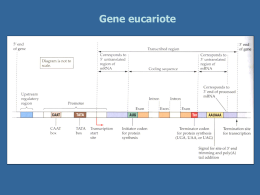
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology Anno I numero 1 - marzo 2009 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali Sindrome del Maschio XX: aspetti clinici e genetici 1 1 1 1 1 1 2 M. C. Cutrupi , R. Civa , A. Giuffrida , P. Vicchio , B. Russo , S. Briuglia , T. Arrigo , C. Salpietro 1 1 2 Dipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, UOC Clinica Pediatrica, Università Messina Abstract The XX male syndrome (OMIM 278850), also known as de la Chapelle syndrome (de la Chapelle A, et al., 1964) from the name of who described it for the first time in 1964, is a rare disorder of the sexual development. We describe the case of a 13 years old boy brought to our attention in order to execute the karyotype exam, motivated by the results of endocrinological investigations that showed an increase in gonadotrophin. The karyotype and the following FISH exam showed results consistent with the framework of the SRY-positive 46, XX male syndrome, condition in many respects similar to the much more common one of the XXY male. The peculiarity of this clinical case lies in the early diagnosis, made possible by a strict collaboration between the pediatric endocrinologist and the genetist. Caso clinico Descriviamo il caso di un ragazzo di 13 anni giunto alla nostra attenzione per eseguire esame del cariotipo in base ai risultati di indagini endocrinologiche che avevano rivelato un aumento delle gonadotropine. Il gentilizio risultava positivo per malattie dismetaboliche. Secondogenito, era nato da gravidanza fisiologica, a termine da parto eutocico. I parametri auxologici neonatali erano nella norma. All’età di 11 anni, ad un controllo auxologico, presentava obesità di grado medio,una statura superiore al target genetico familiare (+ 0.7 SDS vs -0.8 SDS BG) e un’età ossea avanzata di circa 2 anni rispetto all’età cronologica (secondo il metodo di Greulich e Pyle). Una successiva rivalutazione all’età di 12 anni documentava una diminuzione dell’eccesso ponderale dopo opportuno avviamento di trattamento dietetico, una riduzione della velocità di crescita con volume testicolare di tipo prepuberale e pubarca già molto avanzato (dissociazione pubarca -gonadarca). Si eseguiva il test LHRH che mostrava un’inversione del rapporto FSH/LH, segno di pubertà già avviata. Quando è giunto alla nostra osservazione all’età di 13 anni era stato effettuato dosaggio delle gonadotropine che risultavano decisamente elevate (FSH pari a a 44.83 mIU/ml ed LH paria a 15.26mIU/ml). L’esame fenotipico non evidenziava segni dismorfici. Il volume testicolare era di 3 cc a sinistra e 6 cc a destra e l’esame ecografico dei testicoli mostrava caratteristiche morfo-strutturali e vascolari nella norma. L’esecuzione dell’indagine citogenetica eseguita su colture di linfociti di sangue periferico, stimolati con PHA, dopo analisi di 50 metafasi con le tecniche di bandeggio GTG, RBG, mostrava cariotipo 46 xx (Foto 1). Questo risultato è stato approfondito con tecniche di citogenetica molecolare (FISH), per individuare la presenza o l’assenza del gene SRY (SEX determining Region Y). L’esame FISH ha documentato la presenza del gene SRY localizzato sul braccio corto del cromosoma Y, traslocato sulla estremità del braccio corto dell’X con seguente risultato: 46 XX, ish der (X) t(X;Y) (p22.3;p11.3) (SRY+) (Foto 2). Il quadro citogenetico era compatibile con la sindrome del maschio XX, condizione per molto aspetti assimilabile a quella molto più comune del maschio XXY. Si intraprendeva terapia induttiva con testosterone assistendo ad i successivi controlli ad un progressivo aumento del volume testicolare e progressiva crescita staturale. La peculiarità di questo caso clinico risiede nella precocità della diagnosi e quindi del tratttamento terapeutico, resi possibili dalla stretta collaborazione tra il pediatra endocrinologo ed il genetista. Discussione La sindrome del maschio XX (OMIM 278850), anche conosciuta come