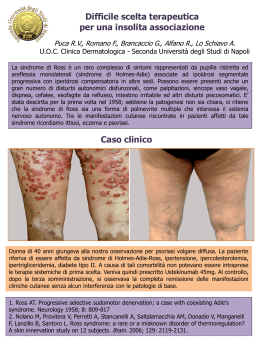
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology Anno III numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali Home page Norme editoriali | Stampa l'articolo 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Numeri precedenti Motore di ricerca ◀ Indietro pagina 8 Avanti ► Sindrome di Alagille: aspetti genetici, clinico-radiologici e terapia Alagille Syndrome: genetics, clinical and radiological aspects and therapy 1 2 E. David , S. Cinconze 1 UOC di Radiodiagnostica Università degli studi di Messina 2 UOC di Cardiologia Università degli studi di Messina Abstract Alagille syndrome (AGS) has an estimated prevalence of about 1/70.000. Classically, the syndrome consists of five signs: (1) chronic cholestasis associated with pruritus, hypercholesterolemia and paucity of interlobular bile ducts on liver biopsy, (2) congenital heart disease (the most common of which is the peripheral pulmonary artery stenosis), (3) bone defects, including butterfly vertebrae, hemivertebrae and lack of progression interpediculare distance, (4) ocular signs (mainly embryotoxon rear (5) typical facies with frontal projections, sunken eyes, rounded tip of the nose, chin appuntito. Una Early detection ensures a better quality of life, this shows the importance of a clarification about the diagnosis. La diagnosis is based on a careful clinical evaluation, a radiologic finding, and ultimately a diagnosis based on genetic research of the most typical mutations (JAG 1 and NOTCH2). Riassunto La sindrome di Alagille (AGS) ha una prevalenza stimata di circa 1/70.000. Classicamente la sindrome comprende cinque segni: (1) colestasi cronica associata a prurito, ipercolesterolemia e scarsità dei dotti biliari interlobulari alla biopsia epatica; (2) patologie cardiache congenite (la più comune delle quali è la stenosi periferica dell'arteria polmonare); (3) difetti ossei, comprese vertebre a farfalla, emivertebre e mancanza di progressione della distanza interpediculare; (4) segni oculari (principalmente embryotoxon posteriore; (5) facies tipica con sporgenze frontali, occhi scavati, punta del naso arrotondata, mento appuntito. Una diagnosi precoce assicura una migliore qualità della vita;da questo si evince l’importanza di un chiarimento diagnostico. La diagnosi si basa su un attenta valutazione clinica, un riscontro radiologico, e in definitiva una diagnosi genetica fondata sulla ricerca delle mutazioni più tipiche (JAG 1 e NOTCH2). Genetica La ALGS è una patologia genetica di tipo autosomico dominante, ma con espressione fenotipica variabile. Esistono due tipi di malattia, il tipo 1 (94% dei casi) è dovuto alle mutazioni del gene JAG1 (20p12) che codifica per un ligando della via di segnalazione Notch[1]; il tipo 2 è dovuto a mutazioni del gene NOTCH2 (1p12). [2]Per i restanti pazienti la causa patogenetica rimane tuttora oscura. Nell’ambito delle mutazioni JAG1, nel 3-7% dei casi sono state riscontrate delezioni totali del gene, mentre il resto dei pazienti presentava mutazioni intrageniche. Il 72% delle mutazioni riportate comportava uno spostamento del frame di lettura determinante la formazione di un codone di stop prematuro. È stata anche riscontrata un'elevata frequenza (60-70%) di nuove mutazioni [3]. Le interazioni fra Jagged e Notch si sono rilevate fondamentali nella determinazione del destino cellulare nelle fasi precoci dello sviluppo. Pazienti aventi delezioni più o meno estese del gene JAGGED1 hanno fenotipo indistinguibile da pazienti aventi mutazioni missenso o nonsenso; ciò porta ad ipotizzare che il meccanismo patogenetico dell’Alagille sia dovuto all’aploinsufficienza di JAGGED1. Nonostante sia una patologia a trasmissione autosomica dominante non sono infrequenti la penetranza ridotta (fino al 50% dei casi) e il mosaicismo somatico (~8%). E’ possibile effettuare una diagnosi genetica prenatale attraverso l'analisi del DNA del tessuto dei villi coriali o degli amniociti in coltura mediante sequenziamento del gene JAG1 o NOTCH2. Oltre l’aspetto molecolare che ci dà la diagnosi definitiva, un sospetto diagnostico può comunque essere avanzato da un determinato quadro clinico ed ecografico (e non solo). L’ecografia fetale può aiutare a identificare eventuali anomalie cardiache e renali. [4] Clinica Non tutti i pazienti presentano i segni classici della sindrome, rendendo la diagnosi difficile in alcuni casi. La malattia epatica associata alla sindrome di Alagille può essere estremamente variabile. I bambini con sindrome di Alagille di solito presentano malattia epatica caratterizzata da una progressiva perdita dei dotti biliari intraepatici nel primo anno di vita ed un restringimento dei dotti biliari extraepatici. Questo provoca un accumulo di bile nel fegato ed un conseguente danno alle cellule epatiche. Ittero ad insorgenza nelle prime sei settimane di vita è la manifestazione più tipica in questo senso. La colestasi può essere così grave che può venire presa in considerazione la presenza di atresia biliare. Col tempo, comunque, l'ittero può migliorare. Tuttavia, in altri casi, può persistere una grave colestasi, accompagnata da prurito intollerabile, infezioni cutanee e lichenificazione della cute come esito. In alcuni pazienti con sindrome di Alagille e cirrosi è stata riportata l'insorgenza di carcinoma epatocellulare. Un gruppo di caratteristiche insolite negli altri organi distingue però la sindrome di Alagille dalle altre malattie epatiche e biliari in età pediatrica. [5] Frequente è l’associazione tra sindrome di Alagille e disordini cardiaci congeniti. I difetti più frequentemente riscontrati sono a carico della valvola polmonare e comprendono l’ostruzione sopravalvolare del tratto di efflusso del ventricolo destro e la stenosi a setto integro o perforato. Frequentemente queste anomalie congenite si presentano in associazione ad altre malformazioni quali tetralogia di Fallot (7-10%), difetti del setto interventricolare, difetti del setto interatriale, stenosi e coartazione aortica. Pur essendo piuttosto frequente l’associazione di difetti multipli, molti si presentano emodinamicamente insignificanti, fatta eccezione per il difetto valvolare polmonare che, seppur asintomatico nelle fasi iniziali, ha un andamento ingravescente e pertanto necessita di essere attenzionato [6]. Da un punto di vista morfologico la valvola polmonare può variare da un’anatomia ben conformata a tre lembi con vari gradi di fusione commissurale a una membrana imperforata. Se è presente stenosi il ventricolo può presentarsi di dimensioni normali o lievemente ipoplasico. La cianosi si manifesta in condizioni di stenosi serrata (infrequente nella presentazione iniziale nella sindrome di Alagille), derivante da uno shunt destrosinistro a livello atriale. I reperti ascoltatori comprendono un secondo tono singolo, assenza di click eiettivo e un soffio che, quando presente, è dovuto ad insufficienza tricuspidalica associata[7]. Di solito tardivamente nel decorso della malattia, si può osservare anche un interessamento renale, presumibilmente secondario all'iperlipemia prolungata e determinante a lungo termine una nefropatia membranosa coinvolgente i glomeruli e i tubuli. Altre alterazioni renali congenite comprendono la duplicazione della pelvi renale, fibrosi interstiziale, nefronoftisi (perdita di parenchima renale), cisti della midollare renale, e rene singolo[8]. Le alterazioni scheletriche associate alla sindrome comprendono le classiche vertebre a farfalla. Altre alterazioni riportate sono: emivertebre, spina bifida incompleta, accorciamento delle falangi distali, ulna corta, mancanza del normale ampliamento della distanza interpediculare lungo il tratto