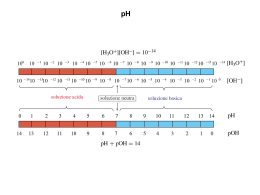
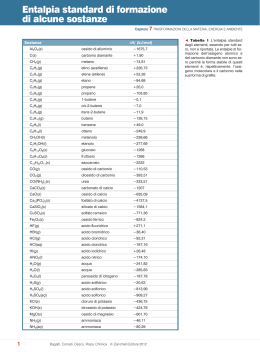
Elenco delle esperienze di laboratorio Equilibri acido-base, pH, idrolisi Blu Preparazione di una soluzione 0,5 M di HCl Preparazione di una soluzione 0,5 M di CH3COOH Preparazione di una soluzione 0,5 M di NaOH Preparazione di una soluzione 0,5 M di NH4OH Costruzione di una scala colorimentrica di pH utilizzando l’indicatore Violetto di metile e Blu di bromotimolo Costruzione di una scala colorimentrica di pH utilizzando l’indicatore Fenolftaleina e di bromotimolo Preparazione di una soluzione 0,1 M di NaCl Preparazione di una soluzione 0,1 M di cloruro di ammonio (NH4Cl) Preparazione di una soluzione 0,1 M di carbonato sodico (Na2CO3) Preparazione di una soluzione tampone acido acetico/acetato di sodio 0,1 M e 0,001 M Preparazione di una soluzione tampone NH4OH /NH4Cl 0,1 M e 0,001 M Equilibri di solubilità (effetto del pH e dello ione comune) Preparazione di una soluzione di PbCl2 satura e verifica dell’effetto dell’aggiunta di NaCl Preparazione di una soluzione di Pb(NO3)2 0,2 M e verifica dell’effetto dell’aggiunta di NaBr 0,2 M Separazione di ioni mediante precipitazione selettiva di sali per superamento del prodotto di solubilità Precipitazione e ridiscioglimento del precipitato (effetto del pH sulla solubilità) Spostamento dell’equilibrio chimico Dipendenza della solubilità dal pH Precipitazione acetato Ag+ (effetto dello ione comune) Determinazione del Kps dell’acetato di Ag+ Reazioni di ossidoriduzione Ossidabilità di metalli (Zn, Fe, Cu) ad opera di HCl e HNO3 Potenziali redox dei metalli: reazioni di ossidoriduzione coinvolgenti coppie di specie metalliche (Cu/Zn, Pb/Cu) Potenziali redox dei non metalli (ossidazione I- e Br-) Costruzione di una pila Daniell Decomposizione termica di solfato di rame pentaidrato Cristallizzazione frazionata di NaCl e KNO3 Preparazione del nitrato di potassio e cloruro di sodio Analisi della purezza dei cristalli Sintesi e reattività di cloruro di calcio Preparazione del cloruro di calcio Reazioni del cloruro di calcio Preparazione dell’allume di cromo e reazioni del cromo e del bicromato Preparazione dell’allume di cromo Reazioni degli ioni Cr3+ Reazioni del dicromato di potassio Sintesi e reattività di cloruro mercurico Preparazione del cloruro mercurico Reazioni del cloruro mercurico 20 Equilibri acido-base, pH, idrolisi Reagenti: HCl, CH3COOH, NaOH, NH4OH, NaCl, NH4Cl, Na2CO3, indicatori (metilarancio, fenolftaleina, blu di bromotimolo) Rischi connessi a quest’esperienza: manipolazione di acidi e basi forti Precauzioni: indossare i guanti, operare sotto cappa quando si adoperino soluzioni ammoniacali Finalità: scopo di quest’esperienza è quello di apprendere a preparare soluzioni a molarità nota, per diluizione di soluzioni più concentrate o per pesata di quantità precise di soluto; verificare le proprietà acido/base di varie sostanze; preparare e verificare le proprietà di soluzioni tampone. A) Preparazione di una soluzione 0,5 M di HCl In un matraccio tarato, preparare 50 ml di una soluzione 0,5 M di HCl a partire dall’acido concentrato, dopo aver calcolato la molarita di quest’ultimo, sulla base dei dati riportati in fondo alle dispense. Calcolare il pH della soluzione e, successivamente, misurarlo utilizzando le apposite cartine indicatrici ed il pH-metro. Annotare il risultato della misura. Conservare la soluzione per le esperienze successive. Le cartine indicatrici non vanno utilizzate immergendole direttamente nella soluzione! Occorre depositare sulla cartina stessa una goccia della soluzione, aiutandosi con una bacchetta di vetro. B) Preparazione di una soluzione 0,5 M di CH3COOH In un matraccio tarato, preparare 50 ml di una soluzione 0,5 M di CH3COOH a partire dall’acido acetico glaciale, dopo aver calcolato la molarita di quest’ultimo, sulla base dei dati riportati in fondo alle dispense. Calcolare il pH della soluzione (Ka=1,76⋅10-5) e, successivamente, misurarlo utilizzando le apposite cartine indicatrici ed il pH-metro. Annotare il risultato della misura. Conservare la soluzione per le esperienze successive. C) Preparazione di una soluzione 0,5 M di NaOH In un matraccio tarato, preparare 50 ml di una soluzione 0,5 M di NaOH (che è un solido e va pesato sulla bilancia tecnica). Calcolare il pH della soluzione e, successivamente, misurarlo utilizzando le apposite cartine indicatrici ed il pH-metro. Annotare il risultato della misura. Conservare la soluzione per le esperienze successive. D) Preparazione di una soluzione 0,5 M di NH4OH In un matraccio tarato, preparare 50 ml di una soluzione 0,5 M di NH4OH a partire da NH4OH concentrata, dopo aver calcolato la molarita di quest’ultima, sulla base dei dati riportati in fondo alle dispense. Calcolare il pH della soluzione (Kb=1,79⋅10-5) e, successivamente, misurarlo utilizzando le apposite cartine indicatrici ed il pH-metro. Annotare il risultato della misura. Conservare la soluzione per le esperienze successive. 21 E) Costruzione di una scala colorimentrica di pH utilizzando l’indicatore metilarancio e blu di bromotimolo A partire dalla soluzione di HCl 0,5 M preparare (per diluizioni successive) 10 ml di: HCl 0,1 M, 0,01 M, 0,001 M, 1x10-4 M, 1x10-5 M. Calcolare il pH di ciascuna soluzione. Suddividere il contenuto di ciascuna provetta in due parti. Alla prima serie di 5 provette addizionare una goccia di indicatore metilarancio. Alla seconda serie di 5 provette addizionare una goccia di indicatore blu di bromotimolo. Agitare ciascuna soluzione con una bacchetta di vetro ed annotare il colore. Disporre le provette in modo da costruire una scala cromatica in funzione del pH. Prelevare in distributorio una soluzione acida a pH ignoto e determinarne il pH utilizzando le due scale colorimetriche appena costruite. Annotare il risultato della misura. F) Costruzione di una scala colorimentrica di pH utilizzando gli indicatori fenolftaleina e blu di bromotimolo A partire dalla soluzione di NaOH 0,5 M preparare (per diluizioni successive) 10 ml di: NaOH 0,1 M, 0,01 M, 0,001 M, 1x10-4 M, 1x10-5 M. Calcolare il pH di ciascuna soluzione. Suddividere il contenuto di ciascuna provetta in due parti. Alla prima serie di 5 provette addizionare una goccia di indicatore fenolftaleina. Alla seconda serie di 5 provette addizionare una goccia di indicatore blu di bromotimolo. Agitare ciascuna soluzione con una bacchetta di vetro ed annotare il colore. Disporre le provette in modo da costruire una scala cromatica in funzione del pH. Prelevare in distributorio una soluzione basica a pH ignoto e determinarne il pH utilizzando le due scale colorimetriche appena costruite. Annotare il risultato della misura. G) Preparazione di una soluzione 0,1 M di NaCl Preparare 50 ml di una soluzione 0,1 M di NaCl. Calcolare il pH della soluzione e, successivamente, misurarlo utilizzando le apposite cartine indicatrici. Annotare il risultato della misura. Addizionare alla soluzione una goccia di indicatore blu di bromotimolo ed annotare il colore osservato. H) Preparazione di una soluzione 0,1 M di cloruro di ammonio (NH4Cl) Preparare 50 ml di una soluzione 0,1 M di NH4Cl. Calcolare il pH della soluzione (Kb=1,79⋅10-5) e, successivamente, misurarlo utilizzando le apposite cartine indicatrici. Annotare il risultato della misura. Addizionare alla soluzione una goccia di indicatore metilarancio ed annotare il colore osservato. I) Preparazione di una soluzione 0,1 M di carbonato sodico (Na2CO3) Preparare 50 ml di una soluzione 0,1 M di Na2CO3. Calcolare il pH della soluzione (k1 = 4,16 x 107 e k2 = 4,84 x 10-11) e, successivamente, misurarlo utilizzando le apposite cartine indicatrici. Annotare il risultato della misura. Addizionare alla soluzione una goccia di indicatore fenolftaleina ed annotare il colore osservato. 