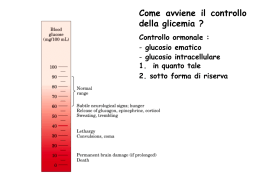
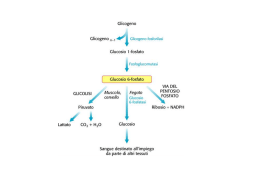
Il Glicogeno ed il suo metabolismo Polimero di unità di D-glucosio legate con legami glucosidici α 1→4 e ramificate con legami α 1→6 ogni 10 unità FUNZIONE DEL GLICOGENO: • Riserva di glucosio (nel muscolo 2/3 e nel fegato 1/3) • Regolazione dei livelli di glucosio nel sangue (nel fegato) costituisce una riserva energetica degli Animali e nei Funghi Il glicogeno ha una struttura molto compatta derivante dall'avvolgimento a spirale delle catene polisaccaridiche Nel muscolo e nel fegato ci sono diverse forme di glicogeno: -MUSCOLO α particelle particelle sferiche che contengono fino a 60.000 residui di glucoso -FEGATO α particelle organelli cellulari: Glicosomi aggregati a rosetta di α particelle contenenti anche una parte proteica (pool enzimatico della glicogenosintesi, della glicogenolisi e di regolazione) Immagazzinare glucosio in forma polimerizzata riduce lo stress osmotico cellulare che si avrebbe con elevate concentrazioni di glucosio Viene accumulato in granuli di 10-40 nm che contengono anche gli enzimi per la sintesi e la degradazione SINTESI DEL GLICOGENO: GLICOGENOSINTESI Il donatore di glucosio nella sintesi del glicogeno è l'UDP-glucosio L' UDP-glucosiltransferasi catalizza il trasferimento di un residuo di glucosio al gruppo ossidrile di una specifica tirosina presente nella proteina Glicogenina La glicogenina catalizza il trasferimento di residui di glucosio fino a formare una catena lineare di circa sette residui legati tra loro con legame a( 1--->4 ) glicosidico La glicogeno sintasi allunga la catena lineare aggiungendo una decina di residui L'enzima ramificante catalizza il trasferimento di un residuo oligosaccaridico (sei o sette molecole di glucosio) dalla parte terminale del polimero in crescita al gruppo ossidrilico in 6 di un residuo di glucosio più vicino alla glicogenina: si forma così il legame a( 1--->6 ) glicosidico L'azione prolungata della glicogeno sintasi e dell'enzima ramificante portano alla formazione della molecola di glicogeno Sintesi del glicogeno La sintesi avviene mediante aggiunta, non di unità di glucosio, ma di un complesso fra glucosio e Uridin-difosfato. La catena del polisaccaride viene allungata grazie all’enzima glicogeno sintasi aggiungendo sequenzialmente UDP-glucosio, a partire da una catena preformata di glicogeno di almeno 8 residui. Il glucosio attivato sotto forma di UDP-glucosio viene trasferito all’estremità non riducente di una catena polisaccaridica, con formazione di un legame 1-4 glicosidico. Le ramificazioni 1-6 sono catalizzate dall’enzima ramificante che lega porzioni della catena lineare 1-4 con legami 1-6 ogni circa 10 unità di glucosio L’UDP-GLUCOSIO L’UDP-GLUCOSIO è il donatore di glucosio nella biosintesi del glicogeno ed è una forma attivata di glucosio. L’atomo di Carbonio C1 dell’unità glicosidica dell’UDPglucosio è attivato in quanto il suo gruppo OH è esterificato all’unità pirofosforica dell’UDP L’UDP-glucosio viene sintetizzato, ad opera della UDP-glucosio pirofosforilasi, dal glucosio 1-fosfato e dall’Uridina Trifosfato (UTP); il Pirofosfato liberato durante la reazione, è dato dai due gruppi fosforici esterni dell’UTP. Sintesi dell’UDP-glucosio La formazione di uno zucchero attivato legato a nucleotidi avviene mediante una reazione di condensazione di un NTP con uno zucchero fosforilato. La carica negativa dell’ossigeno serve da nucleofilo per un attacco sul gruppo fosforico interno del nucleoside trifosfato e libera PPi. L’equilibrio della reazione viene complessivamente spinto verso destra dall’idrolisi del pirofosfato da parte della pirofosfatasi inorganica. La glicogeno sintasi Catalizza l’aggiunta di unità di glucosio (a partire dall’UDPG) ad una catena di glicogeno di almeno 8 residui. Le nuove unità glicosidiche vengono aggiunte al residuo non riducente terminale del glicogeno. L’unità glicosidica attivata dell’UDP-Glucosio viene trasferita al gruppo OH del C4 all’estremità della molecola del glicogeno, formando un legame glicosidico 1-4, reazione catalizzata dalla GLICOGENO SINTASI. La glicogeno sintasi può aggiungere residui glicosidici a catene polisaccaridiche che contengono almeno 8 residui; quindi la sintesi del glicogeno richiede una catena preformata (primer) che in questo