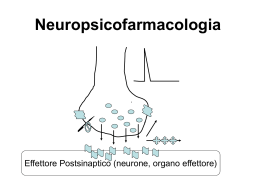
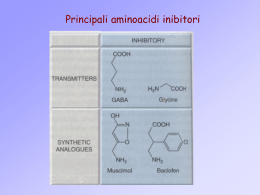
EPILESSIE Prevalenza 0,5 - 2 % abitanti Incidenza 20-70 casi/100.000 50 milioni di pazienti nel mondo CRISI O ACCESSO EPILETTICO • Scarica occasionale, improvvisa, esagerata e ipersincrona di un grande numero di neuroni (J. Hughlings Jackson). • La manifestazione comportamentale dell’accesso epilettico dipende dalla localizzazione dei neuroni coinvolti. • L’accesso può essere convulsivo (cioè accompagnato da manifestazioni motorie) oppure presentarsi con diversi sintomi neurologici. L’epilessia è una sindrome cronica caratterizzata da crisi epilettiche spontanee e ricorrenti. FOCUS EPILETTICO E’ il punto di partenza della scarica elettrica patologica (non sempre individuabile). L’attività elettrica anomala può poi diffondersi a tutta la corteccia. Substrato neuronale dell’epilessia La sinapsi ioni Il cervello Il canale ionico/Recettore Epilessia primaria o idiopatica 70% Epilessia secondaria o sintomatica 30% ORIGINI PATOLOGICHE DEL FOCUS: • • • • • • genetica, spesso nell’ambito di sindromi complesse traumatica flogistica neoplastica ischemica … sconosciuta In alcuni casi, il trattamento della patologia sottostante può risolvere la situazione. Negli altri casi, si deve instaurare una terapia cronica per prevenire le crisi. FATTORI SCATENANTI L’ATTACCO: • variazioni di glicemia, pH plasmatico, equilibrio elettrolitico • anossia • alterazioni metaboliche o endocrine • affaticamento • stress • carenze nutrizionali • interventi farmacologici (intossicazione o brusca sospensione di farmaci) • … indeterminati Storia • • • • • • • • • Phenobarbitone 1912 Phenytoin 1938 Primidone 1952 Ethosuximide 1955 Carbamazepine 1965 Sod. Valproate 1973 Valproic acid 1993 Clonazepam 1974 Clobazam 1979 • • • • • • • • Vigabatrin Lamotrigine Gabapentin Tiagabine Topiramate Levetiracetam Fosphenytoin Zonisamide 1989 1991 1993 1995 2000 2001 2005 Strategie • Modificazioni della conduttanza ionica. • Aumento della trasmissione GABAergica. • Diminuzione dell’attività eccitatoria (glutamatergica) FARMACI ANTIEPILETTICI “CLASSICI” Farmaci efficaci sulle crisi parziali e tonico-cloniche generalizzate IDANTOINE IMINOSTILBENI BARBITURICI DEOSSIBARBITURICI fenitoina carbamazepina fenobarbitale primidone Farmaci con ampio spettro di attività antiepilettica BENZODIAZEPINE ACIDI CARBOSSILICI diazepam, lorazepam, clonazepam, clobazam acido valproico Farmaci efficaci sulle assenze SUCCINIMIDI etosuccimide Struttura chimica X: Barbiturici - C – N IDANTOINE -N– derivati ossazolidinici – O – Succinimidi –C– Fenacemide: E’ attiva in alcune forme di epilessia ma mostra un’elevata tossicità pertanto viene usata solo in caso di crisi epilettiche non controllabili con altri farmaci Acetilurea - NH2 –* *(N connesso al C2) Piccoli cambiamenti alterano l’attività ed il sito d’azione. e.g. R1, un gruppo fenilico (phenytoin) conferisce attività contro l’epilessia ma un Gruppo alchilico (ethosuximide) conferisce attività contro l’assenza. GABAergic SYNAPSE Drugs that Act at the GABAergic Synapse • • • • • GABA agonists GABA antagonists Barbiturates Benzodiazepines GABA uptake inhibitors Goal : GABA Activity BARBITURICI: fenobarbitale 1912 Usi terapeutici E’ oggi scarsamente utilizzato • crisi tonico-cloniche generalizzate, anche secondariamente Effetti collaterali • sedazione • confusione nell’anziano • alterazioni cognitive e irritabilità nell’infanzia • nistagmo, atassia • effetti gravi da sovradosaggio H H Meccanismo d’azione • a basse dosi, potenziamento della trasmissione mediata dal GABA (prolungamento del tempo di apertura del canale GABAA) • ad alte dosi, attivazione diretta del canale GABAA DEOSSIBARBITURICI: primidone Usi terapeutici E’ oggi scarsamente utilizzato • crisi tonico-cloniche generalizzate, anche secondariamente Effetti collaterali • sedazione • nistagmo, atassia, vertigini • reazioni psicotiche • raramente: rash, tossicità midollare H H Viene metabolizzato a feniletilmalondiamide (PEMA) e a fenobarbitale, entrambi metaboliti attivi Meccanismo d’azione • analogo a quello del fenobarbitale Synthetic Routes to Butenyl GABA Uptake Inhibitors: SINTESI TIAGABINA Final stages: Tiagabina H2N COOH GABA H2N COOH VIGABATRIN Anticonvulsants Gamma- Amino- Butyric- Acid (GABA) Before 1883 known as a metabolite of plants and microorganisms (Basidio-, Streptomycetes) 1883 synthesized 1949 identified in animal tissue not incorporated in proteins !!! 