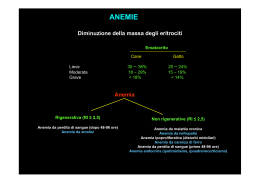
Incontro tra Ematologi di Lodi e Geriatri Sant’Angelo Lodigiano Novembre 2012 G.Nalli La diagnosi iniziale di anemia Dipende dalla vostra attenzione nel valutare un calo, pur lieve, dell’emoglobina (< 13 gr/dl nell’uomo, < 12 gr/dl nella donna) La diagnosi più approfondita richiede un approccio diagnostico sistematico; può infatti trattarsi di anemia come malattia primaria o anemia come segno di malattia generale L’anemia dell’anziano •Il tema che affrontiamo ha grande rilevanza epidemiologica •Ha grande rilevanza clinica •E’ difficile da inquadrare ASH 2010 L’anemia di qualsiasi grado (Hb < 13 nel M e < 12 nella F) è individuata come un significativo fattore indipendente di morbidità, mortalità e fragilità nei pazienti anziani (> 65 anni). Prevalenza circa 10% = o > 85 anni: 20% Stiamo valutando pazienti ambulatoriali 1/3 di questi pazienti hanno deficit nutrizionali 1/3 anemia della malattia infiammatoria 1/3 di origine non chiara Classificazione funzionale delle anemie Alterata produzione Reticolociti ridotti Alterata distruzione (o normali) Reticolociti aumentati Emolisi o emorragia Normocromica microcitica Extraglobulare intraglobulare Es. autoimmune… Coombs positiva Es. sferocitosi Normocitica Aplasia Carenza ferro, talassemia Infiltrazione Infiammazione Insuff renale macrocitica Carenza folati, B12 Farmaci Anemia refrattaria Deficit enzimi Anemia come malattia o segno? Reticolociti > 100.000/mmc: il midollo, nella sua componente eritroide, sta rispondendo in modo appropriato (allo stimolo dell’eritropoietina) al danno ematologico che non coinvolge il midollo. La sede dell’anemia è periferica emorragia acuta emolisi extraglobulare intraglobulare I segni più eclatanti i segni dell’ipovolemia sino allo shock nel caso dell’emorragia acuta (sempre presenti ipotensione e tachicardia, che mancano spesso nell’emorragia cronica) gli stessi segni associati inoltre a cefalea, brividi, (febbre), vomito, dolori addominali… emissione di urine rossastre o nerastre (per emoglobinuria) in caso di emolisi iperacuta Altri segni di emolisi I segni indiretti dell’emolisi come l’ittero, la splenomegalia di grado variabile, la colelitiasi, associati a segni di laboratorio come: l’aumento delle LDH, la diminuzione dell’aptoglobina in caso di emolisi cronica. Un uomo intelligente 64 anni, dirigente aziendale, in viaggio di lavoro nel Salento Ho cominciato a star male d’allora Ho mangiato molte fave: astenia, ittero, vaghi dolori addominali Post hoc propter hoc Post hoc propter hoc Da bambino ho sentito molte volte raccontare: “Gli è venuta l’itterizia per uno spavento” Temevo l’itterizia perché a quei tempi di spaventi se ne provavano tanti (es. il buio, che ha condizionato la mia infanzia….) Mi lascio condizionare dal paziente e doso, con riluttanza, la G6PD Diagnosi: favismo Consegno al paziente un elenco che trovate su mille siti con l’aggiornamento dei farmaci da evitare nel favismo Anemia come segno? Avete aggiunto - il test di Coombs o - le agglutinine a frigore. Anemia emolitica autoimmune Idiopatica (malattia) Secondaria a: - infezione - farmaci - m. neoplastica uomo anziano? - m. linfoproliferativa giovane? - collagenopatia Ieri Ragazza di 24 anni, da 8 giorni febbre In PS per Hb 4 gr/dl Plt < 20.000/mmc GB 18.000 con elementi mielodi immaturi in circolo Diagnosi ipotetica: leucemia acuta esami Midollo: iperplasia eritroblastica Megacariociti numerosi Serie mieloide ipergranulata ma maturante Reticolociti 185.000/mmc Diagnosi: non leucemia acuta Iperemolisi e piastrinopenia da consumo ? Reazione leucemoide da? Anemia della malattia cronica Come orientarsi nella diagnosi (di laboratorio): 2) metabolismo Fe Ferro, transferrina, ferritina, recettore solubile transferrina, epcidina metabolismo del ferro nell’edizione dell’Harrison (2001) (quasi elementare): il Fe si assorbe a livello duodenale, in una quantità media di 1 mg al giorno nel maschio e 1,4 nella femmina Il ferro assorbito viene poi rilasciato nel plasma legato alla molecola di trasporto del ferro, la transferrina. Lo stesso avviene per il ferro rilasciato dai depositi. In sintesi metabolismo Fe sino agli anni 2000 Quantità totale di Fe:. circa 