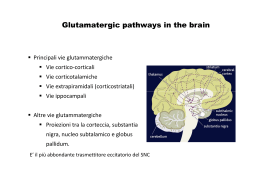
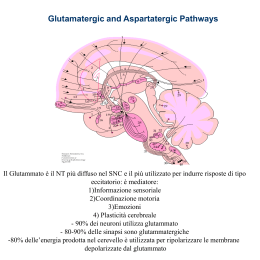
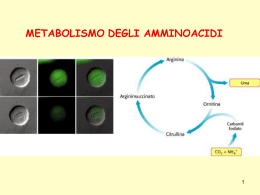
GLUTAMMATO, GLIA e MERCURIO Il glutammato è il principale neurotrasmettitore nel cervello, il suo metabolismo è strettamente legato a quello della glutammina, infatti, il glutammato liberato dalla terminazione nervosa glutammatergica è captato dalle cellule gliali viciniori e convertito in glutammina grazie all’intervento dell’enzima glutammina sintetasi esclusivo di quest’ultimo tipo di cellula. Recentemente si è rivolta più attenzione al ruolo degli astrociti e il rilascio del glutammato, ai pathways capaci di modulare la trasmissione sinaptica, al ruolo del gene Bestrophin 1 e ai canali Ca2+ permeabili al glutammato e al GABA . High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Park H, Han KS, Oh SJ, Jo S, Woo J, Yoon BE, Lee CJ. Mol Brain. 2013 Dec 9;6(1):54. doi: 10.1186/1756-6606-6-54. PMID:24321245[PubMed - in process] Free Article Per la rimozione dell’eccesso di glutammato sono necessari adeguati livelli di ossigeno, in presenza di insulina che diminuisce la quantità di glucosio che a sua volta è importante per eliminare il glutammato, pertanto si deve garantire un giusto equilibrio tra le componenti in gioco. Infatti, una diminuzione del livello di glucosio rende difficoltosa la rimozione del glutammato, che addirittura tende ad aumentare e incide negativamente sulla disponibilità di glutatione, condizione quest’ultima, che aggrava il danno dei neuroni. In questa pathway metabolica è importante la compartecipazione del calcio, che se ad adeguati livelli fisiologici contribuisce al normale funzionamento della cellula nervosa, se in eccesso ne stimolano in continuazione l’attività, fino a provocarne danno irreversibile e quindi la morte. A quanto su esposto va aggiunto che si innesca un meccanismo infiammatorio con conseguente rilascio di molecole infiammatorie, si ha un ulteriore produzione di glutammato e aumento di calcio, in questo circolo vizioso nascono le condizioni per la produzione di NO e perossinitriti, deleteri per la cellula e l’ambiente circostante. In questo caso è importante mantenere livelli ottimali di glicemia somministrando giuste quantità di carboidrati, magnesio e zinco; soprattuto quest’ultimo elemento deve essere somministrato a dosaggi adeguati perché potrebbe influenzare i livellli di glutammato. Sirrieh RE, MacLean DM, Jayaraman V. no-terminal domain tetramer organization and structural effects of zinc binding in the N-methyl-D-aspartate (NMDA) receptor. J Biol Chem. 2013 Aug 2;288(31):22555-64. doi: 10.1074/jbc.M113.482356. Epub 2013 Jun 21. PMID:23792960 Choi UB, Kazi R, Stenzoski N, Wollmuth LP, Uversky VN, Bowen ME. Modulating the intrinsic disorder in the cytoplasmic domain alters the biological activity of the N-methyl-D-aspartate-sensitive glutamate receptor. J Biol Chem. 2013 Aug 2;288(31):22506-15. doi: 10.1074/jbc.M113.477810. Epub 2013 Jun 19. PMID:23782697 Watt NT, Griffiths HH, Hooper NM. Neuronal zinc regulation and the prion protein. Prion. 2013 May-Jun;7(3):203-8. doi: 10.4161/pri.24503. PMID:23764834[PubMed - in process] Free PMC Article Bouron A, Oberwinkler J. Contribution of calcium-conducting channels to the transport of zinc ions. Pflugers Arch. 2013 May 30. [Epub ahead of print] PMID:23719866 Alcune sostanze chimiche rilasciate nell’ambiente, tra i quali mercurio e composti organoclorurati (PCB, 138 , 153 , e 180 , DDT, DDE , esaclorobenzene HCB e β-HCH beta – esaclorocicloesano) possono alterare il livello di glutammato nel cordone ombelicale e creare le condizioni per una disfunzione cerebrale subclinica nell’infanzia. Palou-Serra A, Murcia M, Lopez-Espinosa MJ, Grimalt JO, Rodríguez-Farré E, Ballester F, Suñol C. Influence of prenatal exposure to environmental pollutants on human cord blood levels of glutamate. Neurotoxicology. 