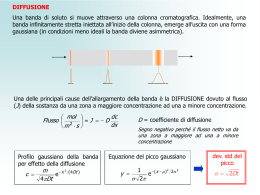
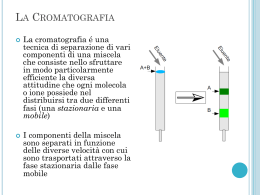
Equilibrio tra le fasi Dc = C1/C2, dove C indica la somma della concentrazione delle varie forme in cui può essere presente la sostanza che si ripartisce tra le fasi. Equilibrio FASE 1 prima dopo 0 V1 C1 V1 C0 V2 C2 V2 Concentrazione Volume FASE 2 Concentrazione Volume 1 p = frazione di soluto nella fase 1 = q = frazione di soluto nella fase 2 = C0 V2 = C1 V1+ C2 V2 Dividendo per C2V2 numeratore e denominatore si ha: p= q= 2 n=0 n=1 Le frazioni di soluto nei recipienti dopo l’equilibrio sono i termini individuali dell’espressione (q+p)n n=2 Se indichiamo con (Fr,n) la frazione di soluto contenuta in r dopo n trasferimenti abbiamo: n=3 n=4 r=0 r=1 r=2 r=3 3 È possibile dimostrare che, matematicamente rmax Il calcolo di F mediante l’espressione fattoriale diventa particolarmente lungo quando n ed r diventano grandi. Fortunatamente, in queste condizioni, la distribuzione binomiale può essere approssimata ad una distribuzione normale (gaussiana) che ha la forma: Per n>24 4 Separazione di due soluti (Dc1 =3, Dc2=1/3) per n=25 ed n=100 5 Lyman C. Craig (1906-1974) A manual version of Lyman C. Craig's CCD machine designed by Erich Hecker 6 TEORIA DEL PIATTO relazione con la distribuzione in controcorrente In cromatografia sia r che n sono molto grandi, ma n>> r, per cui Applicando l’approssimazione di Stirling 7 La frazione di ogni soluto sarà distribuita entro un certo numero di recipienti (piatti teorici) specificato dalla precedente equazione. Se vogliamo calcolare la frazione di soluto per rmax (massimo del picco), ricordando che rmax = np si ha Consideriamo la situazione in cui il soluto costituito da m moli totali, e la sua massima concentrazione si trovi in rmax = N, avremo che la quantità totale Q nel recipiente rmax = N sarà 8 Il soluto si muove lungo la colonna di N piatti in un tempo =tR (tempo impiegato dal massimo della concentrazione ad uscire dalla colonna), per cui la velocità di uscita espressa come piatti/tempo sarà N/tR e la velocità della massima quantità Smax sarà 9 La quantità m è proporzionale all’area del picco, mentre la velocità del massimo di concentrazione di uscita (massimo del picco) è proporzionale alla sua altezza. considerando in prima approssimazione il picco triangolare ed esprimendo l’altezza in unità arbitrarie e la base in unità di tempo avremo e considerando il picco gaussiano (tw = 4τ), dove τ è la deviazione standard espressa in unità di tempo 10 Piatti teorici • Per descrivere il processo cromatografico è utilizzata una similitudine derivante dalla teoria della distillazione. Il sistema cromatografico è immaginato simile ad una colonna di distillazione, cioè composta da una serie di strati sottili chiamati piatti teorici; in ognuno di questi microelementi della colonna si realizza l’equilibrio di distribuzione del soluto tra fase stazionaria e fase mobile. Lo spostamento del soluto lungo la colonna è dovuto all’azione dinamica della fase mobile I termini numero di piatti teorici (N) e altezza del piatto (HETP, Height Equivalent to Theoric Plate) sono comunemente utilizzati in cromatografia per quantificare le prestazioni dei sistemi cromatografici 11 1) 2) Altezza equivalente di piatto teorico H Numero di piatti teorici N N L = lunghezza colonna L H Piatto teorico: sezione della colonna che consente di realizzare un equilibrio reversibile di ripartizione di un componente fra le fasi. Poiché in cromatografia si ha una sequenza continua di stati di equilibrio e non vi è possibilità di realizzare una singola separazione, N ha un significato puramente matematico. Più elevato è il numero di piatti teorici, più grande è la probabilità di una separazione (migliore è la capacità di separazione della colonna). N è proporzionale alla lunghezza della colonna. t N R 2 t N 16 R W 2 W L’altezza equivalente al piatto teorico (H = L/N) consente di confrontare l’efficienza di colonne di differente lunghezza. 