Indice
Chimica
1
Storia della chimica
14
Legame chimico
27
Atomo
32
Molecola
39
Stato della materia
56
Miscela (chimica)
58
Composto chimico
61
Reazione chimica
63
Processo chimico
68
Ossido-riduzione
70
Reazione acido-base
70
Decomposizione (chimica)
73
Metatesi (chimica)
74
Precipitazione (chimica)
75
Complesso (chimica)
76
Reazione organica
83
Reagente
86
Catalizzatore
87
Solubilità
91
Concentrazione
94
Frazione molare
96
Numero di Avogadro
97
Mole
101
Chimica inorganica
104
Chimica organica
109
Sale
112
Ossido
114
Chimica fisica
117
Biochimica
119
Numero atomico
126
Peso atomico
126
Acido
128
Base (chimica)
132
Radicale libero
135
Metallo
142
Non metallo
147
Semimetalli
148
Gas nobili
148
Formula chimica
149
Legame covalente
152
Gas
154
Soluzione (chimica)
157
Dispersione (chimica)
161
Elettrone
162
Isotopo
184
Idrolisi
186
Attinoidi
188
Lantanoidi
190
Stato di ossidazione
191
Equazione chimica
193
Prodotto (chimica)
194
Note
Fonti e autori delle voci
195
Fonti, licenze e autori delle immagini
198
Licenze della voce
Licenza
201
Chimica
1
Chimica
Bottiglie contenenti sostanze ottenute attraverso processi chimici
« Nulla si crea, nulla si distrugge, ma tutto si trasforma. »
[1]
(Antoine-Laurent de Lavoisier )
La chimica (dall'arabo "al kimiaa", e da qui la parola alchimista )ﺍﻟﻜﻴﻤﻴﺎءè la scienza o più precisamente quella branca
delle scienze naturali, che interpreta e razionalizza la struttura, le proprietà e le trasformazioni della materia.
La chimica ha interessato, anche per motivi pratici derivanti dalle sue applicazioni tecnologiche, le varie popolazioni
dell'umanità fin dai tempi antichi. Dal II secolo a.C. si sviluppò, a partire dall'Egitto tolemaico, l'alchimia, un
insieme di conoscenze sulla materia e le sue trasformazioni legate a convinzioni filosofiche ed esoteriche; da essa
derivò la chimica moderna (in seguito alla rivoluzione scientifica, e più precisamente alla rivoluzione chimica alla
fine del XVIII secolo). Anche nel periodo seguente la chimica continuò ad evolversi, perché sempre nuove scoperte
ne ampliarono i campi di interesse e i metodi impiegati.
Oggetto di studio della chimica sono le proprietà e le strutture dei costituenti della materia (atomi, molecole, cristalli
e altri aggregati) e le loro interazioni reciproche, da cui hanno origine gli stati della materia.
Tale studio della materia non è limitato alle sue proprietà e struttura in un dato istante, ma riguarda anche le sue
trasformazioni, dette reazioni chimiche.[2]
Sono studiati anche gli effetti di tali proprietà e interazioni tra i componenti della materia su quelle degli oggetti e
della materia con cui comunemente abbiamo a che fare, e le relazioni tra di essi, il che determina un'ampia
importanza pratica di tali studi. Si tratta quindi di un campo di studi molto vasto, i cui settori sono tradizionalmente
suddivisi in base al tipo di materia di cui si occupano o al tipo di studio.
La conoscenza della struttura elettronica degli atomi è alla base della chimica convenzionale, mentre la conoscenza
della struttura del nucleo atomico e delle sue trasformazioni spontanee ed indotte è alla base della chimica nucleare.
La rottura e la formazione dei legami tra gli atomi e le molecole sono responsabili della trasformazione della materia.
La chimica è anche stata definita come "la scienza centrale" (in inglese central science) perché connette le altre
scienze naturali, come l'astronomia, la fisica, le scienze dei materiali, la biologia e la geologia.[3][4]
Chimica
Storia della chimica
Due erano le principali scuole di pensiero della
filosofia naturale elaborata dai Greci: Democrito
sosteneva che la natura fosse formata da corpuscoli
indivisibili (gli atomi) che si uniscono e separano in
uno spazio vuoto, mentre Aristotele ipotizzava la
struttura continua della materia risultante dalla
combinazione degli elementi acqua, aria, terra e fuoco.
Tra il II e V secolo d.C. si sviluppa ad Alessandria
d'Egitto l'alchimia, che conservava le origini filosofiche
unite a una forte connotazione esoterica. In questo
contesto l'alchimista, o "mago naturale", si poneva
come tramite tra macrocosmo e microcosmo, divino e
Laboratorio alchemico (illustrazione di Pieter Bruegel il Vecchio).
umano. Due erano gli obiettivi fondamentali degli
alchimisti, da realizzare con l'ausilio della pietra filosofale: la trasmutazione dei metalli in oro, che corrispondeva
anche all'elevazione verso la perfezione delle qualità spirituali umane, e la possibilità di curare ogni genere di
malattia e creare la vita. Nel XVI secolo assumeva autonomia propria la branca definita iatrochimica, che ebbe i
maggiori contributori in Paracelso e Jean Baptiste van Helmont e che si prefissava di correlare i processi chimici che
avvengono all'interno dell'organismo umano con gli stati patologici e con i possibili rimedi.
Le basi per lo sviluppo della chimica moderna si pongono nel XVII secolo, con la prima definizione delle reazioni
chimiche (nel Tyrocinium Chymicum di Jean Béguin) e il graduale sviluppo del metodo sperimentale, grazie a diversi
scienziati tra i quali spicca Robert Boyle. Lo spartiacque simbolico tra alchimia e chimica può essere considerato
l'anno 1661, con l'uscita del libro di Boyle Il chimico scettico (The Sceptical Chymist), in cui vengono introdotti i
concetti di elemento chimico e composto chimico.[5]
Successivamente il lavoro di Antoine Lavoisier, che enunciò per primo la legge della conservazione della massa e
confutò la teoria del flogisto, segnò il definitivo superamento dell'alchimia. Nel 1807 Jöns Jacob Berzelius fu uno dei
primi a utilizzare il termine "chimica organica" in riferimento alla chimica che caratterizzava i composti prodotti dal
regno animale, contrapposti a quelli di origine minerale e di pertinenza della chimica inorganica; sarà Friedrich
Wöhler nel 1828 a dimostrare che i composti organici possono essere ottenuti anche da sintesi in laboratorio,
riuscendo a sintetizzare l'urea a partire da sostanze inorganiche.
Nel 1869 Dmitrij Mendeleev e Julius Lothar Meyer ordinarono gli elementi chimici sistemandoli all'interno della
tavola periodica, disposti ordinatamente in base al loro peso atomico. Nel 1937 l'italiano Emilio Segrè scoprì il
tecnezio, primo elemento chimico artificiale, e negli anni seguenti verranno sintetizzati artificialmente molti altri
nuovi elementi che andranno ad arricchire la tavola periodica.
2
Chimica
3
Concetti base
Atomi e molecole
La materia è formata da particelle elementari, chiamate
atomi: in natura ne esistono un centinaio di tipi, e
ognuno di essi ha struttura e proprietà differenti.
Quando gli atomi si combinano fra loro si generano
delle molecole. Queste ultime possono essere costituite
da atomi tutti uguali fra loro, formando quelle che
vengono definite le sostanze semplici (ad esempio N2,
O2 e S8), mentre le molecole costituite da atomi diversi
sono caratteristiche delle sostanze composte (ad
esempio H2O, C12H22O11 e H2SO4).
Per indicare la quantità di sostanza si fa uso della
"mole". Una mole di sostanza risulta costituita da un
numero di Avogadro (6,022 x 1023) di atomi o
molecole. Considerando che una mole di acqua pesa
circa 18 grammi, è facile intuire che la materia che ci
circonda è costituita da un enorme numero di particelle
elementari.
Una singolare forma molecolare del carbonio: il fullerene
I legami chimici e le forze di attrazione
intermolecolare
Gli atomi possono legarsi fra loro, e la forza di natura
elettrostatica che li unisce viene definita legame
chimico. Tale legame, caratterizzato da intensità
differente in relazione al composto a cui dà origine, è
fondamentale nel conferire la particolare reattività e
stabilità del composto stesso, nonché nel determinarne
la struttura e geometria molecolare caratteristica.
Esistono poi forze intermolecolari, di minore intensità
rispetto al legame chimico, che attraggono atomi e
molecole fra di loro. Tali forze originano quello che
viene comunemente definito legame chimico
secondario e hanno un ruolo importante nel
determinare lo stato fisico di una sostanza. Sono inoltre
responsabili anche della struttura secondaria, terziaria e
quaternaria delle proteine.
Cristalli di solfato di rame(II)
Stati e aggregazione della materia
I composti chimici possono presentarsi in diversi stati di aggregazione, tra cui solido, liquido, aeriforme (vapore o
gas) ed infine plasma.
La temperatura di un corpo è direttamente legata al movimento microscopico (o meglio all'energia cinetica
microscopica)[6] delle particelle elementari (molecole): in particolare a bassa temperatura le molecole sono attratte
Chimica
4
fra loro tramite legami più energetici, per cui l'unico moto a cui possono essere sottoposte è quello vibrazionale; lo
stato della materia associato a questa condizione è lo stato solido.
All'aumentare della temperatura, le molecole acquistano energia in quanto sono legate da legami meno energetici,
per cui hanno la capacità di esprimere tre tipologie di moto: traslazionale, rotazionale e vibrazionale; lo stato della
materia associato a questa condizione è lo stato liquido.
Un ulteriore aumento di temperatura indebolisce ulteriormente i legami che intercorrono tra le molecole, per cui
aumentano ulteriormente le distanze tra le molecole e quindi il volume occupato dall'intero sistema;[7] lo stato della
materia associato a questa condizione è lo stato di aeriforme.
Infine, ionizzando un gas, otteniamo il plasma, che si ritiene costituisca il 99% della materia nell'Universo.
Si parla inoltre di "fase" per indicare una porzione omogenea di un sistema termodinamico. A seconda dello stato di
aggregazione, si parla di "fase solida", "fase liquida" o "fase aeriforme". I concetti di "fase" e "stato di aggregazione"
non vanno confusi: infatti un sistema può essere in un determinato stato di aggregazione ma presentare più fasi. Un
esempio è dato dai liquidi immiscibili (come acqua e olio), che condividono lo stesso stato di aggregazione (cioè
liquido) ma sono pertinenti a due fasi distinte (infatti l'olio se versato in un contenitore contenente acqua forma uno
strato sulla superficie del liquido, diviso in maniera netta dall'acqua sottostante).
Un sistema composto da una singola fase è quindi omogeneo, mentre un sistema composto da più fasi è eterogeneo.
I composti chimici e le miscele
Quando gli atomi si legano fra loro in proporzioni definite e costanti si ottengono dei composti chimici (ad esempio
l'acqua, H2O). I composti, oltre ad avere composizione chimica differente rispetto alle sostanze originarie che li
hanno prodotti, hanno anche differenti proprietà chimiche e fisiche rispetto a tali sostanze.
I sistemi formati da più composti chimici sono detti miscele,[8] e possono essere a loro volta omogenei o eterogenei.
Un particolare tipo di miscela omogenea sono le soluzioni, formate da un solvente (composto presente in quantità
maggiore) e da uno o più soluti (composto presente in quantità minore).
Reazioni chimiche
Una reazione chimica è un processo chimico tramite il
quale atomi, ioni o molecole che costituiscono le
sostanze iniziali (chiamate reagenti) si combinano fra
loro originando le sostanze finali (chiamate prodotti).
La composizione e le proprietà chimico-fisiche dei
prodotti sono differenti rispetto ai reagenti.
I reagenti prendono parte alla reazione secondo rapporti
in massa ben stabiliti, in base al loro coefficiente
stechiometrico; la stechiometria di reazione permette di
calcolare il quantitativo teorico di prodotti ottenibili.[9]
Una reazione che avviene producendo calore viene
detta esotermica, mentre una reazione che avviene
assorbendo calore dall'ambiente esterno viene detta
endotermica.
Mentre la termochimica permette di stabilire se una
data reazione può avvenire spontaneamente in
Reazione chimica tra acido cloridrico e ammoniaca, con produzione
di cloruro di ammonio.
Chimica
5
determinate condizioni, la cinetica chimica si occupa di analizzare il meccanismo di reazione e di determinare se una
data reazione chimica possa procedere con una velocità di reazione accettabile. Molte reazioni spontanee non
avrebbero luogo senza la presenza di un catalizzatore, proprio perché presenterebbero altrimenti una velocità
bassissima. La presenza del catalizzatore è necessaria a superare un "muro" energetico che impedisce alla reazione di
avvenire. Una volta che la reazione è iniziata, può in certi casi "autoalimentarsi", per cui la presenza del catalizzatore
non è più necessaria da un certo momento in poi. Un meccanismo simile avviene nelle reazioni di combustione:
queste infatti hanno bisogno di un innesco iniziale per avere luogo (ad esempio una scintilla), ma una volta che la
combustione ha avuto origine, si ha produzione di calore che autoalimenta la reazione stessa.
Alcuni esempi di reazioni chimiche sono:
• Ossido-riduzioni
• Es. K2Cr2O7 + 6 FeSO4 + 7 H2SO4 → Cr2(SO4)3 + K2SO4 + 3 Fe2(SO4)3 + 7 H2O
• Reazioni acido-base
• Es. NaOH + HCl → NaCl + H2O
• Decomposizione
• Es. CaCO3 → CaO + CO2
• Doppio scambio
• Es. KCl + NH4NO3 → KNO3 + NH4Cl
• Precipitazione
• Es. AgNO3 + NaCl → NaNO3 + AgCl↓
• Complessazione
• Es. CuCl2 + NH3 → [Cu(NH3)4]Cl2
• Reazioni organiche
• Es. l'acetilazione dell'acido salicilico con anidride acetica a formare acido acetilsalicilico e acido acetico:
C7H6O3 + C4H6O3 → C9H8O4 + C2H4O2
La freccia verso destra (→) sta indicare il verso in cui la reazione avviene. In questo caso bisogna anche specificare
le condizioni in cui si opera (tra cui temperatura e pressione), in quanto la reazione inversa (cioè da destra verso
sinistra) può essere favorita per talune condizioni. Nel caso più generale, i reagenti (primo membro) e i prodotti
(secondo membro) sono separati dal segno "=".
Il simbolo della freccia verso il basso (↓) indica una sostanza che precipita come corpo di fondo. La precipitazione
però non avviene se le condizioni in cui si opera sono tali che da rendere la solubilità del prodotto nella soluzione
molto elevata. Nella notazione chimica si utilizza talvolta anche il simbolo di una freccia verso l'alto (↑), ad indicare
che il prodotto è gassoso alle condizioni in cui avviene la reazione.
Equilibrio chimico
L'equilibrio chimico è una condizione di equilibrio dinamico che si ha quando i prodotti di una reazione chimica
reagiscono a loro volta fra loro riformando i reagenti di partenza.
Una reazione di equilibrio viene indicata utilizzando le doppie frecce che puntano in verso opposto (
utilizzare la classica freccia che punta dai reagenti verso i prodotti. Un esempio è il seguente:
), invece di
In teoria tutte le reazioni chimiche possono essere considerate di equilibrio, ma nella pratica comune quelle
caratterizzate da valore di costante di equilibrio molto alta sono considerate reazioni "a completamento" (cioè che
avvengono verso una sola direzione). La costante d'equilibrio K è definita dal rapporto dell'operazione di
moltiplicazione delle concentrazioni delle sostanze prodotte, ognuna elevata al proprio coefficiente stechiometrico,
rispetto all'operazione di moltiplicazione delle concentrazioni delle sostanze reagenti, ognuna elevata al proprio
Chimica
6
coefficiente stechiometrico. Considerando l'esempio precedente di due reagenti e due prodotti, vale la relazione:
La costante di equilibrio K è una costante in condizioni di temperatura costante (e pressione costante, nel caso dei
gas). La costante di equilibrio può essere espressa anche in termini di rapporti tra pressioni parziali o anche frazioni
molari.
Leggi della chimica e della fisica
Tutte le reazioni chimiche e le trasformazioni fisiche
avvengono secondo leggi chimico-fisiche. Di seguito viene
presentato un elenco degli enunciati di alcune leggi di
particolare importanza nell'ambito della chimica.
• Leggi sui gas
• Legge dei gas perfetti: mette in relazione fra loro le
funzioni di stato quantità di sostanza, pressione,
volume e temperatura di un gas perfetto.
• Legge delle pressioni parziali: la pressione totale
esercitata da una miscela ideale di gas ideali è uguale
alla somma delle pressioni parziali che sarebbero
esercitate dai gas se fossero presenti da soli in un
eguale volume.
Animazione che spiega la legge di Boyle-Mariotte
• Legge di Boyle-Mariotte: in condizioni di temperatura
costante la pressione di un gas è inversamente
proporzionale al suo volume.
• Prima legge di Gay-Lussac: in condizioni di pressione
costante il volume di un gas aumenta linearmente
all'aumentare della temperatura.
• Seconda legge di Gay-Lussac: in condizioni di
volume costante la pressione di un gas aumenta
linearmente all'aumentare della temperatura.
• Legge di van der Waals: mette in relazione fra loro le
funzioni di stato quantità di sostanza, pressione,
volume e temperatura di un gas reale.
Animazione che spiega la prima legge di Gay-Lussac
• Legge di Henry: a temperatura costante, la quantità di gas che passa in soluzione in un determinato liquido è
direttamente proporzionale alla pressione parziale del gas in equilibrio col liquido stesso.
• Legge di conservazione della massa: in una reazione chimica, la massa dei reagenti è esattamente uguale alla
massa dei prodotti.
• Legge delle proporzioni definite: in un determinato composto chimico allo stato puro gli elementi che lo formano
stanno fra loro in proporzioni di massa definite e costanti.
• Legge delle proporzioni multiple: quando due elementi si combinano in modi diversi per formare diversi
composti, una certa massa di un elemento si combina con masse dell'altro che stanno tra loro in un rapporto che si
può esprimere con frazioni semplici.
• Legge dell'azione di massa: la velocità di una reazione chimica è proporzionale alla concentrazione delle sostanze
che vi partecipano.
Chimica
7
• Principio di Le Chatelier: ogni sistema in equilibrio tende a reagire ad una modifica impostagli dall'esterno
minimizzandone gli effetti.
• Legge di Raoult: mette in relazione la pressione di vapore di un liquido in soluzione con la sua pressione di
vapore allo stato puro e la sua concentrazione in termini di frazione molare.
• Legge di Hess: la variazione di entalpia di una reazione chimica è indipendente dal percorso intermedio attraverso
il quale dai reagenti si ottengono i prodotti.
• Legge di Debye-Hückel: definisce il coefficiente di attività medio delle soluzioni di elettroliti.
• Legge dell'indipendente mobilità degli ioni: la conduttanza equivalente di una soluzione di un elettrolita a
diluizione infinita è uguale alla somma della mobilità del catione e dell'anione dai quali è formato l'elettrolita,
mobilità che a diluizione infinita non si influenzano reciprocamente.
• Legge di Lambert-Beer: mette in relazione la quantità di luce assorbita da una sostanza con la sua natura chimica,
la sua concentrazione e lo spessore del mezzo attraversato.
Meccanica quantistica
La meccanica quantistica è stato il settore della chimica fisica (una disciplina di confine con la fisica) che ha dato
maggior impulso allo sviluppo della chimica moderna, spiegando la struttura e le caratteristiche degli atomi e
creando i presupposti basilari per la trattazione matematica del legame chimico.
Lo spunto iniziale fu dato da De Broglie che nel 1924 ipotizzò la possibilità di associare a una particella in
movimento quale l'elettrone un'onda di lunghezza d'onda ricavabile dalla relazione
dove
rappresenta la costante di Planck mentre il prodotto
è la quantità di moto. Quindi, secondo De Broglie,
una particella in movimento ha una doppia natura corpuscolo-ondulatoria e tanto minore è la massa tanto maggiore
risulterà la lunghezza d'onda dell'onda associata alla massa stessa: a titolo di esempio per un elettrone (massa 9 x
10-31 kg e velocità di rotazione attorno al nucleo di 2 x 106 m/s) si ricava una = 3,68 Å, mentre a un pallone del
peso di 500 g che si muove a velocità di 30 m/s corrisponde un'onda con
= 4,4 x 10-35 m.
Nel 1926 Erwin Schrödinger, basandosi sulla teoria di De Broglie, descrisse un'equazione (l'equazione di
Schrödinger, appunto) che rappresenta la propagazione dell'onda materiale tridimensionale associata a un elettrone
che orbita attorno al nucleo di un atomo idrogenoide. Le soluzioni matematiche di questa equazione costituiscono la
funzione d'onda; sono fisicamente accettabili tutte quelle funzioni d'onda i cui numeri quantici (n, l, m) che le
caratterizzano sottostanno alle regole di quantizzazione dettate dalla meccanica quantistica. L'orbitale è formalmente
definito come la proiezione della funzione d'onda sulla base della posizione, ovvero rappresenta la componente
spaziale della funzione d'onda. In accordo col principio di indeterminazione di Heisenberg, non è possibile conoscere
contemporaneamente con la medesima accuratezza la posizione è la quantità di moto dell'elettrone.
Approssimativamente, l'orbitale viene considerato come regione dello spazio in cui è massima la probabilità (90%)
di rinvenire l'elettrone. Acquisendo o emettendo un quanto di energia l'elettrone è suscettibile di passare a livelli
energetici rispettivamente maggiori o minori.
Discipline fondamentali della chimica
Chimica inorganica
La chimica inorganica si occupa dello studio dei composti inorganici, ovvero dei composti non formati da atomi di
carbonio (anche se in realtà una ristretta classe di composti del carbonio sono considerati inorganici).[10] Essa tratta
lo studio del legame chimico e della simmetria delle molecole; si sofferma sulla caratterizzazione strutturale ed
energetica dei solidi cristallini e di quelli metallici. In modo sistematico viene descritta la chimica degli elementi,
raggruppando gli elementi chimici in base ai gruppi della tavola periodica. Vengono studiate le reazioni di
Chimica
ossido-riduzione, acido-base e la sintesi e caratterizzazione dei composti di coordinazione e dei composti
metallorganici (contenenti un legame metallo-carbonio). Infine la chimica bioinorganica si occupa del ruolo degli
elementi metallici nei processi vitali.
Chimica organica
La chimica organica studia i composti del carbonio. La sistematica raggruppa le classi di composti organici in base
alla presenza di determinati gruppi funzionali, studiandone le proprietà chimico-fisiche, le metodologie di sintesi e le
reazioni caratteristiche. La stereochimica e i meccanismi di reazione sono un ambito di studio fondamentale in
chimica organica. Nell'ambito di questa disciplina rientrano anche i composti aromatici, composti ciclici dotati di
particolare stabilità, e biomolecole quali carboidrati, amminoacidi, proteine, lipidi e acidi nucleici. I polimeri
organici sono una variegata classe di composti di elevato interesse industriale e con diverse applicazioni pratiche. I
metodi fisici applicati alla chimica organica (NMR, spettroscopia IR, spettrometria di massa, spettroscopia UV)
consentono il riconoscimento dei principali gruppi funzionali e della struttura molecolare.
Chimica fisica
La chimica fisica si propone di studiare e descrivere le reazioni e i fenomeni chimici utilizzando le metodologie e gli
strumenti propri della fisica. Vengono studiate le fasi della materia e le transizioni di fase, ponendo enfasi sulle leggi
che governano lo stato gassoso, sulla struttura dei solidi cristallini e sui diagrammi di fase. La termodinamica viene
affrontata in modo dettagliato così come le sue implicazioni nell'ambito delle reazioni chimiche (termochimica),
arrivando a stabilire la spontaneità o meno di una reazione in base al calcolo dell'energia libera di Gibbs di reazione.
Analogamente vengono analizzati i fattori in grado di influenzare l'equilibrio chimico e la termodinamica di miscele
e soluzioni. Partendo dalle basi della meccanica quantistica, si giunge a descrivere il legame chimico in modo
rigoroso su basi matematiche. Appositi modelli risultano utili nello studio del potenziale dovuto alle interazioni
intermolecolari (legami chimici secondari). Dalla struttura atomica si passa alla struttura molecolare, determinata
applicando l'approssimazione di Born-Oppenheimer. La spettroscopia e le varie tecniche spettroscopiche vengono
trattate evidenziandone i fondamenti fisici, piuttosto che le applicazioni pratiche. Altro campo di studio della chimica
fisica è rappresentato dai fenomeni di trasporto. L'elettrochimica si occupa dello studio dell'interconversione tra
energia chimica ed energia elettrica e di tutto ciò che ne viene implicato. La cinetica chimica si occupa del calcolo
della velocità di reazione e della formulazione dei singoli processi elementari di cui si compone una reazione
(meccanismi di reazione), mentre la dinamica molecolare applica i principi della dinamica ai sistemi atomici e
molecolari. Infine la fotochimica studia l'influenza della luce sulla reattività chimica.
Chimica analitica
La chimica analitica applica un insieme di tecniche, strumentali e non, allo scopo di riconoscere e quantificare un
dato analita. Nello specifico l'analisi qualitativa si occupa del riconoscimento della sostanza oggetto di indagine,
mentre l'analisi quantitativa determina la quantità di sostanza presente in un dato campione. In passato l'analisi
qualitativa era condotta manualmente in modo sistematico, sfruttando opportuni reattivi; oggigiorno le tecniche
strumentali quali quelle spettroscopiche hanno soppiantato tale approccio sistematico e puramente manuale da parte
dell'analista. Nell'ambito dell'analisi quantitativa invece convivono tecniche puramente affidate all'operatore, quali le
classiche titolazioni, con svariate tecniche strumentali automatizzate. Quest'ultime, come già detto, possono più
comunemente essere spettroscopiche, cromatografiche, elettroanalitiche, o termiche (come l'analisi termica
differenziale, la calorimetria differenziale a scansione, la termogravimetria). Occorre sottolineare che la chimica
analitica si occupa anche della corretta elaborazione statistica del dato analitico, nonché della qualità e affidabilità di
tale dato.
8
Chimica
9
Biochimica
La biochimica studia i composti e i processi chimici che contraddistinguono gli organismi viventi. Essa si occupa
della biosintesi delle biomolecole, del loro ruolo e funzionalità biologica: acidi nucleici e informazione genetica,
proteine, lipidi e carboidrati. Studia inoltre gli enzimi e la catalisi enzimatica, fino a giungere alla cinetica di
Michaelis-Menten. La biochimica si concentra sugli aspetti chimici del metabolismo, del trasporto di ossigeno
tramite emoglobina e mioglobina, della respirazione cellulare, della fotosintesi clorofilliana, dell'omeostasi e della
trasduzione del segnale all'interno delle cellule. I canali di membrana e le pompe ioniche consentono il passaggio di
ioni e molecole attraverso la membrana cellulare. La biosintesi degli anticorpi e la loro interazione con l'antigene ha
un ruolo fondamentale nell'ambito della risposta immunitaria.
Altre discipline
Esistono numerosissime specializzazioni e discipline della chimica, che possono essere considerate parte delle
discipline fondamentali e spesso anche parte di altre discipline scientifiche affini; ad esempio: la chimica
farmaceutica, la chimica industriale, la chimica dei polimeri e delle macromolecole, la chimica degli alimenti, la
chimica dello stato solido e delle superfici, l'astrochimica, la cosmochimica, l'elettrochimica, la geochimica, la
chimica teorica, la citochimica, l'istochimica, la chimica clinica, la chimica nucleare, la radiochimica, la chimica
delle radiazioni, la chimica metallorganica, la stereochimica, la chimica ambientale, la chimica verde, la fotochimica,
la sonochimica, la chimica del suolo, la chimica dell'atmosfera, la chimica radiofarmaceutica, l'aerotermochimica, la
chimica del restauro, la chimica dei beni culturali, la strutturistica chimica, la magnetochimica, la chimica
quantistica, la femtochimica, la chimica dei colloidi, la chimica delle interfasi, la chimica combinatoria, la chimica
computazionale, la chimica matematica, la chemioinformatica, la chemiometria, la chimica dei materiali, la
merceologia.
Applicazioni della chimica
Chimica e industria
La chimica industriale si occupa della sintesi su vasca scala di
prodotti chimici destinati a vari utilizzi, ottimizzando il ropporto
costi/benefici dell'intero ciclo produttivo chimico. In particolare,
disponendo delle opportune materie prime, tramite un insieme di
processi realizzati all'interno di un impianto chimico, si giunge a
ottenere semilavorati o prodotti finiti in grado di soddisfare le
specifiche e i requisiti tecnici richiesti per il loro utilizzo pratico.
A titolo di esempio, per indicare alcuni dei processi chimici
industriali più noti, si cita il processo Haber-Bosch per la sintesi
dell'ammoniaca e il processo Ostwald per la sintesi dell'acido
nitrico. L'industria petrolchimica e dei polimeri sintetici è un
altro vasto campo molto attivo.
Impianto di distillazione a doppio effetto
Chimica
10
Chimica e medicina
La chimica farmaceutica costituisce il campo di ricerca per la sintesi e
applicazione terapeutica dei nuovi farmaci. Pone le sue basi sullo
studio teorico delle proprietà chimico-fisiche delle molecole e sui
modelli di interazione farmacologica con l'organismo. Si giunge quindi
a formulare una conveniente strategia di sintesi, sfruttando anche
l'approccio della chimica combinatoria, e il nuovo farmaco ottenuto
può iniziare la fase di sperimentazione che se culminerà con esito
positivo potrà permettergli l'immissione sul mercato. Oltre questi
aspetti farmacologici, la chimica risulta un utile ausilio in medicina
diagnostica grazie alla possibilità di effettuare appositi esami chimico
clinici di laboratorio. Isotopi radioattivi vengono utilizzati in medicina
nucleare.
Il principio attivo di un farmaco rappresenta la
molecola che possiede attività biologica
Chimica e tecnologia
L'utilizzo di tecniche chimiche e chimico-fisiche consente in scienza dei materiali di studiare e caratterizzare la
struttura e le proprietà dei materiali, permettendo in questo modo di assicurare l'adeguatezza agli standard di utilizzo,
di sviluppare nuovi materiali o di migliorare quelli già esistenti. La chimica dei polimeri concentra la propria attività
in particolare sui meccanismi di polimerizzazione e sulla relazione esistente tra la struttura e le caratteristiche proprie
dei polimeri. La galvanostegia e la fosfatazione sono degli esempi di processi chimici utilizzati per la protezione
dalla corrosione, così come possono citarsi apposite vernici e rivestimenti in grado di conferire particolari peculiarità
ai materiali. La chimica dello stato solido, tra gli altri campi applicativi, è impegnata attivamente nella sintesi di
semiconduttori innovativi destinati a diverse applicazioni tecnologiche. Lo sviluppo della chimica supramolecolare
riveste un ruolo fondamentale per le nanotecnologie, consentendo la sintesi di dispositivi molecolari come le
nanomacchine.
Chimica e ambiente
La crescente sensibilità verso un basso impatto ambientale e la necessità di applicare politiche di sviluppo sostenibile
hanno condotto alla nascita della cosiddetta chimica verde. Questa disciplina si propone di ridurre l'impatto dei
processi chimici mettendo in pratica concetti quali l'utilizzo di materie prime ricavate da fonti rinnovabili, la
riduzione di reflui e scarti, l'utilizzo di composti biosostenibili ed ecosostenibili. D'altra parte la chimica ambientale è
focalizzata sullo studio del chimismo e biochimismo implicato nell'ambito ambientale: si interessa della chimica
delle acque dolci e marine, della chimica del suolo e dell'atmosfera. Non si limita a comprendere i fondamenti
chimici, ma estende il proprio campo di studio e ricerca ai fenomeni legati all'inquinamento e all'effetto dei tossici
rilasciati in ambiente proponendosi di trovare un rimedio.
Chimica
Chimica e beni culturali
La chimica applicata ai beni culturali si occupa dei materiali utilizzati in ambito artistico e delle tecniche analitiche,
invasive e non, utilizzate per le indagini strumentali sulle opere d'arte. Si interessa inoltre della datazione dei reperti,
dei metodi di restauro e di conservazione. Studia i meccanismi e i fattori che contribuiscono al degrado dei manufatti
artistici cercando di rimediare al loro effetto.
Note
[1] In Histoire et Dictionnaire de la Révolution Française, Parigi, Éditions Robert Laffont, 1998.
[2] Non bisogna confondere le trasformazioni di tipo chimico da quelle di tipo fisico. La differenza principale tra i due tipi di trasformazione
risiede nell'entità delle interazioni che si realizzano tra i costituenti della materia: nel caso di rottura e/o creazione di legami meno energetici
(quali ad esempio legami di van der Waals e forze di London) si parla di trasformazione fisica (ad esempio miscelazione, assorbimento
gas-liquido, distillazione, adsorbimento fisico), mentre nel caso di rottura e/o creazione di legami più energetici (quali ad esempio legami
covalenti e legami ionici) si parla di trasformazione chimica.
[3] Theodore L. Brown, H. Eugene Lemay, Bruce Edward Bursten, H. Lemay. Chemistry: The Central Science. Prentice Hall; 8 edition (1999).
ISBN 0-13-010310-1. Pages 3-4.
[4] Carsten Reinhardt. Chemical Sciences in the 20th Century: Bridging Boundaries. Wiley-VCH, 2001. ISBN 3-527-30271-9. Pages 1-2.
[5] The Cambridge Dictionary of Scientists, op. cit.
[6] Si parla di energia cinetica microscopica per distinguerla dall'energia cinetica macroscopica. La prima compete al movimento di singole
molecole, mentre la seconda compete al movimento del corpo nella sua globalità (ad esempio moto di traslazione e rotazione di un corpo
rigido).
[7] All'aumentare della temperatura, il sistema aumenta il proprio volume, per qualsiasi tipo di stato (solido, liquido o aeriforme). L'aumento del
volume (a parità di pressione e temperatura iniziale e finale) è però molto evidente negli aeriformi rispetto ai liquidi e più evidente nei liquidi
rispetto ai solidi. Dal punto di vista quantitativo, l'aumento del volume può essere espresso dal coefficiente di dilatazione termica.
[8] Esempi di miscele con cui abbiamo spesso a che fare sono: la cioccolata, la birra, l'aria, la benzina e le leghe metalliche.
[9] Si parla di "quantitativo teorico" in quanto si tratta del massimo quantitativo ottenibile dal punto di vista termodinamico, cioè all'equilibrio.
Nella pratica invece intervengono altri fenomeni, che vengono studiati nell'ambito della cinetica chimica (quali ad esempio la presenza di
catalizzatori o inibitori della reazione).
[10] Ad esempio, composti come il solfuro di carbonio, l'anidride carbonica, il monossido di carbonio e i carburi sono considerati inorganici.
Bibliografia
• P. Atkins, L. Jones, Chemical Principles (http://www.whfreeman.com/newcatalog.aspx?disc=Chemistry&
course=General+Chemistry&isbn=1429219556), 5th ed., W.H. Freeman, 2010.
• I. J. Solov'ev, L'Evoluzione del pensiero chimico - Dal '600 ai giorni nostri (http://www.hoepli.it/libro/
l-evoluzione-del-pensiero-chimico.asp?ib=9786001561634&pc=000012013002000), Mondadori, 1976.
• The Cambridge Dictionary of Scientists - "Boyle, Robert (1627 - 1691)" (http://www.credoreference.com/
entry/dicscientist/boyle_robert_1627_1691) (in inglese), Cambridge, Cambridge University Press, 2002.
11
Chimica
12
Voci correlate
Chimici illustri
Composti chimici
Meccanica quantistica
Termodinamica
Elettrochimica
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Premio Nobel per la chimica
Atomi e molecole
•
•
•
•
•
•
•
•
•
•
La struttura dell'atomo
La tavola periodica degli
elementi
Numero atomico, peso atomico,
numero di massa
Elettronegatività, energia di
ionizzazione e affinità
elettronica
Metalli, nonmetalli,
semimetalli, gas nobili
La molecola e la struttura
molecolare
Gli ioni, anioni e cationi
I radicali
Formula chimica, isomeria,
chiralità
IUPAC, nomenclatura chimica
Legami chimici e forze di
attrazione
•
•
•
•
•
•
•
•
•
Legame chimico
Legame ionico
Legame covalente
Legame di coordinazione
Legame metallico
Legame σ
Legame π
Legame idrogeno
Forza di van der Waals
•
•
•
•
•
•
•
•
•
•
•
Composto
chimico
Composto
molecolare
Composto
organico
Composto
inorganico
Composto ionico
Acidi
Basi
Complessi
Cluster
Ossidi
Sali
Polimeri
•
•
•
•
•
•
•
•
•
•
Stati di aggregazione •
•
•
•
•
•
•
•
•
•
•
•
Stati della
materia
Fasi della
materia
Transizioni di
fase
Gas
Solido
Liquido
Plasma
Soluzione
Dispersione
Cristalli liquidi
Colloide
Equilibrio chimico
•
•
•
•
•
•
•
•
Equilibrio
chimico
Costante di
equilibrio
Costante di
dissociazione
Principio di Le
Châtelier
Legge di azione
di massa
Effetto ione
comune
Solubilità
Costante di
solubilità
•
•
•
•
Elettrone
Orbitale
Principio di esclusione
di Pauli
Principio dell'Aufbau
Regola di Hund
Configurazione
elettronica
Meccanica quantistica
Dualismo
onda-particella
Nucleo atomico
Funzione d'onda
Chimica quantistica
Orbitale
Legame di valenza
Combinazione lineare
di orbitali atomici
Equazione di
Schrödinger
Livello energetico
Numero quantico
Teoria degli orbitali
molecolari
•
•
•
•
•
•
•
•
•
•
•
Termodinamica
Termochimica
Funzione di stato
Energia libera
Entalpia
Entropia
Energia interna
Potenziale chimico
Trasformazione
termodinamica
Processo spontaneo
Processo esotermico
Processo endotermico
Entalpia di legame
Entalpia standard di
formazione
Entalpia standard di
reazione
Entropia molare standard
Energia libera di Gibbs
standard di formazione
Legge di Hess
Equazione di Kirchhoff
Equazione di van 't Hoff
•
•
•
•
•
•
•
•
•
•
•
•
•
Storia della chimica
•
•
•
•
Cinetica chimica
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Cinetica chimica
Velocità di reazione
Equazione cinetica
Ordine di reazione
Molecolarità
Costante di velocità
Catalisi
Catalizzatore
Teoria delle collisioni
Equazione di Arrhenius
Equazione di Eyring
stato di transizione
Teoria dello stato di
transizione
Meccanismo di reazione
Energia di attivazione
Effetto isotopico cinetico
Elettrochimica
Elettrochimica
quantistica
Elettrolita
Elettrodo
Catodo
Numero di
ossidazione
Ossidoriduzione
Equazione di Nernst
Cella elettrochimica
Pila
Accumulatore di
carica elettrica
Elettrolisi
Elettrosintesi
Chimica
elettroanalitica
Corrosione
Alchimia
Iatrochimica
Iatromeccanica
Scoperta degli
elementi chimici
Storia della chimica
Storia dell'industria
chimica
Teoria del flogisto
Altre
•
•
Anno Internazionale
della Chimica
Laboratorio chimico
Chimica
13
Altri progetti
•
Wikibooks contiene testi o manuali: http://it.wikibooks.org/wiki/Ripiano:Scienza/Chimica
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Chemistry
Wikinotizie contiene notizie di attualità: http://it.wikinews.org/wiki/Categoria:Chimica
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/chimica
•
Wikiquote contiene citazioni: http://it.wikiquote.org/wiki/Chimica
Collegamenti esterni
•
•
•
•
•
•
•
•
Glossario di chimica (http://www.apertisverbis.it/chimica.htm)
Articoli di chimica (http://www.apertisverbis.it/libri/articoli1.htm)
Società Chimica Italiana (http://www.soc.chim.it/)
Origine della parola "Chimica" (http://www.icapsira.com/Magazine/Chimicacosavuoldire/tabid/111/Default.
aspx)
Come un grande psicologo si è innamorato della Chimica (http://www.ilpalo.com/chimica-scienza-chimici/
index.htm)
Chimica (http://www.med.unipi.it:8080/TESTNET/presentazioni_ppt/pmr42/E018/Chimica 3.pdf)
Raccolta di quiz di autovalutazione (http://www.chimica-online.it/index.htm)
Raccolta di test di autovalutazione (http://www.divini.net/chimica/materiali/test/)
Concetti di base
• Problemi di Chimica 1.0 (http://micvac.dichi.unina.it) Esercizi sui concetti base che seguono possono essere
effettuati utilizzando il software didattico gratuito
Chimica Generale
• (EN) IUPAC Nomenclature Home Page (http://www.chem.qmw.ac.uk/iupac/)
• (EN) Programma didattico di simulazione chimica (http://xeon.concord.org:8080/modeler/)
Storia della chimica
Storia della chimica
« Forse venticinque secoli fa, sulle rive del mare divino, dove il canto degli aedi si era appena spento, qualche filosofo
insegnava già che la mutevole materia è fatta di granelli indistruttibili in continuo movimento, atomi che il caso e il fato
avrebbero raggruppato nel corso dei secoli secondo le forme e i corpi che ci sono familiari. »
(Jean Perrin, Les atomes, 1912)
Le prime teorie che tentavano di spiegare il comportamento
della materia risalgono ai filosofi greci (si pensi all'atomismo di
Democrito), per i quali la scienza e la religione erano ben
distinte. In seguito gli influssi arabi ed egiziani sulla cultura
greca portarono alla nascita dell'alchimia, un'antica pratica
protoscientifica che combinava elementi di chimica, fisica,
astrologia, arte, semiotica, metallurgia, medicina e religione.
La storia della chimica intesa come scienza sperimentale ha
inizio solo nel XVII secolo, quando si cominciò ad analizzare
con metodo scientifico la materia e le sue trasformazioni,
allontanandosi dalle vaghe e misteriose teorie legate al
misticismo dell'alchimia.
Le origini
Nel XVI e XVII secolo moltissimi concetti che successivamente
risulteranno ovvi, quali pressione, temperatura o fasi della
materia, non erano affatto compresi, tantomeno quelli di atomo
o molecola.
L'alchimista in cerca della Pietra Filosofale (1771) di
Joseph
Wright of Derby (Derby Museum and Art Gallery,
Il processo di transizione tra alchimia e chimica avvenne quindi
Derby, Regno Unito).
piuttosto gradualmente.
Le prime reazioni chimiche appaiono nel Tyrocinium Chymicum
dello iatrochimico francese Jean Béguin nel 1610.
Dopo che Evangelista Torricelli scoprì il modo di misurare la pressione atmosferica e formulò il concetto di vuoto si
diede il via a numerosi esperimenti per lo studio dei gas.
L'inglese Robert Boyle fu molto attivo in questo campo, e fu tra i primi ad applicare il metodo scientifico allo studio
della materia e delle sue trasformazioni. La sua opera The Sceptical Chymist ("Il Chimico Scettico") (1661) è
considerata da molti il primo testo scientifico di chimica; in esso Boyle descrive i suoi esperimenti con i gas, e
delinea alcune definizioni (ancora imprecise) di composto chimico.
14
Storia della chimica
Robert Boyle, uno dei padri della chimica moderna
Ritratto di Lavoisier con la moglie. Opera di
Jacques-Louis David, 1818
15
Storia della chimica
16
Una pagina del trattato "Chymical
Nomenclature", A. Lavoisier, 1787
La nascita della chimica
« Nulla si crea, nulla si distrugge »
(Antoine Lavoisier)
Nonostante l'opera di numerosi emeriti studiosi, ancora alla fine del XVIII secolo si consideravano validi alcuni
concetti del tutto errati, come ad esempio la teoria del flogisto.
Nel 1700 emerse la necessità di una teoria che riunisse le varie scoperte nel campo dei gas. L'uomo che fece questo
lavoro fu Antoine Lavoisier, il quale demolì la teoria del flogisto con la sua legge di conservazione della massa nel
1789. Egli è considerato il padre della chimica moderna: fra i suoi meriti vi sono, oltre alla citata legge di
conservazione, il metodo di lavoro (con attenzione alla purezza dei reagenti, e l'uso della bilancia di precisione),
l'opera di nomenclatura di composti binari, la corretta determinazione della composizione dell'aria, l'analisi sulla
composizione di grassi, oli e zuccheri, scoprendo la costante presenza di idrogeno, ossigeno e carbonio (i
"mattoncini" di base di tutte le sostanze organiche).
Inoltre, fino a quel momento, la chimica non possedeva uno status accademico autonomo, ma faceva parte ancora del
piano di studi della medicina.[1]
Storia della chimica
La chimica organica
Nel 1828 Friedrich Wöhler sintetizzò accidentalmente dell'urea partendo da sostanze inorganiche.
Questo fatto fece comprendere che il mondo della chimica organica e della chimica inorganica avevano delle basi in
comune; inoltre aprì degli accesi dibattiti sul vitalismo, teoria che sosteneva una netta demarcazione tra il mondo
della vita (organico) e l'inorganico.
Lo sviluppo della chimica organica compiuto nei secoli successivi, permise di sintetizzare, partendo da molecole più
piccole, innumerevoli sostanze di uso comune, dai coloranti ai medicinali.
Proprio in questi anni, nel 1845, a Londra fu fondato il Royal College of Chemistry e un grande salto di qualità nella
formazione e nel mestiere del chimico si ebbe in terra tedesca, grazie ai laboratori di Justus von Liebig, che
divennero ben presto un modello per l'organizzazione e per la ricerca non solo della chimica, ma un po' di tutta la
scienza in generale.
Nella prima settimana di settembre del 1860, a Karlsruhe si svolse il primo incontro internazionale di chimica
(Congresso di Karlsruhe), al quale confluirono oltre 130 studiosi e ricercatori provenienti da tutta l'Europa, che ebbe
il delicato compito di ridefinire i concetti basilari della chimica, una notazione e una nomenclatura comune, e di
rivedere la suddivisione della chimica in tre branche particolari: la minerale, la vegetale e la animale.
Lo sviluppo industriale
L'ultima parte del XIX secolo segna l'inizio dello
sfruttamento industriale delle nuove conoscenze
chimiche, con la sintesi industriale della soda, lo
sviluppo di nuovi coloranti, dei primi polimeri sintetici,
la petrolchimica ed i farmaci di sintesi, detersivi,
fertilizzanti. I grandi benefici apportati dallo sviluppo
della chimica industriale sono evidenti sotto gli occhi di
tutti, ma è utile ricordare che questo sviluppo ha avuto
anche un costo: da un lato la creazione di armi
distruttive utilizzate nelle due guerre mondiali e anche
Raffigurazione di un impianto chimico a Rostock (1890 ca.)
in seguito, dall'altro incidenti e scarsa sensibilità hanno
causato in passato gravi incidenti ambientali e morti prima che si sviluppasse una sufficiente sensibilità ambientale.
17
Storia della chimica
18
La tavola periodica degli elementi
Per molto tempo l'esistenza stessa degli
elementi chimici fu oggetto di ricerca; la
lista degli elementi si ampliava molto
spesso, ed i chimici stentavano a dare un
senso teorico alle loro scoperte.
Fortunatamente i chimici Dmitrij Mendeleev
e Julius Lothar Meyer ebbero un' intuizione,
sistemando in una tabella gli elementi a
seconda del loro peso atomico e del loro
stato di ossidazione. Più correttamente oggi
si sa che gli elementi sono disposti in ordine
progressivo di numero atomico Z e non di
peso atomico. Malgrado tale inesattezza, che
coinvolge solo pochi elementi (quali Ar e K,
Co e Ni, Te e I, Th e Pa) alcuni dei quali
all'epoca non ancora scoperti, la tavola
periodica permise a Mendeleev di predirre
l'esistenza di vari elementi allora sconosciuti
(germanio, gallio, e scandio, che lui
inizialmente
nominò
ekasilicon,
ekaaluminium, ed ekaboron) nel 1870. In
assenza di una coerente e condivisa teoria
sulla struttura atomica, la comunità
scientifica fu inizialmente scettica, ma in
seguito le sue previsioni furono confermate.
Tavola di Mendeleev
Storia della chimica
19
La chimica moderna
Prima del XX secolo la chimica era considerata una scienza con
pochi punti in comune con la fisica.
Auguste Comte scriveva nel 1830:
Ogni tentativo di utilizzare metodi matematici nello studio
dei problemi chimici è da considerarsi profondamente
irritante e contrario allo spirito della chimica....
Fortunatamente l'atteggiamento cominciò a cambiare dopo la
metà de XIX secolo quando Friedrich August Kekulé scriveva,
nel 1867:
Mi aspetto che un giorno si trovi una spiegazione
matematica e meccanica di ciò che chiamiamo atomi...
In seguito alla scoperta della radioattività da parte di Marie e
Pierre Curie gli scienziati cambiarono drasticamente il loro
punto di vista. Gli atomi rivelarono una struttura complessa e
non più indivisibile.
La teoria atomica
Ritratto di Kekulé nel 1890
Nel XVIII secolo i chimici erano piuttosto favorevoli alla teoria
atomica (ad esempio John Dalton) ma molto più cauti erano i fisici come ad esempio Wilhelm Ostwald, Ernst Mach
e lo stesso Max Planck. Gli atomi potevano essere contati ma nessuno li aveva mai visti! I fautori della teoria come
Amedeo Avogadro, Stanislao Cannizzaro, Svante Arrhenius e Ludwig Boltzmann fecero, grazie ad essa, numerosi
passi in avanti come ad esempio la spiegazione dei comportamenti dei gas o degli acidi e basi in soluzione o la
determinazione dei pesi atomici degli elementi. Ciononostante, la lunga disputa ebbe fine solamente nella prima
decade del XX secolo quando Jean Perrin fece una lunga serie di esperimenti, basati anche sulla teoria che Albert
Einstein aveva formulato per spiegare il moto Browniano, la quale era appunto fondata sulla teoria atomica; Perrin
dimostrò in maniera inoppugnabile l'esistenza degli atomi.
