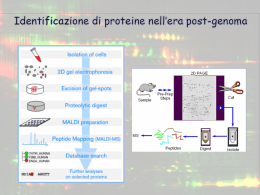
La spettrometria di massa è una metodica che consente l’identificazione e l’analisi quantitativa di una molecola dalla sua massa. Si sfruttano due fenomeni correlati alla massa ed alla carica. 1) la traiettoria di uno ione o di una particella carica in movimento può essere modificata per azione di un campo magnetico od elettrico, e l’entità della deviazione è funzione del rapporto massa/carica della particella: a parità di carica, particelle di massa minore subiranno deviazione maggiore. 2) ioni o particelle cariche, accelerati da un campo elettrico, assumo velocità diverse in dipendenza della loro massa: a parità di carica, particelle di massa maggiore assumono velocità minore. Gli spettrometri di massa di prima generazione sfruttavano unicamente il primo fenomeno; attualmente sono disponibili strumenti che si basano sul primo o sul secondo fenomeno. Spettrometria di massa atomica (analisi qualitativa e quantitativa di sostanze inorganiche). Spettrometria di massa molecolare per l’analisi di sostanze organiche. Attualmente la spettrometria di massa costituisce una metodica largamente diffusa per lo studio di molecole e macromolecole di interesse biologico, quali amminoacidi, glicidi, lipidi, proteine ed acidi nucleici. Uno spettrometro di massa separa gli atomi o le molecole secondo uno dei principi indicati, ma per poter essere separati atomi e molecole devono avere una carica, devono cioè essere ionizzate. Inoltre, devono essere allo stato gassoso. Tappe fondamentali del processo d’analisi: 1. ionizzazione delle molecole in esame, cioè la trasformazione in uno o più ioni, in genere con carica positiva; 2. accelerazione degli ioni per immissione in un campo elettrico; 3. separazione degli ioni con massa diversa; 4. rivelazione dei diversi ioni formatisi e la conseguente determinazione della loro massa. Componenti fondamentali: 1. camera di ionizzazione per produrre ioni; 2. un campo elettrico per accelerare gli ioni prodotti; 3. un analizzatore di massa, che utilizzando un campo magnetico e/o un campo elettrico, separa gli ioni di massa diversa; 4. un rivelatore, che raccoglie gli ioni generando un impulso quantificabile e registrabile. La formazione di ioni di campione in fase gassosa è un pre-requisito essenziale per i processi di separazione e di rivelazione tipici in uno spettrometro di massa. Fino a non molto tempo fa gli spettrometri di massa richiedevano il campione in fase gassosa, ma grazie agli sviluppi più recenti, l’applicabilità della spettrometria di massa è stata estesa fino a includere anche campioni in fase liquida o inglobati in una matrice solida. Il campione, che può essere solido, liquido o gassoso, viene introdotto in una camera da vuoto mediante un opportuno sistema di introduzione. In dipendenza del tipo di sistema di introduzione e della tecnica di ionizzazione utilizzata, il campione può già esistere in forma ionica in soluzione, oppure esso può essere ionizzato di concerto con la sua volatilizzazione o mediante altri metodi nella sorgente ionica. Gli ioni prodotti, che si trovano in fase gassosa, vengono separati nell’analizzatore sulla base del loro rapporto massa/carica (m/z), e vengono raccolti da un rivelatore. Nel rivelatore essi generano un segnale elettrico proporzionale al numero di ioni presenti. Il sistema di elaborazione dati registra questi segnali elettrici in funzione del rapporto m/z e li converte in uno spettro di massa. Sono state sviluppate nel tempo diverse modalità di ionizzazione, che rappresentano il principale elemento di differenziazione tra le varie metodiche e tra le diverse strumentazioni attualmente impiegate. La sorgente di ionizzazione al plasma trova applicazione essenzialmente nella spettrometria di massa atomica, le altre metodiche nella spettrometria molecolare, e si possono suddividere in tecniche hard e soft. Principali modalità di ionizzazione Ionizzazione al plasma ICP Inductively Coupled Plasma Impatto elettronico EI Electronic Impact Ionizzazione chimica CI Chemical Ionization FAB - FIB Desorbimento di ioni PD MALDI Evaporazione ionica Fast Atom/Ion Bombardament Plasma Desorption Matrix Assisted Laser Desorption Ionization TSI Thermo Spray Ionization ESI Electro Spray Ionization APCI Atmospheric Pressure Chemical Ionization La spettrometria di massa MALDI (Matrix assisted laser desorption ionization)- TOF (Time of fly) viene principalmente impiegata per l’analisi di composti organici non volatili che presentano un peso molecolare elevato. Il campo di utilizzo principale riguarda l’analisi di proteine, peptidi, lipoproteine, oligosaccaridi e oligonucleotidi. Rappresenta