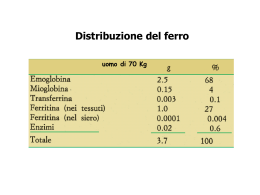
Azienda U.S.L. 6 - Livorno Laboratorio di Patologia Clinica LabNews Anno 8, Numero 32 Settembre 2006 LA LINGUA BATTE DOVE IL DENTE DUOLE Sommario: S Editoriale 1 e è vero che la lingua batte dove il dente duole, il dente dell’appropriatezza deve proprio fare un sacco di male, visto che in ogni momento della nostra pratica professionale ci imbattiamo in esempi diffusi e continuativi di un particolare aspetto della inappropriatezza, quella prescrittiva, che per sua natura coincide con i concetti di diseconomicità e di uso inappropriato delle risorse. Emoglobinopatie 3 Questo pensavo giorni fa dopo la lettura al microscopio a fluorescenza, quando la trascrizione dei risultati mi offriva una visione completa di quanto spesso richiesto da non pochi Colleghi clinici (ospedalieri o MMG). Pancreas: sindrome di Gullo 5 Emocromatosi 6 LabNews foglio di informazione del laboratorio di Patologia Clinica di Livorno direttore: Antonio La Gioia Hanno collaborato a questo numero: Mario Giordano Antonio La Gioia Antonella Leoni Angela Matteucci Patrizia Petricci Fabrizio Torsi Se, infatti, la richiesta isolata di anticorpi anti cellule parietali in un soggetto giovane, non anemico, con MCV “normale” può destare qualche perplessità, gli interrogativi diventano una valanga quando la richiesta “a stecca” di anticorpi antitiroide, ANA, ASMA, LKM, ANCA, APCA, EMA, ci induce al pensiero di segni e sintomi che orientano (?) in maniera indifferenziata verso una tireopatia autoimmune o verso la celiachia, passando per LES, sclerodermia, CREST, epatite autoimmune, cirrosi biliare, vasculite, ecc. Esagerazioni? No, perché se applichiamo un criterio (arbitrario ma verosimilmente… appropriato) di inappropriatezza alla richiesta simultanea di almeno tre autoanticorpi possiamo quantificare l’incidenza del fenomeno nel 35%, fatto cento il numero di richieste alle quali la determinazione di solo unodue autoanticorpi attribuisce un (altrettanto arbitrario e benevolo) giudizio di appropriatezza. Non pare un problema da poco e non è riferito solo ai costi che potrebbero essere ridotti notevolmente in termini percentuali ma trascurabilmente in valore assoluto, stante la complessiva modesta incidenza di tali test sul complesso dei costi generali di laboratorio. Il vero problema, infatti, è rappresentato dalla bassa specificità di alcune di queste determinazioni (70% circa per gli ANA) che, conseguentemente producono una percentuale rilevante di falsi positivi e, inevitabilmente, riducono il valore predittivo positivo. Ed Viale Alfieri, 36 57100 Livorno [email protected] Anno 8, Numero 32 Pagina 1 LabNews (Continua da pagina 1) è su tale sottofondo che si innesca spesso l’avvio di un ulteriore percorso diagnostico che prevedibilmente produrrà un solo risultato certo: quello della medicalizzazione di un soggetto esente da patologie autoimmuni. E’ vero, per alcuni test la specificità è molto più alta e la positività può essere predittiva del successivo sviluppo della specifica patologia, in alcuni casi con anticipazione anche di molti anni. Tale è il caso degli anticorpi anti mitocondrio (AMA) che, se successivamente identificati con specificità M2, sono altamente predittivi dello sviluppo di cirrosi biliare primitiva. In questi casi, tuttavia, sarebbe opportuno aprire un dibattito sull’uso dei test di laboratorio come test di screening o di case finding che, a mio avviso, non possono essere la foglia di fico a copertura dell’uso diagnostico inappropriato. Il linguaggio della politica spesso ci regala stereotipi linguistici (convergenze parallele; opposti estremismi; senza se e senza ma) che, amplificati dai media, inevitabilmente entrano nell’uso comune e, nostro malgrado, spesso li usiamo. Ed è mio malgrado, perché non ho trovato di meglio, che definisco “assordante silenzio” il risultato