UNIVERSITÀ DEGLI STUDI DI CATANIA
DIPARTIMENTO DI SCIENZE DEL FARMACO
DOTTORATO INTERNAZIONALE IN SCIENZE FARMACEUTICHE
XXVI CICLO
Dott.ssa Mariarita Barone
“INFIAMMAZIONE E CARCINOGENESI:
PARADIGMA DI CAUSA-EFFETTO”
SINTESI, STUDI COMPUTAZIONALI E VALUTAZIONE
BIOLOGICA DI NUOVI FLUOROCOXIB A STRUTTURA
BENZOTIENO [3,2-d]PIRIMIDINICA COME POTENZIALI
INIBITORI SELETTIVI DELLA COX-2 E MARKERS
TUMORALI
TESI DI DOTTORATO
Coordinatore:
Tutor:
Chiar.mo Prof. A. Marrazzo
Chiar.mo Prof. A. Santagati
Triennio Accademico 2011-2013
Indice
Abstract
5
1. Infiammazione
1.1 Introduzione
6
1.2 Infiammazione acuta
7
1.3 Infiammazione cronica
13
1.4 Mediatori chimici dell’infiammazione
15
1.5 Cascata dell’acido arachidonico
22
2. Infiammazione e carcinogenesi
2.1 Infiammazione spiega il cancro
28
2.2 Correlazione tra microambiente infiammatorio e tumori
29
2.3 COX-2 e Carcinogenesi
31
2.3.1 COX-2 e angiogenesi tumorale
31
2.3.2 COX-2 e apoptosi
32
3. Cicloossigenasi
3.1 COX-1 e COX-2 a confronto
34
4. Coxib
4.1 Fans e Coxib a confronto
37
4.2 Inibitori selettivi delle COX-2
38
5. Sintesi di nuovi derivati a struttura benzotieno[3,2-d] pirimidin
sulfonammidica
5.1 Introduzione generale
45
2
5.2 Progettazione razionale
45
5.3 Scopo del lavoro
48
Sezione sperimentale
57
5.3 Materiali e metodi
57
6. Molecular Docking
6.1 Studi di molecular docking sui derivati benzotienopirimidinici
98
Materiali e Metodi
102
6.2 Metodi computazionali
102
7. Attività anti-infiammatoria dei derivati
benzotieno[3,2-d]pirimidinici
7.1 Modelli in vitro di infiammazione
103
7.2 I cheratinociti
103
7.3 I macrofagi ( macrofagi murini j774)
105
7.4 Saggi biologici
107
7.4.1 MTT: saggio di vitalità cellulare
107
7.4.2 Valutazione dell’attività anti-infiammatoria dei derivati
benzotienopirimidinici: ICAM-1, iNOS e COX-2
109
7.4.3 Determinazione del rilascio dell’ MCP-1 e dell’ IL-8 dei derivati
benzotienopirimidinici
113
Materiali e Metodi
116
7.5 Colture cellulari
116
7.6 Western blot
116
7.7 E.L.I.S.A.
117
7.8 Saggio di vitalità cellulare: MTT
118
7.9 Analisi statistica
119
3
8. Spettroscopia di Fluorescenza
8.1 La fluorescenza
120
8.2 Diagrammi di Jablonski
123
8.3 Variabili che influenzano la fluorescenza
126
8.4 Fluorofori
130
8.5 Fluorescenza dei derivati benzotienopirimidinici
131
8.6 Saggi in vitro dei derivati fluorocoxib sulla linea cellulare HCA-7 134
Materiali e Metodi
136
8.7 Spettroscopia di fluorescenza
136
8.8 Colture cellulari
137
8.9 Protocollo di imaging cellulare
137
9. Conclusioni
138
Ringraziamenti
139
Bibliografia
141
4
Abstract
The relationship between cyclooxygenase (COX)-2 over-expression and
cancer makes the COX-2 isozyme an attractive molecular target. Accordingly,
the over-expression of COX-2 in cancer cells, relative to normal adjacent
tissues where COX-2 is not over-expressed, constitutes a logical molecular
diagnostic design strategy to discover non-invasive diagnostic agents to detect
tumors. Moreover, COX-2 over-expression can be useful for subsequent
monitoring of disease progression and/or treatment efficacy. Thus, we have
focused our research in the synthesis of new benzo-thieno [3,2-d] pyrimidine
derivatives, potential fluorescent inhibitors of COX-2, possible antiinflammatory agents and tumor markers. The anti-inflammatory activity of the
new derivatives was evaluated in vitro on human keratinocyte cell line NCTC
2544 exposed to interferon (IFN)-γ and histamine, as well as on monocytemacrophage J774 cell line stimulated with bacterial lipopolysaccharide (LPS).
Some derivatives showed effective biological activities inhibiting some
inflammatory parameters in human keratinocytes NCTC 2544 and monocytemacrophages J774.[47] In addition, almost all anti-inflammatory compounds
exhibited good fluorescence in organic solvents displaying peaks with
absorption maxima at two different wavelengths (∼330 and ∼350 nm). For all
the derivatives, the corresponding λem maxima was centered at about 420 nm.
Preliminary fluorescence microscopy experiments were carried out by treating
cancer cell lines with those compounds, which displayed the best COX-2
affinity, selectivity and fluorescence properties. The results suggest that such
derivatives may be used to cancer cell detection and progression.
5
1. Infiammazione
1.1 Introduzione
L'infiammazione (dal latino inflammatio, dal greco phlogosis, incendiare) si
può definire come la risposta dei tessuti vascolarizzati a un insulto endogeno o
esogeno al fine di veicolare nella sede del danno tessutale materiali difensivi
così da neutralizzare ed eliminare lo stimolo dannoso, riparare la struttura e
ripristinare la funzione dei tessuti danneggiati in modo da favorire quella che
viene definita la ΄restitutio ad integrum΄.[1] Le cause che portano a una risposta
infiammatoria sono varie e tra queste si annoverano le infezioni batteriche e
quelle virali, in quanto i batteri rilasciano diverse tossine mentre i virus
penetrano all'interno delle cellule e causano la morte cellulare attraverso
meccanismi di replicazione intracellulare.[1] Alcuni microrganismi, come i
parassiti e il micobatterio della tubercolosi, sono in grado di indurre una
risposta
infiammatoria
immunomediata
attraverso
delle
reazioni
di
ipersensibilità. Diversi agenti fisici (traumi, raggi ultravioletti, radiazioni
ionizzanti e temperature elevate) e alcune sostanze chimiche irritanti e
corrosive (agenti acidi, alcalini e ossidanti) possono causare un processo
infiammatorio.[1] La risposta infiammatoria consiste in una serie di reazioni
protettive essenziali e utili per la sopravvivenza dell’uomo.[1] È costituita da
una componente vascolare e una componente tissutale che si combinano in
varie proporzioni, a seconda del tipo di processo infiammatorio e della causa
che lo ha scatenato, determinando un’alterazione della microcircolazione. La
durata dell'infiammazione dipende dal tempo necessario per eliminare la causa
nociva e per riparare il danno tissutale.[1] Possiamo distinguere due tipi di
processi infiammatori:
flogosi acuta
flogosi cronica
6
1.2 Infiammazione acuta
L’infiammazione acuta o angioflogosi è una risposta immediata e precoce a
uno stimolo lesivo, il suo scopo è quello di recapitare materiali difensivi nella
sede del danno. Non è uno stato, ma un processo dinamico caratterizzato
dall’alterazione della microcircolazione con formazione di mediatori
dell’infiammazione e movimenti di fluidi e leucociti dal sangue al tessuto
extravascolare che si svolgono particolarmente nel tessuto connettivo
circostante i vasi sanguigni della zona colpita (Fig.1).
Figura 1: Infiammazione acuta
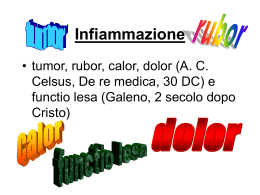
Clinicamente l’infiammazione acuta è caratterizzata da quattro segni cardinali
che furono descritti da Celso nel I sec. d.C. e sono: eritema (rubor), calore
(calor), edema (tumor) e dolore (dolor), ai quali in seguito Virchow ne
aggiunse un quinto: alterazione della funzionalità dell’area colpita (functio
laesa) [1] (Fig.2). Questi aspetti sono presenti a prescindere dalla causa che ha
7
determinato la flogosi e gran parte di essi è riconducibile alla vasodilatazione e
all'aumento della permeabilità capillare.[1]
Rubor è la diretta conseguenza della vasodilatazione, la quale provoca
aumento del flusso ematico nella zona colpita (iperemia attiva), rallentamento
del ritorno venoso (iperemia passiva) con un incremento della quantità di
sangue nella zona dell’infiammazione.[1]
Calor è l’aumento della temperatura nella zona danneggiata dovuto sia
all'iperemia
[1]
che all’aumento del metabolismo cellulare a seguito di un
danno mitocondriale.
Tumor è il gonfiore determinato dall’aumento della permeabilità capillare che
favorisce l’afflusso di liquidi e cellule nell’area danneggiata contribuendo alla
formazione dell'edema infiammatorio.[1]
Dolor è causato da una stimolazione delle terminazioni nervose sensitive in
seguito alle alterazioni biochimiche locali determinate dall’afflusso di cellule
leucocitarie (chemiotassi), dall’elevato metabolismo cellulare e dalla
liberazione di mediatori chimici come le prostaglandine che riducono la soglia
del dolore aumentando la suscettibilità dei nocicettori alla stimolazione da
parte di sostanze algogene liberate durante il processo infiammatorio.
Functio laesa consiste nell’ inibizione della funzionalità dell'area colpita
(specie se si tratta di un'articolazione) a causa del dolore e degli squilibri
indotti dai meccanismi facilitatori dell'infiammazione (es. edema) sull'integrità
delle strutture infiammate; si assume una postura come atteggiamento di difesa
da un insulto che danneggia la normale funzione del tessuto.
8
Figura 2: I segni cardine dell’infiammazione.
Quando uno stimolo infiammatorio colpisce un'area di qualsiasi organo, una
parte delle cellule va in necrosi o viene più o meno gravemente danneggiata
con la conseguenza che i detriti cellulari che si formano costituiscono
anch'essi un'ulteriore stimolazione flogogena per le cellule rimaste indenni. In
conseguenza di ciò si verificano una serie di eventi che coinvolgono il
microcircolo e che rappresentano le fasi dell’infiammazione acuta:
vasocostrizione fugace;
vasodilatazione o iperemia attiva;
essudazione;
diapedesi;
iperemia passiva;
fenomeni produttivi e riparativi.
In seguito all’applicazione dell’insulto si ha la vasocostrizione delle arteriole
della zona colpita, un evento di brevissima durata (circa 10-20 secondi)
mediato dalla branca simpatica del sistema nervoso vegetativo attraverso la
liberazione di catecolamine vasocostrittrici quali adrenalina, noradrenalina e
9
serotonina. In seguito avviene la vasodilatazione delle arteriole conosciuta
come iperemia attiva. Di solito inizia con il rilasciamento delle fibrocellule
muscolari lisce dalla parete delle arteriole terminali ed è successivamente
sostenuta dall'apertura di nuovi letti capillari e dalla chiusura degli shunt
arterovenosi fisiologicamente attivi a riposo. La vasodilatazione può durare da
15 minuti a diverse ore a seconda della gravità del danno, inducendo un
aumento (fino a 10 volte) del flusso ematico locale.[1] La sede della flogosi
diventa rossa ‒ eritema ‒ e calda e proprio per questo è appropriato il termine
di infiammazione che evoca il concetto di fiamma.[1] L'innesco ed il
mantenimento della vasodilatazione sono dovuti al rilascio di mediatori rapidi
come l'istamina o le prostaglandine e successivamente di mediatori lenti come
IFN-γ, TNF-α, IL-1β, LPS, PAF. Quest’ ultimi stimolano l'espressione della
sintetasi inducibile dell'ossido nitrico (iNOs) a livello dell'endotelio vascolare.
Al contrario l'istamina, attraverso un aumento delle concentrazioni di calcio
intracellulare, determina l'attivazione “rapida” della NO-sintetasi costitutiva.
Entrambe le isoforme di quest'ultimo enzima sono responsabili della sintesi di
monossido di azoto (NO), un potentissimo vasodilatatore, che agisce sul
muscolo liscio vascolare provocandone il rilassamento.[2] La vasodilatazione
aumenta la quantità di sangue che rifornisce il tessuto leso (iperemia) e di
conseguenza anche la pressione idrostatica che il sangue stesso esercita sulle
pareti dei vasi; tutto ciò causa dilatazione delle arteriole, apertura del letto
capillare e dilatazione venulare. Per azione di alcuni mediatori chimici
dell’infiammazione si ha il contemporaneo aumento della permeabilità
vascolare; questo fenomeno determina un aumento del flusso non solo di
liquidi e proteine plasmatiche, ma anche di cellule che dal plasma migrano
verso l'area danneggiata formando l'essudato, un liquido a pΗ acido ricco di
proteine plasmatiche e/o cellule, come neutrofili e mononucleati, ma anche
eritrociti, che fuoriuscito dai vasi si raccoglie negli spazi interstiziali dei
tessuti. La diminuzione della pressione colloido-osmotica in seguito a ridotta
concentrazione delle plasmaproteine che si accumulano all'esterno dei vasi,
10
contribuisce all'ulteriore richiamo di acqua nella sede dell’infiammazione e
alla formazione dell’edema infiammatorio. In seguito all'iperemia, il flusso
comincia a rallentare e come accennato in precedenza non si ha solo un flusso
di liquidi verso il compartimento extracellulare ma anche di cellule, fenomeno
conosciuto come diapedesi o migrazione. I leucociti presenti nel sangue si
spostano in prossimità della parete endoteliale, vi aderiscono e fuoriescono dal
compartimento ematico migrando nella sede della flogosi.[1] Questo processo,
consiste in un movimento attivo ameboidale attraverso la parete delle venule e
dei vasi di piccolo calibro ed è dipendente dalla presenza sulle cellule
endoteliali di particolari citochine fornite di attività chemiotattica dette
chemiochine e di fattori chemiotattici.[1] Il processo della migrazione dei
leucociti si articola in diverse fasi (Fig.3):
marginazione: i leucociti che normalmente occupano la parte centrale del
capillare si spostano verso la parete poiché il flusso rallenta;
rotolamento: i leucociti rotolano lungo l’endotelio con minore velocità
finché vi restano invischiati e si arrestano aderendovi transitoriamente.
L’endotelio
viene
poi
rivestito
tramite
un
processo
detto
di
pavimentazione;
adesione: i leucociti rotolano fino a quando non aderiscono saldamente alla
superficie endoteliale;
diapedesi: i leucociti cominciano ad emettere pseudopodi, uno dei quali si
insinua nelle giunzioni tra le cellule endoteliali fino ad attraversare la
parete del capillare. A questo punto il passaggio all’esterno è completato
dal trasferimento del protoplasma, che segue lo pseudopodo riversandosi in
esso;
chemiotassi: i leucociti si dirigono verso gli spazi extracellulari, attirati da
sostanze chemiotattiche (esogene ed endogene) e una volta giunti nel sito
di lesione, svolgono la loro funzione fagocitaria.[1]
11
Figura 3: Diapedesi dei leucociti.
Anche gli eritrociti possono fuoriuscire dai vasi con un meccanismo di
diapedesi; la loro presenza nello spazio extravascolare implica l'esistenza di un
danno endoteliale severo.[1] Dopo la fuoriuscita di liquidi (essudazione) e di
cellule (diapedesi) aumenta la viscosità del sangue, mentre diminuisce la
velocità del flusso sanguigno nel microcircolo determinando iperemia passiva
o stasi. Nella zona infiammata i letti vascolari accolgono (come nelle fasi
iniziali) più sangue della norma, ma ciò è dovuto non all'apertura di nuovi letti
capillari (iperemia attiva) ma al rallentamento del circolo e al diminuito
ritorno venoso (iperemia passiva) causato dalla compressione esercitata sulle
venule da parte dell’ edema, che ostacola così il passaggio di sangue
provocando una riduzione della sua parte liquida ed un aumento relativo delle
componenti solide. L’ultima fase dell’infiammazione consiste nella fagocitosi
dei detriti cellulari e dei microrganismi da parte dei fagociti e nella guarigione
in cui l’essudato viene riassorbito mentre i leucociti vanno incontro a morte
cellulare programmata dopo aver fagocitato e distrutto gli agenti flogogeni.[1]
Durante la guarigione si possono avere: la rigenerazione ossia la sostituzione
12
di cellule e tessuto perduti con nuove cellule e tessuti “restituito ad integrum”
oppure, se questo non è possibile, la riparazione cioè la sostituzione del tessuto
leso con tessuto di granulazione che matura poi in tessuto cicatriziale a cui
segue la risoluzione o cronicizzazione del processo infiammatorio.
1.3 Infiammazione cronica
L’evoluzione dell’ infiammazione verso la cronicizzazione o la risoluzione
dipende dalla persistenza dello stimolo antigenico e dal tipo di citochine che,
prodotte in parte da macrofagi e linfociti T, si accumulano nel tessuto e
condizionano la differenziazzione e l’apoptosi delle cellule del sistema
immunitario. Qualora non venga eliminato efficacemente l’agente flogogeno,
l’intensificarsi del processo infiammatorio conduce alla degranulazione delle
cellule infiammatorie e al rilascio di enzimi lisosomiali che possono
danneggiare le cellule vicine e causare a loro volta la liberazione di acido
arachidonico e di altri precursori che favoriscono la formazione e la
liberazione di mediatori pro-infiammatori e di specie reattive dell’ossigeno
che provocando ulteriore danno, contribuiscono in maniera decisiva
all’infiammazione
cronica.
L’infiammazione cronica
è un
processo
infiammatorio di lunga durata, settimane, mesi o tutta la vita, che si instaura
non solo come evoluzione del processo acuto ma che può insorgere
direttamente come processo cronico ab initio come conseguenza di
un’infezione cellulare persistente, della prolungata esposizione a sostanze
potenzialmente tossiche o irritanti sia esogene che endogene di reazioni
immunitarie,
specie
verso
i
tessuti
dell’organismo
(autoimmunità).
L’infiammazione cronica è caratterizzata dalla contemporanea presenza di:
infiltrazione leucocitaria (linfociti, monociti, plasmacellule) che indica la
continua attività infiammatoria;
distruzione tissutale in seguito alla produzione locale di citochine;
tentativi continui di riparazione (angiogenesi, fibrosi) (Fig.4).
13
Figura 4: Infiammazione cronica
Le infiammazioni croniche si distinguono da quelle acute per la prevalenza dei
fenomeni cellulari su quelli vascolari essudativi. Quando un'infiammazione
acuta si cronicizza, si assiste dapprima ad una progressiva riduzione dei
fenomeni vascolo ematici e della quantità di essudato, come avviene anche nel
processo di guarigione dell’infiammazione acuta; contemporaneamente i
neutrofili
vengono
sostituiti
da
un
infiltrato
cellulare
costituito
prevalentemente da macrofagi, linfociti, plasmacellule e cellule NK che si
dispongono attorno alla parete vascolare come un manicotto che ne induce la
compressione. Successivamente i fibroblasti possono essere stimolati alla
proliferazione con la conseguenza che molte flogosi croniche culminano con
un’ eccessiva formazione di tessuto connettivo che costituisce la cosiddetta
fibrosi o sclerosi rappresentata dal granuloma, espressione dell’infiammazione
cronica. Le flogosi croniche si presentano sotto l'aspetto clinico in due forme
diverse: non granulomatose e granulomatose. Nelle infiammazioni croniche
non granulomatose il quadro morfologico, rappresentato dall'infiltrato
linfomonocitario, si presenta con prevalenza di linfociti e plasmacellule e
mantiene le stesse caratteristiche qualunque sia l'agente eziologico
responsabile del processo. Quelle granulomatose intervengono quando
microrganismi di vario tipo sopravvivono nei fagolisosomi dei macrofagi o
quando in questi rimangono loro prodotti o anche materiali di natura organica
o anche inorganica indigeribili.
14
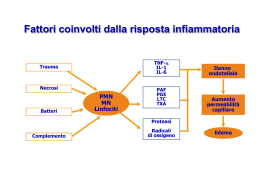
1.4 Mediatori chimici dell’infiammazione
Durante il processo infiammatorio vengono prodotti importanti mediatori
chimici di origine plasmatica e cellulare, che esprimono la loro attività
biologica legandosi a specifici recettori sulle cellule bersaglio.[3] Un mediatore
chimico può stimolare il rilascio di altri mediatori da parte di cellule bersaglio
con effetto di amplificazione, modulazione o regolazione. I mediatori possono
agire su uno o su più tipi cellulari e possono esercitare effetti diversi a seconda
del tipo di tessuto o cellula; una volta attivati o rilasciati dalla cellula hanno
un’emivita breve (Fig.5).
I mediatori possono essere suddivisi in:
mediatori di origine plasmatica
mediatori di origine cellulare
Figura 5: I mediatori chimici dell’infiammazione.
I mediatori di origine plasmatica sono prodotti in forma inattiva dal fegato,
che li mette in circolo e vengono attivati, quando serve, dall’attivazione del
fattore XII (importante nel sistema delle chinine e in quello della
15
coagulazione/fibrinolisi) e dall’attivazione del complemento (fattori che
attivati interagiscono in successione in un sistema a cascata).
Questa classe di mediatori è costituita da diversi elementi:
-il sistema del complemento il quale è formato da più di 30 proteine che si
trovano in alta concentrazione nel plasma in forma inattiva e possono essere
attivate:
dal legame di complessi antigene-anticorpo, prodotti batterici e virali, etc.
(via classica);
dal contatto con componenti della superficie microbica (endotossine),
polisaccaridi, veleno di cobra, virus, etc. (via alternativa);
dalla lectina plasmatica che lega il mannosio (MBL) presente sulle pareti
batteriche (via della lectina).
Durante il processo di attivazione si generano una serie di prodotti che
causano: aumento della permeabilità vascolare, chemiotassi e opsonizzazione
(Fig.6)
Figura 6: Sistema del complemento.
16
La cascata complementare attiva vari componenti tra cui il C3b che ha
funzione di opsonina e il C5a che è un fattore attrattivo per i neutrofili. Se si
genera il complesso C5-9 (MAC, complesso di attacco alla membrana) la
cellula a cui si attacca il complesso verrà lisata tramite la perforazione della
membrana plasmatica. Le anafilotossine (C3a, C5a) aumentano la permeabilità
vasale e causano vasodilatazione inducendo la liberazione di istamina da parte
dei mastociti. Inoltre, C5a attiva la lipossigenasi dei neutrofili e dei monociti.