un metodo d’analisi abbastanza semplice e compatibile con i tamponi utilizzati in laboratorio. L’accuratezza della massa ottenuta dipende dall’analizzatore dello spettrometro di massa, e sulla maggior parte degli strumenti moderni si è in grado di misurare masse con un errore dello 0,01% per masse molecolari fino a 40.000 Da. Un raggio emesso da un laser nell’UV fornisce l’energia per il desorbimento. Si evita l’irradiazione continua che potrebbe decomporre il campione e con impulsi di pochi nanosecondi si ottengono in fase gassosa ioni molecolari, che subiscono scarsissima frammentazione ed il cui rapporto m/z può essere misurato in un analizzatore a tempo di volo, TOF. 1 l della soluzione del campione da analizzare viene aggiunto ad una soluzione satura di un composto organico, la matrice, che sia capace di assorbire luce alla emessa dal laser. La soluzione risultante viene deposta su un’appropriata superficie metallica inerte, definita MALDI spot, ed il solvente fatto evaporare. Si ottiene una matrice solida tra le molecole della quale sono disperse le molecole del campione, cioè matrice e campione sono cocristallizzati in una MALDI spot. La matrice va scelta in funzione del tipo di campione, e deve essere in grado di solubilizzare molecole polari ed apolari. Formula di struttura delle matrici reperibili in commercio. Un raggio emesso ad es a 266 nm da un laser NdYAG od uno a 337 nm da un laser ad azoto, viene inviato al campione e la sua energia verrà prima assorbita dalla matrice e da questa trasferita poi, in parte, alle molecole di campione. Le molecole di campione vengono desorbite, intatte, in forma gassosa, e ionizzate per trasferimento di protoni da parte di ioni derivati dal desorbimento della matrice. Da ciascuna molecola si origina, pertanto, un’unica specie ionica che non subisce ulteriori frammentazioni. I vantaggi dell’utilizzo della matrice sono molteplici: i campioni possono essere analizzati anche in presenza di tamponi, detergenti, chelanti in quanto questi non interferiscono con il processo di desorbimento. MALDI Accoppiando la ionizzazione MALDI ad un analizzatore TOF si possono rapidamente separare ed analizzare i componenti di miscele complesse di macromolecole con masse anche maggiori di 300 kDa, utilizzando solo qualche pmole di campione. Svantaggi: la difficoltà di analizzare composti con P.M. < 600 Da e la difficoltà di usare un collegamento con HPLC od elettroforesi capillare. La peculiarità dell’ESI è il fatto che la ionizzazione avviene a pressione atmosferica e dà origine ad ioni multicarica della stessa molecola. Poiché la separazione avviene in base al rapporto m/z, aumentando il numero delle cariche z di uno ione è possibile che molecole di parecchi milioni di dalton rientrino nell’intervallo di massa rilevabile dalla maggior parte degli analizzatori esistenti. ESI Il campione, sciolto in una miscela acqua/solvente volatile (metanolo, acetonitrile o loro miscele), è introdotto a pressione atmosferica nello spettrometro attraverso un capillare metallico che costituisce il terminale – anado o catodo – di un circuito elettrico cui è applicata una differenza di potenziale di circa 4 – 6 kV, responsabile della ionizzazione della soluzione. L’altro terminale del circuito è dato da un elemento dell’analizzatore, verso il quale saranno attratti gli ioni di carica opposta. Dal capillare la soluzione fuoriesce come uno spray formato da minutissime goccioline cariche che, a seguito dell’evaporazione del solvente provocata da un flusso di azoto o da calore, diminuiscono ulteriormente fino ad esplodere, quando la forza di repulsione supera la tensione superficiale = esplosione coulombiana. Si liberano ioni multicarica dell’analita in fase gassosa. Da un campione è possibile produrre ioni molecolari positivi o negativi a seconda della polarità del voltaggio del capillare e del solvente utilizzato. Da una stessa molecola si formano molteplici ioni molecolari ciascuno recante un diverso numero di cariche, che vengono quindi inviati all’analizzatore sotto vuoto dell’apparecchio: si ottiene così uno spettro in cui i numerosi picchi adiacenti differiscono tra loro per una unità di carica, più comunemente un singolo protone. Il programma computerizzato che gestisce lo spettrometro provvede a calcolare il valore della carica di ogni singolo picco e ciò consente di determinare il valore della massa con una accuratezza superiore a qualsiasi altra tecnica disponibile. Se ne ricava il così detto “spettro deconvoluto” che riporta un unico picco, il cui valore di massa è quello del campione analizzato. La metodica ESI, prestandosi specificamente per la ionizzazione