caso è svolta dalla GLICOGENINA. La glicogenina è una proteina che porta una catena oligosaccaridica con unità di glucosio 1-4. Il C1 della prima unità di questa catena è legato covalentemente al gruppo OH fenolico di una Tirosina specifica nella glicogenina. La glicogenina catalizza l’aggiunta di circa 8 unità di glucosio e l’UDP-glucosio è il donatore di questa autoglicosilazione. A questo punto interviene la glicogeno sintasi, che è strettamente legata alla glicogenina, in particolare la glicogeno sintasi è cataliticamente efficiente solo se legata alla glicogenina. Questa dipendenza ha 2 conseguenze importanti: - Il numero di granuli di glicogeno è determinato dal numero di molecole di glicogenina - L’allungamento si ferma quando la sintasi non è più a contatto con la glicogenina, che forma il centro della particella. L’interazione sintasi-glicogenina limita le dimensioni dei granuli di glicogeno Glicogenina Sezione trasversale di una molecola di glicogeno La proteina glicogenina (Mr 37 284) innesca la sintesi del glicogeno legando al gruppo –OH di un residuo di Tyr del polipeptide un residuo di glucosio. La reazione utilizza UDP-glucosio e avviene grazie all’attività autocatalitica dell’enzima protein-tirosina glicosiltransferasi. All’estremità non riducente di questa prima unità glucosidica legata alla glicogenina vengono aggiunti altri 7 residui di glucosio ad opera della glicogenina associata alla glicogeno sintasi A questo punto, è possibile l’azione diretta della glicogeno sintasi che si dissocia dalla glicogenina e sintetizza una catena lineare. Non appena il numero di residui aggiunti lo permette, abbiamo l’azione dell’enzima ramificante che aggiunge una seconda estremità non riducente sulla quale la glicogeno sintasi può lavorare e così via. L’enzima ramificante (amilo (1,4 1,6 transglicosilasi) Le ramificazioni si formano grazie all’azione dell’enzima ramificante, che lavora trasferendo un segmento terminale di 6-7 residui glucosidici dall’estremità non riducente di una catena poliglucosidica composta da almeno 11 residui… …al gruppo –OH del carbonio C-6 di un residuo di glucosio della stessa catena o di un’altra catena ma localizzato in un punto più interno. L’azione combinata di glicogeno sintasi e dell’enzima ramificante permette la formazione di un polisaccaride a ramifi-cazione crescente che permette una crescita (e una demolizione) molto più rapida di un polisaccaride lineare. 11 7 L’enzima ramificante che catalizza questa reazione è piuttosto esigente: - il blocco di circa sette residui deve comprendere l’estremità non riducente e deve provenire da una catena lunga almeno undici residui; - inoltre il nuovo punto di ramificazione deve distare almeno quattro residui dal punto di ramificazione precedente. 4 residui Struttura della glicogeno transferasi RESA ENERGETICA METABOLISMO GLICOGENO Per incorporare glucosio 6-fosfato nel glicogeno, viene consumato un legame fosforico ad alta energia. La resa energetica della glicogenolisi è molto elevata: circa il 90% dei residui vengono scissi fosforoliticamente in G1-P, che viene poi convertito in G6-P senza ulteriori costi. L’altro 10% del glicogeno è rappresentato dai residui ai punti di ramificazione che vengono invece rimossi idroliticamente. E’ poi necessario ATP per fosforilare queste molecole di glucoso a G6-P. L’ossidazione completa di una molecola di G6-P rende circa 31 molecole di ATP e la sua conservazione consuma poco più di una molecola di ATP… NEL GLICOGENO QUINDI SI HA LA CONSERVAZIONE DI CIRCA IL 97% DELL’ENERGIA GLICOGENO Glicogenolisi Glicogenosintesi GLUCOSIO 6-FOSFATO Shunt G 6-P GLUCOSIO GLUCOSIO 6-FOSFATO Glicolisi Gluconeogenesi LATTATO DEGRADAZIONE DEL GLICOGENO: GLICOGENOLISI La degradazione del glicogeno è rapida e avviene agli estremi non riducenti delle ramificazioni. Interviene l’enzima GLICOGENO FOSFORILASI che rimuove sequenzialmente le unità di glucosio poste ad una delle estremità libere (non riducenti) del glicogeno mediante fosforolisi. Questa reazione può proseguire fino al raggiungimento di una ramificazione, ma essendo specifica per il legame 1-4 non può idrolizzare il legame 1-6 tipico della ramificazione Parte dell’energia del legame glicosidico viene conservata nell’estere fosforico del glucosio 1- fosfato che si forma da questa reazione Intervengono