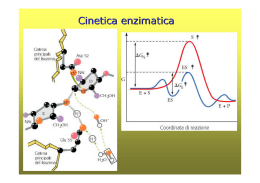
Gli enzimi possono essere inibiti in modo reversibile o irreversibile Inibizione enzimatica irreversibile Chimotripsina + didisopropilfluorofosfato Gli inibitori irreversibili si combinano o distruggono un gruppo funzionale dell’enzima che è essenziale per l’attività catalitica. Inibizione enzimatica reversibile Inibizione competitiva Un inibitore competitivo compete con il substarto per il legame al sito attivo dell’enzima. Un inibitore competitivo riduce la velocità della catalisi riducendo il numero di molecole di enzima in grado di legare il substrato Inibizione competitiva La Vmax von viene alterata da un inibitore competitivo La competizione può essere spostata a favore del substrato aumentando la [S]. La [S] che determina 1/2 Vmax, cioè KM, aumenta in presenza di un inibitore competitivo Il metotrexate è un analogo strutturale del tetraidrofolato, un coenzima per l’enzima diidrofolato riduttasi, che svolge un ruolo importante nella biosintesi delle purine e delle pirimidine. Il metotrexate si lega all’enzima 1000 volte meglio del ligando naturale e ne inibisce la funzione bloccando la sintesi delle basi dei nucleotidi. Il metotrexate è utilizzato nella terapia dei tumori. Inibizione non competitiva Nella competizione non copetitiva il competitore si lega a un sito diverso da quello del substrato Inibizione non competitiva L’inibitore non competitivo agisce riducendo il numero di turnover (kcat) dell’enzima. L’inibitore non competitivo riduce la concentrazione di enzima attivo e quindi abbassa Vmax (Vmax = kcat [Et]) Una inibizione non competitiva non può essere superata aumentando la concentrazione del substrato. Inibizione mista Un tipo particolare di inibizione, detta inibizione mista, si ha quando l’inibitore nasconde in parte il legame del substrato ed allo stesso tempo riduce anche il numero di turnover dell’enzima. Gli analoghi dello stato di transizione sono inibitori potenti degli enzimi Sebbene lo stato di transizione sia per definizione transitorio ed instabile, è possibile in taluni casi costruire molecole simili allo stato di transizione, chiamate analoghi dello stato di transizione. Gli analoghi dello stato di transizione si legano all’enzima più fortemente del substrato nel complesso ES, in quanto si adattano meglio al sito attivo dell’enzima (formano più interazioni deboli dello stesso substrato), e sono pertanto inibitori potenti degli enzimi. Il potere inibitorio degli analoghi dello stato di transizione conferma il principio alla base della catalisi enzimatica: il legame selettivo dell’enzima allo stato di transizione. Questi principi sono utilizzati nell’industria farmaceutica per progettare nuovi farmaci L’isomerizzazione della L-prolina in D-prolina da parte della prolina racemasi, un’enzima batterico, procede attraverso la formazione di uno stato di transizione planare in cui il carbonio α è trigonale invece che tetraedrico. L’acido pirrolo 2-carbossilico, un analogo dello stato di transizione con geometria trigonale, è un potente inibitore della prolina racemasi. Gli anticorpi catalitici dimostrano l’importanza per l’attività catalitica degli enzimi del legame selettivo allo stato di transizione Se è possibile stabilire un analogo dello stato di transizione per la reazione S Â P, un anticorpo che si leghi saldamente all’analogo dello stato di transizione può catalizzare la reazione S Â P. Quando un analogo dello stato di transizione legato ad una proteina viene usato come epitopo per stimolare la produzione di un anticorpo, l’anticorpo diventa un potenziale catalizzatore della reazione corrispondente Sono stati prodotti anticorpi monoclonali che catalizzano l’idrolisi di esteri o di carbonati, utilizzando composti fosforici come analoghi dello stato di transizione. Stato di transizione tetraedrico con una parziale carica negativa sull’ossigeno carbonilico Le analisi strutturali condotte su alcuni anticorpi catalitici hanno confermato che le catene laterali amminoacidiche sono disposte in modo da interagire con il substrato soltanto nello stato di transizione. L’inserimento di uno ione metallico in una porfirina da parte dell’enzima ferrochelatasi procede attraverso uno stato di transizione in cui la porfirina è piegata Porfirine alchilate, come la Nmetilmesoporfirina, che hanno già una struttura ripegata che assomiglia allo stato di transizione della reazione catalizzata dalla ferrochelatasi, sono state utilizzate per produrre anticorpi che sono in grado di catalizzare l’inserimento di uno ione metallico nell’anello porfirinico. Struttura del peptidoglicano del batterio Staphylococcus aureus Il peptidoglicano è costituito da catene lineari di natura saccaridica legate insieme da corti peptidi, formati da una miscela di D ed Lamminoacidi. Struttura del peptidoglicano del batterio Staphylococcus aureus Durante la sintesi del peptidoglicano il gruppo amminico alla fine di una catena di pentaglicina attacca il legame peptidico tra due residui