Il fattore C5a aumenta notevolmente l’interazione fra le integrine dei leucociti
e l’endotelio ed è un potente chemiotattico nei confronti di neutrofili,
monociti, eosinofili e basofili. Il fattore C3b stimola la fagocitosi da parte dei
granulociti, dei neutrofili e dei macrofagi. L’importanza dei fattori C3 e C5 del
complemento è resa ancora maggiore dal fatto che essi possono essere attivati
anche dagli enzimi proteolitici presenti nell’essudato (idrolasi lisosomiali
rilasciate dai neutrofili, plasmina); in tal modo, la risposta infiammatoria si
protrae e si amplifica.
-Il sistema delle chinine genera peptidi vasoattivi a partire da proteine
plasmatiche dette chininogeni, per azione di proteasi specifiche chiamate
Callicreine. Il sistema delle chinine porta al rilascio finale del nonapeptide
vasoattivo bradichinina, potente mediatore in grado di aumentare la
permeabilità vasale. La bradichinina provoca inoltre la contrazione della
muscolatura liscia, vasodilatazione e dolore (effetti simili all’istamina). La
cascata che porta alla produzione della bradichinina viene innescata
dall’attivazione del Fattore di Hageman (Fattore XII della via intrinseca della
coagulazione).
-La cascata della coagulazione e il processo infiammatorio sono intimamente
connessi tra loro; rappresentano due vie convergenti che culminano
nell’attivazione della trombina con conseguente formazione di fibrina. Una
volta attivata, la cascata della coagulazione ha diversi effetti pro-infiammatori:
attivazione del sistema delle chinine, attivazione della plasmina (fibrinolisi),
attivazione del complemento ed effetti flogogeni della trombina (Fig.7).
17
Figura 7: Cascata della coagulazione.
I mediatori di origine cellulare si suddividono in 2 sottogruppi:
1. Mediatori preformati,
2. Mediatori sintetizzati ex novo
1. I mediatori preformati, rappresentati dalle ammine vasoattive: istamina,
serotonina e dagli enzimi lisosomiali, sono presenti in granuli di secrezione
-L’istamina, prodotta da mastociti, basofili e piastrine, viene rilasciata in
seguito a degranulazione e causa nella fase immediata dilatazione delle
arteriole, contrazione delle cellule endoteliali venulari, fenestrazione
endoteliale con conseguente aumento della permeabilità delle venule e
aumento della produzione di prostaglandine. Il rilascio di istamina può essere
altamente nocivo negli organismi sensibilizzati determinando un collasso
circolatorio e respiratorio durante lo shock anafilattico in seguito alla
vasodilatazione sistemica.[3]
-La serotonina, è localizzata nel SNC (mediatore sinaptico), nelle cellule
cromaffini del tratto gastrointestinale e nei granuli densi delle piastrine.
18
Determina un effetto edemigeno per aumento della permeabilità delle venule
post-capillari, per contrazione degli endoteliociti, similmente all’istamina e
aumento dello spazio giunzionale inter-endoteliale.
-Gli enzimi lisosomiali, sono liberati nel focolaio infiammatorio dai fagociti
professionali (granulociti e monociti) che li tengono sequestrati nei granuli
lisosomiali o lisosomi in forma inattiva. Si distinguono tre tipi di lisosomi
ognuno contenente enzimi diversi. I lisosomi primari contengono catepsina,
elastasi, fosfolipasi A2, idrolasi acide, lisozima e mieloperossidasi; i secondari
racchiudono collagenasi, fosfolipasi alcalina e lattoferrina; infine i terziari
includono catepsine e gelatinasi.
2. I mediatori sintetizzati ex novo vengono sintetizzati al momento del
bisogno e comprendono: i metaboliti dell’acido arachidonico, il PAF, le specie
reattive dell’ossigeno, l’ossido di azoto e le citochine.
-I metaboliti dell’acido arachidonico sono: prostaglandine, leucotrieni e
trombossani. Le prostaglandine sono prodotte da leucociti, piastrine e cellule
endoteliali e derivano dal metabolismo dell’acido arachidonico. L’ossidazione
dell’acido arachidonico avviene da parte dell’attività cicloossigenasica della
prostaglandina H sintasi che porta alla formazione di PGG2, successivamente
ridotta dall’attività perossidasica dello stesso enzima a PGH2 . La PGH2, per
azione di sintasi tessuto-specifico, dà vita a prostaciclina, trombossano e
PGD2, PGE2, PGF2α, PGI2.[2] Quest’ ultime sostanze sostengono la
vasodilatazione potenziando l'edema. Inoltre PGE2 e PGF2α agiscono sulle
fibre nervose polimodali di tipo C, in genere in risposta a bradichinina,
abbassandone la soglia di scarica e determinando così i sintomi dolorosi
associati all'infiammazione. PGE2 secreta dall'endotelio dei vasi ipotalamici in
risposta ad alte concentrazioni di TNF, IL-1 e IL-6 agisce sul nucleo
termoregolatore ipotalamico causando la febbre. I leucotrieni sono prodotti da
tutti i leucociti, anch’essi derivano dal metabolismo dell’acido arachidonico e
danno vasocostrizione, broncospasmo, aumento della permeabilità e
chemiotassi.
19
-Il PAF-fattore attivante le piastrine- deriva dai fosfolipidi di membrana dei
neutrofili, delle piastrine, dei monociti, dei basofili, delle cellule endoteliali
per
azione
della
fosfolipasi
A 2.
PAF
determina
vasocostrizione,
broncospasmo, vasodilatazione, aumento della permeabilità vasale e risulta
essere 100-1000 volte più potente dell’istamina. Inoltre, PAF facilita
l’adesione dei leucociti attraverso modificazioni conformazionali delle
integrine, ha azione chemiotattica, induce la degranulazione leucocitaria e il
burst ossidativo. PAF agisce sulle cellule bersaglio interagendo con recettori
specifici e stimolando la produzione di ulteriori mediatori.
-Le specie reattive dell’ossigeno, cioè i radicali liberi dell’ O2, vengono
rilasciati dai leucociti nell’ambiente extracellulare in seguito ad esposizione ad
agenti chemiotattici, immunocomplessi o durante la fagocitosi. Il loro rilascio
può causare seri danni all’ospite a livello endoteliale, con conseguente
aumento
della
permeabilità
vascolare;
possono
determinare
inoltre
inattivazione di inibitori delle proteasi e lesioni ad altre cellule quali eritrociti
e cellule parenchimali.
-L’Ossido di azoto-NO è un gas solubile prodotto a partire dalla L-arginina,
dall’ossigeno molecolare, dal NADPH e dall’enzima ossido nitrico sintetasi
(NOS) (Fig.8).
Figura 8: Ossido d’azoto.
20
Questo enzima è espresso costitutivamente in cellule endoteliali e neuroni,
dove la sua sintesi può essere rapidamente indotta dall’influsso di ioni calcio;
ma in molti tipi di cellule come epatociti, cellule cardiache ed epitelio
respiratorio vi è invece la presenza della NOS inducibile (iNOS), la cui sintesi
è indotta nei macrofagi dalle citochine infiammatorie TNF-α e IFN-γ.
L’attivazione della iNOS stimola la produzione della cicloossigenasi
inducibile (COX-2) e di conseguenza la produzione di prostanoidi. L’NO è un
potente vasodilatatore in quanto induce il rilassamento delle cellule della
muscolatura liscia delle pareti dei vasi; inolte inibisce l’aggregazione
piastrinica, il reclutamento dei leucociti ed ha un effetto antimicrobico.[2]
-Le citochine sono messaggeri polipeptidici prodotti da molti tipi di cellule:
linfociti e macrofagi attivati ma anche cellule endoteliali, epiteliali e del
connettivo.[3] Esse sono in grado di modulare la funzione di altre cellule e sono
coinvolte nelle risposte immunitarie e nell’infiammazione acuta e cronica
(Fig.9). Le citochine prodotte dai fagociti mononucleati sono chiamate
monochine mentre quelle che derivano dai linfociti sono dette linfochine. Tra
le varie citochine abbiamo: i fattori di crescita che determinano la
proliferazione di alcuni tipi cellulari; le interleuchine prodotte dalle cellule
ematopoietiche; le chemiochine ovvero citochine chemiotattiche che hanno la
capacità
attrarre
e
stimolare
il
movimento
dei
leucociti,
specie
nell’infiammazione; gli interferoni e le citochine pro-infiammatorie come il
TNF-α. Le principali citochine infiammatorie sono IL-1 e TNF-α definite
citochine infiammatorie primarie perché in grado di mettere in movimento
l’intera cascata di mediatori caratteristici di una risposta infiammatoria.[3] Esse
sono in grado di agire su tutte le cellule ed i tessuti dell’organismo. A livello
locale, nella sede dell’infiammazione, inducono la produzione di chemochine
e di molecole di adesione sulla superficie cellulare, amplificando il
reclutamento di cellule infiammatorie; inducono l’espressione di enzimi che
portano alla sintesi di prostaciclina e di NO che hanno attività vasodilatatoria;
inoltre modificano le proprietà anticoagulanti dell’endotelio vascolare
21
inducendo la produzione di tissue factor, che ha attività pro-coagulante e
inibiscono l’asse anti-coagulante costituito dalla proteina C e dalla
trombomodulina.[3]
Figura 9: Citochine.
1.5 Cascata dell’acido arachidonico
L’acido arachidonico o acido 5, 8, 11, 14-eicosatetraenoico, è un acido grasso
essenziale a 20 atomi di carbonio, con 4 doppi legami, che non si trova
all’interno della cellula in forma libera, se non in piccolissime concentrazioni,
ma è fortemente concentrato a livello dei fosfolipidi di membrana dove si
ritrova, sottoforma di estere, come fosfatidilinositolo, fosfatidilcolina e
fosfatidiletanolamina.[3] Ѐ sintetizzato dall’organismo a partire dall’acido
linoleico ma il maggior contributo avviene attraverso l’alimentazione. L'acido
arachidonico costituisce il precursore principale degli eicosanoidi, sostanze
coinvolte nella risposta infiammatoria dell'organismo, che vengono prodotte
attraverso reazioni ossidative catalizzate da enzimi specifici o da radicali
dell’ossigeno.[3] L’acido arachidonico esterificato nei fosfolipidi di membrana
può essere liberato in seguito all’interazione di stimoli di diversa natura con la
22
membrana cellulare: stimoli fisiologici (istamina, bradichinina, vasopressina,
IL-1, angiotensina II, fattori di crescita.), stimoli fisici (shear stress, ischemia)
e anche agenti farmacologici (esteri del forbolo come il PMA).[3] Gli stimoli
fisiologici agiscono interagendo con recettori specifici presenti sulla
membrana cellulare, spesso accoppiati a proteine G, che a loro volta attivano
due fosfolipasi cellulari: la fosfolipasi A2 e la fosfolipasi C in grado di liberare
l’acido arachidonico dai fosfolipidi di membrana (Fig.10).[3]
Figura 10: Cascata dell’acido arachidonico.
L’acido arachidonico una volta liberato viene metabolizzato attraverso quattro
vie principali:
1. la via della prostaglandina H sintasi (PGH sintasi) o via cicloossigenasica,
che porta alla formazione di prostanoidi;
2. la via delle lipossigenasi (5-, 12- e 15-lipossigenasi) che porta alla
formazione degli acidi idrossieicosatetraenoici (HETE) e dei leucotrieni;
3. la via che porta alla formazione di acidi epossieicosatetraenoici e
idrossieicosatetraenoici che è catalizzata da enzimi che contengono il
citocromo P450;
23
4. la via della perossidazione lipidica non enzimatica catalizzata dai radicali
dell’ossigeno, la quale porta alla formazione di iso-eicosanoidi.[3]
Le vie metaboliche che hanno maggiore interesse farmacologico nell’ambito
dell’infiammazione sono la via cicloossigenasica, dalla quale sono ottenute le
prostaglandine, prostacicline ed i trombossani e la via lipossigenasica, dalla
quale sono ottenuti i leucotrieni e le lipossine.[3]Tutti questi prodotti vengono
chiamati eicosanoidi per via della struttura a 20 atomi di carbonio che le
caratterizza.
La
via
cicloossigenasica
è
mediata
dall’azione
delle
cicloossigenasi (COX), enzimi presenti a livello microsomiale, che
trasformano l’acido arachidonico in endoperossidi ciclici:
PGG2 e
successivamente in PGH2. Il PGH2 a sua volta viene metabolizzato in:
trombossano, prostaciclina e prostaglandine (Fig.11).
Il trombossano (TXA2) viene prodotto principalmente nelle piastrine ad opera
della trombossano sintetasi; è un potente agente vasocostrittore e promuove
l’aggregazione piastrinica.
La prostaciclina (PGI2) si forma ad opera della prostaciclina sintetasi a livello
vascolare; è un potente vasodilatatore, inibisce l’aggregazione piastrinica ed
aumenta la permeabilità vasale per questo viene considerato l’antagonista
fisiologico del trombossano.
Figura 11: Via cicloossigenasica.
24
Le prostaglandine (PGD2, PGE2, PRG2α) sono delle ammine biogene che
agiscono in vari tessuti (es. app. intestinale, bronchiale, uterino e gastrico);
proteggono la mucosa di rivestimento gastrointestinale (le PGE inibiscono la
produzione di HCl nello stomaco, favoriscono la produzione di muco e la
secrezione di bicarbonato, che contrasta l'acidità gastrica), stimolano
l’aggregazione piastrinica nel mantenere la normale omeostasi, mantengono la
resistenza alla trombosi sulla superficie vascolare delle cellule endoteliali,
sono coinvolte nel dolore e nella febbre.[9]
La via lipossigenasica, mediata dall’azione della lipossogenasi, si ritrova a
livello polmonare, piastrinico e leucocitario. Questo enzima, perossidando
l’acido arachidonico conduce alla formazione di idroperossidi, fra cui il 5HETE (acido idrossieicosatetraenoico) come prodotto principale, che possiede
una spiccata azione chemiotattica per i neutrofili e viene convertito in una
serie di prodotti chiamati leucotrieni (Fig.12)
Figura 12: I leucotrieni
I leucotrieni sono sintetizzati dalla 5-lipossigenasi che dipende da un cofattore
proteico indicato come FLAP (5-Lipoxygenase-Activating Protein) presente
nelle cellule infiammatorie (PMN, basofili, mastociti, eosinofili e macrofagi).
La stimolazione di queste cellule causa l’aumento dei livelli di calcio
intracellulare, la liberazione di arachidonato e l’incorporazione di ossigeno
molecolare da parte della 5-LO con la formazione di un epossido instabile: il
25
leucotriene A4 (LTA4). Questo intermedio è convertito nel diidrossi
leucotriene derivato B4 (LTB4) dalla LTA4 idrolasi, oppure coniugato con il
glutatione ridotto ad opera della LTC4 sintasi a leucotriene C4. L’LTC4 può
subire una degradazione sequenziale della molecola di glutatione per azione
enzimatica di alcune peptidasi, formando l’LTD4 e l’LTE4. Un altro gruppo di
prodotti della 5-LO sono le lipossine LXA e LXB, i cui ruoli biologici non
sono stati ancora chiariti. La capacità di produrre elevate quantità di
leucotrieni dall’acido arachidonico è limitata ai leucociti, sebbene le cellule
non leucocitarie non abbiano sufficiente 5-LO e FLAP per sintetizzare
apprezzabili quantità di leucotrieni, esse esprimono gli enzimi metabolizzanti
l’LTA4 e possono captare quest’ultimo dall’ambiente extracellulare e
metabolizzarlo in leucotrieni bioattivi attraverso un processo biosintetico
definito”transcellulare”. La quantità di leucotrieni sintetizzata è regolata dai
livelli di arachidonato che la PLA2 libera dai fosfolipidi di membrana e dai
fattori che modulano l’attività della 5-LO. Un’altra variabile che influenza la
biosintesi dei leucotrieni è la localizzazione intracellulare della 5-LO. La
trascrizione del gene che codifica per la 5-LO è regolata da citochine, fattore
di crescita e di trasformazione , endorfine e corticosteroidi. I leucotrieni
agiscono legandosi a recettori specifici localizzati sulla membrana cellulare
(recettori a 7 domini transmembrana accoppiati a Gi). Le cellule bersaglio dei
leucotrieni sono leucociti, cellule epiteliali, cellule muscolari lisce e cellule
endoteliali. Il recettore 1 dell’LTB4 (BLT1), è un recettore ad alta affinità che
media a livello delle vie aeree broncocostrizione, secrezione di muco ed
edema. Il recettore 2 dell’LTB4 (BLT2) è un recettore a bassa affinità che lega
anche altri metaboliti della 5-LO. Oltre alle funzioni sulle vie aeree i
leucotrieni svolgono altre azioni biologiche quali contrazione della
muscolatura liscia e chemiotassi. Per quanto concerne quest’ultima funzione è
importante ricordare il ruolo dei leucotrieni nei processi infiammatori, infatti i
leucotrieni agiscono sui leucociti stimolando la crescita dei progenitori dalle
cellule ematopoietiche pluripotenti CD34 positive e la loro successiva
26
migrazione nel torrente circolatorio. I leucotrieni aumentano inoltre
l’espressione delle proteine di adesione (I-CAM, V-CAM) promuovendo la
motilità cellulare.
27
2. Infiammazione e Carcinogenesi
2.1 L’infiammazione spiega il cancro
Gli studi condotti in questi anni hanno messo in evidenza i nessi esistenti fra
gli eventi di alterazione genica che causano il cancro e la corrispondente
risposta infiammatoria che ne deriva. Il primo a descrivere una relazione
funzionale tra infiammazione e cancro è stato Virchow nel 1863 che ha
ipotizzato l’origine del cancro in un ambiente dove persiste uno stimolo
infiammatorio. Questa ipotesi nasce dall’evidenza che sostanze irritanti
insieme alla presenza di una ferita in un tessuto siano in grado di scatenare una
risposta infiammatoria con il conseguente aumento della proliferazione
cellulare.[4] Oggi è chiaro che la proliferazione cellulare da sola non causa il
cancro, ma la proliferazione continua in un ambiente ricco di cellule
infiammatorie, di fattori di crescita, stroma attivato ed agenti che promuovono
danni al DNA, certamente potenzia e/o promuove il rischio neoplastico. Il
processo infiammatorio è un fattore negativo a carico di tutti i tessuti a causa
della continua esposizione agli agenti ambientali. Il microambiente
infiammatorio favorisce infatti l’iniziazione delle cellule normali e la loro
crescita e progressione verso la malignità attraverso la produzione di citochine
pro-infiammatorie, agenti ossidanti ed enzimi litici. E’ ormai un paradigma
accettato il “doppio legame” esistente tra infiammazione e tumore, infatti, i
macrofagi, cellule infiammatorie presenti all’interno dei tumori, non svolgono
come dovrebbero un ruolo di difesa all’interno dell’organismo, ma aiutano lo
sviluppo del cancro. La connessione causa-effetto esistente tra infiammazione
e tumore permetterebbe di spiegare come alcune forme croniche di
infiammazione in determinati organi possano favorire l’insorgere di tumori e
allo stesso tempo come un tumore, indipendentemente dal fatto che sia stato o
meno concausato da un’infiammazione precedente, per svilupparsi crei un
ambiente infiammatorio. Pertanto l’infiammazione è la chiave non solo per
28
comprendere il cancro, ma anche per combatterlo. Da alcuni anni l'approccio
terapeutico ai tumori ha distolto l'attenzione sulle cellule tumorali, spostandola
verso l’angiogenesi, giacchè dall’inibizione di quest’ultima è possibile limitare
la crescita della massa tumorale.
2.2 Correlazione tra microambiente infiammatorio e tumori
La correlazione tra il fenomeno flogistico e quello canceroso ha portato a
considerare l’infiammazione come uno dei segni caratteristici del cancro.[4] I
mediatori e gli effettori cellulari dell’infiammazione sono infatti costituenti
importanti del microambiente tumorale. In alcuni tipi di tumore, i processi
infiammatori sono presenti prima della trasformazione neoplastica delle
cellule. Al contrario, in altri tipi di tumore, un cambiamento oncogenico
induce un microambiente infiammatorio che a sua volta promuove lo sviluppo
del tumore. La componente infiammatoria stimola la proliferazione e la
sopravvivenza
di
cellule
maligne,
promuove
l’
angiogenesi
e
la
metastatizzazione, sovverte le risposte immunitarie adattative e la risposta a
chemioterapici. Infatti, negli ultimi anni è stato messo in evidenza che anche
in tumori non primariamente riconducibili a processi infiammatori, la
componente infiammatoria è presente e costituisce una tappa essenziale nella
formazione del microambiente maligno.[4]
Le caratteristiche di un tumore correlato ad uno stato infiammatorio sono
essenzialmente: l’infiltrazione leucocitaria e macrofagica, la presenza di
citochine, chemochine, interleuchine, TNF-α (Tumor Necrosis Factor-α),
interferoni, fattori di crescita, enzimi proteolitici, proteoglicani, mediatori
lipidici e prostaglandine. Sono state messe in evidenza due vie di attivazione
che legano l’ infiammazione e il cancro:
1. via intrinseca;
2. via estrinseca.
1. La via intrinseca è attivata da eventi genetici capaci di indurre processi
neoplastici. Tali eventi genetici includono: attivazione di vari oncogeni
29
tramite mutazioni, riarrangiamento oppure amplificazione cromosomica ed
inattivazione di geni oncosoppressori. I membri della famiglia RAS sono
gli oncogeni maggiormente coinvolti nello sviluppo dei tumori umani e
attivano la proliferazione attraverso le chinasi RAS-RAF. Le cellule che
subiscono
questo
dell’infiammazione,
tipo
di
generando
trasformazione
in
tal
modo
producono
un
mediatori
microambiente
infiammatorio in tumori che di base non presentano condizioni di
infiammazione. Altri oncogeni che svolgono una funzione simile sono la
proteina tirosin-chinasi RET, coinvolta nello sviluppo del tumore alla
tiroide e MYC, un fattore trascrizionale over-espresso in molti tipi di
tumori umani.
2. Nella via estrinseca, invece, condizioni infiammatorie e/o infettive
aumentano il rischio di progressione del cancro soprattutto in alcuni siti
anatomici (colon, prostata e pancreas).