di molecole polari ad alto peso molecolare, trova larga utilizzazione nell’analisi di polipeptidi e proteine e consente con facilità di mettere in evidenza anche le più piccole variazioni di massa dovute a modificazioni post-traduzionali. Gli analizzatori di massa hanno la funzione di separare ioni di massa diversa utilizzando un campo magnetico e/o campo elettrico. A seconda della modalità con la quale esplicano la funzione si distinguono in analizzatori: - a Settore magnetico; - a Tempo di volo (TOF); - a Quadruplo; - a Trappola ionica; - a Risonanza ciclotronica ionica in trasformata di Fourier FT-ICR. Non esiste un analizzatore di massa adatto per tutte le applicazioni possibili, e la scelta dipende dal tipo di analisi che si effettua, tenendo conto che i vari analizzatori differiscono tra loro principalmente per: il massimo rapporto m/z misurabile; il potere risolutivo; la sensibilità (nmoli e pmoli); la velocità di analisi; la facile utilizzazione se lo spettrometro è interfacciato ad un altro apparecchio (HPLC, GC, EC). Per quanto riguarda il potere risolutivo è da premettere, che in uno spettro di massa, due picchi adiacenti di intensità simile si considerano separati se l’altezza h della valle tra di loro è minore del 10% dell’altezza H del picco maggiore. Il potere risolutivo (R) è quindi la capacità di separare due picchi di massa diversa: R = m2/(m2-m1) dove m2 è il valore m/z maggiore, m1 quello minore. Gli analizzatori si considerano a bassa risoluzione se R è inferiore a 5.000 e ad alta risoluzione se R è superiore a 5.000. All’aumentare del potere risolutivo dell’analizzatore aumenta l’accuratezza della misura che indica di quanto il valore di massa ottenuto o massa misurata si discosta dal valore reale o massa reale, indica cioè l’ampiezza dell’errore insito nella misurazione. L’accuratezza si indica per convenzione come l’inverso del potere risolutivo, e si ricava dall’espressione (m2-m1)/m2, considerando m2 il valore di massa esatta e m1 il valore di massa misurata. Il risultato di questo rapporto viene moltiplicato per 106 per esprimere l’ampiezza dell’errore in ppm (parti per milione = mg per litro). L’accuratezza dipende da molteplici fattori connessi alle metodiche seguite nell’analisi per la ionizzazione, la rilevazione, l’acquisizione dei dati, che possono influire singolarmente o congiuntamente sul risultato finale. Per determinare l’accuratezza di un’analisi si effettua per ogni serie di analisi eseguite nello stesso contesto sperimentale, va effettuata una calibrazione con una molecola di cui si conosce la massa esatta. Analizzatori a settore magnetico; Analizzatori a tempo di volo – TOF; Analizzatori a quadrupolo; Analizzatore a trappola ionica; Analizzatore di risonanza ciclotronica ionica in FIT (FIT-ICR). In questi analizzatori non vi è necessità di un campo magnetico od elettrico per selezionare gli ioni in base ad una diversa traiettoria, perchè si misura la velocità con la quale volano verso il rivelatore. Per poter avere una misura del tempo di volo è indispensabile l’esatta determinazione del momento di inizio della corsa, e vi è quindi la necissità di una produzione pulsata di ioni; l’utilizzazione più diffusa degli analizzatori TOF è perciò in accoppiamento con il sistema di ionizzazione MALDI. Gli ioni provenienti dalla sorgente vengono accelerati da un forte campo elettrico, di 20 kV, all’uscita del quale tutti gli ioni hanno uguale energia cinetica, ma differente velocità a seconda della loro massa. Pertanto lasciandoli correre in una regione libera da campi elettrici o magnetici come un tubo sotto vuoto spinto, gli ioni raggiungeranno il rivelatore in tempi diversi: al collettore situato alla fine del tubo arriveranno prima le particelle più veloci, ossia quelle a massa minore. Wiley and McLaren observed that ions of a particular mass-to-charge ratio would reach the detector with a spread in arrival times, due to the effects of uncertainty in the time of ion formation, location in the extraction field and initial kinetic energy, resulting in reduced resolution. Wiley and McLaren devised an instrument, incorporating a pulsed two-grid ion source, to compensate for temporal, spatial and initial kinetic energy distributions. The basic geometry of the Wiley-McLaren design is shown in the figure below: I primi analizzatori TOF, denominati a “modalità lineare” coprivano un’ampio intervallo di valori di massa con alta sensibilità, ma avevano però una bassa risoluzione. Ciò si deve al fatto che gli ioni contenuti nella nube di materiale desorbito non partono tutti esattamente dalla medesima posizione. Nei più recenti analizzatori TOF effettuando