2013 Dec 19. pii: S0161-813X(13)00184-8. doi: 10.1016/j.neuro.2013.12.003. [Epub ahead of print] PMID:24361731 Il dicloruro di mercurio danneggia i neuroni attraverso una sovra-attivazione dei recettori NMDA, con conseguenze dannose per il citoscheletro cellulare. Xu F, Farkas S, Kortbeek S, Zhang FX, Chen L, Zamponi GW, Syed NI. Mercury-induced toxicity of rat cortical neurons is mediated through N-Methyl-D-Aspartate receptors. Mol Brain. 2012 Sep 14;5:30. doi: 10.1186/1756-6606-5-30. PMID:22980357[PubMed - indexed for MEDLINE] Free PMC Article L’estratto di Ginko biloba, sui neuroni, sembra avere un effetto protettivo da eccitotossicità indotta da un eccesso di attivazione del recettore NMDA, perché l’estratto di Ginko biloba in modo blando blocca il recettore NMDA. Xiao ZY, Sun CK, Xiao XW, Lin YZ, Li S, Ma H, Song GR, Cheng R. Effects of Ginkgo biloba extract against excitotoxicity induced by NMDA receptors and mechanism thereof Zhonghua Yi Xue Za Zhi. 2006 Sep 19;86(35):2479-84. PMID: 17156678 L’esposizione fetale al metilmercurio provoca deficit cognitivo nei bambini nei primi anni di vita, si ipotizza che i livelli di metilmercurio raggiunti dalle mamme che si alimentano regolarmente pesce durante la gravidanza sono associati a deficit neurologici nella loro prole. Anche in questo caso gli astrociti svolgono un ruolo chiave nella eccitotossicità indotta dal metilmercurio: Il metilmercurio si accumula preferenzialmente negli astrociti Il metilmercurio inibisce in maniera specifica l'assorbimento del glutammato negli astrociti La disfunzione neuronale è secondaria a disturbi degli astrociti La co-somministrazione di concentrazioni tossiche di metilmercurio e glutammato porta alla comparsa delle tipiche lesioni neuronali associati alla stimolazione eccitotossica Il metilmercurio induce rigonfiamento degli astrociti Le metallotioneine hanno un ruolo protettivo perché attenuano la citotossicità indotta dal metilmercurio. È da tener presente che le fonti di assunzione del metilmercurio non sono solo quelle alimentari, con il livello d’inquinamento ambientale raggiunto l’esposizione è maggiore, basti pensare all’incenerimento dei rifiuti. Neurochem Int. 2000 Aug-Sep;37(2-3):199-206. Methylmercury alters glutamate transport in astrocytes. Aschner M, Yao CP, Allen JW, Tan KH. PMID:10812205 Non esiste un meccanismo unico che possa spiegare la pletora dei segni osservati nella neurotossicità indotta dal metilmercurio, descriviamo di seguito alcune osservazioni derivanti dallo studio degli astrociti: Il metilmercurio inibisce la cisteina di conseguenza si ha una diminuzione dei livelli di glutatione e ciò si ripercuote su un inadeguato controllo dello stress ossidativo. L’interferenza con le funzioni di trasporto del glutammato ne farà aumentare i livelli, anche di quelli extracellulari. Altri disturbi funzionali si sovrappongono agli effetti del metilmercurio come per esempio la formazione di ROS negli astrociti. È di primaria importanza mantenere adeguati livelli di glutatione all’interno degli astrociti per proteggerli dai danni indotti dallo stress ossidativo, quest’ultimo aggiunto all’accumulo del glutammato amplifica il circolo vizioso sommando danno ossidativo ed eccitotossico. The Role of Glia in Neurotoxicity, Second Edition a cura di Michael Aschner,Lucio G. Costa C’è differenza di risposta all’insulto indotto da metilmercurio tra nevroglia e astrocita, la microglia risponde in maniera più rapida e con più forza; l’astrocita avendo maggior quantità di glutatione forma più complessi GSH-MeHg, da eliminare, nella microglia questi complessi attivano il complesso Keap1-Nrf2 che induce una traslocazione