1956 first indications on an inhibitory activity on nervous tissue main inhibitory fuctions in the vertebrate CNS Gamma Amino Butyric Acid • An inhibitory neurotransmitter of the Central Nervous system. • Binds to GABA receptors on the cell membrane of the post synaptic neuron. • Causes the GABA receptor to open a coupled chloride ion channel. • Chloride enters the neuron causing hyperpolarization and inhibiting transmission of the action potential. Pathophysiology of GABA Reduced GABA-activity in local cell assemblies: Overexcitability / Epilepsy In striatal output neurons: Chorea Huntington Enhanced GABA-activity In local cell assemblies: reduced excitability inhibition of learning anxiolytic anesthetic The GABAa Receptor • Consists of 5 subunits. – 2 alpha units – 2 beta units – 1 gamma units • Functions as a ligand gated chloride channel on the post-synaptic cell. Different Possible States of the GABA Receptor • 5 protein subunits (2 alpha, 2 beta, 1 gamma) 6 different forms of the alpha subunit 4 different forms of the beta subunit 3 different forms of the gamma subunit • Possible number of states of the receptor = 3 X 4 X 6 X 4 X 6 = 1728. • At least 19 different states identified. Transmembrane domains of the subunits Three of the transmembrane domains, (M1, M3, and M4), are identical in their 3dimensional structure The M2 domains ,which form the inner wall of the channel, have a specialized 3-D structure. Mechanism of opening and closing of the channel • When GABA binds to the recognition site, it induces several conformational changes in the receptor molecule. • One of these changes involves the rotation of the five protein subunits such that the diameter of the channel is widened. • The channel opens because the subunit proteins rotate sideways. • The "kinks" that keep the channel closed swivel and open the channel. • The kink is formed at a specific sequence of amino acids on the M2 transmembrane domain. LIGANDS AT GABA-RECEPTORS 1. GABA – site of GABAA-REC. agonist antagonist OH O H2N N O N O MUSCIMOL CH3 O O O pro-convulsive O BICUCULLIN (BARBITURATES) Th: former hypnotics antiepileptic (Cordyalis lutea = Lerchensporn) PICROTOXIN (Anamyrta cocculus = Scheinmyrte) BZD: Mechanism of action α β γ subunits assemble in an uncertain stechiometry to form a pentameric : GABA-A receptor LIGANDS AT GABA-RECEPTORS 2. BDZ – site of GABAA-REC. (BZ1 & BZ2) agonists H3C N Cl O ACTION OF plus GABA N Th: hypnotic (MIDAZOLAM, FLUNITRAZEPAM) antiepileptic / anticonvulsive (CLONAZEPAM, DIA BENZODIAZEPINS anxiolytic (DIAZEPAM) myotonolytic (TETRAZEPAM) IMIDAZOPYRIDINES (Zolpidem) Th: hypnotic anxiolytic LIGANDS AT GABA-RECEPTORS 2. BDZ – site of GABAA-REC. (BZ1 & BZ2) agonists H3C N Cl O antagonist ACTION OF plus GABA N N O O O N pro-convulsive CH3 Th: antidot FLUMAZENIL inverse agonists N H O N F minus BENZODIAZEPINS O N -CARBOLINES (-CCB) LIGANDS AT GABA-RECEPTORS 3. GABAB – receptor agonist antagonist Cl Cl H2N COOH BACLOFEN Th: muscle relaxant H2N SO3H SACLOFEN Zolpidem (Ambien) • Non-benzodiazepine sedative-hypnotic • Acts at benzodiazepine receptor a1 subunit • Used for short-term treatment of insomnia • T1/2 = 2 hours Flumazenil (Romazican) Benzodiazepine antagonist Used in management of suspected benzodiazepine overdose Eliminated by hepatic metabolism after IV