di quanto denunciato nel precedente numero di LabNews a proposito degli orari di prelievo in molti reparti del nostro ospedale, poco attenti alle esigenze di chi forzatamente vi accede. Pensavo che i pochi aventi titolo per intervenire lo facessero, perché, in fondo, basta poco. Forse non ricevono Lab News o forse non lo leggono . Di una cosa, tuttavia, sono contento. Almeno io, il ricorso all’appropriatezza lo posso rivendicare: chi meglio di Fra’ Cristoforo, infatti, poteva rappresentare la tanta pazienza dei nostri pazienti? (alg) Eravamo in tanti, quasi tutti, lunedì 31 luglio a villa Morazzana e proprio l’essere tutti ha un po’ stemperato la commozione che, inevitabilmente, ti prende in questi casi Abbiamo salutato e fatto festa con loro, Roberta (Simonini) e Paolo (Soldatini), che passavano (comincio a provare invidia) tra quelli che non saranno più assillati dalla sveglia mattutina che ti chiama al lavoro. Paolo, per la verità, a giudicare dall’orario con cui arrivava in laboratorio, spesso la sveglia la spengeva e si girava dall’altra parte ed ogni tanto, con scarsa fiducia del risultato, lo riprendevo come lo riprendevo a moderare, a volte, la sua “visceralità”; ma il suo essere una persona onesta legata al suo lavoro e, soprattutto, fortemente consapevole della missione di Servizio pubblico del “suo” laboratorio sono tra le cose che mi piacerà portare con me. Di Roberta, invece, era da pensare che neppure ci andasse a letto o che non avesse bisogno della sveglia: puntuale sempre alle sette in camice e “con le provette in mano”. Non so a quante persone ha fatto da balia nei tanti anni qui ed a quante ha insegnato che anche nell’era delle macchine “che fanno tutto loro”, la manualità, la precisione, l’attenzione e l’ordine sono bagagli che non si disfano mai. Poi sono venuti nuovi compiti e le responsabilità “istituzionali” e forse le ha anche sofferte un po’ perché il cuore era rimasto dall’altra parte ed anche con questo, nell’aver saputo tenere distinti cuore e ragione ha mostrato la sua onestà ed integrità anche a scapito di popolarità e facili consensi. (alg) Anno 8, Numero 32 Pagina 2 LabNews α, β, γ, δ … L’ALFABETO DELL’EMOGLOBINA L ’emoglobina (Hb) è una molecola tetramerica formata da due coppie omologhe di catene globiniche (α, β, γ, δ, ε, ζ) assemblate in combinazioni che caratterizzano sia le emoglobine “normali” diversamente rappresentate nella vita embrionale, fetale ed adulta (vedi figura) sia le c.d. emoglobine patologiche, caratterizzate da assemblamenti non fisiologici di catene globiniche (ad es. tetrameri β4) o dalla presenza di catene globiniche strutturalmente anomale. Nella prassi comune vengono definite per estensione “patologiche” anche le emoglobine fisiologiche presenti in percentuali anomale in stadi cronologicamente inappropriati. L’emoglobina adulta (HbA) è formata da due catene α e due catene β e rappresenta circa il 97% dell’emoglobina nell’adulto. La restante quota di Hb nell’adulto è rappresentata dall’Hb fetale (HbF, α2γ2) e dall’HbA2 (α2δ2). Il gene che codifica per le catene δ ha fisiologicamente una bassa espressione. L’emoglobina fetale si riduce nel primo semestre di vita. Le emoglobinopatie sono alterazioni riguardanti le catene globiniche dell’emoglobina; tra queste possiamo distinguere difetti di sintesi dell’emoglobina, che danno luogo alle talassemie o microcitemie, e difetti di struttura che danno luogo alle varianti emoglobiniche. Talassemie Le talassemie sono le malattie ematologiche caratterizzate dal difetto di sintesi delle catene globiniche. La catena deficitaria dà il nome al tipo di talassemia. β-talassemie I geni che codificano per la catena β si trovano sul braccio corto di ciascun cromosoma 11 (materno e paterno). Ad eccezione dei portatori di mutazioni rare semi- dominanti, i β-talassemici eterozigoti hanno una modesta