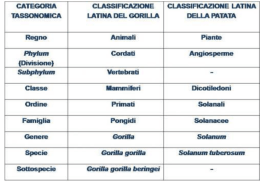
Struttura terziaria α -Elica Foglietto β Nella maggior parte delle proteine le catene di aminoacidi (siano esse organizzati ad α-elica che foglietto β) si ripiegano a gomitolo dando così alla proteina una forma globulare Struttura terziaria delle proteine I ripiegamenti della catena fanno sì che le catene laterali R dei residui amminoacidici possano interagire fra loro formando legami (deboli o forti) che contribuiscono a stabilizzare la conformazione 3D della proteina. ( )44 legami idrogeno legami ionici ponti disolfurici interazioni idrofobiche forze di Van der Waals La struttura terziaria dipende dalla struttura primaria. PONTI DISOLFURICI Oltre al legame peptidico, un altro tipo di legame covalente fra gli amminoacidi presenti nei peptidi e nelle proteine è il legame disolfuro che collega due unità di cisteina. [O] [Rid] CISTEINA CISTINA Comprendere il processo di FOLDING delle proteine, ovvero il meccanismo di ripiegamento grazie al quale queste molecole raggiungono la conformazione più stabile termodinamicamente e biologicamente attiva. Il “folding” di una proteina è il risultato di grandi variazioni entalpiche ed entropiche - 0 + ΔG = ΔH - TΔS Denaturazione Se sottoponiamo una proteina a valori di pH o temperatura molto diversi da quelli normali, la proteina perde la conformazione nativa e perde la sua funzione. Così è stato dimostrato che una proteina possiede nella struttura primaria le istruzioni che deve seguire per ripiegarsi nella sua struttura tridimensionale finale. la sequenza determina la struttura, che determina la funzione Le Proteine “conoscono” il loro ruolo e “sanno” come organizzare la loro architettura molecolare. L’informazione è contenuta nella sequenza degli amminoacidi che è stabilita dal tratto di DNA (gene) il quale contiene il “progetto” di ogni specifica proteina. Molte malattie sono dovute al difettoso ripiegamento (FOLDING) di una proteina A. Alcune patologie derivano da proteine che non sono in grado di raggiungere la loro struttura funzionale e che tendono a formare grossi aggregati (fibrille o forme amiloidi): Alzheimer, encefalopatia spongiforme. B. In altri casi mutazioni del gene generano proteine incomplete che non sono più in grado di svolgere la loro funzione perché incapaci di legare i loro substrati: fibrosi cistica, distrofia muscolare L'encefalopatia spongiforme bovina (Bse) è una malattia neurologica degenerativa che colpisce i bovini E’ causata dall’alterazione proteina, il Prione. Pr = proteica ; i = infettiva ; one = particella di una Il prione 1984 Stanley Prusiner Per anni il prione è stato considerato erroneamente un virus Si trova in tutti gli organi, principalmente sulla membrana dei neuroni Il suo ruolo è quello di coadiuvare la trasmissione dei segnali tra le cellule nervose La conversione da PrPc a PrPSc procede poi con una reazione a catena. Quando viene raggiunta una concentrazione sufficiente di proteine PrPSc, queste si aggregano a formare un lungo filamento che gradualmente danneggia il tessuto neuronale. Una volta mutata la proteina è infettiva: a contatto con la forma normale, la trasforma, rendendola patologica I prioni alterati sono molto resistenti: al calore ai disinfettanti alle proteasi Il salto di specie La Bse tra i bovini nasce, probabilmente, dall'uso di carcasse di animali infetti (pecore) per farne mangimi. NvCJ Farine animali Scrapie Le pecore non sembrano trasmettere la malattia direttamente all’uomo Bse I prioni dei bovini sono in grado di infettare l’uomo STRUTTURA QUATERNARIA Le interazioni che si stabiliscono tra le subunità sono dello stesso tipo di quelle che stabilizzano le strutture terziarie: legami idrogeno, interazioni idrofobiche, ponti disolfurici tra i gruppi R dei residui amminoacidici. Collageno Trp-sintasi Emoglobina Actina-miosina Struttura quaternaria (3D) dell’Emoglobina 50 x 55 x 65 Ǻ Quattro subunità disposte ai vertici di un tetraedro 12 (o 21) e 11 (o 22) (interazioni idrofobiche, legami idrogeno, coppie ioniche); 11, 22 (interazioni polari). LA DISTROFIA MUSCOLARE La più diffusa è la malattia di Duchenne che si trasmette attraverso il cromosoma sessuale X, dalla madre ai figli maschi; le figlie femmine invece possono essere portatrici sane. LA DISTROFIA MUSCOLARE La distrofina, una proteina cruciale per la vita e la funzione dei muscoli La distrofina stabilizza la membrana della cellula muscolare durante i cicli di contrazione-rilassamento La malattia è dovuta alla mancanza della distrofina per un difetto del gene che codifica la proteina (anomalo codone di stop). LA DISTROFIA MUSCOLARE Degenerazione del tessuto muscolare sostituito da tessuto fibroso e adiposo. Progressiva perdita di forza muscolare con conseguente progressiva perdita delle abilità motorie. Insufficienza respiratoria e cardiaca Morbo di Alzheimer 1906 Alois Alzheimer descrisse per primo tale malattia definendola “un processo degenerativo che