22 L) Preparazione di una soluzione tampone acido acetico/acetato di sodio 0,1 M e 0,001 M. Preparare 100 ml di una soluzione tampone acido acetico/acetato di sodio 0,1 M, addizionando 50 ml di soluzione di acido acetico 0,1 M (che si prepara per diluizione della soluzione 0,5 M preparata in precedenza) con 50 ml di soluzione 0,1 M di acetato di sodio (preparata sciogliendo l’acetato in acqua deionizzata). Calcolare il pH della soluzione (Ka=1,76⋅10-5) e, successivamente, misurarlo utilizzando le apposite cartine indicatrici. A partire da questa soluzione preparare, per diluizione, una soluzione tampone 0,001 M. Calcolare il pH della soluzione e, successivamente, misurarlo utilizzando le apposite cartine indicatrici ed il pH-metro. Annotare il risultato della misura. A 25 ml di soluzione tampone 0,1 M addizionare 1 ml di HCl 0,1 M. Come varia il pH? A 25 ml di soluzione tampone 0,001 M addizionare 1 ml di HCl 0,1 M. Come varia il pH? A 25 ml di soluzione tampone 0,1 M addizionare 1 ml di NaOH 0,1 M. Come varia il pH? A 25 ml di soluzione tampone 0,001 M addizionare 1 ml di NaOH 0,1 M. Come varia il pH? Annotare quanto osservato. M) Preparazione di una soluzione tampone NH4OH3/NH4Cl 0,1 M e 0,001 M. Preparare 100 ml di una soluzione tampone NH4OH/NH4Cl 0,1 M addizionando 50 ml di sol 0,1 M di ammoniaca (che si prepara per diluizione della soluzione 0,5 M preparata in precedenza) con 50 ml di soluzione 0,1 M di cloruro d’ammonio (preparata sciogliendo il sale in acqua deionizzata). Calcolare il pH della soluzione (Kb=1,79⋅10-5) e, successivamente, misurarlo utilizzando le apposite cartine indicatrici. A partire da questa soluzione preparare, per diluizione, una soluzione tampone 0,001 M. Calcolare il pH della soluzione e, successivamente, misurarlo utilizzando le apposite cartine indicatrici ed il pH-metro. Annotare il risultato della misura. A 25 ml di soluzione tampone 0,1 M addizionare 1 ml di HCl 0,1 M. Come varia il pH? A 25 ml di soluzione tampone 0,001 M addizionare 1 ml di HCl 0,1 M. Come varia il pH? A 25 ml di soluzione tampone 0,1 M addizionare 1 ml di NaOH 0,1 M. Come varia il pH? A 25 ml di soluzione tampone 0,001 M addizionare 1 ml di NaOH 0,1 M. Come varia il pH? Annotare quanto osservato. 23 Equilibri di solubilità (effetto del pH e dello ione comune) Reagenti: PbCl2, NaCl, Pb(NO3)2, NaBr, Ba(NO3)2, Fe(NO3)3, NaOH, H2SO4, HCl, NaHCO3, CuSO4, CH3COONa, AgNO3, FeCl3, KSCN, KCl, CaCl2, H2C2O4, (NH4)2C2O4, NH4OH, allume ferrico Attrezzatura speciale: agitatore magnetico con ancoretta, buretta Rischi connessi a quest’esperienza: manipolazione di acidi e basi forti, manipolazione di composti potenzialmente tossici per contatto con la pelle Precauzioni: indossare i guanti Finalità: scopo di quest’esperienza è verificare sperimentalmente le proprietà degli equilibri chimici (in fase omogenea ed eterogenea) ed i fattori che li influenzano A) Preparazione di una soluzione di PbCl2 satura e verifica dell’effetto dell’aggiunta di NaCl 1) In una beuta da 100 ml porre 40 ml di H2O (misurati con il cilindro graduato) quindi aggiungere del cloruro di piombo (II), a poco a poco, agitando su agitatore magnetico, sino ad ottenere una soluzione satura. Filtrare la soluzione su filtro di carta piano. 2) Aggiungere al filtrato una punta di spatola di NaCl (solido): si osserva la precipitazione di un sale. Quale? B) Preparazione di una soluzione di Pb(NO3)2 0,25 M e verifica dell’effetto dell’aggiunta di NaBr 0,25 M 1) Preparare 75 ml di soluzione di nitrato di piombo(II) 0,25 M e 125 ml di una soluzione di NaBr 0,25 M (per favorire la dissoluzione dei sali, utilizzare l’agitatore magnetico). 2) In un becker mescolare 50 ml della soluzione di nitrato di piombo(II) e 100 ml della soluzione di NaBr. Si ottiene una soluzione satura di bromuro di piombo e si osserva la precipitazione di un sale: quale? Filtrare la soluzione su filtro di carta a pieghe. 3) a) A 50 ml di filtrato si aggiungono 25 ml di soluzione di nitrato di piombo 0,2 M b) A 50 ml di filtrato si aggiungono 25 ml di soluzione di bromuro di sodio 0,2 M. In entrambi i casi si ha precipitazione di un sale. Quale? C) Separazione di ioni mediante precipitazione selettiva di sali per superamento del prodotto di solubilità 1) Preparare 100 ml di una soluzione contenente la medesima quantità molare (ca. 0,1 M) di ioni Pb2+, Ba2+, Fe3+ utilizzando sali solubili (nitrati). Per favorire la dissoluzione dei sali, utilizzare l’agitatore magnetico. 2) Aggiungere HCl concentrato: si osserverà un precipitato bianco di cloruro di piombo (II), sale poco solubile. Proseguire l’aggiunta di acido fino a quando non si osserva più ulteriore precipitazione. 3) Filtrare su filtro di carta, lavare il residuo solido, essiccarlo in stufa e pesarlo. 4) Al filtrato, aggiungere H2SO4 concentrato: si osserverà un precipitato bianco di solfato di bario. Proseguire l’aggiunta di acido fino a quando non si osserva più ulteriore precipitazione; filtrare su filtro di carta, lavare il residuo solido, essiccarlo in stufa e pesarlo. 24 5) Prelevare qualche ml di filtrato e porlo in una provetta o in una beutina. Aggiungervi alcune gocce di soluzione di NaOH 1 M, agitando il contenuto della provetta con una bacchettina di vetro. Si osserva un precipitato scuro di idrossido di ferro (III). Proseguire l’aggiunta fino ad ottenere un precipitato persistente. 6) Ridisciogliere il precipitato (idrossido di ferro) trattandolo con acido cloridrico concentrato sino a dissoluzione totale. 7) Misurare con le cartine indicatrici il pH raggiunto ed annotare il risultato. 8) Mediante aggiunta successiva di NaOH 1 M (procedere lentamente, addizionando la soluzione basica goccia a goccia) individuare la zona di pH in cui inizia la riprecipitazione dell'idrossido (questa volta occorre sospendere l’aggiunta non appena si osserva la comparsa del precipitato). Annotare il risultato. D) Precipitazione e ridiscioglimento del precipitato (effetto del pH sulla solubilità) 1) A 10 ml di una soluzione di solfato di rame 0,1 M aggiungere 10 ml di una soluzione di idrogenocarbonato di sodio 0,1 M (mescolare in una provetta) 2) Ad avvenuta precipitazione di una polvere verde-azzurra, filtrare su filtro di carta piano e riporre il precipitato in un contenitore 3) Far gocciolare sul precipitato alcune gocce di acido cloridrico concentrato, fino a completa dissoluzione del precipitato 4) Aggiungere goccia a goccia una soluzione acquosa di NaOH 1 M alla soluzione così ottenuta Cosa si osserva? E) Spostamento dell’equilibrio chimico In una beuta mescolare 10 ml di soluzione di cloruro ferrico 0,01 M (FeCl3) con 10 ml di soluzione 0,01 M di tiocianato di potassio (KSCN) (data la esigua quantità di sali necessaria, utilizzare la bilancia analitica); diluire con acqua fino a quando non si osserva una colorazione rossa di media intensità. Ripartire la soluzione su quattro provette ed in ciascuna introdurre 10 ml dei seguenti quattro reagenti: 1) acqua 2) cloruro ferrico 0,01 M 3) tiocianato di potassio 0,01 M 4) cloruro di potassio 0,01 M Cosa si osserva? F) Dipendenza della solubilità dal pH 1) Mescolare 10 ml di una soluzione di cloruro di calcio (CaCl2) 0,1 M con 10 ml di acqua distillata e dividere la soluzione fra due provette; alla prima aggiungere 1 ml di soluzione 0,5 M di acido ossalico (H2C2O4); alla seconda 2 ml di soluzione 0,25 M di ossalato di ammonio (NH4)2C2O4 Cosa si osserva nelle due provette? 2) Alla soluzione in cui è stato aggiunto acido ossalico introdurre 1 o 2 ml di HCl 6 M e mescolare. Cosa si osserva? Interpretare l’esperimento. 3) A questa stessa soluzione addizionare un leggero eccesso di NH4OH 6M e mescolare. Si forma un precipitato. Determinare se il precipitato che si è formato possa essere Ca(OH)2 provando ad aggiungere ad una sol. diluita di CaCl2 un po’ di NH4OH 6M 25 Quale precipitato si forma? G) Precipitazione acetato Ag+ (effetto dello ione comune) 1) Preparare una soluzione di acetato di Ag+ (CH3COOAg) mescolando in una beutina 10 ml di soluzione 3 M di acetato di sodio (CH3COONa) con 25 ml di soluzione 0,1 M di nitrato d’argento (AgNO3), disponibile in reagentario. Lasciare riposare la soluzione per qualche minuto, in modo da completare la precipitazione del sale, poi filtrare su filtro di carta. 2) Fare colare tutta la soluzione dal precipitato, lavarlo con 2 ml di acqua distillata e fare di nuovo colare tutto il liquido. Quando il liquido è stato rimosso, mettere una provetta pulita sotto l’imbuto e pungere il filtro con la punta di una bacchetta di vetro, in modo da forarlo. Lavare il precipitato con un piccolo flusso di acqua, per farlo scivolare dentro la provetta, facendo attenzione a non usare più di 10 ml di acqua (ca. 1/3 del volume della provetta). 3) Scaldare leggermente (su becco Bunsen, reggendo le provetta con le apposite pinze!) la miscela nella provetta (che deve risultare tiepida e non calda) e agitarla delicatamente per almeno 10 min., in modo da stabilire l’equilibrio nella sol. satura CH3COOAg (s) ↔ CH3COO- + Ag+ Successivamente filtrare la soluzione in un filtro asciutto, facendo attenzione a trattenere la maggior parte del precipitato nella provetta di partenza. 