nucleare di Nrf2 e ciò provoca un’aumento dell’espressione degli enzimi di fase 2, con effetti di protezione. Methylmercury and Neurotoxicity a cura di Sandra Ceccatelli,Michael Aschner Alcuni bambini con ASD si mostrano molto intelligenti in ambiti particolari, perché c’è correlazione con una maggiore espressione dei recettori del glutammato e superiore capacità di apprendimento e memoria, a fronte di questo vantaggio però c’è un rischio maggiore di danno eccitotossico. È di fondamentale importanza ridurre l’assunzione di eccitotossine con i cibi e limitare l'assunzione di calcio, perché l’eccesso di glutammato provoca un aumento del flusso di calcio all’interno della cellula, conseguente rilascio di mediatori dell’infiammazione e si innesca un meccanismo di danno neuronale. Una dieta ad alto contenuto di proteine si traduce in un maggior carico eccitotossico, pertanto l’alimentazione va equilibrata, è da tener presente inoltre che i bambini non sono tutti uguali si raccomanda perciò una alimentazione il più possibile personalizzata, soprattutto alla luce delle mutazioni rilevate con lo screening dei SNPs. Le sinapsi sono giunzioni specializzate che permettono, attraverso il rilascio dei neurotrasmettitori, la comunicazione tra cellule nervose o con altre cellule, come per esempio quelle muscolari; le vescicole sinaptiche sono rilasciate (esocitosi) vallo sinaptico e attivano la membrana della cellula destinataria dell’informazione. I neurotrasmettitori hanno elevata affinità per i recettori situati sulla membrana del neurone post-sinaptico, uno dei neurotrasmettitori più importanti è l’acido glutammico che da vita alla neurotrasmissione glutammatergica, che generalmente è eccitatoria. Un ruolo chiave lo hanno i recettori ionotropici rapidi che permettono il passaggio degli ioni Ca++ o Na+ , possono però entrare in gioco i recettori metabotropici che possono rallentarne l’attività. I neuroni glutammatergici sono molto ben rappresentati nel sistema nervoso centrale, ma assume importanza la loro presenza nell’ippocampo deputato alla creazione della memoria. Questo tipo di neurotrasmissione è coinvolta nella percezione, nell’apprendimento e nelle funzioni motorie, pertanto uno squilibrio a questo livello può indurre diverse patologie tra le quali le neurodegenerative, per questo motivo l’industria farmaceutica ha aperto linee di ricerca per trovare farmaci antagonisti o modulatori. vie glutammatergiche Vie cortico-corticali Vie corticotalamiche Vie extrapiramidali (corticostriatali) Vie ippocampali Proiezioni tra la corteccia, substantia nigra, nucleo subtalamico e globus pallidum. Recettori del glutammato Ionotropi (iGlu) Metabotropici (mGlu) AMPA mGlu1 KA mGlu2 NMDA mGlu3 mGlu4 mGlu5 mGlu6 mGlu7 mGlu8 I recettori ionotropici per il glutammato sono complessi polimerici costituiti da quattro o cinque sub unità che formano un canale ionico transmembrana che interagisce con il glutammato, e sono distinti in tre gruppi in in dipendenza dell’agonista selettivo, della cinetica e delle differenze della conduttanza ionica. I recettori AMPA sono localizzati nella membrana postsinaptica, sono oligomeri composti da diverse subunità (iGluR1-4 ) e deputati alla risposta eccitatoria rapida caratteristica delle sinapsi glutammatergiche. I recettori per il kainato (KA) sono localizzati nelle aree pre- e postsinaptiche in determinate aree cerebrali come striato, giro dentato, strati profondi della corteccia cerebrale e regione CA3 dell’ippocampo selettivamente vulnerabili in diverse malattie neurodegenerative I recettori NMDA sono localizzati nelle aree postsinaptica, composti da una subunità NR1 che è comune a tutti i recettori e da almeno una delle subunità NR2A, NR2B, NR2C, NR2D o NR3A e NR3B, hanno una cinetica di attivazione