quindi altri 2 enzimi, una TRANSFERASI e una GLUCOSIDASI che consentono la trasformazione della ramificazione in una struttura lineare suscettibile all’attacco della GLICOGENO FOSFORILASI La FOSFORILASI libera glucosio fosforilato, la GLICOSIDASI glucosio non fosforilato Viene scisso dall’ortofosfato il legame tra l’atomo di Carbonio in C1 e l’Ossigeno Glicosidico e viene mantenuta a livello del C1 la configurazione α La glicogeno fosforilasi • Catalizza la fosforolisi del glicogeno a glucosio-1-Pi • È regolata sia da interazioni allosteriche che da modificazioni covalenti • Utilizza il piridossal-5’-fosfato come cofattore • Non può staccare residui oltre 5 unità da un punto di ramificazione Meccanismo di reazione della glicogeno fosforilasi Regolazione della glicogeno fosforilasi • è un enzima omodimerico che esiste in due stati conformazionali T (meno attiva) ed R (più attiva): il glicogeno si lega preferenzialmente all'enzima nella forma R • Il legame con l’AMP sposta l’equilibrio verso la forma R • La fosforilazione della Ser14 sposta l’equilibrio verso la forma R L’enzima deramificante È una (1,4) glicosiltransferasi e una (1,6) glicosidasi; le 2 attività sono localizzate in 2 siti separati della stessa proteina In una prima fase, l’enzima stacca (attività transferasica) un blocco di tre residui di glucosio dalla ramificazione ad una estremità non riducente vicina, con formazione di un legame ( 1-4) glicosidico. Poi l’enzima idrolizza il legame 1-6 presente nei punti di ramificazione Il residuo coinvolto nella formazione della ramificazione (legame 1-6) viene rilasciato direttamente come glucosio libero (attività glucosidasica). La fosfoglucomutasi •IL G1P,prodotto dalla demolizione del glicogeno ad opera della glicogeno fosforilasi, viene trasformato in G6P dalla fosfoglucomutasi, per entrare nel flusso metabolico principale •La fosfoglucomutasi richiede glucosio 1,6-bisfosfato come cofattore •Secondo l’organo in cui la reazione avviene, i destini del G6P sono diversi; il G6P può entrare nella glicolisi (muscolo) oppure nella via dei pentosi fosfati •Nel fegato la glucosio 6 fosfatasi idrolizza il G6P in glucosio che può così essere esportato ad altri organi La fosfoglucomutasi converte il G-1P in G-6P Il gruppo-fosforico viene trasferito dal residuo di Serina dell’Enzima sul gruppo ossidrilico dell’atomo di carbonio C-6 nella molecola del glucosio 1-fosfato, generando glucosio 1,6bisfosfato. Questo intermedio trasferisce poi il suo gruppo fosforico in posizione C1 sullo stesso residuo di serina, producendo glucosio 6-fosfato e rigenerando il fosfoenzima Il GLUCOSIO FOSFORILATO, prodotto finale della glicogenolisi, al contrario del GLUCOSIO, non può diffondere liberamente dalle cellule. Per questo il FEGATO contiene l’enzima idrolitico GLUCOSO 6-FOSFATASI, che fosforila di nuovo il Glucosio consentendogli di lasciare l’organo. Questo enzima è presente anche nel rene e nell’intestino ma è assente nel MUSCOLO e nel CERVELLO. Quindi il glucosio 6-fosfato rimane intrappolato nel muscolo e nel cervello, tessuti che necessitano di grandi quantità di questo nutriente per generare ATP. Al contrario il glucosio 6fosfato, non è la principale sostanza nutriente del fegato, e può essere immagazzinato o rilasciato (perché defosforilato grazie all’enzima) per le necessità degli altri organi. Il GLUCOSIO può uscire dalla cellula epatica, essere riversato nel sangue ed essere utilizzato da altri organi. Mentre nel MUSCOLO il GLICOGENO è essenzialmente una riserva di energia, che può essere utilizzata solo dalla cellula in cui si trova, Nel FEGATO il GLICOGENO può anche essere una riserva di unità di glucosio che possono essere utilizzate anche da altri organi. IL FEGATO ha perciò la funzione di CONTROLLARE LA GLICEMIA, funzione mancante negli altri organi. La G6Pasi è stabilizzata da una proteina (SP) che lega il Ca2+ Regolazione della sintesi e della degradazione del glicogeno • Sintesi e degradazione del glicogeno sono coregolate in modo da non funzionare simultaneamente • La regolazione comporta sia un controllo allosterico diretto sugli enzimi, sia un controllo ormonale mediato da una modificazione covalente degli enzimi coinvolti • La glicogeno fosforilasi è attivata da AMP e inibita da ATP e G6P • La glicogeno sintasi è attivata dal G6P INSULINA