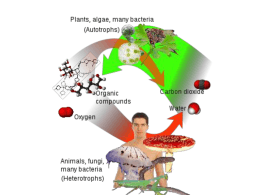
A07 25 Marinella Bosetto Irene Lozzi Elementi di biochimica agraria Copyright © MMVI ARACNE editrice S.r.l. www.aracneeditrice.it [email protected] via Raffaele Garofalo, 133 A/B 00173 Roma (06) 93781065 ISBN 88–548–0724–9 I diritti di traduzione, di memorizzazione elettronica, di riproduzione e di adattamento anche parziale, con qualsiasi mezzo, sono riservati per tutti i Paesi. Non sono assolutamente consentite le fotocopie senza il permesso scritto dell’Editore. I edizione: settembre 2006 Indice Capitolo I Introduzione allo studio della biochimica 7 Capitolo II Gli amminoacidi 17 Capitolo III Le proteine 31 Capitolo IV Gli enzimi 47 Capitolo V Principi di bioenergetica 75 Capitolo VI I carboidrati 95 Capitolo VII Il processo respiratorio 117 Capitolo VIII Il ciclo dell’acido citrico 135 Capitolo IX La fosforilazione ossidativa 153 Capitolo X Gluconeogenesi e via dei pentosi fosfati 167 Capitolo XI La fotosintesi 179 5 6 Indice Capitolo XII I lipidi 225 Capitolo XIII Metabolismo dei lipidi 245 Capitolo XIV Il metabolismo dell’azoto 267 Capitolo XV Nucleotidi ed acidi nucleici 289 Capitolo XVI La sintesi proteica 303 Capitolo XVII Composti fenolici delle piante 315 Capitolo XVIII Terpeni e terpenoidi 329 Capitolo XIX Gli alcaloidi 331 Bibliografia 343 INTRODUZIONE ALLO STUDIO DELLA BIOCHIMICA La biochimica studia la costituzione chimica degli esseri viventi, le funzioni fisiologiche dei loro prodotti e le trasformazioni chimiche che avvengono a carico di questi e che sono alla base della vita. I sistemi viventi si differenziano dal mondo inanimato per certe peculiari proprietà: possono crescere, muoversi, costruire un metabolismo, rispondere a stimoli provenienti dall’ambiente e soprattutto sono in grado di replicare se stessi con eccezionale fedeltà. Gli organismi viventi sono composti da un gran numero di molecole organiche che di per sè stesse sarebbero inerti ma che, riunite in aggregati di un determinato peso molecolare e di idonea complessità, in particolari condizioni possono dare origine a quel complesso sistema di reazioni che prende il nome di “vita”. Nonostante le molteplici forme in cui si manifesta la vita, l’intrico delle strutture biologiche più diverse e la complessità dei meccanismi vitali, le funzioni della vita possono essere interpretate in termini chimici. I costituenti cellulari, o biomolecole, seguono i principi chimici e fisici che governano la materia inanimata. In questo primo capitolo verranno riportate in breve le caratteristiche degli esseri viventi, le principali classi di biomolecole, cioè delle molecole necessarie alla vita e verranno illustrati i fondamenti del metabolismo cellulare. Le singole vie metaboliche, la produzione ed il consumo di energia e le caratteristiche ed il funzionamento dei catalizzatori biologici, gli enzimi, verranno riportati per esteso nei prossimi capitoli. Esaminando attentamente le caratteristiche degli esseri viventi, che li distinguono da un complesso inanimato di molecole come per esempio una roccia, vediamo che: 1) sono formati da cellule, che ne sono le unità costitutive fondamentali. Tutti gli organismi viventi sono organizzati su base cellulare; 2) sono sistemi altamente organizzati ed ordinati, le cui strutture interne sono costituite da molti tipi di molecole diverse; 3) sono capaci di costruire strutture molecolari ordinate a partire da materiali più disordinati; 7 Capitolo I 8 4) possono estrarre energia dall’ambiente, di solito sotto forma di sostanze chimiche nutrienti, e in tal caso si dicono chemiotrofi, oppure utilizzando la luce solare (fototrofi), e sono in grado di utilizzarla trasformandola da una forma in un’altra; 5) si mantengono uguali anche al variare delle condizioni chimiche o ambientali; 6) reagiscono agli stimoli e a perturbazioni provenienti dall’ambiente o prodotte da essi stessi, e possono agire per ripristinare l’equilibrio alterato; 7) sono capaci di riprodursi, cioè di costruire copie di loro stessi che possono essere identiche o diverse; 8) sono capaci di evolversi: i processi di riproduzione, caratterizzati da ricombinazione del materiale genetico, possono dare origine a progenie diversa dai genitori. 9) hanno la capacità di adattarsi all’ambiente; 10) si accrescono e si sviluppano; 11) l’informazione per ogni loro caratteristica ed attività è contenuta all’interno della cellula. Metaboliti e macromolecole I precursori principali per la formazione di biomolecole sono: l’acqua (H2O), l’anidride carbonica (CO2) e tre composti inorganici dell’azoto, lo ione ammonio (NH4+), il nitrato (NO3-) e l’azoto molecolare (N2). I processi metabolici assimilano e trasformano questi precursori inorganici in livelli sempre più complessi di biomolecole. In un primo tempo, i precursori vengono trasformati in metaboliti, composti organici semplici che agiscono come intermedi nelle trasformazioni cellulari dell’energia e nella biosintesi di vari composti complessi come amminoacidi, zuccheri, nucleotidi, acidi grassi, glicerolo. Legando insieme questi composti complessi, che possono essere considerati come blocchi da costruzione, si ottengono le macromolecole: proteine, polisaccaridi, polinucleotidi (DNA, RNA) e lipidi. Interazioni fra queste macromolecole formano dei livelli superiori di organizzazioni strutturali, cioè complessi sopramolecolari, che svolgono importanti funzioni Introduzione 9 cellulari. Un esempio sono i complessi enzimatici, i ribosomi, i cromosomi. Questi complessi sopramolecolari sono tenuti insieme da interazioni non covalenti fra le macromolecole: legami a idrogeno, attrazioni ioniche, forze di Van der Waals e interazioni idrofobiche. Sebbene queste forze non covalenti siano deboli (meno di 40 KJ/mole), i legami sono tanti e quindi tutti insieme riescono a mantenere l’architettura essenziale del complesso sopramolecolare. Ovviamente tutto ciò deve accadere in condizioni di temperatura, pH e forza ionica compatibili con la vita. La cellula è l’unità della vita La cellula è la più piccola entità capace di svolgere le funzioni dell’essere vivente: la crescita, il metabolismo, la risposta agli stimoli e la replicazione. Gli organelli subcellulari sono entità di dimensioni considerevoli, che si trovano solo nelle cellule eucariotiche, cioè appartenenti agli organismi superiori: all’interno di essi si svolgono le reazioni metaboliche. Gli organelli subcellulari che, come dice il nome stesso, sono inclusi nella cellula e sono di solito delimitati da membrane comprendono: il nucleo, i mitocondri, i cloroplasti, i vacuoli ed altri organelli più piccoli, come i lisosomi, i perossisomi e i cromoplasti. Qui di seguito citeremo i più importanti. Il nucleo porta l’informazione genetica, contenuta nelle sequenze lineari di nucleotidi nel DNA dei cromosomi. Nei mitocondri avviene il catabolismo aerobico di carboidrati e lipidi, con rilascio di energia che viene conservata in forme utilizzabili metabolicamente come l’ATP. I cloroplasti sono gli agenti biologici che raccolgono l’energia luminosa e la trasformano in energia chimica utilizzabile nel metabolismo durante la fotosintesi. Le membrane cellulari delimitano i confini delle cellule e degli organelli subcellulari. Sono complessi di lipidi e proteine tenuti insieme da forze non covalenti, fra cui particolarmente importanti sono le interazioni idrofobiche. Queste interazioni riflettono la tendenza delle molecole non polari a riunirsi insieme separandosi dal solvente polare per 10 Capitolo I eccellenza, l’acqua. La riunione spontanea delle membrane in ambiente acquoso dove la vita è nata e viene mantenuta è il naturale risultato del loro carattere idrofobico. Le membrane di nuclei, organelli e cloroplasti sono diverse l’una dall’altra: ciascuna di esse ha una sua propria composizione in proteine e lipidi legata alle funzioni dell’organello. Inoltre la compartimentazione entro le cellule non è solo una conseguenza inevitabile della presenza delle membrane, ma anche una condizione essenziale per il buon funzionamento dell’organello. Proprietà delle biomolecole e loro adattamento alle condizioni della vita Le macromolecole delle cellule viventi sono costituite da unità (amminoacidi nelle proteine, carboidrati nei polisaccaridi) che hanno una polarità strutturale. Cioè le molecole non sono simmetriche e quindi possono essere pensate come composte di una “testa” e di una “coda”. La polimerizzazione che porta a collegare queste unità per formare macromolecole avviene tramite connessioni lineari testa-coda. A causa di ciò, anche i polimeri hanno una testa ed una coda e quindi la molecola ha un “senso” o una direzione nella sua struttura. Da ciò deriva che i blocchi che la compongono, quando vengono letti nella direzione della lunghezza della molecola possono fornire delle informazioni, come le lettere dell’alfabeto possono formare parole quando siano messe una accanto all’altra. Non tutte le macromolecole sono ricche di informazioni: i polisaccaridi sono spesso composti dallo stesso zucchero che si ripete molte volte, come nell’amido e nella cellulosa, che sono omopolimeri costituiti sempre dalla stessa molecola di D-glucosio. Proteine e polinucleotidi invece sono costituiti da blocchi che non costituiscono una struttura ripetitiva: le loro sequenze sono uniche, e nella loro unicità sta il loro significato. Infatti le proteine, sebbene siano costituite da sequenze lineari di amminoacidi legati covalentemente gli uni agli altri, possono torcersi, avvolgersi e disporsi in tre dimensioni costituendo una architettura specifica ed altamente ordinata che è caratteristica di ogni molecola proteica e la identifica fra le altre. Introduzione 11 Le forze che mantengono le strutture biologiche Nelle molecole gli atomi sono in genere tenuti insieme da legami covalenti e da legami ionici. Altre forze attrattive deboli sono i legami a idrogeno, le forze di van der Waals, e le interazioni idrofobiche. Nessuna di queste forze tuttavia riesce da legare stabilmente insieme due atomi. L’energia messa in gioco da queste forze per tenere insieme le molecole è solo di poco superiore alla tendenza che hanno le molecole ad allontanarsi per l’agitazione termica: quindi, nelle condizioni fisiologiche, i legami si potrebbero creare e rompere in continuazione. Tuttavia, se il numero di legami è molto grande, la struttura acquista stabilità per l’azione cumulativa di molte forze deboli che agiscono insieme. Esaminiamo ora queste forze: 1) Forze di attrazione di van der Waals Sono il risultato di interazioni elettrostatiche indotte fra atomi o molecole molto vicini fra loro. Sono dovute alla fluttuazione della nuvola elettronica carica negativamente che ruota intorno a ciascun nucleo. Si formano dei dipoli momentanei che, se di carica opposta, si attraggono. La forza di questi legami è inversamente proporzionale alla sesta potenza della distanza. 2) Legami a idrogeno Si formano fra un atomo di H legato covalentemente ad un atomo elettronegativo (come O o N) e un altro atomo elettronegativo che serve da accettore. Sono legami più forti di quelli di van der Waals e sono anche direzionali: cioè si formano legami lineari fra donatore, idrogeno ed accettore. Sono anche più specifici di quelli di van der Waals perchè richiedono la presenza di gruppi donatori ed accettori complementari. 3) Interazioni idrofobiche Sono dovute alla forte tendenza dell’acqua a escludere gruppi o molecole non polari. Le interazioni idrofobiche prendono origine non tanto da una affinità intrinseca delle sostanze non polari fra loro, ma dal fatto che le molecole d’acqua preferiscono interagire più fortemente fra loro, escludendo i gruppi non polari. Questa esclusione fa sì che 12 Capitolo I le sostanze non polari si aggreghino fra loro formando dei “cluster” in soluzione acquosa. Così, le zone non polari delle macromolecole biologiche stanno confinate all’interno della molecola per escludere il mezzo acquoso. Un esempio sono le goccioline d’olio che si formano e che tendono ad aggregarsi alla superficie dell’acqua. Caratteristiche delle principali classi di biomolecole Le biomolecole sono necessarie per generare, mantenere e perpetuare la vita. Alcune sono più complesse, altre meno, ma tutte ugualmente importanti. La maggior parte dei costituenti molecolari dei sistemi viventi è composta di atomi di carbonio legati con legami covalenti ad altri atomi di carbonio o ad atomi di idrogeno, di ossigeno e di azoto. La materia vivente è infatti composta per il 99% di questi quattro elementi. Idrogeno ed ossigeno sono abbondanti perché sono i costituenti dell’acqua, che è il composto più importante in assoluto in tutti gli organismi. Il restante 1% é formato da Ca, P, K, S, Cl, Na e Mg e meno dello 0.01% da elementi in tracce, che tuttavia assolvono a precise funzioni metaboliche. Le particolari proprietà di legame del carbonio, che è sempre tetravalente ma che può condividere fino a 6 elettroni con se stesso o con un altro atomo, consentono la formazione di un gran numero di molecole. I composti organici con massa molecolare relativamente bassa (inferiore a 500) servono come sub-unità monomeriche che sono costituenti delle macromolecole, composti organici che mantengono in vita le cellule e permettono loro di riprodursi. Le quattro classi principali di biomolecole sono: carboidrati, lipidi, proteine e acidi nucleici. I carboidrati ed i lipidi sono sostanze di riserva che forniscono l’energia necessaria per fare avvenire migliaia di reazioni chimiche e che svolgono anche altre funzioni di primaria importanza. I carboidrati ed i lipidi sono usati principalmente come fonti di energia e solo in casi particolari come materiali da costruzione (ad es. la cellulosa). Proteine ed acidi nucleici invece hanno compiti strutturali e funzionali. E’ questa una divisione di compiti molto importante su cui è bene richiamare l’attenzione. Fra i carboidrati il più importante è il glucosio, fonte di energia per tutti gli organismi. Possiamo quindi dire che il Introduzione 13 glucosio è il carburante per eccellenza della cellula vivente. Come sostanze di riserva costituita da molecole di glucosio troviamo l’amido nelle piante ed il glicogeno negli animali. Carboidrati e lipidi sono dunque riserve energetiche che permettono ai sistemi viventi di compiere trasformazioni che dipendono non solo da quanti materiali nutritivi introducono quotidianamente ma anche dalla capacità di immagazzinare e sfruttare in un secondo tempo quelli non utilizzati immediatamente. Le proteine sono i costituenti strutturali di tutti i sistemi viventi e formano le impalcature che danno ad ogni organismo una forma ed una funzione precisa. Le proteine sono anche i costituenti di una importantissima classe di composti, gli enzimi, che sono insostituibili catalizzatori biologici. Sono gli enzimi, migliaia per ogni cellula, che decidono, con la loro estrema selettività, quale biomolecola deve essere prodotta o eliminata in un certo momento della vita cellulare. Gli acidi nucleici sono le strutture nelle quali sono codificate le caratteristiche genetiche della specie e dell’individuo. In particolare il DNA (acido desossiribonucleico) costituisce i geni, il materiale fondamentale dell’ereditarietà, del processo cioè che trasferisce l’informazione contenuta nei geni da una cellula madre alla cellula figlia. Ognuna di queste classi di composti verrà trattata esaurientemente nel capitolo ad essa dedicato. Il metabolismo cellulare Le trasformazioni realizzate dai sistemi viventi costituiscono il metabolismo, parola greca che significa “cambiamento”. Il metabolismo è un insieme di processi attraverso il quale molecole grandi e complesse sono ridotte a molecole semplici (in tal caso si parla di catabolismo, o distruzione) che, a loro volta, possono essere nuovamente utilizzate per formare altre molecole (anabolismo, o costruzione), che sono alla base del funzionamento dell’intero ciclo vitale. Queste serie di reazioni vengono dette vie metaboliche. Queste vie possono essere lineari, che partono cioè da un composto per giungere ad un prodotto finale diverso da quello di partenza, o cicliche, in cui il 14 Capitolo I prodotto finale è lo stesso di quello iniziale. In ogni caso, a differenza di quanto avviene nelle reazioni organiche o inorganiche in laboratorio, il prodotto di una reazione diventa il reagente della successiva. Inoltre, tutte le parti di un organismo vivente devono operare alla stessa temperatura e alla stessa pressione. Le cellule sono isotermiche, cioè sono sistemi che funzionano a temperatura costante. Anche in questo tipo di reazioni esistono catalizzatori, composti organici di cui parleremo per esteso in seguito, che hanno caratteristiche particolarissime e sono detti enzimi. Produzione e consumo di energia metabolica Le cellule e gli organismi, per funzionare, hanno bisogno di un continuo apporto di energia, senza la quale tenderebbero a decadere verso stati energetici sempre più bassi e disordinati. Tutti i processi di sintesi richiedono energia, sia nel mondo inorganico che negli organismi viventi: in questi ultimi le cellule hanno sviluppato efficientissimi meccanismi per catturare l’energia della luce solare oppure per estrarla dai legami che tengono insieme gli atomi delle sostanze ossidabili. L’energia così ottenuta potrà essere utilizzata per far avvenire processi non spontanei, cioè che richiedono energia. I principi fondamentali che governano le trasformazioni e gli scambi di energia negli organismi viventi costituiscono la bioenergetica. I pilastri fondamentali delle bioenergetica (di cui parleremo in modo dettagliato più avanti, nel capitolo dedicato) sono: • Tutti gli organismi viventi creano e conservano le loro strutture complesse ed ordinate utilizzando l’energia estratta da composti chimici o dalla luce solare. • In ogni modificazione fisica o chimica la quantità totale di energia rimane costante anche se la forma di energia può cambiare. • Le cellule sono motori chimici che operano a temperatura costante. • Le richieste energetiche di quasi tutti gli organismi sono soddisfatte in modo diretto o indiretto dall’energia solare. • Il flusso di elettroni nelle reazioni di ossido-riduzione è alla base della formazione di energia cellulare. Introduzione • 15 Tutti gli organismi viventi dipendono gli uni dagli altri attraverso scambi di energia e materia mediati dall’ambiente. Il punto centrale della bioenergetica è il modo in cui l’energia, liberata dalla combustione di sostanze nutrienti o dalla cattura della luce solare, viene utilizzata dalle reazioni che richiedono energia. Come per la materia inorganica, anche per i sistemi biologici sono valide le leggi della chimica e della fisica. Quindi sarà valido il I° Principio della termodinamica, che dice che, in ogni variazione chimica o fisica, la quantità totale di energia dell’universo resta costante, anche se la forma dell’energia può variare. La quantità di energia immediatamente disponibile per produrre lavoro si chiama energia libera e si indica con la lettera G. Questa energia non rappresenta tutta l’energia messa in gioco nelle trasformazioni dette sopra perché una parte di essa viene dissipata sotto forma di entropia, che aumenterà il disordine del sistema (2° principio della termodinamica) Le reazioni chimiche che avvengono in un sistema chiuso procedono spontaneamente finché non raggiungono l’equilibrio, stato in cui la velocità di formazione dei prodotti diventa uguale a quella in cui i prodotti si ritrasformano nei reagenti. La variazione di energia quando il sistema passa dallo stato iniziale a quello di equilibrio, a pressione e temperatura costanti, si chiama variazione di energia libera ∆G. Il valore di ∆G dipende dalla natura della reazione e da quanto prodotti e reagenti sono lontani dall’equilibrio. Nelle reazioni in cui i prodotti hanno meno energia libera dei reagenti, cioè hanno maggiore stabilità, si avrà un eccesso di energia libera che può essere utilizzata per compiere un lavoro. Tali reazioni si dicono esoergoniche o spontanee e il ∆G acquista valore negativo, ∆G <0. Le reazioni invece in cui i prodotti hanno maggior energia libera dei reagenti e quindi per avvenire necessitano di un rifornimento di energia libera si dicono endoergoniche o non spontanee e il ∆G acquista valore positivo, ∆G >0. Gli organismi viventi hanno superato questo scoglio accoppiando reazioni endoergoniche, che quindi non potrebbero mai avvenire spontaneamente, con reazioni esoergoniche che le rendono possibili. L’accoppiamento di questi due tipi di reazioni è un aspetto essenziale degli scambi energetici delle cellule. La reazione esoergonica che fa- 16 Capitolo I vorisce la maggior parte dei processi endoergonici delle cellule è l’idrolisi di una molecola particolare, l’Adenosin Tri Fosfato (ATP), che è il principale trasportatore di energia nelle cellule ed è il punto di unione fra i processi endo- ed eso-ergonici. L’ATP si scinde in ADP (Adenosin Di Fosfato) e Pi, fosfato inorganico. Il gruppo fosforico terminale dell’ATP viene trasferito ad un gran numero di accettori che vengono così attivati e possono subire quindi una molteplicità di trasformazioni chimiche. Alla luce di queste considerazioni possiamo quindi enunciare altre regole fondamentali della bioenergetica: • • • Le reazioni cellulari endoergoniche sono guidate dall’accoppiamento con processi chimici o fotochimici esoergonici attraverso la formazione di intermedi chimici; L’ATP è il trasportatore universale dell’energia metabolica ed accoppia il catabolismo con l’anabolismo; Le cellule sono motori chimici auto-regolati che procedono secondo il principio della massima economia. GLI AMMINOACIDI Gli amminoacidi sono importanti costituenti delle cellule viventi, sono infatti le unità strutturali di base delle proteine. Sono paragonabili ai mattoni con cui viene costruita una casa: ne sono l’elemento più semplice e fondamentale e le conferiscono le caratteristiche più utili alle esigenze di chi la costruisce. Come si può dedurre dal nome, nella molecola di ogni amminoacido è presente almeno un gruppo amminico (–NH2) e un gruppo acido, in genere carbossilico (–COOH), legati allo stesso atomo di carbonio. Sono inoltre presenti un atomo di idrogeno e un gruppo funzionale di varia natura. L’esistenza di almeno due gruppi fortemente polari e la possibilità di formazione di legami a idrogeno spiega le caratteristiche fisiche degli amminoacidi, che sono tutti solidi cristallini con punto di fusione elevato. Anche la loro solubilità in acqua è elevata, anche se è legata alla natura della catena laterale. Nel corso dell’evoluzione gli esseri viventi hanno selezionato un ristretto gruppo di amminoacidi, costituito da venti elementi, che sono i costituenti delle proteine. Questi composti, definiti amminoacidi proteici, hanno alcune caratteristiche comuni: 1) sono α-amminoacidi, cioè hanno il gruppo amminico ed il gruppo carbossilico legati all’atomo di carbonio in posizione α, cioè sul primo C della catena dopo il gruppo carbossilico. Fa eccezione la prolina perché la catena laterale è legata oltre che al carbonio α anche all’atomo di azoto collegato a quest’ultimo. La prolina è in realtà un imminoacido (–NH–) e non un amminoacido (–NH2). 2) gli amminoacidi differiscono fra loro per la natura chimica del gruppo R, che è legato allo stesso atomo di carbonio α ed è detto catena laterale. Il gruppo R può essere costituito da una catena idrocarburica a cui può essere legato un altro gruppo carbossilico, un altro gruppo amminico, o un gruppo di altra natura. 3) tutti gli amminoacidi hanno quattro sostituenti diversi legati al carbonio α , che è quindi un carbonio asimmetrico o carbonio chirale. 17 18 Capitolo II La formula generale degli amminoacidi è riportata in sintesi in Figura 1 dove a pH 7,0 il gruppo amminico è protonato (–NH3+) e il gruppo carbossilico ha perso l’idrogeno (–COO-). La carica netta è ancora zero, ma la molecola è in realtà uno ione dipolare o zwitterione. Figura 1. Struttura di un amminoacido generico. 4) tutti gli amminoacidi, tranne la glicina che ha due sostituenti uguali (due atomi di H), sono molecole chirali (Figura 2). Cioè possono presentarsi in forme speculari non sovrapponibili, come non lo sono la mano destra e la sinistra. Vengono detti anche isomeri ottici o enantiomeri. Figura 2. Molecole chirali come immagini non sovrapponibili. L’isomeria in chimica organica è il fenomeno per cui due o più composti presentano la stessa formula bruta, sono cioè costituiti dallo stesso tipo e numero di atomi, hanno uguali proprietà chimiche e fisiche, ma differiscono nelle formule di struttura, cioè nella disposizione spaziale degli atomi e dei relativi legami chimici. Gli isomeri ottici Amminoacidi e peptidi 19 hanno inoltre la particolarità di ruotare in modo opposto il piano della luce polarizzata. Tutti i composti che ruotano il piano della luce polarizzata si definiscono otticamente attivi ed il senso di rotazione viene indicato con il segno più (+) o meno (–). Rispetto alla configurazione nello spazio dei vari gruppi intorno al carbonio α, gli amminoacidi sono classificati come D o L (Figura 3). Per convenzione (secondo Fischer) il gruppo più ossidato (–COO-) si scrive in alto con i legami verticali che si allontanano dall’osservatore ed i legami orizzontali che si avvicinano all’osservatore. Se il gruppo amminico si trova a sinistra l’amminoacido è L, se si trova a destra l’amminoacido è D. In realtà gli amminoacidi proteici appartengono sempre alla serie L e, con rare eccezioni, solo questi isomeri partecipano alle reazioni cellulari. Questa particolarità non è stata ancora spiegata in maniera soddisfacente. L’ipotesi più probabile è che la selezione degli isomeri L piuttosto che i D sia avvenuta casualmente all’inizio dell’evoluzione e sia stata mantenuta. Figura 3. Isomeri D e L secondo la convenzione di Fisher. Nelle tabelle 1-3 sono riportate le strutture dei 20 amminoacidi presenti nelle proteine, raggruppati secondo uno dei possibili criteri di classificazione, in base alle diverse proprietà che il gruppo funzionale R apporta alla molecola: Capitolo II 20 • • • gruppi R idrofobici (Tabella 1) gruppi R idrofili non carichi (Tabella 2) gruppi R idrofili carichi, acidi e basici (Tabella 3). Gli amminoacidi vengono spesso indicati con una abbreviazione a tre lettere o mediante il simbolo ad una lettera. Le abbreviazioni sono quasi sempre costituite dalle prime tre lettere del nome inglese (Cys per la cisteina, Ala per l’alanina, Arg per l’arginina e così via). Fanno eccezione l’asparagina (Asn), la glutammina (Gln), la isoeucina (Ile) ed il triptofano (Trp). I simboli ad una lettera derivano per molti amminoacidi dalla prima lettera del loro nome, ma poiché alcuni hanno la stessa iniziale si è ricorsi a convenzioni. Comunque le abbreviazioni a tre lettere sono generalmente da preferirsi perché sono più pratiche e generano meno confusione. Nelle rappresentazioni 3D riportate nelle tabelle con il colore violetto si indica l’atomo di azoto del gruppo amminico (–NH3+), con il colore rosso gli atomi di ossigeno del gruppo carbossilico (–CO2-) e con il giallo l’atomo di zolfo dei gruppi R della cisteina e della metionina. Con il grigio scuro e grigio chiaro si indicano rispettivamente gli atomi di carbonio e di idrogeno. Amminoacidi e peptidi 21 Tabella 1. Amminoacidi con gruppi R idrofobici. Nome Sigla Glicina Gly (G) Alanina Ala (A) Valina Val (V) Leucina Leu (L) Isoleucina Ile (I) Metionina Met (M) Prolina Pro (P) Fenilalanina Phe (F) Triptofano Trp (W) Formula di struttura 3D 22 Tabella 2. Amminoacidi con gruppi R idrofili non carichi. Nome Sigla Serina Ser (S) Treonina Thr (T) Asparagina Asn (N) Glutammina Gln (Q) Tirosina Tyr (Y) Cisteina Cys (C) Formula di struttura 3D Amminoacidi e peptidi 23 Tabella 3. Amminoacidi con gruppi R idrofili carichi (acidi e basici). Nome Sigla Lisina Lys (K) Arginina Arg (R) Istidina His (H) Ac. Aspartico/ Aspartato Asp (D) Ac. Glutammico/ Glutammato Glu (E) Formula di struttura 3D 24 Capitolo II Particolarità di alcuni amminoacidi Amminoacidi con gruppi R idrofobici. La glicina è l’amminoacido strutturalmente più semplice con il gruppo R costituito solamente da un atomo di idrogeno e avendo due sostituenti uguali (due atomi di H) legati al carbonio α, è il solo che non è chirale. Quattro amminoacidi (Ala, Val, Leu, Ile) hanno catene laterali costituite da idrocarburi saturi, cioè che non contengono doppi legami. Sono molecole poco reattive che rivestono un ruolo importante nel determinare e mantenere la struttura tridimensionale delle proteine. Il ripiegamento delle catene polipeptidiche in genere avviene in modo tale che gli amminoacidi idrofobici restino rivolti verso l'interno della molecola e che quelli idrofilici siano, invece, rivolti verso l'esterno, dove sono liberi di interagire con altri composti. L’alanina è l’amminoacido più abbondante nella maggior parte delle proteine. La metionina è uno dei due amminoacidi contenenti zolfo la cui catena laterale R contiene un gruppo metiltioetere: - R = – CH2CH2 – S – CH3 Come ricordato sopra solo la prolina non possiede un gruppo amminico ed uno carbossilico legati al carbonio in α, poiché il gruppo amminico è sostituito da un gruppo –NH imminico nella formula ciclica di questo composto. Spesso la sua presenza condiziona la geometria della proteina di cui fa parte, provocando bruschi cambiamenti di direzione del filamento polipeptidico o interruzioni della catena. La fenilalanina e il triptofano hanno catene laterali aromatiche altamente idrofobiche che spesso si posizionano nell’interno delle proteine globulari. Amminoacidi con gruppi R idrofili non carichi. La catena laterale della serina contiene un gruppo R ossidrilico (– OH) che possiamo ritrovare nei siti catalitici di alcuni enzimi dove si mostra particolarmente reattivo. Amminoacidi e peptidi 25 Anche la treonina ha un ossidrile nella catena laterale, ma non è reattivo come quello della serina. Asparagina e glutammina sono le ammidi di altri due amminoacidi, rispettivamente l’acido aspartico e l’acido glutammico. In questi il gruppo carbossilico (–COO-) è sostituito da un gruppo ammidico terminale (–CO–NH2). La tirosina si forma dalla fenilalanina, per sostituzione di un H con un gruppo ossidrilico (–OH) in posizione para che trasforma la catena laterale R da idrofoba a idrofila. Figura 4. Formazione di un legame disolfuro tra due molecole di Cys. La cisteina è con la metionina il secondo amminoacido che contiene zolfo. Possiede un gruppo solfidrilico (–SH) chimicamente reattivo nella catena laterale. Il suo idrogeno può formare legami a H deboli con l’ossigeno e con l’azoto. Inoltre può anche formare con un’altra molecola di cisteina un legame covalente disolfuro –S–S– costituendo un nuovo amminoacido detto cistina, importante nella struttura primaria delle proteine perché in esse può indurre ripiegamenti o rotture nella catena di amminoacidi. Il legame o ponte disolfuro ha una grande importanza nella struttura delle proteine perché può mettere in connessione catene separate o formare legami crociati tra residui di cisteina della stessa catena (Figura 4). 26 Capitolo II Amminoacidi con gruppi R idrofili carichi. Gli amminoacidi che a pH fisiologico hanno cariche nette positive sono la lisina e l’arginina, che spesso partecipano alla formazione di interazioni elettrostatiche all’interno delle proteine. L’istidina può essere carica positivamente o essere priva di carica in funzione dell’ambiente circostante e per questa caratteristica si trova spesso nel sito attivo degli enzimi, dove gioca un ruolo importante nel catalizzare la formazione e la rottura di legami. La presenza nelle catene laterali di un gruppo imidazolico (Hys) di un gruppo guanidinico (Arg) e di un gruppo amminico (Lys), che sono sempre basi secondo Lewis hanno cioè doppietti elettronici condivisibili con altre molecole e possono essere protonati, fa sì che questi siano chiamati anche amminoacidi basici. Esistono infine due aminoacidi che possiedono catene laterali con un gruppo carbossilico acido, l’acido aspartico e l’acido glutammico, classificati anche come amminoacidi acidi. Questi vengono generalmente chiamati aspartato e glutammato per sottolineare che le loro catene laterali sono quasi sempre cariche negativamente a pH fisiologico. Molte proteine che contengono metalli per funzioni strutturali o funzionali hanno, per questi siti di legame, una o più catene laterali di aspartato e glutammato. Acidità e basicità degli amminoacidi A causa della presenza nella stessa molecola di due gruppi dal comportamento chimico antitetico, come il carbossile che tende alla dissociazione acida ed il gruppo amminico che tende alla protonazione, gli amminoacidi sono da considerarsi composti anfoteri e la loro forma ionica dipende dal pH a cui si trovano. In soluzione neutra (pH 7,0) gli amminoacidi sono presenti in prevalenza come molecole dipolari (dette anche zwitterioni) in cui sia il gruppo amminico (-NH3+) che il gruppo carbossilico (-COO ) legati al carbonio α sono ionizzati. A pH acido o basico tipo e grado di ionizzazione cambiano e varia la forma predominante. Amminoacidi e peptidi 27 Figura 5. Gli stati di ionizzazione dipendono dal pH. Per gli amminoacidi che hanno gruppi ionizzabili nella catena laterale, vanno considerati anche gli equilibri di dissociazione di questi gruppi. Poiché le trasformazioni da catione ad anione e viceversa sono funzione dell’acidità o dell’alcalinità del mezzo, esiste chiaramente un valore di pH intermedio a cui l’amminoacido presenta carica netta complessivamente uguale a zero, avendo assunto la forma di ione bipolare. Tale pH, diverso a seconda delle diverse caratteristiche dei vari amminoacidi, prende il nome di punto isoelettrico. I PEPTIDI Quando due amminoacidi si uniscono in una reazione analoga a quella con cui si formano le proteine, il gruppo amminico in α di un amminoacido reagisce con il gruppo carbossilico in α di un altro amminoacido, con eliminazione di una molecola di acqua. La nuova molecola è un peptide. A seconda che gli amminoacidi siano due, tre, pochi o molti si avranno composti denominati dipeptidi, tripeptidi, oligopeptidi o polipeptidi. Generalmente per catene con più di 100 amminoacidi, si parla di proteine. Il legame –CO-NH- che si forma fra gli amminoacidi in sequenza è un legame ammidico, che è caratteristico dei peptidi e delle proteine e perciò viene anche detto legame peptidico o legame proteico. I gruppi Capitolo II 28 R delle catene laterali, anche se sono a loro volta gruppi amminici o carbossilici, non entrano mai a far parte di questo legame (Figura 6). Figura 6. Reazione di formazione di un legame peptidico Il legame peptidico può avere due forme di risonanza fra due strutture limite, a causa della delocalizzazione della coppia di elettroni π fra il carbonio e l’ossigeno e del doppietto elettronico non condiviso sull’atomo di azoto. Quindi il legame C-N non ha carattere di legame semplice puro, ma ha un parziale carattere di doppio legame (Figura 7). Figura 7. Forme di risonanza del legame peptidico Ciò è stato dimostrato sperimentalmente da misure di lunghezza di legame: infatti mentre un legame C–N semplice misura 1,45 Å1 e quello doppio C=N 1,27 Å, la lunghezza di un legame peptidico trovata è di 1,32 Å, una misura intermedia (Figura 8). 1 L’Angstrom (Å) è un’unità di lunghezza equivalente a 10 –10 m. Amminoacidi e peptidi 29 Figura 8. Dimensioni standard del gruppo peptidico Il legame peptidico, avendo carattere parziale di doppio legame è rigido e planare (piano ammidico). Poiché invece la rotazione dei legami C α–C e C α–N è libera, attorno al legame peptidico sono possibili le due configurazioni cis e trans rappresentate nella Figura 9. Il gruppo amminico –NH è quasi sempre in posizione trans rispetto al gruppo carbonilico –C=O perché esiste una repulsione sterica, o spaziale, che porta la conformazione trans ad essere più stabile della cis. Figura 9. Configurazioni cis-trans del legame peptidico Quando si forma un peptide, o una proteina, la quasi totalità dei gruppi amminici e carbossilici ionizzabili degli amminoacidi sono impegnati nei legami peptidici, per cui i gruppi ionizzabili disponibili e quindi responsabili della carica elettrica complessiva sono quelli pre- 30 Capitolo II senti nelle catene laterali R, oltre ai gruppi amminici e carbossilici liberi alle due estremità di ogni catena peptidica. L’unico gruppo amminico libero è detto terminale amminico e viene sempre considerato l’inizio della catena polipeptidica. L’ultimo amminoacido è il solo ad avere il gruppo carbossilico libero e viene chiamato terminale carbossilico . Gli amminoacidi legati a formare la catena peptidica prendono la desinenza –il se hanno impegnato il gruppo carbossilico: l’ultimo, che ha il gruppo carbossilico libero conserva il proprio nome invariato: Es. il glutatione, un tripeptide, è una glutammil- cisteinil-glicina. Una catena polipeptidica consiste di una parte che si ripete regolarmente, detta catena principale o scheletro covalente, e di una parte variabile che comprende le catene laterali caratteristiche di ciascun amminoacido. Ogni residuo contiene un gruppo C=O, buon accettore di legami a idrogeno, ad eccezione della prolina, e un gruppo –NH- , buon donatore di legami a idrogeno. Questi gruppi interagiscono fra loro e con i gruppi funzionali delle catene laterali per stabilizzare particolari strutture. LE PROTEINE Le proteine costituiscono una classe di sostanze organiche di fondamentale importanza biologica. Sono le molecole funzionali per eccellenza della cellula che, attraverso esse, può svolgere tutte le funzioni alla base della vita. Il loro nome infatti deriva dal greco protos che significa il primo, il principale. Sono molecole di grandi dimensioni, con un peso molecolare che può variare da meno di 10.000 a più di 100.000 dalton1. Nonostante la loro complessità hanno una notevole uniformità nella struttura di base, che è formata dall’unione di un numero più o meno elevato di semplici unità elementari, gli amminoacidi, sempre gli stessi in tutte le proteine di tutti gli organismi conosciuti. Ogni proteina ha un proprio lavoro da svolgere in una cellula, nella matrice extracellulare e nei fluidi corporei degli organismi pluricellulari. Questo lavoro è integrato nella dinamica cellulare e regolato in base alle esigenze della cellula o dell’organismo. Le proteine hanno varie funzioni: a) sono catalizzatori biologici: quasi tutte le reazioni che avvengono a livello cellulare infatti devono essere accelerate e quindi necessitano di catalizzatori. Alla temperatura a cui vivono le cellule le reazioni anche se spontanee, quindi che non necessitano di energia dall’esterno, avverrebbero in modo spontaneo in tempi estremamente lunghi, a causa della loro elevata energia di attivazione. Occorre quindi aumentare in modo significativo la loro velocità. I catalizzatori proteici preposti a questa funzione sono detti enzimi (vedi capitolo Enzimi) ed hanno un ruolo di primaria importanza nella vita cellulare. Esempi: la pepsina, per la degradazione delle proteine, la ribonucleasi, per la degradazione dell’RNA (acido ribonucleico), la catalasi, per trasformare l’ acqua ossigenata in ossigeno ed acqua. b) sono trasmettitori di segnali chimici: gli organismi pluricellulari complessi devono poter regolare l’attività biochimica delle cellule vicine o che appartengono ad organi o tessuti diversi. 1 12 Il dalton è l’unità di massa corrispondente a 1/12 della massa di un atomo di C, praticamente uguale a quella di un atomo di idrogeno. 31 32 Capitolo III L’apparato endocrino è responsabile di questa coordinazione attraverso la produzione di ormoni, molti dei quali sono proteine. Altre proteine costituiscono i recettori, che riconoscono i segnali chimici portati dagli ormoni e da altre sostanze, come i trasmettitori di impulsi nervosi. Esempio: l’insulina, che regola l’utilizzo del glucosio da parte delle cellule. c) servono come protezione: costituiscono cioè sostanze che si oppongono alla penetrazione di agenti estranei per mantenere costante l’ambiente interno. Questa funzione è svolta dagli anticorpi, armi chimiche di natura proteica che servono al sistema immunitario per proteggere l’organismo da sostanze estranee, gli antigeni. Esempio: le immunoglobuline G, anticorpi del sangue, prodotti in seguito all’ingresso di sostanze estranee. d) possono essere contrattili, capaci cioè di allungarsi o contrarsi o di associarsi per poter scorrere le une sulle altre, permettendo quindi alle singole cellule di cambiare forma a seconda delle condizioni o addirittura di spostarsi. Anche a livello delle singole cellule, il movimento è affidato a singole proteine, diffuse nelle cellule eucariotiche, che partecipano alla costituzione di microtubuli, microfilamenti e flagelli, particolarmente abbondanti nel tessuto muscolare. e) hanno funzioni di trasporto: sono necessarie agli organismi pluricellulari per trasferire molecole e ioni da un organo all’altro quando questi devono essere utilizzati in altri compartimenti o dall’esterno all’interno delle cellule. Si tratta di proteine specifiche che si trovano nei liquidi circolanti o sulla membrana cellulare. Esempio: emoglobina e mioglobina, proteine trasportatrici di ossigeno rispettivamente nel sangue e nel muscolo. La stessa molecola porta via l’anidride carbonica di scarto. f) hanno funzioni di riserva: costituiscono materiale da utilizzare in caso di bisogno o necessarie all’embrione per il proprio sviluppo (proteine dell’uovo, dell’endosperma dei semi ecc.). Esempio: l’ovalbumina, nell’albume dell’uovo, per il nutrimento dell’embrione o la ferritina, una proteina che trattiene gli ioni ferrosi nel fegato, nella milza o nel midollo osseo e li rilascia quando siano necessari a specifiche sintesi. g) hanno funzione strutturale, servono cioè per il sostegno di strutture ed organi o per la protezione di superfici delicate, come le muco- Le proteine 33 se. Stanno spesso fuori delle cellule e in molti casi tengono insieme gruppi di cellule negli organi e nei tessuti. Le proteine strutturali sono molte e di vario tipo. Esempio: il collagene e l’elastina, proteine fibrose dei tessuti connettivi che danno resistenza ed elasticità a tendini, ossa e pelle. Cartilagini, capelli e unghie sono costituiti in gran parte da proteine. La funzione strutturale è spesso associata ad altre funzioni. h) hanno funzione di controllo: la trasmissione genetica da un organismo alla sua discendenza è sotto stretto controllo di numerose proteine. i) sono accettori, conduttori e trasformatori di energia: si tratta di attività complesse, molto diversificate, che hanno lo scopo fondamentale di rendere disponibile alle cellule l’energia sotto una forma che esse possano utilizzare. Ad es., quando la luce colpisce la retina dell’occhio è necessaria una proteina che, con la vitamina A ad essa associata, trasforma l’energia luminosa in un impulso elettrico che viene trasferito al cervello. l) possono funzionare come tamponi, possono cioè neutralizzare le basi e gli acidi presenti nell’ambiente biologico mediante i gruppi acidi e basici dei radicali R degli amminoacidi che le compongono. Questa caratteristica è molto importante per alcune proteine che devono tenere il sangue costantemente a pH= 7,35. Infatti una persona non può sopportare che variazioni minime di questo valore. Conformazione delle proteine Le proteine sono macromolecole costituite dall’unione di non più di 20 tipi diversi di amminoacidi, selezionati nel corso dell’evoluzione fra i tanti esistenti (vedi capitolo Amminoacidi). Le proteine sono catene lineari di amminoacidi ripiegate in modo da assumere una conformazione unica e caratteristica per ciascuna proteina. Per classificare una proteina si possono utilizzare vari schemi tra cui il più generale divide le proteine in semplici, cioè formate da soli amminoacidi e coniugate, legate a ioni metallici o a molecole organiche di natura non proteica. Capitolo III 34 Un’altra classificazione tiene conto della conformazione delle proteine, dove per conformazione si intende la disposizione spaziale dei singoli gruppi funzionali presenti nella molecola, in seguito alla libera rotazione di questi intorno ai legami semplici. La conformazione dipende dalle interazioni reciproche di legami non covalenti (ionico, a idrogeno, dipolo-dipolo, idrofobico, di van der Waals) che conferiscono alla proteina una forma tridimensionale unica ed una funzionalità biologica specifica (Tabella 1). Tabella 1. I principali tipi di interazione presenti nelle proteine Nome Tipo di interazione Legame covalente Condivisione di elettroni Legame a idrogeno Condivisione di un atomo di H Legame ionico Attrazione di cariche opposte Distribuzione Forze di van asimmetrica di carica der Waals elettronica Interazione Esclusione del contatidrofobica to con l’acqua Struttura Le proteine 35 La struttura delle proteine può essere suddivisa in quattro livelli di complessità differente schematizzati in Figura 1. Figura 1. Schema dei livelli di organizzazione delle proteine Il primo livello di organizzazione è dato dalla struttura primaria, che rappresenta l’ordine in cui gli amminoacidi si susseguono nella proteina, quindi la sequenza lineare di amminoacidi. La struttura primaria è costituita da legami covalenti, soprattutto legami peptidici e ponti disolfuro che uniscono i singoli amminoacidi in una struttura polipeptidica. 36 Capitolo III In teoria si possono avere infinite proteine con la stessa composizione in amminoacidi ma con sequenza diversa (Figura 2). Figura 2. Struttura primaria: sequenza lineare di residui amminoacidici Molti laboratori biochimici si occupano di determinare le sequenze di amminoacidi di varie proteine e molte oggi sono le sequenze note. Le tecniche impiegate sono diverse e spesso assai sofisticate. Alterazioni anche minime delle sequenza di amminoacidi possono avere conseguenze gravi e essere all’origine di molte malattie, come l’anemia falciforme (o anemia mediterranea), che è caratterizzata dalla presenza nel sangue di globuli rossi a forma di mezzaluna (o di falce) che contengono una forma anomala di emoglobina. In una di queste forme di emoglobina un solo amminoacido, l’acido glutammico, è sostituito con la valina. La sostituzione quindi di un solo amminoacido su 146 basta a provocare una grave malattia. La struttura secondaria è un livello di organizzazione più complesso della struttura primaria e si riferisce a disposizioni particolarmente stabili dei residui di amminoacidi non troppo lontani fra loro, che danno origine ad organizzazioni ricorrenti. E’ costituita dalla formazione di legami a idrogeno fra un gruppo carbonilico (=CO) di un amminoacido della sequenza proteica ed il gruppo amminico (–NH2) di un amminoacido della stessa catena o di una catena vicina. Si possono avere due tipi di struttura: ad α-elica e a foglietto β ripiegato. La struttura ad α-elica è formata da una catena polipeptidica avvolta a spirale su se stessa in senso antiorario e conferisce alla struttura della proteina un’elevata flessibilità ed elasticità. Le proteine 37 Una unità ripetitiva dell’elica è formata da 3,6 residui di amminoacidi che occupano, lungo l’asse principale, uno spazio di 0,54 nm (Figura 3). I residui R dei vari amminoacidi sporgono in fuori della struttura a spirale ed interagiscono fra loro a seconda delle caratteristiche chimiche. La conformazione ad α-elica è quella che ha meno energia, quindi è la più stabile e la più probabile. La stabilità dell’ αelica deriva dalla presenza di moltissimi legami a idrogeno fra l’atomo di idrogeno del gruppo amminico e l’ossigeno del carbonile di due amminoacidi vicini nella sequenza. I legami a idrogeno in sé sarebbero legami deboli, ma diventano forti a causa del loro numero elevato. Figura 3 Struttura secondaria: α-elica e foglietto β Le α-eliche possono aggregarsi fra loro anche per mezzo dei gruppi –SH delle molecole di cisteina formando ponti disolfuro, con formazione di legami covalenti –S–S– molto stabili. Fra le strutture ad αelica la più tipica è quella della cheratina, proteina fibrosa presente nei capelli e nella pelle. Il collagene è la proteina più abbondante nei vertebrati. E’ costituito da fibrille con grande resistenza alla tensione che formano la componente principale del tessuto connettivo e sono formate da tre catene polipeptidiche avvolte su se stesse in modo diverso a seconda della funzione biologica (vedi anche Figura 8). Una tale struttura a volte viene definita come superelica. La proteina elastina, il costituente principale dei tendini, è molto simile al collagene ma a differenza di questo è elastica. La struttura β a foglietto ripiegato è tipica di poche proteine, in particolare della fibroina della seta e della tela di ragno (Figura 3). 38 Capitolo III E’ una struttura a soffietto, costituita da foglietti β disposti parallelamente all’asse della fibra con molti legami a idrogeno intermolecolari, cioè fra catene diverse, che coinvolgono sempre un gruppo amminico e un gruppo carbonilico vicini tra loro. Questi legami si instaurano fra catene parallele di amminoacidi, mentre i radicali R stanno sopra o sotto la struttura a foglietto, alla massima distanza l’uno dall’altro. Nelle proteine globulari spesso si trovano raggruppamenti di strutture secondarie che formano una cosiddetta struttura supersecondaria che è intermedia fra la struttura secondaria e quella terziaria e rappresenta un ulteriore livello di organizzazione della proteina. La forma più comune è l’unità βαβ in cui la connessione tra due catene parallele consecutive è costituita da un’α-elica (Figura 4) Figura 4. Rappresentazione schematica di un'unità βαβ Le proteine 39 Altre strutture supersecondarie comuni sono quella detta a greca perché ricorda motivi geometrici delle decorazioni delle ceramiche greche e il meandro β che è costituito da cinque strutture a foglietto β tra loro collegate e stabilizzate da legami a idrogeno (Figura 5). Figura 5. Strutture supersecondarie a greca e meandro β La struttura terziaria definisce la disposizione di tutti i suoi atomi nello spazio tridimensionale: è presente in tutte le proteine ma ha maggior importanza in quelle globulari (Figura 6). Figura 6. Rappresentazione schematica di struttura terziaria 40 Capitolo III In tutte le zone dove non è possibile la formazione dell’α-elica si instaurano legami non covalenti fra tutti i residui R che si trovano occasionalmente abbastanza vicini da formare un legame. Possono essere legami a idrogeno, ma anche legami ionici fra residui R con cariche opposte, legami dipolo-dipolo e legami idrofobici che coinvolgono una associazione fra catene idrocarburiche non polari. I residui R idrocarburici degli amminoacidi non polari tendono a respingere l’acqua da cui è circondata la maggior parte delle proteine globulari (eccetto quelle nelle membrane cellulari) e quindi formano aggregati associandosi fra loro. Queste regioni non polari all’interno delle proteine globulari determinano la struttura terziaria e spesso sono responsabili di specifiche attività biologiche. La struttura quaternaria si ha quando più proteine interagiscono fra loro riunendosi in aggregati proteici più complessi. Ognuna delle proteine diviene così una sub-unità di una struttura organizzata a livello superiore. Molte proteine che hanno un peso molecolare maggiore di 50000 daltons hanno struttura quaternaria, ma non tutte. Quando più proteine si aggregano insieme dando origine ad una struttura quaternaria, le interazioni fra le sub-unità modificano tutti i legami preesistenti e la molecola risultante acquista una nuova e caratteristica struttura. Proteine fibrose e proteine globulari Le proteine possono essere raggruppate in due categorie principali: le proteine fibrose e le proteine globulari. Nelle fibrose la struttura secondaria è prevalente rispetto alla terziaria, mentre nelle globulari il rapporto si inverte. Una proteina globulare ha nella sua struttura brevi tratti di α-elica e di foglietti β, mentre il resto della molecola è un groviglio della catena polipeptidica casuale che fa assumere alla catena stessa una forma rotondeggiante. Le proteine 41 Le proteine fibrose sono adatte a ruoli strutturali: la cheratina, il collagene, la fibroina della seta (Figura 7) sono esempi della relazione fra la conformazione di una proteina e la sua funzione biologica. L’unità strutturale è un semplice elemento di struttura secondaria ripetuto. L’α-cheratina è strutturata in modo da resistere alla tensione e si trova infatti in: capelli, lana, penne, unghie, artigli, corna, zoccoli e negli strati esterni della pelle. La struttura è un’α-elica destrorsa, superavvolta per aumentare la resistenza. Ponti disolfuro rendono la struttura ancora più stabile. La fibroina è prodotta da insetti e ragni. Le catene polipeptidiche sono quasi esclusivamente in forma di foglietto β ripiegato. La struttura è notevolmente compatta e ciò è dovuto al particolare tipo di amminoacidi presenti nella struttura (principalmente Ala e Gly) e alla molteplicità di legami a H tra i gruppi peptidici. L’assenza di legami covalenti fra i foglietti conferisce nello stesso tempo anche una notevole flessibilità. Figura 7. Architettura tridimensionale della fibroina della seta Il collagene è la proteina più abbondante nei vertebrati e si trova nel tessuto connettivo, come tendini, cartilagini, cornea dell’occhio e parte organica delle ossa. L’elica è una struttura secondaria unica, di- 42 Capitolo III versa dall’α -elica infatti è sinistrorsa ed ha tre residui per giro (Figura 8). Fisicamente il collagene è una struttura primitiva formata da tre unità avvolte su se stesse (tropocollagene) in modo diverso a seconda della funzione biologica che deve svolgere. Questa struttura superelicoidale è destrorsa. Figura 8. Modello di collagene a tripla elica Nelle proteine globulari i segmenti diversi di una catena polipeptidica o di catene polipeptidiche diverse tendono ad avvolgersi gli uni sugli altri, generando una proteina compatta rispetto alla conformazione completamente estesa. Le proteine globulari comprendono gli enzimi, le proteine di trasporto, le proteine motrici, le proteine regolatrici, le immunoglobuline e altre proteine con diverse funzioni. Un esempio di proteina globulare è la mioglobina (Figura 9), proteina relativamente piccola (P.M. 16700) che lega l’ossigeno nelle cellule muscolari, per conservare e diffondere l’ossigeno quando il muscolo si contrae rapidamente. La mioglobina è costituita da una singola catena di 153 residui di amminoacidi, ormai completamente nota, che contiene un gruppo porfirinico, costituito da quattro nuclei pirrolici, detto gruppo eme, che ha al centro un atomo di Fe2+. Il gruppo eme, relativamente piatto, è situato in una infossatura o tasca della molecola della mioglobina. L’atomo di ferro ha quattro posizioni di coordinazione nel piano dell’eme con gli atomi di azoto dell’anello porfirinico e due posizioni di coordinazione perpendicolari ad esso. Le proteine 43 Figura 9. Struttura della mioglobina Di queste una è legata ad un residuo di istidina (His F8) detta anche istidina prossimale e l’altra lega la molecola di ossigeno (Figura 10). Questa sesta posizione di coordinazione dello ione Fe2+ è vuota nella deossimioglobina, mentre è occupata dall’ossigeno nella ossimioglobina. E’ presente anche un’istidina E7, detta istidina distale che ha due funzioni molto importanti, una di prevenire l’ossidazione del Fe2+ a Fe3+ che non sarebbe più in grado di legare l’ossigeno e l’altra di diminuire l’affinità di legame del monossido di carbonio (CO) per il gruppo eme. Figura 10. Modello a sfere e bacchette del gruppo eme 44 Capitolo III Il monossido di carbonio infatti a causa della presenza dell’istidina distale viene legato in modo obliquo sul ferro dell'eme, ma tenderebbe a formare un legame molto più forte con il ferro se potesse legarsi liberamente in modo perpendicolare ad esso. Questa istidina rappresenta quindi una difesa per la mioglobina che altrimenti sarebbe avvelenata anche da minime concentrazioni di monossido di carbonio. L'ossigeno invece si lega spontaneamente in modo obliquo sul ferro e il suo legame non viene ostacolato dall'istidina. Denaturazione delle proteine E’ opportuno anche accennare alla labilità delle proteine, che possono essere facilmente denaturate, cioè alterate da piccole modifiche nell’ambiente che le circonda, ad es. dal calore. L’alterazione riguarda principalmente le interazioni deboli, come i legami a idrogeno. Se il calore viene somministrato molto lentamente la conformazione della proteina non si modifica finché non si ha brusca perdita della struttura e della funzione, che avviene in un intervallo di temperatura molto ristretto. Le proteine possono essere denaturate anche da pH estremi, da miscele di solventi organici, da alcuni soluti, come l’urea, o da detergenti. La denaturazione in genere è irreversibile (si pensi all’albumina dell’uovo con la cottura) ma esistono anche casi di denaturazione reversibile, se la proteina viene riportata alle condizioni in cui la conformazione iniziale è stabile. Una proteina in conformazione stabile viene detta nativa. Un esempio di denaturazione reversibile è presentato dalla ribonucleasi purificata, che può essere denaturata completamente per interazione con l’urea e con un riducente, che rompe i 4 ponti disolfuro ripristinando quattro molecole di cisteina, mentre l’urea scinde le interazioni idrofobiche stabilizzanti. Il processo di ripristino della conformazione nativa viene detto rinaturazione. Proteine coniugate Una proteina che lega a sé una parte non costituita da amminoacidi viene detta proteina coniugata e il gruppo non proteico prende il nome Le proteine 45 di gruppo prostetico (dal greco pròsthesis, aggiunto). Nella mioglobina, come visto precedentemente, il gruppo prostetico è l’eme, grosso anello eterociclico contenente uno ione Fe2+. Molte proteine sono coniugate e il gruppo non proteico può essere una molecola abbastanza complessa, come nel caso dell’eme, o un semplice ione metallico come Cu2+, Zn2+, Mn2+, Mo2+, altrettanto importante per il buon funzionamento della proteina stessa. Sono proteine coniugate anche quelle legate ai lipidi (lipoproteine), ai glucidi (glicoproteine) e acidi nucleici DNA o RNA (ribonucleoproteine). In questi casi le proprietà delle singole parti si sommano, ampliando ancora le possibilità funzionali delle proteine. Le lipoproteine hanno la funzione di spostare i diversi tipi di lipidi, che sono molecole apolari (ad esempio il colesterolo), attraverso un liquido polare come il sangue. La parte proteica, idrofila, della lipoproteina si sistema verso la parte esterna dell’aggregato. La funzione della proteine è quella di far riconoscere ai recettori delle membrane cellulari il tipo di lipide che dovrà essere utilizzato. Le glicoproteine sono associazioni fra proteine e carboidrati semplici o modificati il cui contenuto può variare da meno dell’1% a più del 90% del loro peso. Sono presenti in tutte le forme di vita ed hanno numerose funzioni diverse, ma il gruppo più importante è certamente quello che fa parte delle membrane cellulari. Queste molecole funzionano da recettori virali, individuando le molecole estranee. Le cellule si servono di glicoproteine per riconoscersi tra loro: sono quindi marcatori dell’identità cellulare. In questo caso, all’unicità della proteina nella sua forma a tre dimensioni si aggiunge l’enorme possibilità di ramificazioni che anche pochi monosaccaridi possono formare fra loro, mediante il legame glicosidico. Si ha così in superficie una massa glicoproteica che per ogni tipo di cellula è unica e riconoscibile solo da cellule dello stesso organo o tessuto. GLI ENZIMI Gli enzimi sono catalizzatori biologici, sono cioè molecole che accelerano il raggiungimento dell’equilibrio delle innumerevoli reazioni chimiche che si verificano fuori o dentro le cellule di un organismo, e possono anche mediare l’interconversione fra diverse forme di energia. Ogni cellula contiene alcune migliaia di enzimi diversi, tutti necessari per il suo buon funzionamento. Se viene a mancare anche un solo enzima (caso delle malattie genetiche), se l’enzima è in quantità eccessiva, se la sua attività è bloccata da un agente esterno (avvelenamento) o ancora se vengono introdotti enzimi estranei, un organismo può subire seri danni. L’azione di molti farmaci si basa sull’inibizione o sullo stimolo dell’attività di particolari enzimi. Inoltre, dosando gli enzimi nei flussi biologici si può risalire al buono o cattivo funzionamento di un sistema biologico. Con l’eccezione di piccoli gruppi di molecole di RNA catalitico, gli enzimi sono costituiti da proteine globulari con peso molecolare che varia da circa 10.000 ad 1.000.000 Dalton, formate da una sola catena polipeptidica o da più catene unite insieme da legami generalmente deboli. Molti enzimi sono proteine semplici, cioè non hanno bisogno per la loro attività che della loro catena di amminoacidi. Altri invece sono proteine coniugate, cioè necessitano di composti chimici addizionali detti cofattori. Il cofattore può essere costituito da uno o più ioni inorganici, come Fe2+, Mg2+, Mn2+, Zn2+, oppure da complesse molecole organiche chiamate coenzimi. Un coenzima o uno ione metallico legato alla proteina enzimatica con legami assai stabili, in genere covalenti, viene detto gruppo prostetico. Un enzima con tutti i suoi coenzimi o ioni metallici si dice oloenzima, mentre la sua parte proteica apoenzima o apoproteina. I coenzimi agiscono come trasportatori temporanei di gruppi funzionali e molte vitamine, sostanze organiche necessarie in piccole quantità nella dieta, sono loro precursori. 