12 w½= 2.35σ LIMITI DELLA TEORIA DEL PIATTO • K è costante ed indipendente dalla concentrazione del soluto • L’equilibrio è rapido rispetto alla velocità della fase mobile • Non c’è diffusione longitudinale del soluto (recipienti con pareti) • La fase mobile è aggiunta per incrementi e non in continuo. 14 DI queste assunzioni solo la prima è valida nella cromatografia analitica ed è mantenuta nella teoria dinamica che risulta anche essa valida solo per la cromatografia con isoterma di ripartizione lineare I piatti nella colonna non esistono, sono una approssimazione e non una realtà fisica, l’equilibrio non è istantaneo e la diffusione avviene lungo tutta la colonna, mentre il passaggio da una fase all’altra avviene prima che l’equilibrio sia stabilito. Infine, la larghezza del picco dipende dalla velocità con cui la fase mobile fluisce lungo la colonna. 15 Le molecole di un analita non si muovono lungo la colonna con la stessa velocità: la loro dispersione ha generalmente un profilo Gaussiano. Il centro del profilo (banda di eluizione) rappresenta la velocità media. I fattori che provocano deviazioni dal valore medio sono: 1. diffusione longitudinale (diffusione delle molecole dalla zona a maggiore concentrazione a quella a minore in direzione parallela all’asse della colonna); 2. percorsi multipli; 3. trasferimento di massa tra fase mobile e fase stazionaria. 16 17 18 LA TRATTAZIONE FISICA DI QUESTI FENOMENI HA DATO ORIGINE ALLA TEORIA DINAMICA 19 TEORIA DINAMICA σ2 = Σσj2 H = ΣHj Diffusione longitudinale σmd = (2γm Dm t)½ Ricordando che * N = tr2/σt2 = L2/ σL2 ; H = L/N = σL2/L e t/L =1/u Hmd = 2γm Dm /u ; Hsd = 2k’γs Ds /u Dove γ è il fattore di ostruzione, D il coefficiente di interdiffusione e u la velocità lineare 20 MOLTEPLICITA DEI CAMMINI Detta anche “eddy diffusion” Hfed = 2 λ dp • λ è una misura della non eguaglianza dei flussi vale solo per le colonne impaccate 21 TRASFERIMENTO DI MASSA Colonne impaccate Nella fase mobile Hmfd = ω dp2 u/Dm ω = fattore di impaccamento dp = diametro delle particelle Nella fase stazionaria Hsfd = 2/3(2k’/[1+k’]2) (df2/Ds) u df = spessore del film di fase stazionaria Colonne tubolari Nella fase mobile Hmfd = (1+6k’+11k’2)dt2u/96(1+k’)2Dm dt = diametro del tubo Nella fase stazionaria 22 la formula è come per le impaccate 24 Equazione di Van Deemter L’equazione che meglio riproduce i dati sperimentali è la combinazione di tre equazioni che esprimono i tre fenomeni diffusivi H = A + B/u + Cmu + Cs u 25 L’equazione di Giddings modifica l’equazione di Van Deemter Sources of Extra Column Dispersion 29 Risoluzione cromatografica La possibilità di separare due o più sostanze è descritta dal parametro detto risoluzione, che misura la capacità di un sistema cromatografico di separare due analiti con caratteristiche simili: R 2( tR )B ( t R )A 2Z WA WB WA WB Se la risoluzione non è sufficiente (R < 1), i due picchi non possono essere quantificati in maniera corretta 30 1 1 k IB R N I 4 kB 1 Effetto della selettività, dell’efficienza e del fattore di capacità sulla risoluzione risoluzione scarsa picchi non separati buona risoluzione dovuta a buona efficienza picchi stretti buona risoluzione dovuta a buona selettività picchi distanti risoluzione scarsa dovuta ad un basso fattore di capacità 31 Risoluzione e separazione Capacità di picco 35 36 ELUIZIONE IN GRADIENTE 37 38 39 40
Scaricare