniugato a un enzima (AP o HRP), riconosce specificamente l’an=corpo primario, gia’ legato alla proteina sulla membrana) • Sesta fase: rivelazione o “detec>on”. (L’enzima coniugato all’an=corpo secondario scinde un substrato che, in corrispondenza della proteina specifica, sviluppa precipitato colorato o chemioluminescenza) Western blot: seconda fase Immobilizzazione e trasferimento Le proteine nel gel sono ancora in soluzione – Le bande diffondono e si confondono col tempo E’ necessaria l’immobilizzazione per: – Preservare in maniera permanente l’esperimento di ele9roforesi – Perme9ere il riconoscimento di proteine specifiche La strategia piu’ comune e’ il trasferimento su membrana – Fa9a di Nitrocellulosa, PVDF (Polivinilidene fluoride) o nylon – Si usa l’ele9roforesi, in un processo de9o ‘bloeng’ Western blot: seconda fase Immobilizzazione e trasferimento • Ele9robloeng – Apparato di trasferimento – Il gel e’ messo tra stra= di carta da filtro con la membrana a dire9o conta9o col gel sul lato verso l’ele9rodo posi=vo – Viene applicato un campo ele9rico e le proteine migrano fuori dal gel verso l’ele9rodo posi=vo e si legano alla membrana – Fa9o a 4°C per evitare surriscaldamento, decomposizione del tampone e degradazione delle proteine Apparato per il trasferimento “Semi-dry” Western Blotting - Buffer-soaked filter papers + Nitrocellulose membrane Gel Buffer Electrode SDS-PAGE Direction of transfer Assemble ‘sandwich’ Wet blotting Graphite Electrode Plates - Stained (Red Ponceau) + Semi-dry blotting Nitrocellulose with bound proteins Western blot: terza fase saturazione o “blocking” • Per saturare i si= idrofobici liberi sulla membrana • Per prevenire il legame dell’an=corpo primario alla membrana stessa • La9e scremato o Albumina di Siero Bovino (BSA) Western blot: quarta fase incubazione con anticorpo primario • L’an=corpo primario riconosce la proteina di interesse e non lega le altre proteine immobilizzate sulla membrana • An=corpi come sonde: – Molto sensibili – Possono essere “prodoe” • Immunizzando una specie diversa (an=corpi policlonali) • Generando an=corpi monoclonali (mAb) – Economici Anticorpi Ab + Ag AbAg Kd Kd = [Ab] = 10-9 M [AbAg] An=corpi An=corpi (immunoglobuline, Ig) Una proteina a forma di Y secreta nel sangue in risposta ad uno specifico an=gene, come un ba9erio o un virus, che neutralizza l’an=gene legandosi specificamente ed esso e producendo una risposta immunitaria. An=corpi policlonali Produzione • Immunizzazione ripetuta dell’animale con l’an=gene (pep=de, proteina purificata o ricombinante) • Il sangue e’ prelevato nel momento di picco di produzione dell’an=corpo ed e’ purificato il siero • Il “pool” degli an=corpi riconosce mol= epitopi dell’an=gene usato per l’immunizzazione An=corpi monoclonali Riconoscono solo un epitopo Fusioni tra linfoci= B (milza/ linfonodi) e cellule di mieloma di topo Western blot: quinta fase Incubazione con anticorpo secondario An=corpi • An=corpo primario – Riconosce la proteina • An=corpo secondario – Lega l’an=corpo primario – Generalmente prodo9o in una specie diversa – Coniugato con un enzima – Il substrato dell’enzima sara’ conver=to in un prodo9o colorato – Puo’ anche essere radioaevo o fluorescente Western blot: sesta fase rivelazione o “detection” • Fosfatasi alcalina (AP) o perossidasi del rafano (HRP: horseradish peroxidase) – Conversione di un substrato colorimetrico in un precipitato colorato • Substra= Chemioluminescen= – Eme9ono luce se conver== dall’enzima – Possono essere visualizza= su lastre radiografiche • Marcatura radioaeva • An=corpi secondarii bio=nila= Western blot HRP Anticorpo primario substrato HRP Anticorpo secondario luce Rivelazione Il substrato metabolizzato dalla perossidasi (HRP) emette luce pg proteina Le proteine possono essere modificate dopo la loro sintesi: modificazioni post-traduzionali Fosforilazione Glicosilazione Ubiquitinazione etc……. Tali modificazioni fanno variare il PM della proteina e, quindi, possono essere evidenziabili dopo elettroforesi e western blot La risposta cellulare a danni al DNA Danni al DNA Checkpoints di risposta a danni Apoptosi Ciclo cellulare Replicazione DNA Trascrizione Riparazione e ricombinazione del DNA •Molto conservati negli eucarioti: dal lievito all’uomo •Mutazioni nei geni dei checkpoints causano instabilità genomica e tumori Il checkpoint innescato da danni al DNA è una via di trasduzione dei segnali Lievito Mec1, Tel1, Ddc2, Rad17, Mec3, Ddc1 Rad24,RFC Uomo Proteine sensori Rad9, Rad53, Chk1 Trasduttori RPA, Polα-primase, Cdc28, Swi6, Pds1, Cdc14, ... Effettori ATR, ATM, ATRIP, hRAD1, hHUS1, hRad9, hRAD17, RFC BRCA1?, CHK2, CHK1 RPA, p53, CDC25, CDK, ... DNA damage checkpoint in S. cerevisiae RFC2-5 2 3 4 Rad24 5 Ddc2 Ddc1 P P P P Rad9 P Rad9 P P Rad9 RPA P RPA P P P Ddc2 RPA RPA RPA Mec3 RPA Mec1 PRad53 P P Rad9 Rad53 Targets Rad53 FOSFORILAZIONI !!!!!!
Scaricare