administration Rapid Onset within 2 min Clinical effects persist 30 - 60 minutes Anexate, Lanexat, Mazicon, Romazicon It was introduced in 1987 by Hoffmann-La Roche under the trade name Anexate. Flumazenil, an imidazobenzodiazepine derivative, antagonizes the actions of benzodiazepines on the central nervous system. Flumazenil competitively inhibits the activity at the benzodiazepine recognition site on the GABA / benzodiazepine receptor complex Gerecke, M.; Hunkeler, W.; Kyburz, E.; Mohler, H.; Pieri, L.; Pole, P.; 1982, U.S. Patent 4,316,839. Discovery of BZD: Evolution of Psychopharmacology 1. Prehistoric times: Alcohol and herbs as hypnotic 2. Bromide (first hypnotic), chloral hydrate, paraldehyde 3. 1903 -1960: Barbiturates era, 50 marketed agents – 1930: Separation of anticonvulsant from sedative-hypnoticanesthetic – 1940: Phenytoin – 1950: Taming agents Chlorpromazine and Meprobamate Barbiturates unsafe Discovery of BZD • 1957 accidental discovery of chlordiazepoxide on the heels Chlorpromazine • Wrong structure (ring expansion) and surprisingly low toxicity with remarkable anxiolytic, sedative & anticonvulsant • 1960 Introduction of first BZD to clinic as Librium (Libre French free; Liber Latin freedom) • More than 3000 synthesized & evaluated, 35 in market BZD replaced Barbiturate, 1960 onwards BZD era….. Sternbach L. The BZD story. J Med Chem. 1979; 22:1-7 Meccanismo d’azione • Stimolazione del recettore GABA-A Da parte del GABA • Aumento della permeabilità della membrana neuronale al cloro • Le benzodiazepine agevolano la trasmissione a tutte le sinapsi GABAergiche del SNC The Role of Benzodiazepines • Positive allosteric modulators of the GABAa receptor. • Bind at a site different from that of GABA. • Enhances binding of GABA to its site, increasing chloride conductance. Evidence of Complexity – 3 Broad Types of Ligands • Agonists – bind to BZD site on alpha subunit and increase receptor affinity for GABA resulting in neuroinhibition. (eg. BZD) • Inverse Agonists – bind to BZD site on alpha subunit and decrease receptor affinity for GABA resulting in neurostimulation. • Antagonists – have no effect at receptor but compete with both agonists and inverse agonists and so inhibit their actions. (eg. flumazenil) • Recently partial agonists and partial inverse agonists have also been discovered. Benzodiazepine- Uso • Farmacoterapia dell’ansia e di altri disturbi emotivi • Nell’insonnia • Nell’epilessia e in altre manifestazioni convulsive • Impiegati come miorilassanti centrali in preanestesia o come inducenti in anestesiologia • Le benzodiazepine o i loro metaboliti possono accumularsi nell’organismo Effetti tossici dopo 5-10 gg Ansiolitici • Farmaci impiegati per il controllo diurno degli stati ansiosi, moderati o gravi • Le benzodiazepine sono i più usati • Anche i barbiturici rientrano in questa classe ma presentano effetti secondari molto più gravi Induzione enzimatica Basso indice terapeutico Ansiolitici- Benzodiazepine R O N R2 R1 N X Ansiolitici- Benzodiazepine Impiegato come anticonvulsivante H NHCH3 O N N H3C O N Cl OH N Cl N O Cl N Clordiazepossido Ossazepam Diazepam • Riducono l’ipereccitabilità psicomotoria • Indicati nel trattamento delle crisi di astinenza degli alcolizzati • Molte benzodiazepine sono anche miorilassanti(impiegate nelle Sintesi del nitrazepam e nimetazepam O O NH2 NH N O PhCOCl ZnCl2 O O2N O2N O2N H +/ O O Cl NH2 NH NH2 NH Cl O O O2N O2N + O2N O NH3 O Cl O NH2 O N O H N NH Me2SO4 O MeOH O2N O2N O2N N N Nitrazepam Nimetazepam Relazioni attività-struttura • Sistema ciclico “classico” delle benzodiazepine: 5-Fenil-1,4-benzodiazepin-2-one ‘positive allosteric modulator’ •Anxiolytic •Anti Convulsant •Sedative hypnotic ‘negative allosteric mod •Anxiogenic •Convulsive Haefely WE (1989) In Allosteric Modulation of Amino Acid Receptors: Therapeutic Implications pp 47-69, Raven Press, New York. Relazioni