47 48 Capitolo IV Caratteristiche degli enzimi Le caratteristiche più importanti degli enzimi sono il potere catalitico e la specificità d’azione. Inoltre l’azione di alcuni enzimi è sottoposta a regolazione. Le proteine costituenti la maggior parte degli enzimi possono esplicare una funzione catalitica perché sono capaci di legare in modo specifico una grande varietà di molecole (dette substrati) con un orientamento ottimale, preludio alla formazione o alla rottura di legami chimici. Gli enzimi in sostanza catalizzano le reazioni biologiche perché tengono i substrati orientati in modo che avvenga il massimo numero possibile di urti efficaci con il reagente. In tal modo viene stabilizzato lo stato di transizione, in cui si forma la specie molecolare a più alta energia. Un enzima determina quale fra le innumerevoli reazioni possibili debba realmente avvenire. Può agire inoltre come interruttore molecolare, regolando l’attività catalitica e trasformando l’energia con la sua capacità di accoppiare l’azione di siti diversi di legame. Gli enzimi possono accelerare una reazione anche un milione di volte. Sono altamente specifici sia per la reazione catalizzata che per il tipo di reagenti. In genere un enzima catalizza una sola reazione chimica, generando un ambiente specifico in cui questa reazione è favorita: non ci sono reazioni collaterali o sottoprodotti inutili. Al massimo un enzima può catalizzare una serie di reazioni strettamente correlate. Una caratteristica delle reazioni catalizzate dagli enzimi è quella di avvenire all’interno di una tasca dell’enzima, cioè di una piccola parte della grande molecola proteica, chiamata sito attivo. Il sito attivo è la zona della superficie dell’enzima, in genere di dimensioni assai limitate rispetto a quelle dell’enzima stesso, a cui si legano specificamente la molecola o le molecole di substrato determinando sia la specificità (alta affinità per determinati composti) che la funzione catalitica della molecola proteica attiva. Il sito attivo contiene la catena laterale di almeno tre amminoacidi, in relazione stereochimica fra loro, come conseguenza del ripiegamento della catena polipeptidica. La forma e le capacità catalitiche del sito attivo sono determinate da legami deboli (struttura secondaria e terziaria) fra i resi- Gli enzimi 49 dui di amminoacidi che ne fanno parte e dalla struttura primaria, che come abbiamo visto già in precedenza è la sequenza degli amminoacidi della proteina. Il legame fra sito attivo e substrato può essere descritto come un modello chiave-serratura (Figura 1). In una serratura infatti può girare solo la sua specifica chiave, e parimenti solo una determinata molecola di substrato può avere una forma che le permetta di entrare nella fessura del sito attivo dell’enzima per fare avvenire la reazione. La formazione del complesso enzima-substrato è essenziale per la catalisi ed è il punto di partenza della cinetica di reazione. Figura 1. Rappresentazione schematica del modello chiave-serratura. Esiste anche la possibilità di un adattamento indotto (Figura 2). In questo modello, il sito attivo può essere formato dopo che l’enzima si è legato al substrato. L’enzima si modifica, prendendo una forma adatta a far avvenire la reazione dopo il suo legame con il substrato. Questo modello è più vicino alla realtà rispetto al modello chiaveserratura, ma è ancora notevolmente approssimato. Figura 2. Rappresentazione schematica dell'adattamento indotto Gli enzimi, per essere efficaci devono possedere ben precise caratteristiche: 50 Capitolo IV • pur interagendo con i substrati che generano i prodotti di reazione, al termine della reazione devono ritrovare invariata la propria struttura, pronta per riprendere l’attività catalitica; • devono essere efficaci in piccola quantità, e non devono influenzare l’equilibrio di una reazione chimica reversibile; • devono mostrare specificità nella capacità di accelerare reazioni chimiche. Infatti, la funzione di un enzima, come di qualsiasi altro catalizzatore, è quella di rendere più veloce un processo che altrimenti potrebbe richiedere giorni, mesi o addirittura anni. Ad esempio, consideriamo una reazione tipo A+B=C+D La cui costante di equilibrio è: Keq = [C] [D] / [A] [B]. Supponiamo che, ad equilibrio raggiunto, le concentrazioni dei vari reagenti siano il 30% di A + B e il 70% di C + D. L’enzima renderà più breve il tempo necessario per raggiungere queste concentrazioni sia che si parta dal 100% di A + B sia che si parta dal 100% di C + D. Il rapporto delle concentrazioni di prodotti e reagenti (Keq), si raggiungerebbe comunque indipendentemente dal catalizzatore, ma in presenza di esso viene raggiunto in un tempo inferiore di parecchi ordini di grandezza rispetto al tempo necessario perché la reazione avvenga naturalmente. La maggior parte degli enzimi agisce su un numero assai limitato di composti chimicamente simili o su specifici tipi di reazione (specificità relativa), o su di un singolo substrato (specificità assoluta). La tripsina, ad esempio, catalizza l’idrolisi di un legame peptidico fra residui di lisina e di arginina. La trombina, enzima ancora più specifico, idrolizza il legame peptidico fra residui di arginina e glicina solo in determinate sequenze di peptidi. La specificità relativa di un enzima non è un difetto ma solo un modo fisiologico di fare economia di enzimi con specificità assoluta. Ad esempio, la lipasi idrolizza tutti i legami estere tra glicerolo ed a- Gli enzimi 51 cidi grassi, rendendo così disponibile un substrato comune per successive trasformazioni. Classificazione degli enzimi Conoscere il nome di un enzima è importante, perché spesso si può risalire da esso alla funzione svolta dall’enzima stesso. A causa del grandissimo numero di enzimi conosciuti, già da molti anni si era reso necessario un sistema di nomenclatura e classificazione che desse a ciascun enzima un nome ed una collocazione precisa. Inizialmente il nome dell’enzima veniva fatto terminare con la desinenza “-ina” (pepsina, tripsina, papaina), in cui tale desinenza indicava la sua natura di proteina. In tal modo però non si faceva cenno alla reazione catalizzata né al substrato su cui l’enzima agiva. In seguito, gli enzimi presero i nomi che derivavano dal loro substrato o da una parola che descriveva la loro attività, cui veniva aggiunto il suffissi “–asi” (lattasi, maltasi, saccarasi, lipasi, proteasi), per non parlare che di enzimi che catalizzavano reazioni di idrolisi. Però talvolta lo stesso enzima aveva due o più nomi oppure due enzimi diversi avevano lo stesso nome, e ciò poteva generare ambiguità, soprattutto per il fatto che il numero di enzimi identificati è andato sempre crescendo. Per ovviare a questi inconvenienti, la IUB (International Union of Biochemistry and molecular biology) ha adottato, per convenzione internazionale, un sistema di nomenclatura e di classificazione degli enzimi valido per tutti, basato sul tipo di reazione catalizzata e sul nome del substrato su cui agisce l’enzima. Gli enzimi sono divisi in sei classi principali, numerate da 1 a 6, in base al tipo di reazione catalizzata (Tabella 1). Ciascuna classe è suddivisa in sottoclassi, anch’esse distinte con un numero e suddivise ancora in sotto-sottoclassi, che a loro volta contengono un elenco dei singoli enzimi. Capitolo IV 52 Tabella 1. Classificazione degli enzimi. CLASSI AZIONE CATALITICA 1) Ossidoreduttasi Ossidoriduzione 2) Transferasi Trasferimento di gruppi atomici 3) Idrolasi Rottura di legami per mezzo dell’acqua 4) Liasi Addizione o sottrazione di gruppi con rottura o formazione di doppi legami 5) Isomerasi Reazioni di isomerizzazione 6) Ligasi Formazione di legami accoppiati all’idrolisi dell’ATP Per ogni enzima quindi esiste un nome proposto, in genere breve ed appropriato per l’uso comune, un nome sistematico che individua la reazione catalizzata e un numero di classificazione. Ad esempio, la reazione che trasforma il D-glucosio in D-glucosio-6-fosfato per azione dell’ATP necessita di una fosfotransferasi, che trasforma contemporaneamente l’ATP in ADP. Il nome dell’enzima coinvolto in base alla reazione catalizzata è ATP-D-glucosio-6 fosfato transferasi. Il suo numero di classificazione IUB è 2.7.1.10, dove la cifra 2 rappresenta la classe (transferasi); la cifra 7 la sottoclasse (gruppi fosforici); la cifra 1 la sotto-sottoclasse (fosfotransferasi con gruppo alcolico come accettore); infine il numero 10 il numero progressivo assegnato all’ enzima. Quando tuttavia il nome di un enzima diventa troppo lungo o scomodo da usare se ne può adottare uno più corto: ad esempio, nei trasferimenti di gruppi fosforici che provengono dall’ATP o nell’addizione di gruppi fosforici all’ADP per formare ATP il nome dell’enzima diventa chinasi. Nel caso specifico esochinasi o ancora più specificamente D-glucosio-6-fosfato chinasi. Gli enzimi 53 Nel dettaglio le sei classi di enzimi sono le seguenti: 1. Ossidoreduttasi: è la classe più ampia, e interviene nelle reazioni di ossidoriduzione. A volte questi enzimi prendono il nome di ossidasi (o deidrogenasi) se favoriscono reazioni di ossidazione e reduttasi se la reazione catalizzata è una riduzione. Es. la glucossidasi; 2. Transferasi: catalizzano il trasferimento di un gruppo funzionale da una molecola ad un’altra. Es. la glucochinasi; 3. Idrolasi: catalizzano l’idrolisi di legami con intervento di una molecola d’acqua. Es. le lipasi; 4. Liasi: favoriscono le addizioni di gruppi ad un doppio legame o sottraggono gruppi per formare doppi legami. Es. enoil idratasi. 5. Isomerasi: catalizzano la conversione di un composto nel suo isomero con il trasferimento di gruppi all’interno della molecola. Es.fosfo-trioso-isomerasi; 6. Ligasi: intervengono per generare nuovi legami, creare le molecole strutturali utilizzando l’energia (ATP) proveniente da altre reazioni. Es. acetil-CoA-carbossilasi. Modalita’ d’azione degli enzimi Come detto sopra, gli enzimi sono catalizzatori biologici e come tali hanno caratteristiche in comune con i catalizzatori inorganici: come quelli infatti prendono parte alla reazione ma si ritrovano inalterati alla fine di essa. La catalisi enzimatica, come quella inorganica, aumenta drasticamente la velocità della reazione. Una reazione enzimatica semplice può essere schematizzata così: E+S ES EP E+P dove E, S e P rappresentano rispettivamente l’enzima, il substrato ed il prodotto. ES ed EP sono i complessi dell’enzima con il substrato e con il prodotto. Le frecce a due direzioni dell’equazione scritta sopra indicano che ogni enzima che catalizza la reazione da S a P, catalizza anche quella inversa da P a S. L’enzima non viene consumato durante questo pro- 54 Capitolo IV cesso e il punto di equilibrio resta inalterato. La reazione raggiunge però l’equilibrio molto prima quando è presente l’enzima, perché la velocità di reazione è molto superiore alla norma. La funzione di un catalizzatore, e quindi nel caso specifico dell’enzima, è quella di aumentare la velocità di reazione, senza però modificare gli equilibri della reazione stessa. Qualsiasi reazione può essere rappresentata mediante un diagramma che riporta in ascisse la coordinata di reazione e in ordinate l’energia libera G (si ricorda che l’energia libera è quella parte dell’energia totale di un sistema libera di compiere un lavoro). Quindi l’energia libera viene messa in relazione con il procedere della reazione. Nella sua forma normale stabile o stato basale una molecola contiene una determinata quantità di energia libera. Per descrivere la variazione di energia libera fra lo stato iniziale e lo stato finale sono state stabilite determinate condizioni standard per la cellula (temperatura di 298°K, corrispondente a 25°C; pressione parziale di ogni gas a 1 atmosfera o 101,3 K Pascal; concentrazione di ogni soluto 1 Molare). La variazione di energia libera in queste condizioni si indica con ∆G°, variazione di energia libera standard. Poiché nei sistemi biologici la concentrazione degli ioni H+ è molto lontana da 1M, i biochimici hanno stabilito la costante ∆G°’, cioè la variazione di energia libera standard a pH = 7. L’equilibrio tra S e P è funzione della differenza fra l’E libera dei due composti, cioè fra lo stato finale e lo stato iniziale della reazione, nei rispettivi stati basali. Se l’E libera dello stato basale dei prodotti P è minore di quella dei reagenti iniziali S la differenza fra le energie sarà negativa, quindi ∆G°’<0. La reazione si dirà esoergonica o spontanea (vedi il capitolo sull’energia delle reazioni). Se viceversa l’E libera dello stato basale dei prodotti P è maggiore di quella dei reagenti iniziali S la differenza fra le energie sarà positiva, quindi ∆G°’>0. La reazione si dirà endoergonica o non spontanea. Questi equilibri non vengono modificati da un catalizzatore. Tuttavia, anche nel caso che un equilibrio sia favorevole non significa che la velocità di conversione da S a P sia elevata. Infatti la velocità della reazione dipende da un parametro completamente diverso. Tra i reagenti ed i prodotti esiste una barriera energetica da superare. Gli enzimi 55 Questa energia in più è necessaria per allineare i gruppi che devono reagire fra loro, per formare cariche transitorie instabili, per riorganizzare legami e per compiere altre trasformazioni in modo che la reazione possa procedere verso la formazione dei prodotti. Per far avvenire la reazione le molecole dei reagenti devono superare questa barriera e raggiungere un livello energetico più alto. Il punto più elevato della curva prende il nome di stato di transizione, che è un momento molecolare transitorio, da non confondersi con un intermedio della reazione. La differenza fra lo stato basale dei reagenti indisturbati e lo stato di transizione si dice energia di attivazione (Figura 3). Figura 3. Grafico dell'andamento dell'energia libera durante una reazione. Ovviamente, tanto maggiore sarà l’energia di attivazione, tanto minore sarà il numero di molecole che avranno la possibilità di raggiungere e superare quel punto (esattamente come, se pensiamo a un salto in alto, più elevata sarà l’altezza dell’asticella, tanto minore sarà il numero di persone che avranno la capacità di superarla). Quindi una elevata energia di attivazione corrisponde ad una bassa velocità di reazione. 56 Capitolo IV Fattori che influenzano le reazioni catalizzate dagli enzimi La struttura delle proteine, come si è visto nel capitolo a loro dedicato, può variare in dipendenza di fattori esterni come temperatura, pH, concentrazione salina, presenza di solventi organici ed altro, che possono modificare la loro struttura tridimensionale e causare anche una loro denaturazione irreversibile. Poiché gli enzimi sono proteine, la loro efficacia come catalizzatori può dipendere da questi parametri. 1) Effetto del pH Una variazione nella concentrazione idrogenionica può avere effetto sulla geometria del sito attivo e sulle cariche elettriche dei gruppi coinvolti nel legame con la molecola del substrato. Il pH influenza anche la dissociazione dei gruppi acidi e basici presenti sulle molecole del substrato, che devono avere una ben definita distribuzione di cariche elettriche per poter legare con l’enzima. In genere l’intervallo di pH è abbastanza ristretto. Il pH a cui l’enzima presenta la massima efficienza viene detto pH ottimale. Questo valore può variare anche molto da enzima ad enzima e può anche non essere identico al pH del distretto intracellulare in cui si trova l’enzima. 2) Effetto della temperatura La velocità di una reazione può essere aumentata aumentando la temperatura, e quindi l’energia cinetica delle molecole in modo che un maggior numero di esse possa produrre degli urti efficaci. Un aumento di temperatura troppo elevato però potrebbe portare alla denaturazione delle proteine che costituiscono gli enzimi, quindi deve essere evitato. Le proteine, e quindi anche gli enzimi, sono termolabili. La velocità di denaturazione termica dell’enzima è generalmente molto bassa a 0°C e raddoppia per ogni aumento di 10°C di temperatura. Riportando in diagramma la temperatura contro l’attività massima dell’enzima si ottiene una curva a campana: per temperature superiori a quella ideale si ha la denaturazione della proteina, per temperature inferiori la velocità di reazione diminuisce. A temperature basse tuttavia gli enzimi non vengono denaturati e possono riprendere la loro attività catalitica quando la temperatura venga di nuovo aumentata. 3) Effetto della concentrazione salina Gli enzimi 57 Come per il pH, anche un aumento della concentrazione salina può portare all’alterazione della geometria del sito attivo dell’enzima, salificando ad esempio i gruppi –COOH liberi, facendo variare la distribuzione delle cariche e alterando così i legami enzima-substrato. La catalisi enzimatica Per molti enzimi, la velocità di catalisi (V) varia con la concentrazione del substrato [S]. V è definita come il numero di moli di prodotto che si formano in un secondo. Supponiamo di avere una quantità definita di enzima e che le condizioni ambientali siano ideali per il suo funzionamento (adatta temperatura, adatta concentrazione della soluzione, pH ottimale ecc.). Aggiungendo ad una concentrazione fissa di enzima (E) una piccola quantità di substrato (S) la conversione di S in prodotto (P) avrà una velocità iniziale (V0). In poco tempo l’equilibrio si sposterà completamente a destra e tutto il substrato verrà convertito in prodotto. E+S ES E+P Aggiungendo sempre più substrato la quantità di prodotto ottenuto sarà sempre maggiore, quindi la velocità di trasformazione sarà sempre crescente. In questa fase c’è quindi proporzionalità diretta tra quantità di substrato aggiunto e quantità di prodotto formato. Ad un certo punto però, aggiungendo sempre nuovo substrato, vediamo che la crescita lineare della quantità di prodotto formato si interrompe e la velocità di trasformazione si mantiene costante, per quanto substrato venga aggiunto. La velocità inoltre è la massima possibile (Vmax). La velocità massima viene raggiunta quando tutti i siti catalitici dell’enzima sono saturati dal substrato, cioè quando per ogni molecola di enzima c’è una molecola di substrato. La Vmax è chiamata anche velocità di saturazione. 58 Capitolo IV La relazione matematica che lega la velocità iniziale della reazione con la concentrazione S del substrato è: V0 = Vmax [S] [S] +KM conosciuta come equazione di Michaelis - Menten, dove V è la velocità della reazione, Vmax la velocità massima e KM una costante, detta costante di Michaelis - Menten. Figura 4. Cinetica di Michaelis-Menten. Questa equazione verifica i dati cinetici mostrati in Figura 4 ossia a concentrazioni di substrato molto basse, quando la concentrazione del substrato è molto minore di KM, ([S]<<KM), il termine [S] al denominatore dell’equazione di Michaelis-Menten diviene irrilevante e la velocità della reazione diventa V = [S] * Vmax / KM cioè la velocità della reazione è direttamente proporzionale alla concentrazione del substrato. Per elevate concentrazioni di substrato invece, S è molto maggiore di KM ([S] >> KM) il valore di KM diviene trascurabile rispetto a S e la velocità della reazione diventa uguale alla velocità massima (V0 = Gli enzimi 59 Vmax), cioè la velocità è indipendente dalla concentrazione del substrato. Quando [S] = KM, allora V0 = 1/2 Vmax . Perciò KM corrisponde alla concentrazione di substrato per cui la velocità della reazione è metà della velocità massima. Ma determinare la Vmax non è semplice sperimentalmente, mentre è molto più facile determinare la concentrazione di substrato che corrisponde ad 1/2 della Vmax. Il valore di KM è un parametro molto importante per la caratterizzazione di un enzima, poiché è la misura dell’affinità o dell’attrazione dell’enzima per il substrato. Più alta è la KM, minore è l’affinità dell’enzima per il substrato. Infatti, se è necessaria un’alta concentrazione di substrato per saturare metà delle molecole di enzima in ogni istante, si può dire che l’enzima ed il substrato hanno fra loro una bassa affinità. Se viceversa una bassa concentrazione di substrato è sufficiente per saturare metà delle molecole di enzima in ogni istante si può dire che l’enzima ed il substrato hanno fra loro una elevata affinità. Vmax e KM possono essere misurate variando la concentrazione del substrato. Per questo è conveniente trasformare l’equazione di Michaelis Menten in una nuova equazione che determini una linea retta. Ciò è possibile facendo i reciproci di entrambe le parti dell’equazione: 1 1 KM 1 + [ ] + = Vmax Vmax S V Il grafico di 1/V in funzione di 1/ [S] determina una linea retta con un intercetta pari a 1/Vmax e una pendenza pari a KM / Vmax . Questo viene detto grafico dei doppi reciproci o anche di Lineweaver-Burk. Il valore di KM degli enzimi è piuttosto variabile (da 10-1 a 10-7 ) e dipende dal tipo di substrato e dalle condizioni ambientali (pH, temperatura, forza ionica). La KM corrisponde alla concentrazione di substrato per cui metà dei siti sono occupati (Figura 5). 60 Capitolo IV Figura 5. Grafico dei doppi reciproci o di Lineweaver-Burk. Occorre in questa sede anche far cenno al numero di turnover di un enzima, parametro importante per la valutazione della sua efficacia. Esso rappresenta il numero di molecole di substrato che vengono convertite in prodotto da una molecola di enzima nell’unità di tempo, quando l’enzima è completamente saturato dal substrato. Inibizione dell’attivita’enzimatica Gli enzimi possono essere inattivati o inibiti da alcune sostanze. Si parla di inattivazione se il processo è irreversibile, di inibizione se le sostanze si possono rimuovere. L’inibizione si dice competitiva quando il substrato è spostato dalle sostanze inibenti, cioè se la sostanza che inibisce compete col substrato. Se aumenta la quantità di substrato, questo può competere per il sito attivo e rimuovere la sostanza inibente; si ha inibizione non competitiva quando il substrato si lega nel suo sito e le sostanze inibitorie si fissano altrove. Gli inibitori enzimatici sono molecole che interferiscono con la catalisi, rallentando o bloccando le reazioni catalizzate da enzimi. Poiché la quasi totalità dei processi cellulari dipende da enzimi, non sorprende che gli inibitori enzimatici siano tra i più importanti agenti in farmacologia. Ad esempio l’aspirina (acido acetilsalicilico) inibisce l’enzima cicloossigenasi (COX), fondamentale per la produzione delle prostaglandine coinvolte nei meccanismi dell'infiammazione e del dolore. Gli enzimi 61 L’inibizione reversibile può essere competitiva, non competitiva o mista. Un inibitore competitivo, come dice il nome stesso, compete con il substrato per il legame al sito attivo di un enzima. Quando l’inibitore (I) occupa il sito attivo dell’enzima impedisce il legame col substrato. Gli inibitori competitivi sono spesso composti che hanno struttura simile al substrato e si combinano con l’enzima formando un complesso enzima–substrato (EI) che non subisce la catalisi. Per quanto questi complessi siano di breve durata e abbastanza deboli, tuttavia possono diminuire l’efficacia catalitica dell’enzima. Si può ovviare a questa interferenza semplicemente aggiungendo altro substrato: infatti la geometria molecolare di substrato ed inibitore è molto simile e quindi aumentando la concentrazione del substrato “buono” si riduce la probabilità che il competitore vada a legarsi con l’enzima. Un inibitore non competitivo, a differenza del competitivo, si lega all’enzima in un sito diverso da quello del substrato. Un inibitore misto si lega ad un sito diverso da quello del substrato, ma si può legare sia all’enzima E che al complesso enzima-substrato ES. Inibizione irreversibile. Gli inibitori reversibili si legano all’enzima bersaglio ma possono dissociarsi ripristinando l’attività dell’enzima. Gli inibitori irreversibili invece si combinano o distruggono un gruppo funzionale dell’enzima essenziale per la sua attività catalitica. Si può formare un legame covalente fra un inibitore irreversibile ed un enzima. Gli inibitori irreversibili sono molto utili per lo studio delle reazioni enzimatiche. Gli inattivatori o inibitori suicidi sono composti poco reattivi finché non raggiungono il sito attivo di uno specifico enzima. Un inibitore suicida può subire le prime tappe della reazione enzimatica ma si combina poi in modo irreversibile con l’enzima. Questi composti vengono anche detti inattivatori basati sul meccanismo, perché utilizzano il meccanismo normale della reazione enzimatica per inattivare l’enzima. Enzimi regolatori Una categoria importante di enzimi alla quale occorre far cenno è quella degli enzimi regolatori. Sono enzimi che determinano la veloci- Capitolo IV 62 tà complessiva della sequenza in quanto catalizzano le reazioni più lente o quelle che limitano la velocità. Gli enzimi regolatori variano la loro attività catalitica in risposta a certi segnali. Mediante l’azione di enzimi regolatori la velocità di ogni sequenza metabolica si adegua costantemente alla domanda cellulare di energia e di biomolecole. Esistono due classi di enzimi regolatori: gli enzimi allosterici, che agiscono mediante il legame non covalente e reversibile con composti regolatori, detti modulatori allosterici, che in genere sono metaboliti o piccoli cofattori, e gli enzimi regolati mediante modificazioni covalenti irreversibili. Tutte e due le classi hanno più subunità e in alcuni casi i siti regolatori e i siti attivi si trovano su subunità diverse. In alcuni sistemi multienzimatici l’enzima regolatore viene inibito in modo specifico dal prodotto finale della via, quando questo composto si accumula oltre il normale fabbisogno della cellula. La velocità della produzione del prodotto finale della via metabolica è quindi bilanciata dalle necessità della cellula. Questo tipo di regolazione si dice inibizione a feedback, o inibizione retroattiva. L’accumulo del prodotto finale di una via metabolica inibisce il primo enzima della stessa via. I coenzimi Prima di chiudere il capitolo dedicato agli enzimi, sarà utile accennare alla struttura e alle funzioni dei principali coenzimi, molecole che operano come trasportatori temporanei di specifici gruppi funzionali. Come già ricordato sopra, se il coenzima è legato covalentemente alla proteina viene detto gruppo prostetico. I coenzimi principali che troveremo in molte vie metaboliche sono: 1. 2. Coenzimi delle reazioni di ossido-riduzione. NAD+, FAD, acido lipoico; Coenzimi trasportatori di gruppi. Piridossalfosfato, PLP, per i gruppi amminici; Tetraidrofolato per gruppi contenenti un solo carbonio; Tiamina pirofosfato (TPP) per aldeidi e cheto- Gli enzimi 63 ni “attivati”; Biotina per il gruppo CO2; Coenzima A (coenzima acilante) per il trasporto di gruppi acilici, Adenosin Tri fosfato (ATP) per i gruppi fosfato; 5’ Deossi adenosil cobalammina (Coenzima B12) per atomi di idrogeno e gruppi alchilici. Vedremo di esaminare la struttura e le funzioni dei principali fra questi coenzimi. 1. Coenzimi delle reazioni di ossido-riduzione. La maggior parte delle cellule possiede enzimi che catalizzano l’ossidazione o la riduzione di centinaia di composti diversi. Questi enzimi trasferiscono elettroni dai loro substrati su pochi tipi di trasportatori universali e ioni H+. La loro riduzione durante i processi catabolici consente di conservare l’energia libera rilasciata dall’ossidazione dei substrati. I nucleotidi NAD+ e NADP+ e FMN e FAD sono cofattori solubili in acqua che possono andare incontro ad ossidazioni e riduzioni reversibili in molte reazioni del metabolismo. Mentre i primi si spostano rapidamente da un enzima all’altro, FMN e FAD sono legati saldamente agli enzimi ed agiscono da gruppi prostetici. NAD+ e NADP+ sono le sigle che indicano la forma ossidata di due coenzimi, il primo è la Nicotinammide Adenin Dinucleotide e il secondo la Nicotinammide Adenin Dinucleotide Fosfato. Si tratta di due nucleotidi costituiti da un anello nicotinamidico (A), legato ad una molecola di ribosio (B) e ad un gruppo fosfato (C), a sua volta legato con legame fosfoanidridico ad un altro gruppo fosfato, cui è unita un’altra molecola di ribosio (D) ed un’altra base azotata, l’adenina (E) (Figura 6). 64 Capitolo IV Figura 6. Struttura del NAD+ La parte reattiva del NAD+ è il suo anello nicotinamidico (A) che accetta uno ione idrogeno (H+) e due elettroni, equivalenti ad uno ione idruro dal substrato trasformandosi nella forma ridotta NADH. La struttura del NADP+ è uguale, tranne che per un gruppo fosforico che esterifica l’OH sul C2 del ribosio (D) legato all’adenina. I particolari delle due forme ridotte sono illustrate in Figura 7. Figura 7. Particolari della struttura del NADH e del NADPH. Entrambi i coenzimi possono subire una riduzione reversibile sull’anello nicotinammidico. Quando una molecola di substrato subisce una ossidazione perde due atomi di idrogeno (infatti una reazione Gli enzimi 65 di ossidazione viene detta anche deidrogenazione). La forma ossidata del coenzima accetta uno ione idruro (H-) costituito da un protone e da due elettroni e si trasforma nella forma ridotta. Il secondo idrogeno, sotto forma di H+ viene rimosso dal substrato e rilasciato nell’ambiente acquoso. La semi-reazione per ogni nucleotide è quindi: NAD+ + 2 e- + 2 H+ = NADH + H+ NADP+ + 2 e- + 2 H+ = NADPH + H+ In ambedue le molecole il segno + non indica che ci sono cariche positive nette, ma che l’anello nicotinammidico è nella sua forma ossidata, con una carica positiva sull’azoto. Il NAD+ in genere si trova nelle reazioni cataboliche, mentre il NADPH è di solito il cofattore delle reazioni anaboliche. Pochissimi enzimi possono usare entrambi i coenzimi e quasi sempre hanno una netta preferenza per uno di essi. Ciò consente alla cellula di mantenere due trasportatori di elettroni nello stesso compartimento cellulare. I gruppi prostetici FMN e FAD rappresentano la forma ossidata rispettivamente del Flavin Mono Nucleotide e del Flavin Adenin Dinucleotide (Figura 8). Figura 8. Strutture di FMN e FAD. Le forme ridotte sono FMH2 e FADH2. Gli enzimi che utilizzano questi cofattori sono le flavoproteine, che derivano dalla vitamina ri- 66 Capitolo IV boflavina e catalizzano le reazioni di ossido riduzione. Nelle flavoproteine, la struttura ad anello legata al ribosio (o più esattamente all’alcol ribitolo) detta anello isoallossazinico, subisce riduzioni reversibili accettando uno o due equivalenti riducenti sotto forma di atomi di idrogeno (elettrone + protone) da un substrato riducente. Quando un nucleotide flavinico accetta un solo atomo di idrogeno si genera la forma semichinonica dell’ anello isoallossazinico FADH* e FMNH* con un elettrone spaiato. Nella maggior parte delle flavoproteine i nucleotidi flavinici sono legati saldamente all’enzima e nel caso della succinato deidrogenasi sono veri e propri gruppi prostetici, legati cioè in modo covalente. Le flavoproteine possono trattenere equivalenti riducenti, mentre ne catalizzano il trasferimento da un substrato donatore ad uno accettore. Sono spesso molto complesse, e alcune hanno saldamente legati anche ioni inorganici come Fe e Mo, in grado di partecipare al trasferimento degli elettroni. Il lipoato ha due gruppi tiolici, entrambi essenziali per la sua funzione di cofattore. Allo stato ridotto gli atomi di zolfo sono sotto forma di gruppi –SH, mentre l’ossidazione produce un ponte disolfuro – S-S- simile a quello che si forma tra due residui di cisteina in una proteina. Il lipoato può servire sia come trasportatore di elettroni che come trasportatore di acili. Figura 9. Struttura del lipoato. Nel complesso della piruvato deidrogenasi il lipoato agisce da gruppo prostetico: il legame covalente è un legame ammidico fra l’acido lipoico e la catena laterale di un residuo di lisina. Gli enzimi 67 2. Coenzimi trasportatori di gruppi (coenzimi delle transferasi). 2.1 Piridossalfosfato, PLP, trasportatore di gruppi amminici. Il distacco del gruppo α-amminico, la prima tappa del catabolismo della maggior parte degli amminoacidi è promosso da enzimi detti amminotransferasi o transaminasi che contengono tutte come gruppo prostetico il PLP, che deriva dalla piridossina (vitamina B6) (Figura 10). Nelle reazioni di transaminazione il gruppo amminico α di un amminoacido viene trasferito all’atomo di carbonio α di un chetoacido, generando contemporaneamente l’α-chetoacido corrispondente all’amminoacido. Tutte le amminotransferasi hanno lo stesso gruppo prostetico ed identico meccanismo di reazione. Il piridossalfosfato agisce come trasportatore transitorio di gruppi amminici a livello del sito attivo delle amminotransferasi. Questo cofattore va incontro a trasformazioni reversibili tra la sua forma aldeidica, il piridossalfosfato, che può accettarre un gruppo amminico da un amminoacido e la sua forma amminata, la piridossammina fosfato (PMP) , che dona il suo gruppo amminico ad un α-chetoacido. Figura 10. Struttura del piridossalfosfato (PLP). Capitolo IV 68 NH2 H2 C O HO H3C N CH O P O O Figura 11. Struttura della piridossammina fosfato (PMP) Le amminotransferasi sono esempi di enzimi che catalizzano reazioni bimolecolari a ping-pong. La reazione generale catalizzata dalle transaminasi è la seguente: Amminoacido (1) + α-Chetoacido (1) = α-Chetoacido (2) + Amminoacido (2) Questa reazione totale può essere descritta attraverso una serie di passaggi intermedi. Il primo amminoacido si lega al sito attivo, dona il suo gruppo amminico al PLP, che si trasforma in piridossammina fosfato (Figura 11), e si allontana sotto forma di α-chetoacido. Amminoacido (1) + Enzima-PLP = α-Chetoacido (1) + Enzima-PMP Il primo substrato (amminoacido 1) deve lasciare il sito attivo perché il secondo substrato (amminoacido 2) possa legarsi a sua volta. Si lega poi al sito attivo il secondo substrato, l’α-chetoacido (2), che accetta il gruppo amminico della piridossammina fosfato ed esce sotto forma di amminoacido (2), lasciando di nuovo il piridossal fosfato. α-Chetoacido (2) + Enzima-PMP = Amminoacido (2) + Enzima-PLP Gli enzimi 69 Le transaminasi più note sono: l’aspartato aminotransferasi (AST o glutammato-ossalacetato transaminasi, GOT) e l’alanina aminotransferasi (ALT o glutammato-piruvato transaminasi, GPT). La AST catalizza la seguente reazione: Aspartato + α-Chetoglutarato = Ossalacetato + Glutammato La ALT catalizza quest’altra reazione: Alanina + α-Chetoglutarato = Piruvato + Glutammato Questi enzimi sono ubiquitari nell’organismo e sono usate in medicina soprattutto al fine di evidenziare la presenza o meno di un danno epatico. 2.2 Tetraidrofolato trasportatore altamente versatile di unità monocarboniose attivate. Il tetraidrofolato è costituito da tre gruppi: una pteridina sostituita, il p-amminobenzoato (PABA) e una catena di uno o più residui di glutammato (Figura 12). Il tetraidrofolato interviene in reazioni di trasferimento di unità monocarboniose a diversi livelli di ossidazione e funge da trasportatore intermedio. Le unità monocarboniose possono essere: • gruppo metilico (-CH3); • gruppo metilenico (-CH2-); • gruppo formilico (-CHO); • gruppo formiminico (-CHNH). Il gruppo monocarbonioso si può legare all'azoto N5 della 6metilpterina o a quello N10 del PABA (talvolta ad entrambi). 70 Capitolo IV Figura 12. Struttura del tetraidrofolato. 2.3 Tiamina pirofosfato (TPP) trasportatore di aldeidi e chetoni “attivati”. E’ un derivato della vitamina B1. La TPP si trova legata saldamente (è un gruppo prostetico) a tre importanti enzimi: le transchetolasi, il complesso della piruvato deidrogenasi o la piruvato decarbossilasi della fermentazione alcolica. In ogni caso un gruppo aldeidico o chetonico attivato viene trasferito ad un accettore e in alcuni casi successivamente decarbossilato. In entrambi i casi il sito di addizione del substrato chetonico è l’anello tiazolico del gruppo prostetico. L’atomo di C in posizione 2 della TPP legata, essendo posto fra gli atomi di N ed S si ionizza facilmente per formare un carbanione. L’atomo di C carico negativamente di questo intermedio reattivo può addizionare un gruppo carbonilico. L’atomo di N carico positivamente dell’anello tiazolico funziona da trappola per gli elettroni favorendo la formazione della carica negativa sull’intermedio attivato (Figura 13). Figura 13. Struttura della tiaminapirofosfato (TPP). Gli enzimi 71 2.4 Coenzima A (CoA) trasportatore di gruppi acilici. Il nome Coenzima A sta per Coenzima Acilante, cioè trasportatore di acili (unità bicarboniose). I gruppi acilici sono importanti costituenti dell’ossidazione degli acidi grassi sia della sintesi dei lipidi di membrana. E’ composto di tre parti: una unità di β-mercaptoetilammina, una unità di pantotenato e una adenina legata ad un ribosio fosforilato e a due unità fosfatiche (Figura 14). Il sito reattivo della molecola è il gruppo solfidrilico terminale. Si può rappresentare con la sigla CoA-SH. I gruppi acile si legano al Coenzima A formando un gruppo tioestere e il composto che ne deriva si chiama Acil-CoA, R-CO-S-CoA. Data la loro energia libera di idrolisi, i tioesteri hanno un alto potere di trasferimento del gruppo acilico che può essere ceduto ad una varietà di molecole accettrici. L’idrolisi di un legame tioestere è termodinamicamente più favorevole di quella di un legame estere per il carattere di doppio legame del legame C-O rispetto al C-S. Il gruppo acilico legato al Coenzima A può quindi essere considerato una forma attivata. Il Coenzima A trasporta gruppi acilici attivati, come l’ATP trasporta gruppi fosforici attivati. Figura 14. Struttura del Coenzima A (CoA). 72 Capitolo IV Il gruppo che più spesso si trova legato al Coenzima A è il gruppo acetile, si forma così l’Acetil-CoA. Il ∆G per l’idrolisi di questa molecola è molto negativo, -7,5 Kcal/mole, di conseguenza l’acetil-CoA ha un alto potenziale di trasferimento del gruppo acetile in quanto reazione esoergonica. 2.5 Biotina, trasportatore mobile di CO2 attivata. E’ un gruppo prostetico che ha la funzione di trasportare CO2 attivata (Figura 15). La biotina è una vitamina idrosolubile, vitamina H, coinvolta nel metabolismo di acidi grassi e carboidrati. E’ presente nelle reazioni di carbossilazione, come coenzima ad esempio della piruvato carbossilasi, in cui il gruppo carbossilico terminale della biotina (B) è legato da una catena lunga e flessibile al gruppo amminico e di uno specifico residuo di lisina dell’enzima. La carbossilazione del piruvato avviene in tre fasi: Il gruppo carbossilico nell’intermedio enzima-carbossibiotina (CO2-Enzima-B) è legato all’atomo di azoto N1 dell’anello della biotina risultando così attivato. Il gruppo carbossilico attivato viene poi trasferito alla carbossibiotina al piruvato per formare ossalacetato. Figura 15. Struttura della biotina. Gli enzimi 73 2.6 Adenosin Trifosfato (ATP) trasportatore di gruppi fosfato. L’ATP è un nucleotide costituito da una adenina, un ribosio e una unità trifosfato (Figura 16). La forma attiva dell’ATP è un complesso con ioni Mg2+ o Mn2+ mediante interazioni con gli atomi di ossigeno dei gruppi fosforici. Figura 16. Struttura dell'ATP. L’ATP è una molecola ricca di energia poiché il suo gruppo trifosforico contiene due legami fosfoanidridici. La facilità di idrolisi dell’ATP è dovuta al fatto che l’ATP a pH 7 contiene 4 cariche negative che tendono a respingersi piuttosto fortemente perché sono abbastanza vicine. La repulsione elettrostatica si riduce quando l’ATP viene idrolizzato. Inoltre i prodotti dell’idrolisi dell’ATP, ADP e Pi, hanno una maggiore stabilità per risonanza rispetto alla molecola intera. Nelle condizioni cellulari tipiche il valore di ∆G delle idrolisi è circa – 12 kcal mol-1. L’energia libera rilasciata nell’idrolisi dell’ATP viene utilizzata per favorire le reazioni che richiedono un rifornimento di energia. A sua volta l’ATP si forma a partire da ADP e Pi durante l’ossidazione di sostanze nutrienti o per trasformazione dell’energia solare. Il ciclo ATP-ADP è il sistema fondamentale per lo scambio di energia nei sistemi biologici. E’ importante notare che il potenziale di trasferimento del gruppo fosforico dell’ATP ha un valore intermedio rispetto a quelli delle molecole fosforilate presenti nei sistemi biologici. Questa posizione intermedia consente all’ATP di funzionare in modo efficace come trasportatore di gruppi fosforici. 74 Capitolo IV Nei sistemi biologici vi sono altri composti che hanno un elevato potenziale di trasferimento del gruppo fosforico, come il fosfoenolpiruvato, l’acetil fosfato e la fosfocreatina. Ciò significa che il fosfoenolpiruvato può trasferire il suo gruppo fosforico all’ADP e formare ATP. PRINCIPI DI BIOENERGETICA La bioenergetica è lo studio quantitativo di come i sistemi biologici producono ed usano l'energia. La bioenergetica è un branca della termodinamica ed è essenziale per: • Comprendere come si produce energia a partire dai processi metabolici; • Comprendere la struttura delle macromolecole; • Comprendere come avviene il trasporto nelle membrane. In breve, per la comprensione di tutti i processi fondamentali che definiscono la biochimica. I sistemi biologici e le leggi della termodinamica La prima considerazione da fare quando si parla dell’energia di un sistema biologico è che le cellule non possono sottrarsi alle leggi della chimica e della fisica. Le leggi che governano la conversione dell’energia da una forma in un’altra valgono per una cellula come per una macchina a vapore. Tuttavia, mentre i processi che vediamo nel mondo che ci circonda sono apparentemente irreversibili e portano in definitiva ad un aumento del disordine (pensiamo ad una foresta che brucia, a un palazzo che crolla, a un vegetale che si degrada in putrefazione) sembra invece che gli organismi viventi possano creare ordine dal disordine. Per conservare il suo stato dinamico nel rapporto con le cellule vicine anche la cellula più semplice deve attivare dei processi che ripristinano l’ordine che tende inesorabilmente a decadere verso il disordine. Per comprendere come questo sia possibile occorre studiare da dove viene e come si organizza l’energia necessaria per i processi vitali e come questa energia viene presa dall’ambiente e trasformata durante l’esistenza degli organismi viventi. La bioenergetica è lo studio quantitativo delle conversioni di energia che avvengono nelle cellule e della natura e delle funzioni dei processi chimici che sono alla base di esse. 75 76 Capitolo V Per interpretare e comprendere bene il meccanismo che regola le reazioni chimiche e le trasformazioni energetiche nell’organismo vivente, bisogna avere presenti i concetti fondamentali della termodinamica di equilibrio, cioè dell’insieme di leggi e principi che governano il flusso e lo scambio di energia , calore e materia nei sistemi da studiare. I principi della termodinamica possono essere espressi così in sintesi riconducendoli a tre osservazioni sperimentali fondamentali. Ciascuno di essi porta infatti a definire una funzione di stato fondamentale, nell'ordine: Temperatura (T), Energia Interna (U), Entropia (S). • Principio numero 0: "Se due corpi, A e B, sono entrambi in equilibrio termico con un terzo corpo, C, essi sono in equilibrio termico anche fra loro." Questo enunciato sta alla base del concetto di temperatura e della sua misurazione mediante il termometro. • I Principio: "L'energia dell'universo è costante." È una delle possibili formulazioni del primo principio della termodinamica. Non è altro che l'espressione del concetto di conservazione dell'energia. Si introduce con esso la funzione energia interna di un sistema. Consente di stabilire quali trasformazioni siano possibili. • II Principio: "È impossibile invertire completamente un qualsiasi processo naturale." Questa è una delle possibili formulazioni del secondo principio. Esso pone delle limitazioni alle trasformazioni di calore in lavoro e introduce la funzione entropia. Consente di stabilire quali delle trasformazioni possibili siano spontanee. Col termine di sistema termodinamico s'intende una determinata porzione di spazio delimitata da una superficie, reale o apparente, che racchiude una o più sostanze chimicamente definite che possono costituire l’insieme dei reagenti, dei prodotti, del solvente e dell’atmosfera vicina, tutto ciò in definitiva, che può essere confinato in uno spazio. Il resto della materia che non si identifica con il sistema si chiama ambiente. L'unione di ambiente e sistema viene chiamato universo, ed è ad esso che fa riferimento l'enunciato del primo principio della termodinamica. Un sistema che non ha alcuna comunicazione con l'esterno, che non scambia con l'ambiente né energia, né materia, né informazione, viene detto "sistema chiuso". Un sistema chiuso è dunque Principi di bioenergetica 77 del tutto tagliato fuori dal suo ambiente, non ha né ingressi né uscite. Di converso "aperto" è un sistema che è in relazione con il resto dell'universo. Viene modificato dal suo ambiente e lo modifica. Mentre un sistema isolato può scambiare energia, ma non materia, con l’ambiente. Per quanto riguarda gli scambi energetici si distinguono fondamentalmente due casi: quando l'interazione è attribuibile ad una forza, di varia natura, che sposta nel tempo il suo punto d'applicazione, la quantità di energia meccanica scambiata prende il nome di lavoro termodinamico, mentre si chiama calore la quantità di energia termica trasferita in seguito allo sviluppo di una differenza di temperatura tra sistema ed ambiente. Le cellule e gli organismi viventi sono tipici sistemi aperti, cioè scambiano energia (ad esempio tramite il metabolismo) e materia (nutrienti e materiale di scarto)con il loro ambiente esterno. I sistemi viventi non sono mai in equilibrio con l’ambiente che li circonda e i continui scambi fra sistema e ambiente spiegano come gli organismi possano creare ordine al loro interno pur essendo sfavoriti dal punto di vista energetico. Anche gli aspetti più complicati della termodinamica in fondo si basano su tre principi semplici e lineari che sembrano difficili da capire, ma che una volta ben compresi ed assimilati diventano potenti strumenti per risolvere complicati problemi sia chimici che biochimici. I° Principio della termodinamica Il I° Principio della termodinamica può essere definito il principio della conservazione dell’energia è un assunto fondamentale da cui si diparte gran parte della teoria della termodinamica. Esso non si occupa di considerazioni qualitative, ma affronta i problemi di scambio energetico in un'ottica di bilancio e il postulato di conservazione infatti afferma che l'energia non può essere creata o distrutta, ma può solo essere convertita in un'altra forma. Il primo principio è uno dei concetti fisici che meglio sono entrati nell'immaginario popolare con espressioni come "non ho più energia", "pieno di energie" indicano che questa è una delle idee fisiche meglio comprese dall'uomo della strada. Infatti, in qualsiasi processo o modificazione chimica e fisica la quantità totale di energia del sistema isolato (ovvero dell’Universo) è costante. Durante il processo l’energia può passare da una forma ad 78 Capitolo V stante. Durante il processo l’energia può passare da una forma ad un’altra o essere trasferita da una zona ad un’altra, ma non può venire né creata né distrutta. Le forme dell’energia sono: la luce, il calore, l’energia elettrica, chimica e meccanica, l’energia elastica ecc. Gli studiosi di termodinamica hanno formulato una funzione matematica per indicare i trasferimenti di energia e la dissipazione di energia per compiere un lavoro chiamandola energia interna, comunemente espressa con la lettera E o U. L’energia interna dipende solo dallo stato presente di un sistema ed è quindi una funzione di stato, ed è indipendente dal percorso seguito dalla reazione. Un esempio: se si brucia uno zucchero semplice, ad esempio una pastiglia di glucosio, come si sa si ottiene CO2 ed acqua, e si ha contemporaneamente liberazione di energia sotto forma di calore. Anche il glucosio demolito nelle cellule finisce col formare CO2 ed acqua, ma per un’altra via, che comprende molte trasformazioni e molti composti intermedi. Tuttavia, molecola iniziale e prodotti finali sono gli stessi e la stessa è anche l’energia che viene liberata in questo secondo processo.L’energia interna di un sistema chiuso può cambiare solo se l’energia entra o esce dal sistema sotto forma di calore o di lavoro. Per un processo che converte uno stato (stato 1) in un altro (stato 2) la variazione di energia interna sarà: ∆U = U2 - U1 = Q – L (1) in cui U1 è l’energia del sistema all’inizio del processo, U2 è l’energia alla fine del processo, Q il calore scambiato (positivo se assorbito e negativo se ceduto) e L il lavoro compiuto (positivo se compiuto dal sistema sull'ambiente, negativo se subito). La scelta dei segni dell'espressione precedente deriva da considerazioni di carattere pratico, infatti la termodinamica si è inizialmente sviluppata attorno alle macchine termiche. Queste rappresentavano il sistema, ad esse veniva fornito calore, tramite una combustione, ed in cambio dovevano fornire lavoro. In un sistema isolato, che non può scambiare né lavoro, né calore con l'ambiente, la ∆U è inevitabilmente zero. Pertanto, un'implicazione del Principi di bioenergetica 79 primo principio è la seguente: "L'energia interna di un sistema isolato è costante". Si tenga presente che l'Universo è il più grande dei sistemi isolati. L'equivalenza di calore e lavoro, oggi data per scontata, fu a lungo dibattuta all'alba della scienza. Si può dimostrare tuttavia molto banalmente sfregando le mani e sentendone il riscaldamento (lavoro/calore), o osservando il coperchio di una pentola di acqua bollente che viene sollevato dalla pressione del vapore (calore/lavoro). Il primo principio della termodinamica stabilisce quindi: • • • il principio di conservazione dell’energia; l’esistenza di una funzione di stato detta energia interna; l’equivalenza tra calore e lavoro. Il sistema non fa distinzione fra le due forme di energia, lavoro e calore. Una stessa variazione dell'energia interna si può ottenere trasferendo o solo lavoro o solo calore. Si può dare infatti una definizione meccanica della quantità di calore in termini di lavoro necessario per produrre la stessa variazione dell'energia interna, il cosiddetto equivalente meccanico della caloria (1 caloria = 4.184 Joule) In conclusione, il sistema, nelle varie trasformazioni, oltre al calore può scambiare con l'ambiente anche lavoro meccanico. Comunemente si intende per lavoro meccanico il lavoro compiuto da masse solide in movimento. In questo caso entrano in gioco solo due forme di energia: a) quella potenziale, derivante dalla posizione in un campo gravitazionale, e b) quella cinetica, derivante dal moto vero e proprio della massa. In senso termodinamico ad esempio, se una persona prova ad alzare qualcosa di pesante ma non ce la fa e l’oggetto non si sposta, non viene compiuto alcun lavoro. L'energia di tipo potenziale costringe ad esempio le masse a muoversi lungo una discesa, più o meno rapidamente a seconda della forza d'attrito e della pendenza. L'energia di tipo cinetico consente alle masse in moto di proseguire il movimento anche quando cessa la forza agente, se l'attrito lo consente. Poiché il lavoro è una forma di energia, si usa anche dire energia meccanica anziché lavoro meccanico: in questo modo si distingue Capitolo V 80 questa forma di energia dalle altre, come la termica (calore), l'elettrica, l'idraulica, ecc. In un sistema chimico, se si escludono le celle galvaniche, l'unico modo di produrre lavoro è quello dovuto a variazioni di volume, cioè del tipo P·∆V, dalla relazione precedente (1) sostituendo L otteniamo: ∆U = Q - P·∆V (2) Se la reazione avviene senza variazione di volume, ∆V = 0 allora ∆U rappresenta solo il calore scambiato Q. Quindi ∆U = Q, e questa è una grandezza molto utile per i processi che avvengono a volume costante. Tuttavia i processi biologici avvengono per la maggior parte a pressione costante, quella atmosferica, dove il ∆U non è necessariamente uguale al calore trasferito Q. Per questa ragione i biochimici hanno definito un’altra grandezza, utile soprattutto per i processi a pressione costante, l’entalpia H, definita come: H = U + PV (3) Quindi: ∆H = ∆U + P∆V = Q + L + P ∆V = Q - P ∆V + P ∆V = Q (4) Il ∆H è il calore trasferito nei processi a P costante. Spesso, poiché le reazioni biologiche di solito avvengono in sistemi liquidi o solidi le variazioni di volume sono piccole e quindi trascurabili perché con un ∆V che tende a 0. In biologia infatti la prima parte dell’equazione (4) può essere semplificata nel seguente modo ∆H = ∆U (5) dove entalpia H ed energia interna U sono in pratica equivalenti. L'aver introdotto la funzione di stato entalpia, porta alla conclusione che: "In un sistema chiuso, la variazione di entalpia è pari alla quantità di calore scambiato, poste due restrizioni: l'unica forma di lavoro possibile è il lavoro di espansione e P = costante." Principi di bioenergetica 81 In base alla direzione del flusso di calore (il segno di ∆H), le reazioni chimiche si suddividono in: • • Esotermiche se il sistema libera calore (∆H < 0); Endotermiche se il sistema assorbe calore (∆H > 0). Il I° Principio ci dice soltanto che l’energia totale di un sistema e del suo ambiente è costante, ma non ci dà informazioni sulla possibilità che una reazione possa avvenire spontaneamente o che sia impossibile da realizzare. L'esperienza ci insegna che i processi naturali evolvono spontaneamente (nel tempo) in una determinata direzione. Un gas si espande spontaneamente fino ad occupare tutto il volume a disposizione. Un oggetto caldo si raffredda spontaneamente fino a raggiungere l'equilibrio termico con l'ambiente circostante. Le trasformazioni di questo tipo non possono mai essere invertite completamente e per questo motivo, i processi naturali sono definiti termodinamicamente irreversibili. Dietro al concetto di irreversibilità vi è di fatto "l'impossibilità di riportare un sistema, che ha subìto una trasformazione spontanea, dallo stato finale a quello iniziale, senza provocare mutamenti in qualche parte dell'universo." Questo punto è di fondamentale importanza e fa la differenza fra trasformazione reversibile e irreversibile. A questo punto occorre dare risposta a due quesiti importanti: "Cos'è che determina la direzione di un processo spontaneo?" e inoltre "In che modo si può prevedere se un processo è spontaneo?" La risposta va cercata in una "nuova" funzione di stato, l'entropia (S) trattata all’interno del II principio della termodinamica. Per chiarire meglio il concetto di entropia (di non troppo facile comprensione) facciamo prima un esempio pratico. Noi sappiamo che il vapore prodotto dall’acqua in ebollizione può produrre lavoro (ad es. pensiamo alle prime locomotive). Supponiamo ora di avere un bollitore pieno d’acqua riscaldato a 100°C (il sistema) in una cucina (l’ambiente circostante). Spegniamo il fuoco sotto il bollitore e lasciamolo raffreddare. Durante il raffreddamento non può essere generato un lavoro e il calore passa dal bollitore all’ambiente (la cucina), aumentandone la temperatura finché non viene raggiunto l’equilibrio. 82 Capitolo V A questo punto tutte le parti del bricco e della cucina sono alla stessa temperatura. L’energia libera G (che, come si vedrà più avanti, è la quantità di energia effettivamente disponibile per compiere un lavoro), che una volta era concentrata nel bricco di acqua a 100°C e potenzialmente poteva compiere lavoro è invece stata dispersa nell’ambiente. Una quantità equivalente di energia (termica) è ancora presente nel “bricco + la cucina” (sistema + ambiente) ma ha una distribuzione assolutamente casuale. Questa energia non è più disponibile per compiere un lavoro in quanto non c’è più differenza termica fra il sistema e l’ambiente. Dalla nostra esperienza sappiamo che il calore non tornerà più dalla cucina al bricco per scaldarlo a 100°C. Il disordine e la casualità sono i modi più indicati per rappresentare l’entropia. Per comprendere quindi come avvengono le trasformazioni biologiche bisogna ricorrere all’entropia ed è un modo di esprimere, come vedremo, il disordine del sistema. Uno stato organizzato o ordinato è a bassa entropia, mentre uno stato disordinato è ad alta entropia. A parità di condizioni, le reazioni che portano a grandi e positivi cambiamenti di “disordine” o entropia (∆S) hanno maggior probabilità di avvenire che non le reazioni con variazioni di entropia piccole o addirittura negative ossia che portano ad un maggior ordine. II° Principio della termodinamica Il II° Principio della termodinamica dice in sostanza che l’universo tende ad essere sempre più disordinato e quindi che, in tutti i processi naturali, l’entropia tende ad aumentare. Questo principio può essere espresso in modi diversi: 1) I sistemi tendono a procedere da uno stato ordinato (bassa entropia o bassa probabilità) ad uno stato di disordine (alta entropia o alta probabilità); 2) L’entropia del sistema più ambiente non viene alterata dai processi reversibili, mentre l’entropia del sistema più ambiente aumenta per i processi irreversibili; 3) Tutti i processi che avvengono in natura procedono verso l’equilibrio, che è lo stato a minima energia potenziale. Principi di bioenergetica 83 In pratica, un processo può avvenire spontaneamente solo se la somma delle variazioni di entropia del sistema e dell’ambiente aumentano ∆S sistema + ∆S ambiente > 0. Tuttavia, la cellula è ben lontana dalla situazione ideale in cui tra il sistema e l’universo si scambia solo energia. In realtà come detto sopra le cellule e gli organismi viventi sono dei sistemi aperti, cioè scambiano energia e materia con l’esterno e lo scambio è un flusso continuo perché essi non sono mai in equilibrio con l’ambiente che li circonda. La misura dell’entropia quindi è un problema complesso sia dal punto di vista tecnico che concettuale. Per superare questo scoglio, il chimico Gibbs introdusse una funzione, detta energia libera di Gibbs (G) ricavata dalla prima e dalla seconda legge della termodinamica. Questa funzione viene calcolata come variazione (∆G) ed è molto utile perché tiene conto di vari parametri all’interno del sistema ed esclude l’ambiente. Questi parametri sono: la variazione di energia interna (∆E), di entropia (∆S) e di volume (∆V). Quindi l’energia libera G è la quantità di energia effettivamente spendibile dal sistema, durante una reazione a pressione e temperatura costanti, in grado di produrre un lavoro, cioè che può essere fornita a sistemi vicini perché a loro volta possano utilizzarla per fare avvenire nuove reazioni. ∆G sistema = ∆H sistema – T ∆S sistema (6) Ma, come abbiamo riportato sopra (5), poiché nei sistemi biologici la variazione di volume è praticamente uguale a 0 (∆V=0) e quindi ∆H = ∆U l’equazione (6) può essere scritta come: ∆G sistema = ∆U sistema – T ∆S sistema dove T è la temperatura assoluta espressa in gradi Kelvin (°K). L’energia libera descrive in modo conveniente l’incremento di disordine di un sistema. La differenza di energia libera fra lo stato iniziale e lo stato finale di una reazione ne determina la spontaneità o la non spontaneità. Infatti, quando in una reazione si ha rilascio di energia 84 Capitolo V libera, significa che il sistema si modifica verso uno stato ad energia minore, cioè verso uno stato più stabile. La variazione dell'energia libera (energia dei prodotti - energia dei reagenti) sarà negativa: avremo quindi ∆G<0. La reazione si dice esoergonica, cioè non richiede energia e avviene spontaneamente. Se viceversa il sistema si evolve verso uno stato ad energia maggiore la variazione dell’energia libera sarà positiva. Avremo ∆G >0 e la reazione verrà detta endoergonica o non spontanea, che richiederà energia per avvenire. Se il valore di ∆G = 0 il sistema è in equilibrio termodinamico e non avviene niente. Il valore di ∆G di un sistema che reagisce spontaneamente è sempre negativo. Riprendiamo in esame la combustione del glucosio. La reazione è: C6 H12 O6 + 6 O2 = 6 CO2 + 6 H2O + ENERGIA Con un ∆G°’ = - 686 cal/mole. Si deduce quindi che: 1) la reazione è spontanea perché ∆G è negativo; 2) la reazione avviene: quindi bruciando il glucosio si originano CO2 ed acqua e si libera energia; 3) la reazione avviene indipendentemente dalla via seguita per arrivare dai reagenti ai prodotti, perché nell’equazione non si indica il percorso; 4) il ∆G non ci dice nulla sulla velocità con cui avviene la reazione; 5) il valore di ∆G é espresso come ∆G°’ (energia libera standard per i sistemi biologici) dove l’apice specifica che il valore di ∆G è quello standard, cioè riportato in condizioni di riferimento; Riguardo a quest’ultimo punto occorre precisare che, per paragonare i parametri termodinamici di reazioni diverse bisogna definire lo stato standard. Per soluti in soluzione, lo stato standard è di solito una concentrazione 1M. I parametri termodinamici dati finora se espressi allo stato standard, verranno scritti col segno “ ° “ (∆H°, ∆U° ecc.). Per i sistemi biologici i parametri che identificano lo stato standard sono la concentrazione dei reagenti 1M, la temperatura a 37°C e il Principi di bioenergetica 85 pH=7. L’entalpia, come definito sopra, è il contenuto termico del sistema che sta reagendo e riflette il numero e il tipo di legami chimici dei reagenti e dei prodotti. Quando nelle reazioni chimiche si ha rottura di alcuni legami e se ne formano altri, la differenza fra l’energia fornita dall’ambiente per rompere i legami e quella guadagnata dall’ambiente durante la formazione dei legami viene detta variazione di entalpia (∆H) della reazione. Quando durante una reazione chimica viene rilasciato calore all’ambiente la reazione viene detta esotermica, e la differenza di entalpia fra prodotti e reagenti ha per definizione valore negativo (∆H<0). Se al contrario durante una reazione chimica viene sottratto calore all’ambiente la reazione viene detta endotermica, e la differenza di entalpia ha valore positivo (∆H>0). ∆G e ∆H sono espressi in joule/mole o calorie/mole ( 1 caloria = 4,18 joule) mentre ∆S si esprime in joule/mole gradi Kelvin (K). III° Principio della termodinamica Il III° Principio della termodinamica stabilisce che l’entropia di una sostanza cristallina, perfettamente ordinata tende a zero quando la temperatura assoluta, espressa in gradi Kelvin (°K) si avvicina allo zero assoluto (-273,15 °C ) e che a T=0 l’entropia è esattamente zero. Basandosi su questo principio è possibile stabilire una scala quantitativa assoluta delle entropie per ogni sostanza. Per i processi biologici tuttavia le variazioni di entropia sono molto più utili della conoscenza dei suoi valori assoluti. Le variazioni di entropia possono essere calcolate se sono note le variazioni di entalpia e di energia libera La variazione di energia libera standard è correlata direttamente con la costante di equilibrio. Consideriamo la seguente reazione chimica spontanea (spontanea come tutti i processi naturali): aA + bB = cC + dD 86 Capitolo V dove a,b,c,d rappresentano il numero di molecole dei composti A,B,C,D che partecipano alla reazione. La composizione del sistema che sta reagendo tende a cambiare continuamente finché non si raggiunge l’equilibrio. All’equilibrio, la velocità della reazione che procede da destra a sinistra eguaglia la velocità di quella che procede da sinistra a destra e non vi sono ulteriori cambiamenti del sistema. Le concentrazioni di reagenti e prodotti all’equilibrio definiscono la costante di equilibrio Keq Keq = [C]c . [D]d / [A]a . [B]b dove [A], [B], [C] e [D], sono le concentrazioni molari dei componenti della reazione all’equilibrio. Quando un sistema non è all’equilibrio, la tendenza a spostarsi verso l’equilibrio diventa una forza trainante espressa dalla variazione di energia libera ∆G della reazione. In condizioni standard (298°K, cioè 25°C) quando reagenti e prodotti hanno una concentrazione iniziale 1M la variazione di energia libera si dice ∆G°. Da questa definizione si deduce che lo stato standard quando sono presenti ioni H+ è [H+] = 1M cioè pH=0. La maggior parte delle reazioni biochimiche avviene in ambiente acquoso tamponato a pH circa 7, quindi il pH e la concentrazione dell’acqua 55,5M possono definirsi costanti. I biochimici per facilitare i calcoli hanno definito lo stato standard considerando [H+] = 10-7 M cioè pH=7 e la concentrazione dell’acqua 55,5 M. Quindi quando acqua e [H+] sono reagenti o prodotti le loro concentrazioni sono incorporate nelle costanti ∆G°’ e Keq’. Esiste una relazione semplice fra ∆G°’ e Keq’ ∆G°’= -RT .ln Keq’ dove R è la costante dei gas (8,315 J/mole.