GLUCAGONE Stimola glicogeno sintasi Inibisce glicogeno fosforilasi Inibisce glicogeno sintasi Stimola glicogeno fosforilasi EPINEFRINA Inibisce glicogeno sintasi ADRENALINA Stimola glicogeno fosforilasi Accumulo glucosio sotto forma di glicogeno (ormone ipoglicemizzante) Mobilizzazione delle unità di glucosio dal glicogeno (ormone iperglicemizzante) Ormoni iperglicemizzanti Azione sinergica a quella del glucagone ma agisce con maggior potenza e su tempi più brevi Il bilancio netto tra la sintesi del glicogeno e la sua demolizione viene controllato dai livelli ormonali di insulina e glucagone. Questi, regolando i livelli di cAMP (il secondo messaggero intracellulare), determinano i rapporti tra le forma attive di glicogeno sintasi e glicogeno fosforilasi. Gli stessi ormoni regolano anche i livelli di F2,6BP e dunque il bilancio tra glicolisi e gluconeogenesi. L’adrenalina o epinefrina ha effetti simili a quelli del glucagone ma il tessuto bersaglio di questo ormone è tipicamente il muscolo, mentre il glucagone agisce essenzialmente a livello del fegato. SCHEMA RIASSUNTIVO Glucagone Stimola la gluconeogenesi e la glicogenolisi a livello epatico Insulina Stimola la glicolisi e la glicogenosintesi a livello epatico Adrenalina Stimola la glicogenolisi (fegato e muscolo) e la gluconeogenesi (fegato) CONTROLLO ORMONALE DEL METABOLISMO DEL GLICOGENO Controllo mediante modificazione covalente della glicogeno fosforilasi e della glicogeno sintasi La glicogeno sintasi e la glicogeno fosforilasi sono due enzimi separati che possono esistere in due forme: la forma (a) attiva e la forma (b) inattiva La glicogeno sintasi è nella forma attiva (a) quando defosforilata, mentre la glicogeno fosforilasi risulta attiva (a) quando viene fosforilata. FOSFORILASI DEL MUSCOLO SCHELETRICO E’ un dimero o un tetramero e esiste in 2 forme interconvertibili: -una forma attiva, fosforilata, la FOSFORILASI a -una forma passiva, defosforilata, la FOSFORILASI b La fosforilasi b viene convertita nella fosforilasi a mediante la fosforilazione di un singolo residuo di serina in ogni subunità. L’enzima che catalizza la reazione è la fosforilasi chinasi. La fosforilasi a viene deattivata per idrolisi del residuo di fosfoserina in ciascuna subunità, reazione catalizzata dalla proteina fosfatasi 1. + ATP fosforilasi b Ser OH Enzima inattivo + ADP + H+ fosforilasi a Ser O PO32- Enzima attivo Ognuna delle 2 forme, sia la fosforilasi a che b, a sua volta può adottare: -la conformazione cataliticamente inattiva T (tesa) oppure -la conformazione attiva R (rilassata) L’equilibrio R T della fosforilasi a è spostato verso lo stato attivo R L’equilibrio R T della fosforilasi b è spostato verso lo stato inattivo T La fosforilasi b è inattiva (forma T) in presenza di ATP e GLUCOSIO 6-P che agiscono come effettori allosterici, competendo con l’AMP per il legame all’enzima Invece la fosforilasi b è attiva (forma R) soltanto in presenza di elevate concentrazioni di AMP, che agisce in modo allosterico: si lega al sito per i nucleotidi, alterando la conformazione della fosforilasi b. Nel muscolo a riposo, praticamente tutto l’enzima è nella forma inattiva b; durante l’esercizio muscolare, gli elevati livelli di AMP tendono ad attivare la fosforilasi b. La fosforilasi a è pienamente attiva (forma R), indipendentemente dai livelli di AMP, ATP e GLUCOSIO 6-P FOSFORILASI DEL FEGATO La fosforilasi del fegato differisce da quella del muscolo sotto 2 aspetti principali: -l’AMP non attiva la fosforilasi b del fegato -la fosforilasi a è inattivata dal legame col glucosio, quindi il glucosio sposta l’equilibrio allosterico della forma a da R a T. Lo scopo della degradazione del glicogeno nel fegato è quello di formare glucosio da esportare ad altri tessuti, quando il livello del glucosio ematico è basso Quindi la fosforilasi del fegato risponde al glucosio ma non all’AMP Al contrario, il muscolo necessita del glucosio per attività improvvise; quindi l’attivazione della fosforilasi b del muscolo da parte dell’AMP, che è abbondante durante l’esercizio muscolare, produce la rapida mobilizzazione del glicogeno. La fosforilazione regola la fosforilasi → Inattiva → (defosforilata) Attiva (fosforilata) Inattiva → (defosforilata) Parzialmente attiva → (defosforilata) L'adrenalina controlla l'attività della glicogeno