« La Natura dispiega lo stesso splendore senza limiti nell'atomo come nella nebulosa, ed ogni nuovo mezzo di conoscenza la
mostra più vasta e diversa, più feconda più imprevista, più bella, più ricca d'insondabile immensità. »
(Jean Perrin, Les atomes, 1912)
I primi modelli atomici degni di nota sono quello a "panettone" di Joseph John Thomson (1904) e quello a
"planetario" di Ernest Rutherford (1911), ma, contemporaneamente alle deduzioni di Perrin, la svolta si ebbe con la
teoria di Niels Bohr sulla struttura atomica nel 1912. Bohr spiegò la disposizione delle linee spettrali dell'atomo di
idrogeno introducendo una "quantizzazione".
Finalmente lo studio della chimica non coinvolgeva più solo osservanzioni empiriche sui comportamenti della
materia, ma anche aspetti collegati alla nuvola elettronica che avvolge il nucleo atomico. Si delineava un modo
coerente per spiegare la natura del legame chimico e la disposizione degli elementi nella tavola periodica: in poche
parole aveva inizio la convergenza tra fisica e chimica sollecitata da Kelulé.
Storia della chimica
20
Evoluzione dei modelli atomici
Modello atomico di Thomson
Modello atomico di Rutherford
Modello atomico di Bohr
Il modello di Bohr ottenne degli eccellenti risultati teorici per l'atomo di idrogeno, ma solo con la meccanica
quantistica fu possibile formulare teorie e modelli applicabili ad atomi più complessi.
Chimica quantistica
La nascita della meccanica quantistica è una pietra miliare per la
fisica e per la chimica. La fisica quantistica era di difficile
comprensione per i fisici stessi, e ancora dello scetticismo
aleggiava intorno alle sue applicazioni nella chimica, ma la
storia diede torto agli increduli. Dopo la formulazione
dell'equazione di Schrödinger (1926) si ottennero enormi
progressi nell'analisi della struttura degli atomi, molecole e del
legame chimico in termini fisici. Nel 1927 Walter Heitler e Fritz
London scrissero un articolo in cui si utilizzava per la prima
volta la meccanica quantistica per descrivere la molecola di H2:
era la nascita della chimica quantistica. Negli anni seguenti
molti altri studiosi contribuirono ai progressi; per citarne alcuni:
Robert S. Mulliken, Max Born, Robert Oppenheimer, Linus
Pauling, Erich Hückel, Douglas Hartree, Vladimir
Aleksandrovich Fock. Ecco una breve cronologia dei principali
sviluppi:
• 1924 - Louis de Broglie sostiene che una particella in
movimento possiede doppia natura corpuscolo-ondulatoria.
Erwin Schrödinger nel 1933
• 1925 - principio di esclusione di Pauli.
• 1926 - equazione di Schrödinger e approssimazione di Born-Oppenheimer.
• 1927 - Walter Heitler e Fritz London: analisi quantistica del legame di valenza e della molecola di idrogeno.[2]
• 1927-29 - Friedrich Hund e Robert S. Mulliken descrivono gli orbitali molecolari.
• 1928 - Linus Pauling descrive l'ibridazione degli orbitali di legame.
• 1932 - Henry Eyring e Michael Polanyi analizzano il sistema H2+H.
Prima della metà del XX secolo si era completata l'integrazione fra chimica e fisica, le maggiori proprietà chimiche
potevano essere spiegate in termini di struttura atomica.
Storia della chimica
21
Linus Pauling nel suo libro La natura del legame chimico, pubblicato nel 1937 e considerato una pietra miliare nella
storia della chimica, utilizzò i principi della meccanica quantistica per dedurre angoli di legame ed altre proprietà
molecolari di strutture atomiche complesse.
Biologia molecolare e biochimica
Nonostante i principi dedotti dalla meccanica quantistica avessero
permesso di formulare nuove teorie e comprendere alcuni principi
chimici fondamentali, per le molecole di grandi dimensioni
caratteristiche della biochimica (enzimi, ormoni, vitamine, proteine)
vi erano molte osservazioni empiriche senza una spiegazione
teorica.
Uno dei problemi più dibattuti era la struttura del DNA, una
macromolecola che si sapeva nascondere il "segreto della vita", ma
la cui struttura era un busillis. Grazie agli sviluppi della chimica
organica fisica e dei metodi analitici (come ad esempio la
spettroscopia e la cristallografia a raggi X), nel 1953 viene
finalmente decifrata la struttura a doppia elica del DNA da Francis
Crick e James Watson (peraltro ispirati da ipotesi di Erwin
Schrödinger e Linus Pauling e dalle immagini ai raggi X di
Rosalind Elsie Franklin). Ecco una breve cronologia dei principali
progressi in biochimica di quegli anni:
• 1937 - Hans Adolf Krebs descrive il ciclo di Krebs
• 1950 - Chargaff determina nel DNA il rapporto 1:1 fra adenina e
timina, e fra guanina e citosina (regole di Chargaff).
• 1953 - Watson e Crick propongono la struttura a doppia elica del
DNA
Animazione di un frammento di DNA
• 1953 - Esperimento di Miller-Urey - Stanley Miller e Harold
Clayton Urey ipotizzano e simulano una evoluzione chimica come base dell'origine della vita.
• 1955 - Sanger determina la sequenza dell'insulina
•
•
•
•
•
1956-60 - Perutz determina la struttura tridimensionale dell'emoglobina
1957 - Ingram individua la causa molecolare dell'anemia falciforme
1958-60 - Kendrew determina la struttura tridimensionale della mioglobina
1961 - Braunitzer determina la sequenza dell'emoglobina
1983 - Mullis inventa la metodica nota come PCR
Storia della chimica
I polimeri e le macromolecole
I polimeri sono grossi aggregati molecolari costituiti dalla
ripetizione sistematica di più unità monomere, mentre la
nomenclatura
IUPAC
attuale
utilizza
il
termine
"macromolecola" per indicare singole molecole di grandi
dimensioni.
Nel 1839 Charles Goodyear scoprì il processo di
vulcanizzazione della gomma, sfruttato per aumentarne le
proprietà meccaniche e la resistenza agli agenti chimici. Il primo
polimero sintetico ad essere stato prodotto industrialmente fu la
parkesina, sviluppata da Alexander Parkes nel 1856 e prodotta
Giulio Natta nel 1963
per la prima volta su scala industriale nel 1866. Nel 1963 Giulio
Natta e Karl Ziegler vincono il premio Nobel per la chimica
grazie alla realizzazione della classe di catalizzatori noti come catalizzatori di Ziegler-Natta, che introdussero la
possibilità di sintetizzare industrialmente polimeri stereospecifici (il primo fu il polipropilene isotattico).
• 1839 - Charles Goodyear scopre il processo di vulcanizzazione della gomma.
• 1846 - Christian Schönbein scopre casualmente la nitrocellulosa, che verrà prodotta industrialmente solo nel 1891
a causa delle problematiche legate alla sua esplosività.
• 1856 - Alexander Parkes crea la parkesina, primo polimero sintetico che sarà prodotto su scala industriale.
• 1883 - La viscosa, prima fibra tessile semisintetica, viene inventata da Hilaire de Chardonnet.
• 1907 - Leo Baekeland produce la bachelite.
• 1926 - Lo statunitense Waldo Semon, presso la B.F. Goodrich, mette a punto un metodo di sintesi su larga scala
del PVC consentendo il vasto utilizzo di questo polimero.
• 1929 - Il chimici tedeschi della IG Farben Walter Bock e Eduard Tschunkur sintetizzano la gomma Buna-S.
• 1933 - Sintesi industriale del polietilene.
• 1935 - Prodotto il nylon 6,6, primo fibra tessile totalmente sintetica.
• 1954 - Giulio Natta sintetizza il polipropilene isotattico.
22
Storia della chimica
Le nuove frontiere della chimica
Chimica supramolecolare
La chimica supramolecolare è una branca interdisciplinare,
organizzatasi sistematicamente e razionalmente verso la fine
degli anni sessanta, che riprendendo princìpi e concetti della
chimica moderna rappresenta oggigiorno un campo di ricerca in
forte espansione.
La linea di indagine che sfociò nella nuova disciplina della
chimica supramolecolare ebbe un'origine perfettamente classica,
affondando le sue radici nella chimica organica. Charles
Pedersen, chimico della Du Pont, nel 1967 annunciò che dei
poliesteri macrociclici da lui sintetizzati avevano la curiosa
caratteristica di potersi complessare con ioni sodio e potassio,
una proprietà dovuta alla loro non meno curiosa forma a corona.
Il valore conoscitivo di questa scoperta venne subito reso più
Immagine di una struttura supramolecolare descritta da
intenso dall'entrata nel nuovo campo del francese Jean-Marie
Jean-Marie Lehn e collaboratori in Angew. Chem., Int.
Ed. Engl. 1996, 35, 1838-1840
Lehn, un chimico organico fisico che allora era interessato
principalmente ai meccanismi di trasporto degli ioni alcalini
connessi con i segnali trasmessi nel sistema nervoso. Le ricerche del gruppo di Lehn iniziarono subito, nel 1967, e
con la sintesi di nuove strutture tridimensionali già nel 1969 ottenevano il sequestro degli ioni con la formazione di
criptati. Un secondo gruppo, diretto da Donald Cram, ebbe una falsa partenza, utilizzando i composti di Pedersen
come varianti nelle loro consuete ricerche di chimica organica fisica, ma nel 1973 cominciò a pubblicare un fiume di
lavori su ciò che fu battezzata la chimica ospite/ospitante.
Lehn nutrì fin dall'inizio l'intenzione di comprendere meglio gli eventi fisiologici costruendo molecole modello che
presentassero le stesse caratteristiche dei sistemi naturali, ma nella seconda metà degli anni '70 lo scienziato francese
estese le ricerche sperimentali e le interpretazioni teoriche fino a creare, e a definire, l'ambito della chimica
supramolecolare come quello in cui sono studiati e (ri)prodotti i processi mediante i quali entità di complessità
maggiore risultano da molecole meno complesse a causa dall'azione di forze intermolecolari. Si schiudeva anche da
questo punto di vista classico tutto l'orizzonte dell'auto-organizzazione molecolare, con almeno due finalità ben
visibili: la mimesi di sistemi biologici (viventi, se interviene l'autocatalisi) e la costruzione di vere macchine
molecolari, adatte, ad esempio, al calcolo digitale. Fra le linee di ricerca più attive vi sono: il riconoscimento
molecolare; nel campo dell'auto-replicazione quella di oligonucleotidi e di micelle; nel settore
dell'auto-organizzazione, l'ottenimento di mesofasi tubulari, recettori fotosensibili, interruttori.
Lehn, parafrasando Richard Feynman e il suo noto discorso There's plenty of room at the bottom[3] sulle
nanotecnologie (con l'espressione There's even more room at the top[4]) indicò come la chimica non solo deve
guardare verso l'estremamente piccolo, ma può andare anche al di sopra delle dimensioni molecolari, studiando la
complessità supramolecolare.
Davanti al pubblico mondiale dei chimici, nel Congresso della IUPAC di Tokyo, Lehn propose il termine con cui
correntemente designano la nuova disciplina: chimica supramolecolare.
23
Storia della chimica
24
Chimica combinatoria
Spesso il ricercatore si imbatte in un composto che dimostra una certa attività biologica, che però non è sufficiente
per garantire il successo clinico (e commerciale) del composto. A questo punto inizia un processo di screening "quasi
casuale": vengono preparati e testati tutti i possibili composti che mantengono una analogia strutturale per il nucleo
fondamentale, ma ne differiscono per i sostituenti collegati.
Chimica computazionale
La chimica computazionale è la branca della chimica teorica che si occupa dello sviluppo di modelli matematici,
basati sia sulla meccanica classica che sulla meccanica quantistica, in grado di simulare sistemi chimici, con lo scopo
di calcolarne le grandezze fisiche caratteristiche e prevederne le proprietà chimiche.
Chimica nucleare
La chimica nucleare è un settore della chimica che tratta le reazioni che cambiano la natura del nucleo. Il fenomeno
chimico-fisico studiato dalla chimica nucleare è la radioattività e la grandezza fisica corrispondente nel Sistema
Internazionale è l'attività.
Caos chimico
Per caos chimico si intende quell'insieme di reazioni chimiche
dipendenti da fattori aleatori con apparenza caotica. Il resoconto
di reazioni oscillanti fu pubblicato per la prima volta da Gustav
Theodor Fechner nel 1828. Nel 1833 John Herschel, noto
astronomo e inventore della cianotipia, scoprì una serie di
reazioni periodiche legate al dissolvimento del ferro in acido
nitrico a diversi valori di concentrazione. Le reazioni oscillanti
si incontrano spesso in elettrochimica, come riportato da
Christian Friedrich Schönbein nel 1842 e James Prescott Joule
nel 1844.
Seguirono sul piano sperimentale descrizioni di reazioni
apparentemente caotiche, e la loro interpretazione sulla base di
processi autocatalitici, ma solo con la reazione di
Belousov-Zhabotinsky l'auto-organizzazione nel tempo e nello
spazio di particolari sistemi reagenti divenne un tema accettato
di ricerca.
Schema grafico della complicata reazione di
Belousov-Zhabotinsky
Il chimico sovietico Boris Belousov scoprì la reazione che porta il suo nome mentre cercava di riprodurre in provetta
un insieme di reazioni che avesse qualche analogia con il ciclo di Krebs. La storia dettagliata dei tentativi di
Belousov, sempre frustrati, di pubblicare i suoi risultati (dal 1951 al 1957) entrerà a far parte della leggenda
(negativa) della chimica; maggiore fortuna ebbe il biofisico Anatol Zhabotinsky che rese nota la reazione nel 1964.
Nel frattempo (1952) Alan Turing aveva pubblicato un articolo seminale dal titolo estremamente significativo: La
base chimica della morfogenesi, in cui discuteva in dettaglio gli effetti di meccanismi autocatalitici; inoltre, su un
piano più generale, diversi gruppi di ricercatori, fra cui spiccava quello diretto da Ilya Prigogine, avevano fatto
progredire la termodinamica dei processi irreversibili (gli unici esistenti nella realtà fisica). Nel 1967 Prigogine e
Nicolis proposero il concetto di struttura dissipativa, e avendo dimostrato la relazione fra organizzazione e
dissipazione ne sottolinearono la possibile rilevanza rispetto ai "primi passi biogenetici".
Attualmente la chimica che studia l'origine dell'ordine a partire dal caos molecolare è un campo attivissimo di
ricerca. Nel 1990 i principali temi trattati sulle dinamiche non lineari riguardavano: propagazione di onde e strutture
Storia della chimica
spaziali; oscillazioni in sistemi eterogenei; oscillazioni biologiche; patterns geochimici; proposta e discussione di
sistemi modello. Questo semplice elenco dimostra la pervasività interdisciplinare delle procedure conoscitive della
chimica.
Note
[1] Antonio Di Meo, Storia della chimica, Newton, 1997, Roma, pag.26-28
[2] W. Heitler and F. London, Wechselwirkung neutraler Atome und Homöopolare Bindung nach der Quantenmechanik, Z. Physik, 44, 455
(1927)
[3] Trad. "C'è moltissimo spazio in basso" - dove il termine "in basso" è inteso come il mondo al di sotto delle dimensioni molecolari
[4] Trad. "C'è ancora più spazio in alto" - dove il termine "in alto" è inteso come il mondo al di sopra delle dimensioni molecolari
Bibliografia
• Antonio Di Meo; Luciano Caglioti, Storia della chimica: dalla ceramica del neolitico all'età della plastica (http:/
/books.google.it/books?id=-jtNPQAACAAJ&source=gbs_navlinks_s), 2a ed., Marsilio, 1990. ISBN
8-831-75274-X
• Ernst von Meyer; George McGowan, A history of chemistry from earliest times to the present day: being also an
introduction to the study of the science (http://books.google.it/books?id=sGwtAAAAYAAJ&
source=gbs_navlinks_s) (in inglese), Macmillan and Co., 1891.
• James Riddick Partington, A short history of chemistry (http://books.google.it/books?id=fanHRlU1bSEC&
source=gbs_navlinks_s), 3a ed. (in inglese), Dover Publications, 1989. ISBN 0-486-65977-1
• Henry Marshall Leicester, The historical background of chemistry (http://books.google.it/
books?id=aJZVQnqcwv4C&source=gbs_navlinks_s) (in inglese), Courier Dover Publications, 1971. ISBN
0-486-61053-5
• Aaron John Ihde, The development of modern chemistry (http://books.google.it/books?id=34KwmkU4LG0C&
source=gbs_navlinks_s) (in inglese), Courier Dover Publications, 1984. ISBN 0-486-64235-6
• William Hodson Brock, The chemical tree: a history of chemistry (http://books.google.it/
books?id=vqpuET7q6TgC&source=gbs_navlinks_s) (in inglese), Norton, 2000. ISBN 0-393-32068-5
25
Storia della chimica
26
Voci correlate
Chimici notevoli
Altre voci correlate
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Mikhail Lomonosov, 1711-1765
Joseph Black, 1728-1799
Joseph Priestley, 1733-1804
Carl Wilhelm Scheele, 1742-1786
Antoine-Laurent de Lavoisier, 1743–1794
Alessandro Volta, 1745-1827
Jacques Charles, 1746-1823
Claude Louis Berthollet, 1748-1822
Joseph-Louis Gay-Lussac, 1778-1850
Humphry Davy, 1778-1829
Jöns Jakob Berzelius, 1779-1848
Michael Faraday, 1791-1867
Friedrich Wöhler, 1800-1882
Justus von Liebig, 1803-1873
Louis Pasteur, 1822-1895
Stanislao Cannizzaro, 1826-1910
Friedrich August Kekulé von Stradonitz, 1829-1896
Willard Gibbs, 1839-1903
Jacobus Henricus van 't Hoff, 1852-1911
Marie Curie, 1867-1934
Victor Grignard, 1871-1935
Ernest Rutherford, 1871–1937
Gilbert Newton Lewis, 1875-1946
Otto Hahn, 1879-1968
Alchimia
Chimica
Iatrochimica
Premio Nobel per la chimica
Scoperta degli elementi chimici
Storia dell'elettrochimica
Storia dell'industria chimica
Teoria del flogisto
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:History
of chemistry
Collegamenti esterni
• Minerva: Appunti di storia della chimica (http://www.minerva.unito.it/Storia/AppuntiStoriaChimica/
AppuntiStoria1.htm)
• (EN) Testo completo di The Skeptical Chymist online (http://oldsite.library.upenn.edu/etext/collections/
science/boyle/chymist/index.html?)
• (EN) Carl Schorlemmer The rise and development of organic chemistry (http://www.archive.org/details/
risedevelopmento00schorich) (London: MacMillan, 1894)
• (EN) Karl Hugo Bauer A history of chemistry. Translated by R.V. Stanford (http://www.archive.org/details/
historyofchemist00baueuoft) (London: E. Arnold,1907)
• (EN) Thomas Percy Hilditch A concise history of chemistry (http://www.archive.org/details/
concisehistoryof00hilduoft) (London: Methuen, 1911)
• (EN) James Campbell Brown A history of chemistry from the earliest times. (http://www.archive.org/details/
historyofchemist00browuoft) 2d ed. (London: J. & A. Churchill,1920)
• (EN) William Augustus Tilden Famous chemists, the men and their work (http://www.archive.org/details/
famouschemistsme00tildrich) (London : G. Routledge & Sons, ltd., 1921)
• (EN) Francis Preston Venable History of chemistry (http://www.archive.org/details/
historyofchemist00venarich) (Boston: D.C. Heath & Co, 1922)
• (EN) Scritti selezionati sulla storia della chimica (http://web.lemoyne.edu/~giunta/papers.html)
Legame chimico
27
Legame chimico
Si ha un legame chimico quando una forza di natura elettrostatica tiene uniti più atomi in una molecola o in un
cristallo (legami forti, o intramolecolari) o più molecole in una sostanza allo stato condensato (legami deboli, o
intermolecolari).
I legami chimici "più forti" hanno un contenuto energetico maggiore e sono più difficili da rompere, mentre i legami
"più deboli" hanno un contenuto energetico minore e sono più facili da rompere. Da ciò deriva che le molecole che
hanno al loro interno legami chimici più deboli sono più instabili.[1]
Inoltre tanto più un legame è forte, tanto minore è la lunghezza del legame, essendo la forza che tiene uniti gli atomi
maggiore.[2]
Natura elettrostatica del legame chimico
La natura del legame chimico si può spiegare facilmente osservando le forze coulombiane interagenti tra le
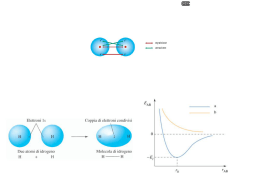
molecole. Prendiamo ad esempio il catione H2+: esso è costituito da due nuclei di H e da un elettrone. Indichiamo
con Ha il primo nucleo di idrogeno e con Hb l'altro nucleo di idrogeno. A ciascuno dei due nuclei è associata una
funzione d'onda elettronica, rispettivamente 1sa e 1sb, la cui combinazione lineare forma l'orbitale molecolare Ψ.
Ψ avrà valori bassi tra i due nuclei, mentre crescerà avvicinandosi ad essi e poi decrescerà allontanandosi
nuovamente da essi. Quindi se si considera un elettrone, ovvero una carica negativa posta tra i due nuclei, esso sarà
sottoposto a forze attrattive da parte dei due nuclei che saranno controbilanciate da quelle repulsive fino a quando
non si sarà raggiunta la stabilità del sistema; quindi l'elettrone sarà caduto in una buca di potenziale dalla quale gli
sarà difficile uscire. In tal modo si è formato un legame chimico.
Legami forti
Lunghezza di legame tipica
[3]
ed energia di legame
Legame Lunghezza Energia
(pm)
(kJ/mol)
H — Idrogeno
H–H
74
436
H–O
96
366
H–F
92
568
H–Cl
127
432
C — Carbonio
C–H
109
413
C–C
154
348
C=C
134
614
C≡C
120
839
C–N
147
308
C–O
143
360
C–F
134
488
C–Cl
177
330
Legame chimico
28
N — Azoto
N–H
101
391
N–N
145
170
N≡N
110
945
O — Ossigeno
O–O
148
145
O=O
121
498
F, Cl, Br, I - Alogeni
F–F
142
158
Cl–Cl
199
243
Br–H
141
366
Br–Br
228
193
I–H
161
298
I–I
267
151
I legami chimici forti sono le forze che tengono uniti gli atomi che formano le molecole. Un legame forte è attuato
dalla condivisione o dal trasferimento di elettroni tra atomi e dall'attrazione elettrostatica tra protoni ed elettroni. Tali
legami generano il trasferimento di un numero intero di elettroni, detto ordine di legame, anche se in alcuni sistemi
vi sono quantità intermedie di carica, come nel benzene, in cui l'ordine di legame è 1,5 per ogni atomo di carbonio. I
legami forti sono generalmente classificati in tre classi, in ordine di polarità crescente:
Legame covalente
Il legame covalente è il legame che si instaura tra due atomi appartenenti alla categoria (degli elementi chimici) dei
non metalli (uguali o aventi differenza di elettronegatività - scala di Pauling - compresa tra 0 e 0,4) che mettono in
compartecipazione una coppia di elettroni (detti coppia di legame) in un orbitale esterno che abbraccia entrambi gli
atomi. Il legame covalente viene rappresentato da un trattino che congiunge i due atomi legati.
Legame covalente puro
Un legame covalente puro (o omopolare) è un legame covalente che s'instaura fra due atomi appartenenti allo stesso
elemento. In pratica si stabilisce una interazione (cioè il legame) tra atomi dello stesso tipo: è il caso tipico
dell'idrogeno, dell'ossigeno, dell'azoto atmosferico, ecc.
Essendo la nube elettronica distribuita simmetricamente, il legame risulta non polarizzato.
I legami covalenti che si formano fra due atomi che condividono due coppie di elettroni prendono il nome di doppio
legame. I legami covalenti che si formano fra due atomi che condividono tre coppie di elettroni prendono il nome di
triplo legame.
Per constatare il numero di legami covalenti formatisi fra due atomi bisogna conoscere la valenza dell'atomo degli
elementi considerati e dopo aver fatto questo scoprire quanti elettroni gli mancano per essere stabili (regola
dell'ottetto).
es. N=azoto V gruppo= 5 elettroni di valenza (+ 3 elettroni per completare l'ottetto)
I legami fra due azoti sono un triplo legame.
Legame chimico
Legame covalente polare
Il legame covalente polare si instaura tra due atomi con differenza di elettronegatività compresa tra 0,4 e 1,7. In
questo caso, gli elettroni coinvolti nel legame risulteranno maggiormente attratti dall'atomo più elettronegativo, il
legame risulterà quindi polarizzato elettricamente, cioè ognuno degli atomi coinvolti nel legame presenterà una
carica elettrica parziale.
Quando una molecola è tenuta coesa da soli legami covalenti puri o possiede una simmetria tale da annullare
reciprocamente le polarità dei suoi legami covalenti risulterà complessivamente apolare. Invece una molecola
costituita da due atomi legati fra loro da un legame covalente polare è polare (o dipolo elettrico); ciò non significa,
in genere, però che la molecola abbia una carica elettrica perché nella sua totalità essa è elettricamente neutra. Si può
prevedere facilmente la struttura polare di una molecola nel caso essa sia biatomica.
Legame di coordinazione
È un tipo particolare di legame covalente detto, in passato, dativo in quanto i due elettroni coinvolti nel legame
provengono da uno solo dei due atomi detto donatore (in sostanza tale atomo "dona" il suo lone pair, cioè entrambi
gli elettroni appaiati presenti in un suo orbitale), mentre l'altro, che deve essere in grado di mettere a disposizione un
orbitale esterno vuoto (cioè con due posti vuoti che possono essere occupati da due elettroni) oppure di riorganizzare
la sua configurazione elettronica per accogliere la coppia di elettroni (cioè ad esempio spostare due elettroni presenti
su di un orbitale dispari su di un altro orbitale dispari, liberando di fatto un orbitale) viene detto accettore. Il legame
dativo può essere rappresentato con una freccia, dal donatore all'accettore, o più impropriamente può essere indicato
con un doppio trattino.
Legami delocalizzati e legame metallico
Alcuni legami covalenti, detti delocalizzati, possono legare insieme tre o più atomi contemporaneamente, come nei
borani e nei composti aromatici.
Legame metallico
La forma più estrema di delocalizzazione del legame covalente si ha nel legame metallico. Secondo questo modello
un metallo può essere rappresentato come un reticolo cristallino di ioni positivi tenuti uniti da una nube di elettroni
condivisi estesa a tutto il reticolo; essendo tali elettroni non legati a nessun atomo particolare, risultano essere
estremamente mobili; tale mobilità è responsabile della elevata conducibilità elettrica dei metalli.
Legame ionico
Il legame ionico è un legame tra ioni con carica di segno opposto. Tali ioni si formano da atomi aventi differenza di
elettronegatività superiore al limite convenzionale di 1,7: in queste condizioni, l'atomo più elettronegativo (quindi
caratterizzato da una elevata energia di ionizzazione ed elevata affinità elettronica) priva l'altro atomo meno
elettronegativo (caratterizzato da una bassa energia di ionizzazione ed una affinità elettronica quasi assente) di un
elettrone; il primo atomo diventa uno ione con carica negativa, il secondo uno ione con carica positiva.
Questo legame è di natura prettamente elettrostatica; l'arrangiamento degli atomi nello spazio non ha la direzionalità
del legame covalente: il campo elettrico generato da ciascuno ione si diffonde simmetricamente nello spazio attorno
ad esso.
29
Legame chimico
30
E' il tipo più semplice di legame chimico, sia dal punto di vista concettuale sia da quello della sua descrizione
analitica, essendo interpretabile in base alle leggi classiche dell'elettrostatica.
Legami deboli (legami chimici secondari)
I dipoli molecolari possono originare delle forze di attrazione intermolecolari.
I legami intermolecolari sono essenzialmente costituiti dalla reciproca attrazione tra dipoli statici - è il caso delle
molecole polari - o tra dipoli ed ioni - è il caso, ad esempio, di un sale che si scioglie in acqua.
Nel caso dei gas nobili o di composti formati da molecole apolari la possibilità di liquefare viene spiegata tramite la
formazione casuale di un dipolo temporaneo quando gli elettroni, nel loro orbitare, si trovino casualmente
concentrati su un lato della molecola; tale dipolo induce nelle molecole vicine a sé uno squilibrio di carica elettrica
(il cosiddetto dipolo indotto) che genera reciproca attrazione e provoca la condensazione del gas. Il legame viene
quindi prodotto da queste particolari forze di attrazione dette forze di dispersione o di Van der Waals.
Un caso particolare di legame intermolecolare, che può anche essere intramolecolare quando la geometria della
molecola lo consente, è il legame idrogeno.
Un atomo di idrogeno legato ad un atomo di ossigeno (o di fluoro), a causa della sua polarizzazione positiva e delle
sue ridotte dimensioni, attrae con un'intensità relativamente elevata gli atomi di ossigeno (e di fluoro e, in misura
minore, di azoto) vicini.
Tale legame, benché debole, è responsabile della conformazione spaziale delle proteine e degli acidi nucleici,
conformazione da cui dipende l'attività biologica dei composti stessi.
Come ordine di grandezza, l'entità delle varie forze di legame può essere indicato dalla seguente tabella:
Forza relativa
Legame ionico
1000
Interazioni dipolari e Legame idrogeno 10 - 100
Forza di van der Waals
1
Note
[1] A tale proposito, un esempio è dato dalla molecola di etilene rispetto alla molecola di etano. Nel caso dell'etilene, i due atomi di carbonio
sono legati da due legami: un legame σ, più forte, e un legame π, più debole. Nella molecola di etano invece i due atomi di carbonio sono
legati da un singolo legame σ. Per questo motivo, la molecola di etilene è più instabile rispetto alla molecola di etano, in quanto in seguito a
riscaldamento si ha l'apertura del legame π, che è più debole.
[2] Infatti il legame chimico funge da forza attrattiva, alla quale è contrapposta una forza di tipo repulsivo (che aumenta di intensità al diminuire
della distanza), per cui la posizione reciproca degli atomi è una posizione di equilibrio data dall'azione contrastante delle due forze. Per
approfondire: potenziale di Lennard-Jones.
[3] La lunghezza di legame è espressa in pm e può essere convertita in Å dividendo per 100 (1 Å = 100 pm). L'energia è espressa in kJ/mol. Dati
presi da (http:/ / www. science. uwaterloo. ca/ ~cchieh/ cact/ c120/ bondel. html).
Legame chimico
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996, pp. 38-97. ISBN 88-408-0998-8
Voci correlate
•
•
•
•
•
Elettronegatività
Forza di van der Waals
Dipolo molecolare
Polarità delle molecole
Scissione
Tipi di legame chimico
•
•
•
•
•
Antilegame
Legame covalente
Legame di coordinazione
Legame ionico
Legame idrogeno
• Legame metallico
Caratteristiche del legame chimico
•
•
•
•
•
Energia di legame
Angolo di legame
Energia di dissociazione di legame
Lunghezza di legame
Regole di Fajans
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Chemical bond
Collegamenti esterni
• Il legame chimico (http://www.itchiavari.org/chimica/lab/legame.html)
31
Atomo
32
Atomo
Proprietà
Massa:
da ≈ 1,67 × 10−27 a 4,52 × 10−25 kg
Carica elettrica: zero (quando numero di elettroni e protoni si equivalgono)
Diametro:
da 100 pm (He) a 670 pm (Cs) [1]
L'atomo (dal greco ἄτομος - àtomos -, indivisibile, unione di ἄ - a - [alfa privativo] + τομή - tomé - [divisione], così
chiamato perché inizialmente considerato l'unità più piccola ed indivisibile della materia, risalente alla dottrina dei
filosofi greci Leucippo, Democrito ed Epicuro, detta teoria dell'"atomismo") è la più piccola parte di ogni elemento
esistente in natura che ne conserva le caratteristiche chimico-fisiche. Verso la fine dell'Ottocento (con la scoperta
dell'elettrone) fu dimostrato che l'atomo era divisibile, essendo a sua volta composto da particelle più piccole (alle
quali ci si riferisce con il termine "subatomiche"). L'atomo risulta infatti costituito da neutroni, elettroni e protoni.
La teoria atomica è la teoria sulla natura della materia la quale afferma che tutta la materia sia costituita da unità
elementari chiamati atomi. La teoria atomica si applica agli stati della materia solido, liquido e gassoso, mentre è
difficilmente correlabile allo stato plasmico, in cui elevati valori di pressione e temperatura impediscono la
formazione di atomi.
Atomo
33
Storia
Il modello atomico oggi riconosciuto è l'ultima tappa di una serie di ipotesi che sono state avanzate nel tempo.
Atomismo
In età antica alcuni filosofi greci, quali Leucippo (V secolo a.C.),
Democrito (V-IV secolo a.C.) ed Epicuro (IV-III secolo a.C.), e romani,
quali Tito Lucrezio Caro (I secolo a.C.), ipotizzarono che la materia non
fosse continua, ma costituita da particelle minuscole e indivisibili,
fondando così la "teoria atomica". Questa corrente filosofica, fondata da
Leucippo, venne chiamata "atomismo"[1]. Si supponeva che i diversi
"atomi" fossero differenti per forma e dimensioni.
Democrito propose la "teoria atomica", secondo cui la materia è
costituita da minuscole particelle, diverse tra loro, chiamate atomi, la cui
unione dà origine a tutte le sostanze conosciute. Queste particelle erano
la più piccola entità esistente e non potevano essere ulteriormente divise:
per questo erano chiamate atomi (da ὰτωμος, in greco "indivisibile").
In contrasto con questa teoria, Aristotele (IV secolo a.C.), nella teoria
della continuità della materia, sostenne che una sostanza può essere
suddivisa all'infinito in particelle sempre più piccole e uguali tra loro.
Queste ipotesi rimasero tali in quanto non suffragate da un approccio
scientifico e non verificate con metodologie basate sull'osservazione e
sull'esperimento.
I diversi ordini di grandezza della materia:
1. Materia (macroscopico)
2.Struttura molecolare (atomi)
3.Atomo (neutrone, protone, elettrone)
4.Elettrone
5.Quark
6.Stringhe
Il corpuscolarismo è il postulato del XIII secolo dell'alchimista Geber,
secondo il quale tutti i corpi fisici posseggono uno strato interno e uno
esterno di particelle minuscole. La differenza con l'atomismo è che i
corpuscoli possono essere divisi. Veniva per questo teorizzato che il
mercurio potesse penetrare nei metalli modificandone la struttura
interna. Il corpuscolarismo rimase la teoria dominante per i secoli
successivi. Tale teoria servì come base a Isaac Newton per sviluppare la
teoria corpuscolare della luce.
In età moderna atomista fu Gassendi per via del suo recupero
dell'epicureismo.
Atomo
34
Origine del modello scientifico.
Solo all'inizio del XIX secolo (più precisamente nel 1808) John Dalton
rielaborò e ripropose la teoria di Democrito fondando la teoria
atomica moderna, con la quale diede una spiegazione ai fenomeni
chimici, affermando che le sostanze sono formate dai loro componenti
secondo rapporti ben precisi fra numeri interi (legge delle proporzioni
multiple), ipotizzando quindi che la materia fosse costituita da atomi.
Nel corso dei suoi studi, Dalton si avvalse delle conoscenze chimiche
che possedeva (la legge della conservazione della massa, formulata da
Antoine Lavoisier, e la legge delle proporzioni definite, formulata da
Joseph Louis Proust) e formulò la sua teoria atomica, che espose nel
libro A New System of Chemical Philosophy (pubblicato nel 1808). La
teoria atomica di Dalton si fondava su cinque punti:
• la materia è formata da piccolissime particelle elementari chiamate
atomi, che sono indivisibili e indistruttibili;
• gli atomi di uno stesso elemento sono tutti uguali tra loro;
• gli atomi di elementi diversi si combinano tra loro (attraverso
reazioni chimiche) in rapporti di numeri interi e generalmente
piccoli, dando così origine a composti;
• gli atomi non possono essere né creati né distrutti;
• gli atomi di un elemento non possono essere convertiti in atomi di
altri elementi.[2]
In definitiva questa è la definizione di atomo per Dalton: "Un atomo è
la più piccola parte di un elemento che mantiene le caratteristiche
fisiche di quell'elemento".
Vari atomi e molecole rappresentati nella prima
pagina di "A New System of Chemical
Philosophy", di John Dalton, pubblicato nel 1808.
Questa viene considerata la prima teoria atomica della materia perché per primo Dalton ricavò le sue ipotesi per via
empirica.
I modelli atomici
L'esperimento di Rutherford: poche particelle alfa
vengono deflesse dal campo elettrico del nucleo, la
maggior parte di esse attraversa lo spazio vuoto
dell'atomo.
Con la scoperta della radioattività naturale, si intuì
successivamente che gli atomi non erano particelle indivisibili,
bensì erano oggetti composti da parti più piccole. Nel 1902,
Joseph John Thomson propose il primo modello fisico
dell'atomo[3]: aveva infatti provato un anno prima l'esistenza
dell'elettrone. Egli immaginò che un atomo fosse costituito da una
sfera fluida di materia caricata positivamente (protoni e neutroni
non erano stati ancora scoperti) in cui gli elettroni (negativi) erano
immersi (modello a panettone, in inglese plum pudding model o
modello ad atomo pieno), rendendo neutro l'atomo nel suo
complesso.
Questo modello fu superato quando furono scoperte da Ernest
Rutherford le particelle che formano il nucleo dell'atomo: il
protone. Nel 1911 Rutherford fece un esperimento cruciale, con lo
Atomo
35
scopo di convalidare il modello di Thomson. Egli bombardò un sottilissimo foglio di oro, posto fra una sorgente di
particelle alfa e uno schermo. Le particelle, attraversando la lamina, lasciarono una traccia del loro passaggio sullo
schermo. L'esperimento portò alla constatazione che i raggi alfa non venivano quasi mai deviati; solo l'1% dei raggi
incidenti era deviato considerevolmente dal foglio di oro (alcuni venivano completamente respinti).
Attraverso questo esperimento, Rutherford propose un modello di atomo in cui quasi tutta la massa dell'atomo fosse
concentrata in una porzione molto piccola, il nucleo (caricato positivamente) e gli elettroni gli ruotassero attorno così
come i pianeti ruotano attorno al Sole (modello planetario).[4] L'atomo era comunque largamente composto da
spazio vuoto, e questo spiegava il perché del passaggio della maggior parte delle particelle alfa attraverso la lamina.
Il nucleo è così concentrato che gli elettroni gli ruotano attorno a distanze relativamente enormi, aventi un diametro
da 10.000 a 100.000 volte maggiore di quello del nucleo. Rutherford intuì che i protoni da soli non bastavano a
giustificare tutta la massa del nucleo e formulò l'ipotesi dell'esistenza di altre particelle, che contribuissero a formare
l'intera massa del nucleo. Nel modello atomico di Rutherford non compaiono i neutroni, perché queste particelle
furono successivamente scoperte da Chadwick nel 1932.
Il modello di Rutherford aveva incontrato una palese contraddizione con le leggi della fisica classica: secondo la
teoria elettromagnetica, una carica che subisce una accelerazione emette energia sotto forma di radiazione
elettromagnetica. Per questo motivo, gli elettroni dell'atomo di Rutherford, che si muovono di moto circolare intorno
al nucleo, avrebbero dovuto emettere onde elettromagnetiche e quindi, perdendo energia, annichilire nel nucleo
stesso (teoria del collasso), cosa che evidentemente non accade.[5] Inoltre un elettrone, nel perdere energia, potrebbe
emettere onde elettromagnetiche di qualsiasi lunghezza d'onda, operazione preclusa nella teoria e nella pratica dagli
studi sul corpo nero di Max Planck (e successivamente di Albert Einstein). Solo la presenza di livelli di energia
quantizzati per quanto riguarda gli stati degli elettroni poteva spiegare i risultati sperimentali: la stabilità degli atomi
rientra nelle proprietà spiegabili mediante la meccanica quantistica.
Elettroni nel nucleo?
Dopo l'esperimento di Rutherford era abbastanza evidente che gli elettroni non potessero trovarsi all'interno del
nucleo. Si può, però, pensare ad una dimostrazione per assurdo: si supponga, per un momento, l'esistenza degli
elettroni nel nucleo. Il suo raggio può essere stimato nell'ordine dei 5 fm.
L'impulso dell'elettrone, nell'atomo, allora sarà:
dove c è la velocità della luce e λ la lunghezza d'onda di de Broglie dell'elettrone.
A questo punto si fissa una lunghezza d'onda massima in 10 fm e si può così calcolare il valore minimo per l'impulso,
che alla fine risulta essere di circa 124 MeV/c. Ora, poiché la massa dell'elettrone è pari a 0,5 MeV/c2, da un semplice
conto relativistico risulta evidente che l'energia totale dell'elettrone è pari a:
E2 = p2c2 + m2c4 = 125 MeV
Quindi, se ci fossero elettroni nel nucleo, la loro energia sarebbe 250 volte maggiore rispetto alla loro intera massa:
elettroni così energetici, però, non sono mai stati emessi da alcun nucleo. L'unico indiziato, l'elettrone emesso nel
decadimento beta dei nuclei, ha un intervallo di energia che va da pochi MeV ad un massimo di 20 MeV.
Bohr e la meccanica ondulatoria: l'atomo oggi
Nel 1913 Niels Bohr propose una modifica concettuale al modello di Rutherford. Pur accettandone l'idea di modello
planetario, postulò che gli elettroni avessero a disposizione orbite fisse, dette anche "orbite quantizzate", queste
orbite possedevano un'energia quantizzata (ossia un'energia già prestabilita identificata da un numero detto numero
quantico principale N) nelle quali gli elettroni non emettevano né assorbivano energia (questa infatti rimaneva
costante): in particolare, un elettrone emetteva o assorbiva energia sotto forma di onde elettromagnetiche solo se
effettuava una transizione da un'orbita all'altra, e quindi passava ad uno stato a energia minore o maggiore.[6] Ciò
nonostante, il modello di Bohr-Sommerfeld si basava ancora su postulati e soprattutto funzionava bene solo per
l'idrogeno: tutto ciò, alla luce anche del principio di indeterminazione introdotto da Heisenberg nel 1927, convinse la
Atomo
36
comunità scientifica che fosse impossibile descrivere esattamente il moto degli elettroni attorno al nucleo, motivo
per cui ai modelli deterministici fino ad allora proposti si preferì ricercare un modello probabilistico, che descrivesse
con buona approssimazione qualsiasi atomo. Ciò fu reso possibile grazie ai successivi risultati della meccanica
ondulatoria. Nel 1932 fu scoperto il neutrone, per cui si pervenne presto ad un modello dell'atomo pressoché
completo, in cui al centro vi è il nucleo, composto di protoni (elettricamente positivi) e neutroni (elettricamente
neutri) ed attorno ruotano gli elettroni (elettricamente negativi).
Fu abbandonato il concetto di orbita e fu introdotto il concetto di orbitale. Secondo la meccanica quantistica non ha
più senso infatti parlare di traiettoria di una particella: da ciò discende che non si può neanche definire con certezza
dove un elettrone si trova in un dato momento. Ciò che si poteva conoscere era la probabilità di trovare l'elettrone in
un certo punto dello spazio in un dato istante di tempo. Un orbitale quindi non è una traiettoria su cui un elettrone
(secondo le idee della fisica classica) poteva muoversi, bensì una porzione di spazio intorno al nucleo definita da una
superficie di equiprobabilità, ossia entro la quale c'è il 95% della probabilità che un elettrone vi si trovi. In termini
più rigorosi, un orbitale è definito da una particolare funzione d'onda, l'equazione di Schrödinger, in tre variabili, i
numeri quantici, ciascuna delle quali è associata rispettivamente all'energia, alla forma e all'orientamento nello
spazio dell'orbitale. Fu Erwin Schrödinger (scopritore dell'Equazione di Schrödinger, per cui ha vinto il premio nobel
per la fisica nel 1933) a ipotizzare la struttura dell'atomo come costituita da un nucleo centrale carico di energia
positiva circondato da una nuvola di elettroni.
Alla luce delle ultime ricerche, sfruttando sofisticate e potenti apparecchiature elettroniche, è stato possibile
determinare in modo più completo anche la struttura del nucleo. In particolare si è scoperto che i protoni e i neutroni
sono a loro volta formati da particelle più piccole: i quark.
Componenti
L'atomo è composto principalmente da tre tipologie di particelle subatomiche (cioè di dimensioni minori dell'atomo):
i protoni, i neutroni e gli elettroni.
In particolare:
• i protoni (carichi positivamente) e i neutroni (privi di carica) formano il "nucleo" (carico positivamente); protoni e
neutroni sono detti quindi "nucleoni";
• gli elettroni (carichi negativamente) sono presenti nello stesso numero dei protoni e ruotano attorno al nucleo
senza seguire un'orbita precisa (l'elettrone si dice quindi "delocalizzato"), rimanendo confinati all'interno dei
cosiddetti "gusci elettronici" (o "livelli energetici").
In proporzione, se il nucleo atomico fosse grande quanto una mela, gli elettroni gli ruoterebbero attorno ad una
distanza pari a circa un chilometro; un nucleone ha massa quasi 1800 volte superiore a quella di un elettrone.
La tabella seguente riassume alcune caratteristiche delle tre particelle subatomiche anzidette:[7]
Particella Simbolo Carica
Massa
Note
Elettrone
e-
-1,6 ×
10−19 C
9,1093826 × 10−31 kg
(0,51099 891 MeV/C²)
Scoperto da Thomson in base alle esperienze sui raggi catodici di William
Crookes. Con l'esperimento della goccia d'olio Millikan ne determinò la carica.
Protone
p+
1,6 ×
10−19 C
1,6726231 × 10−27 kg
(9,3828 × 102 MeV/C²)
Scoperto da Ernest Rutherford con l'esperimento dei raggi alfa, la sua esistenza fu
ipotizzata già da Eugene Goldstein, lavorando con i raggi catodici.
Neutrone
n
0C
1,674 927 29 × 10−27 kg
(9,39565 × 102 MeV/C²)
Scoperto da James Chadwick, la sua esistenza fu desunta a partire da
contraddizioni studiate prima da Walther Bothe, poi da Irène Joliot-Curie e
Frédéric Joliot.
Atomo
Si definiscono due quantità per identificare ogni atomo:
• Numero di massa (A): la somma del numero di neutroni e
protoni nel nucleo
• Numero atomico (Z): il numero dei protoni nel nucleo, che,
allo stato neutro, corrisponde al numero di elettroni esterni
ad esso.[8]
Per ricavare il numero dei neutroni si sottrae al numero di
massa il numero atomico.
Esiste una grandezza che ne quantifica la massa, definita peso
atomico (più correttamente "massa atomica"), espresso nel SI
in unità di massa atomica (o uma), dove una unità di massa
atomica equivale alla dodicesima parte della massa di un
Rappresentazione schematica di un atomo di elio. Attorno al
atomo di carbonio-12 (12C). Il numero degli elettroni che
nucleo, composto da due neutroni (in verde) e due protoni
ruotano attorno al nucleo è uguale al numero dei protoni nel
(in rosso), ruotano gli elettroni (in giallo).
nucleo: essendo le predette cariche di valore assoluto uguale,
un atomo è normalmente elettricamente neutro e pertanto la materia è normalmente elettricamente neutra. Tuttavia
esistono atomi che perdono o acquistano elettroni, ad esempio in virtù di una reazione chimica: la specie che ne
deriva si chiama ione; gli ioni possono essere quindi di carica positiva o negativa.
Gli atomi aventi lo stesso numero atomico hanno le stesse proprietà chimiche: si è dunque convenuto a definirli
appartenenti allo stesso elemento.
Due atomi possono differire anche nell'avere numero atomico uguale ma diverso numero di massa: simili atomi sono
detti isotopi ed hanno medesime proprietà chimiche. Ad esempio l'atomo di idrogeno ha più isotopi: in natura infatti
esso è presente in grande maggioranza come 1H (formato da un protone ed un elettrone) e in minore quantità da 2H
(o deuterio[9], che è formato da un protone, un neutrone ed un elettrone) e 3H (o trizio, estremamente raro, formato
da un protone, due neutroni ed un elettrone). Dal punto di vista chimico, idrogeno, deuterio e trizio presentano
identiche proprietà, anche se recenti ricerche stanno rivelando una maggiore instabilità del deuterio nei composti.
Proprietà
Massa
Poiché la parte principale della massa di un atomo deriva dai protoni e neutroni, la massa totale di tali particelle in un
atomo è chiamato numero di massa. Come unità di massa atomica si usa la dodicesima parte della massa di un atomo
di carbonio-12 (12C); tale unità è chiamata Dalton (Da) e vale approssimativamente 1,66 · 10-27 kg.
Dimensione atomica
Gli atomi non hanno un limite ben definito, per questa ragione le dimensioni sono normalmente descritte in termini
delle distanze che i nuclei hanno quando due atomi sono uniti in un legame chimico. Per questa ragione il raggio
varia con la posizione degli atomi nella tavola periodica degli elementi, il tipo di legame chimico, il numero di atomi
vicini (il numero di coordinazione) e persino lo spin. Nella tavola periodica degli elementi la dimensione degli atomi
tende ad aumentare quando ci si muove in basso lungo le colonne, mentre diminuisce andando da sinistra a destra. Di
conseguenza l'atomo più piccolo è l'elio con un raggio di 32 pm, mentre uno degli elementi più grandi è il cesio con
225 pm di raggio. Queste dimensioni sono migliaia di volte più piccole della lunghezza d'onda della luce (400 –
700 nm) per questa ragione non possono essere visti con un microscopio ottico. Mentre possono essere visti con
microscopi elettronici a trasmissione (TEM) o microscopi tunnel a scansione.
