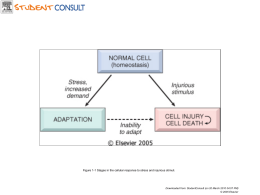
AMILOIDOSI Laurea magistrale in Medicina e Chirurgia Valeria Puglisi Mauro Pavone Gessica Motta Davide Paolillo Prof. Cinzia Di Pietro LE PROTEINE Le proteine sono gli agenti indispensabili per lo svolgimento della funzione biologica e i loro costituenti essenziali sono gli amminoacidi. Anatomia di un amminoacido. La figura mostra la tipica struttura di un aminoacido, avente un estremità ammino-terminale e un estremità carbossi-terminale. La valina e i suoi derivati fanno eccezione. Gli amminoacidi si legano mediante legame peptidico, che si instaura tra L’estremità carbossi-terminale del primo aminoacido e l’estremità Ammino-terminale del secondo. Il ripetersi di questa reazione dà luogo a polipeptidi e proteine. LIVELLI DELLA STRUTTURA PROTEICA 1) Struttura primaria Sequenza di amminoacidi Resa stabile da legami covalenti 2) Struttura secondaria Resa stabile da legami idrogeno che si formano tra residui di amminoacidi adiacenti Conformazioni distinte che può assumere la catena polipeptidica • elica • sheet -elica foglietto Simbologia 3) Struttura terziaria Avvolgimento spaziale di tutte le eliche e le forme determinata dall’interazione dei gruppi laterali della catena principale mediante legami deboli Chimotripsina, modello spaziale Chimotripsina, modello a nastro 4) Struttura quaternaria Resa stabile da legami deboli emoglobina Più strutture terziarie collaborano per formare un unico complesso macromolecolare Folding e misfolding delle proteine. Sebbene la sequenza degli amminoacidi determini la struttura tridimenzionale della proteina sono necessari fattori proteici che agevolino il “folding”. Il continuo monitoraggio del corretto ripiegamento viene effettuato dalle “chaperon”. I prodotti mal ripiegati,frutto del misfolding,sono eliminati da un sistema definito proteosoma-ubiquitina. La mancata eliminazione delle proteine genera l’accumulo di fibrille amiloidi (strutture che sono la causa di patologie che prendono il nome di amiloidosi. Amiloide e Amiloidosi Amiloidosi è un termine generico che si riferisce al deposito extracellulare di proteine sotto forma di fibrille di foglietti B pieghettati. Questo disordine può essere acquisito o ereditario, dove frammenti o intere proteine normalmente solubili,diventano insolubili accumulandosi, distruggono la struttura e funzione di organi e tessuti causandone la malattia. Classificazione • Forma sistemica : presenza di depositi nel parenchima di più tessuti. Si divide in primaria e secondaria. • Forma localizzata : depositi confinati in particolari organi e tessuti. • Forma ereditarie : (febbre mediterranea familiare) Nell’amiloidosi le proteine che normalmente vengono degradate nei proteosomi e nei macrofagi(extracellulare) questo meccanismo fallisce e si verifica l’accumulo di proteine all’esterno della cellula. L’amiloidosi non può essere definita un’unica malattia, ma come un gruppo di malattie che hanno in comune il deposito di proteine simili. Gli organi in cui si è depositato l’amiloide, possono essere : più grandi,più compatti,più pallidi. Microscopia L’amiloide appare come una sostanza extracellulare amorfa eosinofila e ialina Caratteristica reazione rosso Congo Ultrastruttura fibrillare Insolubilità delle fibrille di amiloide nei normali solventi fisioligici Resistenza alla digestione proteolitica B-fibrillosi sistemiche Tipo di b-fibrillosi Tipo delle b-fibrille Eziologia Reattiva AA Processi infiammatori cronici, malettie autoimmuni, neoplasie ( carcinoma al rene e il linfoma di Hodgkin). Senile AA ignota Di origine immunocitica AL Mieloma multiplo e altre gammopatie monoclonali proteine costituite da sole catene leggere Eredofamiliare AA ATTR Tipo portoghese ( dominante), febbre familiare (recessiva), tipo danese Postdialitica AH Colpisce i pazienti con insufficienza renale dopo molti