riduzione della sintesi di catene β che si manifesta come lieve anemia microcitica generalmente asintomatica (talassemia minor). La gravità dei quadri clinici (portatore asintomatico, talassemia intermedia, talassemia maior) dipende dal grado di sbilanciamento della sintesi globinica, e quindi dall’eccesso relativo di catene α, che precipitando nei precursori eritroidi determinano la loro morte prematura (eritropoiesi inefficace). La talassemia maior diviene pienamente manifesta a partire dal secondo semestre di vita quando scompare l’emoglobina fetale. Le diverse forme di β talassemia sono caratterizzate dall’aumento compensatorio della produzione di catene globiniche δ e, quindi, di HbA2 . α-talassemie I geni che codificano per la catena α si trovano sul cromosoma 16 e sono 2 paterni e 2 materni per cui i diversi genotipi possibili sono: αα/αα (normale), -α/αα (α+ thal eterozigote), --/αα (α0 thal eterozigote), -α/-α (α+ thal omozigote) -α/-- (α+/α0 combinazione), --/-- (α0 thal omozigote). Nell’alfa talassemia, come conseguenza della riduzione o assenza di catene alfa si può avere un accumulo di catene β (nell’adulto) e di catene γ (nel neonato) che portano alla formazione di emoglobine patologiche, rispettivamente HbH (β4) ed HbBart (γ4). Gli aspetti clinici dipendono dal numero dei geni colpiti. La maggior parte dei portatori carenti dell’espressione di uno o due geni è asintomatica o presenta una modesta anemia microcitica, mentre quando funziona un solo gene si ha la Malattia da HbH. Piccole quantità (1-3%) di HbH possono essere presenti alla nascita anche in caso di αtalassemia lieve, mentre valori più elevati (10-25%) si ritrovano nella malattia da HbH. L’HbBart (γ4) si trova nel cordone ombelicale in caso di idrope fetale (α0 thal omozigote). Varianti Emoglobiniche Il numero di varianti emoglobiniche osservate e caratterizzate è in continuo aumento (più di 900) (http:// globin.cse.psu.edu). La prevalenza varia considerevolmente in rapporto alla posizione geografica ed al gruppo etnico. Le varianti emoglobiniche possono essere distinte in comuni e rare. Le prime riguardano milioni di persone; sono state per lo più selezionate dalla malaria per il vantaggio selettivo del portatore in ambiente malarico (HbS e C). Sono generalmente silenti, ma possono interagire con altri difetti molecolari più severi. Nei casi di doppia eterozigosi con altra mutazione possono diventare clinicamente evidenti. Le seconde risultano da mutazioni private, sono general(Continua a pagina 4) Anno 8, Numero 32 Pagina 3 (Continua da pagina 3) mente asintomatiche; talora sono associate a condizioni cliniche come emolisi (emoglobine instabili) o, nel caso di emoglobine con alterata affinità per l’ossigeno, a policitemia (aumentata affinità) o cianosi (ridotta affinità). la valutazione delle frazioni emoglobiniche. Per molti anni è stata eseguita l’elettroforesi in ambiente alcalino e acido per la rilevazione di emoglobine patologiche, seguita dalla quantificazione delle frazioni HbA2 e HbF. Attualmente l’elettroforesi è stata sostituita nella maggior parte dei laboratori dalla cromatografia liquida ad alta prestazione (HPLC) che permette una migliore separazione delle diverse emoglobine presenti in un campione (compresa anche l’Hb glicata) e la loro quantificazione espressa come percentuale dell’emoglobina totale. Con tale metodologia le diverse emoglobine sono identificate graficamente nel cromatogramma sulla base della loro permanenza all’interno della colonna di migrazione (tempo di ritenzione). Il tempo di ritenzione è un parametro fisico caratteristico che permette la creazione di specifiche library in cui sono classificate tutte le varianti conosciute ed inserite quelle di nuova scoperta. Nella pratica corrente un ulteriore criterio interpretativo del cromatogramma è rappresentato dalla origine etnica