lombare della colonna vertebrale[9]. In alcuni casi si può anche osservare l'insorgenza di un'artrite sieronegativa poliarticolare e sinostosi radio-ulnari di notevole rilevanza clinica[10]. Molte delle alterazioni ossee osservate nella sindrome di Alagille causano una sintomatologia lieve nei pazienti. Tuttavia, una osteopenia significativa dovuta al ridotto assorbimento di calcio e vitamina D, dovuto alla grave colestasi, può determinare fratture patologiche in molti di questi bambini [11]. Nella sindrome di Alagille si possono riscontrare anche alterazioni oculari. La più comune è l'embriotoxon posteriore, un ispessimento della linea di Schwalbe, che non interferisce comunque con la visione. Un altro segno associato è l'anomalia di Axenfeld (presenza di embriotoxon posteriore, processo dell'iride adeso all'anello di Schwalbe) che può causare glaucoma. Il deficit di vitamina A e/o E può determinare modificazioni della pigmentazione retinica con risultante cecità notturna o visione tubulare (a tunnel), nonché alterazioni neurologiche di vario grado. Si possono avere anche una miopia di grado elevato e cheratocono, e molti di questi bambini possono aver bisogno di lenti correttive[12]. Alcuni pazienti con la sindrome possono presentare un'insufficienza pancreatica. L'eziologia di tale insufficienza non è conosciuta; i bambini con sindrome di Alagille che manifestino diarrea, non dovuta alla colestasi, dovrebbero essere valutati per escludere l'insufficienza pancreatica. Le manifestazioni dermatologiche associate alla sindrome sono principalmente il risultato della grave colestasi. Comunemente si possono osservare lichenificazione della cute, dovuta al prurito cronico e xantomi, dovuti ai livelli elevati di colesterolo. Sono stati anche riportati casi di fotosensibilità e rash porfirici. Sono stati riportati casi di otite media ricorrente e deficit uditivi nei pazienti con la sindrome[13]. Un segno di riscontro non comune è la parziale o completa assenza del canale semicircolare posteriore in alcuni pazienti In alcuni pazienti sono state riportate alterazioni vascolari a livello del sistema nervoso centrale. [14] Tra queste, emorragie intracraniche, malformazione di Arnold-Chiari e occlusione completa delle arterie carotidi interne con formazione di circoli collaterali. Nel 1° anno di vita la facies dei pazienti con S. di Alagille potrebbe non essere distintiva, ma se sono presenti le caratteristiche facciali (sporgenze frontali, occhi scavati, punta del naso arrotondata, mento appuntito), la disfunzione epatica e, in particolare l'ipoplasia dei dotti biliari, la diagnosi può essere fatta precocemente. E' stata anche descritta una S. di Alagille senza coinvolgimento epatico ma con le tipiche caratteristiche facciali, o viceversa. [13] Imaging Radiologico Le tecniche di imaging (ecografia addominale e colangiografia principalmente) aiutano ad identificare l'anatomia biliare e rappresentano un importante ausilio di accuratezza diagnostica. La diagnostica per immagini assume in questa patologia un valore ausiliario di notevole importanza. Ciò è assimilabile alle alterazioni tipiche riscontrabili radiologicamente, che assieme alla clinica sono il punto di partenza per sospettare una sindrome di Alagille e quindi passare a un indagine più specifica come quella di carattere molecolare. Il radiogramma convenzionale del rachide dorsale permette di valutare un certo dimorfismo a livello del soma di D6 caratterizzato da schisi sagittale del corpo vertebrale e conformazione definita tipicamente “ad ali di farfalla. Sempre mediante radiogramma convenzionale sono dimostrabili sinostosi radio ulnari. L’ecografia dimostra assente dilatazione delle vie biliari intraepatiche e mancata evidenziazione di dotto biliare comune e colecisti. Inoltre può dimostrare le tipiche alterazioni cardiovascolari (tetralogia