lenta, sono permeabili al Ca2+ e sono sensibili al blocco del Mg2+ e sono importanti nei processi di apprendimento. L’eccessiva attivazione dei recettori NMDA provoca un eccessivo ingresso di calcio nei neuroni e ciò innesca fenomeni degenerativi e morte neuronale. I recettori metabotropici per il glutammato (mGlu) controllano l’eccitabilità del SNC e i meccannismi di plasticità neuronale e agiscono come modulatori della sinapsi glutammatergica, perché sono reclutati solamente in presenza di elevate concentrazioni extracellulari di glutammato. Gli agonisti dei recettori metabotropici per il glutammato 3 (mGlu3) esercitano effetti di protezione sui neuroni perché stimolano la sintesi del fattore neurotrofico TGF-ß1 da parte della glia. Il glutammato è sintetizzato a livello locale in quanto in condizioni di normalità non attraversa la barriera ematoencefalica ed è opportunamente mantenuto entro livelli omeostatici, per evitare danni di natura eccitotossica. Il glutammato per una quota parte deriva dalla transaminazione dell’acido α-chetoglutarico, intermedio metabolico del ciclo di Krebs, circa 1/5 deriva dalla trasformazione della glutammina in glutammato per opera dell’enzima glutaminasi che fa parte del ciclo metabolico glutammina-glutammato. In questo pathway sono coinvolti gli astrociti vicini alle sinapsi, che inglobano il glutammato rilasciato nello spazio sinaptico e tramite la glutammina sintetasi lo convertono in glutammina che nei neuroni è riconvertita dalla glutaminasi in glutammato. Il ruolo degli astrociti è importante ai fini del controllo dei livelli di glutammato negli spazi presinaptici e di conseguenza la regolazione della trasmissione dell’impulso. Nel catabolismo del glutammato entra in gioco la glutammato deidrogenasi per la formazione di α-chetoglutarato ed ammonio. Molti pathways metabolici tra i quali quelli deputati alla sintesi del Glutatione e dell’ acido γ-aminobutirrico (GABA) utilizzano il glutammato e la glutammina come substrato, restando nell’ambito della biochimica neurologica è importante appuntare l’attenzione al metabolismo dell’ acido γ-aminobutirrico (GABA) strettamente correlato a quello del glutammato; il GABA è sintetizzato per decarbossilazione del glutammato per opera della glutammato decarbossilasi. Sintesi del GABA. Jorgensen, E.M. GABA (August 31, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.14.1, http://www.wormbook.org. Nel pathway catabolico il GABA è trasformato in semialdeide succinica per deaminazione, con l’intervento dell’enzima GABA-T (acido γ-aminobutirrico -α-chetoglutaricotransaminasi), si ha così il trasferimento del gruppo amminico a una molecola di α-chetoglutarato, e si forma acido glutammico che a sua volta può essere riutilizzato per sintetizzare di nuovo acido γ-aminobutirrico. Il glutammato e il GABA devono essere rimossi dallo spazio sinaptico per mezzo dei neurotrasportatori, quelli presenti sulla membrana degli astrociti rimuovono la maggior parte del neurotrasmettitore. Sono altresì importanti le pompe Na+/K+ per il cotrasporto contro gradiente di concentrazione del glutammato, per ogni molecola del neurotrasmettitore sono necessarie due ioni Na+. Le concentrazioni ioniche devono essere ottimali affinché il trasportatore possa funzionare, perché un aumento delle concentrazioni di ioni K+ o una diminuzione degli ioni Na+ del comparto extracellulare può determinare l’inversione del trasporto del glutammato. Una volta che quest’ultimo si libera nello spazio sinaptico non avendo formato le vescicole e non essendo calcio dipendente, aggrava il danno neuronale per le sue proprietà eccitotossiche. I trasportatori EAAT (Excitatory Amino Acid Transporters) assumono un ruolo centrale per il trasporto del glutammato all’interno della cellula nervosa, l’EAAT1 e l’EAAT2 sono di pertinenza gliale, recuperano i neurotrasmettitore in eccesso; l’EAAT3 e l’EAAT4 sono tipici dei neuroni, ma li ritroviamo anche in altri organi tra i quali la placenta e l’intestino. Elevati livelli di glutammato provocano un aumento anomalo dell’attività elettrica che induce alterazioni prolungate nel comportamento, come comunemente si rileva nell’ASD. Leggi articolo intero Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Choudhury PR, Lahiri S, Rajamma U. Pharmacol Biochem Behav. 