del soggetto indagato, essendo numerose varianti emoglobiniche strettamente correlate con specifiche aree geografiche. In alcuni casi, comunque, alla identificazione presuntiva deve necessariamente seguire l’analisi genica eseguibile presso pochi centri di alta specializzazione. Posizione geografica delle varianti emoglobiniche più frequenti HbS (anemia falciforme): Africa centrale e occidentale, Penisola arabica, India; in Italia è frequente nella Sicilia orientale HbC: Africa (in particolare Africa occidentale) HbE: Sud-est asiatico; si è diffuso attraverso la via della seta HbD Punjab: India del nord; diffusione per la via della seta In queste varianti i difetti di struttura riguardano tutti la catena β. I portatori di HbS, C e D sono asintomatici, mentre i portatori di HbE presentano una lieve microcitosi e ipocromia. La doppia eterozigosi HbE / β-talassemia dà luogo ad un fenotipo talassemico da medio a severo. I portatori di HbS sono spesso anche portatori di α-talassemia e questa combinazione può essere associata ad anemia microcitica ipocromica. I portatori di HbS hanno circa il 40% di HbS e il 60% di HbA. La quantità di HbA è sufficiente per avere un fenotipo normale, tuttavia in particolari condizioni (ipossia, disidratazione, sforzi fisici importanti) si possono Nella pagina seguente sono riportati alcuni algoritmi diagnostiverificare crisi anche nei soggetti eterozigoti. co/interpretativi, utili alla identificazione delle più comuni emoglobinopatie. Il ruolo del laboratorio (Continua a pagina 5) La diagnosi delle emoglobinopatie si basa sulla separazione e Anno 8, Numero 32 Pagina 4 LabNews 1. HPLC normale; FeN; MCH < 25 pg; HbA2 ↓ → α thal ? → analisi DNA Nota: Nelle condizioni date la riduzione dell’HbA2 è l’unico segno fortemente orientativo per α thal 2. HPLC normale; FeN; MCH < 27 pg; HbA2 ↑ → portatore β thal Nota: Nelle condizioni date il rilievo di eventuale aumento di HbF è solo criterio aggiuntivo 3. HPLC anormale: a. HbS: b. c. HbC, ed HBE: HbDpunjab: a. b. c. d. e. e. HPLC si per la conferma diagnostica è necessario il test di falcizzazione (le emazie a contatto con una sostanza riducente assumono la caratteristica forma a falce). HPLC si (>90%) con il criterio aggiuntivo della origine etnica; HPLC si (<90%) anche in questo caso con il criterio aggiuntivo dell’etnia. Legenda algoritmi: HPLC “normale”: assenza di frazioni emoglobiniche aggiuntive rispetto a Hb Adulta, HbA2, ed HbF che sono eventualmente segnalate per le loro variazioni quantitative; HPLC “anormale”: presenza di una o più frazioni emoglobiniche aggiuntive.; HPLC si: riconoscibilità HPLC di una variante emoglobinica (l’eventuale % indica la probabilità di identificazione univoca) MCH < 25pg: grado di ipocromia eritrocitaria; MCH < 27pg: grado di ipocromia eritrocitaria; FeN: non iposideremia Note: L’HbH, evidenziabile all’HPLC, può essere sospettata dalla presenza nelle emazie di corpi inclusi dopo colorazione sopravitale: i cosiddetti corpi di Heinz. Utile nella diagnosi differenziale fra talassemia e sideropenia si è rivelato il rapporto fra emazie microcitiche e ipocromiche, aumentato nel primo caso, ridotto nella sideropenia (vedi LabNews n.9) La carenza marziale può abbassare l’HbA2; è buona norma ripetere il dosaggio dopo la correzione terapeutica (am) Bibliografia: Giordano PC.Carrier diagnostics and prevention of hemoglobinopathies using High Performance Liquid Chromatography: Pacini editore 2005. Working Party of the General Haematology Task Force of the British Committee for Standards in Haematology(1998) Guidelines for the laboratory diagnosis of haemoglobinopathies. British Journal of Haematology 1998, 101.783-792. Vinciguerra M. Le Emoglobinopatie: dal Fenotipo al Genotipo- Palermo 21-25 Novembre 2005.Soste.org. C’E’ LA SINDROME DI GULLO, MA IL PANCREAS FUNZIONA L e più comuni malattie del