attività-struttura Presenza di un gruppo elettron – attrattore in posizione 7 attività Relazioni attività-struttura H H O N H O N B B N H N H H L’anello B delle benzodiazepine può adottare due conformazioni e per l’affinità al recettore Lattame aperto Metabolismo Clordiazepossido NH2 NHCH3 N Cl O H N N N N Cl N Cl O O Clordiazepossido O N-desmetilclordiazepossido Demossepam O H N O H N OH N Cl Metaboliti attivi Cl Ossazepam N Idrossilazioni anelli aromatici N-desmetildiazepam BENZODIAZEPINS DIAZEPAM (VALIUM) H3C N O N Cl H N CNS O LIVER N Cl desmethyldiazepam H N H N O O OH Cl N OXAZEPAM O Cl N Glucuronic Acid BENZODIAZEPINS DIAZEPAM (VALIUM) H3C H3C N MIDAZOLAM (DORMICUM) O N Cl N N N Cl F H N CNS O LIVER N Cl OH-mdz mdz-glucuro desmethyldiazepam H N H N O O OH Cl N OXAZEPAM O Cl N Glucuronic Acid BENZODIAZEPINS DIAZEPAM (VALIUM) t1/2=32h H3C H3C N MIDAZOLAM (DORMICUM) O N Cl H N N Cl t1/2=3h ! O t1/2=50-100h desmethyldiazepam H N H N O Cl N t1/2=8h OXAZEPAM O O OH Cl N F N Cl N N Glucuronic Acid t1/2=2h Flunitrazepam (Rohypnol) (roofies, rope, Mexican valium, roach-2, R-2) Illegal in the U.S. (and many other countries) Date rape and robbery Effective within 15-20 minutes Active for 4-6 hours Induces anterograde amnesia Also confusion, sedation, dizziness, visual disturbances AZASPIRODECANDIONI E’ un nuovo tipo di ansiolitico con modesta componente sedativa, efficacia paragonabile al diazepam Attività •Mitiga l’ansietà senza causare sedazione o alterazioni funzionali •Non favorisce l’abuso, né dà dipendenza fisica Pharmacokinetics of Buspirone Rapidly absorbed Extensive first-pass metabolism T1/2 = 2-4 hours Properties of Buspirone Advantages Anxiolytic properties similar to diazepam Tolerance to therapeutic effects does not develop No known interactions with other drugs or alcohol Disadvantages Requires at least two weeks to achieve therapeutic effects Sodium channels • Main target for many drugs • Sodium channels are responsible for the rising phase of the action potential in excitable cells and membranes Potassium channels • • • • Very diverse group of ion channels Responsible for resting potential Influences excitability of neurones Determine potential width RETIGABINA Retigabine (INN) or ezogabine (USAN), codenamed D-23129, is an anticonvulsant being investigated as a possible treatment for partial epilepsies. As of July 2010, several Phase III clinical trials are underway for this indication, and retigabine is being reviewed for approval by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA). The drug is being developed by Valeant Pharmaceuticals and GlaxoSmithKline, with the proposed trade name Potiga. It was approved by the European Medicines Agency under the trade name Trobalt in April 2011. Retigabine works primarily as a potassium channel opener—that is, by activating a certain family of voltage-gated potassium channels in the brain. This mechanism of action is unique among antiepileptic drugs, and may hold promise for the treatment of other neurologic conditions, including migraine and neuropathic pain; a Phase II trial to assess the safety and efficacy of retigabine for treating postherpetic neuralgia is ongoing. Actions of Phenytoin on Na+ Channels Na+ A. Resting State B. Arrival of Action Potential causes depolarization and channel opens allowing sodium to flow in. Na+ Na+ C. Refractory State, Inactivation Sustain channel in this conformation Il felbamato è un farmaco prodotto dalla Schering-Plough, usato come antiepilettico di seconda scelta per le epilessie parziali e generalizzate; appartenente alla seconda generazione di epilettici. Non è raccomandato come prima scelta per i rischi anche mortali di anemie aplastiche [1] e insufficienze epatiche; il suo uso è suggerito quando, in mancanza di alternative valide, i potenziali benefici superano i rischi. È indicato anche come terapia aggiuntiva per il