Le due vie convergono causando nelle cellule tumorali l’attivazione di fattori
di trascrizione, tra cui NF-kB, STAT3 (Signal Transducer and Activator of
Transcription 3) e HIF-1α (Hypoxia-Inducible Factor 1-alpha). L’attivazione
di questi fattori comporta, oltre al reclutamento e l’attivazione di un gran
numero di leucociti, la produzione di numerosi mediatori dell’infiammazione,
comprese citochine e chemochine, e di enzimi quali iNOS (Nitrossido Sintasi
inducibile) e COX-2 (Cicloossigenasi 2), entrambi di fondamentale
importanza nell’infiammazione. Numerosi studi hanno messo in correlazione
l’NO ed il processo neoplastico. L’NO sarebbe coinvolto nell’acquisizione di
proprietà metastatiche da parte di tumori benigni in modelli di progressione
neoplastica su base infiammatoria ed attiverebbe, in maniera indiretta, processi
di invasione e metastatizzazione tramite induzione di MMP e VEGF.[5] La
COX-1 e la COX-2 catalizzano tappe fondamentali nella formazione di
prostanoidi anch’essi centrali mediatori del processo infiammatorio. Mentre la
COX-1 è costitutivamente espressa in molti tessuti, in cui controlla normali
processi fisiologici, la COX-2 non è rilevabile in condizioni normali (tranne
30
nel sistema nervoso centrale, reni e vescicole seminali) ma è indotta da stimoli
infiammatori e mitogeni. La COX-2 viene definita come la isoforma
inducibile, il mediatore che meglio rappresenta le risposte infiammatorie, sia
perché la sua espressione è altamente influenzata da stimoli infiammatori, sia
perché gioca un ruolo chiave nel processo di carcinogenesi a cui partecipa
attivamente con la produzione di sostanze cancerogene capaci di influenzare:
apoptosi e angiogenesi.[6]
2.3 COX-2 e Carcinogenesi
Oggi, impropriamente si afferma che la “COX-2 nutre i tumori”.
Affermazione avvalorata dai vari dati sperimentali che attestano come in molte
forme tumorali sia presente una sovra-espressione dell’ enzima cicloossigenasi
2 (COX-2), responsabile della crescita ed invasività di cellule malate, ciò si
verificherebbe per effetto di un meccanismo prostaglandine-dipendente e
prostaglandine-indipendente.
COX-2
è
coinvolta
nel
processo
di
cancerogenesi, intervenendo nell’angiogenesi apoptosi, invasività e controllo
della proliferazione cellulare.[5] Fattori di crescita, citochine, oncogeni e
oncosoppressori stimolano la trascrizione di COX-2 attraverso la via mediata
da Ras ed attraverso la via della proteina chinasi C (PKC).[6] Numerosi studi
hanno evidenziato che la COX-2 e la PGE2 hanno attività di regolatori positivi
della proliferazione delle cellule tumorali. Questi effetti sono indotti
soprattutto dalle cascate enzimatiche mediate da Ras/Raf/Protein chinasi
mitogena attivata (Ras/Raf/MAPK) e fosfaditilinositolo-3 chinasi (P13K/Akt),
attraverso un meccanismo di feed-back positivo, osservato anche in cellule di
adenoma intestinale e di adenocarcinoma polmonare.
2.3.1 COX-2 e angiogenesi tumorale
In letteratura sono noti i molteplici ruoli che riveste la COX-2 nei confronti
dell’angiogenesi tumorale. La crescita tumorale è strettamente dipendente da
un adeguato apporto ematico, determinato dalla formazione di nuovi capillari,
31
mediante un processo regolato da fattori pro-angiogenetici, come il fattore di
crescita vascolare endoteliale (VEGF). In molti tipi di tumore è stata
dimostrata una correlazione tra attivazione della COX-2 e la neoangiogenesi.
Ad esempio, nelle cellule del carcinoma squamocellulare è stata osservata una
maggiore espressione del VEGF e del proprio mRNA, in risposta
all’induzione di COX-2 con conseguente sintesi di PGE2. Inoltre è stato
dimostrato che la over-espressione di COX-2 nelle cellule del colon-retto
induce la produzione di prostaglandine e fattori pro-angiogenetici.
2.3.2 COX-2 e apoptosi
L’induzione dell’apoptosi sembra essere il principale meccanismo d’azione
anti-neoplastico promosso dai FANS, quindi il potenziale anti-apoptotico della
COX-2 assume notevole rilevanza. Sono state riportate forti correlazioni in
vari tipi di cancro, tra l’attivazione della COX-2 e l’espressione di proteine
anti-apoptiche della famiglia della bcl-2. In molti studi è stato dimostrato che
il trattamento con FANS induce l’ apoptosi delle cellule tumorali, mentre il
trattamento con PGE2 blocca l’apoptosi nelle cellule del cancro del colon,
inoltre la up-regulation di un’altra proteina anti-apoptotica, la Mcl-1, si correla
con l’espressione di COX-2 nel carcinoma basocellulare.
32
3. Cicloossigenasi (COX)
Le COX sono enzimi che catalizzano la conversione dell’acido arachidonico a
prostaglandine biologicamente attive e trombossani, sostanze dotate di
numerose funzioni biologiche: citoprotezione del tratto gastrointestinale,
funzionalità piastrinica, omeostasi renale, funzionalità uterina, impianto
dell’embrione e travaglio del parto, regolazione del ciclo sonno-veglia e della
temperatura corporea. Nei siti in cui insorge uno stato infiammatorio, le COX
(Fig.13) danno origine a prostaglandine responsabili dell’effetto flogogeno; il
blocco della loro biosintesi da parte dei FANS determina scomparsa o
riduzione di tale evento indesiderato. L’inibizione della produzione di
prostaglandine al di fuori dei siti infiammatori può essere clinicamente utile,
ad esempio, per prevenire eventi cardiovascolari indesiderati. L’inibizione
può, tuttavia, dimostrarsi nociva quando la ridotta sintesi di prostaglandine
provoca un deterioramento della normale funzionalità della mucosa
gastrointestinale, con formazione di lesioni più o meno gravi, o di quella
renale.
Figura 13: COX.
33
3.1 COX-1 e COX-2 a confronto
Agli inizi degli anni ‘90 si scoprì l’esistenza di due isoforme dell’enzima
cicloossigenasi: cicloossigenasi-1 (COX-1) e cicloossigenasi-2 (COX-2). Le
due isoforme sono presenti in quantità variabili nei vari distretti
dell’organismo, ove svolgono funzioni biologiche ben distinte l’una dall’altra
La COX-1 è una proteina espressa in modo costitutivo da quasi tutte le
cellule dell’organismo umano comprese le piastrine del sangue, è coinvolta
nella comunicazione intercellulare e nell’omeostasi tissutale; inoltre è
responsabile della produzione delle prostaglandine implicate nella
protezione gastrica, nell’autoregolazione del flusso sanguigno renale e
nell’attivazione del parto;[3]
La COX-2 è una proteina inducibile prodotta da macrofagi, fibroblasti e
cellule endoteliali in seguito a stimoli pro-infiammatori come IL-1, LPS e
fattori di crescita. La produzione di prostanoidi che contribuisce alla
vasodilatazione, all’edema e all’iperalgesia caratteristiche dei processi
infiammatori
è
soprattutto
conseguenza
dell’induzione
locale
dell’espressione della COX-2 nelle cellule infiammatorie;[2]
La COX-3 è una proteina che deriva da un differente splicing dell’mRNA
della COX-1 in cui non viene eliminato l’introne 1. E’ una proteina
enzimatica presente soprattutto a livello del SNC.[3]
L’espressione della COX-1 è regolata in maniera diversa rispetto a quella
della COX-2 (Fig.14). Il gene della COX-1 fa parte degli house-keeping
genes che esprimono per una proteina omodimerica integrata nelle membrane
cellulari. La COX-1 è costituita da 599 amminoacidi e possiede un peso
molecolare di 72 Kda, interviene nella sintesi immediata di prostanoidi che si
verifica entro qualche minuto dalla stimolazione con mobilizzatori del
calcio.[2]
34
Figura 14: COX-1 e COX-2.
La COX-1 è un enzima specifico, metabolizza principalmente l’acido
arachidonico mentre la COX-2 è in grado di accettare un maggior numero di
acidi grassi come substrati.[8]
La COX-2 è codificata da un gene che fa parte degli immediate early genes
cioè di geni che vengono immediatamente espressi in risposta a stimoli
opportuni il cui mRNA è altamente instabile per la presenza di sequenze
AUUUA nella regione 3’ non tradotta.[2] La COX-2 umana è una proteina di
604 amminoacidi, la quale è stata identificata e clonata solo nel 1991.[17]
Contiene vicino all’estremità C-terminale un inserto di 18 amminoacidi che
non sono presenti nella COX-1 mentre tutti i residui essenziali che formano il
canale idrofobico legante il substrato,[13] i siti catalitici e i residui
immediatamente adiacenti a esso sono altamente conservati nelle due
isoforme che presentano infatti un’omologia di struttura proteica primaria di
circa il 63% (Fig.14).[8] Le differenze più rilevanti nella struttura di queste due
isoforme riguarda la sostituzione delle isoleucine in posizione 434 e 523 nella
COX-1 con residui di valine nella COX-2. Il minore ingombro sterico offerto
dalla valina 523 nella COX-2 permette agli inibitori l’accesso ad una cavità
idrofobica adiacente al canale del substrato, accesso che nella COX-1 è
35
inibito dalla maggiore lunghezza della catena laterale dell’isoleucina.[5] La
presenza della tasca idrofobica laterale nella COX-2 aumenta il volume del
sito catalitico di circa il 25% rendendola capace di accogliere molecole
ingombranti come gli inibitori selettivi della COX-2 (COXIB).[2] Un’altra
differenza chiave tra queste due cicloossigenasi è la mutazione dell’His513 in
COX-1 con un’Arg513 in COX-2. Questa sostituzione genera uno specifico
sito di binding per i gruppi sulfonammidici o metilsulfonici che sono presenti
in quasi tutte le molecole che inibiscono selettivamente la COX-2.[8] Entrambi
gli enzimi sono caratterizzata da tre parti fondamentali:
Una sequenza omologa all’Epidermal Growht Factor(EGF)
Una regione che funge da ancoraggio alle membrane
Una regione che contiene i due siti catalitici per l’attività cicloossigenasica
e perossidasica separati dal gruppo prostetico eme.[2]
I FANS bloccano la sintesi dei prostanoidi perché occupano il canale
idrofobico di questi due enzimi impedendo l’interazione con l’acido
arachidonico e la sua trasformazione a PGG2 senza tra l’altro influenzare
l’attività perossidasica dell’enzima.[3] L’inibizione della COX-2 è responsabile
degli effetti terapeutici di questi farmaci mentre l’inibizione della COX-1 è
responsabile delle reazioni avverse a livello del tratto gastrointestinale. Negli
ultimi anni lo scopo della ricerca è stato quello di trovare nuovi farmaci
selettivi per la COX-2 in modo da annullare la tossicità di questa classe di
farmaci.[8]
36
4 Coxib
4.1 Fans e Coxib a confronto
I FANS rappresentano una classe di farmaci estremamente efficaci e
maggiormente prescritti, la cui somministrazione può accompagnarsi
all’insorgenza di eventi avversi. Gli effetti indesiderati dei FANS riguardano
principalmente il tubo gastroenterico e, con minor frequenza, il fegato, il rene
e l’apparato cardiovascolare. Pur mantenendo la stessa efficacia analgesica e
anti-infiammatoria dei FANS tradizionali, gli inibitori selettivi della COX-2
presentano una tollerabilità gastrointestinale nettamente superiore, con
riduzione dell’incidenza di dispepsia, ulcere gastro-duodenali e intestinali e
relative complicanze (emorragia, perforazione e stenosi). Poiché entrambi gli
isoenzimi della COX sono costitutivi a livello renale, la loro inibizione
determina una riduzione di uno o più prostanoidi coinvolti nel mantenimento
dell’integrità funzionale del parenchima. Di conseguenza, gli inibitori selettivi
della COX-2 determinano a livello renale gli stessi effetti indesiderati
(ritenzione idrica, edema e ipertensione) osservabili dopo somministrazione di
FANS non selettivi. Tuttavia, alcuni studi recenti sembrano indicare che
l’incidenza di tali eventi avversi sia significativamente inferiore con gli altri
coxib rispetto al rofecoxib. Lo squilibrio tra prostanoidi ad attività
protrombotica (trombossano) e antitrombotica (prostaciclina) conseguente
all’inibizione della COX-2 ha permesso di ipotizzare un effetto protrombotico
dei coxib. Tuttavia, studi osservazionali pubblicati di recente suggeriscono che
entrambe le classi di FANS (selettivi e non) condividono gli stessi rischi
cardiovascolari: aumento del rischio di infarto del miocardio, di insufficienza
cardiaca congestizia e morte. La mancata evidenziazione dell’effetto
cardioprotettivo del naprossene e la mancata influenza dell’aspirina a basse
dosi sul rischio cardiovascolare dei COX-2 inibitori mette in discussione
l’ipotesi “trombofilica”. Ciò che accomuna maggiormente le due classi di
37
farmaci anti-infiammatori sono gli effetti indesiderati a livello renale:
ritenzione idro-salina, edema periferico e soprattutto ipertensione arteriosa.
Non a caso, tra gli inibitori della COX-2 di prima generazione, il rofecoxib
(che determina un aumento dose-dipendente della pressione sistolica) è stato
sempre associato nei vari studi clinici e di farmacovigilanza ad un maggiore
rischio cardiovascolare. Ogni aumento assoluto del rischio cardiovascolare
con i COXIB sembra comunque essere minimo.
4.2 Inibitori selettivi delle COX-2
Gli inibitori selettivi delle COX-2, conosciuti come COXIB, si possono
definire come inibitori “tempo dipendenti”, poiché inizialmente possiedono un
uguale potenza su entrambe le isoforme COX-1 e COX-2, successivamente la
potenza aumenta selettivamente dopo 10 minuti di incubazione. La rimozione
del farmaco tramite dialisi ripristina l’attività delle COX-1, ma non delle
COX-2, suggerendo l’istaurarsi di un cambiamento irreversibile nei confronti
della COX-2. I primi due composti che hanno mostrato un’attività inibitoria
nei confronti della COX-2 sono stati DUP 697 e NS-398, i quali furono
utilizzati come composti lead per lo sviluppo delle due classi chimiche dei
COXIB:
1. ARILSULFONAMMIDI: NS-398, nimesulide
2. DIARILETEROCICLICI: DuP 697, celecoxib, rofecoxib, valdecoxib,
etoricoxib, parecoxib, SC-560.
1. Le arilsulfonammidi furono sviluppate dal prototipo NS-398 e possono
essere considerate degli inibitori preferenziali della COX-2. A questo gruppo
appartiene anche la Nimesulide, una sulfanilide in grado di inibire entrambe le
ciclossigenasi ma in misura preferenziale la COX-2. E’ stata immessa in
commercio nel 1985 con il nome commerciale di Aulin e svolge un’ attività
analgesica e antipiretica. Il suo utilizzo nel tempo ha destato notevoli
38
preoccupazioni tanto da indurre diversi paesi a ritirare dal commercio tutte le
forme sistemiche di questo farmaco per l’elevato rischio di tossicità epatica.[29]
NIMESULIDE
NS-398
2. I diarileterocicli derivano dall’Indoxolo (Tab.1).
INDOXOLO
Studi relazione struttura-attività (SAR) hanno evidenziato che:
-l’anello eterociclico è importante per incrementare l’attività farmacologica;
-i due gruppi arilici devono essere in posizione 1,2 dell’eterociclo in maniera
tale da poter interagire con l’enzima.
39
Y
X
ETEROCICLO
DUP 697
SC-57666
SC-58125
CELECOXIB
ROFECOXIB
H
Tabella 1: Derivati diarileterociclici.
Il DUP 697 è un agente anti-infiammatorio in grado di inibire la COX-2 con
una selettività 50 volte superiore rispetto alla COX-1. Essenziali per la sua
attività selettiva sembrano essere l’anello centrale che interagisce con la tasca
idrofobica dell’enzima e il gruppo SO2CH3. Studi meccanicistici suggeriscono
che il DUP 697 interagisca reversibilmente con la COX-1 e irreversibilmente
40
con la COX-2. In vitro ha evidenziato un’efficacia anti-infiammatoria,
analgesica e antipiretica paragonabile a quella dell’Indometacina, Piroxicam e
Sulindac. Il DUP-697 è somministrabile per via os, presenta una
biodisponibilità elevata (80%) e scarso metabolismo epatico. L'uso di tale
composto è per ora esclusivamente sperimentale .
Il Celecoxib è stato il primo FANS immesso sul mercato come inibitore
selettivo della COX-2. Il Celecoxib presenta un’affinità per la COX-2 circa
375 volte maggiore che per la COX-1. La molecola dopo somministrazione
orale è ben assorbita dal tratto gastrointestinale e raggiunge la concentrazione
plasmatica massima dopo circa 3 h dall’assunzione. La contemporanea
assunzione di cibo, specialmente un pasto ricco di grassi, ne ritarda
l’assorbimento di circa 1 h. Il legame con le proteine plasmatiche è pari al
97%. Il farmaco presenta un'emivita di circa 8-12 h. Nell'organismo Celecoxib
è metabolizzato principalmente a livello epatico ad opera del CYP2C9
portando a metaboliti farmacologicamente inattivi. Il Celecoxib è indicato per
il sollievo dei sintomi dell’osteoartrite e dell’artrite rumatoide, e può essere un
coadiuvante nella terapia mirata a ridurre il numero di polipi nella poliposi
adenomatosa colon-rettale familiare. A differenza dell’aspirina, il Celecoxib
non
ha
attività
anti-aggregante
piastrinica,
ma
la
contemporanea
somministrazione di aspirina e Celecoxib può aumentare l’incidenza di effetti
collaterali gastrointestinali.[8]
Il Rofecoxib è stato il secondo inibitore selettivo della COX-2 immesso sul
mercato. Rofecoxib in seguito a somministrazione orale è ben assorbito con
picchi plasmatici generalmente ottenibili 2-3 h dopo l’assunzione. La
biodisponibilità media in seguito a somministrazione di una singola dose è del
93%.
Nell'organismo
Rofecoxib
viene
ampiamente
metabolizzato,
prevalentemente a livello epatico attraverso processi di riduzione e di
ossidazione che vedono il coinvolgimento degli enzimi del citocromo P450.
41
Solo l'1% circa di una singola dose somministrata viene ritrovata immodificata
nelle urine. Nell’uomo sono stati identificati 6 principali metaboliti,
caratterizzati da una debole o da nessuna attività misurabile come inibitori
della COX-2. Il 30 settembre 2004, Merck lo ritirò volontariamente dal
mercato a causa del possibile aumento del rischio di infarto del miocardio e
ictus associato ad un suo uso a lungo termine e ad alto dosaggio.[38]
L’Etoricoxib è un inibitore selettivo della COX-2 sviluppato per il trattamento
del dolore post-chirurgico dentale e dell’osteoartrite. Dopo somministrazione
per via orale viene ben assorbito dal tratto gastrointestinale con una
biodisponibilità
del
80-100%.
Dopo
singola
somministrazione
la
concentrazione plasmatica massima è pari a 3,6 mg/ml e viene raggiunta a
circa 1 h dall'assunzione. L’assunzione di cibo rallenta la velocità ma non
l’entità
dell’assorbimento.
Il
legame
con
le
proteine
plasmatiche,
principalmente con l'albumina, è di circa il 92%. Etoricoxib è ampiamente
metabolizzato dall'organismo umano a livello epatico, in particolare grazie al
coinvolgimento dell'isoenzima CYP3A4 che ossida il gruppo metilico in 6’.
Nell’uomo sono stati identificati 5 metaboliti, i quali risultano inattivi o
mostrano solo una debole attività come inibitori della COX-2. L'emivita della
molecola è di 22 h. La maggior parte del farmaco viene eliminato sotto forma
di metaboliti, e meno del 2% è stato escreto come farmaco immodificato.[8]
Il Valdecoxib è un inibitore selettivo della COX-2 usato nel trattamento del
dolore in soggetti affetti da osteoartriti, artrite reumatoide, e nel trattamento
del dolore da dismenorrea primaria. Il farmaco è stato disponibile sul mercato
americano dal 2001 al 2005, quando è stato ritirato per un possibile aumento
del rischio di infarto del miocardio e ictus. In Italia ed Europa la molecola era
venduta dalla società farmaceutica Pharmacia-Pfizer con il nome commerciale
di Bextra, nella forma farmaceutica di compresse rivestite contenenti 20 mg di
principio attivo. Nell'aprile 2005 l'Agenzia Italiana del Farmaco dispose il
42
ritiro dal commercio del farmaco adottando il provvedimento a seguito della
decisione autonoma della casa farmaceutica Pfizer, "a scopo cautelativo" ed in
attesa della conclusione della procedura di rivalutazione dei profili di
sicurezza di tutta la classe degli inibitori della COX-2 in corso da parte di
EMEA. Valdecoxib dopo somministrazione per via orale è rapidamente
assorbito dal tratto gastrointestinale. La concentrazione plasmatica massima
viene raggiunta entro 3 h. La biodisponibilità assoluta del farmaco a seguito di
somministrazione orale si aggira intorno all’83%. L'assunzione del farmaco
con un pasto ricco di grassi non modifica significativamente il picco di
concentrazione plasmatica e neppure il grado di assorbimento della molecola;
il tempo necessario per raggiungere il picco plasmatico risulta ritardato solo di
un paio d'ore. Il farmaco viene metabolizzato principalmente dal CYP2C9 per
ossidrilazione del gruppo metilico in posizione 5’, che viene poi metabolizzato
a carbossilato inattivo. L’emivita è di 8-11 h e circa il 70% del farmaco
metabolizzato viene eliminato per via urinaria mentre meno del 5% viene
escreto immodificato nelle feci e nelle urine. Il Valdecoxib può causare bronco
costrizione o reazioni anafilattiche in pazienti asmatici sensibili all’aspirina o
ad altri FANS, per questo i pazienti devono essere adeguatamente
monitorati.[8]
Il Parecoxib è un profarmaco iniettabile e solubile in acqua che è
metabolizzato in vivo a Valdecoxib. Viene usato in ambito peri-operatorio
quando i pazienti non sono in grado di assumere farmaci per os per il
trattamento del dolore. È disponibile sul mercato europeo ma non negli Stati
Uniti in quanto la Food and Drug Administration, nel 2005, non ne ha
approvato l'immissione in commercio. In Italia è venduto con il nome
commerciale di Dynastat 20, nella forma farmaceutica di polvere per
soluzione iniettabile per via endovenosa oppure intramuscolare. Dopo
somministrazione per via parenterale (endovenosa o intramuscolare),
Parecoxib viene rapidamente trasformato in Valdecoxib, la molecola
43
farmacologicamente attiva, grazie ad un processo di idrolisi enzimatica a
livello epatico. La concentrazione plasmatica massima di Valdecoxib viene
raggiunta in circa mezz’ora (dopo somministrazione endovenosa) e 1 h dopo
somministrazione per via intramuscolare. Il legame con le proteine
plasmatiche si aggira intorno al 98%. L’eliminazione di Valdecoxib
dall'organismo è conseguente ad un importante metabolismo epatico che vede
coinvolto il citocromo P450 e gli isoenzimi CYP3A4 e CYP2C9. Circa il 70%
di una singola dose somministrata è escreto nelle urine in forma di metaboliti
inattivi. Il Parecoxib viene utilizzato nel trattamento a breve termine del dolore
acuto secondario ad interventi chirurgici, sia di chirurgia generale che
ginecologica, ortopedica, maxillo-facciale e cardiochirurgia.[36]
44
5 Sintesi di nuovi derivati a struttura benzotieno[3,2-d]
pirimidin sulfonammidica
5.1 Introduzione generale
I FANS tradizionali, non selettivi, inibiscono sia la COX-1 che la COX-2 ed a
causa di questa marcata selettività verso entrambe le isoforme il loro utilizzo è
spesso compromesso da gravi effetti collaterali a livello del tratto
gastrointestinale.[10,11] Pertanto, sono state ideate nuove strategie di sintesi
basate sulla progettazione di nuovi sistemi eterociclici contenenti un gruppo
funzionale tienopirimidinico capace di inibire selettivamente la COX-2, in
maniera tale da aggirare sia la tossicità gastrica che quella renale.[14] La scelta
di tale sistema eterociclico trova le sue fondamenta in approfondite ricerche di
letteratura riguardanti i vari meccanismi d’azione e gli effetti biologici dei
composti tienopirimidici, i quali hanno suscitato, negli ultimi anni, un grande
interesse nel campo farmacologico.[21] Non a caso, i derivati tienopirimidinici
vengono definiti come delle potenziali molecole bioattive paragonabili
strutturalmente agli analoghi delle purine biogeniche, antimetaboliti dell’acido
nucleico.[22] Negli ultimi due decenni è emerso che molti composti eterociclici
contenenti nel loro scheletro un gruppo tienopirimidinico possono esplicare un
ampio range di attività biologiche quali: anti-infiammatoria, antivirale,
analgesica, antimicotica, antibatterica e ultimamente anche una spiccata azione
antitumorale,[23-25] la quale è stata valutata in vitro su ben tre linee cellulari
tumorali umane, MCF-7 (adenocarcinoma del seno), NCI-H460 (cancro delle
cellule polmonari) e SF-268 (cancro del SNC).[25]
5.2 Progettazione razionale
Approfondite ricerche di letteratura riguardanti gli inibitori selettivi delle
COX-2, affermano la presenza, nel modello farmacoforico, di quattro elementi
strutturali comuni:
45
1) un anello eterociclico o carbociclico in grado di incrementare l’attività
anti-infiammatoria. Studi preliminari inerenti le SAR asseriscono che
l’anello centrale carbociclico riesce a stabilizzare la conformazione della
molecola all’interno della tasca idrofobica delle COX-2;
2) un gruppo sulfonammidico o metilsulfonico in posizione para all’anello
benzenico, il cui ruolo è quello di interagire irreversibilmente con i residui
amminoacidici Arg513 o Arg120 presenti nella tasca laterale idrofobica delle
COX-2;
3) atomi di alogeno o vari sostituenti legati all’anello benzenico. Studi SAR
hanno dimostrato che la selettività e la potenza inibitoria nei confronti delle
COX-2 è strettamente correlata dalla natura del sostituente presente
sull’anello benzenico. L’ordine con cui si attribuisce la maggiore selettività
è il seguente: OH > F > OMe > H > Me > NHCOMe > Cl. Pertanto
derivati che presentano gruppi OH legati in posizione para nell’anello
aromatico esibiscono sia un’ottima attività anti-infiammatoria che una
maggiore selettività verso l’isoforma COX-2;
4) gruppi elettron-attrattori sembrano giocare un ruolo chiave per quanto
concerne la selettività e l’attività anti-infiammatoria. Studi di molecular
docking dimostrano che i gruppi elettron-attrattori interagiscono tramite un
forte legame idrogeno con l’amminoacido Tyr355.