2012 Feb;100(4):841-9. doi: 10.1016/j.pbb.2011.06.023. Epub 2011 Jul 5. Review. PMID:21756930[PubMed - indexed for MEDLINE] In presenza di disfunzioni degli Excitatory Amino Acid Transporters c’è danno eccitotossico che può essere alla base di molte patologie del neurosviluppo e neurodegenerative, negli ultimi tempi è stata prestata molta attenzione alla funzionalità e plasticità delle cellule gliali, in particolare agli astrociti; e stato evidenziato che la clearance del glutammato negli astrociti e nello spazio sinaptico sono alterati nell’ASD. Animal model of autism induced by prenatal exposure to valproate: altered glutamate metabolism in the hippocampus. Bristot Silvestrin R, Bambini-Junior V, Galland F, Daniele Bobermim L, Quincozes-Santos A, Torres Abib R, Zanotto C, Batassini C, Brolese G, Gonçalves CA, Riesgo R, Gottfried C. Brain Res. 2013 Feb 7;1495:52-60. doi: 10.1016/j.brainres.2012.11.048. Epub 2012 Dec 5. PMID:23219577[PubMed - indexed for MEDLINE] Nell’ASD si rilevano problemi sulle sinapsi e sui recettori post-sinaptici AMPA e NMDA legate a mutazioni associate a ProSAP2/Shank3, tali mutazioni possono alterare lo stato funzionale delle sinapsi eccitatorie. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, Boeckers TM, Montgomery JM, Garner CC. J Neurosci. 2012 Oct 24;32(43):14966-78. doi: 10.1523/JNEUROSCI.2215-12.2012. PMID:23100419[PubMed - indexed for MEDLINE] Free PMC Article Sembra esserci un’associazione tra il SNP rs301430 nel gene SLC1A1 (trasportatore del glutammato ) e la gravità dei comportamenti ossessivo-compulsivi, tic e ansia nei bambini con disturbo dello spettro autistico. Pertanto si è ipotizzato che la variazione allelica del gene SLC1A1 può essere un biomarker ma servono studi su più larga scala e su campioni indipendenti più grandi. Glutamate transporter gene (SLC1A1) single nucleotide polymorphism (rs301430) and repetitive behaviors and anxiety in children with autism spectrum disorder. Gadow KD, Roohi J, DeVincent CJ, Kirsch S, Hatchwell E. J Autism Dev Disord. 2010 Sep;40(9):1139-45. doi: 10.1007/s10803-010-0961-7. PMID:20155310[PubMed - indexed for MEDLINE] Family-Based Association Testing of OCD-associated SNPs of SLC1A1 in an autism sample. Brune CW, Kim SJ, Hanna GL, Courchesne E, Lord C, Leventhal BL, Cook EH. Autism Res. 2008 Apr;1(2):108-13. doi: 10.1002/aur.11. PMID:19360657[PubMed - indexed for MEDLINE] Free PMC Article A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. Science. 2007 Oct 5;318(5847):71-6. Epub 2007 Sep 6. PMID:17823315[PubMed - indexed for MEDLINE] Free PMC Article Arch Gen Psychiatry. 2009 Apr;66(4):408-16. doi: 10.1001/archgenpsychiatry.2009.6. A haplotype containing quantitative trait loci for SLC1A1 gene expression and its association with obsessive-compulsive disorder. Wendland JR, Moya PR, Timpano KR, Anavitarte AP, Kruse MR, Wheaton MG, Ren-Patterson RF, Murphy DL. PMID:19349310[PubMed - indexed for MEDLINE] PMCID:PMC2775716Free PMC Article J Autism Dev Disord. 2010 Sep;40(9):1139-45. doi: 10.1007/s10803-010-0961-7. Glutamate transporter gene (SLC1A1) single nucleotide polymorphism (rs301430) and repetitive behaviors and anxiety in children with autism spectrum disorder. Gadow KD, Roohi J, DeVincent CJ, Kirsch S, Hatchwell E. Department of Psychiatry, Stony Brook University, New York, NY, USA. [email protected] PMID:20155310[PubMed - indexed for MEDLINE] Arch Gen Psychiatry. 