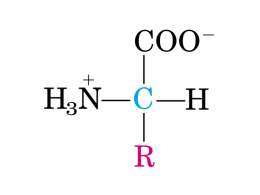
Metabolismo degli amminoacidi 1 2 Glicina, Serina, Alanina e Cisteina sono degradate a piruvato 3 Il glutammato è un amminoacido glucogenico glutammato H O -OOC-C-CH -CH -C-NH 2 2 2 NH3 O H H2O -OOC-C-CH -CH -C-O2 2 NH3 NH3 glutammina NADP+ NADPH + NH3 O -OOC-C-CH -CH -C-O2 2 O a-chetoglutarato 4 L’a-chetoglutarato è il punto di ingresso metabolico di molti amminoacidi che sono inizialmente trasformati in glutammato 5 Nel tessuto nervoso il glutammato per decarbossilazione forma il GABA glutammato decarbossilasi 2 1 GABA Nel tessuto cerebrale questo processo è alternativo al ciclo di Krebs transaminasi deidrogenasi 6 Il GABA è il principale neurotrasmettitore inibitorio del cervello. 7 (nel fegato) S-adenosilmetionina è il principale donatore di metili Degradazione della metionina succinil CoA 8 La S-adenosil-metionina è il principale donatore di gruppi metilici 9 Metilazione del DNA SAM metila le basi dei nucleotidi del DNA formando, nei vertebrati, metilcitosina. I gruppi metilici si proiettano nella scanalatura maggiore del DNA dove interagiscono con le proteine che legano il DNA La loro funzione è quella di spegnere l’espressione genica negli eucarioti. 10 Degradazione degli amminoacidi a catena ramificata: ISOLEUCINA, VALINA, LEUCINA La mancanza dell’enzima a-chetoacido deidrogenasi (decarbossilazione ossidativa) causa la malattia delle urine a sciroppo d’acero (accumulo di a-chetoacidi a catena ramificata nelle urine) isoleucina a-chetoacido deidrogenasisi (unico enzima per i 3 amminoacidi) valina leucina ritardo fisico e mentale 11 Due vie di degradazione del triptofano: 1. formazione di acetoacetato 12 Vie di degradazione del triptofano: 2. Formazione di serotonina * importante neurotrasmettitore nel cervello Morbo di Hartnup: alterato trasporto transmembrana del triptofano melatonina: implicata nella regolazione dei ritmi circadiani. Inibisce la sintesi e la secrezione di altri neurotrasmettitori, quali DOPA e GABA * 13 La mancanza dell’enzima fenilalanina idrossilasi o del coenzima tetraidrobiopterina provoca fenilchetonuria: in questo caso l’unica via degradativa della Phe è quella che porta a 1 fenilpiruvato (2). 2 a b 14 Formazione, utilizzo e rigenerazione della tetraidrobiopterina nella reazione della fenilalanina idrossilasi Phe-idrossilasi= ossidasi a funzione mista poiché utilizza un cofattore e l’ossigeno molecolare per compiere la reazione di idrossilazione. L’ossigeno necessario per la reazione di idrossilazione proviene dalla molecola biatomica dell’O2. L’altro atomo di ossigeno che rimane si unisce ai due atomi di idrogeno portati dalla tetraidrobiopterina formando acqua. * Il ripristino della forma ridotta tetraidrobiopterina si ha con la reazione di riduzione catalizzata dalla diidrobiopterina reduttasi che usa NADH * 15 Fenilchetonuria I soggetti colpiti tendono ad avere: •ritardo mentale •scarsa pigmentazione •andatura e postura insolite •crisi epilettiche Prevenzione per una diagnosi precoce: •screening nei neonati •dieta carente di Phe ma arricchita di Tyr per i primi 4-5 anni •dieta a scarso contenuto proteico per tutta la vita •aspartame = Asp-Phe-metilestere Le madri omozigoti per il difetto hanno una probabilità molto elevata di partorire bambini affetti dall’errore congenito e da ritardo mentale a meno che non venga controllata la loro assunzione di Phe. 16 Esistono casi di fenilalaninemia dovuti ad un difetto a livello della sintesi o della riduzione della bipterina, il cofattore dell’enzima fenilalanina idrossilasi. Il deficit di diidrobiopterina è molto più grave poichè la biopterina è necessaria anche per la biosintesi delle catecolamine e della serotonina che agiscono da neurotrasmettitori Trattamento: •somministrazione di biopterina nella dieta •somministrazione di precursori della serotonina e delle catecolamine 17 Fenilalanina Fenilalnina idrossilasi [PKU] tirosina idrossilasi DOPA