4 gr EPCIDINA e modelli murini •Un eccesso di epcidina a livello fetale fa nascere topini con grave anemia sideropenica, dimostrando che blocca il passaggio placentare del ferro. Successivamente per sopravvivere necessitano di iniezioni di ferro dimostrando che l’eccesso di epcidina blocca l’assorbimento intestinale. •Un topo transgenico privo di epcidina va incontro ad una grave emocromatosi con accumulo di ferro a livello epatico e pancreatico. •Il modello murino ha trovato negli ultimi anni una verifica nell’uomo. Una carenza congenita di epcidina è responsabile di una forma di emocromatosi famigliare Eccesso di epcidina •L’induttore più potente della sintesi di epcidina è l’IL 6, il mediatore più importante del processo infiammatorio. •In modelli murini ma anche in volontari umani la somministrazione di IL6 induce un incremento sino a 7 volte dell’epcidina che molto rapidamente blocca sia l’assorbimento intestinale del ferro che il riciclo del ferro derivante dal catabolismo eritrocitario: si viene quindi a determinare una rapida caduta del compartimento del ferro plasmatico e del suo apporto all’eritrone ed un incarceramento del ferro a livello macrofagico: si spiega così uno stato di anemia con bassa saturazione della transferrina ed un aumento della ferritina (che peraltro è anche un indice di fase acuta) in corso di processi infettivi o infiammatori (quella che va sotto il nome di anemia della malattia infiammatoria). We postulate that the tendency toward a proinflammatory state in the elderly population predisposes them to accentuated debility….. We further propose that genetic factors determining the level of proinflammatory markers contribute to the likelihood of developing features of frailty in response to other stressors. Anemia and inflammation are strongly associated with, and may contribute to, the development of “frailty,” a poorly defined syndrome of the elderly population associated with weight loss, impaired mobility, generalized weakness…. Some studies have suggested that elevated proinflammatory markers are associated with development of frailty. Furthermore, anemia is associated with an increase in nearly all markers of frailty in elderly populations, suggesting that there may be a link between the pathogenesis of the two syndromes • L’organismo ha la necessità di mantenere scorte di ferro per necessità fisiologiche o critiche • difendersi dal ferro libero, in eccesso, altamente tossico • Il ferro viene accumulato in una molecola che forma un guscio entro il quale accumulare il ferro ed impedirne la tossicità (ferritina) •Esiste a livello di quasi tutte le superfici cellulari ed in particolare dell’eritroblasto midollare un recettore della transferrina che lega il complesso ferrotransferrina, per poi internalizzare il Fe, liberarlo dalla transferrina (ed incorporarlo nell’eme a livello eritroblastico).. La maggior parte del ferro necessario per l’eritropoiesi viene dal riciclaggio del ferro contenuto nei globuli rossi senescenti, fagocitati a livello del reticolo endotelio. In caso di carenza di Fe > il recettore solubile della transferrina Anemia sideropenica: alterazioni di laboratorio Segni precoci Ferritina < 40 μg/l Ferro siero < 50 μg/dl saturatione Transferrina < 15 % Total iron binding capac. > 450 μg/dl Recettore solubile transferrina alto GR < 4 × 106/ mm3 Red cell distribution width (RDW) > 14.5 % MCV (volume corpuscolare medio) < 80 Segni fl tardivi Emoglobina < 13 g/dl maschio < 12 g/dl femmina fertile Anemia dell’infiammazione Ferro basso Transferrina bassa Saturazione transferrina > 20% TIBC < 350 Ferritina alta (comunque > 100) I parametri di laboratorio nell’approccio iniziale alla diagnosi di anemia Emocromo e reticolociti Metabolismo del ferro Dosaggio ac folico e vit B12 Bilirubina LDH Terapia dell’anemia sideropenica dell’adulto Am J Med. 2008 November ; 121(11): 943–948 E’ di fondamentale importanza il rendersi conto che i depositi di Fe siano esauriti prima che insorga anemia sideropenica Quindi i parametri di laboratorio che correlano con la deplezione diFe precedono l’insorgere di anemia. 