Le modifiche strutturali effettuate sugli inibitori selettivi delle COX-2
rappresentano un’ interessante strategia per la sintesi di nuovi composti. Infatti
l’introduzione di una base pirimidinica, di un gruppo carbossilico o
metossilico sull’anello benzenico hanno generato dei prototipi di molecole
dalle spiccate attività biologiche. Inoltre un’attenta analisi del sito attivo della
COX-2, il quale si presenta più grande del 60% rispetto a quello della COX-1,
ha generato l’ipotesi che l’introduzione di vincoli sterici nello scaffold dei
coxib potrebbe aumentare la selettività nei confronti dell’enzima stesso
(Fig.15).
46
Figura 15: COX-2 e coxib
In base a tali considerazioni abbiamo progettato una nuova serie di composti
benzotienopirimidinici prendendo come riferimento i seguenti composti:
celecoxib, nimesulide, flosulide, L-745, 337 e NS398 (schema 1) i quali
presentano una spiccata attività anti-infiammatoria accompagnata da minori
effetti gastrolesivi.
Schema 1: Strutture di alcuni coxib e progettazione razionale
47
Pertanto abbiamo sintetizzato delle molecole “ibride” contenenti un anello
pirimidinonico legato ad un benzene o ad un anello eterociclico come il
tiofene o il benzotiofene, con l’atomo di zolfo legato, come tioetere, ad un
gruppo alchilico o arilico con gruppi elettron-attrattori in orto e/o para. La
scelta della struttura benzotienopirimidinica come scaffold comune, è sorta da
attenti studi riguardanti l’ampio range di proprietà biologiche che possiede.
Per cercare di aumentare le interazioni con l’enzima COX-2, abbiamo pensato
di implementare la struttura molecolare introducendo sostituenti arilici o
eterociclici sullo zolfo (Schema 2).
Schema 2: Modello farmacoforico
Sono stati selezionati sostituenti con diverse caratteristiche chimico-fisiche per
poter sondare il sito di legame all’interno delle COX-2. Tra questi abbiamo
introdotto molecole eterocicliche biologicamente attive (antipirina e 1,3
dimetiluracile) in grado di esplicare interessanti attività anti-infiammatorie
all’interno del sistema triciclico.
5.3 Scopo del lavoro
L’attività di ricerca è stata incentrata sulla progettazione e sintesi di una nuova
serie
di
composti
benzo-tio-derivati
con
struttura
tienopirimidinica
sulfonammidica (Fig.16), potenziali inibitori selettivi della COX-2, in grado di
48
esplicare un’efficace azione anti-infiammatoria e allo stesso tempo di ridurre
l’insorgenza di gravi effetti collaterali a livello gastrointestinale. L’obiettivo di
questo studio è stato quello di ottimizzare la sintesi di questi derivati
introducendo un caratteristico gruppo SO2Me o SO2NH2 in maniera tale da
poter interagire irreversibilmente con i residui amminoacidici (Arg513 o Arg120)
presenti nella tasca laterale idrofobica delle COX-2.[33] Queste molecole
tricicliche, così articolate, giocano un ruolo fondamentale sulla selettività della
COX-2.[25,26] Il metile 3-isotiocianato-[1]-benzotiofene-2-carbossilato 2,
composto chiave nella sintesi dei derivati benzotienopirimidinici, è stato
ottenuto tramite una semplice metodica facendo reagire il metile 3aminobenzo[b]tiofene-2-carbossilato 1 con il di-2 piridil-tionocarbonato
(DPT) in CH2Cl2 a temperatura ambiente per 24 h. Il DPT è stato utilizzato in
sostituzione del tiofosgene, un agente altamente tossico per le vie aeree
(schema 3); dati di letteratura asseriscono che le proprietà tossiche del
tiofosgene derivano dalla capacità di trasformarsi per riscaldamento in
fosgene, il quale è in grado di combinarsi con l’acqua contenuta nei tessuti del
tratto respiratorio decomponendosi in CO e HCl, quest’ultimo è in grado di
dissolvere le membrane delle cellule esposte causando il riempimento delle vie
respiratorie di liquido pleurico. Il fosgene è un veleno particolarmente
insidioso, non provoca effetti immediati, infatti, i sintomi si manifestano dopo
circa 72 h dall'esposizione e la morte sopraggiunge per combinazione di
emorragie interne con insufficienza respiratoria. A differenza di altri gas, il
fosgene non viene assorbito attraverso la pelle ma il suo effetto si produce solo
per inalazione. A causa dei suoi effetti letali il fosgene è stato utilizzato come
arma chimica durante la prima guerra mondiale, di conseguenza, possiamo
affermare che i vantaggi dell’uso del DPT in questa reazione sono molteplici:
Semplice metodica (green chemistry) senza l’utilizzo di solventi dry e di
condizioni di reazione drastiche;
il DPT è stabile in condizioni ambientali;
alte rese di reazione.
49
Figura 16: Nuovi tioaril derivati a struttura benzotieno[3,2-d] pirimidin
sulfonammidica
50
La presenza del gruppo –NCS nel composto 2, (il cui atomo di carbonio
centrale è fortemente elettrofilo) è stata di fondamentale aiuto per ottenere in
maniera più agevole i prodotti intermedi: le benzotiosemicarbazidi 3a e 3b. La
reazione è stata condotta a temperatura ambiente in CH2Cl2 facendo reagire il
composto 2 rispettivamente con NH2NH2 e NH2SO2CH3. Successivamente la
ciclizzazione dei derivati 3a e 3b, realizzata con il metodo di Wamhoff
[30,31]
,
ha portato alla formazione dei due derivati: benzo-tiossoammino 4a (resa
60%) e benzo-tiosso-metansulfonammide 4b (resa 80%) (schema 3).
Schema 3: Sintesi degli intermedi benzo-tiossoammino 4a e benzo-tiossometansulfonammide 4b.
I derivati 4a e 4b, isolati mediante tecniche cristallografiche, sono stati
caratterizzati attraverso spettri IR, 1H e 13C NMR e spettrometria di massa. In
51
particolare, lo spettro 1H NMR del composto 4a presenta il singoletto relativo
al gruppo amminico a 6.3 ppm mentre nello spettro 1H NMR del composto 4b
sono evidenti un caratteristico singoletto relativo al protone del gruppo
sulfonammidico a 11 ppm e i multipletti relativi ai protoni aromatici
nell’intervallo tra 7.5-8.8 ppm a conferma della struttura.
Numerosi studi sono stati dedicati alla formazione del legame C-S e all’
interpretazione delle relazioni struttura-attività dei derivati benzo-tieno [3,2-d]
pirimidinici. La formazione del legame C-S rappresenta un passo
fondamentale per la sintesi di nuove molecole implicate nel trattamento non
solo di malattie neurodegenerative come l’Alzheimer e il Parkinson ma anche
per la cura dei tumori. Il protocollo di reazione per la formazione del legame
C-S prevede l’utilizzo di quantità catalitiche di Cu in polvere (~5-10 mol %),
di un ligando (10-20 mol %) e di una base (1.5-2.5 equiv.) in condizioni
relativamente blande (80-100 °C per 5-6 h).
L’ influenza che il catalizzatore di rame CuI può esercitare sulla formazione
del legame C-S è ben documentata in letteratura.[44] Lo ioduro di rame, infatti,
ha dimostrato di avere un’ottima stabilità a contatto con l’aria mentre l’utilizzo
di altri catalizzatori (CuBr2, CuSO4·5H2O, CuCl2) hanno rivelato una minore
efficienza catalitica con rendimenti del 25-30%. Ulteriori esperimenti sono
stati effettuati per trovare la temperatura e il tempo di reazione ottimale. Un
eccellente rendimento ~ 90% è stato ottenuto dopo 6 h e a 100°C. Dai dati
riportati in letteratura è emerso che:
la reazione non ha avuto alcun esito positivo utilizzando altri catalizzatori
che non fossero CuI e Cu in polvere, addirittura con il sale CuCl2 non si è
riscontrata alcuna reazione;
la reazione non ha avuto alcun esito positivo in assenza di quantità
catalitiche di CuI e Cu in polvere;
è importante utilizzare un solvente polare, infatti l’utilizzo di solventi come
il toluene dimezzano la resa di reazione;
52
è interessante notare l’alta resa di reazione che si ottiene (~90%)
utilizzando l’H2O come solvente e l’NaOH come base;
per ottenere una resa ottimale bisogna utilizzare una temperatura di ~
100°C, la diminuzione della stessa anche di solo 15°C ha causato una
significativa diminuzione della resa;
diversi alogenuri arilici sono stati saggiati dimostrando che fluoruri, cloruri
e bromuri arilici hanno mostrato una scarsa reattività in questa reazione,
contrariamente gli ioduri arilici ed eterociclici hanno presentato un’alta
reattività dando luogo alla formazione di derivati benzo pirimidinici con
elevate rese (~ 90%).
Pertanto, le reazioni per ottenere i derivati finali (5-15) sono state condotte
facendo reagire in ambiente basico gli appropriati ioduri arilici e eterociclici
con il derivato 4b in presenza di un opportuno sistema catalitico formato da
Cu in polvere e CuI alla temperatura di ~100°C per 6 h. Nel caso della sintesi
del derivato 13, l’utilizzo di CuI non ha prodotto il risultato sperato; pertanto è
stata tentata una via alternativa trattando il composto 4b con lo ioduro di
cicloesile in DMF a 80 °C per 6 h, in presenza di K2CO3 (schema 4). Dopo
aver effettuato una filtrazione a caldo per eliminare il sistema catalitico
utilizzato ed eventuali impurezze formatesi nel corso della reazione, abbiamo
isolato i derivati in ambiente acido. I derivati (5-15) sono stati ottenuti allo
stato puro mediante cristallizzazione.
Le strutture dei composti isolati sono state confermate attraverso analisi
elementare, spettri IR, 1H e 13C NMR e spettrometria di massa. In particolare
gli spettri
1
H NMR dei derivati (5-15) hanno evidenziato accanto al
caratteristico singoletto relativo al protone del gruppo sulfonammidico a ~ 11
ppm, i multipletti relativi ai protoni aromatici nell’intervallo 7.3-8.3ppm a
conferma della struttura benzotieno sulfonammidica.
53
Schema 4: Sintesi di nuovi derivati benzo-tieno [3, 2-d] pirimidinici.
La fase successiva è stata quella di selezionare i vari ioduri eterociclici da
legare al derivato 4b, tenendo in considerazione dell’influenza che potevano
esercitare una volta che fossero stati incorporati nel sistema triciclico dal
punto di vista farmacologico. A tale scopo, non a caso abbiamo analizzato, in
primis, le caratteristiche dell’antipirina, un analgesico/antipiretico. La sua
azione analgesica è meno intensa di quella della morfina e delle sostanze
morfino-simili, a differenza delle quali, tuttavia, non si manifesta con effetti
collaterali di tipo euforizzante o narcotico. Il meccanismo d'azione antipiretica
interessa i centri nervosi termoregolatori che garantiscono il regolare rapporto
tra la produzione e la dispersione del calore nell'organismo. Tale rapporto,
alterato durante gli stati febbrili, viene ripristinato dall'antipirina grazie
54
all'attivazione centrale di meccanismi (sudorazione, vasodilatazione cutanea)
che compensano a livello periferico il patologico aumento della termogenesi.
L'azione
antireumatica
dell'antipirina
deriva
da
un
aumento
della
concentrazione ematica di idrocortisone attivo. Il farmaco scinde, infatti, tale
corticosteroide dal legame inattivante che esso contrae nel sangue con una
particolare
proteina
plasmatica,
la
transcortina.
L'antipirina
assieme
all’amminofenazone è uno dei farmaci di più comune e vasto impiego, anche
se non privo di effetti collaterali e di alcuni effetti tossici. Con una certa
frequenza determina reazioni di tipo allergico ed episodi di intolleranza
gastrica, più raramente può provocare leucopenia e agranulocitosi. È inoltre
impiegato per via orale e con minore frequenza per via parenterale sotto forma
di sale idrosolubile (per esempio come N-metan-sulfonato) oppure associato al
fenilbutazone. Come antipiretico si usa nelle malattie infettive quali influenza,
scarlattina, reumatismi articolari mentre come analgesico è un forte sedativo
nelle nevralgie e nelle cefalee. L’introduzione dell’antipirina nello scheletro
triciclico benzotieno-pirimidinico, non a caso, ha creato un prototipo di
molecola
con
spiccate
proprietà
analgesiche-anti-infiammatorie.[37]
L’introduzione del gruppo 1,3 dimetiluracile al nostro sistema triciclico ha
avuto origine da un attento studio delle proprietà farmacologiche dell’uracile e
dei suoi derivati. Infatti, il 5–fluoro uracile (5-FU) ha rappresentato, per molti
decenni, la pietra miliare per il trattamento dei tumori colon-rettali. Recenti
studi di letteratura hanno evidenziato anche una spiccata azione antiinfiammatoria di questi composti e in particolare si è evinto che l’introduzione
di 2 gruppi metilici in posizione 1 e 3 all’uracile abbiano aumentato
notevolmente l’attività. Per quanto concerne la scelta dei vari sostituenti
funzionali R1 e R2 all’interno degli ioduri arilici, ci siamo basati su dati
riportati in letteratura secondo i quali i sostituenti anionici, gruppi nitro e la
presenza di alogeni possono interagire con le membrane cellulari e migliorare
l’attività e la potenza anti-infiammatoria del sistema eterociclico.[38] Infatti i
composti n° 12,14,15 che presentano, rispettivamente, un gruppo carbossilico
55
in posizione para, alogeni in orto e para e un gruppo nitro in orto e para
hanno mostrato una capacità inibitoria dei markers anti-infiammatori
paragonabile a quella del Celecoxib. La sintesi di questi derivati (5-15) a
scheletro benzo-tieno [3, 2-d] pirimidinico potrebbero costituire un nuovo
target di rilievo in quanto analoghe strutture sono presenti in composti già
brevettati con proprietà anticoagulanti, antidiabetiche, antistaminiche, antiinfiammatorie e antitumorali.
56
Sezione Sperimentale
5.4 Materiali e metodi
Gli spettri 1H NMR e
13
C NMR sono stati registrati su uno spettrometro
Varian Inova, operante ad una frequenza di 200 o 500 MHz, per gli spettri
protonici, ed a 50 o 125 MHz per gli spettri 13C NMR (Varian, Leini, Italia) e
sono stati eseguiti come soluzioni in dimetilsolfossido deuterato (DMSO). I
dati sono stati riportati come spostamenti chimici o “chemical shifts”, i quali
sono stati indicati con “δ” e misurati in parti per milione (ppm). Tutte le
reazioni sono state monitorate mediante cromatografia su strato sottile (TLC),
la quale è stata eseguita utilizzando lastre di gel di silice dello spessore di 0.2
mm, (Silical gel 60 F254, Merck) su fogli di alluminio. Tali lastre sono state
visualizzate per mezzo di una lampada UV (λ= 254 nm). La purificazione dei
prodotti sintetizzati è stata eseguita tramite cromatografia flash usando
opportune colonne impaccate con gel di silice Merck 60, 230-400 mesh. I
punti di fusione sono stati determinati mediante uno strumento Electrothermal
modello 9100 e sono non corretti. Gli spettri di massa sono stati registrati su
uno Spettrometro di Massa Perkin Elmer Turbo Mass Clarus 560 con una
energia di ionizzazione di 70eV a 250°C ed una corrente di 90μA. Gli spettri
infrarosso (IR) sono stati registrati usando uno spettrofotometro Perkin Elmer
Spectrum RX I FT-IR System. Sono stati eseguiti sia in soluzioni di
diclorometano (CH2Cl2) sia in dischi di KBr. Tutti i prodotti chimici
commerciali sono stati acquistati da Aldrich, Fluka, Merk, Lancaster e Carlo
Erba e sono stati utilizzati senza ulteriore purificazione.
57
Sintesi del Metile 3-isotiocianato-[1]-benzotiofene-2-carbossilato (2)
Una
soluzione
contenente
(3.0
g,
14.4
mmol)
di
metile
3-
aminobenzo[b]tiofene-2-carbossilato 1 in CH2Cl2 (40 mL) è stata aggiunta
goccia a goccia a temperatura ambiente ad una soluzione di di-2-piridil
tionocarbonato (DPT) (3.3 g, 14.4 mmol) in CH2Cl2 (40 mL). La miscela di
reazione è stata lasciata sotto agitazione a temperatura ambiente per 24 h.
Trascorso questo tempo il solvente è stato eliminato a pressione ridotta ed il
residuo ottenuto è stato solubilizzato in CH3COCH3; l’aggiunta di acqua
distillata ha dato origine alla formazione di un precipitato, il quale è stato
raccolto, lavato con H2O dist., asciugato sotto pressione e trattato secondo
Kienzle[23] per dare il composto 2 sotto forma di cristalli gialli.
Metile 3-isotiocianato-[1]-benzotiofene-2-carbossilato
resa: 90%; cristalli gialli; pf: 120 °C; IR (KBr): ν = 2140, 2090, 1710 cm−1;
1
H NMR (500 MHz, DMSO): δ = 3.99 (s, 3H, CH3), 7.40 – 8.15 (m, 4H, Ar-
H);
13
CNMR (125 MHz, DMSO): δ = 14.29, 59.78, 100.24, 128.37, 129.88,
134.78, 163.30, 164.28 ppm; HRMS m/z per C11H7NO2S2 [M]+: 248.9.
58
Sintesi del Metile 3-({[2(metilsulfonil)idrazino]carbotionil}ammino)1benzotiofene-2-carbossilato (3b)
1.1 g (4.1 mmol) di Metile 3-isotiocianato-[1]-benzotiofene-2-carbossilato 2
sono stati aggiunti goccia a goccia ad una soluzione in CH2Cl2 (20 mL) di
NH2NHSO2CH3 (0.48 g, 98%, 4.9 mmol). La miscela di reazione è stata
lasciata sotto agitazione a temperatura ambiente. Dopo 2 h. il precipitato
ottenuto è stato raccolto, lavato in CH2Cl2 e cristallizzato in EtOH per dare il
composto 3b sotto forma di cristalli bianchi.
Metile 3-({[2(metilsulfonil)idrazino]carbotionil}ammino)1-benzotiofene-2carbossilato
resa: 77%; cristalli bianchi; pf: 223 °C dec.; IR (KBr): ν = 3294, 3243, 1657,
1324, e 1146 cm−1; 1H NMR (500 MHz, DMSO): δ = 1.26 (t, J = 7.0Hz, 3H,
CH3), 3.08 (s, 3H, CH3), 7.40 – 8.15 (m, 4H, Ar-H), 9.95, 10.67, 12.55 (s, 1H,
NH); 13C NMR (125 MHz, DMSO): δ = 14.02, 40.92, 60.77, 125.35, 134.67,
140.34, 150.29, 163.34, 177.99 ppm; HRMS m/z per C12H13N3O4S3 [M]+:
359.0.