una “estrazione ritardata” degli ioni ed introducendo nel tubo di volo un “riflettore elettrostatico” il potere risolutivo arriva a 104. L’estrazione ritardata si ottiene applicando il campo elettrico necessario per l’accelerazione degli ioni con un certo ritardo rispetto al desorbimento, quando cioè si hanno a disposizione tutti gli ioni, ed è possibile applicare campi di estrazione differenti a ioni con velocità iniziali diverse in modo da provocare il loro compattamento. Il riflettore elettrostatico posto all’estremità del tubo di volo è composto da una serie di anelli o di griglie alle quali viene applicato un potenziale crescente dello stesso segno della carica degli ioni, che vengono pertanto respinti. Gli ioni penetrano nel riflettore fino a raggiungere energia cinetica pari a zero e vengono poi riflessi e riaccelerati nella direzione opposta. Due ioni aventi lo stesso m/z, ma diversa energia cinetica percorrono spazi diversi nel riflettore: quello con energia cinetica maggiore penetra più in profondità rispetto a quello con energia cinetica minore e quindi viene riflesso dopo. L’analizzatore TOF permette l’analisi di valori di massa molto elevati, e questa caratteristica contribuisce alla sua associazione preferenziale ad una sorgente MALDI, capace di produrre ioni monocarica di elevato peso molecolare. L’analizzatore a quadrupolo o filtro di massa a quadrupolo è formato da quattro barre metalliche parallele di 10 – 20 cm di lunghezza poste in una camera sotto vuoto. Le barre hanno sezione circolare od iperbolica e sono disposte a coppie sovrapposte. Le barre sono collegate sia ad una sorgente di corrente continua, sia ad una sorgente di corrente alternata a radiofrequenza. Ad una coppia di barre diagonalmente opposte è applicato un potenziale positivo, all’altra coppia un potenziale negativo. A tutte e due le coppie è sovrapposta una corrente a RF , ma il voltaggio di RF sovrapposto alle barre negative è tale da trovarsi di 180° fuori fase rispetto alla coppia positiva. La funzione della RF è quella di far variare la polarità ed il voltaggio delle barre, più o meno rapidamente a seconda della frequenza applicata. Il movimento di uno ione che entra nel quadrupolo subisce oscillazioni in quanto viene alternativamente attratto e respinto dalle coppie di barre che variano in continuo il loro potenziale da + a -. Lungo il cammino degli ioni si genera un campo elettrico che varia continuamente, e la traiettoria seguita dagli ioni non è pertanto lineare, ma segue un andamento a spirale. Solo per certi valori di voltaggio e radiofrequenza gli ioni con un dato valore m/z mantengono un’oscillazione stabile, escono dall’analizzatore e giungono fino al rivelatore; gli altri, con diverso rapporto m/z subiranno delle oscillazioni instabili che li porteranno a collidere con le barre del quadrupolo ed ad annullarsi. Una proprietà del quadrupolo è che la massa degli ioni che lo attraversano fino al rivelatore è proporzionale al voltaggio applicato alle barre; pertanto variando voltaggio e RF si ottiene la filtrazione successiva, e quindi l’arrivo al rivelatore, di ioni a massa diversa. Ciò consente la selezione di un particolare ione, oppure la scansione nel campo delle masse tramite la variazione delle tensioni. Se si applica la sola RF tutti gli ioni sono inviati al rivelatore. Il potere risolutivo di questi analizzatori è nell’ordine di 3.000 e quindi il quadrupolo è da considerarsi un analizzatore a bassa risoluzione; è però meno costoso di un analizzatore a settore magnetico e l’acquisizione di uno spettro avviene in pochi secondi. L’analizzatore a quadrupolo è quello più frequentemente accoppiato alla ionizzazione per elettrospray, e trova ampia applicazione come sistema di rivelazione nella cromatografia liquida e nella gas cromatografia accoppiate alla spettrometria di massa, nelle metodiche ifenate. I rivelatori o detectors usati negli spettrometri di massa sono rapportabili a due tipi principali: quelli a misura diretta; quelli a moltiplicatore. Spettro di massa di un peptide La limitata frammentazione dello ione molecolare nelle tecniche di ionizzazione “soft”, MALDI o ESI, che facilita la determinazione della massa, costituisce uno svantaggio quando si vogliono avere informazioni più precise sulla struttura della molecola al fine di un riconoscimento inequivoco. E’ stata messa a punto la Spettrometria di Massa Tandem (MS/MS) nella quale lo ione molecolare stabile, immediatamente dopo la determinazione della sua massa, viene fatto collidere con molecole di gas neutro (elio od argon), acquista ulteriore energia e la dissipa frammentandosi. Il processo è detto Dissociazione