anni di emodialisi B-fibrillosi localizzate Tipo di bfibrillosi Origine e nome Localizzazioni delle b- fibrille cliniche Manifestazioni Encefalopatie spongiformi Prp Encefalo Scarpie (ovini) mucca pazza (bovini),Kuru (uomo) Morbo di alzheimer AB Encefalo Demenza Ghiandole endocrine AC Stroma del Associate alla carcinoma midollare neoplasia della tiroide PRIONE • Prione (dall'inglese prion (acronimo di "PRoteinaceus Infective ONly particle"=particella infettiva solamente proteica) è il nome attribuito da S.B. Prusiner ad un ipotetico "agente infettivo non convenzionale" di natura proteica • La proteina normale è costituita soprattutto da una struttura ad alfa eliche, regioni nelle quali lo scheletro proteico si avvolge in uno specifico tipo di spirale; mentre la struttura che provoca lo scrapie, contiene filamenti beta. La proteina normale, probabilmente, si avvolge in una struttura compatta avente quattro alfa eliche nella parte centrale Encefalopatie Spongiformi Trasmissibili (TSE) • BSE significa letteralmente: Bovine Spongiform Encephalopathy, ma la malattia è universalmente nota come “morbo della mucca pazza” • Scrapie • Malattia di Creutzfeld- Jakob Terapia Non c'è una vera e propria terapia per l'amiloidosi, si tenta di far regredire la malattia tramite la somministrazione di sostanze come colchicina, prednisone e tramite il trapianto di cellule staminali. Nei casi dove i tessuti sono maggiormente danneggiati si effettua il trapianto dell'organo in questione, i pazienti soggetti al trapianto di fegato sopravvivono per circa 10 anni, mentre i pazienti che subiscono un trapianto di fegato in caso di amiloidosi ereditaria possono avere una vita più lunga se viene eliminato il sito dove viene prodotta la proteina mutante. Nell'amiloidosi primaria si cerca di abbassare il livello di immunoglobuline con l'uso della chemioterapia. Morbo di Alzheimer Prende il nome da Alois Alzheimer, neurologo tedesco. Nel 1901 visitò una donna con disturbi del comportamento e registrò questi comportamenti come “disordine da amnesia da scrittura”. Dopo la morte della donna e l’ esecuzione dell’ esame autoptico furono rilevati alcuni segni particolari del tessuto cerebrale. Fu la prima paziente a cui fu diagnosticata la malattia di Alzheimer. La malattia di Alzheimer è oggi definita come “quel processo degenerativo che distrugge progressivamente le cellule cerebrali, rendendo a poco a poco l’individuo che ne è affetto incapace di una vita normale ”. Catalogata anche come demenza essendo un deterioramento cognitivo cronico progressivo. A prevalenza senile. Cervello di paziente affetto da Alzheimer Cervello sano A livello macroscopico la malattia è caratterizzata da una diminuzione del peso e del volume del cervello dovuta ad atrofia corticale, visibile anche in allargamento dei solchi e appiattimento delle circonvolluzioni. A livello microscopico e cellulare sono riscontrabili depauperamento neuronale, placche senili, degenerazione neurofibrillare. Patogenesi Dall’ analisi post-mortem dei tessuti cerebrali di pazienti affetti da Alzheimer, si è potuto riscontrare un accumulo extracellulare di una proteina, chiamata beta-amiloide. Nei soggetti sani la beta-amiloide deriva dalla APP in una reazione catalizzata dall’ alfasecretasi che produce una beta-amiloide con 40 aminoacidi. Per motivi non chiariti, nei soggetti malati l’enzima che interviene sull’ APP è la bet-secretasi che porta alla produzione di una beta-amiloide anomala con 42 aminoacidi. Questa tende a depositarsi in aggregati extracellulari sulla membrana dei neuroni. Tali placche neuronali innescano un processo infiammatorio che richiama macrofagi e neutrofili i quali produrranno citochine, interleuchine e TNF alfa che danneggiano irreversibilmente i neuroni. Ulteriore meccanismo patologico è l’ anomala fosforilazione di una proteina tau, all’ interno