37
Atomo
38
Alcuni esempi mostrano la piccola dimensione di un atomo. Il diametro di un tipico capello umano corrisponde a
circa un milione di atomi di carbonio in fila. Una goccia d'acqua contiene 2 · 1021 atomi di ossigeno e 4 · 1021 atomi di idrogeno. Se una mela diventasse della dimensione della terra, gli atomi nella mela sarebbero
approssimativamente delle dimensioni della mela originale.
Note
[1]
[2]
[3]
[4]
[5]
[6]
L'atomismo era una corrente filosofica e non una teoria scientifica, in quanto queste considerazioni non derivavano da evidenze sperimentali.
Queste ultime due proposizioni verranno smentite in seguito dai risultati della Fisica nucleare e subnucleare.
Caforio e Ferilli, PHYSICA 3, Ed. Le Monnier, pag. 251
Silvestroni, op. cit., p. 2
Il fenomeno dell'annichilazione invece avviene tra particella e antiparticella.
per approfondire si veda l'atomo di Bohr.
Questa idea, non compatibile con le leggi della fisica classica di Newton, si fondava sulle idee dell'allora nascente
meccanica quantistica. Il modello di Bohr spiegava molto bene l'atomo di idrogeno, ma non quelli più complessi.
Sommerfeld propose allora una correzione al modello di Bohr, secondo cui si aveva una buona corrispondenza fra la
teoria e le osservazioni degli spettri degli atomi. Uno spettro è l'insieme delle frequenze delle radiazioni
elettromagnetiche emesse o assorbite dagli elettroni di un atomo.
[7] L'elettrone, il protone e il neutrone non sono le uniche particelle subatomiche; infatti dopo la loro scoperta seguirono le scoperte di molte altre
particelle subatomiche.
[8] Nel suo complesso ogni atomo presenta quindi carica elettrica nulla.
[9] nell'acqua pesante gli atomi di idrogeno sono completamente sostituiti da quelli di deuterio.
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• Isaac Asimov, 5 in Breve storia della chimica - Introduzione alle idee della chimica, Bologna, Zanichelli [1965],
1968. ISBN 88-08-04064-X
Voci correlate
•
•
•
•
•
•
•
•
•
•
Elettrone
Protone
Neutrone
Quark
Numero di Avogadro
Storia della chimica
Atomo di Bohr
Atomismo
Particella elementare
Superatomo
Altri progetti
•
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Atom
Wikiquote contiene citazioni: http://it.wikiquote.org/wiki/Atomo
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Atomo
Molecola
39
Molecola
In fisica, in particolare in fisica dello stato solido, una molecola,
dal latino scientifico molecula, derivato a sua volta da moles, che
significa "mole", "piccola quantità", è un insieme di almeno due
atomi (dello stesso elemento o di elementi diversi) uniti da un
legame chimico covalente.[1][2]
Le molecole sono i costituenti fondamentali della maggior parte
della materia organica presente nell'universo, oltre che degli
oceani e dell'atmosfera terrestre. Nonostante la maggior parte delle
sostanze solide che costituiscono la Terra contengano legami
covalenti, tuttavia, non è possibile identificare singole molecole
all'interno di esse.
Modello della molecola del saccarosio (principale
componente dello zucchero da tavola)
Nella teoria cinetica dei gas il termine "molecola" è spesso usato per indicare qualsiasi particella gassosa
indipendentemente dalla sua composizione; secondo tale definizione i gas nobili sono considerati molecole,
nonostante siano composti da singoli atomi.[3]
Una molecola può essere composta da più atomi di un solo elemento chimico o da atomi di elementi diversi.
Molecole costituite dagli stessi atomi, ma disposti nello spazio in maniera diversa sono dette isomeri. Tale
disposizione definisce anche le proprietà fisiche della sostanza.
Dinamica molecolare
Le proprietà fondamentali delle molecole sono determinate delle leggi che governano il moto dei corpi dai quali sono
composte. Tali leggi consentono di spiegare l'interazione tra gli atomi di una molecola e tra molecole diverse,
permettendo di conoscere la natura dei legami fisici e chimici che stanno alla base dello studio della materia.
Lo studio della dinamica molecolare si basa sull'approssimazione di Born-Oppenheimer, anche detta
approssimazione adiabatica, che permette di poter considerare il moto dei nuclei indipendente da quello degli
elettroni, dal momento che i primi sono estremamente più lenti e pesanti dei secondi. Questo rende possibile la
fattorizzazione della funzione d'onda totale della molecola:[4][5]
dove il pedice e indica la funzione d'onda degli elettroni, il pedice n dei nuclei, ed
e
sono rispettivamente le
posizioni di nuclei ed elettroni.
Tale funzione d'onda soddisfa l'equazione agli autovalori:
dove
è l'energia cinetica degli elettroni,
elettroni,
quella dei nuclei,
l'interazione coulombiana tra gli elettroni e
La precedente espressione è ottenuta grazie al fatto che l'operatore
l'interazione coulombiana tra nuclei ed
quella tra i nuclei.
, contenuto nel termine
, non agisce sulle
coordinate dei nuclei.
La funzione d'onda degli elettroni, nell'approssimazione adiabatica, soddisfa l'equazione agli autovalori:
La funzione d'onda dei nuclei, invece, è ricavata a partire dall'equazione totale, che esplicitanto l'operatore impulso
diventa:
Molecola
40
Essendo che:
Si ottiene:
Andamento del potenziale adiabatico in funzione della
distanza di separazione tra i nuclei in una molecola
biatomica
che, trascurando per l'approssimazione adiabatica il termine:
diventa, inserendo la soluzione
dell'equazione elettronica:
che è l'equazione del moto dei nuclei.
Il potenziale che guida il moto dei nuclei:
è detto potenziale adiabatico, e sta alla base della dinamica della molecola.
Dall'espressione del potenziale adiabatico si evince che la dinamica dei nuclei è guidata dall'energia
fornita
dall'equazione elettronica: questo termine è fondamentale, dal momento che rappresenta il "collante" che tiene uniti i
nuclei degli atomi che compongono la molecola.[6]
Per le molecole biatomiche il potenziale adiabatico è un potenziale armonico, e può essere approssimato dal
potenziale di Morse, che a differenza dell'oscillatore armonico quantistico include esplicitamente gli effetti della
rottura del legame chimico, come l'esistenza di stati non legati.
Molecola
41
Molecole biatomiche
Le molecole diatomiche sono composte da due atomi, e si distinguono in molecole omonucleari, quando gli atomi
sono dello stesso elemento chimico, ed eteronucleari, quando invece gli atomi differiscono.
La molecola H2+
Le molecole diatomiche omonucleari sono composte da due atomi
dello stesso elemento chimico; la più semplice di queste è H2+, per
la quale l'equazione elettronica assume la forma:[7]
Orbitale molecolare di legame
Orbitale molecolare di antilegame
dove
, il secondo ed il terzo termine rappresentano l'attrazione Vne dell'elettrone nei confronti dei
nuclei ed il quarto la repulsione dei due nuclei.
I due protoni formano due buche di potenziale, e la funzione d'onda dell'elettrone è la combinazione lineare di due
funzioni d'onda idrogenoidi
:[8]
La funzione d'onda
[9]
antilegame.
Le funzioni
costituisce l'orbitale molecolare di legame, la funzione
costituisce l'orbitale di
L'orbitale di legame ha energia minore dell'orbitale di antilegame, ed è per questo il più probabile.
, sebbene descrivano bene la distribuzione di probabilità dell'elettrone nello stato fondamentale, non
sono soluzioni esatte dell'equazione elettronica.
La funzione d'onda
, nello spazio tra i due nuclei, è maggiore delle singole funzioni d'onda idrogenoidi
, ed
è questo fatto che genera il legame covalente tra i due nuclei. Si nota infatti che la densità di probabilità associata
alla funzione d'onda:
Molecola
42
contiene un termine di interazione, il doppio prodotto, che rappresenta la sovrapposizione delle due funzioni d'onda:
si tratta di una regione di carica negativa che unisce i due nuclei di carica opposta.
Per quanto riguarda l'orbitale di antilegame
, esso si annulla a metà tra i due nuclei, dove genera una densità di
probabilità minore di quella che avrebbe senza il termine di sovrapposizione.
La molecola H2
Si consideri ora la molecola H2, la più semplice molecola neutra. Avendo due
elettroni, la funzione d'onda elettronica di singoletto è data da:[10]
Orbitale di legame di H2
Orbitale di antilegame di H2
e rappresenta l'orbitale di legame, mentre quella di tripletto da:[11]
che rappresenta l'orbitale di antilegame, dove:
e
sono gli stati di spin, in cui + rappresenta lo spin-up, - lo spin-down.
La densità di probabilità spaziale è:[11]
Anche in questo caso il termine di interferenza rappresenta la sovrapposizione delle funzioni d'onda idrogenoidi nella
regione tra i nuclei, e comporta un aumento di carica nel caso di singoletto (segno +), ed una diminuzione di carica
nel tripletto (segno -).
Molecola
43
Molecole eteronucleari
Nelle molecole eteronucleari la simmetria che caratterizzava le molecole
omonucleari viene a mancare, e gli orbitali non sono una pura
combinazione simmetrica e antisimmetrica degli orbitali atomici. In tali
molecole gli orbitali possono essere approssimati con gli autostati di una
matrice quadrata di dimensione 2:[12]
La molecola biatomica eteronucleare dell'acido
fluoridrico
dove:
è l'effettiva hamiltoniana di singolo elettrone mentre gli stati
e
sono gli orbitali corrispondenti
rispettivamente all'atomo sinistro e destro.
Gli autovalori associati alla matrice sono:
Gli orbitali di legame
e antilegame
sono dati dagli autostati:
con:
per
si ottiene la molecola omonucleare, ed il termine
rappresenta lo splitting tra l'orbitale di legame e di
antilegame di una molecola omonucleare, ovvero lo splitting tra le combinazioni simmetriche ed antisimmetriche.[12]
Al crescere di gli autostati di legame e di antilegame assomigliano sempre più agli orbitali
e
dei
singoli atomi, e lo stesso avviene per i rispettivi autovalori dell'energia.[13] Quando la differenza
da comportare un trasferimento completo di carica tra i due atomi, il legame si dice ionico.
è tale
Molecola
44
Molecole poliatomiche
Le molecole poliatomiche possiedono più di due atomi, che nella maggior
parte dei casi sono diversi fra loro. La loro struttura è estremamente
diversificata poiché le possibili combinazioni tra gli orbitali atomici che
formano gli orbitali molecolari sono estremamente numerose.
Oltre al legame che caratterizza le molecole biatomiche, nelle molecole
poliatomiche gli orbitali atomici s e p si possono combinare fra loro per
formare orbitali detti ibridi.
La molecola dell'acqua
Si riportano di seguito due esempi di molecole poliatomiche, l'acqua ed il
metano:
La molecola H2O
Una delle più semplici molecole poliatomiche è quella dell'acqua, in cui
l'ossigeno ha un orbitale p caratterizzato da una tripla degenerazione sui tre
assi cartesiani, che genera due possibili configurazioni elettroniche: la prima è
il caso in cui i 4 elettroni riempiono completamente due lobi dell'orbitale,
lasciando il terzo vuoto, mentre la seconda è il caso in cui si abbiano due
elettroni su un lobo, ed uno su ognuno dei restanti due. Tale orbitale può
essere quindi scritto come 2pxpypz2, in cui si è supposto che il lobo diretto
lungo l'asse z contenga due elettroni, e questo rende possibile la formazione di
due legami covalenti, in cui ai lobi x e y si legano i due atomi di idrogeno.[14]
Gli orbitali ibridi nella molecola di
metano
La molecola CH4
Il metano è una molecola con un orbitale ibrido. Il carbonio ha configurazione
elettronica 1s22s22p2, e l'orbitale p e nel suo stato fondamentale può quindi
legarsi con solo due atomi di idrogeno. La molecola di metano esiste, tuttavia,
dal momento che un elettrone dell'orbitale 2s2 viene promosso all'orbitale p,
sicché la configurazione elettronica diventa 1s22s2pxpypz, generando quattro
elettroni disaccoppiati che possono legarsi ad altrettanti atomi di idrogeno.
I quatro orbitali molecolari ibridi sono quindi una combinazione lineare degli
stati
,
,
,
della forma:[15]
La struttura della molecola di metano
e formano un tetraedro con l'atomo di carbonio al centro.
Orbitali e legami molecolari
L'orbitale molecolare caratterizza la configurazione elettronica di una molecola, definendo la distribuzione spaziale e
l'energia degli elettroni, ed è stato introdotto da Friedrich Hund[16][17] e Robert S. Mulliken[18][19] nel 1927 e
1928.[20][21]
Un orbitale molecolare è rappresentato da una funzione d'onda il cui quadrato descrive la distribuzione di probabilità
relativa alla posizione dell'elettrone. Tale funzione d'onda si ottiene dall'equazione d'onda che descrive l'intera
molecola, che in generale non è di facile soluzione: questa problematica viene risolta mediante un'approssimazione
che consiste nello scrivere l'orbitale molecolare come combinazione lineare degli orbitali atomici dei singoli atomi.
Molecola
45
Tale approssimazione è descritta dalla teoria degli orbitali molecolari.
L'ordine di legame è inoltre la semidifferenza tra il numero di elettroni leganti e il numero di elettroni antileganti.
L'ordine di legame è un indice della forza del legame stesso e viene utilizzato estensivamente anche nella teoria del
legame di valenza.
Teoria degli orbitali molecolari
La teoria degli orbitali molecolari è una tecnica per determinare la struttura molecolare in cui si pone che gli elettroni
non siano assegnati a particolari legami chimici, ma siano trattati come oggetti che si muovono sotto l'influenza dei
nuclei all'interno dell'intera molecola.[22]
La funzione d'onda totale degli elettroni
combinazione lineare:
dove
è quindi scritta come
[23]
sono gli orbitali atomici, e
i coefficienti della
sommatoria, ricavati risolvendo l'equazione di Schrödinger per
ed applicando il principio variazionale.
Le proprietà principali degli orbitali molecolari così definiti sono:
• Il numero degli orbitali molecolari è pari al numero di orbitali
atomici contenuti nella combinazione lineare dalla quale sono
costituiti, poiché gli stati stazionari non si creano né si
distruggono.[24]
• Se la molecola possiede simmetrie, gli orbitali atomici
degeneri, caratterizzati dalla stessa energia, sono raggruppati in
combinazioni lineari che appartengono alla rappresentazione
del gruppo di simmetria.
Combinazione degli orbitali atomici 1s nella molecola
biatomica omonucleare E2. In alto vi è la combinazioni
antisimmetrica, che costituisce l'orbitale antilegante, in
basso quella simmetrica, meno energetica, che
costituisce l'orbitale legante.
• Il numero di orbitali molecolari appartenenti alla rappresentazione di un gruppo è pari al numero di orbitali
atomici appartenenti a tale rappresentazione.
• All'interno di una particolare rappresentazione, gli orbitali atomici si mischiano maggiormente tanto più i loro
livelli di energia atomici sono vicini.
Rappresentazione degli orbitali molecolari
La nomenclatura degli orbitali molecolari ricalca quella degli orbitali atomici: quando un orbitale ha simmetria
cilindrica rispetto alla congiungente dei due nuclei, detta direzione di legame, viene indicato con la lettera greca ;
quando si trova da parti opposte rispetto alla direzione di legame viene indicato con . Accanto alla lettera si scrive un
indice che indica da quale tipologia di legame atomico è formato l'orbitale molecolare.[25]
Vi è inoltre una terza tipologia di legame, denotato con , ottenuto dalla sovrapposizione di quattro lobi di due orbitali
atomici.
Esistono
in
questo
caso
due
piani
nodali
siti
fra
i
due
Molecola
46
nuclei che contraggono tale legame. Il legame δ è riscontrato nel
legame quadruplo, legame multiplo importante in chimica
inorganica e che caratterizza complessi quale [Re2Cl10]4- o altri
tipi di cluster.
L'orbitale di antilegame si denota inoltre con un asterisco, ad
esempio la molecola H2 possiede un orbitale di legame
ed un
orbitale di antilegame
.
Combinazione degli orbitali atomici 2p e 2s nella
molecola biatomica omonucleare O2. In alto vi sono le
combinazioni degli orbitali atomici 2p, in basso quelle
degli orbitali 2s.
• Nelle molecole biatomiche omonucleari gli elettroni riempiono gli orbitali con lo stesso schema con cui avviene il
riempimento degli orbitali atomici, con l'uinica eccezione che tra gli orbitali derivanti dagli orbitali atomici 2p, gli
orbitali , hanno energia minore degli orbitali a causa del fatto che la repulsione coulombiana degli orbitali
derivati dagli orbitali atomici 1s e 2s aumenta l'energia degli stati . Questo è dovuto al fatto che gli elettroni dei
due legami sono situati nella regione tra i due nuclei, e pertanto si respingono; nelle molecole più pesanti
dell'ossigeno gli orbitali
hanno energia minore e sono situati in prossimità dei nuclei, pertanto il naturale
ordinamento energetico è ristabilito.
La combinazione lineare delle funzioni d'onda che forma l'orbitale molecolare è rappresentata a lato, dove sono
schematizzate la molecola He2 e la molecola O2, la quale ha configurazione elettronica:
.[26]
• Nel caso di molecole biatomiche eteronucleari, se il numero atomico dei due atomi differisce di poco il
procedimento che forma gli orbitali è lo stesso delle molecole omonucleari. Vi è tuttavia una differenza di
elettronegatività tra i due atomi, e ciò implica la presenza di un dipolo elettrico tra di essi dovuto al fatto che gli
elettroni si distribuiscono nelle vicinanze dell'atomo più elettronegativo:[27] il legame che si viene a formare
prende il nome di covalente polare.
Molecola
47
Tale legame viene rappresentato come
in figura a lato, e si può notare che gli
elettroni di
hanno energia
maggiore, e costituiscono un orbitale
detto HOMO (Highest Occupied
Molecular Orbital), mentre gli elettroni
di
e
costituiscono gli
orbitali vuoti a minore energia detti
La molecola del monossido di carbonio CO
LUMO
(Lowest
Unoccupied
Molecular Orbital). L'orbitale LUMO è
il centro in cui la molecola può subire
un attacco nucleofilo di una base di
Lewis, e si tratta quindi del centro di
acidità di Lewis. Viceversa, HOMO è
il centro di basicità di Lewis della
molecola, e può subire un attacco
elettrofilo.
Se la differenza di elettronegatività è maggiore di un valore convenzionale fissato a 1,9 vi è un trasferimento
completo di carica tra i due atomi, cioè la nuvola elettronica può considerarsi come spostata completamente
sull'elemento più elettronegativo. Tale legame prende il nome di legame ionico.
Se il numero atomico dei due atomi differisce di molto accade che gli orbitali molecolari si formino tra orbitali
atomici con energia simile, invece che dello stesso tipo.[28]
• All'aumentare del numero di atomi convolti diventa complessa la caratterizzazione degli orbitali, a nell'ambito
della teoria degli orbitali molecolari sono stati sviluppati diversi metodi di calcolo degli orbitali, tra i quali vi sono
il Metodo di Hückel, proposto da Erich Hückel nel 1930, consiste in un semplice metodo LCAO utilizzato per la
determinazione delle energie degli orbitali molecolari di sistemi π rappresentati da idrocarburi con legami
coniugati, risultando applicabile a molecole quali ad esempio l'etilene, il benzene e il butadiene.[29][30] La nota
regola di Hückel trae origine da queste basi.
Il metodo di Hückel esteso, sviluppato da Roald Hoffmann, rappresenta invece la base delle regole di
Woodward-Hoffmann[31] ed è un'estensione a tutti gli orbitali di valenza. Negli anni successivi il metodo fu reso
applicabile anche agli eterocicli come la piridina, il pirrolo e il furano.[32]
Vi è infine il metodo di Pariser–Parr–Pople, che sfrutta metodi semi-empirici della chimica quantistica
nell'ambito della chimica organica.
Moti interni nelle molecole biatomiche
I nuclei sono soggetti al potenziale adiabatico definito in precedenza, che nelle molecole biatomiche è indipendente
dalla posizione del centro di massa della molecola e dall'orientazione della retta congiungente i due nuclei. Il
potenziale gode quindi di invarianza rispetto alle traslazioni ed alle rotazioni, e il moto dei nuclei può essere studiato
come un problema a due corpi, sicché l'equazione di Schrödinger può essere separata in moto radiale, dipendente
dalla distanza tra i due nuclei, e moto orbitale, dipendente dal numero quantico orbitale. L'equazione di Schrödinger
nel caso di un moto in un campo centrale è:
dove
indica la posizione del centro di massa e
rispettive posizioni.
la posizione relativa dei due nuclei, differenza delle
Molecola
48
Il problema può essere quindi separato in due equazioni, una per il centro di massa ed una per la particella di massa μ
che si muove in un campo centrale rispetto al centro di massa. La funzione d'onda si può quindi fattorizzare nel
seguente modo:
. L'equazione per
, che rappresenta il problema della particella
della molecola. L'equazione per
si può ulteriormente fattorizzare in parte radiale, dipendente da r, e parte angolare,
dipendente dalle coordinate angolari:
La soluzione per
.
sono le armoniche sferiche, ed i rispettivi stati sono autostati del momento angolare orbitale e della
sua componente lungo l'asse z.
L'equazione per
è invece, detta
:[33]
dove il secondo termine rappresenta il contributo energetico rotazionale
, che dipende dal numero quantico
orbitale l.
Il potenziale adiabatico può essere inoltre sviluppato in serie di Taylor, che troncata al secondo ordine è:[5]
dove
è il valore di
che minimizza
, e rappresenta la posizione di equilibrio dei due nuclei. Tale
espressione rappresenta un moto armonico attorno a
dell'equazione elettronica contenuta in
Detta
la lunghezza caratteristica data
dalla relazione
e
detta
,
dell'equazione per
dove
grado
le
che fornisce un contributo energetico dato dall'energia
e dall'energia vibrazionale
.
soluzioni
sono:
è il polinomio di Hermite di
.
Lo spettro energetico contiene in definitiva
tre termini:
Tali termini sono i contributi energetici che
caratterizzano la dinamica della molecola
biatomica, e nello specifico sono:[5][34]
• Il contributo elettronico, dato dal termine
di
, che definisce la profondità
Livelli energetici di una molecola: per ogni livello elettronico, associato ad una
superficie adiabatica, vi sono diversi livelli vibrazionali, e per ogni livello
vibrazionale vi sono diversi livelli rotazionali.
della buca
di potenziale generata dai due nuclei,
responsabile del legame chimico. I livelli energetici associati a questo termine sono detti superfici adiabatiche, e
corrispondono ai diversi stati energetici degli elettroni. Gli elettroni che vengono promossi da un orbitale ad un
altro, ad esempio da un orbitale di legame ad uno di antilegame, effettuano una transizione
Molecola
49
tra due valori
e
del potenziale adiabatico.
Tali transizioni sono dell'ordine di 10 eV,
e a differenti superfici adiabatiche
corrispondono anche diversi valori di
. Le
transizioni elettroniche tra due di tali
superfici sono inoltre accompagnate da
transizioni tra diversi stati vibrazionali e
rotazionali.
• Il contributo vibrazionale, meno
energetico del precedente, che
nell'approssimazione di moto armonico
fornita dall'esclusione dei termini
superiori al secondo ordine nel
precedente sviluppo di
è dato
dagli autovalori dell'oscillatore armonico
quantistico:
dove
è la costante di Planck e
Rappresentazione dei livelli energetici vibrazionali all'interno di una superficie
adiabatica, approssimata dal potenziale di Morse. In verde il potenziale ed i
rispettivi livelli eccitati dell'oscillatore armonico corrispondente.
la frequenza angolare dell'oscillazione
intorno a
.
La frequenza è data da:
con
e
la massa ridotta dell'oscillatore a due corpi, data dal rapporto tra il prodotto e la somma delle masse dei
due nuclei.
Tale contributo descrive il moto armonico dei due nuclei intorno alla posizione di equilibrio, e transizioni tra
due livelli vibrazionali sono dell'ordine del decimo di eV.
• Il contributo rotazionale, il meno energetico dei tre, fornito dall'equazione angolare dell'atomo di idrogeno, pari a:
dove
è il momento angolare orbitale e
il momento d'inerzia.
Tale contributo è generalmente dell'ordine dei meV, ed è calcolato assumendo
In conclusione, quindi, l'energia interna di una molecola biatomica è:
dove i termini sono elencati in ordine di importanza.
.
Molecola
50
Moti interni nelle molecole poliatomiche
Nelle molecole poliatomiche il calcolo dello spettro energetico può essere molto complesso. Le simmetrie della
molecola giocano spesso un ruolo determinante al fine di ottenere gli autovalori dell'energia vibrazionale e
rotazionale.
Moto vibrazionale
Nelle molecole poliatomiche l'energia cinetica data dal moto vibrazionale è espressa come:
dove le coordinate cartesiane sono le posizioni del nucleo α-esimo rispetto alla posizione di equilibrio.
Utilizzando coordinate mass–weighted:
è possibile definire la matrice
di elementi:
E quindi, come nelle molecole biatomiche, l'energia vibrazionale può essere espressa come:
dove
è il vettore che ha per componenti
Le equazioni del moto sono date dal sistema di equazioni
differenziali:
Ogni atomo vibra con la stessa frequenza angolare, e tali frequenze sono dette modi normali di vibrazione, che si
ottengono dalle radici dell'equazione caratteristica per la matrice :
Moto rotazionale
Considerando la moelcola un corpo rigido, è possibile definire il momento d'inerzia attorno a un asse a come:
Gli assi d'inerzia di una molecola sono tre, e i rispettivi momenti d'inerzia sono
Se
, il corpo rigido è detto asymmetrical top, se
,
,
è detto symmetrical top, mentre se
è detto spherical top. All'interno dei corpi rigidi symmetrical top, se
oblato , si tratta di una molecola piatta, come il benzene, se invece
.
il copo è detto
è detto prolato, e si tratta di una
molecola allungata, come il pentacloruro di fosforo.
L'energia cinetica è data da:
dove
,
ed
sono le tre componenti dell'operatore momento angolare totale di rotazione della molecola
lungo gli assi di inerzia a, b e c.
• Nel caso di uno spherical top si ottiene immediatamente che gli autovalori dell'energia rotazionale sono:
e la degenerazione degli autovalori è
.
Molecola
51
• Nel caso di un symmetrical top si ha:
e dal momento che
commuta con ogni sua componente e con
, l'autofunzione associata all'energia
vibrazionale è simultanea a questi tre operatori.
L'energia rotazionale è data allora da:
con degenerazione
se m è diverso da zero,
se è invece nullo.
• Il caso di asymmetrical top è più complesso, ed è necessario diagonalizzare la matrice di
nella basse delle
autofunzioni di L e Lz.
Spettro elettromagnetico molecolare
Lo spettro elettromagnetico molecolare è
generato dalle transizioni tra due autostati
dell'energia totale. Nel caso si studi lo spettro di
emissione la molecola passa da uno stato
eccitato allo stato fondamentale, mentre nel caso
si studi lo spettro di assorbimento si osserva la
transizione inversa. Tale passaggio comporta
l'emissione o l'assorbimento di un fotone, la cui
frequenza è data dalla legge di Planck:
dove
è la differenza di energia tra i due
stati di partenza e arrivo:
Le transizioni elettroniche dallo stato
fondamentale ai primi stati eccitati sono
dell'ordine di alcuni eV, e sono osservate nella
regione del visibile e dell'ultravioletto dello
spettro elettromagnetico, mentre le transizioni
roto-vibrazionali sono osservate nella regione
dell'infrarosso.[35]
Le transizioni tra due autostati dell'energia totale
vengono studiate attraverso le transizioni tra
autostati del momento di dipolo elettrico,
definito come:[5]
con e la carica dell'elettrone.
Tale operatore è esplicitato dall'espressione:
Diagramma delle transizioni energetiche roto-vibrazionali in una molecola tra
due stati vibrazionali. Le transizioni del Q branch non sono permesse, in
quanto
non è permesso, mentre si ha
per il P
branch e
per il Q branch.
Molecola
dove
52
è l'operatore di momento dipolare elettronico della molecola:
Ognuno dei livelli vibrazionali che caratterizzano una superficie adiabatica è associato a diversi stati rotazionali. Nel
diagramma spettroscopico le transizioni rotazionali costituiscono due rami: il primo è detto R Branch, e rappresenta
le transizioni rotazionali tra i numeri quantici
, mentre il secondo, detto P branch, rappresenta le
transizioni
. Tra i due rami vi è un vuoto, motivato dal fatto che la transizione
è proibita
[36]
dalle regole di selezione.
Quando la transizione viene effettuata da un elettrone, essa genera anche transizioni tra autostati dell'energia
roto-vibrazionale dei nuclei: tali transizioni sono dette vibroniche, e sono causate dal fatto che a due differenti
superfici adiabatiche corrispondono geometrie diverse della molecola. In particolare, nelle molecole biatomiche,
corrispondono a distanze internucleari differenti.
Spettro nucleare
Spettro nelle molecole biatomiche
Nel caso di molecole biatomiche
omonucleari il momento di dipolo
elettrico è nullo per motivi di
simmetria,[37] e questo fatto spiega la
trasparenza dell'atmosfera terrestre,
composta prevalentemente da O2 e N2.
Nelle
molecole
biatomiche
eteronucleari, invece, l'elemento di
matrice
della
componente
lungo l'asse z del
Spettro di assorbimento rotovibrazionale della molecola di HCl nella transizione tra lo
stato fondamentale ed il primo stato eccitato: a sinistra l'R Branch e a destra il P branch.
momento di dipolo è:[5]
dove
sono gli autostati simultanei dell'energia vibrazionale e rotazionale. Lo stesso accade per le
componenti x e y.
Dalle proprietà delle armoniche sferiche e dallo sviluppo di
attorno alla distanza di equilibrio si ottengono le
regole di selezione:
che definiscono le transizione permesse tra autostati dell'operatore associato all'osservabile dipolo elettrico.
Spettro nelle molecole poliatomiche
L'operatore di momento dipolare elettronico di una molecola poliatomica è dato da:[5]
in cui
sono i versori degli assi d'inerzia.
Il momento di dipolo elettrico diventa:
Detto
il vettore delle coordinate normali, le cui componenti sono:
Molecola
ed espandendo in serie di Taylor
53
attorno alla posizione di equilibrio:
si ottengono i due termini che generano le transizioni. Le transizioni dovute al primo termine del secondo membro
sono nella regione delle microonde dello spettro, mentre le transizioni dovute al secondo termine nell'infrarosso. Il
secondo termine fornisce inoltre le regole di selezione relative all'osacillatore armonico corrispondente:
.
Per quanto riguarda lo spettro rotazionale, si ha che gli spherical top ed i symmetrical top planari hanno dipolo nullo,
e pertanto non generano transizioni di dipolo. Nel caso di symmetrical top non planari, il dipolo è diretto lungo l'asse
di simmetria, e le transizioni tra autostati degli operatori ,
ed
sono rispettivamente:
e si rilevano nella regione delle microonde dello spettro.
Spettro elettronico
Una transizione elettronica molecolare consiste in
una transizione da parte dell'elettrone tra due
superfici adiabatiche. Tali transizioni sono simili
a quelle atomiche, e consistono nella promozione
di un elettrone da un orbitale molecolare ad un
altro orbitale vuoto.[35]
Le regole di selezione si ricavano osservando che
l'operatore di spin totale:
commuta con l'hamiltoniana elettronica e con
,
l'operatore di dipolo non agisce sullo spin, e
pertanto si ha che
.[5]
Per l'operatore di momento angolare nelle
molecole biatomiche:
Transizioni elettroniche tra due superfici adiabatiche approssimate dal
potenziale di Morse, in cui si evidenzia la sovrapposizione delle funzioni
d'onda che sta alla base del principio di Franck Condon.
solo la componente lungo l'asse z commuta con
, ottenendo che
, mentre per le
altre due componenti si ricava che
.
In definitiva si ha:
Il principio di Franck Condon
Il principio di Franck Condon afferma la probabilità associata ad una transizione vibrazionale, data da:
aumenta all'aumentare della sovrapposizione delle funzioni d'onda dei rispettivi stati iniziale e finale. Questo
comporta che i livelli vibrazionali associati allo stato finale sono favoriti nel momento in cui la transizione comporta
un cambiamento minimo nelle coordinate nucleari. Una conseguenza del principio è che, ad esempio, come mostrato
nella figura a sinistra, se le funzioni d'onda tra lo stato fondamentale della superficie adiabatica iniziale e il secondo
Molecola
stato eccitato della superficie adiabatica finale si sovrappongono, tale transizione è più probabile delle altre dal
momento che minimizza la variazione delle coordinate dei nuclei.
Note
[1]
[2]
[3]
[4]
[5]
Pauling, Linus, op. cit.
Ebbin, Darrell, op. cit.
Sulekh Chandra, Comprehensive Inorganic Chemistry, New Age Publishers. ISBN 8122415121
Manini, op. cit., Pag. 61
Renzo Cimiraglia - Note al corso di Spettroscopia Molecolare (http:/ / chim183. unife. it/ chifi3/ files/ chifi3. pdf). URL consultato il 15
novembre 2010.
[6] Manini, op. cit., Pag. 62
[7] Brehm, Mullins, op. cit., Pag. 503
[8] Brehm, Mullins, op. cit., Pag. 504
[9] Brehm, Mullins, op. cit., Pag. 507
[10] Brehm, Mullins, op. cit., Pag. 509
[11] Brehm, Mullins, op. cit., Pag. 510
[12] Manini, op. cit., Pag. 70
[13] Manini, op. cit., Pag. 71
[14] Brehm, Mullins, op. cit., Pag. 521
[15] Brehm, Mullins, op. cit., Pag. 522
[16] F. Hund, "Zur Deutung einiger Erscheinungen in den Molekelspektren" [On the interpretation of some phenomena in molecular spectra]
Zeitschrift für Physik, vol. 36, pages 657-674 (1926).
[17] F. Hund, "Zur Deutung der Molekelspektren," Zeitschrift für Physik, Part I, vol. 40, pages 742-764 (1927); Part II, vol. 42, pages 93-120
(1927); Part III, vol. 43, pages 805-826 (1927); Part IV, vol. 51, pages 759-795 (1928); Part V, vol. 63, pages 719-751 (1930).
[18] R. S. Mulliken, "Electronic states. IV. Hund's theory; second positive nitrogen and Swan bands; alternate intensities," Physical Review, vol.
29, pages 637 - 649 (1927).
[19] R. S. Mulliken, "The assignment of quantum numbers for electrons in molecules," Physical Review, vol. 32, pages 186 - 222 (1928).
[20] Friedrich Hund and Chemistry, Werner Kutzelnigg, on the occasion of Hund's 100th birthday, Angewandte Chemie, 35, 573 - 586, (1996)
[21] Robert S. Mulliken's Nobel Lecture, Science, 157, no. 3785, 13 - 24, (1967).
[22] Daintith, J., Oxford Dictionary of Chemistry, New York, Oxford University Press, 2004. ISBN 0-19-860918-3
[23] Licker, Mark, J., McGraw-Hill Concise Encyclopedia of Chemistry, New York, McGraw-Hill, 2004. ISBN 0-07-143953-6
[24] Spinicci, op. cit., Pag. 185
[25] Spinicci, op. cit., Pag. 181
[26] Spinicci, op. cit., Pag. 182
[27] Spinicci, op. cit., Pag. 187
[28] Spinicci, op. cit., Pag. 188
[29] E. Hückel, Zeitschrift für Physik, 70, 204, (1931); 72, 310, (1931); 76, 628 (1932); 83, 632, (1933)
[30] Hückel Theory for Organic Chemists, C. A. Coulson, B. O'Leary and R. B. Mallion, Academic Press, 1978
[31] Stereochemistry of Electrocyclic Reactions R. B. Woodward, Roald Hoffmann J. Am. Chem. Soc.; 1965; 87(2); 395-397
[32] Andrew Streitwieser, Molecular Orbital Theory for Organic Chemists, Wiley, New York, 1961
[33] Brehm, Mullins, op. cit., Pag. 523
[34] Manini, op. cit., Pag. 76
[35] Manini, op. cit., Pag. 79
[36] Manini, op. cit., Pag. 78
[37] Brehm, Mullins, op. cit., Pag. 528
54
Molecola
Bibliografia
• (EN) John Brehm; William J. Mullins, Introduction To The Structure Of Matter: A Course In Modern Physics,
Greenville, NC, U.S.A., John Wiley & Sons, 1989. ISBN 978-0-471-60531-7
• (EN) Nicola Manini, Introduction to the Physics of Matter, Milano, CUSL, 2008. ISBN 978-88-8132-552-8
• Roberto Spinicci, Elementi di Chimica, Firenze, Firenze University Press, 2009. ISBN 978-88-6453-062-8
• (EN) Pauling, Linus, General Chemistry, New York, Dover Publications, Inc., 1970. ISBN 0486656225
• (EN) Ebbin, Darrell, D., General Chemistry, 3rd Ed., Boston, Houghton Mifflin Co., 1990. ISBN 0395433029
• (EN) Brown, T.L., Chemistry – the Central Science, 9th Ed., New Jersey, Prentice Hall, 2003. ISBN 0130669970
• (EN) Chang, Raymond, Chemistry, 6th Ed., New York, McGraw Hill, 1998. ISBN 0071152210
• (EN) Zumdahl, Steven S., Chemistry, 4th ed., Boston, Houghton Mifflin, 1997. ISBN 0669417947
Voci correlate
•
•
•
•
Atomo
Composto organico
Formula chimica
Macromolecola
•
•
•
•
•
•
•
Molecola biatomica omonucleare
Interazione debole
Isomeria
Legame chimico
Simmetria molecolare
Storia della chimica
Orbitale molecolare
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/
Category:Molecules
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Molecola
55
Stato della materia
Stato della materia
Con stato della materia o stato di aggregazione si intende una classificazione convenzionale dello stato di
aggregazione della materia a seconda delle sue proprietà meccaniche.
Tre stati classici
La distinzione tra gli stati della materia viene storicamente fatta basandosi sulle seguenti differenze qualitative:
• un materiale allo stato solido ha un volume e una forma propria;
• un materiale allo stato liquido ha un volume proprio, ma acquisisce la forma del recipiente che lo contiene;
• un materiale allo stato gassoso non ha né volume né forma propria, ma si espande fino a occupare tutto lo spazio
disponibile.
Solido
Nello stato solido i costituenti della materia sono legati da forze molto intense che consentono soltanto moti di
vibrazione, nella maggior parte dei casi le molecole si distribuiscono secondo un reticolo cristallino o in maniera
amorfa. L'unico modo per variare la forma di un solido consiste nell'applicazione di forze abbastanza intense da
spezzare i legami, causando però la rottura o il taglio del corpo.
Liquido
Nello stato liquido le forze agenti tra i costituenti sono meno intense ed essi sono liberi di scorrere gli uni sugli altri.
Un liquido va incontro a variazioni di volume molto meno marcate rispetto ai gas [1] e tende ad assumere la forma
del recipiente nel quale è contenuto.
56
Stato della materia
Aeriforme
Nello stato aeriforme le interazioni sono estremamente deboli ed ai costituenti è consentito muoversi
indipendentemente, non hanno dunque forma propria e tendono ad espandersi ed occupare tutto il volume
disponibile, risultando comprimibili.
Particolari aeriformi sono i gas, i vapori e i fluidi supercritici.
Altri stati
Nella scienza moderna in realtà questa semplice classificazione risulta inadeguata a descrivere esaustivamente le
numerose possibilità che ha la materia di organizzarsi. Il plasma è stato probabilmente il primo nuovo stato della
materia ad essere aggiunto a questa catalogazione,[2] ma ce ne sono molti altri, i quali compaiono in condizioni
particolari di temperatura e pressione come i vari tipi di ghiaccio (denominati ghiaccio I, ghiaccio II, ghiaccio III e
così via fino al ghiaccio XII) e lo stato superfluido che l'elio raggiunge a bassissime temperature. Altri stati della
materia di moderna concezione sono lo stato supercritico, superfluido, supersolido, colloidale, neutronio, materia
fortemente simmetrica, materia fortemente asimmetrica, materia strana, condensato chirale, materia degenere,
plasma di quark e gluoni, condensato di Bose-Einstein e lo stato di cristallo liquido.
Cambiamenti di stato
Con i precedenti stati della materia, qui sopra menzionati riscontriamo i passaggi di stato della materia:
•
•
•
•
•
•
sublimazione: passaggio dallo stato solido a quello aeriforme o gassoso;
brinamento: passaggio dallo stato gassoso a quello solido;
fusione: passaggio dallo stato solido a quello liquido;
solidificazione: passaggio dallo stato liquido a quello solido;
evaporazione: passaggio dallo stato liquido a quello aeriforme;
condensazione: passaggio dallo stato aeriforme a quello liquido.
Note
[1] tanto che nel linguaggio comune si dice impropriamente che il volume dei liquidi non varia, ma questo non è vero: piuttosto, a parità di
variazione di temperatura ΔT o variazione di pressione ΔP, la variazione di volume nei liquidi è molto più bassa della variazione di volume
nei gas.
[2] Rolla, op. cit., p. 89
Bibliografia
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori, 29a ed., Dante Alighieri, 1987.
Voci correlate
•
•
•
•
•
•
•
Cristalli liquidi
Fase (chimica)
Aeriforme
Liquido
Solido
Plasma
Fluido
• Superfluido
• Colloide
• Stato supercritico
57
Stato della materia
• Cambiamento di stato
• Entalpia
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:State of
aggregation
Collegamenti esterni
• Esperienze in laboratorio - Gli stati di aggregazione della materia ed i passaggi di stato (http://www.itchiavari.
org/chimica/lab/stataggr.html)
Miscela (chimica)
In chimica si intende per miscela o miscuglio[1] l'insieme di più sostanze chimiche (composti chimici ed elementi
chimici) che insieme conservano comunque inalterate le loro singole caratteristiche (come il colore, il sapore,
l'odore) e lo stato fisico.
Normalmente è assai improbabile che la materia si trovi sotto forma di sostanza pura; il più delle volte si presenta
sotto forma di miscuglio o miscela.
I componenti di un miscuglio possono essere solidi, liquidi o gassosi. Un esempio di miscuglio di sostanze gassose,
liquide e solide è l'aria: i gas sono principalmente l'azoto, l'ossigeno e l'anidride carbonica, le particelle di polvere
costituiscono la parte solida e le goccioline d'acqua costituiscono la parte liquida.
Miscugli omogenei e eterogenei
Un miscuglio è detto omogeneo (o fisico) se i suoi componenti non sono più distinguibili all'osservazione diretta e si
presenta in un'unica fase.[2] Un miscuglio omogeneo in cui una sostanza è in netta prevalenza rispetto alle altre
prende il nome di soluzione. Un esempio di miscugli omogenei è rappresentato dalle leghe. I miscugli omogenei
possono essere separati solo attraverso passaggi di stato che coinvolgano i componenti in maniera differente. Ad
esempio il sale disciolto nell'acqua costituisce un miscuglio omogeneo, e può essere separato riscaldando il
miscuglio: in questa maniera l'acqua evapora, mentre il sale precipita sotto forma di solido (questo processo avviene
nelle saline).
Un miscuglio è eterogeneo (o meccanico) se è costituito da due o più fasi e i suoi componenti sono facilmente
distinguibili.[2] La sospensione è un esempio di miscuglio eterogeneo. I miscugli eterogenei possono essere separati
più facilmente dei miscugli omogenei, anche attraverso metodi meccanici.[2] Quando due o più sostanze si uniscono
per formare un miscuglio, queste non modificano la loro intima struttura, come avviene invece nelle reazioni
chimiche.
Alcuni esempi di miscugli omogenei sono:
• vino e acqua
• acqua e sale.
Alcuni esempi di miscugli eterogenei sono:
• roccia
• sale e pepe
58
Miscela (chimica)
Classificazione delle miscele
• Miscele omogenee: si presentano in un'unica fase (diametro delle particelle <1 nm)
• Soluzioni
• leghe
• Miscele gassose
• Miscele eterogenee: si presentano in più fasi (diametro delle particelle >1 nm)
• Dispersioni (diametro delle particelle >1 μm)
•
•
•
•
•
schiume (fase dispersa:gas; fase continua:liquido)
emulsioni (fase dispersa:liquido; fase continua:liquido)
sospensioni (fase dispersa:solido; fase continua:liquido)
nebbie (fase dispersa:liquido; fase continua:gas)
fumi (fase dispersa:solido; fase continua:gas)
• Colloidi (diametro delle particelle <1 μm)
• emulsione colloidale (fase dispersa:liquido; fase continua:liquido)
• aerosol liquido (fase dispersa:liquido; fase continua:gas)
•
•
•
•
•
•
aerosol solido (fase dispersa:solido; fase continua:gas)
schiuma colloidale (fase dispersa:gas; fase continua:liquido)
sol (fase dispersa:solido; fase continua:liquido)
schiuma solida (fase dispersa:gas; fase continua:solido)
gel (fase dispersa:liquido; fase continua:solido)
sospensione solida colloidale (fase dispersa:solido; fase continua:solido)
Separazione dei componenti di un miscuglio
Per separare due componenti di un miscuglio in laboratorio si possono utilizzare le seguenti tecniche:
• filtrazione
• decantazione
• centrifugazione.
La filtrazione è la separazione dei componenti di un miscuglio per mezzo di un filtro o di un setaccio. Alcuni
particolari filtri servono anche per depurare l'aria; essi sono dotati di piccolissimi fori (detti "pori") che riescono a
trattenere minuscole particelle di polvere. La filtrazione si applica per separare i liquido dai solidi, i solidi dai liquidi
e i solidi dalle sostanze gassose.
La decantazione è la separazione dei componenti di un miscuglio tramite la forza peso: quelli che hanno un peso
specifico maggiore vanno a fondo, quelli che hanno peso specifico minore restano a galla. La decantazione si usa per
separare i solidi dai liquidi, i solidi dai gas, due liquidi immiscibili.
La centrifugazione sfrutta la forza centrifuga. Le fasi del miscuglio vengono separate attraverso la rapida rotazione a
cui è soggetto il miscuglio stesso, dove le fasi di peso maggiore vengono convogliate verso l'esterno.
Esistono anche altri tipi di separazione.
Dal punto di vista industriale, la separazione di una miscela può avvenire seguendo una molteplicità di operazioni
unitarie (ad esempio: distillazione, adsorbimento, assorbimento gas-liquido, estrazione, cristallizzazione).
59
Miscela (chimica)
Note
[1] In genere si preferisce il termine miscuglio quando almeno una delle fasicirconcise è un solido, mentre si preferisce il termine miscela nel
caso di fasi fluide (gas o liquidi).
[2] Rolla, op. cit., p. 10
Bibliografia
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori, 29a ed., Dante Alighieri, 1987.
Voci correlate
•
•
•
•
•
Operazioni unitarie
Soluzione (chimica)
Dispersione (chimica)
Colloide
Miscelazione
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Chemical
mixtures
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Miscela (chimica)
Collegamenti esterni
• I miscugli ed i composti (http://www.itchiavari.org/chimica/lab/miscugli.html)
60
Composto chimico
61
Composto chimico
Un composto chimico è una sostanza
formata da due o più elementi, con un
rapporto fisso tra di loro che ne determina la
composizione (composto stechiometrico).
Generalmente si attribuisce la proprietà di
composto ad un insieme di elementi
sufficientemente stabile da poter essere
isolato o studiato, anche se non raramente si
parla di composto instabile riferendosi a
composti dalla vita estremamente corta,
come può essere per un intermedio di
reazione.
I concetti di "elemento chimico" e
"composto chimico" furono illustrati da
Robert Boyle nel 1661 nel suo libro Il
chimico scettico (The Sceptical Chymist).[1]
Flaconi contenenti vari composti chimici in un laboratorio chimico
Proprietà
In generale, il rapporto fisso deve essere determinato da proprietà chimiche o fisiche, piuttosto che da arbitrarie
selezioni e scelte umane. Per esempio l'acqua è un composto chimico stabile formato da idrogeno e ossigeno in
rapporto di due a uno, mentre sostanze come l'ottone e la cioccolata sono considerate miscugli e non composti
chimici. La proprietà fondamentale del composto chimico è la sua formula chimica. La formula descrive il rapporto
del numero di atomi nell'unità minima della sostanza. Per esempio, nella formula H2O (acqua) ci sono due atomi di
idrogeno per ogni atomo di ossigeno.
Fra le diverse fasi possibili della materia, un composto chimico deve possederne almeno una in cui sia possibile
l'identificazione della struttura chimica.
Tutti i composti chimici si disgregano in composti chimici più semplici od in singoli atomi se vengono riscaldati ad
una temperatura sufficientemente alta. Questa viene chiamata temperatura di decomposizione.
Normativa
Ogni composto chimico che sia stato individuato e descritto nella letteratura scientifica, ha un suo numero di
identificazione univoco detto numero CAS, mentre per l'Unione Europea nel momento in cui un composto viene
immesso sul mercato deve possedere un identificativo detto numero EINECS.
Gradi di purezze dei composti chimici
•
•
•
•
Tecnico
Reagente
Standard primario
Reagente speciale (es: per spettroscopia, per HPLC, ...)
Composto chimico
Tipi di composti chimici
•
•
•
•
•
•
•
•
•
Acidi
Basi
Composti organici
Composti inorganici
Composti ionici
Composti non stechiometrici
Composto idiocromatico
Ossidi
Sali
Note
[1] The Cambridge Dictionary of Scientists, op. cit.
Bibliografia
• The Cambridge Dictionary of Scientists - "Boyle, Robert (1627 - 1691)" (http://www.credoreference.com/
entry/dicscientist/boyle_robert_1627_1691) (in inglese), Cambridge, Cambridge University Press, 2002.