°K, T è la temperatura assoluta 298°K (25°C). La variazione di energia libera standard di una reazione chimica è semplicemente un modo matematico alternativo di esprimere la sua costante di equilibrio. La costante di equilibrio è direttamente proporzionale alla variazione complessiva di energia libera Principi di bioenergetica 87 standard. Un valore di ∆G°’<<0 riflette un equilibrio favorevole, ma non ci dice nulla sulla velocità a cui procede la reazione. Se Keq’ =1 , se Keq’ è >1 , se Keq’ <1, ∆G°’= 0, ∆G°’<0, ∆G°’> 0 Poiché la relazione fra ∆G°’e -RT .ln Keq’ è esponenziale, a variazioni relativamente piccole di ∆G°’ corrispondono grandi variazioni di Keq’. Le reazioni accoppiate Come spiegato sopra, le reazioni che si svolgono con diminuzione di energia libera del sistema sono energeticamente favorite. Al ∆G°’negativo si accompagna la generazione di disordine e quindi la spontaneità. Viceversa, le reazioni che si svolgono con aumento di energia libera sono energeticamente sfavorite e quindi non spontanee. Queste ultime reazioni, per avvenire, possono superare la situazione sfavorevole accoppiando la loro realizzazione ad altre reazioni con ∆G°’> 0. Solo se la somma algebrica dei due processi è <0 si può ipotizzare che la prima reazione (non spontanea) possa realizzarsi. ∆G°’totale = ∆G°’1+ ∆G°’2 Una reazione termodinamicamente sfavorita (endoergonica) può dunque essere guidata mediante il suo accoppiamento con una reazione altamente esoergonica, se è presente un intermedio comune. Per esempio, la sintesi del glucosio-6-fosfato (G-6-P) è la prima tappa nella via di utilizzazione del glucosio in molti organismi. Glucosio + Pi = G-6-P + H2O ∆G°’ = + 13,8 Kcal/mole. Capitolo V 88 In condizioni standard la reazione non procederà spontaneamente verso destra. Ma se viene accoppiata con questa reazione altamente endoergonica: ATP + H2O = ADP + Pi ∆G°’ = -30,5 Kcal/mole Le due reazioni hanno in comune gli intermedi Pi ed H2O e possono diventare sequenziali: ATP + glucosio = ADP + G-6-P ∆G°’ = - 16,7 Kcal/mole La reazione complessiva è esoergonica, quindi la prima reazione potrà avvenire, anche se energeticamente sfavorita. I sistemi viventi richiedono energia libera dall’ambiente per: 1) lavoro meccanico dei muscoli e motilità cellulare; 2) trasferimento dell’informazione genetica; 3) trasporto attivo attraverso le membrane cellulari; 4) sintesi di molecole specifiche del singolo organismo. L’energia libera proveniente dall’ambiente è data dall’energia solare per i fototrofi, mentre per i chemiotrofi dall’ossidazione dei cibi. Riprendiamo ora le due reazioni viste sopra, una esoergonica e l’altra endoergonica appartenenti al medesimo sistema chimico. L’energia prodotta dalla reazione esoergonica in teoria potrebbe passare direttamente a quella endoergonica se vi sono intermedi comuni e se questa necessita della stessa quantità di energia. Questo accoppiamento però è poco probabile perché parte dell’energia liberata dalla reazione esoergonica viene dispersa sotto forma di calore che, in un sistema isotermo, non può compiere un lavoro. In pratica deve essere prodotto un surplus di energia libera da disperdere nell’ambiente. Un sistema che non abbia una utilizzazione diretta dell’energia liberata da una reazione deve ricorrere ad un serbatoio di raccolta di energia. Quindi, la dispersione energetica si avrebbe sempre, ma ci sarebbe il vantaggio di rinviarne l’utilizzo in tempi e modi differenziati, scegliendo quelli più adatti. L’ energia disponibile può essere incorporata Principi di bioenergetica 89 in un composto intermedio che si frappone fra le reazioni esoergoniche e le endoergoniche e funziona da serbatoio di raccolta dell’energia liberata dalle prime e che agisce da distributore per le seconde. L’intermedio spesso detto “carrier” o trasportatore deve prendere parte ad entrambe le reazioni e rimanere inalterato alla fine di esse. Un composto usato per immagazzinare l’energia deve poter essere prodotto in molte reazioni esoergoniche diverse ed essere usato in molti tipi di reazioni endoergoniche. Un composto simile può essere paragonato all’accumulatore di un’automobile che si ricarica man mano che l’auto si muove ma che contemporaneamente distribuisce energia a tutto l’impianto elettrico. Gli organismi viventi possiedono numerose molecole in grado di funzionare come accumulatori di energia e tutte contengono almeno un gruppo fosfato facilmente idrolizzabile, ma la molecola che è più abbondante in ogni forma di vita è il nucleotide adenosin trifosfato, l’ATP. I legami chimici contengono sempre energia. L’energia di legame è l’energia necessaria per formare un legame chimico fra due atomi di una molecola o l’energia che si libera quando questo legame si scinde. In ogni molecola certi legami sono più facili da rompere di altri e questo dipende sia dalle tensioni interne fra i vari gruppi sia dalla stabilità dei composti che derivano dalla rottura della molecola stessa. L’ATP è una molecola abbastanza stabile la cui idrolisi è mediata dall’enzima ATP-asi che abbassa la soglia dell’energia di attivazione, che per l’ATP è abbastanza elevata (Figura 1). Senza l’ATP-asi l’idrolisi dell’ATP è abbastanza difficile. Fra i molti legami dell’ATP solo i due legami fosfoanidridici che tengono uniti i tre gruppi fosfato sono facili da rompere. Figura 1. Struttura dell'ATP. 90 Capitolo V Nell’ATP c’è repulsione fra gli ossigeni dei gruppi fosforici che a pH=7 sono dissociati: tuttavia senza l’ATP-asi come enzima, che avvicina ioni Mg+2 e Mn+2, che sono normali metaboliti cellulari, questa repulsione non sarebbe sufficiente a rompere l’ATP. Infatti uno ione bivalente positivo obbliga due ossigeni carichi negativamente ad avvicinarsi a questi ioni: questo genera una forte tensione nella molecola. Quando lo ione viene allontanato bruscamente induce un’altra violenta repulsione che rompe il legame anidridico.Quando il gruppo fosforico si allontana dall’ATP nell’ambiente acquoso, esso tenderà a stabilizzarsi per risonanza e ad idratarsi liberando una certa quantità di energia (diventa cioè più stabile). La maggiore stabilità di ADP e Pi fanno sì che l’idrolisi dell’ATP sia un processo esoergonico, cioè spontaneo e quindi che si liberi energia. Questa energia non è diversa da quella liberata da altre reazioni di idrolisi, ma la sua importanza sta nel fatto che l’idrolisi dell’ATP avviene in concomitanza con reazioni endoergoniche che possono sfruttare l’energia liberata dal gruppo fosfato per compiere un lavoro, ad esempio per formare un nuovo legame chimico. L’energia è contenuta in prevalenza nei legami fra i gruppi fosfato dell’ATP e quindi potremo indicare i legami ad alta energia col simbolo (segno di “~”) e abbreviare la formula in adenosina (adenina + ribosio) e tre P, dove P sta per PO3 2-. Adenosina - P ~ P ~ P Togliendo un solo gruppo si ha l’adenosin difosfato, l’ADP, che ha un solo gruppo fosfato ad alta energia. Rimuovendo ancora un altro gruppo si ha l’adenosin monofosfato, l’AMP, che non ha nessun gruppo fosfato ad alta energia, poiché il gruppo fosfato è legato al ribosio con un legame estere. In ognuna di queste idrolisi si libera un gruppo fosfato indicato come Pi, fosfato inorganico. Nelle reazioni metaboliche in cui occorre molta energia si può passare direttamente da ATP ad AMP e pirofosfato inorganico, PPi. Questa ultima molecola può idrolizzarsi ancora dando 2 Pi, liberando altra energia e dando luogo ad una reazione fortemente esoergonica. Principi di bioenergetica 91 IL ∆G° di una mole di ATP che si idrolizza libera -7,3 Kcal/mole: in realtà questo è un valore di riferimento per condizioni standard che si discosta molto dal valore nelle condizioni reali. Anche se per praticità di calcolo si fa riferimento a questo valore, il ∆G vero, alle concentrazioni fisiologiche dell’ATP (circa 10-3M) è di circa -13 Kcal/mole, quasi il doppio del valore teorico. L’ATP è un donatore di energia molto rapido, in genere non fa in tempo a formarsi che già si consuma. Una persona mediamente produce e consuma circa 40 Kg (80 moli) di ATP in un solo giorno. Il processo di conversione dell’ATP in ADP normalmente si svolge in due tappe: 1) Il Pi forma un legame covalente col substrato aumentando l’energia libera del sistema; 2) il legame appena formato, instabile, si idrolizza liberando Pi (o AMP). L’ATP quindi partecipa direttamente alla reazione alla quale deve fornire energia. Il ciclo ATP-ADP è il processo fondamentale di scambio dell’energia nei sistemi biologici. Trasportatori di elettroni e ioni idrogeno Come si è visto in precedenza l’uso di un intermedio è la via più seguita per raccordare le reazioni esoergoniche con quelle endoergoniche. Le reazioni di ossido-riduzione implicano la perdita di elettroni da parte di una specie chimica, che diventa quindi ossidata e l’acquisizione di elettroni da parte di un’altra specie chimica che si riduce. Il flusso di elettroni nelle reazioni di ossido-riduzione è responsabile, direttamente o indirettamente, di tutto il lavoro prodotto dagli organismi viventi. Negli organismi non fotosintetici la fonte di elettroni è data dalle sostanze nutrienti ridotte, negli organismi fotosintetici il donatore di elettroni è una specie chimica eccitata dall’assorbimento di luce. Le cellule contengono una grande varietà di molecole che si comportano Capitolo V 92 da trasduttori energetici e che possono convertire l’energia generata dal flusso degli elettroni in lavoro utile. Ogni volta che usiamo un motore, una luce, una stufa elettrica sfruttiamo il flusso di elettroni per compiere un lavoro. La fonte di elettroni può essere una batteria, che contiene due specie chimiche con diversa affinità per gli elettroni. Il flusso di elettroni procederà spontaneamente nel circuito, guidato da una forza proporzionale alla differenza di affinità, detta forza elettromotrice (f.e.m). La f.e.m può generare lavoro se al circuito viene collegato un opportuno trasduttore energetico. In un circuito biologico, la fonte di elettroni è ad es. il glucosio, un composto relativamente ridotto. Quando questo composto viene ossidato enzimaticamente vengono rilasciati elettroni che fluiscono spontaneamente attraverso una serie di trasportatori fino a raggiungere un’altra specie chimica con elevata affinità per gli elettroni come l’ossigeno. Il flusso elettronico è spontaneo ed esoergonico perché l’O2 ha un’affinità per gli elettroni maggiore di tutti gli intermedi che lo precedono nella via metabolica. La forza motrice che si genera fornisce energia a molti trasduttori molecolari (enzimi e proteine) che compiono un lavoro biologico. Ad es. nel mitocondrio i trasduttori molecolari accoppiano il flusso elettronico alla generazione di una differenza di pH tra le due facce della membrana, che poi determina un lavoro osmotico ed elettrico. Le ossidazioni biologiche comportano spesso deidrogenazione. Gli elettroni possono essere trasferiti da una molecola ad un’altra in quattro modi diversi: 1) 2) 3) 4) direttamente come elettroni (Fe+2/ Fe+3 ); sotto forma di atomi di idrogeno (un protone ed un elettrone); sotto forma di ioni idruro (un protone e due elettroni); per combinazione diretta di un riducente organico con l’ossigeno. Tutte queste forme di trasferimento di elettroni possono avvenire nella cellula. Il termine equivalente riducente viene usato per indicare un singolo elettrone che partecipa ad una qualsiasi reazione di ossidoriduzione. La maggior parte delle cellule possiede enzimi che cataliz- Principi di bioenergetica 93 zano l’ossidazione di centinaia di composti diversi. Questi enzimi, detti deidrogenasi, trasferiscono elettroni dai loro substrati su pochi trasportatori universali di elettroni. La loro riduzione nei processi catabolici consente di conservare l’energia libera rilasciata durante l’ossidazione dei substrati. Per queste reazioni di ossido-riduzione è necessario l’intervento di adatti coenzimi (o gruppi prostetici) che esistono in due forme, una ossidata ed una ridotta e che possono passare dall’una all’altra forma per apporto e perdita di elettroni. I più importanti coenzimi delle reazioni di ossido-riduzione sono due nucleotidi, NAD+ e FAD, rispettivamente Nicotinammide Adenin Dinucleotide (forma ossidata NAD+ e ridotta NADH + H+) e il Flavin Adenin Dinucleotide (forma ossidata FAD e ridotta FADH2), per la cui descrizione si rimanda al capitolo sui coenzimi. Questi coenzimi sono cofattori solubili in acqua che possono andare incontro a ossidazioni e riduzioni reversibili in molte reazioni di trasferimento di elettroni del metabolismo. NAD+ (e nella sua forma fosforilata NADP+) si spostano rapidamente da un enzima all’altro mentre i nucleotidi flavinici FMN e FAD sono legati saldamente agli enzimi, chiamati flavoproteine, ed agiscono da gruppi prostetici. Il NAD agisce come ossidante soprattutto sul gruppo funzionale alcolico –OH dando origine a gruppi carbonilici –C=O, ma in certe situazioni metaboliche può agire anche sul gruppo aldeidico trasformandolo in gruppo carbossilico – COOH. Il NADH che si forma è un intermedio riducente che è destinato a cedere l’H, attraverso passaggi successivi all’O2 per formare H2O e liberare energia. Gli enzimi che utilizzano il NAD+ come coenzima sono chiamati deidrogenasi-NAD-dipendenti. Un agente ossidante, per essere un efficace trasportatore di elettroni deve essere in grado di accettare facilmente elettroni e H+. In questo la molecola si riduce ma si deve poi poter riossidare per tornare ad essere l’originario agente ossidante. Per questo motivo NAD e FAD devono poter cedere facilmente gli elettroni appena acquisiti. Nelle biosintesi riduttive, come quelle che si hanno quando si sintetizza una nuova molecola biologica (acidi grassi, amminoacidi) anziché il NADH viene usata una molecola simile, il NADPH (che è un NADH con un gruppo fosfato legato all’ossidrile 2’ del ribosio dell’adenosina. Questa soluzione molecolare permette di operare una distinzione fra i processi ossidativi e quelli riduttivi. Ben pochi enzimi 94 Capitolo V possono utilizzare entrambi i coenzimi NAD+ e NADP+ e quasi sempre presentano una marcata preferenza per uno di essi. Nel FAD la struttura è quella della riboflavina, una delle vitamine del gruppo B che contiene il gruppo reattivo della flavina. Come per il NAD+ viene accettata una coppia di idrogeni dal substrato ridotto per dare la molecola ridotta FADH2 e quest’ultimo riversa gli H prelevati da un substrato a vari accettori. Un coenzima strettamente correlato al FAD è l’FMN, flavinmononucleotide, che svolge funzioni simili a quelle del FAD ma limita la sua azione al trasporto finale di idrogeni nella catena respiratoria. Sia il FAD che l’FMN hanno specificità di substrato, legandosi ad enzimi che si chiamano deidrogenasi FAD o FMN dipendenti. I CARBOIDRATI I carboidrati costituiscono la classe di composti organici più abbondante e diffusa in natura e rappresentano una importantissima fonte energetica per l’uomo, per gli animali e per le piante, anche se altri composti, come i lipidi, forniscono più del doppio di calorie per unità di peso. I carboidrati costituiscono una delle quattro classi principali di biomolecole, insieme alle proteine, agli acidi nucleici ed ai lipidi. Sono sostanze ternarie, costituite cioè da carbonio, idrogeno ed ossigeno ed hanno formula generale (CH2O)n, con C e H2O in rapporto 1:1, da cui l’antico nome idrati di carbonio e più modernamente quello di carboidrati. I carboidrati (o più correttamente glucidi, ma possono anche essere definiti saccaridi o zuccheri) costituiscono la maggior parte della materia organica della biosfera e svolgono un gran numero di funzioni, soprattutto energetiche e strutturali, in tutte le forme di vita. I carboidrati semplici servono innanzitutto come sostanze nutrienti ed intermedi metabolici. Le molecole più complesse, come l’amido nelle piante ed il glicogeno negli animali costituiscono una riserva facilmente mobilizzabile per generare glucosio, la principale sostanza nutriente per la produzione di energia, mentre la cellulosa, la chitina e i glicosamminoglucidi sono fra i costituenti fondamentali dei tessuti di sostegno sia per i vegetali che per gli animali nonché dell’esoscheletro degli artropodi. Fra i carboidrati semplici, il ribosio fa parte di molti coenzimi, e l’ATP (adenosina trifosfato), il trasportatore universale di energia libera, è un derivato di uno zucchero fosforilato. I pentosi ribosio e desossiribosio costituiscono parte della molecola dell’RNA e del DNA. La flessibilità della conformazione di questi zuccheri è importante per la conservazione e per l’espressione dell’informazione genetica. I carboidrati possono trovarsi legati a molte proteine e lipidi e da studi recenti sembra che le unità di carboidrati sulle superfici cellulari possano svolgere un ruolo chiave nei processi di riconoscimento cellulare. 95 96 Capitolo VI In base alla complessità della struttura i carboidrati si possono distinguere in tre gruppi principali: • Monosaccaridi o monosi, detti anche zuccheri semplici • Oligosaccaridi, costituiti da poche molecole di zuccheri semplici legati uno all’altro con legami covalenti • Polisaccaridi o poliosi, formati da lunghe catene di zuccheri semplici, in certi casi di enormi dimensioni, di forma lineare o ramificata. Gli oligo e i polisaccaridi possono essere costituiti da un solo monosaccaride (omopolisaccaridi) o da monosaccaridi diversi (eteropolisaccaridi). Possono essere formati da soli carboidrati (carboidrati semplici) o essere legati ad altre molecole (carboidrati complessi); possono infine contenere elementi chimici addizionali, come fosforo, zolfo e azoto. Monosaccaridi Sono le più semplici molecole di carboidrati e possono esistere in natura liberi o legati a formare molecole di dimensioni maggiori. Sono poliidrossialdeidi e poliidrossichetoni, in quanto sono costituiti da una catena, in genere ciclica, di atomi di carbonio legati ad atomi di idrogeno o ad OH alcolici, con l’eccezione di un atomo di carbonio che porta una funzione aldeidica o chetonica denominata “gruppo anomerico”. Sono diversi come dimensioni e configurazione stereochimica, poiché possono contenere uno o più gruppi chirali. Hanno formula generale (CH2O)n, con n mai minore di 3 e raramente maggiore di 7, e non possono essere demoliti per idrolisi. Si differenziano per il numero di atomi di carbonio contenuti nella molecola e per il gruppo carbonilico che può essere aldeidico, in posizione 1, o chetonico, in posizione 2. Il gruppo anomerico può essere ossidato, da cui il nome di zuccheri riducenti o zuccheri liberi. I monosaccaridi vengono classificati in base al loro gruppo carbonilico e al numero dei loro atomi di carbonio. Nel primo caso se il gruppo carbonilico è un’aldeide, lo zucchero viene detto aldosio, se è un chetone viene detto chetosio. Nel secondo i monosaccaridi più piccoli (con tre atomi di carbonio) vengono detti triosi. Quelli con tre, quattro, cinque, sei e I carboidrati 97 sette atomi di carbonio sono rispettivamente tetrosi, pentosi, esosi e eptosi. Questi termini di classificazione possono essere combinati per definire ad esempio il glucosio come un aldoesosio e il fruttosio come chetoesosio (Figura 1). Figura 1. Esempio di classificazione di due monosaccaridi I monosaccaridi più semplici sono la gliceraldeide ed il diidrossiacetone, due isomeri a tre atomi di carbonio, di cui il primo porta un gruppo aldeidico ed il secondo un gruppo chetonico (Figura 2). Sono importanti perché servono come riferimento per assegnare i simboli della configurazione a tutti i carboidrati. Figura 2. Struttura dei due monosaccaridi più semplici Infatti l’atomo di C in posizione 2 della gliceraldeide è legato a quattro gruppi diversi (-H, -OH, -CHO e –CH2OH). Un atomo di C in 98 Capitolo VI questa situazione viene detto asimmetrico o carbonio chirale. La molecola a cui appartiene presenta un piano di simmetria interno e, seconda della disposizione dei gruppi –H ed –OH, si può presentare in due forme diverse e speculari, una con il gruppo –OH a destra (mettendo sempre il gruppo più ossidato in alto) e una con il gruppo –OH a sinistra. Nel primo caso si avrà la forma D e nel secondo caso la forma L (Figura 3). Figura 3. Proiezioni di Fischer per gli enantiomeri della gliceraldeide Occorre tener presente che si tratta di pure convenzioni, ormai generalmente accettate. La parola chirale deriva dalla parola greca cheir che significa mano, perché i due isomeri sono opposti uno all’altro come le due mani destra e sinistra. Si formano due isomeri ottici, detti enantiomeri, sempre dal greco enantios che vuol dire “contrario” perché le due molecole hanno configurazione opposta, sono immagini speculari uno dell’altro e non sono sovrapponibili. Gli enantiomeri sono un caso particolare della stereoisomeria, cioè dell’isomeria che comporta una diversa disposizione dei sostituenti nello spazio tridimensionale. Inoltre le loro soluzioni possono ruotare in modo opposto il piano della luce polarizzata (luce che vibra soltanto in un piano) che le attraversa. Il diidrossiacetone, non avendo alcun carbonio chirale, si presenta in una sola forma. La maggior parte dei monosaccaridi naturali ha configurazione D ad eccezione dell’arabinosio, che è in configurazione L. Occorre tuttavia ricordare che non c’è relazione fra forma D o L (derivante come si è detto da una convenzione) e senso di rotazione I carboidrati 99 del piano della luce polarizzata, che è invece una proprietà fisica strettamente legata alla configurazione reale della molecola. E’ un puro caso che la D-gliceraldeide ruoti effettivamente il piano della luce polarizzata verso destra. Per i carboidrati a numero maggiore di 3 atomi di carbonio il suffisso da aggiungere è -oso o -osio per gli zuccheri aldeidici (Figura 4) e -uloso o -ulosio per quelli chetonici (Figura 5). Quindi un aldosio con 4 atomi di C verrà detto aldotetrosio (es. eritrosio); un chetosio con 5 atomi di C è un chetopentosio (ad es. il ribulosio), il glucosio è un aldoesosio, il fruttosio un chetoesosio e così via. Figura 4. Relazioni stereochimiche tra i D-aldosi 100 Capitolo VI I triosi sono soltanto i due ricordati sopra: la gliceraldeide ed il diidrossiacetone. Queste molecole sono importanti, oltre che per l’assegnazione della configurazione, anche perché sono i capostipiti degli zuccheri che si formano da essi per aggiunta di atomi di carbonio, idrogeno ed ossidrili. Questi triosi sono anche la forma intermedia con cui i vari carboidrati, anche i più complessi, vengono degradati nelle cellule e da cui i vari organismi ricavano energia. Figura 5. Relazioni stereochimiche tra i D-chetosi I tetrosi importanti sono eritrosio e treosio fra gli aldosi e eritrulosio fra i chetosi. Tra i pentosi gli aldopentosi esistono in otto possibili strutture avendo tre carboni chirali. Il pentosio più importante è senz’altro il ri- I carboidrati 101 bosio, contenuto in ogni cellula e che fa parte di strutture complesse come RNA e, in forma priva di un ossigeno sul C2, del DNA. Arabinosio e xilosio si trovano in certe resine del legno, specialmente nel ciliegio e nel pesco, ma non hanno funzione fisiologica conosciuta. Tra i chetopentosi il ribulosio si forma durante un particolare metabolismo dei carboidrati, la via del pentoso fosfato. Gli esosi sono i più comuni tra i monosaccaridi, tra essi i più importanti sono il glucosio, il galattosio, il mannosio (aldosi) e il fruttosio (chetosio). Configurazioni e conformazioni Per rappresentare le strutture dei monosaccaridi in genere si usano, o meglio si usavano) le proiezioni di Fisher, cioè la struttura a catena aperta, che però contrasta con alcune proprietà chimico-fisiche che essi mostrano in determinate condizioni. Oggi si è certi che esosi e pentosi, quando sono in soluzione, ciclizzano facilmente per formare strutture ad anello (Figura 6) che per gli zuccheri a sei atomi e cinque atomi di carbonio prendono il nome rispettivamente di piranosio e furanosio, ricordando nella struttura il pirano e il furano. Figura 6. Forme cicliche generiche a sei e cinque atomi Generalmente per indicare le forme cicliche dei monosaccaridi si usano le forme di proiezione o formule di Haworth, che rappresentano la molecola come un anello penta- o esa- atomico, in cui il bordo più vicino a chi guarda è rappresentato ingrossato e gli atomi di carbonio sono numerati a partire da quello immediatamente successivo all’atomo di ossigeno (Figura 7). 102 Capitolo VI Queste formule non sono del tutto esatte (gli angoli disegnati non rispettano la forma tetraedrica con cui i vari gruppi sono legati al carbonio, e le molecole non sono planari come appaiono) ma sono entrate nell’uso comune a causa della loro semplicità ed efficacia. Figura 7. Proiezione di Haworth del D-glucosio In generale un’aldeide può reagire con un alcool a formare un emiacetale. Nel caso di un aldoesosio come il glucosio il gruppo aldeidico in C1 reagisce con il gruppo ossidrilico sul carbonio C5 per formare un emiacetale intramolecolare (Figura 8). L’emiacetale ciclico forma un anello a sei termini (piranosio) che è una molecola eterociclica a sei atomi di cui cinque di carbonio e uno di ossigeno. Figura 8. La reazione di ciclizzazione del D-glucosio I carboidrati 103 Analogamente un gruppo chetonico, ad es. il gruppo C2 del fruttosio, può reagire con il gruppo ossidrilico in C5 per formare un emichetale intramolecolare. L’emichetale ciclico forma un anello a cinque atomi (furanosio), molecola eterociclica a cinque atomi, di cui quattro di carbonio e uno di ossigeno. La ciclizzazione di un monosaccaride crea un nuovo centro di asimmetria situato in corrispondenza del C1 o del C2 nel caso dei chetosi. Ad esempio in un piranosio si possono formare due strutture ad anello in cui il gruppo ossidrilico legato al C1 è sotto il piano dell’anello (α) oppure sopra (β) (Figura 9). Il C1 viene chiamato atomo di carbonio anomerico, e quindi le forme α e β sono dette anomeri. La stessa nomenclatura viene applicata alla forma ad anello furanosico eccetto che le forme α e β si riferiscono alla disposizione del gruppo ossidrilico sul C2, che in questo caso è il carbonio anomerico. Figura 9. Gli anomeri di glucopiranosio secondo le proiezioni di Haworth L’uso delle formule di Haworth può generare l’impressione errata che gli anelli piranosici o furanosico siano planari. Queste molecole hanno in realtà una geometria tridimensionale più complessa. Ad esempio l’anello piranosico può assumere la forma di sedia o di barca. La stabilità relativa di queste conformazioni diverse dipende dalle interazioni stereochimiche tra i sostituenti sull’anello. I sostituenti possono essere di due tipi: assiali (a) o equatoriali (e). I legami assiali sono approssimativamente perpendicolari al piano dell’anello, mentre quelli equatoriali sono sfalsati e pressoché paralleli a questo piano. 104 Capitolo VI Nella conformazione a barca i sostituenti assiali si trovano stericamente ammassati e questa risulta meno stabile della conformazione a sedia (Figura 10). Figura 10. Le conformazioni dell'anello di un generico piranosio Reazioni dei monosaccaridi Le reazioni fondamentali dei carboidrati sono legate alla reattività del gruppo aldeidico o chetonico perché la forma ciclica in soluzione è in continuo equilibrio con la struttura aperta, anche se la forma ciclica è preponderante. In un monosaccaride i gruppi ossidabili sono numerosi: infatti oltre al gruppo aldeidico e chetonico si possono ossidare anche i gruppi alcolici primari e secondari. Le reazioni di ossidazione degli aldosi, sia chimiche che enzimatiche, portano alla conversione del gruppo aldeidico in C1 in gruppo carbossilico e all’ottenimento del corrispondente acido aldonico (ad es. dal glucosio l’acido gluconico, dal mannosio l’acido mannonico). I chetosi per ossidazione si spezzano formando acidi con un numero minore di atomi di carbonio. Se a venir ossidata è la funzione alcolica primaria in C6 si parla di acido uronico (ad es. acido glucuronico, mannuronico), queste sono sostanze molto diffuse nelle piante dove costituiscono la base delle pectine. Se vengono ossidati ambedue i carboni terminali (C1 e C6) il composto risultante prende il nome di acido aldarico (ad es. ac. glucarico, mannarico) (Figura 11). I carboidrati 105 Figura 11. Reazioni di ossidazione del D-glucosio I monosaccaridi legandosi l’uno all’altro formano legami acetalici, in genere fra la posizione 1 del primo carbonio se si tratta di un aldosio o con la posizione 2 se si tratta di un chetosio con l’-OH in posizione 4 o 6 del monosaccaride seguente. I composti ottenuti si dicono glicosidi. Se la reazione avviene con il gruppo –OH ( o –NH2 o –SH) di una molecola non glucidica i composti ottenuti vengono definiti eterosidi. Oligosaccaridi Le proprietà sono molto simili a quelle dei monosi: sono solidi bianchi, cristallini, di sapore dolce e facilmente solubili in acqua. Fra essi il saccarosio è il disaccaride più conosciuto come zucchero da tavola. La loro formula generale è C12 H22 O11 e sono costituiti da due monosaccaridi uniti, per condensazione (eliminazione di una molecola d'acqua), da un legame covalente O-glicosidico che si forma tra un gruppo ossidrilico di un monosaccaride e l’OH legato al carbonio anomerico di un altro zucchero. 106 Capitolo VI La rottura del legame O-glicosidico, il processo inverso che avviene nella fase della digestione, è una reazione di idrolisi ossia rottura del legame per aggiunta d'acqua. In pratica le reazioni di condensazione e di idrolisi sono reazioni opposte. I disaccaridi si formano quando due monosaccaridi si legano fra loro con legame acetalico (glucosidico) fra l’ossidrile emiacetalico del primo (scritto in forma chiusa o ciclica) e uno qualsiasi degli ossidrili dell’altro. Il legame acetalico è molto più forte di quello emiacetalico e non si può rompere con i comuni reagenti ossidanti usati per riconoscere i monosaccaridi. I disaccaridi principali sono quattro: maltosio, lattosio, saccarosio e cellobiosio. Il maltosio si trova nell’amido di mais, nel malto e nei semi. E’ formato da due unità di α-glucosio legati con un legame 1,4-αglucosidico (Figura 12). Deriva dall’idrolisi enzimatica dell’amido e può essere idrolizzato a sua volta in due molecole di α-glucosio dall’enzima maltasi. Figura 12. Formula di struttura del maltosio Il saccarosio si estrae dalle barbabietole e dalla canna da zucchero, è il disaccaride più abbondante in natura e si trova nella maggior parte dei frutti e dei vegetali. E’ formato da una unità di α-glucosio legata con un legame glucosidico ad una molecola di β-fruttosio. Il legame è fra la funzione aldeidica in C1 del glucosio e la funzione chetonica in C2 del fruttosio. Poiché entrambi i gruppi anomerici I carboidrati 107 sono impegnati nel legame il saccarosio non ha un’aldeide terminale libera e quindi non è uno zucchero riducente, al contrario della maggior parte degli zuccheri (Figura 13). Figura 13. Formula di struttura del saccarosio Quando il saccarosio viene digerito, si idrolizza ad opera della saccarasi in glucosio e fruttosio. Si ha in questo processo una grande modificazione della rotazione ottica da destrogira a levogira, per cui la saccarasi è chiamata anche invertasi. Il lattosio è formato da una unità di β-glucosio e una unità di βgalattosio legati con un legame 1,4-β-glucosidico (Figura 14). Figura 14. Formula di struttura del lattosio 108 Capitolo VI E’ lo zucchero principale del latte dove la sua concentrazione varia dallo 0 al 7% a seconda della specie, e rispetto al saccarosio è meno solubile in acqua ed è meno dolce. Viene idrolizzato a galattosio e glucosio liberi dalla lattasi nell’uomo e dalla β-galattosidasi nei batteri. La saccarasi, la lattasi e la maltasi sono localizzate sulla superficie esterna delle cellule epiteliali che si trovano nell’intestino tenue. Il cellobiosio si trova raramente libero in natura. E’ molto diffuso nelle piante, in quanto è l’unità strutturale della cellulosa. E’ formato da due unità di β-glucosio legate con un legame β-1,4-glucosidico, che conferisce al cellobiosio proprietà molto diverse da quelle del maltosio (Figura 15). Figura 15. Formula di struttura del cellobiosio In natura sono presenti in quantità significative solo pochissimi trisaccaridi o oligosaccaridi superiori e sono tutti di origine vegetale. Tra questi possiamo citare alcuni zuccheri quali il raffinosio, lo stachiosio ed il verbascosio che consistono di 1, 2 o 3 molecole di alfa-(1-6)-galattosio legate ad una unità terminale saccarosio. Sono contenuti soprattutto nei legumi, nella soia e nei cereali. Questi oligosaccaridi nell’uomo non sono digeribili dagli enzimi presenti nel tratto gastro-intestinale e vengono quindi completamente fermentati dalla flora batterica del colon. Il potere dolcificante è molto più basso di quello del saccarosio o degli zuccheri monomerici. I carboidrati 109 Polisaccaridi Sono i carboidrati più abbondanti in natura: costituiscono dal 50 al 90% del peso secco delle piante. Un polisaccaride è costituito da molti monosaccaridi legati insieme per formare molecole complesse lineari o ramificate. Il peso molecolare varia da decine di migliaia ad oltre un milione. I polisaccaridi in genere sono quasi insolubili in acqua e non hanno sapore dolce. I polisaccaridi naturali possono essere polimeri degli esosi o dei pentosi e prendono quindi il nome di esosani o pentosani I polisaccaridi possono servire come riserva di carboidrati, come struttura di sostegno delle piante o possono costituire polisaccaridi complessi, particolarmente importanti dal punto di vista biochimico (ad es. come costituenti delle membrane cellulari). Polisaccaridi di riserva: amido e glicogeno L’amido è il carboidrato di riserva delle piante che viene depositato nel citoplasma delle cellule vegetali attraverso la polimerizzazione del glucosio prodotto a partire dall’anidride carbonica atmosferica e dall’acqua durante la fotosintesi. L’amido viene immagazzinato come fonte di energia di riserva sotto forma di granuli che differiscono per morfologia e dimensioni a seconda della specie botanica (Figura 16). Figura 16. Fotografia la microscopio elettronico di un granulo di amido 110 Capitolo VI L’amido chimicamente risulta costituito da due polimeri distinti: l’amilosio, non ramificato, e l’amilopectina. L’amido è un polimero molto grande con molte ramificazioni con un peso molecolare di più di 1000 unità. Il contenuto di queste due componenti è di solito del 1530% per l'amilosio e del 70-85% per l'amilopectina, ma può variare nelle diverse piante con conseguente modificazione delle caratteristiche dell'amido. La complessità strutturale dell’amido impedisce una vera e propria solubilità in acqua, ma ad alta temperatura si riesce ad ottenere per rigonfiamento una soluzione colloidale viscosa detta salda d'amido. L’amilosio è costituito da molecole di α-glucosio legate insieme da soli legami 1,4- α-glucosidici che portano all’assunzione di una conformazione a spirale elicoidale (Figura 17). Figura 17. α-amilosio: struttura primaria (a) e modello strutturale (b) L’amilopectina, è costituita da residui di glucosio uniti da legami α-1,4, con punti di ramificazione generati da legami α-1,6 ogni 24-30 residui di media (Figura 18). Le ramificazioni sono dovute alla reazione fra il gruppo aldeidico in C1 di un glucosio e l’ossidrile in C6 di un altro. Sia l’amilopectina che l’amilosio sono facilmente idrolizzati dall’α-amilasi che viene secreta dalle ghiandole salivari e dal pancreas. Per idrolisi l’amilosio dà luogo al disaccaride maltosio e, per idrolisi spinta, ad α-D-glucosio, mentre il meccanismo di rottura dei legami dell’amilopectina è diverso. I carboidrati 111 Figura 18. Amilopectina: struttura primaria (a) e rappresentazione grafica (b) Gli alimenti che contengono grandi quantità di amido sono le patate, i fagioli, il pane e la pasta, che sono costituiti principalmente di amilopectina. Le piante hanno scelto questa struttura perché meno facilmente solubile nell’acqua cellulare. Il glicogeno è il polisaccaride di riserva tipico degli animali, è presente in tutte le cellule, ma è molto più presente nel muscolo scheletrico e nel fegato in forma di granuli nel citosol della cellula. Nel regno vegetale è diffuso in molti funghi superiori e nei tartufi, inoltre è presente anche in alcuni lieviti. E’ costituito da unità di D-glucosio ed è molto simile all’amilopectina sia come struttura che come funzioni. Tuttavia, la molecola di glicogeno è molto più ramificata dell’amilopectina dell’amido avendo punti di ramificazione ogni 8-12 residui di glucosio (Figura 19). Il glicogeno ha un peso molecolare che varia da 1.000.000 (glicogeno muscolare) a 5.000.000 dalton (glicogeno del fegato). Figura 19. Confronto fra le strutture dell'amilopectina (a) e del glicogeno (b) 112 Capitolo VI La formazione del glicogeno nella cellula si dice glicogenesi: quando il livello di glucosio nel sangue è basso il glicogeno di riserva viene demolito per rendere disponibile una maggior quantità di glucosio, il processo viene indicato con il termine glicogenolisi. Polisaccaridi strutturali La cellulosa è il principale componente strutturale della parete cellulare delle piante e rappresenta circa la metà del carbonio presente nella biosfera (Figura 20). Figura 20. Fotografia al microscopio elettronico di fibre di cellulosa E’ un polimero non ramificato costituito da residui di glucosio uniti da legami 1,4- β-glucosidici e, come accade per i grandi polisaccaridi, non ha una grandezza definita (300-15000 unità). La configurazione β permette alla cellulosa di formare catene molto lunghe e dritte con le fibrille formate da catene disposte su piani paralleli. Ogni residuo di glucosio è ruotato di 180 gradi rispetto a quello adiacente e l’atomo di ossigeno dell’anello forma un legame a idrogeno con il gruppo ossidrilico sull’atomo di carbonio C3 del residuo successivo (Figura 21a). I legami α-1,4 del glicogeno e dell’amilosio producono un’architettura molecolare molto diversa. Infatti invece di una catena I carboidrati 113 dritta si forma un’elica. Queste differenze sono determinate dai legami glicosidici α o β che conferiscono caratteristiche biologiche importanti. La catena dritta formata dai legami β permette di costruire fibre ad elevata resistenza stabilizzate ulteriormente dalla formazione di legami a idrogeno sia all’interno della fibra che tra fibre diverse (Figura 21a). Al contrario le eliche aperte formate dai legami α sono facilmente accessibili agli enzimi che idrolizzano il polimero nelle singole unità di glucosio da utilizzare come riserva di energia (Figura 21b). Figura 21. Confronto tra la struttura della cellulosa (a) e dell'amilosio (b) La parete cellulare delle piante è costituita per il 50% da cellulosa, ma nel cotone la cellulosa è presente quasi per il 100%. In particolari condizioni la cellulosa dà origine per idrolisi al disaccaride cellobiosio, che può essere ulteriormente degradato a β-Dglucosio. La cellulosa è importante dal punto di vista commerciale perché è la base di molte fibre artificiali, come il rayon viscosa e di altri composti come la celluloide ed il cellophan. I gruppi OH possono reagire con l’acido nitrico per dare il nitrato di cellulosa, noto come cotone fulminante o fulmicotone. La chitina è il componente principale dell’esoscheletro degli artropodi (crostacei, insetti, ragni) e della parete cellulare della maggior parte dei funghi, ed è il secondo polimero più abbondante sulla terra dopo la cellulosa. E’ un omopolimero di unità di N-acetil-D- 114 Capitolo VI glucosammina legata con legami 1,4 β-glucosidici e differisce dalla cellulosa per avere il gruppo –OH di ogni C2 sostituito da una gruppo amminico acetilato (Figura 22). Figura 22. Struttura primaria della chitina La chitina forma lunghe catene lineari unite tra di loro da legami a idrogeno in modo simile a quello della cellulosa. I glicosamminoglicani sono costituenti fondamentali del materiale extracellulare, cioè del materiale che riempie gli spazi tra le cellule in particolare del tessuto connettivo. In esso sono immersi le cellule, i vasi sanguigni, la cartilagine, i tendini e le terminazioni nervose e attraverso di esso diffondono i segnali chimici endogeni, l’ossigeno, l’anidride carbonica e le altre sostanze di rifiuto dirette verso o provenienti dalle cellule. Questa matrice, simile a un gel, è composta da glicosamminoglicani (o mucopolisaccaridi). I glicosamminoglicani sono polisaccaridi non ramificati costituiti da catene di disaccaridi ripetuti nei quali uno dei monosaccaridi è un’esosammina e l’altro (o entrambi) contiene almeno una carica negativa come gruppo solfato o carbossilato. L’agar, isolato da alghe rosse marine (Rodoficee) è composto da agarosio e agaropectina la cui struttura tridimensionale è una doppia elica al cui interno si possono legare molte molecole di acqua che conferiscono la consistenza di un gel. E’ una sostanza usata in laboratorio come substrato inerte nei terreni di coltura per i batteri. I carboidrati 115 La pectina si trova nella frutta ed è responsabile del mantenimento di una certa rigidità nelle sue pareti cellulari. E’ un polisaccaride composto da una catena lineare di molecole di acido galatturonico. La pectina, quando si disperde in una soluzione acquosa zuccherina forma una gelatina che ingloba grandi quantità di acqua per questo viene aggiunta a molti prodotti commerciali (budini, marmellate) per far assumere loro una struttura più consistente. IL PROCESSO RESPIRATORIO Nel mondo vivente i due processi biochimici fondamentali sono la fotosintesi e la respirazione, senza di essi infatti la vita sulla terra non sarebbe possibile. Mentre nella fotosintesi l’energia solare viene catturata e trattenuta sotto forma di legame chimico, con formazione di carboidrati, che forniranno gli scheletri carboniosi per le altre biomolecole, da CO2 ed acqua, nella respirazione si ha liberazione di tale energia per combustione dei nutrienti, con emissione di CO2 e di acqua. Il primo è un processo anabolico, che quindi richiede energia e il secondo invece è un processo catabolico, che libera energia. In modo estremamente semplificato, il rapporto tra respirazione e fotosintesi, dal punto di vista dei materiali iniziali e finali dei processi può essere rappresentato con uno schema dove nella fotosintesi, CO2 e H2O danno luogo alla formazione di carboidrati e di ossigeno molecolare e nella respirazione, carboidrati e ossigeno riformano CO2 e H2O. Per capire quanto siano invece complessi, bisogna affrontare, nella parte della biochimica del ciclo del carbonio, i loro diversi stadi. Il termine “combustione” non deve ingannare perché, anche se la respirazione può considerarsi un caso particolare della combustione, tuttavia si tratta di due cose diverse: la combustione infatti procede in modo incontrollato, mentre la respirazione può essere suddivisa in vari processi. Inoltre nella combustione il risultato è solo la formazione di CO2 ed acqua, mentre nelle respirazione, cosa importantissima, oltre a CO2 ed acqua vengono prodotti anche intermedi per le altre vie biosintetiche. 117 118 Capitolo VII La respirazione dei carboidrati procede in tre tappe: 1) Glicolisi: trasformazione del glucosio in piruvato (e altri intermedi) che può subire altre trasformazioni nel ciclo seguente. 2) Ciclo dell’acido citrico o ciclo di Krebs, o degli acidi tricarbossilici, in cui si ha la completa demolizione della molecola di piruvato derivato dalla glicolisi, con liberazione di CO2 formazione di ATP e potere riducente (NADH, FADH2). 3) Catena respiratoria di trasporto degli elettroni, in cui tutto il potere riducente viene riportato allo stato ossidato ad opera dell’ossigeno molecolare (O2). Si forma ATP e alla fine l’ossigeno viene ridotto ad H2O. La glicolisi avviene nel citosol della cellula, mentre le altre due fasi della respirazione sono localizzate nei mitocondri (Figura 1). Gli enzimi che partecipano al processo sono distribuiti in compartimenti in modo tale che le tre tappe possano procedere in sequenza. Figura 1. Localizzazione dei processi respiratori In questo capitolo si tratterà della prima parte del processo respiratorio degli organismi aerobi, la glicolisi, che peraltro come vedremo può avvenire anche in assenza di ossigeno. Nella glicolisi si ha la distruzione parziale dei carboidrati, introdotti con l’alimentazione per la maggior parte degli organismi o sintetizzati per fotosintesi sia dalle piante verdi che da alcune alghe e batteri fotosintetizzanti. Glicolisi 119 Durante questa via metabolica si ha la rottura dei legami che tengono insieme gli atomi delle varie molecole e liberazione dell’energia insita in essi. Per poter essere decomposti i carboidrati devono essere idrolizzati se si tratta di poli- o oligosaccaridi, o isomerizzati se sono zuccheri semplici a 6 atomi di carbonio, fino alla formazione del Dglucosio, che è la sostanza nutriente principale per la maggior parte degli organismi ed occupa una posizione centrale nel metabolismo. LA GLICOLISI Il termine glicolisi deriva da due parole greche e precisamente da glykys (dolce, o zuccherino) e lysis (scissione). E’ il processo in cui una molecola di uno zucchero semplice, il D-glucosio o tutto ciò che al D-glucosio è riconducibile, viene scissa in due molecole di una sostanza a tre atomi di carbonio, l’acido piruvico o piruvato (in genere è più corretto usare il termine piruvato, in quanto al pH delle cellule, generalmente gli acidi carbossilici sono in forma dissociata). Si parla di D-glucosio perché in natura i carboidrati semplici sono quasi sempre in forma D, al contrario degli amminoacidi proteici, che si trovano quasi esclusivamente in forma L. Il D-glucosio da scindere può derivare: 1. Da reazioni di idrolisi, ad esempio dall’idrolisi di polisaccaridi come il glicogeno presente negli organismi animali o dall’ amido, e in casi particolari anche dalla cellulosa, che sono caratteristici delle piante. Può derivare anche dall’idrolisi di disaccaridi come il saccarosio, il lattosio, il maltosio ed il cellobiosio. I disaccaridi (e a maggior ragione i polisaccaridi) infatti non possono entrare direttamente nella via glicolitica ma devono essere sempre convertiti in monosaccaridi da processi extracellulari; 2. Da reazioni di isomerizzazione, da fruttosio, mannosio, galattosio. Il fruttosio, presente in forma libera in molti tipi di frutta o proveniente dall’idrolisi del saccarosio può anche essere fosforilato direttamente dall’ATP con l’enzima esochinasi ed essere scisso poi dalla fruttosio-1-fosfato aldolasi. Il galattosio viene dapprima fosforilato a livello del C1 a spese dell’ATP (enzima galattochinasi). Il galattosio-1-fosfato viene poi trasformato in glucosio-1-fosfato, 120 Capitolo VII il suo epimero al C4, mediante una serie di reazioni in cui il trasportatore degli esosi è l’uridina trifosfato (UTP); 3. Da altre vie metaboliche, come la fotosintesi e la gluconeogenesi. La glicolisi è stata la prima via metabolica ad essere identificata per intero ed è a tutt’oggi forse la meglio conosciuta. La glicolisi è il primo passo per la completa distruzione delle molecole di glucosio, si tratta quindi di un processo catabolico, con produzione di energia immediatamente disponibile sotto forma di ATP o di potere riducente (NADH) che potrà in seguito essere convertito in energia, come vedremo più avanti. Nella glicolisi, come in tutte le vie metaboliche, si formeranno anche molti composti intermedi che potranno essere utilizzati o per portare al termine la reazione glicolitica stessa o per la sintesi di numerosi composti in altre vie metaboliche. La glicolisi è la via centrale del catabolismo del glucosio ed è presente praticamente in tutte le cellule. In alcuni tessuti o tipi di cellule di mammiferi (eritrociti, cervello, spermatozoi) la via glicolitica è la sola o la principale fonte di energia metabolica. Poiché la glicolisi può avvenire in condizioni sia a aerobiche che anaerobiche, alcune piante che sono specializzate nella produzione di amido o adattate a crescere in aree sommerse e molti tipi di microrganismi anaerobici possono ottenere da questa via metabolica la maggior parte dell’ energia loro necessaria. La glicolisi converte il glucosio in piruvato e genera due molecole di ATP. Da questo punto in poi se il metabolismo può proseguire in condizioni aerobie il piruvato viene ossidato a CO2 e acqua. Invece in condizioni anaerobiche nel muscolo si svolge la fermentazione omolattica, mentre nei lieviti la fermentazione alcolica (Figura 2). Glicolisi 121 Figura 2. Metabolismo del glucosio La via metabolica La demolizione del glucosio, molecola a sei atomi di carbonio, fino a due molecole a tre atomi di carbonio, l’acido piruvico, avviene attraverso dieci reazioni. Prima di passare alle formule e alla completa descrizione della via metabolica potrà essere utile un quadro riassuntivo delle varie reazioni, che verranno approfondite successivamente. Infatti, nello studio della biochimica è fondamentale formarsi dapprima un quadro sintetico della via metabolica da studiare: una volta memorizzati i composti di iniziali e finali e compreso bene lo scopo della reazione si possono cominciare ad esaminare i vari passaggi, con le for- 122 Capitolo VII mule delle varie molecole, le loro trasformazioni, i prodotti intermedi, le implicazioni energetiche, gli enzimi catalizzatori e tutto quanto è necessario a portare la reazione a buon fine. Va detto, e verrà ripetuto anche in seguito, le reazioni metaboliche sono quasi sempre unidirezionali, cioè non si possono percorrere all’inverso, se non in particolari condizioni. Possiamo separare il processo glicolitico in due fasi, una preparatoria costituita dalle prime cinque reazioni in cui il glucosio viene fosforilato e scisso in due molecole di gliceraldeide-3-fosfato con l’utilizzazione di due molecole di ATP. In una seconda fase le due molecole di gliceraldeide-3-fosfato vengono convertite in due molecole di piruvato con la concomitante produzione di quattro molecole di ATP. La reazione complessiva è: glucosio + 2NAD+ + 2ADP + 2Pi → 2 piruvato + 2ATP + 2NADH + 2H+ + 2H2O Nella prima tappa della prima fase il glucosio viene fosforilato dall’ATP sull’ossidrile legato al carbonio in posizione 6 ottenendo una molecola di glucosio 6-fosfato. Il glucosio 6-fosfato viene isomerizzato a fruttosio 6-fosfato: questo viene a sua volta fosforilato dall’ATP sul C1 generando fruttosio 1, 6- bisfosfato. Il fruttosio 1,6- bisfosfato viene successivamente scisso in due molecole a tre atomi di C, il diidrossiacetonefosfato e la gliceraldeide 3fosfato. Questa è la tappa “litica” o di scissione da cui la via prende il nome. Il diidrossiacetonefosfato (che si forma in quantità preponderante, circa il 96% contro il 4% della gliceraldeide) si isomerizza in una seconda molecola di gliceraldeide 3-fosfato. Questa reazione è necessaria perché è la gliceraldeide che prosegue la reazione e quindi via via che questa viene utilizzata, l’equilibrio si sposta verso destra, consumando il diidrossiacetonefosfato e formando nuova gliceraldeide 3-fosfato. Si completa così la prima fase della glicolisi. Fino a questo punto sono state usate due molecole di ATP e quindi per il momento vi è stato solo un investimento energetico e quindi solo consumo e nessuna produzione di nuova energia. Glicolisi 123 In sostanza, nella prima fase della glicolisi viene spesa energia dell’ATP per aumentare l’energia libera degli intermedi della via metabolica. Le catene carboniose di tutti gli esosi metabolizzati vengono convertite in un prodotto comune, la gliceraldeide 3-fosfato. Nella seconda fase, ogni molecola di gliceraldeide 3-fosfato viene ossidata dal NAD+ e fosforilata da un fosfato inorganico (Pi), formando 1,3 bisfosfoglicerato, molecola ad alta energia di idrolisi (come l’ATP). L’energia verrà rilasciata quando due molecole di 1,3 bisfosfoglicerato verranno trasformate in due molecole di piruvato attraverso la formazione di vari intermedi. La maggior parte di questa energia non verrà dispersa nell’ambiente metabolico, dove potrebbe generare danni irreversibili, ma verrà conservata accoppiandola alla sintesi di quattro molecole di ATP da ADP e fosfato inorganico. La resa netta è quindi di 2 ATP (4 formati e 2 utilizzati) per ogni molecola di glucosio. Un altro modo di conservare l’energia nella fase di recupero energetico si ha mediante la formazione di potere riducente sotto forma di NADH. Il destino del piruvato prodotto, che dipende dal tipo di cellula e dalle necessità metaboliche, verrà esaminato in seguito. 124 Capitolo VII La via metabolica completa della glicolisi è riportata in Figura 3 Figura 3. Le tappe della via glicolitica Glicolisi 125 Le tappe della glicolisi I tipi di reazioni che intervengono durante la demolizione glicolitica sono: 1) trasferimento di gruppi fosforici dall’ATP ad un intermedio glicolitico o viceversa; 2) spostamento di gruppi fosforici all’interno di una molecola da un atomo di ossigeno ad un altro; 3) isomerizzazione: un chetosio viene convertito in aldosio e viceversa; 4) deidratazione: viene eliminata una molecola d’acqua; 5) scissione aldolica: un legame C-C viene scisso con un processo inverso alla condensazione aldolica. Esaminiamo ora in dettaglio i dieci passaggi della glicolisi: 1) Fosforilazione del glucosio Nella prima tappa, il glucosio viene attivato dall’enzima esochinasi mediante fosforilazione sul C6, dando origine a glucosio-6-fosfato (Figura 4). Il donatore del gruppo fosforico è l’ATP. La reazione, irreversibile, è catalizzata dall’enzima esochinasi. Il nome chinasi raggruppa gli enzimi che catalizzano il trasferimento del gruppo fosforico terminale dell’ATP ad un accettore, in questo caso un esosio (il Dglucosio); le chinasi sono una sottoclasse delle transferasi (vedi anche il capitolo sugli Enzimi). L’esochinasi, come tutte le altre chinasi, necessita di ioni Mg2+ come cofattori per l’espressione dell’attività catalitica. Figura 4. Fosforilazione del glucosio a glucosio-6-fosfato 126 Capitolo VII 2) Conversione del glucosio-6-fosfato a fruttosio-6-fosfato L’isomerizzazione reversibile del glucosio-6-fosfato, un aldosio, in fruttosio-6-fosfato, un chetosio, è catalizzata dalla fosfo-glucosioisomerasi (Figura 5). L’anello piranosico del glucosio-6-fosfato viene convertito nell’anello furanosico del fruttosio-6-fosfato, mediante una prima fase di apertura dell’anello, la vera e propria reazione di isomerizzazione, seguita quindi dalla fase di chiusura dell’anello nella nuova configurazione. Questa reazione può procedere in entrambe le direzioni, infatti il ∆G° è basso (1,7 KJ/mole), quindi la transizione non necessita di molta energia. Anche questo enzima richiede ioni Mg2+ ed è specifico per i composti suddetti. Figura 5. Isomerizzazione del glucosio-6-fosfato a fruttosio-6-fosfato 3) Fosforilazione del fruttosio-6-fosfato a fruttosio-1,6-bisfosfato Nella terza reazione, la fosfofruttochinasi-1 catalizza il trasferimento di un gruppo fosforico dall’ATP al fruttosio-6-fosfato con formazione di fruttosio-1,6-bisfosfato (Figura 6). Il termine bisfosfato significa che i due guppi fosforici sono separati, mentre il termine difosfato (come nell’adenosina difosfato, ADP) significa che i due gruppi fosforici sono legati tra loro. Figura 6. Fosforilazione del fruttosio-6-fosfato a fruttosio 1,6-bisfosfato Glicolisi 127 4) Scissione del fruttosio-1,6-bisfosfato L’enzima aldolasi, catalizza una condensazione aldolica reversibile, mediante la quale il fruttosio-1,6-bisfosfato viene scisso in due molecole di triosofosfati, il diidrossiacetone fosfato (DHAP) e la gliceraldeide 3-fosfato (GAP) (Figura 7). Questi composti vengono rapidamente utilizzati nelle reazioni successive e quindi il fruttosio si decompone rapidamente nei due triosi. Le tappe della glicolisi da qui in avanti coinvolgono unità a tre atomi di carbonio invece che a sei atomi. Figura 7. Scissione del fruttosio 1,6-bisfosfato 5) Interconversione dei triosofosfati Poiché soltanto uno dei triosifosfati formati (e precisamente la gliceraldeide 3-fosfato) verrà utilizzata direttamente nelle reazioni successive, il DHAP viene convertito rapidamente nella GAP. Questi composti sono isomeri: il diidrossiacetone è un chetoso, mentre la gliceraldeide 3-fosfato è un aldoso. L’isomerizzazione di questi zuccheri fosforilati è catalizzata dall’enzima triosofosfato isomerasi (Figura 8). Quindi da una molecola di fruttosio 1,6-bisfosfato si formano due molecole di GAP e si completa la fase preparatoria della glicolisi. La fase di recupero, in cui l’energia libera della molecola di glucosio verrà conservata sotto forma di molecole di ATP, inizierà dalla reazione successiva. 128 Capitolo VII Figura 8. Isomerizzazione del DHAP in GAP 6) Ossidazione della gliceraldeide 3-fosfato a 1,3 bifosfoglicerato Le tappe precedenti della glicolisi hanno trasformato una molecola di glucosio in due molecole di gliceraldeide 3-fosfato consumando due molecole di ATP. In questa fase la GAP viene ossidata a 1,3bifosfoglicerato (1,3-BPG) dall’enzima gliceraldeide 3-fosfato deidrogenasi in presenza di NAD+ e di fosfato inorganico (Pi) (Figura 9). E’ la prima delle due reazioni in cui si avrà formazione di ATP da ADP e Pi. La gliceraldeide viene ossidata sul gruppo aldeidico in C1 formando un acido che verrà poi fosforilato. Non si formerà quindi un gruppo carbossilico libero ma un legame anidridico fra due acidi (il gruppo COOH e l’acido fosforico). Si tratta di una anidride mista, chiamata acil-fosfato, che ha una energia libera di idrolisi molto elevata di circa 49 KJ/mole. Una gran parte di questa energia libera viene conservata nel legame. Figura 9. Ossidazione e fosforilazione della GAP a 1,3-BPG Glicolisi 129 Il coenzima NAD+ è l’accettore degli atomi di idrogeno provenienti dall’aldeide e precisamente accetterà uno ione idruro che si legherà all’anello nicotinammidico, generando la forma ridotta del coenzima, NADH. L’altro atomo di idrogeno della molecola del substrato passerà in soluzione sotto forma di ione H+. Il NADH deve essere riossidato a NAD+, poiché le cellule contengono una quantità limitata di NAD+ e la glicolisi si fermerebbe quasi subito se quest’ultimo non venisse velocemente ripristinato. Come vedremo in seguito, in condizioni aerobie il NAD+ verrà ripristinato nella catena respiratoria o catena terminale di trasporto degli elettroni, mentre, in condizioni di anaerobiosi, verrà riformato nella fermentazione. 7) Trasferimento del gruppo fosforico dall’1,3 bisfosfoglicerato all’ATP Questa tappa porta alla formazione della prima molecola di ATP. L’enzima fosfoglicerato chinasi trasferisce il gruppo fosforico ad alta energia dal gruppo carbossilico dell’1,3-bisfosfoglicerato all’ADP, formando ATP e 3-fosfoglicerato (3-PG). Figura 10. Trasferimento del gruppo fosforico dall'1,3BPG all’ADP Queste ultime due reazioni costituiscono un caso di accoppiamento energetico. Infatti, l’1,3 BPG è un intermedio comune che si forma nella prima reazione (endoergonica), mentre nella seconda reazione, fortemente esoegonica, il gruppo fosforico proveniente dall’acilfosfato viene trasferito all’ADP per formare ATP. La somma delle due reazioni è complessivamente esoergonica. Infatti la prima reazione ha un ∆G° di +6,3 KJ/mole, mentre la seconda di –18,8 KJ/mole. La somma algebrica è quindi di –12,5 KJ/mole. 130 Capitolo VII Questa formazione di ATP mediante trasferimento di un gruppo fosforico dal substrato all’ADP viene detta fosforilazione a livello di substrato, per distinguerla dagli altri due modi di formare ATP, e precisamente dalla fosforilazione ossidativa (tappa terminale della respirazione degli organismi aerobi) e dalla fotofosforilazione (nella fotosintesi). 8) Conversione del 3-fosfoglicerato in 2-fosfoglicerato Questa reazione è catalizzata dalla fosfogliceromutasi, che catalizza lo spostamento reversibile del gruppo fosforico dal C3 al C2 (Figura 11). Anche in questo caso sono presenti ioni Mg2+. Figura 11. Conversione del 3-fosfoglicerato in 2-fosfoglicerato 9) Deidratazione del 2-fosfoglicerato a fosfoenolpiruvato Questa reazione catalizzata dall’enzima enolasi genera un enolo per deidratazione del 2-PG (Figura 12), che aumenta significativamente il potenziale di trasferimento del gruppo fosforico del fosfoenolpiruvato (PEP). Lo sviluppo di energia libera che si ha in seguito all’idrolisi del PEP è molto più grande di quanto sarebbe quella legata all’idrolisi del 2-PG. Figura 12. Deidratazione del 2-fosfoglicerato Glicolisi 131 10) Trasferimento del gruppo fosforico del fosfoenolpiruvato all’ADP E’ l’ultimo passaggio della glicolisi, quello in cui un gruppo fosfato (Pi) viene trasferito ad una molecola di ADP con formazione di ATP. L’enzima che catalizza la reazione è la piruvato chinasi. Si ha dapprima la formazione di enolpiruvato, che viene successivamente isomerizzato a piruvato. Il ∆G° di questa reazione è nettamente negativo (31.4 KJ/mole) e ciò è dovuto in gran parte alla conversione spontanea (cioè non mediata da enzimi) della forma enolica nella forma chetonica isomera. Figura 13. Ultima tappa glicolitica con formazione di piruvato e ATP Destino del piruvato prodotto dalla glicolisi Il piruvato, come si è visto, rappresenta il punto di arrivo del processo glicolitico. Il suo destino è però diverso a seconda che il processo avvenga in condizioni aerobiche o anaerobiche. In condizioni aerobiche, il piruvato, come vedremo più avanti, esce dal citosol ed entra nel mitocondrio. Qui viene dapprima ossidato ad acetato e legato ad un particolare coenzima, il coenzima A, che come vedremo ha funzioni di coenzima acilante, cioè trasportatore di gruppi acile (RCO-). Come acetilcoenzima A entra nel ciclo dell’acido citrico (o ciclo di Krebs) e viene ossidato a CO2 ed H2O. Il NADH che si forma nella glicolisi viene riossidato nel processo della respirazione mitocondriale mediante il passaggio dei suoi elettroni all’ossigeno molecolare. In condizioni anaerobiche il NADH non può essere riossidato dall’O2 e quindi la cellula si troverebbe priva di un accettore di elet- Capitolo VII 132 troni necessario per l’ossidazione della gliceraldeide-3-fosfato. La glicolisi si fermerebbe subito. IL NAD+ deve quindi essere rigenerato in un altro modo, e ciò avviene tramite una serie di reazioni che prendono il nome di fermentazioni. LE FERMENTAZIONI Fermentazione omolattica Quando i tessuti animali non vengono riforniti con quantità di ossigeno sufficienti per le ossidazioni aerobiche di piruvato e NADH provenienti dalla glicolisi, il NADH viene ossidato e rigenerato a NAD+ mediante riduzione del piruvato a L-lattato, catalizzata dalla lattato deidrogenasi. Figura 14. Riduzione del piruvato nella fermentazione omolattica Poiché questa riduzione ha un valore del ∆G° molto negativo, l’equilibrio della reazione è fortemente spostato verso destra. Inoltre, poiché nella glicolisi vengono adoperate due molecole di NAD+ e la riduzione di due molecole di piruvato porta alla riossidazione di due molecole di NADH, il processo può continuare indefinitamente. La fermentazione lattica avviene in genere nei tessuti animali, come sopra ricordato: tuttavia, la lattato deidrogenasi è stata trovata anche nelle piante. Sembra che il prodotto della reazione di riduzione del piruvato, l’acido lattico, acidifichi il citoplasma portando il pH a valori ottimali per la fermentazione alcolica. Glicolisi 133 Fermentazione alcolica Il lievito ed altri microrganismi fermentano il glucosio in etanolo e CO2, anzichè a lattato rigenerando NAD+. Il glucosio viene convertito a piruvato nella glicolisi ed il piruvato viene in seguito trasformato in etanolo e CO2 in un processo a due tappe (Figura 15). Inizialmente il piruvato viene decarbossilato con una reazione irreversibile catalizzata dalla piruvato decarbossilasi, enzima che richiede Mg2+ e un gruppo prostetico saldamente legato all’enzima, la tiaminapirofosfato (TPP). La TPP deriva dalla Vitamina B1 e la sua mancanza nella dieta umana provoca una malattia nota col nome di beri-beri. Nella seconda tappa della fermentazione alcolica l’acetaldeide viene ridotta ad etanolo: in questo passaggio viene riossidato il NADH formato dall’ossidazione della gliceraldeide-3-fosfato della glicolisi. L’enzima è l’alcool deidrogenasi. I prodotti finali sono alcool etilico e CO2.E’ presente anche Zn+2 come cofattore. Figura 15. Reazioni nella fermentazione alcolica Fermentazioni microbiche Anche se lattato ed etanolo sono i prodotti comuni delle fermentazioni dei batteri non sono gli unici prodotti possibili. Le fermentazioni sono usate anche a livello industriale per produrre acido formico, acetico, propionico, butirrico e succinico e alcoli come metanolo, glicerolo, isopropanolo, butanolo e butandiolo. Queste fermentazioni vengono fatte avvenire in grandi recipienti chiusi a temperatura ed aereazione controllata per favorire la moltiplicazione dei microrganismi e per escludere la contaminazione da ceppi diversi. IL CICLO DELL’ACIDO CITRICO Il ciclo dell’acido citrico (o ciclo degli acidi tricarbossilici, o ciclo di Krebs, dal nome del suo scopritore) è il punto centrale del sistema metabolico di degradazione ossidativa comune a procarioti ed eucarioti. Aminoacidi, acidi grassi e carboidrati trovano nel ciclo dell’acido citrico la via finale comune per la loro ossidazione, inoltre è sempre a questo livello che vengono generati numerosi precursori per la biosintesi di glucosio, dei lipidi, degli amminoacidi e delle porfirine. Questo significa che alcuni intermedi possono essere sottratti al ciclo per essere utilizzati come precursori per alimentare vie metaboliche biosintetiche fondamentali. Si dice perciò che il ciclo dell'acido citrico è una via anfibolica, che vuol dire che è collocata sia nelle vie cataboliche che in quelle anaboliche e quindi la sua funzione è essenziale anche senza considerare l’importante ruolo nel metabolismo energetico. La maggior parte delle cellule eucariote e molti batteri sono aerobici, cioè hanno bisogno dell’ossigeno per vivere. Gli organismi aerobici sono in grado di ossidare completamente le loro sostanze nutrienti a CO2 ed H2O. In questo caso la glicolisi rappresenta solo il primo passo per la completa ossidazione del glucosio. Il piruvato che si forma non viene ridotto come nel caso della fermentazione, ma viene ossidato a gruppo acetilico e legato al coenzima A, formando l’acetil-CoA. I gruppi acetilici entrano nel ciclo dell’acido citrico dove vengono completamente ossidati a CO2. Il ciclo dell’acido citrico è la via finale comune per l’ossidazione di tutte le sostanze nutrienti. Occupa un posto centrale nel metabolismo perché vi convergono tutte le vie demolitive ed è un punto di partenza per molte reazioni anaboliche (Figura 1). 