4) Il liquido filtrato viene diviso in due porzioni di ca. 4-6 ml dentro due provette: alla prima porzione si aggiungono 10 gocce di acetato di sodio 3 M; alla seconda, 10 gocce di acetato di sodio 6 M. Attendere ca. 10 min e osservare il risultato. Cosa si osserva? H) Determinazione del Kps dell’acetato di Ag+ 1) Prelevare dal reagentario in due becker asciutti ca. 130 ml di AgNO3 0,2 M e 130 ml di CH3COONa 0,2 M. Misurando accuratamente i volumi col cilindro graduato preparare le quattro miscele indicate nella tabella seguente: n° 1 2 3 4 AgNO3 0,2 M 20,0 ml 30,0 ml 35,0 ml 40,0 ml + + + + CH3COONa 0,2 M 40,0 ml 30,0 ml 25,0 ml 20,0 ml 2) Mettere ciascuna soluzione in un recipiente asciutto e pulito da 250 ml che deve venire chiuso, onde evitare l’evaporazione del solvente. Mescolare tutte le soluzioni di tanto in tanto, fino a ca. 30 minuti dopo che la precipitazione dell’acetato di argento sembra completa, in modo da fare stabilire l’equilibrio (è preferibile lasciare riposare le soluzioni molto a lungo, anche una notte, se possibile). 3) Pulire una buretta, riempiendola con acqua, sciacquandola poi con porzioni di 5 ml di acqua distillata e lavandola due volte con 5 ml di soluzione standard 0,100 M di tiocianato di potassio (KSCN), facendola gocciolare dal rubinetto. Riempire quindi la buretta con la soluzione di KSCN. 4) Filtrare la miscela n°1 (usando filtro e imbuto asciutti) in un becker asciutto da 250 ml; con un cilindro graduato da 50 ml misurare accuratamente 25,0 ml di filtrato e porre questa soluzione in un becker asciutto contenente un’ancoretta magnetica per la titolazione. Aggiungere alla soluzione 1 ml di soluzione satura di allume ferrico come indicatore e 1 ml di HNO3 6 M. Qualora il colore rosso dovuto al Fe3+ non sparisca completamente, aggiungere qualche goccia di 26 HNO3. Procedere quindi con la titolazione agitando la soluzione con l’ancoretta magnetica su un agitatore magnetico, aprendo poco il rubinetto della buretta, in modo da far cadere la soluzione titolante di KSCN goccia a goccia nella soluzione di acetato di Ag. L’aggiunta di KSCN deve proseguire fino alla prima comparsa di colore rosso permanente dovuto alla formazione di Fe(SCN)3. Talvolta è necessario aggiungere una o due gocce di titolante in più, dopo il raggiungimento del punto equivalente, poiché gli ioni Ag+ sono parzialmente assorbiti dal precipitato di AgSCN e reagiscono lentamente. Qualora il risultato non fosse convincente, ripetere una seconda volta la titolazione con 25,0 ml della soluzione. Interpretare i fenomeni che avvengono 5) Filtrare ed analizzare per mezzo di titolazione con KSCN le altre tre soluzioni preparate (n° 2,3,4). Essendo la filtrazione delle miscele un’operazione lenta, eseguirla mentre si esegue la titolazione della miscela precedente. 6) Calcolare la conc. degli ioni acetato in ciascuna miscela e, utilizzando le concentrazioni degli ioni Ag+ misurate con la titolazione, calcolare il prodotto di solubilità dell’acetato d’argento. 27 Reazioni di ossidoriduzione Reagenti: Zn, Fe, Cu, Pb, Zn(NO3)2, Cu(NO3)2, Pb(NO3)2, KBr, KI, acqua di cloro, acqua di bromo, soluzione di iodio, HCl, HNO3, CuSO4, ZnSO4, CCl4, Na2S Attrezzatura speciale: tester, tubo a U, carta vetro Rischi connessi a quest’esperienza: manipolazione di acidi e basi forti, sviluppo di gas tossici, manipolazione di sostanze potenzialmente tossiche per contatto Precauzioni: indossare i guanti, svolgere sotto cappa le reazioni suscettibili di sviluppare gas tossici Finalità: scopo di quest’esperienza è verificare sperimentalmente le proprietà redox di vari elementi e porle in relazione fra loro A) Ossidabilità di metalli (Zn, Fe, Cu ) ad opera di HCl e HNO3 1) Introdurre in tre beckerini da 10 ml, rispettivamente, una punta di spatola di zinco in polvere, una punta di spatola di ferro in polvere ed una laminetta di rame. Operando sotto cappa, tentare un attacco acido su questi sistemi metallici allo scopo di portare il metallo in soluzione come ione positivo (catione). Iniziare con HCl concentrato sui tre campioni e registrare quanto osservato. 2) Attaccare, in un secondo tempo, i metalli che non fossero passati in soluzione con HNO3 concentrato e se necessario riscaldare con becco Bunsen. Registrare quanto osservato. 3 Cu(s) + 8 HNO3(aq) → 3 Cu(NO3)2(aq) + 2 NO(g) + 4 H2O(l) 3 Cu + 8 H+ + 2 NO3- → 3 Cu2+ + 2 NO↑ + 4 H2O Cu(s) + 4 HNO3(aq) → Cu(NO3)2(aq) + 2 NO2(g)↑ + 2 H2O(l) Cu + 4 H+ + 2 NO3- → Cu2+ + 2 NO2+ 2 H2O ⇒ NO2↑ è di colore rosso-bruno B) Potenziali redox dei metalli: reazioni di ossidoriduzione coinvolgenti coppie di specie metalliche (Cu/Zn, Pb/Cu) 1) Porre una piccola lamina di rame in 3 ml di soluzione 0,1 M di nitrato di zinco (Zn(NO3)2) ed una piccola lamina di Zn in 3 ml di una soluzione 0,1 M di nitrato di rame(Cu(NO3)2) Interpretare quanto osservato 2) Ripetere l’esperienza immergendo una piccola lamina di Pb (lucidare la superficie con della carta vetro, se necessario) in 3 ml di soluzione 0,1 M di nitrato di rame. Fare anche la prova immergendo una lamina di Zn ed una lamina di Cu in una soluzione di nitrato di piombo 0,1 M. Determinare la posizione della coppia H+/H2 rispetto ai metalli precedenti immergendo le lamine metalliche in una soluzione di HCl concentrato. Interpretare quanto osservato 28 C) Potenziali redox dei non metalli (ossidazione di I- e Br-) 1) In due provette, mettere rispettivamente 3 ml di soluzione 0,1 M di bromuro di potassio (KBr) e 3 ml di soluzione 0,1 M di ioduro di potassio (KI); ad entrambe aggiungere qualche goccia di acqua di cloro (disponibile nel reagentario). L’acqua di cloro è una soluzione satura di cloro in acqua (Cl2 + H2O → HCl + HClO) e ha proprietà ossidanti. L’agente ossidante è il Cl2 stesso. Data la sua instabilità, l’acqua di cloro viene preparata poco prima dell’uso, facendo gocciolare acido cloridrico su permanganato di potassio. Dall’ossidazione di Cl- si sviluppa Cl2 gassoso, che viene fatto gorgogliare in acqua, producendo una soluzione satura. 2) Operando sotto cappa ed indossando i guanti di protezione, aggiungere ad entrambe le provette 1 ml di tetracloruro di carbonio (CCl4). Tappare le provette con un tappo in teflon e agitare bene. Fare stratificare il solvente. Si osserva una colorazione. Quale? Il CCl4 è un solvente organico apolare che ha la funzione di estrarre i prodotti di ossidazione di I- e Br-. Interpretare quanto osservato ed annotarlo. 3) Ripetere l’esperienza mescolando 1 ml di acqua di bromo (disponibile nel reagentario) con 3 ml di soluzione 0,1 M di KI e 1 ml di soluzione di iodio (disponibile nel reagentario) con 3 ml di soluzione 0,1 M di KBr. L’acqua di bromo è una soluzione satura di bromo in acqua (Br2 + H2O) e ha proprietà ossidanti. L’agente ossidante è il Br2 stesso. E’ molto più stabile dell’acqua di cloro e la si prepara per dissoluzione del bromo (che è liquido) in acqua. 4) Operando sotto cappa ed indossando i guanti di protezione, aggiungere ad entrambe le provette 1 ml di tetracloruro di carbonio (CCl4). Tappare le provette con un tappo in teflon e agitare bene. Fare stratificare il solvente. Si osserva una colorazione. Quale? Interpretare quanto osservato ed annotarlo. D) Costruzione di una pila Daniell (esperienza da svolgersi a gruppi di 2 o 3) 1) Preparare 50 ml di soluzione 0,5 M di solfato di rame e 50 ml di soluzione 0,5 M di solfato di zinco. In un becker da 100 ml mettere 35 ml di soluzione di solfato di rame ed introdurvi una lamina di rame che tocchi il fondo del recipiente e rimanga appoggiata al suo bordo. Preparare un’altra semicella usando un secondo becker in cui si introducono 35 ml di soluzione di solfato di zinco e si immerge una lamina di zinco. La soluzione deve avere lo stesso livello nei due becker, per evitare ogni effetto sifone. Le superfici dei due metalli devono essere lucide: se necessario, pulirle con carta vetro. Connettere i due becker con un “tubo a U” (disponibile in distributorio) riempito di una soluzione di solfato di zinco e tappato alle estremità con due batuffoli di cotone imbibiti di soluzione di solfato di zinco. Collegare i due elettrodi con un voltmetro (disponibile in distributorio) e misurare il potenziale e la corrente generate dal sistema. Annotare il risultato. Interpretare quanto osservato. 2) Dopo aver letto il potenziale della cella aggiungere alla semicella contenente gli ioni rame una punta di spatola di solfuro di sodio solido. Mescolare le soluzioni e leggere il voltaggio e la corrente della cella. Se non si ha alcuna variazione, aggiungere ancora un piccolo eccesso di Na2S. Annotare il risultato. Interpretare quanto osservato. 29 Decomposizione termica di solfato di rame pentaidrato Reagenti: CuSO4⋅5H2O, NH4OH, alcol etilico, Na2CO3 Attrezzatura speciale: forno a muffola Rischi connessi a quest’esperienza: ustioni derivanti dalla non corretta manipolazione di oggetti riscaldati ad alta temperatura Precauzioni: manipolare gli oggetti riscaldati ad alta temperatura con le apposite pinze Finalità: scopo di quest’esperienza è verificare sperimentalmente l’effetto della temperatura sulla stabilità di un composto e di saggiare le proprietà chimiche dei prodotti ottenuti 1) In un crogiolo di porcellana porre ca. 4 g di solfato di rame pentaidrato (che contiene 5 molecole di acqua di cristallizzazione e la cui formula è CuSO4⋅5H2O). 