di D-alanina in un’altra unità peptidica. Si forma pertanto un legame peptidico tra la glicina ed uno dei residui di D-alanina, mentre l’altra alanina è eliminata. L’enzima che catalizza questa reazione è la glicopeptide transpeptidasi. Formazione del cross-link durante la sintesi del peptidoglicano in S. aureus La reazione di transpeptidazione prevede la formazione di un intermedio acil-enzima La pennicillina, il primo antibiotico scoperto, è un inibitore dell’enzima glicopeptide transpeptidasi. La pennicillina si lega al sito attivo della transpeptidasi perché ha una struttura simile alla sequenza D-Ala-D-Ala presente nel substrato Una volta legatasi la pennicillina forma un legame covalente con un residuo di serina del sito attivo dell’enzima, inattivandolo in modo irreversibile. La pennicillina è un inibitore suicida. L’anello β-lattamico della pennicillina è particolarmente reattivo. Nel metabolismo cellulare gruppi di enzimi lavorano insieme in vie sequenziali (vie metaboliche) per condurre un determinato processo metabolico. Il prodotto del primo enzima diventa il substrato del secondo enzima e così via. In ogni sistema enzimatico vi è un enzima che determina la velocità complessiva della sequenza di reazioni in quanto catalizza la reazione più lenta o quella che limita la velocità Gli enzimi che occupano queste posizioni chiave nel metabolismo cellulare presentano spesso un’attività catalitica aumentata o diminuita in risposta a segnali cellulari e sono detti enzimi regolatori. Mediante l’azione di questi enzimi regolatori, la velocità di ogni sequenza metabolica si adegua costantemente alle necessità della cellula Nelle vie metaboliche vi sono due classi di enzimi regolatori: 1) Gli enzimi allosterici vengono regolati mediante il legame non covalente e reversibile di composti regolatori, chiamati modulatori allosterici, che in genere sono metaboliti o piccoli cofattori. 2) Gli enzimi regolati mediante modificazioni covalenti reversibili. Sistemi di regolazione dell’attività enzimatica 1. Interazioni allosteriche 2. Modificazioni covalenti reversibili. inoltre 3. Stimolazione ed inibizione da parte di proteine di controllo 4. Attivazione proteolitica La regolazione allosterica (non covalente) permette una modulazione fine delle vie metaboliche. La regolazione da modificazioni covalenti prevede uno switch ON/OFF della via metabolica Inibizione retroattiva (a feedback) della conversione della L-treonina in L-isoleucina, catalizzata da una sequenza di 5 enzimi. La treonina deidratasi (E1) viene inibita allostericamente e in modo specifico dalla Lisoleucina, il prodotto finale della sequenza di reazioni Le proteine allosteriche sono quelle che presentano “altre forme” o conformazioni indotte dal legame di modulatori. Negli enzimi allosterici le modificazioni conformazionali indotte da uno o più modulatori interconvertono forme di enzima più attive in forme meno attive e viceversa I modulatori di un enzima allosterico possono essere sia inibitori che stimolatori Lo stesso substrato può essere un attivatore. Quando il modulatore coincide con il substrato gli enzimi vengono detti omotropici. Quando il modulatore è una molecola diversa dal substrato gli enzimi vengono detti eterotropici. Oltre al sito catalitico gli enzimi allosterici possiedono in genere uno o più siti regolatori o allosterici per il legame del modulatore. La maggior parte degli enzimi allosterici è formato da due o più catene polipeptidiche o subunità Spesso il sito di legame del substrato (sito attivo) e il sito di legame del modulatore (sito allosterico) sono su subunità diverse: a) subunità catalitica (C) b) subunità regolatrice (R). Un tipico modulatore allosterico, l’AMP ciclico Regolazione della protein chinasi A (PKA) Il legame di 4 molecole di cAMP attiva la protein chinasi A inducendo la dissociazione dell’oloenzima non attivo (R2C2) in una subunità regolatrice (R2) e 2 subunità catalitiche. Ciascuna catena R contiene la sequenza Arg-Arg-Gly-Ala-Ile, che è un perfetto sito di riconoscimento per la fosforilazione da parte della chinasi A con la sostituzione della serina bersaglio con un residuo di alanina. Nel complesso R2C2, questa sequenza pseudosubstatrato occupa il sito catalitico delle subunità C impedendo il legame del substrato. In seguito al legame dell’AMPc le catene R subiscono una modificazione conformazionale che allontana la sequenza pseudosubstrato dal sito catalitico e libera le subunità C. Le proprietà cinetiche degli enzimi allosterici non possono essere spiegate dal modello di Michaelis-Menten. Gli enzimi allosterici spesso mostrano una curva della velocità catalitica in funzione della concentrazione del substrato con andamenro sigmoidale invece che iperbolico. Gli enzimi allosterici sono costituiti da più subunità e presentano più siti attivi Negli enzimi allosterici il legame del substrato ad un sito attivo può modificare le proprietà di altri siti attivi nella stessa molecola (enzimi omotropici). E’ possibile