distrugge progressivamente le cellule cerebrali, rendendo a poco a poco l’individuo che ne è affetto incapace di una vita normale" MECCANISMO DEL PROCESSO DI DEGENERAZIONE I neuroni cominciano a produrre una proteina (βamiloide), con formazione di placche (agglomerato di filamenti proteici). Iniziano così una serie di eventi programmati che portano alla morte del neurone (“Suicidio programmato”) L’App (precursore della proteina amiloide) normalmente serve per il normale funzionamento dei neuroni. Fibrosi cistica Malattia autosomica recessiva grave più comune nella popolazione italiana malati 1/3000 portatori 1/28 Difetto nella proteina transmembrana che agisce come un canale degli ioni cloro nelle cellule epiteliali (CFTR: Cystic Fibrosis Transmembrane Conductance Regulator, 1480 amminoacidi). La mutazione più comune è la delezione di un amminoacido (Phe 508) e la proteina mutata non si avvolge correttamente. Le manifestazioni cliniche della malattia sono caratterizzate dalla presenza di secrezioni esocrine mucose dense che portano a una malattia polmonare cronica ostruttiva con evoluzione verso l’insufficienza respiratoria Esempi di proteine fibrose. Funzioni e Meccanismi. …LA CONTRAZIONE MUSCOLARE Muscolo striato: la contrazione si basa sulla struttura del sarcomero, in cui i filamenti di actina e di miosina si intercalano e scivolano gli uni sugli altri. TEORIA DELLO SCORRIMENTO DEI FILAMENTI ACTINA MIOSINA SARCOMERO: unità funzionale del muscolo ACTINA L’actina è presente in tutte le cellule eucariotiche, dove rappresenta il 5% delle proteine cellulari. I microfilamenti, con un diametro di 7 nm, sono polimeri della proteina actina: servono alla struttura della cellula ed al movimento. ACTINA Le singole molecole di actina si chiamano actina G (actina globulare). In condizioni adeguate, le molecole di actina G polimerizzano a formare i microfilamenti; sotto questa forma, l'actina è chiamata actina F (actina filamentosa) Actina G Actina F MIOSINA La miosina è composta da sei catene polipeptidiche: due catene pesanti e due coppie di catene leggere. La testa ha attività ATPasica e motrice. Controlla la contrazione muscolare, la contrattilità nelle fibre da stress e l’anello contrattile durante la citochinesi. INTERAZIONE ACTINA-MIOSINA I PONTI TRASVERSALI sono formati da legami tra l’actina F dei filamenti sottili e le teste di miosina dei filamenti spessi Ogni testa della miosina presente sul filamento va incontro a ripetuti cicli di legame alle subunità di actina sul filamento sottile; subisce un cambiamento conformazionale che richiede energia. La contrazione è controllata dall’ATP e dalla presenza degli ioni calcio La forza che guida la formazione dei ponti trasversi è l’idrolisi dell’ATP, catalizzata da un’ATPasi attivata dall’actina, localizzata nella testa della miosina. Una testa di miosina non legata ad ATP si ancora saldamente all’actina (rigor): stadio breve a cui segue il legame dell’ATP. Quando l’ATP si lega alla testa di miosina, l’associazione actinamiosina si rompe La miosina idrolizza l’ATP: la testa di miosina cambia “posizione” rispetto al resto della molecola La miosina si riattacca debolmente ad un altro monomero di actina, rilascia Pi e questo fa aumentare la sua affinita’ per l’actina Il legame genera un cambiamento conformazionale che sposta la coda della miosina verso il centro del sarcomero Fine del ciclo: si stacca l’ ADP e la testa di miosina si ancora di nuovo saldamente all’actina (rigor) in una posizione diversa sul filamento di actina. La contrazione muscolare richiede tanta energia, e il muscolo scheletrico contiene tanto ATP, che viene prodotto dai mitocondri durante i periodi di riposo. Nel sarcoplasma vi sono anche abbondanti riserve lipidiche e di glicogeno. Se le riserve di ATP finiscono durante I periodi di contrazione prolungata, l’ADP generata dall’idrolisi dell’ATP viene riconvertito in ATP attraverso glicolisi anerobica, che porta all’accumulo di acido lattico. REGOLAZIONE DELLA CONTRAZIONE DA PARTE DEGLI IONI CALCIO Il sito di legame della miosina sul filamento di actina è normalmente mascherato dalla tropomiosina, che deve essere rimossa per permettere l’attacco della miosina. La dipendenza dal calcio è regolata dalla troponina C (TnC), che si associa agli ioni calcio ed, una volta legata, va incontro ad un cambiamento conformazionale che si trasmette alla tropomiosina, inducendola a muoversi verso il centro dell’elica del filamento sottile. In questo modo i siti di legame sull’actina si rendono disponibili al legame con la testa della miosina, permettendo la contrazione. Le cheratine sono un esempio di come la funzione biologica di una proteina riflette la struttura delle catene polipeptidiche Collageno Un altro esempio di come la funzione biologica di una proteina rifletta la sua struttura Responsabile di un’elevata resistenza allo stiramento Sezione di una molecola di tropocollageno OH H 2 idrossiprolina
Scaricare