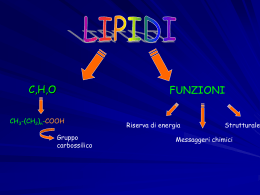
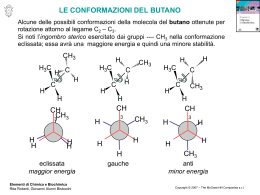
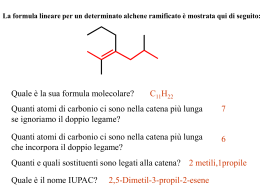
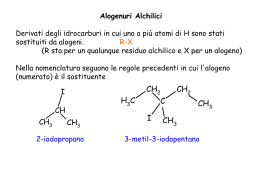
CHIMICA DEI MATERIALI II SECONDO MODULO Prof. Piero Sozzani Università Milano-Bicocca. Dipartimento di Scienza dei Materiali 25 aprile 2006 Dispensa realizzata da Vettigli Marco in riferimento al corso di Chimica dei Materiali II, secondo modulo tenuto dal Prof. Piero Sozzani presso la facoltà di Scienza dei Materiali, Università Milano Bicocca. Per la realizzazione si sono consultati gli appunti di lezione ed i seguenti testi, che restano comunque di riferimento per eventuali approfondimenti e/o delucidazioni: G. Odian P.C. Hiemens Fessenden & Fessenden - Principles of polymerization - Polymer chemistry - Chimica organica INDICE 1. SCIENZA DELLE GRANDI MOLECOLE................................................ 1 1.1. Pesi molecolari medi .......................................................................... 2 1.2. Struttura delle macromolecole .......................................................... 3 1.2.1. Costituzione dei polimeri.......................................................... 3 1.2.2. Configurazione dei polimeri ..................................................... 4 1.3. Chiralità e attività ottica.................................................................... 11 2. POLIMERIZZAZIONE A STADI .............................................................. 12 2.1. Poliammidi......................................................................................... 16 2.2. Polietilentereftalato........................................................................... 17 2.3. Policarbonati ..................................................................................... 18 2.4. Poliuretani ......................................................................................... 20 2.5. Polimerizzazione a stadi polifunzionale.......................................... 20 2.6. Resine fenoliche................................................................................ 22 2.7. Resine amminiche............................................................................. 24 3. POLIMERIZZAZIONE A CATENA ......................................................... 26 3.1. Polimerizzazione radicalica.............................................................. 26 3.1.1. Principi base .......................................................................... 26 3.1.2. Inibizione e ritardo ................................................................. 30 3.1.3. Trasferimento ........................................................................ 31 3.1.4. Cinetica.................................................................................. 32 3.2. Polimerizzazione cationica............................................................... 37 3.3. Polimerizzazione anionica................................................................ 39 3.3.1. Principi base .......................................................................... 39 3.3.2. Copolimeri a blocchi .............................................................. 41 4. POLIMERIZZAZIONE DI COORDINAZIONE ...................................... 43 4.1. Catalizzatori Ziegler-Natta ................................................................ 44 4.1.1. Sviluppo storico ..................................................................... 44 4.1.2. Meccanismo della propagazione stereoselettiva ................... 45 4.1.3. Cinetica.................................................................................. 47 4.2. Iniziatori metallocenici...................................................................... 48 4.3. Analisi della stereoregolarità ........................................................... 51 1. SCIENZA DELLE GRANDI MOLECOLE Una tipologia di materiali che più di ogni altra ha cambiato il vivere quotidiano è la classe dei polimeri. I polimeri vengono solitamente indicati come delle macromolecole, ossia molecole di grandi dimensioni. Bisogna in primo luogo distinguere il concetto di polimero da quello di macromolecola; il primo si definisce come un insieme (dal greco poly, più e meros, parte) di monomeri (dal greco mono, singola e meros, parte) legati tra loro mediante reazioni chimiche, mentre il secondo termine possiede un significato ben più ampio e raggruppa un insieme di molecole complesse come gli stessi polimeri, le proteine, gli enzimi. Il termine unità ripetentesi, che in generale può non concidere con il monomero, indica l’unità base che si ripete all’interno del polimero. Prendiamo come esempio il polietilene, un polimero lineare formato dall’addizione di n unità etileniche. H H C C H H etilene H2 C C H2 n polietilene H2 C n Il monomero è l’etilene mentre l’unità ripetentesi è il metilene poichè esso rappresenta il frammento più piccolo che si ripete all’interno della catena. Sorge spontaneo chiedersi quando si possa iniziare a parlare di polimeri. Non è infatti possibile porre un limite di unità monomeriche oltre il quale la macromolecola possa venir distinta come polimero o molecola semplice. Nel caso del polietilene ad esempio, se n = 2 la molecola che si vuole rappresentare non è altro che il butano, se n = 3 l’esano, n = 4 l’ottano. Se consideriamo il punto di fusione di questi composti, il butano fonde a –138 °C, l’esano a –95 °C, l’ottano a –57°C. Così come il punto di fusione, anche le altre proprietà fisiche di questi composti sono decisamente diverse tra di loro (ad esempio lo stato di aggregazione, il butano a temperatura ambiente è ancora in forma gassosa mentre esano e ottano sono liquidi). Succede però che, al crescere del numero di unità monometriche, le proprietà del composto si stabilizzano. E’ proprio questa la discriminante secondo la quale un composto può essere distinto come polimero od oligomero. Il primo infatti posside proprietà fisiche simili ai suoi omologhi con n leggermente maggiore o minore mentre il secondo possiede poche unità monomeriche e proprietà sensibilmente diverse da quelle del polimero. La questione può essere meglio compresa osservando un diagramma Tfus vs n dove si nota la tendenza asintotica alla temperatura di fusione del polietilene (120 °C) per n Æ ∞. Le proprietà meccaniche (resistenza a trazione, tenacità, ecc..) variano invece sensibilmente tra due campioni di uno stesso tipo di polimero al variare di n. Prendendo sempre ad esempio il polietilene, campioni con n compresi tra 1000 e 5000 si presentano come plastiche rigide e vengono utilizzati nella fabbricazione di taniche e contenitori mentre campioni con n compresi tra 300000 e 600000 presentano proprietà meccaniche superiori e vengono utilizzati nella fabbricazione di giubbotti antiproiettile. Inoltre, anche lo stato di aggregazione delle catene polimeriche gioca un ruolo fondamentale sulle proprietà meccaniche del campione; un polimero cristallino è ben diverso da un polimero amorfo. 1 1.1. Pesi molecolari medi Per caratterizzare un polimero occorre definire alcune grandezze. Bisogna premettere che un polimero reale non si presenta mai come un insieme di catene polimeriche di egual lunghezza, bensì come una miscela di catene più o meno lunghe. Davanti ad una miscela è quindi opportuno specificare alcune grandezze medie che ne definiscono le proprietà complessive. La prima grandezza da introdurre è sicuramente il peso molecolare medio numerale (Mn), definito come media dei pesi delle catene polimeriche. ∑N ⋅M ≡ ∑N i Mn i i i i L’indice i varia tra 0 e +∞ e indica la specie polimerica mentre Ni è il numero di catene di peso molecolare Mi. Il peso molecolare medio ponderale (Mw) è invece definito come: Mw ∑N ⋅M ≡ ∑N ⋅M i 2 i i i i i Questa grandezza è maggiormente influenzata dalla frazione con pesi molecolari più alti e risulta sempre maggiore (o nel caso ideale di un polimero monodisperso uguale) al peso molecolare medio numerale. L’indice di polidispersità (D) definito come: D≡ Mw Mn è una quantità sempre maggiore di (o, nel caso ideale di una miscela polimerica monodispersa, uguale a) uno e indica quanto ampio sia il range di pesi molecolari presenti nella miscela. Un’altra grandezza particolarmente importante è il grado di polimerizzazione (DP), definito come numero medio (ponderale o numerale) di unità monomeriche all’interno delle catene di una miscela polimerica. ∑N ⋅M M ≡ = PM PM ⋅ ∑ N i DP n ≡ x n n i i DP w ≡ x w i ∑i Ni ⋅ Mi2 Mw ≡ = PM PM ⋅ ∑ N i ⋅ Mi i i Esistono altre grandezze che caratterizzano una miscela polimerica, come il peso molecolare medio centrifugo (Mz) o il peso molecolare medio viscosimetrico (Mv), così definite: Mz ∑N ≡ ∑N i i i ⋅ Mi3 i ⋅ Mi2 ⎛ ∑ N i ⋅ Mia +1 ⎜ Mv ≡ ⎜ i ⎜ ∑ N i ⋅ Mi ⎝ i ⎞ ⎟ ⎟ ⎟ ⎠ 1/a con 0 < a < 1 Tuttavia queste grandezze sono piuttosto particolari e servono per caratterizzare proprietà specifiche della miscela (Mv per caratterizzare le proprietà viscose mentre Mz per caratterizzarne le proprietà di sedimentazione). Una tipica distribuzione dei pesi molecolari di una miscela polimerica reale è riportata in figura, nella quale si può osservare la collocazione delle quantità medie sopra definite. 2 1.2. Struttura delle macromolecole Quando si deve studiare un particolare tipo di polimero bisogna tenere a mente due livelli ben distinti di analisi: la struttura chimica del polimero e la sua struttura supramolecolare. Nell’ambito della struttura chimica bisogna specificare costituzione e configurazione. La costituzione del polimero è fondamentalmente la sua chimica, gli elementi da cui è formato, mentre la configurazione riguarda la stereochimica del prodotto, come ad esempio la presenza di isomeria geometrica o di centri stereogenici. La struttura supramolecolare del polimero caratterizza invece il materiale bulk in termini di impacchettamento cristallino o della disposizione delle catene polimeriche nello spazio (detta anche conformazione). Il primo livello strutturale può essere manipolato durante la progettazione e lo sviluppo della sintesi mentre il secondo livello può essere modificato giocando sulle modalità di preparazione del materiale. Sebbene la maggior parte delle proprietà fisiche (solubilità, punto di fusione, ecc..) del prodotto finale sia determinato dalla chimica del polimero, si possono avere consistenti deviazioni giocando sulla chimica supramolecolare del bulk. In questa parte del corso si esaminerà in particolare la struttura chimica dei polimeri. 1.2.1. Costituzione dei polimeri La costituzione di un polimero va ben oltre lo specificare gli elementi chimici di cui è composto. Ad esempio, supponiamo di studiare un particolare polimero; una prima analisi identifica la composizione bruta dell’unità ripetentesi come C2H4O. La determinazione della composizione dell’unità base non ci permette di discriminare il campione tra almeno tre diversi e distinti tipi di polimero: O CH3 CH O poliacetaldeide da polietilenossido da n H2 C C H2 O n C H3C H acetaldeide O H2C CH2 epossido O OH CH C H n O polivinilalcol dall’idrolisi di CH C C H OCH3 n polivinilestere 3 La presenza di diversi composti con medesima composizione è detta isomeria costituzionale. Il precedente esempio mostra che per definire la costituzione di un polimero non è solamente necessario specificare la natura degli atomi che lo compongono, ma anche il suo scheletro molecolare. Per descrivere compiutamente la costituzione di un polimero bisogna dunque studiarne il chimismo, ossia le modalità di preparazione. Tradizionalmente, esistono due definizioni per classificare le polimerizzazioni. La prima distingue due ampie categorie di polimeri, quelli ottenuti per poliaddizione e quelli ottenuti per policondensazione. Esaminiamo la classe delle poliaddizioni. nA A(n) L’addizione di n monomeri di tipo A porta alla formazione di un polimero A(n) contenente n unità monomeriche; in questa categoria ritroviamo tutti i polimeri di tipo vinilico e dienico. La classe delle policondesazioni è invece caratterizzata dalla condensazione di n monomeri di tipo A a dare un polimero di n unità monomeriche, con eventuale espulsione di piccole molecole X, come ad esempio acqua. nA A(n) nX Tra i policondensati più comuni si ritrovano i nylon, i poliesteri, i policarbonati e le resine epossidiche. La seconda definizione si basa sul meccanismo di reazione e distingue tra polimerizzazione a catena (chain polymerization) e polimerizzazione a stadi (stepwise polymerization). La prima prevede la crescita di una catena An* dotata di un centro reattivo (di tipo radicalico, anionico o cationico) per addizione di un’unità monomerica A. La catena risultante An+1* possiede anch’essa un centro reattivo e la polimerizzazione può essere reiterata. A n* An+1* A La polimerizzazione a stadi prevede invece la reazione di una generica catena An con una catena Am mediante funzioni reattive. Il polimero An+m risultante può essere ottenuto con l’eventuale eliminazione di piccole molecole X. An Am An+m (X) Bisogna tenere presente che in questo caso l’eliminazione di molecole non è condizione necessaria alla classificazione del tipo di sintesi. Esistono alcuni tipi di polimerizzazione a stadi in cui l’eliminazione di X non è contemplata (ad esempio nella sintesi dei poliuretani). Le due definizioni sono simili e spesso si parla equivalentemente di poliaddizione e polimerizzazione a catena o di policondensazione e polimerizzazione a stadi, anche se possono esistere delle eccezioni. 