2006 Jul;63(7):778-85. Association testing of the positional and functional candidate gene SLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Dickel DE, Veenstra-VanderWeele J, Cox NJ, Wu X, Fischer DJ, Van Etten-Lee M, Himle JA, Leventhal BL, Cook EH Jr, Hanna GL. PMID:16818867[PubMed - indexed for MEDLINE] Am J Med Genet B Neuropsychiatr Genet. 2007 Dec 5;144B(8):1027-33. Association of the SLC1A1 glutamate transporter gene and obsessive-compulsive disorder. Stewart SE, Fagerness JA, Platko J, Smoller JW, Scharf JM, Illmann C, Jenike E, Chabane N, Leboyer M, Delorme R, Jenike MA, Pauls DL. PMID:17894418[PubMed - indexed for MEDLINE] Autism Res. 2008 Apr;1(2):108-13. doi: 10.1002/aur.11. Family-Based Association Testing of OCD-associated SNPs of SLC1A1 in an autism sample. Brune CW, Kim SJ, Hanna GL, Courchesne E, Lord C, Leventhal BL, Cook EH. PMID:19360657[PubMed - indexed for MEDLINE] PMCID:PMC2688703Free PMC Article L’inibizione selettiva di EAAT1 - 2 e 3 induce l’aumento dei livelli di glutammato extracellulare che probabilmente stimola in maniera eccessiva i recettori, si ha un aumento anomalo di Ca2+ intracellulare e conseguente danno cellulare. Alla base del danno c’è l’attivazione di pathways enzimatici degradativi che portano alla formazione e accumulo di radicali liberi dell’ossigeno. I recettori ionotropici per il glutammato sono canali ligando-dipendenti, tutti derivanti da un unico gene primordiale, appartengono a tre categorie: recettori per l’acido alfa-amino-3-idrossi-5-metil-4-isoxazol-propionico(AMPA) identificate quattro subunità: GluR1, GluR2, GluR3 e GluR4 recettori per il kainato (KA) identificate cinque subunità: GluR5, GluR6, GluR7, KA1 e KA2 recettori per l’N-metil-D-aspartato (NMDA) identificate sette subunità:NR1,NR2A, NR2B, NR2C, NR2D, NR3A e NR3B È importante prestare attenzione al fenomeno della desensitizzazione che è una proprietà intrinseca dei recettori-canale che riduce le capacità del recettore di modificarsi ai fini di un miglior funzionamento. In altre parole, se un ligando non si separa dal recettore in tempi fisiologici si determina desensitizzazione, il legame tra recettore e ligando è avvenuto ma il canale è chiuso. Un esempio classico è quello delle terminazioni della giunzione neuromuscolare, che stimolate ad alta frequenza liberano oltre al neurotrasmettitore anche il CGRP (peptide correlato al gene della calcitonina) che attiva un pathway biochimico che porta all’aumento dei livelli intracellulari di cAMP, in grado di attivare la PKA che promuove la desensitizzazione. http://www.medicinapertutti.altervista.org/argomento/desensitizzazione-dei-recettori-canale I recettori AMPA sono ben rappresentati nel SNC, ma l’area di maggior interesse è l’ippocampo perché in questa regione tali recettori sono importanti per i processi di plasticità sinaptica. I recettori postsinaptici per il kainato sono anch’essi diffusi nelle diverse aree cerebrali, ma assumono rilevanza l’ippocampo, l’amigdala, le connessioni talamo-corticali e le corna dorsali del midollo spinale, hanno la capacità di modulare il rilascio del neurotrasmettitore nelle sinapsi eccitatorie e inibitorie. Nell’ippocampo inibiscono il rilascio di GABA e nel midollo spinale inibiscono il rilascio di glutammato attraverso l’interazione con le proteine G. I recettori NMDA si caratterizzano per avere almeno una subunità NR1 e una NR2, alta conduttanza ed elevata affinità per il glutammato, all’incirca un migliaio di volte rispetto ai recettori AMPA; altra caratteristica è la permeabilità agli ioni Na+, K+ e Ca2+ . Il “blocco da magnesio” è un