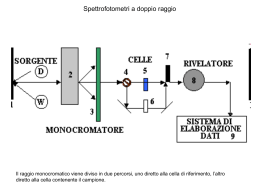
Corso di Chimica Fisica II 2011 Marina Brustolon 17. Le spettroscopie. Generalità Spettroscopie L’interazione radiazione-materia è alla base delle spettroscopie. La materia verrà trattata quantisticamente, la radiazione elettromagnetica in modo semiclassico. L’interazione con la radiazione elettromagnetica si considererà nel seguito come una perturbazione dell’energia delle molecole. Teoria delle perturbazioni E’ uno dei metodi approssimati che si usano in MQ (l’altro è il metodo variazionale). Si supponga di conoscere autostati e autovalori dell’Hamiltoniano H0 di un sistema: H 0 i0 Ei0 i0 H H 0 H ' perturbazione Se il sistema è soggetto ad una perturbazione che può essere rappresentata dall’hamiltoniano H’, molto più piccolo di H0, le proprietà del sistema possono essere descritte adeguatamente apportando correzioni di diverso ordine sia agli autovalori che agli autostati di H0. Teoria delle perturbazioni dipendenti dal tempo H 0 i0 Ei0 i0 H H 0 H ' (t ) La perturbazione può essere: 1. l’interazione tra il campo elettrico E della radiazione elettromagnetica e un momento di dipolo elettrico della molecola oppure E E (t ) E 2 E0 cos t M B (t ) M 2 B0 cos t 2. l’interazione tra il campo magnetico B della radiazione elettromagnetica e un momento di dipolo magnetico della molecola. dipolo elettrico E qi ri i dipolo magnetico H H H ' (t ) 0 e g e B S momento magnetico di spin elettronico N gN N I momento magnetico di spin nucleare H ' (t ) E 2 E0 cos t H ' (t ) M 2 B0 cos t E2 2(r) E1 1(r) L’evoluzione temporale della funzione d’onda (r,t) sotto la perturbazione H’(t) può essere scritta come combinazione lineare delle funzioni d’onda degli stati stazionari (r, t ) c1 (t ) 1 (r, t ) c2 (t ) 2 (r, t ) Al tempo t =0 c1 (0)=1, c2(0)=0. Qual è la probabilità che al tempo t il sistema si trovi descritto da 2 ? Cioè quanto sarà [c2(t )]2? Sviluppando la trattazione con la teoria delle perturbazioni dipendenti dal tempo si trova che, se il sistema è soggetto ad una perturbazione con dipendenza armonica dal tempo, la probabilità di trovare il sistema nello stato 2 cambia nel tempo con velocità costante W12: W12 B B coefficiente di Einstein dell’assorbimento stimolato dalla radiazione densità di energia della radiazione alla frequenza di transizione Frequenza di transizione = E/h E2 E E1 B dipende dagli stati 1 e 2 e dalla perturbazione. Momento di transizione: 21 2 E 1 21 2 M 1 Spettroscopie di dipolo elettrico = spettroscopie ottiche (rotazionale, vibrazionale, elettronica) Spettroscopie magnetiche (NMR, ESR) B=cost (21)2 W12 B W12 ( 21 ) 2 W12 W21 I risultati fondamentali sono quindi: 1. la probabilità di transizione tra due stati 1 e 2 è diversa da zero solo se il momento di transizione tra i due stati è diverso da zero; 2. le probabilità di assorbimento ed emissione indotte dalla radiazione elettromagnetica sono eguali. Probabilità di transizione indotta dalla radiazione elettromagnetica h E2 E2 E1 E1 Assorbimento indotto dalla radiazione W12 h E2 E2 E1 Emissione indotta dalla radiazione W21 E1 h h E2 Probabilità W12 E1 E2 Probabilità W21 E1 W12=W21=W E2 n2 E1 n1 popolazioni degli stati Numero di eventi di assorbimento nell’unità di tempo: n1xW12 Numero di eventi di emissione nell’unità di tempo: n2xW21 All’equilibrio termico: legge di distribuzione di Boltzmann n2 e E / kT n1 Bilancio dell’assorbimento di fotoni dal campo elettromagnetico: n1xW12-n2xW21=W(n1-n2) Emissione spontanea h E2 E1 Un sistema che si trovi in uno stato eccitato può emettere un fotone e tornare allo stato fondamentale anche in assenza di radiazione stimolante. La probabilità di emissione è data dala coefficiente di Einstein A proporzionale a B. 8h 3 A B c3 Le spettroscopie si possono distinguere a seconda dell’interazione tra fotoni e materia in : h Spettroscopia di assorbimento Spettroscopia di emissione h h h h Spettroscopia di scattering (RAMAN) h h h Grandezze e unità di misura in uso nelle h spettroscopie E2 E E in Joules E1 E h E h E 2 c E hc 1 frequenza in Hz (kHz MHz GHz) frequenza angolare in s-1 lunghezza d’onda in m (nm) numero d’onda in cm-1 Velocità dello scambio di energia tra materia e campo elettromagnetico dipende dal dipende dalla momento di preparazione dello transizione stato (da E) dipende dalla radiazione dE (n1 n2 ) B h dt dipende da E Differenze di popolazione n2 e E / kT n1 T 298K kT 200cm1 300 MHz ; 1 m 100 cm 10 2 cm 1 ; E / kT 10 2 / 200 5 10 5 n2 E / kT 5105 e e 0.99995 n1 Zona delle onde radio (NMR) Differenze di popolazione n2 e E / kT n1 1cm T 298K kT 200cm1 1cm 1 E / kT 1 / 200 5 10 3 n2 E / kT 5103 e e 0.995 n1 Zona delle microonde (ESR, spettroscopia rotazionale) Differenze di popolazione n2 e E / kT n1 T 298K kT 200cm1 2000 cm1; E / kT 2000 / 200 10 n2 e E / kT e 10 4.5 10 5 n1 la popolazione dello stato eccitato è trascurabile per la spettroscopia vibrzionale Zona dell’infrarosso (spettroscopia vibrazionale) la popolazione dello stato eccitato è trascurabile anche per visibile e UV Tempo di vita e larghezza delle righe E2 E1 Ci possono essere cause che accorciano il tempo di vita degli stati quantistici, per esempio la collisione tra molecole in gas o in soluzione. Si può dimostrare che stati con tempo di vita hanno un’incertezza sull’energia data da E. E La riga spettrale che ne deriva è quindi allargata.
Scaricare