Voci correlate
• International Uniform Chemical Information Database
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Chemical
compounds
62
Reazione chimica
63
Reazione chimica
Una reazione chimica è una trasformazione della
materia che avviene senza variazioni misurabili di
massa, in cui uno o più reagenti iniziali modificano la
loro struttura e composizione originaria per generare i
prodotti coinvolgendo gli elettroni esterni attraverso la
formazione o la rottura dei cosiddetti legami chimici.
Alcuni processi in cui intervengono reazioni chimiche
sono:
• la corrosione del ferro a ruggine (che è composta da
ossidi di ferro);
• la combustione del metano o altri combustibili (il
metano con l'ossigeno si trasforma in anidride
carbonica e vapore acqueo);
• la digestione (gli alimenti sono decomposti dai
succhi gastrici in sostanze chimiche assimilabili
dall'organismo);
Reazione chimica tra acido cloridrico e ammoniaca, con produzione
di cloruro di ammonio.
• la solubilizzazione e la formaione di miscele negli
stati liquidi e solidi genera reazioni di complessazione e di dissociazione.
Premesse e definizioni della reazione chimica
La materia è composta da atomi. Ogni atomo possiede proprietà peculiari, derivanti dalla sua struttura atomica. Gli
atomi possono legarsi tra loro per formare le molecole. Un composto chimico è un tipo particolare di molecola nella
quale gli atomi sono diversi tra loro.
Ad esempio, l'ossigeno forma una molecola fatta con due atomi di ossigeno, mentre l'acqua è una molecola composta
da due atomi di idrogeno legati ad un atomo di ossigeno, e quindi è anche un composto chimico.
Le molecole si formano attraverso una reazione chimica, che consiste in una rottura e formazione di legami chimici
tra atomi. Più in generale, le reazioni chimiche possono coinvolgere anche altre specie chimiche (ioni, radicali, ecc.)
oltre le molecole.
Le reazioni chimiche non provocano un cambiamento di natura della materia, perché non influenzano i suoi
costituenti fondamentali (gli atomi) ma solo la maniera in cui sono aggregati in molecole; non influenzano nemmeno
l'aggregazione di molecole simili, quindi le trasformazioni puramente fisiche, come i cambiamenti di stato (fusione,
solidificazione, evaporazione, ebollizione, ecc.), l'usura e l'erosione, la frattura, ecc. non sono reazioni chimiche.
Allo stesso modo, non fanno parte delle reazioni chimiche le trasformazioni dei nuclei atomici, cioè le reazioni
nucleari. Purtuttavia tali reazioni assumono anche un certo interesse in chimica e vengono studiate dalla chimica
nucleare.
Le reazioni chimiche, dunque, riguardano esclusivamente le variazioni dei legami tra gli atomi (legame covalente,
legame ionico, legame metallico).
Reazione chimica
Reazioni endotermiche ed esotermiche
Una reazione può sviluppare calore, in tal caso è detta esotermica, o assorbire calore, ed essere quindi endotermica.
Una reazione esotermica è quindi una reazione che comporta un trasferimento di calore dal sistema all'ambiente.
Analogamente una reazione endotermica è una reazione che comporta un trasferimento di calore dall'ambiente al
sistema. Necessita dunque di energia esterna per procedere.
Il sistema è la parte dell'universo oggetto di studio (nel nostro caso sistema chimico, ad es. solvente, reagenti e
prodotti presenti in un becher (che rappresenta il contorno del sistema), mentre l'ambiente è tutto ciò che circonda il
sistema stesso. Sistema + ambiente costituiscono un sistema isolato: l'universo è un sistema isolato.
Reagenti e prodotti
I composti chimici presenti all'inizio della reazione sono detti reagenti, quelli che si ottengono alla fine della
reazione sono invece i prodotti di reazione.
I fenomeni che hanno luogo durante una reazione chimica vengono rappresentati mediante una equazione chimica.
Un'equazione chimica è scritta in maniera simile ad un'equazione matematica, ed in essa compaiono due membri: al
primo membro (cioè a sinistra della freccia o altro simbolo di reazione) compaiono i reagenti, mentre al secondo
membro (cioè a destra della freccia o altro simbolo di reazione) stanno i prodotti.
Una reazione non può avere luogo, o viene rallentata fino a fermarsi o addirittura a regredire se non è soddisfatta una
serie di condizioni, come presenza dei reagenti in misura adeguata e condizioni di temperatura, pressione e luce
adatte alla specifica reazione.
Bilanciamento delle masse
Dal postulato fondamentale di Lavoisier, che dice: nulla si crea, nulla si distrugge, tutto si trasforma ne deriva
necessariamente che la somma delle masse dei reagenti è necessariamente uguale alla somma delle masse dei
prodotti di reazione. Siccome la materia è costituita da atomi, anche il numero degli atomi a destra e a sinistra
dell'equazione deve restare invariato.[1] Ad esempio nell'equazione:
che rappresenta la reazione tra idrossido di sodio ed acido cloridrico per produrre cloruro di sodio, che conosciamo
bene come sale da cucina, troviamo esattamente lo stesso numero di atomi dello stesso tipo sia nella parte sinistra
(reagenti) che nella parte destra (prodotti) della reazione, ma combinati in maniera diversa.
In questo caso, essendo questa una reazione tra un acido (HCl) e una base (NaOH) la reazione procederà verso la
neutralizzazione completa, a meno che uno dei reagenti non sia in eccesso rispetto all'altro: in questo caso, la
soluzione rimarrà acida o basica a seconda del reagente in eccesso.
Bilanciamento delle cariche
Oltre al bilanciamento delle masse, nelle equazioni chimiche deve essere soddisfatto il bilanciamento delle cariche.[1]
Le reazioni chimiche infatti possono avvenire anche tra specie chimiche cariche elettricamente, dette ioni.
Esempio
Un esempio di reazione chimica in cui sono coinvolti ioni è la reazione di autoionizzazione dell'acqua:
In questo caso si ha una carica positiva sulla specie H3O+ e una carica negativa sulla specie OH-, mentre a primo
membro compare una specie neutra (avente carica nulla). Considerando i coefficienti stechiometrici, il bilancio delle
cariche in questo caso può essere scritto come:
64
Reazione chimica
65
2×(0) = (+1) + (-1)
ovvero:
0=0
Per cui si ha l'uguaglianza della somma delle cariche che competono ai reagenti rispetto alla somma delle cariche che
competono ai prodotti, come deve essere.
Reazioni catalizzate
Alcune reazioni per avvenire hanno bisogno, o vengono facilitate, della presenza di una terza sostanza (rispetto a
reagenti e prodotti) detta catalizzatore.
Il catalizzatore permette o facilita la reazione, ma viene ritrovato invariato (o quasi) tra i prodotti di reazione. In
biologia i catalizzatori sono denominati enzimi.
Le trasformazioni che hanno luogo durante una reazione chimica spontanea portano ad una diminuzione dell'energia
totale del sistema. In effetti, in una molecola o in un cristallo, l'organizzazione reciproca degli atomi implica
un'energia, l'energia di legame; perché un legame venga rotto è necessario fornire al sistema una quantità di energia
almeno pari all'energia di legame. Quando gli atomi si ricombinano, formando nuovi legami, tale energia viene
liberata. Al termine di una reazione, l'energia immagazzinata nei legami dei prodotti di reazione è minore di quella
inizialmente presente nei legami dei reagenti iniziali.
Attivazione e velocità di reazione
Durante la reazione, tuttavia, esiste un momento in cui i vecchi legami si sono rotti e quelli nuovi non si sono ancora
formati, è lo stato di transizione dove l'energia del sistema è massima, cosa che costituisce una vera barriera per la
realizzazione della reazione (vedi: energia di attivazione).
Lo studio dell'aspetto energetico delle reazioni chimiche è la termodinamica, che ci permette di verificare se una
reazione può o meno avere luogo e quanta energia è necessario fornire per superare la barriera dell'energia di
attivazione; ma esiste un altro parametro importante: la velocità di reazione.
Alcune reazioni sono molto rapide, addirittura violente, come le esplosioni, altre sono talmente lente che possono
continuare per anni, o secoli. Alcune sono talmente lente che i reagenti coinvolti sembrano in realtà composti stabili,
come nel caso dell'ossidazione dell'alluminio, si parla in tal caso di composti "metastabili" (la forma stabile, in
ambiente con presenza di ossigeno, è l'ossido di alluminio, mentre quella metastabile è l'alluminio metallico); ad
occuparsi di studiare la velocità di reazione è la cinetica chimica.
Per quantificare la velocità di una reazione si utilizza il grado di avanzamento della reazione ξ, definito
globalmente come la proporzione di miscela che ha già reagito (ξ=0 all'inizio della reazione, ξ=1 quando la reazione
è completa). Si può così definire la velocità di reazione come la derivata del grado di avanzamento rispetto al tempo:
.
La reversibilità delle reazioni
Alcune reazioni sono reversibili, cioè il guadagno di energia avuto con la reazione è minimo, in tal modo risulta
possibile anche la reazione inversa; è questo il caso della dissociazione dell'acqua, H2O, negli ioni: H3O+ e OH-. In
questi casi il sistema evolve in generale verso un equilibrio dinamico, ossia il valore di α rimane stabile e compreso
tra 0 e 1, il numero di molecole che reagiscono in un senso è quindi compensato dal numero di molecole che
reagiscono nell'altro.
La cinetica di una reazione dipende da numerosi fattori, il più importante è la temperatura: l'energia termica permette
sia di superare la barriera dell'energia di attivazione più facilmente, sia di avere un numero maggiore di collisioni tra
Reazione chimica
le molecole reagenti.
Reazioni e stati della materia
Un altro parametro importante è la fase in cui si trovano i reagenti. Da questo punto di vista le reazioni
maggiormente favorite sono quelle in fase gassosa o liquida, dove i reagenti sono mescolati tra loro e possono
facilmente venire a contatto.
In tutti gli altri casi, cioè per reazioni tra:
•
•
•
•
•
un solido e un gas;
un solido e un liquido;
un solido e un solido;
un liquido e un gas;
due liquidi immiscibili;
dette reazioni eterogenee, la reazione può aver luogo esclusivamente nei punti di contatto tra le due fasi, quindi sarà
più veloce se i reagenti vengono dispersi l'uno nell'altro come nel caso di:
• aerosol (fini gocce di liquido disperse in un gas);
• emulsioni (dispersioni di gocce di un liquido in un altro immiscibile);
• miscugli di polveri;
• sol (dispersioni di polveri in un liquido);
• schiume (bolle di gas disperse in un liquido).
in questo modo vengono massimizzate le superfici di contatto tra i reagenti e quindi la possibilità di reazione.
Per i solidi questo può esser quantificato misurando la superficie specifica, ossia la superficie esposta per unità di
massa; una polvere o un solido poroso hanno elevati valori di superficie specifica.
Tipi di reazioni chimiche
A seconda del modo in cui si combinano i reagenti per dare luogo ai prodotti, si possono avere le seguenti tipologie
di reazioni chimiche:
•
•
•
•
Decomposizione: un reagente da luogo a più prodotti;
Sintesi: più reagenti danno luogo a un prodotto;
Sostituzione: un gruppo di una specie chimica viene sostituite da un altro gruppo;
Metatesi: scambio di due o più ioni fra elementi e gruppi aventi la stessa valenza.
Una reazione viene detta di ossido-riduzione (o redox) se durante il suo svolgimento alcune specie chimiche
modificano il proprio numero di ossidazione.[2] Le reazioni che non sono di ossido-riduzione sono reazioni
acido-base (ovvero i reagenti di tali reazioni sono un acido e una base).
66
Reazione chimica
Note
[1] Silvestroni, op. cit., p. 636
[2] Silvestroni, op. cit., p. 635
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori, 29a ed., Dante Alighieri, 1987.
Voci correlate
•
•
•
•
•
•
•
Entropia
Entalpia standard di reazione
Legge di Hess
Cinetica chimica
Termochimica
Reazione spontanea
Processo spontaneo
•
•
•
•
•
•
•
•
•
•
•
•
Processo endotermico
Processo esotermico
Processo unitario
Reattore chimico
Reazione di ossido-riduzione
Reazione acido-base
Reazione di neutralizzazione
Reazione organica
Reazione oscillante
Reazione multicomponente
Semireazione
Sintomi di reazione
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Chemical
reactions
Collegamenti esterni
• Le reazioni chimiche (http://www.itchiavari.org/chimica/lab/reazioni.html)
67
Processo chimico
Processo chimico
Il termine processo chimico viene impiegato per indicare una sequenza di una o più operazioni che realizzano la
trasformazione di sostanze chimiche.
I processi chimici si differenzia anzitutto in:
• processi chimici di laboratorio: svolti nell'ambito di un laboratorio chimico;
• processi chimici industriali: svolti su scala industriale
• processi chimici naturali.
I processi chimici naturali si realizzano in natura senza l'azione dell'uomo. A questa categoria appartengono ad
esempio i processi chimici che avvengono naturalmente all'interno degli organismi viventi (tra cui il metabolismo e
la fotosintesi clorofilliana) e le reazioni di biodegradazione. I processi chimici di laboratorio e industriali fanno
invece riferimento a processi chimici messi a punto dall'uomo.
La differenza principale tra processi chimici di laboratorio e processi chimici industriali risiede nelle quantità di
sostanze chimiche reagenti e prodotte. Ad esempio, la produzione annuale di etilene su scala industriale nel 2000 è
stata di circa 100 milioni di tonnellate,[1] mentre nel caso dei processi chimici di laboratorio si possono avere anche
pochi grammi di sostanza prodotta all'anno. Inoltre è differente la destinazione d'uso dei prodotti: le sostanze che
vengono prodotte su scala industriale sono destinate ad essere commercializzate, per cui la quantità prodotta segue la
legge della domanda e dell'offerta, mentre solo una parte delle sostanze prodotte in laboratorio sono destinate al
commercio, infatti molti processi chimici di laboratorio sono svolti per scopi differenti dalla commercializzazione
diretta, ad esempio per scopo analitico, per ricercare nuovi materiali o per fini didattici.
Processi chimici di laboratorio
Esempi
Esempi di processi chimici che avvengono in laboratorio sono:
•
•
•
•
•
Sviluppo fotografico;
Analisi calcimetrica per la determinazione del carbonato di calcio;
Test di Molisch per la determinazione delle aldeidi;
Metodo complessometrico per la determinazione della durezza dell'acqua
Sterilizzazione con ossido di etilene.[2]
68
Processo chimico
69
Processi chimici industriali
I processi chimici industriali che si svolgono in un impianto chimico possono contenere al loro interno, oltre a
trasformazioni di tipo chimico (processi unitari) anche trasformazioni di tipo fisico (operazioni unitarie). Le
apparecchiature in cui si svolgono tali trasformazioni prendono il nome di "apparecchiature chimiche", sebbene in
alcune di esse avvengano esclusivamente trasformazioni di tipo fisico (ad esempio colonna di distillazione non
reattiva).
Rappresentazione di un processo chimico industriale
I processi chimici industriali possono essere
rappresentati in vari modi, ad esempio da un
semplice schema di processo (dove vengono
rappresentate le apparecchiature e le correnti
materiali principali) o da un Piping &
Instrumentation Diagram (dove vengono
messe in evidenza tutte le apparecchiature e
la strumentazione di controllo).
Esempi
Di seguito vengono indicati alcuni processi
chimici industriali:
• Cracking del grezzo per ottenimento di
idrocarburi a catena corta;
•
•
•
•
•
•
•
Esempio di P&ID
Idroformilazione delle olefine per l'ottenimento di aldeidi;
Polimerizzazione dell'etilene per produrre polietilene;
Processo Claus perl'ottenimento di zolfo elementare da acido solfidrico;
Processo Fischer-Tropsch per la produzione di combustibili da gas di sintesi;
Processo Monsanto per la sintesi dell'acido acetico a partire da metanolo;
Processo Wacker per l'ossidazione dell'etilene a acetaldeide;
Vulcanizzazione della gomma.
Note
[1] Weissermel-Arpe, op. cit., p. 63
[2] La sterilizzazione può avvenire anche tramite riscaldamento o per azione di radiazioni: in questi casi non si tratta di un processo chimico, ma
fisico.
Bibliografia
• Klaus Weissermel; Hans-Jürgen Arpe, Charlet R. Lindley, Industrial organic chemistry (http://books.google.
com/books?id=OUGVPYqtnNgC&dq=weissermel+arpe+"industrial+organic+chemistry"&hl=it&
source=gbs_navlinks_s), 4a ed. (in inglese), Wiley-VCH, 2003. ISBN 3-527-30578-5
Processo chimico
70
Voci correlate
• Processo di produzione industriale
• Processo batch
• Sintesi chimica
Ossido-riduzione
1. RINVIA Ossidoriduzione
Reazione acido-base
È detta, in chimica, reazione acido-base una reazione chimica in cui non vi è alcuna variazione dagli stati di
ossidazione degli elementi dei reagenti a quelli dei prodotti.
Il nome deriva dalla partecipazione alla reazione di un reagente, detto
acido e un altro detto base. La definizione di questi due concetti, acido
e base, è diversa a seconda della teoria utilizzata per definire e
modellizzare questo tipo di reazione, teoria che si è evoluta col tempo,
partendo da un approccio empirico e sperimentale fino alle più recenti
definizioni, sempre più generali, legate al modello molecolare ad
orbitali.
Un esempio di reazione acido-base è quella che avviene tra bicarbonato
di sodio e acido acetico, con produzione di acetato di sodio:[1]
CH3COOH + NaHCO3 = CH3COONa + H2O + CO2
Differenza (convenzionale) tra reazioni
acido-base e reazioni di ossido-riduzione
Un campione di acido cloridrico (HCl) che
rilascia vapori che reagiscono con vapori di
ammoniaca (NH3), producendo un fumo bianco
di cloruro d'ammonio (NH4)Cl
Le reazioni acido-base si differenziano quindi da quelle di
ossido-riduzione, in cui invece vi è variazione dello stato di ossidazione di almeno un elemento coinvolto nella
reazione varia.
Poiché l'assegnazione dello stato d'ossidazione (detto anche "numero di ossidazione") teoricamente è convenzionale,
anche la variazione dello stato di ossidazione e quindi la distinzione tra reazioni acido-base e reazioni
d'ossido-riduzione sono convenzionali. Nella pratica però gli stati d'ossidazione vengono assegnati con un unico
metodo convenzionale, e quindi queste due fondamentali tipologie di reazioni chimiche costituiscono una ben noto e
importante metodo di classificazione univoco delle reazioni chimiche.
Reazione acido-base
71
Comuni teorie e definizioni di acido-base
Come detto vi sono varie teorie riguardanti le reazioni acido base (e relative definizioni dei concetti di acido e base),
che si è evoluta col tempo, partendo da un approccio empirico e sperimentale fino alle più recenti definizioni, sempre
più generali, legate al modello atomico ad orbitali.
Tra le più comuni vi sono, in ordine cronologico:
Teoria di Arrhenius
Questa teoria è stata sviluppata, da Svante Arrhenius nel 1884, per le
soluzione acquose e quindi ha in tale ambito la maggiore applicabilità e
utilità.
Secondo la teoria acido-base di Arrhenius,
• un acido è una sostanza che in soluzione acquosa, dà luogo in
qualche modo alla formazione di ione idrogeno ( H+ )
• una base è una sostanza che in soluzione acquosa, dà luogo in
qualche modo alla formazione di ione idrossido ( OH- )
Teoria di Brønsted-Lowry
Questa teoria, sviluppata da Johannes Nicolaus Brønsted e Thomas
Martin Lowry nel 1923, estende le definizioni di acido e base a quelle
sostanze di cui non è possibile o non è pratico valutare il
comportamento in acqua.
Svante Arrhenius
Secondo la teoria acido-base di Brønsted-Lowry,
• un acido è una sostanza capace di donare uno o più ioni idrogeno ( H+ ) accettati da una base
• una base è una sostanza capace di accettare uno o più ioni idrogeno ( H+ ) ceduti da un acido
Una reazione acido-base è quindi una reazione di una specie chimica che trasferisce elettroni ad un'altra specie
capace di accettarli. In tale reazione l'acido si trasforma nella propria base coniugata. Pertanto viene introdotto il
concetto di complementarietà tra acido e base, dato che l'acido non è tale se non in presenza di una controparte cui
donare il proprio ione H+, e la base non è tale se non in presenza di una controparte da cui accettare uno ione H+.
Una sostanza non è quindi acida o basica in assoluto, ma relativamente alla reazione considerata.
Teoria di Lewis
Questa teoria, sviluppata da Gilbert Newton Lewis anch'essa nel 1923, è legata al modello atomico ad orbitali.
Estende ulteriormente le definizioni di acido e base, riuscendo così a spiegare l'acidità di sostanze come ZnCl2, BF3,
AlF3, BH3 e la basicità di sostanze come PCl3 o Br2, che non sono spiegabili con la Teoria di Brønsted-Lowry.
Secondo la teoria acido-base di Lewis,
• un acido è una sostanza capace di accettare un doppietto elettronico (ovvero un orbitale doppiamente occupato,
non impegnato in legame chimico) da un'altra specie chimica.
• una base è una sostanza capace di donare un doppietto elettronico a un'altra specie chimica.
Simile alla teoria di Brønsted-Lowry, sostituisce al trasferimento dello ione H+ il trasferimento in senso inverso di un
doppietto elettronico.
Sono quindi acidi anche composti come il cloruro di alluminio ed il borano, che presentano nella loro struttura un
orbitale vuoto capace di alloggiare un doppietto elettronico proveniente da una molecola donatrice, la base, e legarsi
quindi ad essa tramite un legame dativo. Viceversa sono quindi basi anche composti come il tricloruro di fosforo o la
Reazione acido-base
piridina, che presentano nella loro struttura un doppietto elettronico non condiviso che possono trasferire ad una
molecola accettrice, l'acido, e legarsi quindi ad essa tramite un legame dativo.
Gli acidi di Lewis sono noti in chimica organica anche come reagenti elettrofili, mentre le basi di Lewis anche come
reagenti nucleofili.
Relatività dei concetti di acido e base
Come indicato sopra, secondo la teoria acido-base di Brønsted-Lowry e quella di Lewis, una certa sostanza chimica
non è definita come acida o basica in senso assoluto, ma in relazione alla sostanza con la quale trasferisce uno ione
H+ o un doppietto elettronico e quindi relativamente a una specifica reazione.
Quindi anche se una sostanza ha comportamento acido in alcune reazioni (e magari nel suo nome compare la parola
acido) può avere un comportamento differente (persino uno basico) in altre. Viceversa una sostanza che ha
comportamento basico in alcune reazioni può avere un comportamento differente (persino uno acido) in altre. Un
esempio su tutti è quello relativo alla reazione acido-base sfruttata in chimica organica per effettuare la nitrazione
tramite sostituzione elettrofila aromatica:
H2SO4 + HNO3 → NO2+ + HSO4- + H2O
dove l'acido solforico H2SO4 (acido più forte) protona l'acido nitrico HNO3(acido meno forte che funge da base)
formando lo ione nitronio NO2+, che è la specie nitrante, e acqua H2O.
Note
[1] http:/ / www. funsci. com/ fun3_it/ acidi/ acidi. htm
Voci correlate
•
•
•
•
Acido
Base (chimica)
Nomenclatura chimica
Protonazione
72
Decomposizione (chimica)
73
Decomposizione (chimica)
La decomposizione chimica (o degradazione o reazione di analisi) è una reazione di scissione di un composto
chimico in componenti più piccoli o nei suoi elementi.
È definibile come l'opposto della sintesi chimica e spesso è una reazione indesiderata.
La stabilità di un composto chimico è influenzata dalle condizioni ambientali come calore, radiazioni, umidità o
acidità di un solvente. Generalmente un processo di decomposizione non è molto ben definito, dato che una molecola
può rompersi in una moltitudine di pezzi più piccoli.
La decomposizione chimica è sfruttata in diverse tecniche analitiche, in particolare nella spettrometria di massa,
nell'analisi gravimetrica tradizionale e nella termogravimetria (TGA).
Reazioni
La decomposizione chimica può essere
generalizzata nella formula:
AB → A + B
ad esempio l'elettrolisi dell'acqua nei suoi
componenti gassosi idrogeno e ossigeno
sarà:
2H2O → 2H2 + O2
Un esempio di decomposizione spontanea è
quella dell'acqua ossigenata, che lentamente
si decompone in acqua e ossigeno:
2H2O2 → 2H2O + O2
I carbonati si decompongono quando
vengono
riscaldati
producendo
il
corrispondente ossido metallico e anidride
carbonica. Nell'esempio la decomposizione
del carbonato di calcio:
CaCO3 → CaO + CO2
Grafico che mostra l'andamento della corrente al variare del potenziale di cella
all'interno di una cella elettrolitica. Ogni curva corrisponde ad un sale immerso
nella soluzione elettrolitica e il punto in cui tali curve variano la propria pendenza
corrisponde al "potenziale di decomposizione" del sale, cioè la differenza di
potenziale elettrico oltre la quale il sale subisce una reazione di decomposizione.
Anche i clorati metallici si decompongono
per riscaldamento generando il corrispondente cloruro metallico e ossigeno. Nell'esempio la decomposizione del
clorato di potassio:
2KClO3 → 2KCl + 3 O2
Voci correlate
• Chimica analitica
• Dissociazione (chimica)
• Termolisi
Metatesi (chimica)
Metatesi (chimica)
La metatesi (o doppio scambio o doppia sostituzione) è una reazione chimica basata, appunto, sullo scambio di
due o più ioni fra elementi e gruppi aventi la stessa valenza.
Semplice esempio di reazione di metatesi è il seguente:
Na2SO4 + ZnCl2 → ZnSO4 + 2NaCl
Esempio
Una reazione di metatesi può essere scritta nel modo seguente:
AB + CD = AD + CB
In cui A, B, C e D sono gli ioni di cui sono formati i composti AB, CD (reagenti), AD e CB (prodotti).
La reazione è di doppio scambio in quanto vengono scambiati due ioni tra: AB e CD.
Ad esempio consideriamo come reagenti l'idrossido di calcio e l'acido fluoridrico. La reazione di doppio scambio che
li interessa è del tipo:
Ca(OH)2 + 2 HF ---> CaF2 + 2 H2O
L'idrossido di calcio reagisce con l'acido fluoridrico formando fluoruro di calcio e acqua.
Volendo seguire la simbologia anzidetta, abbiamo:
•
•
•
•
•
•
•
•
A: ione Ca2+
B: 2 ioni OHC: 2 ioni H+
D: 2 ioni FAB: Ca(OH)2
CD: 2 molecole di HF
AD: CaF2
CB: 2 molecole di H2O (oppure HOH evidenziando gli ioni)
Il fluoro presente nell'acido fluoridrico va a sostituire il gruppo ossidrile (OH) formando il bifluoruro di calcio, e a
sua volta il gruppo ossidrile va a sostituire il fluoro nell'acido fluoridrico formando acqua.
Voci correlate
• Metatesi olefinica
74
Precipitazione (chimica)
75
Precipitazione (chimica)
In chimica il termine precipitazione descrive il fenomeno di
separazione di una sostanza solida da una soluzione. Nelle
equazioni chimiche il precipitato, ovvero il composto poco
solubile ottenuto, viene evidenziato con una freccia che punta
verso il basso.
Tale separazione può avvenire a seguito di una reazione chimica o
per una variazione delle condizioni fisiche della soluzione - ad
esempio, la temperatura.
La formazione di precipitati è alla base dei saggi a umido
dell'analisi chimica qualitativa, in cui la verifica della presenza di
un dato ione o gruppo funzionale viene resa evidente dalla
comparsa di un precipitato a seguito del trattamento con un
opportuno reagente - ad esempio, l'uso del nitrato d'argento per
evidenziare la presenza di cloruri, bromuri e ioduri.
AgNO3 + Cl- → AgCl↓ + NO3-
Meccanismo della precipitazione
Il tipico aspetto bianco caseoso del precipitato di AgCl
Le fasi della precipitazione si possono distinguere in nucleazione e
accrescimento. La nucleazione consiste nella formazione di microcristalli di soluto, appunto dei "nuclei" di
cristallizzazione, che tendono ad accrescersi; l’accrescimento consiste invece nell’ingrossamento di questi cristalli ad
opera di altro soluto che attornia il cristallo e stabilisce con esso interazioni di tipo elettrostatico. È possibile che a
causa di una soprassaturazione, la nucleazione prevalga sull’accrescimento, formando in tal caso un colloide, che è
inseparabile mediante ordinari mezzi fisici dal resto della soluzione. Una soluzione colloidale è piuttosto stabile. I
microcristalli di soluto formatisi hanno tutti la stessa carica e poiché si respingono evitano il loro accrescimento. Per
questo motivo non si forma un agglomerato di forma geometrica definita.
Talvolta può capitare che con l’aggiunta di un elettrolita si vada incontro alla formazione di un precipitato fioccoso o
caseoso. La formazione di un precipitato fioccoso viene detta anche coagulazione o flocculazione e avviene quando
si forma una sostanza idrofila, quindi nel reticolo cristallino della sostanza vengono intrappolate fisicamente
molecole d’acqua. La formazione di un precipitato caseoso avviene invece quando si forma una sostanza idrofoba.
Accanto al fenomeno della precipitazione vi è anche il fastidioso fenomeno della cooprecipitazione che determina
una impurità del precipitato, determinando così interferenze quando si effettua un'analisi. La cooprecipitazione può
avvenire per
• adsorbimento di ioni estranei sulla superficie del precipitato principale;
• per inclusione nel precipitato di composti estranei;
• per occlusione in cavità che si formano nel precipitato durante l’accrescimento, di sostanze e ioni estranei.
La postprecipitazione è un altro fenomeno che può interferire nell’analisi e consiste nella precipitazione di sostanze
aventi uno ione in comune col precipitato principale sulla sua superficie.
L’invecchiamento del precipitato si ha quando il precipitato rimane troppo tempo nella soluzione madre oppure
rimane a contatto con sostanze ossidanti o riducenti. Esso consiste nella modificazione della struttura o della
composizione del precipitato.
Precipitazione (chimica)
76
Processo industriale
A livello industriale, la precipitazione è un'operazione unitaria di separazione liquido-solido in presenza di un campo
di forze, che viene svolta all'interno di precipitatori.
Più precisamente, se il campo di forze responsabile della separazione è la forza di gravità, si parla di sedimentazione
o decantazione, mentre se il campo di forze è quello elettrostatico, si parla di precipitazione elettrostatica.
Precipitazione biotica
Quando il processo di precipitazione viene effettuato da un organismo vivente. Un esempio è il carbonato di calcio
nello scheletro dei coralli marini o nel guscio di alcuni molluschi. In questo caso gli ioni di calcio e carbonato
precipitano dall'acqua all'interno dell'organismo a formare la sua parte calcarea.
Voci correlate
• Flocculazione
• Solubilità
• Precipitatore elettrostatico
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Solid
precipitation
Complesso (chimica)
Un complesso in chimica e in biochimica è
il prodotto della formazione, spesso
reversibile, di un legame tra un atomo o ione
centrale (o "ione coordinante") e degli
atomi, ioni o molecole (detti leganti o
ligandi o "ioni coordinati") che circondano
l'atomo centrale.[1]
Una definizione più rigorosa di complesso
chimico può essere: "Un complesso è un
composto chimico in cui un atomo lega un
numero di altre specie chimiche superiore al
suo numero di ossidazione".
Con il termine ione complesso si indica lo
ione che contiene l'atomo centrale e i ligandi
in soluzione acquosa, mentre per composto
di coordinazione si intende il sale secco dello ione complesso.
Il complesso tris(bipiridina)rutenio (II) cloruro
Esistono moltissime tipologie di complessi, che vanno dal semplice metallo in soluzione acquosa (coordinato quindi
da molecole d'acqua) a complessi metallo-enzimi, che prendono parte a svariati processi biochimici.
Complesso (chimica)
77
Struttura dei complessi
Leganti
I leganti possono formare uno o più legami con l'atomo centrale; nel secondo caso si dicono agenti chelanti (per
affinità con le chele di molti crostacei). A seconda dei legami che un legante forma con l'atomo centrale, questo si
chiama monodentato, bidentato o, in generale, polidentato.[2] Esempi di chelanti sono l'EDTA
(etilen-diammino-tetraacetato) o l'en (etilen-diammina), bidentato. I leganti come l'acqua o il cloro formano un solo
collegamento con l'atomo centrale, e sono quindi monodentati. L'EDTA è esadentato, il che spiega la grande stabilità
di molti dei suoi complessi. I leganti polidentati possono formare anche legami con più di uno ione metallico,
formando quindi un unico complesso con più ioni metallici. In questo caso si parla di complesso polimetallico. Se
invece è il metallo a formare un legame con un altro metallo coordinato si parla di complesso a cluster.
Numero di coordinazione e geometria del complesso
L'atomo centrale è spesso, ma non esclusivamente, un metallo di transizione (ovvero un elemento del blocco d della
tavola periodica).[3] L'insieme dei leganti forma la sfera di coordinazione del complesso e il numero di legami
esistenti tra metallo e leganti è detto numero di coordinazione (NC o CN).[2] Tale numero varia, in genere, da 2 a
12; i casi più comuni sono 4 e 6.
Parametri che influenzano il numero di coordinazione sono:
• le dimensioni e la carica dello ione centrale
• il tipo di leganti (leganti molto grandi ed ingombranti riducono il NC)
• le interazioni all'interno del complesso.
A seconda del numero di coordinazione, il complesso può assumere differenti geometrie. Nella tabella seguente sono
illustrate le geometrie più frequentemente osservate. La geometria regolare non è sempre rispettata; ad esempio si
osservano deviazioni quando i leganti non sono tutti uguali e le distanze metallo-legante possono risultare diverse, o
quando i leganti hanno requisiti particolari di ingombro sterico, o per effetti elettronici come l'effetto Jahn-Teller.
Inoltre ci sono casi nei quali due strutture diverse possono interconvertirsi facilmente, perché differiscono di poco in
energia; ad esempio questo è piuttosto comune per le strutture a numero di coordinazione 4 e 5.
Numero di
coordinazione
Forma
Geometria
Esempi e note
2
Lineare
[CuCl2]–, [Ag(NH3)2]+
3
Trigonale planare
molto rara
[HgI3]−
4
Tetraedrica
piuttosto comune
[ReO4]2–, Ni(CO)4
4
Planare quadrata
XeF4, [AuCl4]–, [PtCl4]2–
5
Bipiramidale trigonale
[CdCl5]3–, Fe(CO)5
Complesso (chimica)
78
5
Piramidale a base quadrata
[NbCl4(O)]–, [V(acac)2(O)]
6
Ottaedrica
la più comune
[Cr(H2O)6]3+, [Fe(CN)6]3–
7
Bipiramidale pentagonale
rara
[Nb(O)(ox)3]3–
8
Antiprismatica quadrata
[Mo(CN)8]4–, [ReF8]2–
Sfera esterna e sfera interna
Si dice complesso della sfera esterna (o complesso esterno o complesso di alto spin) il composto ottenuto per
interazione elettrostatica del catione centrale con un anione che viene legato senza però alterare la sfera di
coordinazione. Sono complessi esterni quelli in cui non si ha accoppiamento degli elettroni dispari dell'atomo
centrale.[4] Ad esempio, [Co(NH3)6](NO2)3 è un complesso della sfera esterna.
Di contro, se il metallo centrale si lega a un anione alterando la sua sfera di coordinazione si dice che si è formato un
complesso della sfera interna (o complesso interno o complesso di basso spin). Sono complessi interni quelli in
cui si ha accoppiamento degli elettroni dispari dell'atomo centrale.[4] In analogia con l'esempio precedente, lo ione
nitrito NO2- è anche in grado di formare il nitroso-complesso [Co(NH3)3(NO2)3] che rappresenta un complesso della
sfera interna.
Legame chimico nei complessi
Il legame chimico nei complessi deriva fondamentalmente dalle interazioni tra gli orbitali d dell'atomo centrale e
orbitali s e p dei leganti. I legami risultanti, hanno energie tali che le lunghezze d'onda del visibile causano
transizioni elettroniche; molti ioni complessi (e in genere molti ioni di metalli di transizione) sono per questo motivo
colorati.[3]
La comparsa di effetti cromatici nei composti contenenti metalli di transizione (e nella fattispecie, nei complessi
coordinati) sono spiegati dalla cosiddetta teoria del campo cristallino (o TCC).[5]
Teoria del legame di valenza
Fu opera di Linus Pauling la teoria del legame di valenza, che fu la prima teoria (degli anni trenta) sulla formazione
di un legame legato-legante per sovrapposizione degli orbitali d del metallo e gli orbitali ibridi spn dei leganti.[6] In
questo modo si verrebbero a formare quindi dei legami dativi tali da riempire tutto l'ultimo livello di orbitali
dell'atomo centrale (10d 6p 2s) per un totale di 18 elettroni.
Teoria del campo cristallino
Fu introdotta da Hans Bethe e nel 1929 ed assume che l'interazione metallo-leganti sia di tipo elettrostatico.[6] Le
funzioni d'onda degli orbitali d possono essere graficamente rappresentate come superfici di confine a 4 lobi
orientate rispetto agli assi x,y,z: ciò avviene poiché i leganti assumono una geometria tetraedrica intorno all'atomo
centrale e il campo elettrico generato dai leganti, non avendo simmetria sferica, provoca una separazione dei livelli
energetici degli orbitali d. Tale separazione conduce ad una stabilizzazione del complesso.
Complesso (chimica)
79
Teoria del campo dei leganti
L'evidenza sperimentale mostra che l'interazione legante-legato non può essere spiegata solo attraverso un modello
puramente elettrostatico. Infatti fra i leganti che danno complessi più stabili c'è il monossido di carbonio (ligando a
campo forte) che non ha cariche o dipoli permanenti. Quindi è chiara la necessità di introdurre un certo carattere
covalente nella teoria. Quindi la teoria del campo dei leganti estende quella del campo cristallino descrivendo
l'interazione legante-legato attraverso l'aggiunta di un certo carattere covalente descritto mediante il modello
dell'orbitale molecolare (MO).[6]
Equilibri di complessazione
Indicando con M un generico elemento metallico e con L un ligando, gli equilibri di complessazione sono
usualmente schematizzati così:
M + L
ML + L
ML2 + L
MLn-1 + L
ML ;
ML2 ;
ML3 ;
MLn ;
Sommando membro a membro si ottiene l'espressione totale dell'equilibrio:
M + nL
MLn con costante di formazione
Come tutti gli equilibri multipli è verificata l'uguaglianza Kf = K1 · K2 · K3...Kn da cui pKf = pK1 + pK2 + pK3 + ...
pKn
Il reciproco di Kf esprime la costante di instabilità Kins ed è un altro modo di rappresentare l'equilibrio (in questo
caso in funzione della dissociazione):
Molto spesso, nella pratica di laboratorio, capita di avere
a che fare con leganti suscettibili a variazioni di pH e con metalli che tendono ad essere complessati da altre specie
presenti in soluzione. Tali equilibri sono regolati quantitativamente da un'altra costante che tiene conto di tali fattori:
la costante in oggetto è la costante condizionale di formazione K'f.
K'f è in relazione con Kf tramite l'equazione K'f = Kf · α · β
dove (per un dato pH) α = α-valore, frazione del ligando libero presente in soluzione e β = β-valore, frazione di
metallo non complessato presente in soluzione.
Complesso (chimica)
80
Isomeria dei complessi
Esistono vari tipi di isomeria. I tipi più importanti sono:
• isomeria geometrica
• isomeria ottica
• isomeria di legame.
Isomeria geometrica
Si ha quando gli stessi leganti sono disposti in modo differente attorno al metallo. I casi principali riguardano i
complessi a numero di coordinazione 4 planari quadrati di formula generica ML2X2 e i complessi a numero di
coordinazione 6 ottaedrici di tipo ML4X2 e ML3X3.
Nei complessi planari quadrati si definisce isomero cis il composto di coordinazione che reca leganti identici sui
vertici adiacenti del quadrato. Quando i leganti occupano vertici opposti del quadrato si ha l'isomero trans. Alcuni
complessi del platino sono utilizzati nella chemioterapia antitumorale: solamente gli isomeri di tipo cis del Pt(II)
possono legarsi alle basi del DNA ed esplicare la loro relativa azione farmacologica.
Nei complessi ottaedrici di tipo ML4X2 si possono analogamente avere i due isomeri cis o trans, quando i due leganti
X occupano posizioni rispettivamente adiacenti od opposte.
Anche nei complessi ottaedrici di tipo ML3X3 sono possibili due diversi isomeri geometrici. Quando i tre leganti
dello stesso tipo occupano tre posizioni vicine corrispondenti alla faccia dell'ottaedro si ha l'isomero fac (=facciale).
Quando i tre leganti dello stesso tipo occupano tre posizioni nello stesso piano che contiene anche il metallo si ha
l'isomero mer (=meridionale).
cis-[CoCl2(NH3)4]+
trans-[CoCl2(NH3)4]+
fac-[CoCl3(NH3)3
mer-[CoCl3(NH3)3
Isomeria ottica
L'isomeria ottica genera una coppia di complessi che sono l'uno l'immagine speculare non sovrapponibile dell'altro.
Le due forme isomeriche rappresentano una coppia di enantiomeri ognuno dei quali ruota il piano della luce
polarizzata in un determinato modo (destrorso o sinistrorso). La configurazione assoluta dei complessi chirali si
assegna notando il verso di rotazione assunto dai ligandi lungo un asse ternario di un ottaedro regolare (meccanismo
dell'avvolgimento di una vite). La rotazione sinistrorsa dell'elica costituita dai ligandi indica l'isomero Λ, quella
destrorsa l'isomero Δ.
Complesso (chimica)
Λ-[Fe(ox)3]3−
81
Δ-[Fe(ox)3]3−
Λ-cis-[CoCl2(en)2]+
Δ-cis-[CoCl2(en)2]+
Isomeria di legame
Questo tipo di isomeria si ha con leganti
ambidentati, cioè che possono legarsi in due
modi diversi al metallo. L'esempio più noto
è dato dallo ione nitrito (NO2−) che può
legarsi con l'ossigeno o con l'azoto:
• [Co(NH3)5(NO2)]2+ contiene un legame
Co−NO2 ed è chiamato isomero nitro;
• [Co(NH3)5(ONO)]2+ contiene un legame
Co−ONO ed è chiamato isomero nitrito.
Struttura dei due isomeri di legame [Co(NH3)5(NO2)]2+ e [Co(NH3)5(ONO)]2+.
Altri leganti ambidentati comuni sono lo
ione tiocianato (SCN−) e solfito (SO3−).
Reattività dei complessi
I complessi possono dar luogo a vari tipi di reazioni, che si possono classificare nel modo seguente:
Reazioni di sostituzione dei leganti
In queste reazioni un legante già presente nella sfera di coordinazione viene sostituito da un altro legante presente in
soluzione. Il processo può avvenire con vari meccanismi e velocità. I casi più studiati riguardano i complessi a
numero di coordinazione quattro e sei.
Reazioni di riarrangiamento dei leganti
In questo caso prima e dopo la reazione sono presenti gli stessi leganti coordinati, ma può variare la geometria di
coordinazione o la stereochimica del complesso. Sono possibili vari meccanismi, a seconda che tali reazioni
avvengano con o senza rottura di legami metallo-legante. In quest'ultimo caso si parla di complessi flussionali o
stereochimicamente non-rigidi.
Complesso (chimica)
82
Reazioni sui leganti
In questo caso avviene una reazione chimica sul legante mentre è coordinato al metallo. Queste reazioni sono
comuni soprattutto in chimica metallorganica (ad esempio, reazioni di inserzione), ma ne esistono anche nella
chimica dei complessi classici (ad esempio, reazioni a stampo e sintesi di macrocicli).
Reazioni di trasferimento elettronico
Queste reazioni sono caratterizzate dal trasferimento di uno (o più) elettroni tra due specie chimiche. Sono possibili
due meccanismi: (1) reazioni a sfera esterna, dove durante il trasferimento dell'elettrone la sfera di coordinazione dei
due complessi non viene modificata; (2) reazioni a sfera interna, dove il trasferimento dell'elettrone avviene dopo che
si è formato un legante a ponte tra i due centri metallici.
Nomenclatura dei complessi
Il procedimento di base per denominare un complesso è il seguente:[7]
• Si scrivono i nomi dei ligandi in ordine alfabetico.
• I ligandi monodentati che appaiono più volte ricevono un prefisso secondo il numero di occorrenze: di-, tri-,
tetra-, penta-, o esa-. I ligandi polidentati (per esempio, etilenediamina, ossalato) ricevono i prefissi bis-, tris-,
tetrakis-, e così via.
• Gli anioni finiscono in o. Per esempio: cianuro diventa ciano.
• Ai ligandi neutri si danno i loro soliti nomi, con qualche eccezione: NH3 diventa amino; H2O diventa aquo;
CO diventa carbonile.
• Si scrive il nome dell'atomo/ione centrale. Se il complesso è un anione, il nome dell'atomo centrale finirà in -ato.
• Se lo stato di ossidazione dell'atomo centrale deve essere specificato (quando è uno di vari stati possibili), lo si
scrive come numero romano (o 0) tra parentesi.
Esempi:[7]
Formula chimica
Note
[1]
[2]
[3]
[4]
[5]
[6]
[7]
Silvestroni, op. cit., pp. 863-864
Silvestroni, op. cit., p. 868
Silvestroni, op. cit., p. 861
Silvestroni, op. cit., p. 869
Silvestroni, op. cit., pp. 870-873
Silvestroni, op. cit., p. 864
Silvestroni, op. cit., pp. 865-866
Nome del complesso
[NiCl4]2-
ione tetracloronichelato (II)
[CuNH3Cl5]3-
ione aminopentaclorocuprato (II)
[Cd(en)2(CN)2]
dicianobisetilendiaminocadmio (II)
[Fe(NH3)6]Cl3
esamminoferro (III) cloruro
K3[Fe(CN)6]
potassio esacianoferrato (III)
Complesso (chimica)
Bibliografia
• T. W. Graham Solomons, Chimica organica, 2a ed., Bologna, Zanichelli, 2001, pp. 583-596. ISBN
88-08-09414-6
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
Voci correlate
•
•
•
•
Chimica di coordinazione
Cluster
Curva di Ringbom
Effetto Jahn-Teller
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/
Category:Coordination compounds
Collegamenti esterni
• Le reazioni di complessazione (http://www.itchiavari.org/chimica/lab/comples.html)
Reazione organica
Una reazione organica è il risultato della trasformazione di uno o più composti organici in altri con caratteristiche
chimico-fisiche differenti. Le reazioni organiche sono impiegate per ottenere composti sintetici, artificiali,
trasformare composti economici e largamente disponibili in altri a maggior valor aggiunto, identificare un composto
per reazione con reagenti specifici (saggi e test chimici) o trasformare dei composti in altri meno pericolosi.
Le reazioni organiche più anticamente conosciute sono la combustione e la saponificazione. Attualmente delle
reazioni organiche se ne conoscono qualche migliaio ed aumentano ogni anno con la scoperta di nuove possibili
interazioni fra i composti, nonché prodotti di reazione ottenuti da reagenti e catalizzatori sempre più complessi.
Comunemente ad ogni reazione organica veniva dato il nome dello scopritore o del prodotto ottenuto. Con
l'aumentare delle reazioni e soprattutto con la necessità di indagare il meccanismo di reazione, si è reso
indispensabile organizzarle e classificarle. Per la classificazione si possono adottare differenti metodi, ma il rimando
ad un nome proprio della reazione, spesso riferita ai suoi primi scopritori, rimane una pratica molto diffusa e
comunemente accettata.
83
Reazione organica
Elenco delle reazioni che possiedono un nome proprio
Il termine reazione e sintesi sono spesso usati con lo stesso significato.
Per un elenco vedere la lista nel progetto "Lista di reazioni organiche".
Classificazione in base al meccanismo di reazione
• Reazione di addizione (indicata con A). Reazioni plurireagente con formazione di nuovi legami, si suddividono a
loro volta in
• Addizione elettrofila (AE)
• Addizione nucleofila (AN)
• Addizione radicalica (AR)
• Reazione di eliminazione (indicata con E)
• Reazione di sostituzione (indicata con S). Reazioni bi-reagente con scambio di legami, si suddividono in
•
•
•
•
Sostituzione elettrofila (SE)
Sostituzione elettrofila aromatica (SEA)
Sostituzione nucleofila (SN)
Sostituzione nucleofila aromatica (SNA)
• Sostituzione radicalica (SR)
• Reazione di trasposizione o riarrangiamento (indicata con T). Reazioni mono-reagente con modifica interna alla
molecola di ordine di legami. Si dividono in
• Riarrangiamenti 1,2
• Reazioni pericicliche
• Metatesi
• Reazione di condensazione (indicata con C)
• Reazione di ossido-riduzione (indicata con RedOx)
Classificazione secondo il numero di molecole che determinano la velocità
della reazione
Si dividono in reazioni mono-molecolari, bi-molecolari, tri-molecolari...a molecolarità superiore.
Classificazione secondo la natura del riassestamento dei legami
• radicaliche (R)
• ioniche polari, ulteriormente suddivise in
• nucleofile
• elettrofile
84
Reazione organica
Classificazione in base al gruppo funzionale reagente o al gruppo funzionale
da ottenere
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
acidi carbossilici
aciloine
aldeidi
alcani
alcheni
alchini
alcoli
alogenuri acilici
alogenuri alchilici
alogenuri arilici
amidi
ammine
areni
azidi
aziridine
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
chetoni
cianine
cianidrine
cicloalcani
cicloalcheni
decarbossilazione
dioli
enammine
enoli
epossidi
esteri
eteri
fenoli
immine
isocianati
isotiocianati
lattami
nitrili
nitrosi
ossime
tiocianati
85
Reazione organica
Classificazione in base al tipo di reagente impiegato
Questo approccio è spesso adottato qualora si utilizzi un reagente altamente selettivo, o specifico, come ad esempio
nel caso degli ossidanti inorganici come il tetrossido di osmio, o nel caso di riducenti come il litio alluminio idruro.