dei neuroni, che si accumula in aggregati neurofibrillari o ammassi neurofibrillari. Questo processo colpisce prevalentemente neuroni colinergici di aree corticali e sottocorticali e tre queste le aree ippocampali. La malattia è accompagnata anche da una forte diminuzione di acetilcolina nel cervello Sintomi • Amnesia L’ amnesia anterograda è uno dei sintomi principali. • Aprassia ossia l’incapacità di compiere azioni comuni. • Agnosia che è l’ incapacità di riconoscere cose comuni. • Anomia, l’ incapacità di denominare un oggetto, pur riconoscendolo. • Disorientamento spazio-temporale • Acalculia: il soggetto non sa compiere operazioni matematiche elementari. • Agrafia, difficoltà nella scrittura. • Deficit intellettivi, peggioramento delle capacità di ragionamento, di pianificazione e di giudizio. • Cambiamenti del tono dell’ umore • Sintomi psicotici e modificazioni della personalità Diagnosi Oggi l’ unico modo per fare diagnosi certa di demenza di Alzheimer è attraverso l’ identificazione delle placche amiloidi nel tessuto cerebrale, possibile solo con l’ autopsia dopo la morte del paziente. Questo significa che durante il decorso si può fare soltanto una diagnosi possibile o probabile. I medici si avvalgono di diversi elementi: - Anamnesi (familiare, remota e recente) - Bioimmagini e esami di laboratorio: elettroencefalogramma, esame del sangue, esame del fluido cerebrospinale, TAC - Valutazione neuropsicologica e cognitiva - Diagnosi differenziale Decorso La malattia si può dividere in tre fasi. Nella fase iniziale sono osservabili manifestazioni di deficit di memoria circoscritti a episodi di vita quotidiana (disturbi di quella che viene definita on-going memory) accompagnata da disturbi del linguaggio e anomia. Nella fase intermedia il danno alla memoria aumenta (viene colpita la memoria retrograda e la memoria semantica) e si aggiungono episodi confusionali transitori, errori di valutazione e trascuratezza, aprassia costruttiva e aprassia di abbigliamento. La fare finale è caratterizzata da grave confusione e disorientamento; possono avere luogo allucinazioni, vuoto mentale, perdita totale del controllo delle funzioni corporee. Ai deficit cognitivi si aggiungono complicanze internistiche che portano ad una compromissione insanabile bella salute. Terapia farmacologica • Idea di provare a ripristinare livelli di acetilcolina • • • • FANS (anti-infiammatori non steroidei) Azione protettiva degli estrogeni Azione protettiva di vitamina E Farmaci Nootropi Terapia non farmacologica Morbo di Parkinson Patologia nota anche come paralisi agitante Descritta per la prima volta da James Parkinson 1817 La malattia è dovuta a degenerazione cronica e progressiva a carico di strutture del sistema extrapiramidale : 1. Degenerazione sostanza nera 2. Deficit produzione Dopamina (morte neuroni dopaminergici) 3. L’innervazione eccitatoria prevale su quella inibitoria(neurotrasm. Eccitatori presenti in maggiore %) La dopamina viene prodotta in un nucleo a livello del mesencefalo chiamata substantia nigra (ricca di melanina) Ipotesi cause della genesi della patologia I motivi per cui si verifica un improvviso blocco della produzione di dopamina sono sconosciuti, le principali ipotesi sono: • Ipotesi tossica: (tossina MPTP) • Ipotesi genetica ed ereditaria: Tomografia ad emissione di positroni • Ipotesi legata all’età: elevata incidenza intorno al 60°anno di età • Ipotesi sul catabolismo endogeno: produzione di radicali liberi Meningioma intracranico come causa di sindrome di Parkinson. Anche se raramente un meningioma può essere alla base di un Parkinson. Articolo pubblicato sulla rivista europea Acta Neurologica Belga nel 2008, in cui viene riportato il caso di una donna a cui, in un altro Paese europeo, era stata posta diagnosi di Sindrome di Parkinson ed in cui i sintomi, presenti solo nella metà