1.2.2. Configurazione dei polimeri La presenza di un centro stereogenico può dar luogo a diversi tipi di polimeri di medesima costituzione ma con proprietà drasticamente differenziate. Consideriamo ad esempio il polipropilene, un polimero vinilico formato dall’addizione di n unità propileniche. H H3C C C CH3 H H propilene CH * C H2 n polipropilene 4 Si può riconoscere un centro stereogenico in corrispondenza di ciascun carbonio a cui è legato un metile. Se si rappresenta tridimensionalmente un frammento di catena polimerica, ci si accorge immediatamente della possibilità di avere diverse configurazioni. Una rappresentazione più schematica del frammento può essere ottenuta disponendo la catena principale parallelamente al piano del foglio e disegnando i sostituenti con dei legami pieni o tratteggiati a seconda che questi sporgano in avanti o indietro rispetto al piano. Questa modellizzazione è detta configurazione a catena estesa. H CH3 H CH3 H H H CH3 H H H CH3 H H H CH3 H3C H H H H H polipropilene isotattico H CH3 H3C H H H H H H H polipropilene sindiotattico A seconda della mutua disposizione dei gruppi metilici lungo la catena polimerica si può parlare di isotassia (metili dalla stessa parte) e di sindiotassia (metili da parti opposte). Queste due configurazioni rappresentano due strutture chimicamente diverse in quanto, pur possedendo la stessa costituzione, l’interconversione tra una configurazione e l’altra può aver luogo solamente con la rottura di legami covalenti. Una volta sintetizzato il polimero, la sua configurazione è determinata e non modificabile. In generale il termine tassia indica il tipo di ordinamento configurazionale mentre la tatticità definisce il grado di ordine. Una seconda modalità di rappresentazione della configurazione di un polimero è la cosìdetta Fisher modificata. In questa rappresentazione si disegnano i legami covalenti C-C in maniera lineare. I sostituenti vengono visualizzati perpendicolarmente all’asse della catena e per convenzione essi sono tutti rivolti verso l’osservatore. Tridimensionalmente la catena polimerica assumerebbe una tendenza simile a quella mostrata in figura. Secondo una rappresentazione di Fisher modificata il polipropilene assumerebbe quindi la forma seguente. CH3 H CH3 H CH3 H CH3 H CH3 H H H CH3 H H H H H H H H H H H H H CH3 H H H CH3 H polipropilene isotattico polipropilene sindiotattico 5 Esiste una terza configurazione del polipropilene che in realtà è quella che si ottiene da una sintesi non stereospecifica. Il polipropilene atattico infatti non possiede alcun grado di ordine configurazionale. CH3 H H H CH3 H CH3 H H H CH3 H H H H H polipropilene atattico In generale ottenere catene completamente iso o sindiotattiche è estremamente difficile. Quando la tatticità si mantiene per un certo tratto lungo la catena, intervallata da tratti irregolari o di diversa tassia, ci troviamo di fronte a un polimero a stereoblocchi. Esiste un secondo metodo particolarmente schematico per definire la configurazione di un polimero. Un centro stereogenico da origine a due diversi enantiomorfi. Nella figura si mostra l’enantiomorfo (A) e l’enantiomorfo (B) derivanti dall’esistenza di un carbonio stereogenico nel polipropilene. H3 C C + H H - - C CH3 CH3 + - (A) + (B) I tratti di catena vengono denominati + e – di modo che, eclissando l’idrogeno dietro al carbonio stereogenico, si ottenga la situazione mostrata in figura. L’alternanza di situazioni (A) e (B) definisce la configurazione del polimero. Un polimero isotattico sarà dunque rappresentato nella forma (-+, -+, -+, -+, -+) o (+- ,+- ,+- ,+- ,+- ) mentre la forma sindiotattica sarà rappresentata da (- -, ++, - - , ++, - -). La virgola indica la presenza di un carbonio non stereogenico, in genere un –CH2–. In un generico polimero derivante dalla polimerizzazione di olefine disostituite, la nomenclatura isotattico e sindiotattico non basta per definire compiutamente la loro configurazione. A A H C C C H C B n polimero B monomero Se infatti sia la configurazione del sostituente A che quella del sostituente B lungo la catena è di tipo isotattico, abbiamo due alternative possibili. La prima è che i due gruppi siano disposti su lati diversi nella rappresentazione di Fisher modificata mentre la seconda è che si trovino sullo stesso lato. A seconda dell’una o dell’altra possibilità si utilizzano i prefissi treo ed eritro. Questo problema non esiste se sia il sostituente A che il sostituente B generano configurazioni entrambe sindiotattiche. A A B A B A A B B A B A B A B B treo-di-isotattico eritro-di-isotattico A B B A A B B A di-sindiotattico 6 E’ facile verificare che se la tassia dei due sostituenti è diversa (una sindio e l’altra iso), non vi è possibilità di equivoco; le due forme sono equivalenti e non riconoscibili. A A B A A B B A B B B A A A iso e B sindio B B A A sindio e B iso Alcuni esempi particolarmente rappresentativi sono quelli offerti dalla polimerizzazione delle diolefine coniutate. Il butadiene può subire due tipi di polimerizzazione, la polimerizzazione 1,2 e la polimerizzazione 1,4. Come si evince dalla figura, le due reazioni danno luogo a due polimeri costituzionalmente differenti, pur derivando dal medesimo monomero. 2 H C 1 H2C C H CH2 4 3 1,2 1,4 * CH2 CH CH CH2 CH CH CH2 n n CH2 Il polimero derivante della polimerizzazione 1,2 possiede un centro stereogenico (indicato dall’asterisco) e quindi da origine a diverse configurazioni. In particolare si avrà la configurazione 1,2 isotattica e la 1,2 sindiotattica, in aggiunta all’eventuale configurazione irregolare (atattica). CH2 CH2 CH2 CH2 CH2 CH2 CH CH CH CH CH CH polibutadiene 1,2-isotattico CH CH CH2 CH2 polibutadiene 1,2-sindiotattico Il prodotto derivante dalla polimerizzazione 1,4 non presenta invece alcun carbonio stereogenico ma il legame insaturo da origine a isomeria geometria. Si avrà dunque il polibutatidiene 1,4-cis e il polibutadiene 1,4 trans. n polibutadiene 1,4 cis n polibutadiene 1,4 trans L’isoprene, o 2-metil-1,3-butadiene, è un butadiene a cui è stato sostituito un idrogeno (2) con un gruppo metile. La presenza del gruppo –CH3 distrugge l’equivalenza tra i due doppi legami, e la polimerizzazione che coinvolge un singolo doppio legame porta a due polimeri costituzionalmente diversi. In aggiunta alla polimerizzazione 1,4 avremo dunque polimerizzazione 1,2 e 3,4. 7 CH3 1 CH2 4 2 C H2C C H 3 1,2 1,4 3,4 CH3 CH3 * CH2 C CH CH2 n C CH CH2 n * CH2 CH CH2 C H2C n CH3 Anche in questo caso, le polimerizzazioni 1,2 e 3,4 portano alla formazione di un carbonio stereogenico per ciascun prodotto e dunque diversi tipi di configurazione (1,2-iso, 1,2-sindio e 3,4-iso, 3,4-sindio). Il prodotto della polimerizzazione coniugata 1,4 può presentarsi in forma cis o trans. In natura, il polisoprene 1,4 si ottiene dall’albero della gomma e a seconda che sia di configurazione cis o trans viene chiamato gomma naturale o guttaperca, Le proprietà meccaniche sono enormemente diverse proprio a causa della diversa configurazione; la forma cis è una gomma, morbida al tatto, mentre la forma trans è abbastanza dura da venir utilizzata nella fabbricazione di copertine rigide o palle da golf. guttaperca gomma naturale Il caso del pentadiene è ancora più complicato. Anch’esso possiede un gruppo metilico che rende i due legami coniugati non equivalenti e, per questo, i polimeri derivanti dalla polimerizzazione 1,2 e 3,4 sono costituzionalmente diversi. 1 2 4 H C H C C H H2C CH3 5 3 1,2 1,4 * * CH CH2 CH 3,4 CH2 CH n CH CH CH3 n CH CH3 * CH CH CH3 CH * n CH2 La posizione terminale del sostituente da però luogo ad un maggior numero di diversi polimeri; nel prodotto derivante dalla polimerizzazione 1,2 si ha la presenza simultanea di un carbonio stereogenico e di un doppio legame in grado di portare a isomeri costituzionali. Si ha pertanto la possibilità di avere un polimero cis o trans, 1,2-iso o 1,2-sindio. La polimerizzazione 1,2 porta quindi a quattro polimeri differenti, trascurando la configurazione atattica. 8 trans 1,2-iso cis 1,2-iso trans 1,2-sindio cis 1,2-sindio Anche il prodotto della polimerizzazione 1,4 possiede un carbonio stereogenico e un legame insaturo. Quindi anche in questo caso avremo i prodotti 1,4 cis o trans, iso o sindiotattici. La differenza dalla polimerizzazione 1,2 è che il doppio legame non si trova sul pendaglio laterale ma bensì all’interno della catena e quindi influirà maggiormente sulla struttura macromolecolare del prodotto. Il prodotto della polimerizzazione 3,4, invece, da origine a due centri stereogenici (il doppio legame in questo caso non da origine ad isomeri costituzionali). La configurazione del pentadiene viene dunque trattata alla stregua dei prodotti derivanti dalla polimerizzazione di olefine disostituite. Si hanno dunque le configurazioni 3,4 eritro-di-isotattica, 3,4 treo-diisotattica e 3,4 di-sindiotattica, come mostrato in figura. 3,4 treo-di-iso 3,4 eritro-di-iso 3,4 di-sindio Un ultimo caso da esamirare è la configurazione di polimeri i cui monomeri contengono un’unità ciclica. L’apertura di un anello epossidico variamente sostituito porta a polimeri con diversa configurazione a seconda delle modalità di apertura dell’anello o della stereochimica del monomero. Un esempio è quello del propilenossido. H3C (a) O (b) propilenossido L’apertura ripetuta dell’anello sul legame (a) o (b) porta alla formazione del polimero isotattico ma le catene polimeriche sono tra loro enantiomorfe, nei due casi distinti. Per ottenere un polimero sindiotattico si deve alternare un’apertura di tipo (a) a un’apertura di tipo (b). Si ricorda che l’apertura dell’anello avviene con un’inversione di configurazione. polipropilenossido isotattico da apertura tipo (b) da apertura tipo (a) H3C O H3C O CH3 O O O CH3 CH3 O CH3 CH3 O CH3 O H3C CH3 O O H3C CH3 O O O CH3 O CH3 O CH3 O CH3 polipropilenossido sindiotattico H3C O H3C O O O H3C H3C CH3 O CH3 O O CH3 O CH3 9 Se l’epossido posside due sostituenti, come nel caso del 2,3-epossibutano, è la stereochimica del monomero che determina la configurazione del polimero. cis H3C CH3 CH3 H3C O CH3 CH3 CH3 CH3 CH3 CH3 CH3 CH3 CH3 O CH3 O CH3 O O trans CH3 O O O Se l’apertura dell’anello avviene sempre rompendo il medesimo legame e il monomero è di tipo trans (si ricorda che in un composto ciclico la nomenclatura trans sta ad indicare che i sostituenti giacciono da parti opposte rispetto al piano dell’anello, al contrario della forma cis) si otterrà un polimero eritro-di-isotattico mentre se il monomero è di tipo cis si otterrà il corrispondente polimero treo-di-isotattico. Anche in questo caso, l’apertura dell’anello avviene con l’inversione di configurazione del centro stereogenico attaccato, come si può facilmente notare in figura. Un ultimo esempio è quello della polimerizzazione del ciclopentene. n ciclopentene n policiclopentene Il policiclopentene può assumere quattro diverse configurazioni stereoregolari. A causa del fatto che i sostituenti sui due carboni stereogenici sono vincolati dalla loro forma ciclica, la configurazione di-sindiotattica può assumere due forme diverse. In analogia con il caso diisotattico, esse vengono chiamate treo- ed eritro-. eritro-di-isotattico treo-di-isotattico eritro-di-sindiotattico treo-di-sindiotattico 10 1.3. Chiralità e attività ottica La presenza di centri stereogenici in un composto organico generalmente è causa di attività ottica. Prendiamo come esempio la gliceraldeide. Questa possiede un centro stereogenico ed è quindi possibile scrivere la coppia di enantiomeri, sotto rappresentati in proiezioni di Fisher. O H C OH CH C H2 * C H2 OH H gliceraldeide O O CH CH OH HO CH2OH H CH2OH Se si prepara una soluzione di un singolo enantiomero del composto chirale e lo si lascia attraversare da un fascio di luce polarizzata, questo ha la proprietà di ruotare il piano di polarizzazione della luce. La presenza di centri stereogenici all’interno di un polimero come il polipropilene isotattico potrebbe portare a pensare che questo materiale possegga una certa attività ottica. CH3 CH * CH3 C H2 n catena catena H Questo in realtà è falso in quanto i centri stereogenici del polipropilene non portano a chiralità. Anche se infatti ogni carbonio a cui è legato un metile è un centro stereogenico, le parti laterali di catena sono in realtà indistinguibili tra di loro (questo poichè un polimero è una ripetizione estesa di unità monomeriche). I carboni del polipropilene sono per questo detti enantiotopici o prochirali. L’attività ottica dei polimeri è in genere trascurabile in quanto gli unici centri chirali sono quelli dovuti alle unità vicino ai terminali di catena. 11 2. POLIMERIZZAZIONE A STADI Le polimerizzazioni a stadi sono particolarmente delicate. Al fine di ottenere polimeri ad alti pesi molecolari commercialmente sfruttabili, le reazioni devono essere pulite. Per reazioni pulite si intendono reazioni selettive, ossia che non diano miscele di prodotti diversi, e il cui equilibrio possa essere spinto verso una resa quantitativa del prodotto voluto. Per capire i motivi di queste richieste stringenti, analizziamo una generica policondensazione tra due monomeri, il primo contenente due funzionalità di tipo A e il secondo contenente due funzionalità di tipo B. La reazione delle due diverse funzionalità porta ad un polimero lineare. A A B B A A B B n Ora siano NAA0 e NBB0 le quantità di reagente AA e BB iniziali. Per comodità definiamo la stechiometria di reazione r come la proporzione dei monomeri utilizzati. r= N0AA 0 NBB Il grado di avanzamento p è definito come la quantità di prodotto formatosi in seguito al consumo dei monomeri. Esso dipende dalla costante di equilibrio e dalla cinetica della reazione. Se NA e NB sono le funzionalità A e B non reagite, possiamo calcolarci il grado di avanzamento p come rapporto tra le funzionalità A reagite (2NAA0 - NA) e quelle iniziali 2NAA0. p= 2N 0AA − N A 2N 0AA Da questa relazione esplicitiamo NA, le funzionalità A non reagite, ossia NA=2NAA0(1-p). Le funzionalità B non reagite saranno semplicemente la differenza tra le funzionalità B iniziali 2NBB0 e le funzionalità B reagite (2NAA0 - NA). 