2) Porre il crogiolo nel forno a muffola a 300°C per ca. 20 minuti, fino a completa scomparsa del colore azzurro ∆ CuSO4⋅5H2O(s) → CuSO4(s) + 5 H2O(g) azzurro bianco 3) Estrarre il crogiolo dal forno e attendere che il contenuto sia raffreddato. 4) Pestare la polvere bianca così ottenuta in un mortaio, in modo da renderla il più fine possibile, e trattarla con acqua fino a dissolverla tutta, agitando su piastra magnetica per favorirne la dissoluzione: CuSO4(s) + 6H2O → [Cu(H2O)6]2+ + SO42- la soluzione acquosa appare colorata in azzurro 5) Prelevare una porzione della soluzione acquosa ottenuta (ca. 1/3) precedentemente ed aggiungervi, goccia a goccia, qualche ml di idrossido di ammonio concentrato (NH4OH). Dapprima si noterà l’intorbidimento della soluzione e la formazione di un precipitato verdeazzurro (di che si tratta?) come conseguenza dell’aumento di pH. Proseguendo nell’aggiunta di NH4OH, la soluzione virerà di colore, diventando limpida e di colore blu intenso, in seguito alla formazione del complesso tetra-ammino-rame (II), che riporta lo ione Cu2+ in soluzione: [Cu(H2O)6]2+ + 4 NH3(aq) → [Cu(NH3)4]2+ + 6 H2O(l) blu intenso Porre qualche ml di questa soluzione in una provetta ed addizionare alcol etilico (CH3CH2OH), fino a che non si nota l’intorbidimento della soluzione. L’alcol estrae la coppia di ioni [Cu(NH3)4]2+ e SO42- e ne permette la cristallizzazione: si formano piccoli cristalli di [Cu(NH3)4]SO4 di colore blu intenso. Filtrare su filtro di carta a pieghe, essiccare il residuo solido così ottenuto, ponendolo in stufa (su un vetrino da orologio) per ca. 20 minuti e pesarlo. 6) Prelevare una seconda porzione (ca. 1/3) della soluzione acquosa ottenuta precedentemente (che contiene lo ione esa-aquo-rame(II) [Cu(H2O)6]2+) ed addizionarvi qualche millilitro di una soluzione di carbonato di sodio ca. 1,0 M; precipita il carbonato di rame (II), di colore azzurroverde: 30 CuSO4(aq) + Na2CO3(aq) → CuCO3↓ + Na2SO4(aq) Cu2+ + CO3 2- → CuCO3↓ Dopo aver filtrato il precipitato ottenuto nell’operazione precedente (CuCO3) su filtro di carta a pieghe, prelevarne una punta di spatola e porla in un crogiolo di porcellana e portarlo a una temperatura di circa 600°C in muffola; il precipitato si decompone generando ossido di rame (II), di colore nero: ∆ CuCO3 (s) → CuO (s) + verde-azzurro nero CO2 (g) 31 Cristallizzazione frazionata di NaCl e KNO3 Reagenti: NaNO3, KCl, HCl, HNO3, AgNO3, FeSO4, H2SO4 Attrezzatura speciale: vetro al cobalto, termometro Rischi connessi a quest’esperienza: utilizzo del bunsen, manipolazione di acidi e basi forti Precauzioni: indossare i guanti, manipolare gli oggetti riscaldati ad alta temperatura utilizzando le apposite pinze Finalità: scopo di quest’esperienza è separare i cristalli di due diversi sali a partire da una soluzione che li contiene entrambi, sfruttando la differente dipendenza delle rispettive solubilità dalla temperatura. A)Preparazione del nitrato di potassio e del cloruro di sodio 1) Pesare una quantità in grammi di nitrato di sodio e di cloruro di potassio pari a ½ di mole. In un becker da 400 ml, mescolare i due solidi con 95 ml di acqua e porre il becker sulla piastra riscaldante. Portare la soluzione fino quasi all’ebollizione, agitandola per favorire la dissoluzione dei due sali. Nel frattempo preparare un bagno refrigerante, ponendo in un becker capiente (o in una vaschetta di plastica) una miscela di acqua e ghiaccio e controllare che la temperatura raggiunga ca. gli 0°C. Quando non si osserva ulteriore dissoluzione dei sali, mentre la soluzione è ancora calda, filtrarla con l’imbuto Büchner, facendo il vuoto nella beuta da filtrazione con la pompa ad acqua (vedi figura sottostante e dispense pag. 5 - porre un dischetto di carta da filtro sul setto poroso dell’imbuto, ritagliandolo nelle dimensioni opportune; porre la beuta da vuoto sull’apposito sostegno), in modo da rimuovere ogni impurezza insolubile. 2) Trasferire il filtrato caldo in un becker da 250 ml, metterlo nel bagno di ghiaccio e raffreddarlo, agitando con una bacchetta, in modo da portare la temperatura fino a ca. 5°C. Dalla soluzione satura si separano i cristalli di KNO3. Questa operazione sfrutta la variazione di solubilità del nitrato di potassio al variare della temperatura. Il sale, molto solubile a caldo, lo è meno a freddo. Per questa ragione, raffreddando la soluzione si riesce a provocare la separazione dei cristalli dalla soluzione (che, a bassa temperatura, risulta sovrasatura). Lavare l’imbuto e la beuta da vuoto e filtrare i cristalli alla pompa, pressandoli con un pezzetto di carta da filtro per eliminare l’eccesso di soluzione acquosa (vedi figura sottostante). 32 Conservare sia il precipitato che il filtrato. Porre i cristalli su un vetrino da orologio o una capsula e lasciarli seccare all’aria. Una volta asciutti, pesarli e calcolare la resa della reazione. Su questi cristalli (anche se sono ancora umidi) si eseguono i saggi al punto B. 3) Mettere il filtrato in un becker e portarlo rapidamente ad ebollizione, utilizzando il becco Bunsen. Continuando l’ebollizione, la miscela può iniziare a schizzare violentemente. Ciò può essere minimizzato agitandola con una bacchetta di vetro (vedi figura precedente). 4) L’evaporazione del solvente va proseguita fino a quando il volume del liquido non si è ridotto della metà o a un terzo di quello iniziale ed una discreta quantità di cristalli di NaCl non si sono separati. Prestare attenzione a non ridurre il volume del liquido a meno di un terzo di quello iniziale. Filtrare alla pompa la miscela calda. Il cloruro di sodio è molto più solubile del nitrato di potassio. Per ottenere una soluzione satura in NaCl è ecessario ridurre drasticamente il volume di solvente, concentrando così la soluzione. L’operazione viene svolta scaldando la soluzione stessa su becco Bunsen, perchè ciò consente di ridurre i tempi di evaporazione del solvente. Si otterrebbe lo stesso risultato - ma in tempi molto più lunghi - se si lasciasse evaporare il solvente all’aria. 5) Eliminare l’eccesso di acque madri comprimendo i cristalli con un pezzo di carta da filtro (vedi figura precedente). Porre i cristalli su un vetrino da orologio o una capsula e lasciarli seccare all’aria. Una volta asciutti, pesarli e calcolare la resa della reazione. Su questi cristalli (anche se sono ancora umidi) si eseguono i saggi al punto B. Annotare il peso del KNO3 e del NaCl ottenuti. B) Analisi della purezza dei cristalli Verranno ora eseguiti dei test qualitativi per verificare il grado di purezza dei cristalli. 1) Saggio alla fiamma per lo ione sodio e potassio Prendere il filo al nichel-cromo e una lastrina di vetro al cobalto. Pulire il filo arroventandolo alla fiamma del becco bunsen ed immergendolo caldo in una provetta contenente qualche goccia di HCl concentrato. Proseguire questa operazione fino a quando non si osserva più alcuna particolare colorazione della fiamma. In seguito, prelevare una piccola porzione del sale che si vuole esaminare, porla su un vetrino da orologio, inumidirla e toccarla con il filo metallico in modo da farvi aderire qualche cristallino. Accendere il bunsen con una fiamma bassa, porre il filo metallico con i cristalli sul bordo esterno della fiamma (vedi figura sottostante) e riscaldare fino all’incandescenza. Una fiamma di colore giallo intenso indica la presenza di sodio, mentre una fiamma di colore viola pallido segnala la presenza di potassio. Qualora Na e K siano presenti contemporaneamente, il giallo del sodio maschera il viola del potassio. La colorazione dovuta al 33 sodio può essere schermata osservando la fiamma attraverso la lastrina di vetro al cobalto (vedi figura precedente). La fiamma viola dovuta al potassio permane per poco tempo. 