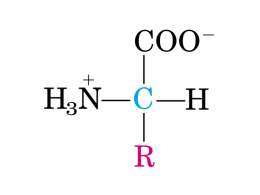
Amminoacidi e Proteine: Ruolo Metabolico e Nutrizionale Il Fabbisogno proteico è costituito da due componenti: Fabbisogno di azoto Fabbisogno di amminoacidi essenziali Overview del catabolismo degli amminoacidi nei mammiferi Overview del catabolismo degli amminoacidi nei mammiferi Ossidazione degli amminoacidi • La degradazione degli amminoacidi è un processo complesso che coinvolge un numero molto grande di intermedi: Ala Ser Cys Gly Thr Trp Piruvato Ile Leu Thr Trp CO2 • Catena Carboniosa • Azoto ammoniacale: – Urea – NH3 • Azoto basi azotate: – Acido urico – Urea – NH3 Acetil-CoA Glucoso Asn Asp Ossalacetato Fumarato Asp Phe Tyr Acetoacetato Citrato KREBS Isocitrato CO2 -chetoglutarato Succinil-CoA Ile Met Val CO2 Leu Lys Phe Tyr Arg Gln Glu Pro His Metabolismo della catena carboniosa degli amminoacidi Lo scheletro carbonioso fornisce energia attraverso l’interazione con la via glicolitica ed il ciclo di Krebs In condizioni fisiologiche esiste una relazione tra il livello dei carboidrati nella dieta ed il metabolismo degli amminoacidi Aumenta quando l'apporto di energia è sufficiente ma viene fornito da proteine a scapito dei carboidrati e dei lipidi La sua efficienza (molecole di ATP prodotte) è inferiore rispetto alla degradazione degli altri due substrati (carboidrati e lipidi): Non tutto lo scheletro carbonioso degli amminoacidi è soggetto ad ossidazione La formazione di urea (il prodotto finale del catabolismo azotato) richiede il consumo di 3 molecole di ATP AMMINOACIDI GLUCOGENICI glicolisi o ciclo di Krebs possono essere usati per la gluconeogenesi AMMINOACIDI CHETOGENICI degradati ad acetil-CoA soltanto LEUCINA e LISINA sono chetogenici puri Amminoacidi più grandi quali ISOLEUCINA FENILALANINA, TIROSINA TRIPTOFANO sono sia glucogenici che chetogenici gli altri sono glucogenici Catabolismo del gruppo amminico • Il primo stadio della degradazione degli amminoacidi è la rimozione del gruppo amminico attraverso le amminotransferasi: -chetoacido1 + AA2 Transaminazione • Le reazione di transaminazione usano come coenzima il piridossal fosfato. • Il piridossal fosfato forma una base di Shiff con un residuo di Lys della transaminasi O OH HO N + O H OH CH3 O NH3+ O OH P O O N + CH3 H H Piridossina (Vit B6) Piridossal fosfato (PLP) O OH P O O N + CH3 H Piridossamina fosfato (PMP) Transaminazioni O O O Asp O Ossalacetato NH3 + O O O O O H3 C NH3 + Ala O H3 C O O O O CH3 NH3 + O O H3 C NH3 + O CH3 O O Leu O + + H3 C O O -chetoglutarato Glu O O OH p-idrossifenilpiruvato NH3 + O O -chetoisocaproato O OH Tyr Piruvato O O O O O O Ruolo metabolico delle transaminasi In aggiunta ad equilibrare il gruppo amminico tra gli -keto acidi disponibili Permettono la sintesi di amminoacidi non essenziali Aiutano a mantenere la varietà degli amminoacidi per la sintesi delle diverse proteine Convogliano il gruppo amminico degli amminoacidi in eccesso (derivanti dalla dieta) verso la formazione del GLUTAMMATO che può essere deaminato Scheletro carbonioso degli amminoacidi deaminati può essere catabolizzato per produrre energia o utilizzato per produrre glucosio o acidi grassi Pochi amminoacidi possono essere direttamente deaminati Glutammato deidrogenasi Glutammato Deidrogenasi catalizza la reazione che porta alla rimozione netta di N dal pool degli amminoacidi E’ uno dei pochi enzimi che utilizza sia NAD+ o NADP+ come accettore di elettroni. Ossidazione