Indotta da Collisione (CID). I frammenti ionici prodotti vengono quindi sottoposti ad una seconda analisi. Spettrometria tandem “nello spazio” e “nel tempo”: MS/MS nello spazio l’apparecchiatura comprende due analizzatori di massa disposti in serie, separati da una cella di collisione: a triplo quadrupolo (QqQ); a quadrupolo accoppiato con TOF (Qq-TOF); con due analizzatori TOF separati (TOF/TOF). Nella spettrometria tandem “nel tempo” non vi è necessità di due analizzatori: passaggi sopra descritti avvengono nello stesso analizzatore, ma in tempi diversi. Viene definita metodica ifenata una tecnica analitica che accoppia una separazione cromatografica od elettroforetica ad una determinazione di massa. GC-MS LC-MS CE-MS RP-HPLC-MS-MS Protein Identification A biological sample contains hundreds or thousands of proteins, all mixed together. The first step is to separate them. One common technique uses two-dimensional gels. The spots on the gel are different proteins, separated horizontally by pH and vertically by mass (the two dimensions). VHLTPEEK SAVTALWGK VNVDEVGGEALGR LLVVYPWTQR FFESFGDLSTPDAVMGNPK VK AHGK K VLGAFSDGLAHLDNLK GTFATLSELHCDK LHVDPENFR LLGNVLVCVLAHHFGK EFTPPVQAAYQK VVAGVANALAHK Mass spectrometers can identify proteins better if they are first broken down into tryptic peptides. Tryptic peptides are chains of amino acids that occur when the proteins are digested with the enzyme trypsin. The tryptic peptides for hemoglobin are shown here. The peptides have to be further sorted before they can be identified. This is often done in the following two steps: HPLC can separate peptides based on how soluble the peptides are — that is, the peptide hydrophobicity. Mass spectrometry separates the peptides based upon their mass. This is the first mass spectrometer of the two in a tandem mass spectrometer instrument. The second part of the tandem mass spectrometer examines the individual peptides in detail for identifying features — the distances between the peaks in the mass spectral graph. The peaks represent the number of ions in the peptide's spectrum that have a certain mass and charge. A protein is identified by matching the identifying features of the peptides to a database of proteins. An identification is more believable if it is based on matching mass spectra from several peptides. Peptide Sequencing by Tandem Mass Spectrometry. The most common usage of MS-MS in biochemical areas is the product or daughter ion scanning experiment which is particularly successful for peptide and nucleotide sequencing. Peptide sequencing: H2N-CH(R')-CO-NH-CH(R")-CO2H There are three different types of bonds that can fragment along the amino acid backbone: the NH-CH, CH-CO, and CO-NH bonds. Each bond breakage gives rise to two species, one neutral and the other one charged, and only the charged species is monitored by the mass spectrometer. The charge can stay on either of the two fragments depending on the chemistry and relative proton affinity of the two species. Hence there are six possible fragment ions for each amino acid residue and these are labelled as in the diagram, with the a, b, and c" ions having the charge retained on the N-terminal fragment, and the x, y", and z ions having the charge retained on the C-terminal fragment. The most common cleavage sites are at the CO-NH bonds which give rise to the b and/or the y" ions. The mass difference between two adjacent b ions, or y"; ions, is indicative of a particular amino acid residue. Peptide sequencing by tandem mass spectrometry backbone cleavages The extent of side-chain fragmentation detected depends on the type of analyser used in the mass spectrometer. A magnetic sector - magnetic sector instrument will give rise to high energy collisions resulting in many different types of sidechain cleavages. Quadrupole - quadrupole and quadrupole time-of-flight mass spectrometers generate low energy fragmentations with fewer types of side-chain fragmentations. Immonium ions (labelled "i") appear in the very low m/z range of the MS-MS spectrum. Each amino acid residue leads to a diagnostic immonium ion, with the exception of the two pairs leucine (L) and iso-leucine (I), and lysine (K) and glutamine (Q), which produce immonium ions with the same m/z ratio, i.e. m/z 86 for I and L, m/z 101 for K and Q. The immonium ions are useful for detecting and confirming many of the amino acid residues in a peptide, although no information regarding the position of these amino acid residues in the peptide sequence can be ascertained from the immonium ions. An example of an MS/MS daughter or product ion spectrum is illustrated below. The molecular