trattamento di bambini (> 4 anni) affetti da Sindrome di Lennox-Gastaut, I nomi con cui il farmaco è commercializzato nel mondo sono: Felbamyl, Felbatol e Taloxa. Agisce sul complesso recettore ionoforo per il NMDA, interagendo specificamente con il sito di riconoscimento per la glicina che non è sensibile alla stricnina. Ha anche un effetto sull’influsso del Na+. Studi suggeriscono una sua duplice azione sia su meccanismi cerebrali eccitatori sia su quelli inibitori GABA-mediati Agisce con azione di tipo inibitorio sulle risposte inibitorie NMDA-evocate e sui potenziali GABA-evocati; L’azione combinata del felbamamto sulla trasmissione eccitatoria ed inibitoria, può contribuire a spiegare le notevoli modalità d’azione del farmaco sui vari modelli di epilessia. Fosphenytoin Fosphenytoin (Cerebyx, Parke-Davis; Prodilantin, Pfizer Holding France) is a water-soluble phenytoin prodrug used only in hospitals for the treatment of epileptic seizures. Phenytoin, in both its acidic and sodium salt forms, is erratically bioavailable whether it is injected or taken orally due to its high melting point, weak acidity, and its being only sparingly soluble in water. Simply putting patients on other drugs is not always an option; this was especially true before 1993, when the number of anticonvulsivant available was much more limited. One solution was to develop a prodrug that did not have these drawbacks. Fosphenytoin was approved by the FDA on August 5, 1996 for use in epilepsy IMINOSTILBENI: carbamazepina 1960: nevralgia del trigemino Usi terapeutici farmaco di prima scelta per: • crisi parziali • crisi tonico-cloniche generalizzate • non è indicata per le assenze! Effetti collaterali • sonnolenza inferiore a quella indotta dalla fenitoina • vertigini, atassia, diplopia • rash cutanei • discrasie ematiche (rare) 1974: efficacia antiepilettica H2 Struttura triciclica. Si trasforma in un epossido, metabolita attivo Meccanismo d’azione • “fenitoino-simile” di blocco dei canali Na+ in modo voltaggioe frequenza- dipendente FDA approves Lamictal®XR™; an extendedrelease once-daily, new generation treatment for epilepsy Patented GlaxoSmithKline Extended-Release Technology Lamictal XR Extended-Release Tablets are enteric-coated and contain a modified release formulation in the center of the tablet. There is a specially designed opening in the enteric coating on both sides of the tablet that utilizes a new technology called DiffCORE™, discovered and developed by GlaxoSmithKline. This allows a controlled release of the medicine in the acidic environment of the stomach, leading to a gradual release of lamotrigine into the bloodstream. Sintesi delle succinimmidi: Etosuccimide O R COOEt HCN + R1 R NC COOEt EtONa/EtOH R1 CN R1 R COOEt NC R1 R H+/ COOH HOOC -CO2 CN COOH R R R1 R1 COOH R2NH2 O Etosuccimide HOOC N R2 O Succinimidi R = CH3 R1 = C2H5 R2 = H Il gabapentin è il nome del principio attivo indicato specificamente nel dolore posterpetico e nell’ EPILESSIA parziale resistente alle terapie standard. Viene commercializzato sotto il nome di Neurontin di Pfizer ed altri nomi ed è un farmaco generico da alcuni anni. Gabapentin è strutturalmente correlato al neurotrasmettitore GABA (acido gammaaminobutirico) ma il suo meccanismo d'azione differisce da quello di altre sostanze che interagiscono sulla sinapsi GABA. A concentrazioni terapeutiche gabapentin non si lega ad altri farmaci noti o a recettori dei neurotrasmettitori cerebrali, quali benzodiazepina, GABAA, GABAB, glutammato, glicina o recettori dell´N-metil-d-aspartato (NMDA). Gabapentin non interagisce con i canali di sodio in vitro, ed in questo si differenzia dalla fenitoina e dalla carbamazepina. Gabapentin provoca una leggera riduzione nel rilascio dei neuro-trasmettitori monoamminici in vitro. Studi nei ratti dimostrano che gabapentin aumenta la sintesi del GABA in diverse aree cerebrali.
Scaricare