meccanismo importante nei recettori NMDA, possiamo considerarlo un meccanismo di “sicurezza” che modula il flusso degli ioni Ca2+ ed evita che elevati livelli di tale ione entri nella cellula nervosa e ne possa provocare la morte per danno eccitotossico. Lo pseudodominio M2 nel sito Q/R determina la permeabilità al calcio, in tale regione è espressa l’asparagina che rende possibile il blocco del magnesio, anche se è presente il fenomeno che va sotto il nome di flickering che può garantire un minimo passaggio di ioni. Vicino al recettore NMDA c’è il recettore AMPA, non soggetto al blocco da magnesio, la stimolazione glutammatergica agisce su tutti e due i recettori, ma la depolarizzazione si ha solo nel canale AMPA perché il canale NMDA è sottoposto al blocco del magnesio. Il blocco del recettore NMDA viene rimosso dalla depolarizzazione indotta dal recettore NMDA dando origine a una sorta di “cooperazione recettoriale” che è importante per i processi di memorizzazione. Si usa il termine “coincidence detector” per definire il recettore NMDA che ha la capacità di depolarizzare la membrana postsinaptica e rilasciare il neurotrasmettitore, questa capacità è utile nei processi di apprendimento e memorizzazione perché il blocco da magnesio è vicariato dal recettore AMPA che depolarizza la cellula nervosa che a quel punto è in grado di rimuovere il blocco da magnesio. Anche i recettori metabotropici per il glutammato accoppiati a proteine G ( mGlu ) sono ben rappresentati nel SNC anch’essi svolgono funzioni eccitatorie e inibitorie, si caratterizzano pper avere una struttura a 7 domini transmembrana, l’N-terminale extracellulare ed è la “binding region”; la parte C-terminale intracellulare con il terzo loop intracellulare forma il sito di legame della proteina G. Sono tre i gruppi dei recettori metabotropici per il glutammato accoppiati a proteine G trimeriche, gruppo 1 mGlu1 e mGlu5 gruppo 2 mGluR2 e mGluR3 gruppo 3 mGluR4, mGluR6, mGluR7 e mGluR8. La protratta attivazione di una sinapsi glutamatergica ha effetti tossici sul neurone fino a causarne la morte, è la cosidetta eccitotossicità da glutammato, si ha per lo più dall’abnorme attivazione dei recettori NMDA. Si ha un’alterazione del metabolismo cellulare e l’eccesso degli ioni Ca2+ attivano il metabolismo degradativo, gli enzimi coinvolti sono: proteasi, peptidasi, fosfolipasi ed endonucleasi, c’è un aumento dei ROS di derivazione mitocondriale. Entra in gioco la neuronal Nitric Oxide Synthase (nNOS) che attiva la sintesi del monossido d’azoto che essendo un messaggero chimico agisce sui terminali presinaptici aumentandone la trasmissione, il monossido d’azoto reagendo con i ROS produce perossinitriti che sono deleteri per la respirazione cellulare. Se questo circolo vizioso non viene antagonizzato conduce i neuroni a morte. Le possibili correlazioni tra la neurotrasmissione glutammatergica e la SLA Diverse mutazioni sono state osservate nei pazienti affetti da SLA, le più studiate sono quelle a carico del gene SOD1 (superossido dismutasi Cu/Zn) Alcune mutazioni riducono l’attività dell’enzima come A4V e G85R), altre la lasciano inalterata G37R e G93A I motoneuroni sono vulnerabili ad alti livelli di glutammato, infatti, un ipertono glutammatergico cronico sembra avere un ruolo importante nella neurodegenerazione di tipo eccitotossico, La degenerazione dei motoneuroni che si osserva nella SLA, è il risultato della sommatoria di più meccanismi come : stress ossidativo, eccitotossicità, problemi che riguardano il citoscheletro e disfunzione mitocondriale, ma non sono ancora del tutto chiari i meccanismi. I motoneuroni subiscono un danno di tipo eccitotossico in seguito all’esposizione prolungata al glutammato o a neurotossine, ne consegue stress ossidativo con produzione