mass of the peptide was measured using standard mass spectrometric techniques and found to be 680.4 Da, the dominant ions in the MS spectrum being the protonated molecular ions (M+H+) at m/z 681.4. These ions were selected for transmission through the first analyser, then fragmented in the collision cell and their fragments analysed by the second analyser to produce the following MS/MS spectrum. The sequence (amino acid backbone) ions have been identified, and in this example the peptide fragmented predominantly at the CO-NH bonds and gave both b and y" ions. (Often either the b series or the y" series predominates, sometimes to the exclusion of the other). The b series ions have been labelled with blue vertical lines and the y" series ions have been labelled with red vertical lines. The mass difference between adjacent members of a series can be calculated e.g. b3-b2 = 391.21 - 262.16 = 129.05 Da which is equivalent to a glutamine (E) amino acid residue; and similarly y4 - y3 = 567.37 - 420.27 = 147.10 Da which is equivalent to a phenylalanine (F) residue. In this way, using either the b series or the y" series, the amino acid sequence of the peptide can be determined and was found to be NFESGK (n.b. the y" series reads from right to left!). The immonium ions at m/z 102 merely confirm the presence of the glutamine (E) residue in the peptide. Peptide sequencing by tandem mass spectrometry - an MS-MS daughter or product ion spectrum. A protein identification study would proceed as follows: •a. The protein under investigation would be analysed by mass spectrometry to generate a molecular mass to within an accuracy of 0.01%. b. The protein would then be digested with a suitable enzyme. Trypsin is useful for mass spectrometric studies because each proteolytic fragment contains a basic arginine (R) or lysine (K) amino acid residue, and thus is eminently suitable for positive ionisation mass spectrometric analysis. The digest mixture is analysed - without prior separation or clean-up - by mass spectrometry to produce a rather complex spectrum from which the molecular weights of all of the proteolytic fragments can be read. This spectrum, with its molecular weight information, is called a peptide map. (If the protein already exists on a database, then the peptide map is often sufficient to confirm the protein.) For these experiments the mass spectrometer would be operated in the "MS" mode, whereby the sample is sprayed and ionised from the nanospray needle and the ions pass through the sampling cone, skimmer lenses, Rf hexapole focusing system, and the first (quadrupole) analyser. The quadrupole in this instance is not used as an analyser, merely as a lens to focus the ion beam into the second (time-of-flight) analyser which separates the ions according to their mass-to-charge ratio. Q-TOF mass spectrometer operating in MS (upper) and MS/MS mode (lower) modes. c. With the digest mixture still spraying into the mass spectrometer, the Q-Tof mass spectrometer is switched into "MS/MS" mode. The protonated molecular ions of each of the digest fragments can be independently selected and transmitted through the quadrupole analyser, which is now used as an analyser to transmit solely the ions of interest into the collision cell which lies in between the first and second analysers. An inert gas such as argon is introduced into the collision cell and the sample ions are bombarded by the collision gas molecules which cause them to fragment. The optimum collision cell conditions vary from peptide to peptide and must be optimised for each one. The fragment (or daughter or product) ions are then analysed by the second (time-of-flight) analyser. In this way an MS/MS spectrum is produced showing all the fragment ions that arise directly from the chosen parent or precursor ions for a given peptide component. An MS/MS daughter (or fragment, or product) ion spectrum is produced for each of the components identified in the proteolytic digest. Varying amounts of sequence information can be gleaned from each fragmentation spectrum, and the spectra need to be interpreted carefully. Some of the processing can be automated, but in general the processing and interpretation of spectra will take longer than the data acquisition if accurate and reliable data are to be generated. The amount of sequence information generated will vary from one peptide to another. Some peptide sequences will be confirmed totally, other may produce a partial sequence of, say, 4 or 5 amino acid residues. Often sequence "tag" of 4 or 5 residues is sufficient to search a protein database and confirm the identity of the protein.
Scaricare