0 NB = 2NBB − (2N0AA − N A ) Le funzionalità B reagite saranno infatti equivalenti al numero di funzionalità A reagite dal momento che ogni A reagisce solo e con una sola B. Sostituendo NA e utilizzando r per eliminare la dipendenza da NBB0 otteniamo: NB = 2 N 0AA ⎛1 ⎞ − 2N 0AA + 2N 0AA (1 − p) = 2N 0AA ⋅ ⎜ − p ⎟ r ⎝r ⎠ Ora, il grado di polimerizzazione xn indica la lunghezza media delle catene polimeriche. A parità di funzionalità A e B iniziali, un maggior numero di funzionalità A e B libere nel polimero finale sarà indice di un basso grado di polimerizzazione. Questo concetto può essere immediatamente compreso osservando la seguente illustrazione. 40 funzionalità libere basso xn 20 funzionalità libere alto xn 10 funzionalità libere 12 Il grado di polimerizzazione può essere quindi espresso come: xn = 0 AA funzionalità iniziali 2N + 2N = funzionalità finali N A + NB 0 BB N 0AA 1+r r = = ⎛1 ⎞ 1 + r − 2pr 2N 0AA (1 − p) + 2N 0AA ⎜ − p ⎟ ⎝r ⎠ 2N 0AA + 2 Otteniamo dunque un’espressione del grado di polimerizzazione xn di una polimerizzazione a stadi in funzione della stechiometria r con cui è stata fatta avvenire la sintesi e del grado di avanzamento p. L’andamento può essere sintetizzato con un grafico, anche se è meglio studiare alcuni casi particolari. Ad esempio per stechiometria r Æ 1 e grado di avanzamento p Æ 1 otteniamo le seguenti relazioni: per r Æ 1 xn = 1 1−p per p Æ 1 xn = 1+r 1−r Per ottenere alti pesi molecolari la stechiometria della reazione e il grado di conversione del monomero in polimero devono dunque essere prossimi ad uno. In pratica la reazione deve essere pulita. Per assicurarsi un alto grado di conversione è necessario spostare l’equilibrio verso i prodotti. Questo può essere ottenuto eliminando dall’ambiente di reazione il sottoprodotto X. 13 Analizziamo ora la distribuzione dei pesi molecolari dei prodotti. Il grado di avanzamento p può essere interpretato anche come la probabilità che un singolo stadio della polimerizzazione abbia luogo al tempo t. Le frazioni molari xi delle catene sintetizzate lunghe i possono essere quindi calcolate utilizzando un metodo statistico. Il monomero x1 presente al tempo t non è nient’altro che il monomero non reagito, ossia (1 – p); x2 è invece pari alla probabilità che avvenga uno stadio di reazione per la quantità di monomero x1; a sua volta x3 è pari alla probabilità che avvenga uno stadio di reazione per la quantità x2, e così via. x1 = 1 − p x 2 = p(1 − p) x 3 = p 2 (1 − p) In generale quindi la frazione xi della catena lunga i sarà uguale a: x i = p i−1 (1 − p) Diagrammando la frazione xi in funzione di i ci si può rendere conto che la distribuzione delle frazioni sarà tanto più dispersa verso catene più lunghe quanto più prossimo ad uno risulta il grado di conversione p. Anche se p ≈ 1 il monomero sarà sempre presente a fine polimerizzazione e la sua frazione molare x1 sarà quella preponderante. Ciò nonostante la sua frazione in peso risulterà nettamente minore rispetto a quella di alti pesi molecolari. Per sincerarsene possiamo calcolarci la distribuzione dei pesi molecolari wi. wi = N i ⋅ Mi = x i (i ⋅ M 0 ) ∝ i ⋅ p i−1 (1 − p) 2 N ∑ i 14 Come si può vedere, anche se il contributo del monomero è grande in termini di frazione molare, il suo contributo alla distribuzione dei pesi è minimo, mentre acquistano valore le frazioni a pesi intermedi. L’andamento del grafico rispecchia molto bene la distribuzione dei pesi molecolari data precedentemente e le grandezze medie si collocano attorno al massimo. L’indice i può essere sostituito al grado di polimerizzazione xn o al peso molecolare. L’indice di polidispersità può essere calcolato come rapporto tra xn medio e xw medio, ossia: ∞ x D= w = xn ∑ i ⋅ wi i=0 ∞ ∑i⋅ x i=0 ∞ = (1 − p) ⋅ ∑i i i=0 ∞ 2 ⋅ pi ∑i⋅p = (1 − p) ⋅ i 1+p =1+p 1−p i=0 Dal momento che p deve essere prossimo ad 1, l’indice di polidispersità dei prodotti di una polimerizzazione a stadi è sempre prossimo a 2. Come già detto, l’indice di polidispersità di un ideale polimero monodisperso è pari ad uno; si vede dunque che già solamente a causa del meccanismo di sintesi, l’indice di polidispersità dei polimeri reali devia dal caso ideale. Diverse importanti classi di polimeri possono essere sintetizzate attraverso una polimerizzazione a stadi. In questo corso verranno trattati solo alcuni tipi di polimeri, approfondendone i principi di sintesi; in particolare si esamineranno le poliammidi, i policarbonati, i poliuretani, le resine amminiche e fenoliche e un tipo di poliestere, il PET. In tabella si riportano le principali classi di polimeri ottenibili mediante polimerizzazione a stadi. Classe poliesteri Polimero O Monomeri O O RO C R' C O HO R OH n O O RO C HO R C OH n poliammidi O HO C R' C OH O O H R C N R C NH n O O H H N R N C R' C O H2N R n poliuretani O O H H O R O C N R' N C NH2 O HO C R' C OH O C N R' N C O HO R OH n policarbonati CH3 CH3 O C C O CH3 polisolfoni O C OH Cl C Cl CH3 n O O S O O polieteri HO Cl S n CH3 CH3 O CH3 Cl O OH CH3 n polisolfuri S Br SH n 15 polisilossani R R HO Si OH O Si R n R resine fenoliche OH O H C H resine amminiche O O H2N C NH2 NH2 H C H N H2N O N N H C H NH2 2.1. Poliammidi Le poliammidi sono una classe di polimeri derivanti dalla condensazione di un diacido e di una diammina. O O HO C H2N NH2 C OH O O C C N N H H n Questi polimeri sono commercialmente chiamati nylon. Le caratteristiche costituzionali del polimero sono identificate da due numeri o da un numero singolo. Quando sono presenti due numeri, il primo di essi identifica il numero di carboni presenti nella diammina mentre il secondo rappresenta il numero di carboni (compresi quelli dei due carbossili) presenti nel diacido. Si riporta l’esempio del nylon 6,6 e del nylon 6,10. O C O C C H2 H2 C C H2 C H2 H2 C H2 C C H2 C H2 H2 C C H N O H2 C C H2 H2 C C H2 C H2 C H N O C H2 C H2 H2 C H2 C C H2 C H2 H2 C H2 C N H C H2 Nylon 6,6 n H2 C N H Nylon 6,10 n Nella sintesi delle poliammidi è molto importante rispettare la stechiometria di reazione. Un modo per ottenere un buon rapporto stechiometrico dei reagenti è quello di cristallizzare il sale dell’acido e dell’ammina. O O C O C O H3N NH3 Se infatti si tratta l’acido dicarbossilico con la diammina in un ambiente a pH opportuno, è possibile ottenere il sale formato dal dicatione e dal dianione. Il sale può essere dunque cristallizzato (e quindi contemporaneamente purificato) così da ottenere un solido in cui sono presenti i monomeri in stechiometria perfetta 1:1. Al fine di ottenere alti pesi molecolari la polimerizzazione deve essere effettuata di modo che p Æ 1; per questa ragione, la reazione viene condotta ad alta temperatura e pressione per aumentarne la cinetica. Se si volesse evitare l’uso del solvente per ottenere prodotti particolarmente puliti si può condurre la reazione utilizzando il sale fuso. L’acqua che si forma dal processo di condensazione va eliminata per spingere la reazione verso completamento. 16 HOOC(CH2)kCOOH O O H H C (CH2)k C N (CH2)m N 275 °C H2N(CH2)mNH2 20 atm N2 n nH2O Per sintetizzare il Nylon 6 si utilizza un precursore ciclico, l’ε-caprolattame. La sua idratazione permette infatti di ottenere il monomero del Nylon 6. NH H2 O O H2N ε-caprolattame C H2 H2 C C H2 H2 C O polimero OH C H2 Naturalmente si possono sintetizzare nylon a catena più lunga. In particolare il Nylon 11 è estremamente insolubile ai derivati del petrolio e viene largamente utilizzato nella fabbricazione di taniche e tubi. Le poliammidi aromatiche vengono comunemente chiamate poliaramidi. Tra gli esempi più importanti di poliaramidi abbiamo il kevlar, derivante dalla polimerizzazione di acido tereftalico e p-fenilendiammina, e il nomex, derivante dalla polimerizzazione di acido isoftalico e m-fenilendiammina. HO H2N NH2 O C O p-fenilendiammina H2N NH2 O H N C H N C C OH n KEVLAR acido tereftalico HO O O O C C O H N C H N OH O C n m-fenilendiammina acido isoftalico NOMEX Le poliaramidi sono materiali particolarmente rigidi e vengono utilizzate in applicazioni che sfruttano le loro proprietà meccaniche superiori, come nella produzione di fibre per la fabbricazione di giubbotti antiproiettile. 2.2. Polietilentereftalato La sintesi del polietilentereftalato (PET) viene normalmente raggiunta attraverso due diversi processi. Il primo schema di sintesi prevede l’utilizzo dell’acido tereftalico (TA) e del glicole etilenico (EG), mentre nel secondo si utilizza, al posto dell’acido, un suo estere, il dimetiltereftalato (DMT). O C HO OH HO H2 C C O TA C H2 OH EG O C MeO OMe HO C O DMT H2 C C H2 OH EG 17 In presenza di acetati di Zn, Ca o Mn si può far reagire il DMT con il glicole etilenico a formare il bis(2-idrossietil)tereftalato, più comunemente indicato con BHET. La reazione di transesterificazione ha come sottoprodotto l’alcool metilico, che viene allontanato dal sito di reazione per spostare l’equilibrio a favore del prodotto. Il rapporto tra i reagenti EG/DMT è leggermente maggiore di 2, per assicurarsi che tutto il DMT reagisca. O C MeO OMe HO H2 C C C H2 OH 180°C 1 atm O DMT EG O C HO H2 C C H2 O O H2 C C H2 OH 2CH3OH C O BHET Questo primo stadio di prepolimerizzazione permette di ottenere il monomero da utilizzare nella vera e propria fase di polimerizzazione. Trattando il BHET a 300°C in presenza di Sb2O3 come catalizzatore, si ottiene la policondensazione del reagente. Il sottoprodotto della reazione, il glicole etilenico, viene recuperato e purificato per essere poi riutilizzato in un nuovo ciclo di sintesi. O C HO H2 C C H2 O O H2 C C H2 OH 300°C vuoto C O O C O O H2 C C H2 n C O HO H2 C C H2 OH n Grazie alla sua forte impermeabilità ai gas, il PET viene largamente utilizzato nell’industria dei contenitori di bibite gasate. 2.3. Policarbonati Il policarbonato più comune è quello che deriva dal bisfenolo A ossia: O CH3 C O C CH3 O O C n Il bisfenolo A viene preparato a partire dal fenolo e dall’acetone. L’anello elettronricco del fenolo (la posizione para viene attivata dalla presenza dell’idrossile) attacca il carbonile elettronpovero dell’acetone due volte, portando alla formazione del bisfenolo A e all’espulsione di acqua. 18 CH3 H3C C O HO HO C OH H3C OH CH3 CH3 HO C H2 O OH CH3 bisfenolo A La sintesi del policarbonato prevede l’utilizzo del fosgene. La maggior parte dei processi industriali che coinvolgono reazioni con il fosgene sono condotte mediante sintesi interfacciale. Nel caso del policarbonato si tratta il reagente da funzionalizzare (bisfenolo A) con soda (NaOH) di modo da ottenerne il sale sodico. Dal momento che il sale è solubile in fase acquosa, si separa il bisfenolo A dal fosgene in due fasi distinte: nella fase acquosa il sale sodico del bisfenolo A, mentre in fase organica di CH2Cl2 il fosgene. La reazione di polimerizzazione avviene all’interfaccia con produzione di cloruro di sodio (NaCl). CH3 HO C OH NaOH CH3 CH3 Na Cl O O C O CH3 O CH3 C O C CH3 O C CH3 O C CH3 Cl O x O OLIGOMERI O C Cl CH3 Na C Cl O C Cl CH3 Na Na y O CH3 O C O z C ONa CH3 La temperatura a cui avviene il processo è attorno ai 35 °C. Per facilitare il trasferimento dei reagenti all’interfaccia, si possono usare piccole quantità di catalizzatori, come sali di ammonio quaternario o eteri corona. Le catene ottenute sono in genere abbastanza corte per gli utilizzi pratici del polimero e vengono dunque ritrattate una seconda volta con ammine terziarie che catalizzano le reazioni che portano ad un polimero con più alto peso molecolare. L’alternativa alla sintesi interfacciale è un processo simile a quello della produzione del PET, in cui si utilizzano il bisfenolo A e il fosgene allo stato fuso. La sintesi all’interfaccia è da preferirsi rispetto a quest’ultima alternativa per il maggior controllo sul peso molecolare del prodotto e per il minor costo di produzione. Il policarbonato derivato dal bisfenolo A viene solitamente indicato come policarbonato o PC. Questo tipo di polimero possiede una buona resistenza contro acidi e ossidanti e trova applicazioni pratiche soprattutto grazie alla sua eccezionale trasparenza e durezza. 19 2.4. Poliuretani I poliuretani furono per la prima volta sintetizzati da Bayer nel 1937, come risultato della reazione di diisocianati con dioli. Dal 1945 in avanti, vennero proposte diverse nuove sintesi, come ad esempio la reazione di diammine con biscloroformiati a basse temperature. HO R' OH OCN R NCO O Cl H2N R NH2 O O H H C N R N C O R' O O n C OR'O C Cl Il metodo di sintesi che coinvolge la reazione di disiocianati con dialcoli ha avuto maggior successo rispetto al metodo bis(cloroformiato)-diammina poichè consiste in una semplice reazione di addizione, senza formazione di sottoprodotti. La sintesi dei poliuretani è infatti un esempio di poliaddizione che avviene con un meccanismo a stadi. Il meccanismo è quello di una condensazione di un gruppo isocianato con un alcolo ma nella polimerizzazione non si ha eliminazione di piccole molecole; i monomeri si addizionano l’uno all’altro. Di seguito si mostra il meccanismo di condensazione. O R N C O gruppo isocianato R R' OH gruppo alcolico N C O O H R' R N H C O R' I poliuretani possono essere visti come un insieme di ammidi ed esteri ed hanno quindi proprietà intermedie tra quelle dei poliesteri e delle poliammidi. I poliuretani normalmente si decompongono a temperature maggiori di 220 °C a dare isocianati, alcoli, ammine, anidride carbonica ed olefine. I poliuretani hanno trovato varie applicazioni nel campo degli adesivi, rivestimenti, materiali di riempimento, elastomeri e fibre. 