59
Sintesi del 3-Ammino-4-osso-3,4-diidrobenzo[4,5]tieno[3,2-d]pirimidinico-2tiolato di sodio (4)
Una soluzione in CH2Cl2 (30 ml) contenente 2.5 g (10.0 mmol) di metile 3isotiocianato-[1]-benzotiofene-2-carbossilato 2 (2.5 g, 10.0 mmol) è stata
aggiunta goccia a goccia a temperatura ambiente ad una soluzione di NH2NH2
(0.5 mL, 10.0 mmol) in CH2Cl2 (50 mL). La miscela di reazione è stata
lasciata sotto agitazione per 2 h. Il precipitato ottenuto (miscela di più derivati
tra cui il composto 3a non separabile cromatograficamente) è stato filtrato,
raccolto, lavato con CH2Cl2 e lasciato asciugare fino ad ottenere una polvere
bianca (2.3 g). Quest’ultima successivamente è stata riscaldata per 1 h a
ricadere in una soluzione 0.1 M di NaOH (0.53 g, 9.5 mmol) in EtOH (190
mL). Il precipitato ottenuto è stato raccolto, lavato a caldo con diossano e
lasciato asciugare per dare il composto 4 sotto forma di polvere bianca.
3-Ammino-4-osso-3,4-diidrobenzo[4,5]tieno[3,2-d]pirimidinico-2-tiolato
di
sodio
resa: 67%; polvere bianca; pf: > 310 °C dec.; IR (KBr): ν = 3230, 3135, 1640
cm−1; 1H NMR (500 MHz, DMSO): δ = 6.33 (s, 2H, NH2), 7.40 – 8.15 (m, 4H,
Ar-H);
13
C NMR (125 MHz, DMSO): δ = 115.32, 129.45, 130.90, 154.87,
161.85, 168.80 ppm.
60
Sintesi del 3-Ammino-2-tiosso-[1]benzotieno[3,2-d]pirimidino-4(1H)-one (4a)
Il
composto
4
(3-ammino-4-osso-3,4-diidrobenzo[4,5]tieno[3,2-
d]pirimidinico-2-tiolate di sodio) (1.98 g, 7.0 mmol) è stato solubilizzato in
200 mL di H2O dist., la soluzione ottenuta è stata acidificata con HCl conc.
fino a raggiungere un pH ~ 3 – 4; si ottiene un precipitato che è stato raccolto,
lavato con H2O dist., essiccato e cristallizzato con diossano per dare il derivato
ammino-tiosso 4a sotto forma di polvere bianca.
3-Ammino-2-tiosso-[1]benzotieno[3,2-d]pirimidino-4(1H)-one
resa: 60%; polvere bianca; pf: > 280 °C; IR (KBr): ν = 3230, 3135, 1640 cm−1;
1
H NMR (500 MHz, DMSO): δ = 6.33 (s, 2H, NH2), 7.40 – 8.15 (m, 4H, Ar-
H); 13C NMR (125 MHz, DMSO): δ = 115.32, 129.45, 130.90, 154.87, 161.85,
168.80 ppm.
61
Sintesi del N-(4-osso-2-tiosso-1,4-diidro[1]benzotieno[3,2-d]pirimidin3(2H)il-metanosulfonammide (4b)
Una soluzione di mesil-tiosemicarbazide (1.0 g, 2.65 mmol) e NaOH (0.24 g,
6.0 mmol) in H2O dist. (40 mL) è stata lasciata a ricadere per 3 h. Trascorso
questo tempo la soluzione contenente sale sodico è stata filtrata, raffreddata e
acidificata con HCl conc. fino a pH 3 – 4; il precipitato ottenuto è stato
raccolto, lavato con H2O, cristallizzato in HCON(CH3)2/H2O per dare il
composto 4b sotto forma di cristalli bianchi.
N-(4-osso-2-tiosso-1,4-diidro[1]benzotieno[3,2-d]pirimidin-3(2H)ilmetanosulfonammide
resa: 80%; cristalli bianchi; pf: 270 °C dec.; IR (KBr): ν = 3214, 1708, 1345,
1158cm−1; 1H NMR (500 MHz, DMSO): δ = 3.26 (s, 3H, CH3), 7.56 – 8.87
(m, 4H, Ar-H), 11 (s, 1H, NH);
13
C NMR(125 MHz, DMSO): δ = 44.51,
115.28, 129.38, 131.10, 149.00, 155.50, 174.53 ppm; HRMS m/z per
C11H9N3O3S3 [M]+: 326.8.
62
Sintesi dei derivati benzotienopirimidinici (5-12, 14, 15).
Procedura generale:
Ad una soluzione contenente la benzo-tiosso-metansulfonammide 4b (1 eq.) e
2 eq. di KOH in una miscela 1:1 di EtOH/H2O (in un volume tale da avere una
soluzione circa 8.5 M), sono stati aggiunti gli appropriati ioduri arilici o
eterociclici (1 eq.), Cu in polvere (0.5 eq.) e CuI (0.3 eq.). La sospensione
ottenuta è stata riscaldata a 80 °C a ricadere per 5 h e successivamente filtrata
a caldo. Dopo aver raffreddato a temperatura ambiente la soluzione risultante,
è stata acidificata con HCl conc. fino ad un pH 3 – 4; l’aggiunta dell’acido ha
portato alla formazione di un precipitato, che è stato raccolto, lavato con H2O
distillata, asciugato e cristallizzato con EtOH/H2O per dare i derivati (5-12, 14,
15) sotto forma di solidi cristallini.
Nel caso della sintesi del derivato 13, dal momento che la procedura
precedentemente descritta ha prodotto il composto 13 con bassa resa, è stata
utilizzata una metodica alternativa, facendo reagire 0.180 g (0.55 mmol) del
composto 4b, 0.115 g (0.55 mmol) di iodocicloesile e 0.076 g (0.55mmol) di
K2CO3 in 2 mL di HCON(CH3)2. La sospensione è stata riscaldata a 80 °C a
ricadere per 12 h. Dopo aver raffreddato a temperatura ambiente la miscela di
reazione, è stata acidificata con HCl conc.. Il precipitato ottenuto è stato
filtrato, raccolto e lavato con H2O distillata, essiccato e cristallizzato con etere
di petrolio per dare il composto 13 sotto forma di cristalli bianchi.
63
N-[2-[(4-nitrofenil)tio]-4-osso[1]benzotieno[3,2-d]pirimidin-3(4H) il]
metansulfonammide (5)
resa: 90%; cristalli gialli; pf: 255 °C; IR (KBr): ν = 3310, 1690, 1340, 1150
cm−1; 1H NMR (500 MHz, DMSO): δ = 3.41(s, 3H, CH3), 7.50 – 8.59 (m, 8H,
Ar-H), 11.63 (s, 1H, NH);
13
C NMR (125 MHz, DMSO): δ = 43.83, 119.66,
120.64, 127.34, 130.91, 131.14, 133.96, 138.36, 148.07, 150.80, 155.68,
157.015, 159.53 ppm; HRMS m/z per C17H12N4O5S3 [M]+: 448.9.
64
N-[2-[(2-nitrofenil)tio]-4-osso[1]benzotieno[3,2-d]pirimidin-3(4H) il]
metansulfonammide (6)
resa: 90%; cristalli gialli; pf: 235 °C; IR (KBr): ν = 3310, 1690, 1340, 1150
cm−1; 1H NMR (500 MHz, DMSO): δ = 3.41 (s, 3H, CH3), 7.50 – 8.59 (m, 8H,
Ar-H), 11.63 (s, 1H, NH);
13
C NMR (125 MHz, DMSO): δ = 43.83, 119.66,
120.64, 127.34, 130.91, 131.14, 133.96, 138.36, 148.07, 150.80, 155.68,
157.015, 159.53 ppm; HRMS m/z per C17H12N4O5S3 [M]+: 448.9.
65
N-[2-[(1,3-dimetil-2,4-diosso-1,2,3,4-tetraidropirimidin-5-il)tio]-4osso[1]benzotieno[3,2-d]pirimidin-3(4H)il]metansulfonammide (7)
resa: 90%; cristalli marroni; pf: 275 °C; IR (KBr): ν = 3059, 1690, 1640, 1600,
1351, 1154 cm−1; 1H NMR (500 MHz, DMSO): δ = 3.25 (s, 3H, CH3), 3.39 (s,
3H, CH3), 3.43 (s, 3H, CH3), 7.56–8.42 (m, 5H, Ar-H), 11.58 (s, 1H, NH); 13C
NMR (125 MHz, DMSO): δ = 42.30, 119.15, 125.17, 125.99, 130.83, 131.14,
133.96, 138.67, 148.34, 154.10, 156.64, 156.83, 157.42, 159.33 ppm; HRMS
m/z calcolato per C17H15N5O5S3 [M]+: 465.1.
66
N-[2-{[4-nitro-2-(trifluorometil)fenil]tio}-4-osso[1]benzotieno[3,2d]pirimidin-3(4H)il]metansulfonammide (8)
resa: 80%; cristalli arancioni; pf: 125°C; IR (KBr): ν = 3227, 1700, 1350,
1150cm−1; 1H NMR (500 MHz, DMSO): δ = 3.41 (s, 3H, CH3), 7.52 – 8.46
(m, 7H, Ar-H), 11.69 (s, 1H, NH);
13
C NMR (125 MHz, DMSO): δ = 44.12,
115.12, 125.16, 128.33, 130.01, 132.01, 133.44, 135.01, 135.66, 140.01,
158.22, 163.27, 168.01, 169.79 ppm; HRMS m/z per C18H11F3N4O5S3 [M]+:
516.5.
67
Acido 4-({3-[(metilsulfonil)ammino]-4-osso-3,4-diidro[1]benzotieno[3,2d]pirimidin-2-il}tio)benzoico (9)
resa: 95%; cristalli bianchi; pf: 238 °C; IR (KBr): ν = 3494, 1695, 1350, 1156
cm−1; 1H NMR (500 MHz, DMSO): δ = 3.28 (s, 3H, CH3), 7.21 – 8.05 (m, 8H,
Ar-H), 11.37 (s, 1H, NH), 13.01 (s, 1H, COOH);
13
C NMR (125 MHz,
DMSO): δ = 44.12, 115.12, 125.16, 128.33, 130.01, 132.01, 133.44, 135.01,
135.66, 140.01, 158.22, 163.27, 168.01, 169.79 ppm; HRMS m/z per
C18H13N3O5S3 [M]+: 447.9.
68
N-[2-[(1,5-dimetil-3-osso-2-fenil-2,3-diidro-1H pirazol-4-il)tio]-4osso[1]benzotieno[3,2-d]pirimidin-3(4H)il]metansulfonammide (10)
resa: 95%; cristalli verdi; pf: 190 °C; IR (KBr): ν = 3224, 3038, 1694, 1650,
1344, 1155 cm−1; 1H NMR (500 MHz, DMSO): δ = 2.36 (s, 3H, CH3), 3.38 (s,
3H, CH3), 3.81 (s, 3H, CH3), 7.39–8.14 (m, 9H, Ar-H), 11.51 (s, 1H, NH); 13C
NMR (125 MHz, DMSO): δ = 42.30, 99.83, 119.15, 121.72, 124.08, 125.17,
125.99, 128.29, 130.01, 132.83, 134.21, 135.01, 138.67, 143.05, 154.61,
156.80, 157.10, 167.37 ppm; HRMS m/z per C22H19N5O4S3[M]+: 513.1.
69
Acido 4-({3-[(metilsulfonil)ammino]-4-osso-3,4 diidro[1]benzotieno[3,2d]pirimidin-2-il}tio)-3-nitrobenzoico (11)
resa: 80%; cristalli gialli; pf: 215 °C; IR (KBr): ν = 3225, 1695, 1345, 1155
cm−1; 1H NMR (500 MHz, DMSO): δ = 3.42 (s, 3H, CH3), 7.53–8.62 (m, 7H,
Ar-H), 11.69 (s, 1H, NH);
13
C NMR (125 MHz, DMSO): δ = 44.12, 115.12,
125.16, 128.33, 130.01, 132.01, 133.44, 135.01, 135.66, 140.01, 158.22,
163.27, 168.01, 169.79 ppm; HRMS m/z per C18H12N4O7S3 [M]+: 492.5.
70
N-[2-[(2,4-nitrofenil)tio]-4-osso[1]benzotieno[3,2-d]pirimidin3(4H)il]metansulfonammide (12)
resa: 79%; cristalli gialli; pf: 126 °C; IR (KBr): ν=3220,1700, 1343, 1156
cm−1; 1H NMR (500 MHz, DMSO): δ=3.39 (s, 3H, CH3), 7.26 – 7.80 (m, 7H,
Ar-H), 11.43 (s, 1H, NH).
13
C NMR (125 MHz, DMSO): δ = 43.83, 119.66,
120.64, 127.34, 130.91, 131.14, 133.96, 138.36, 148.07,150.80, 155.68,
157.015, 159.53 ppm; HRMS m/z per C17H11N5O7S3 [M]+: 449.6.
71
Sintesi del N-[2-(cicloesil tio)-4-osso[1]benzotieno[3,2-d]pirimidin3(4H)il]metansulfonammide (13)
resa: 86%; cristalli bianchi; pf: 108 °C; IR (KBr): ν = 3195, 1695, 1350, 1150
cm−1; 1H NMR (500 MHz, DMSO): δ = 1.45 - 2.05 (m, 10H, cycloexyl), 3.29
(s, 3H, CH3), 3.72 (s, 1H, CH), 7.38 - 8.10 (m, 4H, Ar-H), 11.14 (s br, 1H,
NH);
13
C NMR (125 MHz, DMSO): δ = 43.83, 119.66, 120.64, 127.34,
130.91, 131.14, 133.96, 138.36, 148.07, 150.80, 155.68, 157.015,159.53 ppm;
HRMS m/z per C17H19N3O3S3 [M]+: 409.09.
72
N-[2-[(2,4-difluorofenil)tio]-4-osso[1]benzotieno[3,2d]pirimidin(4H)il]metansulfonammide (14)
resa: 85%; cristalli argentati; pf: 236 °C; IR (KBr): ν = 3200, 3095, 1695,
1350, 1155 cm−1; 1H NMR (500 MHz, DMSO): δ = 3,37 (s, 3H, CH3), 7.2 –
8.10 (m, 7H, Ar-H ), 11.46 (s, 1H, NH);
13
C NMR (125 MHz, DMSO): δ =
43.97, 105.12, 112.68, 130.88, 132.70, 138.77, 139.21, 160.22, 161.50,
165.51, 166.50,184.99, 188.44. HRMS m/z per C17H11F2N3O3S3 [M]+: 438.6.
73
Acido 2-({3-[(metilsulfonil)ammino]-4-osso-3,4-diidro[1]benzotieno[3,2d]pirimidin-2-il}tio)benzoico (15)
resa: 85%; cristalli bianchi; pf: 238 °C. IR (KBr): ν = 3494, 1695, 1350,
1156cm−1; 1H NMR (500 MHz, DMSO): δ = 3,28 (s, 3H, CH3), 7.21 – 8.05
(m, 8H, Ar-H), 11.37 (s, 1H, NH), 13.01 (s, 1H, COOH);
13
C NMR (125
MHz, DMSO): δ = 44.12, 115.12, 125.16, 128.33, 130.01, 132.01, 133.44,
135.01, 135.66, 140.01, 158.22, 163.27, 168.01, 169,79 ppm; HRMS m/z per
C18H13N3O5S3 [M]+: 447.9.
74
Spettro 1H NMR del derivato 5
75
Spettro 13C NMR del derivato 5
76
Spettro 1H NMR del derivato 6
77
Spettro 13C NMR del derivato 6
78
Spettro 1H NMR del derivato 7
79
Spettro 13C NMR del derivato 7
80
Spettro 1H NMR del derivato 8
81
Spettro 13C NMR del derivato 8
82
Spettro 1H NMR del derivato 9
83
Spettro 13C NMR del derivato 9
84
Spettro 1H NMR del derivato 10
85
Spettro 13C NMR del derivato 10
86
Spettro 1H NMR del derivato 11
87
Spettro 13C NMR del derivato 11
88
Spettro 1H NMR del derivato 12
89
Spettro 13C NMR del derivato 12
90
Spettro 1H NMR del derivato 13
91
Spettro 13C NMR del derivato 13
92
Spettro 1H NMR del derivato 14
93
Spettro 13C NMR del derivato 14
94
Spettro 1H NMR del derivato 15
95
Spettro 13C NMR del derivato 15
96
6 Molecular Docking
Il riconoscimento molecolare è un fenomeno molto importante in biochimica,
infatti, l’elevata specificità di riconoscimento all’interno dei sistemi biologici
tra gli enzimi e i loro substrati, tra i recettori e i ligandi che inducono un
determinato segnale o ancora tra gli antigeni ed i corrispondenti anticorpi, è
ciò che rende possibile il complesso meccanismo che regola la vita degli
organismi
viventi.
Una
conoscenza
dettagliata
dei
meccanismi
di
riconoscimento molecolare è di particolare interesse nella scoperta di nuovi
farmaci, poiché la maggior parte di essi interagisce con proteine target come
enzimi o recettori. Per riuscire a capire nel dettaglio quali siano le basi
energetiche dell’interazione tra una proteina ed un certo ligando, che potrebbe
rappresentare un potenziale farmaco, occorre prima di tutto conoscere la
struttura tridimensionale del complesso proteina-ligando che viene in generale
ricavata attraverso cristallografia a raggi-X o NMR. Di notevole interesse sono
anche gli approcci computazionali che sono stati sviluppati negli ultimi anni e
che si pongono come possibile alternativa ai metodi sperimentali sopra citati
nel tentativo di predire la struttura tridimensionale di un complesso proteinaligando. Questi metodi vengono chiamati complessivamente molecular
docking. Le strutture proteiche possono essere utilizzate per posizionare i
ligandi all’interno del sito attivo della proteina e per studiare le interazioni
intermolecolari. Il poter predire quali siano le modalità di binding di un
ligando nei confronti di una proteina target può essere di grande aiuto nello
stabilire delle relazioni tra attività e struttura nella fase di sviluppo di nuovi
farmaci (metodologie QSAR). Quindi, i ligandi sono dei composti che
mostrano una determinata attività biologica e che possono essere modificati
strutturalmente per migliorarne la bioattività. Il docking applicato a questo
scopo rappresenta uno strumento per il disegno di nuove e potenziali molecole
con attività farmacologica. La qualità di una struttura proteica, sia che sia stata
97
ricavata sperimentalmente o che sia stata ottenuta tramite tecniche
computazionali, è comunque di cruciale importanza, poiché anche piccoli
cambiamenti in essa possono influenzare enormemente il risultato di un
esperimento di docking. Idealmente, la risoluzione atomica di una struttura
dovrebbe essere al di sotto di 2.5Ǻ. Per trovare la corretta modalità di legame
di un certo ligando nel sito attivo di un recettore è inoltre necessario fare un
adeguato campionamento dello spazio conformazionale disponibile per una
molecola di ligando flessibile all’interno della tasca di binding della proteina
stessa.
6.1 Studi di molecular docking sui derivati benzotienopirimidinici
Al fine di analizzare le possibili interazioni dei derivati benzo-tieno [3, 2-d]
pirimidinici con i residui amminoacidi presenti nel sito binding della COX-2,
sono stati effettuati degli studi simulativi di “molecular docking”. Il motivo
per cui questi derivati dovrebbero presentare una certa specificità nei confronti
dell’enzima COX-2 si può attribuire alla sostituzione delle due Isoleucine
(Ile523, Ile434) presenti nella COX-1 con due Valine (nella COX-2), ciò ha
aumentato di circa il 25% il sito di binding rendendo accessibile la tasca
idrofobica nella COX-2. Un’altra differenza chiave tra queste due
cicloossigenasi è la mutazione dell’His513 nella COX-1 con un’Arg513 nella
COX-2. Questa sostituzione ha generato uno specifico sito di binding per i
gruppi sulfonammidici o metilsulfonici che sono presenti in quasi tutte le
molecole che inibiscono in maniera selettiva l’enzima COX-2. Non è
comunque possibile stabilire delle regole empiriche che valgano per tutti i
farmaci esistenti anche qualora essi abbiano uno scheletro strutturale comune.
Questo suggerisce il fatto che debbano verificarsi sottili cambiamenti
strutturali a livello del sito di binding della COX-2 perché l’enzima possa
adattare la sua struttura a quella dell’inibitore. Il sito binding dell’enzima
COX-2 è un lungo e stretto canale idrofobico che si estende a partire dalla
regione di legame alla membrana della proteina. All’ingresso del canale gli
98
amminoacidi Arg120, Glu524, Tyr355 ed Arg513 formano una rete di legami
idrogeno che facilitano l’ingresso per il sito di binding. Llorens et al.
recentemente hanno affermato che la capacità dei differenti ligandi di
perturbare questa rete sia determinante per le cinetiche e per i contributi della
selettività dell’inibizione.[45] Un ruolo importante sembra che venga svolto
anche dalle molecole di acqua che partecipano alla dinamica di questa rete di
legami idrogeno posta proprio all’ingresso del sito attivo. In base a tali
considerazioni abbiamo applicato il docking proteina-ligando in modo da
poter predire in maniera ottimale le posizioni, gli orientamenti e le energie di
interazione dei derivati benzotienopirimidinici nel sito binding della COX-2. I
risultati ottenuti hanno mostrato che i composti 7, 10, 12, 13, 14 e 15
interagiscono con i seguenti residui amminoacidici: Lys83, Pro84, Val89,
Leu93, Ile112, Tyr115, Val116, Ser119, Arg120, Tyr122, Tyr355 e Ser471. In
particolare il gruppo metansulfonammidico presente nelle loro strutture si
colloca nella tasca idrofobica laterale andando così a formare dei forti legami
idrogeno con il gruppo idrossilico della Tyr355 e con il gruppo amminico
dell’Arg120. Fa eccezione il composto 10 poiché interagisce con i seguenti
residui amminoacidici: Asn382, His386, Thr212. Dati di letteratura
asseriscono che gli amminoacidi Asn382, His386 e Thr212 giochino un ruolo
fondamentale per quel che concerne la selettività verso la COX-2, in quanto
riescono a stabilizzare la conformazione delle molecole nella tasca idrofobica
(Fig.17-19).[46] Inoltre i derivati 7, 10, 12, 13, 14 e 15 hanno mostrato delle
forti interazioni idrofobiche di Van der Waals con differenti amminoacidi
presenti nel sito binding della COX-2. A completamento di questo studio è
stata determinata l’energia di binding dei derivati benzotienopirimidinici.
99
Figura 17: Sito binding della COX-2 con i derivati 7 e 10
Figura 18: Sito binding della COX-2 con i derivati 12 e 13
Figura 19: Sito binding della COX-2 con i derivati 14 e 15
100
Solamente i derivati 7, 10, 12, 13, 14 e 15, indipendentemente dalla presenza
di diversi gruppi funzionali nella loro struttura, hanno mostrato dei legami
favorevoli con il sito attivo della COX-2 presentando alti valori di energia di
binding. In particolare il composto 10 è risultato il miglior candidato per
inibire la COX-2, poichè la sua energia di binding (ΔG = 9,4 kcal/mol) risulta
essere il valore più alto tra tutti i derivati (Tab.2).