135 136 Capitolo VIII Figura 1. Principali interconnessioni metaboliche con la respirazione Ciclo dell’acido citrico e del gliossilato 137 Decarbossilazione ossidativa del piruvato e formazione di AcetilCoA e CO2. Il piruvato formato dalla glicolisi nel citoplasma entra nei mitocondri, dove avverrà la sua ossidazione e decarbossilazione ad acetil-CoA prima di entrare nel ciclo dell’acido citrico. I mitocondri sono organelli subcellulari in cui si svolgono tutte le reazioni che producono energia: possono essere definiti le centrali energetiche delle cellule eucariotiche aerobiche. Figura 2. Schema della struttura di un mitocondrio Ogni mitocondrio ha due membrane: una esterna e liscia che lo circonda completamente ed è in contatto col citoplasma cellulare ed una interna, sollevata in numerosi ripiegamenti chiamati creste, che ne aumentano considerevolmente la superficie specifica e che è in contatto con la parte interna contenente materiale amorfo (Figura 2). Questa viene detta matrice, ed è una soluzione acquosa molto concentrata di enzimi e di intermedi chimici del metabolismo energetico. Gli enzimi contenuti nel mitocondrio catalizzano l’ossidazione dei nutrienti organici da parte dell’O2 molecolare, alcuni di questi enzimi si trovano all’interno della matrice, altri nelle membrana mitocondriale interna. L’energia chimica rilasciata nelle ossidazioni mitocondriali viene usata per generare ATP, la principale molecola trasportatrice di energia. L’ATP formato nei mitocondri diffonde poi in tutte le altre parti della cellula rifornendola di energia. 138 Capitolo VIII Le reazioni di deidrogenazione e decarbossilazione del piruvato ad Acetil-CoA avvengono nella matrice mitocondriale e sono catalizzate dal complesso multienzimatico della piruvato deidrogenasi. Questa serie di reazioni è il collegamento tra la glicolisi e il ciclo dell’acido citrico vero e proprio. Si tratta di una serie di cinque reazioni sequenziali la cui stechiometria complessiva porta ad una decarbossilazione ossidativa ossia un processo di ossidazione irreversibile in cui una molecola di CO2 viene rimossa dal piruvato e i due atomi di carbonio che restano diventano un gruppo acetilico legato al Coenzima A. Il NAD+ in questa reazione accetta dal piruvato uno ione idruro, i cui due elettroni verranno poi ceduti alla catena respiratoria che li porterà fino all’O2. Piruvato + CoA + NAD+ → acetil-CoA + CO2 + NADH Il complesso della piruvato deidrogenasi è uno dei sistemi multienzimatici più grandi e complessi la cui struttura è costituita da copie multiple di tre diversi enzimi e cinque tra coenzimi e gruppi prostetici, per un totale di 60 subunità e un peso molecolare che a seconda dei casi può variare tra 4 e 10 milioni di dalton. Gli enzimi che la costituiscono sono: la piruvato deidrogenasi (E1), la diidrolipoil transacetilasi (E2) e la diidrolipoil deidrogenasi (E3). Questi hanno rispettivamente come gruppo prostetico: la tiamina pirofosfato (TPP), la lipoamide, e il flavin adenin dinucleotide (FAD). I coenzimi coinvolti sono: il coenzima A (CoA) e la nicotinammide adenin dinucleotide (NAD+). Il complesso multienzimatico della piruvato deidrogenasi porta avanti cinque reazioni consecutive che vediamo di seguito più nel dettaglio (Figura 3): 1. La reazione è essenzialmente identica alla reazione catalizzata dalla piruvato decarbossilasi. Il piruvato reagisce con la TPP legata alla piruvato deidrogenasi (E1). L’atomo C1 del piruvato viene rilasciato sotto forma di CO2 e l’atomo C2, che nel piruvato ha lo stato di ossidazione di un’aldeide, viene legato alla TPP come gruppo idrossietilico. 2. Il gruppo idrossietilico viene ossidato ad acido carbossilico (acetato) a spese della forma ossidata del lipoato. I due elettroni rimossi Ciclo dell’acido citrico e del gliossilato 139 durante l’ossidazione vanno a ridurre il ponte -S-S- del gruppo lipoilico sull’E2 (l’enzima è la diidrolipoiltransacetilasi), formando due gruppi –SH. 3. L’acetato prodotto durante l’ossidazione del gruppo idrossietilico viene dapprima esterificato con uno dei gruppi –SH del gruppo lipoilico e poi transesterificato a CoA formando acetil-CoA. Quindi l’energia dell’ossidazione guida la formazione di un tioestere dell’acetato ad alta energia (infatti l’aldeide o il chetone sono ad un livello energetico più alto che non l’acido carbossilico). 4. La parte rimanente della reazione catalizzata dalla piruvato deidrogenasi consiste in una serie di reazioni che rigenerano la forma ossidata disolfuro del lipoato. Questo in un primo momento viene ossidato dal FAD che diventa FADH2. Qui entra in gioco il terzo enzima, E3, che è la diidro-lipoil-deidrogenasi. 5. Nell’ultima tappa della reazione il FADH2 ridotto sull’enzima E3 trasferisce uno ione idruro al NAD+, formando NADH + H+. Il complesso enzimatico è ora pronto per un altro ciclo. Figura 3. Reazioni del complesso della piruvato deidrogenasi La regolazione del complesso della piruvato deidrogenasi nei mammiferi, in cui la formazione di acetil-CoA è irreversibile, avviene in due modalità principali qui riportate sinteticamente: 140 Capitolo VIII 1. Inibizione da prodotto operata da NADH sulla subunità E3 e acetil-CoA sulla subunità E2; 2. Modificazione covalente per fosforilazione reversibile della subunità E1 che viene inattivata da una chinasi specifica che utilizza ATP come donatore di gruppi fosforici. L’idrolisi del gruppo fosforico e il ripristino dell’attività avviene ad opera di una fosfatasi specifica. Perciò la piruvato deidrogenasi è inattiva quando la carica energetica complessiva è elevata e gli intermedi biosintetici sono abbondanti, dall’altro lato l’attivazione avviene in presenza di piruvato e ADP. Il ciclo dell’acido citrico (ciclo di Krebs) Il ciclo dell’acido citrico, come dice il nome, è una via metabolica ciclica, cioè che parte da un composto che è lo stesso che si riformerà nella reazione finale. Potrebbe sembrare che si trattasse di un ciclo futile, cioè fine a se stesso, ma vedremo presto che non è così. Per iniziare un giro del ciclo, l’acetil-CoA, precedentemente ottenuto dalle reazioni a carico del complesso della piruvato deidrogenasi, dona il suo gruppo acetilico a due atomi di carbonio (C2) alla molecola di partenza, l’ossalacetato, un chetoacido a 4 atomi di carbonio (C4). Si forma il citrato, composto a 6 atomi di carbonio (C6). Il citrato viene poi trasformato in isocitrato, un idrossiacido isomero sempre a 6 atomi di carbonio, che verrà poi deidrogenato, subendo una decarbossilazione ossidativa, con perdita di una molecola di CO2 e formazione di una molecola a 5 atomi di carbonio (C5), l’αchetoglutarato. L’ α-chetoglutarato perde a sua volta una molecola di CO2 per formare il succinato composto a 4 atomi di carbonio (C4), che sarà poi convertito enzimaticamente in 3 tappe ad ossalacetato, il composto di partenza. I composti intermedi sono il fumarato ed il malato. In tutto il ciclo vengono utilizzati 3 NAD+ e 1 FAD In ogni giro del ciclo entra un gruppo acetilico a 2 atomi di C ed escono 2 molecole di CO2. Viene utilizzata una molecola di ossalacetato che però, dopo una serie di reazioni viene rigenerata. Non vi è quindi consumo netto di ossalacetato, e quindi questa molecola potrà servire per l’ossidazione di un numero quasi infinito di gruppi acetili- Ciclo dell’acido citrico e del gliossilato 141 ci. Il ruolo del ciclo di Krebs tuttavia non è solo quello di costituire il nucleo centrale del metabolismo volto alla produzione di energia. Gli intermedi del ciclo a 4 e 5 atomi di C sono anche precursori per le biosintesi che portano alla formazione di moltissimi composti. Per rimpiazzare gli intermedi utilizzati, le cellule possiedono delle reazioni di riempimento, dette anaplerotiche. La reazione complessiva del ciclo dell’acido citrico è: Acetil-CoA + 3 NAD+ + FAD + GDP + Pi + H2O → → 2 CO2 + 3 NADH + FADH2 + GTP + 2H+ + CoA Le reazioni e gli enzimi coinvolti sono riassunti in Tabella 1 Tabella 1. Il ciclo dell'acido citrico Reazione Enzima 1 Ac-CoA + ossalacetato + H2O → citrato + CoA + H+ Citrato sintasi 2 Citrato ↔ isocitrato Aconitasi 3 Isocitrato + NAD+ ↔ α-chetoglutarato + CO2 + NADH Isocitrato deidrogenasi 4 α-chetoglutarato + NAD+ + CoA → succinil-CoA + CO2 + NADH α-chetoglutarato deidrogenasi 5 succinil-CoA + Pi + GDP ↔ succinato + GTP + CoA Succinil-CoA sintetasi Succinato + FAD ↔ fumarato + FADH2 Succinato deidrogenasi 7 Fumarato + H2O ↔ L-malato Fumarasi 8 L-malato + NADH ↔ ossalacetato Malato deidrogenasi 6 142 Capitolo VIII Il ciclo dell’acido citrico è quindi costituito da otto tappe che vediamo di seguito più in dettaglio. 1) Formazione del citrato Nella prima reazione del ciclo, l’acetil-CoA condensa con l’ossalacetato formando citrato (Figura 4). L’enzima catalizzante è la citrato sintasi (nota bene: sintasi e non sintetasi: il termine sintetasi infatti indica che la reazione di sintesi avviene con consumo di ATP. In questa reazione invece non entra l’ATP). In questo primo passaggio, l’atomo di C metilico del gruppo acetile si lega al gruppo carbonilico C2 dell’ossalacetato. Si tratta di una condensazione aldolica. Si ottiene il citril-CoA, un intermedio transitorio che si forma sul sito attivo dell’enzima e che si idrolizza rapidamente dando CoA libero e citrato, che vengono poi rilasciati dal sito attivo. L’idrolisi dell’intermedio tioestere ad alta energia rende la reazione fortemente esoergonica. Il valore negativo del ∆G è essenziale per l’attività del ciclo. Figura 4. Reazione della citrato sintasi 2) Formazione dell’isocitrato attraverso il cis-aconitato L’isomerizzazione reversibile del citrato in isocitrato è catalizzata dall’enzima aconitasi (Figura 5). La reazione procede in due fasi consecutive mediante una prima tappa di deidratazione del citrato che porta alla formazione dell’intermedio cis-aconitato, che normalmente non si dissocia dal sito attivo dell’enzima. Segue quindi una seconda tappa di addizione di una molecola d’acqua al doppio legame dell’intermedio legato all’enzima portando all’isocitrato. Il risultato è Ciclo dell’acido citrico e del gliossilato 143 uno scambio di posizione tra un atomo di idrogeno ed un gruppo ossidrilico. Nelle condizioni intracellulari la reazione è reversibile. Figura 5. Reazione dell'aconitasi 3) Ossidazione dell’isocitrato ad α-chetoglutarato e CO2 Nella tappa successiva la isocitrato deidrogenasi catalizza la decarbossilazione ossidativa dell’isocitrato per formare α-chetoglutarato (Figura 6). Il coenzima usato per questa reazione di ossidazione è il NAD+, e come cofattore troviamo il Mg2+. Si forma dapprima un intermedio, l’ossalsuccinato, che viene poi decarbossilato ad αchetoglutarato, liberando in questo modo la prima molecola di CO2. e contemporaneamente la prima molecola di NADH. Figura 6. Reazione dell'isocitrato deidrogenasi 4) Ossidazione dell’ α-chetoglutarato a succinil-CoA e CO2 A questo punto avviene un’altra decarbossilazione ossidativa, in cui l’α-chetoglutarato viene trasformato in succinil-CoA e CO2 dal 144 Capitolo VIII complesso dell’ α-chetoglutarato deidrogenasi con produzione di una molecola di NADH. L’accettore finale degli elettroni è il NAD+ e il CoA trasporta il gruppo succinile. L’energia che si libera dall’ossidazione dell’ αchetoglutarato viene conservata nel legame tioestere del succinile con il CoA. Il complesso dell’ α-chetoglutarato deidrogenasi è una struttura omologa al complesso della piruvato deidrogenasi, descritta precedentemente, perciò la reazione è praticamente identica. Troviamo anche qui tre enzimi E1, E2 ed E3 e cinque coenzimi legati allo stesso modo come per la piruvato deidrogenasi. Anche questo enzima richiede Mg2+ come cofattore. 5) Conversione del succinil-CoA a succinato Il legame tioestere del succinil-CoA, come per l’acetil-CoA, è un legame ad alta energia. L’energia rilasciata dall’idrolisi di questo legame viene usata per la sintesi di un legame fosfoanidridico sotto forma di ATP o GTP che sono energeticamente equivalenti. La reazione catalizzata dalla succinil-CoA sintetasi avviene in tre tappe (Figura 7): 1. il succinil-CoA reagisce con il fosfato formando succinil fosfato che è un’anidride mista ad alta energia; 2. il gruppo fosforico del succinil fosfato viene trasferito temporaneamente ad un sito di attacco dell’enzima e il succinato viene rilasciato; 3. il gruppo fosforico sull’enzima viene trasferito al GDP formando GTP. La formazione di GTP (nei mammiferi) o ATP (nelle piante e nei batteri) sono reazioni praticamente equivalenti in quanto il GTP formato dalla succinil-CoA sintetasi può trasferire a sua volta il suo gruppo fosforico terminale all’ADP per formare ATP tramite l’enzima nucleoside trifosfato chinasi. Questa reazione è importante perché è il solo punto del ciclo dove viene prodotto ATP direttamente dal substrato senza coinvolgere la catena respiratoria. Questa formazione di ATP prende il nome di fosforilazione a livello di substrato. Ciclo dell’acido citrico e del gliossilato 145 Figura 7. Reazione della succinil-CoA sintetasi A questo punto le ultime tre reazioni che seguono, hanno la funzione di convertire il succinato in ossalacetato e permettere un nuovo inizio del ciclo. Fino a questo punto sono state generate due molecole di NADH e una di GTP. Il succinato viene convertito in ossalacetato in tre tappe: una ossidazione, una idratazione e una seconda ossidazione. 6) Ossidazione del succinato a fumarato Il succinato viene ossidato a fumarato dalla flavoproteina succinato deidrogenasi, che ha come gruppo prostetico il FAD ed è l’unico enzima legato alla membrana interna dei mitocondri (Figura 8). Questo è anche il solo punto del ciclo dove si ha l’intervento del FAD in quanto la variazione di energia in gioco in questo passaggio è troppo bassa per ridurre il NAD. Infatti il FAD funziona come accettore di elettroni nelle reazioni di ossidazione biochimica di alcani (es. succinato) ad alcheni (es. fumarato), mentre il NAD ossida alcoli (es. malato) ad aldeidi o chetoni (es. ossalacetato). Gli elettroni del succinato passano attraverso il FAD, che si riduce a FADH2 ed entrano nella catena di trasporto degli elettroni sulla membrana mitocondriale interna. Figura 8. Reazione della succinato deidrogenasi 146 Capitolo VIII 7) Idratazione del fumarato per produrre malato L’idratazione reversibile del doppio legame del fumarato ad Lmalato è catalizzata dalla fumarasi (Figura 9). L’enzima catalizza l’idratazione stereospecifica di idrogeno e gruppo OH trans del fumarato, ma non agisce sull’isomero cis (il maleato). Figura 9. Reazione della fumarasi 8) Ossidazione del malato a ossalacetato E’ l’ultima reazione del ciclo. La L-malato deidrogenasi, NAD+ dipendente, catalizza l’ossidazione del malato ad ossalacetato (Figura 10). L’equilibrio sarebbe molto spostato a sinistra (il ∆G°’ = 29,7 KJ/mole) ma l’ossalacetato viene rimosso continuamente dalla successiva reazione esoergonica della citrato sintasi, che mantiene sempre bassa la concentrazione dell’ossalacetato. Figura 10. Reazione della malato deidrogenasi Ciclo dell’acido citrico e del gliossilato 147 La Figura 11 riassume le varie tappe del ciclo dell’acido citrico: Figura 11. Le reazioni del ciclo dell'acido citrico Regolazione del ciclo dell’acido citrico Il ciclo dell’acido citrico si svolge esclusivamente in presenza di ossigeno e la sua velocità è regolata in modo tale da soddisfare il fabbisogno energetico di ATP da parte delle cellule. I punti di regolazione fondamentali sono tre: la citrato sintasi, l’isocitrato deidrogenasi e l’α-chetoglutarato deidrogenasi. 148 Capitolo VIII La loro attività viene regolata dalla rispettiva disponibilità di substrati, dall’inibizione da prodotto, dall’inibizione da intermedi del ciclo. In modo sintetico il meccanismo viene rappresentato in Figura 12. Figura 12. Regolazione del ciclo dell'acido citrico Energia liberata nel ciclo dell’acido citrico Come abbiamo visto, nel ciclo dell’acido citrico il gruppo acetilico a due atomi di carbonio dell’Ac-CoA si è combinato con l’ossalacetato. I due atomi di carbonio sono stati poi rilasciati sotto Ciclo dell’acido citrico e del gliossilato 149 forma di due molecole di CO2 nell’ossidazione dell’isocitrato e dell’αchetoglutarato. L’energia liberata da queste due ossidazioni è stata poi conservata nei tre NADH e in un FADH2 prodotti . Il ciclo dell’acido citrico produce direttamente solo un GTP (o ATP nelle piante e nei batteri) per giro, ma nelle quattro reazioni di ossidazione si genera un flusso di elettroni che verranno convogliati poi sulla catena respiratoria di trasporto degli elettroni, portando alla formazione di un gran numero di molecole di ATP. Questo processo che vedremo nel capitolo successivo è detto fosforilazione ossidativa. I componenti del ciclo dell’acido citrico sono precursori biosintetici Negli organismi aerobi, il ciclo dell’acido citrico è una via anfibolica, che cioè serve sia ai processi anabolici che a quelli catabolici. Non solo si trova nel catabolismo dei carboidrati, degli amminoacidi e degli acidi grassi ma produce anche precursori per molte vie biosintetiche: 1. l’α-chetoglutarato e l’ossalacetato sono precursori di glutammato e aspartato (per transaminazione) che possono essere usati per costruire altri amminoacidi o nucleotidi purinici e pirimidinici; 2. l’ossalacetato viene convertito in glucosio nel processo della gluconeogenesi; 3. il succinil-CoA è un intermedio della sintesi del gruppo eme e della clorofilla. Quando al ciclo dell’acido citrico vengono sottratti intermedi per utilizzarli come precursori per altre vie, essi possono essere rimpiazzati da reazioni anaplerotiche (di riempimento). Ad esempio la formazione di ossalacetato per carbossilazione reversibile del piruvato ad opera della piruvato carbossilasi quando il ciclo dell’acido citrico è povero di ossalacetato. Quando l’acetil-CoA è in eccesso, stimola la reazione della piruvato carbossilasi a produrre più ossalacetato, aumentando la reazione della citrato sintasi. Capitolo VIII 150 Un’altra reazione anaplerotica che ha luogo nelle piante superiori, nel lievito e nei batteri è la carbossilazione del fosfoenolpiruvato ad ossalacetato tramite la PEP-carbossilasi. Il ciclo del gliossilato I vertebrati non possono convertire gli acidi grassi in carboidrati. La conversione del fosfoenolpiruvato a piruvato e quella del piruvato ad acetil-CoA sono tanto esoergoniche da essere praticamente irreversibili. Quindi questi organismi non possono trasformare in carboidrati i nutrienti degradati ad acetato. Molti altri organismi (ad es. batteri e piante) invece convertono l’acetato in carboidrati mediante il ciclo del gliossilato. Questa via metabolica, studiata per la prima volta nell’endosperma di semi di ricino, si svolge in organelli subcellulari detti gliossisomi. Il suo ruolo fondamentale nei tessuti di riserva è la mobilizzazione dei trigliceridi durante la germinazione dei semi oleosi. I trigliceridi subiscono la β-ossidazione (Vedi il Metabolismo dei lipidi) che porta alla formazione di acetil-CoA. L’acetil-CoA entra nel ciclo del gliossilato e fornisce il substrato necessario per le reazioni di gluconeogenesi che producono saccarosio a partire da molecole lipidiche. Si generano così i carboidrati che nel seme, sprovvisto di clorofilla, non potrebbero essere formati per via fotosintetica. Il ciclo del gliossilato come il ciclo dell’acido citrico inizia dalla condensazione dell’acetil-CoA e dell’ossalacetato per formare citrato, che viene isomerizzato a isocitrato. A questo punto a differenza di quanto accade nel ciclo dell’acido citrico, invece di venire decarbossilato l’isocitrato viene scisso in succinato e gliossilato. Le tappe successive rigenerano l’ossalacetato a partire dal gliossilato. Oltre all’assenza delle reazioni di decarbossilazione un’altra differenza rispetto al ciclo dell’acido citrico è l’ingresso di due molecole di acetil-CoA ad ogni ciclo invece di una sola. La sintesi di queste reazioni è: 2 Acetil-CoA + NAD+ + 2 H2O → → succinato + NADH + + 2 H+ + 2 CoA Ciclo dell’acido citrico e del gliossilato 151 Il ciclo del gliossilato è costituito dalle cinque tappe che seguono: 1. l’ossalacetato condensa con una molecola di acetil-CoA in una reazione catalizzata dalla citrato sintasi, formando una molecola di citrato; 2. questa viene utilizzata dall’aconitasi che lo trasforma in isocitrato; 3. a questo punto interviene non l’isocitrato deidrogenasi come nel ciclo di Krebs, ma l’isocitrato liasi, che catalizza la scomposizione reversibile dell’isocitrato in succinato e gliossilato. Il succinato prodotto da questa reazione esce dal gliossisoma ed entra nel mitocondrio dove viene trasformato in acido fumarico e poi in malico dagli enzimi del ciclo di Krebs; 4. il gliossilato reagisce con un’altra molecola di acetil-CoA tramite la malato sintasi e dà origine al malato; 5. il malato viene poi deidrogenato ad ossalacetato tramite la malato deidrogenasi. I cinque enzimi coinvolti sono localizzati nei gliossisomi, ad eccezione dell’aconitasi che si trova nel citosol. L’ossalacetato prodotto è il substrato dell’enzima fosfoenolpiruvato carbossi-chinasi che opera la decarbossilazione e la fosforilazione del substrato, che diventa acido fosfoenolpiruvico (PEP). Questo composto ripercorre in senso inverso le tappe della glicolisi portando alla produzione di triosofosfati, esosi difosfati ed infine saccarosio. Tale processo di sintesi non fotosintetica dei carboidrati si dice gluconeogenesi. 152 Capitolo VIII Figura 13. Il ciclo del gliossilato 153 LA FOSFORILAZIONE OSSIDATIVA La fosforilazione ossidativa, cioè la sintesi di ATP guidata dal trasferimento di elettroni all’O2 e la fotofosforilazione, che è la sintesi di ATP guidata dall’energia luminosa, sono i due sistemi di trasduzione dell’energia più importanti della biosfera. La fosforilazione ossidativa è il culmine del metabolismo energetico degli organismi aerobici, che va sotto il nome di respirazione. Respirare significa quindi in biochimica degradare in ambiente aerobico ciò che è stato sintetizzato per altre vie o assunto dall’esterno (ricordiamo gli organismi fototrofi e chemiotrofi) in modo da produrre energia per tutte le funzioni vitali. Le tappe della degradazione ossidativa dei principali nutrienti degli organismi aerobici (carboidrati, grassi, proteine) convergono nella tappa finale in cui viene generato ATP. Nel corso di reazioni cataboliche come la glicolisi, il ciclo dell’acido citrico e l’ossidazione degli acidi grassi si formano NADH e FADH2, molecole ricche di energia in quanto contengono elettroni con elevato potenziale di trasferimento. Quando questi elettroni arrivano all’ossigeno molecolare (O2) questo viene ridotto, si forma acqua e viene liberata una grande quantità di energia, che può essere usata per generare ATP. La fosforilazione ossidativa, che è la principale fonte di ATP negli organismi aerobici, è quindi il processo in cui viene formato ATP mentre gli elettroni sono trasferiti dal NADH o dal FADH2 all’ossigeno attraverso una serie di trasportatori di elettroni. NADH e FADH2 costituiscono il potere riducente che deve venir ossidato nuovamente durante la fosforilazione ossidativa per poter continuare le reazioni cataboliche. Il flusso di elettroni passa attraverso complessi proteici localizzati nella membrana mitocondriale interna e causa la fuoruscita di protoni dalla matrice mitocondriale. Si crea una forza motrice generata da protoni formata da un gradiente di pH e da un potenziale elettrico transmembrana. L’ATP viene sintetizzato quando i protoni rientrano nella matrice mitocondriale attraverso un complesso enzimatico. Pertanto, l’ossidazione e la fosforilazione sono accoppiate da un gradiente di protoni attraverso la membrana mitocondriale interna. 1 154 Capitolo IX In pratica, la forza motrice generata da elettroni viene convertita in forza motrice generata da protoni e poi in potenziale di fosforilazione. Vi sono tre pompe protoniche, dipendenti da elettroni, dette rispettivamente NADH-Q reduttasi, citocromo reduttasi e citocromo ossidasi. Questi grandi complessi transmembrana contengono vari centri di ossido-riduzione come flavine, chinoni, centri ferro-zolfo, gruppi eme e ioni rame. La seconda fase della fosforilazione ossidativa viene eseguita dall’ATP sintasi, un complesso che sintetizza l’ATP, attivato dal flusso di protoni che rientra nella matrice mitocondriale. Vediamo ora più da vicino cosa avviene durante queste due fasi della fosforilazione ossidativa che, negli eucarioti, avviene nei mitocondri. I mitocondri, che sono stati già descritti nel capitolo dedicato al ciclo dell’acido citrico, sono organelli di forma ovale, lunghi circa 2 mm e con un diametro di circa 0,5 mm, che contengono le strutture respiratorie, gli enzimi del ciclo dell’acido citrico e dell’ossidazione degli acidi grassi. I mitocondri hanno due sistemi di membrane, una membrana esterna e una membrana interna. Quindi nei mitocondri esistono due scompartimenti: lo spazio intermembrana (fra la membrana esterna e quella interna) e la matrice, lo spazio delimitato dalla membrana interna, che presenta una serie di pieghe chiamate creste. La fosforilazione ossidativa avviene nella membrana mitocondriale interna, mentre la maggior parte delle reazioni del ciclo di Krebs e dell’ossidazione degli acidi grassi avviene nella matrice. La membrana esterna è permeabile alla maggior parte delle piccole molecole e degli ioni perché contiene la porina, una proteina transmembrana che forma un grande canale detto poro. La membrana interna invece è impermeabile alla maggior parte degli ioni e alle molecole polari. Un gruppo di trasportatori porta metaboliti, come ATP e citrato, attraverso la membrana mitocondriale interna le cui due facce di questa membrana sono dette versante della matrice e versante citosolico. La matrice mitocondriale contiene il complesso della piruvato deidrogenasi e gli enzimi del ciclo dell’acido citrico, delle vie della βossidazione degli acidi grassi e dell’ossidazione degli amminoacidi, cioè tutte le vie di ossidazione delle sostanze nutrienti, eccetto la glicolisi, che avviene nel citosol. Fosforilazione ossidativa 155 Poiché la membrana mitocondriale interna è selettivamente permeabile, si determina una separazione netta fra gli intermedi e gli enzimi del citosol e quelli dei mitocondri. Trasportatori specifici consentono il passaggio del piruvato, degli acidi grassi e degli amminoacidi o degli α-chetoacidi da loro derivati, nella matrice mitocondriale, dove vengono degradati dal ciclo dell’acido citrico. Anche l’ADP ed il Pi sono trasportati all’interno del mitocondrio da specifici trasportatori e contemporaneamente l’ATP percorre la via inversa. La fosforilazione ossidativa ha inizio con l’ingresso degli elettroni nella catena respiratoria di trasporto degli elettroni, o ETC (Electron Transport Chain). La maggior parte di questi elettroni viene dall’azione delle deidrogenasi che li raccolgono dai processi catabolici per incanalarli verso nucleotidi piridinici (NAD+, NADP+) o nucleotidi flavinici (FMN, FAD) che sono accettori universali di elettroni. Le deidrogenasi catalizzano reazioni del tipo: Substrato ridotto + NAD+ = Substrato ossidato + NADH + H+ Le deidrogenasi NAD+ dipendenti rimuovono due atomi di idrogeno dai substrati. Uno di questi viene trasferito come ione idruro (H-) al NAD+, mentre l’altro atomo di idrogeno, H+, viene rilasciato nell’ambiente. Il NAD+ può raccogliere elettroni anche da substrati che hanno donato i loro elettroni ad una deidrogenasi NADP+ dipendente, tramite l’enzima piridina nucleotide transidrogenasi. Il NADH è un trasportatore di elettroni solubile in acqua che si associa reversibilmente alla deidrogenasi. Il NADH non può attraversare la membrana mitocondriale interna ma lo possono fare gli elettroni mediante trasportatori. Le flavoproteine contengono un cofattore flavinico (FMN o FAD) legato saldamente alla proteina, in qualche caso anche in modo covalente. Il coenzima flavinico ossidato può accettare sia un elettrone (si genera così una forma semichinonica) o due elettroni (generando FADH2 o FMNH2). 156 Capitolo IX La catena respiratoria La catena respiratoria mitocondriale è costituita da una serie di trasportatori di elettroni, la maggior parte dei quali sono proteine integrali di membrana, contenenti gruppi prostetici in grado di accettare o donare uno o due elettroni. Oltre al NAD ed alle flavoproteine operano altri tre gruppi di trasportatori di elettroni: un benzochinone idrofobico, l’ubichinone, e due diversi tipi di proteine contenenti ferro, i citocromi e le proteine ferro-zolfo (Fe-S). L’ubichinone, detto anche coenzima Q o semplicemente Q, è un benzochinone con una catena laterale isoprenoide molto lunga (simile al plastochinone della fotosintesi). Esso può accettare un solo elettrone trasformandosi in un radicale semichinonico QH* o due elettroni, trasformandosi nella forma completamente ridotta ubichinolo QH2. Poiché l’ubichinone è di piccole dimensioni ed idrofobico, può diffondere liberamente nel doppio strato lipidico della membrana mitocondriale interna e può agire da ponte fra trasportatori meno mobili nella stessa membrana. I citocromi sono proteine contenenti un gruppo Fe-porfirinico simile a quello dell’emoglobina e della mioglobina, che trasferiscono elettroni. Il Fe passa da Fe3+ a Fe2+ e viceversa. Nei mitocondri ci sono tre classi di citocromi, citocromi a, citocromi b e citocromi c. Per distinguere citocromi molto simili che appartengono alla stessa classe viene spesso usata la lunghezza d’onda a cui presentano il massimo di assorbimento della luce (es. il citocromob562). Il gruppo eme dei citocromi a e b è saldamente legato alla proteina, ma con legami non covalenti. Il gruppo eme dei citocromi c invece è legato covalentemente attraverso i residui di cisteina della proteina. I citocromi a e b e alcuni del tipo c sono proteine integrali della membrana mitocondriale interna. Una eccezione è il citocromo c dei mitocondri, una proteina solubile che si lega mediante interazioni elettrostatiche sulla superficie esterna della membrana mitocondriale interna. Nelle proteine Fe-S il Fe non è presente all’interno di un gruppo eme ma è associato ad atomi di zolfo inorganico e/o ad atomi di zolfo di residui di cisteina della proteina. Queste proteine partecipano a reazioni redox in cui viene trasferito un elettrone per volta, utilizzando le modificazioni dello stato di ossidazione dell’atomo di Fe viste sopra. Fosforilazione ossidativa 157 Questi centri Fe-S possono avere strutture semplici (un atomo di ferro coordina 4 atomi di zolfo di catene laterali di residui di cisteina), oppure molto complesse e possono contenere 2 o 4 atomi di Fe. Le proteine Fe-S hanno potenziali di riduzione standard piuttosto bassi e sono quindi buoni donatori di elettroni. I trasportatori di elettroni funzionano in complessi multienzimatici ordinati, infatti nella catena respiratoria mitocondriale gli elettroni passano dal NADH, dal succinato o da qualche altro donatore di elettroni attraverso flavoproteine, proteine Fe-S e citocromi, tutti immersi nella membrana mitocondriale, alla molecola di ossigeno. La sequenza dipende dai potenziali standard dei trasportatori, dalla cinetica di ossidazione e da eventuali inibitori che possono bloccare a vari livelli il trasporto elettronico. I trasportatori di elettroni della catena respiratoria sono organizzati in quattro complessi intramembrana (Figura 1). Ognuno di questi complessi è una frazione della catena respiratoria, ha una ben determinata composizione e catalizza il trasporto degli elettroni in una zona specifica della catena. Figura 1. Schema della localizzazione della catena respiratoria. 158 Capitolo IX I complessi I e II trasferiscono elettroni rispettivamente dal NADH e dal succinato all’ubichinone. Il complesso III trasporta gli elettroni dall’ubichinone al citocromo c e il complesso IV trasferisce gli elettroni all’O2 molecolare, completando così le tappe della catena respiratoria. 1) Complesso I detto anche NADH-Q ossidoreduttasi, è un enzima complesso che contiene più di 42 catene polipeptidiche, fra cui una flavoproteina contenente FMN e 6 centri Fe-S. Questo complesso è immerso nella membrana mitocondriale interna e orientato con il sito che lega il NADH verso la matrice, in modo da poter interagire con il NADH proveniente dalle diverse deidrogenasi. Il complesso I catalizza due processi accoppiati e simultanei: il trasferimento (esoergonico) all’ubichinone di uno ione idruro dal NADH e di un protone dal solvente acquoso della matrice. La reazione completa è: NADH + H+ + Q → NAD+ + QH2 Il complesso I trasferisce dapprima una coppia di equivalenti di riduzione dal NADH al suo gruppo prostetico FMN e poi all’ubichinone attraverso sette centri Fe-S di almeno due tipi diversi. Il complesso I può quindi essere definito una pompa protonica guidata dall’energia del trasferimento degli elettroni. L’ubichinolo QH2 completamente ridotto diffonde dal complesso I al complesso III dove viene riossidato a Q, con spostamento di protoni dalla matrice alla faccia esterna citosolica della membrana mitocondriale interna. 2) Complesso II detto anche succinato deidrogenasi, è il solo enzima del ciclo dell’acido citrico ad essere legato alla membrana mitocondriale interna. Contiene due tipi di gruppi prostetici e almeno quattro proteine diverse: una proteina che ha legato a sé un FAD in modo covalente e un centro Fe-S con quattro atomi di ferro, oltre ad una seconda proteina Fe-S. Si pensa che gli elettroni passino dal succinato al FAD e poi attraverso centri Fe-S arrivino all’ubichinone. Altri substrati di deidrogenasi mitocondriali passano elettroni all’ubichinone, ma non attraverso il complesso II. Ad esempio è la flavoproteina acilCoA deidrogenasi nella prima tappa della β-ossidazione degli acidi Fosforilazione ossidativa 159 grassi, che trasferisce due elettroni dal substrato al FAD della deidrogenasi e poi alla flavoproteina che trasferisce gli elettroni (ETF) e infine alla ETF- ubichinone ossidoreduttasi, una proteina Fe-S con un coenzima flavinico legato saldamente, che trasferisce elettroni alla catena respiratoria. Un altro esempio è la glicerolo-3-fosfato deidrogenasi che catalizza l’ossidazione del glicerolo (rilasciato dalla degradazione degli acidi grassi e successivamente fosforilato) a diidrossiacetonefosfato. Questo enzima è una flavoproteina sulla faccia esterna della membrana mitocondriale interna che invia i suoi elettroni alla catena respiratoria riducendo l’ubichinone. 3) Complesso III è detto anche ubichinone-citocromo c reduttasi e accoppia il trasferimento degli elettroni dall’ubichinolo al citocromo c con il trasporto di protoni dalla matrice allo spazio intermembrana. Contiene i citocromi b562, b566, c1, una proteina Fe-S e almeno sei subunità proteiche E’ stato proposto un modello sul passaggio di elettroni e protoni (ciclo dell’ubichinone): QH2 + 2 Cit c1 (oss.) + 2H+n = Q + 2 Cit c1 (rid.) + 4 H+p dove n è il lato affacciato sulla matrice e p è il lato affacciato sullo spazio intermembrana. Il citocromo c è una proteina solubile dello spazio intermembrana. Il suo gruppo eme accetta un elettrone dal complesso III e lo passa al complesso IV. Il complesso III è una pompa protonica. A causa dell’asimmetria del complesso, i protoni liberati dall’ossidazione del QH2 vengono rilasciati nello spazio intermembrana generando una differenza nella concentrazione protonica transmembrana, quindi un gradiente protonico. 4) Complesso IV è detto anche citocromo ossidasi. Il complesso IV trasporta elettroni dal citocromo c all’ossigeno molecolare riducendolo ad H2O. E’ un grosso enzima della membrana mitocondriale interna, costituito dai citocromi a1 ed a3. Questi citocromi sono in realtà due gruppi eme legati alla stessa proteina in punti diversi. La citocromo ossidasi contiene anche due ioni rame, CuA e CuB.che partecipano al trasferimento degli elettroni all’O2. Il trasferimento degli elet- 160 Capitolo IX troni attraverso il complesso IV procede dal citocromo c al centro CuA, al gruppo eme a, al centro a3-CuB e finalmente all’O2. Ogni volta che quattro elettroni attraversano questo complesso l’enzima utilizza quattro ioni H+ dalla matrice per convertire l’O2 in due molecole di H2O. Il complesso utilizza anche l’energia di questa reazione redox per pompare un protone nello spazio intermembrana. L’intera reazione è: 4 Cit c (rid.) + 8 H+n + O2 = 4 Cit c (oss.) + 4 H+ p + 2 H2O Nella fosforilazione ossidativa due elettroni passano attraverso la catena respiratoria dal NADH all’O2 e questo trasferimento degli elettroni all’O2 è altamente esorgonico. Per la coppia redox NAD+ / NADH il valore di E°’ è = -0,32 V, mentre quello della coppia O2/H2O è = + 0,816 V. Il ∆E°’ di questa reazione è quindi = + 1,14 V e la variazione di energia libera standard (∆G°’) è: ∆G°’ = -n F ∆E°’ = (-2) (95,5 kJ/V mole) (1,14 V) = - 220 kJ/mole Nel mitocondrio quindi l’azione combinata dei complessi I, III e IV porta gli elettroni dal NADH all’O2, mentre i complessi II, III e IV trasferiscono elettroni dal succinato all’O2. L’energia rilasciata durante il trasferimento degli elettroni viene conservata in un gradiente protonico. Una gran parte dell’energia rilasciata durante il trasferimento degli elettroni viene usata per pompare protoni fuori dalla matrice. Per ogni coppia di elettroni trasferita all’O2, quattro protoni sono pompati fuori del complesso I, altri quattro dal complesso III e due dal complesso IV. La reazione è: NADH + 11 H+n + ½ O2 = NAD+ + 10 H+p + H2O L’energia elettrochimica contenuta in questa differenza di concentrazione protonica viene conservata temporaneamente in un gradiente, detto forza motrice protonica. Questa forza motrice è formata da due componenti: Fosforilazione ossidativa 161 1) l’energia potenziale chimica dovuta alla differenza di concentrazione di ioni H+ nelle due regioni separate dalla membrana; 2) l’energia del potenziale elettrico dovuto alla separazione di cariche prodotta da uno ione che attraversa la membrana senza un flusso contrario di pari carica. Si può calcolare la variazione di energia libera: ∆G = RT ln (C2/C1) + Z ∆Ψ dove C2/C1 è il rapporto fra le concentrazioni dello ione, Z è il valore assoluto della sua carica elettrica (1 per un protone) e ∆Ψ è la differenza di potenziale elettrico transmembrana, misurata in volt. L’energia rilasciata dal trasferimento di due elettroni dal NADH all’ossigeno accompagnata dal pompaggio di 10 H+ è quindi conservata nel gradiente protonico. La sintesi dell’ATP Il trasferimento degli elettroni durante la catena respiratoria rilascia una quantità di energia pari a 200 kJ/mole, più che sufficiente per la sintesi di una mole di ATP, che viene conservata nella forza motrice protonica. L’ATP viene sintetizzato da un complesso enzimatico, l’ATPsintasi, detto anche Complesso V, costituito da due componenti: F1, un complesso proteico periferico di membrana, unità catalitica che sintetizza l’ATP e Fo, una unità inserita nella membrana, conduttrice di protoni. Il pedice “o” indica la porzione dell’ATP sintasi che conferisce al complesso la sensibilità all’oligomicina, un inibitore della fosforilazione ossidativa. L’unità F1 è costituita da 5 tipi di catene polipeptidiche, mentre l’unità Fo è un segmento idrofobico che attraversa la membrana mitocondriale interna. Fo è il canale per i protoni del complesso ed è formato da 4 tipi di catene polipeptidiche. Fotografie al microscopio elettronico ad alta risoluzione hanno mostrato che il complesso F1-Fo è costituito ha la forma di un pomello, dove F1 è la testa globulare e Fo è la base, normalmente inserita nella 162 Capitolo IX membrana. Lo stelo che congiunge F1 e Fo è costituito da vari tipi di proteine. L’ATP sintasi è detta anche Fo F1-ATP asi. Il componente F1 è stato il primo ad essere identificato come essenziale per la fosforilazione ossidativa. Un F1 isolato non può sintetizzare l’ATP da ADP e Pi, ma catalizza invece l’idrolisi dell’ATP per cui è stato chiamato F1-ATP-asi. Quando F1 si riassocia a F0 la membrana riacquista la capacità di accoppiare il trasferimento protonico alla sintesi di ATP. L’ATP- sintasi catalizza la formazione di ATP da ADP ed ortofosfato: ADP3- + Pi2- = ATP4- + H2O I reali substrati dell’ATP-sintasi sono i complessi dell’ATP e dell’ADP con gli ioni Mg2+, come in tutte le reazioni conosciute di trasferimento dei gruppi fosforici. Meccanismo della reazione: anche se ormai è chiaro che il gradiente di protoni transmembrana fornisce l’energia per la sintesi di ATP, non è stato ancora chiarito come questa energia viene trasmessa all’ATP-sintasi. Questo enzima catalizza la conversione di ADP e Pi legati all’enzima in ATP, anch’esso legato all’enzima, in assenza di gradiente protonico. Sembra quindi che la reazione ADP + Pi = ATP + H2O sia facilmente reversibile, cioè che la variazione di energia libera della sintesi di ATP sulla superficie dell’enzima sia molto vicina allo zero. Poiché l’ATP resta saldamente legato al sito attivo, impedendo qualsiasi altro processo, la forza motrice protonica sembra fornire l’energia necessaria a dissociare l’ATP dall’enzima, consentendo di ricominciare un altro ciclo catalitico. Come meccanismo di reazione, un atomo di ossigeno terminale dell’ADP attacca l’atomo di fosforo dell’ortofosfato formando un intermedio pentacovalente che si dissocia poi in ATP ed acqua. Vediamo ora il meccanismo chimico che accoppia la il flusso protonico con la fosforilazione. Alla base sta il modello chemiosmotico, secondo il quale il trasferimento di elettroni lungo la catena respiratoria è accompagnato da un pompaggio di protoni attraverso la membrana mitocondriale interna, che porta ad una differenza di concentrazione di protoni e quindi ad un aumento del pH. Quindi la faccia che Fosforilazione ossidativa 163 guarda verso la matrice diventa alcalina rispetto al lato citosolico della membrana stressa. L’energia elettrochimica contenuta in questa differenza di concentrazione protonica e in questa separazione di carica, costituisce la forza motrice protonica che rappresenta la conservazione di parte dell’energia liberata dalle ossidazioni. Per pompare ioni contro il gradiente elettrochimico bisogna spendere energia e quindi il trasporto avrà un ∆G positivo. Quando i protoni fluiscono spontaneamente nella direzione del gradiente elettrochimico si avrà a disposizione una quantità di energia pari al ∆G consumato per il trasporto in senso contrario: questa è la forza elettromotrice protonica. La sintesi di ATP si ha quando il flusso protonico inverte la sua direzione e i protoni rientrano nella matrice attraverso un canale protonico associato all’ATP sintasi. In questa teoria, applicata ai mitocondri, gli elettroni provenienti dal NADH o da altri substrati ossidabili, passano attraverso una catena di trasportatori disposti in modo asimmetrico sulla membrana interna. Il flusso di elettroni è accompagnato da una traslocazione di protoni attraverso la membrana mitocondriale, che produce un gradiente chimico (∆pH) e un gradiente elettrico (∆ψ) . La membrana mitocondriale interna è impermeabile ai protoni e questi per rientrare devono attraversare i canali proteici specifici del complesso Fo. La forza motrice protonica che spinge i protoni verso la matrice fornisce l'energia necessaria per la sintesi di ATP catalizzata dal complesso F1 associato ad Fo. Ossidazione completa di una molecola di glucosio Vediamo ora quante molecole di ATP si formano quando il glucosio viene completamente ossidato a CO2. Durante la glicolisi ed il ciclo di Krebs si sono formate 2 molecole di ATP durante la glicolisi (4 formate – 2 consumate) e 2 durante il ciclo di Krebs (1 per 2 acetilCoA). Si ha quindi per ogni glucosio un guadagno netto di 4 ATP, risultato della fosforilazione a livello di substrato. La quantità di ATP prodotto durante la fosforilazione ossidativa è meno certa, perché più difficile da quantificare: infatti le stechiometrie della traslocazione dei protoni, della sintesi di ATP e del trasporto dei 164 Capitolo IX metaboliti non corrispondono necessariamente a numeri interi, né a valori fissi. E’ stato calcolato che durante il flusso di una coppia di elettroni dal NADH all’O2 si formano circa 2,5 ATP citosolici. Per gli elettroni che entrano a livello della citocromo reduttasi, quelli che provengono dall’ossidazione del succinato o dal NADH citosolico la resa è di circa 1,5 ATP per coppia di elettroni. Quindi, quando il glucosio viene completamente ossidato a CO2, si formano circa 30 ATP, anziché i tradizionali 36. La maggior parte dell’ATP (26 molecole su 30) viene generato dalla fosforilazione ossidativa. In condizioni fisiologiche standard il trasporto degli elettroni è strettamente accoppiato alla fosforilazione ossidativa. Normalmente gli elettroni non scorrono attraverso la catena di trasporto degli elettroni fino all’O2 se contemporaneamente l’ADP non viene fosforilato ad ATP. Occorre un rifornimento costante di NADH, di O2, di ADP e di Pi. La fosforilazione ossidativa quindi è accoppiata all’utilizzo dell’ATP. Gli elettroni non possono essere trasferiti dalle molecole di sostanze nutrienti all’O2 se non è richiesta la sintesi di ATP. Regolazione della fosforilazione ossidativa La velocità della respirazione (consumo di ossigeno) nei mitocondri è strettamente controllata; in generale è limitata dalla disponibilità di ATP come substrato per la fosforilazione. La dipendenza della velocità di consumo di O2 dalla concentrazione dell’accettore del gruppo fosforico, l’ ADP, chiamato controllo dell’accettore della respirazione, può essere quasi assoluta. La concentrazione intracellulare di ADP è una misura dello stato energetico della cellula. Un altro modo di valutare lo stato energetico della cellula è il rapporto di azione di massa del sistema ATP-ADP. Il rapporto è: ATP / [ADP] [Pi] Questo rapporto è normalmente molto alto e quindi il sistema ATPADP è generalmente fosforilato. La velocità di ossidazione delle sostanze nutrienti è regolata ed il rapporto d’azione di massa si modifica Fosforilazione ossidativa 165 di poco nella maggior parte dei tessuti, anche se la domanda energetica della cellula varia notevolmente. In sostanza, l’ATP viene formato solo alla velocità con cui viene consumato per le attività cellulari che necessitano di energia. Esiste comunque un’eccezione alla regola generale: la respirazione rallenta quando il rifornimento di ATP della cellula è sufficiente. In presenza di disaccoppianti, può darsi il caso che il trasporto di elettroni dal NADH all’ossigeno proceda normalmente, ma che non si formi ATP ad opera dell’ATP-asi mitocondriale perché la forza motrice protonica attraverso la membrana mitocondriale viene dissipata. Il disaccoppiamento della fosforilazione ossidativa può essere biologicamente importante perché ha la funzione di produrre calore per mantenere costante la temperatura del corpo di animali in ibernazione, in alcuni tipi di animali appena nati (incluso l’uomo) e nei mammiferi adattati ai climi freddi. La termogenina, proteina disaccoppiante presente nei mitocondri di un particolare tipo di tessuto adiposo (grasso bruno), forma una via per il passaggio di elettroni dallo spazio intermemebrana alla matrice cortocircuitando la batteria protonica dei mitocondri. La termogenina ossidando le sostanze nutrienti non produce ATP, ma calore che serve a mantenere il corpo a temperatura costante (ad es. neonati e animali in letargo). GLUCONEOGENESI E VIA DEI PENTOSI FOSFATI Il metabolismo del glucosio è essenziale per la produzione di energia nella cellula, pertanto è di fondamentale importanza che le concentrazioni di questo zucchero siano sempre a livelli sufficienti. Negli animali questa condizione è mantenuta mediante la sintesi e la degradazione del glicogeno, un polisaccaride di riserva costituito da glucosio. Nelle piante la funzione di riserva è compiuta dall’amido, anch’esso un polisaccaride costituito da molecole di α-glucosio legate fra loro con legami α-glucosidici. Le necessità del metabolismo cellulare possono però richiedere anche la biosintesi di nuovo glucosio proveniente da precursori non saccaridici, come: il piruvato, il lattato, il glicerolo, alcuni intermedi del ciclo dell’acido citrico e alcuni amminoacidi detti appunto gluconogenetici. Il processo che porta a questi composti di neoformazione si dice gluconeogenesi. Si tratta di un processo citosolico che ha una funzione essenziale per la sintesi del glucosio e dei carboidrati in genere (compreso il glicogeno) negli organismi non fotosintetizzanti. L’amido, presente nelle cellule vegetali è costituito da amilosio, un polisaccaride lineare (cioè non ramificato) e amilopectina, un polisaccaride ramificato. L’amilosio è solubile in acqua ed è costituito da circa 100 - 300 residui di glucosio connessi tra loro con legami 1,4 αglucosidici. Ha conformazione ad elica e forma un complesso di colore blu intenso con lo iodio. L’amilopectina è insolubile in acqua e contiene migliaia di residui di glucosio che nella catena principale sono connessi tra loro con legami 1,4 α-glucosidici e nelle catene laterali con legami 1,6 α-glucosidici. Il glicogeno è simile nella struttura all’amilopectina ma la sua molecola è più grande, più ramificata e più compatta. E’ il polisaccaride di riserva degli animali e costituisce il 10% della massa del fegato e l’1% della massa muscolare. La catena principale anche in questo caso è costituita da residui di glucosio connessi tra loro con legami 1,4 αglucosidici, con ramificazioni costituite da residui di glucosio legati con legami 1,6 α-glucosidici. 167 168 Capitolo X Il fabbisogno giornaliero di glicogeno per l’uomo adulto viene soddisfatto per la maggior parte dall’amido, abbondante ad esempio nelle patate, nei cereali e in alcuni frutti come le banane. Un’altra importante via del metabolismo del glucosio è la via dei pentosi-fosfati, che produce, come vedremo più avanti, sia monosaccaridi a cinque atomi di carbonio, che potere riducente sotto forma di NADPH che alimenta i processi biosintetici riduttivi. Si ricorda qui che il NADPH, distinto dal NADH per avere un gruppo fosforico sul C2 di una delle unità di ribosio, è il trasportatore di potere riducente nelle cellule. Nella maggior parte delle reazioni esiste una differenza fondamentale fra NADH e NADPH. Il NADH viene ossidato nella catena respiratoria di trasporto degli elettroni per generare ATP, mentre il NADPH serve come donatore di elettroni nelle biosintesi riduttive. La via dei pentosi fosfati produce, partendo dal glucosio, NADPH e ribosio-5-fosfato, quest’ultimo è il precursore per la biosintesi di acidi nucleici e nucleotidi. Altri zuccheri a cinque atomi di carbonio, e i loro polimeri, possono essere prodotti a partire dal ribosio tramite reazioni di isomerizzazione ed epimerizzazione. Esaminiamo ora in dettaglio le vie metaboliche alternative collegate al glucosio. Metabolismo del glicogeno Quando con la dieta non si assume abbastanza glucosio il glicogeno, accumulato come sostanza di riserva nel fegato e nei muscoli, viene idrolizzato e fornisce il glucosio necessario al metabolismo cellulare. La sintesi del glicogeno avviene praticamente in tutti i tessuti animali ma soprattutto nel fegato e nel muscolo scheletrico. Il glicogeno, nelle cellule animali, può essere sintetizzato soltanto tramite la via gluconogenetica ed è sempre a questa via che bisogna fare riferimento. Il punto di partenza è il glucosio-6-fosfato. La sintesi del glicogeno passa attraverso tre reazioni enzimatiche: 1. La fosfoglucomutasi trasforma il glucosio-6-fosfato in glucosio-1fosfato, molecola glucidica attivata. 2. Il glucosio-1-fosfato reagisce con l’UTP (Uridin Tri Fosfato) per dare UDP-glucosio e pirofosfato inorganico PPi, che si idrolizzerà Gluconeogenesi e via dei pentosi fosfati 169 a sua volta in due molecole di Pi. L’enzima è l’UDP-glucosio pirofosforilasi (Figura 1). Figura 1. Reazione di formazione di UDP-glucosio. 3. Altri residui glucidici provenienti dall’UDP-glucosio vengono aggiunti alle estremità non riducenti della catena. L’enzima è la glicogeno sintasi. La glicogeno sintasi ha bisogno come innesco (primer) di una catena di poliglucosio formata da circa sette residui. Questo primer viene prodotto da un enzima, la glicogenina, che lega il primo residuo di glucosio e funziona anche da catalizzatore per la glicogeno sintasi. Le catene ramificate del glicogeno vengono poi inserite con l’intervento dell’enzima ramificante, l’amilo (1, 4 – 1,6) transglicosilasi. 170 Capitolo X Figura 2. Fasi della biosintesi del glicogeno. La degradazione del glicogeno inizia da una fosforilasi, che catalizza l’attacco nucleofilo di un fosfato inorganico sui legami glicosidici α-1,4 che congiungono i residui di glucosio del glicogeno. Si libera così glucosio 1-fosfato e la catena si accorcia di un residuo. Questa reazione viene detta fosforolisi in quanto utilizza un fosfato nel meccanismo di rottura. La glicogeno fosforilasi non è però in grado di eliminare i residui di glucosio dalle ramificazioni con legami α−1,6. Interviene allora un enzima deramificante in grado di scindere specificatamente i legami α-1,6. Il glucosio 1-fosfato, prodotto terminale delle reazioni della glicogeno fosforilasi viene convertito in glucosio 6fosfato dalla fosfoglucomutasi. L’amido, nelle cellule vegetali, viene demolito da enzimi simili che catalizzano un processo analogo. La glicogeno-fosforilasi e l’amidofosforilasi nelle cellule delle piante sono gli enzimi che regolano il flusso della rimozione di unità di glucosio dal corrispondente polisaccaride. Gluconeogenesi e via dei pentosi fosfati 171 La gluconeogenesi Come ricordato sopra, la gluconeogenesi è la via attraverso la quale il glucosio può essere generato da precursori non glucidici, per esempio il piruvato, intermedi del ciclo di Krebs, alcuni amminoacidi, acido lattico e glicerolo. Il 90% della gluconeogenesi nei mammiferi avviene nelle cellule del fegato, il resto nelle cellule renali. Il glucosio è la fonte principale di energia per le cellule cerebrali e muscolari e la fonte primaria di energia per i globuli rossi. Il glucosio, introdotto con la dieta o proveniente dalle riserve di glicogeno del fegato si esaurisce dopo circa diciotto ore. A questo punto la gluconeogenesi diventa essenziale per la sopravvivenza. La gluconeogenesi può avvenire anche in alcuni microrganismi sviluppati in assenza di glucosio. Nelle piante la gluconeogenesi si trova principalmente nel ciclo del gliossilato e nella via metabolica delle piante CAM. La gluconeogenesi trasforma il piruvato in glucosio, mentre la glicolisi compie il cammino inverso, trasformando il glucosio in piruvato. La gluconeogenesi non è però la via inversa della glicolisi, infatti l’equilibrio netto nella glicolisi favorisce la formazione di piruvato. Delle dieci tappe che trasformano il glucosio in piruvato nella glicolisi, sette fanno parte, in senso inverso, della gluconeogenesi. Le altre tre sono irreversibili e sono quelle catalizzate da: esochinasi che converte il glucosio in glucosio 6-fosfato; fosfofruttochinasi per la fosforilazione del fruttosio 6-fosfato in fruttosio 1,6-bisfosfato e piruvato chinasi che converte il fosfoenolpiruvato in piruvato. Queste tappe vengono aggirate dalla gluconeogenesi con un diverso pool di enzimi. Il confronto tra glicolisi e gluconeogenesi è illustrato in Figura 3 dove le reazioni e gli enzimi caratteristici della gluconeogenesi sono illustrati in rosso, mentre quelli della glicolisi in nero. Le reazioni reversibili che rimangono in comune sono indicate in verde. 172 Capitolo X Figura 3. Glicolisi e gluconeogenesi a confronto. Gluconeogenesi e via dei pentosi fosfati 173 Gli enzimi che sono propri ed esclusivi della gluconeogenesi sono quattro: 1. La piruvato carbossilasi, che trasforma il piruvato in ossalacetato; 2. La fosfoenolpiruvato carbossichinasi (PEP carbossichinasi), che trasforma l’ossalacetato in fosfoenolpiruvato; 3. La fruttosio 1,6-bisfosfatasi, che trasforma il fruttosio 1,6bisfosfato (F1,6BP) in fruttosio 6–fosfato (F6P); 4. La glucosio 6-fosfatasi che trasforma il glucosio 6-fosfato (G6P) in glucosio. La gluconeogenesi richiede 4 ATP, 2 GTP e 2 NADH per la resintesi del glucosio. La stechiometria della gluconeogenesi è: 2 piruvato + 4 ATP + 2 GTP + 2 NADH + 6 H2O → glucosio + 4 ADP + 2 GDP + 6 Pi + 2 NAD+ + 2 H+ E’ importante notare che nella gluconeogenesi vengono idrolizzate sei molecole di nucleoside trifosfato (ATP e GTP), mentre nella glicolisi vengono prodotte soltanto due molecole di ATP nella conversione di glucosio a piruvato. Perciò il costo energetico della biosintesi di glucosio è pari a quattro molecole ad alto potenziale di trasferimento del gruppo fosforico. Vediamo ora in particolare le singole reazioni: 1) La piruvato carbossilasi trasforma il piruvato in ossalacetato. Questa trasformazione avviene nei mitocondri ed è catalizzata dalla piruvato carbossilasi, che ha come coenzima la biotina. La reazione si può dividere in due fasi: nella prima la biotina reagisce con la CO2 o meglio col bicarbonato attivo (che non è altro che HCO3- che deriva dalla CO2 sciolta in acqua) e forma la carbossibiotina. In questa fase il trasferimento di un gruppo fosfato dall’ATP al bicarbonato produce un intermedio, un carbossifosfato attivo, e ADP. Nella seconda fase, il trasferimento del gruppo carbossilico dalla carbossibiotina al C2 del piruvato produce ossalacetato. piruvato + HCO3- + ATP → ossalacetato + ADP + Pi 174 Capitolo X 2) L’ossalacetato viene trasportato all’esterno del mitocondrio da una coppia di deidrogenasi. La trasformazione del piruvato in ossalacetato avviene infatti nei mitocondri, mentre gli altri enzimi della gluconeogenesi stanno nel citosol. La PEP-carbossichinasi è citosolica in alcune specie e mitocondriale in altre. La membrana mitocondriale interna invece è impermeabile all’ossalacetato. Essa può essere superata solo dall’azione della malato deidrogenasi mitocondriale e della malato deidrogenasi citosolica. La prima trasforma l’ossalacetato in malato a spese del NADH e lo trasporta all’esterno del mitocondrio. ossalacetato + NADH + H+ = malato + NAD+ La seconda malato deidrogenasi rigenera quindi l’ossalacetato che può essere usato successivamente nella gluconeogenesi, con la contemporanea produzione di NADH citosolico. malato + NAD+ = ossalacetato + NADH + H+ 3) La PEP-carbossichinasi trasforma l’ossalacetato in fosfoenolpiruvato (PEP). Quando l’ossalacetato è stato rigenerato nel citosol la PEP-carbossichinasi catalizza la trasformazione, GTP-dipendente, dell’ossalacetato in piruvato. Questo passaggio richiede l’idrolisi netta di una molecola di ATP e di una di GTP. piruvato + ATP + GTP + H2O = PEP + ADP + GDP + Pi Da qui la reazione prosegue con le reazioni inverse della glicolisi e con gli stessi enzimi, fino al fruttosio 1,6 bisfosfato. 