pancreas esocrino, per le quali è rilevante il contributo diagnostico delle indagini di Laboratorio, sono rappresentate dai processi infiammatori acuti e cronici (pancreatiti) e dalle forme neoplastiche. In generale un aumento dei valori sierici degli enzimi pancreatici è legato alla presenza di una pancreatite acuta o si verifica durante le fasi dolorose di una forma cronica; talvolta può riscontrarsi anche in corso di altre malattie come: la fibrosi cistica, il morbo celiaco, l’infarto o perforazione intestinale, l’epatite, la cirrosi, le malattie infiammatorie croniche intestinali, l’ischemia pancreatica acuta dopo interventi sul cuore o sull’aorta toracica. Oltre a queste forme, vi è un gruppo di “iperenzimemie pancreatiche non patologiche”; la più nota delle quali è la “Iperamilasemia legata alla presenza di macroamilasi.” Si intende per Macroamilasi un complesso molecolare formato da amilasi-pancreatica ed IgG o IgA; il conseguente aumen- Anno 8, Numero 32 to di dimensioni del complesso riduce notevolmente l’eliminazione renale dell’amilasi determinandone un accumulo nel siero. In questo caso, oltre all’assenza di sintomatologia, il soggetto presenta valori di amilasuria normali o ridotti. Recentemente il Prof. Lucio Gullo direttore dell’Istituto di Medicina Interna dell’Università di Bologna (Ospedale S.Orsola) ha descritto una forma di Iperenzimemia Pancreatica Benigna cui è stato attribuito il nome di “Sindrome di Gullo”. Viene riportato il lavoro del Professore estratto da un suo articolo pubblicato su Leadership Medica: “L’Iperenzimemia Pancreatica Benigna” è una Sindrome caratterizzata da un abnorme aumento degli enzimi pancreatici in assenza di malattia pancreatica. I pazienti seguiti nello studio (sia di sesso maschile che femminile) erano in (Continua a pagina 8) Pagina 5 LabNews QUANDO IL FERRO E’ TROPPO: EMOCROMATOSI L a ferritina è una proteina globulare formata da 24 subunità polipeptidiche che possono essere di due tipi, H (P.M. 21 KDa) ed L (P.M. 19 KDa). Secondo il rapporto H/L si hanno diverse isoferritine: quelle più ricche di subunità L si trovano nei tessuti che immagazzinano il ferro più a lungo ed in maggiore concentrazione (siero, epatociti), mentre le isoferritine ricche di subunità H si ritrovano in tessuti a rapido scambio di ferro, contengono meno metallo ed esplicano funzione ossidante nei confronti del dannoso Fe(II). La ferritina extracellulare sierica è caratterizzata da una specifica subunità G, glicosilata, (P.M. 23 KDa), immunologicamente simile alla subunità L, assente nelle isoforme intracellulari. Una particolare isoferritina è l'emosiderina, proteina insolubile, ad alta concentrazione di ferro, presente in molti quadri di sovraccarico cellulare. La ferritina riveste un ruolo essenziale nel metabolismo marziale: è una molecola implicata nell'accumulo del ferro intracellulare (può contenere il metallo fino al 23% in peso), manifesta proprietà ferrossidasica, ed è presente nel siero di individui sani con valori compresi tra 20 e 280 ng/mL. Il sovraccarico di ferro con valori di ferritina sierica, che possono superare i 2000 ng/mL, è un fattore di rischio per malattie proliferative e degenerative, per l'infarto del miocardio e nei decorsi post trapianto. L'infiammazione, per azione di varie citochine, provoca riduzione dell'assorbimento del ferro, e determina iposideremia ed aumento della ferritinemia. L'epatocita è normalmente ricco di ferritina, e l'aumento di ferritina sierica è spesso associato a citolisi epatica ed a degenerazione cellulare, che mette così in circolo la ferritina intracellulare. Per quanto la ferritina sierica sia minore dell'1% rispetto a quella intracellulare, il suo dosaggio è significativo nella valutazione del ferro di deposito. Il sovraccarico (escludendo quello di origine genetica) è legato all'abuso di alcol, ed a