di fallot) ed epatiche (carcinoma). L’indagine strumentale può evidenziare deviazione assiale sinistra e ventricolo di sinistra dominante all’ecg di superficie in pazienti con ventricolo destro ipoplasico, l’esame ecocardiografico può essere dirimente evidenziando l’anatomia valvolare e le diverse anomalie associate, in particolar modo per i difetti settali e le alterazioni aortiche. La colangiografia percutanea rivela ridotta arborizzazione delle vie biliari intraepatiche spesso accompagnata da ipoplasia della colecisti;talvolta sono evidenti stenosi segmentali delle vie biliari intra ed extra epatiche visualizzabili anche con indagini non invasive come la colangio RM. Tale metodica rientra nelle tecniche di ” idrografia con RM”.Vengono acquisite immagini con sequenze fortemente pesate in T2 che esaltano il segnale dei fluidi stazionari come la bile con abbattimento del segnale dei parenchimi e delle strutture vascolari. L’Angiografia può essere di notevole ausilio per la ricerca di condizioni di morbosità che spesso si associano alla patologia di Alagille; è possibile infatti dimostrare mediante questa tecnica diagnostica una stenosi dell’arteria polmonare periferica, della celiaca o della mesenterica superiore, o ancora, un aneurisma dell’arteria epatica. [15] La TC può essere d’ausilio soprattutto per la ricerca di un segno, che associato ad altre condizioni tipiche, può essere patognomonico, e cioè la parziale assenza dei canali semicircolari, grave alterazione spesso riscontrabile[16]. Ci si può servire della TC per dimostrare calcificazioni aortiche, non infrequenti, o cisti renali, generalmente a riscontro tardivo. [17] La CEUS, la TC con mdc (trifasica) e la RM sono indicate per una attenta valutazione del fegato ed eventualmente la dimostrazione di una iperplasia nodulare epatica [18]. Fig. 2 Valvola Polmonare Stenotica e Ispessita [7] Fig. 3 Ecografia: Arteria Polmonare Principale notevolmente slargata [7] Fig. 4 Rx Torace che mostra la fusione dell'arco anteriore di T8 (vertebra a farfalla) [9] Fig. 5 Visione in M-Mode con evidente rigidità del setto interventricolare [7] Fig. 6 Insufficienza Polmonare in asse corto [7] Fig. 1 Ecografia: Tetralogia di Fallot [15] Fig. 7 Ecografia epatica: assente dilatazione delle vie biliari intraepatiche e mancata evidenzazione dotto biliare [15] Fig. 8- 9:RMN, ERCP vie biliari [15] Terapia La terapia non è specifica e include una dieta ricca di carboidrati, di trigliceridi a catena media e di integratori vitaminici. Il prurito può essere ridotto con la colestiramina . Il trapianto di fegato è necessario nelle fasi avanzate della malattia. La prognosi è di solito buona, ma possono verificarsi complicanze come cirrosi, emorragia delle varici, asciti refrattarie e peritoniti batteriche spontanee. [19]Gli interventi cardiochirurgici o vascolari possono essere necessari in caso di lesioni sintomatiche importanti. La scelta terapeutica viene valutata da caso a caso con particolare attenzione al trattamento chirurgico, che viene generalmente consigliato quando il gradiente di picco attraverso il tratto di flusso supera i 50mmHg a riposo . In attesa del trattamento risolutivo, misure generali terapeutiche comprendono il mantenimento della pervietà del dotto mediante prostaglandine nelle forme dotto dipendenti, successivamente si esegue dilatazione con palloncino nel tentativo di ridurre il gradiente presente mentre in pazienti con atresia valvolare può essere tentata perforazione con radiofrequenza insieme a dilatazione. La malattia si stabilizza di solito tra i 4 e i 10 anni d'età. In presenza di insufficienza epatica e di lesioni cardiache aumenta il rischio di morte. La maggior parte delle terapie mirano in ogni caso, a controllare le complicanze della colestasi. Il malassorbimento dei grassi e delle vitamine liposolubili richiede spesso una