4) Interconversione del fruttosio 6-fosfato e del fruttosio 1,6bisfosfato è il punto più importante e lo stadio chiave della regolazione della gluconeogenesi. Gli enzimi che catalizzano questa reazione sono la fruttosio 1,6-bisfosfatasi e la fosfofruttochinasi (PFK 1). L’attività delle PFK1 e della fruttosio 1,6-bisfosfatasi sono regolate dall’effettore allosterico (cioè lontano dal sito attivo dell’enzima) fruttosio 2,6-bisfosfato (F 2,6 BP), che nella glicolisi Gluconeogenesi e via dei pentosi fosfati 175 attiva la PFK1 e inibisce la fruttosio-1,6-bifosfatasi, favorendo così la prima rispetto alla seconda, mentre nella gluconeogenesi prevale la fruttosio-1,6-bifosfatasi, che catalizza l’idrolisi, essenzialmente irreversibile, del gruppo fosforico sul C1: fruttosio 1,6-bisfosfato + H2O = fruttosio 6-fosfato + Pi 5) La glucosio 6-fosfatasi trasforma il glucosio 6-fosfato in glucosio libero e Pi (fosfato inorganico). glucosio 6-fosfato + H2O = glucosio + Pi L’insieme delle reazioni biosintetiche che portano dal piruvato al glucosio, è un processo costoso e molta dell’energia libera necessaria per rendere possibile la gluconeogenesi rende la stessa un processo irreversibile. La via biosintetica della gluconeogenesi consente una sintesi netta di glucosio non solo dal piruvato ma anche dagli intermedi del ciclo dell’acido citrico: citrato, isocitrato, α-chetoglutarato, succinil-CoA, succinato, fumarato e malato. Tutti questi composti vengono ossidati nel ciclo dell’acido citrico e trasformati in ossalacetato. Nei semi in germinazione la gluconeogenesi converte grassi e proteine in glucosio. Abbiamo già trattato questo argomento nella via del gliossilato nel capitolo dedicato al ciclo di Krebs, tuttavia non sarà inutile qualche altra precisazione. Al contrario degli animali, le piante ed alcuni microrganismi possono convertire in glucosio l’acetil-CoA che proviene dall’ossidazione degli acidi grassi. Parte degli enzimi essenziali per questa trasformazione sono raggruppati nei gliossisomi, organelli in cui gli acidi grassi sono ossidati ad acetil-CoA mediante enzimi della β-ossidazione specifici dei gliossisomi. Attraverso il ciclo del gliossilato l’acetil-CoA viene convertito in succinato, che si trasferisce nella matrice mitocondriale e diventa ossalacetato nelle reazioni del ciclo dell’acido citrico. L’ossalacetato, una volta entrato nel citosol, viene trasformato in fruttosio 6-fosfato, precursore diretto del saccarosio, zucchero utilizzato come sistema di trasporto per gli atomi di carbonio nelle piante. Capitolo X 176 L’idrolisi dei triacilgliceroli di deposito produce anche glicerolo 3fosfato che può entrare nella via gluconogenetica dopo essere stato ossidato a diidrossiacetonefosfato e trasformato in fruttosio 6-fosfato oppure in saccarosio. Via del pentosio fosfato La via del pentosio fosfato, detta anche via del gluconolattone o via del fosfogluconato, produce NADPH e ribosio 5-fosfato. Il NADPH è un trasportatore di energia chimica sotto forma di potere riducente e rappresenta il riducente universale nelle vie anaboliche. Una seconda funzione di questa via è quella di generare pentosi essenziali, in particolare il D-ribosio, usati nella biosintesi degli acidi nucleici. La via dei pentosi fosfati può essere suddivisa in due stadi: uno ossidativo e uno non ossidativo. Nello stadio ossidativo, la prima reazione della via del pentosio fosfato è la deidrogenazione enzimatica del glucosio 6-fosfato ad opera della glucosio 6-fosfato deidrogenasi per dare 6-fosfogluconolattone, un estere intramolecolare. L’accettore degli elettroni è il NADP+ e l’equilibrio complessivo è spostato a destra. Il lattone viene quindi idrolizzato a 6-fosfogluconato ad opera di una specifica lattonasi. Nella tappa successiva il 6-fosfogluconato viene ancora deidrogenato e decarbossilato ad opera della 6-fosfogluconato deidrogenasi con produzione del chetopentosio ribulosio 5-fosfato, reazione che produce una seconda molecola di NADPH. Da questa tappa in poi inizia la fase non ossidativa. La fosfopentosio isomerasi converte il ribulosio 5-fosfato nell’isomero ribosio 5fosfato (un aldosio). In alcuni tessuti la reazione termina qui e l’equazione complessiva è: glucosio 6-fosfato + 2 NADP+ + H2O → ribosio 5-fosfato + CO2 + NADPH + 2 H+ Il risultato netto è la produzione di NADPH necessario per le reazioni di sintesi e di ribosio-5-fosfato, un precursore della sintesi dei nucleotidi. Gluconeogenesi e via dei pentosi fosfati 177 Nella parte non ossidativa della via del pentosio fosfato la transchetolasi, un enzima TPP-(tiamina pirofosfato) dipendente catalizza il trasferimento di un frammento a due atomi di carbonio (gli atomi C1 e C2) dello xilulosio 5-fosfato al ribosio 5-fosfato, formando un prodotto a sette atomi di carbonio, il sedoeptulosio 7-fosfato. I restanti tre atomi di carbonio dello scheletro dello xilulosio escono sotto forma di gliceraldeide-3-fosfato. La transaldolasi catalizza poi una reazione simile a quella dell’aldolasi nella glicolisi: dal sedoeptulosio 7-fosfato viene rimosso un frammento a tre atomi di carbonio e condensato con la gliceraldeide 3-fosfato, formando fruttosio 6-fosfato. I rimanenti quattro atomi di carbonio del sedoeptulosio 7-fosfato escono sotto forma di eritrosio 4-fosfato. La transchetolasi agisce ancora formando fruttosio 6-fosfato e gliceraldeide 3-fosfato, partendo da eritrosio 4fosfato e xilulosio 5- fosfato. Due molecole di gliceraldeide ricavata dalla ripetizione di queste reazioni possono essere convertite in fruttosio bisfosfato attraverso l’intermedio fruttosio 1,6 bisfosfato. Tutte le reazioni della parte non ossidativa della via del pentosio fosfato sono facilmente reversibili e quindi rappresentano anche un modo di convertire esosi fosfati in pentosi fosfati. I fabbisogni relativi di NADPH, di ribosio-5-fosfato e di ATP determinano il destino del glucosio-6-fosfato La glucosio 6-fosfato deidrogenasi è l’enzima chiave che regola la via del pentosio fosfato. Una delle sue funzioni metaboliche principali è la produzione di NADPH che viene utilizzato nei processi biosintetici. Il NADPH è un potente inibitore della glucosio 6-fosfato deidrogenasi, un suo lieve aumento nella concentrazione cellulare rallenta notevolmente la velocità delle reazioni della via dei pentosi fosfati. La biosintesi degli acidi grassi utilizza NADPH e quindi la glucosio 6fosfato deidrogenasi diventa più attiva. Invece, i prodotti intermedi della biosintesi degli acidi grassi , come le molecole di acil-CoA inibiscono la glucosio 6-fosfato deidrogenasi, legandosi a questo dimero e trasformandolo in due monomeri inattivi. Quando il fabbisogno di NADPH è maggiore della domanda di ribulosio 5-fosfato, il glucosio 6-fosfato può essere completamente ossidato a CO2. Per ogni molecola di glucosio 6-fosfato che viene trasformata in ribulosio 5-fosfato lo stadio ossidativo della via dei pentosi fosfati produce 2 NADPH. A loro volta, 6 molecole di ribulosio 6- 178 Capitolo X fosfato (prodotto da 6 molecole di glucosio 6-fosfato) possono essere ritrasformate in cinque molecole di glucosio 6-fosfato attraverso una combinazione di reazioni non ossidative della via del pentosio fosfato e di reazioni della gluconeogenesi a partire dalla reazione catalizzata dalla triosofosfato isomerasi. Se il fabbisogno di NADPH e di ATP è elevato, il NADPH viene prodotto nello lo stadio ossidativo della via del pentosio fosfato. Il fruttosio 6-fosfato e la gliceraldeide 3-fosfato prodotti nello stadio non ossidativo della stessa via vengono trasformati in piruvato attraverso le reazioni della glicolisi, con produzione di ATP. Quando due moli di NADPH per ogni mole di ribosio 5-fosfato bastano a soddisfare il bisogno cellulare il glucosio 6-fosfato viene trasformato in ribosio 5-fosfato attraverso lo stadio ossidativo della via del pentosio fosfato, con produzione di NADPH. Se il NADPH non è necessario, attraverso la glicolisi il glucosio 6fosfato viene trasformato in fruttosio 6-fosfato e gliceraldeide 3fosfato. La transchetolasi e la transaldolasi catalizzano reazioni facilmente reversibili e quindi fruttosio 6-fosfato e gliceraldeide 3-fosfato possono essere trasformati in ribosio 5-fosfato e xilulosio 5-fosfato. Quest’ultimo può essere a sua volta trasformato in ribosio-5-fosfato attraverso l’azione della ribulosio 5-fosfato epimerasi e della ribulosio 5-fosfato isomerasi. La reazione netta è dunque: 5 G6P + ATP = 6 Ribosio-5P + ADP LA FOTOSINTESI Molti processi cellulari producono energia libera attraverso l’ossidazione dei carboidrati, come glucosio e glicogeno, tramite reazioni esoergoniche. La sintesi dei carboidrati invece è endoergonica, cioè richiede energia. La fonte prima dell’energia libera per la sintesi dei carboidrati è il sole, che irradia energia, poi trasformata in energia libera immagazzinata nei carboidrati attraverso la fotosintesi. La cattura dell’energia solare da parte di organismi fotosintetici (piante, alghe, cianobatteri, batteri purpurei e batteri verdi) e la sua conversione nell’energia chimica di composti organici ridotti è la fonte di tutta l’energia biologica. Gli organismi fotosintetici intrappolano l’energia solare e formano ATP e NADPH, che vengono poi utilizzati per la produzione di carboidrati ed altri composti organici da CO2 ed acqua, rilasciando contemporaneamente O2 nell’atmosfera. Gli organismi eterotrofi aerobici useranno poi l’ossigeno per degradare i prodotti organici ricchi di energia generati nella fotosintesi, producendo CO2, acqua e l’ATP necessario alle loro varie attività. La CO2 formata dagli eterotrofi durante la respirazione ritorna così nell’atmosfera e potrà essere utilizzata dagli organismi fotosintetici. L’energia solare fornisce quindi la forza trainante per il continuo riciclaggio della CO2 e dell’ O2 nella biosfera e produce substrati ridotti, sostanze nutrienti come il glucosio, da cui dipendono gli organismi non fotosintetici. Si suppone che la proliferazione dei cianobatteri capaci di produrre O2 attraverso la fotosintesi abbia portato alla trasformazione dell’atmosfera terrestre da riducente ad ossidante. L’evoluzione di questi batteri, fra i 2,5 ed i 3,2 miliardi di anni fa è stato uno degli avvenimenti più importanti nella storia della vita sulla Terra. L’equazione complessiva della fotosintesi è una reazione di ossidoriduzione in cui l’acqua dona elettroni e atomi di H per ridurre la CO2 a carboidrati (CH2O)n, principalmente saccarosio ed amido. La reazione può essere così schematizzata: n CO2+ n H2O + Energia della luce = n O2 + (CH2O)n 179 180 Capitolo XI Il meccanismo della fotosintesi è complesso e richiede l’interazione di molte proteine e piccole molecole. Nelle piante verdi la fotosintesi si svolge in particolari strutture intracellulari dette cloroplasti (Figura 1). L’energia catturata da molecole di pigmenti, le clorofille, viene utilizzata nei cloroplasti per generare elettroni ad alta energia, cioè con elevato potere riducente. Figura 1. Cloroplasti al microscopio Questi elettroni vengono usati per produrre ATP e NADPH in una serie di reazioni dette reazioni alla luce o fase luminosa della fotosintesi. Questi composti riducono poi il biossido di carbonio e lo convertono dapprima in 3-fosfoglicerato e successivamente in due triosi fosfati isomeri, che daranno luogo alla formazione di fruttosio-6-fosfato da cui deriveranno gli altri mono-, di- e poli-saccaridi. Questa serie di reazioni è nota come Ciclo di Calvin-Benson, e le reazioni prendono il nome di reazioni al buio o fase oscura. La quantità di energia immagazzinata dalla fotosintesi è enorme: sulla terra vengono immagazzinate annualmente dalla fotosintesi più di 1017 Kcal di energia libera, che corrispondono all’assimilazione di più di 1010 tonnellate di carbonio in carboidrati e in altre forme di materia organica. La fotosintesi 181 La fotosintesi è la fonte di quasi tutti i composti del carbonio e di tutto l’ossigeno che rende possibile il metabolismo aerobico. Inoltre esistono paralleli sia in termini di meccanismi che di evoluzione fra le reazioni alla luce delle fotosintesi e le tappe della fosforilazione ossidativa. Il processo fotosintetico si svolge in due fasi distinte: 1- Reazioni alla luce. L’energia della luce viene catturata e conservata temporaneamente sotto forma di ATP e NADPH (Fotofosforilazione ciclica e non ciclica). Le molecole di acqua vengono scisse in O2, elettroni e ioni H+ (Fotolisi dell’acqua); 2– Reazioni al buio o ciclo di Calvin . L’energia chimica sottoforma di ATP e NADPH viene utilizzata per la sintesi di carboidrati a partire da CO2 e H2O. Nelle cellule eucariotiche fotosintetiche sia le reazioni della fase luminosa sia l’assimilazione del carbonio della fase oscura, avvengono nei cloroplasti. Il cloroplasto, come il mitocondrio, ha una membrana esterna, una membrana interna ed uno spazio intermembrana. La membrana interna racchiude lo stroma, il sito in cui avvengono le reazioni della fase oscura della fotosintesi. Nello stroma sono presenti strutture membranose dette tilacoidi, che hanno la forma di sacchi appiattiti o dischi, impilati a formare delle strutture chiamate grani. I vari grani sono collegati fra loro da regioni della membrana tilacoidale dette lamelle stromali (Figura 2). Figura 2. Struttura schematica di un cloroplasto. 182 Capitolo XI Le membrane tilacoidali separano lo spazio tilacoidale da quello dello stroma. Quindi i cloroplasti hanno tre tipi di membrana: la membrana esterna, la membrana interna e la membrana tilacoidale. Conseguentemente si possono individuare tre spazi distinti: lo spazio intermembrana, lo spazio stromale e lo spazio tilacoidale. Si pensa che nei cloroplasti i tilacoidi si originino da invaginazioni della membrana interna, analogamente alla formazione delle creste nei mitocondri. Come le creste mitocondriali i tilacoidi sono il sito di reazioni di ossidoriduzione accoppiate, che generano una forza motrice protonica. Le membrane tilacoidali contengono il meccanismo di trasduzione dell’energia, che è composto da: complessi proteine-pigmenti per la cattura della luce, centri di reazione, catene di trasporto degli elettroni e ATP-sintasi. Sono costituite da proteine e da lipidi, in massima parte galattolipidi (45%), per il 10% fosfolipidi e per il 4% solfolipidi. A differenza della membrana esterna, sia la membrana tilacoidale che quella interna sono impermeabili alla maggior parte delle molecole e degli ioni. Nello stroma si trovano gli enzimi che convertono la CO2 in zucchero. Le cellule foliari delle piante contengono da 1 a 100 cloroplasti, a seconda della specie,del tipo di cellula e delle condizioni di crescita. I pigmenti fotosintetici Lo strato di ozono, nell’atmosfera, è in grado di assorbire quasi tutte le radiazioni ultraviolette emesse dal sole; pertanto, le radiazioni luminose che raggiungono la crosta terrestre si trovano per la maggior parte nella regione visibile dello spettro elettromagnetico (400-700 nm) e si estendono nell’infrarosso (700-106 nm). Oltre la metà dell’energia di origine solare che raggiunge la crosta terrestre si trova in questa parte dello spettro. La cattura dell’energia luminosa è essenziale per la fotosintesi. Il primo passo è l’assorbimento della luce da parte di pigmenti, localizzati nelle membrane tilacoidali delle piante superiori e nelle membrane plasmatiche dei batteri fotosintetizzanti. I due pigmenti principali nelle piante superiori, nelle alghe e nei cianobatteri sono la clorofilla a (Cl a), più abbondante, e la clorofilla b (Cl b). La Cl b e differisce dalla a per la presenza di un gruppo formile (-CHO) al posto del gruppo metile (-CH3). Nei batteri fotosintetizzanti La fotosintesi 183 (ad eccezione dei cianobatteri) i principali pigmenti sono la batterioclorofilla a (BCl a) e la batterioclorofilla b (BCl b). Figura 3. Struttura base della clorofilla. La clorofilla è costituita da un sistema policiclico porfirinico e può anche essere descritta come un tetrapirrolo sostituito con al centro uno ione Mg+2 (Figura 3). Questa struttura ha come precursore la protoporfirina IX e quindi presenta delle analogie con le strutture dell’emoglobina e della mioglobina. Caratteristica della clorofilla è la presenza del fitolo, un alcol a 20 atomi di carbonio molto idrofobico, esterificato con una catena laterale acida. La lunga coda alifatica del fitolo favorisce l’ancoraggio della clorofilla nell’ambiente idrofobico della membrana tilacoidale in cui si trova, mentre l’anello tetrapirrolico è polare. 184 Capitolo XI Le clorofille sono molto efficienti perché contengono molti polieni, una serie di doppi legami coniugati (legami singoli e doppi alternati), che hanno bande di assorbimento molto elevate nella regione visibile dello spettro. Oltre alle clorofille gli organismi fotosintetizzanti contengono altri pigmenti, i pigmenti accessori, che sono anch’essi in grado di assorbire la luce: i carotenoidi e le ficobiline. I carotenoidi si distinguono in caroteni e xantofille e sono sempre presenti. I caroteni sono idrocarburi di tipo isoprenoide, a 40 atomi di C, costituiti da otto molecole di isoprene (2-metil-1,3-butadiene). Poiché due molecole di isoprene costituiscono un monoterpene, i caroteni possono dunque essere definiti anche come tetraterpeni. Si diversificano l’uno dall’altro soprattutto nelle parti terminali, costituite da anelli cicloesenici. Le xantofille sono analoghe ai caroteni, ma a differenza di essi contengono atomi di ossigeno negli anelli terminali. Alcune alghe e i cianobatteri contengono anche le ficobiline, che hanno struttura tetrapirrolica lineare e i cui costituenti principali sono la ficoeritrina, il pigmento delle alghe rosse e la ficocianina, pigmento delle alghe blu-verdi. Le clorofille a e b sono entrambe verdi, assorbono entrambe le radiazioni luminose nelle regioni del blu (λ max 424-491 nm) e del rosso (λ max 647-700 nm) (Figura 4). Figura 4. Assorbimento della luce visibile da parte di fotopigmenti. La fotosintesi 185 I loro massimi di assorbimento non sono uguali perché le loro strutture sono leggermente diverse. Gli spettri di assorbimento dei pigmenti accessori coprono invece tutto lo spettro del visibile. Il numero, il tipo e la quantità dei pigmenti con i loro diversi assorbimenti rispetto alla clorofilla conferiscono il colore caratteristico agli organismi fotosintetizzanti. I carotenoidi inoltre, oltre ad assorbire la luce hanno funzione protettiva verso la clorofilla. Durante la fotosintesi infatti, per assorbimento di energia, l’O2 molecolare può essere trasformato in una forma molto reattiva, l’ossigeno singoletto che, se persistesse nel cloroplasto, ossiderebbe i lipidi di membrana e altre molecole di pigmenti. I carotenoidi assorbono la sua energia, dissipandola poi sotto forma di calore. Le reazioni alla luce Fotofosforilazione non ciclica I pigmenti fotosintetici che assorbono la luce sono disposti in gruppi funzionali detti fotosistemi, che catalizzano la conversione dell’energia contenuta nella luce (fotoni) in forme utili di energia. I fotosistemi sono formati principalmente da due componenti: i complessi antenna e i centri di reazione fotochimici. Questi fotosistemi non sono uguali e lavorano in serie. I complessi antenna, detti anche LHC (Light Harvesting Complex). sono costituiti da pigmenti in grado di assorbire la luce e trasferire l’energia verso il centro di reazione. In Figura 5 viene schematizzato il trasferimento dell’energia luminosa dai pigmenti accessori al centro di reazione, i cerchi verdi rappresentano molecole di clorofilla e i cerchi arancio molecole di carotenoidi. 186 Capitolo XI Figura 5. Flusso di energia in un complesso antenna. Il centro di reazione è la parte fondamentale del fotosistema ed è costituito da un complesso di proteine, molecole specializzate per il trasporto di elettroni e da una coppia di molecole di clorofilla a, dette doppietto speciale. Tutti i pigmenti di un fotosistema possono assorbire la luce (sotto forma di fotoni, cioè di quanti di energia luminosa), ma solo il doppietto speciale può partecipare direttamente alla trasformazione di energia fotochimica in energia chimica. Così perché la fotosintesi possa svolgersi, i pigmenti del complesso antenna devono assorbire energia luminosa e trasferirla al dimero di clorofilla a presente nel centro di reazione. Nel primo stadio del processo, un pigmento del complesso antenna assorbe un fotone e passa così molto rapidamente nello stato eccitato (eccitone). L’energia dell’eccitone è poi trasferita in modo casuale ad altri pigmenti del complesso antenna e procede lungo il gradiente di potenziale fino al doppietto speciale costituito dalle due molecole di clorofilla a del centro di reazione. Una volta raggiunto il centro di reazione il processo di trasferimento dell’eccitazione viene interrotto, in quanto le molecole di clorofilla qui presenti, anche se sono chimicamente identiche alle molecole di clorofilla antenna, hanno un’energia dello stato eccitato leggermente più bassa, per il diverso ambiente molecolare in cui sono immerse. La fotosintesi 187 Uno schema del trasferimento di energia di un fotone assorbito dalle clorofille antenna è riportato in Figura 6. Le fasi di questo processo secondo quanto riportato nello schema sono le seguenti: 1) Un fotone colpisce una molecola di clorofilla (o di pigmento accessorio) portando un elettrone a un livello energetico più alto (*); 2) La molecola antenna eccitata passa l’energia a una molecola di clorofilla vicina, eccitandola. La prima torna allo stato basale pronta ad assorbire un altro fotone; 3) Questi due primi passaggi si possono ripetere diverse volte fino a che l’energia raggiunge il doppietto speciale del centro di reazione che viene eccitato; 4) La clorofilla eccitata del centro di reazione trasferisce un elettrone ad un accettore vicino che fa parte della catena di trasporto degli elettroni dei cloroplasti. La molecola di clorofilla rimane quindi con un orbitale vuoto e l’accettore acquista una carica negativa; 5) L’elettrone perso dalla clorofilla viene rimpiazzato da un elettrone fornito da un donatore di elettroni che diviene carico positivamente. In questo modo l’eccitazione prodotta dalla luce sugli elettroni delle clorofille antenna causa una separazione di carica a livello del centro di reazione e dà inizio ad una serie di reazioni di ossidoriduzione che vedremo in seguito. Nelle piante verdi la fotosintesi è mediata da due tipi di complessi fotosensibili all’interno della membrana tilacoidale: il fotosistema II (PSII) ed il fotosistema I (PSI). Ognuno con il proprio tipo di centro di reazione e il proprio insieme di molecole antenna. Il rapporto numerico tra PSI e PSII varia a seconda della pianta e della quantità di luce che riceve, ma si ritiene che il PSII sia più abbondante del PSI. La membrana tilacoidale contiene, oltre ai PSII e PSI, due altri complessi multiproteici di membrana importanti per la fotosintesi, il complesso del citocromob6f che pompa protoni durante il trasporto degli elettroni e l’ATP sintetasi del cloroplasto, che catalizza la sintesi di ATP durante la fotofosforilazione ciclica. 188 Capitolo XI Figura 6. Schema del trasferimento di energia di un fotone assorbito. I due fotosistemi sono localizzati in regioni diverse della membrana tilacoidale e sono collegati tra loro da una catena di trasporto di elettroni. Il processo di trasporto degli elettroni richiede una serie di reazioni di ossido-riduzione ogni volta che un elettrone (o uno ione idruro, H-) viene scambiato. La tendenza in una coppia redox ad acquistare o cedere elettroni è in relazione ai relativi valori del potenziale redox standard (Eo’). Un composto è un forte ossidante se presenta un valore alto e positivo di Eo’, valori alti e negativi contraddistinguono i forti riducenti. Anche la variazione di energia libera e di conseguenza la spontaneità di una reazione, è in relazione con il potenziale redox. Sa- La fotosintesi 189 ranno spontanee quelle reazioni in cui gli elettroni vengono trasferiti da coppie con valori di potenziale redox più negativi a valori più positivi (o meno negativi). Nelle piante i due fotosistemi agiscono in coppia per catalizzare il trasporto, indotto dalla luce, degli elettroni dall’H2O al NADP+, permettendo la riduzione del NADP+ a NADPH + H+ , la produzione di ossigeno e la formazione di un gradiente di protoni attraverso la membrana tilacoidale che darà origine all’ATP. 2 H2O + 2 NADP+ + 8 fotoni → O2 + 2 NADPH + 2 H+ Il potenziale di riduzione del centro di reazione del PSII è più positivo di quello del PSI e quindi gli elettroni non possono passare spontaneamente dal fotosistema I al fotosistema II. Allo stesso modo gli elettroni non possono fluire spontaneamente dal centro di reazione del PSI non eccitato al NADP+ che si trova ad un livello più elettronegativo. Per superare questa barriera energetica le clorofille dei centri di reazione passano gli elettroni da un potenziale di riduzione più positivo ad uno più negativo (reazione non spontanea) tramite la conversione di energia luminosa in energia chimica. Per rimpiazzare gli elettroni che si muovono dal PSII attraverso il PSI fino al NADP+, i cianobatteri e le piante superiori ossidano H2O producendo O2. Questo processo si chiama fotosintesi ossigenica per distinguerlo dalla fotosintesi non ossigenica dei rodobatteri e dei solfobatteri verdi che ossidano H2S. Tutte le cellule fotosintetiche che sviluppano O2 (quelle delle piante superiori, delle alghe e dei cianobatteri) contengono sia il PSII che il PSI. Gli organismi che non sviluppano ossigeno hanno un solo fotosistema. Diamo adesso una descrizione del processo complessivo iniziando dall’assorbimento di un fotone da parte del PSII. Il fotosistema II è un complesso pigmento-proteina posto nella membrana tilacoidale e localizzato quasi esclusivamente nelle lamelle dei grani (quindi non in contatto con lo stroma) e contiene approssimativamente quantità equivalenti di clorofille a e b (LHCII). Le due molecole di clorofilla del suo centro di reazione (il doppietto speciale) hanno un massimo di assorbimento a 680 nm e quindi viene abbrevia- 190 Capitolo XI to in P680. L’energia convogliata dalle clorofille antenna sul P680 viene utilizzata per catalizzare una serie di reazioni di trasferimento di elettroni, che vengono poi rimpiazzati dalla scissione dell’acqua in ossigeno molecolare (O2) e protoni (H+). Sempre associato con il PSII, sul lato della membrana tilacoidale rivolto verso il lume, troviamo un complesso di tre proteine estrinseche che, insieme ad un complesso a quattro atomi di manganese (il centro manganese), è coinvolto nella reazione di scissione dell’acqua. L’elettrone viene quindi trasferito dal P680* ad una molecola di feofitina (Pheo) dandole una carica negativa (Pheo-). La feofitina chimicamente è uguale alla clorofilla, ma con due protoni al posto dell’atomo di magnesio. Dalla Pheo- l’elettrone viene trasferito rapidamente ad una molecola di plastochinone (QA), che diviene un semichinone QA-. Questo non viene ulteriormente ridotto, ma trasferisce l’elettrone ad una seconda molecola di plastochinone (QB). P680* Pheo → P680+ Pheo- → P680+ Pheo- + QA QB → P680+ Pheo + QA- QB → QAQBAl contrario di QA- , il semichinone QB- può accettare un secondo elettrone e con l’arrivo di due protoni (H+) dallo stroma, il plastochinone viene ridotto a QBH2 (plastochinolo). QA-QB- → QAQB2- + 2 H+ → QA QBH2 La molecola di plastochinolo quindi diffonde nella regione idrofobica della membrana dove cede i due elettroni e i due protoni ad un pool di plastochinoni mobili (PQ). Con questa reazione di formazione del PQH2 lo schema di trasporto non ciclico degli elettroni lascia il PSII, ma prima di passare alla descrizione del percorso verso il PSI occorre fare un passo indietro per vedere più nel dettaglio la razione di fotolisi dell’acqua. La reazione globale del PSII è: 4 P680 + 4 H+ + 2 PQB + 4 fotoni = 4 P680+ + 2 PQH2. La fotosintesi 191 A questo si aggiunge la contemporanea reazione di scissione di due molecole di acqua che porta alla produzione di quattro ioni idrogeno e una molecola di ossigeno, la reazione di fotolisi dell’acqua. Fotolisi dell’acqua Come abbiamo già visto, il P680+ del PSII dopo aver ceduto un elettrone alla Pheo, deve acquistare un elettrone per ritornare allo stato iniziale ed essere pronto per ricevere un altro fotone. Per poter utilizzare l’energia della luce, si è dovuto evolvere un sistema capace di ottenere elettroni da un donatore facile da reperire e da scomporre, cioè l’acqua. Nel processo vengono scisse due molecole d’acqua liberando 4 protoni, 4 elettroni e una molecola di ossigeno. 2 H2O = 4H+ + 4 e- + O2 Poiché un solo fotone non ha abbastanza energia per rompere i legami dell’acqua, per questa reazione di fotolisi (cioè di rottura della molecola d’acqua sotto l’azione della luce) sono necessari 4 fotoni. I 4 elettroni provenienti dall’acqua non passano direttamente al P680+, che può accettare solo un elettrone per volta, ma occorre la presenza di un complesso che libera ossigeno (Il centro manganese o complesso che scinde l’acqua) che trasferisce gli elettroni, uno alla volta, al P680+. Il centro manganese, detto anche OEC (Oxygen Evolving Complex), contiene quattro ioni manganese, uno ione calcio, uno ione cloruro e un residuo di tirosina, ma la sua la struttura dettagliata non è ancora conosciuta. Il manganese forse è stato selezionato nel corso dell’evoluzione per la capacità di esistere in molteplici stati di ossidazione (Mn2+, Mn3+ Mn4+ Mn5+) e di formare forti legami con specie contenenti ossigeno. Il donatore di elettroni al P680+. è un residuo di tirosina (Yz) di un polipeptide (la proteina D1), che si trova nel centro di reazione del PSII. A questo punto Yz+ recupera l’elettrone ceduto tramite l’ossidazione in serie dei 4 ioni Mn, presenti nel complesso OEC (M). Dopo ogni trasferimento di un elettrone il centro Mn diventa sempre più ossidato, e alla fine del trasferimento dei 4 elettroni (ognuno dei 192 Capitolo XI quali corrisponde all’assorbimento di un fotone) si genera una carica elettrica +4 sul complesso M. P680++ Yz → P680 + Yz+ Yz+ + M → Yz + M+ M4+ + 2H2O → M + O2 + 4H+ E’ necessario l’accumulo di tutte e le quattro cariche positive sul centro Mn prima del rilascio della molecola di ossigeno. I quattro protoni (H+) rilasciati dall’acqua vanno nel lume del tilacoidale, il complesso che libera ossigeno agisce da pompa protonica, che a sua volta è guidata dal trasferimento di elettroni (Figura 7). Figura 7. Schema delle reazioni del complesso OEC. Il complesso del citocromo b6f Con la formazione del PQH2, gli elettroni abbandonano il PSII e iniziano il passaggio attraverso un complesso proteico integrale di membrana che serve di collegamento fra PSII e PSI, il complesso del citocromo b6f. Il complesso del citocromo b6f comprende quattro subunità: due citocromi (b6 e f), una proteina Fe-S e ed infine la subunità IV che non ha una funzione nota. Questo complesso multienzimatico catalizza il trasferimento degli elettroni dal PQH2 alla plastocianina La fotosintesi 193 (PC) ed ha un ruolo anche nel trasporto di protoni attraverso la membrana (dallo stroma al lume) e conseguentemente nella formazione di un potenziale elettrochimico. La plastocianina è una proteina contenente rame, localizzata sulla superficie luminale della membrana tilacoidale che trasferisce a sua volta elettroni al fotosistema I. Il ciclo catalitico del complesso del citocromo b6f prende il nome di ciclo Q che è il mezzo principale di generazione del gradiente protonico attraverso la membrana, utilizzato per produrre ATP. Nella prima metà del ciclo Q il plastochinolo (PQH2) viene ossidato a plastochinone. Gli elettroni provenienti dal plastochinolo fluiscono attraverso la proteina Fe-S per convertire la plastocianina ossidata (Cu2+) nella forma ridotta (Cu+).I due protoni prodotti vengono rilasciati nel lume tilacoidale. PQH2 + 2 PC(Cu2+) → PQ + 2 PC(Cu+) + 2 H+ Nella seconda metà del ciclo Q il citocromo b6f riduce una seconda molecola di plastochinone (PQ), proveniente dal pool, a plastochinolo che viene quindi riossidato con il conseguente rilascio di altri due H+ nel lume. In ciascun ciclo Q vengono rilasciati nel lume tilacoidale quattro protoni. Il trasporto degli elettroni dal complesso del citocromo b6f al P700+ del PSI è mediato dalla plastocianina, che funzione come un trasportatore mobile. Gli eventi fotochimici per il PSI sono analoghi a quelli del PSII. Il fotosistema I è un complesso multiproteico di membrana localizzato quasi esclusivamente nelle lamelle dello stroma. Anche in questo caso sono presenti una serie di complessi antenna (LHCI) che raccolgono l’energia dei fotoni e la convogliano al centro di reazione. Il doppietto speciale del centro di reazione ha un massimo di assorbimento a 700 nm, per cui viene denominato P700. Il PSI contiene una maggior quantità di clorofilla a rispetto alla clorofilla b. Il P700 eccitato (P700*) perde un elettrone in favore di un accettore, la clorofilla A0, un agente riducente estremamente forte, che passa il suo elettrone ad una catena di trasportatori che termina con il NADP+. Questa catena di trasporto è esoergonica in quanto trasferisce elettroni da potenziali di riduzione più negativi a potenziali meno negativi. A0 a sua vol- 194 Capitolo XI ta lo cede ad un secondo trasportatore il fillochinone A1. Quindi l’elettrone passa attraverso tre centri Fe-S (FX, FB , FA) P700* A0 A1 FX FB FA → P700+ A0- A1 FX FB FA → → P700+ A0 A1- FX FB FA → P700+ A0 A1 FX- FB FA → → P700+ A0 A1 FX FB- FA P700+ A0 A1 FX FB FAIl P700+ è un forte agente ossidante, che prende velocemente un elettrone dalla plastocianina. Il trasferimento di elettroni dalla PC al P700+ colma così il buco elettronico originato dall’eccitazione fotonica convogliata dagli LHCI al centro di reazione, come già visto per il PSII nello schema illustrato in Figura 6. L’elettrone, al termine della catena dei tre centri Fe-S (FA-), viene trasferito ad una ferredossina (Fd), una proteina estrinseca debolmente legata alla membrana tilacoide. La Fd a sua volta cede un elettrone, in due fasi successive, all’ultimo trasportatore di elettroni, una ferredossina-NAD+ reduttasi (FNR), che a sua volta riduce il NADP+ a NADPH + H+. La reazione complessiva di questi passaggi è: 2 Fdrid + 2H+ + NADP+. = 2Fdoss + NADPH + H+ L’insieme delle reazioni della fotofosforilazione non ciclica fin qui illustrate, possono essere riassunte in uno schema, detto schema a Z per la sua forma complessiva che assomiglia alla lettera Z (Figura 8). Questo schema descrive il percorso del flusso elettronico tra i due fotosistemi dall’H2O al NADP+ e le relazioni energetiche nelle reazioni alla luce. La fotosintesi 195 Figura 8. Schema Z del trasporto degli elettroni. Come abbiamo visto le attività combinate dei due fotosistemi trasferiscono gli elettroni dall’acqua al NADP+ conservando sotto forma di NADPH parte dell’energia derivata dall’assorbimento della luce. Questo flusso unidirezionale viene chiamato flusso elettronico non ciclico. Per ogni coppia di fotoni assorbiti (uno per ogni fotosistema) un elettrone viene trasferito dall’H2O al NADP+. Per formare una molecola di ossigeno che comporta il trasferimento di 4 elettroni da due molecole di H2O a due molecole di NADP+ devono essere assorbiti in totale 8 fotoni, quattro per ogni fotosistema. Adesso siamo in grado di stimare la stechiometria complessiva delle reazioni alla luce. L’assorbimento di quattro fotoni sul PSII genera una molecola di O2 e rilascia quattro protoni nel lume tilacoidale. Nel ciclo Q del complesso citocromo b6f vengono rilasciati otto protoni nel lume. Infine altri quattro fotoni convogliano gli elettroni dalla PC alla FNR che produce due molecole di NADPH. Perciò la reazione complessiva della fotofosforilazione non ciclica è: 2 H2O + 2 NADP+ + 10 H+ stroma → O2 + 2 NADPH + 12 H+ lume 196 Capitolo XI Dal trasporto non ciclico degli elettroni si producono circa due molecole di ATP e due di NADPH. Una terza molecola di ATP si può formare grazie al flusso ciclico di elettroni tra il fotosistema I, il complesso citocromo b6f e il pool di plastochinoni. Fotofosforilazione ciclica Le molecole che trasferiscono gli elettroni nella catena di connessione fra il PSII ed il PSI sono orientate asimmetricamente nella membrana tilacoidale, cosicché il flusso elettronico indotto dalla luce porta ad un movimento di protoni attraverso la membrana, dall’esterno (stroma) verso l’interno (lume) e l’energia viene conservata sotto forma di potenziale elettrochimico dovuto al gradiente protonico. Dati sperimentali hanno mostrato che per ogni molecola di ossigeno prodotta vengono prodotte tre molecole di ATP. Almeno 8 fotoni devono essere assorbiti per muovere 4 elettroni dall’acqua al NADPH (1 fotone per ogni elettrone del centro di reazione). L’energia di otto fotoni della luce visibile è più che sufficiente per la sintesi di tre ATP. La fotofosforilazione ciclica è una via alternativa al flusso di elettroni indotto dalla luce, che consente di variare le quantità di ATP e di NADPH prodotti durante il periodo di illuminazione. Questa via è definita “ciclica” per distinguerla dal flusso elettronico non ciclico, che procede dall’acqua al NADP+. Al flusso elettronico ciclico partecipa solo il PSI, gli elettroni che passano dal P700 alla ferredossina non continuano fino al NADP+, ma tornano indietro alla plastocianina attraverso il complesso di citocromi b6f (Figura 9). La plastocianina restituisce elettroni al P700 (PSI), che li aveva prima ceduti, uno alla volta, alla ferredossina. Quindi il PSI, quando viene illuminato, porta elettroni in una via ciclica, in cui un elettrone esce dal centro di reazione per poi ritornarvi dopo essere passato per un certo numero di trasportatori. Il flusso elettronico ciclico non porta alla formazione di NADPH o allo sviluppo di ossigeno, però è accompagnato dal pompaggio di protoni e di conseguenza permette, mediante il gradiente di potenziale chimico protonico, la formazione di ATP a partire da ADP e fosfato inorganico (Pi). La fotosintesi 197 Figura 9. Schema della fotofosforilazione ciclica. Formazione dell’ATP La formazione di ATP nei cloroplasti è catalizzata da una proteina complessa detta ATP sintasi o anche ATP-asi (perché può catalizzare anche la reazione di scissione dell’ATP). Questo grande complesso è costituito da due componenti funzionali: il CFo e il CF1. L’indice “o” dell’unità CFo deriva dall’iniziale di “oligomicina” che è un antibiotico capace di legarsi al canale del complesso proteico e di impedire il passaggio di protoni inibendo anche l’attività del CF1. Il componente CFo è un poro protonico transmembrana che ha natura idrofobica ed è simile all’Fo mitocondriale. E’ costituito da quattro diversi polipeptidi identificati con le lettere a, b, b’ e c, presenti probabilmente con la seguente stechiometria, abb’c12 Le subunità c e a sono direttamente coinvolte nel trasporto transmembrana dei protoni. Le subunità b e b’ costituiscono probabilmente un legame che ha lo scopo di mantenere la posizione tra CFo e CF1. Il CF1 è un complesso periferico di membrana di struttura sferica con nove subunità di cinque tipi diversi, con la seguente composizione: α3β3γδε.Le tre subunità α e β sono disposte alternativamente a 198 Capitolo XI spicchio d’arancia attorno ad un asse centrale costituito dalla subunità γ. Sia la subunità δ che la ε sembrano necessarie per mantenere l’integrità e/o la posizione rispettivamente delle due subunità b e della subunità γ (Figura 10) durante l'attività di sintesi dell'ATP. Figura 10. Il complesso dell'ATP sintasi. Tuttavia la loro mancanza non altera l’attività ATP-asica (scissione dell’ATP). Quando il flusso protonico attraversa la corona costituita dalla subunità c12 dell’Fo, imprime a questa una rotazione che provoca a sua volta la rotazione della subunità γ che fuoriesce dal complesso F1 ed è legata al c12. Le tre subunità α e β dell’F1 però non ruotano perché sono ancorate in una posizione fissa dalle due subunità b dell’Fo che a loro volta sono ancorate alla membrana. In questo modo la subunità γ può ruotare all’interno del cilindro formato dalle subunità α3β3. Il modello del meccanismo di azione più largamente accettato fino ad oggi è quello del meccanismo a tre siti secondo il quale i tre siti catalitici ruotano in sequenza attraverso tre stati distinti. Dal mo- La fotosintesi 199 mento che la subunità γ è asimmetrica, durante la sua rotazione provoca a turno delle modificazioni conformazionali in ciascuno dei tre eterodimeri αβ che provocano una loro diversa affinità di legame per l’ATP e l’ADP. All’interno dell’eterodimero sembra che solo la componente β abbia proprietà catalitiche. I tre livelli di conformazione/affinità sono generalmente indicati come: semichiusa (L), chiusa (T) e aperta (O). La conformazione L lega debolmente il substrato (ADP + Pi). La conformazione T lega il substrato così saldamente che si ha la formazione di ATP. La conformazione O, che ha l’affinità più bassa per il substrato, rilascia l’ATP che si era formata nello stato precedente (T) (Figura 11). Figura 11. Rappresentazione grafica del modello a tre siti. E’ quindi la rotazione della subunità γ che, provocando al interconversione degli stati delle subunità β, permette la produzione in continuo di ATP. Tutti e tre i siti passano sequenzialmente attraverso le tre conformazioni al rientro dei protoni nel lune tilacoidale. La conformazione chiusa (T) diventa un sito aperto (O) con la liberazione di ATP; il sito L si converte in sito T, dove il legame forte di ADP + Pi porta alla formazione di ATP. Contemporaneamente il terzo sito si converte dalla forma O, con affinità molto bassa per i nucleotidi, alla foma L capace di legare debolmente ADP e Pi. 200 Capitolo XI Le reazioni al buio La sintesi dei carboidrati nelle cellule animali necessita sempre di precursori con almeno tre atomi di carbonio (vedi la gluconeogenesi), tutti in uno stato di ossidazione più basso dell’atomo di carbonio nella CO2. Gli organismi fotosintetici invece possono produrre carboidrati da CO2 ed acqua, riducendo la CO2 a spese dell’energia dell’ATP e del NADPH generati nel trasferimento fotosintetico degli elettroni durante l’esposizione alla luce. Le reazioni al buio della fotosintesi comportano la fissazione della CO2 e la sua riduzione a carboidrati attraverso un processo chiamato ciclo della riduzione fotosintetica del carbonio. Queste reazioni sono dette reazioni al buio in quanto non dipendono direttamente dalla presenza di luce e utilizzano il potere riducente (NADPH) e l’energia (ATP) prodotti durante la fase luminosa. Le piante verdi contengono nei cloroplasti un sistema enzimatico che catalizza la conversione della CO2 in composti organici semplici ridotti. Il processo si dice anche assimilazione della CO2. o fissazione (organicazione) del carbonio. In questa parte della descrizione della fotosintesi si considera però solo la specifica reazione con cui la CO2. viene incorporata, o fissata, nella molecola di 3-fosfoglicerato. Il 3-fosfoglicerato è il precursore di molecole ben più complesse, come zuccheri, polisaccaridi ed altri derivati, usando vie metaboliche simili a quelle degli animali. La fotosintesi fornisce inoltre lo scheletro carbonioso per quasi tutti i composti organici conosciuti. Le reazioni che portano all’assimilazione della CO2 costituiscono una via ciclica in cui gli intermedi chiave sono continuamente rigenerati. Questo meccanismo fu identificato da Melvin Calvin e collaboratori nel 1950 viene spesso chiamata Ciclo di Calvin-Benson, Ciclo delle piante C3 o Ciclo fotosintetico principale. Le reazioni del ciclo di Calvin-Benson avvengono nello stroma del cloroplasto. Il ciclo di Calvin-Benson La prima fase del ciclo è costituita dalla condensazione della CO2 con un accettore a 5 atomi di C, il ribulosio 1,5-bifosfato per formare La fotosintesi 201 2 molecole di 3-fosfoglicerato. Nella seconda il 3-fosfoglicerato viene ridotto a gliceraldeide-3-fosfato (G3P), in equilibrio con il di-idrossi aceton fosfato (DHAP). Nella terza fase parte delle molecole di triosofosfati viene usata per rigenerare il ribulosio 1,5-bifosfato., il composto da cui ha preso il via l’intero processo. Ne risulta quindi una via ciclica che consente l’assimilazione continua di CO2 sotto forma di triosi ed esosi fosfato. Le singole reazioni che costituiscono del ciclo di Calvin sono qui descritte più in dettaglio e successivamente riassunte in Tabella 1 secondo la numerazione riportata nel testo: Fase 1: Fissazione della CO2 nel 3-fosfoglicerato. Questa fase comporta la fissazione del carbonio mediante la carbossilazione del ribulosio 1,5-bifosfato. L’enzima che catalizza l’incorporazione della CO2 nei composti organici si chiama ribulosio 1,5 bifosfato carbossilasi o RuBP carbossilasi/ossigenasi, chiamata spesso semplicemente RuBisCO, Ru (ribulosio), Bis (bifosfato), C (carbossilasi), O (ossigenasi), o ancora più semplicemente rubisco. La rubisco catalizza la formazione di un legame covalente della CO2 con lo zucchero a cinque atomi di carbonio, ribulosio 1,5 bifosfato (RuBP), e la scissione dell’intermedio instabile a sei atomi di carbonio così ottenuto, in due molecole di 3-fosfoglicerato (3PG), una delle quali contiene il gruppo carbossilico appena incorporato. La reazione procede attraverso la formazione di una serie di intermedi, qui non riportati nel dettaglio, che portano alla rottura di un legame C-C con formazione di due C3 (3-fosfoglicerato) (Figura 12). 202 Capitolo XI Figura 12. Reazione di fissazione della CO2 ad opera dell'enzima RuBisCO. Fase 2: Conversione del 3-fosfoglicerato in gliceraldeide-3-fosfato. Questa fase porta alla riduzione del carbonio fissato per iniziare la sintesi di esosio. Il 3-fosfoglicerato (3PG) formato nella fase 1 viene ridotto dal NADPH formato nella fase luminosa e trasformato in gliceraldeide-3-fosfato (G3P) in due tappe, che sono l’inverso delle tappe corrispondenti della glicolisi. Nella prima tappa (2.1) la 3-fosfoglicerato chinasi dello stroma catalizza il trasferimento di un gruppo fosforico dall’ATP al 3fosfoglicerato, formando 1,3-bisfosfoglicerato (BPG) e ADP. Il NADPH nella seconda tappa (2.2) dona elettroni in una reazione di riduzione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi formando gliceraldeide-3-fosfato (G3P) (Figura 13). I triosi fosfati possono poi essere convertiti in amido nei cloroplasti e conservati per essere usati in un secondo tempo, oppure possono essere immediatamente esportati nel citosol e convertiti in saccarosio da trasportare nelle zone di crescita della pianta. La fotosintesi 203 Figura 13. Reazioni di riduzione del 3PG a G3P. Fase 3: Rigenerazione del ribulosio 1,5 bisfosfato. Questa fase porta alla rigenerazione del composto di partenza, il ribulosio 1,5 bisfosfato, che è l’accettore della CO2. Come visto sopra, la prima reazione del ciclo di Calvin consuma ribulosio 1,5 bisfosfato. Per avere una assimilazione continua della CO2, il ribulosio 1,5 bisfosfato deve essere continuamente rigenerato. Per fare questo le cellule delle piante ricorrono ad una serie di reazioni che formano, insieme alla fase 1 e 2 una via ciclica. In questa via, il prodotto della prima reazione (3PG) attraverso una serie di trasformazioni rigenera il materiale di partenza, il ribulosio 1,5 bisfosfato. La formazione di ribulosio 1,5 bisfosfato necessita di un riarrangiamento degli scheletri carboniosi della gliceraldeide-3-fosfato e del di-idrossiacetonefosfato (DHAP) prodotti nella prima fase dell’assimilazione del carbonio. Gli intermedi di questa via comprendono zuccheri a tre, quattro, cinque, sei e sette atomi di carbonio. Nella prima reazione di questa fase (3.1) la triosofosfato isomerasi converte reversibilmente alcune molecole di G3P in diidrossiacetonefosfato (DHAP). Successivamente (3.2) un’aldolasi condensa G3P e DHAP a formare un esosiofosfato, il fruttosio 1,6-bisfosfato (FBP) (Figura 14). 204 Capitolo XI Figura 14. Reazioni dell’aldolasi della triosofosfato isomerasi. Il FBP viene defosforilato ad opera della fruttosio 1,6-bisfosfatasi a formare fruttosio 6-fosfato (F6P) (3.3). Quindi il F6P insieme ad un’altra molecola di G3P viene utilizzato dalla transchetolasi (3.4) per formare xilulosio 5-fosfato (X5P) ed eritrosio 4-fosfato (E4P) (Figura 15). Figura 15. Reazione della transchetolasi. La fotosintesi 205 L’E4P e una molecola di DHAP sono condensati in sedoeptulosio 1,7-bisfosfato(SBP) dall’aldolasi (3.5) (Figura 16). Figura 16. Reazione dell'aldolasi per formare SBP. Il SBP viene defosforilato dalla sedoeptulosio 1,7-bisfosfatasi (3.6) formando il sedoeptulosio 7-fosfato (S7P) che partecipa ad un’altra reazione ad opera della transchetolasi con G3P per produrre ribosio 5fosfato e xilulosio 5-fosfato (Figura 17). Figura 17. Reazione della transchetolasi con formazione di R5P e Xu5P. 206 Capitolo XI La conversione del R5P in Ru5P (3.8) è una reazione di isomerizzazione che procede attraverso la formazione di un intermedio enediolico ad opera della ribosio fosfato isomerasi. Figura 18. Reazione di isomerizzazione di R5P e Ru5P. La reazione procede con una epimerizzazione (3.9), ossia uno scambio di gruppi sullo stesso atomo di carbonio, che dopo la formazione di un intermedio porta alla formazione di Ru5P. Figura 19. Reazione di epimerizzazione tra Ru5P e Xu5P. In fine il Ru5P proveniente da questa reazione e dalla reazione precedente portano alla rigenerazione del ribulosio 1,5-bisfosfato in una La fotosintesi 207 reazione catalizzata dalla fosforibulochinasi, un enzima che utilizza ATP (3.10). In Figura 20 viene riportato uno schema riassuntivo della fase 3 del Ciclo di Calvin, che illustra le interconversioni dei triosi fosfato (3C) e dei pentosi fosfato (5C). Le molecole di partenza sono: la gliceraldeide 3-fosfato e il diidrossiacetone fosfato. Le reazioni catalizzate dalla transaldolasi (tappe 1 e 4) e dalla transchetolasi (tappe 3 e 6) producono pentosi fosfato. Questi sono poi convertiti in ribulosio 5-fosfato (tappe 7 e 8). Nelle tappe 9 il ribulosio 5-fosfato viene fosforilato con consumo di ATP rigenerando il ribulosio 1,5 bisfosfato. Figura 20. Reazioni della terza fase dell'assimilazione della CO2. In Tabella 1 sono riportate tre fasi del Ciclo di Calvin con le tredici reazioni catalizzate dagli undici enzimi in esso coinvolti. Capitolo XI 208 Tabella 1. Reazioni del ciclo di Calvin-Benson Fase Reazione Enzima 1 3 RuBP + 3 CO2 → 6 3PG Ribulosio 1,5-bifosfatocarbossilasi 2.1 3PG + 6 ATP → 6 BPG + 6 ATP 3 fosfoglicerato chinasi 2.2 6 BPG + 6 NADPH + 6 H+ → G3P + 6 NADP+ + 6Pi Gliceraldeide-3-fosfato deidrogenasi 3.1 G3P ' DHAP Triosofosfato isomerasi 3.2 G3P + DHAP ↔ FBP Aldolasi 3.3 FBP → F6P Fruttosio 1,6-bisfosfatasi 3.4 F6P + G3P ' E4P + Xu5P Transchetolasi 3.5 E4P + DHAP ' SBP Aldolasi 3.6 SBP → S7P+ Pi Sedoeptulosio 1,7bisfosfatasi 3.7 S7P + G3P ' Xu5P + R5P Transchetolasi 3.8 R5P ' Ru5P Ribosio 5-fosfatoisomerasi 3.9 2 Xu5P ' 2 Ru5P Fosfopentosio epimerasi 3.10 3 Ru5P + 3 ATP → RuBP + 3 ADP Fosforibulo chinasi Reazione netta 3 CO2 + 9 ATP + 6 NADPH + 6 H+ → 9 ADP + 8 Pi + 6 NADP+ + Triosofosfato ( G3P o DHAP) La fotosintesi 209 Costo metabolico del Ciclo di Calvin Il risultato del ciclo di Calvin è la conversione di 3 molecole di CO2 e di una molecola di fosfato in una molecola di trioso fosfato. La reazione complessiva da CO2 a trioso fosfato con la rigenerazione del ribulosio 1,5 bisfosfato è riportata qui di seguito. Figura 21. Stechiometria dell'assimilazione della CO2 nel ciclo di Calvin. Come si vede, 3 molecole di ribulosio 1,5 bifosfato (in totale 15 atomi di carbonio) si condensano con 3 molecole di CO2 (3 atomi di carbonio), formando 6 molecole di 3-fosfoglicerato (18 atomi di C), che poi verranno ridotte a 6 molecole di gliceraldeide-3-fosfato, in e- 210 Capitolo XI quilibrio con altrettante molecole di diidrossiacetone fosfato. Verranno spese 6 molecole di ATP nella sintesi del 3-fosfoglicerato e consumate 6 molecole di NADPH nella riduzione dell’1,3bisfosfoglicerato a gliceraldeide-3-fosfato. Il prodotto netto della reazione è una molecola di gliceraldeide-3-fosfato. Le altre 5 molecole di trioso-fosfato sono impiegate nella rigenerazione di 3 molecole di ribulosio 1,5 bifosfato (in tutto 15 atomi di C) dal ribulosio 5 fosfato, processo che richiede altre 3 molecole di ATP. In conclusione, per ogni molecola di triosofosfato prodotta dall’assimilazione fotosintetica di una molecola di CO2 sono necessarie 6 molecole di NADPH e 9 di ATP. Fotorespirazione La rubisco non ha specificità assoluta di substrato per la CO2, perchè l’O2 compete con essa per il legame al sito attivo, quindi la rubisco catalizza non solo la carbossilazione, ma anche l’ossigenazione del ribulosio 1,5 bisfosfato (infatti il suo nome completo contiene anche la parola ossigenasi). Questa reazione procede nello stesso sito attivo della carbossilazione e produce, anziché due molecole di 3-fosfo glicerato (3PG), una molecola di 3-fosfoglicerato ed una di fosfoglicolato, un composto a due atomi di carbonio. La rubisco viene classificata come monoossigenasi perché introduce un atomo di ossigeno nell’acido fosfoglicolico e uno nell’acqua, come è stato dimostrato usando isotopi radioattivi marcati sull’ossigeno. Sebbene l’affinità della rubisco per la CO2 sia molto più elevata che non quella per l’O2, il fatto che la concentrazione atmosferica dell’ossigeno (21%) sia molto più elevata di quella della CO2 (0,03%) fa sì che le reazioni, in condizioni normali, praticamente si equivalgano, e l’interazione di CO2 e O2 sia di natura competitiva. Il rapporto carbossilazione-ossigenazione del ribulosio 1,5 bifosfato varia, in condizioni normali, da 3:1 a 4:1. L’assorbimento di O2 e la formazione di fosfoglicolato durante l’ossigenazione del ribulosio 1,5 bifosfato sono alla base della fotorespirazione. Questa via metabolica, detta anche ciclo C2, è costituita da una sequenza di reazioni in cui due molecole di acido fosfoglicolico La fotosintesi 211 vengono metabolizzate per formare una molecola di acido 3fosfoglicerico ed una di CO2. La capacità di fissare O2 in presenza di luce e di liberare CO2 conferisce a questo processo il nome di fotorespirazione. Le reazioni della fotorespirazione avvengono in tre diversi compartimenti cellulari: il cloroplasto, il perossisoma ed il mitocondrio (Figura 22). Nella prima reazione il fosfoglicolato viene defosforilato da un enzima specifico con formazione di glicolato, un intermedio C2. Questo lascia il cloroplasto ed entra nel perossisoma dove viene ossidato a gliossilato, reazione catalizzata dalla glicolico ossidasi. Il perossido di idrogeno liberato durante questa reazione viene rapidamente trasformato in acqua dall’enzima catalasi. Il gliossilato viene transamminato e trasformato in glicina, sempre nel perossisoma. La glicina lascia poi il perossisoma ed entra nel mitocondrio, dove viene decarbossilata ossidativamente, liberando NH3 e CO2. Un gruppo metilenico rimasto viene trasferito sul tetraidrofolato (un coenzima delle transferasi) e in seguito ceduto ad un’altra molecola di glicina, formando un altro amminoacido, la serina. La serina passa poi dal mitocondrio al perossisoma, dove viene transamminata dall’a-chetoglutarato, formando idrossipiruvato e glutammato. La reazione finale che avviene nel perossisoma è la riduzione dell’idrossipiruvato in glicerato. Quest’ultimo lascia il perossisoma ed entra nel cloroplasto, dove viene fosforilato da una gliceratochinasi ATP- dipendente con formazione di 3-fosfoglicerato che entra, nello stroma, nel ciclo di Calvin. Le funzioni metaboliche di questa reazione non sono chiare, infatti il processo non porta alla fissazione del carbonio e non sembra portare vantaggi alla cellula. Il recupero degli atomi di carbonio dal fosfoglicolato costa energia in più, sotto forma di ulteriore richiesta di ATP. Inoltre, le perdite di resa fotosintetica per fotorespirazione sono dell’ordine 20-40%. 212 Capitolo XI Figura 22. Reazioni della fotorespirazione. La fotosintesi 213 La condensazione dell’ ossigeno con il ribulosio 1,5 bifosfato avviene insieme alla fissazione della CO2, ma a velocità tre volte più bassa. Apparentemente l’evoluzione della rubisco ha prodotto un sito attivo che non riesce a discriminare tra ossigeno e CO2, la spiegazione più attendibile è che questa evoluzione sia avvenuta prima che la vita si sviluppasse in aerobiosi. La fotorespirazione tuttavia, nonostante possa sembrare una sorta di spreco, permette di recuperare il carbonio perduto in seguito alla formazione del fosfoglicolato. E’ inoltre uno strumento efficace per metabolizzare i prodotti della reazione di ossigenazione, catalizzata dalla rubisco. Un’altra ipotesi sulla funzione della fotorespirazione è che l’ossigenazione del ribulosio 1,5 bifosfato e la fotorespirazione possano fornire un mezzo per dissipare l’energia fotochimica in condizioni di bassa concentrazione di CO2 quando l’ATP e il NADPH non vengono consumati rapidamente nel ciclo di Calvin-Benson. Il trasporto di elettroni, in assenza di NADP+ e di ADP provoca un’eccessiva riduzione nei trasportatori e la formazione di un gradiente di protoni attraverso la membrana tilacoidale potrebbe arrecare danno ai pigmenti fotosintetici. Poiché la fotorespirazione consuma sia ATP che NADPH il metabolismo del fosfoglicolato potrebbe proteggere i pigmenti che catturano la luce, fornendo ADP e NADP+ per un continuo flusso di elettroni nel cloroplasto. La fotosintesi delle piante C4 La maggior parte delle piante tropicali o di origine tropicale, come il mais, il sorgo, la canna da zucchero, anche se coltivate nelle zone temperate, hanno sviluppato un meccanismo per superare il problema della fotorespirazione. In una prima fase, anziché fissare la CO2 sul ribulosio 1,5-bisfosfato, e quindi non utilizzando la rubisco, queste piante usano un composto diverso, il fosfoenolpiruvato (PEP), ottenendo una molecola a 4 atomi di carbonio. Da qui il nome di piante C4, mentre il processo di assimilazione della CO2 prende il nome di metabolismo del C4 o via del C4. 214 Capitolo XI Le piante invece che fissano la CO2 sul ribulosio 1,5-bisfosfato per dare 3-fosfoglicerato vengono chiamate piante C3, in quanto il composto principale di questa via metabolica è a 3 atomi di carbonio. Nella via di assimilazione C4 del carbonio, le piante possono eliminare l’attività ossigenasica della rubisco concentrando la CO2 nel sito di questo enzima. La via C4 non sostituisce il ciclo di Calvin-Benson ma è piuttosto l’introduzione ad esso. Le piante C4 procedono alla fissazione del carbonio in due tipi di cellule, le cellule del mesofillo e quelle della guaina del fascio vascolare. Le cellule della guaina del fascio nelle piante C4, a differenza delle C3, contengono cloroplasti e sono circondate dalle cellule del mesofillo. Le cellule del mesofillo hanno nel citoplasma la fosfoenolpiruvato carbossilasi (PEPcarbossilasi) ma non la rubisco, mentre le cellule della guaina del fascio hanno la rubisco, ma non la PEP-carbossilasi, che, a differenza della rubisco, non possiede attività ossigenasica. Uno schema semplificato della via C4 è illustrato nella Figura 23. Nelle cellule del mesofillo la PEP-carbossilasi condensa la CO2 con il PEP, formando ossalacetato. Il substrato della PEP-carbossilasi è in realtà HCO3-, che è in equilibrio con la CO2. L’ossalacetato viene poi trasformato in malato o aspartato a seconda della decarbossilasi presente nelle cellule della guaina del fascio. Malato e aspartato vengono poi trasportati dalle cellule del mesofillo alle cellule della guaina del fascio adiacenti, dove una decarbossilasi specifica libera la CO2 che viene fissata dalla rubisco ed entra nel ciclo di Calvin-Benson. La decarbossilazione degli acidi C4 fa aumentare significativamente la concentrazione di CO2 nelle cellule della guaina del fascio e quindi l’attività carbossilasica della rubisco prevale su quella ossigenasica. Il composto C3 rimasto dopo la decarbossilazione viene trasportato nelle cellule del mesofillo e trasformato in PEP, il substrato della reazione iniziale di carbossilazione. La fotosintesi 215 Figura 23. Schema generale della fissazione C4 del carbonio. Poiché nelle cellule della guaina del fascio sono presenti 3 diversi enzimi, le piante C4 sono classificate in 3 sottogruppi in base all’enzima che in esse prevale e precisamente: 1. enzima malico NADP+ dipendente; 2. enzima malico NAD+ dipendente; 3. PEP-carbossichinasi. 1. Nelle cellule del mesofillo delle specie che possiedono l’enzima malico NADP+ dipendente l’ossalacetato viene ridotto a malato poi trasferito nelle cellule della guaina del fascio e decarbossilato dall’ enzima malico NADP+ dipendente. Viene prodotto NADPH, H+, piruva- 216 Capitolo XI to e CO2. Il piruvato viene trasportato alle cellule del mesofillo e la CO2 viene poi fissata nel ciclo di Calvin-Benson (Figura 24). Figura 24. Via C4 nella specie con enzima malico NADP+ dipendente. 2. Nelle specie che possiedono l’enzima malico NAD+ dipendente l’ossalacetato viene transamminato ad aspartato (trasformando contemporaneamente glutammato in α-chetoglutarato). L’aspartato viene poi trasferito nelle cellule della guaina del fascio e deamminato in ossalacetato. Questo poi viene ridotto a malato dall’enzima malico NAD+ dipendente. Viene prodotto NADH, H+, piruvato e CO2. Il piruvato viene transamminato in alanina, poi trasportata nelle cellule del mesofillo dove viene anch’essa transamminata in piruvato. La CO2 viene fissata nel ciclo di Calvin-Benson (Figura 25). La fotosintesi 217 Figura 25. Via C4 nelle specie con l'enzima malico NAD+ dipendente. 