Energie di
N° di
Donatore/accettore
Distanza
binding
legami
legami idrogeno
legame
(ΔG)
idrogeno
idrogeno
(kcal/mol)
(H)
(Å)
naprossene
-9.0
2
ARG-120:NH
TYR-355:OH
1.863
2.165
nimesulide
-7.3
2
ARG-120:NH
TYR-355:OH
3.088
2.737
7
-8.1
2
ARG-120:NH
TYR-112:OH
3.002
2.788
10
-9.4
3
ASN-382:NH
HIS-386:NH
THR-212:OH
2.867
2.945
2.692
12
-9.1
2
TYR-355:OH
ARG-120:NH
2.875
2.876
13
-7.9
2
ARG-120:NH
TYR-112:OH
3.149
2.806
14
-8.9
1
ARG-120:NH
2.882
15
-8.2
2
ARG-120:NH
TYR-355:OH
2.970
3.120
Derivati
Tabella 2: Energie di binding.
I dati ottenuti dagli studi docking sono perfettamente in linea con i risultati
farmacologici.
101
Materiali e Metodi
6.2 Metodi computazionali
La realizzazione di un adeguato protocollo ha richiesto la valutazione, in
successione, di diverse metodiche di docking. Attraverso questo approccio è
stato possibile definire con precisione numerosi parametri. Il criterio
principale con cui è stata stabilita l’adeguatezza di una procedura si è basata
sul confronto dell’ orientamento e della posizione dei ligandi presenti nel
complesso dockato rispetto a quelli analoghi per il complesso cristallografico.
L’ analisi è stata realizzata mediante il calcolo della Root Mean Square
Deviation (RMSD), previa sovrapposizione dei residui amminoacidici del
backbone proteico. Uno studio condotto da Cole e Murray, in cui si voleva
verificare la riproducibilità di alcuni complessi sui principali algoritmi di
docking attualmente in uso, raccomandava di mantenere la struttura del
complesso entro il valore soglia di RMSD pari a 2,00 Å; consigliando, inoltre,
di ridurre ulteriormente questa soglia a 1,50 Å nel caso venissero messe a
confronto le coordinate atomiche di piccoli ligandi. Il valore di RMSD della
cicloossigenasi-2 murina (mCOX-2) è di 0.910 Å (gli studi docking sono stati
effettuati prendendo come riferimento la struttura della COX-2 murina poiché
presenta le stesse caratteristiche di quella umana). Gli studi computazionali di
docking sono stati condotti applicando l’algoritmo genetico di Lamarckian
(LGA), implementato in AutoDock 4.0. AutoDock è costituito da due
programmi: il motore di docking (AutoDock) che esegue il docking del
ligando basandosi su un set di griglie descriventi la proteina target ed
AutoGrid, che pre-calcola queste griglie. Questo programma è capace di
tenere in considerazione, oltre la flessibilità del ligando anche la flessibilità
della componente proteica e questo consente ad AutoDock 4.0 di effettuare
studi di interazione proteina-proteina.
102
7 Attività anti-infiammatoria dei derivati
benzotieno[3,2-d] pirimidinici
7.1 Modelli in vitro di infiammazione
I progressi nelle colture cellulari hanno fornito modelli in vitro di eccezionale
versatilità e utilità al fine di valutare i diversi aspetti connessi con la biologia
molecolare e cellulare, la virologia e l’immunologia. In molti casi
rappresentano il materiale di partenza per l’estrazione di proteine o acidi
nucleici, mentre in altri casi le cellule cresciute in vitro sono utilizzate per
analizzare un determinato comportamento biologico (proliferazione, capacità
di organizzare tessuti, proprietà adesive o di migrazione, risposta immunitaria,
risposta infiammatoria). Le colture cellulari possono anche essere usate per
eseguire test diagnostici o per rigenerare in vitro tessuti e organi. Uno dei
modelli più usati per lo studio delle patologie infiammatorie, in particolare a
carico della pelle, è costituito da cellule isolate dall’epidermide umana
mediante biopsia. Le cellule più largamente diffuse nell’epidermide sono i
cheratinociti, melanociti, cellule di Langerhans (immunocompetenti), cellule
di Merkel (sensori del tatto), insieme alle cellule dendritiche, endoteliali, i
linfociti, i mastociti, le terminazioni nervose peptidergiche formanti il SIS
(Skin Immune System).
7.2 I cheratinociti
I cheratinociti sono stratificati in un epitelio squamoso e costituiscono la
maggiore popolazione di cellule epidermiche della pelle (Fig.20), ma si
trovano anche a livello delle mucose, degli epiteli orali, corneale,
congiuntivale e genitale. Le caratteristiche della superficie cutanea dipendono
essenzialmente da una corretta proliferazione e differenziazione dei
cheratinociti, che è soggetta a fine regolazione. Il risultato terminale della
differenziazione dei cheratinociti è la formazione dello strato corneo in cui i
103
cheratinociti anucleati (corneociti), ricchi di componenti proteiche altamente
stabilizzate, sono immersi e “cementati” in una matrice lipidica estremamente
compatta. Per queste caratteristiche lo strato corneo costituisce una
formidabile barriera chimico-fisica che regola le perdite di acqua e impedisce
alle sostanze esogene di penetrare la cute. I cheratinociti hanno la proprietà di
iniziare e regolare le risposte infiammatorie cutanee, in quanto sono in grado
di produrre in maniera altamente controllata una grande varietà di molecole
pro - e anti-infiammatorie in risposta ad una vasta serie di stimoli sia esogeni
che endogeni. In presenza di una barriera epidermica impropria i cheratinociti
sono indotti a produrre e rilasciare nel distretto epidermico, una serie di
mediatori chimici che tendono a ripristinare uno strato corneo perfettamente
funzionale, stimolando la proliferazione cellulare e le sintesi lipidiche. Tra
queste sostanze vi sono fattori di crescita quali il “nerve growth factor”, il
“trasforming growth factor-”, l’anfiregulina; citochine come l’interleuchina1 (IL-1), il “tumor necrosis factor-” (TNF-), il “granulocyte macrophage
colony-stimulating factor” (GMCSF); chemochine come l’interleuchina-8 (IL8). Alcune di queste sostanze (IL-1, TNF-e GM-CSF) sono potenti iniziatori
dell’infiammazione e, insieme alle chemochine, forniscono un valido richiamo
per molte popolazioni di leucociti dal sangue periferico. I cheratinociti
giocano un ruolo importante nell’indurre e nel perpetuare le reazioni
infiammatorie della pelle attraverso il rilascio di citochine e la risposta ad esse.
Una varietà di stimoli ambientali, come ad esempio promotori tumorali, UV,
agenti chimici, possono indurre nei cheratinociti il rilascio di citochine
infiammatorie (IL-1 e TNF-), chemiochine e citochine che promuovono la
crescita (IL-6; GM-CSF, TGF-).[37-38] Di tutte le citochine prodotte dai
cheratinociti solo IL-1e TNF-attivano un sufficiente numero di
meccanismi effettori in grado di indurre un’infiammazione cutanea
indipendente.
104
Figura 20: Linea cellulare di cheratinociti umani (NCTC 2544)
7.3 I macrofagi ( macrofagi murini j774)
I macrofagi sono una popolazione cellulare che presenta una marcata
eterogeneità, dovuta sia alla loro diffusa distribuzione tissutale che alla
capacità di rispondere a diversi stimoli, sia endogeni che esogeni (Fig.21).
Infatti l’interazione con le cellule circostanti o con molecole alterate ed agenti
esogeni influisce sulla funzionalità di queste cellule. I ligandi vengono
riconosciuti da diversi tipi di recettori, innescando cosi diverse risposte
cellulari, tra cui fagocitosi o endocitosi, attivazione o repressione di geni. Le
funzioni cellulari indotte includono i processi di adesione e migrazione
cellulare, secrezione di citochine, processazione e presentazione dell’antigene
e attivazione delle funzione effettrici nella risposta immunitaria. I macrofagi
presentano, inoltre, la capacità di produrre diversi tipi di sostanze, quali gli
enzimi
idrolitici
(idrolasi,
proteasi,
lisozima),
proteine
ad
attività
immunostimolante (interferoni, transferrina, interleuchina I), un fattore
stimolante la formazione di colonie (CSF), componenti del complemento (C2,
C3, C4 e C5) e prostaglandine. Nell'ambito del sistema immunitario, i
macrofagi sono coinvolti in quattro attività funzionali fondamentali:
interagiscono e degradano qualsiasi tipo di materiale estraneo;
iniziano e potenziano l'attivazione dei linfociti;
105
svolgono la funzione di cellule effettrici;
esplicano una attività regolatrice sulla risposta immunitaria.
Queste funzioni non sono svolte da tutti i macrofagi contemporaneamente, né
tutti i macrofagi posseggono tali funzioni. L' attività di degradazione del
materiale estraneo è dovuta fondamentalmente ai recettori di superficie,
all’attività fagocitaria ed agli enzimi idrolitici lisosomiali, e rappresenta la
funzione posseduta, sia pure in grado variabile, da quasi tutti i macrofagi. La
funzione effettrice dei macrofagi è legata all' azione delle linfochine prodotte
dai linfociti T attivati. Le linfochine determinano a loro volta uno stato di
attivazione dei macrofagi, i quali possono acquisire la capacità di svolgere
azione citotossica sia nei confronti di cellule neoplastiche, sia di
microrganismi intracellulari (killing). I macrofagi, infine, possono svolgere
una attività soppressiva nei confronti dei linfociti attivati. Questa funzione
regolatrice può verificarsi sia mediante un contatto diretto tra macrofagi e
linfociti, sia tramite la liberazione di fattori solubili linfochino-simili (soluble
immune response suppressor - SIRS) o di altre molecole pure dotate di azione
soppressiva, quali l'interferone, il perossido d' idrogeno, l' anione superossido
e le prostaglandine E2.
Figura 21: Linea cellulare di macrofagi murini J774
106
7.4 Saggi biologici
L’ attività anti-infiammatoria dei derivati benzo-tieno[3, 2-d] pirimidinici è
stata effettuata determinando sia l’espressione della molecola di adesione
intercellulare (ICAM-1), ossido nitrico sintasi inducibile (iNOS) e
cicloossigenasi-2 (COX-2) mediante analisi Western Blot che la modulazione
del rilascio della proteina chemiotattica per i monociti (MCP-1) e
interleuchina 8 (IL-8) tramite test E.L.I.S.A. in due linee cellulari: i
cheratinociti umani normali (NCTC 2544) e i macrofagi murini J774.[32,33]
7.4.1 MTT: saggio di vitalità cellulare
Il test MTT prevede la valutazione della vitalità e della proliferazione cellulare
mediante la misurazione dell’ attività dell’ enzima deidrogenasi mitocondriale.
Il test MTT è una metodica semplice, accurata e fornisce risultati riproducibili.
Questo metodo è stato sviluppato originariamente da Mossman. Il reagente
chiave è il bromuro di 3-(4,5-dimetiltiazol-2-il)-2,5-difenil tetrazolio o MTT,
sostanza che dà un colore giallo in soluzione acquosa. La deidrogenasi
mitocondriale delle cellule vitali taglia l’anello tetrazolico, portando alla
formazione di cristalli di formazano color viola-porpora insolubili in acqua. I
cristalli vengono sciolti con una soluzione solubilizzante l’MTT. La soluzione
viola risultante viene misurata spettrofotometricamente. Un aumento o
diminuzione delle cellule vitali ha per risultato un cambiamento concomitante
nella quantità di formazano che si forma e che può essere considerato come un
indicatore del grado di citotossicità causato dall’esposizione alle sostanze
irritanti. Il saggio MTT, pertanto, è stato effettuato per verificare l’eventuale
tossicità
dei
derivati
benzo-tieno[3,
2-d]
pirimidinici
attraverso
la
compromissione della vitalità delle cellule nei cheratinociti (NCTC 2544) e
nei macrofagi J774. Dai grafici si evince chiaramente che le sostanze in
esame, alla concentrazione 10μM, non risultano essere citotossiche quando
lasciate a contatto con le cellule NCTC 2544 e J774 per 48 ore (Fig.22).
107
Figura 22: Risultati del test MTT
108
7.4.2 Valutazione dell’attività anti-infiammatoria dei derivati
benzotienopirimidinici: ICAM-1, iNOS e COX-2
Allo scopo di simulare in vitro un modello infiammatorio, le cellule NCTC
2544 sono state esposte a interferone-γ (IFN-γ), citochina essenziale
nell’amplificare le reazioni infiammatorie, e istamina, che potenzia l’azione
pro-infiammatoria dell’IFN-γ. IFN-γ è il più potente stimolo per l’ espressione
della molecola di membrana ICAM-1 che è coinvolta nella lisi dei
cheratinociti da parte dei linfociti T citotossici. IFN-γ e istamina utilizzano
distinte vie di trasduzione del segnale, provocando una maggior attivazione
dei
geni
infiammatori.
In
particolare,
l’istamina
aumenta
l’attività
trascrizionale sia della proteina di attivazione -1 (AP-1) che del fattore
nucleare kB (NF-kB), mentre l’IFN- γ usa i fattori di trascrizione STAT1 e
STAT3. Il celecoxib è stato utilizzato come farmaco anti-infiammatorio di
riferimento. La linea cellulare dei macrofagi murini J774, invece, è stata
esposta all’endotossina batterica lipopolisaccaride (LPS), il più potente
stimolo per i macrofagi. Le cellule NCTC 2544 e J774 non trattate presentano
quantità non rilevabili di ICAM-1, iNOS, e COX-2 mentre l'incubazione delle
colture cellulari con IFN-γ e istamina per i cheratinociti e LPS per i macrofagi
per ~ 48 h ha indotto una forte espressione di ICAM-1, iNOS, e COX-2.
L’aggiunta delle sostanze in esame alla concentrazione 10μM insieme ad IFNγ e istamina per le cellule NCTC2544 e LPS per le cellule J774 ha prodotto
una significativa inibizione dell’espressione di ICAM-1 (Fig.23), iNOS
(Fig.24) e COX-2 (Fig.25). Il composto 10 è risultato il più potente tra i
derivati nell’inibizione dei tre parametri infiammatori mostrando una potenza
superiore al celecoxib mentre i derivati 5, 6, 8, 9 e 11 non hanno mostrato
alcuna attività anti-infiammatoria nelle cellule NCTC 2544 e J774 trattate. Dai
risultati ottenuti si evince chiaramente che i composti 7, 10, 12, 13, 14 e 15,
insieme ad IFN-γ e istamina per le cellule NCTC 2544 e LPS per i macrofagi
J774 hanno indotto una rilevante inibizione dell’espressione di ICAM-1, iNOS
e COX-2.
109
Figura 23: Effetti delle sostanze sull’espressione di ICAM-1 nei cheratinociti
umani normali NCTC 2544 e nei macrofagi J744.
110
Figura 24: Effetti delle sostanze sull’espressione delle iNOS nei cheratinociti
umani normali NCTC 2544 e nei macrofagi J744.
111
Figura 25: Effetti delle sostanze sull’espressione delle COX-2 nei cheratinociti
umani normali NCTC 2544 e nei macrofagi J744.
112
7.4.3 Determinazione del rilascio dell’ MCP-1 e dell’ IL-8 dei
derivati benzotienopirimidinici
Nell’infiammazione il principale ruolo dell’IL-8 è quello di dirigere in loco il
reclutamento di neutrofili e basofili e di promuovere il danno tissutale. Una
sua inibizione potrebbe quindi avere un effetto protettivo sul danno prodotto
dall’infiammazione. Dati di letteratura asseriscono la forte sinergia esistente
tra l’IL-8 e l’ MCP-1, un’altra chemochina implicata nei processi
infiammatori. L’MCP-1 ha un ruolo fondamentale nell’attrazione dei
monociti, delle cellule T e delle cellule NK, inoltre è implicata nelle malattie
caratterizzate da infiltrazione monocitica. La sua espressione è stata
documentata in molte malattie, come l’aterosclerosi, la sclerosi multipla,
l’artrite reumatoide e la nefrite. Inoltre l’up-regulation di MCP-1/CCL2 è stata
osservata in associazione con risposte neuroinfiammatorie in modelli animali
di ischemia cerebrale e nei tessuti del miocardio. Quindi a completamento del
nostro studio abbiamo analizzato la modulazione del rilascio della proteina
chemiotattica per i monociti (MCP-1) e per l’interleuchina 8 (IL-8) tramite test
E.L.I.S.A. I cheratinociti normali umani NCTC 2544 e i macrofagi J774 non
rilasciano costitutivamente MCP-1; nessuna quantità di MCP-1 è stata, infatti,
prodotta dalle cellule non stimolate. Dai grafici ottenuti si evince che i derivati
7, 10, 12, 13, 14 e 15 hanno inibito notevolmente il rilascio di MCP-1 indotto
da IFN-γ e istamina alla concentrazione di 10μM (Fig.26,27). Per quanto
concerne il rilascio dell’IL-8, trascurabili quantità di IL-8 sono state prodotte
dai cheratinociti NCTC 2544 e dai macrofagi J774 non stimolati. IFN-γ e
istamina per le cellule cheratinocitiche e LPS per i macrofagi hanno indotto un
notevole rilascio di IL-8. Pertanto, i derivati benzo-tieno [3, 2-d] pirimidinici
7, 10, 12, 13, 14 e 15 inibiscono il rilascio di MCP-1 e di IL-8, le quali
svolgono un’importante ruolo nel corso dell’infiammazione.
113
Figura 26: Effetti dei composti benzo-tieno [3, 2-d] pirimidinici sul rilascio di
MCP-1
114
Figura 27: Effetti dei composti benzo-tieno [3, 2-d] pirimidinici sul rilascio di
IL-8
115
Materiali e Metodi
7.5 Colture cellulari
La linea cellulare di cheratinociti umani NCTC 2544 è stata fornita dalla
Interlab Cell Line Collection (Genova, Italia) ed è stata mantenuta in Minimun
Essential Medium (MEM) (Sigma Aldrich, Italia) con l’aggiunta del 10% di
siero fetale di vitello, 100 U/ml di penicillina e 100 μg/ml di streptomicina e
tenuta a 37°C in un incubatore con un’ atmosfera umidificata al 95% di aria e
al 5% di CO2. La linea cellulare dei macrofagi murini J774 è stata fornita dall’
American Type Culture Collection (Rockville, MD, USA). Le cellule sono
state coltivate in DMEM contenente il 10% di siero fetale di vitello, 4.5 g/l di
glucosio, 1mM di piruvato di sodio, 100 U/ml di penicillina, 100 μg/ml di
streptomicina e 25 μg/ml di fungizone (Invitrogen, UK) sotto incubazione a
37°C in un’ atmosfera umidificata al 95% di aria e al 5% di CO2. Il terreno di
coltura è stato cambiato ogni 2-3 giorni. Ventiquattro ore prima
dell’esperimento, le cellule sono state tripsinizzate, contate in un
emocitometro e seminate o in piastre da 96 pozzetti (per il test MTT) o in
piastre di Petri da 100 mm (per Western blot). I cheratinociti sono stati
stimolati, tranne le cellule di controllo, con 200U/ml di IFN-γ e istamina 10-4
M, mentre i macrofagi con LPS (1 mg/mL) in maniera tale da poter riprodurre
i meccanismi coinvolti nella patogenesi dei processi infiammatori, in assenza
o presenza di un unica concentrazione dei composti benzotienopirimidinici 515 (10μM) o celecoxib (10 μgM), quest’ultimo usato come farmaco antiinfiammatorio di riferimento. Dopo 48 h ogni campione è stato testato per
determinare l’espressione dell’ iNOS, ICAM-1 e COX-2 e il rilascio dell’
MCP-1 e dell’ IL-8.
7.6 Western blot
L’espressione di ICAM-1, iNOS e COX-2 è stata valutata mediante analisi
116
Western blot. In breve, i cheratinociti e i macrofagi trattati e non trattati sono
stati lavati due volte con PBS a freddo e sono stati raccolti con un tampone di
lisi contenente 10 mM di Tris-HCl, 10 mM di KCl, 2mM di MgCl2, 0,6mM di
PMSF, e 1% di SDS a pH 7,4. Dopo averle lasciate raffreddare per 30 min a
0°C, le cellule sono state sonicate. Sessanta microgrammi di proteine totali,
presenti nel surnatante, sono state caricate in ogni “lane” e poi separate tramite
un gel di poliacrilammide, Bis-Tris 4-12% Novex per elettroforesi (NuPAGE,
Invitrogen). Le proteine sono state quindi trasferite in una membrana di
nitrocellulosa (Invitrogen, Italia) in ambiente umido. Il trasferimento delle
proteine è stato verificato colorando le membrane di nitrocellulosa con il
Ponceau S e il gel Bis-Tris Novex con il Brillant blue R. Le membrane sono
state messe in blocking con un tampone salino contenente 0,1 % di Tween-20
(TBST) e 5 % di latte in polvere senza grassi a 4°C per tutta la notte.
L’anticorpo
monoclonale
anti-ICAM-1
(1H4:sc-51632,
Santa
Cruz
Biotecnology) (diluizione 1:200), anti-NOS2 (N-20, sc-651, Santa Cruz
Biotechnology) (diluizione 1:300), anti-COX-2 (N-20, sc-1746, Santa Cruz
Biotechnology) (diluizione 1:100) e anti-α-tubulina (Sigma, Milano, Italia)
(diluizione 1:5000) sono stati diluiti con TBST e le membrane sono state
incubate per 2 h a temperatura ambiente. Gli anticorpi sono stati rivelati con
un anticorpo secondario coniugato con la perossidasi usando per la chemioluminescenza il substrato Supersignal West Pico Chemiluminescent Substrate
(Pierce Chemical Co., Rockford, IL). L’ espressione della proteina è stata
quantificata per mezzo dell’ analisi densitometrica delle autoradiografie. La
densità delle singole bande per ogni campione è stata messa in relazione a
quelle dell’α-tubulina, presa come proteina di riferimento, e i valori riportati
(corrispondenti all’intensità di segnale) sono stati espressi come unità
densitometriche arbitrarie.
