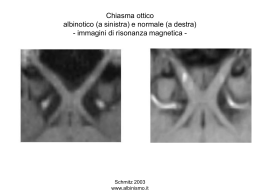
AIIC associazione italiana ingegneri clinici I Quaderni dell’AIIC REALIZZAZIONE E GESTIONE DI UN SITO PER DIAGNOSTICA CON RISONANZA MAGNETICA “aspetti tecnici” Appunti del Corso N. 4 del XIV Congresso Nazionale AIIC VENEZIA - anno 2014 - Vol 4 SOMMARIO Prefazione .............................................................................4 CAPITOLO 1: Principi di funzionamento ed applicazioni in ambito medico ..................................................5 CAPITOLO 2: Information Technology e integrazione dati/rete..............................................................12 CAPITOLO 3: Normativa di Sicurezza e Prevenzione del Rischio in Risonanza Magnetica............15 CAPITOLO 4: Progettazione di un sito RM.....................20 RACCOLTA SLIDES DEL CORSO................................................23 Bibliografia essenziale....................................................147 I DOCENTI DEL CORSO..............................................................149 GLI AUTORI .........................................................................150 Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici PREFAZIONE CAPITOLO 1 Principi di funzionamento ed applicazioni in ambito medico La realizzazione di immagini biomedicali mediante l’uso di sistemi a risonanza magnetica (RM) avviene sfruttando le proprietà magnetiche dei protoni, e, in particolare, del protone del nucleo di idrogeno (H), preferito in questo tipo di applicazione diagnostica in quanto: E’ con estrema soddisfazione che mi accingo a presentarvi questo lavoro, risultato tangibile della fortunata esperienza dei corsi di approfondimento che l’AIIC organizza in occasione del suo Convegno Nazionale. Questo “libretto” (così mi piace chiamarlo) è da intendersi come una vera e propria dispensa che raccoglie, nella maniera più fedele possibile, i contenuti divulgati durante il corso specifico cui è dedicato. Non è e non può essere un’opera esaustiva ma piuttosto uno strumento rivolto a tutti coloro, soci e non, che, interessati agli argomenti trattati, vogliano arricchire le proprie conoscenze e, soprattutto, trovare spunti di approfondimento. - è l’elemento prevalente nel corpo umano; - fornisce un segnale di risonanza a maggiore intensità rispetto agli altri elementi chimici. Il protone è una particella con caratteristiche magnetiche che, ruotando attorno al proprio asse, si comporta come un sistema dipolare magnetico, il cui momento di dipolo, definito SPIN, è il generatore del segnale di risonanza. Lo spin è una grandezza quantistica costante, assume, cioè, valori discreti che non variano nel tempo; fisicamente è un vettore, ed è possibile pertanto considerare la somma in senso vettoriale dei contributi di ciascun nucleo H in un elemento di volume del corpo, detto VOXEL. In assenza di campi magnetici esterni gli spin dei nuclei H sono orientati in modo tale che statisticamente si ritrova una somma vettoriale nulla (figura 1). L’auspicio è che sia uno strumento semplicemente utile, gelosamente custodito nella propria libreria. L’AIIC intende realizzare un libretto per ciascuno dei corsi realizzati, dando vita ad una vera e propria Collana. Un forte e sentito ringraziamento a tutti coloro che mossi unicamente dalla comune passione per la professione, hanno lavorato duramente e gratuitamente, alla realizzazione del presente volume (libretto). Il PRESIDENTE NAZIONALE AIIC Ing. Lorenzo Leogrande AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Figura 1: rappresentazione degli spin in assenza del campo B Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici In presenza di un campo magnetico uniforme esterno B, invece, la popolazione di spin all’interno del voxel si orienta parallelamente ad esso, assumendo lo stesso verso (SPIN UP o configurazione parallela) o verso discorde (SPIN DOWN o configurazione antiparallela) a quello di B1. Il livello energetico assunto dagli spin in configurazione parallela è leggermente inferiore (e pertanto favorito) a quello antiparallelo, per cui esiste un leggero eccesso di spin nella direzione UP tale da indurre una magnetizzazione netta M (figura 2). Il moto di precessione degli spin è statisticamente casuale, quindi questi, pur ruotando tutti con la stessa frequenza, sono completamente sfasati fra loro (figura 3). Figura 3: sfasamento degli spin in precessione Scomponendo, infatti, il vettore M nelle due componenti, l’una longitudinale (lungo l’asse z) e l’altra sul piano trasversale xy, notiamo che mentre la componente Mz è costante per tutti gli spin, Mxy è variabile e mediamente nulla. Figura 2: rappresentazione degli spin e del vettore M in presenza del campo B Come conseguenza della loro natura quantistica, gli spin non si allineano perfettamente nella direzione del campo B, ma ruotano attorno ad esso, seguendo, cioè, un moto di PRECESSIONE attorno a B. La velocità, e quindi la frequenza, della precessione rivestono un ruolo importante nell’imaging RM, e dipendono fortemente dal tipo di nucleo e dall’intensità del campo B applicato. Se siamo interessati a rilevare un segnale dagli spin che precedono, dovremo sintonizzare il nostro sistema di ricezione esattamente alla frequenza di precessione, che è nota come frequenza di Larmor wL: È, dunque, indispensabile stimolare gli spin in modo da alterare la loro condizione naturale di sfasamento, cedendo energia elettromagnetica mediante stimoli esterni RF alla frequenza di Larmor. Al termine dell’impulso gli spin si riportano nella condizione iniziale (configurazione energetica minima), emettendo radiazione elettromagnetica nella banda delle radiofrequenze (il segnale registrato dalle bobine prende il nome di FID (Free Induction Decay)). L’utilizzo di questi impulsi a radiofrequenza deviano il vettore M dal proprio asse di precessione; l’angolo che M forma col piano xy (flip angle) aumenta con l’energia dell’impulso. Una delle sequenze di impulsi RF maggiormente utilizzate è lo SPIN ECHO e consta prima di un impulso a 90° che ribalta M completamente sul piano xy (Mz=0), poi, al fine di rendere coerente il segnale sul piano xy e riallineare gli spin (in modo da ottenere l’informazione utile), è applicato un impulso a 180°, che inverte la magnetizzazione nel verso opposto rispetto all’asse z. Questi processi di rilassamento hanno un andamento temporale di tipo esponenziale: T1 è la costante di tempo che caratterizza il recupero di Mz, mentre T2, molto più breve di T1, è quella relativa alla diminuzione del valore di Mxy (figura 4). wL = γ x B dove γ = 42 MHz/T è detto rapporto giromagnetico; nei sistemi RM γ cade nello spettro delle radiofrequenze (RF): a 1,5 T wL = 63 MHz. ___________________ In realtà ciascuno spin occupa uno stato energetico intermedio, che è combinazione lineare delle due configurazioni UP e DOWN; tuttavia, sulla base del principio di indeterminazione di Heisenberg, è impossibile determinare la posizione esatta dello spin, se nonché la sua appartenenza ad uno dei due stati di cui sopra. 1 AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Figura 4: tempi di rilassamento T1 e T2 in funzione dei differenti tessuti illustrati Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici T1 dipende da: - la composizione del tessuto: il rilassamento di Mz comporta cessione di energia dagli spin al reticolo, cioè alla materia circostante; tessuti differenti hanno valori di T1 diversi, per cui esso può essere utilizzato per esaltare il contrasto in una immagine RM; - il valore di B: campi B elevati comportano un T1 elevato e, quindi, maggiore contrasto nell’immagine. In una immagine dell’encefalo, dove viene esaltato il valore di T1 (T1-pesata), il fluido cerebrospinale (CSF) appare di colore scuro (figura 5). Figura 6: visualizzazione del fluido cerebro-spinale mediante immagine T2-pesata Secondo quanto visto finora, applicando un campo statico B ed usando opportuni impulsi RF, ritroveremo un segnale somma dei singoli segnali provenienti dai voxel dell’intero corpo umano; se intendiamo, invece, localizzare un segnale risonante proveniente da un singolo distretto anatomico occorre fare utilizzo dei GRADIENTI DI CAMPO (campi magnetici lentamente variabili) attraverso la visualizzazione di immagini “a fette”, o slice. Figura 5: visualizzazione del fluido cerebro-spinale mediante immagine T1-pesata I gradienti sono generati da bobine (figura 7) che producono campi elettromagnetici lentamente variabili per brevi intervalli di tempo sia lungo la direzione longitudinale z (figura 8) sia sul piano trasversale xy che su piani obliqui. Anche T2 dipende dal tessuto circostante e può essere utilizzato per differenziare i tessuti in termini di contrasto; tuttavia dipende fortemente dalla interazione fra spin e spin. Nella stessa immagine di prima, dove, però, viene esaltato il valore di T2 (immagine T2-pesata), il fluido cerebro-spinale (CSF) appare di colore più chiaro (figura 6). Figura 7: bobine di gradiente AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici Una problematica relativa all’installazione di un sistema RM risiede nella dispersione del campo magnetico statico B, che non è confinato all’interno della regione in cui è collocato il paziente ma si estende anche a zone limitrofe, rendendo necessaria l’applicazione degli aspetti regolatori e tecnici legati alla sicurezza del personale tecnico che vi opera al proprio interno. Effetti fisici reversibili, inoltre, sono causati dal rilascio di energia da parte degli impulsi a radiofrequenza che innalzano la temperatura del paziente. Figura 8: applicazione del gradiente lungo z per la selezione della slice a z=Z0 I valori dei segnali di quel distretto corporeo in risonanza saranno, infine, convertiti in tonalità di grigio differenti, in modo da costruire l’immagine desiderata. SISTEMI CHIUSI SISTEMI APERTI SISTEMI SPECIALI Il campo B è generato all’interno di un tunnel (o BORE), che permette la scansione di tutti i distretti corporei (WHOLE BODY system); l’asse z del campo B è allineato con l’asse del tunnel, in direzione piedi-testa del paziente. Permettono scansioni WHOLE BODY; l’accesso del paziente è reso molto più agevole, allo scopo di ovviare ai problemi sperimentati con i sistemi chiusi. Permettono lo studio delle estremità e delle articolazioni; esistono anche sistemi speciali di ricerca, ad alto campo e bore stretto ad es. per studi su animali SVANTAGGI VANTAGGI DESCRIZIONE I sistemi RM si differenziano in base al loro design in: sistemi chiusi, sistemi aperti e sistemi speciali (o settoriali), le cui caratteristiche sono meglio descritte nella tabella seguente. AIIC • Elevato campo magnetico B • Non necessaria la rimozione del lettino • Elevata omogeneità di campo • alta omogeneità di campo • studio delle articolazioni • limitazione di campo all’area di applicazione • possibili effetti di claustrofobia o altri disagi • campi B meno elevati (fino a 1,2 T) • in genere bassi campi magnetici • omogeneità di campo minore • associazione italiana ingegneri clinici per eseguire procedure interventistiche occorre estrarre temporaneamente il lettino dal tunnel Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” La riduzione dell’intensità del campo magnetico disperso avviene sia mediante l’uso di schermature passive con materiali ad alta permeabilità magnetica µ sia tramite la schermatura attiva (active shielding), realizzata con un contro-campo magnetico generato con avvolgimenti superconduttivi, in direzione opposta a quella delle bobine che danno luogo al campo principale. Un’altra distinzione dei sistemi RM consiste nel tipo di magnete che genera il campo statico B, ed in particolare si distinguono magneti permanenti da quelli superconduttori. I primi, aventi forma a ferro di cavallo, sono costituiti da grossi blocchi di lega ferromagnetica (NeodimioFerro-Boro) e producono un campo magnetico B che può spaziare tra 0,01 T e 0,35 T; i loro costi di gestione sono bassi, anche se riescono a mantenere un’elevata uniformità del campo statico a patto che la temperatura della lega resistiva sia stabile. Nei magneti superconduttori (più utilizzati), invece, il campo B, generato da una corrente indotta per superconduzione in un avvolgimento realizzato tipicamente in niobio/titanio ed annegato nel rame, assume valori almeno pari a 0,5 T. I vantaggi riscontrabili fanno riferimento ad una migliore qualità dell’immagine grazie all’alto rapporto S/N, minori tempi di acquisizione dell’immagine, minori tempi di esecuzione dell’indagine, migliore risoluzione spaziale e alla possibilità di visualizzare processi a livello molecolare (imaging molecolare). Sebbene le potenzialità di questi strumenti siano innegabili, altrettanto palese è l’aumento del rischio globale connesso alla presenza di elio, fondamentale per il raffreddamento del magnete, che presenta proprietà superconduttive a bassissime temperature (4 K): il passaggio dalla fase liquida (a -273°C) a quella gassosa (a -270°C) comporta una espansione adiabatica che determina un aumento del volume occupato dall’elio di circa 760 volte; questo fenomeno, accompagnato da pressioni significative, è molto pericoloso per il paziente. Al fine di evitare ciò, i sistemi RM superconduttivi devono essere dotati di impianti per il convogliamento dell’elio post-quench in atmosfera (tubo di quench), le cui caratteristiche verranno discusse meglio nell’ultimo paragrafo. Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici CAPITOLO 2 Information Technology e Integrazione dati/rete Negli ultimi 20 anni la sostituzione dell’utilizzo di sistemi di dispositivi medici isolati con quello di sistemi integrati ha messo in risalto potenziali rischi che discendono da una cattiva integrazione dei device fisici, quali perdita, sincronizzazione e non protezione dei dati da accessi non autorizzati. L’art. 14 della Parte 1 (Prescrizioni generali relative alla sicurezza fondamentale e alle prestazioni essenziali) della CEI EN 60601-1 (05/2007) in materia di Apparecchi Elettromedicali fa riferimento alla programmabilità di quest’ultimi (SEMP: systèmes électromédicaux programmables); in particolare viene esplicitato come il fabbricante, non più autorizzato a gestire l’accoppiamento rete/device, possa non essere in grado di seguire tutti i processi per ciascun componente costituente il SEMP; deve, così, tener conto della necessità di misure supplementari di controllo del rischio (perdita dati, mancata sincronizzazione dei dati, disaccoppiamento rete/dati) mediante un piano di gestione del rischio, che deve contenere un riferimento al piano di validazione del SEMP ed al rispettivo ciclo di vita. Si riporta nella parte 6 del detto articolo l’obbligo dell’esistenza di una specifica documentata delle prescrizioni per il SEMP e per ciascun suo sottosistema (SSEP) ed un’architettura che soddisfi tale specifica. Nel caso di qualunque modifica al progetto originario occorre revisionare l’intero progetto al fine di evitare degli effetti collaterali che possono invalidare l’implementazione del progetto stesso. All’interno della norma si parla anche dell’identificazione delle responsabilità in materia di integrazione dei sistemi: qualora il fabbricante non sia a conoscenza di tali requisiti, si limita a fornire le informazioni richieste sui propri apparecchi; in tal caso si fa riferimento ad un’organizzazione responsabile, con al centro la figura dell’assemblatore del sistema, che deve avere la competenza per valutare e affrontare i pericoli che possono presentarsi nell’integrare un sistema, e per garantire che i rischi residui del SEMP non aumentino, pianificando l’integrazione di un qualunque dispositivo da integrare sulla base delle istruzioni fornite dal fabbricante, eseguendo la gestione del rischio sul sistema integrato e passando le eventuali istruzioni del fabbricante all’organizzazione responsabile. La norma CEI EN 60601-1 classifica le reti in funzione delle applicazioni software implementate sulla rete, delle conseguenze che possono esserci per il paziente e sulla base dei tempi di reazione, dove il tempo di reazione è il tempo di ritardo tra un guasto dell’accoppiamento rete/dati e il verificarsi di un danno per il paziente. AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Una più recente norma in materia della gestione del rischio per reti IT che incorporano dispositivi medicali è la CEI EN 80001-1 (03/2011). La norma indica la presenza di un responsabile dell’organizzazione, solitamente il direttore generale (DG), per la redazione del documento di gestione del rischio della rete IT medicale che fa riferimento alla pianificazione, connessione, configurazione, mantenimento e dismissione dei dispositivi. L’applicazione del documento di gestione del rischio deve assicurare il soddisfacimento di tre parametri: safety (assenza di rischi non accettabili), effectiveness (capacità di produrre il risultato voluto), data and systems security (protezione da accessi non autorizzati, integrità e disponibilità dei dati). Il DG deve così nominare il responsabile del rischio per le reti IT medicali, persona che deve avere le necessarie qualifiche, conoscenze e competenze. Al fine di attuare quanto stabilito nel documento di gestione del rischio nel migliore dei modi, il responsabile del rischio coopera con quelle figure professionali appartenenti allo staff d’ingegneria clinica, del SIS (sistema informativo sanitario) etc; si preoccupa, quindi, di avere un inventario dei sistemi connessi alla rete, una tracciabilità dei costruttori e fornitori e di chi è coinvolto nei processi e di sottoscrivere accordi di responsabilità (prima del collaudo) con i fabbricanti in modo che siano resi espliciti a ciascun attore del processo i propri compiti e responsabilità. Un altro aspetto di fondamentale importanza è la privacy dei dati sensibili (origine razziale ed etnica, credo religioso, opinioni politiche, salute e vita sessuale), che devono essere riservati (confidenzialità), integri (correttezza dei dati) e disponibili (la garanzia per gli utenti di poter disporre dei dati, delle informazioni e dei servizi). I dati personali devono essere trattati nel rispetto dei criteri di pertinenza (solo per raggiungere le finalità per le quali è stato acquisito il consenso) e non eccedenza (esclusivamente per quanto strettamente necessario al raggiungimento delle finalità per le quali è stato acquisito il consenso). Uno dei problemi che la privacy ci pone è capire come gestire la riservatezza e le credenziali di accesso alle informazioni di ciascuna figura autorizzata. Esistono pertanto delle misure minime di sicurezza da rispettare che fanno riferimento all’autenticazione informatica, alle credenziali di autenticazione, al sistema di autorizzazioni. È necessaria, inoltre, una tracciabilità degli accessi e delle azioni svolte sui dati. L’applicazione di tali misure non è sempre facile in quanto molto spesso i fabbricanti sono aziende multinazionali ignare della normativa italiana in materia di privacy e dati sensibili. Per la gestione della privacy si ritrova un modello gerarchico piramidale, dove all’apice è posto il titolare del trattamento, segue poi il responsabile del trattamento e l’incaricato del trattamento. Circa il Comitato Elettrotecnico Italiano (CEI) sono in via di sviluppo delle linee guida sulla gestione del software; in particolare esse introducono un FASCICOLO DI PRODOTTO/ SISTEMA, dove vengono registrate le attività inerenti la sicurezza di un prodotto/sistema software inteso o come dispositivo medico oppure come software in contesto sanitario, collocato nel contesto di organizzazione responsabile. Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici La prima fase della valutazione del processo consiste nell’analisi del workflow; in particolare, constatato che l’implementazione dello standard DICOM da solo non garantisce il pieno successo dell’integrazione dei sistemi, l’IHE, associazione no-profit dedicata alla interoperabilità nell’Health Information Technology, ha progettato ed implementato documenti (o profili) di integrazione attraverso cui applicare in modo univoco gli svariati standard utilizzati e integrarli. L’IHE Scheduled Workflow Profile (figura 9) rappresenta uno dei possibili punti di partenza necessari per stilare un CAPITOLATO tecnico sulle specifiche degli interfacciamenti/ integrazioni nel caso dell’acquisizione di una nuova tecnologia, che dovrebbero portare ad una riduzione di tempi e costi e ad un aumento di efficienza in termini di produttività e di efficacia. Nel capitolato tecnico si descrivono i profili di integrazione da utilizzare che il fabbricante della tecnologia da acquisire deve rispettare. CAPITOLO 3 Normativa di Sicurezza e Prevenzione del Rischio in Risonanza Magnetica Il quadro normativo italiano in materia di sicurezza e prevenzione del rischio in RM essenzialmente si compone di 4 decreti legislativi (figura 10): Figura 10: il quadro normativo italiano in materia di sicurezza e prevenzione del rischio in RM Figura 9: IHE Scheduled Workflow Profile Nel caso di profili “sofisticati”, al fine di non escludere costruttori su aspetti tecnici di poco conto (a meno che non siano aspetti legati al DICOM), ci sono dei margini di accettazione del progetto. Nell’ultima fase, quella di COLLAUDO, avviene la reale implementazione dei profili IHE richiesti all’interno del sistema IT ospedaliero preesistente in termini di attori e transazioni, si identificano le problematiche di integrazione eventuali emergenti e vengono fornite indicazioni agli utilizzatori sul corretto uso dei sistemi e sul comportamento da mantenere di fronte a possibili errori. AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Al fine di agevolare l’utenza a gestire un quadro normativo così frammentario e composito, l’ISPESL (oggi INAIL) nel 2004 ha pubblicato sul proprio sito le indicazioni operative che raccolgono in un unico testo una serie di metodi, suggerimenti, consigli ed esperienze per ottimizzare la gestione della sicurezza in un sito di Risonanza Magnetica; tali indicazioni non hanno valore specifico di legge, ma sono oggi considerate una linea guida di riferimento in materia. Gli standard di sicurezza in RM previsti dal quadro normativo italiano sono ritrovati all’interno degli all. 1 e 4 del DM 2/8/1991 e degli all. A e B del DM 3/8/1993 e stabiliscono: - i requisiti minimi di carattere strutturale, tecnologico ed organizzativo; - le procedure di gestione dell’attività diagnostica; - il ruolo ed i compiti sia del Medico Responsabile del sito RM che dell’Esperto Responsabile per la Sicurezza dell’apparecchiatura RM; - i limiti di esposizione per pazienti ed operatori. Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici Le criticità riscontrabili nell’attuazione di questi decreti, oramai vetusti, fanno riferimento, in particolare, ai limiti di esposizione per pazienti (limiti di esposizione ai campi elettromagnetici a radiofrequenza) e per operatori (limiti di esposizione al campo magnetico statico), definiti in una fase in cui c’era ancora poca sensibilità a riguardo. Tali criticità sono messe in risalto sulla base di quanto contenuto nelle Linee Guida ICNIRP 2009-2010 e nella Norma di Buona Tecnica Internazionale IEC 60601-2-33 (3° edizione 2010) che evidenzia delle soglie-limite meno stringenti di quelle indicate nel D.M. 3/8/1993; nonostante l’affidabilità di questi standard di sicurezza, l’installazione e l’utilizzo di un tomografo RM devono far fede sempre al quadro normativo italiano. Inoltre, a differenza di altri paesi europei che aggiornano ed integrano lo stato legislativo con le norme sancite dagli Organismi di “buona tecnica”, la tecnica normativa italiana non permette l’autoaggiornamento delle leggi. Ci sono, tuttavia, fattori di allineamento tra Italia ed Europa: prima di tutto il marchio CE, che è obbligatorio per immettere sul mercato comunitario un dispositivo medico (come la RM) e garantisce il rispetto dei requisiti essenziali di sicurezza (R.E.S.) (Direttiva Dispositivi Medici, 93/42/CE come emendata dalla Direttiva 2007/47/CE), indipendentemente dallo scenario in cui avviene la messa in esercizio del medesimo. Per gli impianti RM con magnete superconduttore la marcatura CE da sola non basta a garantire un livello di sicurezza appropriato alla messa in esercizio dello strumento, ma per il suo utilizzo negli Stati Membri, è necessario un valido accoppiamento fra dispositivo medico e l’impianto di sicurezza ad esso asservito (tubo di QUENCH), nonché si deve garantire il rispetto dei requisiti essenziali di sicurezza (R.E.S.) contenuti nell’art. 3 punto 3 della Direttiva Impianti a Pressione, 97/23/CE (PED) per quanto applicabile. Il compimento di una corretta prassi costruttiva (ossia rispettare i R.E.S. nell’art. 3 punto 3 della 97/23/CE) si concretizza col rilascio della certificazione di conformità della parte impiantistica (tubo di QUENCH, impianto di ventilazione ed impianto elettrico), ai sensi del DM 37/08. La certificazione si compone di: 1. la dichiarazione di conformità (rilasciata dalla azienda abilitata), che, come tale, deve riportare l’indicazione da parte del soggetto installatore di aver rispettato il progetto redatto per l’installazione, di aver seguito le norme di buona tecnica applicabili all’impiego dell’installazione, di aver installato componenti e materiali adatti al luogo d’installazione e controllato l’impianto ai fini della sicurezza e della funzionalità con esito positivo, avendo eseguito le verifiche richieste dalle norme e dalle disposizioni di legge (figura 11); 2. altri allegati, quali il progetto dell’impianto, la relazione con tipologie dei materiali utilizzati, lo schema dell’impianto realizzato, il riferimento a dichiarazioni di conformità precedenti, la copia del certificato di riconoscimento dei requisiti tecnico – professionali per le imprese abilitate. AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Figura 11: certificazione del tubo di quench ai sensi del D.M. 37/2008 Nel caso di sostituzione della sola risonanza magnetica e nell’ipotesi in cui l’impianto è antecedente al 2008, o in corrispondenza di qualunque modifica apportata al sistema, occorre verificare che l’impianto e l’accoppiamento risonanza/impianto rispettino sempre i requisiti minimi di sicurezza attraverso la dichiarazione di conformità ai sensi DM 37/08. La progettazione, costruzione o modifica del sito RM dovrà essere poi comunicata all’Autorità Competente (ISPESL), inviando ad esso copia delle dichiarazioni di conformità. In RM i fattori di rischio vengono classificati in rischi costanti (campo magnetico statico B, criogeni per i superconduttori) e rischi in corso di esame diagnostico (campi a RF, gradienti di campo). Il campo magnetico statico, per applicazioni di diagnosi variabile tra 0.15 T e 3 T, è pericoloso per l’effetto di attrazione, sotto la cui azione oggetti metallici possono acquisire una considerevole velocità (effetto proiettile). D’altra parte, corpi metallici in movimento in vicinanza al sito RM possono provocare delle interferenze sovrapponibili all’informazione diagnostica. Al fine di evitare i rischi legati all’effetto proiettile provocato dal campo statico, all’interno del sito RM si è soliti individuare le cosiddette zone di rischio: - zona ad accesso controllato: zona in cui si registra un rischio reale per coloro che sono portatori di pacemaker; il valore di campo magnetico statico mediato nel tempo è almeno pari a 0.5 mT e interessa tipicamente la sala magnete (zona controllata) e i locali strettamente dedicati alla gestione in sicurezza delle attività e dei pazienti; - zona di rispetto: non si ritrova un rischio fisico concreto, tuttavia ci sono delle soglie di attenzione da rispettare. Il valore di B varia tra 0,1 mT e 0,5 mT; non è necessariamente tutta contenuta all’interno del sito RM, ma comunque deve essere necessariamente contenuta nella proprietà del presidio; - zona ad accesso libero: valore di rischio basso o trascurabile con valore di B<0,1 mT. Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici Nella progettazione di un sito RM occorre non solo contenere la zona ad accesso controllato, ma anche quella di rispetto. Solitamente a terra, oltre ad evidenziare la linea che delimita la zona ad accesso controllato, viene segnata anche la linea corrispondente a 200 mT, che indica, secondo le linee guida ICNIRP, il massimo valore di campo (calcolato su una media temporale di un’ora al giorno), a cui il corpo può essere esposto. Un altro rischio, proveniente stavolta non dal campo magnetico statico, riguarda i sistemi RM con magnete superconduttore, ed in particolare il tubo di quench, che rappresenta il tubo attraverso cui avviene la fuoriuscita all’esterno dei fluidi criogenici gassosi a seguito di un quench. Le valvole sulla testa del magnete e la flangia di raccordo della tubazione di quench con la macchina del magnete rappresentano i principali punti critici connessi ad un’eventuale fuoriuscita di gas criogenico in sala RM. Qualora l’elio dovesse liberarsi, essendo più leggero dell’aria, si stratificherebbe verso l’alto, invadendo l’ambiente interno della sala; per questo motivo viene posto sulla prima flangia un sensore di ossigeno, che misura la diminuzione della concentrazione di O2 dovuta alla presenza dell’elio e permette l’attivazione dell’impianto di ventilazione in modo che l’elio possa essere direttamente ripreso e buttato fuori prima che invada il resto dell’ambiente. La taratura, il cui procedimento è previsto dalle ditte costruttrici e chiaramente indicato nel libretto di “Istruzioni per l’uso” del sensore, deve prevedere la possibilità di settare una soglia di pre-allarme al 19% di O2 in corrispondenza della quale si attiva l’avvisatore sonoro-luminoso collegato alla centralina, e una soglia di allarme rigorosamente al 18% di O2 che attiva automaticamente un sistema di ventilazione di emergenza, capace di implementare notevolmente l’efficienza del ricambio d’aria nella sala magnete. Le modalità di taratura (figura 12) del sensore e dell’elettronica ad esso associata sono sancite dalla norma CEI EN 50104, come indicato dalle stesse ditte costruttrici. Il sensore deve essere tarato ogni 6 mesi. Figura 12: certificato di taratura del sensore ossigeno Tipicamente il tubo di quench viene coibentato termicamente e fatto uscire esternamente dal punto più alto della struttura. I raccordi tra le diverse parti di tubo possono avvenire mediante flangiatura, saldatura (in tal caso non deve essere visibile la saldatura) oppure mediante utilizzo di colla stycast, che resiste alla temperatura di -270°C. L’impianto di ventilazione nella sala magnete costituisce, col sensore ossigeno, la catena dei sistemi di sicurezza asserviti al magnete superconduttore: la sua corretta realizzazione è fondamentale per evitare le problematiche connesse all’eventuale rientro di elio in fase gassosa all’interno della sala magnete durante un quench. In condizioni di normale funzionamento deve: AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” - mantenere la temperatura 22±2 °C e l’umidità compresa tra 40% e 60% al fine di contenere i livelli del SAR e di salvaguardare il benessere del paziente; - garantire 6-8 ricambi/h nel locale esame; - assicurare una leggera pressione sulla porta della gabbia per non aspirare la polvere dall’esterno (ΔP > 0). Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici In condizioni di emergenza, invece, deve: - garantire 18-22 ricambi/h di ventilazione - assicurare una leggera depressione sul locale esami (Δp<0), per facilitare l’apertura della porta e, una volta aperta, evitare che fuoriesca elio all’esterno della sala; - prevedere al di sopra della torretta del tomografo un canale per l’eventuale aspirazione diretta dell’ elio gassoso che dovesse fuoriuscire (ripresa supplementare d’emergenza). CAPITOLO 4 Progettazione di un sito RM Un presidio di RM deve avere un unico accesso rigidamente controllato e riservato al solo personale autorizzato e a pazienti da esso accompagnati, previo preventivo consenso. L’accesso controllato è garantito, oltre dalla cartellonistica, attraverso l’utilizzo di un dispositivo di accesso (chiave, codice numerico, scheda a banda magnetica, etc.) all’interno del sito. La linea di campo dei 5 Gauss (che circoscrive la zona controllata) deve necessariamente essere contenuta all’interno della zona ad accesso controllato, e per lo più è solita essere confinata all’interno della sala magnete. Le zone esterne alla sala magnete eventualmente interessate vanno interdette con barriere fisse ed identificate con cartellonistica che ne indichi i rischi all’esposizione ai campi magnetici (presenti all’interno) e le restrizioni di accesso. La zona di rispetto deve essere completamente contenuta all’interno della proprietà di pertinenza del datore di lavoro, possessore del tomografo RM e deve avere al proprio interno dotazioni che tengano conto delle problematiche esistenti connesse alla compatibilità elettromagnetica con apparecchi elettronici e della possibile magnetizzazione di apparati ferromagnetici. La sala di attesa per i pazienti deambulanti deve essere all’esterno del sito RM, ed è auspicabile averla nei suoi immediati pressi; i pazienti barellati devono avere una sala di attesa “dedicata” che ne garantisca la privacy, tranne nei casi in cui siano solo pazienti ricoverati (e quindi programmabili) o quando le procedure siano tali da garantire l’assenza di tempi di attesa (pronto soccorso, etc.). AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” L’accettazione, specifica per l’RM o in comune con altre tecniche diagnostiche, deve essere esterna o con sportello afferente all’esterno; essa può rilasciare il modulo di anamnesi da effettuarsi all’interno del sito RM o nei suoi immediati pressi, o comunque in un locale dedicato nel quale sia presente la dotazione minima per una visita medica. L’anamnesi consiste nell’accertamento da parte del medico responsabile dell’esecuzione dell’esame, della assenza di controindicazioni all’esame RM, in riferimento alla sua esposizione ai campi magnetici utilizzati dall’apparecchiatura RM e ai rischi ad essi connessi. Il questionario anamnestico viene compilato sempre prima dell’esame dal medico, interrogando il paziente e firmandolo in calce, ai sensi di legge; l’atto di eventuale delega non è prevista per legge. Il paziente, a sua volta, firma il consenso informato all’indagine e all’eventuale somministrazione del mezzo di contrasto. In aggiunta all’anamnesi, la legislazione vigente prevede la presenza di un metal detector (generalmente portatile) nel sito RM al fine di individuare protesi interne, oggetti sul corpo, parti metalliche negli indumenti che non vengono tolti. All’interno del sito RM devono essere previsti, a seconda delle modalità di gestione dell’attività diagnostica, uno o più locali adibiti a spogliatoio pazienti, con dotazione minima adatta allo scopo; è buona norma includere una cassetta di sicurezza ad uso dei pazienti. Lo spogliatoio del personale non è richiesto, ma, se ritenuto necessario, può essere locato sia all’interno che all’esterno del sito RM; se all’interno, deve essere considerato ad uso esclusivo. All’interno o negli immediati pressi del sito RM deve essere previsto almeno un servizio igienico ad uso dei pazienti, attrezzati anche per portatori di handicap. La zona Preparazione è un locale o area delimitata da barriere fisse o mobili (che garantiscano la privacy del paziente trattato), destinata a trattamenti medici sul paziente che precedono l’esame RM (sedazione, somministrazione di liquidi di contrasto,etc.); essa deve essere attrezzata di cabinet per i farmaci, lettino/barella amagnetica, disponibilità di gas anestetici e dispositivi medici specifici, nonché della dotazione minima prevista dal Medico Responsabile del sito RM. La zona Emergenza è un locale o area destinata per un eventuale primo soccorso medico sul paziente che dovesse necessitare di un primo intervento, anche per motivi non strettamente