così un legame di tipo cooperativo. Curva sigmoide di un enzima omotropico, in cui il substrato serve anche da modulatore positivo (stimolatore). Legame cooperativo. Curva simile a quella di ossigenazione dell’emoglobina Effetti di un modulatore positivo (+) e di un modulatore negativo (-) su un enzima allosterico in cui la K0,5 viene modulata senza variazioni nella Vmax (situazione più comune) Un tipo meno comune di modulazione in cui la Vmax viene modulata e la K0,5 resta quasi costante. I modelli per spiegare il funzionamento degli enzimi allosterici si basano sul presupposto che ciascuna subunità possa esistere in due conformazioni interconvertibili: Forma R (Rilasciata) = alta affinità per il substrato Forma T (Tesa) = bassa affinità per il substrato Modello sequenziale per il legame del substrato ad un enzima allosterico (D. Korshland jr.) 1) Esistono solo due stati conformazionali possibili per ciascuna subunità: Forma R (Rilasciata) = alta affinità per il substrato Forma T (Tesa) = bassa affinità per il substrato 2) Il legame del substrato modifica la forma delle subunità a cui è legato. La conformazione delle altre subunità non viene alterata in modo apprezzabile. Modello sequenziale per il legame del substrato ad un enzima allosterico (D. Korshland jr.) 3) Le modificazioni conformazionali determinate dal legame del substrato in una subunità possono aumentare o diminuire l’affinità per il substrato delle altre subunità Il legame è cooperativo se il sito vuoto nel dimero RT ha una affinità per il substrato più elevata di quella dei siti del dimero TT Modello concertato per il legame cooperativo del substrato ad un enzima allosterico (Monod, Wyman, Changeux, 1965) 1) Tutte le subunità devono avere lo stesso stato conformazionale. TT e RR sono conformazioni permesse, RT no 2) In assenza di substrato tutte le molecole sono in forma T. 3) L’aggiunta di substrato sposta l’equilibrio verso la forma R Quando il substrato si lega ad un sito, anche l’altro sito assume la conformazione R 4) La proporzione di molecole di enzima presenti nello stato R aumenta progressivamente con l’aumento della concentrazione del substrato Il legame del substrato risulta cooperativo. Il modello concertato può facilmente spiegare gli effetti di attivatori ed inibitori allosterici. Un inibitore allosterico si lega preferenzialmente alla forma T inattiva, stabilizzandola e spostando l’equilibrio conformazionale R'T verso T Un attivatore allosterico si lega preferenzialmente alla forma R attiva, stabilizzandola e spostando l’equilibrio conformazionale R'T verso R Stimolazione ed inibizione da parte di proteine di controllo La calmodulina, una proteina regolatrice ubiquitaria negli eucarioti, “sente” il livello di calcio intracellulare ed attiva molti enzimi legandosi ad essi quando i suoi siti sono occupati dal calcio. Motivo helix-loop-helix che lega il calcio presente in molte proteine che legano ioni calcio in seguito ad un aumento della loro concentrazione nella cellula. La calmodulina interagisce con un dominio elicoidale amfipatico di uno dei suoi molti enzimi bersaglio, la proteina chinasi I calmodulina-dipendente. Il lungo segmento centrale ad α-elica della calmodulina si ripiega su se stesso quando la calmodulina lega il substrato. Il legame della Ca2+-calmodulina attiva l’enzima permettendo che quest’ultimo assuma una conformazione cataliticamente attiva. Regolazione dell’attività delle proteine mediante modificazione covalente. La fosforilazione è una modificazione molto utilizzata per regolare l’attività di enzimi e di altre proteine Le fosfatasi catalizzano la rimozione mediante idrolisi del gruppo fosforico legato alle proteine da parte delle chinasi La fosforilazione e la defosforilazione non sono l’uno l’inverso dell’altro; ciascuna reazione è di fatto irreversibile in condizioni fisiologiche Un esempio di regolazione mediante modificazione covalente: la glicogeno fosforilasi Reazione catalizzata: (glucosio)n + Pi ¿ (glucosio)n-1 + glucosio -1-P glicogeno Alcuni enzimi utilizzano molti meccanismi di regolazione La glicogeno fosforilasi, un enzima chiave del metabolismo intermedio, è soggetto sia a regolazione mediante modificazione covalente (fosforilazione del residuo Ser14, in giallo), sia ad attivazione allosterica da parte dell’AMP (in blu) L’acetilazione dei residui di lisina: una modificazione covalente delle proteine L’attivazione di un enzima mediante proteolisi è un altro meccanismo di regolazione Un precursore inattivo di un enzima (zimogeno) viene scisso in modo da generare la forma attiva dell’enzima. L’attivazione mediante proteolisi è un meccanismo di attivazione enzimatica comune a molti importanti processi biologici: Gli enzimi digestivi che idrolizzano le proteine sono sintetizzati sotto forma di zimogeni nello stomaco e nel pancreas. La coagulazione del sangue è mediata da una cascata di attivazioni proteolitiche che assicura una rapida risposta ad un trauma. Alcuni ormoni sono sintetizzati come