del carbonio è seguita da idrolisi che porta al rilascio di NH4+. + H2O NH4 H2O HO CH2 H C COO H2C COO H3C C COO NH3+ NH3+ serine C O aminoacrylate pyruvate Serine Dehydratase Altri processi di deamminazione: 1. Serina Deidratasi catalizza: serina piruvato + NH4+ 2. Perossisomi L- e D-amminoacidi ossidasi catalizzano: amminoacido + FAD + H2O -chetoacido + NH4+ + FADH2 FADH2 + O2 FAD + H2O2 Catalasi catalizza: 2 H2O2 2 H2O + O2 Trasporto dello ione ammonio sotto forma di Glutammina L’ecceso di ione ammonio nei tessuti viene convertito in Glutammina, un processo catalizzato dalla GLUTAMMINA SINTETASI. Attraverso il flusso sanguigno la Glutammina raggiunge il fegato e NH4+ è liberato nei mitocondri dall’enzima GLUTAMMINASI Ciclo Glucosio-Alanina Un altro modo di trasportare lo ione ammonio dai tessuti muscolari al fegato O H 2N C NH2 urea La maggior parte degli animali terrestri convertono l’eccesso di ammonio in urea, prima dell’escrezione. Urea è meno tossica dello ione ammonio. Il ciclo dell’ Urea avviene prevalentemente nel fegato. I due gruppi amminici dell’urea derivano da NH3 (prodotto principalmente dalla Glutammato Deidrogenasi) e dal gruppo N dell’aspartato. L’ NH3 e HCO3- (gruppo carbonilico) sono dapprima trasformati in carbamil fosfato. Ciclo dell’urea Complicanze metaboliche Carenze ereditarie di qualsiasi enzima del ciclo dell’urea porta a Iperammoniemia - elevata [ammonio] nel sangue. Mancanza totale di ogni enzima del ciclo dell’Urea è letale. Elevate concentrazioni di ammoniaca sono tossiche specialmente per il cervello. Disturbi se non trattati immediatamente dopo la nascita portano a gravi alterazioni a livello dello sviluppo cognitivo. Meccanismi di tossicità per [ammoniaca]: 1. Alta [NH3] attiva Glutammina Sintasi: glutammato + ATP + NH3 glutammina+ ADP + Pi Porta a deplezione di glutammato – un neurotrasmettitore e precursore dei GABA. 2. Deplezione di glutammato e alta ammoniaca portano la Glutammato Deidrogenasi a funzionare nel verso contrario: glutammato + NAD(P)+ -chetoglutarato + NAD(P)H + NH4+ La deplezione di -chetoglutarato, un intermedio essenziale del Ciclo di Krebs altera il metabolismo energetico nel cervello. Ammoniaca può essere in parte riutilizzata -chetoglutarato + NH4+ glutammato glutammato + NH4+ + ATP glutammina + ADP + Pi NH4+ + CO2 + folato glicina Urea può essere ritrasformata in ammoniaca dalla microflora intestinale: non è chiaro l’eventuale contributo “perdita obbligatoria di azoto” con le urine Urea 10 - 30 g/die dipende quantità proteine alimentari NH3 0,4-1,2 g/die dipende equilibrio acido-base Amminoacidi 0,3 -1,2 g/die Acido urico 0,2-0,7 g/die dipende dalla dieta Creatinina 0,3-0,8 g/die dipende dalla massa muscolare (indice del turnover proteico del muscolo) secrezioni intestinali, turnover enterociti, desquamazione pelle, … NH3 deriva dal catabolismo degli amminoacidi basi puriniche (tramite deaminasi) basi pirimidiniche Formazione di acidi non volatili dal catabolismo di a.a. solforati metionina, cisteina SO42fosfoproteine fosfati fosfolipidi acidi grassi e glucosio acetoacetato, -idrossibutirrato, lattato acidi nucleici basi puriniche urati nutrienti con anioni inorganici (superiori alla quantità di cationi inorganici) Richiede l’escrezione di un catione NH4+ (fornito dalla glutammina) per permettere all’organismo di conservare cationi quali Na+, K+, Ca2+, dieta ricca in proteine genera acidi non volatili