correlati all’esecuzione dell’esame; tale zona non deve essere delimitata da porte o altra barriera fissa che possa in qualche modo creare impedimento alle procedure di soccorso e deve essere quanto più vicino alla porta della sala esami per consentire un tempestivo intervento. Le postazioni di emergenza attrezzate devono essere tante quante sono le apparecchiature RM presenti nel sito. Qualora la stessa postazione sia adibita sia a preparazione sia ad emergenza, occorre definire e formalmente istituire delle procedure restrittive di esecuzione degli esami che consentano la presenza di un solo paziente alla volta all’interno del sito RM Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici e che facciano riferimento ad una allocazione individuata per quanto attiene i farmaci ed i dispositivi medici. Nel caso di più apparecchiature RM presenti, occorre sempre avere tante postazioni P/E quante sono le apparecchiature, con la restrizione di un numero di pazienti all’interno del sito sempre pari al numero di macchine RM operanti. Le strumentazioni di supporto devono essere amagnetiche (in realtà le strumentazioni non sono mai del tutto amagnetiche, ma deve essere il costruttore a garantire il loro corretto funzionamento se mantenute al di là di un certo campo). La ventilazione e la climatizzazione della sala magnete devono garantire una temperatura costante di 22 +/- 2°C ed un’umidità relativa del 40-60%, al fine di contenere eccessivi aumenti del SAR e di salvaguardare il benessere del paziente. Le curve isomagnetiche teoriche, fornite in fase di progetto dalla casa costruttrice all’esperto responsabile dell’impianto RM, consentono di valutare a priori l’impatto ambientale dell’apparecchiatura nel sito di installazione, da cui prevedere, nel caso sia necessario, schermature aggiuntive e limitazioni di utilizzo dei locali attigui, mentre a posteriori (in sede di collaudo) l’esperto responsabile deve mappare le linee di isocampo reali a conferma delle scelte protezionistiche fatte a priori. Il fine è la corretta delimitazione della zona controllata, l’opportuna individuazione della zona di rispetto, la puntuale definizione della destinazione d’uso degli ambienti che circondano la sala magnete. RACCOLTA SLIDE DEL CORSO Occorre, inoltre, prevedere un sistema di schermatura per evitare le interferenze provocate dall’esterno rispetto al segnale a radiofrequenza generato all’interno della sala RM (gabbia di Faraday); le uniche aperture sono quelle per il passaggio degli impianti (pannello di controllo), per la finestra della consolle di comando (dove si ritrovano i pulsanti di sicurezza e dei display dei sensori ossigeno e termoigrometri), per la porta di accesso, che, grazie alla presenza di appositi fingers di rame, garantiscono la tenuta della gabbia. La schermatura è funzione del tempo e dell’uso, per cui va periodicamente controllata (su base annuale) ai fini di preservare opportuni standard di qualità. La sala dedicata alla refertazione delle immagini RM ottenute può essere allocata all’interno del sito RM (ma a suo uso esclusivo) o all’esterno (con possibilità di refertare in condivisione con altre tecniche diagnostiche), con la possibilità di utilizzare a tale scopo aree o altre sale già dedicate ad altra attività; l’archivio può trovarsi sia all’interno che all’esterno del sito RM. AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici Lo SPIN Lo spin è una grandezza di natura quantistica, intuitivamente facile da comprendere se pensiamo al protone come ad una pallina magnetica in rotazione. In meccanica quantistica, lo spin rimane costante. Può variare solo la direzione dell’asse di rotazione. spin Osserviamo che un nucleo dotato di spin è sempre magnetico. north Nell’imaging RM si ricorre alle caratteristiche collettive degli spin di un distretto corporeo di interesse. RISONANZA MAGNETICA Principi di funzionamento ed applicazioni in ambito medico Fisicamente lo spin è un vettore, ed è possibile pertanto considerarne la somma in senso vettoriale, estesa ad una popolazione come l’insieme dei nuclei di H in un distretto corporeo, o meglio ancora in un elemento di volume del corpo, detto VOXEL south Francesco Lisciandro Principi di funzionamento dei tomografi RM La tomografia RM utilizza le caratteristiche magnetiche di alcune particelle atomiche per ottenere informazioni (comunemente, immagini) cliniche. Si ricorre principalmente alle proprietà magnetiche del protone o, più correttamente, del nucleo di idrogeno (H). Perché l’idrogeno? •E’ largamente presente nell’acqua e nel grasso, ed è quindi l’elemento prevalente nel corpo umano •I nuclei di H forniscono il segnale di risonanza a maggiore intensità, rispetto agli altri elementi chimici La grandezza fisica che ci permette di utilizzare in RM il protone dell’idrogeno è il suo momento di dipolo magnetico, detto comunemente SPIN. In assenza di campi magnetici B esterni, gli spin dei nuclei H sono orientati in modo casuale ed i loro effetti magnetici si annullano statisticamente Summed X In presenza di un campo B, la popolazione del voxel si allinea parallelamente ad esso, con quasi la stessa probabilità di orientarsi in ciascuno dei due versi opposti: SPIN UP o SPIN DOWN. Il rapporto SPIN UP/SPIN DOWN non è esattamente pari a 1: esiste un leggero eccesso di spin nella direzione UP. Si determina così una magnetizzazione netta M. Z Y NET Summed Z MAGNETISATION B X Y Come conseguenza della loro natura quantistica, gli spin non si allineano perfettamente nella direzione del campo B e sono costretti ad un moto di PRECESSIONE La velocità, e quindi la FREQUENZA, della precessione rivestono un ruolo importante nell’imaging RM. In particolare dipendono: •Dal tipo di nucleo •Dall’intensità del campo B applicato Con un po’ di intuizione possiamo comprendere che se siamo interessati a rilevare un segnale dagli spin che precedono, dovremo sintonizzare il nostro sistema di ricezione esattamente alla frequenza di precessione, che è nota come frequenza di Larmor ωL: Con opportuni impulsi RF è possibile modificare la condizione degli spin, alterandone temporaneamente lo stato energetico. Al termine dell’impulso gli spin si riportano nella condizione iniziale (configurazione energetica minima) emettendo radiazione elettromagnetica (RF). Il flip angle aumenta con l’energia dell’impulso RF: Un impulso a 180° inverte la magnetizzazione nella direzione opposta lungo l’asse z Un impulso a 90°ribalta la magnetizzazione sul piano xy ωL = γ x B γ è detto RAPPORTO GIROMAGNETICO = 42 MHz/T Nei sistemi RM imaging (RMI) γ cade nello spettro della radiofrequenza (RF). A 1,5 T, ωL = 63 MHz Anche il moto di precessione degli spin è statisticamente casuale, quindi gli spin, pur ruotando tutti con la stessa frequenza, sono completamente sfasati fra loro. Graficamente la situazione è la seguente: Il segnale viene generato dal movimento della magnetizzazione M sul piano xy, poiché questa è la sola componente magnetica in movimento. Nel piano xy, tuttavia,le componenti trasversali della magnetizzazione M si annullano fra loro, mentre lungo la direzione z è presente una magnetizzazione netta Mz Se vogliamo ricevere un segnale RF è pertanto indispensabile stimolare gli spin in modo da alterare la loro condizione naturale di sfasamento. Possiamo quindi modificare lo stato degli spin utilizzando un’onda magnetica, o meglio, un impulso RF. L’impulso deve però avere una frequenza appropriata per interagire con gli spin in precessione: deve cioè essere in RISONANZA. In altre parole, la radiofrequenza deve avere una frequenza pari alla frequenza di Larmor. Come si genera il segnale RM? Innanzitutto, osserviamo che sia Mz che Mxy precedono alla frequenza γL. Mxy può essere analizzato come se si trattasse di un magnete rotante. Se inseriamo una bobina conduttrice all’interno di un campo magnetico rotante, generiamo in essa una tensione indotta: ecco il segnale MR! Ovviamente l’intensità del segnale MR è proporzionale a Mxy, e tale segnale decade molto rapidamente. Poiché Mxy: •Precede liberamente •Induce un segnale RF •Decade liberamente Il segnale prende il nome di FID (Free Induction Decay) T2 Tempi di rilassamento T1 e T2 Il FID decade rapidamente nel tempo perché dopo l’impulso RF a 90°, la magnetizzazione torna a riallinearsi lungo z: si ha un processo cosiddetto di rilassamento. Si osserva inoltre che magnetizzazione trasversa Mxy decade più rapidamente di quanto la Mz (magnetizzazione longitudinale, o ML) impiega a ricostituirsi. Questi processi di rilassamento hanno un andamento temporale di tipo esponenziale. T1 è la costante di tempo che caratterizza il recupero di Mz, mentre T2, molto più breve di T1, è quella relativa alla diminuzione del valore di Mxy. Dopo un impulso RF a 90°, la Mxy genera il segnale di FID che decade rapidamente, poiché gli spin tornano i brevissimo tempo a precedere fuori fase. Il rapido sfasamento è dovuto alla interazione fra spin e spin, ed il rilassamento è caratterizzato da una costante di tempo T2, che non dipende da B ma dipende fortemente dal tessuto circostante. Anche il T2 può essere utilizzato per differenziare i tessuti in termini di contrasto In una immagine dove viene esaltato il valore di T2 (T2-pesata), il fluido cerebro-spinale (CSF) appare di colore più chiaro. T1 T1 non è uguale in ogni punto del corpo umano: dipende dalla composizione del tessuto. Infatti il rilassamento di Mz comporta cessione di energia dagli spin al reticolo, cioè all amateria circostante. Tessuti differenti hanno valori di T1 diversi: il tempo T1 può quindi essere utilizzato per esaltare il CONTRASTO in una immagine RM. Inoltre T1 è influenzato anche dal valore di B: campi B elevati comportano valori elevati per T1 e quindi maggiore contrasto nella immagine. I tessuti patologici mostrano differenti concentrazioni di H2O rispetto a quelli sani; avranno quindi tempi T1 diversi e la loro differenza può essere visualizzata in termini di contrasto in una immagine RM. In una immagine dove viene esaltato il valore di T1 (T1-pesata), il fluido cerebro-spinale (CSF) appare di colore scuro. I gradienti di campo magnetico Secondo quanto visto finora, applicando un campo statico B ed usando opportuni impulsi RF è possibile indurre un segnale RM nel corpo umano. L’impiego clinico di tale tecnica, tuttavia, è finalizzato alla visualizzazione le differenze nelle strutture tissutali, all’interno di regioni corporee di interesse. E’ necessario pertanto provocare il fenomeno fisico della risonanza magnetica in termini locali, e di conseguenza localizzare spazialmente i singoli segnali RM ricevuti dalle singole strutture anatomiche. La soluzione a questo problema consiste nel differenziare spazialmente il valore del campo magnetico statico B. In questo modo posizioni differenti avranno frequenze di precessione diverse e la risonanza magnetica risulterà spazialmente differenziata L’imaging tomografico prevede la visualizzazione clinica delle regioni di interesse attraverso immagini “a fette”, o slice. Queste slice vengono ottenute accendendo dei GRADIENTI DI CAMPO MAGNETICO I gradienti sono generati per mezzo di apposite bobine, le quali agiscono sia sul piano xy, sia lungo l’asse z. In questo modo i gradienti determinano delle variazioni sul valore del campo B, nell’intervallo di tempo durante il quale essi vengono accesi. Una considerazione importante: Abbiamo visto che l’imaging RM è ottenuto tramite l’impiego di tre distinti agenti fisici: •Il campo magnetico B0 (campo statico) •La radiazione RF (campi elettromagnetici variabili) •I gradienti (campi magnetici lentamente variabili) E’ importante osservare che sono tutti agenti di rischio fisico! Come agiscono i gradienti? Immaginiamo, come esempio, di accendere un gradiente nella direzione z simultaneamente all’impulso RF: avremo una unica posizione z0 dove avverrà il fenomeno della risonanza magnetica (ωL = γ x B0): solamente nel piano (x,y,z0) sarà originato un impulso RM Il gradiente impiegato viene detto di SLICE SELECTION Analogamente si può intervenire sul piano xy perpendicolare a z, ed anche su piani obliqui z0 B0 ωL Impiego clinico delle RM Normalmente i sistemi RM sono differenziati in base al loro design. Vengono così distinti fra: •Sistemi chiusi (a tunnel) •Sistemi aperti •Sistemi speciali (e settoriali) Sistemi chiusi Il campo B è generato all’interno di un tunnel (o BORE). Si tratta di sistemi WHOLE-BODY e permettono scansioni di tutti i distretti corporei. L’asse z del campo B è allineato con l’asse del tunnel, in direzione piedi-testa del paziente. VANTAGGI •Si tratta di sistemi ad alto B •Realizzano una alta omogeneità di campo Sistemi speciali (e settoriali) L’impiego clinico prevalente è lo studio delle estremità e delle articolazioni I sistemi sono caratterizzati da bassi campi magnetici, limitati all’area di applicazione. Esistono anche sistemi speciali di ricerca, ad alto campo e bore stretto ad es. per studi su animali . SVANTAGGI •Il paziente viene posizionato all’interno di un tunnel •Possibili effetti di claustrofobia o di altri disagi •Per eseguire procedure interventistiche occorre estrarre temporaneamente il lettino dal tunnel Sistemi aperti In questi sistemi l’accesso del paziente è reso molto più agevole, allo scopo di ovviare ai problemi sperimentati con i sistemi chiusi. VANTAGGI Permettono scansioni WHOLE-BODY Le procedure interventistiche non richiedono la rimozione del lettino Permettono lo studio delle articolazioni SVANTAGGI Operano a campi B meno elevati L’omogeneità di campo è inferiore Il tipo di magnete e l’intensità di campo magnetico B influenzano fortemente le problematiche relative alla installazione ed al posizionamento delle apparecchiature. Occorre tenere presente che il campo magnetico non è confinato all’interno della zona riservata al paziente, ma si estende in tutte le direzioni spazio (campo magnetico DISPERSO), rendendo necessario porre molta attenzione agli ambienti circostanti, alle persone che li frequentano ed alle attrezzature installate al loro interno. Anche l’impiego della RF comporta la necessità di tenere in opportuna considerazione gli aspetti legati alla sicurezza. Normalmente si rende necessario realizzare adeguate schermature di contenimento per il campo magnetico e per la RF Magneti permanenti e superconduttori E’ possibile distinguere i sistemi RM anche in base alla modalità con cui viene instaurato il campo magnetico statico B Esistono infatti: Andamento tipico delle linee di campo B per un sistema aperto da 0,4 T a magnete permanente. Campo Bz ortogonale al lettino Particolare attenzione va prestata alla posizione della linea da 0,5 mT (5 Gauss) •MAGNETI PERMANENTI con B = da 0,01 a 0,35 T •MAGNETI SUPERCONDUTTORI con B da 0,5 a 3 T (o più…) (i magneti resistivi sono sempre meno utilizzati e realizzati) Magneti permanenti Sono costituiti da grossi blocchi di lega ferromagnetica (Neodimio-Ferro-Boro), con forma ad U (a ferro di cavallo) I poli magnetici sono solitamente posti sopra e sotto al lettino: il campo B risulta quindi ortogonale al paziente. In altri sistemi (generalmente quelli settoriali), il campo B può anche essere diretto parallelamente al pavimento •Posizionando i poli in prossimità del paziente, è possibile ottenere una alta uniformità di campo, a condizione di avere una alta stabilità della temperatura •I costi di esercizio sono bassi •Si realizzano campi ≤ 0,5 T Andamento tipico delle linee di campo B per un sistema aperto da 0,3 T a magnete permanente Per i magneti superconduttori si evidenziano principalmente due aspetti problematici: Magneti superconduttori In questi magneti il campo B è generato da una corrente indotta per superconduzione in un avvolgimento realizzato tipicamente in niobio/titanio ed annegato nel rame. •La gestione dei criogeni (recupero dell’elio, tubo di quench, rivelatore ossigeno, ventilazione d’emergenza) •E’ possibile raggiungere elevati valori di campo magnetico, anche > 7T •Le proprietà superconduttive del materiale delle bobine si manifesta solo a temperature estremamente basse, pari a ~ 4° K Attualmente i sistemi più diffusi sono quelli a 1,5 T, ma sono sempre più utilizzati, nell’ambito della ricerca clinica, magneti superconduttivi da 3 T •La presenza di campi magnetici dispersi a distanze notevoli dall’isocentro I vantaggi principali sono: •Migliore qualità dell’immagine a causa dell’alto rapporto S/N •Minori tempi di acquisizione dell’immagine •Minori tempi di esecuzione dell’indagine •Migliore risoluzione spaziale •Possibilità di visualizzare processi a livello molecolare (imaging molecolare) Gestione dei criogeni e QUENCH Schema del sistema di raffreddamento Outer vacuum can •L’elio è il fluido criogenico che consente di raffreddare il magnete e di garantire quindi la “superconduzione”. Tipicamente 1200 litri di elio in fase liquida permettono di raffreddare un magnete da 1,5 tesla . Il problema di sicurezza principale è legato al fatto che l’elio – elemento tossico inodore e incolore - è liquido a -273 e bolle a 270: un piccolo surriscaldamento del sistema è potenzialmente pericoloso perché potrebbe comportare un improvviso e violento passaggio dell’elio dalla fase liquida a quella vapore, comportando una espansione adiabatica che porta ad aumentare il volume occupato dall’elio di circa 760 volte. •Al fine di evitare che tale fenomeno, associato peraltro a pressioni significative, comporti una evaporazione che possa anche solo parzialmente interessare la sala RM, i sistemi RM superconduttivi devono essere dotati di impianti per il convogliamento dell’elio post-quench in atmosfera, ed in condizioni di sicurezza (tubo di quench) . •Le cause generalmente più frequenti di sopravvenuto quench sono: •Cause naturali: aumenti anche lievi della temperatura del criogeno; spegnimento controllato del magnete in occasione di particolari interventi di manutenzione o decommissioning •Cause accidentali: terremoti, abbattimento forzato del campo magnetico a causa di un incidente (incendio, etc.) Main Magnet Coils Superconductor NbTi Helium container Liquid Helium at 4.2K •Delle caratteristiche del tubo di quench e dei dispositivi di sicurezza associati alla gestione dei fluidi criogenici si occuperà la prossima relazione. Campo magnetico disperso (Stray field o Fringe field) Linee di campo B per un tipico impianto da3 T superconduttore Per ridurre l’intensità del campo magnetico disperso, una prima soluzione è il ricorso a schermature passive con materiali ad alta permeabilità magnetica µ. In aggiunta a ciò, nei moderni sistemi RM, il campo disperso viene contenuto principalmente tramite la schermatura attiva (active shielding), realizzata con un contro-campo magnetico generato con avvolgimenti superconduttivi in direzione opposta a quella delle bobine che danno luogo al campo principale. Le bobine di schermatura attiva (in blu) sono poste in serie a quelle principali (in rosso), ma sono attraversate da una corrente in direzione opposta Linee di campo B per un tipico impianto da1,5 T superconduttore Linee di campo B per un sistema da 1,2 T superconduttivo aperto indice CORSO:REALIZZAZIONE E GESTIONE IN SICUREZZA DI UN SITO PER DIAGNOSTICA CON RISONANZA MAGNETICA - ASPETTI TECNICI E OPERATIVI responsabile scientifico: Ing. Alessandro REOLON docente: Ing. Maurizio RIZZETTO • Contesto normativo – CEI EN 60601-1 – il SEMP – IEC 80001-1 – Linea guida CEI – Privacy • Acquisizione modalità – specifiche Interoperabilità – Capitolato – interfacciamenti/integrazioni – Capitolato – sicurezza informatica e reti • Configurazione e processo di implementazione Maurizio Rizzetto CONTESTO NORMATIVO CEI EN 60601-1 - Apparecchi elettromedicali Parte 1: Prescrizioni generali relative alla sicurezza fondamentale e alle prestazioni essenziali CEI EN 60601-1 • Art. 14 SISTEMI ELETTROMEDICALI PROGRAMMABILI (SEMP) • NOTA 2 E` noto come il FABBRICANTE possa non essere in grado di seguire tutti i PROCESSI identificati nell’art. 14 per ciascun componente costituente il SEMP, come il software a disposizione immediata (OTS), i sottosistemi di origine non medicale, e i dispositivi a configurazione manuale. In questo caso il FABBRICANTE dovrebbe tener conto in modo particolare della necessita` di misure supplementari di CONTROLLO DEL RISCHIO. 