varie forme di anemia, in particolare all'anemia sideroblastica, mentre la carenza è da associare in primis all'ipoalimentazione ed al malassorbimento del ferro. Un discorso a parte merita l'anemia associata ad attività sportive intense, in cui una bassa ferritinemia va compensata con un opportuno regime alimentare. Al di sotto di 10 ng/mL la ferritinemia è indice di deplezione di ferro ed una tempestiva rilevazione di questa carenza può prevenire lo sviluppo dell’anemia. Casi di iperferritinemia di origine genetica sono stati osservati in soggetti affetti da cataratta congenita, dove alte concentrazioni di subunità L sono connesse con una mutazione del DNA codificante per una regione dell'mRNA nota come IRE (Iron Responsive Element), Anno 8, Numero 32 ed in soggetti affetti da emocromatosi dove il dosaggio della ferritina non ha valore specifico, ma conferma il sovraccarico. L'emocromatosi è una malattia ereditaria caratterizzata da un progressivo accumulo di ferro nell'organismo. Si conoscono oggi 4 forme distinte di emocromatosi, 3 di esse sono a trasmissione autosomica recessiva ed una dominante. La forma più comune (emocromatosi tipo 1), è dovuta a due mutazioni del gene HFE situato nel cromosoma 6p: la mutazione C282Y (sostituzione di una cisteina con tirosina) e la H63D (sostituzione di una istidina con aspartato). Le altre tre forme sono meno frequenti: A. l’emocromatosi tipo 2, essenzialmente giovanile B. l’emocromatosi tipo 3, una forma dell'adulto causata da mutazioni del gene del recettore 2 della transferrina C. l’emocromatosi di tipo dominante dovuta a mutazioni del gene della ferroportina Oltre a queste sono descritte altre forme ancor più rare (ipotransferrinemia ed aceruloplasminemia congenite), alcune ad ereditarietà diaginica. Un capitolo a parte è rappresentato dalle forme secondarie o acquisite (talassemia, anemie diseritropoietiche congenite, anemia sideroblastica) in cui il sovraccarico di ferro si sviluppa in conseguenza dell'eritropoiesi inefficace e conduce a complicanze simili a quelle dell'emocromatosi idiopatica. Il difetto ereditario responsabile della forma idiopatica determina un'alterata regolazione dell'assorbimento intestinale del ferro in rapporto al fabbisogno dell'organismo ed alla disponibilità dei depositi: mentre in un soggetto adulto normale l'assorbimento intestinale di questo elemento è pari circa ad 1 mg/die nell'uomo e a 1,52,0 mg/die nella donna, in presenza di emocromatosi l'assorbimento intestinale del ferro supera mediamente i 4 mg/die. Il ferro in eccesso si accumula a livello di vari organi come il fegato, il pancreas, il cuore, le articolazioni e le ghiandole endocrine. L'emocromatosi classica è una malattia: -relativamente comune, la cui frequenza nella popolazione è stata stimata da 1 a 3 casi ogni 1000 individui, -con elevata variabilità di espressione fenotipica, da lieve a molto severa, dovuta ad una diversa penetranza del difetto genetico e all'interazione con geni modulatori e fattori ambientali, -con elevata morbilità (se non diagnosticata e trattata in tempo conduce allo sviluppo di danni gravi a carico di vari organi: cirrosi epatica, diabete mellito, cardiopatia, (Continua a pagina 7) Pagina 6 LabNews (Continua da pagina 6) ipogonadismo, artropatia), -potenzialmente mortale (in genere per epatocarcinoma e insufficienza cardiaca) -prevenibile (la diagnosi e la terapia precoce impediscono lo sviluppo delle complicanze e conferiscono una normale aspettativa ai pazienti). Le altre forme sono più rare, ma presentano quadri clinici simili all'emocromatosi classica a parte l'emocromatosi giovanile, che si manifesta in genere prima dei trent'anni e con complicanze più severe soprattutto a carico dell'asse ipofisi-gonadi e del cuore. Per quanto riguarda la diagnosi, l'obiettivo è quello di identificare i soggetti affetti prima che si