fosforilasi anche attraverso i recettori α-adrenergici mediante il meccanismo calciocalmodulina dipendente. Questo meccanismo dipendente di attivazione della fosforilasi cinasi è funzionante anche in assenza dello stimolo ormonale come conseguenza della liberazione del calcio durante l'esercizio muscolare. GLICOGENO SINTASI E’ un tetramero e esiste in 2 forme interconvertibili: una forma attiva, defosforilata, la SINTETASI a una forma inattiva, fosforilata, la SINTETASI b La sintetasi a (attiva) viene convertita nella sintetasi b (inattiva) mediante la fosforilazione di un singolo residuo di serina in ogni subunità. L’enzima che catalizza la reazione è la glicogeno sintetasi chinasi, dipendente dal cAMP. Più precisamente l’enzima è chiamato sintetasi-fosforilasi chinasi, in quanto capace di catalizzare anche la fosforilazione della glicogeno fosforilasi. La sintetasi b (inattiva) viene convertita nella sintetasi a (attiva) per idrolisi del residuo di fosfoserina in ciascuna subunità, reazione catalizzata dalla glicogeno sintetasi fosfatasi, denominata anche fosfoproteina fosfatasi. GLICOGENO SINTASI NEL FEGATO La sintetasi b (inattiva, fosforilata), è attivata da un accesso di GLUCOSIO 6-P, che agisce come effettore allosterico positivo. La sintetasi a (attiva, defosforilata), è invece insensibile al GLUCOSIO 6-P e quindi attiva anche in sua assenza. La glicogeno sintetasi del fegato è quindi un enzima regolabile sia per modificazione covalente della sua molecola (fosforilazione-defosforilazione, forma a-forma b), sia per meccanismo allosterico (da parte del G6-P). Questa duplice possibilità di regolazione consente l’adeguamento della sintesi del glicogeno sia alle sollecitazioni ormonali mediate dal cAMP, sia alla disponibilità di G6-P che riflette la ricchezza di glucosio nel tessuto. GLICOGENO SINTASI NEL MUSCOLO Nel muscolo l’attività della glicogeno sintasi non risente, come quella del fegato, dell’effetto allosterico esplicato dal G6-P, presente nel muscolo sempre in basse concentrazioni; risente invece della concentrazione di glicogeno che la inibisce con “meccanismo a feedback”. Inoltre il muscolo dispone di tre glicogeno sintetasi chinasi (GSK) che fosforilano differente residui serinici della glicogeno sintetasi, producendo forme diverse dell’enzima fosforilato, dotate di attività differente. La GSK1 ad esempio, è regolata da cAMP e la GSK2 dalla concentrazione del Ca2+. Si comprende così perché, sia l’adrenalina, che induce un aumento di cAMP, sia la contrazione muscolare, che si accompagna all’aumento del Ca2+, inducano entrambe una diminuzione dell’attività della glicogeno sintasi nel mscolo. Controllo della glicogeno fosforilasi e della glicogeno sintasi La fosforilazione di un residuo di Tyr attiva la glicogeno fosforilasi (b a) e inattiva la glicogeno sintasi (a b) Questo sistema risponde ad un numero più elevato di effettori rispetto ai sistemi regolati allostericamente Il meccanismo a cascata amplifica enormemente il segnale. In questo meccanismo sono coinvolti 3 enzimi: 1. La fosforilasi chinasi 2. La proteina chinasi cAMP dipendente 3. La fosfoproteina fosfatasi-1 Regolazione coordinata del metabolismo del glicogeno e Glucagone e Glucagone Forma inattiva Forma attiva PKA PKA Disttivazione sintasi Attivazione fosforilasi STIMOLO DEMOLIZIONE GLICOGENO INIBIZIONE SINTESI GLICOGENO ADRENALINA e GLUCAGONE attivano la demolizione del glicogeno e inibiscono la sua sintesi; il fegato è più sensibile al glucagone, secreto dalle cellule α del pancreas quando i livelli di glucoso nel sangue sono bassi, il muscolo è sensibile all’adrenalina, secreta durante l’attività muscolare. 1. Adrenalina e glucagone si legano a recettori a sette eliche presenti sulla mambrana plasmatica delle cellule bersaglio e innescano l’attivazione della proteina G stimolatoria (Gs) 2. La forma legata al GTP della subunità α di Gs, attiva l’adenilato ciclasi, un enzimatransmembrana che catalizza la formazione di cAMP a partire da ATP 3. Elevati livelli di cAMP citosolico attivano la proteina chinasi A (PKA). Questa chinasi è inattiva in assenza di cAMP a causa dell’azione inibitoria delle sue subunità regolatorie; il legame di cAMP alle subunità regolatorie libera le subunità catalitiche 4. La PKA fosforila sia la fosforilasi chinasi che la glicogeno sintasi; la