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Organic
reactions
Collegamenti esterni
• list of named reactions from UConn [1]
• organic reactions [2]
Bibliografia
•
•
•
•
Nomenclature for organic chemical transformations (Pure Appl. Chem., 1989, 61, 725).
System for symbolic representation of reaction mechanisms (Pure Appl. Chem., 1989, 61, 23).
Detailed linear representation of reaction mechanisms (Pure Appl. Chem., 1989, 61, 57).
Reazioni Organiche a cura di D. Pocar, Casa Ed. Ambrosiana, Milano 1966.
Reagente
Si definisce reagente qualsiasi sostanza che prende parte ad una reazione chimica. i reagenti sono sostanze originarie
nella composizione chimica.
Col procedere della reazione, i reagenti - scritti solitamente nella parte sinistra di un'equazione chimica - si
trasformano nei "prodotti di reazione" - scritti solitamente nella parte destra dell'equazione chimica.
Spesso i prodotti di una reazione chimica a loro volta possono essere reagenti di altre reazioni, anche concomitanti.
Esempio: nella reazione
A + B →C + D
A e B sono i reagenti, C e D sono i prodotti di reazione.
Affinché la reazione abbia luogo, le molecole dei reagenti devono urtarsi con un'energia, detta energia di attivazione,
sufficiente per produrre uno stato di transizione che evolve successivamente nei prodotti di reazione.Zanini
Voci correlate
•
•
•
•
Velocità di reazione
Catalizzatore
Reazione chimica
Prodotto (chimica)
86
Catalizzatore
Catalizzatore
Un catalizzatore è una sostanza, fonte o dispositivo che interviene in una reazione chimica aumentandone la
velocità ma rimanendo inalterato al termine della stessa.[1]
L'aumento di velocità viene reso possibile grazie alla diminuzione dell'energia di attivazione (energia potenziale),
che deve essere raggiunta per far sì che i reagenti evolvano poi spontaneamente verso il prodotto/i. L'effetto è tale da
rendere possibili reazioni che in condizioni normali non procederebbero in maniera apprezzabile: i casi più eclatanti
si hanno in biochimica sia in laboratorio che nella ingegneria biochimica, dove gli enzimi aumentano la velocità
delle reazioni anche di 1020 volte.
Azione
Un catalizzatore, in generale, modifica
il "meccanismo di reazione" della
reazione a cui partecipa tramite un
percorso reattivo alternativo al quale
compete una minore energia di
attivazione.
Lo schema più semplice di intervento
di un catalizzatore C nella reazione fra
due composti A e B è:
A + C → AC
AC + B → AB + C
La reazione netta è sempre A + B →
AB , mentre C viene rigenerato alla
fine di ogni ciclo e non si consuma.
Nel caso in cui un composto presente
Diagramma di una reazione catalitica che mostra l'energia richiesta a vari stadi lungo
nell'ambiente di reazione (prodotto,
l'asse del tempo (coordinate di reazione). I substrati normalmente necessitano di una
solvente, ecc.) si leghi al catalizzatore
notevole quantità di energia (picco rosso) per giungere allo stato di transizione, onde
in modo permanente, si parla di
reagire per formare il prodotto. La presenza di un catalizzatore (come un enzima) crea un
microambiente nel quale i substrati possono raggiungere lo stato di transizione (picco blu)
avvelenamento del catalizzatore (o
più facilmente, riducendo così la quantità d'energia richiesta. Essendo più facile arrivare a
disattivazione), che perde così la sua
uno stato energetico minore la reazione può avere luogo più frequentemente e di
efficacia. In alcuni casi si avvelena
conseguenza la velocità di reazione sarà maggiore.
volontariamente parte del catalizzatore
per modularne l'efficacia, consentendo così l'ottenimento di intermedi di reazione altrimenti non sintetizzabili.
La frequenza di turnover definisce il rendimento di un catalizzatore ed è data dalla formula
dove v è la velocità di reazione e [Q] la concentrazione molare del catalizzatore omogeneo. In caso di catalisi
eterogenea, al denominatore compare la massa del catalizzatore o la sua estensione superficiale.
Una classe particolare di catalizzatori è rappresentata dai catalizzatori per trasferimento di fase, come ad esempio gli
eteri corona,che permettono la reazione fra composti in fasi distinte, che non potrebbero reagire altrimenti.
Ci sono sostanze che invece di aumentare la velocità di reazione, la diminuiscono. Questi composti vengono definiti
catalizzatori negativi[2] o inibitori.[3]
87
Catalizzatore
Catalizzatori omogenei ed eterogenei
Catalizzatori omogenei
Un catalizzatore è detto omogeneo se
si trova nella stessa fase dei reagenti. Il
vantaggio dei catalizzatori omogenei
sta nel miglior contatto con i reagenti;
questo è al tempo stesso uno
svantaggio, perché è difficile separare
e recuperare il catalizzatore alla fine
della reazione.
Siccome la molecola che costituisce il
catalizzatore
omogeneo
è
completamente esposta ai reagenti, essi
presenterebbero (se usati tal quali)
un'elevata attività catalitica e una
selettività bassa. Per ovviare a questo
inconveniente, spesso si uniscono ai
Struttura di un tipico catalizzatore omogeneo al rodio impiegato nel processo di
catalizzatori dei leganti, che sono
idroformilazione. In questo caso il legante è costituito da tre gruppi di trifenilfosfina
costituiti da gruppi stericamente
solfonata.
ingombranti, che diminuiscono il
numero di siti attivi ma ne aumentano la selettività.
Un esempio di catalizzatore omogeneo è dato dalla molecola cloro-tris(trifenilfosfina)-rodio(I) (avente formula
RhCl(PPh3)3), detto anche catalizzatore di Wilkinson e usato per l'idrogenazione in soluzione degli alcheni. Nel caso
del catalizzatore di Wilkinson, l'azione di legante è svolto dai gruppi di trifenilfosfina.
88
Catalizzatore
89
Catalizzatori eterogenei
Un catalizzatore è detto eterogeneo se non si trova nella stessa fase in cui
sono presenti i reagenti. Un catalizzatore eterogeneo è in genere formato
da un supporto (inerte o reattivo) su cui sono posizionati il catalizzatore
vero e proprio, ed eventualmente composti per prevenire la sinterizzazione,
oltre ad eventuali promotori (sostanze che agiscono in modo particolare
migliorando o modulando la performance catalitica).
Le particelle di catalizzatore eterogeneo presentano una struttura porosa,
quindi la catalisi avviene sia sulla superficie esterna del catalizzatore sia
sulla superficie interna. Questo fa sì che la superficie disponibile allo
scambio di materia sia di diversi ordini di grandezza maggiore di quella
che si avrebbe se la struttura del catalizzatore eterogeneo fosse compatta.
Siccome la superficie interna di un catalizzatore eterogeneo è molto più
estesa della sua superficie esterna, in fase di progettazione bisogna tenere
conto del trasporto di materia all'interno dei pori del catalizzatore.
I catalizzatori eterogenei sono più vulnerabili all'avvelenamento rispetto
alla catalisi omogenea, in quanto è sufficiente che la superficie esterna del
catalizzatore sia avvelenata (per esempio a causa di fouling) per rendere
inservibile l'intera particella di catalizzatore.
Catalizzatori di interesse industriale
Dal punto di vista pratico, l'uso principale dei catalizzatori nell'industria
chimica consente condizioni di reazione meno drastiche per fare procedere
velocemente reazioni di sintesi. Si stima che almeno il 60% di tutte le
sostanze commercializzate oggi richiedano l'uso di catalizzatori in qualche
stadio della loro sintesi.
Idrogenazione dell'etene su un catalizzatore
eterogeneo
Dal punto di vista chimico, i catalizzatori eterogenei possono essere raggruppati come segue:
•
•
•
•
metalli: ferro, platino, argento, rutenio, rodio (idrogenazione e deidrogenazione)
ossidi isolanti: ossido di alluminio, silice, ossido di magnesio (disidratazione)
ossidi semiconduttori: ossido di zinco, ossido di nichel (ossidazione)
acidi: ossido di alluminio su silice, zeoliti (polimerizzazione, cracking, alchilazione)
Alcuni fra i più importanti catalizzatori eterogenei usati nell'industria chimica sono:
• platino con 10% rodio (processo Ostwald, produzione di acido nitrico)
• tetracloruro di titanio e un composto organometallico di alluminio (processo Ziegler-Natta, polimerizzazione di
vari polimeri)
• ossido di cromo (processo Phillips, polimerizzazione del polietilene)
• la zeolite ZSM-5 (conversione di idrocarburi,decomposizione NOx)
• i silicoalluminofosfati SAPO (conversione di idrocarburi)
• pentossido di vanadio (produzione di anidride ftalica)
Alcuni esempi di catalizzatori omogenei d'interesse industriale:
• nichel(IV) acetilacetonato (sintesi del benzene)
• dicarbonildiiodo-iridio(III) (processo Cativa, sintesi dell'acido acetico)
• ottacarbonilcobalto(II) (idroformilazione, sintesi di aldeidi)
• cloruro di alluminio(III) (reazione di Friedel-Crafts, sintesi dell'etilbenzene)
Catalizzatore
Biocatalizzatori
Come biocatalizzatori si intendono i catalizzatori che agiscono in reazioni biochimiche, di solito proteine (enzimi,[4]
a volte abzimi), raramente RNA (ribozimi). Anche nel caso dei biocatalizzatori si può usare un promotore di catalisi
che si chiama cofattore di tipo effettore, dei substrati di supporto e si hanno le varie tecniche di immobilizzazione
delle cellule o i catalizzatori possono essere avvelenati da un cofattore di tipo inibitore enzimatico. Gli enzimi
possono catalizzare molti tipi di reazioni chimiche, e ciascun tipo di enzima è specifico per un tipo di reazione. Le
reazioni avvengono con grande velocità proprio grazie alla specificità degli enzimi, alcuni enzimi sono vicini alla
perfezione catalitica. La parte della molecola reagente con cui questi catalizzatori enzimatici hanno specificità è
chiamata substrato. Si forma quindi un complesso enzima-substrato, la cui formazione è dovuta a interazioni deboli
di tipo elettrostatico o legami covalenti. Non tutto l'enzima è interessato alla formazione del complesso
enzima-substrato, ma solo una parte detta sito attivo. A seconda delle condizioni di flessibilità tra enzima e substrato
si avranno diversi gradi di specificità: assoluta, di gruppo, di legame, stereochimica.
Catalisi ambientale
I catalizzatori usati nelle marmitte delle automobili sono formati da metalli nobili (generalmente platino e rodio)
dispersi su un supporto ceramico, formato da ossido di cerio e ossido di zirconio. Promuovono la contemporanea
ossidazione del carburante incombusto e del monossido di carbonio ad anidride carbonica e acqua, e la riduzione
degli ossidi di azoto ad azoto e acqua. Data la contemporanea attività su tre reazioni, sono detti catalizzatori a tre vie
(TWC).
Note
[1] D'Ischia, op. cit., p. 375
[2] http:/ / www. google. it/ search?hl=& q=%22catalizzatore+ negativo%22& sourceid=navclient-ff& rlz=1B3MOZA_itIT362IT362&
ie=UTF-8
[3] Silvestroni, op. cit., p.364
[4] Silvestroni, op. cit., p. 369
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• Marco D'Ischia, La chimica organica in laboratorio (http://books.google.com/books?id=XVPt6yF9yDMC&
hl=it&source=gbs_navlinks_s), Piccin, 2002. ISBN 88-299-1621-8
Voci correlate
•
•
•
•
•
•
•
•
Attività catalitica
Catalisi
Catalisi enzimatica
Catalisi eterogenea
Disattivazione dei catalizzatori
Enzima
Fotocatalisi
Supporto catalitico
90
Catalizzatore
91
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Catalysts
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/catalizzatore
Solubilità
In chimica si definisce solubilità di un soluto in un solvente, a una data temperatura, la massima quantità di un
soluto che a quella temperatura si scioglie in una data quantità di solvente. Una soluzione si dice satura quando, in
una data quantità di solvente a una certa temperatura, non è possibile sciogliere ulteriore soluto.
Il rapporto tra soluto e solvente per unità di soluzione è espressa dalla concentrazione.
La solubilità di un certo soluto in un certo solvente dipende, oltre che dalle caratteristiche delle due sostanze, anche
dalla temperatura e dalla pressione.
La solubilità
in cui
e
si esprime come:
sono le concentrazioni attuale e a saturazione, e
è la frazione di sovrassaturazione.
La solubilità in genere viene espressa come grammi di soluto (per esempio NaCl) disciolti in 100 grammi di solvente
(per esempio acqua) per una data temperatura.[1][2]
Regole di solubilità di alcuni soluti solidi
Le regole di solubilità ci permettono di sapere quali composti sono solubili in acqua e le loro eccezioni (casi in cui
non si sciolgono).
Le regole di solubilità sono utili per determinare quale prodotto di una reazione di doppio scambio (a cui è associato
un fenomeno di precipitazione), con i reagenti in soluzione acquosa, non è solubile in acqua e quindi formerà un
precipitato solido dentro la soluzione stessa.
Sono solubili:
•
•
•
•
•
•
•
•
•
Tutti i sali di litio, sodio, potassio e ammonio;
I cloruri, i bromuri e gli ioduri (tranne HgI, AgCl, AgBr, AgI, PbCl2 e HgCl)
Gli acetati (tranne CH3COOAg);
Tutti i nitrati;
I sali di bario (tranne BaSO4 e BaCO3);
Tutti gli idrossidi (tranne quelli di calcio e dei metalli di transizione);
I solfuri dei metalli alcalini, dei metalli alcalino terrosi e dell'ammonio;
I fosfati, i cromati e i carbonati dei metalli alcalini e dello ione ammonio;
I solfati, tranne quelli di bario, mercurio, piombo (assolutamente insolubili), calcio e argento (un poco più solubili
dei precedenti).
Solubilità
92
Sistemi a solubilità diretta e a solubilità inversa
La curva di saturazione esprime il valore della
concentrazione del soluto in condizioni di
saturazione (ovvero la sua solubilità in funzione
della temperatura). Se la solubilità cresce con la
temperatura il sistema solvente-soluto è detto a
solubilità diretta, mentre se al crescere della
temperatura la solubilità diminuisce, il sistema
è detto a solubilità inversa.
In generale all'aumento della temperatura
aumenta la solubilità delle sostanze solide (ad
esempio il cloruro di sodio[1]), mentre
diminuisce quella delle sostanze gassose.
È però da rilevare che non tutte le sostanze
hanno comportamenti analoghi riguardo alla
dipendenza della solubilità dalla temperatura ad
esempio la solubilità del carbonato di litio in
acqua diminuisce con l'aumentare della
temperatura.
Le tabelle seguenti indicano la solubilità di
alcune sostanze in acqua:[2]
Curve di solubilità per sistemi a solubilità diretta e inversa.
Solubilità del cloruro di sodio in acqua
(grammi di soluto in 100 grammi di acqua)
Temperatura (°C) 0
Solubilità
10
20
30
40
50
60
70
80
90
100
35,7 35,8 36,0 36,3 36,6 37,0 37,3 37,8 38,4 39,0 39,8
Solubilità del diossido di zolfo in acqua
(grammi di soluto in 100 cm3 di soluzione H2O-SO2, a P = 1 atm)
Temperatura (°C) 0
10
20
30
40
50
Solubilità
16,21
11,29
7,81
5,41
4,5
22,83
Si può notare dalle tabelle precedenti che la solubilità del cloruro di sodio (solido) aumenta con la temperatura,
mentre quella del diossido di zolfo (gas) diminuisce.
Anche l'aumento di pressione provoca un aumento della solubilità. Esempio di ciò è la solubilità dell'anidride
carbonica nelle bevande gassate: fino a quando la bottiglia è sigillata la maggior pressione interna permette
all'anidride di rimanere in soluzione, quando la bottiglia viene aperta la diminuzione di pressione comporta la rapida
gassificazione dell'anidride disciolta con la conseguente formazione delle bollicine. La solubilità dei gas in acqua è
ben descritta la legge di Henry p=k*C ove p è la pressione parziale del gas sulla soluzione, C è la concentrazione del
gas nella soluzione e k è una costante tipica di ciascun gas che correla la pressione del gas sulla soluzione e la sua
concentrazione; K come tutte le costanti di equilibrio dipende dalla temperatura. Si deve considerare però che CO2,
Solubilità
SO2 ed NH3,per quanto siano spesso indicate, non sono degli esempi ottimali in quanto queste specie chimiche
formano dei composti con il solvente e quindi presentano delle solubilità anomale.
L'aumento della solubilità dipendentemente dalla pressione è un fenomeno apprezzabile solamente nei gas.
Solubilità nello stato solido
La solubilità in fase solida è fondamentale per capire il comportamento di alcune leghe. Alcuni materiali metallici
(per esempio Au e Cu o, entro certi limiti, Fe e Cr) quando solidificano formano una sola fase, in cui un elemento è
indistinguibile dall'altro. In questo caso si dice che i due metalli formano una soluzione solida. La solubilità degli
elementi nella matrice cristallina dell'elemento base può essere totale (vedi i casi indicati sopra), in generale in
questo caso si ha una lega di sostituzione, cioè gli atomi di un materiale sostituiscono quelli dell'altro nel reticolo
cristallino, ne consegue che, per avere questo tipo di lega, i due materiali devono avere lo stesso reticolo cristallino.
In altri casi la solubilità non è totale, ma solo parziale, cioè quando si supera una certa percentuale del materiale
soluto nella matrice del materiale solvente (per soluto si intende il materiale in percentuale atomica più bassa e
solvente quello con percentuale atomica più alta) supera un certo valore, si forma una fase diversa formata solo dal
soluto. Il caso più noto è quello del C nel Fe (vedi Diagramma ferro-carbonio), in cui il C è solubile nel Fe per un
massimo di circa il 2% a 1150 °C, mentre a temperatura ambiente è praticamente insolubile. In genere questa
solubilità forma leghe interstiziali, in cui gli atomi del soluto (che devono avere un raggio atomico molto più basso
di quello del solvente) occupano gli spazi lasciati liberi dentro il reticolo dagli atomi di dimensioni maggiori.
Naturalmente la presenza di corpi estranei modifica sensibilmente i parametri reticolari.
Note
[1] Chemistry 30 Solution Chemistry - Solubility Curves (http:/ / www. saskschools. ca/ curr_content/ chem30_05/ 4_solutions/ solution3_1.
htm)
[2] Perry's Chemical Engineers' Handbook, cap.2, p.120
Bibliografia
• Robert Perry; Don W. Green, Perry's Chemical Engineers' Handbook, 8a ed. (in inglese), McGraw-Hill, 2007.
ISBN 0-07-142294-3
Voci correlate
•
•
•
•
•
Concentrazione
Prodotto di solubilità
Saturazione chimica
Solvente
Parametro di solubilità di Hansen
93
Concentrazione
Concentrazione
La concentrazione di un componente in una
miscela è una grandezza che esprime il
rapporto tra la quantità del componente
rispetto alla quantità totale di tutti i
componenti della miscela (compreso il
suddetto componente), o, in alcuni modi di
esprimerla, del componente più abbondante.
Nel caso specifico di una soluzione (che è
Esempi di soluzioni diluite e concentrate. La concentrazione è evidenziata
un tipo particolare di miscela), la
qualitativamente dalla colorazione data dall'inchiostro.
concentrazione di un determinato soluto
nella soluzione esprime il rapporto tra la
quantità del soluto rispetto alla quantità totale di soluzione, o, in alcuni modi di esprimerla, del solo solvente (ad
esempio molalità).
Quando la sostanza in esame ha una concentrazione molto elevata nella miscela, si parla in genere di purezza; se
non è diversamente specificato, la purezza viene intesa come la percentuale in peso della sostanza in esame rispetto
al peso totale della miscela. Ad esempio se un campione di 100 grammi di argento presenta una purezza del 99,9%
vuol dire che tale campione contiene 99,9 grammi di argento e 0,1 grammi di altre sostanze (dette impurezze).
Modi per esprimere la concentrazione
Esistono diversi modi per esprimere la concentrazione di una soluzione, a seconda che ci si riferisca a misure di
volume, di peso, o di mole del soluto, della soluzione o del solvente. Per indicare la concentrazione di una soluzione
ci si può riferire a:
• concentrazione in massa di un componente i è data dal rapporto tra la massa del componente i rispetto al volume
della soluzione.[1]
• concentrazione molare di un componente i è data dal rapporto tra il numero di moli del componente i rispetto al
volume della soluzione.[1]
Grandezze relative
• percentuale in peso (%P/P m/m: massa soluto / massa soluzione x 100)
Indica quanti grammi di soluto sono sciolti in 100g di soluzione.
• percentuale peso/volume (% P/V )
Indica quanti grammi di soluto sono sciolti in 100 cm³ di soluzione
• percentuale in volume (C % V/V: volume soluto / volume soluzione x 100)
Indica quanti cm³ di un soluto liquido sono sciolti in 100 cm³ di soluzione
• percentuale mista (C % m/V: massa soluto / volume soluzione x 100)
• parti per milione (ppm)
Indica quanti milligrammi di soluto sono sciolti in 1 dm³ di soluzione
• molarità (M = moli soluto / litri soluzione)
Indica quante moli di soluto sono sciolte in 1 dm³ (1 L) di soluzione
• molalità (m = moli soluto / kg solvente)
94
Concentrazione
Indica quante moli di soluto sono state aggiunte a 1000 grammi di solvente
• normalità (N =equivalenti / litri soluzione)
• frazione molare (x = moli soluto / moli soluto+solvente)
Indica il rapporto tra il numero di moli di un componente della soluzione e il numero di moli totali. Può essere
anche espressa come il rapporto tra la concentrazione molare del componente i rispetto alla densità molare
della soluzione.[1]
• La frazione in massa di un componente i è data dal rapporto tra la concentrazione in massa del componente i
rispetto alla densità (di massa in questo caso) della soluzione.[1]
La concentrazione viene utilizzata nei calcoli relativi agli equilibri chimici e per ricavare svariate grandezze
termodinamiche.
In alcuni casi (in particolare per sistemi che deviano dall'idealità) si rende necessario utilizzare il concetto di attività
al posto della concentrazione.
Note
[1] Bird, op. cit., p. 506
Bibliografia
• R. Byron Bird; Warren E. Stewart, Edwin N. Lightfoot, Enzo Sebastiani (a cura di), Fenomeni di trasporto (http:/
/www.libreriauniversitaria.it/fenomeni-trasporto-bird-byron-cea/libro/9788840800516), Milano, Casa editrice
ambrosiana, 1979. ISBN 88-408-0051-4
Voci correlate
• Diluizione infinita
• Composizione chimica
Altri progetti
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Concentrazione
95
Frazione molare
96
Frazione molare
La frazione molare è una grandezza che viene impiegata per esprimere la concentrazione di una specie chimica in
una miscela omogenea, sia che si tratti di una soluzione liquida, di una miscela solida o di una miscela gassosa.
La frazione molare della specie j in una miscela è definita come il rapporto delle moli di j e le moli di tutte le specie
presenti nella miscela.[1]
Poiché le moli di ciascuna specie nella miscela è direttamente proporzionale al numero di entità (molecole, atomi,
ioni, zwitterioni) di tale specie (più precisamente è dato dal numero di entità chimiche diviso per il numero di
Avogadro), si può anche definire la frazione molare della specie j come il rapporto tra le moli di j e le moli totali
presenti nella miscela.
Dalla definizione si deduce immediatamente che xj sarà uguale a 0 nel caso in cui la specie j non sia presente nella
miscela e sarà uguale a 1 quando j è il solo costituente del sistema. Dalla definizione segue anche che la somma delle
frazioni molari di tutte le specie chimiche presenti nella miscela sarà uguale a 1.
Il rapporto molare, inteso come rapporto tra le moli
di una j-esima specie e le moli
di una k-esima specie,
si può porre in relazione con la frazione molare tramite l'equazione
Il termine
rappresenta il rapporto adimensionale fra le moli nj/nk.
Moltiplicando il valore della frazione molare per 100, si ottiene il valore della percentuale molare (o percentuale in
moli).
Note
[1] Silvestroni, op. cit., p. 235
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 8840809988
Voci correlate
•
•
•
•
Composizione chimica
Concentrazione
Frazione ponderale
Percentuale in volume
Numero di Avogadro
97
Numero di Avogadro
Il numero di Avogadro, chiamato così in onore di Amedeo Avogadro e denotato dal simbolo
o
, è il
numero di particelle (solitamente atomi, molecole o ioni) contenute in una mole. Tale numero di particelle è pari a
circa 6,022·1023. Viene formalmente definito come il numero di atomi di carbonio isotopo 12 presenti in 12 grammi
di tale sostanza.
Significato fisico del numero di Avogadro
Anche se definito con riferimento al carbonio-12, il numero di Avogadro si applica a qualsiasi sostanza. Corrisponde
al numero di atomi o molecole necessario a formare una massa pari numericamente al peso atomico o al peso
molecolare in grammi rispettivamente della sostanza. Ad esempio, il peso atomico del ferro è 55,847, quindi un
numero di Avogadro di atomi di ferro (ovvero, una mole di atomi di ferro) ha una massa di 55,847 g. Viceversa,
55,847 g di ferro, contengono un numero di Avogadro di atomi di ferro. Quindi il numero di Avogadro corrisponde
anche al fattore di conversione tra grammi (g) e unità di massa atomica (u):
Poiché l'unità di massa atomica viene definita facendo riferimento alla massa del carbonio-12 anche la definizione di
numero di Avogadro si riferisce a questo isotopo. L'altra unità di misura che appare nella definizione, cioè il kg, è
arbitraria e viene definita con una massa campione che si trova a Sèvres.
Di conseguenza, essendo un fattore di conversione tra due unità di misura non-omogenee,
è completamente
arbitrario e non viene considerato una costante fondamentale. Per la sua importanza e diffusione viene comunque
tabulato in ogni tabella di costanti fisiche.
Il numero di Avogadro compare anche in altre relazioni fisiche, come fattore di scala tra costanti microscopiche e
macroscopiche:
• la costante universale dei gas
• la costante di Faraday
e la costante di Boltzmann
e la carica elementare
:
:
Valore numerico
Al momento non è tecnologicamente possibile contare il numero esatto di atomi in 0,012 kg di carbonio-12, quindi il
valore preciso del numero di Avogadro è sconosciuto. Il valore raccomandato dal CODATA [1] del 2010 per il
numero di Avogadro è
,
dove il numero tra parentesi rappresenta la deviazione standard sulle ultime due cifre del valore (ovvero queste
possono passare da 29-27=02 a 29+27=56). A scopo di semplificazione, il numero di Avogadro viene spesso
approssimato a:
,
che è sufficientemente accurato per la maggior parte delle applicazioni.
Numero di Avogadro
98
Connessione tra massa dei protoni e dei neutroni
Un atomo di carbonio-12 consiste di 6 protoni e 6 neutroni (che hanno approssimativamente la stessa massa) e da 6
elettroni (la cui massa è in prima approssimazione trascurabile al confronto essendo a riposo 1836 volte inferiore a
quella del protone). Si potrebbe quindi pensare che un
di protoni o neutroni abbia massa 1 grammo. Anche se
questo è approssimativamente corretto, la massa di un protone libero a riposo è di 1,00727 u, quindi una mole di
protoni ha una massa di 1,00727 g. Similarmente, una mole di neutroni a riposo ha massa pari a 1,00866 g.
Chiaramente, 6 moli di protoni combinate con 6 moli di neutroni dovrebbero avere massa superiore a 12 g. Ci si
potrebbe chiedere quindi come è possibile che una mole di atomi di carbonio-12, che deve consistere di 6 moli di
neutroni, 6 di protoni e 6 di elettroni, possa avere una massa di appena 12 g.
Cosa ne è della massa in eccesso?
La risposta è legata all'equivalenza massa-energia, derivata dalla teoria della relatività ristretta. Nella struttura del
nucleo, i protoni e i neutroni sono tenuti assieme dalla forza nucleare forte. I legami corrispondono a stati di energia
potenziale minore rispetto ai protoni e neutroni liberi e isolati. In altre parole durante la formazione del nucleo
atomico viene liberata una grande quantità di energia e, poiché la massa è equivalente all'energia, si ha una "perdita
di massa" del nucleo rispetto alla semplice somma delle masse dei protoni e dei neutroni liberi. La differenza tra
massa del nucleo e la somma delle masse dei suoi nucleoni, o numero di massa A, non è costante e dipende dalla
forza dei legami. È massima per gli elementi più stabili (in particolare l'elio-4, nonché Fe, Co e Ni) ed è minore per
gli elementi meno stabili, cioè con legami nucleari più deboli (come il deuterio e gli isotopi radioattivi degli
elementi). Per il carbonio-12 la differenza è all'incirca dello 0,7% e rende conto, per definizione, della massa
"mancante" in una mole dell'elemento (difetto di massa).
Si può quindi dire che
è il rapporto tra massa in grammi di una mole di elemento e la sua massa nucleare in u,
tenendo però conto che è un'approssimazione, anche se molto precisa; perché la massa di un nucleo atomico non
dipende solo dal numero di protoni e neutroni che lo compongono ma anche dalla sua struttura.
Misurazione sperimentale del numero di Avogadro
Numerosi metodi possono essere usati per misurare il numero di Avogadro, a seconda delle conoscenze che si danno
per note all'atto della misurazione.
Un metodo moderno è quello di calcolarlo dalla densità di un cristallo, la sua massa atomica relativa e dalla
lunghezza della singola cella determinata tramite cristallografia a raggi X. Valori molto accurati di queste quantità,
dai quali deriva la attuale stima numerica di
, sono stati misurati per il silicio al National Institute of Standards
and Technology (NIST).
Numero di Avogadro
99
Tuttavia non è necessario ricorrere alla cristallografia:
nota la carica dell'elettrone, la formula chimica
dell'idrogeno gassoso molecolare e l'equazione di stato
dei gas perfetti si può misurare
con un semplice
esperimento di elettrolisi dell'acqua.
Nella figura a
rappresentazione
sperimentale:
destra si può
schematica
vedere una
dell'apparato
1. In un contenitore pieno d'acqua (distillata per
maggiore precisione) sono immersi due elettrodi,
uno dei quali è coperto con un contenitore graduato
rovesciato anch'esso pieno d'acqua.
2. I due elettrodi sono collegati a un amperometro e un
generatore di corrente orientato in modo che
l'elettrodo coperto diventi il catodo.
3. Viene fatta circolare della corrente attraverso il
circuito, l'elettrolisi dell'acqua provoca la liberazione
di idrogeno sul catodo e ossigeno sull'anodo.
4. L'ossigeno e l'idrogeno si combinano
immediatamente in molecole di H2 e O2, ma mentre
l'ossigeno può sfuggire dal contenitore, l'idrogeno
gassoso, rimane intrappolato nel contenitore
graduato.
Diagramma dell'apparato sperimentale.
5. Dopo un certo tempo, durante il quale la corrente
deve rimanere costante, il circuito viene aperto.
Si possono misurare due quantità:
1. Il volume di idrogeno prodotto
2. La carica totale transitata nel circuito
dove
è l'intensità di corrente e
il tempo
trascorso.
da queste due quantità se ne possono ricavare
direttamente altre due:
1. Le moli di idrogeno, tramite l'equazione di stato dei
gas perfetti:
2. Il numero di elettroni transitati nel circuito
in cui
è la carica dell'elettrone, nella
stessa unità di misura di
.
Per motivi pratici si può supporre la pressione e la
temperatura interne del contenitore graduato pari alla
pressione atmosferica e alla temperatura atmosferica.
Disegno di una tipica ampolla per
apparato sperimentale didattico.
Numero di Avogadro
100
Come ultima considerazione osserviamo che a due elettroni transitati nel circuito corrisponde l'elettrolisi di una
molecola d'acqua, con la conseguente liberazione di due atomi di idrogeno e la formazione di una molecola di H2.
Tenendo a mente che il numero
di molecole di H2 è pari ad
moli per
ricaviamo:
,
e, infine:
.
Rappresentazioni del numero di Avogadro
Per avere un'idea di quanto sia grande il numero di Avogadro, possiamo servirci delle seguenti visualizzazioni: Se si
prendesse un numero di palle da tennis pari a quello di Avogadro (quindi una "mole" di palle da tennis) e le si
disponesse in modo omogeneo su tutta la superficie terrestre, si raggiungerebbe un'altezza di cinquanta chilometri,
ovvero più di sei volte l'altezza del monte Everest. Ancora: se si disponessero tali palle in un'unica fila essa avrebbe
una lunghezza pari a circa 20 128 000 000 volte la larghezza di tutto il Sistema solare. Il numero di tazzine d'acqua
contenute nell'Oceano atlantico è dell'ordine di grandezza del numero di Avogadro, così come il numero di molecole
d'acqua in una tazzina. Se la stessa quantità di centesimi di euro fosse distribuita uniformemente tra la popolazione
mondiale, ogni abitante della Terra avrebbe mille miliardi di euro.
Curiosità
• NA ≈ 279, corretto allo 0.4% circa. È una delle più note coincidenze matematiche.
Note
[1] Fundamental Physical Constants (http:/ / physics. nist. gov/ constants) in The NIST Reference on Constants, Units, and Uncertainty. NIST,
2010
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996, pp. 166-167. ISBN 88-408-0998-8
Voci correlate
• Mole
• Giorno della mole
Collegamenti esterni
• Avogadro and molar Planck constants for the redefinition of the kilogram (http://www.inrim.it/Nah/
Web_Nah/home.htm)
Mole
101
Mole
La mole (ex grammomole, simbolo mol; il simbolo della grandezza quantità è n) è una delle sette unità di misura
fondamentali del Sistema internazionale. Misura la quantità delle sostanze; essa contiene tante entità elementari
quante sono gli atomi contenuti in 12 grammi dell'isotopo 12 del carbonio.
La mole è definita come la quantità di sostanza di un sistema che contiene un numero di unità interagenti[1]
pari al numero degli atomi presenti in 12 grammi di carbonio-12.[2]
Tale numero è noto come numero di Avogadro, dal matematico italiano Amedeo Avogadro, ed è pari a
6,02214179(30) · 1023 .[3] Una mole è quindi associata ad un numero enorme di entità o particelle (più di
seicentomila miliardi di miliardi).
Nel caso di un composto chimico, si può definire la mole come il valore della quantità (ad esempio espressa in
grammi) di particelle uguale al peso molecolare di ogni singola molecola. Tale quantità in realtà varia a seconda
della miscela isotopica che prendiamo in considerazione, ma la sua variazione può in genere assumersi trascurabile.
Il concetto di mole fu introdotto da Wilhelm Ostwald nel 1896.[3]
Rappresentazioni del numero di Avogadro
Per avere un'idea di quanto sia grande il numero di Avogadro, possiamo servirci delle seguenti visualizzazioni:
• Se si prendesse un numero di palle da tennis pari a quello di Avogadro (quindi una "mole" di palle da tennis) e le
si disponesse in modo omogeneo su tutta la superficie terrestre, si raggiungerebbe un'altezza di cinquanta
kilometri, ovvero più di sei volte l'altezza del monte Everest.
• Ancora: se si disponessero tali palle in un'unica fila essa avrebbe una lunghezza pari a circa 20 128 000 000 volte
la larghezza di tutto il Sistema solare.
• Il numero di tazzine d'acqua contenute nell'Oceano atlantico è dell'ordine di grandezza del numero di Avogadro,
così come il numero di molecole d'acqua in una tazzina.
• Se la stessa quantità di centesimi di euro fosse distribuita uniformemente tra la popolazione mondiale, ogni
abitante della Terra avrebbe mille miliardi di euro.
Definizioni aggiornate di mole
La grammomole e la grammomolecola sono state eliminate nel 1963 dal XIII CGPM e sostituite dalla "mole di
sostanza". Dal 1972 la mole fa parte del SI e in Italia il SI è diventato, per legge, l'unico sistema ufficiale di unità di
misura. Dal 2002 il SI è in vigore in tutto il mondo (ultimi arrivati: gli USA, che mantengono in vigore il vecchio
sistema per gli usi interni).
Dalla definizione segue che una quantità di sostanza è pari a una mole quando contiene un numero di particelle
uguale al numero di Avogadro. Una mole della sostanza B contiene 6,022 1023 particelle di B. Il rapporto fra il
numero delle particelle considerate e la quantità di B (in moli) si chiama costante di Avogadro e vale NA = 6,022
1023 molB-1 cioè il numero di Avogadro moltiplicato per il fattore di conversione (numero/molB)
Normalmente la sostanza B è una sostanza pura o una miscela ben definita (l'aria, per esempio, contiene 4 molecole
di azoto e 1 molecola di ossigeno, in prima approssimazione). La "quantità della sostanza B" diventa la "quantità di
B" quando la sostanza viene esplicitata; ad esempio "la quantità dell'aria" o "la quantità dell'ossigeno").
La quantità di B è il rapporto fra il numero delle particelle considerate e la costante di Avogadro:
nB = N°B / NA
in cui:
• n è espresso in moli
Mole
102
• NA in mol-1
• N° è un numero adimensionale.
La massa molare di una sostanza B (MB) è data dal rapporto fra la massa e la sostanza di un corpo.
Ad esempio,la massa atomica del sodio è pari a 22,99 u; una mole di sodio ( cioè un numero di atomi di sodio pari al
numero di Avogadro) corrisponde a 22,99 grammi di sostanza. La massa molare del sodio è 22,99 g/molNa.
Analogamente, nel caso dell'acqua (H2O), la massa molecolare è pari a 18,016 u; una mole di questa sostanza è pari
a 18,016 grammi. La massa molare dell'acqua 18,016 g/molH2O.
Nel caso del metano (CH4), la cui massa molecolare è 16,04, mezza mole (quindi mezzo numero di Avogadro di
molecole) corrisponde a 8 grammi. Talvolta si preferisce esplicitare i due casi usando le denominazioni ormai
obsolete di grammoatomo (mole di un elemento) e grammomolecola (mole di un composto).[4]
Nei paesi anglosassoni vengono inoltre utilizzate le definizioni di libbramolecola e libbramole, che sono simili alle
definizioni di grammomolecola e grammomole, tranne per il fatto che ci si riferisce alla libbra per la misura della
massa.
Non ci si può riferire alla mole di atomi o molecole come massa molare perché la massa molare in grammi/mole è
una grandezza diversa. Indicando con le moli e con
la massa molare (mole di entità), abbiamo:
Alcune applicazioni del concetto di mole
Il concetto di mole è utilizzato spesso in chimica, in quanto permette di paragonare particelle di massa differente.
Inoltre, riferendoci alle moli anziché al numero di entità, ci divincoliamo dall'uso di numeri molto grandi.
La mole è utilizzata anche nelle definizioni di altre unità di misura; ad esempio la carica di una mole di elettroni è
chiamata costante di Faraday[5], pari a 96 485 coulomb, mentre una mole di fotoni è detta einstein.
Il concetto di mole è utilizzato anche nelle equazioni di stato dei gas ideali; si ha che una mole di molecole di un
qualunque gas ideale, in condizioni normali (temperatura di 0 °C e pressione 101 325 Pa = 1 atm) occupa un volume
di 22,414 L per la legge di Avogadro. Così è possibile calcolare il numero di molecole presenti in un dato volume di
gas, e quindi la sua massa.
Esempio - calcoli stechiometrici
Nel seguente esempio, le moli sono usate per calcolare la massa di CO2 emessa, quando è bruciato 1 g di etano. La
formula coinvolta è:
3,5 O2 + C2H6 → 2 CO2 + 3 H2O
Qui, 3,5 moli di ossigeno reagiscono con 1 mole di etano, per produrre 2 moli di CO2 e 3 moli di H2O. Si noti che la
quantità di molecole non necessita di essere bilanciata su ambo i lati dell'equazione: da 4,5 moli di gas si passa a 5
mol di gas. Questo perché la quantità delle molecole di gas non conta la massa o il numero di atomi coinvolti, ma
semplicemente il numero di particelle individuali. Nel nostro calcolo è prima di tutto necessario calcolare la quantità
dell'etano che è stato bruciato. La massa di una mole di sostanza è definita come pari alla sua massa atomica o
molecolare, moltiplicata per il numero di Avogadro. La massa atomica dell'idrogeno è pari a 1 u, mentre la massa
molare di H è pari a 1 g/molH; la massa atomica del carbonio è pari a 12 u, la sua massa molare a 12 g/molC; quindi
la massa molare del C2H6 è: 2×12 + 6×1 = 30 g/molC2H6. Una mole di etano pesa 30 g. La massa dell'etano bruciato
era di 1 g, o 1/30 di mole. La massa molare della CO2 (con massa atomica del carbonio 12 u e dell'ossigeno 16 u) è:
2×16 u + 12 u = 44 u, quindi una mole di diossido di carbonio ha una massa di 44 g. Dalla formula sappiamo che:
• 1 mole di etano produce 2 moli di diossido di carbonio.
Conosciamo anche la massa dell'etano e del diossido di carbonio, quindi:
Mole
• 30 g di etano producono 2×44 g di diossido di carbonio.
È necessario moltiplicare per due la massa del diossido di carbonio perché ne vengono prodotte due moli .
Comunque, sappiamo anche che solo 1/30 dell'etano è stato bruciato. E di nuovo:
• 1/30 di mole di etano produce 2 × 1/30 di mole di biossido di carbonio.
E infine:
• 30 × 1/30 g di etano producono 44 × 2/30 g di biossido di carbonio = 2,93 g
Note
[1] Le entità chimiche e fisiche a cui si fa riferimento nella definizione di mole possono essere atomi, molecole, ioni, radicali, elettroni, fotoni, e
altre particelle o raggruppamenti specifici di queste entità. Si veda anche lista delle particelle.
[2] (EN) Unit of amount of substance (mole) (http:/ / www. bipm. org/ en/ si/ si_brochure/ chapter2/ 2-1/ mole. html) in SI brochure, Section
2.1.1.6. BIPM. URL consultato il 10 marzo 2011.
[3] Silvestroni, op. cit., p. 157
[4] Silvestroni, op. cit., p. 156
[5] da non confondere con l'unità della capacità elettrica, il farad
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• Silvio Gori, Chimica fisica, 1a ed., Padova, PICCIN, 1999.
• IUPAC,IUPAP,ISO, "Green Book", 1a ed., Londra, Blackwell, 1993.
Voci correlate
• Equivalente
• Frazione molare
• Molarità
103
Chimica inorganica
Chimica inorganica
ATTENZIONE: l'interpretazione dell'articolo non può essere effettuata - è mostrato come testo semplice.
Le possibili cause del problema sono (a) un errore nel software che crea il file PDF (b) un errore nella codifica
MediaWiki (c) una tabella troppo grande
La chimica inorganica è quella branca della chimica che studia gli elementi e la sintesi e caratterizzazione dei
composto inorganicocomposti inorganici.I composti inorganici possono essere di tipi molto diversi:A: il diborano
possiede legami molto inusualiB: il cloruro di cesio è un prototipo di Sistema cristallinostruttura cristallinaC: il
tetracarbonilbis(ciclopentadienil)diferroFp2 è un complesso Chimica metallorganicaorganometallicoD: il silicone ha
numerose applicazioni, da adesivi a protesi mammarieE: il catalizzatore di Grubbs ha fatto vincere nel 2005 il
Premio Nobel per la chimica al Robert H. Grubbssuo scopritoreF: le zeoliti sono molto usate come setacci
molecolariG: l'acetato di rame(II) sorprese i Chimica teoricachimici teorici per il suo diamagnetismoDefinizione
Secondo la definizione storica la chimica inorganica si occupa delle sostanze non prodotte dalla materia vivente,
oggetto di studio della chimica organica, quali sono i composti derivati dal mineraleregno minerale che non possiede
forza vitale, in accordo con la teoria vitalismovitalista. Caratteristica delle sostanze inorganiche era considerata
anche la loro possibilità di sintesi in laboratorio. Dopo la sintesi dell'urea effettuata da Friedrich Wöhler nel 1828,
utilizzando il composto inorganico cianato d'ammonio, i confini tra le due branche della chimica si sono ristretti
essendo dimostrato che anche i composto organicocomposti organici sono in realtà sintetizzabili in laboratorio.
Successivamente è stato constatato che gli organismo viventeorganismi viventi sono anch'essi in grado di sintetizzare
composti inorganici e con l'ulteriore affinarsi di conoscenze e tecniche sono addirittura fioriti campi di studio "ibridi"
come quelli della chimica metallorganica e della chimica supramolecolare. Tuttavia permane una certa differenza
fondata sulla diversa e caratteristica reattività della quale godono i composti organici e che si evidenzia in
meccanismo di reazionemeccanismi di reazione del tutto peculiari. Concetti chiave Struttura cristallina dell'ossido di
potassio, K2O, un composto ionicoMolti composti inorganici sono composti ionici, sali costituiti da cationi e anioni
tenuti assieme da legame ionico. Esempi di sali sono: il cloruro di magnesio MgCl2, costituito da cationi magnesio
Mg2+ e anioni cloruro Cl–, e l'ossido di sodio Na2O, costituito da cationi sodio Na+ e anioni ossido O2–. In tutti i
sali la carica totale dei cationi è bilanciata da quella degli anioni, in modo che il composto solido è elettricamente
neutro. La facilità di formazione di ioni monoatomici può essere valutata dal potenziale di ionizzazione (per i
cationi) o dall'affinità elettronica (per gli anioni) dell'elemento corrispondente. Ossidi, carbonati, solfati e alogenuri
costituiscono importanti famiglie di sali inorganici. Molti composti inorganici hanno punto di fusione elevato. Allo
stato solido i sali inorganici in genere sono cattivi Resistività elettricaconduttori di elettricità. Una caratteristica
abbastanza comune è la solubilità in acqua e la facilità di cristallizzazione (ad es. NaCl), ma molti sono invece
insolubili (ad es. SiO2SiO2). La più semplice reazione chimica inorganica è il doppio scambio, dove mettendo
assieme due sali gli ioni si scambiano senza variazioni del numero di ossidazione. Nelle reazioni di ossidoriduzione
un reagente, detto ossidante, acquista elettroni e diminuisce il proprio stato di ossidazione, mentre l'altro, detto
riducente, cede elettroni e aumenta il proprio stato di ossidazione. Scambi di elettroni possono avvenire anche
indirettamente, ad esempio nelle Pila (elettrotecnica)pile, che sono dispositivi Elettrochimicaelettrochimici.Quando
uno dei reagenti contiene atomi di idrogeno si può avere una reazione acido-base, dove vengono trasferiti protoni. In
una definizione più generale, qualsiasi specie chimica capace di legarsi ad una coppia di elettroni è detta acido di
Lewis; per contro, ogni specie che tende a cedere una coppia di elettroni è detta base di Lewis. Un trattamento più
raffinato delle interazioni acido-base è la teoria HSAB, che tiene conto della polarizzabilità e della dimensione degli
ioni. I composti inorganici si trovano in natura come minerali. Il suolo può contenere solfuro di ferro sotto forma di
104
Chimica inorganica
pirite o solfato di calcio sotto forma di Gesso (minerale)gesso. I composti inorganici possono svolgere molte
funzioni anche in campo biologico: come elettroliti (cloruro di sodio), come riserva di energia (Adenosina
trifosfatoATP), come materiali strutturali (i gruppi fosfato che formano lo scheletro del DNA). Il primo composto
inorganico importante prodotto artificialmente è stato il nitrato d'ammonio, un fertilizzante ottenuto con il processo
Haber-Bosch. Alcuni composti inorganici si sintetizzano per utilizzarli come catalizzatori, come V2O5V2O5 e
TiCl3TiCl3, altri servono come reagenti in chimica organica, come LiAlH4LiAlH4. Chimica inorganica industriale
La chimica inorganica è un ambito scientifico con molte applicazioni industriali. La quantità di acido solforico
prodotto è un parametro utile per valutare l'economia di una nazione. Nel 2005 i primi 20 composti chimici
inorganici prodotti in Canada, Cina, Europa, Giappone e Stati Uniti sono stati (in ordine alfabetico):(lingua
ingleseEN) (10 luglio 2006) Facts & figures of the chemical industry. Chem. Eng. News. acido cloridrico, acido
fosforico, acido nitrico, acido solforico, ammoniaca, azoto, carbonato di sodio, clorato di sodio, cloro, diossido di
titanio, idrogeno, idrossido di sodio, ossigeno, nero di carbone, nitrato d'ammonio, perossido di idrogeno, silicato di
sodio, solfato d'ammonio, solfato di alluminio, solfato di sodio. Un'altra applicazione pratica della chimica
inorganica industriale è la produzione di fertilizzanti. Chimica inorganica descrittiva La chimica inorganica
descrittiva si occupa di classificare i composti in base alle loro proprietà. La classificazione è basata in parte sulla
posizione occupata nella tavola periodica dall'elemento con il più alto numero atomico presente nel composto, e in
parte raggruppando i composti che presentano somiglianze strutturali. Succede spesso che un certo composto possa
essere classificato in più categorie. Ad esempio, un composto organometallico può essere anche un composto di
coordinazione, e può avere proprietà interessanti allo stato solido. Una serie di usuali classificazioni è riportata nel
seguito. Composti di coordinazione L'EDTA Chelazionechela uno ione Co3+ formando il complesso ottaedrico
[Co(EDTA)]– Nei composti di coordinazione o complessi classici un metallo è legato tramite una coppia di elettroni
ad un atomo di un ligandolegante come H2O, NH3, Cl– e CN–. Nella chimica di coordinazione moderna quasi tutti i
composti organici e inorganici possono essere usati come leganti. Il "metallo" appartiene in genere ai gruppi 3–13,
oppure ai lantanoidi o attinoidi, ma da un certo punto di vista tutti i composti chimici possono essere descritti come
complessi. La stereochimica dei complessi può essere molto ricca, come già osservato da Alfred WernerWerner nel
1914 con la separazione dei due enantiomeri di Exolo[Co{(OH)2Co(NH3)4}3]6+, che dimostrò per la prima volta
che la Chiralità (chimica)chiralità non era proprietà esclusiva dei composti organici. Un argomento di attualità
all'interno di questa specializzazione è la chimica di coordinazione supramolecolare.(lingua ingleseEN)J.-M. Lehn,
Supramolecular chemistry: Concepts and perspectives, Weinheim, Wiley-VCH, 1995. ISBN
978-3527293117Esempi:
[Co(EDTA)]–,
Cloruro
di
esamminocobalto(III)[Co(NH3)6]3+,
TiCl4TiCl4(TetraidrofuranoTHF)2. Composti dei gruppi principali Il tetranitruro di tetrazolfo, S4N4, un composto
dei gruppi principali che continua a incuriosire i chimici Queste specie contengono elementi dei Gruppo della tavola
periodicagruppi 1, 2 e 13–18 della tavola periodica, escludendo l'idrogeno. Spesso si aggiungono anche gli elementi
dei gruppi 3 (ScandioSc, IttrioY e LantanioLa) e 12 (ZincoZn, CadmioCd e Mercurio (elemento)Hg) perché hanno
una reattività simile.(lingua ingleseEN)N. N. Greenwood; A. Earnshaw, Chemistry of the elements, 2a ed., Oxford,
Butterworth-Heinemann, 1997. ISBN 0-7506-3365-4A questo gruppo si possono aggiungere specie note sin dagli
albori della chimica, ad esempio lo zolfo elementare S8 e il fosforo bianco P4. Antoine LavoisierLavoisier e Joseph
PriestleyPriestley con i loro esperimenti sull'ossigeno, OssigenoO2, non solo identificarono un importante gas
Molecola biatomicadiatomico, ma permisero di descrivere composti e reazioni in base a rapporti
Stechiometriastechiometrici. All'inizio del 900 Carl Bosch e Fritz Haber resero possibile la sintesi dell'ammoniaca
usando catalizzatori a base di ferro, una scoperta che ebbe un enorme impatto sulla storia dell'umanità, dimostrando
l'importanza della sintesi di composti inorganici. Esempi tipici di composti dei gruppi principali sono SiO2SiO2,
SnCl4SnCl4 e N2ON2O. Molti composti dei gruppi principali si possono anche considerare organometallici, dato
che contengono gruppi organici, come ad esempio TetrametilsilanoSi(CH3)4. Composti dei gruppi principali sono
reperibili anche in natura, ad esempio i fosfati nel DNA e nelle Ossoossa, e quindi si possono classificare come
bioinorganici. Viceversa possono essere considerati "inorganici" i composti organici senza (molti) idrogeni, come ad
esempio i Fullerenefullereni, i nanotubi di carbonio, e gli ossidi di carbonio Composto binariobinari. Esempi:
105
Chimica inorganica
tetranitruro di tetrazolfo S4N4, diborano B2H6, siliconi, fullerene C60. Composti dei metalli di transizione Il
cisplatino è un complesso dei metalli di transizione usato nella chemioterapia di alcuni tumori I composti che
contengono metalli dei Gruppo della tavola periodicagruppi 4–11 sono considerati composti dei metalli di
transizione. I composti con metalli dei gruppi 3 e 12 sono a volte inclusi in questa categoria, ma spesso sono
considerati composti dei gruppi principali. I composti dei metalli di transizione hanno una chimica di coordinazione
ricca, spaziando da composti tetraedrici come TiCl4TiCl4 a composti planari quadrati come i complessi di platino, e
a composti ottaedrici per i complessi del cobalto. Vari metalli di transizione si trovano in composti di importanza
biologica, come il ferro nell'emoglobina. Esempi: tetracloruro di titanio, ferrocianuro di potassio, cisplatinoComposti
organometallici Struttura di n-butillitio; i composti organometallici del litio esistono spesso in forma polimerica I
composti organometallici sono quelli dove dove un metallo è legato covalentemente ad uno o più atomi di carbonio
di un gruppo organico. Il metallo M di queste specie può appartenere ai gruppi principali o ai metalli di transizione.