3 anni di dieta povera di fe portano ad una deplezione dei depositi di Fe (normalmente 1 gr di Fe in forma di ferritina) The loss of iron stores is reflected in the blood by a reduction in ferritin and reduced levels of iron bound to transferrin. As the iron stores become more severely depleted, availability of transferrin-bound iron to the erythroid precursor causes reduced heme and hemoglobin production. This is reflected in a drop in the red blood cell count and mean corpuscular volume. The red cell count usually becomes abnormal before the mean corpuscular volume, which remains within the normal range until the hemoglobin reaches about 10 g/dl. Ferro alimentare E’ importante un counseling sulla dieta, ma… La dieta da sola è in genere insufficiente a rimpiazzare le scorte di Fe nella quasi totalità dei pazienti con anemia sideropenica esempi Le cpr di Fe: Meglio assumerlo lontano dai pasti o prima di andare a letto per evitare l’effetto alcalinizzante dei pasti e per favorire l’effetto del picco di acidità gastrica nella notte Assuming that 10% of the iron is absorbed, the hemoglobin concentration may fully correct after 4 weeks in patients with moderate, uncomplicated iron deficiency (about 500–800 mg of iron, enough for 500 to 800 mL of packed red blood cells, or enough to raise the whole blood hemoglobin 2–3 g/dL) Dosi stimate di ferro per correggere anemia In one study in 111 hospitalized patients ≥75 years of age who were found to have iron deficiency anemia, 92 percent of whom underwent endoscopy and 82 percent of whom underwent colonoscopy, 68 percent were found to have a bleeding source, and 11 percent had synchronous lesions [the 43 patients found to have a colorectal source of bleeding, 31 (72 percent) had colon cancer; of the 44 patient found to have an upper gastrointestinal source of bleeding, 6 (14 percent) had a malignancy. A proposito di Vit B12 Il fabbisogno quotidiano è di 3-7 microgrammi I depositi, prevalentemente epatici, raggiungono i 5 mg Quindi sono necessari anni per arrivare ad esaurimento dei depositi pur con una assenza di assorbimento quotidiano Sintomi neurologici da deficit di Vit B12 Lesione tipica: degenerazione combinata subacuta di cordoni posteriori e laterali del midollo spinale This lesion, specific for Cbl deficiency, is due to a defect in myelin formation. The neuropathy is symmetrical and affects the legs more than the arms. It Neurologic problems, when present, consist of the classic picture of subacute begins with paresthesias and ataxia associated with loss of vibration and combined degeneration of the dorsal (posterior) and lateral spinal columns [ position sense, and can progress to severe weakness, spasticity, clonus, paraplegia, and even fecal and urinary incontinence Deficit di B12 … neurologic abnormalities….include cerebellar ataxia, ……central nervous system symptoms including memory loss, irritability, dementia, and extrapyramidal signs. importante in clinica: Non tutti i pazienti con deficit di B12 e quadro neurologico sono anemici ed hanno la macrocitosi; Iin oltre il 10% dei casi Hb e MCV sono normali i Terapia sostitutiva con B12 Vit B12 parenterale — L’anemia perniciosa deve essere curata con B12 IM in dosi di 1000 microgrammi al giorno per la prima settimana, seguita da 1 mg alla settimana per 4 settimane e in seguito 1 mg al mese per la vita Vitamin B12 and/or folate deficiency should be suspected in patients with one or more of the following clinical or laboratory findings O m acrocytic red blood cells w ith or w ithout anem ia Tthe presence of hypersegm ented neutrophils on the peripheral blood sm ear (ie, >5 percent of neutrophils w ith ≥5 lobes or ≥1 percent of neutrophils w ith ≥6 lobes) Pancytopenia (ie, the com bination of anem ia, throm bocytopenia, and neutropenia) of uncertain cause Unexplained neurologic signs and sym ptom s, especially dem entia or w eakness, sensory ataxia, and paresthesias Special populations, such as older adults, alcoholics, and patients w ith m alnutrition are at high risk for the developm ent of folic and/ or Cbl deficiency. Patients w ho have undergone bariatric surgery Anemia perniciosa Ricordare che AP è malattia autoimmune. Anti F intrinseco Anti cellule parietali gastriche Funzione tiroidea ed anti tireoperossidasi