2.5. Polimerizzazione a stadi polifunzionale Se almeno uno dei monomeri impiegati in una polimerizzazione a stadi possiede un numero di funzionalità f maggiore di 2, si ha la formazione di molecole non lineali. La polimerizzazione viene chiamata polimerizzazione polifunzionale o tridimensionale. In questo caso il polimero che si ottiene non può essere semplicemente descritto attraverso il numero di unità monomeriche da cui è composto ma bisogna specificare anche il grado di ramificazione, ossia il numero di unità di ramificazione per molecola. Analizziamo il sistema ottenibile dalla polimerizzazione di due unità monomeriche (1) e (2). B x A A A y B B (1) (2) BA AB BA AB AB BA A BA A BA A BA A La prima molecola possiede due funzionalità di tipo A mentre la seconda possiede tre funzionalità di tipo B. Un esempio di polimero ottenibile da questo tipo di unità monomeriche è il poliestere derivante dalla condensazione di acido tereftalico e glicerina. 20 COO φ O C HO OOC HO CH2 HO CH CH2 HO OH C O COO OOC φ H2C CH H2C OOC φ COO COO CH2 CH OOC COO CH2 CH2 φ COO CH CH2 φ COO OOC La macromolecola polifunzionale continua a crescere alle estermità, creando un numero sempre più grande di ramificazioni. Il prodotto che si ottiene viene in genere chiamato reticolo infinito. Il numero di funzionalità terminali in grado di reagire aumenta all’avanzare della reazione e questo porta ad una maggiore viscosità del prodotto che sfocia in proprietà simili a quelle di un gel. Per mezzo di trattamenti termici è possibile reticolare il polimero, ovvero formare un network tridimensionale, come illustrato in figura. A A A B B A B B A B A A A B A B B A B A B A B A B A La possibilità di reticolare il polimero porta all’indurimento del materiale e, se avviene in maniera controllata, questo processo può essere sfruttato in applicazioni pratiche. Esempi di polimeri reticolati (o cross-linked) sono le resine fenoliche o le amminoplastiche. Naturalmente la polifunzionalità di un monomero non porta necessariamente alla reticolazione; nel caso seguente si mostra la polimerizzazione del monomero (3) che porterà a prodotti ramificati privi della possibilità di reticolare. BA B A x A B (3) BA BA B BA BA B BA Solitamente la ramificazione termina al quarto o quindi livello ma se si aggiunge un monomero del tipo AB è possibile allungare i rami ed estendere ulteriormente la ramificazione. In assenza di AB si ottiene un sistema molto compatto che viene solitamente chiamato dendrimero. Spesso un dendrimero porta alla formazione di sistemi sferulitici come indicato in figura. All’interno dello sferulita è presente una bassa densità di ramificazioni mentre, via via che ci si avvicina alla superficie, la densità aumenta. Si ottiene dunque una cavità artificiale in cui poter ospitare altre molecole, come ad esempio additivi o farmaci. Un altro tipo di polimero derivante da monomeri polifunzionali sono gli stellati, o polimeri a stella. Per sintetizzare uno stellato si utilizza un monomero polifunzionale dotato di un solo tipo di terminale B, detto regolatore, e un monomero bifunzionale del tipo A-B. 21 I gruppi A e B sono reattivi e quindi i pendagli del monomero polifunzionale si accrescono, formando la tipica struttura a stella da cui deriva in nome della classe. B x B BABABAB y A B B BABABAB B BABABAB BABABAB (4) (5) Le lunghezze dei pendagli dello stellato non sono necessariamente uguali all’interno della medesima macromolecola. Il rapporto x/y è arbitrario e una sua accurata scelta permette di modulare la lunghezza dei bracci dello stellato (solitamente non si necessità di bracci troppo lunghi). Si può dimostrare che il grado di polimerizzazione xn e l’indice di polidispersità D di una miscela di stellato con un regolatore avente f funzionalità è pari a: xn = 1 ⋅f 1−p D= xw xn =1+ 1 f Dunque all’aumentare del numero di funzionalità f, l’indice di polidispersità D tende al limite di quello di un ideale polimero monodisperso. Nell’altro caso limite (f = 1), la funzione del regolatore sarebbe semplicemente quella di terminare la polimerizzazione del monomero (5) e il prodotto risultante possiederebbe un indice di polidispersità D pari a 2, come in una normale policondensazione. 2.6. Resine fenoliche Le resine fenoliche sono ottenute dalla polimerizzazione del fenolo e della formaldeide. Come indicato in figura, le funzionalità reattive del fenolo sono tre (f = 3) mentre le funzionalità della formaldeide sono due (f = 2). OH O H C H x2 fenolo formaldeide Anche se ad una prima impressione sembra che la formaldeide possieda solamente una funzionalità reattiva (quella carbonilica), la presenza di una seconda funzionalità può essere meglio compresa valutando il seguente equilibrio: O H C H2O H formaldeide HO H2 C OH glicole metilico La prima funzionalità è l’evidente funzionalità carbonilica mentre la seconda è associata al gruppo alcolico che si viene a formare in seguito all’attacco nucleofilo al carbonile. In generale, la velocità di polimerizzazione di fenolo e formaldeide dipende dal pH. In particolare, si hanno situazioni ottimali a valori di pH alto e basso. La polimerizzazione può essere dunque catalizzata sia da basi che da acidi; esaminiamo inizialmente la catalisi basica. La presenza di un composto alcalino promuove la formazione del fenolato, il cui anello elettronricco attacca la funzione carbonilica della formaldeide. Questa reazione porta alla formazione di fenoli sostituiti da gruppi metilolici, tra cui il resolo. 22 OH OH OH CH2OH O C H CH2OH OH60-100°C H CH2OH OH HOH2C CH2OH CH2OH resolo Il meccanismo di reazione si può così sintetizzare: O O O H C O CH2 H O CH2OH H Oltre ai prodotti sopra elencati, nella sintesi si forma anche una miscela di oligomeri, dovuta alle ulteriori reazioni dei fenolati sui carboni alcolici. Si riporta il meccanismo di reazione: O H2 C OH O -OH- O CH2 O O C H2 Durante questa prima fase si formano dunque dei prepolimeri, che in genere non possiedono grande estensione. La prepolimerizzazione è condotta utilizzando un rapporto molare formaldeide/fenolo pari circa a 1,2. La formaldeide utilizzata viene aggiunta sotto forma di formalina (formaldeide idrata). Per la catalisi basica si utilizzano idrossidi di Na, Ba o Ca. In generale, la composizione ed il peso molecolare del prodotto dipendono dal rapporto fenolo/formaldeide, dal pH e dalla temperatura a cui avviene la prepolimerizzazione. Il polimero vero e proprio si forma a seguito della reticolazione dei prepolimeri sopra ottenuti. Riscaldando ad una temperatura di circa 180 °C si ha la formazione di ponti metilenici (ponti –CH2-) o di ponti eterei (-CH2-O-CH2-). ponte metilenico OH OH O OH O OH ponte etereo 23 I sistemi ottenuti dalla prepolimerizzazione sotto catalisi basica vengono detti resoli o resine monostadio. La catalisi basica infatti forma prodotti con gruppi metilolici terminali e, se la reazione non viene fermata (neutralizzando l’ambiente di reazione), la reticolazione avviene spontaneamente. La catalisi acida porta invece alla formazione delle novolacche, o resine bistadio. L’acido utilizzato è di solito l’acido ossalico e possiede il compito di esaltare il carattere elettrofilo del carbonile. O H O H C OH O O H OH OH H C H H C H acido ossalico Anche in questo caso il fenolo compie un attacco nucleofilo ai danni del carbonile della formaldeide. A differenza della sintesi dei resoli, in questo caso il rapporto formaldeide/fenolo è 0,8 in modo da assicurarsi che al termine della prepolimerizzazione, gli oligomeri non abbiano alcun gruppo metilolico libero. OH OH O H C H2 C (COOH)2 H H2 C H2 C OH H2 C 100 °C HO OH Questo impedisce al prepolimero di reticolare con la semplice presenza di una fonte di calore poichè non vi sono funzionalità metiloliche in grado di formare ponti tra gli oligomeri. Per poter reticolare la novolacca si aggiunge un additivo (detto anche reticolante), l’hexa (esametilentetrammina). N N N N hexa Questo composto deriva dalla reazione di 6 moli di formaldeide e 4 moli di ammoniaca. La sua funzione è appunto quella di fornire la formaldeide necessaria per la reticolazione. 2.7. Resine amminiche Le amminoresine (o amminoplastiche), strettamente collegate alle resine fenoliche nei principi di sintesi, vengono convenzionalmente ottenute dalla polimerizzazione della formaldeide con l’urea o con la melammina. NH2 O H2N C urea N NH2 H2N N N NH2 melammina La sintesi della amminoplastiche può essere condotta sia in condizioni acide che in condizioni basiche. Il controllo della reazione di polimerizzazione viene raggiunta operando sulla temperatura e sul rapporto tra i reagenti. In una prima fase, si sintetizza il prepolimero ad un pH acido o basico (il tipico rapporto molare formaldeide/urea è 1,4:1). La condensazione della formaldeide e dell’urea porta alla formazione di varie metiloluree e 24 miscele di oligomeri. Il numero di funzionalità reattive a disposizione della formaldeide è, come già sappiamo, pari a due mentre le funzionalità dell’urea sono quattro. O H2N HO H2 C O N H C N H H2 C C O NH2 H C H O OH (HOH2C)2N HO H2 C O N H C N H H2 C C N(CH2OH)2 OH n In una seconda fase si raffredda il prodotto e si riporta il pH ai livelli di neutralità per reticolare il prepolimero. La reticolazione, ad opera di un moderato riscaldamento sotto catalisi acida, coinvolge la formazione di ponti metilenici, eterei e ciclici, come schematizzato di seguito. O O N C CH2 N C N N H H N O C CH2 N H O CH2 N C H N O N H C N H2C O H2 C N O N C CH2 N H C O ponte metilico ponte etereo ponte ciclico La formazione del prepolimero formaldeide-melammina è simile al processo appena descritto, anche se bisogna tenere presente che la melammina possiede ben sei funzionalità reattive. Le amminoplastiche possiedono proprietà simili alle resine fenoliche, ma sono più trasparenti; le resine melamminiche possiedono una maggiore durezza e resistenza al calore rispetto alla resine ureiche. Le loro proprietà di trasparenza le rendono particolarmente adatte a funzioni di rivestimento. Esse, inoltre, possiedono proprietà adesive e vengono largamente utilizzate nel settore delle vernici e come smalti. 25 3. POLIMERIZZAZIONE A CATENA Come già introdotto in precedenza, una polimerizzazione a catena prevede la crescita di una catena An*, dotata di un centro reattivo, per successive addizioni di unità monomeriche. La catena in accrescimento An+1* possiede sempre il medesimo centro reattivo e continua crescere indefinitamente fino a quando la sua reattività non viene spenta. A n* An+1* A Tra le principali polimerizzazioni a catena troviamo le polimerizzazioni radicaliche, in cui il centro reattivo di propagazione è un radicale, e le polimerizzazioni a catena ioniche. In questo ultimo tipo di polimerizzazione, il centro reattivo non è più un radicale ma bensì un centro carico (positivamente o negativamente), ossia un carbocatione o un carbanione. A seconda che si abbia a che fare con l’uno o l’altro tipo, si parla di polimerizzazione cationica o di polimerizzazione anionica. Queste classi di polimerizzazioni sono particolarmente importanti perchè portano a prodotti unici, in generale non ottenibili attraverso nessun altro tipo di sintesi. Per formare il centro reattivo sui monomeri si utilizzano degli iniziatori che donano o privano di elettroni il carbonio. La presenza di gruppi elettronattrattori (Electron WithDrawing) o elettrondonatori (Electron Donor), favorisce energeticamente la formazione della specie ionica, depauperando o arricchendo la densità elettronica sul carbonio. H2C H C EWD H2C H C EWD influenza di un gruppo elettronattrattore H2C H C ED H2C H C ED influenza di un gruppo elettrondonatore Nella cationica è anche possibile utilizzare gruppi alchilici come sostituenti poichè questi stabilizzano la carica sul carbocatione per effetto induttivo. Inoltre, l’aggiunta di sostituenti al centro reattivo aumenta la stabilità del carbocatione (si ricorda che la stabilità dei carbocationi e R+ primario < R+ secondario < R+ terziario). 3.1. Polimerizzazione radicalica 3.1.1. Principi base Tra le polimerizzazioni a catena si parla di polimerizzazione radicalica se i centri reattivi sono radicali liberi. Una polimerizzazione radicalica può essere suddivisa in tre fase distinte: l’inizio, la propagazione e la fase di terminazione. La fase di inizio consiste nella formazione del primo radicale libero che andrà ad iniziare la polimerizzazione. I + (A) R + (B) Per ottenere un radicale libero bisogna utilizzare un iniziatore (I), ossia un composto instabile che decomponga in specie radicaliche. La formazione del radicale primario (in questo caso con primario indichiamo il fatto che il radicale in questione è quello che da origine alla polimerizzazione) può avvenire principalmente per dissociazione termica o fotochimica di iniziatori o mediante reazioni di ossido-riduzione. Tipici iniziatori termici sono i perossidi organici o gli azocomposti. Ad esempio nella sintesi di polimeri vinilici si utilizza spesso l’AIBN (2,2-azo-bis-isobutirronitrile). 