3. Nelle specie che possiedono l’enzima PEP-carbossichinasi l’ossalacetato viene transamminato ad aspartato a livello delle cellule del mesofillo, trasformando contemporaneamente il glutammato in αchetoglutarato. L’aspartato viene poi trasferito nelle cellule della guaina del fascio e deamminato nuovamente ad ossalacetato, poi decarbossilato dall’ enzima PEP-carbossichinasi ATP dipendente, con forma- 218 Capitolo XI zione di ADP, PEP e CO2. Anche in questo caso la CO2 viene utilizzata nel ciclo di Calvin-Benson mentre il PEP viene trasportato alle cellule del mesofillo. Poiché l’aspartato viene trasportato nelle cellule delle guaina del fascio e il PEP viene successivamente esportato dalle stesse cellule è necessario un ulteriore trasporto di piruvato e di alanina alle cellule del mesofillo perché possano disporre della quantità di glutammato necessaria alla transamminazione dell’ossalacetato. La via di assimilazione del C4 richiede un maggior dispendio di energia che non del ciclo di Calvin-Benson nelle piante C3. Nelle piante C3 per ogni molecola di CO2 fissata sono richiesti tre ATP e due NADPH. Nelle specie con enzima malico NADP+ o NAD+ dipendente sono necessarie altre due molecole di ATP per trasformare l’acido piruvico in PEP tramite la piruvico fosfatodichinasi (PPDK). Nelle specie con la con la PEP carbossichinasi serve solo una molecola di ATP per la stessa trasformazione. Nonostante questo dispendio supplementare di energia, il ciclo C4 è così efficace nell’inibire l’ossigenazione del ribulosio 1,5-bisfosfato che in condizioni ottimali, le piante C4 hanno una resa in moli di CO2 fissate per moli di fotoni assorbiti superiori alle piante C3 in cui si svolge la fotorespirazione. Metabolismo acido delle crassulacee (piante CAM) In particolari situazioni ambientali il costo che le piante pagano per poter fotosintetizzare è troppo elevato in termini di quantità di acqua traspirata. Così in particolari ambienti esiste un’altra via ausiliaria della fissazione del carbonio: si tratta del metabolismo acido delle Crassulacee, (Crassulacean Acid Metabolism, in sigla CAM), un adattamento della fotosintesi di alcune piante, che vivono in situazioni di carenza idrica. L’efficienza di utilizzo dell’acqua aumenta con la possibilità di variare il ritmo circadiano di apertura e chiusura degli stomi e contemporaneamente adattare il meccanismo di carbossilazione della CO2 atmosferica. Queste piante sono le Crassulacee, che comprendono tutti i cactus e molte epifite, fra le quali le Orchidee e le Bromeliacee. Queste ultime sono piante parassite che, specie nei climi tropicali, vivono su altre piante a spese delle loro sostanze nutritive. Nei nostri La fotosintesi 219 climi un esempio può essere rappresentato dal vischio. Le piante CAM si trovano in prevalenza in ambienti desertici e in microclimi aridi. La via metabolica della fissazione del carbonio è analoga a quella delle piante C4: vi è una reazione iniziale di carbossilazione catalizzata dalla PEP-carbossilasi ed una successiva decarbossilazione degli acidi C4, che fa aumentare la concentrazione interna di CO2. Tuttavia, a differenza della via C4, il metabolismo degli acidi nelle crassulacee si svolge solo a livello delle cellule del mesofillo e le reazioni di decarbossilazione e di carbossilazione avvengono in tempi diversi, durante il ciclo giorno/notte. Carbossilazione e decarbossilazione sono separate temporalmente nelle CAM, mentre sono separate spazialmente nelle piante C4 (Figura 26). Figura 26. Schema generale del metabolismo CAM. 220 Capitolo XI Durante la notte, gli stomi delle piante con metabolismo CAM si aprono e assorbono notevoli quantità di CO2 atmosferica che, dopo essersi equilibrata col bicarbonato si combina con acido fosfoenolpiruvico (PEP) e forma acido ossalacetico in una reazione catalizzata dalla fosfoenolpiruvatocarbossilasi. L’acido ossalacetico viene poi ridotto dalla NAD+-malicodeidrogenasi in acido malico. Tutti e due questi enzimi stanno nel citosol, ma l’acido malico non può accumularsi in esso perché in alta concentrazione abbasserebbe troppo il pH. Così l’acido malico attraversa il tonoplasto (membrana del vacuolo) e viene conservato nel vacuolo per la notte. I vacuoli delle cellule delle piante CAM sono molto grandi e occupano più del 95% del volume cellulare. Al termine della notte nel vacuolo c’è una concentrazione di acido malico elevata, maggiore di 0,2 M. Qui termina la fase carbossilativa delle piante CAM. Il giorno successivo, l’acido malico viene liberato dai vacuoli e decarbossilato in acido piruvico, tramite gli stessi enzimi delle piante C4, enzima malico NADP+ dipendente e PEP carbossichinasi. Di conseguenza, anche le piante con metabolismo CAM sono classificate in 3 sottogruppi a seconda della decarbossilasi predominante. La CO2 prodotta durante la decarbossilazione dell’acido malico viene poi nuovamente fissata dalla RuBisCO e incorporata nel ciclo di Calvin. L’acido piruvico prodotto dalla decarbossilazione dell’acido malico viene trasformato in fosfoenolpiruvico dalla piruvatofosfato dichinasi. Il PEP viene poi metabolizzato attraverso la gluconeogenesi e forma carboidrati di riserva, generalmente amido. Di notte, quando ricomincia la fase carbossilativa delle piante CAM, l’amido viene idrolizzato in unità di esosio e poi trasformato, attraverso la glicolisi, in acido fosfoenolpiruvico. In questo modo, nell’arco di un ciclo giorno/notte si hanno variazioni nelle riserve di acido malico e amido nelle foglie delle piante CAM. Dato che la decarbossilazione dell’acido malico è una fonte interna di CO2, gli stomi delle piante CAM possono restare chiusi durante il giorno, mentre si svolge fase di fissazione del carbonio. La fotosintesi 221 Biosintesi di saccarosio ed amido La maggior parte delle piante superiori, le C4, le C3 o le CAM sintetizzano il saccarosio e lo trasportano dal sito di sintesi alle regioni non fotosintetizzanti della pianta, come radici, semi e foglie giovani, a seconda delle necessità metaboliche. Durante un intero ciclo giorno/notte questi organi hanno a disposizione un apporto relativamente costante di saccarosio. Infatti, oltre a quello prodotto durante il giorno dalla fissazione della CO2 le cellule fotosintetizzanti producono e conservano nei cloroplasti l’amido, che può poi essere trasformato in saccarosio ed esportato durante la notte. La sintesi del saccarosio, che avviene nel citosol, parte dai triosifosfati che provengono durante il giorno dal ciclo di Calvin e durante la notte dall’idrolisi dell’amido. I triosifosfati necessari sono trasportati dallo stroma del cloroplasto allo spazio intermembrana da un trasportatore di fosfati situato nelle membrana interna del cloroplasto. Questo trasportatore funziona a scambio una molecola di triosofosfato viene portata dal cloroplasto verso il citosol mentre una molecola di fosfato inorganico migra dal citosol verso il cloroplasto. I triosifosfati, presenti nello spazio intermembrana, attraversano la membrana interna permeabile e giungono al citosol. Nella sintesi del saccarosio, a partire dai triosifosfati la prima reazione, catalizzata dall’aldolasi, è reversibile quindi la seconda reazione è la tappa obbligata della sintesi ed è catalizzata dalla fruttosio-1,6bisfosfatasi che, avendo questa importante funzione, è soggetta a regolazione da parte del fruttosio-2,6-bisfosfato, che funziona da regolatore glucidico sia negli gli animali che nelle piante. Per ogni molecola di saccarosio sintetizzata vengono consumate quattro molecole di triosofosfato di cui due sono necessarie nella prima reazione del ciclo e altre due per formare il fruttosio-6-fosfato che partecipa alla reazione catalizzata dalla saccarosio-6-fosfato-sintetasi. Un legame ad alta energia viene scisso nella formazione dell’UDPglucosio a partire dall’UTP e dal glucosio-1-fosfato. Inoltre vengono prodotte quattro molecole di Pi: una nella reazione catalizzata dalla fruttosio-1,6-bisfosfatasi, due nella reazione catalizzata dalla UDP- 222 Capitolo XI glucosio-pirofosforilasi e dalla pirofosfatasi ed una nella reazione catalizzata dalla saccarosio-fosfato-fosfatasi (Figura 27). Il Pi liberato in queste reazioni può essere trasferito nel cloroplasto dal trasportatore di fosfati in uno scambio col triosofosfato che viene trasferito al citosol. Figura 27. Sequenza di reazioni nella biosintesi del saccarosio. Quando i triosofosfati vengono trattenuti nel cloroplasto vengono metabolizzati, attraverso il ciclo di Calvin, in fruttosio-6-fosfato ad opera dell’aldolasi e della fruttosio-1,6-bifosfatasi. Il fruttosio-6fosfato (F6P) può continuare la sua strada nel ciclo di Calvin-Benson, rigenerando ribulosio-1,5-bifosfato o essere deviato dal ciclo verso la produzione di amido, forma di riserva del glucosio. Le vie che portano al saccarosio e all’amido hanno molte reazioni in comune (Figura 28). Ad esempio, l’aldolasi e la fruttosio-1,6-bifosfatasi catalizzano la trasformazione di triosofosfati in F6P in entrambi i casi. Di notte, quando non ha luogo la fotosintesi, l’amido viene mobilizzato e fornisce il carbonio necessario per il metabolismo delle piante (regolazione e crescita). La fotosintesi 223 L’enzima nella via che porta alla sintesi dell’amido è l’ADPglucosio pirofosforilasi. Figura 28. Sequenza di reazioni nella biosintesi dell'amido. Esistono due possibili vie per il catabolismo dell’amido: 1) l’amido viene scisso dall’amilasi in polimeri di glucosio a catena corta, le destrine, successivamente idrolizzate in glucosio dall’aglucosidasi. Quindi l’esochinasi fosforila il glucosio che diventa glucosio-fosfato che viene poi trasformato in triosofosfato dalla glicolisi, trasportato nel citosol ed utilizzato per la sintesi del saccarosio. 2) l’amido viene direttamente degradato in unità di glucosio-fosfato dall’amido-fosforilasi in presenza di Pi in una reazione simile a quella catalizzata dalla glicogeno-fosforilasi. I LIPIDI Negli organismi viventi è presente una classe di sostanze organiche, i lipidi, molto eterogenea dal punto di vista chimico, con caratteristiche sostanzialmente diverse da quelle dei carboidrati, delle proteine e degli acidi nucleici, ma che ha ugualmente un ruolo fondamentale ed insostituibile nel metabolismo cellulare. Poiché le cellule viventi sono prevalentemente costituite di acqua, ci si potrebbe aspettare che tutti i loro componenti fossero solubili in essa o quanto meno idrofili: la proprietà più rilevante nei lipidi biologici invece è l’insolubilità in acqua o in liquidi fortemente polari, mentre sono assai solubili in solventi organici non polari come cloroformio, acetone, etere di petrolio, benzene. A differenza di altre classi di composti, si tratta di molecole piuttosto piccole. Anche le funzioni biologiche sono molto diverse: i grassi e gli oli sono le principali forme di conservazione dell’energia in molti tipi di organismi, mentre fosfolipidi e steroli formano più della metà della massa delle membrane biologiche. Altri lipidi, presenti in quantità minore, possono agire da cofattori, trasportatori di elettroni, pigmenti per l’assorbimento della luce, punti di formazione di interazioni idrofobiche o agenti emulsionanti. Fra i lipidi si trovano anche molti ormoni, le vitamine liposolubili, le prostaglandine e le cere: queste ultime sono sostanze di rivestimento della superficie di molti organismi, soprattutto vegetali. Molti lipidi derivano dagli acidi grassi, cioè da acidi carbossilici alifatici, la maggior parte dei quali a catena idrocarburica lineare con numero pari di atomi di carbonio (da 4 a 24) . Possono essere saturi, cioè non portare doppi legami, o insaturi, con un numero di doppi legami che può arrivare a 6. I lipidi che sono esteri degli acidi grassi hanno due ruoli . -1 fondamentali: servono come fonte di energia (9 Kcal g ) e sono componenti delle membrane cellulari. I lipidi che non si possono mettere in relazione con gli acidi grassi sono ormoni o vitamine. 225 226 Capitolo XII Classificazione dei lipidi I lipidi possono essere divisi in due categorie principali: 1) lipidi semplici o non saponificabili1 non idrolizzabili, che comprendono terpeni, steroidi, vitamine, ormoni. 2) lipidi complessi, detti anche lipidi saponificabili. Le classi principali dei lipidi complessi sono gli acilgliceroli, i glicerofosfolipidi, gli sfingolipidi e le cere. In questa sede verranno trattati principalmente i lipidi complessi, perché sono molto importanti per la vita e le funzioni cellulari. Tuttavia occorre fare un cenno anche sui lipidi semplici o non saponificabili, riservandosi di ampliare la trattazione delle loro caratteristiche e delle loro funzioni eventualmente in un altro capitolo. Lipidi semplici In questi composti si nota una grande eterogeneità di strutture in cui tuttavia si può notare la presenza di un precursore comune, l’isoprene. L’isoprene (2-metil-1,3-butadiene) è un idrocarburo alifatico a 5 atomi di carbonio che non si trova libero in natura perché molto instabile, ma il suo scheletro carbonioso si ritrova in un gran numero di molecole dette appunto isoprenoidi. Figura 1. Formula di struttura dell'isoprene (2-metil-1,3-butadiene) 1 Il termine saponificazione è comunemente utilizzato in riferimento alla reazione di un idrossido di un metallo alcalino (base) con un grasso o un olio che dà origine al sapone. La saponificazione è l'idrolisi di un estere in condizioni basiche e provoca la formazione di un alcool e del sale dell'acido corrispondente. I saponi sono di solito sali di sodio di acidi carbossilici a lunga catena. Lipidi e membrane cellulari 227 A questa struttura isoprenoide si riconduce una importante gruppo di lipidi, quella dei terpeni, una classe di idrocarburi insaturi costituiti da multipli dell'unità isoprenica (C5) che possono essere lineari, ciclici o misti. I terpeni sono costituiti da un numero generalmente pari di molecole di isoprene che risultano legate quasi sempre “testa-coda” , ma talvolta anche “coda-coda” (Figura 2). Nella reazione di condensazione delle unità isopreniche si possono verificare reazioni di idrogenazione, deidrogenazione e formazione di derivati ciclici. Figura 2. Schema delle modalità di condensazione di unità isopreniche Un monoterpene (C10) è formato da due unità isopreniche, i sesquiterpeni (C15) sono formati da tre, un diterpene (C20) da quattro, un triterpene (C30) da sei, un tetraterpene (C40) da otto. I monoterpeni sono i componenti principali degli oli essenziali delle piante, miscele di composti che danno alle piante officinali il caratteristico aroma. Sono da annoverarsi fra questi il geraniolo, la canfora, il mentolo, il limonene. I sesquiterpeni sono generalmente poco studiati e si trovano negli olii essenziali come i monoterpeni. Il loro significato fisiologico forse risiede nel fatto che sono precursori di sostanze ormonali vegetali o antifungali. I diterpeni includono il fitolo che è un costituente essenziale della clorofilla (Figura 3a) e le gibberelline, ormoni prodotti dai tessuti vegetali in accrescimento e trasportati dal sistema vascolare delle piante, che hanno lo scopo di favorirne la crescita. Tra i triterpeni sono inclusi lo squalene (Figura 3b) e il lanosterolo che sono precursori del colesterolo e di altri steroidi. 228 Capitolo XII Sono sempre sostanze isoprenoidi, e precisamente tetraterpeni, i carotenoidi e le xantofille, importantissimi pigmenti accessori che fiancheggiano la clorofilla nella cattura della luce per la fotosintesi. Figura 3. Struttura del fitolo (a), dello squalene (b) e della vitamina A (c) I terpeni possono contenere sostanze isoprenoidi più o meno modificate (terpenoidi) fra i quali rivestono particolare importanza le vitamine liposolubili come la A (Figura 3c), la D, la E e la K. Vengono così chiamate, perché possono sciogliersi nei grassi e la loro assunzione è vincolata dalla presenza dei grassi nella nostra alimentazione. Infatti, è grazie a loro che vengono introdotte nel nostro corpo e assorbite a livello dell'intestino. Queste molecole sono indispensabili per controllare e regolare molti processi metabolici. La vitamina A si chiama anche retinolo e il nostro corpo la ricava anche da provitamine (cioè sostanze che possono essere convertite in vitamine), come il carotene nelle sue varie forme, che è il pigmento giallo arancio di frutta e verdura. Questa vitamina ha un ruolo molto importante nella formazione di una sostanza che ci permette di vedere, la rodopsina. La vitamina D o calciferolo regola il metabolismo del calcio e la sua carenza provoca rachitismo nei bambini e osteomalacia nell'adulto. La vitamina E detta anche tocoferolo, è la sostanza che protegge i lipidi dall'ossidazione e impedisce la formazione dei radicali liberi, che causano l'invecchiamento delle cellule. Mantiene inoltre l'integrità dei globuli rossi, che in carenza di questa vitamina diventano fragili. Infine la vitamina K viene prodotta principalmente dalla Lipidi e membrane cellulari 229 flora batterica intestinale, anche se può essere assunta con il cibo. La sua carenza provoca emorragie, poiché la sua funzione è quella di presiedere alla formazione delle sostanze coagulanti del sangue, che avviene nel fegato. Gli steroidi hanno struttura a quattro anelli condensati che conferiscono loro l’apolarità, la struttura base è quella del ciclopentanoperidrofenantrene (Figura 4). Figura 4. Struttura schematica dell'unità di base degli steroidi Quando uno steroide contiene uno o più gruppi ossidrilici e non ha gruppi carbonilici o carbossilici viene detto sterolo. Un tipico rappresentante di questa categoria è il colesterolo che è precursore di alcuni ormoni steroidei, quali l'aldosterone, il cortisolo, il progesterone, ecc. e della vitamina D. E’ inoltre uno dei componenti principali delle membrane plasmatiche delle cellule animali, ma meno rappresentato nelle membrane degli organelli sub-cellulari. Gli ormoni sono sostanze che non danno luogo a nuovi processi biochimici ma modificano quelli già esistenti nell’organismo. Alcuni di essi, che hanno struttura proteica o che derivano dagli amminoacidi, sono idrofili e quindi non riescono neppure a superare la membrana cellulare. Gli ormoni steroidei invece sono liposolubili e riescono a passare nel citoplasma cellulare, ma tutti poi si legano a recettori specifici localizzati sulla membrana, sulla parte esterna i primi e sulla interna i secondi. Gli ormoni sono importanti perché regolano le reazioni dell’organismo. In pratica inducono, mediante un segnale di natura chimica, una enorme amplificazione di specifiche reazioni cellulari. 230 Capitolo XII Lipidi complessi Le principali classi di lipidi complessi sono due: gli acilgliceroli ed i fosfogliceridi. Questi due tipi di composti sono assai simili dal punto di vista chimico. Sono entrambi costituiti da una molecola di glicerolo a cui sono legate con legame estere nel primo caso soltanto molecole di acidi grassi e nel secondo due molecole di acidi grassi e una molecola di acido fosforico, che costituisce un acido fosfatidico. A sua volta, un –OH acido dell’acido fosforico può essere esterificato con un gruppo alcolico di un’altra molecola organica. Acilgliceroli e fosfogliceridi sono i lipidi più rappresentati nel mondo vivente, e sono importanti dal punto di vista biologico perché hanno una funzione sia energetica che strutturale. Prima tuttavia di esaminare queste molecole è necessario riportare le proprietà degli acidi grassi, costituenti fondamentali di questa classe di composti. Gli acidi grassi Gli acidi grassi sono lipidi semplici e sono i costituenti più importanti dei lipidi saponificabili. In natura si trovano liberi solo occasionalmente, mentre è normale trovarli sotto forma di esteri o di ammidi. Sono acidi monocarbossilici, cioè che contengono un solo gruppo – COOH a cui è legata una catena alifatica con un numero di atomi di carbonio, in genere pari, da 4 a 24. La lunghezza della catena li rende più o meno fortemente idrofobici. Hanno come formula generale: CH3(CH2)n COOH dove il numero n di gruppi –CH2- caratterizza il tipo di acido. Poiché le caratteristiche dei lipidi dipendono soprattutto dagli acidi che li costituiscono, è importante conoscerne struttura e proprietà. I più comuni acidi grassi naturali hanno un numero pari di atomi di C perché vengono sintetizzati da unità bicarboniose, derivate dall’acetil coenzima A, un composto presente in tutte le cellule. Gli acidi grassi possono essere saturi, cioè senza doppi legami tra atomi di carbonio e non possono (come dice il nome) aggiungere altri Lipidi e membrane cellulari 231 atomi di idrogeno. Gli acidi grassi insaturi, mono- o poli-insaturi, hanno uno o più legami doppi fra gli atomi di carbonio (fino a un massimo di 6) che possono rompersi e legare altri atomi di idrogeno (Figura 5). Figura 5. Rappresentazione schematica di un acido grasso saturo e insaturo E’ importante ricordare che in tutti i principali acidi grassi insaturi presenti negli esseri viventi i sostituenti legati ai carboni che portano il doppio legame sono in posizione cis, cioè dalla stessa parte rispetto al piano del doppio legame (Figura 6). Figura 6. Schema di doppi legami in configurazione cis o trans Gli acidi grassi saturi hanno una configurazione spaziale lineare che consente loro di disporsi in modo ordinato nei trigliceridi, questa disposizione facilita le interazioni molecolari (ponti di idrogeno) e di conseguenza porta ad un punto di fusione elevato. Capitolo XII 232 Gli acidi grassi mono e polinsaturi, invece, hanno le molecole "piegate" che non riescono a disporsi in modo ordinato, per cui i legami fra le catene idrocarburiche sono in numero inferiore rispetto ai saturi e la temperatura di fusione è più bassa. Maggiore infatti è il numero di doppi legami minore è la temperatura di fusione. Questa caratteristica ci consente di distinguere facilmente i diversi tipi di acidi grassi: i saturi sono solidi a temperatura ambiente (pensiamo al burro o al grasso della carne), i monoinsaturi e i polinsaturi sono liquidi. Se raffreddiamo un alimento grasso allo stato liquido, il primo a solidificare sarà costituito da monoinsaturi. Infatti se mettiamo in frigorifero l'olio di oliva solidifica (è costituito principalmente da monoinsaturi), mentre l'olio di girasole (costituito principalmente da polinsaturi) rimane liquido. Ovviamente non mancano le eccezioni: esistono grassi saturi liquidi a temperatura ambiente e grassi polinsaturi solidi. I principali acidi grassi presenti in natura sono riportati in Tabella 1. Tabella 1. Gli acidi grassi più comuni in natura Numero carboni Nome comune Struttura Acidi grassi saturi 12 14 16 18 20 22 24 Acido laurico Acido miristico Acido palmitico Acido stearico Acido arachidico Acido beenico Acido lignocerico CH3(CH2)10COOH CH3(CH2)12COOH CH3(CH2)14COOH CH3(CH2)16COOH CH3(CH2)18COOH CH3(CH2)20COOH CH3(CH2)22COOH Acidi grassi in saturi (tutti i doppi legami hanno configurazione cis) 16 18 18 18 18 20 24 Acido palmitoleico Acido oleico Acido linoleico Acido α-linolenico Acido γ-linolenico Acido arachidonico Acido nervonico CH3(CH2)5CH=CH(CH2)7COOH CH3(CH2)7CH=CH(CH2)7COOH CH3(CH2)4(CH=CHCH2)2COOH CH3CH2(CH=CHCH2)3 (CH2)6COOH CH3(CH2)4(CH=CHCH2)3COOH CH3(CH2)4(CH=CHCH2)4(CH2)2COOH CH3(CH2)7CH=CH(CH2)13COOH Lipidi e membrane cellulari 233 Alcuni acidi grassi, detti essenziali, non possono essere sintetizzati dal nostro organismo ed è perciò indispensabile che essi siano forniti con l’alimentazione. Essi sono: l’acido linoleico, l’acido linolenico, l’acido arachidonico. Acidi grassi saturi a basso numero di atomi di C si trovano in piccole quantità in alcuni grassi e oli, specialmente nel burro; altri in oli vegetali, sotto forma di esteri. Fra gli acidi grassi più importanti e abbondanti in natura sono gli acidi miristico, palmitico e stearico (rispettivamente a 14, 16 e 18 atomi di C), che si trovano come esteri in tutti gli oli vegetali e animali. Acidi grassi saturi a catena molto lunga si trovano anche nelle cere, di cui si parlerà in seguito. In tutti gli esseri viventi sono pochi gli acidi grassi a numero dispari di atomi di carbonio, mentre nei batteri non ci sono in genere acidi grassi con più di un doppio legame. Fra i monoinsaturi il più importante è l’acido oleico, la cui formula è: CH3 (CH2)7 CH=CH (CH2)7 COOH L’acido oleico si trova nella maggior parte dei grassi e degli oli sia di origine animale che vegetale e probabilmente è l’acido insaturo più diffuso in natura. Tra gli acidi grassi polinsaturi essenziali sono presenti gli omega-3 (acido α-linolenico) e gli omega-6 (acido linoleico). Il numero dopo la parola omega indica la posizione del primo doppio legame a partire dall’atomo di carbonio più lontano dal gruppo carbossilico (che è per questo denominato carbonio omega, l'ultima lettera dell'alfabeto greco). Gli omega-3 sono contenuti soprattutto nei grassi del pesce (salmone, sgombri, acciughe ecc.) e nell'olio di pesce. Gli omega-6 sono contenuti soprattutto negli oli vegetali (oli di girasole e di mais che però non devono essere cotti), ma anche in cibi proteici, nelle verdure e nei cereali. L’insieme degli acidi grassi essenziali linoleico, αlinolenico e arachidonico è definito vitamina F. Gli acilgliceroli Gli acilgliceroli costituiscono la classe di lipidi più rappresentata negli organismi e una delle maggiori fonti di energia. Sono i lipidi di 234 Capitolo XII deposito nei vegetali, soprattutto in forma di oli e negli animali come grassi nelle cellule adipose. Gli acilgliceroli sono molecole che derivano dall’esterificazione del glicerolo con uno, due, o tre molecole di acidi grassi, uguali o diversi tra loro (R1, R2, R3) con l’eliminazione di una, due o tre molecole d’acqua (Figura 7). A seconda del numero di molecole di acidi grassi presenti si chiamano rispettivamente monoacilgliceroli (monogliceridi), diacilgliceroli (digliceridi) o triacilgliceroli (trigliceridi). Figura 7. Reazione di esterificazione tra glicerolo e acidi grassi I trigliceridi differiscono per il tipo e la posizione degli acidi grassi legati. Nei trigliceridi semplici gli acidi grassi sono tutti uguali, mentre i trigliceridi misti contengono più di un tipo di acido grasso. La maggior parte dei grassi naturali è costituita da trigliceridi semplici e misti. I trigliceridi sono sempre insolubili in acqua e in tutti i solventi polari. A temperatura ambiente i trigliceridi possono essere sia solidi che liquidi, nel primo caso vengono detti grassi, nel secondo oli. Questa differenza dipende dal grado di interazione delle molecole. Infatti i grassi contengono acidi grassi saturi, in cui le lunghe catene idrocarburiche sono lineari e quindi possono legarsi in modo regolare con legami idrofobici o di van der Waals, formando strutture compatte che danno origine ad uno stato solido. Viceversa, se gli acidi grassi costituenti sono insaturi, e le catene sono dalla stessa parte del piano individuato dal doppio legame o dai doppi legami (forme cis), come av- Lipidi e membrane cellulari 235 viene molto spesso in natura, si avrà maggior disordine e minore impaccamento delle varie catene, con conseguente stato fisico meno ordinato e quindi si avrà la forma liquida. Ovviamente, cambiando le condizioni di temperatura lo stato fisico può cambiare (col freddo gli oli si rapprendono, mentre col caldo i grassi liquefanno). In acqua i trigliceridi liquidi formano uno strato superficiale, limitando così la zona di contatto. I triacilgliceroli hanno un punto di fusione un poco più elevato degli acidi grassi che li costituiscono e ciò è dovuto in parte ad un maggior peso molecolare e in parte a una diversa struttura tridimensionale assunta dalle molecole cioè a una diversa forma dei cristalli. Proprietà chimiche degli acilgliceroli: Le reazioni più importanti a cui possono essere sottoposti gli acidi grassi sono: 1) Idrolisi del legame estere: Gli acilgliceroli possono essere idrolizzati da alcuni batteri ed altri microrganismi, con produzione di acidi grassi liberi e glicerolo. Questa idrolisi spesso è spontanea, non esiste grasso o olio naturale che non contenga anche in piccola percentuale, acidi grassi liberi. 2) Idrogenazione dei doppi legami degli acidi grassi: Gli oli commerciali possono essere convertiti in grassi per idrogenazione del doppio legame, a drastiche temperature e pressioni. I nutrizionisti raccomandano l’uso di oli ricchi di acidi grassi insaturi di origine vegetale perché non contengono colesterolo. 3) Ossidazione dei doppi legami degli acidi grassi: Questo fenomeno è indicato in genere col nome di irrancidimento che consiste nell’attacco dell’ossigeno atmosferico sul C in α del doppio legame. Questo fenomeno è facilitato dalla luce, dal calore, dall’umidità, da agitazioni meccaniche, da tracce di metalli come ferro e rame e chiaramente dalla presenza di ossigeno. Poiché le reazioni che portano all’irrancidimento sono in massima parte radicaliche, un modo di contrastarle è quello di aggiungere antiossidanti. I glicerofosfolipidi Sono i lipidi più abbondanti in natura dopo oli e grassi: sono presenti in tutte le cellule vegetali ed animali e sono i costituenti principali delle membrane biologiche. Capitolo XII 236 La molecola di partenza è il glicerolo-3-fosfato, in cui un –OH del glicerolo è esterificato in C1 e C2 con acidi grassi e in C3 con una molecola di acido fosforico. L’acido fosforico dell’acido fosfatidico può essere esterificato a sua volta con un altro gruppo derivato di un alcool polare (X-OH) (Figura 8). Figura 8. Formula generale del glicerolo-3-fosfato e di un glicerofosfolipide I glicerofosfolipidi più semplici, in cui X è un atomo di idrogeno, sono gli acidi fosfatidici. Nei glicerofosfolipidi più comuni le teste polari sono costituite da alcol polari per formare classi di sostanze diverse (Tabella 2). Tabella 2. Le classi più comuni di glicerofosfolipidi Nome di X-OH Nome del fosfolipide Acqua Acido fosfatidico Etanolammina Fosfatidiletanolammina Colina Fosfatidilcolina Serina Fosfatidilserina Mio-inositolo Fosfatidilinositolo Glicerolo Fosfatidilglicerolo Fosfatidilglicerolo Difosfatidilglicerolo Lipidi e membrane cellulari 237 Quando l’amminoalcol è la colina si hanno le lecitine, quando è l’etanolammina si hanno le cefaline, che sono i più importanti componenti delle membrane della maggior parte delle cellule animali. Le lecitine si trovano nel cervello, nel tessuto nervoso e nel citoplasma di tutte le cellule, nel rosso d’uovo e sono abbondanti nei semi di soia. Sono eccellenti agenti emulsionanti largamente usati dalle industrie alimentari (maionese, cioccolata). Questa loro caratteristica rende le lecitine un importante mezzo di trasporto dei grassi da un tessuto ad un altro. Gli stessi triacilgliceroli, per superare le barriera delle membrane cellulari devono essere convertiti in lecitine. Alcuni morsi di serpente (specie del cobra) sono spesso fatali perché il veleno contiene un enzima che demolisce le lecitine della membrana dei globuli rossi e promuove quindi l’emolisi, con dispersione del materiale cellulare contenuto nei globuli. La presenza del fosfato e di una molecola organica polare conferisce ai fosfogliceridi caratteristiche particolari assai diverse da quelle degli acilgliceroli. Infatti parte della molecola dei fosfolipidi è altamente polare ed idrofila (gruppo fosforil-X), al contrario della parte rimanente che è nettamente apolare e quindi idrofoba. Le molecole dei fosfogliceridi sono quindi anfipatiche ed in acqua hanno comportamento caratteristico. Le molecole del lipide infatti, in soluzione acquosa, tendono spontaneamente ad orientarsi in modo da presentare al solvente le teste polari, mentre le lunghe code apolari interagiscono fra loro. Si formano così aggregati molecolari di forma rotonda, detti micelle, in cui le teste polari sono all’esterno e le code apolari all’interno (Figura 9). In questo modo viene ad annullarsi l’interazione con le molecole di acqua. 238 Capitolo XII Figura 9. Sezione di una micella In mezzo acquoso esiste un’altro tipo di disposizione delle molecole di fosfogliceridi che possono aggregarsi in modo da formare foglietti costituiti da un doppio strato di molecole, disposte parallelamente le une alle altre e orientate in modo da esporre al solvente solo le teste polari e da mantenere le code apolari a contatto fra loro all’interno del foglietto (Figura 10). Questa disposizione si trova nelle membrane cellulari ed è di enorme importanza biologica, infatti essa permette la formazione di aggregati molecolari di dimensioni molto superiori a quelle delle micelle, che in genere sono inferiori a 200 Å. Figura 10. Le due strutture principali dei lipidi in soluzione acquosa Lipidi e membrane cellulari 239 Gli sfingolipidi Gli sfingolipidi si dividono in due categorie principali: le sfingomieline ed i glicosfingolipidi. Le sfingomieline sono simili ai fosfogliceridi in quanto contengono sia il gruppo fosfato che un amminoalcol, come la colina o la etanolammina ma, anziché contenere una molecola di glicerolo hanno un amminoalcol insaturo a lunga catena, chiamato sfingosina (o diidrosfingosina). Gli acidi grassi sono molto più lunghi che non nei trigliceridi e formano la base strutturale di tutti gli sfingolipidi: la ceramide. Le sfingomieline si trovano nella guaina mielinica che avvolge le fibre nervose e costituiscono un isolante elettrico molto efficace. In alcuni casi, un difetto nella struttura della guaina mielinica provoca una scarsa protezione della fibra nervosa: ciò può causare la demielinizzazione della sclerosi multipla. I glicosfingolipidi sono sfingolipidi che contengono residui di carboidrati (glucosio e lattosio fra i più comuni). La parte glucidica ha un ruolo importante nella funzionalità delle membrane plasmatiche. Le cere Le cere sono lipidi solidi a temperatura ambiente e sono esteri di un acido grasso con un alcol monovalente alifatico a lunga catena (di solito con numero di C da 26 a 34) o con uno sterolo. Sono molto più resistenti alla saponificazione dei trigliceridi e, poiché sono formati da alcoli ed acidi carbossilici saturi, sono molto resistenti all’ossidazione. Le cere hanno in natura funzioni di protezione e ricoprono con un velo idrorepellente la pelle, i peli, le foglie, la frutta, impedendo una eccessiva traspirazione e quindi eccessive perdite di acqua dai tessuti. Le cere hanno una certa importanza commerciale perché possono venir usate nella fabbricazione di candele, lucido da scarpe e pavimenti, vernici, cosmetici. Le membrane cellulari La cellula dei sistemi viventi deve contenere tutto quanto è necessario per potersi riprodurre e per garantire la sua sopravvivenza: deve prelevare ossigeno dall’esterno per le proprie reazioni metaboliche ed 240 Capitolo XII eliminare l’anidride carbonica e i materiali di scarto in genere, e deve saper selezionare i prodotti giusti tra quelli che ha a disposizione. La comunicazione con l’esterno e fra le cellule e lo scambio di materiali si realizza attraverso le membrane cellulari che delimitano la cellula e gli organelli subcellulari. Le membrane cellulari espletano funzioni indispensabili alla vita: infatti i due processi principali di conversione dell’energia nei sistemi biologici sono situati in sistemi di membrane che contengono strutture ordinate di enzimi e di altre proteine. La fotosintesi, processo nel quale l’energia luminosa viene convertita in energia chimica ha luogo nelle membrane tilacoidi del cloroplasto, mentre la fosforilazione ossidativa, in cui viene formato ATP dall’ossidazione di sostanze nutrienti ha luogo nella membrana interna dei mitocondri. L’architettura di base con cui sono costruite tutte le membrane cellulari e quelle che delimitano le formazioni endocellulari (mitocondri, cloroplasti, gliossisomi ecc.) si avvale sempre degli stessi elementi strutturali, foglietti composti principalmente da proteine e fosfolipidi, generalmente sistemati in un doppio strato. Si hanno lamine molecolari anche di notevoli dimensioni che possono ripiegarsi su se stesse circoscrivendo uno spazio cavo ed isolandolo dall’esterno. Questa formazione è stata verificata mediante studi ai raggi X. Un fosfolipide di membrana si può rappresentare schematicamente con una porzione polare, la parte contenente il gruppo fosfato legato all’amminoalcol e indicato come “testa” per la sua forma a globo e una porzione non polare contenente le lunghe catene idrocarburiche degli acidi grassi, indicata cone “coda”. Quando le molecole fosfolipidiche si trovano circondate da molecole di acqua le code idrofobe tendono a fuggirla mentre le teste idrofile rimangono esposte all’acqua formando il doppio strato fosfolipidico caratteristico delle membrane cellulari. In realtà la struttura delle membrane cellulari è molto più complessa: non è infatti un semplice rivestimento che richiude tutte le formazioni cellulari ma ha compiti assai più importanti e diversificati. Infatti, sebbene i lipidi svolgano un ruolo determinante nel caratterizzare funzioni e struttura delle membrane cellulari è evidente che il doppio strato lipidico può isolare perfettamente la cellula dall’ambiente circostante ma non le permette di comunicare con l’esterno. In pratica nes- Lipidi e membrane cellulari 241 sun tipo di molecole potrebbe attraversare questa barriera, tranne O2 ed N2 fra le molecole idrofobe e H2O, CO2, urea e glicerolo fra le piccole molecole polari e neutre. La cellula o l’organello subcellulare diventerebbero così impermeabili agli ioni ed alla maggior parte delle molecole polari. Le membrane cellulari devono isolare la cellula dall’ambiente o un compartimento sub-cellulare dall’altro ma non sono barriere invalicabili: hanno una permeabilità altamente selettiva, dovuta alla presenza di canali e di pompe specifici, per permettere gli scambi e le interazioni con le sostanze necessarie al funzionamento delle strutture che racchiudono. In vista di tutte queste funzioni, le membrane cellulari consistono, oltre che di fosfolipidi, anche di altri tipi di lipidi e alcuni glicolipidi, di proteine semplici o coniugate, spesso glicoproteine, legate più o meno strettamente al doppio strato lipidico. Hanno uno spessore che oscilla fra i 5 e i 10 nm. Pur variando da cellula a cellula mediamente una membrana cellulare è costituita da circa il 40% di lipidi e per il 60% di proteine. Alcune membrane cellulari, come quelle della cellula nervosa contengono più del 75% di lipidi e sono questi a caratterizzare le proprietà strutturali e funzionali di esse. La struttura molecolare della membrana cellulare oggi più comunemente accettata è il modello a mosaico fluido di Singer e Nicolson (Figura 11). Le membrane cellulari, secondo questo modello, sono soluzioni di proteine globulari in una lamina bidimensionale di lipidi orientati. La struttura base è il doppio strato fosfolipidico, in cui si trovano anche altri lipidi, come il colesterolo, e proteine globulari che interagiscono con il doppio strato come fossero in soluzione. Ciò permette a molte proteine di membrana di spostarsi lungo il doppio strato lipidico. 242 Capitolo XII Figura 11. Modello a mosaico fluido di una membrana Secondo questo modello, le membrane cellulari non avrebbero struttura rigida ma semifluida, controllata dalla presenza di acidi grassi insaturi le cui molecole non sono lineari ma angolari, in quanto i sostituenti sui doppi legami in natura in genere si trovano in posizione cis rispetto al doppio legame, con aumento del disordine molecolare e diminuzione dell’impaccamento delle code idrofobiche con conseguente aumento della fluidità. Il fatto che il doppio strato sia fluido è molto importante dal punto di vista fisiologico: infatti una eccessiva rigidità delle membrane cellulari comporterebbe la rottura di alcune cellule a seguito di determinati movimenti. Nonostante la fluidità delle membrane ognuno dei due strati ha una composizione lipidica diversa: nello strato esterno prevale la fosfatidilcolina, in quello interno Lipidi e membrane cellulari 243 la fosfatidilserina, la fosfatidiletanolammina ed il fosfatidilinositolo. La membrana perciò risulta asimmetrica. Le proteine di membrana hanno funzione di supporto, cioè di tenere insieme le piccole molecole di fosfolipidi, o di recettore, cioè possono interagire in modo specifico con i segnali chimici di alcune sostanze, come regolatori di crescita, ormoni, prostaglandine, che possono essere libere o legate alla superficie di altre cellule. Altre proteine controllano l’entrata e l’uscita nella cellula di determinate specie chimiche, in modo che la concentrazione intracellulare rimanga ottimale. Altre ancora sono enzimi che svolgono la loro azione ancorati alla membrana. Le modalità del trasporto transmembrana dei materiali nutritivi o di scarto di una cellula dipendono dalla natura chimica della molecola trasportata. Ossigeno, azoto, anidride carbonica e acqua si muovono per diffusione semplice, secondo un gradiente di concentrazione (cioè dal più concentrato al meno concentrato). Tutte le altre molecole necessitano di una proteina trasportatrice, detta permeasi. Se il trasporto avviene secondo gradiente di concentrazione, cioè dalle zone a maggior concentrazione a quelle a concentrazione minore la proteina serve solo per abbassare l’energia di attivazione del processo di trasferimento, comportandosi quindi come un enzima. E’ un trasporto passivo che, come la semplice diffusione, non necessita di energia, a parte una energia di attivazione iniziale. Se però il trasporto di una molecola deve avvenire contro gradiente, allora le molecole devono essere pompate nella direzione voluta, cosa che richiede dispendio di energia. In genere, l’energia per questo trasporto attivo è fornita da enzimi che idrolizzano l’ATP, detti ATP-asi. I soluti in questo modo sono messi in grado di attraversare la membrana. Il trasporto in gradiente di concentrazione si può realizzare anche attraverso canali ionici, strutture proteiche colme d’acqua, che forniscono il mezzo in cui le sostanze ioniche possono muoversi. METABOLISMO DEI LIPIDI Come già detto nel capitolo dedicato alla struttura dei lipidi, sotto questo nome si intendono compresi i lipidi semplici, non saponificabili e non idrolizzabili, che comprendono terpeni, steroidi, vitamine, ormoni, ecc. ed i lipidi complessi, detti anche lipidi saponificabili, che comprendono gli acilgliceroli, i fosfogliceridi, gli sfingolipidi e le cere. La maggior parte dei grassi è costituita da triacilgliceroli, detti anche trigliceridi, e in misura minore da fosfolipidi. Ricordiamo che, come già visto, un trigliceride è composto da glicerolo esterificato con tre molecole di acidi grassi, uguali o diversi, saturi o insaturi. Le lunghe catene alchiliche di acidi grassi sono essenzialmente idrocarburi, strutture altamente ridotte, con una elevata energia per la completa ossidazione (38 KJ/g), più del doppio di quella di carboidrati o di proteine dello stesso peso. Un fosfolipide invece è formato da due catene di acidi grassi legati al glicerolo 3-fosfato, mentre l’acido fosforico che esterifica il terzo OH del glicerolo è a sua volta esterificato con diversi tipi di ammino alcoli (colina, colammina, serina ecc.). La maggior parte dei lipidi delle piante è formata da glicerolo e acidi grassi. I lipidi di membrana sono glicosilgliceroli o fosfogliceridi, mentre il materiale di riserva è costituito per lo più da triacilgliceroli. Per questo motivo in questa sede ci occuperemo principalmente del catabolismo e della biosintesi degli acilgliceroli, dei fosfogliceridi, e degli altri lipidi di membrana, rimandando al capitolo dei lipidi per la struttura di sfingolipidi e cere. Reazioni del catabolismo dei trigliceridi Il primo passaggio nell’utilizzazione dei grassi come fonte di energia è l’idrolisi dei trigliceridi da parte di una lipasi (che appartiene al gruppo delle esterasi, parte a sua volta di una categoria più vasta, quella delle idrolasi.) Il trigliceride viene scomposto in glicerolo e tre molecole di acidi grassi, uguali o diversi fra loro. Negli animali l’attività della lipasi della cellula adiposa è regolata da ormoni, come adrenalina, noradrenalina, glucagone ed ormone adrenocorticotropo. 245 246 Capitolo XIII L’aumento dei livelli di adenosina monofosfato ciclico (AMP ciclico) stimola anche una proteina chinasi che attiva la lipasi fosforilandola, e provocando così la lipolisi. Al contrario, l’insulina inibisce la lipolisi. Il glicerolo formato nella lipolisi viene fosforilato dall’ATP ed ossidato a diidrossiaceton fosfato (DHAP), che a sua volta viene isomerizzato a gliceraldeide-3-fosfato, un intermedio sia della via glicolitica che della gluconeogenesi. Quindi il glicerolo può essere convertito in piruvato o in glucosio nell’organismo che contenga gli enzimi necessari (ad es. nel fegato). Il processo inverso può avvenire mediante riduzione del diidrossi acetonfosfato in glicerolo-3-fosfato: l’idrolisi da parte di una fosfatasi produce infine glicerolo. Quindi glicerolo ed intermedi glicolitici sono facilmente interconvertibili. Catabolismo degli acidi grassi (β-ossidazione) Gli acidi grassi vengono degradati mediante rimozione sequenziale di unità bicarboniose. Il processo prende il nome di β-ossidazione. In esperimenti condotti all’inizio del secolo scorso da Franz Knoop, si era trovato che, quando venivano somministrati ad alcuni cani cibi contenenti acidi grassi a catena lineare con l’ultimo atomo di C legato ad un gruppo fenilico, nelle loro urine veniva trovato un composto simile ma con due atomi di carbonio in meno. Ne fu dedotto che gli acidi grassi venivano degradati mediante ossidazione e rottura del legame fra il 2° e il 3° atomo di C dopo il gruppo –COOH, da qui il nome di β-ossidazione, perché la rottura avviene a livello del carbonio β. Gli acidi grassi vengono ossidati nei mitocondri e devono essere attivati prima di entrare nella matrice mitocondriale, poiché le stesse proprietà che rendono i triacilgliceroli ottime sostanze di riserva a causa della loro relativa stabilità, rappresentano un problema per la loro funzione di combustibili intracellulari. Infatti la difficoltà a rompere i legami carbonio-carbonio degli acidi grassi deve essere superata dall’attivazione del gruppo –COOH sul C1. Questa reazione di attivazione si svolge sulla membrana mitocondriale esterna, ed è catalizzata dalla acil-CoA sintetasi, o acido grasso tiochinasi, che lega il –COOH al Coenzima A (CoA), formando un a- Metabolismo dei lipidi 247 cil-CoA. Ciò consente poi l’ossidazione a tappe del gruppo acilico in posizione 3. E’ stato dimostrato che l’attivazione di un acido grasso si svolge in due tappe: 1. l’acido grasso reagisce con l’ATP per dare un acil-adenilato, una anidride mista in cui il gruppo –COOH di un acido grasso è legato al gruppo fosforico di un AMP. (Si ricordi che acile è la parte di un acido grasso priva del gruppo –OH, R-CO, dove R sta per una catena idrocarburica qualunque). Gli altri due gruppi fosforici vengono rilasciati come pirofosfato, PPi. R-COO- + ATP R-CO-AMP + PPi 2. il gruppo solfidrilico del CoA attacca l’acil-adenilato, che è fortemente legato all’enzima e si formano acil-CoA e AMP. RCO-AMP + HS–CoA R-COO-S-CoA + AMP Il pirofosfato potrà poi venir idrolizzato a sua volta da una fosfatasi, rendendo la reazione irreversibile. Queste reazioni sono liberamente reversibili, infatti la costante di equilibrio per la somma di queste reazioni è vicino a 1. RCOO- + CoA + ATP RCO-CoA + AMP + PPi Viene scisso un legame ad alta energia (fra PPi e AMP) e si forma un legame ad alta energia, il legame tioestere nell’acil-CoA. La reazione viene spostata verso destra perché il pirofosfato viene rapidamente idrolizzato da una pirofosfatasi. La reazione completa sarà: RCOO- + CoA + ATP + H2O RCO-CoA + AMP + 2 Pi + 2H+ Questo rende la reazione favorevole ed irreversibile perché viene idrolizzato l’equivalente di due molecole di ATP, quindi vengono consumati due legami ad alta energia mentre viene formato un solo composto ad alto potere di trasferimento. In biochimica, molte reazioni biosintetiche sono rese irreversibili dalla idrolisi del pirofosfato inorganico. L’intermedio acil-adenilato 248 Capitolo XIII legato all’enzima non è esclusivo della sintesi dell’acil-CoA. Gli aciladenilati si formano spesso quando nelle reazioni biochimiche vengono attivati gruppi carbossilici. Ad esempio, gli amminoacidi vengono attivati per la sintesi delle proteine con un meccanismo simile. Gli acidi grassi vengono attivati sulla membrana mitocondriale esterna mentre la loro ossidazione avviene nella matrice mitocondriale. Le molecole di acil-CoA a catena lunga non attraversano facilmente la membrana mitocondriale interna per cui necessitano di un meccanismo di trasporto. Il trasportatore è la carnitina, un composto zwitterionico che deriva dalla lisina. Il gruppo acile viene trasferito dall’atomo di zolfo del CoA al gruppo -OH della carnitina, formando acil-carnitina. La reazione è catalizzata dalla carnitina aciltransferasi I, localizzata sulla faccia esterna della membrana mitocondriale interna (verso lo spazio intermembrana). L’acil-carnitina viene quindi trasportata attraverso la membrana mitocondriale interna da una traslocasi (trasportatore acilcarnitina/carnitina). Il gruppo acile viene poi nuovamente trasportato sul CoA con una reazione catalizzata dalla carnitina aciltransferasi II, che è posta sul lato interno della membrana interna (verso la matrice mitocondriale). Questo enzima rigenera l’acil-CoA che viene rilasciato insieme alla carnitina libera nella matrice. La carnitina rientra nello spazio intermembrana tramite il trasportatore acil-carnitina/carnitina scambiandosi con una acil-carnitina che entra (Figura 1). La reazione è termodinamicamente possibile perché il legame tra il gruppo -OH della carnitina e l’acile ha un alto potenziale di trasferimento del gruppo. L’acil-CoA può così andare incontro alle reazioni della β-ossidazione. L’ossidazione mitocondriale degli acidi grassi ha luogo in tre fasi. Nella prima fase (la β-ossidazione) gli acidi grassi vanno incontro al distacco ossidativo ripetuto di unità bicarboniose nella forma di acetilCoA, iniziando dall’estremità carbossilica della catena idrocarburica. Metabolismo dei lipidi 249 Figura 1. Ingresso degli acidi grassi nella matrice mitocondriale. Nella seconda fase l’unità acetilica dell’acetil-CoA viene ossidata a CO2 nel ciclo dell’acido citrico, che è anche esso localizzato nella matrice mitocondriale. L’acetil-CoA prodotto dall’ossidazione degli acidi grassi entra quindi nella via di ossidazione dell’acetil-CoA derivato anche dal glucosio ad opera dalla glicolisi e dell’ossidazione del piruvato. Le prime due fasi dell’ossidazione degli acidi grassi convertono i trasportatori di elettroni NAD+ e FAD nella loro forma ridotta NADH e FADH2, i quali nella terza fase donano gli elettroni ricevuti alla catena respiratoria dei mitocondri; gli elettroni arrivano così all’ossigeno con la concomitante fosforilazione di ADP ad ATP (Figura 2). L’energia rilasciata dall’ossidazione degli acidi grassi viene così conservata sotto forma di ATP. 250 Capitolo XIII Figura 2. Fasi dell'ossidazione degli acidi grassi. La β-ossidazione, ossia la prima fase dell’ossidazione degli acidi grassi saturi, avviene in quattro reazioni fondamentali: 1. 2. 3. 4. una reazione di ossidazione che dipende dal FAD; una reazione di idratazione; una reazione di ossidazione che dipende dal NAD+; una reazione di tiolisi che dipende dal CoA. Di conseguenza, la catena dell’acido grasso viene accorciata di due atomi di carbonio alla volta e si generano FADH2, NADH e acetilCoA. Metabolismo dei lipidi 251 La prima reazione in ciascun ciclo di degradazione è l’ossidazione dell’acil-CoA ad opera di una acil-CoA deidrogenasi che forma un enoil-CoA con un doppio legame trans fra il C-2 ed il C-3. acil-CoA+ E-FAD → trans-∆2–enoil-CoA + E-FADH2 (con E-FAD si indica che il FAD è un gruppo prostetico, cioè un coenzima legato covalentemente all’enzima deidrogenasi). L’accettore di elettroni in questo caso è il FAD e non il NAD+ perché il ∆G di questa reazione è insufficiente a ridurre il NAD+. Il FADH2 , gruppo prostetico dell’Acil-CoA deidrogenasi ridotta, verrà riossidato nella catena respiratoria di trasporto degli elettroni. Gli elettroni del FADH2 infatti vengono trasferiti ad una flavoproteina di trasferimento degli elettroni (ETF) che a sua volta li passerà a una proteina Fe-S detta ETF-ubichinone reduttasi e infine all’ubichinone. L’ubichinone, ridotto ad ubichinolo, dona i suoi elettroni ad alto potenziale al secondo sito di pompaggio di protoni della catena respiratoria. Per ogni FADH2 prodotto in questa deidrogenazione verranno generate due molecole di ATP, come nell’ossidazione da succinato a fumarato del ciclo di Krebs. La seconda reazione è l’idratazione del doppio legame trans fra C2 e C-3 ad opera della enoil-CoA-idratasi. L’idratazione è stereospecifica, come quella del fumarato e dell’aconitato nel ciclo di Krebs. L-3-idrossiacil-CoA trans-∆2–enoil-CoA + H2O La terza reazione è una ossidazione che converte l’OH sul C-3 a gruppo chetonico, generando NADH. L’enzima è la L-3-idrossiacilCoA deidrogenasi, specifica per l’isomero L dell’idrossiacile. L-3-idrossiacil-CoA + NAD+ 3-chetoacil-CoA + NADH + H+ La quarta e ultima reazione è la scissione del L-3 chetoacil-CoA che dà acetil-CoA e acil-CoA (n-2) accorciato di due atomi di carbonio. Questa scissione tiolitica è catalizzata dalla β-chetotiolasi. 252 Capitolo XIII 3-chetoacil-CoA(n) + HS-CoA acetil- CoA + acil-CoA (n-2) L’acil-CoA accorciato subisce poi un altro ciclo di ossidazione a partire dalla reazione catalizzata dalla acil-CoA deidrogenasi (Figura 3). Figura 3. β-ossidazione degli acidi grassi saturi. Le catene di acido grasso contenenti da 12 a 18 atomi di carbonio vengono ossidate dalle acil-CoA deidrogenasi specifiche per le catene lunghe. Le acil-CoA deidrogenasi specifiche per le catene medie ossidano le catene aciliche da 4 a 14 atomi di carbonio, mentre le acilCoA deidrogenasi specifiche per le catene corte agiscono solo sulle catene aciliche lunghe 4 e 6 atomi di carbonio. Gli altri 3 enzimi hanno larga specificità rispetto alla lunghezza delle catene. Metabolismo dei lipidi 253 Le reazioni dell’ossidazione degli acidi grassi, a partire dall’attivazione fino alla reazione conclusiva della β-ossidazione, sono riassunte in Tabella 1. Tabella 1. Principali reazioni nell'ossidazione degli acidi grassi. Reazione Ac. grasso + ATP Carnitina + acil-CoA Enzima R-CO-AMP + PPi acil carnitina + CoA Acil-Co-A sintetasi Carnitina acil tranferasi acil-CoA+ E-FAD → trans-∆2–enoil-CoA + EFADH2 Acil-CoA genasi trans- ∆2 –enoil-CoA + H2O CoA L-3 idrossiacil- Enoil-CoA idratasi L-3idrossiacil-CoA + NAD+ + NADH + H+ 3-chetoacil-CoA 3-chetoacil-CoA(n) + HS-CoA acetil- CoA deidro- L-3-idrossiacilCoA deidrogenasi β-chetotiolasi + acil-CoA (n-2) Calcoliamo ora, come esempio, la resa energetica dell’ossidazione completa di un acido grasso. In ciascun ciclo vengono formate una molecola di FADH2, una di NADH e una di acetil-CoA. La degradazione completa del palmitil-CoA (C16 Acil-CoA) richiede sette cicli di reazioni. Nel 7° ciclo, il C4-chetoacil-CoA viene scisso in due molecole di acetil-CoA. La reazione complessiva è quindi: palmitil-CoA + 7 FAD + 7 NAD+ + 7 CoA + 7 H2O → 8 acetil-CoA + 7 FADH2 + 7 NADH + 7H+ 254 Capitolo XIII Ogni molecola di FADH2 formata durante l’ossidazione degli acidi grassi dona una coppia di elettroni alla catena respiratoria; circa 1,5 molecole di ATP sono generate nel successivo trasferimento degli elettroni all’ossigeno accoppiato alla fosforilazione ossidativa. Il trasferimento di una coppia di elettroni dal NADH all’O2 produce circa 2,5 molecole di ATP (alcuni ricercatori arrotondano questa cifra a 3). Quindi per ogni unità bicarboniosa staccata dall’acido grasso durante la β-ossidazione, si formano quattro molecole di ATP. L’acetil-CoA prodotto nella fase 1 della sequenza di reazioni di ossidazione, può essere ossidato a CO2 e H2O nel ciclo dell’acido citrico. Sommando i bilanci della fase 2 e 3 (vedi anche Figura 2) si ottiene la seguente reazione: 8 acetil-CoA + 16 O2 + 80 Pi + 80 ADP → 8 CoA + 80 ATP + 16 H2O + 16 CO2 Considerando anche gli ATP che si formano nella riossidazione dei coenzimi ridotti durante la β-ossidazione si ottiene la reazione complessiva di ossidazione della nostra molecola di esempio ( il palmitilCoA) ad anidride carbonica e acqua. palmitil-CoA + 23 O2 + 108 Pi + 108 ADP → CoA + 108 ATP + 23 H2O + 16 CO2 Poiché il processo di attivazione ha richiesto energia equivalente a due molecole di ATP, il guadagno netto che si ha per la molecola di acido palmitico è di 106 molecole di ATP. La β-ossidazione compie la degradazione completa degli acidi grassi saturi a numero pari di atomi di carbonio. L’ossidazione degli acidi grassi insaturi richiede ulteriori tappe. Invece gli acidi grassi a numero dispari di atomi di carbonio formano, nella tappa finale della tiolisi un propionil-CoA che deve essere convertito in una forma facilmente utilizzabile mediante reazioni enzimatiche addizionali. Per quanto riguarda gli acidi grassi insaturi la maggior parte delle reazioni è identica a quelle degli acidi grassi saturi, sono però necessari due enzimi in più, una isomerasi ed una reduttasi. Metabolismo dei lipidi 255 Prendiamo ad esempio il palmitoleato, un acido grasso insaturo a 16 atomi di carbonio che ha un doppio legame fra il C9 e il C10. Il palmitoleil-CoA subisce tre cicli di degradazione, svolti dagli stessi enzimi che degradano gli acidi grassi saturi. Tuttavia il cis-∆3-enoilCoA del terzo ciclo non è il substrato per l’acil-CoA deidrogenasi. La presenza di un doppio legame fra il C3 e il C4 impedisce che se ne formi un altro fra il C2 e il C3. Questo ostacolo viene superato da un’altra reazione che sposta la posizione e la configurazione del doppio legame cis ∆3, che una isomerasi converte poi in un trans- ∆2. Le reazioni successive sono quelle dell’ossidazione degli acidi grassi saturi, in cui il trans-∆2-enoil-CoA è un substrato ordinario. Per l’ossidazione di qualsiasi acido grasso polinsaturo sono sufficienti due enzimi addizionali. I doppi legami in posizione dispari sono convertiti dall’isomerasi, quelli in posizione pari sono convertiti dalla reduttasi e dalla isomerasi. Gli acidi grassi a numero dispari di atomi di carbonio sono una specie meno comune e vengono ossidati allo stesso modo degli acidi grassi a numero pari, tranne che, nel ciclo finale di degradazione, invece di due molecole di acetil-CoA vengono prodotti propionil-CoA ed acetil-CoA. Il propionil-CoA viene convertito in succinil-CoA tramite un riarrangiamento molecolare che richiede come coenzima un derivato della vitamina B12 ed entra nel ciclo dell’acido citrico. Il propionil-CoA viene carbossilato a spese di una molecola di ATP per dare l’isomero D del metil-malonil-CoA. L’enzima è la propionil-CoA carbossilasi, un enzima biotina-dipendente. L’isomero D del metilmalonil-CoA viene racemizzato al suo isomero L e in seguito una mutasi lo converte in succinil-CoA mediante un riarrangiamento intramolecolare. Il gruppo –CO-S-CoA migra dal C2 al C3 scambiandosi con un atomo di idrogeno. L’enzima è una metil-malonil-CoA mutasi, che contiene un derivato della vitamina B12 (cobalammina) come coenzima. Va notato inoltre che gli animali non sono capaci di effettuare la sintesi netta di glucosio a partire da acidi grassi, quindi, l’acetil-CoA non può essere convertito in piruvato o ossalacetato. I due atomi di carbonio dell’acetil-CoA entrano nel ciclo dell’acido citrico, ma due di essi escono dal ciclo durante le decarbossilazioni catalizzate dalla 256 Capitolo XIII citrato deidrogenasi o dall’α-chetoglutarato deidrogenasi. L’ossalacetato quindi viene rigenerato ma non viene formato ex-novo quando il gruppo acetile dell’acetil-CoA viene ossidato nel ciclo dell’acido citrico. Le piante invece hanno due enzimi aggiuntivi che permettono loro di convertire gli atomi di carbonio dell'acetil-CoA in ossalacetato (Vedi “Il ciclo del gliossilato” nel capitolo dedicato al ciclo di Krebs). Biosintesi degli acidi grassi Anzitutto va notato che gli acidi grassi vengono sintetizzati e degradati in comparti cellulari diversi e attraverso vie differenti. La sintesi degli acidi grassi non è semplicemente l’inverso della via degradativa, ma è costituita da una serie di reazioni che rispettano alcuni principi: 1. la sintesi ha luogo nel citosol, mentre la degradazione avviene nella matrice mitocondriale; 2. gli intermedi nella biosintesi degli acidi grassi sono legati covalentemente a gruppi solfidrilici di una proteina trasportatrice di acili ACP (Acyl Carrier Protein); 3. negli organismi superiori gli enzimi della sintesi degli acidi grassi sono uniti in una singola catena polipeptidica detta acido grasso sintasi, mentre gli enzimi degradativi non sembrano essere associati; 4. l’allungamento della catena avviene ad opera di una addizione sequenziale di unità bicarboniose derivate dall’acetil-CoA. Il donatore attivato è la malonil-ACP. La reazione è favorita dal rilascio di CO2. 