precursori inattivi (es.: insulina), che vengono attivati mediante proteolisi. Numerosi processi dello sviluppo sono regolati dall’attivazione specifica di zimogeni (es. metamorfosi del girino in rana). L’attivazione della procollagenasi (uno zimogeno) in collagenasi (l’enzima attivo) è finemente regolata nei processi di rimodellamento dei tessuti. La morte cellulare programmata, o apoptosi, è mediata dall’azione di enzimi proteolitici, detti caspasi, che sono sintetizzati come precursori inattivi, le procaspasi. Attivazione di zimogeni mediante proteolisi L’attivazione mediante proteolisi, a differenza del controllo allosterico o delle modificazioni covalenti reversibili, avviene solo una volta nella vita di un enzima. Gli enzimi digestivi che idrolizzano le proteine sono sintetizzati come zimogeni nel pancreas o nello stomaco Molti degli enzimi digestivi vengono sintetizzati nel pancreas sotto forma di zimogeni ed accumulati in granuli di secrezione. Quando le cellule vengono stimolate il contenuto di questi granuli viene rilasciato nel dotto pancreatico che li porta nel duodeno. Il chimotripsinogeno viene attivato mediante taglio specifico di un singolo legame peptidico L’attivazione proteolitica del chimotripsinogeno porta alla formazione del sito di legame per il substrato Il nuovo gruppo amminoterminale della isoleucina 16 può formare un legame ionico con il residuo di acido spartico in posizione 194. Questa interazione stabilizza la conformazione attiva della chimotripsina L’enteropeptidasi, secreta dalle cellule della parete del duodeno, inizia l’attivazione degli zimogeni pancreatici attivando la tripsina che a sua volta attiva gli altri zimogeni Attivazione di zimogeni mediante proteolisi Inibitori specifici bloccano l’azione delle proteasi legandosi all’enzima in maniera molto forte L’attivazione di un enzima mediante proteolisi è irreversibile. Per essere inattivato l’enzima deve essere degradato o bloccato da inibitori specifici. L’inibitore pancreatico della tripsina impedisce che anche piccolissime quantità di tripsina possano essere attive nel pancreas o nei dotti pancreatici portando ad un’attivazione prematura della cascata delle proteasi e conseguente danno tissutale (pancreatite acuta) . α1-antitripsina L’inibitore pancreatico della tripsina non è l’unico importante inibitore delle proteasi. L’α1-antitripsina è un inibitore dell’elastasi. Disordini genetici che portano a deficit di questo inibitore si associano a danni degli alveoli polmonari per una digestione delle fibre elastiche da parte dell’elastasi (enfisema). Il fumo di sigaretta ossida il residuo metionina 358 dell’α1-antitripsina, un inibitore dell’elastasi, aumentando il rischio di enfisema. R - CH2 - CH2- S - CH3 Metionina 358 Ossidazione R - CH2 - CH2- S - CH3 O Derivato sulfossido L’attività enzimatica viene modificata dal pH Il pH ottimale di un’attività enzimatica si adatta di solito molto bene alle condizioni intracellulari in cui l’enzima deve operare. La pepsina, che idrolizza i legami peptidici delle proteine durante la digestione nello stomaco, ha un pH ottimale di circa 1,6. Il pH del succo gastrico è normalmente tra 1 e 2. La glucosio 6-fosfatasi epatica ha un pH ottimale di circa 7,8 ed è responsabile del rilascio di glucosio nel sangue. Il pH del citosol di un epatocita è di circa 7,2. Le vitamine Numerosi enzimi richiedono dei cofattori per poter funzionare. Alcuni cofattori sono piccole molecole organiche (coenzimi) e spesso sono derivati dalle vitamine. Le vitamine sono piccole molecole organiche che sono necessarie in piccole quantità nella dieta degli animali superiori. Le vitamine sono necessarie e hanno la stessa funzione in quasi tutte le forme di vita, ma gli animali superiori hanno perso la capacità di sintetizzarle durante l’evoluzione. Mentre E. coli può sopravvivere solo con il glucosio e sali organici, gli esseri umani hanno bisogno di almeno 12 vitamine. Le vie biosintetiche delle vitamine sono complesse e pertanto è più efficiente ingerire tali molecole con la dieta che produrre tutti gli enzimi necessari per la sintesi di queste molecole da precursori più semplici. Questo risparmio ha un costo: la dipendenza da altri animali per alcuni composti essenziali. Un deficit di vitamine può portare all’insorgenza di patologie negli animali che ne hanno bisogno. Le reazioni che avvengono nelle cellule sono divisibili in cinque categorie: 1) Ossido-riduzione 2) Scissione e formazione di legami carbonio-carbonio 3) Riarrangiamenti interni 4) Trasferimento di gruppi 5) Reazioni di condensazione (due unità monomeriche si uniscono eliminando una molecola di acqua) Le reazioni che fanno parte della stessa categoria hanno di solito lo stesso meccanismo chimico In tutte le reazioni di ossido-riduzione si ha un trasferimento di elettroni. Quando due atomi condividono elettroni in un legame covalente, essi hanno la stessa elettronegatività, come nel caso di due atomi di C. Quando due atomi hanno un’elettronegatività