di radicali ossidrilici e perossinitriti in grado di danneggiare strutture cellulari nobili come DNA, membrana cellulare e proteine. In patologie dove si rinviene elevato stress ossidativo sono rinvenibili elevati livelli di 3nitrotirosina, sostanza che proviene dalla nitrazione della tirosina considerata un marker di stress ossidativo dipendente da NO. http://www.eurekaone.com/attachments/258_z61010.pdf Un farmaco che la FDA ha approvato per la SLA (il riluzolo,) un benzotiazolo ha la funzione di ridurre il rilascio di glutammato attraverso il blocco dei canali del sodio, sebbene il farmaco sia poco efficace in termini di aspettativa di vita, serve a farci capire il suo meccanismo d’azione. L’EAAT2 neurotrasportatore gliale per il glutammato viene attaccato dai ROS si ha un ipertono glutammatergico, perché viene a mancare un meccanismo di regolazione della durata della trasmissione glutammatergica, il danno all’EAAT2 è confinato nella zone interessate dal fenomeno e coinvolge solo gli astrociti viciniori. Quando il glutammato interagisce con i recettori AMPA privi di GluR2 innesca uno stress ossidativo, in questo caso è necessario l’influsso di calcio nei motoneuroni ma è inibito da bloccanti della catena di trasporto degli elettroni e questo ci fa pensare a un cinvolgimento dei mitocondri, perché è il calcio entrato nei motoneuroni che innesca la produzione dei ROS. I ROS oltrepassano la membrana cellulare e vanno nella matrice extracellulare e danneggiano gli astrociti molto facilmente, in vitro si è osservato che l astrociti trattati con SOD non vanno incontro a danneggiamento. Per concludere, vi presento brevemente uno studio tutto italiano del 2011, pubblicato su Neurobiology of Disease e concernente l’azione neuroprotettiva di un farmaco antagonista dei recettori metabotropici mGlu5. Si tratta di una ricerca molto interessante i cui risultati potrebbero promettere lo sviluppo di una nuova strategia terapeutica contro la SLA. Bibliografia: Shin Kwak, Takuto Hideyama, Takenari Yamashita and Hitoshi Aizawa. 2010: AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology, 30, 182–188. Shyam D. Rao, Hong Z. Yin, and John H. Weiss. 2003: Disruption of Glial Glutamate Transport by Reactive Oxygen Species Produced in Motor Neurons. The Journal of Neuroscience, 23(7):2627–2633. Shyam D. Rao and John H. Weiss. 2004: Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. TRENDS in Neurosciences, Vol.27 No.1. Siân C. Barber, Pamela J. Shaw. 2010: Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radical Biology & Medicine 48: 629–641. Simona D’Antoni, Antonio Berretta, Giovanna Seminara, Patrizia Longone, Anna Maria Giuffrida-Stella, Giuseppe Battaglia, Maria A. Sortino, Ferdinando Nicoletti, Maria V. Catania. 2011: A prolonged pharmacological blockade of type-5 metabotropic glutamate receptors protects cultured spinal cord motor neurons against excitotoxic death. Neurobiology of Disease 42: 252–264 Potrebbero interessarti A tre biologi il premio Nobel 2013 per la Fisiologia o Medicina - ott 7, 2013 La neurotrasmissione glutammatergica 5/ Eccitotossicità nella patogenesi della SLA - giu 15, 2012 La neurotrasmissione glutammatergica 4/ Ruolo dello ione zinco - apr 5, 2012 La neurotrasmissione glutammatergica 3/ Recettori metabotropici ed eccitotossicità - mar 11, 2012 La neurotrasmissione glutammatergica 2/ I recettori ionotropici - feb 4, 2012 http://www.inbiochem.com/la-neurotrasmissione-glutamatergica-prima-parte/ A loss of astrocytes can result in not only a massive release of stored glutamate upon astrocyte cell death, but also a long-term loss of one of the brain's major regulators of extracellular glutamate. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3784951/#ref49" \t "_top ///////
Scaricare