e tireoglobulina Se indicato cortisolo e ACTH vitiligine Sindromi mielodisplastiche gruppo di disordini primitivi del midollo osseo che interessano tipicamente, ma non esclusivamente, soggetti anziani e coinvolgono la cellula staminale emopoietica (cioè la cellula midollare progenitrice da cui derivano le cellule che circolano nel sangue periferico: globuli bianchi, globuli rossi e piastrine). Nelle sindromi mielodisplastiche, la cellula staminale emopoietica matura in modo disordinato (alterazioni morfologiche: dismielopoiesi) e inefficace (emopoiesi inefficace). Le sindromi mielodisplastiche sono malattie clonali in quanto gli elementi delle tre serie maturative midollari derivano tutte dalla stessa cellula progenitrice la quale ha acquisito una alterazione genetica ad impronta displastica. Il difetto maturativo midollare determina tipicamente anemia (refrattaria al trattamento), neutropenia, e piastrinopenia persistenti (o varie combinazione delle stesse). La storia naturale della malattia, in assenza di trattamento, è caratterizzata da un progressivo aggravamento dell'emopoiesi inefficace e da un rischio di evoluzione in leucemia acuta mieloide. In Europa, l'incidenza complessiva è di circa 8 casi ogni 100.000 persone per anno. Nei soggetti di età inferiore a 30 anni è di 1 caso ogni 100.000 persone per anno, mentre oltre i 70 anni di età l'incidenza è di 35 casi ogni 100.000 persone per anno. Quando richiedere visita ematologica In presenza di citopenia uni-bi-o trilineare (Hb < 13 gr/dl nel M e 12 gr/dl nelle F) GB < 3000/mmc e N < 2000/mmc) Piastrine < 100.000/mmc Domande e risposte Dottore, mi dica la verità! Ho un tumore? Perché ho letto che ho una malattia clonale. La risposta non può essere di paternalismo consolatorio Note di storia recente sulle CM Nel 1960 Waldenstrom descrisse una serie di pazienti che presentavano una banda stretta in zona gamma all’elettroforesi e nei quali escludeva mieloma multiplo o m. di Waldenstrom. Li definì: Gammopatie monoclonali benigne Basandosi sull’osservazione che individui genericamente sani con CM avevano rischio eccessivo di sviluppare mieloma multiplo, m. di Waldenstrom, amilodosi…. Robert Kyle nel 1978 coniò il termine usato in tutto il mondo di “monoclonal gammopathy of undetermined significance” (MGUS) Schematismo da bigino. Chi vede la CM: Voi Ematologo… per molte ragioni Dermatologo……..vasculiti Neurologo……neuropatie periferiche ..MAG Oncologo…..d’accompagnamento Epatologo…. Crioglobuline Nefrologo……IR MM e amilodosi Reumatologo.. Schnitzler syndrome (orticaria, IgM, artralgie) …………………. International Myeloma Working Group 2010 Tutti e 3 i criteri devono essere rispettati: 1) La CM < 3 g/dL 2) plasmacellule midollari clonali < 10% 3) assenza di danno di organo come ipercalcemia, insufficienza renale, anemia, lesioni ossee attribuibili a proliferazione plasmacellulare Etiopatogenesi 1)Sembra coinvolto l’uso di pesticidi 2) In grandi studi di popolazione risulta che i parenti di primo grado di MM hanno rischio doppio di sviluppare MM 3) I parenti di primo grado di MGUS hanno rischio triplo di sviluppare sia MGUS che MM Epidemiologia 3,5% della popolazione > 50 anni è portatrice di MGUS Due tipi di MGUS Circa il 15-20% secernono IgM ed hanno un fenotipo midollare linfoide o linfo-plasmocitoide. La loro evoluzione è verso la m. di Waldenstrom, o altri linfomi maligni 80% secernono IgG>IgA> catene leggere o IgD ed IgE ed evolvono verso il MM Da MGUS a smoldering MM Definizione: International Myeloma Working Group 2010 CM IgG o IgA > 3 gr/dl Plasmacellule clonali > 10% In assenza di danni d’organo In questi casi la presenza alla RMN della colonna di lesioni occulte è predittiva di rapida evoluzione verso MM Il dilemma clinico: da MGUS a neoplasie linfoproliferative Follow-up a lungo termine mostra che MGUS ha un rischio annuale di evolvere in neoplasia di 1% Smoldering MM ha un rischio annuale del 10% Mayo Clinic proceedings March 2011; 86 (3) Recentemente è stata definita una nuova entità, : MGUS a catene leggere; it rappresenta il precursore del MM a catene leggere, che rappresenta quasi il 20% di tutti i casi di mieloma. Esiste pertanto anche l’equivalente idiopathic Bence