Proteolisi • Idrolisi del legame peptidico – Nell’intestino dell’uomo sono presenti diverse proteasi secrete da diversi organi digestivi: • Dallo stomaco: pepsina • Dal pancreas: chimotripsina, tripsina, carbossipeptidasi • Dall’intestino tenue: peptidasi intestinali, aminopeptidasi Con l’eccezione della lipasi linguale e dell’amilasi salivare tutti gli enzimi digestivi vengono secreti in forma di zimogeni Digestione delle proteine L’assorbimento di molecole proteiche intatte avviene raramente nell’adulto ma è più frequente nel neonato. Gli anticorpi materni arrivano intatti grazie anche alla presenza nel latte di inibitori di peptidasi e vengono assorbiti. Questo spiega i fenomeni allergici a cui vanno incontro i neonati non nutriti con latte materno e il meccanismo di trasferimento delle difese anticorpali dalle nutrici al neonato. orletto a spazzola 1 3 5 2 6 4 Digestione delle proteine: 6 fasi 1. Idrolisi gastrica del legame peptidico; 2. Digestione da parte delle proteasi pancreatiche nel lume dell’intestino tenue; 3. Idrolisi degli oligopeptidi da parte di peptidasi dell’orletto a spazzola degli enterociti; 4. Ulteriore digestione dei di- e tri-peptidi da peptidasi citoplasmatiche nell’enterocita; 5. Metabolismo degli AA negli enterociti; 6.Trasporto degli AA attraverso la membrana basolaterale e invio al sangue portale e quindi al fegato Alcuni alimenti contengono inibitori della tripsina (piselli,fagioli,lenticchie,soia) diminuendo in tal modo il valore nutrizionale delle proteine Origine Zimogeno/ Enzima Attivazione Substrato Prodotto finale Stomaco Pepsinogeno/pepsina pH 1-2, autoattivazione Proteine Peptidi Pancreas Tripsinogeno/tripsina Enteropeptidasi Proteine, peptidi Peptidi, dipeptidi Tripsina Proteine, peptidi Peptidi, dipeptidi Tripsina Proteine, peptidi Peptidi, dipeptidi Tripsina Estremità Cterminale polipeptidi Peptidi, aminoacidi Estremità Nterminale di oligopeptidi Peptidi, aminoacidi (membrana enterociti duodenali) Chimotripsinogeno/ chimotripsina Pro-elastasi/elastasi Pro-carbossipeptidasi Intestino tenue (membrana e citoplasma) Aminopeptidasi Assente Contenuto in proteine di un UOMO ADULTO: circa 12 kg/70 kg peso actina, miosina, collagene ed emoglobina costituiscono circa la metà di tutte le proteine 40% nel muscolo: possono diventare fonte di amminoacidi in condizioni di stress, si perdono però proteine funzionali 10% tessuti viscerali (fegato, intestino): scarsamente mobilizzate in condizioni di stress 30% nella pelle e nel sangue: diventano fonte di amminoacidi in deficit di proteine alimentari CONTINUO RICAMBIO PROTEICO Serve energia sia per la sintesi che per la degradazione: 15-20 % del bilancio energetico La continua demolizione e sintesi è fondamentale per degradare e rimpiazzare proteine danneggiate modificare la quantità relativa di differenti proteine in base alle necessità nutrizionali e fisiologiche rapido adattamento metabolico La regolazione del turnover proteico è influenzata da: stato nutrizionale (energetico e proteico) da alcuni ormoni (insulina, glucocorticoidi, ormoni tiroidei, ormone della crescita, citochine) ORGANISMO Ricambio giornaliero 1-2% proteine totali Amminoacidi 70-80% riutilizzati 20-30% metabolizzati Proteine dalla dieta Proteine metabolizzate 70 grammi/giorno 250 grammi/giorno % ricambio muscolo 30-50% fegato 25% leucociti emoglobina diversa emivita pochi minuti: proteine regolatorie 300 giorni: collageno
Scaricare