dx del corpo, erano provocati dalla presenza di un voluminoso meningioma frontale posteriore sinistro. Dopo l'asportazione della neoplasia benigna, la paziente è tornata ad una vita del tutto normale, con completa regressione della sintomatologia simil-parkinsoniana Sintomatologia Disturbi Motori: Impaccio motorio,senso di rigidezza,acinesia, bradicinesia, tremore,scarsa mimica facciale,festinazione,alterazioni posturali, ridotta velocità dei movimenti oculari,scialorrea,turbe dell’affettività,alterazione capacità cognitive Diagnosi La diagnosi della malattia del Parkinson si basa essenzialmente sull’esame clinico,a questo scopo è stato deciso di classificare la diagnosi in: • Possibile • Probabile • Certa La diagnosi in vivo è comunque soltanto presuntiva Condizioni Generali per la Diagnosi • Degenerazione del SN: Senilità e perdita progressiva parafisiologica dei neuroni sostanza nera • della Paralisi sopranucleare progressiva: paralisi sopranucleare dello sguardo verticale, instabilità posturale e ipertono assiale con frequenti cadute all’indietro • Parkinsonismo Vascolare Fattori degenerativi multipli a carico della sostanza bianca e dei nuclei della base,non risponde alla L-DOPA Diagnosi strumentale La medicina nucleare permette uno studio accurato della patologia dal punto di vista anatomico e funzionale: essa sfrutta l'uso di traccianti radioattivi iniettati nell'organismo, i quali vanno a depositarsi nei distretti corporei oggetto di studio, evidenziandone il metabolismo, e quindi in maniera diretta o indiretta, caratteristiche come la vitalità o l'attività. Essendo la malattia di Parkinson una patologia a carico del sistema dopaminergico, i traccianti sono diretti verso: • • • il trasportatore della dopamina il trasportatore vescicolare delle monoamine di tipo 2 l'enzima DOPA decarbossilasi La PET con 18F mostra la riduzione dell'attività dopaminergica a livello dei gangli della base. Decorso • Il decorso della patologia è variabile ma nella maggior parte dei casi si ha una lenta ed inarrestabile progressione. Si possono distinguere due forme di evoluzione: • Forma Ipercinetica • Forma acinetico-ipertonica Le Terapie La terapia con levodopa è la più diffusa ma ha molti limiti ed uno dei problemi è la così detta “sindrome da trattamento con levodopa” • Levodopa/carbidopa (Sinemet) • Levodopa/benserazide (Madopar) Complicazioni e fenomeni clinici che insorgono dopo alcuni anni di terapia con levodopa: • Fenomeno del “wearing off” (effetto fine dose) • Fasi “on-off” • Turbe neuropsichiatriche Terapia chirurgica La tecnica più utilizzata è la chirurgia stereotassica che utilizza una tecnica,chiamata Deep Brain Stimulation (DPS) , che diminuisce sensibilmente la dipendenza da levodopa. Terapia Genica Sperimentata negli Stati Uniti,prevede l’iniezione di un virus,contenente un vettore il per il gene di GABA,in una zona preciso del cervello. Il neurotrasmettitore GABA “calma” i neuroni iperattivi,si è riscontrato essere deficitario nei pazienti affetti. La tecnica non ha effetti collaterali ma è ancora in fase di sperimentazione Iniezione intracelebrale • Iniezione FATTORI TROFICI: BDNF e GDNF. Sono sostanze prodotte dal cervello o da altri organi, in grado di riparare e rigenerare le cellule danneggiate, prima della loro morte. Purtroppo necessitano di impianto diretto sul tessuto (zona profonda del cervello), non per via orale o endovenosa. Grazie Per la vostra attenzione… Fine Bibliografia sezione Parkinson: • Langston JW The promise of stem cells in Parkinson disease J Clin Invest 115 23-25 (2005) • Fazio C, Loeb C, Favale E: Neurologia. Società Editrice Universo (2003) • Cancelliere M: Miss Parkinson. Società Editrice San Paolo (2007) • Douglas M. Anderson; A. Elliot Michelle, Mosby’s medical, nursing, & Allied Health Dictionary sesta edizione, New York, Piccin, 2004.
Scaricare