Noradrenalina Adrenalina [albinismo] Melanina tetraidro biopterina diiidro biopterina Dopamina Tirosina Tirosinasi Omogentisato omogentisato ossidasi [ALCAPTONURIA] Maleilacetoacetato Fumarilacetoacetato Fumarato Acetoacetato 18 In condizioni normali la Phe viene idrossilata a tirosina 1 2 a b 19 Catabolismo della tirosina (a) Nella porzione midollare della ghiandola surrenale: tirosina idrossilasi tetraidrobiopterina SAM DOPA decarbossilasi Piridossalfosfato (prodotto finale nella substantia nigra) 20 Morbo di Parkinson (1817) •tremori sempre più intensi che interferiscono con la funzionalità motoria di vari gruppi muscolari •causa: degenerazione delle cellule presenti in piccoli nuclei cerebrali noti collettivamente come “substantia nigra” e “locus coeruleus” •presente nei tossicodipendenti che utillizzavano MTPT (metil-fenil-tetraidropiridina) tossico per le cellule produttrici di dopamina •trattamento con L-DOPA, il precursore della dopamina: effetti collaterali quali nausea, vomito, ipotensione, aritmia cardiaca e vari sintomi a carico del SNC dovuti alla trasformazione della DOPA in dopamina in compartimenti diversi dal SNC •gli effetti collaterali scompaiono con la contemporanea somministrazione di analoghi della DOPA che inibiscono l’enzima DOPA decarbossilasi ma che non sono in grado di oltrepassare la BBB 21 Catabolismo della tirosina (b) 1 2 a b 22 b omogentisato ossidasi: la sua deficienza provoca alcaptonuria 23 Alcaptonuria •soggetti con un deficit di omogentisato ossidasi eliminano quasi tutta la tirosina sotto forma di acido omogentisico, idrochinone incolore che tende ad autoossidarsi nel corrispondente chinone che polimerizza producendo una sostanza di colore bruno intenso •l’eliminazione di urine nerastre è la sola conseguenza di questa patologia nei primi anni di vita •l’acido omogentisico forma con gli anni pigmenti che si depositano nelle ossa, nel tessuto connettivo e in vari organi (OCRONOSI) e ciò rappresenta la causa delle complicazioni artritiche riscontrate in questi pazienti 24 b I prodotti del metabolismo della Phe entrano nel ciclo di Krebs 25 Fenilalanina Fenilalnina idrossilasi [PKU] Tirosina Tirosinasi [albinismo] Melanina tetraidro biopterina DOPA diiidro biopterina Dopamina Noradrenalina Adrenalina Omogentisato omogentisato ossidasi [ALCAPTONURIA] Maleilacetoacetato Fumarilacetoacetato Fumarato Acetoacetato 26 BIOSINTESI DEGLI AMMINOACIDI 27 Principali precursori degli amminoacidi 28 Sintesi di alanina, aspartato, glutammato, asparagina e glutammina 29 30 La cisteina si trova nella molecola del glutatione: g-glutammil-cisteinilglicina 31 Il glutatione è presente in grande quantità negli eritrociti dove ha la funzione di eliminare l’H2O2 ed i perossidi organici che possono danneggiare irreversibilmente l’emoglobina 2 GSH + R-O-O-H GSSG + ROH + H2O E’ di vitale importanza per l’integrità degli eritrociti un rifornimento continuo di NADPH che è prodotto dall’attività dell’enzima G6PD (via del pentoso fosfato) 32 33 34 L’amminoacido lisina è il precursore della carnitina, composto che lega l’acil CoA e ne permette l’entrata all’interno del mitocondrio dove viene ossidato con liberazione di energia. 35 Gli amminoacidi arginina e glicina sono i precursori della creatina 36 La creatina che si forma nel fegato, viene fosforilata nel mucolo. La fosfocreatina rappresenta un deposito di energia immediatamente utilizzabile. 37 nel muscolo: 38 L’amminoacido ornitina è il precursore delle poliamine: ORNITINA esse svolgono un importante ruolo nel controllo della sintesi del DNA e dell’RNA ornitina decarbossilasi PLP dipendente PUTRESCINA SAM spermidina sintetasi SPERMIDINA SAM spermina sintetasi In queste reazioni SAM è donatore di un gruppo amminopropile H3N-(CH2)3 SPERMINA 39 Biosintesi degli amminoacidi della famiglia del glutammato 40 Conversione del 3-fosfoglicerato in serina 41 Biosintesi degli amminoacidi della famiglia del piruvato 42
Scaricare