diversa (es. C e O), essi formano un legame covalente polarizzato; gli elettroni condivisi hanno più probabilità di trovarsi nella regione dell’atomo più elettronegativo (in questo caso O) che in quella dell’atomo meno elettronegativo (in questo caso C). Quando due atomi hanno un’elettronegatività molto diversa (es. Na e Cl), un atomo dona elettroni all’altro, generando specie ioniche che interagiscono tra loro, come nell’NaCl solido. Stati di ossidazione del carbonio nelle biomolecole Nei legami carbonio-idrogeno, il C più elettronegativo “si appropria” dei due elettroni condivisi con l’idrogeno. Nei legami carbonio-ossigeno la condivisione degli elettroni è tutta a favore dell’ossigeno. Passando dal gruppo -CH3, (un alcano) al gruppo -CH2OH (un alcol), l’atomo di carbonio tende a perdere elettroni; avviene cioè un’ossidazione. In molte ossidazioni biologiche un composto cede due elettroni e due ioni idrogeno (cioè due atomi di idrogeno). Queste reazioni sono comunemente chiamate deidrogenazioni e gli enzimi che le catalizzano sono deidrogenasi. In alcune ossidazioni biologiche un atomo di carbonio si lega covalentemente a un atomo di ossigeno. Gli enzimi che catalizzano queste ossidazioni sono in genere detti ossidasi oppure, se gli atomi di ossigeno derivano dall’ossigeno molecolare (O2) ossigenasi. Ogni ossidazione è accompagnata da una riduzione, in cui un accettore di elettroni acquista gli elettroni rimossi dall’ossidazione. Le reazioni di ossidazione rilasciano in genere energia. Reazioni di scissione e formazione di legami carbonio-carbonio I due meccanismi per la rottura di un legame C - C Scissione omolitica Radicali del carbonio Scissione eterolitica Carboanione Carbocatione Scissioni omolitiche (rare negli organismi viventi): Ciascun atomo lascia il legame sotto forma di radicale, trasportando uno dei due elettroni che erano condivisi. Scissioni eterolitiche: Uno degli atomi trattiene entrambi gli elettroni impegnati nel legame covalente (e si genera un anione), lasciando l’altro atomo privo di un elettrone (si genera un catione) Nelle scissioni eterolitiche, quando un secondo gruppo ricco di elettroni va a prendere il posto dell’anione che è stato eliminato, si ha una sostituzione nucleofilica. nucleofilica - Gruppo Nucleofilo uscente Molte delle reazioni biochimiche avvengono mediante interazioni tra Nucleofili = gruppi funzionali ricchi di elettroni capaci di donarli e Elettrofili = gruppi funzionali poveri di elettroni che cercano di legarli I nucleofili si combinano con gli elettrofili donando loro elettroni. Alcuni gruppi funzionali che si comportano da nucleofili nelle cellule I gruppi funzionali contenenti ossigeno, azoto e zolfo sono di solito importanti nucleofili biologici. Gli atomi di idrogeno carichi positivamente (ioni idrogeno o protoni) e i metalli carichi positivamente (cationi) sono spesso utilizzati come elettrofili. Un atomo di carbonio si può comportare sia da nucleofilo sia da elettrofilo a seconda del tipo di legame e di sostituenti che lo circondano. L’elenco è in ordine di forza decrescente. I nucleofili deboli sono gruppi uscenti migliori Due diversi modi con cui un nucleofilo può sostituirne un altro durante la formazione di un legame carbonio - carbonio 1) Reazione SN1 (Sostituzione nucleofilica unimolecolare) Carbocatione intermedio W: = gruppo uscente Z:-= nucleofilo entrante Conservazione della configurazione 2) Reazione SN2 (Sostituzione nucleofilica bimolecolare) Intermedio pentacovalente W: = gruppo uscente Z:-= nucleofilo entrante Inversione della configurazione I nucleofili deboli sono buoni gruppi uscenti, mentre i nucleofili forti sono buoni gruppi sostituenti. Il trasferimento di elettroni all’interno di una molecola produce un riarrangiamento molecolare Il C-1 viene ridotto (da aldeide a alcol) e il C-2 viene ossidato (da alcol a chetone) Nel riarrangiamento molecolare, una ridistribuzione degli elettroni porta a isomerizzazione, trasposizione o riarrangiamento cis-trans di doppi legami. Meccanismo di azione della reazione di conversione del glucosio 6-fosfato in fruttosio 6-fosfato Elettrofilo Nucleofilo B1 e B2 sono i gruppi basici dell’enzima: essi sono in grado di donare e di accettare ioni idrogeno (protoni) mentre la reazione va a compimento Le reazioni di trasferimento di gruppi attivano gli intermedi metabolici. Uno dei temi fondamentali del metabolismo è l’attacco di un buon gruppo uscente a un intermedio metabolico, in modo da “attivarlo” per le successive reazioni. Tra i migliori gruppi uscenti nelle reazioni di sostituzione nucleofilica vi è l’ortofosfato inorganico. Le sostituzioni nucleofiliche in cui il gruppo fosforico (-PO32-) funziona da gruppo uscente avvengono molto speso nelle reazioni metaboliche Le reazioni di trasferimento di gruppi attivano gli intermedi metabolici. L’ortofosfato inorganico (Pi) •Il fosforo può formare 5 legami covalenti. •La rappresentazione convenzionale del Pi con 3 legami P -O e un legame P = O non è corretta. •Nel