5. il riducente nella sintesi degli acidi grassi è il NADPH, mentre gli ossidanti della degradazione sono NAD+ e FAD; 6. l’allungamento della catena ad opera dell’acido grasso sintasi si ferma al palmitato (C16). Un ulteriore allungamento e l’inserimento di doppi legami sono catalizzati da altri sistemi enzimatici. L’insieme delle reazioni di questa via metabolica viene riportato in (Figura 4) Metabolismo dei lipidi 257 Figura 4. Reazioni della biosintesi degli acidi grassi. Vediamo ora in dettaglio le reazioni per la sintesi degli acidi grassi. La premessa essenziale è la carbossilazione dell’acetil-CoA a malo- 258 Capitolo XIII nil-CoA. La reazione è irreversibile ed è la tappa principale nella biosintesi degli acidi grassi. E’ catalizzata dall’acetil-CoA carbossilasi, che ha come gruppo prostetico la biotina. Si forma un intermedio carbossi biotina a spese di un ATP. Il gruppo CO2 attivato (HCO3-) viene poi trasferito all’acetil-CoA per dare malonil-CoA. Le reazioni sono: biotina-enzima + ATP + HCO3- CO2-biotina-enzima + ADP + Pi Quindi il gruppo CO2 attivato viene trasferito all’acetil-CoA per formare malonil-CoA CO2-biotina-enzima + acetil-CoA → malonil-CoA + biotina-enzima Gli intermedi della sintesi degli acidi grassi sono legati all’ACP, e precisamente al terminale SH di un gruppo di fosfopantoteina a sua volta legata all’ACP (nel catabolismo invece è sempre presente una fosfopantoteina ma è legata al gruppo SH del CoA). Il sistema enzimatico che catalizza la sintesi degli acidi grassi saturi a catena lunga a partire da acetil-CoA, malonil-CoA e NADPH si dice acido grasso sintasi. La fase di allungamento inizia con la formazione di acetil-ACP e malonil-ACP catalizzate rispettivamente dall’acetil transacilasi e la malonil transacilasi. acetil-CoA + ACP malonil-CoA + ACP acetil-ACP + CoA malonil-ACP + CoA La malonil-transacilasi è assolutamente specifica, mentre l’ acetil transacilasi può trasferire altri gruppi acile oltre l’acetile, anche se a velocità più bassa. L’ acetil-ACP ed il malonil-ACP reagiscono tra loro per formare acetoacetil-ACP. La reazione di condensazione è catalizzata dall’acil-malonil-ACP enzima condensante. acetil-ACP + malonil-ACP → acetoacetil-ACP + ACP + CO2 Nella reazione si forma una unità a quattro atomi di carbonio a partire da una unità a due atomi e una unità a tre atomi di carbonio e vie- Metabolismo dei lipidi 259 ne rilasciata CO2. Questo passaggio è necessario perché la sintesi diretta di acetoacetil-ACP da due molecole di acetil-ACP è altamente sfavorevole. Invece l’equilibrio è favorevole passando dal malonilACP perché la sua decarbossilazione diminuisce notevolmente l’energia libera. Infatti, quando l’acetil-CoA viene carbossilato a malonil-CoA viene utilizzato ATP la cui energia si accumula nel malonilCoA e viene rilasciata nella reazione di decarbossilazione che accompagna la formazione di acetoacetil-ACP. Le tre reazioni successive della sintesi degli acidi grassi sono le seguenti. Nella prima reazione il gruppo chetonico in C3 viene ridotto a gruppo metilenico e l’acetoacetil-ACP diviene D-3-idrossibutirril.ACP. A differenza della β-ossidazione, qui si forma l’isomero D anziché l’isomero L, ed interviene il NADPH (prodotto dalle piante nella fotosintesi e nella via dei pentosi-fosfati e negli animali dalla via dei pentosi-fosfati) anziché il NAD+. Questa differenza è un esempio del principio generale secondo cui il NADPH è consumato nelle reazioni di biosintesi, mentre il NADH è generato nelle reazioni che producono energia. Il NADH a sua volta viene riossidato negli organismi aerobi nella catena respiratoria di trasporto degli elettroni, negli organismi anaerobi nelle vie di fermentazione. Nella seconda tappa il D-3-idrossibutirril-ACP viene deidratato e si forma il crotonil-ACP, che è un trans-∆2- enoil-ACP. La terza tappa finale del ciclo riduce il crotonil-ACP a butirrilACP, per azione del NADH, mentre nella β-ossidazione l’ossidante corrispondente era il FAD. L’enzima è l’enoil-ACP-reduttasi. Queste tre reazioni (una riduzione, una deidratazione e una seconda riduzione) convertono l’acetoacetil-ACP in butirril-ACP completando il primo ciclo di allungamento (Tabella 2). Capitolo XIII 260 Tabella 2. Principali reazioni della sintesi degli acidi grassi. Reazione Enzima acetil-CoA + HCO3- + ATP → malonilCoA + ADP + Pi + H+ acetil-CoA carbossilasi acetil-CoA + ACP acetil transacilasi malonil-CoA + ACP CoA acetil-ACP + CoA malonil-ACP + acetil-ACP + malonil-ACP acetoacetil-ACP + ACP + CO2 acetoacetil-ACP + NADPH + H+ idrossibutirril-ACP + NAD+ D-3-idrossibutirril-ACP + H2 O malonil transacilasi enzima condensante D-3- β-chetoacil-ACP reduttasi crotonil-ACP 3-idrossiacil-ACP deidratasi crotonil-ACP +NADPH + H+ → butirrilACP + NAD+ enoil-ACP reduttasi Nel secondo ciclo di allungamento, il butirril-ACP condensa con il malonil-ACP per formare un C6-β-chetoacil-ACP. Questa reazione è simile a quella del primo ciclo in cui una molecola di acetil-ACP condensa con una molecola di malonil-ACP formando una molecola a quattro atomi di carbonio. Una riduzione, una deidratazione e una riduzione convertono il C6-β-chetoacil-ACP in un C6-β−acil-ACP pronto per il terzo ciclo di allungamento. I cicli di allungamento proseguono fino alla formazione di un C16-acil-ACP, che non è più un buon substrato per l’enzima condensante, ma viene idrolizzato a palmitato e ACP. L’enzima condensante agisce come un “metro” per determinare la lunghezza della catena dell’acido grasso. La stechiometria della sintesi del palmitato è: acetil-CoA + 7 malonil-CoA + 14 NADPH + 7 H+ → palmitato + 7 CO2 + 14 NADP+ + 8 CoA + 6 H2O Metabolismo dei lipidi 261 L’equazione per la sintesi del malonil-CoA usato nella reazione precedente è: 7 acetil-CoA + 7 CO2 + 7 ATP → 7 malonil-CoA + 7 ADP + 7 Pi + 7 H+ Quindi la stechiometria totale per la sintesi del palmitato è: 8 acetil-CoA + 7 ATP + 14 NADPH → palmitato + 14 NADP + 8 CoA + 6 H2O + 7 ADP + 7 Pi E’ da notare che gli acidi grassi vengono sintetizzati nel citosol, mentre l’acetil-CoA viene formato dal piruvato nei mitocondri, quindi l’acetil-CoA deve essere trasferito dai mitocondri nel citosol. I mitocondri però non sono molto facilmente permeabili all’acetil-CoA, e la carnitina trasporta solamente acidi grassi a catena lunga. A questo punto interviene il citrato, che si forma nella matrice dei mitocondri da acetil-CoA ed acido ossalacetico e che trasporta i gruppi acetile attraverso la membrana mitocondriale interna. Il citrato a sua volta, se presente in concentrazioni troppo alte viene trasportato nel citosol, dove viene scisso dalla citrato liasi. citrato + ATP + CoA + H2O → acetil-CoA + ADP + Pi + ossalacetato Quindi acetil-CoA ed ossalacetato sono trasferiti dai mitocondri al citosol a spese di una molecola di ATP. L’ossalacetato formato nel trasferimento del gruppo acetile al citosol deve tornare ai mitocondri utilizzando una serie di reazioni. La membrana mitocondriale interna è impermeabile all’ossalacetato quindi sono necessarie una serie di reazioni che inoltre producono la maggior parte del NADPH necessario alla sintesi degli acidi grassi. La prima è la riduzione dell’ossalacetato a malato operata dal NADH. L’enzima è la malato deidrogenasi del citoplasma. ossalacetato + NADH + H+ malato + NAD+ 262 Capitolo XIII Poi il malato viene decarbossilato ossidativamente ad opera di un enzima malato NADP+ dipendente (chiamato anche enzima malico) malato + NADP+ → piruvato + CO2 + NADPH Il piruvato diffonde facilmente nei mitocondri dove viene carbossilato ad ossalacetato dalla piruvato carbossilasi. piruvato + CO2 + ATP + H2O = ossalacetato + ADP + Pi + 2 H+ La somma delle tre reazioni è: NADP+ + NADH + ATP + H2O → NADPH + NAD+ + ADP + Pi + H+ Quindi per ogni acetil-CoA trasferito dai mitocondri al citosol viene prodotta una molecola di NADPH. Quando otto molecole di acetilCoA sono trasportate dai mitocondri al citosol per la sintesi del palmitato, si formano otto molecole di NADPH. Le altre sei molecole di NADPH necessarie per questo processo vengono dalla via del pentoso fosfato. Il prodotto principale dell’acido grasso sintasi è il palmitato. Negli eucarioti gli acidi grassi più lunghi vengono formati da reazioni di allungamento catalizzate da enzimi che stanno sul versante citosolico della membrana del reticolo endoplasmatico. Queste reazioni addizionano in sequenza unità bicarboniose alle estremità carbossiliche dei substrati acil-CoA sia saturi che insaturi. Il donatore di unità bicarboniose è il malonil-CoA e di nuovo la condensazione è favorita dalla decarbossilazione del malonil-CoA. Per inserire doppi legami negli acil-CoA a catena lunga vengono utilizzati degli enzimi legati alla membrana. Per esempio, per convertire lo stearoil-CoA ad oleoil-CoA viene inserito un doppio legame cis-D9 da parte di una ossidasi che impiega ossigeno molecolare O2 e NADH o NADPH. stearoil-CoA + NADH + H+ = oleoil-CoA + NAD+ + 2 H2O Metabolismo dei lipidi 263 La reazione è catalizzata da una catena di trasporto degli elettroni legati alla membrana composta dalla NADH-citocromo b5-reduttasi, dal citocromo b5 e una desaturasi. Dapprima vengono trasferiti elettroni dal NADH alla porzione FAD della NADH-citocromo b5-reduttasi. Poi l’atomo di ferro dell’eme del citocromo b5 viene ridotto da 3+ a 2+. Poi l’atomo di ferro non emico della desaturasi viene anch’esso convertito da 3+ a 2+, cosa che gli permette di interagire con l’O2 e con il substrato acil-CoA saturo. Si forma un doppio legame e vengono rilasciate due molecole di H2O. Due elettroni provengono dal NADH e due dal legame singolo del substrato acile. Dall’oleato poi si possono formare moltissimi acidi grassi insaturi mediante reazioni di allungamento e di desaturazione. I mammiferi sono privi degli enzimi necessari per introdurre nella catena di un acido grasso doppi legami a livello di atomi di carbonio oltre il C9, che devono pertanto essere introdotti con la dieta, e prendono il nome di acidi grassi essenziali. Biosintesi dei lipidi di membrana e degli ormoni steroidei In questo paragrafo verrà trattata brevemente anche la sintesi di tre importanti componenti delle membrane biologiche: i fosfogliceridi, gli sfingolipidi ed il colesterolo. Il colesterolo presenta interesse sia come componente delle membrane che come precursore di molte molecole biologiche, infatti ormoni sterodei come il progesterone ed il testosterone sono suoi derivati. La sintesi dei fosfogliceridi utilizza la maggior parte delle vie di sintesi dei triacilgliceroli. Il fosfatidato (diacilglicerolo-3-fosfato) infatti è un intermedio comune nella sintesi dei fosfogliceridi e dei triacilgliceroli. Il punto di partenza è il glicerolo-3-fosfato che si forma principalmente dalla riduzione del diidrossi acetone fosfato e, in misura minore, dalla fosforilazione del glicerolo. Il glicerolo-3-fosfato viene acilato dall’acilCoA per formare lisofosfatidato, in cui solo due gruppi OH del glicerolo sono esterificati, uno con un acido carbossilico R-COOH, generalmente insaturo, e l’altro con acido fosforico. Il lisofosfatidato viene nuovamente acilato 264 Capitolo XIII dall’acilCoA per dare fosfatidato, questa volta con un acido che generalmente è saturo. Queste acilazioni sono catalizzate dalla glicerofosfato acil transferasi (Figura 5). Figura 5. Reazioni catalizzate dalla glicerofosfato acil transferasi. Le vie divergono a livello del fosfatidato. Nella sintesi dei triacilgliceroli il fosfatidato viene idrolizzato da una fosfatidasi specifica per produrre un diacilglicerolo (DAG), che verrà acilato a triacilglicerolo da una reazione catalizzata dalla digliceride acil transferasi. Questi enzimi sono associati nel complesso della triacilglicerolo sintetasi. I fosfogliceridi possono essere sintetizzati in modi diversi. Una via parte dalla formazione di citidina difosfo diacilglicerolo (CDPdiacilglicerolo) da fosfatidato e citidina trifosfato (CTP). Questa reazione, come molte altre biosintesi, è resa irreversibile dall’idrolisi del pirofosfato. L’unità fosfatidica attivata reagisce poi con il gruppo ossidrilico di un alcool polare. Se l’alcool è la serina i prodotti sono fosfatidilserina e citidina monofosfato, CMP. In modo analogo, il fosfatidil inositolo si forma dal trasferimento di una unità di diacilglicerolo fosfato da CDP-diacilglicerolo all’inositolo. Nella sintesi di questi fosfogliceridi il CTP gioca lo stesso ruolo dell’UTP nella formazione del glicogeno. In entrambe le biosintesi si forma un intermedio attivato (UDP-glucosio o CDP-diacilglicerolo) da un substrato fosforilato (glucosio-1-fosfato o fosfatidato) e da un nucleotide trifosfato, UTP o CTP. L’intermedio attivato reagisce poi con un intermedio ossidrilico, il 4-OH terminale del glucosio o la catena laterale ossidrilica della serina. Fosfatidil-etanolammina e fosfatidil-colina si possono formare dalla fosfatidil-serina. Nei batteri infatti la decarbossilazione della fosfa- Metabolismo dei lipidi 265 tidil-serina da parte di un enzima piridossal fosfato (PLP) dipendente forma una fosfatidil-etanolammina. Il gruppo amminico di questo fosfogliceride viene poi metilato tre volte per dare fosfatidil-colina. Nei mammiferi, la fosfatidilcolina viene sintetizzata da una via che utilizza la colina ottenuta dalla dieta, questa viene fosforilata dall’ATP a fosforilcolina che reagisce poi col CTP per formare CDP-colina.L’unità fosforilcolina della CDP-colina viene poi trasferita ad un diacil glicerolo per formare fosfatidil-colina (Figura 6). Figura 6. Reazioni della sintesi di fosfatidil colina. In modo analogo si forma la fosfatidil-etanolammina partendo dall’etanolammina attraverso un intermedio CDP-etanolammina. Gli sfingolipidi hanno come scheletro la sfingosina, un alcool a lunga catena che si trova al posto del glicerolo. Il palmitoil-CoA (acido palmitico attivato) e la serina condensano per formare deidrosfinganina, che a sua volta viene convertita in sfingosina attraverso due ossidazioni operate prima dal NAD+ e poi dal FAD. Un acil-CoA a catena lunga reagisce con la sfingosina formando il ceramide (N-acilsfingosina). Nella sfingomielina il sostituente è la fosforilcolina che deriva da CDP-colina. In un cerebroside, il gruppo ossidrilico terminale del cerammide è legato ad un glucosio o ad un galattosio. Nella sintesi dei cerebrosidi i donatori di zuccheri sono UDP-glucosio o UDP-galattosio. 266 Capitolo XIII Il colesterolo è uno steroide che modula la fluidità delle membrane degli eucarioti. E’ precursore degli ormoni steroidei e viene trasportato nel sangue verso bersagli specifici da una serie di lipoproteine. Il colesterolo viene sintetizzato a partire dall’acetil-CoA. Tutti e ventisette gli atomi di C del colesterolo derivano dall’acetil-CoA. La tappa chiave nella biosintesi del colesterolo è la formazione di mevalonato dal 3-idrossi-3-metil-glutaril-CoA, che a sua volta è un derivato dall’acetil CoA e dall’acetoacetilCoA (vedi biosintesi degli acidi grassi). Il mevalonato viene convertito ad isopentenil pirofosfato, IPPP, (5 C) che condensa con il suo isomero dimeti-allil pirofosfato (C5) per formare il geranil-pirofosfato (C10). L’addizione di un’altra molecola di IPPP forma farnesil pirofosfato (C15) che condensa con se stesso per formare squalene (C30). Questo intermedio ciclizza a lanosterolo (C30) dalla cui modificazione si forma il colesterolo (C27). Acetato (C2); Isoprene (C5); Squalene (C30); Colesterolo (C27) Per l’intera via biosintetica e suoi approfondimenti si rimanda a libri di Biochimica non di base. IL METABOLISMO DELL’AZOTO Il ciclo dell’azoto Tutti gli organismi viventi necessitano di azoto, sia per la sintesi degli amminoacidi, e quindi delle proteine, che per la formazione di altri composti molto importanti, quali i nucleotidi e gli acidi nucleici. L’azoto è presente in tutti gli elementi della biosfera, acqua, aria e suolo, in varie forme e vari stati di ossidazione, ma la maggior parte deriva dall’azoto atmosferico N2 (detto anche azoto elementare o azoto molecolare), che ha grado di ossidazione zero. Circa l’80% dell’atmosfera è rappresentato da questa forma. L’azoto elementare, ad opera della fissazione biologica, può venir ridotto ad ammoniaca (NH3) o a ione ammonio (NH4+), ambedue a grado di ossidazione –3. Queste forme possono poi essere ossidate, passando allo stato di ossidazione +3 dei nitriti e +5 dei nitrati. La denitrificazione può trasformare ancora i nitrati in ossido nitroso N2O (+1) e quindi di nuovo in N2. I processi metabolici di organismi diversi operano in modo interdipendente per salvare e riutilizzare l’azoto disponibile per i processi biologici, creando un grande ciclo dell’azoto, la cui prima tappa è la fissazione (o riduzione) dell’azoto atmosferico da parte di batteri che formano ammoniaca o ione ammonio. Solo un numero relativamente piccolo di specie può convertire l’azoto atmosferico in forme utilizzabili dagli organismi viventi. Infatti l’N2 non può essere assimilato direttamente dalle piante ma piuttosto indirettamente tramite alcuni microrganismi che vivono in simbiosi con esse. Esistono anche microrganismi autotrofi che compiono questa assimilazione. L’ammoniaca così formata può essere convertita in altre forme azotate o venir organicata, cioè inserita nella molecola degli amminoacidi delle piante. Infatti, mentre l’azoto nell’atmosfera è sotto forma di N2, nei vegetali, e in genere in tutto il materiale biologico, invece è quasi tutto sotto forma ammoniacale (amminoacidi, proteine, altri composti azotati come gli alcaloidi, composti eterociclici ecc.). Gli animali utilizzano poi le piante come fonte di amminoacidi essenziali e non essenziali per costruire le loro proteine. 267 268 Capitolo XIV L’azoto organico viene poi restituito al suolo nella stessa forma ammoniacale quando gli organismi muoiono e vengono decomposti dai batteri ammonificatori (Figura 1). Figura 1. Il ciclo dell'azoto. Le piante superiori e la maggior parte dei vegetali inferiori assimilano l’azoto presente nel suolo sotto forma di NO3–, NH4+e in casi particolari di qualche sostanza organica non molto complessa, come l’urea o anche sotto forma di purine e di pirimidine. Comunque, la fonte principale è costituita da NO3– che, essendo solubile, si trova nella soluzione circolante del terreno ed è quindi facilmente assunto dai vegetali superiori attraverso l’apparato radicale. Lo ione NH4+, che può anch’esso essere preso facilmente dalle radici, è meno disponibile, in quanto adsorbito per scambio e trattenuto dal complesso adsorbente del terreno. Inoltre, anche se l’ammoniaca può essere utilizzata dalla maggioranza degli organismi viventi, i batteri del suolo, che ottengono l’energia di cui hanno bisogno dall’ossidazione dell’ammoniaca prima a nitrito (NO2-) e poi a nitrato (NO3-), sono così abbondanti che praticamente tutta l’ammoniaca prodotta nel suolo Il metabolismo dell’azoto 269 viene trasformata in nitrato. Questo processo è noto col nome di nitrificazione. Si ricordi inoltre che alcuni composti azotati (in genere ossidi) possono formarsi durante i temporali e, una volta portati verso il basso dalle piogge, reagiscono con l’acqua formando acidi che possono essere assorbiti attraverso l’apparato fogliare. Il ruolo delle piante è quindi di vitale importanza nella conversione dell’azoto inorganico in azoto organico. Infatti esse, e solo esse, possono catturare azoto dall’atmosfera, assumerlo dal suolo sotto forma di ione ammonio e/o assimilare e ridurre il nitrito e il nitrato del suolo ad opera della nitrito e nitrato reduttasi. Nel prossimo paragrafo verrà descritto il processo di fissazione dell’azoto atmosferico, principale sorgente di azoto per tutti gli organismi. Fissazione dell’azoto La disponibilità di azoto per le piante dipende dallo stato di ossidazione dell’elemento. Purtroppo la maggior parte dell’ N2 disponibile nella biosfera non è direttamente accessibile per le piante perché esse sono incapaci di spezzare il triplo legame della molecola biatomica dell’azoto elementare. Le piante devono quindi utilizzare una fonte non atmosferica di azoto, come i nitrati o l’ammonio per poter soddisfare le esigenze metaboliche legate a questo elemento. Fino al 1915 la fonte principale di azoto da usare come fertilizzante era lo stallatico o il guano estratto dai depositi millenari del Cile, ma quando per necessità belliche Haber e Bosch misero a punto un processo industriale capace di ridurre l’N2 atmosferico e di produrre ammoniaca, indispensabile per produrre esplosivi a base di azoto, si pensò che questo elemento, sotto opportune forme, poteva venir impiegato anche come fertilizzante. La sintesi chimica industriale dell’ammoniaca o processo Haber-Bosch si basa sulla reazione seguente: N2 + 3 H2 → 2 NH3 Il processo richiede energia per vincere l’energia di attivazione necessaria a far avvenire la reazione, di per sé spontanea ma lenta, tem- 270 Capitolo XIV perature elevate (200-500°C) e alte pressioni (150-200 atm). L’industria fissa ogni anno circa 60 milioni di m3 di azoto, valori consistenti ma nettamente inferiori a quelli della fissazione biologica (100-180 milioni di m3). Anche la fissazione biologica dell’azoto necessita di una adeguata quantità di energia, che viene fornita dalla moneta energetica cellulare, l’ATP. Nella produzione di ammoniaca la fissazione biologica è un guadagno perché permette di ridurre l’uso dei combustibili fossili, in quanto la fonte principale dell’ energia è quella solare (l’ATP infatti si forma in grande quantità durante la fotosintesi). L’azoto dell’atmosfera può essere assimilato solamente da alcuni microrganismi procarioti che vivono liberi o in associazione simbiotica con piante superiori, alghe e licheni, e che comprendono batteri liberi, alghe blu-verdi e batteri simbionti. Si stima che i batteri azotofissatori riducano oltre 1010 Kg di azoto per anno. Fra le più importanti piante alimentari solo le leguminose sono in grado di utilizzare l’azoto così fissato e perciò ogni anno vengono spese ingenti somme per fornire alle piante coltivate la necessaria quantità di fertilizzante azotato indispensabile per la loro crescita e sviluppo. Si stanno quindi facendo grandi sforzi per comprendere i meccanismi biologici della fissazione dell’azoto, con l’intento di trasferirli alle piante coltivate. Oltre al vantaggio economico, si ridurrebbe drasticamente l’inquinamento delle falde acquifere e l’eutrofizzazione di fiumi, laghi e mari. La reazione di fissazione biologica dell’azoto può essere così schematizzata: N2 + 8 H+ + 8 e- → 2 NH4+ + H2 Come si nota vengono utilizzati protoni e non idrogeno, quindi occorre fornire elettroni. Il fabbisogno elettronico è di sei elettroni, più altri due che vengono utilizzati per la riduzione degli H+. Per ogni mole di N2 si forma infatti una mole di H2 molecolare. La reazione è esoergonica, con ∆G°’ = -33.5 kJ/mole. La fissazione dell’azoto ha una energia di attivazione molto elevata perché l’azoto atmosferico, come si è detto, è praticamente inerte nelle condizioni normali. La barriera Il metabolismo dell’azoto 271 di attivazione viene superata almeno in parte dalla rottura del legame e dall’idrolisi dell’ATP. La reazione complessiva quindi può essere riscritta: N2 + 10 H+ + 8 e- + 16 ATP → 2 NH4+ + 16 ADP + 16 Pi + H2 Come detto sopra, l’input energetico per il procedere della reazione è fornito dall’ATP. Per ogni elettrone trasferito occorrono due ATP e quindi occorrono in totale sedici molecole di ATP. In realtà in condizioni naturali il fabbisogno di ATP risulta maggiore (da 20 a 30) in quanto il processo sembra essere meno efficiente di quanto non sia in condizioni sperimentali. Nel conteggio generale si deve poi tener conto delle molecole utilizzate per la produzione di H2 (altri quattro ATP) che dissipano il 25% dell’energia disponibile per l’N2. La ricerca di un composto intermedio ha dato risultati contrastanti. Con l’uso di radioisotopi tuttavia si è potuto dimostrare che dalla riduzione dell’N2 si ottiene lo ione NH4+ come intermedio chiave della reazione. Questo viene successivamente e rapidamente convertito in forma organica. Classificazione dei microrganismi azotofissatori Batteri liberi: Clostridium pasteurianum (anaerobico eterotrofo), quasi universalmente distribuito nel terreno; Azotobacter (aerobico eterotrofo); Rhodospirillum rubrum (anaerobico eterotrofo). A volte questi batteri possono vivere in simbiosi con le alghe blu-verdi, che riforniscono di azoto i campi coltivati a riso. Batteri simbionti: Rhizobium questi possono vivere liberi nel terreno o in simbiosi nei tubercoli (noduli) dell’apparato radicale delle Leguminose (piselli, fagioli, soia, trifoglio). Possono essere in simbiosi con alcuni alberi (ontani). I Rhizobium sono aerobici. La fissazione dell’azoto avviene nei centri dei noduli radicali. La pianta rifornisce il batterio di carboidrati, che questi non possono produrre. I carboidrati vengono poi ossidati durante la respirazione e gli elettroni prodotti vengono usati dai batteri per ridurre l’azoto a ione ammonio, la forma assimilabile, per dare amminoacidi e proteine. Una parte di questi amminoacidi viene poi ceduta alle cellule che circondano i tubercoli e da qui traslocata in altre zone della pianta. Altri 272 Capitolo XIV amminoacidi vengono escreti dai noduli direttamente nel terreno, dove quasi sempre vengono deamminati. L’azoto liberato viene poi trasformato in NO3– ad opera dei batteri nitrificanti. Così i batteri simbionti riforniscono di azoto anche altre piante (non leguminose) che circondano le leguminose. La quantità di N2 fissata dai Rhizobium può variare da un minimo di 45 Kg/ha per il Rhizobium del fagiolo, ad un massimo di 179 Kg/ha per il Rhizobium dell’erba medica. Meccanismo di fissazione dell’azoto Allo stato attuale delle conoscenze la fissazione dell’azoto è un processo riduttivo ed i fattori che controllano questo processo sono: 1. una sostanza riducente (che quindi si ossida); 2. una sorgente di energia; 3. un prodotto che trasferisce elettroni; 4. un enzima, la nitrogenasi. Il complesso della nitrogenasi, che catalizza il processo di fissazione dell’azoto, fornisce elettroni ad elevato potere riducente che vengono utilizzati per ridurre N2 a NH4+. Contemporaneamente si libera H2 dalla riduzione di H+. La reazione catalizzata dall’enzima nitrogenasi, come visto sopra, è: N2 + 8 H+ + 8 e- → 2 NH4+ + H2 Ogni trasferimento di un elettrone è accompagnato dall’idrolisi di due molecole di ATP. Quindi saranno necessari sedici ATP La maggior parte degli studi sulla nitrogenasi è stata condotta sulla Klebsiella pneumoniae, in cui è stato dimostrato che la nitrogenasi contiene molibdeno (Mo). Alcuni altri batteri possono utilizzare anche enzimi contenenti vanadio (V) se il molibdeno non è disponibile. Altri ancora oltre alle Mo- e V-deidrogenasi possono utilizzare anche una terza nitrogenasi contenente Fe, se i livelli di molibdeno e vanadio sono molto bassi. Tutte le nitrogenasi contengono una Fe-proteina composta di due subunità sensibili all’ossigeno con peso molecolare di circa 60 dalton e con 4 Fe e 4 S per dimero. Il centro Fe-S ha un potenziale medio di –350 mV. Quando si lega l’ATP la Fe- proteina su- Il metabolismo dell’azoto 273 bisce un cambiamento di conformazione e il potenziale diminuisce. E’ stato dimostrato che la Fe-proteina è ATP-dipendente ed è un donatore di elettroni specifico per la proteina che contiene Mo. La parte dell’enzima nitrogenasi che contiene Mo si chiama proteina Mo-Fe. Un cofattore Fe-Mo è legato alla subunità α della proteina Mo-Fe. Meccanismo d’azione della nitrogenasi Inizialmente il complesso ridotto Fe-proteina-ATP si lega alla proteina Mo-Fe. Poi l’elettrone viene trasferito, con la concomitante idrolisi di ATP. Ci sono otto passaggi successivi, ognuno dei quali corrisponde al trasferimento di un elettrone dalla Fe-proteina alla proteina Mo-Fe. A seguito dello svolgimento di H2 dopo il trasferimento di tre elettroni, l’azoto viene ridotto dall’addizione consecutiva di elettroni e protoni fino al rilascio di due molecole di NH3. Un intermedio probabile è il complesso di tipo idrazide Enzima=N-NH2 ed è probabile che tale complesso liberi idrazina. Sia la Fe- che la Fe-Mo proteina della nitrogenasi sono inattivate dall’ossigeno, che cambia lo stato di ossidazione dei centri Fe. Dopo una lunga esposizione si hanno perdite di Fe, S e Mo dalla proteina., per cui gli organismi azotofissatori hanno dovuto sviluppare strategie per permettere una fissazione aerobia. Ricapitolando, la riduzione dell’N2 richiede tre tipi principali di trasferimento di elettroni: 1) una riduzione della Fe-proteina per opera di un donatore di elettroni come la ferredossina o la flavodossina; 2) il trasferimento di un singolo elettrone dalla Fe-proteina alla Fe-Mo proteina, reazione associata all’idrolisi di due molecole di ATP per ogni elettrone trasferito; 3) il trasferimento di elettroni e protoni al substrato (N2) che si ritiene sia legato al cofattore Fe-Mo della Fe-Mo proteina. 274 Capitolo XIV Figura 2. Riduzione dell'N2 ad opera della nitrogenasi. Questa riduzione tuttavia non è un processo immediato e sono molto probabilmente coinvolti alcuni passaggi intermedi. Nel meccanismo di reazione la presenza dell’ATP è sicura ma la sua funzione non è certa. L’ATP può fornire energia per mantenere la ferredossina in stato stazionario (in equilibrio dinamico) o attivare la formazione del complesso enzima-N2 o partecipare alle reazioni di riduzione del complesso. Questo schema viene proposto per la fissazione di batteri liberi e per le alghe, in quanto nei batteri simbionti si trova una cromoproteina rossa cui è stato dato il nome di leghemoglobina, che contiene quindi Fe2+ e Fe3+, che sembra controlli il flusso dell’ossigeno nel citoplasma della pianta. La funzione precisa della leghemoglobina non è conosciuta, ma probabilmente essa partecipa al trasferimento di elettroni. Assimilazione dei composti organici Solo i composti quaternari più semplici possono essere assimilati dalle piante. Alcune piante (pomodoro, tabacco, trifoglio) possono assimilare amminoacidi. Anche l’urea può essere assimilata direttamente dalle piante senza subire alcuna modifica. In altre piante viene scissa in CO e NH3, che viene poi assimilata nel modo consueto. Il metabolismo dell’azoto 275 Assimilazione di NO3– e NH4+ L’assimilazione dei nitrati (importantissima nei vegetali superiori e possibile anche negli inferiori) necessita della riduzione di NO3– a NH3. Lo ione NO3– entra nella pianta attraverso l’apparato radicale e, come gli altri ioni che entrano nella pianta, è soggetto al fenomeno di trasporto passivo ed attivo. Quest’ultimo è regolato a sua volta dall’energia che si produce durante i processi catabolici (respirazione). Attraverso lo xilema l’NO3– è trasportato dalle radici agli altri organi del vegetale. Anche l’età della pianta può influire sull’adsorbimento di NO3– e NH4+: le plantule giovani di riso, frumento ed avena assorbono meglio NH4+che non NO3–, mentre alla maturità assumono meglio NO3–. La patata cresce meglio su NH4+ che non su NO3–. Comunque la riduzione di NO3– ad esempio nel frumento, avviene sia nelle foglie che nelle radici. Lo schema è il seguente: NO3– → NO2– → X1 → X2 → NH3 1° 2° 3° La riduzione dei nitrati avviene in diversi stadi, in ciascuno dei quali si ha la perdita di due elettroni. Il primo stadio consiste nella riduzione dei nitrati a nitriti ad opera della nitrato reduttasi. Il secondo consiste in riduzioni (nitrito reduttasi) con formazione di composti non ben definiti ed identificati (X1 e X2). L’ultima riduzione porta ad ammoniaca. Il primo stadio, cioè la riduzione di NO3– a NO2– è il più studiato. L’enzima implicato è la nitrato reduttasi, un complesso flavoproteico contenente Mo. Il donatore di idrogeno (o di elettroni) è normalmente il NADH + H+ ma in alcuni casi può essere utilizzato anche il NADPH + H+: Quest’ultimo può essere fornito dal processo primario della fotosintesi e allora si parlerà di riduzione fotochimica dei nitrati. La reazione è: NO3– + 2 H+ + 2e- → NO2- + H2O La riduzione dei nitrati avviene nelle radici e dipende dal tipo di pianta, dallo stadio di sviluppo e dalla disponibilità di nitrati. 276 Capitolo XIV La restante parte del processo riduttivo fino ad ammoniaca è catalizzata dalla nitrito reduttasi, un enzima contenente ferredossina, Cu+, FAD ed ATP, la cui funzione non è ancora ben conosciuta. La reazione è: NO2- + 8 H+ + 6 e- → NH4+ + H2O L’enzima è presente nelle foglie e nelle radici ed è collocato esclusivamente nei cloroplasti o nei plastidi. Il potere riducente è dovuto alla ferredossina ridotta, che proviene direttamente dalle reazioni alla luce nelle foglie o dal metabolismo dei carboidrati nelle radici. La presenza di altri intermedi è incerta e dibattuta, a causa delle difficoltà di carattere sperimentale incontrate nelle ricerche. Un tempo si riteneva che il composto NH2OH (idrossilammina) fosse un punto chiave nella riduzione dei nitrati, tale importanza non è però stata confermata negli ultimi anni. Nelle foglie di soia comunque è stata trovata una reduttasi dell’idrossilammina contenente NADH + H+ e Mn2+. La riduzione di NO3– a NO2–, non implica necessariamente una “attivazione” della riduzione di NO3– (come accade per lo ione SO42-). La reazione ha un ∆G negativo, ossia è esoergonica e procede quindi con sviluppo di energia. La tappa successiva all’assimilazione dell’azoto nelle biomolecole è l’ingresso dell’NH4+ negli amminoacidi e in seguito in altre biomolecole azotate. In questo processo il glutammato e la glutammina hanno un ruolo centrale. L’ammoniaca infatti viene incorporata direttamente in questi due composti, che sono poi utilizzati come fonte di azoto in molte vie biosintetiche. La maggior parte degli amminoacidi riceve il gruppo amminico dall’acido glutammico attraverso reazioni di transamminazione. La glutammina fornisce l’azoto della sua catena laterale per la biosintesi di molti metaboliti, compresi molti amminoacidi. Le vie biosintetiche che producono glutammato e glutammina sono simili in tutte le forme di vita. Il glutammato viene sintetizzato a partire da NH4+ e α-chetoglutarato, che è un intermedio del ciclo dell’acido citrico, mediante l’azione della glutammato deidrogenasi NADPH-dipendente. Il metabolismo dell’azoto 277 Il NADPH infatti è il riducente per questa biosintesi. La reazione, che avviene quando la concentrazione di NH3 nell’ambiente è elevata è: NH4+ + α-chetoglutarato + NADPH + H+ → glutammato + NADP+ + H2O Lo ione NH4+ viene incorporato nella glutammina per azione della glutammina sintetasi sul glutammato, la reazione richiede l’idrolisi di ATP. La reazione avviene in due tappe: 1) glutammato + ATP → γ-glutammil fosfato + ADP 2) γ-glutammil fosfato + NH4+ → glutammina + Pi + H+ Il γ-glutammil fosfato che si forma nella prima tappa resta legato all’enzima. Ambedue gli enzimi suddetti sono presenti in tutti gli organismi. La maggior parte dei procarioti possiede anche la glutammato sintasi che catalizza l’amminazione riduttiva dell’α-chetoglutarato utilizzando la glutammina come donatore di gruppi amminici. Si formano due molecole di glutammato secondo la seguente reazione: α-chetoglutarato + glutammina + NADPH + H+ → 2 glutammato + NADP+ Regolazione del metabolismo dell’azoto Quando l’NH3 nell’ambiente è limitante, la maggior parte del glutammato si forma per azione sequenziale della glutammina sintetasi e della glutammato sintasi. NH4+ + α-chetoglutarato + NADPH + H+ + ATP → L-glutammato + NADP+ + ADP + Pi 278 Capitolo XIV L’attività della glutammina sintetasi è critica nel controllo del metabolismo dell’azoto e l’enzima è regolato allostericamente mediante modificazioni covalenti. Almeno sei prodotti finali del metabolismo della glutammina, oltre all’alanina e alla glicina, sono inibitori allosterici dell’enzima. Ogni inibitore promuove solo un’inibizione parziale, mentre l’effetto combinato di inibitori diversi blocca praticamente l’attività dell’enzima. Il risultato è che l’attività delle glutammina sintetasi diminuisce quando i livelli di glutammina sono elevati, mentre un aumento dell’attività dell’enzima si ha quando i livelli di glutammina sono bassi, ma sono disponibili sia ATP che αchetoglutarato. Biosintesi degli altri amminoacidi I batteri come l’Escherichia coli possono sintetizzare tutti e venti gli amminoacidi di base, mentre gli esseri umani non sono in grado di sintetizzarne nove, detti amminoacidi essenziali, che debbono essere assunti con l’alimentazione. Questi amminoacidi essenziali sono: istidina, leucina, isoleucina, lisina, metionina, fenilalanina, treonina, triptofano e valina. La mancanza anche di un solo amminoacido determina un bilancio negativo dell’azoto. In questa condizione vengono degradate più proteine di quante ne vengono sintetizzate per cui viene escreto più azoto di quanto ne venga ingerito. Le vie biosintetiche degli amminoacidi sono diverse ma hanno una proprietà in comune molto importante, il loro scheletro carbonioso deriva da intermedi delle seguenti vie metaboliche: 1) la glicolisi; 2) il ciclo dell’acido citrico; 3) la via dei pentosi-fosfati. L’azoto entra in queste vie sotto forma di glutammato o di glutammina. Alcune vie sono relativamente semplici altre invece sono piuttosto complesse. Dieci amminoacidi sono sintetizzati dai loro precursori mediante vie con una o poche tappe enzimatiche. La sintesi di altri amminoacidi invece, in particolare di quelli aromatici, è più comples- Il metabolismo dell’azoto 279 sa. Un modo molto utile di organizzare le vie biosintetiche degli amminoacidi è quello di raggrupparle in sei gruppi biosintetici, a seconda del loro precursore metabolico. Questi sei gruppi biosintetici, che esamineremo in dettaglio, sono: 1) 2) 3) 4) 5) 6) ossalacetato piruvato ribosio-5-fosfato fosfoenolpiruvato + eritrosio-4-fosfato α-chetoglutarato 3-fosfoglicerato. In Figura 3 sono riportati i gruppi biosintetici degli amminoacidi nei batteri e nelle piante. I principali precursori metabolici, numerati secondo quanto indicato nell’elenco sopra riportato, sono in blu. Gli amminoacidi che danno origine ad altri amminoacidi sono indicati in giallo. Figura 3. Gruppi biosintetici degli amminoacidi in batteri e piante. Oltre ai sei precursori occorre tener presente che diverse di queste vie di sintesi hanno a comune un intermedio, il 5-fosforibosil-1pirofosfato (PRPP), che viene sintetizzato dal ribosio-5-fosfato, in una 280 Capitolo XIV reazione catalizzata dalla ribosio-fosfato pirofosfatochinasi, la cui reazione è: ribosio-5-fosfato + ATP = 5-fosforibosil-1-pirofosfato + AMP Vediamo ora in dettaglio la sintesi dei principali amminoacidi: 1) Il gruppo dell’ossalacetato, che dà origine ad aspartato, asparagina, metionina, treonina, lisina ed isoleucina. L’aspartato viene sintetizzato direttamente dall’ossalacetato per transamminazione con il glutammato. L’asparagina si forma per ammidazione dell’aspartato e lo ione ammonio di questa reazione deriva dalla glutammina. Aspartato ed asparagina non sono amminoacidi essenziali e le loro vie biosintetiche, piuttosto semplici, sono presenti in tutti gli organismi. Gli amminoacidi metionina, treonina, lisina, valina (che si forma dal piruvato, mentre l’isoleucina può derivare sia dal piruvato che dalla treonina), leucina ed isoleucina sono invece essenziali e le loro vie biosintetiche sono complesse ed interconnesse. In alcuni casi vi sono significative differenze tra le vie presenti nei batteri e quelle nei funghi e nelle piante. 2) Il gruppo del piruvato, che dà origine ad alanina, valina e leucina. Anche l’alanina, come l’aspartato, si forma direttamente dal piruvato per transamminazione mediante il glutammato. Le altre hanno vie biosintetiche complesse, come ricordato sopra. 3) Il gruppo del ribosio-5-fosfato, che dà origine all’istidina. L’istidina deriva da tre precursori: il PRPP (5-fosforibosil-1pirofosfato) che fornisce cinque atomi di carbonio, l’anello purinico dell’ATP, che fornisce un atomo di azoto ed uno di carbonio e dalla glutammina, che fornisce un altro atomo di azoto. L’uso di ATP come metabolita anziché come cofattore ad alta energia è insolito, ma non vi sono sprechi perché questa via metabolica va ad intersecarsi con la via biosintetica delle purine. 4) Il gruppo del fosfoenolpiruvato + eritrosio-4-fosfato che dà origine a fenilalanina, tirosina e triptofano. Gli anelli aromatici non sono facilmente disponibili in natura, anche se l’anello benzenico è mol- Il metabolismo dell’azoto 281 to stabile. Nei batteri e nelle piante la via ramificata che porta alla formazione di fenilalanina, tirosina e triptofano è la strada principale per la formazione degli anelli aromatici e viene chiamata via dell’acido scichimico. La via inizia con la chiusura dell’anello a partire da un precursore alifatico. Il punto di ramificazione è l’acido corismico, da cui si dipartono due vie: la prima arriva al triptofano e la seconda porta alla formazione di fenilalanina e tirosina. 5) Il gruppo dell’α-chetoglutarato, che dà origine a glutammato, glutammina, prolina ed arginina. Della biosintesi di glutammato e glutammina si è detto sopra. La prolina è un derivato ciclico del glutammato. Dapprima il glutammato viene ridotto a glutammato-γsemialdeide, che poi ciclizza e viene ulteriormente ridotto a prolina. L’arginina viene sintetizzata dal glutammato attraverso l’intermedio ornitina ed il ciclo dell’urea. Le vie che portano alla sintesi di prolina ed arginina nei mammiferi sono un pò diverse. La prolina si può formare dall’arginina dalla dieta o da proteine dei tessuti. L’arginasi, un enzima del ciclo dell’urea, converte l’arginina in ornitina ed urea. L’ornitina viene convertita in glutammato-γ-semialdeide dall’enzima ornitina-δ-amminotransferasi e poi in D1-pirrolina-5carbossilato, che viene convertito in prolina. 6) Il gruppo del 3-fosfoglicerato, che dà origine a serina, cisteina, metionina e glicina. La via principale di formazione della serina è la stessa in tutti gli organismi. Nella prima tappa il 3-fosfoglicerato viene ossidato a 3-fosfoidrossipiruvato. La transamminazione, che avviene ad opera del glutammato, produce 3-fosfoserina, che verrà poi defosforilata dalla fosfoserina fosfatasi formando serina libera. La serina è il precursore della glicina. Le piante ed i batteri producono S ridotto necessario per la sintesi della cisteina e della metionina dai solfati presi dall’ambiente. Nei mammiferi la cisteina viene prodotta partendo dalla metionina (che fornisce l’atomo di zolfo) e dalla serina (che fornisce lo scheletro carbonioso). 282 Capitolo XIV Regolazione della biosintesi degli amminoacidi La velocità di sintesi degli amminoacidi dipende principalmente dalla quantità di enzimi biosintetici e dalla loro attività. Il sito di regolazione dell’attività di un enzima è di solito la prima reazione irreversibile della via biosintetica. Il prodotto finale inibisce spesso l’enzima che catalizza la tappa che dà inizio alla via. Questo meccanismo di controllo prende il nome di retroinibizione o inibizione a feedback. La prima reazione in queste sequenze è di solito una reazione irreversibile catalizzata da un enzima allosterico. Questo tipo di controllo è essenziale per la conservazione delle molecole dei precursori e dell’energia metabolica. Ossidazione degli amminoacidi e ciclo dell’urea Gli amminoacidi in eccesso rispetto alle necessità della sintesi di proteine e di altre macromolecole non possono essere conservati, contrariamente a quanto accade per gli acidi grassi e per il glucosio, né essere escreti. Essi vengono invece utilizzati come materiale energetico. I gruppi amminici in alfa vengono rimossi e lo scheletro di atomi di C che rimane viene trasformato in importanti intermedi metabolici. La maggior parte dei gruppi amminici viene convertita in urea, mentre lo scheletro carbonioso viene trasformato in acetil-CoA, aceto acetil-CoA, piruvato oppure in uno degli intermedi del ciclo dell’acido citrico. Quindi dagli amminoacidi si possono formare acidi grassi e glucosio. Consideriamo per prima cosa il destino del gruppo amminico degli amminoacidi, quindi quello della parte carboniosa. Il gruppo amminico in posizione “alfa” di molti amminoacidi è trasferito all’achetoglutarato con formazione di glutammato, che a sua volta può essere deamminato ossidativamente per dare NH4+. Gli enzimi che catalizzano il trasferimento di un gruppo amminico α da un α-amminoacido ad un α-chetoacido sono le amminotransferasi ( o transamminasi). Questi enzimi incanalano gruppi α-amminici provenienti da vari amminoacidi verso l’α-chetoglutarato per formare NH4+. L’aspartato amminotransferasi, uno degli enzimi più importan- Il metabolismo dell’azoto 283 ti, catalizza il trasferimento del gruppo amminico dall’aspartato all’αchetoglutarato: aspartato + α-chetoglutarato → ossalacetato + glutammato L’alanina amminotransferasi, l’enzima più rappresentato nei tessuti dei mammiferi, catalizza il trasferimento del gruppo amminico dall’alanina all’α−chetoglutarato. alanina + α−chetoglutarato → piruvato + glutammato Lo ione ammonio si forma dal glutammato mediante deamminazione ossidativa. La reazione è catalizzata dalla glutammato deidrogenasi che, a differenza delle altre deidrogenasi, può utilizzare sia NAD+ che NADP+. L’attività della glutammato deidrogenasi è regolata allostericamente. Il GTP e l’ATP sono inibitori allosterici, mentre GDP e l’ADP sono attivatori allosterici. Quindi, una diminuzione della carica energetica accelera l’ossidazione degli amminoacidi. La somma delle reazioni catalizzate dalla amminotransferasi e dalla glutammato deidrogenasi è: α-ammin. + NAD+ + H2O = α-chetoacido + NH4+ + NADH + H+ Nei vertebrati terrestri l’NH4+ è convertito in urea, che viene poi escreta. Il ciclo dell’urea Una parte degli ioni NH4+ formati nella demolizione degli amminoacidi viene consumata nella biosintesi dei composti azotati. Nella maggior parte dei vertebrati terrestri, l’NH4+ in eccesso viene convertito in urea e quindi escreto. In molti animali acquatici invece viene escreto direttamente sotto forma di ione NH4+, mentre negli uccelli e nei rettili terrestri sotto forma di acido urico. Queste tre classi di organismi vengono detti rispettivamente ureotelici, uricotelici e ammoniotelici. 284 Capitolo XIV Nel ciclo dell’urea (Figura 4) uno degli atomi di azoto dell’urea deriva dall’aspartato, mentre l’altro azoto ed il carbonio derivano dall’NH4+ e dalla CO2. Il trasportatore di questo atomo di carbonio e dei due atomi di azoto è l’ornitina. Figura 4. Il ciclo dell'urea. Il precursore immediato dell’urea è l’arginina, che viene idrolizzata ad urea ed ornitina dall’arginasi. Le altre reazioni del ciclo portano alla sintesi dell’arginina dall’ornitina. Dapprima un gruppo carbammilico viene trasferito sull’ornitina per formare citrullina. L’enzima è l’ornitina transcarbammilasi. Il donatore del gruppo carbammilico è il carbammilfosfato che ha alto potenziale di trasferimento dovuto al suo legame anidridico. L’arginin-succinato sintetasi catalizza poi la condensazione della citrullina con l’aspartato. Questa sintesi di arginosuccinato è resa possibile dalla scissione di ATP in AMP e pirofosfa- Il metabolismo dell’azoto 285 to, seguita dall’idrolisi del pirofosfato. Infine l’arginino-succinasi scinde l’arginino-succinato in arginina e fumarato. Da notare che queste reazioni, che trasferiscono il gruppo amminico dall’aspartato formando arginina, conservano lo scheletro carbonioso dell’aspartato. Il carbammilfosfato viene sintetizzato da NH4+, CO2, ATP e H2O in una reazione complessa catalizzata dalla carbammilfosfato sintetasi: CO2 + NH4+ + 2ATP + H2O → H2N–COO–PO3 + 2 ADP + Pi + 3H+ Il consumo di 2 ATP rende questa sintesi praticamente irreversibile. La stechiometria della sintesi dell’urea è: CO2 + NH4+ + 3 ATP + aspartato + 2 H2O → urea + 2 ADP + 2Pi + PPi + fumarato Il PPi viene rapidamente idrolizzato, quindi in questa reazione per sintetizzare una molecola di urea vengono consumati quattro legami fosforici ad alta energia. La sintesi del fumarato nel ciclo dell’urea è importante perché lega il ciclo dell’urea al ciclo dell’acido citrico. Il fumarato viene idratato a malato, che a sua volta viene ossidato ad ossalacetato. L’ossalacetato, a sua volta, può: 1) 2) 3) 4) essere transaminato ad aspartato; essere convertito in glucosio attraverso la gluconeogenesi; condensare con l’acetil-CoA per sintetizzare il citrato; convertirsi in piruvato. Nel ciclo dell’urea la formazione di ione ammonio da parte della glutammato deidrogenasi, la sua incorporazione nel carbammil fosfato e la sintesi di citrullina avvengono nella matrice mitocondriale, mentre le tre successive reazioni che portano all’urea hanno luogo nel citosol. Fino ad ora abbiamo considerato una serie di reazioni che rimuovono il gruppo α-aminico dagli aminoacidi e lo convertono in urea, ora ci occuperemo del destino dello scheletro di atomi di carbonio. La strategia della degradazione degli amminoacidi è quella di formare i 286 Capitolo XIV più importanti intermedi metabolici che possano essere convertiti i glucosio oppure essere ossidati nel ciclo dell’acido citrico. Gli scheletri carboniosi provenienti dalla rimozione del gruppo amminico dagli amminoacidi possono essere convertiti in glucosio, o ossidati attraverso il ciclo di Krebs. Infatti gli scheletri carboniosi dei venti amminoacidi naturali possono essere trasformati solo in sette molecole: piruvato, acetil-CoA, acetoacetil-CoA, α-chetoglutarato, succinil-CoA, fumarato e ossalacetato. Gli aminoacidi che vengono degradati ad acetil-CoA o acetoacetil-CoA sono chiamati chetogenici (indicati in giallo in Figura 5) perché danno origina a corpi chetonici. Figura 5. Destino dello scheletro carbonioso degli aminoacidi. Questi sono tre composti che sono normalmente presenti nel sangue in piccole quantità: l'acetone, l'acido acetacetico, e l'acido betaidrossibutirrico. La produzione del corpi chetonici avviene in deficienza di glucosio quando l'organismo mobilizza grassi dai tessuti adiposi, ricavandone acidi grassi, che diventano una fonte energetica, diretta- Il metabolismo dell’azoto 287 mente o attraverso la loro trasformazione da parte del fegato in corpi chetonici. Costituiscono quindi una fonte di energia alternativa al glucosio per i tessuti periferici. Gli aminoacidi che sono degradati a piruvato, α-chetoglutarato, succinil-CoA, fumarato, ossalacetato sono chiamati glucogenici (indicati in rosa sopra in Figura 5). Da questi composti è possibile la sintesi di glucosio perché questi intermedi del ciclo dell’acido citrico possono essere convertiti in fosfoenolpiruvato e poi in glucosio. Nei mammiferi la capacità di sintesi del glucosio a partire da acetil-CoA o acetoacetil-CoA è stata persa. Solo leucina e lisina sono esclusivamente chetogenici. L’isoleucina, la fenilalanina, il triptofano e la tirosina sono sia chetogenici che glucogenici. Gli altri quattordici aminoacidi sono classificati come esclusivamente glucogenici. NUCLEOTIDI ED ACIDI NUCLEICI Gli acidi nucleici, acido desossiribonucleico, DNA e acido ribonucleico, RNA, sono i portatori chimici dell’informazione genetica della cellula. Nel DNA cellulare si trova infatti codificata tutta l’informazione che determina la natura della cellula, ne controlla la crescita e la divisione e dirige la biosintesi degli enzimi e delle altre proteine necessarie alle funzioni cellulari. La struttura di ogni proteina, e in ultima analisi quindi di ogni componente cellulare, è il risultato di una informazione programmata nella sequenza nucleotidica degli acidi nucleici di una cellula. Come le proteine sono polimeri costituiti da singole unità di amminoacidi, così gli acidi nucleici sono polimeri costituiti da singoli blocchi detti nucleotidi, che sono composti ricchi di energia che guidano i processi metabolici, in particolare le biosintesi, in tutte le cellule. I nucleotidi sono formati da tre componenti: una base contenente azoto (base azotata), uno zucchero riducente a 5 atomi di carbonio (un pentosio) ed un gruppo fosforico (costituito cioè da acido fosforico, H3PO4). La stessa molecola, in assenza di gruppo fosforico, prende il nome di nucleoside (Figura 1). Figura 1. Struttura schematica di nucleotide e nucleoside. 289 290 Capitolo XV Occorre qui ricordare che quando si parla di “base azotata” si intende sempre una base di Lewis, che dispone cioè di una coppia elettronica che può essere condivisa con un altro composto. Le basi azotate di DNA e RNA sono derivate da due composti, la pirimidina e la purina. La prima è un anello eterociclico, la seconda è costituita da due anelli eterociclici. La base azotata di ogni nucleotide è unita con legame covalente tra un atomo di azoto e l’atomo di carbonio 1 del pentosio. Si tratta di un legame N-β-glicosidico, che si forma per eliminazione di una molecola di acqua fra un gruppo –OH del pentosio ed un -NH della base, come nella formazione dei legami O-glicosidici. Il gruppo fosforico è invece esterificato sul carbonio 5 del pentosio. I nucleotidi possono avere diversi ruoli: • costituiscono le subunità degli acidi nucleici. Sono infatti i precursori attivati del DNA e dell’RNA; • sono intermedi attivati in molte biosintesi. Ad es. l’UDP-glucosio (uridin tri-fosfato) è il precursore del glicogeno e il CDPdiacilglicerolo (citidin di-fosfato) è precursore dei fosfogliceridi; • l’ATP, un nucleotide adeninico, è il trasportatore universale di energia nei sistemi biologici. Il GTP (guanidin tri-fosfato) fornisce l’energia per i movimenti di molte macromolecole; • i nucleotidi adeninici possono essere cofattori enzimatici: sono infatti componenti dei tre coenzimi più importanti, NAD+, FAD e Coenzima A; • possono essere messaggeri chimici e regolatori metabolici. L’AMP (adenosin monofosfato) ciclico è un mediatore dell’azione di molti ormoni. Gli acidi nucleici Il componente zuccherino dell’RNA è il ribosio, quello del DNA il 2’desossiribosio. Il prefisso 2’-desossi indica la perdita di un ossigeno nella posizione 2’ del ribosio. I numeri seguiti dall’apice indicano le posizioni dei sostituenti sullo zucchero, mentre quelli senza apice si riferiscono alle posizioni dei sostituenti della base azotata. Nel DNA si hanno quattro diverse basi eterocicliche: due sono purine sostituite (adenina e guanina) e le altre due sono pirimidine sostituite (citosina Nucleotidi e acidi nucleici 291 e timina). Adenina, guanina e citosina si trovano anche nell’RNA, dove però al posto della timina troviamo una pirimidina detta uracile (Figura 2). Figura 2. Struttura della basi puriniche e pirimidiniche. I nucleotidi sono i monomeri degli acidi nucleici, cioè i costituenti elementari con cui questi acidi vengono costruiti. I nucleotidi si concatenano sia nell’RNA che nel DNA formando “ponti” covalenti tra gruppi fosforici mediante legami fosfodiestere fra il componente 5’ fosfato di un nucleotide e l’ossidrile 3’ dello zucchero di un altro nucleotide. Una estremità del polimero risultante ha un –OH libero in C3’, l’altra un residuo acido fosforico in C5’. Lo scheletro degli acidi nucleici è costituito dall’alternanza di gruppi fosforici e residui di pentosio, mentre le basi azotate possono essere considerate come gruppi laterali uniti allo scheletro ad intervalli regolari ed in ordine non casuale. Si può dire infatti che i nucleotidi rappresentano l’alfabeto della vita sulla Terra, infatti il corredo genetico delle cellule è espresso come una sequenza di nucleotidi. La sequenza delle basi lungo la catena costituisce l’informazione genetica contenente ad esempio le istruzio- 292 Capitolo XV ni per la sintesi delle proteine. Una successione sufficientemente lunga di combinazioni dei quattro nucleotidi è in grado di codificare un intero patrimonio genetico di una specie. Figura 3. Legame fosfodiestere nello scheletro di DNA e RNA. Benché chimicamente simili, DNA e RNA hanno dimensioni e funzioni diverse. Le molecole del DNA sono molto grandi, con lunghezza fino a 12 cm, e si trovano principalmente nel nucleo della cellula. Le molecole dell’RNA sono molto più piccole, e si trovano principalmente fuori del nucleo. La successione dei nucleotidi di una catena si descrive partendo dall’estremità 5’ ed identificando le basi nell’ordine in cui si presentano, abbreviando i nomi delle basi in A per adenina, T per timina, G per guanina, C per citosina e U per uracile. L’appaiamento delle basi nel DNA: il modello di Watson e Crick Prima di parlare dei risultati dei celeberrimi lavori di Watson e Crick sulla struttura del DNA, è doveroso ricordare che alcune impor- Nucleotidi e acidi nucleici 293 tanti caratteristiche del DNA erano state già riportate da Chargaff e altri alla fine degli anni 40. Questi ricercatori avevano notato che: • • • • la composizione del DNA varia in genere da una specie all’altra; le molecole di DNA in tessuti diversi della stessa specie hanno uguale composizione in basi; la composizione in basi del DNA di una data specie non si modifica con l’età dell’organismo, con lo stato nutrizionale o in seguito a variazioni nell’ambiente; in tutte le molecole del DNA, indipendentemente dalla specie a cui appartengono, il numero di residui di adenina è uguale al numero di residui di timina (cioè A= T) ed il numero di residui di guanina è uguale al numero di residui di citosina (cioè G= C). Quindi la somma dei residui purinici è uguale alla somma dei residui pirimidinici, A + G = T + C. Queste relazioni, chiamate “Regole di Chargaff” rappresentarono la chiave per la comprensione della struttura tridimensionale del DNA e dei sistemi con i quali l’informazione genetica del DNA viene codificata e trasferita da una generazione all’altra. E’ stato in seguito confermato che campioni di DNA provenienti da vari tessuti della medesima specie presentano la stessa proporzione fra le basi eterocicliche, mentre quelli che provengono da specie diverse possono differire molto. Ad esempio nel DNA umano si trova un 30% di adenina e timina e circa il 20% di guanina e citosina, in altre specie la proporzione è diversa. Come già accennato sopra, nel DNA le basi vanno a coppie. Per capire il modo e le cause di questo appaiamento bisogna riferirsi al modello della doppia elica, formulato nel 1953 da James Watson e Francis Crick. Essi scoprirono che il DNA è formato da due filamenti polinucleotidici elicoidali avvolti intorno allo stesso asse a formare una doppia elica destrorsa. Lo scheletro covalente idrofilo, composto da un’alternanza di ribosio e gruppi fosforici è all’esterno della doppia elica, in contatto con l’ambiente circostante. Le basi puriniche e pirimidiniche sono impilate all’interno della doppia elica con le loro strutture planari ad anello poste in posizione quasi perpendicolare al lungo asse longitudinale della molecola. I due filamenti decorrono in senso 294 Capitolo XV opposto e sono collegati mediante legami a idrogeno fra determinate coppie di basi. Adenina e timina possono formare due legami a idrogeno una con l’altra, mentre guanina e citosina possono legarsi con tre legami a idrogeno soltanto fra loro. Nell’RNA dove non è presente la timina, l’adenina forma due legami a idrogeno con l’uracile. Figura 4. Disposizione dei legami a idrogeno nelle coppie di basi. Quando in un filamento si trova la base citosina, sul filamento parallelo si trova la guanina, mentre all’adenina corrisponde la timina. Questo appaiamento spiega perché le basi affacciate sono sempre in quantità uguale. Ogni base è sfalsata di 3,4 Å lungo l’asse dell’elica. Un giro della doppia elica contiene 10,5 coppie di basi con una periodicità di circa 36 Å. Misure eseguite con i raggi X hanno mostrato che la doppia elica del DNA è larga circa 20 Å e che ci sono 10 coppie di basi per ogni giro. Inoltre, i due filamenti della doppia elica si avvolgono in modo da formare due tipi di solco, una scanalatura maggiore, o solco principale, ampio 12 Å e una scanalatura minore, o solco se- Nucleotidi e acidi nucleici 295 condario di 6 Å, entrambi sulla superficie della doppia elica (Figura 5). Il primo è leggermente più profondo dell’altro ed entrambi portano potenziali donatori ed accettori di legami a idrogeno. Per questo esiste una serie di molecole che possono inserirsi in uno dei solchi fra i filamenti. Si pensa che molti agenti cancerogeni o che prevengono la degenerazione dovuta al cancro si possano intercalare nel DNA in questo modo. Le due catene polinucleotidiche del DNA non hanno sequenze di base o composizioni identiche, ma sono complementari. Le due catene del DNA sono antiparallele, cioè i loro legami fosfodiestere corrono in posizioni opposte. Figura 5. Modello di Watson e Crick della struttura del DNA. Acidi nucleici e trasmissione dell’informazione genetica La molecola del DNA è la depositaria chimica dell’informazione genetica dell’organismo a cui appartiene, che risiede nella successione di desossiribonucleotidi concatenati. Il compito del DNA consiste nel conservare questa informazione e nel trasmetterla al momento oppor- 296 Capitolo XV tuno, all’RNA. A sua volta l’RNA dovrà leggere, decodificare e applicare l’informazione ricevuta allo scopo di sintetizzare le proteine. Ciascuna delle migliaia di geni presenti in un cromosoma contiene le istruzioni necessarie per fabbricare una determinata proteina, che deve assolvere uno specifico compito biologico. Decodificando i geni giusti, al momento opportuno e nel giusto sito, l’organismo sintetizzerà le molte migliaia di proteine per le reazioni biochimiche necessarie al suo funzionamento. I processi fondamentali del trasferimento dell’informazione genetica dal DNA sono tre: 1. Replicazione E’ il processo di riproduzione identica del DNA, che permette di conservare e di trasmettere l’informazione genetica alla progenie; 2. Trascrizione E’ il processo mediante il quale i messaggi genetici contenuti nel DNA vengono letti e copiati esattamente nell’RNA; 3. Traduzione E’ il processo di decodificazione dei messaggi genetici contenuti nell’RNA che viene tradotto nei ribosomi in specifiche sequenze di amminoacidi, ossia le proteine. Figura 6. Schema del trasferimento dell'informazione genetica. La replicazione del DNA La replicazione del DNA è caratterizzata da un elevato grado di accuratezza e si verifica in un dato periodo del ciclo cellulare. E’ un processo detto “semiconservativo” in quanto ogni catena funziona da Nucleotidi e acidi nucleici 297 stampo per la catena nuova. L’inizio si ha con lo srotolamento parziale della doppia elica paragonabile all’apertura di una cerniera lampo. Man mano che i filamenti si separano ed espongono le proprie basi, i nuovi nucleotidi si allineano lungo il filamento in modo esattamente complementare, A con T e C con G e incominciano a crescere nuovi filamenti. Ognuno dei filamenti è complementare al vecchio stampo per cui da una doppia elica hanno origine due doppie eliche identiche (Figura 7). La DNA-polimerasi catalizza l’addizione dei nuovi nucleotidi alla catena,sommando un 5’mononucleotide trifosfato all’ossidrile libero in 3’ della catena esistente Figura 7. Processo di replicazione del DNA. Il nucleo di una cellula umana contiene 46 cromosomi (23 coppie) costituiti ciascuno da una grandissima molecola di DNA. Ogni cromosoma a sua volta è costituito da parecchie migliaia di segmenti di DNA detti geni e la somma di tutti i geni delle cellula (il genoma) 298 Capitolo XV contiene circa 3 miliardi di coppie di basi (Figura 8). Una catena di DNA può arrivare a 12 cm di lunghezza e contenere fino a 250 milioni di coppie di basi. Anche se sono molecole così grandi, la sequenza delle loro basi viene fedelmente ricopiata. Il processo si svolge in pochi minuti e non comporta più di un errore in 10-100 miliardi di basi. Figura 8. Dalla cellula alle singole basi del DNA. Nucleotidi e acidi nucleici 299 L’RNA: la trascrizione Replicazione e trascrizione differiscono per un aspetto importante, durante la replicazione viene copiato l’intero cromosoma per dare origine a due copie identiche; al contrario la trascrizione è un processo selettivo in quanto vengono trascritti solo quei gruppi di geni necessari in ogni data circostanza in modo tale che questa possa poi trasformare queste informazioni in molecole proteiche. Esistono 3 tipi diversi di acido ribonucleico, ciascuno con una ben precisa funzione. 1) RNA messaggero (mRNA): Trasporta i messaggi genetici dal DNA ai ribosomi, minuscole particelle granulari del citoplasma cellulare, in cui ha luogo la sintesi delle proteine. E’ circa il 5% dell’RNA totale. 2) RNA ribosomiale (rRNA): complessato con materiale proteico assicura la costruzione dei ribosomi. E’ circa il 70-80 % dell’RNA totale. 3) RNA transfer (tRNA): Trasporta sui ribosomi determinati amminoacidi, che vengono concatenati in modo da formare le proteine. E’ circa il 10-20 % dell’RNA totale. L'RNA transfer è costituito da piccole molecole a forma di trifoglio, ciascuna delle quali porta uno specifico amminoacido. A ogni codone dell'mRNA (un codone è la sequenza di tre basi azotate, specifica per ogni amminoacido) corrisponde un anticodone del tRNA. La trascrizione richiede l’enzima RNA polimerasi, enzima che ha il compito fondamentale di individuare, sulla catena del DNA il punto esatto in cui iniziare il processo. La traduzione in proteina dell’informazione contenuta in un segmento di DNA inizia con la sintesi di molecole di mRNA, che contengono un numero di nucleotidi adatto alla proteina da costruire. Nell’ mRNA si trascrive però solo un filamento del DNA. Quello che contiene il gene si dice filamento dell’informazione, mentre quello complementare è il filamento stampo. Solo il filamento stampo viene trascritto, per cui risulta una copia del filamento dell’informazione. L’unica differenza è la sostituzione della timina (DNA) con l’uracile (RNA). 300 Capitolo XV La traduzione e la biosintesi delle proteine La traduzione è il processo mediante il quale diversi amminoacidi vengono uniti per formare una nuova proteina, mediante le istruzioni contenute in un filamento di RNA messaggero. Poiché tale filamento si forma sullo stampo del DNA, in realtà è questo acido nucleico a presiedere alla sintesi proteica. Mentre sta ancora avvenendo la trascrizione di un filamento di RNA messaggero, mRNA, questo inizia a staccarsi dal filamento stampo. Al termine di tale processo, un’estremità del filamento della nuova molecola si inserisce in un ribosoma. Questo organulo "scorre" lungo la molecola di mRNA; in tal modo, il ribosoma "legge" la sequenza delle basi azotate sull’mRNA. Questo processo prende il nome di traduzione e coinvolge un terzo tipo di molecola di RNA, chiamata RNA transfer (tRNA), che da una parte porta una tripletta di nucleotidi e dall’altra un amminoacido specifico, corrispondente alla tripletta. La tripletta di ciascun tRNA aderisce alla molecola di mRNA quando si trova una tripletta complementare. Ad esempio, la sequenza uracile - citosina - uracile (UCU) sul filamento dell’mRNA viene occupata dal tRNA contenente la tripletta adenina - guanina - adenina (AGA). In contrapposizione alla tripletta dell’mRNA, che si chiama codone, quella del tRNA prende il nome di anticodone. Gli amminoacidi portati dal tRNA nella sequenza specificata dall’mRNA vengono, quindi, legati l’uno all’altro sul ribosoma, a formare una nuova catena polipeptica. Completata la sintesi della proteina un codone di arresto segnala il termine dell’operazione e la proteina abbandona il ribosomi (Figura 9). Una volta terminata, la catena polipeptidica si libera dal ribosoma e assume la sua forma tridimensionale specifica, determinata dalla sequenza degli amminoacidi. Nucleotidi e acidi nucleici Figura 9. Sequenza di eventi durante la sintesi proteica. 