Jones proteinuria American Society of Hematology 2010 In breve 2010 IMWG guidelines suggeriscono che “low-risk MGUS” (CM < 1,5 gr/dl, isotipo IgG, normale rapporto di catene leggere libere) ripetano l’elettroforesi ogni 6 mesi e se stabili rivalutati ogni 2 anni For “intermediate-/high-risk MGUS” (CM > 1.5 g/dL, isotipo IgA or IgM, o abnorme FLC ratio), prevede BOM e aspirato midollare baseline ripetizione dell’elettroforesi delle sieroproteine ed esami ematici ogni 6 mesi Test ematici routinari per MGUS e SMM Elettroforesi sieroproteine e identificazione e quantificazione della CM Emocromo Creatinina Calcio IF proteine urinarie 2 studi recenti (USA) in MM Pazienti con MM che per varie ragioni avevano campioni di sangue congelati TUTTI I CASI di MM erano stati preceduti da MGUS Infine Mancano markers citogenetici, molecolari, immunofnotipici per predire nel singolo paziente l’evoluzione da MGUS a SMM a MM Am.Soc.Hem.2010 L a leucemia linfatica cronica (LLC) rimane una malattia enigmatica. Benchè la prima descrizione clinica sia stata pubblicata 150 anni fa, l’etiologia della LLC è ancora ignota, così come la cellula dalla quale origina. LLC rimane incurabile al di fuori del trapianto allogenico Using the Biology of Chronic Lymphocytic Leukemia to Choose Treatment ASH 2011 The notion that the disease was characterized by the accumulation of quiescent terminally differentiated “memory” lymphocytes has now been shattered The peripheral blood CLL cells are only part of the life cycle of the CLL cell. From the peripheral blood, the cells migrate into the tissue compartment (BM, lymph nodes, and spleen), where they receive a proliferative stimulus. It appears that this is at least in part driven by antigen that is presumably presented to the CLL cells by components of the The cell then migrates from the proliferating centers back to the peripheral blood, where the cell stops dividing. The CLL cell then either undergoes apoptosis or continues to cycle back into the proliferation centers so that it is continuously cycling between a proliferative state in the tissue compartment and a quiescent state in the periphery. The BCR of CLL cells is not random; different patients have socalled stereotyped receptors, meaning that there is a highly selective use of a small number of immunoglobulin genes. In some cases, identical immunoglobulin receptors are observed in different patients with CLL and this could not happen by chance.4 Therefore, these receptors are preferentially used in CLL and must have been positively selected for due to their specificity to a particular antigen. Therefore, if there are a limited number of antigens responsible for the development of CLL, then by “chance” different clones might be selected for due to their antigen specificity. This receptor/antigen interaction almost certainly drives the survival and/or proliferation of the CLL cell. Up to 30% of patients have stereotyped receptors, and the antigen is probably an autoantigen such as the myosin heavy chain or a common alloantigen …demonstrated that the “birth rate” (the proportion of CLL cells produced each day) in CLL is very high, with up to 2% of CLL cells being “born” each day in some patients. The immunophenotype suggesting proliferation of CLL cells is uncommonly expressed by the CLL cells in the peripheral blood, and it seems that the small number of CLL cells in the peripheral blood with a proliferative phenotype probably represents the cells exiting the tissue compartment before they become quiescent again Response rates in CLL have improved significantly with the use of combination immunochemotherapy. The combination of fludarabine with cyclophosphamide and rituximab (FCR) is associated with higher complete remission rates (between 44% and 72%), with approximately 90% of patients achieving a response and improved overall survival (OS) compared with fludarabine plus cyclophosphamide (FC).10 However, even with FCR given as the initial therapy, 10% of patients fail to respond and therefore have Il nuovo standard di terapia Response rates in CLL have improved significantly with the use of combination immunochemotherapy. The combination of fludarabine with