2) Saggio per lo ione cloruro Sciogliere una piccola quantità di sale da analizzare (0,01 g) in 2 ml di acqua, acidificare con 2 gocce di acido nitrico (HNO3) concentrato e aggiungere una goccia di nitrato di argento (AgNO3) 0,1 M (disponibile in reagentario). Un precipitato lattiginoso di AgCl indica la presenza dello ione cloruro. AgNO3 + NaCl → AgCl↓ + NaNO3 3) Saggio per lo ione nitrato Sciogliere 0,01 g di sale in 2 ml di acqua e aggiungervi 2 ml di soluzione satura di solfato ferroso (preparata sciogliendo una punta di spatola di solfato ferroso in pochi ml di acqua in una provetta, subito prima di utilizzarla). Inclinare la provetta contenente il campione ed aggiungervi delicatamente una piccola quantità di acido solforico concentrato, in modo da stratificarlo sotto la soluzione da analizzare. La formazione di un anello marrone sulla superficie di separazione delle due soluzioni indica la presenza di nitrato. Lo ione nitrato, in ambiente acido, è in grado di ossidare il Fe(II): 2 KNO3 + 6 FeSO4 + 4 H2SO4 → 3 Fe2(SO4)3 + K2SO4 + 2NO + 4H2O L’ossido di azoto (NO) forma a sua volta un complesso colorato con il Fe(II) presente in eccesso: FeSO4 + NO → [FeNO]SO4 marrone 34 Sintesi e reattività di cloruro di calcio Reagenti: CaCO3, HCl, acqua di cloro, CaO, (NH4)2CO3, NH4Cl, Na2CO3, Ba(OH)2, H2SO4, NH4OH, (NH4)2C2O4, CH3COOH, fenolftaleina Rischi connessi a quest’esperienza: utilizzo del bunsen e della soffieria, manipolazione di acidi e basi forti, sviluppo di gas tossici Precauzioni: indossare i guanti, manipolare gli oggetti riscaldati ad alta temperatura utilizzando le apposite pinze, svolgere sotto cappa le reazioni suscettibili di sviluppare gas tossici Finalità: scopo di quest’esperienza è la preparazione di un sale di un tipico metallo alcalinoterroso, seguita dall’esame delle sue proprietà chimiche. A) Preparazione del cloruro di calcio 1) In una capsula di porcellana mescolare 30 ml di acqua con 30 ml di acido cloridrico concentrato e scaldare leggermente, operando sotto cappa ed utilizzando il becco Bunsen, con fiamma diretta e senza retina spargifuoco; aggiungere a poco a poco ed agitando 20 g carbonato di calcio (CaCO3) impuro. Il carbonato viene decomposto dall’acido e portato in soluzione. Si riscalda ancora per qualche minuto all’ebollizione, sostituendo man mano l’acqua che vaporizza (attenzione a non aggiungerne troppa, altrimenti il carbonato non passerà in soluzione! La soluzione finale deve risultare quasi limpida e giallina: se necessario, addizionare ancora acido), dopo di che si filtra a caldo su filtro di carta piano, in una beuta da 100 ml e si lascia raffreddare. CaCO3 + 2 HCl → CaCl2 + CO2↑ + H2O 2) La soluzione così ottenuta contiene cloruro di calcio e tracce di cloruro di magnesio (MgCl2), di cloruro manganoso (MnCl2) e cloruro ferroso (FeCl2); si aggiungono 5 ml di acqua di cloro (Cl2 + H2O → HClO + HCl) in modo da ossidare Fe2+ a Fe3+ (2 FeCl2 + HClO + HCl → 2 FeCl3 + H2O). Si lascia a riposo per ca. mezz’ora, quindi si fa bollire il liquido per alcuni minuti (sempre operando sotto cappa, utilizzando ancora il Bunsen, ma ponendo la beuta sulla retina spargifuoco), in modo da eliminare il cloro che non ha reagito. La beuta viene tolta dalla fiamma e vi si addizionano 4 g di ossido di calcio (CaO) per precipitare le impurezze di Mg, Fe e Mn. CaO + H2O → Ca(OH)2 Ca(OH)2 → Ca2+ + 2 OHFe3+, Mn2+ e Mg2+ formano idrossidi poco solubili, che precipitano. 3) Raffreddare la beuta sotto acqua corrente e filtrare su filtro a pieghe per eliminare i precipitati insolubili e l’eccesso di idrossido di calcio. La soluzione filtrata, che è basica, viene neutralizzata aggiungendo goccia a goccia HCl 2 M e controllando il viraggio alla neutralità con la cartina indicatrice (attenzione a non aggiungere troppo acido; se ciò accadesse, addizionare qualche goccia di una soluzione diluita di CaO - preparata sciogliendo una punta di spatola di CaO in acqua, in una provetta - fino a neutralizzare la soluzione). Il liquido così ottenuto si travasa in un matraccio tarato e si porta al volume di 200 ml, mediante aggiunta di acqua, in modo da ottenere una soluzione ca. 1 M CaCl2. Conservare la soluzione per i saggi successivi. B) Reazioni del CaCl2 35 1) In una piccola beuta diluire 10 ml di soluzione di CaCl2 con 10 ml di acqua; scaldare a 70-80°C (senza far bollire) e aggiungere goccia a goccia 10 ml di soluzione di carbonato di ammonio 1,0 M. Si forma un precipitato bianco. Raccogliere il precipitato, filtrando la soluzione alla pompa con imbuto Buchner coperto con un disco di carta da filtro e, dopo averlo lavato bene con acqua bollente, introdurlo in una beuta e, dopo aver aggiunto 20 ml di cloruro di ammonio 4 M (NH4Cl), scaldarlo a moderata ebollizione, sostituendo ogni tanto l’acqua che evapora. Dopo ca. 15 minuti di ebollizione, il precipitato si scioglie; odorare il vapore che esce dalla beuta (avvicinando i vapori al naso con il palmo della mano) che ha un odore caratteristico; immergere poi l’estremità di un bastoncino di vetro in una soluzione di idrossido di bario (disponibile in reagentario) ed avvicinarla ai vapori; la goccia si intorbidirà, in seguito alla formazione di carbonato di bario insolubile. I sali dell’acido carbonico e dell’ammoniaca hanno, rispettivamente, comportamento basico e acido. Il carbonato di ammonio è in grado di precipitare il sale poco solubile CaCO3: CaCl2 + (NH4)2CO3 → CaCO3↓ + 2 NH4Cl Un eccesso di ione NH4+ è in grado di liberare H2CO3 che, scaldato, si trasforma in CO2: CaCO3 + 2 NH4Cl → CaCl2 + 2 NH3↑ + CO2↑ + H2O La presenza di CO2 viene rilevata per reazione con lo ione bario che, analogamente al Ca2+, forma carbonati insolubili: CO2 + Ba(OH)2 → BaCO3↓ + H2O 2) In una piccola beuta introdurre 3 ml di soluzione di CaCl2, 3 gocce di acido cloridrico 2 M e 4 ml di soluzione 1,0 M di carbonato di sodio (Na2CO3). Dapprima si ha effervescenza ed in seguito si osserva la formazione di un precipitato bianco. Si filtra la soluzione su filtro di carta piano dentro una piccola beuta e si scalda all’ebollizione il filtrato limpido: si osserva un intorbidimento della soluzione, dovuto alla formazione di carbonato di calcio. L’acido carbonico è un acido debole biprotico che, in soluzione acquosa, dà origine ai seguenti equilibri : H2CO3 + H2O ↔ HCO3- + H3O+ HCO3- + H3O+↔ CO2 + H3O+ I sali di calcio di HCO3-, a differenza di quelli di CO32-, sono solubili, ma termicamente instabili: 2CaCl2 + 3 Na2CO3 + 2 HCl → CaCO3↓ + Ca(HCO3)2 + 6 NaCl CaCO3 ↓ + 2 HCl → CaCl2 + CO2↑ + H2O ∆ Ca(HCO3)2 → CaCO3↓ + CO2 + H2O 3) In una provetta, ad 1 ml di soluzione di CaCl2 aggiungere 1 ml di acido solforico 1 M (H2SO4). Si forma un precipitato bianco. I metalli del II gruppo formano sali insolubili con i gruppi: CO32-, SO42-, PO43-: CaCl2 + H2SO4 → CaSO4↓ + HCl 4) In una beuta da 100 ml introdurre 10 cc di soluzione di CaCl2 e 4 ml di soluzione 2 M di idrossido di ammonio (NH4OH); si scalda a 80-90°C, senza far bollire e quindi si tratta il liquido con 40 ml di soluzione 0,25 M di ossalato di ammonio ((NH4)2C2O4), agitando con un bastoncino di vetro. Si lascia riposare la soluzione e si ha la formazione di un precipitato bianco. 36 Si filtra la soluzione su filtro di carta piano e si lava il precipitato con acqua. Il precipitato viene diviso in tre porzioni, due piccole ed una grande. - La prima porzione piccola viene introdotta in una provetta e si aggiunge 1 ml di acido acetico (CH3COOH). Il precipitato non si scioglie. - La seconda porzione piccola viene introdotta in una provetta e si aggiunge 1 ml di acido cloridrico 2 M. Il precipitato si scioglie. - La porzione più grande di precipitato viene introdotta in un crogiolo di porcellana e riscaldata dapprima leggermente con precauzione e poi fortemente con un bunsen. Una volta che il precipitato è completamente essiccato, si prosegue il riscaldamento alla soffieria (rivolgersi al tecnico di laboratorio). Si fa quindi raffreddare completamente il crogiolo e si versano in esso 2 ml di acqua, rimescolando con una bacchetta di vetro. Si aggiunge infine una goccia di soluzione di fenolftaleina e la soluzione si colora di rosa. L’acido ossalico è un acido organico debole, che forma sali insolubili con il calcio. I suoi sali perciò idrolizzano e l’acido viene ripristinato, spostandolo da questi con un acido forte (a differenza degli acidi deboli, quali l’acido acetico, che non riescono a spostarlo dai suoi sali): C2O42-+ H2O ↔ H2C2O4 + 2 OHCaCl2 + (NH4)2C2O4 → CaC2O4 ↓ + 2 NH4Cl CaC2O4 + 2 HCl → H2C2O4 + CaCl2 Molte sostanze, sottoposte all’azione del calore, si trasformano per decomposizione. L’ossalato di calcio forma l’ossido di calcio, le cui soluzioni sono basiche. ∆ CaC2O4 → CaO + CO2↑ + CO↑ CaO + H2O → Ca(OH)2 5) Immergere la punta di un filo al nichel-cromo nella soluzione di CaCl2 e scaldare nella parte esterna della fiamma di un becco Bunsen. La fiamma si colora di rosso-arancio. 