26 CH3 CH3 H3C C N N C CH3 CN CH3 CH3 ∆ H3C C CN N N C CH3 CN CN AIBN I legami C-N sono instabili e sotto riscaldamento si scindono omoliticamente a dare una più stabile molecola d’azoto (che si allontana dal sito di reazione sotto forma gassosa) e due radicali liberi che andranno ad iniziare la reazione di polimerizzazione. CH3 H H H3C C CN (R) H H R C C C C H H con X= H; CH3; C6H5; Cl X X Nel caso specifico si ha l’attacco del doppio legame vinilico e la formazione di un nuovo sito reattivo radicalico. La reazione del radicale primario con il monomero può avvenire con due regiochimiche differenti: H H (A) R C C H H2C R CHX X H H (B) R C C X H In linea di principio si può ottenere sia il prodotto (A) che il prodotto (B). In realtà, tra i due possibili meccanismi di attivazione, prevale quello cineticamente favorito, in generale quello che porta alla formazione di un radicale termodinamicamente più stabile. L’ordine di stabilità dei radicali è: R• terziario > R• secondario R R' C R'' R' > R• primario R R C H C H H Ovvero, il radicale più sostituito è quello termodinamicamente più stabile. Nell’esempio riportato, dunque, il prodotto (A) predomina abbondantemente rispetto al prodotto (B) proprio a causa della maggiore stabilità termodinamica del prodotto. energia (B) (A) reagenti grado di avanzamento 27 Nel caso dello stirene, il radicale sul sito benzilico risulta maggiormente stabilizzato a causa delle diverse formule mesomere che è possibile riportare. H2 C R CH R H2 C CH H2 C R CH R H2 C CH2 Un secondo esempio di iniziatore termico è il perossido di benzoile, un perossido organico. Sotto riscaldamento in benzene, questo decompone sviluppando anidride carbonica e liberando radicali fenilici. Un iniziatore inorganico è invece il persolfato che, se riscaldato, decompone in due ioni bisolfato e due radicali idrossili. O O C O O 60°C C O O O S S O O in benzene O O O 60°C O S O C O O O O C in benzene O 2HSO4- 2H2O 2OH O Come esempio di iniziatore redox si può riportare l’ossidazione del ferro da parte dell’acqua ossigenata. In questa reazione il metallo si ossida da Fe2+ a Fe3+ e l’acqua ossigenata si scinde in uno ione e un radicale ossidrile. Fe2+ Fe3+ H2O2 OH OH Durante la fase di propagazione, il polimero si accresce per successive reazioni del centro reattivo (radicale libero) con il legame insaturo di altri monomeri. Il processo di propagazione può essere schematizzato nel seguente modo: RMi RM(i+1) M dove RM•(i+1) è il radicale polimerico in accrescimento. Ad esempio, la polimerizzazione del monomero vinilico precedentemente iniziata dall’AIBN si propaga in questo modo: H H R C C H H2C X H C H H H H R X C C C C H X H X Naturalmente, anche in questo caso, la regiochimica della propagazione segue la via che porta a prodotti termodinamicamente più stabili. Ecco le possibili regiochimiche che si possono presentare a seguito di una singola reazione di propagazione: H H H H R H H H H C C C C H H X X R C C C C H X H testa-coda X H H H H R C C C C H X testa-testa X H 28 H H H H R H H H H R C C C C X H H X coda-coda C C C C X H H X H H H H R coda-testa C C C C X H X H Secondo le regole di stabilità prima enunciate, la crescita avverrà prevalentemente secondo un meccanismo testa-coda, mentre la propagazione testa-testa e coda-coda costituirà la parte difettuale della catena. La quantità di difetti può essere valutata attraverso delle analisi 13 C-NMR. Queste analisi permettono infatti di discriminare l’intorno chimico (ossia la natura dei legami) degli atomi di carbonio della catena polimerica e di stimare dunque la percentuale di giunzioni testa-testa o coda-coda presenti nel campione. Il processo di polimerizzazione termina quando il centro reattivo sulla catena in accrescimento si spegne. Questo può aver luogo principalmente mediante due processi: • terminazione per accoppiamento: due catene polimeriche in accrescimento reagiscono a formare una singola catena inattiva. • RM(i+j)R' R'Mj RMi H H H H H H H H C C C C C C C C H X H H X X X H terminazione per disproporzionamento: si ha estrazione di un atomo di idrogeno da una catena, con conseguente formazione di due catene inattive, di cui una insatura: R'Mj RMi H H C C H X R'Mj RMi H H H H C C C C H C C H H H X X saturo H X insaturo Chiaramente i due meccanismi portano a due prodotti differenti. Se domina l’accoppiamento dei radicali si avranno polimeri con un alto numero di difetti testa-testa mentre se domina il processo di disproporzionamento di avrà un alto numero di terminali insaturi. Il processo di terminazione influenza naturalmente anche la distribuzione dei pesi molecolari del prodotto, come si vedrà in seguito. 29 3.1.2. Inibizione e ritardo L’inizio della polimerizzazione può essere inibita grazie all’aggiunta di additivi detti inibitori, in pratica molecole più reattive del monomero che consumano i radicali liberi. La reazione dell’inibitore con il centro radicalico sulla catena in accrescimento porta a prodotti inattivi e dunque blocca la polimerizzazione. RMi RMi Z Z L’aggiunta di un inibitore permette di ritardare l’inizio della polimerizzazione poiché questa inizierà quando tutto l’inibitore verrà consumato. Un tipico inibitore è il DPPH (2,2-difenil-1picralidrazile), sotto mostrato. O2N N N NO2 O2N DPPH Il composto è relativamente stabile (e dunque utilizzabile) perchè l’elettrone spaiato viene delocalizzato sul sistema aromatico elettronpovero. Altri inibitori utilizzati sono i chinoni, i fenoli e lo zolfo. Per rallentare la polimerizzazione si fa invece uso di ritardanti. Le molecole utilizzate sono, in questo caso, meno reattive delle molecole inibitrici precedentemente mostrate e consentono una più lenta crescita del polimero. I ritardanti permettono inoltre di aumentare il numero di terminazioni e dunque riducono i pesi molecolari. Una molecola di ritardante ZH reagisce con un radicale polimerico in accrescimento per trasferimento di un atomo di idrogeno: RMi RMiH ZH Z Il radicale Z• è abbastanza stabile da non iniziare una nuova polimerizzazione anche se ha la possibilità di terminarne una in corso, spegnendo la reattività del centro radicalico su di un’altra catena in propagazione. RMi Z RMiZ Le molecole utilizzate come ritardanti sono in genere fenoli sostituiti, chinoni, nitroderivati e N-ossidi. Il seguente diagramma mostra l’andamento della polimerizzazione in assenza di additivi (A), in presenza di un inibitore (B), di un ritardante (C) o di una molecola con proprietà intermedie (D). 30 3.1.3. Trasferimento Il trasferimento è un processo parallelo al processo di polimerizzazione grazie al quale il centro reattivo di una macromolecola in accrescimento viene trasferito ad un’altra macromolecola. In generale questo porta all’interruzione della crescita di una catena e all’inizio di una nuova. RMi M X X RMi X XM Il processo di trasferimento può essere promosso dal solvente in cui avviene la polimerizzazione o dall’aggiunta di molecole dette agenti trasferitori. Grazie ad un accurato utilizzo di questo processo è possibile regolare il peso molecolare del prodotto di sintesi. Tipici agenti trasferitori sono i composti polialogenati (come ad esempio il tetraclorometano), i tioli (composti contenenti gruppi –SH) o i solfuri (composti contenenti gruppi -SS-). Il meccanismo di trasferimento è illustrato prendendo come esempio un tiolo; questo cede un protone alla catena in accrescimento e si trasforma in un radicale libero RS•. H H C C H X H H H S C C H R H RS X Il radicale formatosi andrà quindi ad iniziare un nuovo processo di polimerizzazione attaccando un altro monomero presente in soluzione. RS H H H H C C RS C C H X H X Tra i vari processi di trasferimento, è bene sottolinearne uno in particolare. Nel trasferimento intramolecolare non esiste alcuna molecola esterna che partecipi al processo; il processo è in realtà monomolecolare, poiché il centro reattivo si trasferisce dall’estremità della catena in accrescimento all’interno della stessa catena. Questa alternativa porta a problemi inaspettati durante la sintesi dei polimeri. Ad esempio, il LDPE (Low Density Polyethylene) viene sintetizzato per via radicalica ad alte temperature e pressioni. Sotto queste condizioni si creano spesso lunghe ramificazioni, a causa del trasferimento di catena con del polimero già cresciuto, e ramificazioni corte (C2 o C4), dovute al trasferimento intramolecolare di un atomo di idrogeno. I pendagli butilici si formano a causa della relativa stabilità di un intermedio ciclico a sei atomi grazie al quale avviene il trasferimento intramolecolare: H2 C C H2 H2 C C H2 H2 C H2 H2 C C CH CH2 CH2 H C H2 H2 C CH2 C H H2 C C H2 H2 C CH3 Il centro reattivo trasferitosi all’interno della catena continua la sua polimerizzazione, portando alla ramificazione della macromolecola. Se il trasferimento si ripete dopo una singola addizione di un monomero, si formano due ramificazioni etiliche ravvicinate. H2 C C H H2 C C H2 H2 C CH3 H2C CH2 H2 H2 H2 C C C CH CH CH3 H2C C H2 H 31 H2 H2 C C CH CH CH2 CH2 CH3 CH3 Nonostante questi inconvenienti, i processi di trasferimento possono essere vantaggiosamente utilizzati per modulare il peso molecolare del prodotto. Come si vedrà nella sezione della cinetica, la legge di Mayo permette di correlare il grado di polimerizzazione del prodotto con il rapporto tra le concentrazioni del trasferitore e del monomero utilizzato nella sintesi. 3.1.4. Cinetica Abbiamo detto che una polimerizzazione a catena di tipo radicalico può essere suddivisa in tre fasi distinte: l’inizio, la propagazione e la fase di termine. La fase di inizio in realtà coinvolge due reazioni distinte, ossia la formazione del radicale iniziale per dissociazione omolitica dell’iniziatore I e la successiva formazione del germe di catena RM1• mediante addizione del radicale alla prima molecola di monomero. kd I + ( A ) ⎯⎯ ⎯→ n R • + (B ) k i R • + M ⎯⎯→ ⎯ RM1• Ognuno di questi due processi possiede una costante di equilibrio; nel caso della dissociazione dell’iniziatore, la costante verrà chiamata kd mentre nel caso dell’iniziazione vera e propria, ki. La propagazione consiste invece in una serie di ripetute reazioni di addizione del monomero al centro radicalico della catena in accrescimento. Il primo germe di catena RM1• addiziona dunque un monomero per divenire RM2•, e così via fino a quando la catena raggiunge una lunghezza imprecisata RMn+1•. k p RM1• + M ⎯⎯ ⎯ → RM•2 k p RM•2 + M ⎯⎯ ⎯ → RM•3 M k p RMn• + M ⎯⎯ ⎯ → RMn• +1 Si suppone che la costante di equilibrio per ciascuna singola reazione di accrescimento sia costante ed uguale a kp. Ad un certo punto la propagazione della catena polimerica verrà interrotta da uno dei molteplici processi di terminazione possibili all’interno dell’ambiente di reazione. Per semplicità, possiamo raggruppare i processi di terminazione in accoppiamento e disproporzionamento, definendo dunque kt,a e kt,d le costanti di equilibrio dei due processi. k RMn• + RMm• ,a ⎯⎯t⎯ → RM(n + m) k ,d ⎯⎯t⎯ → Rn + Rm Per ottenere un’espressione cinetica della velocità di polimerizzazione è indispensabile l’aver considerato kp costante per ciascuna reazione di propagazione. In realtà, la reattività della specie radicalica cambia a seconda della lunghezza della catena (i radicali di dimensioni ridotte sono ben più reattivi) e dunque cambia anche la costante di equilibrio. Il monomero sparisce ogni volta che reagisce con una specie radicalica. Dunque ogni volta che avviene un’iniziazione o una propagazione, parte del monomero iniziale scompare. Questo fatto può essere ben descritto attraverso la seguente equazione. 32 v pol = − d[M] = vi + vp dt Infatti, la velocità di polimerizzazione vpol non è nient’altro che la velocità con cui si riduce la concentrazione di monomero nell’ambiente di reazione; se il monomero scompare, si forma polimero. Essendo l’iniziazione una reazione del secondo ordine, la sua velocità è pari a: v pol = k i [R • ][M] + v p In prima approssimazione, il numero di monomeri che reagisce a causa del processo di iniziazione è molto minore rispetto a quello dovuto al processo di propagazione. Nell’ipotesi che vp >> vi, abbiamo: d[M] = v p = k p [RM• ][M] dt v pol = − La velocità di propagazione vp è proporzionale alle velocità di parecchie reazioni successive ma, dal momento che kp non è influenzata dalla reazione specifica, abbiamo potuto utilizzare la concentrazione totale di radicali [RM•], ossia: [RM• ] = ∑ [RM ] • n n Se consideriamo la variazione della concentrazione di radicali primari d[R•]/dt, essa sarà uguale alla differenza tra la velocità di produzione (velocità di dissociazione dell’iniziatore vd) e la velocità di consumo (velocità di iniziazione vi) di quest’ultimi. d[R • ] = v d − v i = v d − k i [R • ][M] dt Così anche per la variazione della concentrazione di specie radicaliche estese d[RM•]/dt che sarà uguale alla differenza tra la velocità di formazione (dovuta all’iniziazione, quindi vi) e di terminazione (sia per accoppiamento che per disproporzionamento, quindi vt,a e vt,d). Per semplicità consideriamo k t = k t ,a + k t ,d , così che: d[RM• ] = v i − v t = k i [R • ][M] − k t [RM• ]2 dt Per poterci inoltrare nella discussione dobbiamo a questo punto fare un’assunzione. Si assume che la concentrazione di radicali aumenti inizialmente per poi raggiungere un valore costante, stazionario. Questa approssimazione è detta approssimazione dello stato pseudo-stazionario. La concentrazione dei radicali dunque attraversa una fase di veloce transiente iniziale per poi diventare costante per tutto il resto della polimerizzazione. Le variazioni della concentrazione delle specie radicaliche [R•] e [RM•] nel tempo sono dunque nulle. d[R • ] = v d − k i [R • ][M] = 0 dt d[RM • ] = k i [R • ][M] − k t [RM • ]2 = 0 dt Da queste condizioni otteniamo le seguenti relazioni: [R • ] = vd k i [M] [RM• ] = vd kt 33 Possiamo a questo punto calcolarci la velocità di propagazione vp e, nel limite per vi << vp, la velocità di polimerizzazione vpol: v pol ≈ v p = − v d[M] = k p [RM • ][M] = k p [M] d dt kt Abbiamo dunque un’espressione che lega la velocità di propagazione (o di polimerizzazione) a delle costanti di equilibrio kp e kt, alla concentrazione di monomero [M] e alla velocità di dissociazione dell’iniziatore. Si può dimostrare che la velocità di produzione di radicali primari (e dunque la velocità di dissociazione) dalla scissione omolitica di iniziatori termici è data da: v d = 2f ⋅ k d [I] dove [I] è la concentrazione e f il fattore di efficienza dell’iniziatore. L’efficienza dell’iniziatore è definita come la frazione di radicali prodotti dalla scissione dell’iniziatore che effettivamente andranno ad iniziare la polimerizzazione. In genere il suo valore è minore dell’unità a causa di reazioni indesiderate che consumano radicali prima che questi vadano ad iniziare il processo. Uno dei motivi principali del numero ridotto di specie radicaliche che riescono ad iniziare realmente la polimerizzazione è l’effetto gabbia del solvente. Quando si formano i radicali primari, (negli esempi si riporta la decomposizione di benzoilperossido e AIBN) questi restano intrappolati tra le molecole di solvente e non riescono a raggiungere il monomero. All’interno della gabbia creata dal solvente, i radicali hanno modo di ricombinarsi o evolvere verso specie non reattive. O C O O C O O C O O C O O O C O C O Benzoilperossido N N CH3 H3C C CH3 C CH3 CN CN CH3 CH3 H3C C N N C CH3 CN CN N N CH3 CH3 H3C C C CH3 CN CN tetrametil-succino dinitrile AIBN Altre cause che portano ad una diminuzione del fattore di efficienza dell’iniziatore sono le reazioni di termine con le macromolecole in accrescimento, reazioni con il solvente e reazioni con le stesse molecole di iniziatore. 