cyclophosphamide and rituximab (FCR) is associated with higher complete remission rates (between 44% and 72%), with approximately 90% of patients achieving a response epidemiologia LLC è la forma di leucemia più diffusa soprattutto nei paesi occidentali con maggiore diffusione nel sesso maschile M/F: 2/1. L'incidenza è calcolata 5-15 su 100.000 persone. è definita "la leucemia dell'anziano". Mediana di età alla diagnosi 65 anni 15% < 50 anni Fattori di rischio Il più importante fattore di rischio è la famigliarità Tra i pazienti con una nuova diagnosi di LLC 8-10% hanno una storia familiare di malattia Famigliarità LLC Un recente studio condotto su 101 nuovi casi familiari ha identificato il cromosoma 2q21 come associato alla trasmissione. Tuttavia non si è ancora riusciti ad identificare a questo locus il gene causativo Fattori genetici acquisiti La trisomia 12 (16% dei casi), la delezione 17p13 (17%) e la delezione 11q23 (18%) hanno significato prognostico sfavorevole. La delezione 13q14 (55%) è invece favorevole (sopravvivenza simile a quella dei pazienti con cariotipo normale). Ad oggi la delezione 17p e la delezione11q appaiono i più potenti fattori prognostici,in quanto influenzano in modo statisticamente significativo sia l’ottenimento della risposta alla terapia che la durata della risposta e la sopravvivenza globale stato mutazionale dei geni IgVH (regione variabile delle catene pesanti delle immunoglobuline). In base allo stato mutazionale si distinguono oggi due sottotipi di Leucemia Linfatica Cronica-B: una frazione di casi (50% circa) con IgVH in stato non mutato, cioè senza mutazioni somatiche, ed una frazione con mutazioni somatiche (stato mutato). La situazione non mutata si associa ad una malattia più estesa (stadio più avanzato) e comporta una prognosi più sfavorevole. L'impatto prognostico negativo dello stato non mutato è evidente anche nei pazienti in stadio clinico iniziale. Altri marcatori biologici L'espressione della proteina ZAP70 da parte dei linfociti. I linfociti B normali non esprimono questa proteina. All’analisi in citofluorimetria a flusso circa il 40% dei pazienti con leucemia linfatica cronica esprime invece ZAP70. L’espressione correla con lo stato IgVH non mutato e dal punto di vista prognostico ha un significato sfavorevole. L'espressione dell'antigene CD38 da parte dei linfociti. In presenza di una percentuale di linfociti CD38-positivi superiore al 30% l'andamento clinico è sfavorevole. Criteri diagnostici (National Cancer Institute Working Group (NCI-WG). Linfociti nel sangue periferico pari o superiori a 5.000/µL. tipizzazione immunofenotipica: i linfociti patologici sono positivi per gli antigeni CD5, CD19, CD20, CD23, debolmente positivi per CD22, generalmente negativi per FMC7 e CD79b; esprimono immunoglobuline di superficie (SmIg) a bassa densità con restrizione monoclonale, k o λ, della catena leggera. All'esame del midollo osseo l'infiltrato linfatico deve essere pari o superiore al 30%. I fattori prognostici La variabilità del decorso clinico rende necessaria lo studio alla diagnosi dei parametri clinici e biologici di significato prognostico. Ciò serve per adeguare il trattamento alla severità della malattia. Il primo parametro prognostico è lo stadio clinico. Vi sono due differenti sistemi di stadiazione: il sistema di Rai (1975) ed il sistema di Binet (1977) Stadiazione della LLC La Classificazione RAI modificata : a) Pazienti a basso rischio: solo con linfocitosi nel sangue periferico e nel midollo osseo (stadio 0); b) Pazienti a rischio intermedio: con linfocitosi e linfoadenopatia (stadio I) e/o epatosplenomegalia (stadio II); c) Pazienti ad alto rischio: con linfocitosi e anemia (Hb<11g/dL) (stadio III) e/o trombocitopenia (Plt <100,000/mm3) (stadio IV) Stadiazione LLC La Classificazione di Binet si basa sul numero di aree linfonodali interessate dalla malattia e il grado di anemia e piastrinopenia: a) Stadio A: include i pazienti con Hb >10g/dL, PLt>100,000/mm3 e fino a 2 aree linfonodali interessate; b) Stadio B: include i pazienti con Hb >10g/dL, PLt>100,000/mm3 e più di 2 aree linfonodali interessate; c) Stadio C: include i pazienti con Hb <10g/dL e/o PLt<100,000/mm3 a