14.3 PIANO DI GESTIONE DEL RISCHIO • Il piano di GESTIONE DEL RISCHIO richiesto in 3.5 della ISO 14971 deve comprendere anche un riferimento al piano di VALIDAZIONE DEL SEMP (vedi 14.11). 14.4 CICLO DI VITA DEL SEMP • 14.7 * Specifica per le prescrizioni • Per il SEMP e ciascun suo sottosistema (ad esempio un SSEP) deve esistere una specifica documentata delle prescrizioni. • 14.8 * Architettura • Per il SEMP e ciascun suo sottosistema, deve essere specificata un’architettura che soddisfila specifica per le prescrizioni. • 14.12 * Modifiche • Se uno o tutti i risultati del progetto portano ad una modifica rispetto al progetto originario, si applicano tutte le prescrizioni del presente articolo come se si trattasse di un nuovo progetto, oppure la continuita` della validita` della documentazione del precedente progetto deve essere valutata da una PROCEDURA di modifica/cambiamento documentata. • Il CICLO DI SVILUPPO DI UN SEMP deve essere documentato. • per lo sviluppo del software. • Il CICLO DI SVILUPPO DI UN SEMP deve includere una serie di punti di riferimento. (milestone) • In ciascun punto di riferimento, devono essere definite le attivita` che devono essere completate e i metodi di VERIFICA da applicare a tali attivita`. • Ciascuna attivita` deve essere definita, comprese le informazioni in ingresso e uscita. • Ciascun punto di riferimento deve identificare le attivita` della GESTIONE DEL RISCHIO che devono essere completate prima di tale punto di riferimento. • Il CICLO DI SVILUPPO DI UN SEMP deve essere ritagliato su uno sviluppo specifico, redigendo piani che dettaglino le attivita`, i punti di riferimento e i programmi. • Il CICLO DI SVILUPPO DI UN SEMP deve comprendere le prescrizioni relative alla documentazione. 14.6 PROCESSO GESTIONE DEL RISCHIO • • • • • • • • • • • • • 14.6.1 * Identificazione di PERICOLI noti e prevedibili Nel compilare l’elenco dei PERICOLI noti e prevedibili, il FABBRICANTE deve considerare i PERICOLI associati agli aspetti del software e dell’hardware del SEMP, compresi quelli associati all’ACCOPPIAMENTO RETE/DATI, ai componenti forniti da terze parti e dei sottosistemi con componenti hardware obsoleti o non conformi. NOTA Oltre al materiale fornito nell’Allegato D della ISO 14971, l’elenco delle possibili cause di PERICOLI associati al SEMP dovrebbe includere: – il guasto dell’ACCOPPIAMENTO RETE/DATI che fornisce le necessarie caratteristiche di SICUREZZA FONDAMENTALE o di PRESTAZIONI ESSENZIALI del SEMP; – le retroazioni impreviste [fisiche o di dati] (le possibilita` comprendono, segnali di ingresso non richiesti, il superamento dei valori limite o segnali di ingresso non congrui o generati da interferenze elettromagnetiche); – i dati non disponibili; – la perdita di integrita` dei dati; – i dati errati; – l’ errata sincronizzazione dei dati. – le interazioni involontarie all’interno e tra i SSEP; – gli aspetti o le caratteristiche sconosciute del software fornito da terze parti; – gli aspetti o le caratteristiche sconosciute di un SSEP fornito da terze parti; – la perdita di sicurezza dei dati, in particolare la vulnerabilita` a manomissioni, interazioni involontarie con altri programmi e virus. 14.13 * Connessione del SEMP ad altri apparecchi attraverso un ACCOPPIAMENTO RETE/DATI • • • • • • • • • • • • • Se e` previsto che il SEMP sia collegato attraverso l’ACCOPPIAMENTO RETE/DATI ad altri apparecchi posti fuori del controllo del FABBRICANTE del SEMP, la descrizione tecnica deve: a) specificare le caratteristiche dell’ACCOPPIAMENTO RETE/DATI necessarie per la DESTINAZIONE D’USO del SEMP; b) elencare le SITUAZIONI DI PERICOLO conseguente ad un guasto dell’ACCOPPIAMENTO RETE/DATI per fornire le caratteristiche specificate; c) informare l’ORGANIZZAZIONE RESPONSABILE che: – la connessione del SEMP attraverso un ACCOPPIAMENTO RETE/DATI che includa altri apparecchi potrebbe portare a RISCHI non identificabili in anticipo per i PAZIENTI, gli OPERATORI e terze parti; – l’ORGANIZZAZIONE RESPONSABILE dovrebbe identificare, analizzare, valutare e controllare tali RISCHI; – le modifiche successive all’ACCOPPIAMENTO RETE/DATI potrebbero introdurre nuovi RISCHI e richiedere un’ulteriore analisi; e – le modifiche all’ACCOPPIAMENTO RETE/DATI comprendono: – modifiche alla configurazione dell’ACCOPPIAMENTO RETE/DATI; – la connessione di ulteriori elementi all’ACCOPPIAMENTO RETE/DATI; – lo scollegamento di elementi dall’ACCOPPIAMENTO RETE/DATI; – l’aggiornamento degli apparecchi collegati all’ACCOPPIAMENTO RETE/DATI; – il miglioramento degli apparecchi collegati all’ACCOPPIAMENTO RETE/DATI. H.3.1 CICLO DI SVILUPPO DEL SEMP • Un CICLO DI SVILUPPO DEL SEMP, come quello illustrato nella Figura H.2, consiste in un certo numero dei PROCESSI composti da attivita`. Ciascuna attivita` viene svolta per realizzare uno scopo specifico. Per applicare la GESTIONE DEL RISCHIO, e` richiesta l’affidabilita` delle attivita` di progettazione su cui si basa la GESTIONE DEL RISCHIO. In particolare, questa e` una prescrizione riferita al ciclo di vita del software. • La IEC 62304 descrive i processi che devono essere inclusi nel ciclo di vita di sviluppo del software di un dispositivo medico sicuro. H.6 ACCOPPIAMENTO RETE/DATI H.6.1 Generalità • Nel contesto della presente Norma, le informazioni trasmesse come parte dell’ACCOPPIAMENTO RETE/DATI, sono quelle previste dal FABBRICANTE come trasmissibili (vale a dire non a seguito di azioni illegali o illecite da parte di persone non autorizzate). • L’ACCOPPIAMENTO RETE/DATI nel senso della presente Norma non include le informazioni trasferite attraverso le interfacce utente. Il FABBRICANTE definisce i tipi possibili di informazione e i loro protocolli di trasmissione nella descrizione tecnica (vedi 14.13). H.6.2 Responsabilità nell’integrazione dei sistemi • Per poter svolgere la propria funzione, l’assemblatore del sistema deve conoscere: – come e` previsto che il sistema integrato venga impiegato; – le prestazioni richieste al sistema integrato; – la configurazione prevista del sistema; – i vincoli sull’estendibilita` del sistema; – le specifiche di tutti gli APPARECCHI EM e degli altri apparecchi che devono essere integrati; – le prestazioni di ciascun APPARECCHIO EM e degli altri apparecchi; e – il flusso delle informazioni all’interno e attorno al sistema. • Queste informazioni non saranno disponibili ai singoli FABBRICANTI, e per questo motivo ciascun singolo FABBRICANTE non puo` rivestire il ruolo dell’assemblatore del sistema. In qualche caso questa figura deve essere una persona singola od un’organizzazione che ha l’intera responsabilita`, la quale non puo` essere condivisa tra i molti e diversi FABBRICANTI. La responsabilita` di un FABBRICANTE si limita a fornire le informazioni richieste sui propri apparecchi (vedi 14.13). • Evidentemente un’ORGANIZZAZIONE RESPONSABILE puo` incaricare un FABBRICANTE di integrare il suo sistema. In questo caso l’intero sistema puo` diventare un SISTEMA EM, e sara` responsabilita` del FABBRICANTE fornire un sistema correttamente integrato. In questo caso, il sistema puo` essere regolato in modo indipendente. • L’assemblatore del sistema deve avere la competenza per valutare e affrontare i pericoli che può presentarsi nell’integrare un sistema, e per garantire che i RISCHI RESIDUI del singolo SEMP non aumentino. • Normalmente un assemblatore di sistema dovrebbe: – pianificare l’integrazione di un qualsiasi APPARECCHIO EM o SISTEMA EM con apparecchi non medicali, rispettando le istruzioni fornite dai diversi COSTRUTTORI; – eseguire la GESTIONE DEL RISCHIO sul sistema integrato; e – passare le eventuali istruzioni del FABBRICANTE all’ORGANIZZAZIONE RESPONSABILE quando H.7 Considerazioni sul progetto per l’ACCOPPIAMENTO RETE/DATI H.7.2 Cause di PERICOLI associati all’ACCOPPIAMENTO RETE/DATI • Nei sistemi di RETE/DATI ACCOPPIATI, le probabili cause di PERICOLI sono costituite da: perdita di dati; interscambio non appropriato di dati; dati corrotti; sincronizzazione inappropriata nello scambio di dati; ricezione imprevista di dati; accesso non autorizzato ai dati. – – – – – – • nell’identificare le cause dei PERICOLI associati all’ACCOPPIAMENTO RETE/DATI, si dovrebbe prendere in considerazione almeno quanto segue: – – – – l’assistenza a distanza (accesso esterno alla rete); il sistema operativo (compatibilita` tra i sistemi operativi); modifica/aggiornamenti del software (sistemi operativi, applicazioni, etc.); compatibilita` delle interfacce (collisioni di dati, formato dei dati): • • • – – – – – – – – – – connessioni (modifica dell’hardware, connettori di rete); schede di rete (compatibilita`); protocolli di rete (DICOM, HL7, etc.); struttura/sincronizzazione dei pacchetti di dati; carichi/larghezza di banda normali della rete; carico di picco della rete; supporto dei dati (longevita` e rintracciabilita`); sicurezza (virus, bachi, aggiornamenti o miglioramenti del software non autorizzati); tempo massimo di risposta accettabile; tasso accettabile di guasti di rete; disponibilita` della rete (manutenzione pianificata e non pianificata); incompatibilita` tra le interfacce/formati che portano ad una perdita di affidabilita` durante il trasferimento delle informazioni; tipologie eterogenee di reti. Nell’analisi delle cause potenziali dei PERICOLI sopra elencati, si dovrebbero prendere in considerazione le seguenti questioni: • a) Usi impropri ragionevolmente prevedibili – La connessione alla rete e` compatibile con la DESTINAZIONE D’USO di ciascun componente del SEMP? • b) Circolazione non corretta di dati da e verso ciascun componente del SEMP – Quali sono i dati trasferiti attraverso la rete utilizzata e a quali compiti si riferiscono? Quali sono le conseguenze di un’interruzione dell’ACCOPPIAMENTO RETE/DATI? • c) Lo scostamento dalle caratteristiche di funzionamento specificate di tutti i componenti del SEMP – Quali sono le caratteristiche di funzionamento del SEMP, e a che livello sono influenzate dall’ACCOPPIAMENTO RETE/DATI? • d) Caratterizzazione incompleta dei parametri dell’ACCOPPIAMENTO RETE/DATI – La tipologia della rete, la configurazione, i parametri (ad esempio aperta, chiusa, larghezza di banda, protocollo di trasmissione) sono completamente definiti? Esistono caratteristiche/principi sul guasto della rete e quali sono? • e) Utilizzo/carico eccessivo dell’ACCOPPIAMENTO RETE/DATI da parte dei nodi della rete – Qual e` in numero previsto di nodi di rete e il loro grado di utilizzo previsto? Le risorse sono sufficienti a soddisfare le necessita` sia dell’ACCOPPIAMENTO RETE/DATI stesso che dei dispositivi ad esso collegati? • f) Errori nell’utilizzo – Quali esperienze sono richieste all’OPERATORE per il corretto funzionamento del sistema? • g) Gestione inadeguata della configurazione – L’esecuzione di compiti periodici di assistenza influisce sulle caratteristiche della rete (ad esempio dopo l’accesso remoto, aggiornamenti o miglioramenti)? L’ORGANIZZAZIONE RESPONSABILE e` in grado di garantire che le modifiche di ciascun componente del SEMP sono state esaminate ed approvate? • h) Informazioni nel punto sbagliato – I dati sono pervenuti nel punto giusto e previsto? Sono accompagnati da dati ininfluenti che potrebbero confondere l’OPERATORE o nascondere i dati richiesti? In caso affermativo, la sorgente e` specificata in modo adeguato? H7.3 Classificazione della rete sulla base delle conseguenze per il PAZIENTE H.7.3.1 Conseguenze per il PAZIENTE • Per correlare le cause indicate in H.7.2 alle conseguenze per il PAZIENTE, puo` essere utile classificare gli ACCOPPIAMENTI RETE/DATI sia dal punto di vista delle conseguenze che dei tempi di reazione, dove il tempo di reazione e` il tempo di ritardo tra un guasto dell’ACCOPPIAMENTO RETE/DATI e il verificarsi di un DANNO per il PAZIENTE. IEC 80001 RUOLI E RESPONSABILITA’ - ORGANIZZAZIONE RESPONSABILE RO – 80001-1 IEC 80001 • Responsabilità complessiva della gestione del rischio per ogni specifica rete IT medicale. • Standard internazionale • Si rivolge a – Strutture sanitarie – Fabbricanti dispositivi medici – Fornitori di soluzioni IT • Risk managment per il “ciclo di vita” dei medical IT network KEY PROPERTIES • Redazione del documento di gestione del rischio della rete IT medicale (MEDICAL IT-NETWORK RISK MANAGEMENT FILE) che deve contenere tutta la documentazione relativa ai requisiti richiesti dallo standard e la descrizione della configurazione esistente della rete IT medicale. • Definire il processo di gestione del rischio per reti IT medicali, con riferimento a pianificazione, connessione dei dispositivi, configurazioni, mantenimento e dismissione. KEY PROPERTIES: tre caratteristiche fondamentali per la gestione del rischio SAFETY EFFECTIVENESS DATA AND SYSTEM SECURITY – SAFETY ( assenza di rischi non accettabili), – EFFECTIVENESS (capacità di produrre il risultato voluto), – DATA AND SYSTEMS SECURITY ( le risorse informatiche sono ragionevolmente protette da accessi non autorizzati, integrità e disponibilità dei dati). Responsabilità del TOP MANAGMENT • Nominare/incaricare il responsabile del rischio per le reti IT-medicali, persona che deve avere le necessarie qualifiche, conoscenze e competenze • Individuare (assicurandosi che cooperino) le persone responsabili di seguenti : – Analisi, valutazione e archiviazione delle informazioni necessarie alla gestione del rischio – Gestione del ciclo di vita dei DM incorporati nelle reti IT – Revisione delle regole di accettazione dei rischi residui – Manutenzione delle reti IT – Scelta e acquisto dei DM Responsabilità del TOP MANAGMENT – Definire le regole per la gestione del rischio nel caso di introduzione di nuovi medical device nella rete. – definire le regole per la determinazione del rischio accettabile, tenendo conto degli standard internazionali, nazionali e locali. – assicurare la fornitura di risorse adeguate. – assicurare l’assegnazione di personale qualificato per la gestione, l’esecuzione delle attività operative e per le attività di valutazione. – rivedere i risultati delle attività di gestione del rischio a intervalli definiti, per assicurare la continua adeguatezza e l’efficacia del processo di risk managment. Quanto suddetto deve essere documentato nel MEDICAL IT-NETWORK RISK MANAGEMENT FILE. Responsabilità del TOP MANAGMENT • assicurarsi che la partecipazione al processo di gestione del rischio per le reti IT medicali comprenda la gestione di: – Reti IT medicali (MEDICAL IT-NETWORK) – Attività IT in generale – Gestione del ciclo di vita dei DM inseriti nella rete IT – Uso dei DM – Manutenzione e supporto tecnico per i DM Responsabilità del TOP MANAGMENT assicurare: – Che tutte le operazioni, installazione e manutenzioni delle reti IT medicali siano fatte in accordo con il piano di gestione del rischio – Che tutte le componenti dell’organizzazione preposte alla vigilanza, all’installazione, ai servizi, alla manutenzione di reti IT medicali siano adeguatamente informate riguardo alle proprie responsabilità. Le ISO/IEC 80001 • ISO/IEC 80001 consists of the following parts, under the general title Application of risk management for IT-networks incorporation medical devices: • Part 1: Roles, responsibilities and activities • Part 2-1: Step-by-step risk management of medical ITnetworks – Practical applications and examples • Part 2-2: Guidance for the disclosure and communication of medical device security needs, risks and controls • Part 2-3: Guidance for wireless networks • Part 2-4: Application guidance – General implementation guidance for Healthcare Delivery Organizations • Part 2-5: Application guidance – Guidance on distributed alarm systems LE ISO/IEC 80001 • ISO/TR 80001-2-x, Application of risk management for IT-networks incorporating medical devices – Part 2-x: Application Guidance – Guidance for Healthcare Delivery Organizations (HDOs) on how to self-assess their conformance with IEC 80001-1 • ISO/TR 80001-2-x: Application of risk management for IT-networks incorporating medical devices – Part 2-x Guidance for responsibility agreements RUOLI E RESPONSABILITA’ - ORGANIZZAZIONE RESPONSABILE RO – TR 80001-2-4 • Gruppo di lavoro multidisciplinare (referenti clinici, referenti IT, referenti IC) • Definire le politiche di gestione del rischio con particolare riferimento alle Key properties KEY PROPERTIES: tre caratteristiche fondamentali per la gestione del rischio, – SAFETY (2. - assenza di rischi non accettabili), – EFFECTIVENESS (2.6 - capacità di produrre il risultato voluto), – DATA AND SYSTEMS SECURITY (2.5 - le risorse informatiche sono ragionevolmente protette da accessi non autorizzati, integrità e disponibilità dei dati). • Definire criteri di accettabilità del rischio tenuto conto di norme e standards • Prevedere un processo di revisione RUOLI E RESPONSABILITA’ - ORGANIZZAZIONE RESPONSABILE RO – TR 80001-2-4 • Piccole organizzazioni – Esistono sistemi interfacciati con MD? Siamo nelle condizioni di applicabilità della 80001? Esistono programmi futuri di integrazione di MD sulla rete IT? – Esiste la possibilità di pianificare nel tempo queste attività – Sono presenti organizzazioni simili alla ns. nelle vicinanze con le quali collaborare? Sono presenti organizzazioni simili che stanno facendo e possono condividere la loro esperienza? – È presente una conoscenza dettagliata della propria rete? – Sono presenti all’interno dell’organizzazione esperienze di risk management? Se si possono collaborare o assumersi le nuove responsabilità riferite alle reti IT MD? IMPLEMENTAZIONE DEL RISK MANAGEMENT– TR 80001-2-4 • Definire il contesto clinico – Bisogni del paziente per i quali l’organizzazione fornisce risposte/servizi – Tipologia dei servizi forniti e relativi processi – Staff clinico e competenze • Definire il contesto dei rischi considerando che – L’attività di risk management non sia essa stessa un fonte di rischio ad esempio distraendo i clinici con nuovi oneri. – Non creare una sovra-burocratizzazione – Vengano coinvolte le persone giuste in grado di valutare i singoli rischi – Conformità con stadard clinici di settore, norme e regolamenti – Valutazione del rischio netto. L’introduzione di un nuovo sistema elimina alcuni rischi e ne introduce altri RISK ASSESSMENT– TR 80001-2-4 Per una buona valutazione dei rischi è necessaria una dettagliata conoscenza su come la rete IT medicale viene usata per erogare i servizi • Una rete IT medicale può essere schematizzata in tre principali tipologie – Singolo MD collegato alla rete IT – Multipli MD collegati direttamente alla rete IT – Una specifica rete IT medicale connessa alla rete IT • Nel revisionare una rete IT Medicale è necessario: – – – – – – Definire la configurazione della rete con tutte le sue componenti Lo stato di sviluppo della stessa Individuare chi fornisce le apparecchiature valutare il livello di assistenza fornito Decidere dove fare il pilota di test del processo Criteri per il recupero delle informazioni – preaccordi su chi collaborerà. configurazione di una rete IT Medicale Conoscenza della configurazione della rete IT medicale dal punto di vista: • Fisico (limiti della rete medicale) • Stand alone • Multi centro (due o piu’ organizzazioni responsabili collaborano) • Centralizzata (grandi strutture) • Flusso di dati • Processi (lista dei servizi implementati) Esempio configurazione rete Medicale IT -1Rete medicale isolata Esempio configurazione rete Medicale IT -3Rete IT medicale multi centro Servizi Diagnostici distribuiti Rete non soggetta a 80001-1 Esempio configurazione rete Medicale IT -2Rete medicale Standalone Piccola struttura diagnostica Esempio configurazione rete Medicale IT -4Rete IT medicale centralizzata Ospedale per acuti di grandi dimensioni Caso – Organizzazioni di piccole dimensioni Rischio di essere “sommersi” da informazioni e di generare sovrastrutture complesse • Esiste un inventario delle apparecchiature collegate alla rete? • Sono specificate le apparecchiature collegate che sono dispositivi medici? • Sono disponibili tutti i documenti relativi ai MD: certificati di conformità, manuali ecc? STATO DI REALIZZAZIONE DELLA RETE IT MEDICALE La valutazione dei rischi deve tener conto dello stato di fatto secondo la seguente classificazione: • Esistente in modo stabile • Stabile ma soggetta a modifiche (valutazione dell’impatto dei cambiamenti sull’analisi dei rischi) • In fase di sviluppo o nuova • Quali sono le interfacce attive tra dispositivi medici ed altre componenti del sistema IT? Caso – Organizzazioni di grandi dimensioni In aggiunta ai punti di cui sopra: • Valutare il possibile contributo dei referenti clinici • Valutare il possibile contributo di tutte le altre componenti tecniche dell’organizzazione (IT e IC) • Valutare il contributo di chi ha responsabilità della Clinical Governance (DS) • Valutare il contributo dei servizi di risk management (SPP) • valutare la separazione dei MD in specifici domini di competenza clinica • Valutare se vi sono MD il cui uso è comune in più siti clinici Identificazione del costruttore • Fase di identificazione dei costruttori/fornitori delle varie componenti della rete IT • Definizione delle responsabilità dei fornitori • Costruttore e fornitore sono gli stessi? • Le regole di acquisto garantiscono l’accessibilità a tutte le informazioni tecniche necessarie? • Chi è titolato a chiedere informazioni ai fornitori? • Chi può costringere un costruttore a mettere in atto azioni per la mitigazione del rischio? • Qual’è la complessità della catena di fornitura? Ci sono sub-fornitori? Supporto dell’IT • Chi si occupa di fornire supporto IT? • Esistono contratti di supporto IT? • Sono chiare le regole di intervento stabilite dai contratti IT? • Il supporto IT è adeguato alle esigenze? Accordi di responsabilità • Il responsabile della gestione del rischio deve poter contare su continuo informazioni ed aggiornamenti provenienti dai fornitori esterni • Sono in essere specifici accordi scritti che obbligano i fornitori a fornire queste informazioni? • Sulle di sottoscrizione di accordi di responsabilità esiste uno specifico TR 80001 - Impatto strutture sanitarie • Se un DM è incorporato in una rete ospedaliera, la rete stessa diventa una parte del DM • Presa d’atto delle nuove responsabilità che si assumono insieme al costruttore • Nuove politiche di gestione • Nuove strategie di acquisizioni GESTIONE DELLE TECNOLOGIE – NUOVO PARADIGMA • Prima – Singole apparecchiature con accessori • dopo – Software – Sw upgrade – Accesso remoto (anche per le manutenzioni) – Diffusa interoperabilità – Collaborazione multidisciplinare Temi aperti • • • • • • Costi Analisi dei bisogni reali Ritorno dell’investimento (difficile da valutare) Impatto sul workflow (si spera in meglio) Impatto sul QC Nuovi ruoli – necessità di multidisciplinare Codice in materia di dati personali Il Codice definisce in maniera chiara gli elementi indispensabili per garantire la protezione dei dati e la fiducia degli utenti Riservatezza (Confidenzialità) la protezione delle informazioni mediante l’accesso consentito soltanto agli autorizzati, la protezione delle trasmissioni, il controllo degli accessi, ... Integrità la salvaguardia della correttezza dei dati, la difesa dalle manomissioni e da modifiche non autorizzate, il monitoraggio automatico degli accessi, … Disponibilità la garanzia per gli utenti di poter disporre dei dati, delle informazioni e dei servizi, evitando la loro perdita o riduzione I due principi fondamentali I dati personali devono essere trattati nel rispetto dei criteri di: PRIVACY Pertinenza: solo per raggiungere le finalità per le quali è stato acquisito il consenso Non eccedenza: esclusivamente per quanto strettamente necessario al raggiungimento delle finalità per le quali è stato acquisito il consenso NB: le restrizioni previste dal Codice Privacy non si applicano sulle attività obbligatorie per legge dati personali e dati sensibili I dati “sensibili” sono quelli relativi a: ►Origine razziale ed etnica ►Convinzioni religiose e filosofiche ►Opinioni politiche ►Appartenenza a partiti e sindacati ►Salute e vita sessuale Codice in materia di dati personali Art. 31: Obblighi di sicurezza ► I dati personali oggetto di trattamento devono essere custoditi e controllati, anche in relazione alle conoscenze acquisite in base al progresso tecnico, alla natura dei dati e alle specifiche caratteristiche del trattamento, in modo da ridurre al minimo, mediante l'adozione di idonee misure di sicurezza preventive, i rischi di: – – – – distruzione o perdita, anche accidentale accesso non autorizzato trattamento non consentito trattamento non conforme alle finalità della raccolta Codice in materia di dati personali le misure minime di sicurezza ►Art.4, comma 3, lettera a – il complesso delle misure tecniche, informatiche, organizzative, logistiche e procedurali di sicurezza che configurano il livello minimo di protezione richiesto in relazione ai rischi previsti nell’articolo 31 e nell’allegato B. ma le misure debbono essere anche adeguate allo stato delle conoscenze tecnologiche. Codice in materia di dati personali le misure minime ► il controllo dell’accesso ai dati dev’essere garantito da – autenticazione informatica – credenziali di autenticazione – sistema di autorizzazioni ► Altre misure – adozione di procedure per la generazione e la custodia di copie di sicurezza dei dati – ripristino della disponibilità dei dati e dei sistemi – protezione dei dati sensibili o giudiziari contro l’accesso abusivo – aggiornamento periodico dei programmi per elaboratore volti a prevenire la vulnerabilità degli strumenti elettronici e alla correzione dei difetti del software da Ricordare Quindi i dati personali sono protetti dalla Legge! e i dati sanitari sono dati sensibili Amministratore di Sistema Decreto legislativo 196/03 “Codice in materia di protezione dei dati personali” violarli è un reato penale e sono previste severe sanzioni da Ricordare Nome Utente e Password sono personali Non devono essere divulgati a nessuno, per nessun motivo Art. 615 ter del Codice Penale "accesso abusivo ad un sistema informatico o telematico“ Art. 615 quater del Codice Penale "detenzione e diffusione abusiva di codici di accesso a sistemi informatici o telematici“ Vanno utilizzati per accedere ai dati dei pazienti in cura e non ad altri E’ responsabilità personale di ogni Incaricato non allontanarsi dalla sessione di lavoro senza averla chiusa. Art. 640-ter. Frode informatica. Chiunque, alterando in qualsiasi modo il funzionamento di un sistema informatico o telematico o intervenendo senza diritto con qualsiasi modalità su dati, informazioni o programmi contenuti in un sistema informatico o telematico o ad esso pertinenti, procura a sé o ad altri un ingiusto profitto con altrui danno, è punito con la reclusione da sei mesi a tre anni e con la multa da euro 51 a euro 1.032. La pena è della reclusione da uno a cinque anni e della multa da euro 309 a euro 1.549 se ricorre una delle circostanze previste dal numero 1) del secondo comma dell'articolo 640, ovvero se il fatto è commesso con abuso della qualità di operatore del sistema. La pena è della reclusione da due a sei anni e della multa da euro 600 a euro 3.000 se il fatto è commesso con furto o indebito utilizzo dell'identità digitale in danno di uno o più soggetti. (1) Il delitto è punibile a querela della persona offesa salvo che ricorra taluna delle circostanze di cui al secondo e terzo comma o un'altra circostanza aggravante. (2) (1) Comma inserito dall’art. 9, comma 1, lett. a), D.L. 14 agosto 2013, n. 933, convertito, con modificazioni, dalla L. 15 ottobre 2013, n. 119. (2) Comma così modificato dall’art. 9, comma 1, lett. b), D.L. 14 agosto 2013, n. 933, convertito, con modificazioni, dalla L. 15 ottobre 2013, n. 119. ►I Regolamenti sul trattamento dei dati sensibili sono come le norme dell’igiene ►Rispettarli evita spiacevoli conseguenze ►Cambiare abitudini richiede uno sforzo, ma poi la nuova abitudine diviene consuetudine ►La Sicurezza è prima di tutto un atteggiamento mentale ►Buoni risultati si ottengono soltanto quando ognuno è consapevole dell’importanza del proprio comportamento e del proprio ruolo ORGANIZZAZIONE RESPONSABILE Guida alla gestione del software e delle reti IT-medicali nel contesto sanitario PARTE PRIMA: Gestione del Software (in fase di sviluppo) FASCICOLO DI PRODOTTO/SISTEMA • Fascicolo riguardante il prodotto/sistema che comprende o è esso stesso software DISPOSITIVO MEDICO oppure software in CONTESTO SANITARIO, dove vengono registrate le attività inerenti la sicurezza di un prodotto/sistema e comunque quello derivante dagli obblighi ex DPR 14 Gennaio 1997 • Ente responsabile dell’uso e della manutenzione di un apparecchio elettromedicale (EM) o di un sistema EM , della rete IT medicale, e del software sanitario. • Nota 1. La definizione è derivata da IEC 60601-1:Ente responsabile dell’uso e della manutenzione di un apparecchio EM o di un sistema EM , e da IEC 80001-1: Ente responsabile dell’uso e della manutenzione della rete IT Medicale • Nota 2 L’ORGANIZZAZIONE RESPONSABILE può essere, per esempio, un ospedale, un singolo medico o una persona inesperta. Nelle applicazioni domiciliari, il paziente, l’operatore e l’ORGANIZZAZIONE RESPONSABILE possono coincidere. RETI • RETE INFORMATICA (RETE IT) • Sistema o sistemi costituiti da nodi di comunicazione e collegamenti di trasmissione per permettere la trasmissione attraverso un collegamento fisico o senza fili, tra due o più nodi di comunicazione specificati. [nella IEC 80001-1:2010] • Nota: Lo scopo della rete informatica ai fini della salute è definito dall’ORGANIZZAZIONE RESPONSABILE, in funzione di dove il software sanitario è collocato all’interno della rete informatica sanitaria e dall’uso definito della rete informatica stessa. Essa può contenere un’infrastruttura informatica, componenti o sistemi informatici anche per la cura a domicilio per scopi generali e non previsti dal progetto per essere utilizzati in un contesto sanitario. • RETE IT MEDICALE • rete IT che collega almeno un DISPOSITIVO MEDICO. [nella IEC 80001-1 Ed. 1.0:2010] SISTEMA SOFTWARE • Raccolta integrata di elementi software organizzati per svolgere una funzione o un insieme di funzioni specifiche. [nella IEC 62304:2006, 3.27] INTERFACCIAMENTO • L’operazione d’interfacciamento, assai comune nelle strutture sanitarie, che porta ad usare dati provenienti da dispositivi medici in altri sistemi è un’operazione che di per sé comporta rischi potenzialmente rilevanti e di diversa natura, specialmente nel caso di sistemi eterogenei. Particolarmente critici sono i sistemi nei quali l’intera catena di elaborazione del dato non sia stata completamente prevista dai fabbricanti di DISPOSITIVI MEDICI coinvolti, tramite una caratterizzazione delle singole interfacce presenti SISTEMA COMBINAZIONI • Struttura composta integrata costituita da uno o più processi, da hardware, software, dispositivi e persone, in grado di soddisfare un bisogno o realizzare un obiettivo dichiarato. [nella IEC 62304:2006 – Armonizzata. • Nota esplicativa : il sistema può essere costituito da un software DISPOSITIVO MEDICO in combinazione con software e hardware OTS (all’interno di uno stesso apparato/apparecchiatura) o quando si ha a che fare con un sistema di software e hardware con una combinazione di dispositivi medici e non (le componenti del sistema non sono all’interno di un unico apparato/apparecchiatura ma il sistema è costituito da più apparati). Sistemi sono anche tutti i sistemi assemblati e/o comunque creati ad hoc e che possono costituire un pericolo per il paziente. La presenza di un fascicolo di prodotto/sistema è sempre obbligatoria. • Si ricorda che i fabbricanti di DISPOSITIVI MEDICI destinati ad essere combinati con altri software o dispositivi devono anche considerare la compatibilità dei loro prodotti verso altri moduli; secondo il DLGS 46-97 e ss.m.ii infatti, “se un dispositivo è destinato ad essere utilizzato insieme ad altri dispositivi o impianti, l'insieme risultante, compreso il sistema di connessione deve essere sicuro e non deve nuocere alle prestazioni previste per i singoli dispositivi. Ogni eventuale restrizione di utilizzazione deve figurare sulla etichetta o nelle istruzioni per l'uso” RESPONSABILITA’ FABBRICANTE • E’ responsabilità del fabbricante analizzare e documentare le specifiche di interfacciamento che possano avere delle ricadute in termini di sicurezza dell’intero sistema ed è responsabilità dell’ORGANIZZAZIONE RESPONSABILE rispettare quanto stabilito dal fabbricante Ma…. • Nella realtà è assai frequente rilevare casi di sistemi in cui il puntuale rispetto di quanto indicato dai fabbricanti delle componenti del sistema non è sufficiente a garantire una assoluta garanzia di sicurezza dell’intero sistema, sia perché non è stato possibile, da parte dei fabbricanti, fornire delle specifiche di sistema completamente esaustive, sia per via del fatto che in molti casi è necessario ed inevitabile un certo grado di personalizzazione nella interconnessione di sistemi OBBLIGHI DEL OR 1/2 • L’ ORGANIZZAZIONE RESPONSABILE deve evitare che siano resi disponibili al personale sanitario dati o informazioni, sui quali potrebbero essere prese decisioni mediche/diagnostiche, derivanti dai suddetti sistemi, senza aver prima condotto una gestione suppletiva dei rischi associati. OBBLIGHI DEL OR 2/2 • L’ORGANIZZAZIONE RESPONSABILE dovrà garantire il mantenimento nel tempo della sicurezza dei sistemi interconnessi tramite la revisione • I criteri per la revisione periodica ed i relativi risultati dovranno essere documentati nel fascicolo di prodotto/sistema in cui saranno contenute anche le misure per mitigare gli eventuali rischi e la loro verifica SISTEMI INTERCONNESSI Rischi