37 Preparazione dell’allume di cromo e reazioni di cromato e bicromato Reagenti: K2Cr2O7, H2SO4, alcol etilico, NaOH, HCl, NH4OH Attrezzatura speciale: imbuto gocciolatore, cristallizzatore con ghiaccio, termometro Rischi connessi a quest’esperienza: utilizzo del bunsen, manipolazione di acidi e basi forti, sviluppo di gas tossici, manipolazione di sali potenzialmente tossici per contatto Precauzioni: indossare i guanti, manipolare gli oggetti riscaldati ad alta temperatura utilizzando le apposite pinze, svolgere sotto cappa le reazioni suscettibili di sviluppare gas tossici Finalità: scopo di quest’esperienza è la preparazione di un sale doppio, attraverso un processo redox che richiede un accurato controllo della temperatura (affinchè la reazione avvenga nel verso desiderato). Vengono inoltre esaminate alcune proprietà di un tipico metallo di transizione, il cromo. A) Preparazione dell’allume di cromo Cr2(SO4)3⋅K2SO4⋅24H2O 1) In una beuta da 500 ml si pongono 200 ml di soluzione di dicromato di potassio (K2Cr2O7) 0,4 M (disponibile in reagentario). Ad essi si aggiungono goccia a goccia (con una pipetta Pasteur) 20 ml di acido solforico concentrato, mantenendo la beuta sotto acqua corrente, in modo da impedire che la temperatura superi i 30-40°C. Il liquido ottenuto (detto miscela cromica) si versa in una capsula di porcellana che viene posta in un cristallizzatore (disponibile in distributorio) contenente acqua e ghiaccio in pezzi. Da qui in poi si procede, operando sotto cappa: la soluzione va rimescolata con un termometro fino a che la temperatura scende al di sotto di 10°C. Continuando a rimescolare, si fanno gocciolare lentamente mediante un imbuto a rubinetto (imbuto gocciolatore disponibile in distributorio) 20 ml di alcol etilico (CH3CH2OH), regolando la velocità di caduta delle gocce in modo che la temperatura non oltrepassi mai i 20°C. La soluzione cambia colore e si ha liberazione di acetaldeide (CH3CHO) gassosa (tossica! operare sotto cappa!), dall’odore caratteristico. K2Cr2O7 + 4 H2SO4 + 3 CH3CH2OH → K2SO4 + Cr2(SO4)3 + 3 CH3CHO↑ + 7 H2O Il dicromato di potassio in ambiente acido è un ottimo ossidante e ossida l’alcol etilico ad acetaldeide, riducendosi a Cr(III). L’utilizzo di etanolo è vantaggioso poichè il suo prodotto di ossidazione, l’acetaldeide, è volatile e può essere facilmente rimossa dalla miscela di reazione. 2) Terminata l’aggiunta di alcol, si agita ancora un po’ per facilitare l’eliminazione dell’acetaldeide rimasta indisciolta, quindi si lascia a riposo. Il solfato di potassio ed il solfato cromico si combinano e si separa solfato doppio icosatetraidrato Cr2(SO4)3⋅K2SO4⋅24H2O più o meno impuro, in cristalli grigio-violacei. La miscela viene lasciata a riposo per 24 ore, per consentire ai cristalli di separarsi ed accrescersi (una volta che si è raffreddata, la si pone nell’armadietto e si copre la capsula con un vetro). 3) I cristalli grigio-violacei vengono frantumati con una bacchetta di vetro e raccolti su filtro di carta a pieghe. Si lavano più volte con poca acqua fredda, fino a quando non assumono un colore violetto e l’acqua di lavaggio non passi più colorata in verde, ma sia leggermente violacea. 4) I cristalli ottenuti devono ora essere purificati per ricristallizzazione. Si introduce il prodotto così ottenuto in un pallone da 500 ml nel quale viene posta anche un’ancoretta magnetica e lo si immerge in un becker grande, pieno per 1/4 di acqua riscaldata a 40-50°C (si opera sul bancone, 38 utilizzando la piastra riscaldante ed agitante). Quindi si versa nel pallone, a poco a poco, la quantità di acqua necessaria perchè essa, raggiunta tale temperatura, possa sciogliere quasi tutta la sostanza dopo forte e prolungata agitazione (attenzione a non utilizzare troppa acqua e lasciare un po’ di sostanza indisciolta, per essere sicuri di avere una soluzione satura a 40°C). Si filtra quindi dentro una capsula di porcellana (con un filtro di carta a pieghe) e si lascia raffreddare la soluzione, dopo averla coperta con una lastra di vetro. Dopo un po’ di tempo, dal liquido cominciano a separarsi cristalli violetto-scuri di forma ottaedrica. I cristalli che si separano da una soluzione satura trascinano con sé delle impurezze (ad esempio, le specie non reagite) presenti nella miscela di reazione. E’ perciò necessario purificarli e ciò può essere fatto mediante ricristallizzazione. I cristalli vengono ridisciolti in poco solvente, a caldo, in modo da ottenere una soluzione satura a quella temperatura. Raffreddandosi, la soluzione diventa soprasatura e da essa si separa nuovamente la fase solida, mentre i contaminanti restano in soluzione e vengono allontanati per filtrazione. 5) Terminata la cristallizzazione, i cristalli vengono filtrati alla pompa su imbuto Buchner con disco di carta, si lavano con pochissima acqua e si lasciano seccare all’aria. B) Reazioni degli ioni Cr3+ 1) Pesare l’allume di cromo e calcolare il volume di acqua necessaria per preparare una soluzione 0,08 M del sale. In una beuta, scaldare l’acqua necessaria portandola alla temperatura di 40-50°C (attenzione a non superare tale temperatura!); nel frattempo, polverizzare l’allume di cromo in un mortaio, introdurlo in un pallone e aggiungere subito l’acqua calda. Agitare fino a completa dissoluzione. 2) In una provetta, a 4 ml di soluzione di allume di cromo aggiungere 1 ml di idrossido di sodio (NaOH) 2 M ed agitare. Si forma un precipitato grigio-verde 3) Dividere la soluzione con il precipitato in due provette: alla prima aggiungere alcune gocce di HCl concentrato; alla seconda aggiungere qualche goccia di NaOH 5 M. In entrambi i casi il precipitato si scioglie. Il Cr(III) forma idrossidi poco solubili: Cr2(SO4)3 + 6NaOH → 2 Cr(OH)3↓ + 3 Na2SO4 L’idrossido di Cr(III) ha proprietà anfotere: è solubile sia in acidi che in basi. In presenza di un eccesso di base, esso passa in soluzione, generando la specie: Cr(OH)4Cr(OH)3 + 3 HCl → CrCl3 + 3 H2O Cr(OH)3 + NaOH → Na[Cr(OH)4] → NaCrO2+ 2 H2O 4) In una beutina a 2 ml di soluzione di allume di cromo aggiungere (sotto cappa) 8 ml di soluzione di idrossido di ammonio (NH4OH) 6 M. Si forma un precipitato grigio-verde. Agitare la soluzione a lungo e filtrarla: si ottiene un liquido rosso-violetto. In seguito fare bollire tale liquido, ponendo la beuta su becco Bunsen, con retina spargifiamma: si forma un precipitato grigio-verde e si ha liberazione di NH3. Come molti altri metalli di transizione, il cromo forma degli aminocomplessi solubili, ma instabili termicamente; trattando il solfato cromico con ammoniaca, si forma inizialmente l’idrossido, che precipita e viene in seguito disciolto a causa della formazione del complesso [Cr(NH3)6]3+ Cr2(SO4)3 + 6NH4OH → 2 Cr(OH)3↓ + 3 (NH4)2SO4 Cr(OH)3 + 6NH3 → [Cr(NH3)6]3+ + 3 OH[Cr(NH3)6]3+ + 3 H2O → Cr(OH)3↓ + 3NH3↑ + 3 NH4+ 39 C) Reazioni del K2Cr2O7 1) In una beuta da 50 ml, porre ca. 10 ml di soluzione di dicromato di potassio 0,4 M (K2Cr2O7). Trattare il liquido con una quantità di acido solforico concentrato pari ad una volta e mezza il suo volume (l’acido deve essere aggiunto poco alla volta e con precauzione, mantenendo la beuta sotto acqua corrente per ovviare al surriscaldamento). 2) Si separano cristalli rossi. Quando il liquido è freddo, filtrare alla pompa su imbuto Buchner senza disco di carta. Il prodotto viene raccolto per le successive reazioni. Acidificando il dicromato di potassio, si ha la formazione reversibile dell’acido da cui esso deriva: K2Cr2O7 + H2SO4 → H2Cr2O7 + K2SO4 H2Cr2O7 + H2O → 2 H2CrO4 Il prodotto finale è l’anidride cromica: H2CrO4 → CrO3 + H2O 3) Sciogliere una piccola parte di cristalli rossi in acqua dentro un piccolo becker. La soluzione si colora di rosso-arancio. Aggiungere goccia a goccia una soluzione di idrossido di sodio 5 M. La soluzione si colora di giallo. Neutralizzando l’acido cromico con una base, si formano i cromati gialli: 2 CrO3 + H2O → H2Cr2O7 H2Cr2O7 + 4 NaOH → 2 Na2CrO4 + 3 H2O 4) Mettere alcuni cristalli rossi in una provetta e, operando sotto cappa, aggiungere 2 ml di acido cloridrico concentrato. Si libera un gas verde-pallido. Il triossido di cromo è un potente ossidante: 2 CrO3 + 12 HCl → 3 CrCl3 + 6 H2O + 3 Cl2↑ 40 Sintesi e reattività di cloruro mercurico (esperienza da svolgersi a coppie) Reagenti: Hg, Cu, HCl, HNO3, Na2CO3 anidro, NaOH, KOH H2O2, NH4OH, KI, CuSO4, SO2, H2SO3 Attrezzatura speciale: apparecchio per lo sviluppo di anidride solforosa Rischi connessi a quest’esperienza: manipolazione di acidi e basi forti, utilizzo del bunsen, manipolazione di sostanze volatili o gassose potenzialmente tossiche Precauzioni: indossare i guanti, manipolare gli oggetti riscaldati ad alta temperatura utilizzando le apposite pinze, svolgere sotto cappa tutte le manipolazioni di Hg metallico e le reazioni suscettibili di sviluppare gas tossici Finalità: scopo di quest’esperienza è la preparazione di un sale di mercurio a partire dal Hg metallico e lo studio delle sue proprietà chimiche, che riflettono la natura parzialmente covalente del legame chimico in esso presente. A) Preparazione del cloruro mercurico HgCl2 1) Preparare l’acqua regia: sotto cappa, indossando guanti di protezione, mescolare in un becker 15 ml di acido nitrico concentrato (HNO3) con 45 ml di acido cloridrico concentrato (HCl). Successivamente, sempre operando sotto cappa, introdurre 10 g di mercurio (densità = 13,6 g/ml) in un pallone da 100 ml e aggiungervi lentamente 50 ml della miscela dell’acqua regia (operare con cautela: l’acqua regia è fortemente corrosiva!). Riscaldare su piccola fiamma, agitando continuamente, sino a che tutto il mercurio non si è sciolto. Si ha evoluzione di vapori rosso-bruni. L’acqua regia è un potente ossidante, in grado di sciogliere anche i metalli più “nobili”: HNO3 + 3 HCl → Cl2 + NOCl + 2 H2O acqua regia HNO3 + 3 HCl + Hg → HgCl2 + NOCl ↑ + 2 H2O Si versa la soluzione in una capsula di porcellana e, sempre operando sotto cappa, si concentra a fuoco diretto fino a quando non rimangono pochi ml di liquido; si tira quindi a secco, ponendo la capsula su un bagnomaria con acqua bollente. Si pesa il cloruro mercurico così ottenuto. 