Pi vi sono 4 legami P-O che condividono un carattere di parziale doppio legame, e l’anione viene ad avere una struttura tetraedrica. •Dato che l’ossigeno è più elettronegativo del fosforo, gli elettroni non sono ugualmente distribuiti; il nucleo centrale costituito dal fosforo ha una carica parzialmente positiva e può agire da elettrofilo. Intermedio pentacovalente transitorio Quando un nucleofilo Z (in questo caso il gruppo -OH del C-6 del glucosio) attacca l’ATP, viene spiazzato l’ADP (W) e in questa reazione SN2, si forma un intermedio pentacovalente transitorio. Gli enzimi che catalizzano trasferimenti di gruppi fosforici con l’ATP in qualità di donatore sono detti chinasi (es.: esochinasi). Reazioni di condensazione Le reazioni di condensazione sono alla base della formazione dei biopolimeri. Le subunità che costituiscono le proteine, gli acidi nucleici e i polisaccaridi sono unite tra loro da reazioni di condensazione che avvengono mediante spiazzamento nucleofilo con sostituzione di gruppo uscente. Il gruppo OH è un gruppo uscente povero. La reazione non avviene in questo modo! Il tRNA è un gruppo uscente migliore. Gli amminoacidi devono essere attivati mediante il legame ai tRNA! Le macromolecole possono essere demolite da reazioni di idrolisi, in cui l’acqua diventa il nucleofilo attaccante, spiazzando un’unità monomerica o un piccolo frammento di polimero. Specifici gruppi catalitici contribuiscono alla catalisi Una volta che il substrato si è legato, un enzima può utilizzare diversi meccanismi di catalisi per facilitare la rottura o la formazione di un legame, sfruttando i suoi gruppi funzionali catalitici opportunamente disposti . Tra i meccanismi meglio caratterizzati vi sono : La catalisi covalente. La catalisi acido-base. La catalisi da ioni metallici. La catalisi da avvicinamento dei substrati. Gli enzimi utilizzano una o più di questi meccanismi per accellerare le velocità delle reazioni. Catalisi covalente Nella catalisi covalente il sito attivo presenta un gruppo reattivo, generalmente un forte nucleofilo che viene temporaneamente modificato covalentemente durante la reazione. Catalisi acido-base Molte reazioni biochimiche comprendono la formazione di intermedi instabili carichi che tendono a degradarsi rapidamente nelle loro specie costituenti, impedendo quindi alla reazione di arrivare a compimento. Lo sviluppo della carica sfavoreviole può essere superato mediante il coinvolgimento di accettori o donatori di protoni . La catalisi a cui partecipano ioni H+ (H3O+) oppure OH- presenti nell’acqua viene chiamata catalisi acido-base specifica . Nella catalisi acido-base generale una molecola diversa dall’acqua svolge il ruolo di donatore o accettore di protoni. Nel sito attivo di un enzima vi possono essere catene laterali di amminoacidi capaci di svolgere la funzione di accettore o donatore di protoni. Esempio di catalisi acido-base: l’idrolisi di un legame amidico Lo sviluppo della carica è sfavorevole e può essere superato mediante la donazione di un protone da parte di H3O+ (catalisi acida specifica) oppure di HA (catalisi acida generale), dove HA rappresenta un qualsiasi acido Analogamente, la carica può essere neutralizzata dalla sottrazione di un protone da parte di OH- (catalisi basica specifica) oppure di B: (catalisi basica generale), dove B: rappresenta qualsiasi base. Gli amminoacidi nella catalisi acido-base generale Catalisi da ioni metallici Gli ioni metallici possono funzionare nella catalisi in diversi modi: - Uno ione metallo può funzionare stabilizzando una carica negativa su di un intermedio di reazione. - Uno ione metallico può generare un forte nucleofilo aumentando l’acidità di una molecola vicina. - Uno ione metallico può legare il substrato, aumentando il numero di interazioni tra quest’ultimo e l’enzima e quindi l’energia di legame . Catalisi da avvicinamento dei substrati Molte reazioni prevedono due distinti substrati. In questi casi la velocità della reazione può essere accellerata anche dal solo avvicinamento dei due substrati nel sito attivo dell’enzima Chimotripsina La chimotripsina è un enzima digestivo nei mammiferi che catalizza l’idrolisi di proteine nell’intestino tenue. La chimotripsina appartiene alla famiglia delle serina proteasi. proteasi Chimotripsina La chimotripsina ha una massa di 25 kd ed è costituita da 3 catene polipeptidiche unite da ponti disolfuro. Struttura tridimensionale della chimotripsina (David Blow, 1967) Nella struttura tridimensionale della chimotripsina tutti i residui carichi e/o polari sono sulla superficie della molecola, eccetto tre residui che hanno un ruolo fondamentale nella catalisi La chimotripsina idrolizza in modo selettivo i legami peptidici dalla parte carbossilica di amminoacidi con catene laterali aromatiche (tirosina, triptofano, e fenilalanina) o con lunghe catene laterali idrofobiche come la metionina. La chimotripsina idrolizza anche legami estere. Reazione catalizzate dalla chimotripsina O R1 - C - N - R2 + H2O R1 - C O = O- + +H3N - R2 H Peptide Acido Ammina O R1 - C - O - R2 + H2O R1 - C H Estere Acido O = O- + HO- R2 + H+ Alcool Le proteasi Le proteasi catalizzano la scissione idrolitica di un legame peptidico (l’addizione di una molecola di acqua a un legame peptidico) O R1 - C - N - R2 + H2O R1 - C =O O- + +H3N - R2 H Peptide Acido Ammina Anche se questa reazione è termodinamicamente favorevole, è estremamente lenta (in assenza di un catalizzatore l’emivita di un legame peptidico a pH neutro è tra i 10 e i 1000 anni) Il legame peptidico è particolarmente resistente. Il parziale carattere di doppio legame del legame peptidico lo rende particolarmente resistente. Inoltre l’atomo di carbonio carbonilico del legame peptidico è meno elettrofilico e meno suscettibile ad attacco nucleofilico degli atomi di carbonio carbonilici presenti in altri composti (es.: esteri carbossilici). Per permettere il taglio del legame peptidico, un enzima deve facilitare l’attacco nucleofilico ad un gruppo carbonilico normalmente poco reattivo Chimotripsina La chimotripsina è un ottimo esempio dell’uso della catalisi covalente come strategia catalitica. L’enzima utilizza un potente nucleofilo per attaccare il gruppo carbonilico poco reattivo. La chimotripsina catalizza l’idrolisi di legami peptidici e di legami estere in due fasi, che prevedono un intermedio di reazione in cui il substrato è legato covalentemente al gruppo nucleofilo dell’enzima. Il comportamento cinetico di un enzima può essere studiato facilmente se si conosce un analogo del substrato che viene trasformato in un prodotto colorato. Nel caso della chimotripsina, il substrato cromogenico è l’N-acetil-L-fenilalanina p-nitrofenil estere. Uno dei prodotti formati in seguito al taglio di questo composto da parte della chimotripsina è il p-nitrofenolo, che ha un colore giallo. Cinetica della catalisi mediata dalla chimotripsina Il meccanismo catalitico della chimotripsina Fase veloce acilazione E + S ' ES Fase lenta deacilazione E - P2 P1 E P2 P1 = componente aminico (o alcolico) del substrato E - P2 = intermedio covalente enzima-acile P2 = componente acido del substrato Il meccanismo della catalisi prevede la formazione di un intermedio covalente (catalisi covalente) Ser195 Fase veloce acilazione Fase lenta deacilazione Il gruppo acile si lega transitoriamente all’atomo di ossigeno di uno specifico residuo di serina, la serina 195. La serina 195 è il nucleofilo che la chimotripsina utilizza per attaccare il gruppo carbonilico del substrato La serina 195 è particolarmente disopropilfluorofosfato (DIFP). reattiva ed interagisce con il Gli altri 27 residui di serina presenti nella chimotripsina non reagiscono con il DIFP L‘attività catalitica della chimotripsina dipende dall’insolita reattività della serina 195 La presenza di un residuo di serina iperreattivo coinvolto nella catalisi è tipico delle serina proteasi (es.: chimotripsina, tripsina, elastasi, trombina, subtilisina, acetilcolinesterasi, etc…) Struttura tridimensionale della chimotripsina. Gli amminoacidi della triade catalitica sono vicini nella struttura tridimensionale dell’enzima La serina 195, Ser195,è parte di una triade catalitica che comprende anche un residuo di istidina, His57, ed un residuo di acido aspartico, Asp102. La triade catalitica converte la serina 195 in un potente nucleofilo. •Il residuo di istidina serve per posizionare la catena laterale della serina e per polarizzare il gruppo ossidrilico (agisce come un catalizzatore basico generale, accettando uno ione idrogeno). •La rimozione del protone dal gruppo ossidrilico genera uno ione alcossido, che è un nucleofilo molto più potente di un alcol. •Il ruolo del gruppo carbossilico dell’aspartato 102 è quello di orientare il residuo di istidina e di renderlo un migliore accettore di protoni mediante effetti elettrostatici (la carica negativa dell’Asp102 stabilizza la carica positiva che si viene a creare sul residuo di His). Meccanismo di azione della chimotripsina Legame del substrato Il gruppo ossidrilico della Ser195 procede con un attacco nucleofilo verso il carbonio carbonilico del substrato Il buco dell’ossanione stabilizza la struttura dell’intermedio tetraedico che si forma durante la reazione della chimotripsina I gruppi NH della catena peptidica formano legami idrogeno con con l’ossigeno carico negativamente. Queste interazioni contribuiscono a stabilizzare lo stato di transizione Dall’intermedio tetraedrico instabile si forma il complesso covalente acil-enzima Questo passaggio è facilitato dal trasferimento di un protone dall’istidina carica positivamente al gruppo amminico che si forma dalla rottura del legame peptidico. La componente amminica è libera di lasciare l’enzima ……. ……. ed è rimpiazzata da una molecola di acqua Il gruppo estere del complesso acil-enzima viene quindi idrolizzato mediante un processo che essenzialmente è una ripetizione delle tappe precedenti. La molecola di acqua attacca il gruppo carbonilico mentre un