specifici • Nuove funzionalità possono essere aggiunte, intenzionalmente o non, quando diversi dispositivi sono interconnessi in un sistema (o all’interno di un singolo computer) e nuovi rischi inaspettati possono verificarsi. Le operazioni del sistema sono difficili da monitorare dalle singole persone e diverse attività possono essere eseguite contemporaneamente in un modo che possono coinvolgere sia la sicurezza del paziente che dell’operatore. SISTEMI INTERCONNESSI Rischi specifici • L’interconnessione può essere garantita a sua volta da un software stand-alone con sua specificità e la sua introduzione deve essere considerata nell’analisi del rischio complessiva. SISTEMI INTERCONNESSI Rischi specifici SISTEMI INTERCONNESSI Rischi specifici • Poiché i componenti individuali in un sistema interconnesso possono avere la destinazione d’uso individuale difforme (ad es qualificate come dispositivi medici e non), i requisiti formali possono differire tra le differenti parti del sistema (es requisiti di sistema). Questo causa dei rischi per il paziente. • Le interfacce utente su diversi dispositivi appartenenti al sistema, possono avere un design non coerente e usare termini/funzionalità diverse per funzioni simili che possono confondere l’utente. SISTEMI INTERCONNESSI Rischi specifici • I dati clinici possono essere visualizzati da dispositivi non medici • I dati non clinici possono interferire con la sicurezza dei dispositivi medici • L’aggiornamento di alcuni componenti del sistema possono causare problemi alle parti che sono DISPOSITIVO MEDICO SISTEMI INTERCONNESSI Rischi specifici • L’uso di dispositivi non medici può richiedere formazione diversa da quella dei dispositivi medici • Gli operatori possono trovarsi con condizione di errore non pensate per l’ambiente medico che non indicano cosa fare (es. Errore di sistema 0x000444) generando gravi pericoli per il paziente. Cosa deve fare l’operatore? Non usare il dispositivo? Spegnerlo? SISTEMI INTERCONNESSI Rischi specifici • La manutenzione dei dispositivi non medici può compromettere la sicurezza dei dispositivi medici (es aggiornamento VM Java o DB che usa il DB. Cosa fare?) • Prestazioni della rete dati possono non essere sufficienti compatibili con i dispositivi • Degrado di prestazioni degli apparati (es dovute alla frammentazione del disco) SISTEMI INTERCONNESSI Rischi specifici • Grado d’invecchiamento del sistema (es per parti meccaniche o elettroniche con vita limitata come i flash disk , batterie , lampade) • Valutazione della sicurezza in termini di hardware/software (antivirus, firewall, patch disponibili ) che di invecchiamento relativamente allo stato dell’arte • Aggiornamento dei sistemi operativi e patch IHE Scheduled Workflow Profile SISTEMI INTERCONNESSI Rischi specifici • In generale verifiche periodiche sulle componenti hardware e software (es sostituzione HD, Sostituzione RAM, aggiornamento Sistema Operativo) • Verifiche periodiche sulla funzionalità del DISPOSITIVO MEDICO report report Registration Report Repository report HIS patient Diagnostic Workstation information PACS images retrieved Orders Placed examination orders RIS Orders Filled procedure scheduled Image Manager Prefetch any relevant & Archive prior studies Acquisition modality images Modality worklist stored acquisition completed acquisition acquisition images completed in-progress printed 87 SPECIFICHE INTEROPERABILITA’ Film Lightbox Modality Film Folder Film CAPITOLATO Specifiche interfacciamenti/integrazioni STANDARDS Approccio IHE • L’acquisizione di una nuova modalità radiologica o software medicale, tenuto conto dei sistemi (PACS, RIS, workstation, ecc.) e delle configurazioni applicative (DICOM, Modality working list, ecc.) già presenti in ospedale, può trarre significativi vantaggi dall’adozione dalle peculiarità di integrazione offerte da IHE Obiettivi e profili d’integrazione IHE Radiology User’s Handbook Obiettivi di integrazione • La corretta scelta degli strumenti di integrazione IHE dipende fortemente dagli obiettivi organizzativi che l’amministrazione acquirente intende perseguire. • Ridurre errori e migliorare l’assistenza al paziente • Incrementare la produttività • Ridurre la dose erogata al paziente • Aumentare la qualità delle immagini • Ridurre costi e tempi Riduzione errori e miglioramento assistenza al paziente • La presenza del profilo SWF consente: – – – – La riduzione degli errori di data entry La disponibiltà di informazioni cliniche prioritarie La riduzioni dei tempi di erogazione del servizio La riduzione delle perdite di informazioni cliniche • La presenza del profilo CPI: – Aumenta la qualità delle immagini e riduce gli errori di visualizzazione • La presenza del profilo PPS: – Riduce gli errori di assegnazione dell’anagrafica dalla working list Riduzione della dose erogata • La presenza del profilo SWF consente di: – Prevenire erogazione di dose causata da esami ripetuti in seguito a perdita dell’esame – Prevenire erogazione di dose causata da esami ripetuti in seguito a errata applicazione del protocollo d’esame (il protocollo proviene dalla Working list) • La presenza del profilo PGP consente la: – Riduzione della dose causata da richieste multiple non ottimizzate con l’esecuzione di un singolo esame Aumentare la qualità delle immagini • La presenza del profilo CPI consente di: – Ridurre errate impostazioni dei monitor grazie alle informazioni provenienti della modalità – Ridurre errate impostazioni dei monitor grazie alle funzioni di calibrazione automatica • La presenza del profilo KIN consente: – La trasmissioni al radiologo di utili informazioni relative allo svolgimento dell’esame da parte del tecnico. – La trasmissioni al tecnico di utili informazioni relative alla corretta applicazione del protocollo. Riduzione di costi e tempi • In generale i profili IHE consentono la riduzione dei costi e tempi attraverso: – Riduzioni di esami inutili – Aumento della produttività – Riduzione della stampa su film – Riduzione di costi per la costruzione ed il mantenimento di interfacce informatiche proprietarie Incremento della produttività • La presenza del profilo SWF : – Riduce il tempo impiegato per il data entry manuale – Previene perdite di tempo per la ricerca di esami persi – Riduce il tempo di gestione manuale degli archivi immagini – Riduce il tempo di produzione referto – Riduce errori di errata imputazione paziente – Riduce il tempo per la riconciliazione del paziente ignoto – Semplifica la programmazione del protocollo d’esame Incremento della produttività • La presenza del profilo CPI: – Riduce il tempo di preparazione delle immagini • La presenza del profilo PPS: – Semplifica la fase di riprogrammazione esame (in caso di abort) • La presenza del profilo PGP: – Riduce il tempo impiegato dal radiologo per preparare la visulizzazione di studi comparativi con la corretta finestratura Quali profili? • Nella pratica non è possibile raggiungere tutti gli obiettivi d’integrazione in quanto il sistema con cui interagirà la nuova modalità non è (o è solo parzialmente) IHE • Più spesso è possibile individuare uno o più attori e transizioni di uno specifico profilo che sono presenti nel sistema dove verrà inserita la modalità. Successivamente potrà essere programmato un graduale incremento del processo d’integrazione Selezione di profili ed attori: fasi • Selezione del profilo di integrazione • Selezione degli attori che si vuole siano implementati • Selezioni delle eventuali ulteriori opzioni (funzioni addizionali) Come si formula una richiesta d’offerta • In modo molto semplice si riportano: – Profilo IHE seguito dagli attori che dovranno essere supportati – La complessità resta nel “Technical Framework” Come si formula una richiesta d’offerta esempio risultati connectathon • Esempi: – La nuova Modalità dovrà supportare il profilo d’integrazione IHE “Scheduled Worflow” attraverso gli attori “Acquisition Modality” e “Image Display” – La nuova Modalità dovrà supportare il profilo d’integrazione IHE “Consistent presentation of images” attraverso gli attori “Acquisition Modality” e “Print Composer” Come si individuano i prodotti di mercato idonei • Attraverso informazioni di dominio pubblico – Esiti dei “Connectathon” informazioni indicative su profili ed attori presentati dai Vendor partecipanti • www.ihe.net/ • www.ihe-europe.org/con_result – Disponibilita dei documenti di “Integration Statement” • link dalle webpages dei singoli vendor • www.ihe.net/Resources/ihe_integration_statements.cfm • www.ihe-europe.org esempio di Integration Statement E per le specifiche opzionali ? Nel CASO di una Risonanza Magnetica • Nel caso di indisponibilità del profilo “consistent presentation of image” Il sistema offerto dovrà disporre delle seguenti classi DICOM: – Print Managment- Modalità SCU Specifiche Interfacciamenti secondo IHE • per le Modalità – scheduled workflow SWR (attori: Acquisition Modality) – patient information reconciliation (attori: Acquisition Modality) – preferibilmente con “consistent presentation of image” (attori: Evidence Creator, Image Display, Print Composer) – preferibilmente con “Key Image Note” (attori acquisition modality) • per le workstation – scheduled workflow SWR (attori: Image Display, Image (or Evidence) Creator) – preferibilmente con “consistent presentation of image” (attori: Evidence Creator, Image Display, Print Composer) – preferibilmente con “Key Image Note” (attori acquisition modality Interfacciamento vs sw IHE complaint attori e delle transazioni disponibili sul RIS da interfacciare secondo la terminologia IHE Modulo Software e sicurezza informatica 2/3 • Documentazione – – – – – CAPITOLATO Sicurezza Informatica e rete Modulo Software e sicurezza informatica 1/3 • Trattamento dati personali D Lgs 196/2003 (dati sensibili) – – – – – Misure minime di sicurezza (art. 33-26) Autenticazione informatica Sistema autorizzazioni Protezione dati e sistemi Misure per garantire il ripristino – – – – • Relativa al software proprietario e di base Manutenzione del sw, recupero dati, interfacciamenti Credenziali per accesso a livello di amministratore Inserimento nel dominio aziendale e gestione della password locale Richiesta del fornitore di disporre di password nominative per amministratori di sistema Descrizione moduli funzionali, interazioni con S.O. e con la rete, protocolli di integrazione con altri sistemi Procedure di ripristino e password Presenza di specifici tools per il recupero dati Caso della protezione per privativa industriale Software installati e sicurezza – Descrizione del sistema operativo utilizzato – Gestione delle patch – Disattivazione sw non strettamente necessari (giochi, salva schermi, navigazione internet) – Presenza di antivirus, antintrusione (descrizione) – Coordinamento fornitore-cliente sulla gestione e aggiornamento sicurezze – Asset managment, antivirus aziendali, software delivery Modulo Software e sicurezza informatica 3/3 • Collegamento in rete – – – – – – Necessità di un collegamento in rete Necessità di uniformarsi alle policy aziendali ASL Utilizzo di protocolli di rete (preferibilmente solo IP) Necessità di condividere file o cartelle in rete Utilizzo di specifiche VLAN Sicurezza elettrica e sistemi medicali CEI 62.51 • Collegamento remoto • Protezione del software – Illeciti impieghi del software (chiavi HW/SW, numero licenza) – Gestione della chiave – – – – Accesso remoto a scopo di teleassistenza e supporto utenti Utilizzo delle risorse che mette a disposizione l’ASL Divieto ad utilizzare soluzioni non condivise Sottoscrizione delle policy Processo di configurazione di un sistema IT • Acquisire una modalità diagnostica Configurazione e processo di implementazione degli Integration Profiles in un sistema IT ospedaliero Approccio IHE in fase di collaudo • Necessità di verificare con particolare attenzione la reale implementazione dei profili IHE richiesti all'interno del sistema IT ospedaliero preesistente in termini di attori e transazioni • Individuare all'interno del workflow di reparto (SWF) attori e transazioni IHE (chi fa che cosa) -> ad esempio la modalità va interfacciata al server worklist per la lista pazienti e al server Pacs per l’archiviazione delle immagini Assegnare a ciascun sistema coinvolto i parametri necessari al collegamento in rete (ind. IP AE TITLE, porta) Processo di verifica delle interfacce • Simulare le transazioni tra i vari attori secondo gli standard implementati (DICOM, HL7 etc.) Obiettivo: Garanzia di corretto trasferimento dati anagrafici e ordini utilizzando le workilist, gestione delle anagrafiche e dei pazienti ignoti o non programmati, certezza della corretta archiviazione di tutte le immagini -> per SWF: Query Modality Worklist, Modality PS, Modality Image Stored,Storage Commitment… 01/04/14 Processo di verifica delle interfacce • Identificare le problematiche di integrazione eventuali emergenti -> fare riferimento al DICOM Conformance Statement fornito dai produttori per individuare le SOP class supportate da ciascun sistema • Fornire indicazioni agli utilizzatori sul corretto uso dei sistemi e sul comportamento da mantenere di fronte a possibili errori NORMATIVA DI SICUREZZA E PREVENZIONE DEL RISCHIO IN RISONANZA MAGNETICA Francesco Campanella, INAIL 01/04/14 Premessa you are "safe" in a place that is “secure” I dispositivi di legge relativi alla Risonanza Magnetica sono: tanti, datati di almeno 15 anni, alcuni solo parzialmente in vigore, a volte poco circostanziati proprio perché scritti in una fase che allora era “sperimentale” l’osservanza degli obblighi risulta a volte di difficile attuazione, e conseguentemente, “operare in sicurezza”, e nel rispetto delle norme, non è sempre impresa agevole QUADRO NORMATIVO ITALIANO • Al fine di agevolare l’utenza a gestire un quadro normativo così frammentario e composito, l’ISPESL nel 2004 ha pubblicato sul proprio sito le “INDICAZIONI OPERATIVE“ che raccolgono – in un unico testo – una serie di metodi, suggerimenti, consigli, esperienze per ottimizzare la gestione della sicurezza in un sito di Risonanza Magnetica. • Tali indicazioni non hanno valore specifico di legge, ma comunque sono oggi unanimemente considerate una LINEA GUIDA di riferimento in materia. LE QUESTIONI IRRISOLTE GLI STANDARD DI SICUREZZA IN RISONANZA MAGNETICA L’ Art. 2 – comma 2 del DPR 542/94 stabilisce che gli “standards” di sicurezza sono quelli previsti nei: DM 2/8/1991 - all. 1 e 4 e DM 3/8/1993 - all. A e B essi stabiliscono: 1. Requisiti minimi di carattere strutturale, tecnologico ed organizzativo; 2. Procedure di gestione dell’attività diagnostica; 3. Ruolo e compiti del Medico Responsabile del sito RM e dell’Esperto per la Sicurezza dell’apparecchiatura RM. 4. Limiti di esposizione per pazienti ed operatori; Responsabile Rappresentano condizioni ineludibili ed indifferibili che è necessario attuare e garantire, con carattere di permanenza, all’interno dei siti nei quali viene svolta un’attività di diagnostica per immagini a scopo medico tramite apparecchiature di risonanza magnetica. E’ possibile utilizzare in Italia un’apparecchiatura senza rispettare i limiti di esposizione occupazionale al campo magnetico statico di cui al D.M. 2/8/1991 allegati 1 e 4 , sebbene questi ultimi possano risultare ormai “vetusti” rispetto allo stato dell’arte relativo alle conoscenze e magari superati (Linee Guida ICNIRP 2009-2010) ? La conformità agli STANDARD è obbligatoria La verifica della conformità è il fine della vigilanza INAIL La struttura degli “standard” italiani Dotazioni del Servizio di Diagnostica per Immagini Prestazioni tecniche di impianto Idoneità edilizia e strutturazione delle installazioni fisse Idoneità di approntamento per le installazioni mobili Controlli, misure e responsabilità finalizzate alla sicurezza Limiti – esposizione al campo magnetico statico per gli operatori, – esposizione ai campi elettromagnetici a radiofrequenza per pazienti [valori limite del rateo di assorbimento specifico medio (SAR) a corpo intero, valori limite di esposizione ai campi magnetici variabili nel tempo (dB/dt)] Intervallo di tempo t < 15 min 15 min ≤ t <30 min t ≥ 30 min SAR (W/Kg) a corpo intero SAR (W/Kg) nella testa SAR (W/Kg) nel tronco SAR (W/Kg) negli arti 2 4 8 12 30/t (min) 60/t (min) 120/t (min) 180/t (min) 1 2 4 6 E’ possibile installare in Italia un’apparecchiatura che rispetti i limiti di esposizione ai campi elettromagnetici a radiofrequenza (e quindi …..di SAR), ed i limiti di esposizione ai campi magnetici variabili nel tempo (dB/dt) indicati nella Norma di Buona Tecnica Internazionale IEC 60601-2-33 (3° edizione 2010) - “Prescrizioni particolari di Sicurezza relative agli apparecchi a Risonanza Magnetica per diagnostica medica” e non quelli – più restrittivi - indicati nel D.M. 3/8/1993 ancora in vigore ?? Le indicazioni europee relative ai requisiti minimi di sicurezza Non è MAI possibile installare ed utilizzare un tomografo RM che non rispetti gli standard di sicurezza vigenti in ITALIA, come sanciti in basi al disposto di cui all’articolo 2 del DPR 542/94: gli allegati 1 e 4 del D.M. 2/8/91 gli allegati A e B del D.M. 3/8/93 UE SCENARIO NORMATIVO EUROPEO • LINEE GUIDA ICNIRP (2009-2010) Esposizioni professionali al campo magnetico statico ed ai campi e.m. variabili • NORMA DI BUONA TECNICA IEC 60601-2-33 (2010) Problematiche di sicurezza, compresa quella relativa ad esposizioni su pazienti vs NAZIONE RIFERIMENTO GERMANIA IEC 60601-2-33 LINEE GUIDA ICNIRP 2009 OLANDA DANIMARCA FRANCIA INGHILTERRA ITALIA TIPO CAMPO DI APPLICAZIONE 2010 Indicazione di riferimento Quello definito dalle Norme di buona tecnica internazionali Using MRI safely IEC 60601-2-33 LINEE GUIDA ICNIRP 1994,98 2008 Raccomandazione Regole pratiche per operatori Technical report IEC 60601-2-33 LINEE GUIDA ICNIRP 2009 2010 Raccomandazione Prescrizioni particolari di Sicurezza relative agli apparecchi a Risonanza Magnetica ed indicazioni per le donne in stato di gravidanza 2011 Norma di buona tecnica Agence Regionale de Santè