sindrome de la Chapelle (de la Chapelle A, et al., 1964) dal nome di colui che la descrisse per la prima volta nel 1964, è un raro disordine dello sviluppo sessuale. Il tasso di incidenza è di circa 1: 20 000-25 000 (Rajender et al., 2006) e rappresenta circa il 2% delle cause di infertlità maschile. I sintomi clinici dei pazienti, spesso mostrano un certo grado di eterogeneità (Ergün-Longmire et al., 2005). Si può osservare uno spettro piuttosto variabile di anomalie, dalla presenza di genitali ambigui sin dalla nascita fino ad un fenotipo maschile normale, tanto che in alcuni casi l'anomalia può essere scoperta soltanto in età adulta, a seguito di esami finalizzati ad individuare possibili cause genetiche di infertilità. Fenotipicamente si possono dividere 3 gruppi; il primo gruppo include soggetti fenotipicamente maschi, il secondo gruppo comprende soggetti con ambiguità dei genitali, il terzo gruppo è formato dai soggetti con ermafroditismo vero (Boucekkine C, et al., 1994). I soggetti fenotipicamente maschi sono circa l’80% dei casi e oltre alla infertilità, dovuta probabilmente ad un doppio dosaggio di X oppure all’assenza di geni della spermatogenesi presenti sul cromosoma Y, questi soggetti possono presentare ipogonadismo ipergonadotropo,bassa statura, mancato aumento del volume testicolare, pubertà normale, ritardata o assente, talvolta ginecomastia, criptorchidismo nel 15% dei casi, ipospadia nel 10% circa dei casi (Boucekkine et al 1994). Circa il 20% dei soggetti affetti presenta al momento della nascita genitali ambigui, circa il 10% sono ermafroditi veri. La maggior parte dei casi di sindrome di inversione sessuale sono sporadici, tuttavia, sono stati pubblicati casi familiari. Dorsey et al (2009) ha descritto due fratelli 46 XX SRY negativi affetti da ermafroditismo vero. Ciò suggerisce che i 46 XX fenotipicamente maschi e 46 XX affetti da ermafroditismo vero sono in realtà diverse manifestazioni dello stesso disordine di sviluppo gonadico, probabilmente uno trasmissione autosomica dominante con variabile penetranza o una trasmissione X-linked. In generale, la presenza del gene SRY è spesso associata alla presenza di maschi con normali genitali esterni,bassa statura, lenta crescita della barba, distribuzione femminile dei peli pubici, normale lunghezza e calibro del pene, mentre le persone senza SRY più spesso hanno genitali ambigui, ipospadia, criptorchidismo e ginecomastia [Grigorescu-Sido et al 2005]. La storia naturale del maschio 46 XX, se non trattata, è analoga a quella caratterizzata dalle tipiche conseguenze di deficit di testosterone: bassa libido e possibile disfunzione erettile, diminuzione dei caratteri sessuali secondari, ridotta massa muscolare, aumento della massa grassa con minore forza muscolare e aumentato rischio di osteopenia. La diagnosi differenziale si pone con la Sindrome di Klinefelter con la quale la sindrome del maschio XX presenta notevoli analogie. In genere i maschi XXY presentano una statura superiore, mentre la ginecomastia e il criptorchidismo si presentano con frequenza minore (Vorona et al.,2007). Altre sindromi sono il mosaicismo 46, XX/46, XY e il mosaicismo 45, X/46, XY. Attualmente, la patogenesi della sindrome del maschio 46 XX non è chiara. La formazione sessuale di un individuo è determinata in primis dal sesso genetico che si determina durante la fertilizzazione, cioe l’assetto cromosomico 46 XX o 46 XY. Ciò condiziona una successiva cascata di eventi genetici che portano alla formazione del sesso gonadico (gonadi maschili o femminili). Le gonadi a loro volta secernono ormoni essenziali allo sviluppo dei genitali interni ed esterni (sesso fenotipico). Il principale fattore che influenza la determinazione del sesso di un embrione è il sesso genetico determinato dalla presenza o assenza del cromosoma Y. Tuttavia, alcune persone portano un cromosoma Y, ma sono fenotipicamente femmine (46, XY femmine) mentre altre hanno un cariotipo femminile, ma sono fenotipicamente maschi (46, XX maschi).Nel 1990 il gene SRY (sex-determining region of the Y chromosome),che codifica per il fattore determinante lo sviluppo del testicolo è stato isolato nell'uomo e nel topo. Questo gene si identifica con il TDF (Testis determining factor) e mappa sul braccio corto del cromosoma Y in prossimità della regione pseudoautosomale 1 (PAR1). Si presenta molto conservato. Esso agisce da interruttore molecolare ed è necessario e sufficiente ad attivare il differenziamento maschile. Sry presenta un Open Reading frame (ORF) non interrotto da introni che codifica per una proteina di 204 aminoacidi all’interno della quale è presente un dominio noto come HMG-box che codifica per una sequenza di 79 basi altamente conservata che lega in DNA. La presenza di un dominio di DNA-binding nella proteina SRY ne conferma il ruolo di fattore trascrizionale. L’80% dei casi di inversione sessuale completa nei maschi 46 XX è dovuta ad una traslocazione del gene SRY sulla estremità del braccio corto del cromosoma X o in una minoranza dei casi su un autosoma. La maggior parte del cromosoma Y non ricombina con X, ma due regioni limitate e con sequenze indentiche alla X, situate alle estremità distali del braccio corto e del braccio lungo della Y (PAR: Pseuso Autosomal Region) permettono l’accoppiamento durante la meiosi. (A. Cao 2004). Infatti mutazioni di questo gene nell'uomo o la sua delezione nel topo determinano sviluppo del fenotipo sessuale femminile in individui XY. Viceversa la presenza di SRY in topi transgenici XX porta allo sviluppo dei testicoli e alla completa inversione di sesso. Molti autori ritengono che l'espressione di SRY in embrioni XY interrompa lo sviluppo in senso femminile e inizi quello in senso maschile, in modo che se SRY non è espresso lo sviluppo continua secondo la linea femminile. Infine, esperimenti condotti su topi transgenici XX femmine esprimenti SRY hanno dimostrato che SRY agisce come inibitore di DAX1 (Swain A, et al., 1998) a sua volta repressore dei geni della differenziazione maschile. La regolazione di SRY richiede con tutta probabilità la partecipazione di altre proteine. Anche se è ormai chiaro che SRY gioca un ruolo centrale nella determinazione sessuale, la presenza di maschi XX in cui SRY non è presente indica che vi sono altri geni non situati sulla Y coinvolti nella differenziazione sessuale. Huang et al. (1999) ha segnalato un caso 46, XX, negativo per il gene SRY, in cui era presente duplicazione di SOX9. Il gene SOX9 mappa sul locus SRA1 nella regione distale del braccio lungo del cromosoma 17 e fa parte della famiglia denominata SOX (SRY-like HMG-box). Codifica per una proteina di 509 aminoacidi che mostra caratterische di fattore trascrizionale. per la presenza di HMG-box e di una zona ricca di prolina e glutamina nella regione C-terminale. E’ trascritto soprattutto dopo dopo espressione di SRY nelle stutture gonadiche maschili. (Lim HN e Hawkins JR, 1998). La proteina è espressa in numerosi tessuti dell’adulto, nei testicoli e nel tessuto condrogenico del feto (Wagner T et al.,1994).Nel topo la proteina SOX9 è espressa nelle gonadi indifferenziate di entrambi i sessi, ma persiste solamente nelle cellule del Sertoli dopo che l’espressione di SRY si è innalzata, confermando così la sua implicazione nella differenziazione gonadica maschile. L’overespressione di SOX9 durante le fasi precoci dell’embriogenesi in topi XX transgenici, induce la differenziazione testicolare. E’ quindi probabile che SOX9 agisca molto precocemente nello sviluppo gonadico Le mutazioni sono associate a displasia campomelica (OMIM 211970), malattia autosomica dominante caratterizzata da malformazioni osteo-cartilaginee gravi associate a malformazioni genitali e disgenesia gonadica. I soggetti con cariotipo maschile hanno nel 75% dei casi gonadi disgenetiche che danno luogo a genitali ambigui. I soggetti con cariotipo 46 XX hanno solo anomalie scheletriche per cui SOX9 coopererebbe con SRY nella determinazione sessuale maschile