7.7 E.L.I.S.A.
L’ MCP-1 e l’ IL-8 sono state misurate nel surnatante libero da cellule
117
raccolte 48 h dopo il trattamento per mezzo di kit E.L.I.S.A. (enzyme-linked
immunosorbent assay) (Amersham Biosciences, Svizzera), in grado di
riconoscere in modo specifico le proteine mediante il legame antigeneanticorpo monoclonale. Il complesso è riconosciuto e legato da un anticorpo
policlonale marcato con un enzima. Calcolando l’attività enzimatica, dopo
aggiunta del substrato, si ha una stima direttamente proporzionale alla quantità
di antigene in esame. Le colture dei cheratinociti sono state eseguite in
triplicato per ogni condizione. Tutti i saggi sono stati eseguiti come specificato
dai produttori dei rispettivi kits. Gli standards, i controlli e i surnatanti sono
stati aggiunti ai pozzetti della micropiastra. Ogni pozzetto contiene un
anticorpo monoclonale anti-MCP1 o anti-IL8 e una soluzione liofilizzata di
anticorpo policlonale secondario HRP-coniugato (perossidasi di rafano). Ad
ogni pozzetto sono stati aggiunti 130 l di acqua e 20 l di campione. Dopo
una incubazione di 3 h a temperatura ambiente sono stati effettuati 3 lavaggi
con il wash buffer fornito dal kit per eliminare l’anticorpo HRP-coniugato che
non si è legato, quindi è stata aggiunta la soluzione substrato TMB
(tetramethyl-benzidina) per 15 minuti al buio. La reazione enzimatica è stata
bloccata con la Stop solution (acido fosforico 1M). La lettura dell’assorbanza
a 450 nm è stata effettuata mediante uno spettrofotometro (microlettore di
piastre). Per ogni saggio è stata utilizzata una curva standard usando
concentrazioni note di MCP-1 e di IL-8. La sensibilità del kit E.L.I.S.A. per
MCP-1 era 3,5 pg/ml e per IL-8 era <5 pg/ml. I risultati sono stati espressi
come pg/ml ±SEM.
7.8 Saggio di vitalità cellulare: MTT
La vitalità cellulare è stata misurata attraverso il saggio colorimetrico ai sali di
tetrazolio, che valuta la capacità delle cellule di ridurre, per mezzo della
succinato deidrogenasi mitocondriale, il bromuro di 3-(4,5-dimetiltiazol-2-il)2,5-difenil tetrazolio (MTT). L’MTT entra nelle cellule e passa nei mitocondri
dove viene ridotto in un prodotto colorato ed insolubile, il formazano. Per
118
rendere visibile il colore si solubilizzano i granuli colorati di formazano
tramite l’aggiunta di DMSO. La reazione MTT-cleavage richiede la completa
integrità della cellula ed è proporzionale al grado di attività metabolica della
stessa. Dopo incubazione delle cellule per 24 h, in 5% CO2 a 37°C, con 20 μl
della soluzione del sale di tetrazolio, solubilizzato in medium (5 mg/ml), e 180
μl di medium, si rimuove il sovranatante e si aggiungono 100 μl di DMSO.
Per ogni campione si allestiscono prove in triplicato e su ognuna viene
misurata la densità ottica a λ=550 nm con uno spettrofotometro per
micropiastre (Titertek Multiscan, Flow Laboratories). La vitalità cellulare
viene espressa in % Abs rispetto a quella del controllo non trattato.
7.9 Analisi statistica
L’analisi statistica è stata eseguita mediante il software SYSTAT, versione 11
(Systat Inc., Evanston IL, USA). Ogni risultato è stato riportato negli
istogrammi rappresentanti le medie ± l’errore standard di tre esperimenti
eseguiti in triplicato. La significatività statistica è stata valutata mediante il test
di Student.
119
8 Spettroscopia di fluorescenza
8.1 La fluorescenza
In natura gli elettroni delle molecole si trovano principalmente nel più basso
livello vibrazionale dello stato elettronico fondamentale, S0. In queste
condizioni una molecola è in grado di assorbire radiazioni elettromagnetiche
nell’ultravioletto o, meno frequentemente, nella regione del visibile e passare
ad uno stato eccitato. Quando una molecola assorbe un fotone di appropriata
lunghezza d’onda uno dei suoi elettroni dello stato fondamentale viene
promosso in un orbitale a più alto contenuto energetico (Fig.28).
Figura 28: Assorbimento ed emissione di una radiazione elettromagnetica.
Una molecola eccitata a livelli superiori al primo ritorna al livello più basso
dello stato eccitato, S1, cedendo la sua energia attraverso un processo definito
di rilassamento vibrazionale.[9] L’energia vibrazionale, persa durante il
rilassamento, viene trasferita ad altre molecole (per esempio il solvente),
attraverso collisioni. L’effetto è quello di convertire parte dell’energia del
fotone assorbito in calore distribuito in tutto il mezzo. A partire dallo stato
120
eccitato S1 la molecola può perdere l’eccesso di energia e tornare allo stato
fondamentale S0 emettendo un fotone e determinando la fluorescenza (Fig.29).
Figura 29: Componenti vibrazionali della fluorescenza.
La fluorescenza è un fenomeno che si sviluppa in tempi brevissimi (10-9,10-8
secondi), dando luogo ad una luce intensa e di breve durata. La fosforescenza,
altro fenomeno che comporta l'emissione di luce, invece si sviluppa in tempi
molto più lunghi, nell'ordine dei millisecondi, e dà origine ad una luce più
debole ma di lunga durata. I materiali fluorescenti cessano di essere luminosi
al cessare dello stimolo che ne determina la luminosità, invece nei materiali
fosforescenti la luce continua ad essere emessa per un certo periodo dopo la
fine dello stimolo. Nella fluorescenza la radiazione è generata in virtù di
transizioni tra stati con la stessa molteplicità di spin, singoletto-singoletto (per
esempio S1→S0), mentre nella fosforescenza la transizione coinvolta comporta
variazione della molteplicità di spin: il caso più frequente sono transizioni
tripletto-singoletto (T1→S0). La maggior parte delle molecole in condizioni
normali ritorna allo stato fondamentale attraverso processi che non
comportano emissione di radiazione e che hanno soltanto l’effetto di
convertire l’energia in calore.
121
Questi processi sono:
Conversione interna
As*→ A + calore
Intersistem crossing
As*→ At
Trasferimento di energia
La conversione interna consiste nel ritorno allo stato fondamentale
accompagnato da emissione di calore. In genere si tratta di un processo poco
efficiente che rende conto solo di una piccola parte dell’energia restituita dalla
molecola eccitata all’ambiente.
L’intersystem crossing consiste nel passaggio delle molecole eccitate da uno
stato di singoletto ad uno di tripletto; ciò implica ovviamente un cambiamento
di molteplicità e quindi si tratta di una transizione fondamentalmente proibita.
Tuttavia, spesso, gli stati di tripletti (e quelli di singoletti) nelle molecole non
sono puri cioè possono avere una certa percentuale dell’altro stato a causa
dell’accoppiamento spin-orbital. Tale accoppiamento è dato dall’influenza
esercitata dal momento magnetico dovuto all’orbitazione dell’elettrone intorno
al nucleo e sul momento magnetico dovuto all’ orbita di rotazione
dell’elettrone sul proprio asse. In seguito all’inversione di spin, le molecole,
che in questo modo occupano lo stato di tripletto, possono emettere una
radiazione luminosa che non sarà più di fluorescenza ma di fosforescenza.[9] A
questo proposito è opportuno ricordare che la presenza in vicinanza
dell’elettrone coinvolto nel processo, di un nucleo di metallo pesante produce
un aumento dell’accoppiamento spin-orbital, con aumento, quindi, dello
scambio intersistemico (effetto da metallo pesante).
Il trasferimento di energia può essere di due tipi: collisionale e non
collisionale. Nel primo caso il processo è bimolecolare, coinvolge un inibitore
e dipende dalla sua concentrazione. Il processo può essere descritto così:
A*+ I → A + I
La molecola eccitata ritorna così allo stato fondamentale senza emissione di
radiazione.
122
Il processo non collisionale avviene invece tra un donatore D e un accettore A
secondo questo schema:
D + hv → D*
D* + A → D + A*
A* → A + hv
I tre processi appena descritti possono competere con la fluorescenza ma,
affinché ciò possa accadere, devono possedere costanti cinetiche maggiori e
quindi avvenire in un tempo minore. Si osserverà quindi il fenomeno della
fluorescenza quando questo processo è più rapido degli altri. Sarà, infatti, il
rapporto tra la fluorescenza e tutti questi altri processi a determinare la resa
quantica, ed, in definitiva, il fatto che una molecola sia, o meno,
fluorescente.[9]
8.2 Diagrammi di Jablonski
I diagrammi di Jablonski sono usati per illustrare i processi di luminescenza.
Tipicamente riportano una struttura energetica principale dovuta ai livelli
elettronici più una struttura energetica secondaria dovuta ai livelli vibrazionali
del fluoroforo. Il diagramma di Jablonski riassume le trasformazioni
energetiche che si verificano per assorbimento di una radiazione di sufficiente
energia in una molecola (Fig.30). Lo stato fondamentale viene indicato come
S0 mentre quelli a energia superiore come S1, S2, … Sn rispettivamente. Le
transizioni tra gli stati sono rappresentate da linee verticali e corrispondono
all’assorbimento o all’emissione di luce. In un fluoroforo l’assorbimento di
energia luminosa porta alla promozione di un elettrone a uno dei livelli
vibrazionali eccitati (S1 o S2).
123
Figura 30: Diagrammi di Jablonski
Le molecole a questo punto rilassano rapidamente e raggiungono il più basso
livello vibrazionale dello stato S1. Questo processo, che si realizza in un tempo
relativamente breve (10-12 s), viene denominato internal conversion (IC) e si
completa prima dell’emissione di fluorescenza che ha dei tempi di vita
nell’ordine di 10-8 s. Il ritorno avviene raggiungendo uno stato vibrazionale
eccitato dello stato elettronico fondamentale, che evolve rapidamente verso
l’equilibrio. Le molecole nello stato S1 possono inoltre passare allo stato di
tripletto (T1) subendo un’inversione dello spin. L’emissione dallo stato T1 è
chiamata fosforescenza ed è generalmente spostata verso lunghezze d’onda
maggiori (energie minori) rispetto alla fluorescenza. La conversione da S1 a T1
è detta intersystem crossing, conversione intersistema (ISC). La transizione da
T1 allo stato fondamentale di singoletto è proibita e, come conseguenza, la
velocità risultante per il tripletto è di molti ordini di grandezza minore rispetto
al singoletto. L’esame del diagramma di Jabłoński mostra che l’energia
dell’emissione è minore di quella assorbita, pertanto la fluorescenza avviene a
energie minori (shift batocromico), corrispondenti a lunghezze d’onda
maggiori. Questo fenomeno è stato osservato per la prima volta da Sir G.G.
Stokes nel 1852 presso l’università di Cambridge. La causa comune del
124
fenomeno, denominato Stokes’s shift (Fig.31), è il rapido decadimento allo
stato vibrazionale più basso compreso nello stato elettronico S1. Oltre a quanto
descritto si possono verificare Stokes’s shifts dovuti al solvente, agli urti tra le
molecole e alla temperatura.
Figura 31: Spettro di eccitazione e di emissione di un fluoroforo con relativo
Stoke’s shitf.
Un’altra generale caratteristica della fluorescenza è la seguente: lo spettro di
emissione osservato è generalmente indipendente dalla lunghezza d’onda di
eccitazione. Questo fenomeno è conosciuto come regola di Kasha.
L’eccitazione porta alla promozione di un elettrone nello stato eccitato S1 in
un livello vibrazionale che non è quello a più bassa energia. Questo eccesso di
energia viene rapidamente dissipato per internal conversion lasciando il
fluoroforo nello stato eccitato, ma più basso dal punto di vista vibrazionale
(Fig.32). Poiché il rilassamento allo stato più basso vibrazionale avviene in
tempi molto brevi (10-12 s) e la differenza di energia tra lo stato eccitato e
quello fondamentale è costante, l’emissione di fluorescenza non dipende dalla
lunghezza d’onda di eccitazione.
125
Figura 32: Zoom del diagramma di Jabłoński
Altra conseguenza di questo fenomeno è la generale simmetria degli spettri di
fluorescenza. Dal momento che i livelli vibrazionali dello stato fondamentale e
del primo eccitato sono simili e dal fatto che il salto energetico è il medesimo,
lo spettro di assorbimento e di emissione risulteranno pressoché identici.
Nonostante la specularità degli spettri di fluorescenza sia mantenuta nella
maggior parte dei casi, esistono delle eccezioni dovute al fatto che in alcune
molecole gli stati eccitati portano a un riarrangiamento dei nuclei con
conseguente variazione dei livelli energetici vibrazionali dello stato S1.
8.3 Variabili che influenzano la fluorescenza
Diversi sono i fattori che influenzano la fluorescenza:
1. Resa quantica: intesa come il rapporto tra il numero di fotoni emessi in un
secondo e il numero di fotoni assorbiti in un secondo.
126
2. Solvente: in funzione della sua polarità si può avere uno spostamento dei
massimi di emissione. Il solvente può interagire sia con lo stato
fondamentale ma ancor più con lo stato eccitato. Nei composti aromatici
un solvente polare stabilizza lo stato eccitato essendo quest’ultimo
generalmente più polare dello stato fondamentale, ciò comporta uno
spostamento batocromico, cioè a lunghezze d’onda superiori, nel relativo
spettro di emissione. Un solvente apolare garantisce l'emissione di un
quanto di energia in più rispetto ad un solvente polare. Solventi come il
CBr4 o EtI riducono la fluorescenza di un determinato soluto per la
presenza di atomi pesanti. Un effetto analogo si riscontra in presenza di O2
in soluzione.
3. pH: può influenzare l’assetto elettronico della molecola, soprattutto se
possiede idrogeni a carattere acido. Sia la lunghezza d’onda che l’intensità
dell’emissione sono diverse per la forma dissociata e indissociata e questo
dipende dalla possibilità di un numero diverso di forme di risonanza
associate con le forme acide o basiche delle molecole. Molti fenoli sono
fluorescenti a pH neutro o acido e non lo sono a pH alcalini.
4. Temperatura: incide sulla viscosità della matrice e quindi può favorire o
meno le collisioni con le particelle del solvente e degli altri soluti presenti
in soluzione che circondano la molecola; ciò comporta la riduzione del
fenomeno della fluorescenza perché aumenta la possibilità di trasferimento
dell’energia per conversione interna. E' importante sapere che l'aumento
della temperatura di 1°C porta alla diminuzione dell'1% della fluorescenza
per questo in genere si opera a temperatura ambiente proteggendo il
campione dal calore generato dalle lampade con un buon sistema di
termostatazione.
127
5. Matrici: possono contenere sostanze che assorbono parte della radiazione
eccitante o emessa.
6. Smorzamento o quenching: è il fenomeno per cui una quota di energia che
potrebbe essere emessa come fluorescenza, viene invece trasferita ad
un'altra molecola determinando un’ attenuazione del segnale. A volte il
quenching è il risultato di trasferimento di energia ad altre molecole
accettori che risiedono vicino al fluorocromo eccitato; questo particolare
fenomeno prende il nome di trasferimento di energia per risonanza (FRET)
e consiste nell’interazione fra gli stati eccitati di due molecole di
fluorocromo, in cui lʹeccitazione di una molecola donatore è trasferita ad
una molecola accettore, senza emissione di fotoni. Questo fenomeno
dipende dalla distanza intermolecolare (R), diventando molto evidente a
distanze fra donatore e accettore dellʹordine di 40 ‐ 100 Å.
7. Photobleaching è la scomparsa irreversibile della fluorescenza a causa di
un’eccessiva intensità della luce stimolante che degrada il fluorocromo.
Questo fenomeno viene sfruttato in una tecnica nota come il recupero di
fluorescenza dopo photobleaching (FRAP) per indagare sulla diffusione e
sul movimento delle macromolecole biologiche. Il metodo si basa sul
photobleaching di una regione ben definita del campione esposta ad
un’intensa luce laser. Successivamente vengono analizzati i tassi e le
modalità di recupero della fluorescenza nell’area interessata. Una tecnica
correlata, nota come la perdita di fluorescenza in photobleaching (FLIP), è
impiegata per monitorare la diminuzione della fluorescenza in una regione
adiacente a quella dove si è verificato il photobleaching. Simile al FRAP,
quest'ultima tecnica è utile per lo studio della mobilità molecolare e delle
dinamiche delle cellule viventi.
8. Concentrazione: quando la soluzione in esame è molto concentrata si
sviluppa quello che viene chiamato "effetto filtro", per cui la radiazione
viene assorbita interamente dalle molecole di superficie e non è in grado di
raggiungere la profondità adatta all'emissione del fotone a 90°. Si osserva
128
sperimentalmente che una soluzione molto concentrata, che dovrebbe avere
il massimo di fluorescenza, rivela allo spettrofluorimetro assenza completa
di essa. I fotoni in realtà vengono emessi, ma in quantità minore a quelli
aspettati e con una angolazione diversa per cui non raggiungono il
rivelatore. Man mano che la soluzione viene diluita si osserva, prima un
aumento della fluorescenza proporzionalmente alla diminuzione dell'effetto
filtro, e, conseguentemente, una diminuzione di questa giustificata
dall'aumento della diluizione. L’emissione fluorescente aumenta con la
concentrazione in modo lineare solo per valori bassi di quest’ultima.
9. Rigidità interna: la rigidità strutturale determina un aumento della
fluorescenza; nelle molecole flessibili c’è una maggiore probabilità che
avvenga un processo di conversione interna con conseguente disattivazione
non radiante. Per aumentare la rigidità strutturale di una molecola flessibile
si può farla adsorbire su una superficie solida oppure formare chelati a
struttura rigida.
10. Struttura della molecola: i composti che presentano sistemi di doppi
legami coniugati, si prestano molto bene alla fluorescenza; in particolare
molecole aromatiche, nelle quali, per il fenomeno di risonanza, i doppi
legami risultano sparsi per tutta la struttura, se eccitate danno luogo a
transizioni π→π* favorendo la fluorescenza.
11. Tipi di transizione: per la maggior parte delle molecole organiche sono
possibili quattro transizioni. La fluorescenza è legata principalmente a
transizioni π–π* o n–π*, a seconda di quale delle due abbia energia minore.
Empiricamente si osserva che la fluorescenza si presenta più comunemente
in composti nei quali la transizione ad energia più bassa è del tipo π–π*. La
resa quantica di questo tipo di transizione è maggiore sia perché c’è una
più alta probabilità che avvenga (la vita dello stato eccitato di una
transizione π–π* è di 10-9-10-7 secondi contro i 10-7-10-5 secondi dello stato
eccitato di una transizione n–π*), sia perché i processi che competono con
la fluorescenza avvengono con minore facilità.
129
8.4 Fluorofori
Esistono migliaia e migliaia di fluorofori, alcuni costituiti da molecole
organiche ed altri da molecole inorganiche. In ambito biochimico, è possibile
dividere i fluorofori organici in due grandi categorie, gli intrinseci e gli
estrinseci, corrispondenti rispettivamente a quelli che si trovano in natura e a
quelli di sintesi. Tra gli intrinseci rientrano, ad esempio, gli aminoacidi
aromatici, il NADH, le flavine e le clorofille. Alcune proteine mostrano
fluorescenza instrinseca dovuta agli aminoacidi triptofano, tirosina e
fenilalanina. Il gruppo indolo del triptofano è la fonte dominante di
assorbimento nell’UV e di emissione delle proteine. L’emissione del
triptofano è molto sensibile per quanto riguardano i parametri chimico-fisico
ed è pertanto sfruttato per ottenere informazioni sulla conformazione delle
proteine. Un esempio importante di fluorofori intrinseci è la green fluorescent
protein (GFP) che è prodotta naturalmente dalla medusa Aquorea victoria. La
GFP contiene un gruppo altamente fluorescente in una regione interna della
sua struttura. La caratteristica più importante della GFP è che il gene per la sua
produzione può essere inserito in cellule di vario tipo mediante ingegneria
genetica, allo scopo di rendere fluorescenti le cellule, i tessuti da esse
composte o addirittura interi organismi. Tra i fluorofori estrinseci vi sono ad
esempio i dansili, la fluoresceina, le rodamine e le cianine. Questi fluorofori
sono spesso utilizzati per rendere fluorescenti delle biomolecole di interesse
che non hanno fluorescenza intrinseca, come il DNA e i lipidi. Nel caso delle
proteine, spesso è necessario conferire fluorescenza a lunghezze d’onda
maggiori di quelle a cui emettono gli aminoacidi, per evitare il fondo di
fluorescenza delle matrici biologiche. Il numero di fluorofori per il
biolabelling (bio-etichettatura) è cresciuto in modo significativo negli ultimi
anni ed esistono degli handbook dei label fluorescenti dove è possibile
selezionare i fluorofori di interesse. In particolare, molti fluorofori sono
disponibili con funzioni chimiche adatte alla formazione di legami covalenti o
non covalenti con le biomolecole di interesse. I più comuni sono la
130
fluoresceina e le rodamine, che hanno massimi di assorbimento intorno a 490
e 570 nm e massimi di emissione intorno a 520 e 600 nm rispettivamente,
hanno elevate rese quantiche di fluorescenza, hanno coefficienti di
assorbimento molare molto elevati (~ 80000 M-1cm-1) e sono disponibili in
un’ampia varietà di derivati come isotiocianati (reattivi verso le ammine),
iodoacetamidi e maleimidi (reattivi verso i tioli). Una delle caratteristiche più
importanti di un fluoroforo è la fotostabilità. Tutti i fluorofori organici tendono
a danneggiarsi sotto irraggiamento continuo ed intenso (photobleaching). In
particolare, la fluoresceina è uno dei label meno fotostabili. Esistono altri tipi
di sostanze luminescenti che presentano photobleaching minore, come
nanoparticelle di semiconduttori, composti a base di lantanidi e complessi
metallorganici con metalli di transizione.
8.5 Fluorescenza dei derivati benzotienopirimidinici
Le proprietà fluorescenti dei composti benzotienopirimidinici (5-15) sono state
opportunamente analizzate in modo da poterli utilizzare come potenziali
rivelatori di tumori colon-rettali. Uno degli ostacoli più grandi nella lotta al
cancro è l'impossibilità di delimitare con certezza i confini della massa
tumorale, rendendo difficili eventuali interventi chirurgici. Chi opera è spesso
costretto ad asportare, qualora se ne ravveda il bisogno, anche i tessuti non
direttamente colpiti dalla malattia. Per contrastare questo e altri problemi, un
gruppo di ricerca della Vanderbilt University (Tennessee) ha sviluppato dei
composti in grado di “accendere la luce” all’interno delle cellule tumorali.
L'illuminazione delle cellule tumorali avverrebbe grazie a degli inibitori
fluorescenti dell'enzima COX-2, una proteina che, se presente in grandi
quantità, può rappresentare il campanello d'allarme di un tumore. Questa
caratteristica lo rende particolarmente adatto per le tecniche di imaging
molecolare, il cui obiettivo è ricavare una “fotografia” di ciò che avviene a
livello cellulare. COX-2 compare già nelle prime fasi della crescita tumorale,
per poi aumentare con la malignità del tumore. Assodato che l'enzima non è
131
presente nei tessuti sani, la sua presenza può essere utilizzata come un vero e
proprio “faro” nella ricerca delle cellule maligne. In base a tali considerazioni,
sono state analizzate le proprietà fluorescenti dei sei derivati che hanno
presentato una spiccata attività anti-infiammatoria. Gli spettri d’assorbimento,
eccitazione e emissione sono stati eseguiti in DMSO ad una concentrazione
dell’ordine di 10-5 M. Negli spettri d’assorbimento UV-vis i derivati hanno
presentato due picchi con un massimo d’assorbimento in corrispondenza di
due diverse lunghezze d’onda (∼330 e ∼350 nm) (Fig.33).