2) Per cristallizzarlo, occorre preparare una soluzione di HgCl2 quasi satura. Sapendo che la sua solubilità a 100°C è 55 g su 100 g di acqua, calcolare la quantità di acqua necessaria per scioglierlo a quella temperatura e se ne aggiunge un eccesso del 10%. Si scioglie il sale all’ebollizione nella quantità di acqua calcolata e si lascia raffreddare a temperatura ambiente; si raccolgono i cristalli ottenuti, filtrando alla pompa su imbuto Buchner coperto con un disco di carta. Si lavano con poca acqua e si lasciano seccare all’aria. B) Reazioni di HgCl2 Per via secca 1) Mettere 0,5 g di HgCl2 in una provetta. Sotto cappa, scaldare alla fiamma del bunsen. Il sale fonde (200°C) e poi dà un vapore (302°C) che si condensa in polvere bianca sulle pareti fredde del tubo da saggio. Il cloruro mercurico presenta un legame con spiccato carattere covalente: ciò giustifica il suo basso punto di fusione e la facilità con cui sublima. 41 2) In un mortaio di porcellana mescolare 0,5 g di HgCl2 con 1,5 g di carbonato di sodio anidro (Na2CO3). Introdurre la miscela in una provetta asciutta e, tenendola inclinata, scaldare al becco bunsen sotto cappa. Si forma uno specchio metallico con delle goccioline sulle pareti. Il carattere “nobile” del mercurio viene evidenziato dalla reazione con il carbonato di sodio: scaldando energicamente il Hg(II), si riduce a spese dell’ossigeno: ∆ 2 HgCl2 + 2 Na2CO3 → 2 Hg + 4 NaCl + 2 CO2 + O2 Per via umida 1) Sciogliere il rimanente cloruro mercurico in acqua (scaldando leggermente) dentro una beuta, in modo da preparare una soluzione 0,5 N. 2) Preparare in una piccola beuta beuta 8 ml di soluzione di idrossido di sodio 5 M (NaOH) e versarvi a poco a poco 8 ml di soluzione di cloruro mercurico. Si forma un precipitato giallo. Lasciare decantare la soluzione e rimuovere il liquido soprastante il precipitato. Al precipitato giallo aggiungere alcuni ml di acqua ossigenata (H2O2). Osservare la reazione che avviene. Il mercurio, come l’argento, non forma idrossidi stabili: HgCl2 + 2 NaOH → Hg(OH)2 + 2 NaCl Hg(OH)2 → HgO↓ + H2O giallo Ed anche l’ossido può essere facilmente ridotto: HgO + H2O2 → Hg + H2O + O2 3) In una provetta, mescolare 2 ml di idrossido di ammonio 2 M (NH4OH) con 2 ml di soluzione di HgCl2. Si forma un precipitato bianco. Aggiungere alla soluzione qualche goccia di HCl concentrato. Il precipitato si scioglie. La tendenza del Hg a formare legami con carattere covalente è dimostrata dal particolare tipo di aminocomplesso che forma: HgCl2 + 2 NH3 → [HgNH2]Cl↓ + NH4Cl precipitato bianco infusibile [HgNH2]Cl + HCl → HgCl2 + NH3 4) In un piccolo becker mescolare 40 cc di soluzione di HgCl2 con 10 ml di soluzione 0,5 M di ioduro di potassio (KI). Si forma un precipitato rosso. Filtrare su carta il precipitato e lavarlo bene con acqua. Con la punta di una bacchetta di vetro prelevare una piccola quantità di precipitato rosso ed introdurlo in un tubo da saggio asciutto; tenendo inclinato il tubo, riscaldarlo alla fiamma del bunsen. Si ha fusione del sale (223°C) e formazione di cristalli rombici gialli. A temperatura ambiente si riottengono lentamente cristalli rossi (la trasformazione è più rapida se si sfregano i cristalli con una bacchetta di vetro). Con lo iodio, Hg(II) forma un precipitato: HgCl2 + 2 KI → HgI2↓ + 2 KCl ∆ HgI2 ←→ HgI2 Rosso Giallo 42 5) Il rimanente precipitato rosso viene trattato in una piccola beuta con alcuni ml di soluzione 0,5 M di ioduro di potassio (KI) e 0,5 g di KI cristallino. Il precipitato si scioglie. Si preleva 1 ml di questa soluzione, si basifica con qualche goccia di KOH 0,5 M e si aggiunge una goccia di soluzione di idrossido di ammonio 2 M. Si forma un precipitato bruno. Il solido rosso può essere disciolto attraverso la formazione di complessi: HgI2 + KI ↔ K[HgI3] K[HgI3] + KI ↔ K2[HgI4] K[HgI3] e K2[HgI4] sono incolori. Mescolando K[HgI3] con KOH si ottiene il reattivo di Nessler, sensibilissimo all’ammoniaca: 2 K[HgI3] + 3 KOH + NH3 → OHg2NH2I ↓ + 5 KI + 2 H2O giallo 6) Al resto della soluzione nella beutina si aggiungono 2 ml di soluzione 0,5 M di solfato di rame (CuSO4). Si addiziona ca. 1 ml di soluzione di SO2 in acqua (H2SO3), disponibile in reagentario. Si forma un precipitato rosso Con alcuni cationi dei metalli di transizione, gli iodomercurati formano complessi mercurometallici, come ad es., con il Cu(I). Lo ione rameoso, molto instabile all’ossidazione, viene generato sfruttando l’azione riducente della SO2 (che viene ossidata a H2SO4) ed utilizzato immediatamente: 2 CuSO4 + SO2 + 2 H2O → Cu2SO4 + 2 H2SO4 2 Cu+ + [HgI4]2- → Cu2[HgI4] ↓ lacca rossa 7) In una piccola beuta si mescolano 5 ml di acido solforoso (H2SO3, disponibile in reagentario) con due gocce di soluzione di HgCl2. Si scalda a lungo su bagno di acqua bollente. Si forma un precipitato bianco. L’acido solforoso è instabile e le sue soluzioni devono essere preparate poco prima dell’uso, facendo gocciolare acido solforico su solfito di sodio, in modo da spostare l’acido dal suo sale. Si sviluppa SO2, che viene fatta gorgogliare in acqua e genera l’acido corrispondente. Esso riduce Hg(II) a Hg(I), che forma sali meno solubili: 2 HgCl2 + H2SO3+ H2O → Hg2Cl2 ↓ + H2SO4 + 2 HCl Calomelano 8) In 10 ml di soluzione di HgCl2 si immerge una lamina di rame ben pulita: si deposita una patina nera che, sfregata con carta da filtro, forma uno specchio lucente. Il mercurio è più nobile del rame, quindi i suoi sali vengono ridotti da quest’ultimo: Cu + HgCl2 → CuCl2 + Hg Il mercurio, così prodotto, è in grado di formare una lega (amalgama) con il rame metallico, da cui la formazione di uno specchio argenteo per sfregamento della superficie della lamina di Cu. 43 Reagentario Acidi e basi Acido cloridrico Acido nitrico Acido solforico Acido solforoso Acido acetico glaciale Acido ossalico Idrossido di potassio Idrossido di sodio Idrossido di ammonio Idrossido di bario Ossido di calcio Anidride solforosa Indicatori HCl HNO3 H2SO4 H2SO3 CH3COOH H2C2O4 KOH NaOH NH4OH Ba(OH)2 CaO SO2 metilarancio fenolftaleina blu di bromotimolo allume ferrico Soluzioni tampone acide e basiche Sali Cloruro di sodio Cloruro di potassio Cloruro di calcio Cloruro di piombo Cloruro ferrino Cloruro d’ammonio Bromuro di sodio Bromuro di potassio Ioduro di potassio Nitrato di bario Nitrato ferrino Nitrato di piombo(II) Nitrato di sodio Nitrato d’argento Nitrato di rame(II) Nitrato di zinco Bicarbonato di sodio Carbonato di sodio Carbonato d’ammonio Carbonato di calcio Solfato di zinco Solfato ferroso Solfato di rame(II) Tiocianato di potassio NaCl KCl CaCl2 PbCl2 FeCl3 NH4Cl NaBr KBr KI Ba(NO3)2 Fe(NO3)3 Pb(NO3)2 NaNO3 AgNO3 Cu(NO3)2 Zn(NO3)2 NaHCO3 Na2CO3 (NH4)2CO3 CaCO3 ZnSO4 FeSO4 CuSO4 KSCN 44 Solfuro di sodio Acetato di sodio Ossalato di ammonio Na2S CH3COONa (NH4)2C2O4 Ossidanti Dicromato di potassio Acqua di bromo Acqua di cloro Iodio Perossido di idrogeno K2Cr2O7 I2 H2O2 Metalli Pb metallico Fe metallico Cu metallico Hg metallico Zn metallico Composti organici Tetracloruro di carbonio Alcol etilico CCl4 CH3CH2OH 45 Soluzioni presenti nel reagentario Acido cloridrico concentrato Soluzione d =1,189 g/ml al 37,9 % in peso Acido nitrico concentrato Soluzione d =1,386 g/ml al 62,44 % in peso Acido solforico concentrato Soluzione d =1,845 g/ml al 96 % in peso Acido acetico glaciale Soluzione d =1,05 g/ml al 100 % in peso Acido solforoso Soluzione ca. 1,8 N di H2SO3 (ca. 152 g/l). Si prepara saturando acqua distillata con anidride