In pratica è comunemente usata una definizione più ampia di composto organometallico, includendo i
Metallocarbonilemetallocarbonili e anche i metallo alcossidi. I composti organometallici sono di solito considerati
una categoria particolare perché i leganti organici sono spesso sensibili all'idrolisi e all'ossidazione, e quindi la
preparazione di composti organometallici richiede tecniche più sofisticate rispetto ai tradizionali complessi di
Werner. Le nuove procedure di sintesi, e in particolare la capacità di manipolare i complessi in solventi poco
coordinanti, hanno permesso di utilizzare leganti scarsamente coordinanti come gli idrocarburi, H2 e N2. La chimica
organometallica ha tratto molti vantaggi dal fatto che questi leganti sono in un certo qual modo prodotti dell'industria
petrolifera. Esempi: ferrocene Fe(C5H5)2, molibdeno esacarbonile Mo(CO)6, clorotris(trifenilfosfina)rodio(I)
RhCl(PPh3)3Composti cluster Il decaborano è un Cluster (chimica)cluster del boro estremamente tossicoI centri di
ferro-zolfo sono i componenti centrali di proteine essenziali per il metabolismo umano Composti cluster si possono
trovare in tutte le classi di composti chimici. A rigore, un cluster prevede più centri metallici legati tra loro con
legami covalenti, ma si considerano cluster anche quelli formati da non metalli come il boro. Esistono cluster
puramente inorganici, ma anche organometallici o bioinorganici. La distinzione tra cluster molto grandi e solidi
estesi è sempre più sfumata. A questo livello di dimensioni si parla di nanoscienza e nanotecnologia, e sono
importanti gli studi di effetti quantici. I cluster di grandi dimensioni possono essere considerati come un insieme di
atomi legati tra loro, con caratteristiche intermedie tra una molecola e un solido. Esempi: Triferro
dodecacarbonileFe3(CO)12, DecaboranoB10H14, Cloruro di molibdeno(II)[Mo6Cl14]2−, Centri di
ferro-zolfo4Fe-4SComposti bioinorganici Il centro di cobalto ottaedrico della CobalaminaVitamina B12 Questi
composti sono presenti in natura per definizione, ma questa categoria comprende anche specie antropogeniche come
inquinanti (ad es. metilmercurio) e farmaci (ad es. cisplatino).(lingua ingleseEN)S. J. Lippard, J. M. Berg, Principles
of bioinorganic chemistry, Mill Valley, CA, University Science Books, 1994. ISBN 0-935702-73-3 In questo campo
si incontrano molti aspetti della biochimica e molti tipi di composti, ad esempio i fosfati nel DNA, complessi
metallici con leganti che spaziano da macromolecole biologiche come i peptidi a specie poco definite come gli acidi
umici, all'acqua coordinata in complessi di gadolinio (usati per imaging a risonanza magnetica). Un campo
d'indagine tradizionale della chimica bioinorganica riguarda i processi di trasferimento di elettroni e di energia nelle
proteine che servono nella respirazione. La chimica inorganica in campo medico studia anche quali siano gli Sali
mineralielementi essenziali costituenti delle biomolecole, con relativa applicazione a diagnosi e terapia. Esempi:
emoglobina, metilmercurio, carbossipeptidasi, ferritinaComposti allo stato solido YBa2Cu3O7, abbreviato come
YBCO, è un superconduttore capace di Effetto Meissner-Ochsenfeldlevitare sopra un magnete a temperature
inferiori alla sua temperatura critica, circa 90 K (−183°C) Questa area della chimica inorganica si focalizza su
struttura,(lingua ingleseEN)U. Müller, Inorganic structural chemistry, 2a ed., Chichester, Wiley, 2006. ISBN
978-0470018651 legami e proprietà fisiche dei materiali. In pratica la chimica inorganica dello stato solido usa
tecniche come la cristallografia per comprendere le proprietà che si generano dall'insieme di interazioni tra i
componenti presenti nel solido. Questa area si interessa anche di metalli, Lega (metallurgia)leghe e derivati
intermetallici. Campi di studio correlati sono la fisica della materia condensata, la mineralogia e la scienza dei
materiali. Esempi: zeoliti, YBCOYBa2Cu3O7, semiconduttoriChimica inorganica teorica Un altro modo di
106
Chimica inorganica
avvicinarsi alla chimica inorganica è partire dal modello atomico di Bohr, e usare gli strumenti e i modelli della
chimica teorica e della chimica computazionale per spiegare i legami in molecole semplici, per poi passare a specie
più complesse. I composti inorganici contengono molti elettroni, ed è quindi arduo descriverli accuratamente con i
metodi della meccanica quantistica. Per affrontare queste difficoltà sono stati inventati molti approcci
semiquantitativi o semiempirici, tra i quali la teoria degli orbitali molecolari e la teoria del campo dei leganti. Oltre a
queste descrizioni teoriche si usano anche metodi approssimati, come la teoria del funzionale della densità. I
composti che si comportano in modo inspiegabile per le teorie, sia in senso qualitativo che quantitativo, sono molto
importanti per l'avanzamento delle conoscenze. Ad esempio, Acetato di rame(II)CuII2(OAc)4(H2O)2 è quasi
diamagnetico al di sotto della temperatura ambiente, mentre la teoria del campo cristallino predice che la molecola
abbia due elettroni spaiati. Il disaccordo tra teoria (paramagnetico) e osservazione sperimentale (diamagnetico) ha
portato allo sviluppo di modelli di "accoppiamento magnetico", che a loro volta hanno generato nuove tecnologie e
nuovi materiali magnetici. Teorie qualitative La teoria del campo cristallino spiega perché Ferricianuro di
potassio[FeIII(CN)6]3− ha un solo elettrone spaiato La chimica inorganica ha tratto grandi vantaggi dalle teorie
qualitative, che sono più facili da comprendere perché richiedono poche conoscenze di chimica quantistica. La teoria
VSEPR è in grado di razionalizzare e predire le strutture di molti composti dei gruppi principali; ad esempio spiega
perché NH3NH3 è piramidale mentre ClF3ClF3 ha forma a T. Nei metalli di transizione la teoria del campo
cristallino permette di interpretare le proprietà magnetiche di molti complessi; ad esempio Ferricianuro di
potassio[FeIII(CN)6]3− ha un solo elettrone spaiato, mentre [FeIII(H2O)6]3+ ne ha cinque. Simmetria molecolare e
teoria dei gruppi Il diossido di azoto, NO2, ha simmetria molecolaresimmetria C2v Un concetto molto utile in
chimica inorganica è quello di simmetria molecolare.(lingua ingleseEN)F. A. Cotton, Chemical applications of group
theory, New York, John Wiley & Sons, 1990. ISBN 0471510947 In matematica, la teoria dei gruppi fornisce il
formalismo adatto a descrivere la forma delle molecole a seconda del gruppo puntuale cui appartengono. La teoria
dei gruppi permette anche di semplificare i calcoli teorici. In spettroscopia, le applicazioni più comuni della teoria
dei gruppi riguardano spettri vibrazionali ed elettronici, perché conoscendo le proprietà di simmetria dello stato
fondamentale e degli stati eccitati di una specie chimica si può prevedere numero e intensità delle bande di
assorbimento. Una applicazione comune della teoria dei gruppi è la previsione del numero di vibrazioni C–O in
complessi Metallocarbonilemetallocarbonilici sostituiti. La teoria dei gruppi è anche uno strumento didattico per
evidenziare somiglianze e differenze nelle proprietà di legame di specie diversissime come Esafluoruro di
tungstenoWF6 e Molibdeno esacarbonileMo(CO)6, o Diossido di carbonioCO2 e Diossido di azotoNO2.
Termodinamica e chimica inorganica Un differente approccio quantitativo alla chimica inorganica considera
l'energia scambiata durante le reazioni. Questo approccio è molto tradizionale ed Ricerca empiricaempirico, ma di
grande utilità. Potenziale standard di riduzionePotenziale redox, acidoacidità e Fase (chimica)transizioni di fase sono
alcuni dei concetti che si possono esprimere in termini termodinamici. Un altro concetto classico della
termodinamica inorganica è il ciclo di Born-Haber, usato per determinare l'energia di processi elementari che non
possono essere misurati direttamente, come l'affinità elettronica. Meccanismi in chimica inorganica Un altro aspetto
della chimica inorganica è lo studio dei meccanismi di reazione, che vengono in genere discussi in base alle
differenti categorie di composti. Elementi dei gruppi principali e lantanoidi Nel discutere i meccanismi di composti
dei gruppi principali (soprattutto 13-18) si utilizzano in genere gli approcci della chimica organica, dato che in fin
dei conti anche i composti organici fanno parte dei gruppi principali. Gli elementi più pesanti di CarbonioC, AzotoN,
OssigenoO e FluoroF possono formare composti con più elettroni di quanti previsti dalla regola dell'ottetto, e per
questo motivo possono avere meccanismi di reazione diversi da quelli dei composti organici. Elementi più leggeri
del carbonio (BoroB, BerillioBe, LitioLi) nonché AlluminioAl e MagnesioMg formano spesso strutture elettron
deficienti in qualche modo simili ai carbocationi, che tendono a reagire con meccanismi associativi. La chimica dei
lantanoidi rispecchia per molti aspetti quella dell'alluminio. Complessi dei metalli di transizione I meccanismi di
reazione coinvolgenti metalli di transizione sono discussi in modi diversi da quelli degli elementi dei gruppi
principali, perché la presenza degli orbitali d influenza notevolmente la loro reattività. Alcuni tipi di reazione
osservabili nei complessi sono i seguenti. Reazioni di sostituzione dei leganti La presenza degli orbitali d è
107
Chimica inorganica
determinante nell'influenzare velocità e meccanismo delle reazioni di sostituzione e di dissociazione dei leganti.
Queste reazioni possono avvenire con meccanismi associativi o dissociativi. Un aspetto generale della chimica dei
metalli di transizione è la labilità cinetica degli ioni metallici, come si osserva nei tipici complessi [M(H2O)6]n+ che
scambiano l'acqua coordinata con quella del solvente: [M(H2O)6]n+ + 6H2O* → [M(H2O*)6]n+ + 6H2O dove
H2O* denota acqua isotopoisotopicamente arricchita, cioè H217O Le velocità di scambio dell'acqua variano di 20
ordini di grandezza nella tavola periodica; i lantanoidi sono i più veloci e i composti di Ir(III) sono i più lenti.
Reazioni redox Le reazioni redox sono comuni per i metalli di transizione. Si possono suddividere in due classi: le
reazioni di trasferimento di atomi, come le reazioni di addizione ossidativa/eliminazione riduttiva, e le reazioni di
trasferimento di elettroni. Una reazione fondamentale è la reazione di scambio, dove i reagenti sono uguali ai
prodotti. Ad esempio gli ioni permanganato e manganato reagiscono scambiandosi un elettrone:[MnO4]− +
[Mn*O4]2− ⇄ [MnO4]2− + [Mn*O4]−Reazioni sui leganti I leganti coordinati reagiscono in modo diverso dai
leganti liberi. Ad esempio, l'acidità dei leganti ammoniaca in Cloruro di esamminocobalto(III)[Co(NH3)6]3+ è
maggiore rispetto a NH3 non coordinata. Gli alcheni legati a cationi metallici reagiscono con nucleofili, mentre gli
alcheni liberi di solito non lo fanno. La Catalizzatorecatalisi è un'area importante per l'industria, e si basa sulla
capacità dei metalli di modificare la reattività di leganti organici. La catalisi omogenea è condotta in soluzione, e la
catalisi eterogenea si ha quando substrati gassosi o disciolti reagiscono con superfici solide. Tradizionalmente, la
catalisi omogenea è considerata parte della chimica organometallica mentre la catalisi eterogenea è parte della
scienza delle superfici, un settore della chimica dello stato solido, ma i principi basilari di chimica inorganica sono
gli stessi. Alcuni composti dei metalli di transizione hanno la peculiarità di reagire con piccole molecole come
Monossido di carbonioCO, IdrogenoH2, OssigenoO2 ed EtileneC2H4. L'importanza industriale di queste materie
prime traina lo sviluppo della catalisi. Caratterizzazione dei composti inorganici I composti chimici inorganici
possono contenere praticamente tutti gli elementi della tavola periodica e avere proprietà diversissime, quindi la loro
caratterizzazione può richiedere i metodi di analisi più disparati. I metodi più vecchi tendevano ad esaminare
proprietà generali come punto di fusione, solubilità, acidità e conducibilità elettrica in soluzione. Successivamente,
l'avvento della meccanica quantistica e il perfezionamento delle apparecchiature elettroniche hanno reso disponibili
nuovi strumenti per studiare le proprietà elettroniche di molecole e solidi inorganici. I dati così ottenuti hanno spesso
permesso di perfezionare i modelli teorici. Ad esempio, la determinazione dello Spettroscopia fotoelettronica
ultraviolettaspettro di fotoelettroni del metano ha dimostrato non è del tutto appropriata la descrizione della molecola
in base alla teoria del legame di valenza, che prevede legami a due centri e due elettroni tra carbonio e idrogeno.
Risultati come questo hanno favorito la diffusione della teoria degli orbitali molecolari, basta su orbitali totalmente
delocalizzati, che permettono di descrivere più accuratamente cosa succede rimuovendo un elettrone. Tecniche
comunemente utilizzate: CristallografiaCristallografia a raggi X, per determinare la struttura tridimensionale delle
molecole. Interferometria a doppia polarizzazione, per determinare conformazione e variazioni di conformazione
nelle molecole SpettroscopiaMetodi spettroscopici di vario tipo: Spettroscopia ultravioletta/visibile (UV/Vis),
storicamente importante, dato che molti composti inorganici sono fortemente colorati Spettroscopia di risonanza
magnetica nucleare (NMR); oltre a 1idrogenoH e 13CarbonioC, molti altri nuclidi sono adatti per la spettroscopia
NMR (ad es. 11BoroB, 19FluoroF, 31FosforoP e 195PlatinoPt) e forniscono informazioni sulla struttura dei
composti. Anche la spettroscopia NMR di sostanze paramagnetiche può fornire informazioni strutturali. L'analisi
NMR 1idrogenoH è importante anche perché il nucleo dell'idrogeno è molto leggero ed è difficile da localizzare con
la cristallografia a raggi X Spettroscopia infrarossa (IR), usata soprattutto per i Metallocarbonilecomplessi
metallocarboniliciSpettroscopia ENDORSpettroscopia Mössbauer Spettroscopia di risonanza paramagnetica
elettronica (EPR), permette di osservare l'intorno di centri metallici Paramagnetismoparamagnetici Metodi
Elettrochimicaelettrochimici come la ciclovoltammetria e tecniche simili forniscono informazioni sulle proprietà
redox dei composti. Chimica inorganica sintetica Le specie inorganiche che si possono trovare pure in natura sono
poche; tutte le altre devono essere sintetizzate in impianti e laboratori chimici. I metodi di sintesi della chimica
inorganica si possono classificare grossolanamente a seconda della volatilità o della solubilità dei reagenti
utilizzati.(lingua ingleseEN)G. S. Girolami, T. B. Rauchfuss, R. J. Angelici, Synthesis and technique in inorganic
108
Chimica inorganica
109
chemistry, Mill Valley, CA, University Science Books, 1999. ISBN 0935702482 I composti inorganici solubili si
preparano con metodi analoghi a quelli della sintesi organica. Composti che reagiscono con l'aria richiedono tecniche
tipo linea Schlenk e glove box. Composti volatili e gas sono manipolati in linee da vuoto, costituite da tubi di vetro
connessi tramite valvole, entro cui si può fare un Vuoto (fisica)vuoto di 0,1 Pascal (unità di misura)Pa o meno; i
composti vengono condensati usando azoto liquido, che ha punto di ebollizione 77 K, o altri Liquido criogenoliquidi
criogeni. I solidi sono in genere preparati usando forni tubolari: i reagenti sono chiusi in contenitori, spesso fatti di
silice fusa (SiO2 amorfo), ma a volte sono necessari materiali speciali come tubi saldati di tantalio e "navicelle" di
platino. Aree Principali aree di interesse della chimica inorganica sono: Chimica degli elemento
(chimico)elementiChimica
nucleareChimica
di
coordinazioneMetallorganicaChimica
dello
stato
solidoElettrochimicaDiffrazione
dei
raggi
XCristallografiaChimica
bioinorganicaChimica
supramolecolareGeochimicaMineralogiaAstrochimicaSpettroscopia molecolareChimica teoricaMetallurgiaChimica
ambientaleStrutturistica chimicaSintesi e tecniche speciali inorganicheNote Altri progetti Wikimedia Commons
contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Inorganic compounds
Chimica organica
La chimica organica si occupa delle caratteristiche chimiche e fisiche delle molecole organiche. Si definiscono
convenzionalmente composti organici i composti del carbonio con eccezione degli ossidi, monossido e diossido, e
dei sali di quest'ultimo: anione idrogenocarbonato ed anione carbonato rispettivamente, derivati solo formalmente
dall'acido carbonico (in realtà inesistente in soluzione acquosa), oltre ad altre piccole eccezioni.
Storia
Il termine "chimica organica" fu adottato per la prima volta nel 1807 da Jöns Jacob
Berzelius.[1] L'aggettivo "organica" fu inizialmente legato al fatto che questa branca
della chimica studiava composti più o meno complessi estratti da organismi viventi,
vegetali o animali, o dai loro metaboliti. Tale definizione fu abbandonata a favore di
quella sopra esposta nel 1828, quando il chimico tedesco Friedrich Wöhler per primo
riuscì nella sintesi di un composto organico (l'urea) a partire da composti inorganici,[2]
dimostrando che le sostanze prodotte in laboratorio a partire da composti inorganici
erano in tutto identiche a quelle aventi la medesima struttura isolate da organismi viventi,
e confutando quindi l'ipotesi vitalistica che voleva le sostanze "organiche" in qualche
modo peculiari per via della loro origine biologica.
Jöns Jacob Berzelius
Nel 1861 August Kekulè identificò la chimica organica come "lo studio dei composti del carbonio".[3]
Idrocarburi ed eteroatomi
I composti organici costituiti solo da atomi di idrogeno (H) e carbonio (C) sono detti idrocarburi; ad esempio il
metano, avente formula chimica CH4, è il più semplice degli idrocarburi.
Chimica organica
110
Altri elementi, spesso presenti nelle molecole organiche, sono denominati
collettivamente "eteroatomi" e sono l'ossigeno, l'azoto, il fosforo, lo zolfo, il boro, gli
alogeni (fluoro, cloro, bromo e iodio), ed anche altri elementi semimetallici, nonché
alcuni metalli in grado di formare composti di coordinazione col carbonio stesso. In
particolare i composti organici contenenti atomi metallici direttamente legati ad atomi di
carbonio sono detti metallorganici od organo-metallici; tra i metallorganici si
annoverano gli organo-litio, -sodio, -magnesio, -manganese, -mercurio, -piombo, -tallio,
-zinco. I composti ciclici il cui anello contiene uno o più eteroatomi sono invece definiti
"eterociclici".
Friedrich Wöhler
Sistematica organica
L'approccio più classico allo studio della chimica organica consiste nel raggruppare i composti in classi di sostanze
che presentano un medesimo gruppo funzionale, definendo così una serie omologa. I composti che fanno parte di una
stessa classe possiedono la stessa composizione e le stesse proprietà chimiche, mentre le proprietà chimico-fisiche
(come punto di fusione, tensione di vapore etc.) variano in funzione del peso molecolare.
All'interno di questa classificazione gli alcani rappresentano la famiglia di composti più semplice, essendo formati
solamente da atomi di carbonio e idrogeno che instaurano tra loro un legame semplice. Alcheni e alchini sono simili
agli alcani, ma presentano rispettivamente doppi e tripli legami. Queste tre classi di composti rappresentano gli
idrocarburi alifatici, che si differenziano dagli idrocarburi aromatici (come il benzene) per il fatto di non possedere
aromaticità. Man mano che si vanno aggiungendo altri elementi chimici differenti dal carbonio e dall'idrogeno si
tende ad ottenere molecole più complesse. Gli alogenuri alchilici sono derivati direttamente dagli idrocarburi alifatici
aggiungendo atomi di alogeno, allo stesso modo dagli idrocarburi aromatici si ottengono gli alogenuri arilici.
Carbonio, idrogeno e ossigeno possono formare due classi di composti caratterizzati dal gruppo ossidrilico (-OH): gli
alcoli e i fenoli (composti aromatici). Questi tre elementi possono formare anche gli eteri, composti caratterizzati da
un legame R-O-R', e composti ciclici noti come epossidi. L'ossigeno può anche legarsi al carbonio con un doppio
legame formando aldeidi (R-CHO) e chetoni (R-CO-R'); la contemporanea presenza di un gruppo -OH porta inoltre
alla formazione degli acidi carbossilici (R-COOH). Negli alogenuri acilici, che sono dei derivati degli acidi
carbossilici, il carbonile (C=O) è legato a un atomo di alogeno. Altri importanti derivati degli acidi carbossilici sono
gli esteri, composti caratterizzati dalla presenza del gruppo estereo -COOR.
Con l'azoto, un altro importante eteroatomo, si possono ottenere i nitrili (R-C≡N), le ammidi (R-CONH2), i
nitrocomposti (R-NO2) e le ammine (R-NH2), le basi della chimica organica. I composti che presentano sia il gruppo
amminico che quello carbossilico sono definiti amminoacidi.
Tra le biomolecole si hanno infine i carboidrati, le proteine, i lipidi e gli acidi nucleici.
Chimica organica
Meccanismi di reazione in chimica organica
Le sostanze organiche reagiscono in modo caratteristico in base alla loro natura e alla presenza di determinati gruppi
funzionali, oltre che in funzione delle condizioni di reazione (tipo di solvente usato, temperatura, pH etc.). I
meccanismi di reazione maggiormente diffusi in chimica organica sono i seguenti:
•
•
•
•
•
sostituzione radicalica;
sostituzione nucleofila (alifatica e aromatica);
reazione di addizione (elettrofila, nucleofila, radicalica, periciclica);
reazione di eliminazione;
reazione di riarrangiamento.
Note
[1] (EN) Carbon Chemistry: The Breakdown of Vitalism (http:/ / www. 3rd1000. com/ history/ carbon. htm)
[2] Reazione di metatesi del cianato d'ammonio, ottenuto a sua volta per reazione fra cianato d'argento e cloruro d'ammonio, o fra cianato di
piombo ed ammoniaca acquosa. Solomons, op. cit., p. 2
[3] Solomons, op. cit., p. 3
Bibliografia
• T. W. Graham Solomons, Chimica organica, 2a ed., Bologna, Zanichelli, 2001. ISBN 88-08-09414-6
• (EN) Andrew Streitwieser; Clayton H. Heathcock, Edward M. Kosower, Introduction to Organic Chemistry , 4a
ed., New York, Macmillan Publishing Company, 1992. ISBN 0-02-418170-6
• Paula Y. Bruice, Chimica organica, 4a ed., Napoli, Edises, 2004. ISBN 88-08-09414-6
Voci correlate
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Chimica organica fisica
Chimica
Chimica inorganica
Chimica fisica
Chimica farmaceutica
Sistematica
Stereochimica
Sintesi organica
Meccanismi di reazione
Chimica combinatoria
Chimica biorganica
Chimica dei composti eterociclici
Geochimica organica
NMR
Spettrometria di massa
Petrolchimica
Chimica dei polimeri e delle macromolecole
Chimica supramolecolare
111
Chimica organica
112
Altri progetti
•
Wikibooks contiene testi o manuali: http://it.wikibooks.org/wiki/Chimica organica
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Organic
compounds
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/chimica organica
Collegamenti esterni
• chimicaorganica.net (http://www.chimicaorganica.net)
• Chimica organica (http://www.chem4tech.it/index.php?option=com_docman&task=doc_download&
gid=76&Itemid=11&lang=en)
• Documenti di sintesi di composti con meccanismi di reazione e procedura di laboratorio (http://www.
itiskennedy.it/modules.php?op=modload&name=Downloads&file=index&req=viewsdownload&sid=35)
• (EN) organicworldwide.net (http://www.organicworldwide.net)
Sale
In chimica, un sale è un composto chimico ottenuto sostituendo gli atomi di idrogeno di un acido con atomi metallici
o con un gruppo funzionale che abbia comportamento metallico (per esempio il gruppo ammonio, NH4)
Un sale può essere organico o inorganico.
In generale, i sali sono composti ionici costituiti da atomi appartenenti ad un reticolo cristallino. Presentano
caratteristiche esteriori variabili e diverse (colore, odore, sapore, trasparenza) a seconda della loro composizione.
Possono essere molto solubili o completamente insolubili in acqua, dove i due gruppi uniti da legame ionico si
dissociano. Sono altresì solubili in altri solventi, e tipicamente i sali formati da elettroliti forti si sciolgono bene in
solventi polari. I sali hanno punto di fusione variabile, spesso bassa durezza, e bassa comprimibilità. Se fusi o
dissolti in acqua son detti elettroliti e conducono elettricità proporzionalmente all'elettronegatività degli atomi
costituenti, comportandosi da conduttori di seconda specie.
I due elementi di un sale binario (come ad esempio il cloruro di sodio) possono essere separati per elettrolisi del sale
fuso.
Ciò che comunemente viene chiamato sale o sale da cucina è in realtà uno dei tanti sali possibili, cioè il cloruro di
sodio (NaCl).
Reazioni che producono sali
Reagenti
Risultato
Equazione di esempio
idrossido + acido
sale + acqua
2NaOH + H2SO4
Na2SO4 + 2H2O
(solfato di sodio)
metallo + acido
sale + idrogeno
Zn + H2SO4
ZnSO4 + H2
(solfato di zinco)
anidride + ossido basico
sale
CO2 + CaO
CaCO3
(carbonato di calcio)
ossido basico + acido
sale + acqua
metallo + non metallo (del VI o del VII gruppo A della Tavola
Periodica)
sale (non
ossigenato)
Fe2O3 + 6 HNO2
2 Fe(NO2)3 + 3
H2O
(nitrito ferrico)
Zn + Cl2
ZnCl2
(cloruro di zinco)
Sale
113
idrossido + anidride
sale + acqua
2NaOH + SO2
Na2SO3 + H2O
(solfito di sodio)
NB: ci sono altri modi per ottenere sali partendo da sali: sale + acido, sale + non-metallo, sale + idrossido, sale +
sale.
Nomenclatura
Le regole di nomenclatura usate in chimica assegnano ai sali nomi a
partire dagli ioni che li costituiscono. Frequentemente nomi dei
componenti cationici, spesso ioni metallici o ammonici, sono dati per
primi, seguiti dai nomi del componente anionico. Gli anioni sono
spesso chiamati in accordo al loro acido coniugato, sostituendo il
suffisso -idrico con il suffisso -uro, il suffisso -oso con il suffisso -ito
ed il suffisso -ico con il suffisso -ato; alcuni esempi:
• acetati sono sali dell'acido acetico
• carbonati sono sali dell'acido carbonico
Cristalli di cloruro di sodio
• cloruri sono sali dell'acido cloridrico
• cianuri sono sali dell'acido cianidrico
• nitrati sono sali dell'acido nitrico
•
•
•
•
nitriti sono sali dell'acido nitroso
fosfati sono i sali dell'acido fosforico
solfati sono sali dell'acido solforico
fluoruri sono i sali dell'acido fluoridrico
Sali acidi
Nelle reazioni di preparazione dei sali con acidi con più di un atomo di idrogeno può accadere in determinate
condizioni che al radicale acido rimangano uno o più atomi di idrogeno, quindi legandosi al metallo si ottiene un sale
acido.
2H2SO4 + 2Na → 2NaHSO4 + H2 (solfato acido di sodio o bisolfato di sodio)
Sali basici
Nelle reazioni di preparazione dei sali partendo dagli idrossidi con più di un gruppo idrossido in determinate
condizioni può accadere che dall'idrossido non si stacchino tutti i suddetti gruppi ed uno (o più d'uno) rimanga legato
al sale. Si ha così un sale basico.
Es. Ca(OH)2 + HCl → Ca(OH)Cl + H2O (cloruro basico di calcio o idrossicloruro di calcio)
Sapore dei sali
I sali possono avere due sapori: amaro o salato. Se l'uno o l'altro dipende dalla somma dei diametri delle specie
ioniche.[1]
Come riferimento viene preso il bromuro di potassio che ha un sapore intermedio e la somma dei diametri ionici è
pari a 0,658 nm. Sali che hanno un valore minore di questa somma sono salati (il cloruro di sodio ha 0,556 nm);
valori superiori (come quello del cloruro di magnesio, 0,850 nm) conferiscono il sapore amaro.
Sale
114
Note
[1] P.T. Coultate, La chimica degli alimenti, Zanichelli, pag 209
Bibliografia
• I. Bertini, C. Luchinat, F. Mani, Chimica, CEA, ISBN 88-408-1285-7
Voci correlate
•
•
•
•
•
•
Legame chimico
Igroscopia
Deliquescenza
Efflorescenza
Acque salse
Salinità
Altri progetti
•
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Salt
Wikiquote contiene citazioni: http://it.wikiquote.org/wiki/Sale
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Sale
Ossido
Un ossido è un composto chimico binario
che si ottiene dall'ossidazione dell'ossigeno
su di un altro elemento. Nel XVII secolo
erano compresi nelle arie, nel XVIII secolo
erano conosciuti genericamente come calci,
mentre si è passati al termine attuale dopo
Lavoiser e la scoperta dell'ossigeno. Gli
ossidi sono estremamente diffusi sulla
superficie terrestre, e sono i costituenti base
di molti minerali: ad esempio, la magnetite è
un ossido misto di ferro, e la silice è un
ossido di silicio.
Ossidi di terre rare
Ossidi basici
Gli ossidi basici vengono formati dal legame tra un elemento e l'ossigeno quando reagendo con l'acqua danno luogo
a idrossidi, comportandosi come basi.
Tutti
i
metalli
alcalini
e
i
metalli
alcalino
terrosi
hanno
solo
un
numero
di
ossidazione,
Ossido
cioè formano esclusivamente un ossido
ciascuno ed esso è sempre basico: perciò
secondo la nomenclatura IUPAC, questi
ossidi vengono identificati come "ossido
di..." seguito dal nome dell'elemento. Ad
esempio Na2O è un ossido basico e secondo
la nomenclatura IUPAC sarà chiamato
semplicemente ossido di sodio.
Se invece l'elemento ha più numeri di
ossidazione, come nel caso dei metalli di
transizione e dei non metalli soprattutto dal
terzo periodo in poi, di norma esso si
comporta come basico se il suo numero di
ossidazione è compreso tra 0 e +3: per
Ossido di ferro, comunemente conosciuto con il nome di "ruggine"
esempio il cromo, il manganese possono
anche formare ossidi acidi pur essendo
metalli, mentre il carbonio e lo zolfo possono formare ossidi basici pur essendo non metalli. Si noti però che con
ciascun numero di ossidazione questi quattro elementi hanno un unico comportamento possibile: diverso è il caso
degli ossidi anfoteri.
Ossidi acidi
Gli ossidi acidi sono ossidi che reagendo con l'acqua formano un ossiacido, comportandosi quindi in soluzione come
un acido di Arrhenius.
In prima approssimazione si formano dalla ossidazione di un non metallo; in realtà ciò contraddice il comportamento
basico osservato in particolari ossidi di non metalli come il monossido di carbonio, gli ossidi basici dello zolfo, il
biossido di stagno, e non spiegherebbe il fatto che anche alcuni ossidi di metalli di transizione come l'ossido di
zirconio e gli ossidi di molibdeno hanno comportamento acido.
Si arriva perciò parallelamente a quanto stabilito per gli ossidi basici al criterio empirico che per gli ossidi acidi il
numero di ossidazione sia maggiore o uguale a +3, tenendo però presente che esistono ancora eccezioni come il
monossido di dicloro. Il comportamento acido è comunque per ogni elemento in generale tanto più marcato e puro
quanto più è alto il numero di ossidazione, come nel caso del cloro.
Ossidi anfoteri
Gli ossidi possono avere in realtà comportamento anfotero anche secondo Arrhenius, specie quando il numero di
ossidazione è nell'intorno di +3: è il caso in particolare dell'ossido di zinco[1], che reagisce in modo differente in base
al pH della soluzione,
• Soluzione acida: ZnO + 2HCl → ZnCl2 + H2O
• Soluzione basica: ZnO + 2NaOH + H2O → Na22+[Zn(OH)4]2come anche dell'ossido di piombo:
• Soluzione acida: PbO + 2HCl → PbCl2 + H2O
• Soluzione basica: PbO + Ca(OH)2 +H2O → Ca2+[Pb(OH)4]2e anche notevolmente dell'ossido di alluminio. Altri elementi che formano ossidi anfoteri sono silicio, titanio,
vanadio, ferro, cobalto, germanio, zirconio, argento, stagno, oro[2]. Per convenzione i composti che formano
reagendo con l'acqua si inseriscono negli idrossidi, poiché esistono maggiori analogie come la solubilità che è
115
Ossido
116
piuttosto bassa. Anche questi però, non bisogna dimenticare, hanno carattere anfotero: per esempio si osservi come
l'Idrossido di alluminio reagisca in
• Soluzione acida: Al(OH)3 + 3HCl → AlCl3 + 3H2O
• Soluzione basica: Al(OH)3 + NaOH → Na[Al(OH)4]
o anche l'idrossido di berillio:
• Soluzione acida: Be(OH)2 + 2HCl → BeCl2 + 2H2O
• Soluzione basica: Be(OH)2 + 2NaOH → Na2Be(OH)4
Nomenclatura attuale
La nomenclatura IUPAC è molto semplice e dipende unicamente da tre fattori: dalla quantità di atomi di ossigeno e
dell'elemento nella formula bruta, inserita sotto forma di prefissi derivanti dai numeri in greco, e dal numero di
ossidazione dell'elemento, che si scriverà tra parentesi in caratteri romani al termine. Il comportamento acido o
basico potendo dipendere in generale come si è visto nel paragrafo precedente dall'ambiente, non rientra nella
nomenclatura, anche se per il criterio precedentemente illustrato sul numero di ossidazione esso di fatto ci indirizza
verso la previsione del comportamento secondo Arrhenius in generale dell'ossido.
Ad esempio il rame, ha come numeri di ossidazione +1 e +2. Avremo di conseguenza per Cu2O il nome monossido
di dirame (I) e per CuO il nome monossido di rame (II), che prevederemmo a ragione avere carattere basico, mentre
per N2O3 il nome è triossido di diazoto (III), per N2O5 il nome è pentaossido di diazoto (V), ed entrambi
prevederemmo (a ragione) avere carattere acido secondo Arrhenius.
Nomenclatura classica
Nella nomenclatura classica ormai in disuso gli ossidi erano molto più macchinosamente e arbitrariamente distinti in
base al comportamento: quelli reputati sempre basici erano detti propriamente ossidi mentre quelli reputati
generalmente acidi erano detti anidridi, termine che invece viene oggi utilizzato in senso più specifico.[3]
• Gli ossidi erano nominati in funzione del numero di atomi di ossigeno della molecola e della valenza dell'atomo
del metallo ossidato. La nomenclatura non era rigidamente definita: nel caso di elementi a con diversi numeri di
ossidazione basici, che quindi possono dare diversi ossidi, si usava la desinenza -oso per il più basso, e quello -ico
per il più alto.[3]. Però era anche possibile utilizzare prefissi [3] come: sottossido con un atomo di ossigeno con
metallo monovalente, nessun suffisso per un atomo di ossigeno con metallo bivalente, sesquiossido con tre atomi
di ossigeno e metallo trivalente, biossido con due atomi di ossigeno e metallo tetravalente. Ad esempio:
•
•
•
•
FeO → ossido ferroso o ossido di ferro
Fe2O3 → ossido ferrico o sesquiossido di ferro
Cu2O → ossido rameoso o sottossido di rame
CuO → ossido rameico o ossido di rame
• Per le anidridi di elementi con un solo numero di ossidazione acido, il termine era seguito dal nome dell'elemento
e dalla desinenza -ica. Ad esempio l'ossido di boro B2O3 era chiamato anidride borica, e il biossido di carbonio,
in cui il carbonio ha ossidazione +4, era chiamato anidride carbonica mentre nel CO il carbonio avendo
ossidazione +2 basica si chiamava monossido di carbonio.[3]
Se i numeri di ossidazione acidi sono due, si usava il suffisso -osa per quello minore[3]. Come nel caso dello zolfo:
• SO → monossido di zolfo (+2)
• SO2 → anidride solforosa (+4)
• SO3 → anidride solforica (+6)
Se fossero stati tre numeri di ossidazione acidi, per il minore avremmo introdotto anche il prefisso ipo- oltre al
suffisso -osa; con quattro, avremmo introdotto una quarta forma con prefisso per- oltre al suffisso -ica.[3] È il caso
Ossido
117
del manganese e del cloro:
•
•
•
•
Cl2O → anidride ipoclorosa (+1)
Cl2O3 → anidride clorosa (+3)
Cl2O5 → anidride clorica (+5)
Cl2O7 → anidride perclorica (+7)
Note
[1] C.E. Housecroft and A.G. Sharpe, "Inorganic Chemistry" (2nd edn, Pearson 2005), p.173-4
[2] CHEMIX School & Lab - Software for Chemistry Learning, by Arne Standnes (http:/ / home. c2i. net/ astandne/ ) (program download
required)
[3] Anna Guglielmi, Chimica e mineralogia per le scuole medie superiori, Signorelli Editore, Milano, 1949
Voci correlate
•
•
•
•
Anidride
Ossidazione
Ossigeno
Idrossido
• Base
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Oxides
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/ossido
Chimica fisica
La chimica fisica è la branca della chimica che più si avvicina alla fisica, adottandone il rigore metodologico sia a
livello sperimentale che teorico e adoperando un significativo formalismo matematico, nel tentativo di descrivere
nella maniera più accurata possibile i fenomeni fondamentali che stanno alla base dei sistemi chimici.
Storia
Il termine "chimica fisica" è stato probabilmente utilizzato per la prima volta da Michail Lomonosov nel 1752,
quando davanti agli studenti dell'Università Statale di San Pietroburgo presentò il corso intitolato <<Corso di vera
chimica fisica>>.[1]
Fondatore della moderna chimica fisica viene considerato il chimico statunitente Willard Gibbs, che con la sua
pubblicazione "On the Equilibrium of Heterogeneous Substances" (Sull'equilibrio delle sostanze eterogenee) del
1876 introdusse alcuni concetti quali quelli di energia libera, potenziale chimico e regola delle fasi che risulteranno
tra i principali fondamenti di questa disciplina. Il successivo sviluppo fu dato dal contributo di chimici quali Svante
Arrhenius, Jacobus Henricus van 't Hoff, Wilhelm Ostwald e Walther Nernst verso la fine degli anni 1800.
Gli sviluppi del ventesimo secolo comprendono l'applicazione della meccanica statistica ai sistemi chimici e lavori
riguardanti i colloidi e la chimica delle superfici, dove Irving Langmuir diede un notevole contributo. Altri
importanti sviluppi riguardarono la nascita della chimica quantistica, evolutasi dalla meccanica quantistica negli anni
1930, dove Linus Pauling fu uno dei maggiori contributori. Gli sviluppi teorici sono andati di pari passo con
l'evoluzione dei metodi sperimentali e l'utilizzo delle varie forme di spettroscopia è tra uno dei più importanti
progressi del ventesimo secolo.
Chimica fisica
Discipline
La chimica fisica applica la termodinamica allo studio dei gas, delle soluzioni e delle reazioni chimiche,
quantificando gli aspetti energetici di quest'ultime e arrivandone a prevedere l'eventuale sponteneità o le condizioni
di spontaneità teoriche. La termodinamica consente anche di trattare l'equilibrio chimico. L'uso della meccanica
quantistica non solo permette di interpretare gli spettri atomici e molecolari, ma facendo uso del suo rigoroso
formalismo matematico permette anche di descrivere il legame chimico e di predire importanti proprietà delle
molecole quali la loro stabilità e reattività. La spettroscopia permette di determinare sperimentalmente la struttura e
composizione delle molecole, mentre la cinetica chimica studia la velocità delle reazioni e l'insieme di processi
elementari che intercorrono durante una reazione chimica quando a partire dai reagenti si ottengono i prodotti finali.
L'elettrochimica è un'altra importante area della chimica fisica che si occupa delle implicazioni dei fenomeni elettrici
in ambito chimico.
Riassumendo, le principali aree di interesse della chimica fisica si possono così elencare:
•
•
•
•
Meccanica quantistica e chimica quantistica
Chimica computazionale
Termodinamica e termochimica
Meccanica statistica
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Cinetica chimica
Dinamica molecolare
Elettrochimica
Spettroscopia molecolare
Fenomeni di trasporto
Chimica dello stato solido e delle superfici
Chimica delle interfasi
Chimica dei colloidi
Fotochimica
Femtochimica
Chimica supramolecolare
Chimica nucleare
Sonochimica
Astrochimica
Strutturistica chimica
Transizioni di fase
Magnetochimica
Note
[1] Galina Evgenevna Pavlova, Aleksandr Sergeevich Fedorov, Mikhail Vasilevich Lomonosov: his life and work, Mir Publishers, 1984
Bibliografia
• P. Atkins, J. De Paula, "Physical Chemistry", Oxford University Press, 2006 (ottava ed.), ISBN
978-0-19-870072-2
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Physical
chemistry
118
Biochimica
Biochimica
La biochimica è lo studio della chimica della vita, un ponte fra la biologia e la chimica che studia le reazioni
chimiche complesse che danno origine alla vita: oggetto di studio sono la struttura e le trasformazioni dei
componenti delle cellule, come proteine, carboidrati, lipidi, acidi nucleici e altre biomolecole.
Sebbene vi sia un grande numero di diverse biomolecole, sono tutte essenzialmente composte dagli stessi costituenti
di base (genericamente chiamati monomeri), posizionate in ordini diversi. Ogni classe di biomolecole ha un set di
differenti subunità. Recentemente, la biochimica si è concentrata specificatamente sulla catalisi di reazioni da parte
degli enzimi e sulle proprietà delle proteine.
La biochimica del metabolismo cellulare e del sistema endocrino è già stata ampiamente descritta. Altre aree della
biochimica includono lo studio del codice genetico (DNA, RNA), la sintesi proteica, il meccanismo di trasporto della
membrana cellulare e la trasduzione del segnale.
Sviluppo della biochimica
Originariamente si credeva che la vita non fosse soggetta alle normali leggi della chimica, contrariamente agli
oggetti inanimati. Si pensava che soltanto gli esseri viventi potessero produrre le molecole della vita (da altre
biomolecole preesistenti). Ma già verso la fine del Settecento si stabilirono i principi di partenza della biochimica,
grazie alle ricerche di Lavoisier e di Spallanzani sulla respirazione degli organismi viventi.[1]
Poi, nel 1828, Friedrick Wöhler pubblicò una ricerca sulla sintesi dell'urea, provando che i composti organici
possono essere creati artificialmente, seguito pochi anni dopo dalle analisi e sintesi di Justus von Liebig che
consentirono le prime applicazioni pratiche della nuova disciplina, tra le quali la fertilizzazione con concimi
inorganici.
L'alba della biochimica può essere considerata la scoperta del primo enzima, la diastasi, nel 1833, da parte di
Anselme Payen. Nonostante il termine biochimica" sembri essere stato usato per la prima volta nel 1881 (la parola
chimica biologica invece risale al 1826 ed è attribuibile a Huenefeld), è generalmente accettato che la parola sia
stata coniata formalmente nel 1903 da Carl Neuber, un chimico tedesco. Da allora la biochimica ha fatto grandi
passi in avanti, specialmente a partire dalla metà del XX secolo, con lo sviluppo di tecniche come la cromatografia,
la diffrattometria a raggi X, la spettroscopia NMR e simulazioni delle dinamiche molecolari. Queste tecniche
permisero la scoperta e l'analisi dettagliata di numerose molecole e delle sequenze metaboliche delle cellule, come
la glicolisi ed il Ciclo di Krebs (o ciclo dell'acido citrico).
Al giorno d'oggi le scoperte della biochimica vengono applicate in molte aree, dalla genetica alla biologia
molecolare, dall'agricoltura alla medicina.
119
Biochimica
I carboidrati
La funzione dei carboidrati è duplice: strutturale e di riserva
energetica. Gli zuccheri sono carboidrati, anche se ci sono carboidrati
che non sono zuccheri. Sulla Terra esistono molti più carboidrati di
qualsiasi altro tipo di biomolecola. Il più semplice tipo di carboidrato è
un monosaccaride, che tra le altre proprietà contiene carbonio,
idrogeno e ossigeno nella proporzione 1:2:1 (formula generale
Lo zucchero più usato in alimentazione è senza
CnH2nOn, dove n vale almeno 3). Il glucosio, uno dei più importanti
dubbio il disaccaride saccarosio
carboidrati, è un esempio di monosaccaride. Così come il fruttosio, lo
zucchero che dà alla frutta il suo caratteristico sapore dolce. Due
monosaccaridi possono essere uniti assieme usando la sintesi per disidratazione, nella quale un atomo di idrogeno
viene rimosso dalla fine di una molecola ed un gruppo ossidrile (-OH) viene rimosso dall'altra. Il gruppo H-OH o
H2O viene poi rilasciato come una molecola di acqua, da cui il nome disidratazione. La nuova molecola, consistente
in due monosaccaridi, viene chiamata disaccaride ed è tenuta insieme da un legame glicosidico oppure da altri tipi di
legame. Può anche verificarsi la reazione opposta, chiamata idrolisi, usando una molecola di acqua per scindere il
disaccaride mediante rottura del legame glicosidico. Il disaccaride più conosciuto è il saccarosio (il comune zucchero
da tavola o zucchero di canna), che è composto da una molecola di glucosio ed una di fruttosio legate assieme. Un
altro importante disaccaride è il lattosio, che consiste in una molecola di glucosio ed una di galattosio. In età
avanzata la produzione di lattasi (l'enzima che idrolizza il lattosio in glucosio e galattosio) tipicamente decresce,
determinando una deficienza di lattasi, che conduce alla cosiddetta "intolleranza al lattosio".