34 Conoscendo un’espressione della velocità di dissociazione dell’iniziatore e sapendo l’espressione della velocità di polimerizzazione, otteniamo la seguente relazione. v pol ≈ v p = k p 2f ⋅ k d [M] ⋅ [I]1 / 2 = k gl [M][I]1 / 2 kt il coefficiente kgl dipende dalle costanti di equilibrio e dal fattore di efficienza. Una volta determinato il sistema, tale fattore rimane costante. Mostriamo due grafici sperimentali dell’andamento della velocità di polimerizzazione dello stirene in funzione (A) della radice quadrata della concentrazione di iniziatore e (B) della concentrazione di monomero iniziale per sincerarsi dell’esattezza della relazione. (A) Velocità di polimerizzazione dello stirene a T = 60 °C in funzione di [I]1/2 in presenza di diversi iniziatori: AIBN, benzoilperossido (BP), cumilidroperossido (CHP) e t-butilperossido (t-BHP) (B) Velocità di polimerizzazione dello stirene in bromo-benzene a T = 50 °C in funzione di [M} in presenza di AIBN a diverse concentrazioni. Rimane da fare un’ultima considerazione sul grado di polimerizzazione del prodotto di una polimerizzazione radicalica. In assenza di trasferimento e per alti pesi molecolari, il grado di polimerizzazione DPn può essere messo in relazione con le velocità della reazione di propagazione e delle reazioni di terminazione. DP n = v t ,d vp vp =α 1 vt + v t ,a 2 Se la velocità di propagazione è infatti molto più grande delle velocità di terminazione si otterranno catene mediamente più lunghe, come è facile intuire. Il coefficiente ½ davanti a vt,a è giustificato dal fatto che, nelle terminazioni per accoppiamento, due catene in accrescimento dimerizzano, formando una catena lunga mediamente il doppio della lunghezza delle catene iniziali. Il coefficiente α introdotto è definito come: α= k t ,d kt 1 + k t ,a 2 con k t = k t ,d + k t ,a 35 ed è un indice di quanto prevalga una reazione di terminazione rispetto all’altra. Per α = 1 la terminazione avviene esclusivamente per disproporzionamento mentre per α = 2 si ha solo terminazione per accoppiamento (in ogni caso 1 ≤ α ≤ 2). Dall’espressione della velocità di propagazione sappiamo comunque che DP n ∝ [M] ⋅ [I]1 / 2 . In presenza di trasferimento, possiamo raggruppare tutti i processi in uno singolo e dunque sommare tutte le loro velocità. Otteniamo un termine vtr che dipende da processi di trasferimento al monomero, solvente, trasferitore, iniziatore o quant’altro possa provocare trasferimento. Il grado di polimerizzazione DPn sarà dunque uguale a: DP n = v t ,d vp vp = 1 vt + v t ,a + v tr + v tr 2 α Se le uniche forme di trasferimento possibile sono ad opera del solvente e del monomero, vtr = vtrS + vtrM. Sappiamo che la velocità di propagazione vp è proporzionale alla concentrazione di monomero e che v trS ∝ [S] e v trM ∝ [M] . Scrivendo il reciproco dell’equazione, 1 DP n = 1 DP 0 + CM + CS [S] [M] otteniamo l’equazione di Mayo. Questa relazione ci permette di conosce, dato DP0, ossia il grado di polimerizzazione in assenza di trasferimento, la riduzione del grado di polimerizzazione dovuto al trasferimento con il monomero e con il solvente. I coefficienti CM e CS sono dette costanti di trasferimento e sono definite attraverso le costanti di equilibrio delle reazioni di trasferimento e propagazione. Il seguente grafico mostra il reciproco del grado di polimerizzazione di una miscela di stirene a 100 °C in funzione del rapporto [S]/[M] con diversi solventi. 36 3.2. Polimerizzazione cationica La polimerizzazione cationica non è molto diffusa industrialmente anche se permette di ottenere alcuni polimeri interessanti. Alcuni comuni monomeri utilizzati nelle cationiche sono i vinileteri, l’isobutene e il benzofurano. H2C H C CH3 OR viniletere H2C C CH3 isobutene O benzofurano Da questi monomeri si ottengono il poli(iso)butene, il poliviniletere e il polibenzofurano. CH3 H2 C C H2 H C C OR n poliviniletere CH3 n poli(iso)butene O n polibenzofurano I cationi vengono solitamente generati attraverso l’addizione di un protone al legame insaturo. La presenza di sostituenti elettrondonatori o alchilici stabilizza il carbocatione rendendo questo processo possibile. Come iniziatori si utilizzano dunque acidi di Bronsted come H2SO4, H3PO4 e HF, o acidi di Lewis come gli alogenuri metallici (AlCl3, BF3, SnCl4, SbCl5, ZnCl2, TiCl4) e i loro derivati organometallici (RAlCl2, R2AlCl, R3Cl). L’iniziazione ad opera di acidi di Lewis richiede o, più in generale procede più velocemente, in presenza di un donatore di protoni (come acqua, HX, alcoli e acidi carbossilici) o di un donatore di carbocationi (come un alogenuro alchilico, esteri, eteri o anidridi). Secondo la terminologia normalmente usata, il donatore (di protoni o carbocationi) è detto iniziatore, mentre l’acido di Lewis è detto coiniziatore. I due composti (iniziatore e coiniziatore) reagiscono a formare un complesso, il quale dona il protone (o il carbocatione) al monomero e permette di iniziare la propagazione. Ad esempio, il BF3 o AlCl3 complessano l’iniziatore come mostrato in figura: F BF3 H2O BF3 H2O F F Cl AlCl3 Ph3CCl AlCl3 Ph3CCl Cl H B O H Ph Al Cl C Ph Cl Ph Il legame C-Cl del sale di carbenio utilizzato come iniziatore con AlCl3 possiede un considerevole carattere ionico dal momento che il catione Ph3C+ è particolarmente stabile (questo poichè il composto possiede parecchie forme mesomere derivanti dalla delocalizzazione della carica sugli anelli benzenici). Ph Ph C C Ph Ph3CCl Ph -Cl Ph Ph C C Ph Ph 37 Il legame insaturo presente nel monomero addiziona il protone o in generale il catione dell’iniziatore, generando un carbocatione. R H H H H C C H R C C ED H ED Ad esempio, il monomero isobutene addiziona un protone in presenza di H2SO4, generando un carbocatione terziario, quindi particolarmente stabile. H+ H CH3 H2SO4 C C H H CH3 R C C CH3 H CH3 La propagazione avviene dunque per continua addizione di monomeri alla macromolecola in accrescimento. Il sito reattivo (cationico) si sposta durante la propagazione al termine della catena. H CH3 R CH3 C C H CH3 H C C H CH3 R CH2 C CH2 C CH3 CH3 CH3 CH3 In teoria il carbocatione può presentare diversi riarrangiamenti, ma la specie predominante è sempre quella energicamente più stabile. Nel caso dell’isobutene, il carbocatione terziario è sempre preferito all’altra alternativa, il carbocatione primario. Un altro esempio di polimerizzazione cationica è quello della polimerizzazione del THF (tetraidrofurano). Normalmente il THF è utilizzato come solvente ma, in condizione acide, esso apre l’anello generando un carbocatione contenente un gruppo etereo. R O RO H2 C C H2 H2 C O CH2 H2 C C H2 H2 C C H2 n La polimerizzazione del THF non porta però a polimeri con alti pesi molecolari e dunque non viene normalmente utilizzata. Sono varie le reazioni che portano alla terminazione della crescita della catena nella polimerizzazione cationica. La maggior parte di queste reazioni comunque non interrompono la cinetica della polimerizzazione poichè si genera una nuova specie cationica che continua a propagarsi. Il processo di trasferimento più comune è quello del trasferimento di un protone in posizione β rispetto al centro cationico. Il legame insaturo del monomero, invece di attaccare il centro reattivo della catena in propagazione, attacca il β-protone. H CH3 C C H CH3 H CH3 C C H H CH3 C C CH3 H CH3 H C C H CH3 CH3 Si ha dunque la terminazione della catena, con formazione di un legame insaturo, e la generazione di un nuovo centro reattivo che continua la crescita. Alternativamente, il 38 trasferimento di un protone può avvenire anche ad opera del solvente come mostrato nel caso del toluene. H H CH3 CH2 CH2 H CH3 C C C C H H CH3 H CH3 Il trasferimento è reso possibile dalla buona stabilità del catione benzilico. Da notare che in questo caso la catena termina con un legame saturo, a differenza del trasferimento precedente. Si possono presentare processi di trasferimento anche con il controione. In generale questi processi sono molto rari e portano alla terminazione della catena senza inizio di una nuova propagazione. Il controione può ricombinarsi con il carbocatione o strappare un β-protone alla catena come mostrato nei seguenti esempi. H CH3 H CH3 C C ricombinazione del controione C C A A H CH3 H CH3 H CH3 H CH3 A C C C C H CH3 HA eliminazione dell’idrogeno CH3 3.3. Polimerizzazione anionica 3.3.1. Principi base La polimerizzazione anionica presenta molte delle caratteristiche della polimerizzazione cationica appena illustrata, sebbene vi siano alcune differenze fondamentali. La specie reattiva in questo caso è un centro anionico e, a differenza della controparte positiva, la sua reattività dipende fortemente dalle condizioni dell’intorno. A seconda della nudità del centro reattivo, la polimerizzazione avviene più o meno velocemente. In soluzione, il centro anionico ed il controione possono infatti possedere diversi gradi di associazione con il solvente, a seconda della polarità di quest’ultimo. I solventi che possiedono una maggiore associazione con il controione, lasciano il centro anionico libero di reagire, aumentandone la cinetica. Data la forte reattività del centro anionico, bisogna evitare qualsiasi possibilità di attacco nucleofilo indesiderato. Composti elettronpoveri come i gruppi carbonilici o le specie acide come l’acqua sono severamente vietate in una polimerizzazione anionica, poichè spegnerebbero la reattività dell’anione. I solventi utilizzati si limitano quindi agli idrocarburi alifatici (esano e cicloesano), aromatici (benzene) e agli eteri (THF, Et2O). I solventi alogenati, largamente utilizzati nelle polimerizzazioni radicaliche, non sono adatti alle sintesi anioniche poichè porterebbero a facili sostituzioni nucleofile. Nelle polimerizzazioni anioniche, l’iniziatore deve riuscire a formare un carbanione. L’acidità di un idrocarburo è molto bassa (pKa ≈ 50) ed è per questo che per poter creare il carbanione bisogna utilizzare composti altamente reattivi. Un esempio di iniziatore è un metallo alcalino (litio o sodio), il cui potenziale di ossido-riduzione è abbastanza elevato da poter strappare un protone ad un idrocarburo. Li H3C H2 C C H2 CH3 Li C H2 H2 C C H2 CH3 H2 In questo esempio si mostra la formazione di un carbanione butilico dalla reazione di litio metallico con il butano. Il legame metallo-carbonio non è propriamente un legame ionico, ma 39 possiede bensì un certo grado di proprietà covalenti. Il solvente etereo permette la solvatazione della specie, rendendo il centro anionico più libero, e dunque più reattivo. L’aggiunta di una piccola quantità di etere corona permette addirittura di isolare il controione metallico. O O + Li O O Il catione infatti si coordina con gli ossigeni dell’etere corona e il centro anionico rimane “nudo” e libero di reagire. Altri iniziatori nelle polimerizzazioni radicaliche possono essere la sodio ammide (NaNH2), idrossidi ed alcossidi e alcuni composti particolari, come ad esempio il litiofluorene, derivante dalla reazione del litio metallico con il fluorene. H2 Li Li fluorene litiofluorene L’idrogeno del fluorene è particolarmente acido poichè, senza di esso, il composto acquista un certo grado di aromaticità. Quando reagisce a formare il carbanione, il litiofluorene acquista l’idrogeno e torna nella forma di fluorene. La particolarità di questo iniziatore è che il litiofluorene assorbe nel visibile (e quindi appare colorato) mentre il fluorene assorbe nell’ultravioletto (e dunque appare incolore). La perdita di colorazione diventa quindi indice dell’avvenuto inizio della polimerizzazione. Quasi tutti i polimeri vinilici possono essere ottenuti via anionica. Il meccanismo di inizio è l’attacco nucleofilo del carbanione al legame vinilico, con formazione di un nuovo centro reattivo sulla testa del monomero. Ad