301 LA SINTESI PROTEICA Concetti generali Le proteine sono il prodotto finale della maggior parte delle vie dell’informazione. Una cellula ha bisogno di migliaia di proteine che devono essere sintetizzate a seconda delle necessità, trasportate nella appropriata sede cellulare e degradate quando non servono più. Il processo di formazione delle proteine, che ha un meccanismo biosintetico molto complesso, forse il più complesso di tutta la biochimica, è ancor oggi meglio conosciuto che non quello della loro degradazione. In parole semplici, le proteine vengono sintetizzate in modo da avere una particolare sequenza di amminoacidi, che le caratterizza, e che deriva dalla traduzione dell’informazione, proveniente dal DNA, codificata dall’RNA messaggero (mRNA) in un complesso di RNA e proteine chiamato ribosoma. Gli amminoacidi sono specificati da unità dell’mRNA dette codon. Per la traduzione sono necessari degli adattatori, gli RNA transfer (tRNA) che riconoscono i codon e inseriscono gli amminoacidi nelle loro corrette posizioni sequenziali nel polipeptide che si sta formando. I codon sono costituiti da specifiche triplette di nucleotidi. Le sequenze di base dei codon sono state dedotte per mezzo di esperimenti che utilizzavano mRNA sintetici a sequenza nota. Il codice genetico è “degenerato” ossia presenta più codici per quasi tutti gli amminoacidi. Come si vedrà in seguito, la terza posizione è molto meno specifica della prima e della seconda ed è detta oscillante. Le parole standard del codice genetico sono probabilmente universali per tutte le specie, sebbene esistano piccole differenze nei mitocondri e in alcuni organismi unicellulari. L’amminoacido che segnala l’inizio delle catene polipeptidiche in tutte le cellule, la metionina, è codificato dalla tripletta AUG. Altre triplette (UAA, UAG e UGA)non codificano amminoacidi ma sono segnali di terminazione della catena. La sintesi delle proteine, nelle cellule eucariotiche, richiede la partecipazione di più di 70 proteine ribosomiali, di più di 20 enzimi che attivano i precursori degli amminoacidi e di una dozzina di enzimi 303 304 Capitolo XVI ausiliari e di altri fattori proteici, specifici per l’inizio, l’allungamento e la terminazione dei polipeptidi. Sono necessari inoltre quasi cento enzimi per le modificazioni finali dei diversi tipi di proteine e più di quaranta tipi di RNA transfer e ribosomiali (t-RNA e r-RNA). Per la descrizione dei vari tipi di RNA e le informazioni principali sul DNA si rimanda al capitolo “Nucleotidi ed acidi nucleici”. La sintesi proteica può utilizzare fino al 90% dell’energia chimica necessaria alla cellula per tutte le reazioni biosintetiche. Nonostante la complessità del processo, le proteine vengono sintetizzate a velocità abbastanza elevate: una catena polipeptidica completa composta di circa cento residui viene sintetizzata in una cellula di Escherichia coli a 37°C in circa 5 secondi. La sintesi delle tante e diverse proteine in ogni cellula è rigidamente regolata, in modo che , in determinate condizioni metaboliche, vengono prodotte solo le molecole necessarie. Il processo di sintesi proteica ed il codice genetico Vediamo ora più in dettaglio il processo della biosintesi proteica. A partire dagli anni’50 tre grandi scoperte hanno contribuito a far luce sul meccanismo che porta alla biosintesi delle proteine. 1) Il sito dove avviene la sintesi proteica è situato in una frazione che contiene piccole particelle ribonucleoproteiche, già scoperte in precedenza nei tessuti animali mediante la microscopia elettronica, dette in seguito ribosomi. 2) Gli amminoacidi vengono “attivati” se incubati con ATP e con la frazione citosolica delle cellule del fegato. Gli amminoacidi sono uniti ad una forma speciale di RNA solubile, stabile al calore, detto RNA transfer (tRNA), formando un complesso amminoacil-tRNA. Gli enzimi che catalizzano questo processo sono le amminoacil-tRNA sintetasi. 3) Watson e Crick, spiegarono il meccanismo secondo il quale l’informazione genetica, codificata dal linguaggio a quattro lettere degli acidi nucleici, viene tradotta nel linguaggio a venti lettere delle proteine. Crick intuì che il tRNA serviva da adattatore (o da traduttore), con una parte della molecola che lega l’amminoacido e l’altra par- La sintesi proteica 305 te che riconosce una breve sequenza del mRNA che codifica quell’amminoacido. Infatti, il tRNA adattatore traduce la sequenza nucleotidica del mRNA nella sequenza amminoacidica del polipeptide. Il processo complessivo della sintesi proteica guidata dal mRNA viene detto traduzione. Il codice genetico Già dagli anni ’60 si era visto che per codificare un amminoacido erano necessari almeno 3 residui nucleotidici del DNA. Si ricordi che un nucleotide è costituito da una base azotata, uno zucchero, generalmente il ribosio, o il desossiribosio nel caso del DNA, e un gruppo fosforico H3PO4 e che le basi azotate nel DNA sono adenina, guanina, timina e citosina (rappresentate rispettivamente con le lettere A,T,G e C), mentre nell’RNA la base timina è sostituita dall’uracile (U). Il codice a quattro lettere del DNA (A,T,G,C,) preso a gruppi di due può dare solo 42 = 16 combinazioni diverse, non sufficienti a codificare per venti amminoacidi, ma se preso a gruppi di tre può dare 43 = 64 combinazioni. Esperimenti precedenti avevano mostrato che le parole del codice genetico, i codon, che codificano per gli amminoacidi sono triplette di nucleotidi. Inoltre fu trovato che i codon non si sovrappongono e che tra loro non esistono intervalli tra residui amminoacidici consecutivi. La sequenza degli amminoacidi di una proteina è dunque definita da una sequenza lineare di triplette codificanti contigue. Il primo codon della sequenza determina un quadro di lettura, in base al quale ogni tre residui nucleotidici inizia un nuovo codon. Secondo questo schema ci sono tre possibili quadri di lettura per ogni sequenza di DNA e ognuno determina tre diverse sequenze di codon. Sono stati identificati 61 dei 64 possibili codon. Gli altri tre sono codon di terminazione, perché interrompono la sequenza degli amminoacidi, quando vengano inseriti nella sequenza di un polimero di RNA sintetico. Quindi queste triplette non hanno praticamente senso e quindi non corrispondono ad alcun amminoacido. Il “dizionario” completo dei codon per i 20 amminoacidi proteici, così come si trova nel mRNA, è riportato in Figura 1. 306 Capitolo XVI Figura 1. Il dizionario del codice genetico. La terza base di ogni codon ha un ruolo meno importante rispetto alle altre. La tripletta AUG è il codon di inizio, che segnala l’inizio della catena polipeptidica, sia nei procarioti che negli eucarioti. Inoltre, AUG codifica anche i residui di metionina all’interno dei polipeptidi. Tre delle possibili triplette nucleotidiche, e precisamente UAA, UAG e UGA non codificano nessun amminoacido noto e sono dette codon di terminazione, o stop codon o codon nonsenso. La sintesi proteica 307 Va notato che tutti gli amminoacidi, con l’eccezione di metionina e triptofano, hanno più di un codon ma nessun codon è specifico per più di un amminoacido. Leucina e serina hanno sei codon, glicina e alanina quattro, glutammato, tirosina ed istidina due. In genere, i codon che specificano per lo stesso amminoacido differiscono solo per la terza base. Ad esempio, l’alanina è codificata dalle triplette CGU, GCC, GCA e GCG. Le prime due lettere sono quindi i principali determinanti della specificità dell’amminoacido. Esaminiamo ora le fasi principali delle sintesi proteica. Il processo può essere diviso in cinque stadi, di cui in un primo momento verrà presentato un sommario. I vari processi relativi ai cinque stadi verranno poi descritti per esteso. Stadio 1: attivazione degli amminoacidi La sintesi di un polipeptide i cui amminoacidi sono allineati in una determinata sequenza deve avere due requisiti fondamentali: • il gruppo carbossilico di ciascun amminoacido deve essere attivato per facilitare la formazione del legame peptidico; • ciascun amminoacido deve corrispondere al codon sull’mRNA. Questo si realizza con l’attacco dell’amminoacido in questione al tRNA nel primo stadio della sintesi proteica. Stadio 2: inizio L’mRNA che contiene il codice per il polipeptide da sintetizzare si lega alla subunità minore del ribosoma e all’amminoacil-tRNA di inizio. A questo punto si lega la subunità ribosomiale maggiore e si ha un complesso di inizio. Stadio 3: allungamento La catena polipeptidica nascente si allunga mediante l’unione con legami covalenti fra unità successive di amminoacidi, ciascuna trasportata verso il ribosoma e correttamente posizionata dal rispettivo tRNA, che si appaia al codon corrispondente nell’mRNA. Stadio 4: termine e rilascio La fine della catena polipeptidica è segnalata da un codon di terminazione nell’mRNA. La nuova catena polipeptidica viene quindi rilasciata dal ribosoma, con l’aiuto di proteine chiamate fattori di rilascio. 308 Capitolo XVI Stadio 5: ripiegamento e modificazioni post-traduzione Per ottenere la sua forma biologicamente attiva il nuovo polipeptide deve ripiegarsi nella sua conformazione tridimensionale. Il nuovo polipeptide può subire modificazioni mediate da enzimi sia prima che dopo il suo ripiegamento. Queste modificazioni possono essere: • la rimozione di uno o più amminoacidi all’estremità amminoterminale; • l’aggiunta a determinati residui amminoacidici di gruppi acetile, gruppi fosfato, metilici, carbossilici; • la scissione della proteina mediante proteasi; • l’attacco di polisaccaridi; • l’attacco di gruppi prostetici. Vediamo ora più in dettaglio questi processi: Stadio 1 In questa prima fase, che ha luogo nel citosol, i 20 diversi amminoacidi vengono legati con legame estere al loro tRNA ad opera di amminoacil-tRNA sintetasi, ciascuna delle quali è specifica per un amminoacido e per uno o più tRNA corrispondenti. L’amminoacilazione del tRNA ha due obiettivi: 1) attivare ogni amminoacido per la formazione di un legame peptidico. 2) attaccare l’amminoacido ad un tRNA adattatore, che lo porta nella giusta posizione nel peptide in formazione. L’identità dell’amminoacido legato al tRNA non viene controllata a livello del ribosoma, quindi l’attacco del corretto amminoacido dipende dalla specificità dell’amminoacil-tRNA sintetasi. Nella maggior parte degli organismi esiste una amminoacil-tRNA sintetasi per ciascun amminoacido. La reazione di attivazione avviene in due passaggi separati nel sito attivo dell’enzima. Nel primo passaggio si forma un intermedio legato all’enzima, l’amminoacil-adenilato (amminoacil-AMP). Amminoacido + ATP → Amminoacil-AMP + PPi In questa reazione il gruppo carbossilico dell’amminoacido si lega con legame anidridico al gruppo fosforico dell’ATP con liberazione di La sintesi proteica 309 pirofosfato. Il legame estere fra l’amminoacido ed il tRNA ha alta energia libera standard di idrolisi (∆G°’= –29 kJ/ mole). Il pirofosfato liberato viene idrolizzato a fosfato dalla pirofosfatasi inorganica. Quindi per ogni molecola di amminoacido attivata sono due i legami fosforici ad alta energia utilizzati, il che rende la reazione praticamente irreversibile. Nel secondo passaggio, il gruppo amminoacilico viene trasferito dall’amminoacil-AMP legato all’enzima al tRNA specifico corrispondente (Figura 2). La reazione è: Amminoacil-AMP + tRNA → Amminoacil-tRNA + AMP Figura 2. Struttura generale di un amminoacil-tRNA. Il legame estere che provoca sia l’attivazione dell’amminoacido sia il suo attacco al tRNA è evidenziato in giallo. La reazione complessiva è: Amminoacido + tRNA + ATP → Amminoacil-tRNA + AMP + 2Pi (∆G°’= –29 kJ/ mole) 310 Capitolo XVI La presenza degli errori nella sintesi proteica (uno ogni 104 amminoacidi) non è così rara come per il DNA, forse perché un eventuale errore in una proteina viene eliminato distruggendo la proteina stessa e quindi non viene trasmesso alle generazioni future. Stadio 2 La sintesi proteica nelle cellule eucariotiche inizia dall’estremità ammino-terminale e procede con l’aggiunta graduale di altri amminoacidi all’estremità carbossi-terminale di un polipeptide. Nelle cellule eucariotiche tutti i polipeptidi sintetizzati dai ribosomi del citosol iniziano con un residuo di metionina. Il codon di inizio AUG specifica infatti un residuo di metionina ammino-terminale. Sebbene esista un solo codon per la metionina tutti gli organismi possiedono due tRNA per questo amminoacido. Un tRNA viene usato quando AUG è il codon di inizio della sintesi proteica, l’altro quando nel polipeptide la metionina viene aggiunta in una posizione intermedia. Nei batteri, l’amminoacil-tRNA di inizio di tutte le proteine è la Nformilmetionil-tRNA. Si forma un complesso, detto complesso di inizio, fra la subunità ribosomiale 30S, l’mRNA, il GTP, la tNformilmetionil-tRNA due fattori d’inizio e la subunità 50S. Il GTP viene idrolizzato a GDP e Pi. Stadio 3: Il terzo stadio della sintesi proteica è l’allungamento. Questo richiede : • il complesso d’inizio descritto nello stadio 2; • gli amminoacil-tRNA; • un gruppo di tre proteine solubili del citosol dette fattori di allungamento; • il GTP Durante l’aggiunta di ciascun amminoacido si verificano tre fasi e questo ciclo viene ripetuto tante volte quanti sono gli amminoacidi che vengono aggiunti alla catena polipeptidica. Nella prima fase del ciclo di allungamento l’amminoacil-tRNA successivo viene inizialmente legato ad una delle proteine di allunga- La sintesi proteica 311 mento che contiene a sua volta una molecola di GTP. Il complesso risultante si lega a sua volta al complesso di inizio. Il GTP viene idrolizzato e il complesso modificato viene poi rilasciato dal ribosoma. Il complesso iniziale viene poi rigenerato. Nella seconda fase si forma un nuovo legame peptidico fra i due amminoacidi ancora legati ai loro tRNA nei siti A e P del ribosoma (Figura 3). Il sito peptidilico (sito P) trattiene la catena polipeptidica in fase di allungamento ancora attaccata mediante un legame aminoacilico al t-RNA dell’ultimo aminoacido aggiunto. Esiste anche un sito E o sito di uscita in cui i tRNA scarichi, che hanno fornito alla catena l’aminoacido, escono dal ribosoma. Nel ribosoma i siti A, P, E sono distanziati in modo da portare ogni tRNA sul codone corrispondente senza sovrapposizioni né spazi. Figura 3. Schema dei siti A, P, ed E nella subunità ribosomale. Il sito accettore (sito A) lega l’aminoacil-tRNA del successivo aminoacido che deve essere aggiunto alla catena. Questa reazione produce un dipeptidil-tRNA nel sito A e il tRNA che portava la metionina, ora “scarico” (cioè deacilato) rimane legato al sito contiguo P. La formazione del legame peptidico è catalizzata da una peptidil transferasi. Nella fase finale del ciclo di allungamento, chiamata traslocazione, il ribosoma si muove di una lunghezza pari ad un codon verso l’estremità del mRNA. Poiché il dipeptidil-tRNA è ancora attaccato al 312 Capitolo XVI secondo codon dell’mRNA il movimento del ribosoma sposta il dipeptidil-tRNA dal sito A al sito P. Questo spostamento richiede una traslocasi ed energia fornita da un’altra molecola di GTP. Il ribosoma, con il dipeptidil-tRNA e l’mRNA ad esso legati è ora pronto per un altro giro di allungamento che porta a legare il terzo residuo amminoacidico. Stadio 4: La quarta fase della sintesi polipeptidica è segnalata da uno dei tre codon di terminazione dell’mRNA (UAA,UAG, UGA) che segue immediatamente il codon dell’ultimo amminoacido. Nei batteri, quando un codon di terminazione occupa il sito A sul ribosoma, tre fattori di terminazione o fattori di rilascio (tre proteine RF1, RF2 e RF3) provvedono ad effettuare: 1) l’idrolisi del legame terminale del peptidil-tRNA; 2) il rilascio del polipeptide libero e dell’ultimo tRNA, ora scarico, dal sito P; 3) la dissociazione del ribosoma nelle sue subunità. Si rende così possibile l’inizio di un nuovo ciclo di sintesi polipeptidica. L’RF1 riconosce i codon di terminazione UAG e UAA, mentre RF2 riconosce UGA e UAA. Poi una delle proteine (a seconda del codon presente) si lega al codon di terminazione e induce la peptidil transferasi a trasferire la catena polipeptidica ad una molecola d’acqua anziché ad un altro amminoacido. Negli eucarioti esiste un solo fattore di rilascio, eRF. Stadio 5: Nella quinta ed ultima fase della sintesi proteica la catena polipeptidica in formazione viene ripiegata su se stessa e modificata per essere alla fine biologicamente attiva. Prima o dopo la sua sintesi, la catena polipeptidica assume spontaneamente la sua conformazione nativa, quella che permette la formazione del massimo numero di legami a idrogeno, interazioni di Van der Waals, di interazioni ioniche ed idrofobiche. In tal modo, il messaggio genetico viene convertito nella struttura tridimensionale della proteina. La sintesi proteica 313 Alcune proteine appena sintetizzate, sia nei procarioti che negli eucarioti, non raggiungono la loro conformazione biologicamente attiva finché non sono state modificate in una o più reazioni dette appunto modificazioni post-traduzionali. Le modificazioni possibili sono: 1) modificazioni ammino-terminali o carbossil-terminali: alcuni residui possono essere rimossi enzimaticamente e pertanto non compaiono nel prodotto finale e funzionale. Nel 50% delle proteine degli eucarioti il gruppo amminico terminale viene acetilato. 2) modificazioni di singoli amminoacidi: i gruppi OH ossidrilici di alcuni amminoacidi possono essere fosforilati enzimaticamente dall’ATP. I gruppi fosforici aggiungono cariche negative ai polipeptidi e possono regolare l’attività di molte proteine mediante cicli di fosforilazione e defosforilazione. Altri gruppi carbossilici possono essere aggiunti al glutammato. Questi legano il Ca2+ indispensabile per le reazioni di coagulazione. In altre proteine i –COOH del glutammato vengono metilati perdendo così la carica negativa. 3) Aggiunta di catene laterali di carboidrati : le catene laterali di carboidrati delle glicoproteine vengono unite in modo covalente durante o dopo la sintesi della catena polipeptidica. Ad es. in alcune glicoproteine la catena laterale di carboidrati viene unita enzimaticamente a residui di Asparagina, in altre a residui di Serina o Treonina. 4) Aggiunta di gruppi isoprenilici: un certo numero di proteine eucariotiche vengono modificate tramite l’aggiunta di gruppi derivati dall’isoprene. Si forma un legame tioetere fra un gruppo isoprenilico e un residuo di cisteina della proteina. In alcuni casi il gruppo isoprenilico serve ad ancorare la proteina alla membrana. 5) Aggiunta di gruppi prostetici: molte proteine hanno bisogno di gruppi prostetici per svolgere la loro attività: esempi sono dati dalla molecola di biotina legata covalentemente all’acetil-CoA carbossilasi e il gruppo eme del citocromo c. 6) Modificazioni proteolitiche: molte proteine come l’insulina e le proteasi (tripsina, chimotripsina) sono precursori inattivi di dimensioni più grandi: questi vengono modificati proteoliticamente per produrre le forme attive finali più piccole. 7) Formazione di legami disolfuro: le proteine che devono essere secrete dalle cellule eucariotiche dopo essersi spontaneamente ripiega- 314 Capitolo XVI te nelle loro conformazioni native vengono spesso legate mediante ponti disolfuro intra- o intercatena tra residui di Cisteina. Questi legami servono a proteggere la conformazione nativa della proteina dalla denaturazione nell’ambiente extra-cellulare. Trasporto a destinazione e degradazione delle proteine Dopo la sintesi, molte proteine vengono indirizzate a particolari destinazioni nella cellula. Un meccanismo di trasporto a destinazione si basa sulla presenza di sequenze peptidiche segnale, che si trovano in genere alle estremità ammino-terminali delle proteine neosintetizzate. Le proteine vengono alla fine degradate da sistemi proteolitici specializzati presenti in tutte le cellule. Le proteine difettose e quelle destinate ad un rapido ricambio sono generalmente degradate da un sistema proteolitico ATP-dipendente. Negli eucarioti, le proteine che devono essere degradate da tale sistema vengono prima contrassegnate dal legame di una proteina altamente conservata chiamata ubiquitina. 315 COMPOSTI FENOLICI DELLE PIANTE I composti fenolici rappresentano una delle principali classi di metaboliti secondari, che comprende un ampio spettro di sostanze molto eterogenee ma tutte caratterizzate dalla presenza di un anello aromatico con uno o più sostituenti ossidrilici. Sebbene alcune sostanze fenoliche siano presenti anche in organismi animali, la presenza di composti fenolici è una caratteristica specifica dei tessuti vegetali. I fenoli sono particolarmente importanti nei prodotti ortofrutticoli in cui determinano colore e sapore. In particolare si associa alla presenza di acidi fenolici il sapore acidulo, ai tannini l’astringenza, mentre il sapore amaro è spesso associato ad alcuni flavonoidi. Il colore, infine, viene determinato dalla presenza degli antociani. Il contenuto di composti fenolici nei tessuti vegetali varia in funzione della specie, della varietà, dell’organo considerato, così come dello stadio fisiologico e delle condizioni climatiche. Le piante producono un grandissimo numero di composti contenenti uno o più residui fenolici. Questi composti possono essere riuniti in vari gruppi, a seconda del numero di atomi di carbonio che costituiscono il loro scheletro (Tabella 1). Queste strutture derivano tutte, ad eccezione dei flavonoidi, da un intermedio biosintetico comune, la fenilalanina o dal suo immediato precursore, l’acido scichimico (Figura 1). Figura 1. Formule di struttura di fenilalanina e acido scichimico. 1 Capitolo XVII 316 Tabella 1. Le principali classi di composti fenolici vegetali. Atomi di carbonio Scheletro carbonioso Classe 6 7 8 9 C6 C6 - C1 C6 - C 2 C 6 - C3 10 13 14 15 18 30 C 6 - C4 C6 - C 1 - C 6 C6 - C 2 - C 6 C 6 - C3 - C6 (C6 - C3)2 (C6 - C3 - C6)2 Fenoli Acidi fenolici Acidi fenilacetici Acidi idrossicinnamici, polipropeni, curarine, isocoumarine Naftochinoni Xantoni Stilbeni, antrachinoni Flavonoidi, isoflavonoidi Lignani, neolignani Biflavonoidi n (C6 - C3)n (C6)n (C6 - C3 - C6)n Lignine Melanine Tannini condensati Non è ancora stato completamente chiarito se i derivati fenolici abbiano o no un ruolo chiave nella crescita e nello sviluppo delle piante, sebbene vi siano alcune indicazioni in questo senso. A parte l’acido lunularico, che sembra sostituire l’acido abscissico come fitormone, nessun fenolo ha un ruolo importante come ormone vegetale. Ci sono molte ricerche, tuttavia, che mostrano il loro effetto in vitro su certi ormoni o sistemi enzimatici collegati alla sintesi o all’azione degli ormoni. Se questi effetti esistano anche in vivo, resta da chiarire. Forse la loro funzione più probabile è la produzione di etilene dalla metionina (un amminoacido solforato) in cui un estere dell’acido pcumarico è un cofattore necessario. Vediamo ora in dettaglio i composti fenolici più rappresentativi. Composti fenolici delle piante 317 I fenoli Tra i fenoli semplici il più comune è l’idrochinone, mentre il suo isomero, il catecolo non è molto diffuso (Figura 2). Figura 2. Formule di struttura di idrochinone e catecolo Tra i derivati può essere interessante l’urusciolo, il principio tossico dell’edera velenosa, (Toxicodendon radicans) e del tetraidrocannabinolo, un derivato del resorcinolo che è il principio allucinogeno della Cannabis sativa (Figura 3). Figura 3. Struttura di urusciolo e tetraidrocannabinolo. Anche le melanine contenute nelle piante sono probabili polimeri del catecolo. Tutti questi composti derivano dalla fenilalanina, attraverso la formazione dell’acido p-cumarico, il quale viene convertito ad acido benzoico, successivamente decarbossilato fino a fenolo (vedi più avanti). 318 Capitolo XVII Acidi fenolici Gli acidi fenolici sono ampiamente diffusi nel regno vegetale. Fra questi i più importanti sono riportati in Figura 4: a) l’acido paraidrossibenzoico, b) il protocatechico, c) il vanillico, d) il siringico, sempre presenti nelle angiosperme. L’acido gallico (e) è presente in forme polimere come i tannini solubili. Figura 4. Formule di struttura di alcuni acidi fenolici Si conoscono anche le aldeidi e gli alcoli corrispondenti agli acidi carbossilici sopra indicati, come la vanillina, nei baccelli di vaniglia e l’alcol salicilico, nel salice. I gruppi carbossilici e fenolici possono essere metilati, con formazione di esteri o di gruppi metossilici. Figura 5. Formule di struttura di vanillina e alcol salicilico. Acidi idrossicinnamici, cumarine, cromoni Questi acidi, come il p-cumarico e il caffeico (Figura 6) e i loro derivati metilati come l’acido ferulico e il sinapico sono sempre presenti nelle piante superiori, liberi o sotto forma di una grande varietà di esteri. Composti fenolici delle piante 319 Anche gli zuccheri sono legati a queste molecole in forma di esteri e non di glucosidi. Le cumarine sono lattoni (esteri interni di idrossiacidi) che derivano dalla reazione fra l’-OH in orto ed il gruppo carbossilico dell’acido della catena laterale dell’acido o-idrossicinnamico. Probabilmente non esistono libere nelle foglie fresche. Le idrossicumarine, che derivano dall’acido p-cumarico, dal caffeico e dal ferulico sono molto diffuse. Alcuni esempi sono l’umbrelliferone, l’esculetina e la scopoletina. Hanno intensa fluorescenza in soluzione, cosa che facilita il loro riconoscimento. I cromoni e le isocumarine sono isomeri delle curarine che non hanno vasta diffusione, tranne l’eugenina (cromone) e l’idrangenolo, una isocumarina, trovata nell’Hydrangea macrophylla. Figura 6. Formule di struttura di ac. cumarico (A) e ac. caffeico (B). Chinoni Fra i chinoni, il benzochinone non ha ampia diffusione, al contrario del suo prodotto di riduzione, l’idrochinone. Derivati del benzochinone, come il plastochinone e l’ubichinone (Figura 7) hanno importanti ruoli funzionali rispettivamente nella fase luminosa della fotosintesi e nel trasporto elettronico mitocondriale della catena respiratoria. I naftochinoni si trovano generalmente in forma ridotta e legati nei glucosidi. Durante l’estrazione vengono spesso ossidati e risultano colorati. Un derivato importante dei naftochinoni è il glucoside ridotto dello iuglone, presente nelle noci. Più di 200 antrachinoni sono stati trovati nelle piante da fiore. 320 Capitolo XVII Figura 7. Formule di struttura di ubichinone (A) e plastochinone (B). Flavonoidi I flavonoidi costituiscono un gruppo molto ampio di derivati fenolici solubili in acqua, molti dei quali hanno colori brillanti (rosso, cremisi, porpora, giallo). In genere stanno nei vacuoli, sebbene possano trovarsi anche nei cromoplasti e nei cloroplasti. I flavonoidi sono glucosidi e i loro agliconi (parte non glucosidica) hanno la struttura di base del flavano. Il flavano è formato da due anelli aromatici legati fra loro mediante una catena a 3 atomi di C, quindi sono derivati fenilpropanici, C6-C3-C6. Figura 8. Struttura di un flavano. Gli agliconi (parte non glucidica dei glucosidi) liberi sono stati riscontrati in tessuti lignei morti e in apparenza si sono formati per idrolisi enzimatica dei flavonoidi. Molti dei colori brillanti del mondo vegetale sono dovuti ai flavonoidi che hanno in comune una struttura fondamentale dei fenilpropani, costituita da due anelli benzenici e da un terzo anello con un atomo Composti fenolici delle piante 321 di ossigeno. La maggior parte di questi composti si trova in combinazione con una molecola di zucchero (componente glucidico). Le classi principali di flavonoidi che contribuiscono al colore delle piante sono: 1. Antocianidine Una delle classi principali di flavonoidi è costituita dalle antocianidine. In combinazione con le molecole glucidiche come il glucosio, le antocianidine diventano antocianine (dal greco: fiore blu). Le antocianine sono responsabili di molti dei colori rossi, porpora e blu che si trovano in natura. Sebbene si conoscano almeno 22 tipi di antocianidine, le tre più comuni sono la pelargonidina, la delfinidina e la cianidina che differiscono soltanto per il numero di gruppi OH sull’anello B. A causa del loro carattere ionico, sia l’intensità che la tonalità di colore delle antocianidine varia col pH. In soluzione acida (HClmetanolo) il colore varia dal rosso-arancio della pelargonidina al magenta della cianidina al malva della delfinidina. La differenza dipende dal numero di gruppi OH sull’anello B. Se si alza il pH fino alla neutralità, la soluzione rosso-arancio della pelargonidina diventa incolore perché si forma una pseudobase. Sopra pH 7 si forma una anidrobase blu e a pH molto più alti avvengono cambiamenti irreversibili a cominciare dalla ionizzazione degli OH fenolici. La reazione delle antocianidine con il glucosio (glucosilazione) sul C3 produce le antocianine, senza però variazioni di colore perché in genere tutte le antocianidine sono glicosilate sul C3. La glicosilazione su altre posizioni, più rara, ha solo debole effetto sul colore. Colore dei petali Le variazioni di pH nella linfa delle piante non sono importanti e non costituiscono il principale fattore a cui è dovuta la colorazione dei petali di queste, che si basa invece sui pigmenti antocianici e/o su fenomeni di co-pigmentazione e chelazione di metalli. La copigmentazione avviene fra una antocianina ed un glucoside flavonico o un tannino idrolizzabile, e la differenza di colore si deve allo spostamento dei massimi delle bande spettrali di assorbimento. La copigmentazione è il risultato di legami a H fra il gruppo carbonile della anidrobase e i gruppi OH aromatici dei flavonoidi. L’effetto della chelazione di metalli sul colore dei petali si può vedere paragonando il colore blu del fiordaliso con il colore rosso di una 322 Capitolo XVII rosa, in cui la principale antocianidina, la cianidina, è la stessa nei due casi. Nel caso del fiordaliso però c’è stata una chelazione col Fe ed una co-pigmentazione fra la cianina (glucoside della cianidina) ed un flavone, mentre nel caso della rosa c’è l’antocianina pura senza metalli. Colore delle foglie Le antocianine sono i soli flavonoidi che impartiscono il colore alle foglie, essi contribuiscono al colore passeggero delle foglie giovani e alla colorazione permanente dell’autunno. In quasi tutti i casi il pigmrento è la cianidin-3-glucoside. La ragione del flusso passeggero di antocianine nei primi stadi di formazione della foglia non è nota ma la colorazione permenente è controllata geneticamente. Le piante in cui si nota meglio la presenza di antocianine sono quelle del pero e dell’acero, intensamente rosse in autunno. Il colore giallo o marrone delle foglie autunnali è dovuto principalmente a carotenoidi e tannini. Colore dei frutti Anche nel caso dei frutti, sono principalmente le antocianine i flavonoidi che impartiscono il colore a molti frutti eduli, come mele, ciliegie, lamponi, mirtilli rossi, melagrane, melanzane, mirtilli neri, arance tarocchi eccetera. Le variazioni di colore, per esempio nelle ciliegie rosse e bianche è dovuto più a differenze in quantità che a differenze nella natura dei pigmenti. Il colore blu di molti frutti è probabilmente il risultato della formazione di complessi con metalli e proteine e non dovuto a co-pigmentazione come nei petali. 2. Flavonoli e flavoni I più comuni flavonoli e flavoni, sebbene diffusi nei petali dei fiori, non contribuiscono al loro colore, a meno che non siano metilati o legati in insoliti legami glucosidici. Tuttavia, sebbene i flavoni non contribuiscano direttamente al colore dei fiori possono agire come copigmenti intensificando il colore giallo di flavonoli, calconi ed auroni. Flavonoli e flavoni incolori contribuiscono a creare i colori bianco, avorio e crema di molti fiori. In genere flavonoli e flavoni non contribuiscono al colore dei frutti sebbene siano ampiamente distribuiti in questi. Composti fenolici delle piante 323 3. Calconi ed auroni La presenza di questi pigmenti gialli può facilmente essere rilevata nei petali esponendoli a vapori di ammoniaca che provocano la variazione del colore nettamente da giallo a rosso. Per questa ragione calconi ed auroni si chiamano anche pigmenti antocoloranti. Non si trovano in più di 9 famiglie di fiori, mentre nelle foglie e nei frutti solo occasionalmente. Non sono a stretto rigore dei flavonoidi, ma sono strettamente collegati sia per le proprietà chimiche che per le vie di biosintesi. Importanza dei pigmenti flavonoidi nell’ambiente E’ ben nota l’importanza dei composti flavonoidi nei fiori e nei frutti, infatti i loro colori vivaci attraggono insetti ed uccelli, favorendo l’impollinazione e la dispersione dei semi. Tuttavia, anche flavoni e flavonoli incolori sono sempre presenti nei fiori e hanno un ruolo importante nell’attrarre gli insetti perché possono assorbire le radiazioni nel vicino UV che possono essere percepite dagli insetti. In alcuni fiori, come la Rudbeckia hirta ad esempio, i flavonoidi sono presenti solo alla base dei petali ma il loro forte assorbimento nella regione dello spettro fra 240 e 380 nm fa sì che insetti impollinatori come le api possano localizzare con sicurezza il centro del fiore che contiene polline e nettare. Altre funzioni le loro attività come agenti allelopatici (prodotti di escrezione che possono essere tossici per la pianta stessa o colpire le piante vicine), per es. l’acido salicilico nella Quercus falcata; come deterrenti sui predatori come ad esempio i tannini; come agenti antimicrobici, come il luteone nel Lupino; come fitoalessine, agendo cioè come ormoni vegetali. Biosintesi dei composti fenolici Molti esperimenti hanno mostrato che i fenoli derivano tutti dalla fenilalanina passando per l’acido p-cumarico, che viene poi convertito in acido benzoico, che viene a sua volta decarbossilato ossidativamente per dare il fenolo. Una visione d’insieme della biosintesi dei composti fenolici delle piante parte dalla via dell’acido scichimico. Va notato che plastochinone, fillochinone, ubichinone e tocoferoli sono di solito classificati 324 Capitolo XVII tra i chinoni terpenoidi o cromanoli piuttosto che come composti fenolici ma sono compresi nella figura per paragonarli con altri chinoni, che invece sono classificati tra i composti fenolici. I tannini Il termine tannini indica un vasto gruppo di composti delle piante che possono conciare la pelle degli animali, rendendendola imputrescibile, impermeabile ed elastica e trasformandola così in cuoio. Il termine deriva dall’inglese “tan”, forma latina di una parola celtica che significava “quercia”, in quanto un estratto di corteccia di quercia costituisce in genere un ottimo conciante. I tannini sono composti polifenolici a carattere colloidale, amorfi, astringenti, di colorazione variabile dal giallo al rossastro. Danno colorazione con i sali ferrici e precipitano le proteine. Piccole quantità di tannini sono diffuse in molte piante, ma grandi quantità si riscontrano nel rivestimento dei semi di Caesapinia brevifolia e Caesalpinia coriaria, nella corteccia di eucalipto e nelle mangrovie. Concentrazioni anche maggiori si trovano in tessuti patologici, come le galle prodotte dalle foglie di Rhus semialata, che sono costituite da tannini per il 64%. Un buon conciante deve avere peso molecolare fra 500 e 3000 e avere gruppi fenolici in quantità sufficiente a formare complessi con le proteine. Composti fenolici semplici sono troppo piccoli, sebbene possano essere adsorbiti sulle proteine, mentre composti troppo grandi hanno difficoltà a penetrare fra le fibrille di collagene della pelle degli animali. I tannini si dividono in due gruppi principali, i tannini idrolizzabili e i tannini condensati. 1) I tannini idrolizzabili contengono un nucleo formato da un alcool poliossidrilato, di solito, ma non sempre, il glucosio, che è esterificato o con l’acido gallico per formare i gallotannini o con l’acido esaidrossidifenico (acido ellagico) per formare gli ellagitannini. Questo nome è abbastanza diffuso, tuttavia studi recenti hanno mostrato che l’acido ellagico in realtà è un artefatto derivante dalla lattonizzazione dell’acido esaidrossidifenico, cioè si forma non un vero e proprio Composti fenolici delle piante 325 nuovo composto ma un estere interno dell’acido esaidrossidifenico. Quindi il nome ellagitannini non è appropriato. I gallotannini, detti anche acidi tannici, si trovano nelle escrescenze (galle) che si originano sulle piante di sommacco cinese per reazione della pianta alla puntura di un afide. Si riscontrano anche nelle nostre piante nelle querce e nei cerri. Per idrolisi enzimatica si ottiene acido gallico e acido m-digallico. Nel passato l’acido tannico è stato usato ampiamente nelle preparazione degli inchiostri fluidi (con sali di Fe) e viene usato in medicina come antidoto per gli avvelenamenti da alcaloidi e da sali metallici. Alcuni derivati vengono impiegati come astringenti. L’acido tannico viene inoltre impiegato come mordente in tintoria e nella chiarificazione della birra. Gli ellagitannini contengono diversi acidi fenolici, liberi o legati come glucosidi o esteri. 2) I tannini condensati sono formati soltanto da fenoli del tipo del flavone e sono polimeri di flavani come il l flavan-3.olo o il flavan3,4-diolo. A differenza dei tannini idrolizzabili non contengono residui di zuccheri. Un tipico tannino condensato può essere rappresentato da un dimero della procianidina a cui si aggiungono altre molecole di flavano. Un trattamento con agenti idrolizzanti ha mostrato che i tannini condensati non contengono quantità significative di composti a basso peso molecolare, ma tendono a polimerizzare, particolarmente in ambiente acido, per dare composti amorfi detti flobafeni, spesso di colore rosso. Non sono noti i dettagli sulle reazioni coinvolte nella biosintesi dei tannini. La lignina La composizione del legno è assai variabile e complessa, a parte l’acqua che nel legno fresco si aggira sul 40% mentre è del 15% nel legno stagionato. Il legno è costituito da tre componenti fondamentali: la cellulosa, la lignina e le emicellulose. Vi sono inoltre sostanze secondarie come cere, resine, sali minerali e sostanze accessorie, come tannini, pigmenti, alcaloidi, glucidi, acidi grassi. Nel 1838 Payern, trattando il legno alternativamente con acido nitrico e alcali, separò le cellulosa da altre sostanze residue che chiamò 326 Capitolo XVII sostanze incrostanti. A questo materiale incrostante fu dato poi il nome di lignina. Oggi si sa che la lignina incrosta la cellulosa a cui è talvolta chimicamente legata con legami eterei, formando complessi ligno-cellulosici. In generale, la presenza della lignina nel mondo vegetale è associata alla presenza di tessuti di supporto e trasporto, come ad es. lo xilema. La lignina si ritrova nelle piante vascolari come Angiosperme, Gimnosperme, licopodi, felci, ma è assente nelle piante non vascolari come funghi ed alghe. Comunque la lignina è presente non solo nelle pareti cellulari dei tessuti lignificati (piante perenni) , ma anche, seppur in minima quantità, nelle pareti cellulari di radici, nella buccia e nei noccioli di frutta, nelle gemme ecc. La lignina è un composto molto complesso, ben riconoscibile al microscopio, abbastanza resistente ai mezzi chimici, infatti resiste anche all’azione ossidante del bicromato, e non è separabile con mezzi blandi. E’ più opportuno parlare non di lignina ma di lignine, in quanto la lignina si diversifica nelle diverse specie vegetali e nei diversi tessuti della stessa pianta. Estrarre la lignina dalla parete cellulare è un processo complesso, possibile solo con l’impiego di reagenti chimici aggressivi come HCl 40%, una miscela di HCl ed acido fosforico in rapporto 3:1, acido solforico e CuO ammoniacale a temperatura elevata. Questi procedimenti portano alla separazione di lignine diverse e modificate rispetto alla lignina originale. Si parla quindi di HCllignina, lignina cupro-ammoniacale ecc. Le difficoltà di estrazione furono parzialmente superate da Brauns il quale trovò che il 5% della lignina totale del legno poteva essere estratto usando etanolo al 95% a temperatura ambiente per diversi giorni. Questa blanda procedura di estrazione apparentemente non causa modificazioni nella struttura della lignina. Questa lignina così estratta si chiama “lignina di Brauns” ed è considerata praticamente identica alla protolignina, che è la lignina delle pareti cellulari. Dal punto di vista chimico la lignina è una sostanza ternaria ( composta cioè da C, H, O) di natura non glucidica e l’unità costituente è un residuo di fenil-propano (C6-C3). Il processo di polimerizzazione avviene mediante una condensazione casuale di radicali fenilpropanici, per cui non esiste una formula o una struttura unica. Si può però disegnare una ipotetica e parziale struttura che mette in evidenza i diffe- Composti fenolici delle piante 327 renti tipi dell’unità fenil-propanica ed i diversi modi in cui sono legati insieme. Inoltre, nella lignina, oltre ai gruppi aromatici con catene propaniche, si trovano anche gruppi OH alcolici e fenolici, gruppi metossilici –OCH3, gruppi chetonici C=O, ponti eterei, anelli furanici. La biosintesi della lignina inizia dagli acidi p-cumarico, ferulico e sinapico (acidi cinnamici). L’acido p-cumarico si forma dalla fenilalanina in tutte le piante e dalla tirosina nelle erbe. Successivamente l’acido p-cumarico si trasforma in acido ferulico ed acido sinapico: i tre acidi vengono poi ridotti agli alcoli corrispondenti, con consumo di NADPH , dopo che sono stati convertiti nei corrispondenti CoA-Sderivati. Gli alcoli sono poi glucosilati, per reazione del glucosio con gli OH fenolici ad alcol glucocumarilico, coniferina e siringina. In tale forma possono essere trasportati nelle cellule del cambio, dove una βglucosidasi ripristina gli alcoli. Questi sono poi ridotti enzimaticamente da fenolossidasi e perossidasi a radicali organici che poi polimerizzano dando luogo alla lignina. TERPENI E TERPENOIDI I prodotti naturali delle piante noti come terpeni sono estremamente importanti per le enormi possibilità biosintetiche che li caratterizzano. In genere il termine terpene indica composti a numero di atomi di carbonio multiplo di cinque, infatti si tratta di lipidi costituiti da multipli dell’unità isoprenica, come si vedrà più avanti. I terpeni sono i componenti principali degli oli essenziali delle piante, miscele di sostanze che conferiscono ad ogni fiore o pianta il caratteristico profumo o aroma. Sono terpeni il geraniolo, il mentolo, la canfora, il limonene e lo squalene. I terpenoidi possono contenere unità isopreniche più o meno modificate, fra questi ci sono composti che hanno rilevanza particolare, come le vitamine liposolubili A,D,E e K. I terpeni sono classificati come composti secondari delle piante, insieme agli alcaloidi ed ai derivati fenolici, tuttavia oggi è chiaro che sono composti essenziali per la vita delle piante. La fotosintesi dipende infatti oltre che dalla clorofilla anche dalla presenza di terpeni e terpenoidi, come i carotenoidi e le xantofille. Come già accennato, chimicamente terpeni e terpenoidi possono considerarsi derivati da una unità ramificata a cinque atomi di carbonio, l’isoprene (2 metil-1,3-butadiene) (Figura 1) e vengono classificati in base al numero di tali unità presente nella molecola (Tabella 1). Figura 1. Formula di struttura dell'isoprene. Tranne gli emiterpeni, i sesquiterpeni e le gomme i composti in tabella sono importanti intermedi biosintetici, sotto forma di pirofosfati. Talvolta il residuo isoprenoide viene indicato con “ip”. 329 Capitolo XVIII 330 Il farnesolo viene indicato con ip- ip- ip, e ciò indica che è formato da tre residui collegati in forma testa-coda, in cui la parte finale ramificata dell’unità isoprenica rappresenta la testa e la parte opposta non ramificata rappresenta la coda. Sebbene questo sia il tipo di legame più comune per legare insieme unità isopreniche, esiste anche il legame in forma coda-coda, indicato come ip-pi, che si riscontra al centro di molecole di tri- e tetra-terpeni: ad esempio, lo squalene è composto da due residui di farnesolo legati coda a coda e rappresentato da ip-ip ip-pi-pi-pi. Tabella 1. Classificazione generale dei terpeni. Tipo N° di atoStruttura mi di C Nome Emiterpene 5 Isoprene Monoterpene 10 Geraniolo Sesquiterpene 15 Farnesolo Diterpene 20 Geranilgeraniolo Triterpene 30 Squalene Tetraterpene 40 Fitoene Politerpene ~7,5 x 103 fino a ~3 x 105 Gomma Emiterpeni Il solo emiterpene libero rappresentato nelle piante, anche se in piccola quantità, è l’isoprene. Gli emiterpeni si trovano di frequente Terpeni e terpenoidi 331 combinati con parti non terpenoidi. Alcuni composti a cinque atomi di carbonio a catena ramificata non sono in origine degli isoprenoidi, ad esempio l’amminoacido valina, che richiama la struttura isoprenica ma non ha la stessa via biosintetica. L’isoprene si forma nel cloroplasto, ma la via metabolica non è chiara. Composti simili, come l’alcol isoamilico o l’aldeide isovalerianica si formano direttamente nel metabolismo della valina e non attraverso la via dell’acido mevalonico. Monoterpeni Sono composti da due unità isopreniche, e si trovano in tre formeprincipali: aciclica, cicloesanoide ciclopentanoide. I monoterpeni sono ampiamente diffusi nelle piante superiori e il loro forte odore li ha resi assai importanti per l’industria cosmetica e profumiera. Si trovano spesso entro speciali ghiandole secretorie (come nel caso della menta) come componenti importanti degli oli essenziali, che vengono prodotti sottoponendo le piante a distillazione in corrente di vapore. Gli oli essenziali si trovano principalmente nelle Labiate, nelle Pinacee e nelle Umbrellifere. Sembra che le sole piante che non producono significative quantità di monoterpeni siano quelle del tipo delle Viole. Sono assai diffusi anche i glicosidi dei monoterpeni, che però non sono volatili e non vengono estratti in corrente di vapore. Fra i monoterpeni troviamo il γ-terpinene, l’ α-tuiene, il p-cimene, il timolo, la canfora, il borneolo, il mentolo, l’iso-mentolo, l’α-pinene, il β-pinene, ed altri. Il precursore è il geranil-pirofosfato. Sesquiterpeni Sono composti a quindici atomi di carbonio (tre unità isopreniche) che si originano da varie ciclizzazzioni del farnesilpirofosfato (FPP). Rappresentano il gruppo più ampio dei terpeni, non solo per il numero di composti reperibili in natura, ma per la variabilità delle strutture: si contano almeno duecento scheletri carboniosi diversi. Spesso si trovano insieme ai monoterpeni negli oli essenziali, in questi casi si trovano all’interno di particolari strutture. Le piante del tipo delle Magnolie 332 Capitolo XVIII sono assai ricche in sesquiterpeni, mentre le Ranuncolacee sembrano esserne prive. Diterpeni Sono composti che contengono venti atomi di carbonio (quattro unità isopreniche). Ne esistono oltre cinquecento varietà. Si trovano due gruppi tetraciclici con la stessa struttura di base del gruppo del kaurano, da cui derivano i fitormoni gibberelline. Triterpeni e triterpenoidi I triterpeni e i loro derivati, come gli steroli, rappresentano un altro vasto gruppo di isoprenoidi, che contengono trenta atomi di carbonio (sei unità isopreniche). Il gruppo principale consiste di derivati tetraciclici, alcuni dei quali però sono raramente presenti negli organismi fotosintetici. Un gruppo importante che si pensa derivi dai triterpeni tetraciclici è costituito dagli steroli. Gli steroli delle piante, in generale, che si possono trovare sia nelle piante superiori che nei funghi o nelle alghe, sono caratterizzati dalla presenza di uno o due sostituenti nella catena laterale a livello del C24, anche se nelle piante sono stati trovati anche steroli privi di sostituenti. Ad esempio, il colesterolo, il principale steroide dei mammiferi è presente in tracce nelle piante superiori e largamente diffuso in alcune alghe rosse. Tetraterpeni – Carotenoidi I tetraterpeni constano di un solo gruppo, i pigmenti carotenoidi. Sono note circa cinquecento strutture, ma solo un numero limitato è stato trovato nelle foglie verdi. I carotenoidi sono localizzati nei cloroplasti. La loro funzione protettiva è essenziale alla sopravvivenza dei cloroplasti in condizioni di luce e di aerobiosi. Inoltre contribuiscono alla raccolta della luce nella fotosintesi. Il colore dei carotenoidi è dovuto alla presenza di una lunga serie di doppi legami coniugati presenti nella molecola. Altri pigmenti delle piante superiori sono il Terpeni e terpenoidi 333 risultato di variazioni della formula del licopene, il caratteristico pigmento carotenoide rosso del pomodoro. La ciclizzazione nella parte finale del licopene forma un anello β come nel γ-carotene, e un anello ε come nel δ-carotene. La ciclizzazione in tutte e due le parti finali del licopene può produrre pigmenti come β- carotene, α-carotene o εcarotene. Il gruppo aciclico finale, come nel licopene, si chiama ψ−carotene. I tetraterpeni idrocarburici costituiscono i caroteni, quelli contenenti funzioni ossigenate, le xantofille. Le funzioni ossigenate comprendono gruppi –OH (luteina), gruppi epossido (violaxantina e zeaxantina), mentre la neoxantina contiene gruppi allenici e l’alloxantina gruppi acetilenici. Oltre ad essere presenti in tutti i tessuti fotosintetici, comprese le alghe e i batteri fotosintetici, i carotenoidi sono ampiamente diffusi negli organismi non fotosintetici delle piante superiori, nei funghi e nei i batteri non fotosintetici. Il gruppo dei carotenoidi presenti nelle foglie verdi è lo stesso in tutte le piante superiori (soprattutto β-caroteni, luteina, violaxantina e neoxantina). Tuttavia, in organi che producono caroteni, come petali e frutti, si riscontrano ampie variazioni strutturali. I pigmenti si accumulano nei cromoplasti, che si sviluppano dai cloroplasti, sia nei fiori che nei frutti. La carota domestica, la classica fonte di caroteni, è una miscela di caroteni, ma il β-carotene è presente al 90%. Poli-isoprenoidi E’ noto da tempo che l’alcol diterpenoide non ciclico fitolo si trova esterificato con la clorofilla. Altri derivati simili, ma più grandi, sono stati trovati in seguito liberi nei tessuti vegetali, come il solanesolo isolato nelle foglie di tabacco, che ha tutti i suoi doppi legami in configurazione trans. Esistono anche famiglie di poli-isoprenoidi con doppi legami in configurazione sia trans che cis, come i castaprenoli degli ippocastani (Aesculus Hyppocastanum). Tutti i poli-isoprenoidi trans sono attaccati a chinoni importanti metabolicamente, come plastochinoni, ubichinoni e fillochinone (Vitamina K1). Nei tocoferoli (Vitamina E) le unità isopreniche della catena laterale sono sature. 334 Capitolo XVIII Gomma, gutta e chicle La gomma è un poli-isoprene ad alto peso molecolare che si trova nel lattice di circa 300 generi di Angiosperme. Di queste, solo l’Hevea Brasiliensis ha valore commerciale e suoi cloni ad alta resa sono stati sviluppati in Malesia, paese che produce almeno 700 Kg di gomma per acro per anno. I coltivatori di questa pianta hanno migliorato molto il meccanismo di regolazione che controlla la trasformazione del fotosintetato in gomma, portando così la resa a livelli più alti del naturale. L’effetto di questa super-produzione di gomma sulla crescita è notevole nei giovani alberi prodotti da cloni ad alta resa. La riduzione dell’aumento di circonferenza nei primi 8 mesi di incisione è anche del 70%. Il lattice che contiene particelle di gomma si accumula in speciali cellule dette lattifere. Nell’Hevea la gomma viene formata e immagazzinata negli anelli delle cellule lattifere nella corteccia. La messa in comunicazione di cellule adiacenti in un anello permette al lattice di vaste zone della corteccia di colare attraverso l’incisione. Il lattice oltre alle particelle di gomma contiene molti organelli, come mitocondri, nuclei, frammenti del reticolo endoplasmatico, ribosomi e inoltre due particelle specializzate, i lutoidi e le particelle di FreyWyssling. I lutoidi (parola che significa “di colore giallo”), principali componenti del lattice, sono sfere osmoticamente attive ricche di acido fosfatidico e di residui di acili di acidi grassi saturi. La gutta è un poli-isoprene trans con peso molecolare più basso di quello della gomma. Viene prodotta da varie piante che crescono in Malesia: il lattice non fluisce facilmente come il lattice della gomma e l’estrazione prevede la distruzione della pianta, cosa che ha portato alla virtuale estinzione della pianta che la produce, Palaquium gutta. Oggi viene prodotta su piccola scala. La gutta è una sostanza termoplastica, un solido compatto, duro, non elastico che riscaldato diventa morbida, pieghevole ma sempre non elastica. Il chicle, che è la base della gomma da masticare, è una miscela di poli-isoprenoidi cis e trans con resine solubili in acetone. Il lattice secreto dall’albero viene raccolto praticando delle incisioni multiple sul tronco ed essiccato mediante un´attente bollitura, per rimuovere l’eccesso di acqua. Il chicle grezzo viene quindi purificato mediante ripetuti lavaggi con una soluzione fortemente alcalina, seguito da neu- Terpeni e terpenoidi 335 tralizzazione, infine si procede all’essiccazione e alla polverizzazione. Il prodotto risultante è una polvere insolubile in acqua ed amorfa che si ammorbidisce con il calore. Il chicle raffinato che si usa per i chewingum non contiene i costituenti solubili in acqua presenti in quello grezzo, tuttavia i dati riguardanti la sua precisa composizione chimica sono piuttosto limitati. Biosintesi dei terpenoidi Dopo aver identificato i terpenoidi che è più frequente incontrare nelle piante, verranno dati qui alcuni accenni sulla via metabolica di formazione di questi composti. Già nel 1887 Wallach aveva proposto la “regola dell’isoprene”. Egli aveva trovato che molte sostanze naturali erano formate dallo scheletro dell’isoprene e quindi probabilmente derivavano dalla sua polimerizzazione. Questa idea fu ripresa più tardi da Ruzicka, che sviluppò la regola biogenetica dell’isoprene, e ipotizzò che tutti i terpenoidi venissero sintetizzati da un precursore comune, detto isoprene attivo, in seguito identificato come il ∆3 isopentenil pirofosfato, IPP (il ∆3 indica la presenza di un doppio legame in posizione 3 della catena). Figura 2. Struttura del ∆3-isopentenil pirofosfato. L’IPP deriva da un altro precursore base della biosintesi dei terpenoidi, l’acetil-CoA attraverso una via metabolica piuttosto complessa che ha come intermedio l’acido mevalonico (MVA). L’IPP viene dapprima isomerizzato a dimetil-allil-pirofosfato (DMAPP), poi convertito in emiterpeni. Il DMAPP è di nuovo il composto di partenza per l’allungamento della catena e si condensa con altre molecole di IPP per formare il geranil-pirofosfato (GPP), che a sua volta, per reazione 336 Capitolo XVIII con un altro IPP, può essere incanalato nella biosintesi dei monoterpeni. Lo schema di tutte queste reazioni è riportato in Figura 3: Figura 3. Schema generale della biosintesi dei terpenoidi. ALCALOIDI Gli alcaloidi sono un importante gruppo di prodotti secondari delle piante, possiedono uno o più atomi di azoto (di solito in un anello eterociclico) e possiedono forti azioni fisiologiche su uomini e animali. Sono fortemente basici, da cui il nome di alcaloidi, spesso molto velenosi e/o psicoattivi. Molti di essi, ma non tutti, hanno importanza farmacologica e alcuni sono noti da secoli. Per esempio gli alcaloidi presenti nella corteccia di piante del genere Cinchona. Il nome deriva da Ana de Osorio, contessa di Chincon e moglie del viceré del Perù, che scoprì su se stessa le virtù della corteccia di china, guarendo da febbri malariche e decidendo l'importazione in Europa. Tuttavia, solo negli ultimi 100 anni sono stati isolati oltre 2000 alcaloidi nelle piante e sono state determinate le loro strutture. Quando si pensi che è stato esaminato meno del 5% delle piante esistenti al mondo si ha un’idea di quanto vasto sia il campo ancora da esplorare. Una stessa pianta può contenere una grande varietà di alcaloidi. Il papavero da oppio contiene nel suo lattice circa 25 alcaloidi differenti. Inversamente uno stesso alcaloide può trovarsi in piante appartenenti a Famiglie molto differenti. L'alcaloide caffeina è presente nel caffè, nella noce di cola, nel tè e nel guaranà. Quasi tutti gli alcaloidi desumono la denominazione da quelle piante da cui furono estratti la prima volta. Gli alcaloidi sono caratterizzati da una notevole farmacodinamica con azione fisiologica varia che interessa più organi e apparati. Dei duemila alcaloidi noti ricordiamo la morfina ad azione sedativa, la codeina ad azione calmante della tosse, l'atropina come parasimpaticolitico e la chinina ad azione antimalarica. La nomenclatura degli alcaloidi non è molto precisa ma quelli che contengono un anello eterociclico in genere si dicono veri alcaloidi, mentre quelli privi di tale anello, protoalcaloidi. I precursori sono quasi sempre amminoacidi, mentre altre unità a più atomi di C, come l’acetato, sono incorporati nella struttura. Gli alcaloidi, con o senza anelli eterociclici, che non derivano dagli amminoacidi sono detti pseudoalcaloidi, e quasi sempre il loro scheletro carbonioso deriva dall’isoprene. 337 338 Capitolo XIX Fra gli alcaloidi più noti ricordiamo: 1) nicotina (nelle piante Nicotiana tabacum, N. rustica, N. glutinosa), un derivato piridinico; 2) chinina (Cinchona e Cinchona Officinalis), derivata della chinolina; 3) morfina (Papaver somniferum) un derivato isochinolinico; 4) iosciamina (Atropa Belladonna, Datura Stramonium, altre Solanacee) che deriva dal tropano; 5) cocaina (Erythroxolon Coca) che deriva dal tropano; 6) scopolamina (Datura metel, Scopolia Carniolica) che deriva dal tropano; Gli alcaloidi si accumulano soprattutto in 4 tipi di tessuti: • tessuto che si sta attivamente accrescendo; • cellule epidermiche e ipodermiche; • fasci vascolari; • cellule contenenti lattice. Ci sono tre tipi principali di alcaloidi: • alcaloidi non eterociclici, o alcaloidi atipici (protoalcaloidi, ammine biologiche) come per esempio la colchicina; • pseudo alcaloidi, derivati da terpenoidi o purine; • alcaloidi eterociclici o alcaloidi tipici. Gli alcaloidi sono sostanze altamente reattive con attività biologiche anche a dosaggi bassi. Ci limiteremo ad esaminarne alcuni tra i più importanti. Il gruppo del tropano che comprende atropina, iosciamina e ioscina: presenti nelle Solanaceae (Atropa, Datura, Hyosciamus, Mandragora). Bloccano l'attività del parasimpatico. Il gruppo della piridina comprendente piperidina, derivati dall'acido nicotinico (vitamina B3), lobelina presente nella Lobelia inflata, nicotina ricavabile dal tabacco, piperina nella piante della specie Piper. Stimolano e poi bloccano tutte le attività del sistema nervoso autonomo. Gli alcaloidi 339 Il gruppo della pirrolizidina comprendente composti molto tossici, soprattutto a livello epatico. Si trovano ad esempio nelle piante di Crotalaria e di Senecio contenute nei cespugli del tè. Il gruppo dell'indolo è un gruppo complesso, comprende gli "alcaloidi animali" come adrenalina, noradrenalina e serotonina (5idrossitriptamina - 5HT), quelli tranquillizzanti derivati dalla Passiflora, gli stimolanti uterini come l'ergotamina ed ergometrina ricavabli da piante della specie Claviceps e il suo derivato dietilammide dell'acido lisergico (LSD). Gli alcaloidi di Rauwolfia serpentaria, ipotensivi e depressivi, e molti stimolanti del sistema nervoso centrale come stricnina, johimbina e psilocibina. Il gruppo della purina come caffeina, teobromina, teofillina e aminofillina presenti nel cacao, caffè e tè. Prolungano l’emivita di molti ormoni, tra cui l'adrenalina. I terpenoidi come l'estratto da Aconitum napellus (tossico) e la valeriana (tranquillizzante). Gli alcaloidi si trovano nei vacuoli e quindi non appaiono nelle cellule giovani finché non si formano i vacuoli. Raramente si trovano nei tessuti morti, anche nella corteccia di Cinchona, che contiene circa il 12% di alcaloidi, sono presenti solo nel parenchima. Gli alcaloidi vengono spesso immagazzinati nei tessuti oltre a quelli del sito di sintesi. L’esempio più noto è la nicotina che viene sintetizzata nelle radici delle piante di tabacco ma che viene traslocata alle foglie e lì immagazzinata. Gli alcaloidi della Datura spp. vengono prodotti nelle radici e poi trasportati alle foglie, dove subiscono notevoli modificazioni. Biosintesi degli alcaloidi Non è il caso in questa sede di occuparci per esteso della biosintesi dei singoli alcaloidi, che ovviamente dipende dal tipo di struttura del composto da cui derivano. Basterà accennare ai tre tipi principali di reazioni che portano in generale alla biosintesi di questo tipo di composti. 1) accoppiamento ossidativo dei fenoli. L’idrogeno del gruppo OH di un fenolo può venir facilmente rimosso mediante trasferimento di 340 Capitolo XIX un elettrone ad un agente ossidante come FeCl3 o ferricianuro di K, per dare un radicale libero fenolato con un elettrone spaiato. Questi radicali si accoppiano facilmente fra loro producendo un dimero. 2) reazione di Mannich. Si tratta della reazione fra un composto carbonilico, spesso ma non necessariamente una aldeide, una ammina e un composto che contenga un carbanione, spesso per perdita di un H+. 3) la formazione di una base di Schiff. E’ la reazione, talvolta spontanea, fra composti con un gruppo amminico primario (-NH2) con composti carbonilici. Funzioni biologiche degli alcaloidi Non è ancora chiaro quali siano il ruolo biologico degli alcaloidi e perché vengano prodotti. L’ampia diffusione degli alcaloidi in tutte le parti della pianta ha stimolato l’attività dei ricercatori a trovare la funzione di questi composti nel metabolismo generale della pianta. Alcune delle motivazioni, che meglio lo spiegano, sono : • difesa chimica contro attacchi di predatori; • riserva di azoto ma non è stato dimostrato che vengano utilizzati in caso di deficienza di questo elemento; • servono come prodotti escretori di N, come l’urea e l’acido urico lo sono per gli animali; • regolatori di crescita, in particolare come inibitori della germinazione; • aiutano a mantenere il bilancio ionico, in virtù del loro potere chelante. La difesa chimica contro gli attacchi di predatori è sicuramente uno dei motivi, per i quali alcuni organismi producono gli alcaloidi. Infatti gli alcaloidi vengono prodotti in maggior quantità nelle piante erbacee rispetto alle arboree, si accumulano o si formano in quelle parti con maggiore valenza alimentare come semi, gemme, e foglie giovani. Spesso in risposta ad un attacco di insetti la produzione di alcaloidi Gli alcaloidi 341 viene marcatamente aumentata e si ha accumulo di questi composti nelle piante. Gli alcaloidi hanno notevole effetto sugli animali e sui loro tessuti e sono usati ( e talvolta abusati) come agenti neurotrofici. Per approfondimenti si rimanda a testi di Farmacologia. Alcaloidi negli alimenti Gli alcaloidi che spesso sono utilizzati come farmaci, possono anche diventare veleni, per l'uomo e gli animali, che li ingeriscono durante l'alimentazione, sia perché presenti negli alimenti di origine vegetale, sia perché liberati da funghi o batteri che infestano gli alimenti stessi. Problema che maggiormente coinvolge gli animali erbivori ma che spesso tocca anche l'uomo. Come tutte le sostanze ad attività farmacologica, gli alcaloidi esplicano un azione tossica sui vertebrati, che è funzione della loro attività farmacologica e della dose ingerita dai soggetti esposti. Quindi la quantità di alcaloide presente nel vegetale e il vegetale ingerito, fanno si che alcuni composti, a parità di attività, siano più tossici di altri. Per gli animali il problema è sempre connesso all'ingestione di foraggio contaminato, mentre per l'uomo la possibilità di intossicazione è collegata anche all'uso di medicine o quant'altro derivato dal mondo vegetale. BIBLIOGRAFIA • • • • • • • • • J.M. BERG, J.L. TIMOCZKO e L. STRAYER, Biochimica (V° Ed.), Zanichelli, Bologna 2003. D.L. NELSON e M.M. COX, Introduzione alla biochimica di Lehninger (III° Ed.), Zanichelli, Bologna 2003. L. SCARPONI, Biochimica Agraria, Patron editore, Bologna 2003. D. VOET e J.G. VOET, Biochimica, Zanichelli, Bologna 1993. A.L. LEHNINGER, D.L: NELSON e M.M. COX, Biochimica (II° Edizione Italiana) , Zanichelli, Bologna 1994. L. STRAYER, Biochimica (IV° Ed.), Zanichelli, Bologna 1996. P..M. DEY e J.B. HARBORNE, Plant Biochemistry, Academic Press, London (U.K.) 1997. T.W. GOODWIN e E.I. MERCER, Introduction to plant biochemistry (II° Ed.), Pergamon Press, Oxford (U.K.), 1990. M. MAFFEI, Biochimica Vegetale, Piccin Nuova Libraria, Padova 1998. 343 Finito di stampare nel mese di giugno del dalla «ERMES. Servizi Editoriali Integrati S.r.l.» Ariccia (RM) – via Quarto Negroni, per conto della «Aracne editrice S.r.l.» di Roma
Scaricare