Figura 33: Spettri d’assorbimento
Per evitare fenomeni di riassorbimento abbiamo eccitato i composti a 368 nm
(nella parte terminale dello spettro). Gli spettri di emissione come chiaramente
si può notare nel grafico hanno mostrato un λem massima intorno a 415-450
nm (Fig.34). Tutti gli spettri sono stati normalizzati alla stessa assorbanza di
A368= 0.074e alla lunghezza d'onda di eccitazione di 368 nm.
132
Figura 34: Spettri di emissione
Per valutare la fluorescenza di ogni singolo derivato abbiamo misurato la resa
quantica, quest’ultima è stata calcolata prendendo come composto di
riferimento il solfato di chinino disciolto in H2SO4 0.5 M ed effettuando varie
diluizioni tali che l’assorbanza, nella lunghezza d’onda di eccitazione,
corrispondesse a quella dei composti da testare (Tab.3)
Composti
ε (λmax)
ε (λmax)
Φfl
7
8,304(338)
7,500(352)
0.013
10
9,784(338)
8,905(352)
0.055
12
10,773(338)
9.855(352)
0.007
13
10,991(340)
9,916(354)
0.080
14
9,662(338)
8,199(350)
0.032
15
9,435(334)
8,643(346)
0.087
Tabella 3: Resa quantica dei derivati benzotienopirimidinici
133
Tutti i composti hanno mostrato una buona fluorescenza, in particolare i
composti 15 e 13 hanno presentato i più alti valori di resa quantica. La
fluorescenza dei derivati benzotienopirimidinici è dovuta al gruppo
benzotienico mentre la presenza di diversi sostituenti (-COOH, -NO2, F)
nell’anello aromatico può aumentare, diminuire o addirittura spegnere tale
fluorescenza. Dati riportati in letteratura a tal proposito asseriscono che:
gli atomi pesanti (in particolare gli alogeni come Cl, Br, F, I) riducono la
resa quantica di fluorescenza poiché facilitano il decadimento del
singoletto eccitato S1 allo stato di Tripletto T1 mediante conversione
intersistema;
i gruppi –COOH possiedono un comportamento molto complesso, legato
all’ orientamento del gruppo carbossilico rispetto al nucleo aromatico;
i gruppi –NO2 elettron-attrattori diminuiscono la delocalizzazione degli
elettroni π e quindi indeboliscono o spengono la fluorescenza. Inoltre
anche l’uso di solventi contenenti sostituenti –NO2 generano dei campi
magnetici attorno a questi nuclei che possono provocare disaccoppiamento
di spin paralleli e favorire la formazione dello stato di tripletto con
spegnimento della fluorescenza.
In base a tali considerazioni si può affermare che le misure quantiche di
fluorescenza ottenute dai sei derivati risultano essere in linea con i dati
riportati in letteratura.
8.6 Saggi in vitro dei derivati fluorocoxib sulla linea cellulare HCA-7
Studi clinici hanno dimostrato che la cicloossigenasi-2 è un potenziale
bersaglio per la rivelazione di tumori colon-rettali. Pertanto, la caratteristica
dei derivati 7, 10, 12, 13, 14 e 15, battezzati “fluorocoxib”, dovrebbe essere
quella di identificare lesioni precancerose nell’intestino, quelle lesioni cioè
che non sono ancora a tutti gli effetti dei tumori maligni, ma lo stanno per
diventare. Una volta identificate tali lesioni, queste sostanze si “attaccano” alle
cellule alterate “illuminandole” permettendo così di poter prevenire lo
134
sviluppo di un vero e proprio tumore. Esperimenti di immunostaining sono
stati effettuati in vitro su una linea cellulare tumorale HCA-7 con i derivati 7,
10, 12, 13, 14 e 15. I composti sono stati incubati per 12 h alla concentrazione
di 100μM, come si può notare dalle immagini ottenute solo i composti 10 e 15
sono riusciti ad attraversare la membrana cellulare delle cellule HCA-7 tramite
diffusione passiva e ad emettere la propria autofluorescenza (Fig.35) mentre
gli altri derivati pur presentando una discreta fluorescenza non sono riusciti ad
entrare nelle cellule tumorali.
Figura 35: Esperimenti al microscopio a fluorescenza
Dalle immagini si evince che i derivati 10 e 15 riescono ad attraversare la
membrana cellulare ma non quella nucleare, infatti, i nuclei cellulari si
presentano di color nero. Questi dati preliminari saranno in futuro supportati
da esperimenti in vivo su topi da laboratorio, in maniera tale da poter
impiegare questi derivati come possibili markers tumorali in diagnostica
biomedica basata su tecniche spettrofluorimetriche e citofluorimetriche, in
modo da rimpiazzare i coloranti convenzionali attualmente in uso.
135
Materiali e Metodi
8.7 Spettroscopia di fluorescenza
Gli spettri di assorbimento sono stati registrati con uno spettrofotometro UVVIS HP (8452A), e gli spettri di fluorescenza sono stati ottenuti con una
Spettrofluorimetro Spex Fluorolog-2 (Mod. F-111). I coefficienti di estinzione
molare (ε) sono stati determinati solubilizzando i sei derivati in DMSO, ad una
concentrazione variabile che va da 1 a 50 μM e ad un’assorbanza
nell’intervallo tra 200 e 800 nm in cuvette di quarzo standard. I valori di
epsilon sono stati determinati applicando la legge di Lambert-Beer:
A=εdc
dove (A) è l'assorbanza; (c) è la concentrazione molare della soluzione e (d) è
la lunghezza del cammino ottico. Le misure sono state ripetute per ben tre
volte. Gli spettri di emissione dei derivati 7, 10, 12, 13, 14 e 15 sono stati
registrati in DMSO. Gli spettri di emissione sono stati ottenuti nell’intervallo
che va da 388 a 700 nm, ad una lunghezza d'onda di eccitazione di 368 nm.
Gli spettri di emissione ottenuti hanno presentato un λem massima intorno a
415-450 nm. Tutti gli spettri sono stati normalizzati alla stessa assorbanza di
A368= 0.074e alla lunghezza d'onda di eccitazione di 368 nm. La resa quantica,
quest’ultima è stata calcolata prendendo come composto di riferimento il
solfato di chinino disciolto in H2SO4 0.5 M ed effettuando varie diluizioni tali
che l’assorbanza, nella lunghezza d’onda di eccitazione, corrispondesse a
quella dei composti da testare. Il Φx rendimento quantico per ogni campione è
stata calcolata secondo la seguente equazione:
Φx = ΦS(AS/Ax)(FX/FS) (nx/ns)2
dove (Φx) è il rendimento quantico di emissione, (A) è l'assorbanza alla
lunghezza d'onda di eccitazione, (F) è l'area sotto la curva di emissione
corretta, (n) è l'indice di rifrazione del solvente per il campione (X) e per lo
standard (S).
136
8.8 Colture cellulari
La linea cellulare è stata acquistata dall’American Type Culture Collection
(Manassas, VA, USA) e messa in coltura in terreno Dulbecco’S Modified
Eagle’s Medium (DMEM, Cambrex Bio Science, Bergamo, Italia)
supplementato del 10% di siero fetale bovino (FBS, Euroclone, Milano, Italia),
dell’1% di glutammina (Euroclone, Milano, Italia) e dell’1% di antibiotici
(penicillina e streptomicina, Euroclone, Milano, Italia). La linea è stata
mantenuta a 37°C in atmosfera umidificata al 5% di CO2.
8.9 Protocollo di imaging cellulare
Le cellule sono state raccolte utilizzando 0,25% tripsina-EDTA (GIBCO,
25200), piastrate in una piastra da 6 pozzetti ad una densità di 200,000 cellule
a pozzetto. Le cellule sono state incubate per 24h. Prima della
permeabilizzazione sono state lavate due volte con PBS e dopo sono state
sospese in PBS contenente lo 0,1 % di triton X-100 per 5 min. Fissate le
cellule, quest’ultime sono state lavate 3 volte con una soluzione di PBS e
disposte in un vetrino portaoggetti aggiungendo 4 mg/mL di anticorpo
monoclonale anti-COX-2 (Santa Cruz Biotechnology, sc-166475), 2 mg/ml di
Texas-Red anticorpo secondario
monoclonale anti-topo (Santa Cruz
Biotechnology, sc-2781) e una concentrazione di 100μM dei derivati
benzotienopirimidinici, il tutto è stato ricoperto con un vetrino copri oggetto.
Sono stati tenuti al buio per 16h a temperatura ambiente. Successivamente le
cellule sono state lavate con PBS e sono stati posti 10μL su dei vetrini da
microscopia per poter essere visualizzati mediante un microscopio a Scansione
Laser Confocale Zeiss (Germania)
137
9. Conclusioni
I risultati ottenuti hanno dimostrato l’efficacia anti-infiammatoria delle
sostanze in esame su due modelli di linee cellulari: i cheratinociti NCTC 2544
e i macrofagi murini J774. Le sostanze esaminate sono risultate efficaci nel
ridurre notevolmente l’espressione di ICAM-1, iNOS, COX-2 e la
modulazione del rilascio di MCP-1 e IL-8. I dati ottenuti dalla valutazione
biologica hanno indicato che queste sostanze hanno manifestato proprietà antiinfiammatorie estremamente interessanti, poiché sono in grado di contrastare
alcuni effetti pro-infiammatori nella linea cellulare dei cheratinociti umani
normali NCTC 2544 e dei macrofagi murini J774. Inoltre, in base a tali
considerazioni, sono state analizzate le proprietà fluorescenti dei sei derivati
che hanno presentato una spiccata attività anti-infiammatoria. Quasi tutti i
derivati hanno mostrato una buona fluorescenza, in particolare i composti 15 e
13 hanno presentato i più alti valori di resa quantica. Tali risultati hanno
incentivato la nostra ricerca verso un ulteriore studio in vitro di imaging per
verificare la fluorescenza all’interno delle cellule tumorali HCA-7. Gli esiti
hanno confermato che soltanto i derivati 10 e 15 riescono ad attraversare la
membrana cellulare e ad emettere la propria fluorescenza. Questi dati
preliminari saranno in futuro supportati da esperimenti in vivo su topi da
laboratorio, in maniera tale da poter impiegare tali derivati come possibili
markers
tumorali
in
diagnostica
biomedica
basata
su
tecniche
spettrofluorimetriche e citofluorimetriche. In definitiva, lo scopo delle nostre
future attività di ricerca sarà quello di progettare nuovi composti dalle elevate
proprietà fluorescenti, anti-infiammatorie e antineoplastiche.
138
Ringraziamenti
In queste pagine colgo l’occasione per ringraziare tutte le persone che mi sono
state vicine e che hanno fatto parte di questo lungo percorso della mia vita.
un grazie sincero,
alla Prof.ssa Venera Cardile
per aver creduto sin dall’inizio nelle mie potenzialità e per avermi
pazientemente seguito nella realizzazione del mio lavoro.
al Prof. Luigi Casella
per l’imprescindibile supporto morale e per avermi trasmesso il suo amore e la
sua dedizione verso la ricerca scientifica.
al Prof. Venerando Pistarà
per l’infinita disponibilità, il prezioso aiuto scientifico e la grande umanità che
lo contraddistingue.
al Prof. Salvatore Pistarà
andrà per sempre il mio grazie…. per esser stato un “Professore-amico”,
offrendomi nel buio lo spiraglio di luce che talvolta non riuscivo a vedere, per
avermi sostenuta fin qui accompagnandomi con dedizione profonda e per
avermi insegnato la dignità di vivere rifiutando il compromesso.
al Dott. Alfio Catalfo e al Prof. Guido De Guidi
per la straordinaria disponibilità dimostrata nel fornirmi sempre un valido
supporto tecnico-scientifico per quel che concerne gli aspetti di spettrometria e
di fluorescenza.
139
al Prof. Salvatore Sortino,
andrà sempre un ringraziamento speciale e la mia riconoscenza per avermi
dato l’ opportunità di iniziare questo percorso formativo nel mondo della
ricerca universitaria, trasmettendomi la sua enorme passione, conoscenza
scientifica ed esperienza professionale.
al Prof. Andrea Santagati
in qualità di tutor.
140
Bibliografia
[1] Frati L, Russo M. (2000) Universo del corpo. Istituto dell’ enciclopedia TRECCANI s.p.a.
[2] Rossi F, Cuomo V, Riccardi C. (2011) Farmacologia principi di base e
applicazioni terapeutiche. MINERVA MEDICA seconda edizione.
[3] Clementi F, Fumagalli G. (2012) Farmacologia generale e molecolare.
UTET Scienze mediche, quarta edizione.
[4] Balkwill F, Mantovani A. (2001) Inflammation and cancer: back to
Virchow? Lancet. 17:539-545.
[5] Dannenberg AJ, Altorki NK, Boyle JO, et al. (2001) Cyclo-oxigenase 2: a
pharmacological target for prevention of cancer. Lancet Oncology 2:544-551.
[6] Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, Dubois RN.
(2005)
Cyclooxygenase
2
and
epidermal
growth
factor
receptor:
pharmacological targets for chemoprevention. J. Clin. Oncol. 23:254-66.
[7] Rang HP, Maureen Dale M, Ritter JM, Flower RJ, Henderson G. (2012)
Farmacologia . ELSEVIER, settima edizione.
[8] Lemke TL, Williams DA, Foye WO. (2011) Principi Di Chimica
Farmaceutica. PICCIN quinta edizione.
[9] Mondovì B. (1977) Biochimica Applicata. UTET.
141
[10] Howe LR, Subbaramaiah K, Chung WJ, Dannenberg AJ, Brown AMC.
(1999) Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed
mouse mammary epithelial cells. Cancer Res. 59:1572-1577.
[11] Vadlamudi R, Mandal M, Adam L, Steinbach G, Mendelsohn J, Kumar
K. (1999) Regulation of cyclooxygenase-2 pathway by HER2 receptor.
Oncogene. 18:305-314.
[12] Rocca B, Spain LM, Pure E, Langenbach R, Patrono C, FitzGerald G.
(1999) Distinct roles of prostaglandin H synthases 1 and 2 in T-cell
development. J. Clin. Invest. 103:1469-1477.
[13] Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF.
(1993) Expression of a mitogen-inducible cyclooxygenase in brain neurons:
regulation by synaptic activity and glucocorticoids. Neuron. 11:371-386.
[14] Kniss DA. (1999) Cyclooxygenases in reproductive medicine and
biology. J. Soc. Gynecol. Invest. 6:285-292.
[15] Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD.
(1994) Cyclooxygenase-2 is associated with the macula densa of rat kidney
and increases with salt restriction. J. Clin. Invest. 94:2504-2510.
[16] Vio CP, Cespedes C, Gallardo P, Masferrer JL. (1997) Renal
identification of cyclooxygenase-2 in a subset of thick ascending limb cells.
Hypertension. 30:687-692.
[17] Ferreri NR, An SJ, McGiff JC. (1999) Cyclooxygenase-2 expression and
function in the medullary thick ascending limb. Am. J. Physiol. 277:F360F368.
142
[18] Lanza A. (1998) A guideline for the treatment and prevention of NSAIDinduced ulcers. Members of the Ad Hoc Committee on Practice Parameters of
the American College of Gastroenterology. Am. J. Gastroenterol. 93:20372046.
[19] Singh G, Ramey DR. (1998) NSAID induced gastrointestinal
complications: the ARAMIS perspective. J. Rheumatol. 25:8-16.
[20] Talley JJ. (1999) Selective inhibitors of cyclooxygenase-2 (COX-2). Prog.
Med. Chem. 36:201-234.
[21] Litvinov VP. (2004) Thienopyrimidines: synthesis, properties, and
biological activity. Russ. Chem. Bull. 53:487–516.
[22] Alagarsamy V, Vijayakumar S, Solomon VR. (2007) Synthesis of 2mercapto-3-substituted-5,6-dimethylthieno[2,3-d]
pyrimidin-4(3H)-ones as
new analgesic, anti-inflammatory agents. Biomed. Phamacol. 61:285-291.
[23] Bhutyan M, Rahman M, Abdurrahim K, Hossain M, Abunaser M. (2006)
Synthesis and anti microbial evaluation of some new thienopyrimidine
derivatives. Acta Pharm. 56:411-450.
[24] El-Sherbeny MA, El-Ashmawy MB, El-Subbagh HI, El-Emam AA.
(1995) Synthesis, antimicrobial and antiviral evaluation of certain
thienopyrimidine derivatives. Eur. J. Med. Chem. 30:445-449.
[25] Alagarsamy V, Meena S, Ramseshu KV, Solomon VR, Thirumuruganb
K, Dhanabala K, Murugan M. (2006) Synthesis, analgesic, anti-inflammatory,
ucerogenic index and antibacterial activities of novel 2-methylthio-3-
143
substituted-5,6,7,8-tetrahydrobenzo (b) thieno[2,3-d]pyrimidin-4-(3H)-ones.
Eur. J. Med. Chem. 41:1293-1300.
[26] Santagati A, Modica M, Santagati M, Cutuli V, Amore D, Caruso A.
(1995) Synthesis and pharmacological properties of thieno[2',3':4,5]pyrimido
[2,1-b][1,3,4]thiadiazine derivatives. Farmaco. 50:605-609.
[27] Santagati A, Granata G, Marrazzo A, Santagati M. (2003) Synthesis and
effects on the COX-1 and COX-2 activity in human whole blood ex vivo of
derivatives
containing
the
[1]benzothienol-[3,
2-d]pyrimidin-4-one
heterocyclic system. Arch. Pharm. Med. Chem. 336:429-435.
[28] DeWitt DL. (1999) Cox-2-selective inhibitors: the new super aspirins.
Mol. Pharmacol. 55:625-631.
[29] Dannhart G, Kiefer W. (2001) Cyclooxygenase inhibitors-current status
and future prospects. Eur. J. Med. Chem. 36:109-126.
[30] Wamhoff H, Lichtenthaler L. (1978) Heterocyclische β-enaminoester, 22:
pyrido[2,3-d]pyrimidine
tetrahydropyridin
und
aus
isocyanaten,
2-amino-3-ethoxycarbonyl-1,4,5,6isothiocyanaten,
imidsäureestern,
formamid und lactimethern. Chem. Ber. 111:2297-2306.
[31] Wamhoff H. (1985) Advances in Heterocycles Chemistry: Heterocycles,
b-enamino esters, versatile synthons in heterocyclic synthesis. Academic
Press, Orlando.
[32] Cardile V, Lombardo L, Granata G, Perdicaro A, Balazy M, Santagati A.
(2009) Inhibition of iNOS and COX-2 in human whole blood ex vivo and
144
monocyte-macrophage J774 cells by a new group of aminothiopyrimidone
derivatives. Bioorg. Med. Chem. 17:1991-1996.
[33] Cardile V, Frasca G, Rizza L, Rapisarda P, Bonina F. (2010)
Antiinflammatory effects of a red orange extract in human keratinocytes
treated with interferon-gamma and histamine. Phytother. Res. 24:414-418.
[34] Gewald K, Schinke E, Bottcher H. (1996) 2-Amino-thiophene aus
methylenaktiven nitrilen, carbonylverbindungen und scwefel. Chem. Ber.
99:94-100.
[35] Elslager E, Jacob P, Werbel L. (1972) Folate antagonists. 6. synthesis
and antimalarial effects of fused 2,4-diaminothieno[2,3-d] pyrimidine(1-3). J.
Heterocycl. Chem. 11:775-782.
[36] Hayashi S, Sumi, Ueno N, Murase A, Takada J. (2011) Discovery of a
novel COX-2 inhibitor as an orally potent anti-pyretic and anti-inflammatory
drug: Design, synthesis, and structure-activity relationship. Biochem.
Pharmacol. 82:755-768.
[37] Bondock S, Rabie R, Etman HA, Fadda AA. (2008) Synthesis and
antimicrobial activity of some new heterocycles incorporating antipyrine
moiety. Eur. J. Med. Chem. 43:2122-2129.
[38] Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA,
Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC,
Stallings
WC.
(1996)
Structural
basis
for
selective
inhibition
of
cyclooxygenase-2 by anti-inflammatory agents. Nature. 384:644-648.
145
[39] Meade EA, Smith WL, DeWitt DL. (1993) Differential inhibition of
prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by
aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem.
268:6610-6614.
[40] Kock A, Schwarz T, Kirnbauer R, Urbanski A, Perry P, Ansel JC, Luger
TA. (1990) Human keratinocytes are a source for tumor necrosis factor
alpha: evidence for synthesisand release upon stimulation with endotoxin or
ultraviolet light. J. Exp. Med. 172:1609-1614.
[41] Kupper TS, Ballard DW, Chua AO, McGuire JS, Flood PM, Horowitz
MC, Langdon R, Lightfoot L, Gubler U. (1986) Human keratinocytes contain
mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA.
Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to
interleukin 1. J. Exp. Med. 164:2095-2100.
[42] Steinberg TH, Silverstein SC. (1987) Extracellular ATP4- promotes
cation fluxes in the J774 mouse macrophage cell line. J. Biol. Chem.
262:3684-3688.
[43] Greenberg SS, Zhao X, Wang JF, Hua L, Ouyang J. (1997) cAMP and
purinergic P2y receptors upregulate and enhance inducible NO synthase
mRNA and protein in vivo. Am. J. Physiol. 273:L967-L979.
[44] Palomo C, Oiarbide M, Lo´pez R, Go´mez-Bengoa E. (2000)
Phosphazene bases for the preparation of biaryl thioethers from aryl iodides
and arenethiols. Tetrahedron Lett. 41:1283–1286.
[45] Plount Price LP, Jorgensen WL. (2000) Analysis of binding affinities for
Celecoxib analogues with COX-1 and COX-2 from combined docking and
146
Monte Carlo simulations and insight into the COX-1/COX-2 selectivity. J. Am.
Chem. Soc. 122:9455-9466.
[46] Singh P, Sharma P, Bisetty K, Mahaian MP. (2011) Synthesis and
docking studies of thiophene scaffolds in COX-2. Arkivoc. 11:55-70.
[47] Barone M, Graziano ACE, Marrazzo A, Gemmellaro P, Santagati A,
Cardile V. (2013) Synthesis and biological evaluation of new benzothieno[3,2-d]pyrimidin-4-one sulphonamide thio-derivatives as potential
selective cyclooxygenase-2 inhibitors. Molecular Diversity. 17:445-458.
147
Scaricare