esempio, nel caso della polimerizzazione anionica dello stirene con LiBu come iniziatore: H3C C H2 H2 C H3C CH2 C H2 H2 C C H2 H2 C Li CH Li La propagazione si ha per successivi attacchi del centro anionico sui legami insaturi dei monomeri. Li H3C C H2 H2 C C H2 H2 C CH H3C C H2 H2 C H2 H2 C C C CH CH H2 Ph Ph Li La particolarità della polimerizzazione anionica è che gli anioni non possono dimerizzare ne subire processi di trasferimento con il solvente. Se la reazione viene eseguita con molta accortezza, evitando quindi qualsiasi impurità nociva per la reattività del carbanione, la polimerizzazione non termina mai. Si parla in questo caso di polimerizzazione vivente. Questa peculiarità permette di ottenere polimeri con una distribuzione dei pesi molecolari particolarmente ristretta dal momento che, se non esistono reazioni di terminazione, l’accrescimento è lineare nel tempo e la reattività non si spegne mai. 40 Per spegnere la polimerizzazione è sufficiente introdurre dell’anidride carbonica (CO2) o quasiasi altro composto con un centro elettronpovero. O H C O H2C CH2 H2 C C O H2 H H2 C C C O H2 H2 CH2 CH2 O C O O C C O H2 CH2 I terminali di catena possono essere quindi funzionalizzati a seconda delle esigenze. Per terminare la polimerizzazione senza funzionalizzare il terminali di catena, è sufficiente introdurre nell’ambiente di reazione un composto che contiene idrogeni acidi, come ad esempio l’acqua. Nel 1968 Szwarc e I suoi collaboratori studiarono alcune interessanti polimerizzazioni iniziate dalla presenza di anioni-radicali aromatici come il sistema sodio naftalene. L’iniziatore attivo della polimerizzazione è l’anione-radicale naftalene: Na Na La reazione coinvolge il trasferimento di un elettrone dal metallo alcalino al naftalene. Questo sistema si forma quantitativamente se viene generato in THF e permette di sintetizzare catene polimeriche che si propagano da ambo i terminali. Se consideriamo lo stirene, l’iniziatore trasferisce un elettrone al monomero in modo da formare l’aniore-radicale stirile: H2C CH H2C CH Na Na Questo reagisce con un secondo anione-radicale stirile, dimerizzando. Il prodotto della dimerizzazione è un dicarbanione che può accrescere da entrambi i terminali. HC CH2 H2C CH H2 H2 HC C C CH 3.3.2. Copolimeri a blocchi La polimerizzazione anionica offre la possibilità di creare dei copolimeri a blocchi. AAAAAAAAA-BBBBBBBBB (1) BBBBBB-AAAAAA-BBBBBB (2) Un copolimero è un polimero formato dalla ripetizione di blocchi di monomeri chimicamente diversi. Gli esempi mostrano due diversi tipi di copolimeri a blocchi; in particolare (1) è 41 formato da due blocchi, uno di monomeri di tipo A e uno di monomeri di tipo B, mentre (2) è formato da tre blocchi, due composti da monomeri di tipo B e uno da monomeri di tipo A. I copolimeri possiedono proprietà significativamente diverse sia da quelle polimeri semplici che da quelle di miscele di diversi polimeri. Un esempio di copolimero a blocchi particolarmente diffuso nell’industria delle gomme (in particolare nella fabbricazione dei pneumatici) e apprezzato per le sue proprietà elastomeriche è l’SBR (Styrene-Butadiene Rubber). La polimerizzazione anionica vivente permette la realizzazione di questi sistemi complessi. Se infatti la reattività del centro anionico non si spegne mai, una volta esaurito il monomero A, è possibile inserire all’interno dell’ambiente di reazione un diverso monomero B e continuare l’accrescimento della catena. Il prodotto risultante è un copolimero tipo (1): B AAAAA AAAAABBBBB Se il blocco iniziale possiede reattività anioniche su entrambi i terminali, è possibile accrescere il copolimero (2) mediante l’introduzione del monomero B. B AAAAA BBBBBAAAAABBBBB In generale ci sono dei limiti al numero e tipo di blocchi che è possibile copolimerizzare via anionica. Il centro anionico deve infatti essere abbastanza reattivo da poter attaccare la funzionalità del monomero. In generale la scala di reattività delle specie anioniche viene valutata confrontando la reattività dei monomeri da cui derivano. Se il monomero è stabile, il suo carbanione è in generale molto reattivo perché tende a ritornare alla più stabile forma neutra. Al contrario, se il monomero è reattivo, la specie anionica è relativamente più stabile. In questa tabella si riporta una scala delle reattività dei monomeri e dei corrispondenti anioni. MONOMERO CH2 HC H2C H C H C C H CH2 butadiene stirene H2C ANIONE DERIVATO C OR REATTIVITA' O acrilati O H2C CH2 epossidi CN acrilonitrile Quindi se nell’esempio precedente, il monomero A è un acrilato, il monomero B potrà essere un acrilonitrile, ma non lo stirene o il butadiene. La polimerizzazione anionica permette inoltre di ottenere copolimeri o omopolimeri a stella. Se il centro reattivo viene spento con un sistema che può subire più di una sostituzione nucleofila come ad esempio un tetraclorosilano, si ottengono degli stellati. Cl 4 CH2 Cl Si Cl Cl CH2 H2 C Si C H2 CH2 4Cl- 42 4. POLIMERIZZAZIONE DI COORDINAZIONE La polimerizzazione stereoselettiva venne introdotta durante la metà degli anni cinquanta grazie al lavoro separato di Ziegler in Germania e di Natta in Italia. In quel periodo, Ziegler stava studiando le reazioni dell’etilene catalizzate da trialchilalluminio ad alta temperatura e pressione. Con i primi tentativi, ottenne solamente miscele di oligomeri e polimeri a basso peso molecolare (il peso molecolare più alto si accostava attorno alle 5000 unità ripetenti). L’aggiunta di un composto contenente un metallo di transizione al trialchilalluminio ebbe effetti sconvolgenti. La sintesi, a basse temperature (50-100°C) e pressioni, portò infatti alla formazione di polietilene ad alto peso molecolare, con un basso grado di ramificazioni e proprietà meccaniche superiori rispetto al normale polietilene sintetizzato fino ad allora per via radicalica. In Italia, Natta utilizzò i catalizzatori sperimentati da Ziegler per ottenere polimerizzazioni stereoselettive (sia iso che sindioselettive) del propilene ed altre α-olefine. Il risultato fu di grande successo poichè fino ad allora le poli-αolefine prodotte per via radicalica o ionica non possedevano alti pesi molecolari. Per l’enorme importanza scientifica ed applicativa del loro lavoro, nel 1963 Ziegler e Natta ottennero il premo Nobel per la chimica. I risultati ottenuti da Ziegler e da Natta portarono allo sviluppo di un’ampia classe di sistemi per la catalisi polimerica eterogenea a due componenti, come ad esempio composti organometallici o alogenuri di metalli del I-III gruppo assieme a derivati contenenti un metallo di transizione del IV-VIII gruppo. Il componente più importante del sistema è il composto contenente il metallo di transizione; alcuni esempi sono TiCl3, TiCl4 e VCl3. La funzione del composto contenente il metallo del I o III gruppo è invece quello di attivare il composto contenente il metallo di transizione e per questo vengono detti cocatalizzatori; alcuni dei cocatalizzatori più comuni sono AlR3, AlR2Cl e ZnEt2. Questi sistemi catalitici sono comunemente conosciuti come catalizzatori Ziegler-Natta. Nella seconda metà degli anni ottanta ci fu un’altra rivoluzione nel settore delle polimerizzazioni stereoselettive: l’introduzione dei metalloceni come catalizzatori. Alcuni esempi di metalloceni industrialmente utilizzati sono il bis(ciclopentadienil)titanio dicloruro e il bis(indenil)zirconio dicloruro. Cl Ti Cl Cl Zr Cl Alcuni metalloceni più complessi possiedono sostituenti sui liganti e un ponte metilenico che li raccorda. H2C Ti Cl H2C Zn Cl Diversamente dai tradizionali catalizzatori Ziegler-Natta, i sistemi metallocenici possono essere dispersi in soluzione e dare quindi luogo a catalisi omogenea. Modificando la chimica dei liganti è possibile modulare la stereoselettività della catalisi ed ottenere prodotti altamente stereospecifici. Anche i metalloceni, così come i tradizionali catalizzatori Ziegler-Natta, necessitano di un composto contenente un metallo del I o III gruppo per poter essere attivati. 43 Sia i metalloceni che i catalizzatori di Ziegler-Natta tradizionali possono essere raggruppati nella più ampia classe dei catalizzatori per coordinazione. Le polimerizzazioni che permettono di ottenere sono dette polimerizzazioni di coordinazione. Il termine coordinazione sottolinea il meccanismo con cui viene spiegata la stereospecificità della polimerizzazione. 4.1. Catalizzatori Ziegler-Natta 4.1.1. Sviluppo storico Prima di procedere oltre ed esaminare il meccanismo della polimerizzazione di coordinazione, ci soffermiamo sull’evoluzione delle varie generazioni dei più tradizionali catalizzatori Ziegler-Natta. Il primo sistema studiato da Ziegler (e successivamente da Natta) per ottenere polimerizzazioni stereoselettive è il sistema Ti-Al, ossia un composto contenente titanio, metallo di transizione del IV gruppo, e un composto contenente alluminio, metallo del III gruppo. Il catalizzatore vero e proprio è dunque il composto contenete il titanio mentre il cocatalizzatore è il composto contenente l’alluminio. Per catalizzare la polimerizzazione dell’etilene, Ziegler utilizzo originariamente TiCl4 e AlEt3 in solvente idrocarburico. La reazione del TiCl4 con AlEt3 porta allo scambio dei sostituenti, in particolare di un sostituente etilico dal cocatalizzatore al TiCl4. TiCl4 AlEt3 TiCl3Et AlEt2Cl La vera specie attiva è in realtà il TiCl3 in forma β poichè il sistema evolve attraverso una serie di reazioni complesse che portano alla formazione di β-TiCl3 e di oligomeri di polietilene. TiCl3Et Et Et β-TiCl3 CH3(CH2)2CH3 Et polietilene (oligomeri) Natta si accorse della forte presenza di β-TiCl3 (di colore marrore) e cerco di migliorare l’efficienza del sistema, per esempio riducendo il TiCl4 con idrogeno prima di utilizzarlo nella catalisi. La stereoselettività di questi primi iniziatori si accostava attorno al 20-40% di percentuale di polipropilene isotattico in miscela. Successivamente, l’utilizzo dei polimorfi cristallini α-, δ- e γ- del TiCl3 portarono ad un aumento della stereoselettività della polimerizzazione. Alla fine degli anni cinquanta, la percentuale di polipropilene isotattico sintetizzato mediante questi catalizzatori raggiunse il 90% in miscela. La tecnica di ballmilling, ossia la deformazione plastica della superficie del catalizzatore ad opera di piccole sfere di acciaio, portò ad un miglioramento dell’efficienza della catalisi. Questo risultato è da attribuirsi al fatto che la deformazione superficiale porta alla formazione di difetti e coordinazioni insature del titanio, e quindi fornisce un maggior numero di siti attivi per la polimerizzazione. In ogni caso, l’attività del catalizzatore risultava ancora troppo bassa per permettersi sintesi senza una successiva purificazione del prodotto (eliminazione della parte atattica del polimero e eventuali parti di catalizzatore disperse nella miscela). Le successive generazioni di cataliizatori migliorarono l’efficienza della catalisi senza rinunciare alla stereoselettività. La superficie attiva fu approssimativamente raddoppiata utilizzato un supporto di MgCl2 sul quale veniva finemente disperso TiCl4. L’alta attività del catalizzatore non solo ridusse i costi della produzione, ma eliminò anche l’operazione di purificazione. Infatti, la percentuale di isoselettività raggiunse il 98% rendendo inutile anche l’eliminazione della parte atattica del prodotto. 44 4.1.2. Meccanismo della propagazione stereoselettiva Per spiegare la polimerizzazione stereoselettiva catalizzata dai sistemi Ziegler-Natta sono state proposte varie teorie. Secondo la teoria più accreditata, la specie attiva che catalizza il processo è il metallo di transizione (nel caso specifico del sistema Ti-Al, il titanio dunque). La polimerizzazione dipende intimamente dalla struttura cristallina della superficie del catalizzatore. Un reticolo di coordinazione, come quello del β-TiCl3, possiede un certo numero di siti vacanti per assicurarsi la complessiva elettroneutralità del cristallo. In particolare, il cristallo di α-TiCl3 possiede strati di titanio che si alternano a strati di cloro, come mostrato in figura. Gli atomi di titanio sono al centro di ottaedri, ai cui vertici si trovano atomi di cloro. Ogni due atomi di titanio all’interno dello strato, il terzo è vacante ( ). La polimerizzazione ha luogo sui siti attivi alla superficie del cristallo; in particolare essa avviene su dei titani coordinati con solamente 5 atomi di cloro (il sesto sito è vacante perchè sia mantenuta l’elettroneutralità del cristallo). Quattro di questi atomi sono fortemente legati al metallo di transizione perchè fanno da ponte con altri titani più interni. Il quinto atomo di cloro è solo debolmente legato e può essere quindi sostituito da un gruppo alchilico quando il cocatalizzatore reagisce con il metallo di transizione. Ti Ti Ti Ti Ti Ti Ti Ti R Ti Ti Ti AlR3 Ti Ti Ti Ti R Ti Ti Ti Ti Ti Ti Cl Cl Ti Cl Cl Ti Il simbolo rappresenta un sito non coordinato del complesso ottaedrico del titanio attivo. Il frammento sopra mostrato rappresenta un intorno dei titani alla superficie del cristallo di TiCl3 dopo la reazione con il cocatalizzatore (trialchilalluminio). I siti attivi, ossia gli atomi di titanio superficiali, condividono quindi quattro leganti cloro con gli atomi di titanio vicini, un gruppo alchilico R e un orbitale vuoto. 