Ile-de-France Prescrizioni autorizzative e di sicurezza 2008 Linea Guida (Medical and Healthcare products Regulatory Agency) Sicurezza per l’utilizzo clinico e per la sicurezza degli operatori Dossieur Promoteur pour l’autorisation de IRM LINEE GUIDA ICNIRP 2009 IEC 60601-2-33 Safety Guidelines for Magnetic Resonance Imaing Equpment in Clinical Use IEC 60601-2-33 LINEE GUIDA ICNIRP 1994,98 ANNO FINLANDIA Technical Report IEC 60601-2-33 LINEE GUIDA ICNIRP 2009 2010 Linea Guida (Radiation Safety Center) Prescrizioni di Sicurezza per l’uso AUSTRIA Önorms S 1125-1 IEC 60601-2-33 LINEE GUIDA ICNIRP 2009 2009 Standard (Austrian Standards Institute) Linee guida per l’esposizione a Campi Magnetici statici ed ai campi elettromagnetici variabili ITALIA Standard di sicurezza DM 2/8/1991 DM 3/8/1993 1994 Disposizione di legge Sicurezza, prevenzione e protezione in Risonanza Magnetica I PROBLEMI RISOLTI Cos’è la marcatura CE? • La Marcatura CE è un’ “attestazione di conformità di prodotto” ai requisiti di sicurezza previsti da una o più direttive comunitarie europee che ne disciplinano l’analisi e la gestione del rischio. • La finalità della marcatura è pertanto esclusivamente connessa alla sicurezza nell’uso del dispositivo così come fabbricato, prescindendo dalla tipologia di installazione, ovvero dallo scenario in cui avviene la messa in esercizio del medesimo ALLINEAMENTO ITALIA-EUROPA Un dispositivo medico ( ad esempio, un tomografo RM) può circolare liberamente all’interno degli Stati Membri dell’Unione Europea solo se provvisto di marcatura CE, la cui apposizione garantisce da parte del fabbricante il rispetto dei requisiti essenziali di sicurezza (R.E.S.)di cui alla Direttiva Dispositivi Medici,93/42/CE come emendata dalla Direttiva 2007/47/CE Per gli impianti RM con magnete superconduttore • Le caratteristiche della “messa in esercizio”, ovvero le modalità di accoppiamento fra dispositivo medico e l’impianto di sicurezza ad esso asservito (tubo di quench), senza il quale il dispositivo medico stesso non potrebbe essere utilizzato, fanno la differenza in termini di SICUREZZA, al di là della marcatura CE comunque preesistente La direttiva Una Risonanza Magnetica con MAGNETE SUPERCONDUTTORE può essere utilizzata liberamente all’interno degli Stati Membri dell’Unione Europea solo se nella messa in esercizio si garantisce il rispetto dei requisiti essenziali di sicurezza (R.E.S.) contenuti nella Direttiva Impianti a Pressione, 97/23/CE (PED), lì ove applicabile : Il Magnete superconduttore è “recipiente a pressione” che contiene l’Elio liquido in equilibrio con la sua fase gassosa Un recipiente a pressione è definito tecnicamente come un “… alloggiamento progettato e costruito per contenere fluidi pressurizzati comprendente gli elementi annessi diretti sino al punto di accoppiamento con altre attrezzature…” La particolare forma toroidale del recipiente è legata alla finalità clinica dell’apparecchio RM che deve consentire l’introduzione del paziente al suo interno ove viene instaurato il campo statico raggiunta la soglia di super conduttività I tomografi di Risonanza Magnetica rientrano nell’applicazione della direttiva 97/23/CE, denominata PED. Per il il tubo di quench delle apparecchiature RM (sistema di sicurezza asserviti al tomografo RM), è infatti necessario che il fabbricante garantisca l’applicazione dell’art. 3 punto 3 della Direttiva (camini di evacuazione) Queste attrezzature “… devono essere progettate e fabbricate secondo una corretta prassi costruttiva in uso in uno degli stati membri, che assicuri la sicurezza di utilizzazione … Modelli di certificazione degli impianti D.M. n. 37 del 22.01.2008 La certificazione d’installazione alla “regola d’arte” La certificazione si compone di due parti: la dichiarazione di conformità e come tale deve riportare l’indicazione da parte del soggetto installatore di aver: 1. o rispettato il progetto redatto per l’installazione o seguito le norme di buona tecnica applicabili all’impiego dell’installazione, o installato componenti e materiali adatti al luogo d’installazione, o controllato l’impianto ai fini della sicurezza e della funzionalità con esito positivo, avendo eseguito le verifiche richieste dalle norme e dalle disposizioni di legge. Allegati alla certificazione 2. alla dichiarazione di conformità vanno allegati OBBLIGATORIAMENTE: • il progetto dell’impianto • la relazione con tipologie dei materiali utilizzati • lo schema dell’impianto realizzato • il riferimento a dichiarazioni di conformità precedenti o parziali, già esistenti e, per le imprese abilitate anche: • copia del certificato di riconoscimento dei requisiti tecnico - professionali Cosa va certificato ai sensi del D.M. 37/08 TUBO DI QUENCH IMPIANTO DI VENTILAZIONE IMPIANTO ELETTRICO Le apparecchiature diagnostiche a Risonanza Magnetica possono essere installate ed utilizzate in Italia solo se risultano rispondenti : requisiti di corretta fabbricazione previsti all’interno della Direttiva Europea sui Dispositivi medici (Direttiva 93/42/CEE, recepita in Italia dal D. Lgs. 24 Febbraio 1997, n.46 ed emendato col D. Lgs. 25.01.2010 n. 37, in recepimento della nuova Direttiva 2007/47/CE - art.1), ovvero nel rispetto del Requisiti Essenziali di Sicurezza (R.E.S.) della Direttiva medesima. requisiti di corretta installazione e messa in esercizio previsti per quanto applicabile, dalla Europea Direttiva PED (Direttiva 97/23/CE ), standard di sicurezza vigenti in Italia e sanciti dall’art.2 del DPR 542/1994, ovvero dagli allegati 1 e 4 del D.M. 2/8/1991 e dagli allegati A e B del D.M. 3/8/1993. Essenziali di Sicurezza (R.E.S.) della Direttiva medesima. per la parte impiantistica, ai requisti del D.M. 37/2008 Autorità Competenti : La Direzione Generale dei Dispositivi Medici del Ministero della Salute è depositaria del “Repertorio dei Dispositivi Medici”, ove tutti i dispositivi medici circolanti in Italia, ivi compresi le apparecchiature diagnostiche a Risonanza Magnetica , sono registrati e dichiarati “CONFORMI” ( e ove se ne possono constatare caratteristiche tecniche e prestazionali): Rischi legati al campo magnetico statico della Risonanza Magnetica I rischi specifici (ma “giustificati”) per i pazienti sono diagnosti: legati al sistema di Campi magnetici variabili nel tempo Campi elettromagnetici a radiofrequenza marcate CE, Allineate ai R.E.S. di tutte le Direttive Comunitarie applicabili, rispettose dei dettami legislativi previsti dalle vigenti normative nazionali. I rischi per i lavoratori sono invece legati nella maggior parte dei casi alla sola esposizione al campo magnetico statico, a meno di particolari (e non convenzionali) procedure operative (risonanza pediatrica, pazienti critici) Pazienti e Lavoratori sono comunque esposti ai rischi legati all’uso del fluido criogenico di raffreddamento atto a garantire il corretto funzionamento dei magneti superconduttori Fattori di rischio tipici della RN Rischi costanti Campo magnetico statico (B0) (ATTRAZIONE ) Criogeni (solo superconduttori) Rischi in corso di esame diagnostico Campi magnetici variabili nel tempo (dB/dt) Campi e.m. a RF ( MHz/T) Livelli di campo statico Tesla 3 TOMOGRAFIA A RISONANZA MAGNETICA 0.15 0.01 INTERNO DI UN TRENO CAMPO MAGNETICO TERRESTRE 0.0002 SMAGNETIZZAZIONE DI MEMORIE MAGNETICHE DISTORSIONI NEI MONITOR A COLORI 0.00005 30 Interazioni RM - Ambiente Monitor / Televisori Zone di rischio ed individuazione sito Lettini Orologi Zona ad accesso controllato: zona di rischio (B ≥ 0,5 mT, tipicamente la sala magnete) + locali strettamente dedicati alla gestione in sicurezza delle attività e dei pazienti. (Nel D. M. 2/8/91 in realtà ZAC ≡ zona controllata ≡ zona di rischio) TC Zona di rispetto: 0,1 mT ≤ B ≤ 0,5 mT. Non è necessariamente tutta contenuta all’interno del sito RM, ma comunque deve essere necessariamente contenuta nella proprietà del presidio Supporti magnetici Veicoli Computer Pace-maker Ascensori ZL Bisturi Carte di credito ZR Zona ad accesso Controllato: B ≥ 0.5 mT (5 G) Zona di Rispetto: 0.1 mT (1 G) ≤ B < 0.5 mT (5 G Zona Libera: B < 0.1 mT Attrezzi 31 Misure generali di prevenzione Rischi legati al campo magnetico statico della Risonanza Magnetica •Interdizione degli accessi, ove solo il personale autorizzato può entrare liberamente •Segnaletica di rischio apposta sul lato esterno delle porte di accesso al sito RM e sulla porta della sala magnete 0.5 mT 200 mT Rischi legati alla presenza di fluidi criogenici sotto pressione all’interno dei magneti superconduttori: L’uscita del tubo di quench Fuoriuscita all’esterno dei fluidi criogenici gassosi a seguito di un quench: importanza di una corretta progettazione e realizzazione a regola d’arte della tubazione Il quench Il tubo necessita di coibentazione se è raggiungibile con le mani da (rischio di ustione della popolazione durante un quench) Il problema del tubo di quench Modelli di certificazione degli impianti D.M. n. 37 del 22.01.2008 Caratteristiche del Sensore Ossigeno Certificazione del tubo di quench • Il sensore ossigeno è direttamente collegato ad una centralina di comando dotata di avvisatore luminoso e sonoro capace di segnalare eventuali situazioni anomale. • La taratura del sensore deve prevedere la possibilità di settare una soglia di pre-allarme (19 %) in corrispondenza della quale si attiva l’avvisatore sonoro-luminoso collegato alla centralina, e una soglia di allarme ( rigorosamente al 18%) che implichi l’attivazione automatica di un sistema di ventilazione di emergenza capace di implementare notevolmente l’efficienza del ricambio d’aria nella sala magnete • Le modalità di taratura del sensore e dell’elettronica ad esso associata sono sancite dalla norma CEI EN 50104, come indicato dalle stesse ditte costruttrici Il sensore ossigeno Esso è capace di rilevare l’eventuale presenza di elio gassoso in sala magnete criogeno mediante la rilevazione dell’abbassamento della concentrazione di ossigeno in sala RM. la condizione ideale è rappresentata da una quota di circa 2.5 metri da terra, sulla torretta della macchina RM, in prossimità della prima flangia di raccordo del tubo del quench di dotazione sull’apparecchiatura, ovvero nella posizione di massima tempestività d’intervento in caso di fuoriuscita di elio. La verifica periodica del corretto funzionamento del sistema di rilevazione dell’ossigeno è fondamentale per garantire il mantenimento delle condizioni di sicurezza nella sala esame cui sia installato un tomografo con magnete superconduttore la taratura periodica del sensore garantisce l’attivazione della ventilazione di emergenza alla soglia di allarme del 18% di ossigeno. Centralina e Display del sensore O 2 • Il display del monitoraggio in continuo della % O2 rilevata in sala magnete e i segnali di allarme acustico- luminoso devono essere disponibili o ripetuti in console N E / E N Ripresa Mandata LA TARATURA DEL SENSORE OSSIGENO Il procedimento di taratura, confacente a quanto sancito dalla norma CEI EN 50104 , è previsto dalle ditte costruttrici e chiaramente indicato nel libretto di “Istruzioni per l’uso”. In alcuni casi sono le stesse ditte che sono in grado fornire un “kit” dedicato per svolgere le operazioni, a cui bisogna affiancare delle miscele di gas tecnici di riferimento nelle % N2/O2 richieste. Una ditta specializzata o lo stesso Esperto Responsabile può dotarsi di un proprio sistema di Taratura, propriamente realizzato, purchè confacente alle caratteristiche tecniche richieste dal costruttore e a quanto previsto dalla norma di riferimento. Il Certificato di Taratura L’impianto di ventilazione nella sala magnete costituisce col sensore ossigeno la catena dei sistemi di sicurezza asserviti al magnete superconduttore : la sua corretta realizzazione è fondamentale per evitare le problematiche connesse con l’eventuale rientro di elio in fase gassosa all’interno della sala magnete durante un quench. emergenza normale 1. 1. Mantenere la temperatura 22±2 °C e l’umidità max 60% 2. Garantire 6-8 ricambi/h nel locale esame 3. Assicurare una leggera pressione sulla porta della gabbia per non aspirare la polvere dall’esterno (ΔP > 0) 2. 3. Garantire 18-22 ric/h in emergenza Assicurare una leggera depressione sul locale esami (Δp<0), per facilitare l’apertura della porta e, una volta aperta, evitare che fuoriesca elio all’esterno della sala Prevedere al di sopra della torretta del tomografo un canale per l’eventuale aspirazione diretta dell’ elio gassoso che dovesse fuoriuscire (RIPRESA SUPPLEMENTARE d’EMERGENZA) Layout impianto di ventilazione in sala RM Preallarme (19%) Allarme (18%) Mandate anteriori alla macchina Tubo di ripresa supplementare di emergenza nel controsoffitto Riprese ai lati posteriori e dietro alla macchina ULTERIORI MISURE DI SICUREZZA IN RISONANZA MAGNETICA Monitoraggio del microclima in sala RM La ventilazione e la climatizzazione della sala magnete devono garantire una temperatura costante di 22 +/- 2°C ed un’umidità relativa del 40-60%, al fine di “mantenere” i livelli del SAR e di salvaguardare il benessere del paziente Termoigrometro Le figure professionali alle quali la normativa vigente assegna il compito di garantire - in fase di installazione e messa in esercizio il rispetto degli standard di sicurezza nazionali nonché dei requisiti essenziali di sicurezza eventualmente previsti dalle Direttive Europee di riferimento applicabili sono i Responsabili della Sicurezza Nominati dall’esercente, devono accettare esplicitamente l’incarico ed ottemperare ai compiti puntualmente definiti negli allegati del D.M. 2/8/1991 53 Gestione sicurezza in zona comandi • Riassumendo, in consolle vanno posizionati: Responsabili per la sicurezza • I pulsanti di sicurezza: - ventilazione di emergenza - sgancio elettrico - quench Medico Responsabile della sicurezza (per gli aspetti di gestione clinica dell’apparecchiatura) • I Display: - sensore ossigeno - termoigrometro sala magnete - termoigrometro locale tecnico (non sempre necessario, mai superfluo) Esperto Responsabile della Sicurezza (per gli aspetti di sorveglianza fisica e di garanzia della qualità) Qualificazione professionale ai sensi di legge dei Responsabili per la Sicurezza: medico responsabile per la sicurezza in RM: laurea in medicina e chirurgia ed esperienza nella diagnostica per immagini + curriculum professionale specifico che comprovi la competenza esperto responsabile per la sicurezza in RM: laurea + curriculum professionale specifico che comprovi la competenza Il quadro autorizzativo: DPR 542/94 TIPOLOGIA CAMPO MAGNETICO STATICO MAGNETE FINALITA’ DI UTILIZZO AUTORITA’ COMPETENTE NOTE Settoriale < 0.5 T Resistivo o permanente Diagnostica sulle estremità Nessuna Articolari aperte ? A corpo intero gruppo A <2T Resistivo, permanente, superconduttore Diagnostica su qualunque distretto corporeo Regione A corpo intero gruppo B >2T superconduttore Ricerca Ministero della Salute (ISS, INAIL) Sopra 4 tesla solo su arti, manca la marcatura CE Nessuno può entrare liberamente nel sito RM se non è autorizzato, e quindi se non ha si ha il dispositivo per accedere (chiave, codice numerico, scheda a banda magnetica, etc.). Il paziente una volta all’interno del sito non è lasciato mai solo dal personale interno Francesco Campanella, INAIL Zona controllata Zona ad Accesso Controllato Un Presidio di Risonanza Magnetica deve : 1. essere confinato nel suo perimetro 2. avere un unico accesso rigidamente controllato e “riservato al solo personale autorizzato e a pazienti da esso accompagnati”. 