fosforilazione attiva la fosforilasi (attivando la fosforilasi chinasi) e simultaneamente disattiva la glicogeno sintasi (in maniera diretta). La glicogeno sintasi viene anche fosforilata dalla fosforilasi chinasi, assicurando che il glicogeno non venga sintetizzato mentre è in corso la sua degradazione. Quindi la PKA controlla in modo coordinato degradazione e sintesi del glicogeno 5. Gli effetti degli ormoni sono molto amplificati dalla cascata del cAMP, infatti il legame di poche molecole ormonali a recettori cellulari porta al rilascio di molte unità di glucosio. Inoltre… La PKA regola anche la fosforilazione dell' inibitore della fosfoproteina fosfatasi (PPI): nella sua forma fosforilata il PPI inibisce la fosfoproteina fosfatasi, mantenendo quindi la glicogeno sintasi nella forma inattiva (e la glicogeno fosforilasi nella forma attiva). La PKA induce la fosforilazione della subunità Rgl, impedendo il legame di Rgl alla subunità catalitica di PP1 che rimane inattiva perché essa non può legare i suoi substrati Con queste azioni la PKA contribuisce a mantenere attiva la demolizione di glicogeno e inattiva la sua sintesi La proteina fosfatasi 1 (PP1) La PP1 inattiva la fosforilasi chinasi e la fosforilasi a defosforilando questi enzimi, quindi fa diminuire la velocità di demolizione del glicogeno; Rimuove il gruppo fosforico dalla glicogeno sintasi b convertendola nella forma a molto più attiva e accelerando così la sintesi di glicogeno. L’attività fosfatasica di PP1 è controllata rigorosamente. La subunità catalitica di PP1di per sé ha bassa affinità per le particelle di glicogeno ma l’alta affinità gli viene conferita dall’associazione con una subunità Rgl “riconoscimento glicogeno” La subunità Rgl attira PP1 verso le particelle di glicogeno, dove può rimuovere in modo efficiente i gruppi fosforici dalla glicogeno sintasi e dalla fosforilasi chinasi. Però l’attività della PP1 è regolata in due modi… 1. La fosforilazione della subunità Rgl da parte della PKA, impedisce il legame di Rgl alla subunità catalitica di PP1 L’attivazione della cascata del cAMP (e quindi della PKA) da parte dell’adrenalina porta all’inattivazione di PP1 perché essa non può legare i suoi substrati 2. La forma fosforilata dell’inibitore1, una piccola proteina, inibisce attività di PP1, mentre la forma defosforilata no. Il grado di fosforilazione dell’inibitore1 è, a sua volta, sotto controllo ormonale. Il cAMP, che agisce tramite la PKA, blocca PP1, fosforilando l’inibitore1. La fosforilazione di Rgl e dell’inibitore1, indotte dall’adrenalina, inattivano PP1, così che viene mantenuta attiva la demolizione del glicogeno. Subunità G fosforilata dalla proteina fosfatasi 1 Adrenalina Proteina chinasi attivata Proteina fosfatasi inattivata Inibitore 1 fosforilato Una fosfatasi PP1 inverte gli effetti inibitori delle chinasi Rgl=Subunità per il riconoscimento del glicogeno Inibitore Regolazione della proteina fosfatasi 1 (PP1) Il legame dell’insulina al suo recettore, porta all’attivazione di una proteina chinasi sensibile all’insulina che fosforila la subunità Rgl della PP1 in un sito diverso da quello modificato dalla PKA. L’insulina, contrariamente all’adrenalina, rende la PP1 più attiva. In questo modo, la PP1 più attiva, defosforilando la glicogeno sintasi la fosforilasi chinasi e la fosforilasi induce la sintesi del glicogeno e inibisce la sua demolizione L’insulina attiva la fosfatasi PP1 che inverte gli effetti inibitori delle chinasi Attivazione sintesi glicogeno L’insulina attiva la proteina fosfatasi 1 Il metabolismo del glicogeno nel fegato regola i livelli di glucoso nel sangue Il fegato infatti “sente” la concentrazione di glucosio nel sangue e conseguentemente lo assume o lo rilascia La quantità di fosforilasi a nel fegato diminuisce rapidamente quando viene infuso glucosio e, dopo un certo periodo dall’infusione, la quantità di glicogeno sintasi a aumenta, e si ha l’inizio della sintesi di glicogeno La fosforilasi a è il sensore di glucosio nelle cellule epatiche Come fa il glucosio ad attivare la sintasi? La fosforilasi b, al contrario della a, non si lega alla fosfatasi. Di conseguenza, la conversione di a in b è accompagnata dal rilascio della PP1 che è quindi libera di attivare la glicogeno sintasi. L’attività della sintasi inizia ad aumentare solo dopo che la