solforosa. Il reattivo deve essere di recente preparazione Idrossido di ammonio concentrato Soluzione d =0,892 g/ml al 30% in peso Acqua ossigenata Soluzione al 3% in peso di perossido di idrogeno H2O2 Acqua di bromo Soluzione satura (a 25°C 3,4 g Br2 su 100 g di soluzione) Acqua di cloro Soluzione satura (a 25°C 0,64 g Cl2 su 100 g di soluzione) Soluzione di iodio Soluzione 0,1N di iodio in soluzione al 2% di ioduro potassico (12,7 g di I2 e 20 g KI per litro) Soluzione 0,1 M di AgNO3 Soluzione satura di Ba(OH)2 Soluzione di K2Cr2O7 0,4 M Soluzioni da preparare Acqua regia Acido cloridrico diluito Acido nitrico diluito Acido solforico diluito Acido acetico diluito Idrossido di ammonio diluito Miscela di acido cloridrico e acido nitrico nel rapporto di 3 parti di HCl conc. per 1 parte di HNO3 conc. Soluzione HCl 2 M Soluzione HNO3 4 M Soluzione H2SO4 1 M Soluzione CH3COOH 2 M Soluzione NH4OH 2 M 46 Pesi atomici e molecolari delle sostanze in uso HCl HNO3 CH3COOH H2SO4 H2SO3 H2C2O4 NaOH NH3 SO2 NaCl NaBr NaHCO3 Na2CO3 Na2S NaNO3 CH3COONa KSCN KCl KBr KI KOH CaCl2 CaO Ba(NO3)2 Ba(OH)2 Pb PbCl2 Pb(NO3) 2 Fe Fe(NO3) 3⋅ 9H2O FeCl3 FeSO4⋅ 7H2O K2Cr2O7 AgNO3 Cu CuSO4⋅ 5H2O Cu(NO3) 2⋅ 3H2O (NH4)2C2O4⋅ H2O (NH4)2CO3 NH4Cl Hg Zn Zn(NO3) 2⋅ 6H2O ZnSO4 H2O2 36,46 63,01 60,05 98,08 82,08 90,04 40,00 17,03 64,06 58,44 102,9 84,01 106,0 78,05 85,00 82,04 97,18 74,55 119,0 166,0 56,11 111,0 56,08 261,3 315,5 207,2 278,1 331,2 55,84 404.00 162,2 151,9 294,2 170,0 63,55 249,7 241,60 142,12 94.08 53,49 200,6 65,39 297,48 161,5 34,02 47 Gruppi di lavoro e assegnazione posti I TURNO NOMINATIVO 1 AGRAMONTE SANCHEZ 2 AGU' MASIEL Posto n° 1 Gruppo 26 CAVALLO ELISA 40 A 27 CAVANNA DANIELE 41 F 28 CECCARINI CARLOTTA 43 “ MAURIZIO 2 “ 29 CHIONETTI FABRIZIO 44 “ 3 AVATANEO OTTAVIA 3 “ 30 COLANGELO 45 “ 4 BADOLATO FABRIZIO 6 “ 31 CORONA ELENA LAURA LUISA ALESSANDRA 47 “ 5 BALDASSARR MICHELE 8 “ 32 CRAVANZOL SARA 48 G DANIELE 9 B 33 CURCIO MARCO 92 “ 7 BARDELLI LORENZO 10 “ 34 D'ALOIA DEBORAH 94 “ 8 BARLETTA SIMONE 11 “ 35 D'AMELIO 49 “ 9 BELLONIO LORENA 13 “ 36 DEGENNARO MARIA GRAZIA FABIO 50 “ 10 BELTRAME VALENTINA 15 “ 37 DI PUMPO ANTONIETTA 52 H 11 BENEDETTINI FRANCESCA 18 C 38 D'IMPERIO VERONICA 54 “ 12 BERARDO ENRICO 19 “ 39 DIVOTTI FEDERICA 55 “ 13 BERGOGLIO MONICA 21 “ 40 DURANDO ANDREA 56 “ 14 BIGINELLI IVAN 23 “ 41 FERRERO CINZIA 59 “ 15 BISSACCA EUGENIO 24 “ 42 60 I 16 BOBBIO MARCO 26 D 43 62 “ 17 BOSCO MARCO 27 “ 44 63 “ 18 BUNINO CHIARA 28 “ 45 74 “ 19 CAGNINA STEFANIA 30 “ 46 75 “ 20 CAMBARERI SERENA 31 “ 47 67 “ 21 CAPUTO MARIA PIA 32 E 48 71 22 CARDALANA PAOLO 34 “ 49 72 23 CARINGELLA ROSALINDA 36 “ 50 73 24 CASTRONUO FRANCESCO 38 “ GIORGIA 39 “ E 6 BALZANO A VO 25 CAVAGNERO 48 Gruppi di lavoro e assegnazione posti II TURNO NOMINATIVO 25 MERALDI CHIARA 39 A 26 MESSA MANUELA 40 ALESSANDR A MARCO 41 F 43 “ Posto n° 1 Gruppo “ 1 FERRERO FEDERICA 2 FERRO VALENTINA 2 “ 27 MIRACCO 3 FULGHESU FEDERICA 3 “ 28 MUROLO 4 GALLO ANNA 6 “ 29 NILO ANDREA 44 “ 5 GALLO BARBARA 8 “ 30 NUCERA MATTEO 45 “ 6 GAUDINO JESSICA 9 B 31 OLIVERO FABIO 47 “ 7 GAVRIS ANISOARA RODICA MARIA 10 “ 32 OPPEDISANO ALBERTO 48 G 11 “ 33 ORSALLA MICHELE 92 “ DAVOOD 13 “ 34 PADOAN ELIO 94 “ 10 GIORGIO VALERIA 15 “ 35 PAGLIA FABRIZIO 49 “ 11 GIOVANDO ELENA 18 C 36 PAOLINI EMANUELE 50 “ 12 GULINO ALESSIA 19 “ 37 PELLEGRINO MARCELLO 52 H 13 HABIB HAJAR 21 “ 38 PERSICO DAVIDE 54 “ 39 PISANI ILARIA 55 “ 8 GENTILE 9 GHANI EDDINE 14 LAGANA' ALESSIA FEDERICA ANDREA 23 “ 40 RAVERA STEFANO 56 “ 24 “ 41 REGINATO 59 “ 16 LENTI MUSIO SARA 26 D NOEMI BARBARA 42 60 I 17 LILA ERIOLA 27 “ 43 62 “ 18 LIZZI FEDERICO 28 “ 44 63 “ 19 MANSUETO BATTISTA GIUSEPPE NOEMI 30 “ 45 74 “ 31 “ 46 75 “ 21 MARINACCIO ELISABETTA 32 E 47 67 “ 22 MARSICOVET ILARIA 34 “ 48 71 CHIARA 36 “ 49 72 ARIANNA 38 “ 50 73 15 LAMBERTO 20 MARENGO ERE 23 MARTANO 24 MASSAIOLI Gruppi di lavoro e assegnazione posti III TURNO NOMINATIVO 25 TORTELLO DARIO 39 A 26 TOSCANI ANITA 40 Posto n° 1 Gruppo “ 1 REGOLO SALVATORE 2 RIBERO MICHELE 2 “ 27 TOSCO ANDREA 41 F 3 RINALDI LAURA 3 “ 28 TRENTO 43 “ 4 RIZZO ANDREA 6 “ 29 TURONE VIRGINIA BRUNA VALENTINA 44 “ 5 ROLANDO MANUELA 8 “ 30 VIGNOLO CHIARA 45 “ 6 ROLLE RICCARDO 9 B 31 VINDROLA STEFANO 47 “ 7 ROMANIELL FRANCESCO 10 “ 32 VIOLA ANDREA 48 G 33 VIRARDI ROBERTO 92 “ O 8 ROSATO VALENTINA 11 “ 34 VOTA FEDERICO 94 “ DEBORAH 13 “ 35 ZAMBITO ELENA 49 “ 10 RUBINO ILARIA 15 “ 36 VERTOLO LILIANA 50 “ 11 RUCCI MARTA 18 C 37 SGARBOSSA SPERANZA 52 H 12 SARACINO STEFANO 19 “ 38 KUKAJ EDISONA 54 “ 13 SARTI ROBERTA 21 “ 39 CACA' LUIS 55 “ 14 SCAGLIONE MARTA 23 “ 40 BORASO ANDREA 56 “ 15 SCHIVO ALESSANDRO 24 “ 41 MADDALEN ANNA 59 “ 16 SERRA FEDERICA 26 D 42 60 I 43 62 “ 44 63 “ 45 74 “ 9 ROVICONE 17 SESTI ANDREA 27 “ 18 SODANO MARCELLA 28 “ 19 SPINAZZOL ANTONELLA 30 “ A 20 STANESCU O ANGELICA RAMONA ALMA 31 “ 46 75 “ 32 E 47 67 “ CRISTINA 34 “ 48 71 23 TOMASELLI ELISA 36 “ 49 72 24 TOMASSONI ALBERTO 38 “ 50 73 21 TERRANEGR A 22 TESIO Calendario del laboratorio Lunedì 29 gennaio alle ore 14 in Magna: lezione sulla sicurezza in laboratorio (obbligatoria per tutti) I TURNO - Consegne: 29 gennaio 2007 h.16 Laboratorio: dal 30 gennaio al 7 febbraio 2007 – tutti i pomeriggi dalle 14.00 alle 18.00 – Consegne finali: giovedì 8 febbraio 2007 h.14 II TURNO - Consegne: giovedì 8 febbraio 2007 h.16 Laboratorio: dal 9 al 19 febbraio 2007 – tutti i pomeriggi dalle 14.00 alle 18.00 – Consegne finali: martedi 20 febbraio 2007 h.14 III TURNO - Consegne: martedi 20 febbraio 2007 h.16 Laboratorio: dal 21 febbraio al 1 marzo 2007 – tutti i pomeriggi dalle 14.00 alle 18.00 – Consegne finali: venerdi 2 marzo h.16 Venerdì 2 marzo alle ore 14 in Aula Magna: correzione schede didattiche (obbligatoria per tutti) I TURNO Lunedi 29/1 Martedi 30/1 Mercoledì 31/1 Giovedì 1/02 Venerdi 2/2 Lunedi 5/2 Martedì 6/2 Mercoledì 7/2 Giovedì 8/2 II TURNO Giovedì 8/2 Venerdi 9/2 Lunedì 12/2 Martedì 13/2 Mercoledì 14/2 Giovedi 15/2 Venerdi 16/2 Lunedì 19/2 Martedì 20/2 II TURNO Martedì 20/2 Mercoledì 21/2 Giovedì 22/2 Venerdi 23/2 Lunedì 26/2 Gruppi A-D-G Gruppi B-E-H Gruppi C-F-I LEZIONE SICUREZZA h.14 + CONSEGNE I turno h.16 Equilibri acido-base (A-D) Equilibri solubilità (A-D) Equilibri acido-base (A-D) Equilibri acido-base (G-M) Equilibri acido-base (A-D) Equilibri acido-base (G-M) Equilibri solubilità (A-D) Equilibri acido-base (G-M) Equilibri solubilità (A-D) Reazioni di ossidoriduzione Preparazione dell’allume di Reazioni di ossidoriduzione (A-D) cromo (A(I parte) e C) (A-D) Cristallizzazione frazionata Preparazione dell’allume di Preparazione dell’allume di cromo (A(I parte) e C) cromo (A(II parte) e B) Preparazione dell’allume di Cristallizzazione frazionata Preparazione dell’allume di cromo (A(II parte) e B) cromo (A(I parte) e C) Preparazione dell’allume di Reazioni di ossidoriduzione Cristallizzazione frazionata cromo (A(II parte) e B) (A-D) CONSEGNE I turno h.14 Gruppi A-D-G Gruppi B-E-H Gruppi C-F-I CONSEGNE II turno h.16 Equilibri acido-base (A-D) Equilibri solubilità (A-D) Equilibri acido-base (A-D) Equilibri acido-base (G-M) Equilibri acido-base (A-D) Equilibri acido-base (G-M) Equilibri solubilità (A-D) Equilibri acido-base (G-M) Equilibri solubilità (A-D) Reazioni di ossidoriduzione Preparazione dell’allume di Reazioni di ossidoriduzione (A-D) (A-D) cromo (A(I parte) e C) Cristallizzazione frazionata Preparazione dell’allume di Preparazione dell’allume di cromo (A(II parte) e B) cromo (A(I parte) e C) Preparazione dell’allume di Cristallizzazione frazionata Preparazione dell’allume di cromo (A(I parte) e C) cromo (A(II parte) e B) Preparazione dell’allume di Reazioni di ossidoriduzione Cristallizzazione frazionata cromo (A(II parte) e B) (A-D) CONSEGNE II turno h.14 Gruppi A-D-G Gruppi B-E-H Gruppi C-F-I CONSEGNE III turno h.16 Equilibri acido-base (A-D) Equilibri solubilità (A-D) Equilibri acido-base (A-D) Equilibri acido-base (G-M) Equilibri acido-base (A-D) Equilibri acido-base (G-M) Equilibri solubilità (A-D) Equilibri acido-base (G-M) Equilibri solubilità (A-D) Reazioni di ossidoriduzione Preparazione dell’allume di Reazioni di ossidoriduzione 51 Martedi 27/2 Mercoledì 28/2 Giovedì 1/3 Venerdi 2/3 (A-D) cromo (A(I parte) e C) (A-D) Cristallizzazione frazionata Preparazione dell’allume di Preparazione dell’allume di cromo (A(II parte) e B) cromo (A(I parte) e C) Preparazione dell’allume di Cristallizzazione frazionata Preparazione dell’allume di cromo (A(I parte) e C) cromo (A(II parte) e B) Preparazione dell’allume di Reazioni di ossidoriduzione Cristallizzazione frazionata cromo (A(II parte) e B) (A-D) CORREZIONE SCHEDE h.14 + CONSEGNE III turno h.16 52 53 54 55 56 57 58 59 60
Scaricare