3. consentire l’accesso previo preventivo consenso * La zona controllata è quella in cui il campo magnetico disperso è uguale o superiore a 0.5 mT (5 Gauss). La linea di campo dei 5 gauss deve necessariamente essere contenuta all’interno della zona ad accesso controllato, e per lo più si riscontra essere confinata all’interno della sala magnete. Opportuna segnaletica identificatrice apposta sull’esterno delle porte deve indicare sia i rischi all’esposizione ai campi magnetici presenti all’interno e sia le opportune restrizioni di accesso e di gestione * Zone esterne alla sala magnete eventualmente interessate vanno interdette con barriere fisse ed identificate con cartellonistica che ne indichi i rischi all’esposizione ai campi magnetici presenti all’interno e le restrizioni di accesso Zona di rispetto La zona di rispetto si definisce come quella in cui il campo magnetico disperso va da 0.1 mT (1 Gauss) a 0.5 mT (5 Gauss) Deve essere completamente contenuta all’interno della proprietà di pertinenza del datore di lavoro possessore del tomografo RM Deve preferibilmente non essere utilizzata per scopi o finalità che prevedano postazioni di lavoro fisse Deve avere al proprio interno dotazioni che tengano conto 1. delle problematiche esistenti connesse alla compatibilità elettromagnetica con apparecchi elettronici, 2.della possibile magnetizzazione di apparati ferromagnetici. Sale di attesa pazienti La sala di attesa per i pazienti deambulanti deve essere all’esterno del sito RM, ed è auspicabile averla nei suoi immediati pressi I pazienti barellati devono avere una sala di attesa “dedicata” che ne garantisca la privacy, tranne nei casi in cui siano solo pazienti ricoverati,e quindi programmabili le procedure siano tali da garantire l’assenza di tempi di attesa (pronto soccorso, ecc…..) Accettazione amministrativa L’accettazione, specifica per l’RM o in comune con altre tecniche diagnostiche, deve essere esterna o con sportello afferente all’esterno Il personale amministrativo non può essere in linea di principio autorizzato ad entrare liberamente nel sito RM, ovvero ad avvicinarsi alle zone di rischio Nel caso di confinamento della zona controllata in sala RM, e se previsto nel Regolamento di Sicurezza dall’ER (sulla base del rapporto costo/beneficio), il personale amministrativo può accedere occasionalmente al sito RM, esclusa la sala magnete, senza attivare la sorveglianza medica Anamnesi L’anamnesi del paziente viene effettuata all’interno del sito RM o nei suoi immediati pressi, e comunque in un locale dedicato nel quale sia presente la dotazione minima per una visita medica. Essa consiste nell’ accertamento da parte del medico responsabile dell’esecuzione dell’esame della assenza di controindicazioni all’esame RM, in riferimento alla sua esposizione ai campi magnetici utilizzati dall’apparecchiatura RM e ai rischi ad essi connessi Il medico compila SEMPRE il questionario anamnestico PRIMA dell’esame, interrogando il paziente e firmandolo in calce, ai sensi di legge. Il paziente firma il consenso informato all’indagine e all’eventuale somministrazione del mezzo di contrasto. Anamnesi L’anamnesi del paziente viene effettuata all’interno del sito RM o nei suoi immediati pressi, e comunque in un locale dedicato nel quale sia presente la dotazione minima per una visita medica. Essa consiste nell’ accertamento da parte del medico responsabile dell’esecuzione dell’esame della assenza di controindicazioni all’esame RM, in riferimento alla sua esposizione ai campi magnetici utilizzati dall’apparecchiatura RM e ai rischi ad essi connessi Il medico compila SEMPRE il questionario anamnestico PRIMA dell’esame, interrogando il paziente e firmandolo in calce, ai sensi di legge. Il paziente firma il consenso informato all’indagine e all’eventuale somministrazione del mezzo di contrasto. Disponibilità di un Metal Detector La legislazione vigente prevede che un metal detector (generalmente portatile) sia sempre disponibile nel sito RM al fine di individuare: protesi interne (rilevabili) oggetti sul corpo (piercing) Parti metalliche negli indumenti che non vengono tolti IL METAL DETECTOR NON SI SOSTITUISCE MAI ALL’ANAMNESI Spogliatoi pazienti All’interno del sito RM devono essere previsti – a seconda dei casi, ovvero delle modalità di gestione dell’attività diagnostica - uno o più locali adibiti a spogliatoio pazienti, con dotazione minima adatta allo scopo È buona norma dotare lo spogliatoio di una cassetta di sicurezza ad uso dei pazienti. Lo spogliatoio del personale non è richiesto, ma se ritenuto necessarioinvece può essere locato sia all’interno che all’esterno del sito RM; se all’interno, deve essere considerato ad uso esclusivo Servizi igienici All’interno o negli immediati pressi del sito RM deve essere previsto almeno un servizio igienico ad uso dei pazienti, possibilmente accessibile anche ai disabili Sul piano del sito di Risonanza Magnetica deve essere presente un locale WC attrezzato per portatori di handicap. SPECIFICHE DI UN WCH •Il WCH deve garantire al suo interno la movimentazione intorno a se stessa della carrozzella del disabile. •Le specifiche di realizzazione sono normate dal DPR 503/96 Dotazioni fondamentali: •Sanitari di tipo “dedicato” •Illuminazione supplementare di emergenza •Maniglioni lato water •Sistema di allarme sonoro con interuttore a filo e tacitazione interna al locale •Apertura della porta agevole, verso l’esterno •Ricambio d’aria Preparazione La “Zona Preparazione” è un locale o area attrezzata destinata a trattamenti medici sul paziente che precedono l’esame RM (es.sedazione, somministrazione di liquidi di contrasto,etc.) Deve essere dotata della dotazione minima prevista dal Medico Responsabile del sito RM, custodita ed organizzata in modo razionale, efficace e sicuro Va delimitata da barriere fisse o mobili che garantiscano la privacy del paziente trattato. Dotazione minima: cabinet per i farmaci, lettino/barella amagnetica, disponibilità di gas anestetici, dispositivi medici specifici Emergenza La “Zona Emergenza” è un locale o area - all’uopo attrezzata - destinata per un eventuale primo soccorso medico sul paziente che dovesse necessitare di un primo intervento “anche per motivi non strettamente correlati all’esecuzione dell’esame”. Deve essere dotata della dotazione minima prevista dal Medico Responsabile del sito RM, custodita ed organizzata in modo razionale, efficace e sicuro Tale zona non deve essere delimitata da porte o altra barriera fissa che possa in qualche modo creare impedimento alle procedure di soccorso e deve essere quanto più vicino alla porta della sala esami per un tempestivo intervento Le postazioni di emergenza attrezzate devono essere tante quante sono le apparecchiature RM presenti nel sito. Preparazione - emergenza insieme Qualora la stessa postazione sia adibita sia a “preparazione” e sia ad “emergenza”, occorre definire e formalmente istituire delle procedure restrittive di esecuzione degli esami che dispongano: 1. di consentire la presenza di un solo paziente alla volta all’interno del sito RM di mantenere una allocazione comunque non ambigua e razionalmente individuata per quanto attiene i farmaci ed i dispositivi medici utili alla bisogna 2. Nel caso di più apparecchiature RM presenti, occorre sempre avere tante postazioni P/E quante sono le apparecchiature, con la restrizione di un numero di pazienti all’interno del sito sempre pari al numero di macchine RM operanti Sala magnete : dimensioni Sala Magnete La superficie della sala magnete deve essere tale da consentire l’efficienza delle procedure, soprattutto in caso di emergenza (movimentazione del paziente, ingresso e movimentazione di una barella per le emergenze, spazi minimi di manutenzione). magnete permanente magnete superconduttore Sala magnete: cartellonistica All’interno della sala magnete si accede ad una zona nella quale il campo magnetico è superiore a 5 mT (5 Gauss): una chiara cartellonistica posta sull’esterno della porta deve indicare l’ultima soglia di avvertimento all’esposizione ad alti campi magnetici e i rischi ad essa legati, un esplicito richiamo all’attenzione al divieto assoluto di introduzione - anche accidentale - di materiale ferromagnetico. Sala magnete: interno E’ consentito lungo il perimetro della sala sia la presenza di scaffalature atte alla custodia del materiale tecnico di pertinenza dell’apparecchiatura (ex. le bobine per gli esami), e sia la realizzazione, se le procedure lo prevedono, se gli spazi e lo dotazioni lo consentono, e se le strumentazioni di supporto sono amagnetiche, della postazione di primo intervento per il paziente N.B. In verità le strumentazioni non sono mai del tutto amagnetiche: il costruttore garantisce il loro corretto funzionamento se mantenute al di là di un certo campo Sala magnete: Condizioni ambientali Termoigrometro Sala magnete: Gabbia di Faraday Sistema di scheramtura per veitare interferenze provocate dall’esterno rispetto al segnale a radiofrequenza generato all’interno della sala RM. La ventilazione e la climatizzazione della sala magnete devono garantire una temperatura costante di 22 +/- 2°C ed un’umidità relativa del 40-60%, al fine di contenere eccessivi aumenti del SAR e di salvaguardare il benessere del paziente Esempio di gabbia di tipo “chiuso”, ovvero rivestita di pannelli di rame o di acciaio Sala magnete: Curve isomagnetiche Le curve isomangetiche teoriche fornite in fase di progetto dalla casa costruttrice all’Esperto responsabile dell’impianto RM consentono di valutare a priori l’impatto ambientale dell’apparecchiatura nel sito di installazione, da cui prevedere, se del caso, schermature aggiuntive e limitazioni di utilizzo dei locali attigui A posteriori, ovvero dopo l’installazione ed in sede di collaudo, e quindi prima dell’avvio dell’attività diagnostica, l’E.R. deve mappare le linee di isocampo reali a conferma delle scelte protezionistiche fatte a priori. Il fine è la corretta delimitazione della zona controllata (da 5 gauss in su), l’opportuna individuazione della zona di rispetto (da 5 a 1 gauss), la puntuale definizione della destinazione d’uso degli ambienti che circondano la sala magnete Sala magnete: Gabbia di Faraday Le uniche aperture sono quelle: per il passaggio degli impianti (pannello di controllo) per la finestra della consolle, realizzata comunque in in materiali speciali Per la porta di accesso che, grazie alla presenza di appositi fingers di rame, garantisce – se chiusa - la necessaria continuità per la tenuta della gabbia. Lo scadimento della schermatura è funzione del tempo e dell’uso: essa va quindi periodicamente controllata – preferibilmente su base annuale - ai fini di preservare opportuni standard di qualità Zona Comandi La console è la postazione di comando dell’apparecchiatura RM è allocata davanti ad una finestra della gabbia di faraday, detta “visiva”, che consente di sorvegliare il paziente posto in esame: tale postazione di lavoro è sempre presidiata dagli addetti alla RM (TSRM) durante l’esecuzione degli esami Rappresenta la posizione dalla quale viene sia gestita l’attività medica e sia monitorati i parametri di sicurezza Locale tecnico Il locale tecnico contiene tutta l’elettronica di supporto dell’apparecchiatura RM: opportuni sistemi di ventilazione devono garantire al suo interno una temperatura pressochè costante prevenendo un eccessivo surriscaldamento dell’ambiente. In particolare, Materiale infiammabile, ma anche tutto quanto non di pertinenza rispetto alla destinazione d’uso del locale medesimo, deve essere allontanato in via definitiva dal suo interno Console di comando In consolle devono essere presenti: I pulsanti di sicurezza: - ventilazione di emergenza - sgancio elettrico - quench (preferibilmente) I Display: - sensore ossigeno - termoigrometro sala magnete - termoigrometro locale tecnico se locato in posizione remota Refertazione La sala dedicata alla refertazione delle immagini RM ottenute può essere allocata all’interno del sito RM (ma a suo uso esclusivo) o all’esterno (con possibilità di refertare in condivisione con altre tecniche diagnostiche), Non è possibile utilizzare a tale scopo aree o altre sale già dedicate - all’interno del sito RM - ad altra attività. Eventuale Termoigrometro Archivio L’archivio può trovarsi sia all’interno che all’esterno del sito RM. In caso di archivio cartaceo, è buona prassi che la sua struttura preveda cartelle che, per ogni esame effettuato, contengano: ESEMPI PRATICI DI PROGETTAZIONE DI UN SITO DI RISONANZA MAGNETICA l’immagine RM il referto medico il questionario anamnestico compilato l’eventuale consenso informato del paziente Estintori Ferromagnetico Tutti gli estintori all’interno del reparto RM devono essere di tipo amagnetico, appesi ed identificati come per legge Amagnetico Esempio di un sito RM ove poter gestire 2 pazienti contemporaneamente Sito RM in grado di gestire 4 pazienti alla volta Bibliografia essenziale Principi di risonanza magnetica − Scott A. Huettel, Allen W. Song, Gregory McCarthy. Functional Magnetic Resonance Imaging. Sinauer Associates, 2004 − A. L. Baert, K. Sartor, J. E. Youker. Functional MRI. Springer-Verlag Berlin Heidelberg, 2000. − Bloch F, Hanson WW, Packard M. Nuclear Induction. (1946) Phys Rev; 69: 127. − Abragam A. The principles of nuclear magnetism. (1961).London, Oxford University Press. − Allen PS.: Some fundamental principles of nuclear magnetic resonance. In medical Physics monograph No. 21: The Physics of MRI: (1992) AAPM Summer School Proceedings, American Institute of Physics, 21:15. − Lauterbur PC. Image formation by induced local interactions: examples employing nuclear magnetic resonance. (1974) Pure Appl. Chem. − Mansfield P, Pykett IL. Biological and Medical imaging by NMR. (1978) Journal of Magn. Reson.. − Hedelstein WA, Hutchinson JMS, Johnson G et al. Spin-Warp NMR imaging and applications to human whole body imaging, Phys. (1980) Med. Biol. − Maudsley A. Multiple line-scanning spin-density imaging. (1980) Journal of Magn. Reson. − Makovski A. Volumetric NMR imaging with time-varying gradients. (1985) Magn. Reson. Med. − Mansfield P. Proton spin imaging by nuclear magnetic resonance. (1976) Contemp. Physics. − P. Rinck - Magnetic Resonance in Medicine. The basic Textbook of the European Magnetic Resonance Forum. (1993). Third Edition. Oxford, UK: Blackwell Scientific Pubblications,. − Stark D.D, Bradley W.G. Magnetic Resonance Imaging (1999). Mosby Edts. Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici Information Technology − Campanella F., Mattozzi M., et al.: Indicazioni operative per la valutazione del rischio all’esposizione professionale ai campi magnetici statici nella risonanza magnetica ad alto campo. 2009. www.ispesl.it − http://www.ispesl.it/settore/dispositivi%20medici%20dotati%20di%20impianti%20 a%20pressione_RM%20a%20magnete%20superconduttore.pdf − M. Mattozzi - Dalla fabbricazione all’installazione: il quadro delle regole in Europa ed in Italia per i magneti superconduttori - Convegno Nazionale INAIL 16 Dicembre 2011 − Campanella F., Mattozzi M. - Realizzazione alla regola dell’arte degli impianti di ventilazione nelle sale di Risonanza Magnetica. Indicazioni operative, esperienze, criticità. 2012 − Campanella F., Mattozzi M., Moretti L., D’Avanzo M. A.: Soluzioni strutturali per la progettazione e realizzazione a regola d’arte di un sito di Risonanza Magnetica: indicazioni operative – Ed. 2013 - http://www.ispesl.it − E. Coiera - Guida all’informatica medica, Internet e telemedicina - Il Pensiero Scientifico Editore, 1999 − Autonomic Computing: IBM Perspective on the State of Information Technology, Armonk, NY: International Business Machines, Oct. 2001. − “To err is human: building a safer health system,” Committee on Quality Health Care in America, Institute of Medicine, 2000. − CEI EN 60601-1:2007: Apparecchi elettromedicali - Parte 1: Prescrizioni generali relative alla sicurezza fondamentale e alle prestazioni essenziali – Par. 14: Apparecchi elettromedicali programmabili (PEMS) − CEI 62-233 CEI EN 80001-1:2012-01: applicazione della gestione del rischio per reti IT che incorporano dispositivi medicali - Parte 1: Ruoli, responsabilità e attività − ISO 14971:2007 - Application of risk management to medical devices − P. Pallaro, Libertà della persona e trattamento dei dati personali nell’Unione Europea, Giuffrè, Milano, 2002 − M. Migliazza, Profili internazionali ed europei del diritto all’informazione e alla riservatezza, Giuffrè, Milano, 2004 − E. Barilà - C. Caputo, La tutela della privacy nella pubblica amministrazione, Giuffrè, Milano, 2000 − IHE technical framework and its application (Article in Chinese), Feng JY, et al., Chinese Journal of Medical Instrumentation, 2005, May, Vol. 29, Num. 3, pp. 199-201 Dott. Francesco Campanella Moore, Stephen M. - Using the IHE Scheduled Work Flow Integration Profile to Drive Modality Efficiency, 2003, Radiographics, Vol. 23, pp 523-529 INAIL, Dipartimento di Igiene del Lavoro, Radiazioni Ionizzanti e Non Ionizzanti – Settore Radiazioni Ionizzanti e Risonanza Magnetica − I DOCENTI DEL CORSO Normativa di sicurezza e prevenzione del rischio in risonanza magnetica − F. Campanella, M. Mattozzi, A.S. Panebianco, C. Petrucci, M. Marchetti, G. Spagnoli: Indicazioni operative dell’Ispesl: procedure autorizzative e gestionali relative all’installazione ed uso di apparecchiature diagnostiche a risonanza magnetica. 31 maggio 2004. Internet: http://www.ispesl.it − Giannelli M., Mascalchi M., Mattozzi M, Campanella F.: Standard di Sicurezza in Risonanza Magnetica: Il Regolamento di Sicurezza (2008). www.ispesl.it − Campanella F., Mattozzi M., et al. Indicazioni operative per l’installazione e l’utilizzo di apparecchiature di risonanza magnetica a scopo medico. 2004. www.ispesl.it − Campanella F., Mattozzi M., et al. Sicurezza e qualità nelle apparecchiature di Risonanza Magnetica a basso campo. 2008. www.ispesl.it AIIC associazione italiana ingegneri clinici Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” Dott. Francesco Lisciandro I.R.C.C.S. Policlinico San Matteo Pavia – Fisica Sanitaria Ing. Maurizio Rizzetto A.O. di Pordenone – IT e Ingegneria Clinica Realizzazione e Gestione di un Sito per Diagnostica con Risonanza Magnetica “aspetti tecnici” AIIC associazione italiana ingegneri clinici GLI AUTORI GLI AUTORI Ing. Giovanni Poggialini Responsabile Scientifico generale dei corsi del XIV Congresso Nazionale AIIC – Venezia 2014 Ing. Aessandro Reolon Responsabile Scientifico del Corso Direttore della Struttura Complessa di Ingegneria Clinica dell’ALSS di Belluno, oltre ad essere Iscritto all’albo dell’Ordine degli Ingegneri della provincia di Belluno è esperto responsabile per le installazioni di apparecchiature a risonanza magnetica (D.M. 2/8/91). Socio AIIC dal 1997 e già membro del consiglio direttivo della medesima, a tutt’oggi collabora con l’associazione in qualità di socio ordinario. Laureato in Ingegneria Biomedica presso il Politecnico di Milano, inizia immediatamente a lavorare come ingegnere clinico e biomedico consulente, ruolo che tutt’ora ricopre per diverse aziende, sanitarie e non. E’ Iscritto all’Albo degli Ingegneri della Provincia di Biella dall’anno 2005, socio ordinario AIIC dall’anno 2005, referente regionale AIIC per le regioni Piemonte e Valle d’Aosta dall’anno 2010, componente delle commissione sanità della FIOPA dall’anno 2012, socio ordinario SIHTA dall’anno 2013. - e-mail: [email protected] - CV: it.linkedin.com/in/gpoggialini/ - SKYPE: giovanni.poggialini - Twitter: @gpoggialini Ing. Salvatore Caccavale Collaboratore AIIC per la stesura del volume Laureato in Ingegneria Biomedica presso l’Università degli Studi di Napoli “Federico II” collabora come Ingegnere Clinico con il Servizio di Ingegneria Clinica dell’A.O.U. “Federico II” di Napoli e come docente del corso di “Informatica in riabilitazione” dell’Università degli Studi di Napoli “Federico II”. Iscritto all’Ordine degli Ingegneri della Provincia di Napoli, dove ricopre il ruolo di “segretario” della commissione Biomedica, è socio AIIC dall’anno 2013. AIIC associazione italiana ingegneri clinici REAlIZZAZIoNE E GESTIoNE DI UN SITo PER DIAGNoSTICA CoN RISoNANZA MAGNETICA “ASPETTI TECNICI” REAlIZZAZIoNE E GESTIoNE DI UN SITo PER DIAGNoSTICA CoN RISoNANZA MAGNETICA “ASPETTI TECNICI” AIIC associazione italiana ingegneri clinici [email protected] AIIC associazione italiana ingegneri clinici www.aiic.it tel: 333.8060876 - fax: 06.96681277 Twitter: @AIIC_IngClinici
Scaricare