supplementazione di vitamine A, D, E e K. I bambini possono richiedere formule speciali con trigliceridi a catena Home page intermedia come principale fonte di grassi. Alcuni bambini con la sindrome beneficiano della nutrizione continua per via nasogastrica o attraverso la gastrostomia. Questo permette di aumentare l'introito calorico in bambini che possono avere difficoltà ad assumere le calorie necessarie per la crescita a causa della presenza di malassorbimento.Il prurito associato con la sindrome può essere di difficile gestione. L'intervento chirurgico di diversione biliare, usato spesso per altre sindromi con colestasi intraepatica, non è efficace nella sindrome di Alagille. Una piccola percentuale di bambini con la sindrome ha una progressione verso l'insufficienza epatica grave e necessita del trapianto di fegato. Il trapianto di fegato può anche essere indicato nei pazienti con prurito intrattabile, non controllabile con la terapia medica, nei pazienti con ipertensione portale e con scarsa crescita. [20] BIibliografia [1]A novel JAG1 mutation in a patient with Alagille's syndrome. Wang Y, Yu Y, Wang J, Tsuei SH, Zaho L, Fu Q. [2]Analysis of liver repair mechanisms in Alagille syndrome and biliary atresia reveals a role for notch signaling. Fabris L, Cadamuro M, Guido M, Spirli C, Fiorotto R, Colledan M, Torre G, Alberti D, Sonzogni A, Okolicsanyi L, Strazzabosco M. [3]Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. [4]Novel human pathological mutations. Gene symbol: JAG1. Disease: Alagille syndrome. Marchetti D, Iascone MR, Pezzoli L. [5] Metabolic liver disease in the pediatric patient. Kelly DA, McKiernan PJ. [6]Diagnosis of Alagille syndrome-25 years of experience at King's College Hospital. Subramaniam P, Knisely A, Portmann B, Qureshi SA, Aclimandos WA, Karani JB, Baker AJ. [7]Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, Goldmuntz E. [8]Renal abnormalities in a family with Alagille syndrome. Yucel H, Hoorntje SJ, Bravenboer B. [9]Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Sanderson E, Newman V, Haigh SF, Baker A, Sidhu PS. [10]Alagille syndrome: case report with bilateral radio-ulnar synostosis and a literature review. Ryan RS, Myckatyn SO, Reid GD, Munk P. [11]Notch and the skeleton. Zanotti S, Canalis E. [12]The genetics and ocular findings of Alagille syndrome. Kim BJ, Fulton AB. [13]Alagille Syndrome. Spinner NB, Hutchinson AL, Krantz ID, Kamath BM. [14]Multiple abdominal vascular anomalies in a patient with Alagille syndrome. Kayhan A, Ilkhchoui Y, Venu N, Jensen DM, Oto A. [15]Syndrome of Alagille: radiological and sonographic findings. A review of 37 cases. Berrocal T, Gamo E, Navalón J, Prieto C, Al-Assir I, Cortés P, Pastor I, Hierro L. [16]Partial absence of the posterior semicircular canal in Alagille syndrome: CT findings. Koch B, Goold A, Egelhoff J, Benton C [17]Hepatic nodular hyperplasia in a boy with Alagille syndrome: CT and MR appearances. Tajima T, Honda H, Yanaga K, Kuroiwa T, Yoshimitsu K, Irie H, Kuwabara Y, Taguchi K, Masuda K. [18]Aortic calcification and renal cysts demonstrated by CT in a teenager with Alagille syndrome. Pombo F, Isla C, Gayol A, Bargiela A. [19]Medical management of Alagille syndrome. Kamath BM, Loomes KM, Piccoli DA. [20]Alagille syndrome and liver transplantation. Kamath BM, Schwarz KB, Hadzić N. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 ◀ Indietro pagina 8 Avanti ► Scarica l'articolo: pagina 8.pdf Sommario 20 pagine Direttore scientifico Trimestrale di divulgazione scientifica dell'Associazione Pediatrica di Immunologia e Genetica Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009 Carmelo Salpietro - Direttore responsabile Giuseppe Micali - Segreteria redazione Basilia Piraino - Piera Vicchio Direzione-Redazione: UOC Genetica e Immunologia Pediatrica - AOU Policlicnico Messina
Scaricare