protone viene rimosso dal residuo di istidina, e si forma pertanto un nuovo intermedio tetraedrico. L’intermedio tetraedrico si rompe e si forma il prodotto acido carbossilico Il rilascio dell’acido carbossilico rende disponibile l’enzima per una nuova reazione Immagine del sito attivo della chimotripsina con il substrato (verde) legato. La Ser195 attacca il gruppo carbonilico del substrato (il cui ossigeno è il viola); la carica negativa che si genera è stabilizzata da buco dell’ossanione, i cui azoti amidici sono in colore arancione. Nel substrato sono indicate in azzurro la catena laterale aromatica amminoacidica e l’azoto amidico del legame peptidico che deve essere rotto. Studi cristallografici di complessi della chimotripsina con analoghi del substrato, hanno chiarito il meccanismo della specificità del riconoscimento del substrato La chimotripsina presenta una tasca idrofobica in cui si adattano le catene laterali aromatiche e alifatiche lunghe dei substrati. Il legame della giusta catena laterale in questa tasca idrofobica posiziona il legame peptidico adiacente sul lato carbossiterminale nel sito attivo dell’enzima in posizione corretta per il taglio. Chimotripsina = tasca in cui si lega la catena laterale aromatica del substrato = I residui chiave del sito attivo, Ser195, His57 e Asp102 Interazioni tra le proteasi e il substrato I siti potenziali di interazione del substrato con l’enzima sono indicati come P (in rosso) e i siti di legame corrispondenti sull’enzima sono indicati con le S. Similarità strutturali della tripsina e della chimotripsina = chimotripsina = tripsina Le sequenze amminoacidiche della tripsina, chimotripsina ed elastasi sono identiche per circa il 40% e le loro strutture sono molto simili. Tutte queste proteine sono serin-proteasi ed operano con un meccanismo simile a quello descritto per la chimotripsina I tre enzimi chimotripsina, tripsina ed elastasi sono simili nella struttura e nel meccanismo d’azione, ma differiscono per specificità di substrato. La chimotripsina richiede una grossa catena aromatica o alifatica sul lato carbonilico del legame peptidico suscettibile La tripsina richiede una lisina o una arginina sul lato carbonilico del legame peptidico suscettibile L’ elastasi richiede piccole catene laterali non cariche sul lato carbonilico del legame peptidico suscettibile Studi ai raggi X hanno chiarito che la diversa specificità è dovuta a piccole differenze nella struttura del sito di legame del substrato. Tasca idrofobica Tirosina Triptofano Fenilalanina Metionina Lisina Arginina Glicina e altri amminoacidi con catene laterali poco ingombranti La triade catalitica ed il buco dell’ossanione della subtilisina La subtilisina è una proteasi batterica non omologa alla chimotripsina ma che presenta un sito attivo simile La carbossipeptidasi II, nonostante abbia una struttura tridimensionale molto diversa da quella della chimotripsina, presenta una triade catalitica composta degli stessi residui amminoacidici. In questo enzima è presente anche il buco dell’ossanione (in giallo) Mutagenesi sito-specifica della triade catalitica nella subtilisina Non tutte le proteasi utilizzano strategie catalitiche basate su residui di serina attivati Le cistein-proteasi, le aspartilproteasi e le metalloproteasi sono altre classi principali di enzimi che tagliano catene polipeptidiche Queste classi di proteasi, anche se non usano un residuo di serina attivato, comunque hanno una strategia catalitica che prevede un nucleofilo che attacca il carbonile del legame peptidico da idrolizzare. Le strategie di attivazione delle tre classi di proteasi Il gruppo carbonilico peptidico è attaccato da: a) Una cisteina attivata da un’istidina nelle cistein proteasi b) Una molecola di acqua attivata da una coppia di residui di acido aspartico nelle aspartil proteasi c) Una molecola di acqua attivata da un metallo, quasi sempre zinco, nelle metallo proteasi Gli inibitori di proteasi possono essere utilizzati come farmaci. Struttura della proteasi di HIV La proteasi di HIV, dimerica, è necessaria per tagliare le proteine virali multidominio e trasformarle nelle forme attive. Bloccando la proteasi di HIV si blocca l’infettività del virus. Un inibitore di proteasi utilizzato come farmaco deve essere specifico per un enzima e non inibire altre proteine presenti nell’organismo Il crixivan, un inibitore della proteasi dell’HIV La struttura del crixivan è confrontata con quella di un peptide substrato della proteasi di HIV. Struttura del complesso proteasi di HIV - crixivan Il crixivan si lega nel sito attivo dell’enzima, inducendo, così come il naturale substrato, una modificazione conformazionale della proteina. Struttura della proteasi di HIV Struttura del crixivan che mette in evidenza la conformazione approssimativamente simmetrica che ricorda la struttura simmetrica dell’enzima. Il crixivan è in grado di interagire specificamente con la proteasi di HIV ma non con le aspartil-proteasi cellulari. FINE
Scaricare