e non sarebbe implicato in quella femminile. La mutazione che determina la sindrome del maschio XX origina de novo al momento del concepimento e non ha nessuna implicazione sugli altri familiari. La prognosi dei soggetti affetti è eccellente con sviluppo intellettivo normale. Il probando e genitori di un soggetto affetto devono essere incoraggiati a richiedere l’aiuto professionale di psicologi e delle Associazioni che accolgono il bambino e i familiari che vivono questa condizione. Nel nostro caso, abbiamo cercato di tranquillizzare il paziente ed i genitori sulla assenza di una condizione “patologica”, tranne che per il problema della infertilità. Abbiamo riscontrato nei genitori del ragazzo una grande difficoltà nel comprendere il messaggio della consulenza e nell’accettare tale diagnosi. L’outcome della consulenza genetica è influenzato da molteplici e complessi fattori legati all’attività prettamente medico-genetica ed ai risvolti psicologici. Il ragazzo ed i genitori potranno essere inseriti in un follow up a lungo termine affiancato dall’endocrinologo e dallo psicologo. Bibliografia 1. Boucekkine C, Toublanc JE, Abbas N, Chaabouni S, Ouahid S, Semrouni M, Jaubert F, Toublanc M, McElreavey K, Vilain E 1994 Clinical and anatomical spectrum in XX sex reversed patients. Relationship to the presence of Y specific DNA-sequences. Clin Endocrinol (Oxf) 40:733-742 2. Cao Antonio; Dalla Piccola Bruno; Notarangelo Luigi D. Malattie genetiche. Molecole e geni. Diagnosi, prevenzione e terapia 2004 Piccin-Nuova Libraria 3. De la Chapelle A, Hortling H, Niemi M, Wennstroem J 1964 XX sex chromosomes in a human male. first case. Acta Med Scand 175(Suppl 412):25-28 4. Dorsey FY, Hsieh MH, Roth DR.46,XX SRY-negative true hermaphrodite siblings. Urology. 2009 Mar;73(3):529-31. 5. Ergun-Longmire B, Vinci G, Alonso L, Matthew S, Tansil S, Lin-Su K, McElreavey K, New MI (2005) Clinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literature. J Pediatr Endocrinol Metab 18:739-748. 6. Grigorescu-Sido A, Heinrich U, Grigorescu-Sido P, Jauch A, Hager HD, Vogt PH, Duncea I, Bettendorf M. Three new 46,XX male patients: a clinical, cytogenetic and molecular analysis. J Pediatr Endocrinol Metab. 2005; 18: 197-203 7. Huang B, Wang S, Ning Y, Lamb AN, Bartley J (1999) Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet 87:349-353. 8. Lim HN, Hawkins JR.Genetic control of gonadal differentiation.Baillieres Clin Endocrinol Metab. 1998 Apr;12(1):1-16. Review. 9. Rajender S, Rajani V, Gupta NJ, Chakravarty B, Singh L,Thangaraj K (2006) SRY-negative 46, XX male with normal genitals, complete masculinization and infertility. Mol Hum Reprod 12:341-346. 10. Ramos ES, Moreira-Filho CA, Vicente YA, Llorach-Velludo MA, Tucci S Jr, Duarte MH, Araujo AG, Martelli L. SRY-negative true hermaphrodites and an XX male in two generations of the same family. Hum Genet. 1996; 97: 596-8. 11. Swain A, Narvaez V, Burgoyne P, Camerino G, Lovell-Badge R. Dax1 antagonizes Sry action in mammalian sex determination. Nature. 1998 Feb 19;391(6669):761-7. 12. Vorona, E.; Zitzmann, M.; Gromoll, J.; Schuring, A. N.; Nieschlag, E.: Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J. Clin. Endocr. Metab. 92: 3458-3465, 2007. 13. Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9.Cell. 1994 Dec 16;79(6):1111-20 Corrispondenza: Maria Concetta Cutrupi, UOC di Genetica ed Immunologia Pediatrica, AOU G.Martino, Dipartimento di Scienze Pediatriche Mediche e Chirurgiche, pad. NI, Policlinico Universitario, Messina Tel. 0902213114- Fax 0902213788; e-mail: [email protected] Trimestrale di divulgazione scientifica dell'Associazione Pediatrica di Immunologia e Genetica Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. in corso di registrazione Direttore scientifico Carmelo Salpietro - Direttore responsabile Giuseppe Micali - Segretaria di Redazione Direzione-Redazione: UOC Genetica e Immunologia Pediatrica - AOU Policlicnico Messina www.geneticapediatrica.it Basilia Piraino
Scaricare