prescindere dal numero di aree linfonodali interessate. sopravvivenza Da oltre 10 anni per gli stadi iniziali Ai 2 anni per gli stadi avanzati Altri criteri clinici di prognosi tempo di raddoppio dei linfociti inferiore a 6 mesi o aumento della linfocitosi superiore al 50% in meno di due mesi una morfologia dei linfociti "variante" valori elevati di beta2-microglobulina e di LDH (indici di massa di malattia e di rapida proliferazione cellulare) un assetto immunofenotipico non tipico Grande novità Linfocitosi Monoclonale delle Cellule B (LMB) le cellule B monoclonali CD5 positive, che manifestano il fenotipo distintivo della LLC, possono essere identificate tramite citometria di flusso nel 3.5% di pazienti, altrimenti ritenuti individui sani con parametri ematologici nella norma Questo fattore è correlato all’età: é, infatti, presente in più del 7% degli individui con età superiore a 70 anni, ed è più frequente nei familiari di primo grado dei pazienti con LLC Questo disordine è denominato linfocitosi monoclonale delle cellule B (analogo ad MGUS rispetto al MM?) In breve: i nuovi criteri Utilizzare la conta dei B-linfociti piuttosto che la conta dei linfociti come base per la diagnosi di LLC La soglia è definita come B-cell di 5.0 x109/L per distinguere LLC da monoclonal B-cell linfocitosi Differentiating chronic lymphocytic leukemia from monoclonal Blymphocytosis according to clinical outcome: on behalf of the GIMEMA chronic lymphoproliferative diseases working grouphaematologica | 2011; 96(2) Il lavoro retrospettivo del GIMEMA identifica un numero di linfociti di 11.5 x109/L e di cellule B di 10.0 x109/L come la miglior soglia per predire quali pazienti richiederanno terapia o avranno malattia stabile. In chi ha < 10 x 109/L B-cell il tasso di progressione verso una LLC che richiederà terapia è di 2,3%per anno. Il tasso è doppio per chi ha > 10 x 109/l Per questa categoria di pazienti possiamo utilizzare termini meno pesanti di leucemia linfatica cronica e gravidi di conseguenze psicologiche per un paziente che ha poche probabilità e remote di evolvere verso una neoplasia che richiederà terapia. Iperferritinemia Iperferritinemie Ferritina > 300 µg/L in maschi > 200 µg/L in femmine Cause di iperferritinemia Aumentata sintesi (per fattori genetici o acquisiti, con o senza sovraccarico di Fe) Aumentata liberazione di ferritina conseguente a danno cellulare Table 1. Mechanism of hyperferritinemia in selected disorders Aumentata sintesi/secrezione: ethanol ingestion; malignancy (malignant histiocytosis; carcinomas of lung, breast, ovary, and kidney; lymphoma;liposarcoma); Gaucher disease; reactive histiocytosis; hereditary hyperferritinemia-cataract syndrome Aumentata liberazione da danno cellulare:: hepatic steatosis and steatohepatitis; chronic viral hepatitis; massive liver necrosis due to sepsis, acute hepatitis, or toxic injury; autoimmune disorders; acute and chronic infections; acute myocardial infarction; splenic infarct Aumentata sintesi da sovraccarico di Fe: HFE and other types of hemochromatosis; heritable and acquired anemias associated with ineffective erythropoiesis; increased iron absorption from supplemental iron or ingestion of traditional beer (sub-Saharan African Natives); transfusion iron overload; parenteral iron overload; aceruloplasminemia Importante il setting di presentazione . Per es. è frequnte una iperferritinemia < 1000 con normale saturazione della transferrina nel contesto di assunzione quotidiana di alcol o in un paziente obeso Usare la saturazione della transferrina (ST) per interpretare livelli elevati di ferritina Il loro screening di massa 64,230 participanti ST > 45 % in femmine ST > 50% in maschi Sensibilità del 75 % Specificità 95 % nel rivelare C282Y omozigoti Riepilogo di alcuni concetti All’interno della popolazione con Carcinoma Colon Retto quanti hanno SOF positivo? (definisce la sensibilità del test) 23% per neoplasia avanzata 11% per qualsiasi tumore N Engl J Med 23 Agosto 2001 la specificità del test Tra coloro che non hanno tumore quanti hanno un test negativo? Si deve partire da un lavoro di screening che comporti la colonscopia per essere certi di chi non ha il tumore. 