sviluppino i danni conseguenti all'accumulo di ferro e si basa su: Test Biochimici: 1. percentuale di saturazione della transferrina: esame di primo livello, semplice, poco costoso e sensibile. In condizioni normali circa un terzo della transferrina è legata al ferro; valori elevati si riscontrano in presenza di sovraccarico di ferro e valori bassi nel caso di carenze di ferro. Secondo le linee guida della British Columbia Medical Association i valori discriminanti per lo screening dell'emocromatosi sono i seguenti: a. <= 45%: normale, non c'è necessità di ulteriori indagini b. 45% - 60%: controllo dopo un mese c. >60% : viene consigliata l'esecuzione del test genetico 2. dosaggio della ferritina sierica che non è un vero test di screening in quanto aumenta in modo aspecifico in varie condizioni come infiammazioni, neoplasie, necrosi epatocellulare, ma è utile per confermare un sovraccarico di ferro rilevato con l'aumentata percentuale di saturazione della transferrina ed è inoltre necessaria per monitorare la terapia Test Genetici: analisi molecolare del gene HFE ed in particolare della mutazione C282Y che è la più frequente presente in omozigosi Anno 8, Numero 32 nell'80-100% dei casi di emocromatosi nelle popolazioni nord europee, ma solo nel 65% di quelli in Italia. Nei soggetti eterozigoti e negativi per la mutazione C282Y, dovranno essere analizzate la altre mutazioni. L'analisi di tutte le mutazioni del gene HFE conferma la diagnosi nell'80% dei casi . Tra le metodiche emergenti che permettono l'analisi molecolare in tempi rapidi troviamo la PCR (Polymerasi Chain Reaction) real-time in cui l'amplificazione ed il rilevamento dell'amplificato avvengono nello stesso momento; con questa tecnica l'accumulo logaritmico degli acidi nucleici amplificati enzimaticamente può essere monitorato in tempo reale attraverso la determinazione della fluorescenza dei prodotti della reazione. E' importante ricordare che l'emocromatosi non dovrebbe essere diagnosticata od esclusa solo sulla base del risultato del test genetico. Questa considerazione si basa sulla dimostrata esistenza di individui con genotipo a rischio senza espressione di sovraccarico di ferro e di altre forme di emocromatosi non correlate al gene HFE, alcune delle quali non definibili da punto di vista genetico. Nell'iter diagnostico dell'emocromatosi è spesso necessaria la conoscenza delle altre cause di sovraccarico di ferro per condurre un'adeguata anamnesi e gli esami opportuni per la diagnosi specifica. Determinazione del Ferro Epatico con metodi non invasivi o con biopsia epatica. Nei casi non definiti dal punto di vista genetico con valori di ferritina elevati (>1000 ng/mL) o con alterazione degli indici di integrità epatica (transaminasi) è necessario ricorrere alla biopsia epatica. La terapia consiste nel rimuovere il ferro in eccesso. Nota: a partire dal prossimo mese di ottobre e, comunque, dopo specifica nota informativa, applicheremo nel nostro laboratorio la tecnica di PCR in real time anche per la ricerca della mutazione C282Y riservandola, ovviamente, ai casi con appropriato indice di saturazione della transferrina. (mg-pp) Sito web http://www.emocromatosi.it Orari per il pubblico Prelievi Poliambulatorio: Tutti i giorni dalle 7.30 alle 12.00 Consegna campioni Poliambulatorio: Tutti i giorni dalle 7.30 alle 12.00 Appuntamenti per prelievo al Poliambulatorio Giorni feriali: dalle 11.00 alle 18.15 Sabato: 11.00 - 12.30 Consegna risposte: Giorni feriali: dalle 8.30 alle 18.15 Sabato: 8.30 - 13.15 Prelievi Distretto Via E.Rossi: Tutti i giorni dalle 7.30 alle 9.15 Consegna risposte: 11.30-12.45 Prelievi Distretto Fiorentina: Tutti i giorni dalle 7.30 alle 9.15 Consegna risposte: 11.30-12.45 Prelievi Distretto Via del Mare: Da Lun a Ven dalle 7.30 alle 9.00 Consegna risposte: 11.30-12.30 Prelievi Distretto Collesalvetti: Lun-Mer-Sab* dalle 7.30 alle 8.30 Prelievi Distretto Stagno Venerdì* dalle 7.30 alle 8.30 *su prenotazione Pagina 7 LabNews (Continua da pagina 5) ottima salute, privi di qualsiasi malattia e tutti gli accertamenti diagnostici come l’ecografia, la TAC e la Wirsungrafia erano risultati negativi. Durante il successivo follow-up,durato più di 5 anni, gli enzimi si sono mantenuti elevati, anche se con ampie fluttuazioni. Nessuno dei pazienti ha presentato sintomi o segni di malattia pancreatica. Esiste una forma sporadica e una forma familiare. La forma familiare può interessare più membri della stessa famiglia. Il meccanismo che è alla base di questo abnorme passaggio di enzimi dalla cellula pancreatica nel sangue non è conosciuto. Il fatto che il difetto è stato riscontrato in più membri della stessa famiglia solleva la possibilità che vi sia una base genetica.” Iperenzimemia pancreatica benigna o sindrome di Gullo. Leadership Medica (2- 2006) Tale condizione non è poi così rara: a noi è capitato di riscontrare qualche anno fa in una paziente in ottima salute valori degli enzimi pancreatici sierici abnormemente elevati. Tutti gli esami eseguiti bioumorali e strumentali erano normali. Nessuna malattia pancreatica è mai comparsa, il soggetto ha continuato a stare bene. A conferma che la forma può essere ereditaria sono stati effettuati controlli anche ai familiari ed alcuni di essi hanno presentato la stessa alterazione. Conclusione Per un corretto riconoscimento della Sindrome è importante tenere presenti la seguenti considerazioni: 1) E’ una forma che appare in soggetti sani e quindi non collegati ad episodi di “colica addominale”. Si mantiene nel corso del tempo, con ampie oscillazioni, in assenza di qualsiasi evidenza clinica e morfologica di malattia del pancreas. Può presentarsi in forma sporadica o familiare. 2) Deve trascorrere almeno un anno dal primo riscontro della iperenzimemia prima di considerarla con sufficiente certezza espressione di questa nuova Sindrome. E’ importante ricordare che, anche se raramente, un aumento degli enzimi pancreatici, soprattutto in un soggetto con più di 50-60 anni, può essere la prima manifestazione clinica di un tumore pancreatico. 3) Nella quasi totalità dei casi sono aumentati i livelli sierici di tutti gli enzimi pancreatici. 4) La diagnosi corretta di questa Sindrome è importante perché serve ad assicurare i portatori del difetto enzimatico che non vi è una malattia pancreatica ed ad evitare ricoveri e terapie inutili. (al) Informazioni al pubblico Segreteria Laboratorio Analisi Tel 223014 Tel 223355 Fax 223378 Dalle 10.30 alle 18.30 Distretto via E. Rossi Tel 223610 Distretto Fiorentina Tel 223522 Distretto via del Mare Tel 223194 Distretto Collesalvetti Tel 962978 Distretto Stagno Tel 941291 E’ stato introdotto in laboratorio un test immunoenzimatico (immunoblotting) di 3° generazione su striscia, che presenta AgHCV derivati dalla regione del Core (C1-C2), dalla regione ipervariabile E2(HVR), dalla regione Elicasi (NS3) e dalle regioni NS4 e NS5. E’ un test ad alta sensibilità per i differenti genotipi (prossima al 100%) ed ad alta specificità (94,5%). La presenza di anticorpi nel siero viene evidenziata sotto forma di bande distinte che consentono la loro identificazione in modo differenziato in base alla reattività antigenica. Il test di immunoblotting permette, quindi, di identificare i falsi positivi, che in alcuni casi, nei test di screening, sono causati dalla interferenza di IgG aspecifiche (per HCV). Laboratorio Analisi Direzione Batteriologia Biochimica Ematologia Tossicologia Sistema qualità Il test viene eseguito su specifica richiesta del medico di base o di reparto che necessiti di approfondimenti diagnostici. CODICE DI INSERIMENTO: “INNO” TICKET: € 69.70 Anno 8, Numero 32 Pagina 8 223207 223299 223084 223087 223085 223086
Scaricare