maggiorparte delle molecole di fosforilasi a è stata convertita in b Benchè l’insulina sia il principale segnale per la sintesi di glicogeno, il Fegato è sensibile alla concentrazione di glucosio nel sangue e capta o rilascia glucosio a seconda delle necessità. La fosforilasi è il sensore epatico per il glucosio ematico Il glucosio attiva la fosfatasi nel fegato Nel fegato operano altri meccanismi non ormonali si lega attiva si dissocia Regolazione del metabolismo del glicogeno epatico ad opera del glucosio. MALATTIE DA ACCUMULO DI GLICOGENO: LE GLICOGENOSI Insieme di patologie ereditarie caratterizzate da un difetto di funzione di uno degli enzimi implicati nel metabolismo del glicogeno. Ne consegue un anomalo accumulo in tessuti diversi del glicogeno non utilizzato. A seconda di quale proteina è alterata, si hanno vari tipi di glicogenosi che differiscono per la sede di accumulo, per la quantità e la qualità di glicogeno accumulato, per la gravità, per la sintomatologia e per l'evoluzione. Sono contraddistinte da un numero romano o dal nome di chi le ha descritte. Le malattie da accumulo di Glicogeno hanno una spiegazione biochimica La Glicogenosi di tipo I, o malattia di von Gierke, ad esempio, è imputabile ad una carenza ereditaria dell’enzima epatico Glucosio-6-fosfatasi. - Il glicogeno epatico di questi pazienti presenta una struttura normale, ma è presente in quantità enormemente grandi. - L’assenza di questo enzima nel fegato causa ipoglicemia. - La presenza di un eccesso di G6P fa aumentare la glicolisi epatica e di conseguenza determina una elevata concentrazione di piruvato e lattato nel sangue. - I pazienti affetti da questa patologia presentano anche una elevata dipendenza dal metabolismo dei grassi. Questa malattia può essere causata -anche da una mutazione del gene che codifica il trasportatore (T1) di G6P o -da mutazioni dei geni che codificano le altre tre proteine (T2, T3, SP) del RE implicate nella sintesi di Glc a partire da G6P. Glicogenosi tipo I Glicogenosi tipo I è causata dalla mancanza dell'attività della glucosio-6-fosfatasi nel fegato e nel rene. In tali organi il glicogeno si accumula in quanto, per la mancanza dell'enzima, non viene trasformato in glucosio e rilasciato nel sangue. Questo determina un aumento del volume del fegato (epatomegalia) che è la causa dell'addome prominente e dell'ipoglicemia (basso glucosio nel sangue). Accanto all'ipoglicemia abbiamo anche acidosi lattica, iperuricemia (aumento di acido urico nel sangue) e iperlipidemia. I pazienti con questo tipo di glicogenosi possono esordire nel periodo neonatale con ipoglicemia e acidosi lattica; tuttavia, più comunemente l'esordio clinico è a 3-4 mesi con epatomegalia ed ipoglicemia. Questi bambini spesso presentano una faccia da "bambola" per un eccesso di tessuto adiposo nelle guance, estremità sottili, un addome prominente e bassa statura. Rari casi possono essere asintomatici ad esclusione dell'epatomegalia. Nel tipo 1b si osservano caratteristiche molto simili alla 1a. Sebbene il difetto sia associato ad un'altra proteina (trasportatore del glucosio-6-fosfato), si ottiene lo stesso risultato finale. Infatti, la mancanza del trasportatore che fornisce il substrato all'enzima glucosio-6-fosfatasi, impedisce la formazione del glucosio. A questo tipo di glicogenosi sono associate inoltre, quasi sempre, anche la neutropenia (diminuzione dei globuli bianchi) e un deficit delle funzioni leucocitarie, che determinano infezioni batteriche ricorrenti ed ulcerazioni della mucosa orale ed intestinale. Il tipo 1c non è stato ancora ben caratterizzato, potrebbe essere uguale alla 1b, ma senza le alterazioni leucocitarie. Glicogenosi tipo II Glicogenosi tipo II (malattia di Pompe) è causata dalla mancanza della a-glicosidasi o maltasi acida nei lisosomi. La forma più grave è quella infantile, caratterizzata da cardiomegalia, grave ipotonia muscolare e morte per insufficienza respiratoria nei primi 2 anni di vita. La forma più lieve è quella che si manifesta tra la seconda e la sesta decade, con coinvolgimento muscolare scheletrico progressivo lento. Tra questi due estremi, c'è un gruppo eterogeneo con coinvolgimento del muscolo scheletrico senza implicazioni cardiache, con esordio infantile tardivo o adulto a decorso lento. Fotografia al microscopio elettronico