Quando più di due (solitamente da tre a dieci) monosaccaridi sono legati assieme, viene a formarsi un oligosaccaride
(dal greco, la radice oligo- significa "poco"). Queste molecole di solito vengono usate come marcatori e segnali, ma
possono avere anche altri tipi di funzioni.
Più monosaccaridi legati assieme formato un polisaccaride. I monosaccaridi possono essere legati in una lunga
catena lineare oppure in una catena ramificata. I tre più comuni polisaccaridi sono la cellulosa, l'amido ed il
glicogeno, formati dalla ripetizione di monomeri di glucosio. La cellulosa è prodotta dai vegetali ed è un importante
componente strutturale delle loro cellule. Gli esseri umani non possono produrre cellulosa, né digerirla poiché sono
privi dell'enzima β-glicosidasi adibito proprio alla scissone di questa. L'amido è un polisaccaride di deposito del
glucosio. Il glicogeno, invece, è un carboidrato animale; viene usato dall'uomo come deposito di energia.
Il glucosio è la più importante fonte di energia per la maggior parte delle forme di vita ed un gran numero di vie
cataboliche convergono su di esso. Ad esempio, i polisaccaridi vengono spezzati nei loro monomeri (la glicogeno
fosforilasi rimuove i residui di glucosio dal glicogeno). I disaccaridi come il lattosio o il saccarosio vengono scissi
nei due monosaccaridi che li compongono. Il glucosio è metabolizzato da un importantissimo processo, ampiamente
conservato nelle specie viventi, composto di dieci tappe e chiamato glicolisi, il cui risultato finale è la rottura di una
molecola di glucosio in due molecole di piruvato; questo produce anche due molecole di adenosintrifosfato (ATP),
l'energia utilizzata dalle cellule, assieme a due equivalenti ridotti convertendo il NAD in NADH. Questo processo
non richiede ossigeno; quando l'ossigeno non è disponibile (o le cellule non possono utilizzarlo), il NAD viene
ripristinato convertendo il piruvato in lattato (accade ad esempio nel'uomo), oppure in etanolo (nel lievito). Altri
monosaccaridi come il galattosio ed il fruttosio possono essere convertiti nei composti intermedi della glicolisi. Nelle
cellule aerobiche con sufficiente ossigeno, come la maggior parte delle cellule umane, il piruvato può essere
metabolizzato ulteriormente. Esso viene convertito irreversibilmente in acetil-CoA, dando come prodotto di scarto
un atomo di carbonio sotto forma di biossido di carbonio, generando un ulteriore equivalente riducente di NADH. Le
due molecole di acetil-CoA (da una molecola di glucosio) entrano poi nel ciclo di Krebs, producendo altre due
molecole di ATP, sei di NADH e due molecole legate di FADH2, e rilasciando il carbonio restante come biossido di
carbonio. Le molecole ridotte di NADH e FADH2 entrano poi nel sistema di trasporto elettronico, nel quale gli
elettroni vengono trasferiti ad una molecola di ossigeno, producendo acqua, e gli originari NAD+ e FAD vengono
120
Biochimica
rigenerati. Per questo motivo gli esseri umani inspirano ossigeno ed espirano biossido di carbonio. L'energia di
trasferimento degli elettroni dagli stati di alta energia di NADH e FADH2 viene utilizzata per generare altre 28
molecole di ATP (soltanto due erano state prodotte nella glicolisi), per un totale di 32 molecole di ATP. È chiaro che
usare ossigeno per ossidare completamente il glucosio, fornisce all'organismo una grande energia, ed è per questo
che le forme di vita complesse comparvero sulla Terra soltanto quando nell'atmosfera si accumularono forti quantità
di ossigeno.
Nei vertebrati, durante la contrazione vigorosa (nel sollevamento pesi o nello sprint, ad esempio) i muscoli
scheletrici non ricevono abbastanza ossigeno per sviluppare l'energia richiesta, e così viene impiegato il metabolismo
anaerobico, convertendo il glucosio in lattato (acido lattico). Il fegato può rigenerare il glucosio mediante la
gluconeogenesi. Questo processo non è esattamente l'opposto della glicolisi e richiede una quantità tripla di energia
guadagnata con la glicolisi (vengono utilizzate sei molecole di ATP, mentre nella glicolisi ne vengono prodotte due).
Il glicogeno (così come l'amido nelle piante) può essere considerato una riserva di glucosio per le necessità
dell'organismo. Il glicogeno conservato nel fegato, infatti, viene utilizzato per riportare alla normalità i livelli ematici
di glucosio durante il digiuno. Il glicogeno presente nei muscoli, invece, viene usato esclusivamente per i muscoli
stessi, nel corso di impegnativi sforzi contrattili.
Le proteine
Allo stesso modo dei carboidrati, alcune proteine hanno una funzione
strutturale. Ad esempio i movimenti delle proteine actina e miosina sono
responsabili della contrazione dei muscoli scheletrici. Una proprietà che
molte proteine possiedono è quella di legarsi a particolari molecole o classi di
molecole; possono essere estremamente selettive in ciò che legano. Gli
anticorpi sono un esempio di proteine che si attaccano ad un tipo specifico di
molecole. Infatti l'ELISA (acronimo dell'espressione inglese Enzyme-Linked
ImmunoSorbent Assay), che fa uso di anticorpi, è attualmente uno dei più
sensibili test che la medicina moderna usa per identificare varie biomolecole.
Rappresentazione schematica
Probabilmente le più importanti proteine sono gli enzimi. Queste sorprendenti
dell'emoglobina. Le parti a forma di
molecole riconoscono specifiche molecole reagenti chiamate substrati e sono
nastro rappresentano la proteina globina;
in grado di catalizzare le reazioni in cui essi sono coinvolti. Abbassando
le quattro parti in verde sono i gruppi
l'energia di attivazione, un enzima riesce a velocizzare una reazione di un
eme.
tasso di circa 1011 e oltre: una reazione che in condizioni normali
impiegherebbe 3000 anni per completarsi spontaneamente, in presenza di enzimi può impiegare meno di un secondo.
L'enzima non viene utilizzato nel processo ed è libero di catalizzare la stessa reazione con un nuovo set di substrati.
Usando vari agenti modificatori, l'attività dell'enzima può essere regolata, permettendo il controllo della biochimica
cellulare nel suo insieme.
121
Biochimica
Composizione
Essenzialmente le proteine sono catene di amminoacidi. Un amminoacido è costituito da un atomo di carbonio legato
a quattro gruppi:
• Un gruppo amminico, -NH2;
• Un gruppo carbossilico, -COOH;
(Questi gruppi in particolari condizioni fisiologiche esistono come -NH3+ e -COO−)
• Un atomo di idrogeno disposto sopra al carbonio (C);
• Un gruppo -R (radicale), differente per ogni amminoacido. In funzione delle proprietà chimiche di tale gruppo, un
amminoacido viene classificato come acido, basico, idrofilo (o polare) e idrofobo (o apolare).
Esistono venti amminoacidi standard. Alcuni di questi hanno funzioni così come sono oppure in una forma
modificata. Il glutammato, ad esempio, è un importante neurotrasmettitore.
Gli amminoacidi possono essere legati
assieme tramite un legame peptidico.
In questa sintesi per disidratazione
viene rimossa una molecola di acqua
ed il legame peptidico lega l'azoto del
gruppo amminico di un amminoacido
Amminoacidi generici (1) in una forma neutrale, (2) nella forma in cui esistono
con il carbonio del gruppo carbossilico
fisiologicamente, e (3) legati assieme a formare un dipeptide.
dell'amminoacido
contiguo.
La
molecola risultante viene chiamata
dipeptide, mentre le corte catene di amminoacidi (di solito meno di 30) prendono il nome di peptidi o polipeptidi e le
catene più lunghe sono le proteine. Ad esempio l'albumina, presente nel siero, contiene 585 residui amminoacidici.
Struttura
La struttura delle proteine è tradizionalmente descritta come una gerarchia a quattro livelli.
• La struttura primaria di una proteina semplice consiste nella sua sequenza lineare di amminoacidi (ad esempio
"alanina-glicina-triptofano-serina-glutammato-asparagina-glicina-lisina-...").
• La struttura secondaria consiste nella morfologia locale. Alcune combinazioni di amminoacidi tendono a
ripiegarsi in una spirale chiamata α-elica (alcune sono facilmente osservabili nell'immagine schematica più in
alto).
• La struttura terziaria è l'intera struttura tridimensionale della proteina, determinata dalla sequenza degli
amminoacidi. Infatti una singola sostituzione può cambiare l'intera struttura. I foglietti β dell'emoglobina
contengono 146 residui amminacidici; la sostituzione del glutammato in posizione 6 con una valina ne cambia il
comportamento, provocando l'anemia falciforme.
• Infine la struttura quaternaria riguarda la struttura delle proteine con più subunità peptidiche, come l'emoglobina
che ne possiede 4. Non tutte le proteine hanno più di una subunità.
Metabolismo delle proteine
Le proteine ingerite vengono di solito scisse in singoli amminoacidi o in dipeptidi nell'intestino tenue e in seguito
assorbiti. Possono poi essere ancora legati assieme a formare nuove proteine. I prodotti intermedi della glicolisi, del
ciclo di Krebs e della via dei pentoso fosfati possono essere utilizzati per produrre tutti i 20 amminoacidi, e molti
batteri e piante posseggono tutti gli enzimi necessari a sintetizzarli. L'uomo ed altri mammiferi invece non possono
sintetizzarne la metà: isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptofano e valina, ovvero gli
amminoacidi essenziali che devono essere assunti con la dieta. I mammiferi posseggono invece gli enzimi necessari
alla sintesi di alanina, asparagina, aspartato, cisteina, glutammato (o acido glutammico), glutammina, glicina,
122
Biochimica
prolina, serina e tirosina. Possono produrre anche arginina ed istidina, ma non in quantità sufficienti per i giovani
animali in crescita, quindi anche questi ultimi due amminoacidi vengono di solito considerati essenziali.
Se da un amminoacido viene rimosso un gruppo amminico, questo lascia dietro di sé uno scheletro di carbonio
chiamato α-chetoacido. Gli enzimi transaminasi possono facilmente trasferire un gruppo amminico da un
amminoacido (trasformando quest'ultimo in α-chetoacido) ad un altro α-chetoacido, trasformandolo in amminoacido.
Questo processo è molto importante nella biosintesi degli amminoacidi, come per molte altre trasformazioni
biochimiche.
Gli amminoacidi possono essere anche utilizzati per produrre energia. Tale processo porta l'amminoacido ad essere
scisso in una molecola di ammoniaca ed in uno scheletro carbonioso. L'ammoniaca (NH3, esistente in forma di ione
ammonio NH4+ nel sangue), ad alte concentrazioni è tossica. Per questo in tutti gli animali si è sviluppato il metodo
più adatto alla sua escrezione. Gli organismi unicellulari rilasciano semplicemente l'ammoniaca nell'ambiente.
Similmente, i pesci della classe Osteichthyes, o pesci ossei, possono rilasciarla nell'acqua dove è facilmente diluita.
In generale i mammiferi convertono l'ammoniaca in urea tramite il ciclo dell'urea, prima della sua espulsione.
I lipidi
Il termine lipide comprende un gran numero di molecole ed in senso esteso comprende tutti i composti di origine
biologica insolubili in acqua o apolari, incluse le cere, gli acidi grassi, i fosfolipidi derivati degli acidi grassi, gli
sfingolipidi, i glicolipidi ed i terpenoidi, come retinoidi e steroidi. Alcuni lipidi sono molecole lineari alifatiche,
mentre altri hanno una struttura ad anello. Possono essere flessibili oppure rigidi.
La maggior parte dei lipidi ha una zona a carattere polare, pur essendo in gran parte apolare. Generalmente il corpo è
apolare o idrofobico, cioè non interagisce bene con solventi polari come l'acqua. La testa è invece polare, o idrofilica
e tende ad associarsi ai solventi polari. Nel caso del colesterolo la parte polare è il gruppo -OH (idrossile o alcol); nel
caso dei fosfolipidi i gruppi polari sono più ingombranti e più polari.
Gli acidi nucleici
Un acido nucleico è una molecola complessa ad alto peso molecolare, composta da catene di nucleotidi, che
trasmettono le informazioni genetiche. I più noti acidi nucleici sono il DNA e l'RNA, presenti in tutte le cellule
viventi e nei virus.
Gli acidi nucleici, chiamati in questo modo per via della loro presenza nel nucleo delle cellule, sono anche detti
biopolimeri. I monomeri di cui sono formati sono chiamati nucleotidi, ognuno dei quali ha tre componenti:
• Una base azotata (purinica o pirimidinica);
• Uno zucchero pentoso;
• Un gruppo fosfato
Gli acidi nucleici differiscono tra loro per lo zucchero contenuto nelle loro catene. Il DNA (o acido
desossiribonucleico), ad esempio, contiene desossiribosio. Anche le basi azotate differiscono nei due tipi di acidi
nucleici (DNA ed RNA): adenina, citosina e guanina sono presenti in entrambi, mentre la timina presente nel DNA è
sostituita nell'RNA dall'uracile.
123
Biochimica
124
Relazioni con altre scienze biologiche a scala molecolare
I ricercatori in biochimica utilizzano tecniche di ricerca
originali, ma combinano sempre più queste nuove
tecniche con altre prese in prestito dalla genetica, dalla
biologia molecolare e dalla biofisica. Non è mai esistita
una precisa linea di demarcazione tra queste discipline
in termini di tecniche e contenuti, ma i membri di
ognuna di esse sono stati in passato molto "territoriali".
Al giorno d'oggi i termini biologia molecolare e
biochimica
sono
praticamente
intercambiabili.
L'immagine seguente è uno schema che illustra le
possibili relazioni tra queste discipline.
• La biochimica è lo studio delle sostanze chimiche e
dei processi vitali degli organismi viventi.
Relazione tra biologia molecolare, genetica e biochimica in
• La genetica è lo studio dell'effetto delle differenze
un'accezione classica dei relativi campi di studio
genetiche sugli organismi, che spesso possono
essere causate dall'assenza di un normale componente (ad esempio un gene); lo studio degli organismi mutanti,
mancanti di uno o più componenti funzionali, con riferimento al cosiddetto "wild-type" o al normale fenotipo.
• La biologia molecolare è lo studio dei sostegni molecolari del processo di replicazione, trascrizione e traduzione
del materiale genetico. Il dogma centrale della biologia molecolare, secondo cui il materiale genetico è trascritto
nell'RNA e poi tradotto in proteine, oltre ad essere un modello molto semplificato, può ancora essere considerato
un buon punto di partenza per la comprensione in questo campo di indagine. Questo modello, comunque, è in via
di revisione, alla luce di nuovi ruoli riguardanti l'RNA.
• La biologia chimica punta a sviluppare nuovi strumenti basati su piccole molecole che permettono di raccogliere
dettagliate informazioni sui sistemi biologici provocando loro minime perturbazioni. Infine la biologia chimica
impiega sistemi biologici per creare ibridi artificiali tre biomolecole ed elementi sintetici (ad esempio capsidi
virali svuotati che possono diventare vettori per terapie geniche o altri medicinali).
Metodologie biochimiche
Nell'ultimo ventennio ha assunto sempre maggiore importanza l'analisi chimico-fisica delle biomolecole, con
particolare accento alla loro struttura al fine di correlare quest'ultima alla funzione delle molecole stesse. In
particolare, tre tecniche sono utilizzate con questi fini:
• Spettroscopia di risonanza magnetica nucleare
• Diffrattometria a raggi X
• Spettrometria di massa
È sempre crescente, inoltre, l'interesse verso studi computazionali/statistici di biomolecole attraverso tre importanti
metodologie computazionali:
• Dinamica molecolare
• Meccanica quantistica
• Bioinformatica
Biochimica
125
Note
[1] Universo, De Agostini, Novara, Vol. II, pag.304-305
Bibliografia
• Graeme K. Hunter. Vital Forces. The discovery of the molecular basis of life. San Diego, Academic press, 2000.
ISBN 0-12-361810-X.
• David L. Nelson e Michael M. Cox. I principi di biochimica di Lehninger. IV edizione. Bologna, Zanichelli,
2006. ISBN 978-88-08-19774-0.
• Lubert Stryer. Biochimica. Bologna, Zanichelli, 1996. ISBN 88-08-09806-0.
• Donald Voet e Judith G. Voet. Biochimica. Bologna, Zanichelli, 1995. ISBN 88-08-10538-5.
• Keith Wilson e John Walker (a cura di). Metodologia biochimica. Milano, Raffaello Cortina Editore, 2001. ISBN
88-7078-687-0.
Voci correlate
Chimica
Biologia
Applicazioni
Altro
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Chimica
Chimica organica
Chimica bioinorganica
Chimica alimentare
Biochimica clinica
Biochimica applicata
Citochimica
Istochimica
Biologia
Biologia molecolare
Biologia computazionale
Bioinformatica
Biologia cellulare
Farmacologia
Biotecnologia
Ingegneria biochimica
Proteomica
Genetica
Ingegneria genetica
Genetica molecolare
Psichiatria biologica
Biofisica
Medicina
Bioetica
Biologia e chimica organica (cronologia)
Bios
Zoé
Altri progetti
•
Wikibooks contiene testi o manuali: http://it.wikibooks.org/wiki/Biochimica
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/
Category:Biochemistry
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Biochimica
Collegamenti esterni
• Biblioteca virtuale di Biochimica e Biologia della cellula (http://www.biochemweb.org/)
• Biochemistry, 5th ed. (http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowTOC&rid=stryer.
TOC&depth=2) Testi completi di Berg, Tymoczko e Stryer, per gentile concessione del NCBI.
• Società biotecnologica del Costa Rica (http://www.biotecnologia.co.cr/)
Numero atomico
126
Numero atomico
Il numero atomico (indicato solitamente con Z , dal tedesco Zahl, e detto anche numero protonico) corrisponde al
numero di protoni contenuti in un nucleo atomico. In un atomo neutro il numero atomico è pari anche al numero di
elettroni; in caso contrario l'atomo è detto ione. Si usa scrivere questo numero come pedice sinistro del simbolo
dell'elemento chimico in questione: per esempio 6C, poiché il carbonio ha sei protoni.
A ogni numero atomico corrisponde un diverso elemento chimico, il quale viene collocato nella tavola periodica
proprio in funzione del relativo valore di Z. La legge di Moseley permette di ricavare il numero atomico di un dato
elemento misurando la frequenza della riga caratteristica corrispondente all'emissione di raggi X.
Atomi aventi stesso numero atomico ma diverso numero di neutroni sono detti isotopi.
Voci correlate
•
•
•
•
Tavola periodica degli elementi
Elementi per numero atomico
Peso atomico
Numero di massa
• Numero neutronico
Altri progetti
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/numero atomico
Peso atomico
Il peso atomico (da non confondere col numero di massa
) o massa atomica
è la massa di un atomo di un
dato elemento. In questo caso si parla spesso impropriamente di peso atomico assoluto e viene espresso in grammi o
chilogrammi: l'ordine di grandezza dei valori è tra i 10-25 kg e i 10-27 kg. Per ovviare alla scomodità di avere nei
calcoli numeri così piccoli, si è convenuto di esprimere la massa atomica in rapporto al peso atomico assoluto di 1/12
dell'atomo 12C, il cui valore è adottato nel SI quale unità di massa atomica (u o uma): sperimentalmente si è ricavato
che equivale a 1,660 538 921(73)x 10-27 kg, secondo i dati CODATA del 2010. [1] Questa notazione della massa è
nota come peso atomico relativo (o massa atomica relativa, spesso abbreviata in massa relativa) e si può ottenere
dalla formula:
Da ciò si evince che la massa atomica relativa è un numero adimensionale e non è espresso in ragione di una unità di
misura di massa. Se si esprime in u una data massa atomica relativa, tale valore corrisponde a quello della massa
assoluta (espressa in grammi) di una mole dell'elemento. La massa relativa di un dato elemento chimico è una media
ponderata delle masse relative dei suoi isotopi: in particolare è la sommatoria del prodotto tra la massa relativa di
ciascun isotopo e la relativa abbondanza isotopica.
In prima approssimazione, il peso atomico è legato al numero totale di nucleoni presenti nel nucleo considerato. Il
peso reale è leggermente inferiore alla somma dei pesi dei differenti componenti perché protoni e neutroni hanno
massa diversa (anche se solo del 2 per mille) e perché parte della massa delle particelle costituenti il nucleo viene
ceduta sotto forma di energia di legame nella fase di nucleosintesi, riducendo il peso totale (difetto di massa). Il peso
degli elettroni modifica solo leggermente il totale, perché la massa di un elettrone è pari a 1/1836 quella di un
protone, se considerati entrambi a riposo. Si noti che il peso atomico non ha relazione alcuna con la nozione di peso
Peso atomico
degli oggetti ordinari, che è una misura di forza: è invece una misura del peso relativo tra atomi diversi, tale
denominazione è di derivazione storica ed è tuttora utilizzata, benché scorretta.
La massa atomica assoluta (espressa in grammi) è pari alla massa atomica relativa divisa per il numero di Avogadro
(6,022 x 1023).
Relazioni con il concetto di mole
Una considerazione a latere correla il valore ottenuto con il concetto di mole, la settima grandezza fondamentale
del SI: in 12 g [esattamente] di carbonio-12 (che ha massa assoluta pari a 1,992 65 x 10-26), si hanno 6,022 x 1023
atomi, che è il numero di Avogadro. Siccome 12 u è la massa atomica del carbonio-12, si deduce che il peso atomico
relativo di un elemento o di un'altra specie chimica è numericamente (ma non dimensionalmente) uguale alla massa
molare[2], che si esprime quindi g/mol. Nel SI le moli di entità (molecole, ioni, radicali, zwitterioni, elettroni, fotoni,
...) si indicano con e la massa molare (la massa di una mole di entità) con
.
La seguente formula correla il peso molecolare alle moli di una entità di data massa:
Note
[1] Fundamental Physical Constants (http:/ / physics. nist. gov/ constants) in The NIST Reference on Constants, Units, and Uncertainty. NIST,
2010
[2] nel caso di un composto, la massa molare è pari al peso molecolare.
Voci correlate
•
•
•
•
•
•
Congresso di Karlsruhe
Numero atomico
Numero di massa
Numero di Avogadro
Massa molecolare
Mole
• Unità di massa atomica
• Legge di Dulong e Petit
Altri progetti
•
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Peso atomico
127
Acido
128
Acido
In chimica, le definizioni di acido e base hanno subìto diverse modifiche nel tempo, partendo da un approccio
empirico e sperimentale fino alle più recenti definizioni, sempre più generali, legate al modello molecolare ad
orbitali,divisibili in acidi forti e acidi deboli.
Nella quotidianità, il termine "acido" identifica sostanze generalmente irritanti e corrosive, capaci di intaccare i
metalli e il marmo (sviluppando rispettivamente idrogeno e anidride carbonica) e di far virare al rosso una cartina al
tornasole.
Esempi di sostanze acide sono l'aceto, l'acido muriatico e il succo di limone.
Un indice della forza di un acido, funzione della sua natura e della sua concentrazione, è il pH.
Nell'ambito della chimica inorganica di base, per rimarcare la differenza tra le due tipologie di acidi inorganici
(ossiacidi e idracidi) si fa spesso ricorso a schemi semplificati del tipo:[1]
anidride + acqua → ossiacido
non-metallo + idrogeno → idracido
Ovvero un ossiacido si forma facendo reagire l'anidride corrispondente con acqua (ad esempio l'anidride solforica
combinandosi con acqua forma acido solforico), mentre dalla reazione chimica tra un non-metallo e idrogeno si
forma l'idracido corrispondente (ad esempio l'acido fluoridrico si forma dalla combinazione del fluoro con
l'idrogeno).
Definizioni di acido
Di seguito vengono elencate le definizioni di
"acido" più diffuse, in ordine cronologico.
Definizione di acido secondo la
teoria di Arrhenius
Secondo la teoria di Arrhenius, un acido è
una sostanza che dissociandosi in acqua
libera ioni H+.[2] Una base è invece una
sostanza che dissociandosi in acqua libera
ioni OH-.[2]
Rientrano in questa definizione tutti i
composti che identifichiamo come acidi
Gocce di acido solforico concentrato al 98% carbonizzano la carta.
nell'uso comune, sia per la loro azione
irritante sui tessuti viventi e corrosiva sui
metalli, sia per la loro capacità di far virare opportunamente sostanze indicatrici.
Sono acidi secondo Arrhenius, per esempio, acidi inorganici forti come l'acido solforico e l'acido cloridrico ed acidi
deboli come l'acido acetico e l'acido citrico.
La "forza" di un acido, e con essa anche i suoi effetti corrosivi e irritanti, è misurata per il tramite della costante di
dissociazione acida.
Se la dissociazione completa di una molecola di acido fornisce uno ione idrogeno, l'acido in questione è detto
monoprotico (o monobasico), mentre se la sua dissociazione fornisce più ioni idrogeno si dirà poliprotico (o
polibasico).[2]
Acido
129
Definizione di acido secondo la teoria di Brønsted-Lowry
Secondo la teoria di Brønsted-Lowry, un acido è una sostanza capace di cedere ioni H+ ad un'altra specie chimica
detta base.[3][4]
La teoria di Brønsted-Lowry estende la definizione di acido a quelle sostanze di cui non è possibile o non è pratico
valutare il comportamento in acqua, come de facto succede nella definizione data da Arrhenius. Introduce anche il
concetto di complementarità tra acido e base, dato che l'acido non è tale se non in presenza di una controparte cui
cedere il proprio ione H+.
Secondo Brønsted e Lowry, quindi, anche composti che non presentano un carattere evidentemente acido nella
quotidianità, come per esempio gli alcoli, possono avere un comportamento acido quando sono in presenza di una
base sufficientemente forte. Un esempio è la reazione tra metanolo e idruro di sodio, in cui il metanolo si comporta
da acido, secondo la definizione di Brønsted e Lowry, cedendo allo ione idruro (la base) uno ione H+
CH3OH + NaH → CH3O-Na+ + H2
Secondo questa teoria non esistono quindi acidi e basi a sé stanti, ma solo coppie di acido e base coniugati. Una
coppia acido/base coniugata è una coppia di specie chimiche che differiscono soltanto per uno ione H+. Quando un
acido cede uno ione H+ si trasforma nella sua base coniugata; quando una base acquista uno ione H+ si trasforma nel
suo acido coniugato.
Qualunque reazione che comporta il trasferimento di uno ione H+ da un acido a una base è una reazione acido-base
secondo Brønsted e Lowry. Un acido può, in determinate circostanze, comportarsi da base e viceversa.
Definizione di acido secondo la teoria di Lewis
Secondo la cosiddetta teoria di Lewis, un acido è una sostanza capace di accettare un doppietto elettronico da
un'altra specie chimica capace di donarli (detta base).[5][6]
Simile alla teoria di Brønsted-Lowry, sostituisce al trasferimento dello ione H+ il trasferimento in senso inverso di un
doppietto elettronico. Secondo Lewis sono quindi acidi anche composti come il cloruro di alluminio ed il borano,
che presentano nella loro struttura un orbitale vuoto capace di alloggiare un doppietto elettronico proveniente da una
molecola donatrice, la base, e legarsi quindi ad essa tramite un legame dativo. Nell'esempio qui riportato,
l'ammoniaca è la base ed il trifluoruro di boro è l'acido, secondo Lewis
H3N: + BF3 → H3N→BF3
Gli acidi di Lewis si comportano da reagenti elettrofili, mentre le basi di Lewis si comportano da reagenti nucleofili.
La differenza tra le definizioni di "acido di Lewis" e "elettrofilo" sta nel fatto che il carattere di un acido di Lewis è
legato alla termodinamica della reazione, infatti un composto si comporta tanto più da acido di Lewis quanto più
tende ad attirare a sé i doppietti elettronici (in condizioni di equilibrio), mentre il carattere elettrofilo è legato alla
cinetica della reazione, infatti un composto si comporta tanto più da elettrofilo quanto più velocemente attira a sé i
doppietti elettronici.
Acido
130
Classificazione degli acidi
Di seguito sono indicate alcune classi di acidi di Arrhenius descritte nell'ambito della chimica inorganica e
organica.[7]
Acidi inorganici
Tra gli acidi inorganici si annoverano:
• idracidi: sono composti da idrogeno e alogeni, zolfo, selenio, azoto o ione cianuro (CN-);[8]
• ossiacidi: sono composti da idrogeno, un non metallo e ossigeno. Si formano facendo reagire un'anidride con
l'acqua.
Acidi organici
Secondo la sistematica organica, agli acidi organici appartengono:[9]
• acidi carbossilici: contengono uno o più gruppi carbossilici (-COOH), e in particolare:
• acidi monocarbossilici (tra cui si annoverano gli acidi grassi): con un gruppo carbossilico
• acidi dicarbossilici: con due gruppi carbossilici
• perossiacidi (o peracidi): con uno o più gruppi -COOOH
•
•
•
•
•
•
•
•
•
•
•
•
acidi solfonici: con uno o più gruppi -SO3H
acidi solfinici: con uno o più gruppi -SO2H
acidi sulfenici: con uno o più gruppi -SOH
acidi selenonici: con uno o più gruppi -SeO3H[10]
acidi seleninici: con uno o più gruppi -SeO2H
acidi selenenici: con uno o più gruppi -SeOH
acidi telluronici: con uno o più gruppi -TeO3H
acidi tellurinici: con uno o più gruppi -TeO2H
acidi tellurenici: con uno o più gruppi -TeOH
acidi fosfonici: con uno o più gruppi -PO(OH)2
acidi arsonici: con uno o più gruppi -AsO(OH)2
tioacidi
• O-tioacidi: con uno o più gruppi -CS(OH)
• S-tioacidi: con uno o più gruppi -CO(SH)
• ditioacidi: con uno o più gruppi -CS(SH)
• seleneoacidi
• O-selenoacidi: con uno o più gruppi -CSe(OH)
• S-selenoacidi: con uno o più gruppi -CO(SeH)
• diselenoacidi: con uno o più gruppi -CSe(SeH)
• telluroacidi
• O-telluroacidi: con uno o più gruppi -CTe(OH)
• S-telluroacidi: con uno o più gruppi -CO(TeH)
• ditelluroacidi: con uno o più gruppi -CTe(TeH).
Sono classificati come acidi anche composti che presentano più gruppi funzionali (di cui uno acido), come gli
amminoacidi.
Acido
131
Rischi
Il contatto della pelle (o di qualunque altra parte del corpo) con un acido produce generalmente un'irritazione; se
l'acido è particolarmente forte o concentrato può prodursi anche una ustione. Entità e gravità degli effetti dipendono
dalla forza dell'acido e dalla sua concentrazione, nonché dalle modalità e dai tempi di contatto.
Note
[1] In realtà, le reazioni chimiche descritte risultano approssimative; ad esempio gli idracidi non si formano da qualsiasi non-metallo, ma solo a
partire dagli alogeni o dallo zolfo, oltre che dallo ione cianidrico, come verrà spiegato più avanti.
[2] Silvestroni, op. cit., p.407
[3] Solomons, op. cit., p. 58
[4] Silvestroni, op. cit., p.408
[5] Solomons, op. cit., p. 65
[6] Silvestroni, op. cit., p.411
[7] Gli acidi di Brønsted-Lowry e gli acidi di Lewis comprendono invece un insieme molto più ampio ed eterogeneo di composti chimici, per cui
non può essere data qui una loro classificazione sommaria.
[8] A. Post Baracchi; A. Tagliabue, Chimica per le scuole medie superiori, Torino, Lattes, 1988
[9] Dispensa di chimica organica (http:/ / www. dispenseagrariatorino. it/ forestale/ difesadelsuolo/ difesadelsuolo1livello/ 1anno/
Chimicaorganica/ Organica. pdf)
[10] IUPAC - Gold book (http:/ / goldbook. iupac. org/ S05576. html)
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• T. W. Graham Solomons, Chimica organica, 2a ed., Bologna, Zanichelli, 2001, pp. 58-66. ISBN 88-08-09414-6
Voci correlate
•
•
•
•
•
•
•
•
•
•
•
Protonazione
Reazione di neutralizzazione
Base
Costante di dissociazione acida
pH
Reazione acido-base
Superacido
Nomenclatura chimica
Acido salicidico
Sostanze corrosive
Ustione
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Acids
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/acido
Collegamenti esterni
• Fun Science Gallery - Esperimenti con acidi e basi (http://www.funsci.com/fun3_it/acidi/acidi.htm)
• Calcolo di pH di acidi e basi e curve di titolazione con Excel – programma in inglese (http://www2.iq.usp.br/
docente/gutz/Curtipot_.html) o in portoghese (http://www2.iq.usp.br/docente/gutz/Curtipot.html)
Base (chimica)
132
Base (chimica)
In chimica, le definizioni di acido e base hanno subìto diverse modifiche nel tempo, partendo da un approccio
empirico e sperimentale fino alle più recenti definizioni, sempre più generali, legate al modello molecolare ad
orbitali.
Nella quotidianità, il termine "base" identifica sostanze generalmente caustiche e corrosive, capaci di intaccare i
tessuti organici e di far virare al blu una cartina al tornasole.
Esempi di sostanze basiche sono l'ammoniaca, la soda caustica e i più comuni tipi di sapone.
Un indice della forza di una base, funzione della sua natura e della sua concentrazione, è il pH.
Di seguito vengono elencate le definizioni più diffuse, in ordine cronologico.
Definizione di base secondo la teoria di Arrhenius
Secondo la teoria di Arrhenius, una base è una sostanza che dissociandosi in acqua produce ioni OH-.[1] Un acido ,
invece, è una sostanza che dissociandosi in acqua produce ioni H+.[1]
Rientrano in questa definizione tutti i composti che identifichiamo come basi (o alcali) nell'uso comune, sia per la
loro azione irritante sui tessuti viventi (ed il tipico gusto amaro), sia per la loro capacità di far virare opportunamente
sostanze indicatrici.
Sono basi secondo Arrhenius, per esempio, basi inorganiche forti come l'idrossido di sodio e l'idrossido di potassio.
La "forza" di una base, e con essa anche i suoi effetti corrosivi ed irritanti, è misurata tramite la costante di
dissociazione basica.
Una base viene detta monoacida o poliacida se libera uno o più ioni ossidrile.[1]
Definizione di base secondo la teoria di Brønsted-Lowry
Secondo la teoria di Brønsted-Lowry, una base è una sostanza capace di acquisire ioni H+ da un'altra specie
chimica, detta acido.[2][3]
La teoria di Brønsted-Lowry estende la definizione di base a quelle sostanze di cui non è possibile o non è pratico
valutare il comportamento in acqua, come de facto succede nella definizione data da Arrhenius. Introduce anche il
concetto di complementarietà tra acido e base, dato che la base non è tale se non in presenza di una controparte a cui
strappare uno ione H+, e viceversa.
L'ammoniaca, ad esempio, si comporta come una base secondo Brønsted e Lowry quando è sciolta in acqua perché è
in grado di sottrarre all'acqua uno ione H+
NH3 + H2O
→ NH4+ + OH-
Secondo questa teoria non esistono quindi acidi e basi a sé stanti, ma solo coppie di acido e base coniugati. Una
coppia acido/base coniugata è una coppia di specie chimiche che differiscono soltanto per uno ione H+. Quando un
acido cede uno ione H+ si trasforma nella sua base coniugata; quando una base acquista uno ione H+ si trasforma nel
suo acido coniugato.
Qualunque reazione che comporta il trasferimento di uno ione H+ da un acido a una base è una reazione acido-base
secondo Brønsted e Lowry. Un acido può, in determinate circostanze, comportarsi da base e viceversa.
Base (chimica)
133
Definizione di base secondo la teoria di Lewis
Secondo la teoria di Lewis, una base è una sostanza capace di donare un doppietto elettronico ad un'altra specie
chimica (detta acido).[4][5]
Simile alla teoria di Brønsted-Lowry, sostituisce al trasferimento dello ione H+ il trasferimento in senso inverso di un
doppietto elettronico. Secondo Lewis sono quindi basi anche composti come il tricloruro di fosforo o la piridina, che
presentano nella loro struttura un doppietto elettronico non condiviso che possono trasferire tramite un legame dativo
ad un accettore, un acido di Lewis. Nell'esempio qui riportato, l'ammoniaca è la base ed il trifluoruro di boro è
l'acido, secondo Lewis
H3N: + BF3
→ H3N→BF3
Una reazione acido-base secondo Lewis comporta la formazione di un legame dativo tra una base di Lewis
(donatore) e un acido di Lewis (accettore).
Le basi di Lewis si comportano da reagenti nucleofili, mentre gli acidi di Lewis si comportano da reagenti elettrofili.
La differenza tra le definizioni di "base di Lewis" e "nucleofilo" sta nel fatto che il carattere di una base di Lewis è
legato alla termodinamica della reazione, infatti un composto si comporta tanto più da base di Lewis quanto più
tende a donare i doppietti elettronici (in condizioni di equilibrio), mentre il carattere nucleofilo è legato alla cinetica
della reazione, infatti un composto si comporta tanto più da nucleofilo quanto più velocemente dona i doppietti
elettronici.
Basi forti
Una base è forte quando è completamente dissociata, e cioè dà luogo ad una ionizzazione totale. Le principali basi
forti sono:
•
•
•
•
•
idrossido di sodio: NaOH
idrossido di potassio: KOH
idrossido di calcio: Ca(OH)2
idrossido di bario: Ba(OH)2
idrossido di magnesio: Mg(OH)2.
Solubilità delle basi
Vi sono basi che si sciolgono e liberano ioni OH- producendo soluzioni basiche, ma vi sono anche basi, come
Cu(OH)2, Fe(OH)2, Fe(OH)3, Zn(OH)2, che sono poco, o per nulla solubili, quindi non riescono a formare una
soluzione basica.
È possibile che le basi derivanti da metalli anfoteri, in soluzioni concentrate, diano segni di solubilità a causa della
formazione di ioni complessi (ad esempio, Al(OH)3, che in soluzioni alcaline concentrate reagisce a dare lo ione
Al(OH)4-, solubile).
Base (chimica)
Note
[1]
[2]
[3]
[4]
[5]
Silvestroni, op. cit., p.407
Solomons, op. cit., p. 58
Silvestroni, op. cit., p.408
Solomons, op. cit., p. 65
Silvestroni, op. cit., p.411
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• T. W. Graham Solomons, Chimica organica, 2a ed., Bologna, Zanichelli, 2001, pp. 58-66. ISBN 88-08-09414-6
Voci correlate
•
•
•
•
•
Protonazione
Reazione di neutralizzazione
Acido
Costante di dissociazione basica
pH
•
•
•
•
•
Reazione acido-base
Superbase
Base insolubile
Nomenclatura chimica
Sostanze corrosive
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Bases
Collegamenti esterni
• Fun Science Gallery - Esperimenti con acidi e basi (http://www.funsci.com/fun3_it/acidi/acidi.htm)
134
Radicale libero
Radicale libero
Si definisce radicale (o radicale libero[1]) una specie chimica molto reattiva avente vita media di norma brevissima,
costituita da un atomo o una molecola formata da più atomi che presenta un elettrone spaiato: tale elettrone rende il
radicale estremamente reattivo, in grado di legarsi ad altri radicali o di sottrarre un elettrone ad altre molecole
vicine.[2]
I radicali giocano un ruolo importante in fenomeni come la combustione, la polimerizzazione e nella fotochimica, e
molti altri processi chimici, compresi quelli che riguardano la fisiologia umana. In questo ultimo caso il superossido
e il monossido di azoto hanno una funzione importantissima nel regolare molti processi biologici, come il controllo
del tono vascolare. (Attenzione: il monossido di azoto non deve essere considerato un radicale libero)
Il termine radicale e radicale libero sono spesso utilizzati con lo stesso significato. Il primo radicale libero stabile, il
trifenilmetile, è stato individuato da Moses Gomberg nel 1900 alla University of Michigan.
Nonostante la loro reattività, la maggior parte di essi ha una vita sufficientemente lunga da permetterne
l'osservazione tramite metodi spettroscopici.
Si formano, spontaneamente in natura o in laboratorio, per azione della luce o del calore in seguito alla scissione
omolitica di un legame covalente.
L'esempio sotto riportato illustra la formazione di due radicali metile a partire da una molecola di etano; in questo
caso si richiedono 88 kcal/mol di energia, che rappresenta l'energia di dissociazione del legame C-C:
CH3-CH3 → CH3• + CH3•
Radicali, ioni e gruppi funzionali
Il concetto di "radicale" ricorda i concetti di "ione" e "gruppo funzionale"; di seguito vengono indicate le analogie e
le differenze tra questi concetti:
• gli ioni e i radicali sono specie chimiche, mentre i gruppi funzionali non sono specie chimiche, bensì parti di altre
specie chimiche (ad esempio parti di molecole);
• i radicali e gli ioni possono combinarsi con altre specie chimiche diventando gruppi funzionali; i radicali sono più
reattivi degli ioni, quindi si combinano più facilmente con altre specie chimiche, mentre per i gruppi funzionali
non ha senso parlare di reattività;
• i radicali sono specie chimiche neutre (escludendo il caso particolare degli ioni radicalici), mentre gli ioni sono
specie chimiche cariche (positivamente o negativamente); per i singoli gruppi funzionali, essendo parte di una
specie chimica, non è possibile invece definire una "carica";
• i radicali contengono un elettrone spaiato, mentre gli ioni possono avere l'ottetto completo (ad esempio Cl-)
oppure no, mentre i gruppi funzionali, essendo parte di una specie chimica, possono avere solo elettroni condivisi;
• la funzione radicalica viene indicata con il simbolo "•" (che indica l'elettrone spaiato), gli ioni vengono
accompagnati nella rappresentazione dal numero di cariche positive (indicate da un numero seguito dal simbolo
"+" o da tanti segni "+" quanto sono le cariche positive) o negative (indicate da un numero seguito dal simbolo "-"
o da tanti segni "-" quanto sono le cariche negative), mentre i gruppi funzionali sono rappresentati indicando i
legami che presentano con il resto della specie chimica (ad esempio il -COOH presenta un legame semplice,
mentre il =CO può presentare un doppio legame o due legami semplici).
Esistono inoltre particolari specie chimiche che hanno entrambe le proprietà caratteristiche degli ioni e dei radicali:
tali specie chimiche sono dette "ioni radicalici".
135
Radicale libero
Meccanismi di formazione di ioni e radicali
A parte il caso degli ioni radicalici, che sono dotati carica (positiva o negativa), i radicali sono specie chimiche
neutre.
Siccome il radicale presenta un elettrone spaiato, si potrebbe incorrere nell'errore di considerare il radicale come una
specie carica negativamente. Tale equivoco nasce nel momento in cui si pensa che il radicale derivi da una specie
chimica neutra a cui sia stato aggiunto un elettrone spaiato, per cui avrebbe carica negativa; in realtà il radicale non
nasce dall'aggiunta di un elettrone ad una specie neutra (come avviene invece nel caso della "ionizzazione" di specie
neutre, che dà luogo appunto a ioni), bensì dalla "scissione" di una specie chimica neutra, quindi se la specie chimica
di partenza è neutra, scindendosi in due parti distinte, darà origine a due radicali neutri: infatti se per assurdo una
delle due specie fosse caricata negativamente, per la conservazione della carica, l'altra dovrebbe essere caricata
positivamente, ma in quest'ultimo caso non si avrebbe più il meccanismo di "scissione omolitica" (da cui si originano
i radicali), bensì "scissione eterolitica" (da cui si originano ioni).
Considerando una specie chimica neutra, in particolare una molecola biatomica A:B (dove i due punti rappresentano
un legame singolo tra A e B), i meccanismi di scissione omolitica e eterolitica a cui può essere soggetta la molecola
possono essere rappresentati rispettivamente nel seguente modo:
• Scissione omolitica: A:B → A• + B•
• Scissione eterolitica: A:B → A+ + B:Esempio
Si consideri una molecola di cloro. La formula bruta del cloro è Cl2 e la sua molecola è costituita quindi da due atomi
di cloro (Cl).
Con il termine "cloro" si intende sia la molecola Cl2 sia l'elemento chimico Cl, ma mentre la molecola Cl2 è una
specie chimica stabile, il cloro come elemento chimico (Cl) non può essere definito una "specie chimica".
Nonostante ciò, possiamo definire una configurazione elettronica dell'atomo di cloro, che è la seguente:
1s22s22p63s23p5
L'atomo di cloro presenta quindi 17 elettroni (2 elettroni nel livello 1, 8 elettroni nel livello 2 e 7 elettroni nel livello
3); l'atomo di cloro presenta inoltre 17 protoni e un numero variabile di neutroni (18, 19 o 20), a seconda dell'isotopo
considerato. Siccome il numero di elettroni in un atomo di cloro è pari al numero di protoni (come per tutti gli altri
elementi chimici), l'atomo di cloro ha carica neutra. Nonostante l'atomo di cloro abbia carica neutra, esso però
presenta un "elettrone spaiato", in quanto per raggiungere l'ottetto ha bisogno di formare un legame singolo con un
altro atomo.
Una molecola di cloro Cl2 può dare luogo per scissione omolitica a due radicali Cl•, secondo il seguente
meccanismo:
Cl:Cl → Cl• + Cl•
Ogni radicale è in questo caso costituito da un singolo atomo di cloro, che come detto in precedenza è neutro. Il
simbolo posto tra i due atomi di cloro ":" indica la coppia di elettroni condivisa, che costituisce il legame covalente,
mentre il simbolo "•" non indica uno scompenso di carica, bensì la presenza di un elettrone spaiato, il quale è
responsabile del carattere altamente energetico del radicale.
136
Radicale libero
137
Stabilità dei radicali
Nel caso di radicali alchilici, si ha questo ordine di stabilità:
terziario > secondario > primario
Un radicale alchilico terziario è quindi più stabile del corrispondente radicale alchilico secondario, che a sua volta è
più stabile del corrispondente radicale alchilico primario.
I radicali possono essere anche stabilizzati per risonanza, quando sono coniugati a sistemi π quali doppi legami o
anelli aromatici.
Formule di risonanza del radicale allile
Formule di risonanza del radicale benzile
I radicali sono comunque in genere specie molto reattive e quindi a vita corta. Esistono però radicali a vita lunga, che
si possono categorizzare nel modo seguente:
Radicali stabili
Il primo esempio di radicale stabile è l'ossigeno molecolare, O2. I radicali organici possono avere vita lunga se fanno
parte di un sistema π coniugato, come il radicale che deriva dal α-tocoferolo (vitamina E). Ci sono anche centinaia di
radicali tiazilici, che hanno una notevole stabilità cinetica e termodinamica pur con una stabilizzazione di risonanza
π molto limitata.[3][4]
Radicali persistenti
I radicali persistenti sono specie che possono vivere a lungo perché attorno al centro radicale esiste un notevole
impedimento sterico; la reazione del radicale con altre specie è di conseguenza fisicamente difficile.[5] Alcuni esempi
sono il radicale trifenilmetile scoperto da Gomberg, il sale di Fremy (nitrosodisulfonato di potassio, (KSO3)2NO•),
gli ossidi amminici (formula generale R2NO•) come TEMPO e TEMPOL. Durante i processi di combustione si
generano grandi quantità di radicali persistenti, che "possono essere responsabili di stress ossidativo con conseguenti
malattie cardio-polmonari e, probabilmente, il cancro che è stato attribuito all'esposizione a polveri sottili presenti
nell'aria."[6]
Radicale libero
Fisiopatologia
I radicali liberi sono uno dei meccanismi di danno cellulare più importante, sebbene assolvano a molte funzioni
fondamentali dell'organismo quando controllati. Sono molecole che posseggono un elettrone spaiato sull'orbitale più
esterno e questa configurazione elettronica le rende altamente instabili e particolarmente reattive. I radicali liberi
reagiscono facilmente con una qualsiasi molecola si trovi in loro prossimità (carboidrati, lipidi, proteine, acidi
nucleici) danneggiandola e spesso compromettendone la funzione. Inoltre, reagendo con altre molecole, hanno la
capacità di autopropagarsi trasformando i loro bersagli in radicali liberi e scatenando così reazioni a catena che
possono provocare estesi danni nella cellula. In condizioni normali, ciascuna cellula produce radicali liberi tramite
vari processi, come reazioni enzimatiche (ad esempio la xantina ossidasi o la NO sintasi), fosforilazione ossidativa,
difesa immunitaria (granulociti neutrofili e macrofagi). Queste piccole quantità sono tollerate e vengono inattivate da
sistemi enzimatici come il glutatione ed altri antiossidanti detti scavenger per la loro capacità di neutralizzare i
radicali liberi. Quando la produzione di radicali liberi è eccessiva si genera ciò che viene chiamato stress ossidativo.
I sistemi enzimatici e gli antiossidanti intracellulari non riescono più a far fronte alla sovraproduzione e i radicali
liberi generano danno cellulare che può essere sia reversibile, in tal caso la cellula torna alle condizioni normali, o
irreversibile, con conseguente morte cellulare per apoptosi o per necrosi. Lo stress ossidativo è imputato quale causa
o concausa di patologie quali il cancro, l'invecchiamento cellulare e malattie degenerative. Le specie reattive
dell'ossigeno possono essere classificate come ROS (da Reactive Oxygen Species) o alternativamente come ROI (da
Reactive Oxygen Intermediate). Allo stesso modo, le specie reattive dell'azoto possono essere nominate RNS
(Reactive Nitrogen Species) o RNI.
ROS
Le specie reattive dell'ossigeno, i ROS, sono i radicali liberi a maggior diffusione. I più importanti ROS sono l'anione
superossido O2-, il perossido d'idrogeno H2O2 e il radicale ossidrilico •OH.