45 La chiave della polimerizzazione stereospecifica catalizzata dagli iniziatori ZieglerNatta è la chiralità del sito attivo. Tale proprietà può essere facilmente intuita osservando la figura riportata sopra in cui si mostrano i due differenti enantiomeri del centro stereogenico. Il meccanismo per razionalizzare la propagazione stereoselettiva fu proposto da Arlman e Cossee nel 1964. PROPAGAZIONE ISOSELETTIVA H2C Cl Cl Ti Cl Cl H2 C H2C CHCH3 Cl Ti Cl Cl H2C Cl CHCH3 Cl Ti Cl Cl CH2 Cl CH2CH3 CH3 CH2 CHCH3 H2C Cl Cl Ti Cl Cl CH2 Cl Cl Ti Cl Cl CH2 CHCH3 Il monomero si coordina con il titanio nel sito vacante, formando uno stato di transizione a quattro membri. L’inserzione del monomero sposta l’estremità della catena in accrescimento nel sito originariamente occupato dal monomero; è da sottolineare il fatto che non è l’intera macromolecola a muoversi, ma solamente la parte terminale. Per spiegare l’isoselettività della sintesi, la catena in accrescimento deve obbligatoriamente ritornare nel sito iniziale; così facendo, si ripresenta dunque il metallo di transizione coordinato con il gruppo alchilico in accrescimento, pronto per una nuova inserzione isoselettiva. La catena dunque migra due volte per ogni monomero inserito perchè si possa ottenere un prodotto isotattico. PROPAGAZIONE SINDIOSELETTIVA H2C Cl Cl V Cl Cl H2C H2C CHCH3 Cl V Cl Cl H2C Cl CHCH3 Cl V Cl Cl CH2 Cl CH2CH3 CH3 CH2 CHCH3 CH2 CH3HC H2C Cl Cl V Cl Cl CH3 H3C CH2 Cl Cl V Cl Cl CH2 C CHCH3 H3 H 2C CHCH3 Cl Cl V Cl Cl CH2 CH2 CHCH3 46 Al contrario, nel caso di una polimerizzazione sindioselettiva, la catena non migra dopo l’inserimento del monomero. La ripetuta coordinazione di nuovi monomeri al sito vacante che si forma dopo l’inserzione porta ad un prodotto sindiotattico. Nell’esempio si mostra la sintesi sindioselettiva del polipropilene catalizzata da VCl4 e AlEt3. Nella propagazione, il polimero che si forma è solitamente un polimero testa-coda con alta percentuale iso(o sindio)-tattica. L’inserzione del monomero che porta ad un polimero così fatto è detta inserzione primaria. Anche se molto meno probabile, è possibile avere un’inserzione che porta a difettualità testa-testa. Tale tipo di inserzione è detta inserzione secondaria. Cat H2 Cat CH C H2 H C C CH3 CH3 inserzione secondaria inserzione primaria L’apertura del legame insaturo può avvenire in due modi. Si può infatti avere addizione cis o addizione trans. Se si polimerizzano delle olefine disostituite si otterranno rispettivamente prodotti treo ed eritro. cis R' R' R' treo H R' C C R R R R H trans eritro R' R R' R R' R Analisi NMR in soluzione hanno permesso di verificare che i catalizzatori Ziegler-Natta operano solamente addizioni di tipo cis e il prodotto della polimerizzazione di olefine disostituite è sempre quello con configurazione treo. Tra i polimeri più importanti che possono essere ottenuti da una sintesi stereospecifica mediante catalizatori Ziegler-Natta abbiamo naturamente il polipropilene, il polistirene, e i polimeri derivanti da dieni coniugati come il polibutadiene e il polisoprene. La polimerizzazione dello stirene confuta l’ipotesi di un meccanismo di tipo anionico poichè il processo non è in generale influenzato dalla natura dei sostituenti del monomero. La polimerizzazione dei dieni coniugati porta a prodotti 1,4-cis e non si presenta mai il prodotto trans. 4.1.3. Cinetica Le cinetiche delle polimerizzazioni catalizzate da iniziatori Ziegler-Natta in fase eterogenea sono complesse e diversificate. Nel grafico si mostrano tre tipici andamenti della velocità di polimerizzazione in funzione del tempo. Sia il comportamento (1) che il comportamento (2) prevedono un tempo iniziale di attivazione dei siti catalitici, dovuto per la maggior parte alla reazione del cocatalizzatore con il metallo di transizione. L’andamento descritto dalla curva (2) mostra un tempo di attivazione minore; questo è infatti il caso di superfici trattate con un processo di ball-milling che permette di creare un maggior numero di siti catalitici attivi ed accessibili al cocatalizzatore. In entrambi i casi, dopo un periodo di attivazione iniziale, la velocità di polimerizzazione raggiunge uno stato stazionario, stabilizzandosi. Altri sistemi mostrano un andamento simile a quello mostrato dalla curva (3). L’attività dei siti catalitici viene velocemente promossa per poi decadere verso lo stato stazionario. Questo è in parte dovuto a siti catalitici con diversa attività e alla degradazione nel tempo di alcuni di questi. In ogni caso, dopo un certo tempo di vita, l’attività dei catalizzatori Ziegler-Natta inizia a decadere, a causa del loro avvelenamento. 47 L’avvelenamento è dovuto alla coordinazione dei siti attivi con impurezze, alla loro occlusione da parte di sostanza polimerica sintetizzata o al degradamento termico. 4.2. Iniziatori metallocenici I metalloceni sono composti di formula generale LL’MtX2 e hanno ricevuto enorme attenzione nell’ambito della catalisi per polimerizzazioni stereoselettive. Il simbolo L o (L’) sta ad indicare la presenza di un ligante, ossia un composto elettronricco che si coordina al metallo di transizione (Mt) del IV gruppo grazie a due suoi orbitali non occupati. Alcuni dei liganti più comunemente utilizzati sono gli η5-ciclopedienili, η5-indenili o η5-fluorenili, sotto rappresentati. η5-pentadienile η5-indenile η5-fluorenile Si ricorda che il numero che segue il simbolo η (eta) indica il numero di elettroni del ligante che partecipano alla complessazione del metallo. I metalli solitamente utilizzati sono zirconio (Zr), titanio (Ti) o più raramente afnio (Hf), mentre i gruppi X (solitamente Cl e Br, ma anche gruppi metilici CH3) sono direttamente legati al metallo. L’interesse iniziale nei composti metallocenici era quello di studiare dei modelli per meglio comprendere il meccanismo di funzionamento degli iniziatori isoselettivi in fase omogenea, ma ben presto ci si rese conto che questi offrivano nuove opportunità rispetto ai tradizionali catalizzatori Ziegler-Natta. Ad esempio, parecchi composti metallocenici possiedono attività catalitiche ben maggiori rispetto ai tradizionali catalizzatori eterogenei proprio perché, essendo in fase omogenea, ogni metallo di transizione si comporta da sito attivo. Inoltre, la stereoselettività può essere controllata mediante la giusta scelta dei sostituenti da posizionare sui liganti e questo porta alla sintesi di prodotti configurazionalmente unici, non raggiungibili mediante catalisi eterogenea. I primi metalloceni studiati furono il diclorotitanocene e il diclorozirconiocene, sotto rappresentati. Cl Ti Cl dicloro-titanocene Cl Zr Cl dicloro-zirconiocene 48 Così come i tradizionali catalizzatori Ziegler-Natta, anche i metalloceni necessitano di un coiniziatore per poter avviare la polimerizzazione. Anche in questo caso si utilizzano degli acidi di Lewis, più comunemente AlRCl2 o AlR3. L’azione del coiniziatore è quello di strappare un gruppo X dal metallo di transizione e di rimpiazzare l’altro gruppo X con un gruppo alchilico R. Il composto attivo è dunque: R Mt catalizzatore attivo Come si deduce dalla figura, il metallo di transizione possiede un sito vacante e una piccola catena alchilica. Il meccanismo di accrescimento della catena polimerica è simile al modello alternante di Cossee presentato per la catalisi eterogenea. Cp Cp Mt Cp H2C Mt R Cp CH2 R Cp = Cp Cp H2 R C C H2 Mt H2 C Cp Mt Cp CH2 R Questi tipi di metalloceni portarono alla polimerizzazione di varie α-olefine come ad esempio il propilene. Purtroppo la cinetica della sintesi era ancora abbastanza ridotta ma, soprattutto, i prodotti ottenuti possedevano bassi pesi molecolari ed erano atattici. La polimerizzazione stereoselettiva che portava a prodotti con alti pesi molecolari poteva essere raggiunta solamente utilizzando un iniziatore metallocenico che fosse sia chirale che stereorigido. Nel dicloro-titanocene, ad esempio, i due leganti ciclopentadienili non possiedono alcuna ragione per non muoversi nello spazio, e anche se così fosse, non rendono affatto chirale il sistema. Il problema della stereorigidità è stato risolto creando un ponte tra i due liganti, solitamente un ponte metilenico –CH2-, di modo che la struttura sia rigida, priva della possibilità di muoversi liberamente. I liganti planari si dispongono nello spazio con una certa inclinazione esponendo il metallo di transizione ed i suoi siti attivi all’ambiente esterno. La forma caratteristica di questi metalloceni ne determinò la scelta del nome, ossia metalloceni ad ansa. H2C Mt X X metallocene ad ansa Anche in questo modo, però, la polimerizzazione non è assolutamente stereospecifica (tranne per alcuni casi particolari di scarso interesse applicativo in cui si produce una piccola quantità di prodotto stereoselettivo) e questo è dovuto al fatto che il sistema risulta di per sé achirale. La necessità di fornire un qualche tipo di chiralità ai siti attivi del catalizzatore porta all’introduzione di sostituenti sui liganti. Se infatti si modifica l’impalcatura dell’iniziatore in 49 maniera ragionata è possibile rendere stereospecifica la polimerizzazione. Se, ad esempio, si introducono due anelli benzenici sui ciclopentadienili come mostrato in figura, si ottiene un sistema con simmetria C2 (l’unico elemento di simmetria del complesso è un asse di rotazione binario). I due spazi ai lati del metallo di transizione possiedono ora una propria simmetria; in questa particolare situazione gli spazi vengono detti omotopici poiché possiedono la medesima simmetria. H2 C Mt X X Catalizzatori omotopici portano alla formazione di prodotti isotattici. Per ottenere polimeri sindiotattici si deve invece creare un catalizzatore con simmetria enantiotopica. Il seguente metallocene ne è un esempio: H2C X Mt X In questo caso i due spazi ai lati del metallo di transizione possiedono simmetria enantiomerica, ossia opposta. La simmetria complessiva del complesso è CS poiché esso possiede solamente un piano di simmetria. Basta aggiungere un sostituente (nell’esempio un metile) sul ciclopentadienile superiore per distruggere completamente la simmetria del metallocene (quindi avere simmetria C1). H3C H2C X Mt X Gli spazi ai lati del metallo, in questo caso, non possiedono né uguale simmetria né simmetria opposta. Essi possiedono semplicemente una diversa simmetria e per questo vengono chiamati spazi diastereotopici. Un catalizzatore diastereotopico porta alla formazione di prodotti emisotattici. Il polimero in questo caso possiede una stereoregolarità diversa da quella del polimero isotattico o sindiotattico; in successione il polimero possiede siti con una definita tassia (isotattica) alternati a siti atattici, come mostrato in figura. CH3 H CH3 H CH3 H H H CH3 H H H H H H H H H CH3 H H H CH3 H Le unità monomeriche in grassetto sono quelle che possiedono configurazione isotattica mentre quelli in corsivo possiedono configurazione atattica. 50 4.3. Analisi della stereoregolarità Per studiare la stereoregolarità di un campione polimerico si utilizza la spettroscopia di risonanza magnetica nucleare (NMR, Nuclear Magnetic Resonance). Le spettroscopie ad alta risoluzione 1H-NMR e 13C-NMR permettono di ottenere un gran numero di informazioni sulla distribuzione sequenziale delle unità stereoisomeriche all’interno della catena polimerica. m A r A A A Consideriamo il prodotto della polimerizzazione di una α-olefina; all’interno della catena possiamo riconoscere alcuni frammenti che distinguono la configurazione isotattica e la configurazione sindiotattica. Il caso più semplice distingue le diadi, ossia frammenti di catena formati da due unità ripetenti. Il frammento [m] (meso) corrisponde ad una configurazione isotattica mentre il frammento [r] (racemic) ad una configurazione sindiotattica. m r isotattico sindiotattico La spettroscopia NMR è sensibile all’intorno chimico degli atomi su cui viene eseguita e dunque distingue i contributi delle due diverse diadi. Nella situazione più banale si avranno due picchi, uno corrispondente alla frazione di catena che possiede una configurazione tipo [m] e l’altro che corrisponde ai frammenti [r]. A seconda dell’abbondanza dei frammenti nel campione, l’intensità dei picchi varierà. Per un polimero altamente isotattico il picco relativo alla diade [m] sarà particolarmente marcato mentre quello relativo alla diade [r] quasi inesistente, per un polimero altamente sindiotattico accadrà l’opposto. Un polimero atattico presenterà statisticamente un egual numero di contributi dovuti alle diadi [m] e [r] e le intensità dei due picchi sarà pressoché uguale. Se consideriamo le triadi la faccenda si complica. Abbiamo quattro combinazioni possibili, ossia il caso isotattico [mm], quello sindiotattico [rr] e due eterotattici [mr] [rm]. Così come per le diadi, si avrà un picco per ciascun diverso frammento. Il picco dovuto alla triade eterotattica avrà sempre un’intensità superiore rispetto agli altri due perchè è la somma di due contributi equivalenti. Il caso di un polimero altamente iso o sindiotattico è praticamente uguale alla situazione illustrata nel caso delle diade, predomineranno o solamente i picchi [mm] o solamente i picchi [rr]. Nel caso di un campione atattico i picchi [mm] e [rr] avranno uguale intensità mentre il picco [mr] avrà intensità doppia. Se il picco [mr] predomina (in proporzione) rispetto agli altri due, allora il campione può presentare un certo grado di emitatticità. m m isotattico r r sindiotattico m r eterotattico L’avvento della spettroscopia 13C-NMR ad alta risoluzione permette la distinzione di frammenti sempre più lunghi e consente quindi di studiare le tetradi o le pentadi. Aumentando la lunghezza del frammento base la trattazione si complica ma si riesce ad ottenere una sempre più accurata analisi della stereoregolarità del campione polimerico. 51
Scaricare