di tessuto muscolare scheletrico di un neonato affetto da glicogenosi di tipo II (malattia di Pompe). I lisosoma sono pieni di glicogeno a causa di una carenza di -1,4 glucosidasi, un enzima idrolitico confinato nei lisosomi. Glicogenosi tipo III Glicogenosi tipo III è causata dalla carenza dell'enzima deramificante con accumulo di glicogeno molto più ramificato. Le caratteristiche cliniche sono simili al tipo 1°, ma generalmente senza iperuricemia ed acidosi lattica. In alcuni casi (tipo IIIb) c'è solo il coinvolgimento epatico. I sintomi epatici, compresa la fibrosi, generalmente scompaiono dopo la pubertà. In molti pazienti (tipo IIIa) c'è anche interessamento muscolare, che è scarso durante l'infanzia ed aumenta nella terza e quarta decade Glicogenosi tipo IV Glicogenosi tipo IV è causata dalla mancanza dell'enzima ramificante con accumulo di glicogeno non ramificato in molti tessuti. Si presenta nei primi mesi di vita con epatomegalia e deficit della crescita, seguita da cirrosi epatica progressiva con ipertensione portale, ascite (accumulo di liquidi nell’addome) e morte entro i primi anni di vita. L'ipoglicemia è rara, ma può instaurarsi in seguito alla cirrosi. Può esserci debolezza muscolare e grave cardiomiopatia. E' stata descritta anche una forma ad interessamento Glicogenosi tipo V Glicogenosi tipo V provocata dalla carenza di fosforilasi muscolare che impedisce l'utilizzazione del glicogeno durante l'esercizio fisico. Compaiono fatica, dolore e, se lo sforzo fisico continua, seguono crampi e danno muscolare, evidenziato da una colorazione borgogna delle urine, dovuta alla mioglobulina rilasciata durante la lesione della fibre muscolari. Si manifesta in genere nella seconda e terza decade di vita, ma può manifestarsi anche durante l'infanzia. I sintomi possono essere causati o da sforzi brevi ma intensi oppure da sforzi meno intensi ma prolungati Glicogenosi tipo VI Glicogenosi tipo VI è dovuta ad un difettoso funzionamento della fosforilasi epatica. E' una forma non grave nella quale, all'epatomegalia e ad un difetto nella crescita, è associata una modesta ipoglicemia e chetosi. L'epatomegalia si risolve in genere con la pubertà e la crescita è normale. Il cuore ed il muscolo non sono interessati. Glicogenosi tipo VII Glicogenosi tipo VII è associata alla mancanza della fosfofruttochinasi muscolare. Le caratteristiche cliniche sono molto simili al tipo V, tuttavia può essere differenziata dalla V perché si manifesta più precocemente, gli attacchi possono essere accompagnati da nausea e vomito, si può avere anemia emolitica ed iperuricemia. Glicogenosi tipo VIII Glicogenosi tipo VIII è dovuta alla carenza di fosforilasi chinasi e presenta le stesse caratteristiche cliniche molto simili al tipo VI, anche se si descrivono altre varianti. Molti casi di questo colpiscono i maschi perché il gene si trova nel cromosoma X Glicogenosi tipo IX Glicogenosi tipo XI (Fanconi-Bickel) è causata, anche se c'è qualche perplessità, da alterazioni del trasportatore del glucosio (GLUT2) all'interno degli epatociti, cellule beta-pancreatiche, enterociti e cellule tubulari renali. E' caratterizzata da nefropatia tubulare (glucosuria, fosfaturia e aminoaciduria), epatomegalia ed intolleranza al glucosio e galattoso. Glicogenosi tipo 0 Glicogenosi tipo 0 (aglicogenosi) La glicogenosi tipo 0 o deficit di glicogeno-sintetasi non è una glicogenosi propriamente detta, in quanto il deficit enzimatico comporta una diminuzione delle riserve di glicogeno. Si tratta di un deficit genetico molto raro a trasmissione autosomica recessiva. I sintomi clinici comprendono l'ipoglicemia a digiuno (senza epatomegalia), associata ad affaticamento mattutino e, sul piano biochimico, iperchetonemia senza iperalaninemia, né iperlattacidemia. Dopo i pasti compaiono sia una importante iperglicemia, sia un aumento dei lattati e dell'alanina. La diagnosi di laboratorio, sospetta dopo prove da carico di glucosio, richiede una biopsia epatica, che dimostra una concentrazione di glicogeno normale o poco diminuita e permette di mettere in evidenza il deficit enzimatico (che non è espresso nel muscolo, negli eritrociti, nei leucociti o nei fibroblasti). Il gene è stato localizzato sul cromosoma 12p22.2. La sua struttura è conosciuta e sono state identificate diverse mutazioni.
Scaricare