93% Potere predittivo potere predittivo positivo Tra coloro che hanno SOF positivo quanti hanno il tumore? Esprime nel contempo un valore di quanti sono i falsi positivi (tanti) 39,7% potere predittivo negativo Tra coloro che hanno SOF negativo quanti non hanno il tumore? 63% se si considera l’assenza di qualsiasi tumore. (E’ correlato alla % di falsi negativi) Un caso particolare A volte una epatopatia avanzata porta ad un difetto di sintesi di transferrina e dunque ad un aumento della ST senza sovraccarico di Fe Rapporto tra ST e concentrazione epatica di Fe Pazienti divisi in 2 gruppi: • HFE C282Y omozigoti • Altri casi: hemochromatosis, non-HFE iron overload, juvenile hemochromatosis, alcoholic liver disease, fatty liver, hepatitis C, and hepatitis B. Figure 1. The relationship between transferrin saturation and hepatic iron overload in C282Y homozygotes (●) and non-homozygotes (○). 1) ST ha un potere predittivo positivo migliore in C282Y homozygotes che in nonhomozygotes 2) Alcuni pazienti con ST normale e ferritina elevata hanno un sovraccarico di Fe (documentato da biopsia epatica) Valutazione di pazienti con ferritina elevata e ST normale In questo setting è utile valutare: AST, ALT, alkaline phosphatase, gammaglutamyl transpeptidase (GGT), HBsAg, and anti-HCV. Valutare l’assunzione di alcol Eseguire una ecografia epatica Se un paziente ha elevate concentrazioni di enzimi epatici il quesito è: I livelli di ferritina elevati conseguono alla necrosi epatocitaria? O, meno frequentemente, il sovraccarico di Fe determina il danno epatico? Obesity or fatty liver detected by ultrasonography suggest the presence of steatohepatitis. Chronic alcohol consumption may be suspected based on the clinical history and an elevated level of GGT. • In pazienti con epatite cronica da HBv ed HCV i livelli di ferritina sono spesso elevati ma in genere non indicano un sovraccarico di Fe. • I pazienti con sindrome dismetabolica e resistenza insulinica ( spesso associati ad obesità, ipertensione arteriosa che pure non definiscono la s. dismetabolica) hanno in genere livelli di ferritina elevata e mormale ST. • La biopsia epatica in questi pazienti in genere dimostra la presenza di Fe nelle cellule di Kuppfer e steatosi.. •Dopo salasso terapia la resistenza insulinica può diminuire . Hereditary hyperferritinemia-cataract syndrome (HHCS) is caused by heterogeneous mutations in the iron-responsive element (IRE) of L-ferritin that reduce the binding affinity of ironresponsive proteins (IRPs) to IREs and thereby diminish the negative control of L-ferritin (but not H-ferritin) synthesis. This leads to the constitutive upregulation of ferritin L-chain synthesis characteristic of HHCS. Serum ferritin concentrations are elevated and relatively constant; TS is normal. Aceruloplasminemia is a rare genetic syndrome due to mutations in the CP gene that encodes ceruloplasmin. This condition is associated with normal TS, hyperferritinemia, and iron overload; retinal abnormalities; and prominent neurological problems. Anemia is uncommon. Un tentativo di osservazione clinica • In molti casi l’osservazione clinica e il follow-up possono essere più appropriate di un’indagine invasiva (biopsia epatica) o un tentativo con salasso terapia. • Può essere utile osservare se i livelli di ferritina sono stabili nel tempo o tendono ad aumentare. Questo accade in genere nell’emocromatosi HFE correlata e comunque nei sovraccarichi di Fe. • In pazienti con steatosi epatica o steato epatite o con assunzione di alcol i livelli di ferritina fluttuano grandemente. Circa il 90% dei pazienti con iperferritinemia incontrati nella pratica medica routinaria NON hanno sovraccarico di Fe Un tentativo con salasso terapia • E’ il metodo più remoto tra i tanti pensati. • Si è accertato che un paziente con sovraccarico di Fe possa tollerare sino a 20 salassi settimanali senza sviluppare anemia • Un salasso di 500 ml rimuove circa 0,25 gr di Fe • La salasso terapia permette pertanto di quantificare il Fe rimosso L’analisi mutazionale per rivelare òle più comuni mutazioni HFE sono ormai disponibili in commercio e questo test è molto utile per valutare la popolazione bianca europea con iperferritinemia
Scaricare