• L'anione superossido (O2-) è prodotto dalla riduzione incompleta di O2 durante la fosforilazione ossidativa, da
alcuni enzimi (xantina ossidasi) e dai leucociti. Viene inattivato dalle superossido dismutasi (SOD) che,
combinandolo con 2H+ e catalizzando la reazione tramite il suo cofattore metallico (Fe, Mn, Cu, Zn o Ni) lo
converte in H2O2 e O2. Se non viene inattivato danneggia i lipidi di membrana, proteine e DNA, può inoltre
stimolare la produzione di enzimi nei leucociti. Generalmente ha un raggio d'azione limitato.
• Il perossido d'idrogeno (H2O2) è spesso il prodotto della superossido dismutasi (SOD) o da alcune ossidasi
contenute nei perossisomi. Viene metabolizzato dalla catalasi dei perossisomi in H2O e O2 che catalizza la
reazione tramite il suo gruppo eme e dalla glutatione perossidasi nel citosol e nei mitocondri.
• Il radicale ossidrilico (•OH) è generalmente un prodotto dell'idrolisi dell'acqua da parte di radiazioni, oppure è un
prodotto della reazione di Fenton a partire dal perossido d'idrogeno (con lo ione ferroso Fe2+ quale catalizzatore).
E' il ROS più reattivo ed è prodotto dai leucociti a partire dal perossido d'idrogeno per distruggere patogeni, ma se
in eccesso provoca danni alla membrana plasmatica, alle proteine e agli acidi nucleici. Viene inattivato per
conversione in H2O da parte della glutatione perossidasi.
RNS
Le specie reattive derivate dall'azoto (RNS) di maggior interesse sono l'ossido nitrico (NO) ed il perossinitrito
(ONOO-).
• L'ossido nitrico è prodotto dalle NO sintasi di cui esistono, nell'uomo, tre tipi: NO sintasi neuronale (nNOS),
presente nei neuroni e nel muscolo scheletrico, NO sintasi inducibile (iNOS) presente nel sistema cardiovascolare
e nelle cellule del sistema immunitario e NO sintasi endoteliale (eNOS), presente nell'endotelio. L'ossido di azoto
è un neurotrasmettitore, è coinvolto nella risposta immunitaria, è un potente vasodilatatore, un secondo
messaggero e partecipa all'erezione del pene.
138
Radicale libero
• Il perossinitrito (ONOO-) è formato dalla reazione tra ossido nitrico e ione superossido. Viene convertito in
HNO2 dalle perossiredossine presenti nel citosol e nei mitocondri. Può danneggiare lipidi, proteine e DNA.
Generazione di ROS e RNS
All'interno della cellula i radicali liberi possono essere generati in vari modi.
• Le radiazioni ionizzanti idrolizzano l'acqua (H2O) a idrogeno (H) e radicale ossidrilico (•OH). Fanno parte di
questa categoria i raggi ultravioletti, i raggi X e i raggi gamma.
• Le infiammazioni sono processi che scatenano la produzione di ROS da parte della NADPH ossidasi dei leucociti
al fine di sbarazzarsi di organismi patogeni; talvolta però i radicali liberi prodotti danneggiano anche cellule sane.
• Alcuni enzimi come la xantina ossidasi che genera O2-, la NO sintasi che genera NO, la superossido dismutasi che
genera H2O2, oppure a partire da enzimi che metabolizzano farmaci o altre sostanze chimiche esogene.
• La fosforilazione ossidativa che si verifica durante la respirazione cellulare e che genera piccole quantità di
ciascuno dei tre più importanti ROS.
• I metalli di transizione fungono da catalizzatori nelle reazioni che portano alla produzione di radicali liberi. Il più
comune è il Fe2+ tramite la reazione di Fenton, seguito dal rame (Cu).
• Altri radicali liberi possono concorrere alla formazione di ulteriori radicali liberi, per esempio quando NO e O2reagiscono per formare il perossinitrito ONOO-.
Rimozione di ROS e RNS
La cellula possiede diversi metodi per metabolizzare i ROS.
• Il sistema più comune è quello che utilizza enzimi deputati alla conversione delle specie reattive dell'ossigeno in
prodotti meno reattivi e tossici per la cellula. Sono state citate la superossido dismutasi (ne esistono almeno tre
tipi) che agisce su O2- tramite la reazione 2O2- + 2H+ --> H2O2 + O2, la catalasi che opera sul perossido
d'idrogeno tramite la reazione 2H2O2 --> 2H2O + O2 e la glutatione perossidasi che agisce sia sul perossido
d'idrogeno che sul radicale ossidrilico tramite le reazioni H2O2 + 2GSH --> GSSG + 2H2O e 2OH + 2GSH -->
GSSG + 2H2O. Il rapporto tra il glutatione ridotto (GSH) e il glutatione ossidato (GSSG) viene analizzato per
valutare la capacità della cellula di eliminare i ROS ed è un indice del suo stato ossidativo.
• La cellula controlla il livello di metalli di transizione al suo interno, particolarmente quelli del ferro e del rame. Il
ferro è infatti sempre legato ad una proteina e tendenzialmente mantenuto allo stato ferrico Fe3+. Nel sangue è
legato alla trasferrina, la proteina con la maggiore affinità per il suo substrato conosciuta, è immagazzinato nella
ferritina, ma è anche utilizzato nel gruppo eme di molte metalloproteine e ferrossidasi a diverso significato. Il
rame è legato prevalentemente alla ceruloplasmina e all'efestina.
• La cellula possiede antiossidanti deputati alla neutralizzazione di radicali liberi, gli scavenger. Ne fanno parte il
glutatione, la vitamina A (retinolo, retinale, acido retinoico), la vitamina C (acido ascorbico) e la vitamina E
(tocoferolo).
Effetti dei radicali liberi
I radicali liberi tendono a danneggiare particolarmente tre componenti della cellula: i lipidi, le proteine e gli acidi
nucleici.
• La perossidazione lipidica, in particolare della membrana plasmatica e delle membrane degli organelli
intracellulari è un danno cellulare comune dovuto ai ROS e agli RNS. I radicali liberi, in presenza di ossigeno,
reagiscono con i doppi legami dei lipidi di membrana generando dei perossidi lipidici che, essendo reattivi, si
propagano determinando un danno esteso alle membrane. Il ROS più temibile in questo caso è •OH. Negli
eritrociti possono provocare quindi emolisi. La degradazione dei lipidi operata dai radicali liberi è riscontrabile
139
Radicale libero
tramite la presenza di prodotti terminali di lipossilazione avanzata (ALEs, Advanced Lipoxylation End-products)
quali il 4-idrossi-nonenale (4) HNE) e la malonil-dialdeide (MDA). Sono stati sviluppati dei dosaggi colorimetrici
molto sensibili (metodo ELISA) che permettono di rilevare 4-HNE ed MDA a concentrazioni tissutali inferiori al
micromolare.
• L'ossidazione delle proteine, in particolare i radicali liberi agiscono ossidando i gruppi laterali degli
amminoacidi, danneggiando la funzione della proteina, promuovono la formazione di legami crociati come il
legame disolfuro, alterandone la struttura o il ripiegamento. Possono anche dare origine ad amminoacidi
modificati (diidrossifenilalanina, ditirosina...).
• Il danno al DNA, dal momento che i radicali liberi possono determinare mutazioni o danneggiare
macroscopicamente lo stesso DNA e alterare la struttura chimica delle basi azotate formandone di nuove come
8-ossiguanina o 5-idrossimetiluracile. Tramite questo tipo di danno sono concausa dell'invecchiamento cellulare e
promuovono il cancro.
Misurazione dello stress ossidativo
È possibile misurare sia la concentrazione di sostanze ossidanti (ROS: radicali liberi dell'ossigeno) sia di quelle
antiossidanti grazie ad un test di semplice esecuzione, da cui si ottiene un valore detto "indice di stress ossidativo".
Lo stress ossidativo è definito come la mancanza di equilibrio tra lo stato ossidante (danni da radicali liberi) e lo
stato antiossidante (difese anti-radicaliche).
Il test può essere particolarmente utile per la seguenti categorie di persone:
• Adulti sani che vogliano fare un "check-up" in chiave preventiva (famigliarità per malattie cardiovascolari,
diabete, dislipidemie, malattie atrosiche, ecc.)
• Sportivi, per monitorare l'efficacia di allenamenti e metodologie di scarico e di recupero dopo sforzi od attività
agonistica.
• Donne, per migliorare i consigli dermocosmetici di anti-invecchiamento della pelle.
Inoltre, grazie ai parametri del test, è possibile valutare meglio l'azione a livello cellulare e l'eventuale riduzione del
danno ossidativo durante:
•
•
•
•
Diete
Attività fisica
Trattamenti cosmetici e nutrizionali
Modifiche dello stile di vita (es. riduzione o abolizione del fumo di sigaretta).
Modalità di esecuzione
• Il prelievo è sul sangue capillare (puntura del polpastrello).
• È preferibile essere riposati e non in fase di stress recente.
• È consigliabile non aver fumato almeno mezz'ora prima del test.
Il test è basato sul rapporto tra la valutazione della concentrazione di ROS (FORT TEST) e la capacità antiossidante
totale (FORD TEST)[7]
140
Radicale libero
Risultati
Più elevato è il valore del FORT TEST maggiore è il rischio di danni da stress ossidativo. Il risultato è legato al
livello delle difese (FORD TEST): più alte sono le difese, minore è il rischio generale. Eventualmente le difese
possono essere stimolate e potenziate/integrate qualora risultino sotto i livelli usuali. Naturalmente ogni persona ha
un suo valore di partenza riguardo a questi parametri.
È consigliabile eseguire un primo test di controllo per conoscere i propri parametri in un momento in cui si è “sani”.
Controlli successivi ci diranno se c'è un miglioramento o peggioramento in termini di stress ossidativo con una
diminuzione o un aumento potenziale del rischio patologico generale. In tal caso potrebbe essere utile consultare il
proprio medico per eventuali controlli diagnostici mirati.
Note
[1] Solomons, op. cit., p. 122
[2] P. Silvestroni, "Fondamenti di chimica", nota 22 pag. 362, ed. Cea-Zanichelli
[3] (EN)R. T. Oakley (1998). Cyclic and heterocyclic thiazenes. Prog. Inorg. Chem. 36: 299-391. DOI: 10.1002/9780470166376.ch4 (http:/ / dx.
doi. org/ 10. 1002/ 9780470166376. ch4). URL consultato il 24-12-2010.
[4] (EN)J. M. Rawson, A. J. Banister e I. Lavender (1995). Chemistry of dithiadiazolydinium and dithiadiazolyl rings. Adv. Hetero. Chem. 62:
137-247. DOI: 10.1016/S0065-2725(08)60422-5 (http:/ / dx. doi. org/ 10. 1016/ S0065-2725(08)60422-5). URL consultato il 24-12-2010.
[5] (EN)D. Griller e K. U. Ingold (1976). Persistent carbon-centered radicals. Acc. Chem. Res. 9 (1): 13-19. DOI: 10.1021/ar50097a003 (http:/ /
dx. doi. org/ 10. 1021/ ar50097a003). URL consultato il 24-12-2010.
[6] (EN)S. Lomnicki, H. Truong, E. Vejerano e B. Dellinger (2008). Copper oxide-based model of persistent free radical formation on
combustion-derived particulate matter. Environ. Sci. Technol. 42 (13): 4982–4988. DOI: 10.1021/es071708h (http:/ / dx. doi. org/ 10. 1021/
es071708h). URL consultato il 24-12-2010.
[7] FORT and FORD: two simple and rapid assays in the ... [Metabolism. 2009] - PubMed result (http:/ / www. ncbi. nlm. nih. gov/ pubmed/
19604518)
Bibliografia
• Muller, F. L., Lustgarten, M. S., Jang, Y., Richardson, A., Van Remmen, H., Trends in oxidative aging theories,
Free Radic. Biol. Med. 43, 477-503 (2007)
• T. W. Graham Solomons, G. Ortaggi, D. Misiti (a cura di), Chimica organica (http://www.catalogo.zanichelli.
it/Pages/Opera?siteLang=IT&id_opera=534962), 2a ed., Zanichelli, 1988. ISBN 88-080-9414-6
Voci correlate
•
•
•
•
•
•
•
•
•
Alcani
Alchile
Alterazione omeostasi del calcio
Arile
Carbocatione
Ione radicalico
Legame chimico
Sistematica organica
Sostituzione radicalica
141
Metallo
142
Metallo
Il metallo è un materiale che riflette la luce
conferendole una particolare tonalità (detta
appunto lucentezza metallica), un ottimo
conduttore di calore e di elettricità,[1] che
può essere attaccato dagli acidi (con
sviluppo di idrogeno) e dalle basi, spesso
con buone caratteristiche di resistenza
meccanica. L'acqua può attaccare i metalli
(soprattutto del primo e secondo gruppo), ai
quali strappa gli elettroni di valenza per dare
appunto l'idrogeno attraverso una reazione
particolarmente esotermica.
Metallo, nello specifico ferro.
In base alle proprietà chimiche i metalli
danno luogo ad ossidi basici (es: Na2O, CaO).
Sono elementi chimici (si tratta di una delle tre categorie in cui gli elementi chimici sono suddivisi, insieme ai
semimetalli e ai non metalli) oppure leghe.
Esistono vari tipi di metalli; questi furono scoperti in epoche distanti nel tempo, perché ben pochi metalli sono
reperibili in natura allo stato nativo; inoltre ogni metallo ha una sua temperatura di fusione: più bassa è tale
temperatura, più è facile l’estrazione del metallo dalle rocce che lo contengono. In particolare i primi metalli lavorati
(il rame e lo stagno) hanno una temperatura di fusione relativamente bassa, ottenibile con gli antichi forni di circa
10.000 anni fa (epoca in cui, presumibilmente, iniziò la lavorazione del rame).
Con l'espressione materiale metallico si fa riferimento in generale ai metalli e alle loro leghe.
Caratteristiche
Numero di atomi per cella
unitaria
Numero di
coordinazione
Fattore di impacchettamento
atomico
Reticolo cubico a corpo
centrato
(CCC)
9
8
0,68
Reticolo cubico a facce
centrato
(CFC)
14
12
0,72
Esagonale compatto
(EC)
17
12
0,72
Metallo
143
Caratteristica essenziale del metallo è la sua struttura regolare, basata
sulla ripetizione di una cella elementare. Le più comuni celle sono la
CCC (cubica corpo centrato; esempi: ferro, tungsteno e molibdeno),
CFC (cubica facce centrate; esempi: rame e alcune sue leghe, acciaio
austenitico, leghe di alluminio o di nichel, piombo, oro e argento) ed
EC (esagonale compatta; esempi: magnesio, cadmio e zinco).[2]
Sottoraffreddato dallo stato liquido un metallo si solidifica in grani, le
cui dimensioni sono immagine della temperatura a cui avviene il
processo e i cui bordi rappresentano un'importante zona di
Cristalli di gallio
discontinuità
della
struttura
metallica.
Maggiore
è
il
sottoraffreddamento, minore sarà il raggio critico al di sotto del quale si decostruiscono gli embrioni dallo stato
solido; inoltre per un maggior numero di embrioni che diventano grani risulterà minore la dimensione del grano
metallico. Quest'ultimo aspetto ha fondamentale importanza nello studio della resistenza a deformazione: a
temperatura ambiente la frattura a trazione avviene per rottura dei grani e non per distacco tra essi, a causa del
maggior contenuto energetico, e quindi migliore coesione, associato alla distorsione dei giunti cristallini. Per contro
all'aumentare della temperatura la maggiore mobilità dei difetti, concentrati nei giunti, ne abbassa notevolmente la
coesione (frattura intercristallina).
I metalli tendono a cedere con facilità i propri elettroni di valenza a non tenersi quelli in eccesso per raggiungere la
configurazione elettronica dei gas nobili: hanno cioè una bassa energia di ionizzazione e una scarsa affinità
elettronica.[1] Il contrario accade per i semimetalli ed a maggior ragione per i non metalli. Quando più atomi
metallici si aggregano a formare una struttura cristallina quindi, gli elettroni di legame vengono condivisi tra tutti i
partecipanti dando luogo ad orbitali molecolari delocalizzati in tutto il solido. La delocalizzazione elettronica e
l'elevato numero di oggetti presenti contribuisce a tenere insieme gli ioni costituenti, anche se l'energia di legame per
atomo non è molto elevata; nel contempo, essa dà luogo alla sovrapposizione delle bande di energia, permettendo di
conseguenza alle cariche di muoversi liberamente all'interno del metallo. Si parla per questo di gas di elettroni. La
disponibilità di tante cariche libere spiega bene l'ottima conducibilità elettrica e termica, insieme alla proprietà di
assorbire e/o riflettere la luce totalmente anche in strati sottilissimi, di poche decine di atomi.
Generalmente gli elementi chimici metallici sono quasi tutti nella zona di transizione centrale della tavola periodica,
fra i alcalino-terrosi e gli alogeni; sono quasi tutti di peso atomico medio o medio-alto; gli elementi metallici più
leggeri possono essere portati allo stato metallico solo con difficoltà.
I metalli (nome esteso minuscolo secondo IUPAC) e le loro leghe
più comuni sono:
• l'argento (Ag)
• l'alluminio (Al)
• il ferro (Fe)
• il rame (Cu)
• l'oro (Au)
• lo zinco (Zn)
Il ferro, uno dei metalli più noti.
• il platino (Pt)
•
•
•
•
il piombo (Pb)
lo stagno (Sn)
il titanio (Ti)
il mercurio (Hg)
• il bronzo (lega rame-stagno, ma anche -alluminio, -nichel, -berillio)
• l'ottone (lega rame-zinco, con aggiunta di Fe, As, Sn, Sb, Al, ed altri metalli e semimetalli)
Metallo
144
• gli acciai (leghe ferro-carbonio-cromo-nichel-molibdeno ed altri metalli cobalto, vanadio).
Estrazione e lavorazione dei metalli
Il minerale che contiene il metallo viene prelevato dalle miniere. In
seguito avviene l’estrazione della materia prima (i minerali).
L'estrazione consiste nel separare il metallo dalle altre sostanze. Quindi
il metallo viene fuso e passa allo stato liquido per le successive
lavorazioni. Dopo l’estrazione, il metallo viene raffinato, cioè vengono
eliminate le impurità fino ad ottenere la percentuale di purezza
desiderata. A questo punto il metallo viene colato, cioè viene estratto
dal forno come metallo fuso. I metalli fusi possono quindi essere
modellati, messi in appositi stampi e quindi assumere la forma data.
Metallo lavorato a caldo
Per ottenere i prodotti metallici finiti bisogna prima passare alla
produzione dei semilavorati, cioè dove si producono le lastre, i lingotti, le bramme, i blumi o bilette. Infine avviene
la lavorazione meccanica, che consiste nella laminazione e stampaggi dei semilavorati.
Metalli pesanti
Non esiste una definizione ufficiale di metallo leggero o pesante da parte della IUPAC, l'autorità internazionale che
fissa e aggiorna la nomenclatura e la terminologia degli elementi e composti chimici, o da parte di organismi
simili.[3] Nonostante questo, numerosi articoli e pubblicazioni parlano genericamente di "metalli pesanti" e "leggeri"
omettendo una chiara definizione o dando definizioni in contrasto tra loro basate sulla densità, sul peso atomico o
altre proprietà chimiche.[3]
Spesso all'aggettivo pesante è associato il concetto di tossicità anche se di per sé la densità di un metallo non ha un
legame diretto con effetti sul corpo umano. La tossicità di una qualunque sostanza dipende dalla sua natura (esatto
composto chimico) e dalla sua quantità. Un composto chimico può essere tossico pur essendo formato da atomi di
elementi chimici che presi singolarmente non sono tali, e viceversa. Inoltre una certa sostanza può essere ben
tollerata o addirittura necessaria se al di sotto di una certa quantità. Infine la tossicità dipende dalla combinazione e
sinergia con altri elementi.
Metalli indicati come "pesanti" messi tipicamente in correlazione alla loro tossicità e bioaccumulazione nella catena
alimentare sono: mercurio, cromo, cadmio, arsenico, piombo[3] e recentemente uranio[4].
Difetti del cristallo
La ripetitività della struttura cristallina è interrotta localmente da difetti che possono essere di vario genere.
• Difetti di punto: sono le occasionali vacanze (la cui concentrazione dipende dalla temperatura secondo un legame
esponenziale), gli atomi sostituzionali o interstiziali (specie quando nelle soluzioni solide è notevole la differenza
nelle dimensioni degli elementi componenti), gli atomi autointerstiziali (nel caso di atomi uguali a quelli del
reticolo), i difetti di Frenkel (uno ione positivo lascia la sua posizione reticolare, creando così una vacanza
cationica, per andare a formare uno ione interstiziale) e di Schottky (coppia di una vacanza anionica e cationica in
un solido ionico).
• Difetti di linea: separano parti che hanno subito lo slittamento da altre che non lo hanno subito. Accumulano
tensione e per effetto dell'applicazione di sforzi tendono a moltiplicarsi. Sono detti dislocazioni.
• Difetti di superficie: ovvero i bordi di grano presso i quali cambia interamente l'orientamento dei piani di reticolo
da un grano all'altro.
• Difetti di volume: le irregolarità nella sequenza ordinata dei piani cristallini nel metallo.
Metallo
Poiché questi difetti influenzano enormemente il comportamento metallico sono estremamente importanti per la
metallurgia.
Elasticità e plasticità del metallo
Sottoposto ad uno sforzo crescente, il metallo in un primo tempo si deformerà linearmente secondo la legge di
Hooke in maniera elastica, e reversibile una volta cessato il carico.
L'aumentare dello sforzo oltre un certo limite imporrà in seguito una irreversibile deformazione plastica
accompagnata dall'incrudimento, cioè un aumento progressivo del limite elastico del materiale e del valore della
tensione di rottura. Se il valore teorico di energia necessario per deformare plasticamente un campione è
notevolmente maggiore rispetto a quello in effetti necessario, ciò è dovuto alla presenza di dislocazioni, ossia
discontinuità di linea nella struttura cristallina che a seconda della forma sono dette a vite, a spigolo o mista.
La frattura si distingue a seconda della natura del metallo in duttile o fragile. Nel primo caso il metallo si deforma
sensibilmente nel campo plastico, si verifica uno strizzamento a causa dei microvuoti venutisi a creare e la superficie
di frattura ha la caratteristica forma di coppa cono. Nel secondo caso la frattura è improvvisa, subito oltrepassato il
limite elastico e la superficie è perpendicolare alla direzione dello sforzo, di aspetto brillante e cristallino.
Fenomeni degenerativi
Un particolare tipo di frattura fragile è il cosiddetto clivaggio, tipico della struttura cubica a corpo centrato (CCC),
esagonale compatta (EC) più raramente. Il clivaggio è frutto di sforzi elevati condotti a bassa temperatura. Il
clivaggio è in genere transgranulare ma può essere anche intergranulare se a bordo grano sono presenti particolari
precipitati o impurezze.
Il creep è invece un fenomeno che avviene ad alte temperature che in funzione del tempo vede prima l'aumento delle
dislocazioni e l'incrudimento, fenomeno non attivato termicamente (creep primario) quindi il disancoramento delle
dislocazioni (fenomeno questo si attivato termicamente) che comporterà la rottura dopo aver pareggiato l'intensità
dell'incrudimento (nel creep secondario la velocità di creep diventa stazionaria) la supera, accelera la velocità di
deformazione ( creep Terziario) e induce la rottura.
Creep dei metalli: Deformazione plastica dipendente dal tempo detta anche scorrimento viscoso a caldo che avviene
quando un materiale metallico è sottoposto ad una sollecitazione costante a temperatura elevata. Il meccanismo del
creep viene illustrato da curve che riportano la deformazione in funzione del tempo e si può suddividere in varie fasi:
• Allungamento elastico istantaneo
• Creep primario: La velocità di deformazione decresce con il tempo, a causa del blocco delle dislocazione e
conseguente incrudimento.
• Creep secondario: A tempi maggiori, la diffusione degli atomi, permette un parziale sblocco delle dislocazioni
rendendo nuovamente possibile il loro scorrimento. Il blocco e lo sblocco si equilibrano e la velocità di
deformazione rimane pressoché costante.
• Creep terziario: La velocità di deformazione aumenta rapidamente e in breve il materiale arriva a rottura, in
seguito alla formazione di microvuoti al bordo grano ed il successivo scorrimento dei grani tra di loro.
Nota: L'aumento della temperatura provoca l'innalzamento della curva di creep e la diminuzione della durata delle
varie fasi (il materiale si rompe più velocemente).
La fatica è quel fenomeno per il quale un metallo sottoposto ad uno sforzo ciclico può pervenire a rottura anche per
valori dello sforzo molto al di sotto del suo limite di snervamento. Ad una prima fase di incrudimento (hardering)
segue l'assestamento microstrutturale (softering), l'orientarsi delle dislocazioni presso precise bande di slittamento, il
presentarsi presso la superficie di caratteristiche microintrusioni e microestrusioni. È lungo le bande di slittamento,
che si presentano dopo appena il 5% della vita utile del campione, che avrà luogo la rottura il cui punto di innesco è
145
Metallo
appena al di sotto della superficie. La rugosità superficiale è un parametro importantissimo per quel che riguarda la
resistenza a fatica di un metallo.
Fatica dei metalli: I materiali vengono sottoposti a sollecitazioni cicliche che possono portare a rottura il componente
anche per carichi inferiori al carico di rottura; queste prove avvengono in prevalenza su componenti in movimento e
si articolano in 3 fasi:
• Innesco della cricca: in un punto in cui la geometria del componente permette una concentrazione degli sforzi
oppure in corrispondenza di difetti.
• Propagazione della cricca: avviene per effetto dell'applicazione ciclica dello sforzo e provoca una riduzione della
sezione resistente.
• Rottura finale: Avviene in corrispondenza del raggiungimento delle dimensioni critiche da parte della cricca.
Lo studio della resistenza a fatica dei materiali viene effettuato con prove accelerate su provini già dotati di intaglio
(pre – ciccati) e i risultati vengono riportati in grafici “sforzo-numero di cicli a rottura” [σ-N]. Alcuni materiali
presentano un limite di fatica, ovvero un asintoto della curva [σ-N], al di sotto del quale non si ha più diminuzione
della resistenza a fatica all'aumentare di N (es. Acciaio 1047). su provini già dotati di intagli (pre – ciccati)
La corrosione nasce dalle iterazioni di ossidoriduzione con l'ambiente e naturalmente è particolarmente dannosa per i
metalli. Si cercano espedienti per prevenire come un rivestimento in PVC, la verniciatura o utilizzare un anodo
sacrificale. Variegata la casistica: la corrosione può avvenire in fessura o per aerazione differenziata, intergranulare,
per 'pitting (superato in un punto il film protettivo), esaltata da un ambiente galvanico o dalle forti tensioni cui il
pezzo è soggetto.
L'usura infine distrugge il metallo in presenza di un ambiente tribologico dove cioè vi è attrito tra il pezzo e altre
componenti. L'usura può essere dovuta alle forze fluidodinamiche, è detta tribossidazione in un ambiente
particolarmente aggressivo, si dice adesiva, quando è determinata da microgiunzioni venutesi a creare tra le creste di
rugosità di due corpi in mutuo slittamento l'uno sull'altro, o erosiva quando semplicemente una superficie è in moto
relativo contro particelle particolarmente dure. Un caso particolare risulta la corrosione-erosione, in cui un'usura
superficiale non eccessiva è però sufficiente ad asportare lo strato superficiale passivato, ripresentando quindi
metallo vivo agli agenti corrosivi.
Note
[1] Rolla, op. cit., pp. 43-44
[2] Arduino, op. cit., p. 316
[3] "Heavy metals" - A meaningless term? (IUPAC Technical Report). Pure Appl. Chem. vol 74. no 5, pp.793-807, 2002 (http:/ / old. iupac. org/
publications/ pac/ 2002/ pdf/ 7405x0793. pdf)
[4] What is DUF6? Is it dangerous and what should we do with it? (http:/ / www. ieer. org/ sdafiles/ vol_5/ 5-2/ deararj. html)
Bibliografia
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori (http://books.google.it/
books?id=Y83UPAAACAAJ&source=gbs_navlinks_s), 29a ed., Dante Alighieri, 1987.
• Gianni Arduino; Renata Moggi, Educazione tecnica, 1a ed., Lattes, 1990.
146
Metallo
147
Voci correlate
•
•
•
•
•
•
•
•
Tavola periodica
Metalli nobili
Nonmetallo
Semimetallo
Metallurgia
Fosfatazione
Materiale da costruzione
Schiuma metallica
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Metals
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/metallo
Non metallo
I non metalli sono cattivi conduttori di calore ed elettricità. Insieme con i metalli e i semimetalli, i non metalli (a
volte indicati anche come non-metalli o nonmetalli) sono una delle tre categorie in cui si suddividono gli elementi
chimici secondo le loro proprietà di ionizzazione e legame.
Caratteristiche
Tali proprietà nascono dal fatto che i non metalli sono tutti molto elettronegativi, cioè guadagnano elettroni di
valenza da altri atomi più facilmente di quanto li cedano.
Diversamente dai metalli, un non metallo può essere o un isolante o un semiconduttore; i non metalli possono
formare legami ionici con i metalli, guadagnando elettroni, o legami covalenti con altri non metalli. In base alle
proprietà chimiche i non metalli danno luogo ad anidridi acidi (es: SO2, SO3, CO2).
Non metalli
In ordine di numero atomico, i non metalli sono:
•
•
•
•
•
•
•
•
•
•
•
Carbonio (C)
Azoto (N)
Ossigeno (O)
Fluoro (F) (alogeno)
Fosforo (P)
Zolfo (S)
Cloro (Cl) (alogeno)
Selenio (Se)
Bromo (Br) (alogeno)
Iodio (I) (alogeno)
Astato (At) (alogeno, ma possiede anche caratteristiche metalliche e per questo viene considerato da alcuni un
semimetallo)
Tutti i non metalli si trovano nell'angolo in alto a destra della tavola periodica (con l'eccezione dell'Idrogeno, che si
pone di norma insieme ai metalli alcalini, ma si comporta di solito come un nonmetallo). Anche i gas nobili sono da
considerarsi non metalli.
Non metallo
Esistono solo dodici non metalli noti, a fronte di ottanta e più metalli; però i non metalli costituiscono la maggior
parte della massa della Terra, soprattutto negli strati più esterni, e gli organismi viventi sono composti quasi
interamente di non metalli. L'idrogeno, l'azoto, l'ossigeno, il fluoro, il cloro, il bromo e lo iodio sono elementi
biatomici che nella tavola periodica di Dmitri Mendeleev occupano posizioni tali da raffigurare una 'L' capovolta
puntata.
Voci correlate
• Metallo
• Semimetallo
• Tavola periodica
Semimetalli
1. RINVIA Semimetallo
Gas nobili
I gas nobili sono dei gas inerti che costituiscono il diciottesimo[1] gruppo della tavola periodica degli elementi, cioè
la colonna più a destra. Sono costituiti da atomi con gusci elettronici completi. Ne fanno parte i seguenti elementi:
•
•
•
•
•
•
•
Elio
Neon
Argon
Kripton
Xeno
Radon
Ununoctio
Il termine gas nobili deriva dal fatto che essi evitano di reagire con gli elementi "comuni", esibendo un
atteggiamento attribuito comunemente alla "nobiltà". I gas nobili venivano anche chiamati gas inerti, ma il termine
non era accurato, in quanto alcuni di essi hanno mostrato di prendere parte in reazioni chimiche.
A causa della loro non-reattività, i gas nobili non furono scoperti fin quando l'esistenza dell'elio non fu dedotta
ipoteticamente da un'analisi spettrografica del Sole, e successivamente provata quando William Ramsay riuscì a
isolarlo. I gas nobili hanno inoltre forze di attrazione interatomica molto deboli e conseguentemente punti di fusione
ed ebollizione molto bassi.
Gli atomi più grossi della serie sono leggermente più reattivi, e lo xeno è stato indotto a formare un numero di
composti con il fluoro e con l'ossigeno. Nel 1962, Neil Bartlett, mentre lavorava alla University of British Columbia,
fece reagire lo xeno con il fluoro ottenendo XeF2, XeF4, e XeF6. Il radon reagisce con il fluoro formando fluoruro di
radon, RnF, e il composto, allo stato solido, emette una luce gialla. Anche il kripton reagisce con il fluoro formando
KrF2.Nel 2003 è stato scoperto che anche l'argon forma composti come ad esempio il fluoruro di argo ArF2.
Nel 2002, vennero scoperti composti dove l'uranio formava molecole con argon, kripton, o xeno. Ciò suggerisce che
i gas nobili potrebbero formare composti anche con altri metalli.
148
Gas nobili
149
Note
[1] I gas nobili appartengono al diciottesimo gruppo della tavola periodica secondo l'attuale nomenclatura IUPAC, mentre secondo la vecchia
nomenclatura appartengono al gruppo VIIIA.
Voci correlate
•
•
•
•
•
•
•
Tavola periodica
Elemento
Atomo
Nonmetallo
Metallo
Metalloide
Composto di gas nobili
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Noble
gases
Formula chimica
Una formula chimica è una rappresentazione sintetica che descrive quali e quanti atomi vanno a comporre una
molecola (o una unità minima) di una sostanza (formula bruta), nonché la loro disposizione nello spazio (formula di
struttura).
Formula grezza o bruta
Nella formula bruta ogni tipo di elemento chimico è identificato attraverso il suo simbolo chimico. Il numero di
atomi di ogni elemento presente nella molecola viene indicato con un numero subscritto se è diverso da uno,
altrimenti viene omesso.
Per esempio il metano, una molecola semplice che consiste in un atomo di carbonio legato a quattro atomi di
idrogeno ha formula bruta:
CH4
Il glucosio, con sei atomi di carbonio, dodici di idrogeno e sei di ossigeno ha formula bruta:
C6H12O6
Formula di struttura
Mentre l'unica informazione trasmessa dalla formula bruta riguarda gli elementi e le loro proporzioni, una formula di
struttura fornisce anche informazioni sui tipi di legami e la disposizione spaziale degli atomi della molecola.
Gli atomi vengono rappresentati dai loro simboli ed i legami tra essi tramite tratti semplici, doppi o tripli. In funzione
dell'informazione che la formula deve trasmettere, questa può rappresentare più o meno fedelmente l'esatta geometria
della molecola.
Una formula di struttura permette di distinguere due o più isomeri, cioè due o più sostanze composte dai medesimi
elementi nelle medesime proporzioni, ma i cui atomi sono spazialmente arrangiati in modo diverso.
Un esempio di formula di struttura è rappresentato dall'etano, che consiste di due atomi di carbonio legati tramite
legame singolo fra loro ed in cui ognuno è legato a tre atomi di idrogeno. Una possibile formula di struttura può
Formula chimica
150
essere pertanto scritta come:
CH3−CH3
dove il tratto di unione (che raramente può trovarsi sostituito da due punti :) rappresenta un legame chimico
covalente semplice. Un legame covalente doppio tra due atomi viene invece rappresentato da un doppio tratto = o
raramente da una coppia di due punti :: ,come nel caso dell'etene (o etilene):
CH2=CH2
Analogamente, un triplo tratto ≡ (o raramente una tripletta di due punti :::) rappresenta un legame covalente triplo.
Un esempio è l'etino (o "Acetilene"):
CH≡CH
Gruppi funzionali multipli uguali possono essere raggruppati nel modo seguente:
(CH3)3CH
Cis-2-butene
Trans-2-butene
Le formule di struttura brevi fin qui descritte possono essere insufficienti nel caso di composti che presentano
isomeria geometrica. Un esempio è l'alchene 2-butene:
CH3CH=CHCH3
Dal momento che la rotazione attorno ad un doppio legame è impedita, i due gruppi metile (CH3) possono trovarsi
sul medesimo lato del doppio legame oppure ognuno su un lato diverso. Questo fa sì che esistono due diverse
molecole di 2-butene, aventi caratteristiche chimiche e fisiche diverse.
La distinzione dei due composti a livello di nomenclatura viene fatta utilizzando i prefissi cis- (o Z-) per quei
composti che hanno i gruppi più voluminosi sullo stesso lato del doppio legame e trans- (o E-) per i composti che
hanno i gruppi più voluminosi su lati opposti. A livello di formula, l'ambiguità viene risolta disegnando in maniera
lievemente più esplicita la geometria della molecola (struttura di Lewis).
Esempi di formule di struttura semplificate.
Cis-2-butene, trans-2-butene, cicloesanolo
di idrogeno legati agli atomi di carbonio.
Infine, in special modo per la
rappresentazione della struttura di
molecole relativamente complesse, una
formula può essere ulteriormente
semplificata sostituendo una catena di
atomi di con una linea spezzata in cui
ogni vertice rappresenta un atomo di
carbonio (un poligono nel caso di
composti ciclici) e omettendo gli atomi
Formula chimica
Formula minima
La formula minima di un composto indica gli elementi che lo costituiscono e i loro rapporti numerici minimi
(espressi come valori interi) all'interno del composto stesso.
Alcuni esempi:
• NaCl: composto formato da atomi di sodio e cloro in rapporto 1:1.
• Al2O3 : composto formato da atomi di alluminio e ossigeno in rapporto 2:3.
• NH3 : composto formato da atomi di azoto e idrogeno in rapporto 1:3.
Formula molecolare
La formula molecolare di un composto molecolare ne indica gli elementi e il numero effettivo di atomi di ciascun
elemento.
Alcuni esempi:
• NH3 : composto formato da 1 atomo di azoto e 3 atomi di idrogeno (anche formula minima).
• C6H6 : composto formato da 6 atomi di carbonio e 6 di idrogeno.
• C6H12O6 : composto formato da 6 atomi di carbonio, 12 di idrogeno e 6 di ossigeno.
La formula molecolare è ricavabile dalla formula minima a partire dal peso molecolare.
Unità di formula
L'unità di formula di un composto ionico ne indica gli elementi e il numero effettivo di atomi di ciascun elemento.
Alcuni esempi:
• NaCl (sale)
• Na2S (sale)
• Na2O2
Voci correlate
•
•
•
•
•
Formula di struttura
Formula bruta
Formula minima
Formula molecolare
Unità di formula
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/
Category:Chemistry diagrams
151
Legame covalente
152
Legame covalente
Un legame covalente polare si viene a instaurare quando avviene una sovrapposizione degli orbitali atomici di due
atomi con una differenza di elettronegatività minore o uguale a 1,9.[1] Ciò avviene per una ragione ben precisa: gli
atomi tendono al minor dispendio energetico possibile ottenibile con la stabilità della loro configurazione elettronica
(ad esempio l'ottetto). Gli orbitali atomici contenenti gli elettroni spaiati si sovrappongono, costituendo appunto,
nuovi orbitali molecolari. Le sostanze così ottenute sono dette molecole.
Un legame covalente omopolare o puro si ha quando la differenza di elettronegatività tra due elementi è minore di
0,4.
Δe<0,4
0,4<Δe<1,9
Δe>1,9
Legame Covalente Puro (il doppietto elettronico appartiene in egual misura ai due elementi coinvolti);
Legame Covalente Polare (il doppietto elettronico appartiene maggiormente all'elemento più elettronegativo);
Legame Ionico.
La molecola biatomica omonucleare dell'idrogeno
Un tipico esempio è fornito dalla combinazione di due atomi di idrogeno, che porta alla struttura covalente:
H· + ·H --> H:H
Nella molecola finale, H2, i due atomi sono tenuti assieme da una coppia di elettroni (carichi negativamente)
condivisi, i quali attirano a sé i rispettivi nuclei (carichi positivamente). Un legame covalente è quindi il risultato di
un'interazione elettrostatica che coinvolge i nuclei. Quando la nube elettronica è distribuita simmetricamente il
legame risulta non polarizzato. In questo caso si parla di legame covalente omopolare o puro.
Nel caso in cui vi sia un dipolo molecolare permanente, gli elettroni saranno maggiormente attratti dall'atomo più
elettronegativo ed il legame risulterà quindi polarizzato elettricamente determinando quindi uno sbilanciamento della
nuvola elettronica. In questo caso si parla di legame covalente eteropolare o più semplicemente polare.
Quando entrambi gli elettroni coinvolti nel legame provengono da uno solo dei due atomi, mentre l'altro fornisce un
orbitale vuoto in cui allocarli, si parla di legame dativo.
Legame monovalente e polivalente
Un legame covalente in cui viene condivisa una sola coppia di elettroni viene detto legame monovalente (legame
semplice), se vengono condivise due coppie di elettroni viene detto bivalente (doppio legame) e se le coppie
condivise sono tre, si dice legame trivalente (triplo legame). Esistono anche legami tetravalenti, ampiamente studiati
in chimica inorganica, e nel 2005 è stata dimostrata l'esistenza di legami quintupli in molecole stabili.[2] Il legame
monovalente si esprime con un trattino tra i simboli dei due atomi che vi sono coinvolti, nel caso del legame
bivalente il trattino è doppio e triplo nel caso del legame trivalente e così via.
Legame covalente
La molecola dell'acqua
La molecola dell'acqua è un legame covalente polare tra ossigeno ed idrogeno. L'elettronegatività dell'ossigeno (3,52
ca) prevale su quella dell'idrogeno (2,11 ca) attirando verso di sé gli elettroni dei due atomi H. Se è pur vero che nel
legame a idrogeno la molecola dell'acqua ha un angolo di legame di 108°3', nel legame covalente la molecola tende
ad essere disposta elettronicamente a 90°. La molecola dell'acqua diventa cosi un dipolo magnetico. Un altro
esempio è l'acido cloridrico (HCl).
Note
[1] Alberto Costanzo, Esercitazioni di chimica. Compendio teorico ed esercizi di chimica per ingegneria, 2a ed., Esculapio, 2010, p.16. ISBN
9788874883776
[2] Synthesis of a Stable Compound with Fivefold Bonding Between Two Chromium(I) Centers Tailuan Nguyen, Andrew D. Sutton, Marcin
Brynda, James C. Fettinger, Gary J. Long, and Philip P. Power Pubblicato online il 22 settembre 2005; 10.1126/Science.1116789 Support info
(http:/ / www. sciencemag. org/ cgi/ data/ 1116789/ DC1/ 1)
Bibliografia
• T. W. Graham Solomons, Chimica organica, 2a ed., Bologna, Zanichelli, 2001, pp. 6-7. ISBN 8-808-09414-6
Voci correlate
•
•
•
•
•
•
•
•
Legame chimico
Legame σ
Legame π
Legame δ
Legame di coordinazione
Legame ionico
Polarità delle molecole
Risonanza (chimica)
153
Gas
154
Gas
Un gas è un aeriforme caratterizzato da una temperatura, detta
temperatura critica, inferiore alla temperatura ambiente; gli
aeriformi per cui ciò non avviene si trovano nello stato di vapore.
Un gas può anche essere definito come un aeriforme non
condensabile a temperatura ambiente.
Inoltre, per estensione, tutti gli aeriformi che si trovano ad una
temperatura superiore a quella critica vengono detti gas: un
esempio è dato dal vapore d'acqua, caratterizzato da una
temperatura critica superiore a quella ambiente (374 °C, ovvero
647 K), viene definito come "gas d'acqua" solo quando viene
portato a superare questa temperatura (temperatura critica).
Il gas, come tutti gli aeriformi, rappresenta lo stato della materia in
Rappresentazione di un sistema gassoso secondo la
cui le forze interatomiche e intermolecolari tra le singole particelle
teoria cinetica dei gas.
di una sostanza sono così piccole che non c'è più un'effettiva
coesione tra di esse. Gli atomi o le molecole del gas sono liberi di
muoversi assumendo ciascuno una certa velocità: le particelle atomiche o molecolari del gas quindi interagiscono
urtandosi continuamente l'un l'altra. Per questo un gas non ha un volume definito ma tende ad occupare tutto lo
spazio a sua disposizione, e assume la forma del contenitore che lo contiene, riempiendolo. Un altro vincolo che può
limitare il volume di un gas è un campo gravitazionale, come nel caso dell'atmosfera terrestre.
Nel linguaggio corrente si dice che una data sostanza "è un gas" quando la sua temperatura di ebollizione è molto al
di sotto della temperatura ambiente, cioè quando si trova normalmente allo stato di gas sulla Terra. Per esempio è
normale dire che "il metano è un gas mentre il ferro non lo è", sebbene il metano possa benissimo trovarsi allo stato
liquido (raffreddato al di sotto di -161 °C, ovvero 112 K) e il ferro allo stato gassoso (riscaldato oltre i 2750 °C,
ovvero 3023 K).
Un gas può essere approssimato ad un gas ideale quando si trova ad una temperatura "molto maggiore" della sua
[1]
temperatura critica, ossia che
e convenzionalmente si intende che i due termini devono differire di
almeno un ordine di grandezza). Ciò equivale a chiedere che
.
La temperatura critica è la temperatura corrispondente al punto di massimo della curva (a forma di campana)
liquido-vapore. All'interno della campana, il fluido cambia di fase, all'esterno resta allo stato gassoso qualunque sia
la sua pressione. Imponendo che
, la curva del liquido-vapore può non essere rappresentata nel
diagramma di Andrews (diagramma pressione-volume), non è visibile se si adotta una scala normale.
Etimologia e storia del termine gas
Il termine "gas" fu coniato da un chimico fiammingo belga Jean Baptiste van Helmont nel 1630. Sembra derivi,
come spiegò Leo Meyer, dalla trascrizione della sua pronuncia della parola greca Χαος (chaos) che lui fece
diventare geist; ma Weigand e Scheler fecero risalire l'origine etimologica al tedesco gascht (fermentazione): quindi
sarebbe, secondo loro, inizialmente usata dal chimico van Helmont per indicare la fermentazione vinosa.
Tralasciando l'etimologia, sappiamo per certo che il chimico di Bruxelles van Helmont all'età di 63 anni fu il primo a
postulare l'esistenza di sostanze distinte nell'aria che così chiamò nei suoi saggi pubblicati dal figlio Mercurio van
Helmont. Pochi anni dopo l'irlandese chimico Robert Boyle enunciò che l'aria era costituita da atomi e da vuoto e
solo dopo 140 anni le affermazioni di Boyle e di van Helmont si dimostreranno vere.
Gas
155
I gas perfetti
In fisica e in termodinamica si usa generalmente l'approssimazione detta dei gas perfetti: il gas cioè viene
considerato costituito da atomi puntiformi, che si muovono liberi da forze di attrazione o repulsione fra loro e le
pareti del contenitore: questa approssimazione conduce a formulare la legge nota come equazione di stato dei gas
perfetti, che descrive, in condizioni di equilibrio termodinamico, la relazione fra pressione, volume e temperatura del
gas:
dove p è la pressione, V il volume occupato dal gas, n il numero di moli del gas, R la costante universale dei gas
perfetti e T è la temperatura. Per esempio, una mole di gas perfetto occupa 22,4 litri a temperatura di 0 °C e
pressione di 1 atmosfera.
Da questa legge discendono:
• la legge di Boyle;
• la prima legge di Gay Lussac;
• la seconda legge di Gay Lussac.
Oltre alle leggi summenzionate, per i gas perfetti vale anche la legge di Avogadro: a pari condizioni di temperatura e
pressione, se due gas occupano lo stesso volume hanno lo stesso numero di molecole.
La legge di Boyle
Per una certa massa di gas a temperatura costante, il prodotto del volume del gas V per la sua pressione p è costante.
Cioè per una certa massa di gas a temperatura costante, le pressioni sono inversamente proporzionali ai volumi. La
curva nel piano cartesiano pressione-volume che ha per equazione l'equazione sopra riportata è un'iperbole
equilatera. La legge di Boyle è una legge limite, vale cioè con buona approssimazione, non in modo assoluto per tutti
i gas. Un gas perfetto o gas ideale che segua perfettamente la legge di Boyle non esiste. Le deviazioni dei gas reali
dal comportamento del gas perfetto sono piccole per un gas che si trovi a bassa pressione e ad una temperatura
nettamente al di sopra di quella liquefazione.
Una variazione del volume e della pressione che lasci invariata la temperatura è detta trasformazione isoterma.
La prima legge di Gay Lussac
Un gas perfetto che alla temperatura di 0 °C occupa un volume V0 e che viene riscaldato mantenendo costante la
pressione occupa alla temperatura T un volume VT espresso dalla legge
in cui
è il volume occupato dal gas a 0 °C (ovvero 273,15 K) e
è pari a 1/273,15. La temperatura è espressa
in gradi Celsius. La trasformazione isobara avviene a pressione costante, mentre si ha una variazione del volume e
della temperatura. Tale trasformazione nel diagramma pressione-volume è rappresentata da un segmento parallelo
all'asse dei volumi. Quindi la variazione di volume che subisce un gas per la variazione di temperatura di ogni grado
centigrado ammonta a 1/273 del volume che il gas occupa a 0 °C.
Gas
156
La seconda legge di Gay Lussac
Dalle relazioni che intercorrono tra pressione e volume e tra temperatura e volume si ricava la relazione tra pressione
e temperatura a volume costante. Un gas perfetto che alla temperatura di 0 °C ha una pressione p0 e che viene
scaldato mantenendo costante il volume si trova, alla temperatura T, a una pressione pT espressa dalla legge:
La trasformazione isocora è una variazione della pressione e della temperatura a volume costante.
I gas reali
Un tentativo di produrre un'equazione che descriva il comportamento dei gas in modo più realistico è rappresentato
dall'equazione dei gas reali.
Le correzioni apportate all'equazione dei gas perfetti sono due: si tiene conto del volume proprio delle molecole, che
non sono quindi più considerate puntiformi, e si considerano le interazioni tra molecole che venivano trascurate nel
caso dei gas perfetti.
La prima correzione ha l'effetto di rendere non indefinitamente comprimibile il gas; il suo riscontro empirico è la
liquefazione cui vanno soggetti i gas reali se compressi (e raffreddati) a sufficienza.
L'altra correzione fa sì che i gas reali non si espandano infinitamente ma arrivino ad un punto in cui non possono
occupare più volume (questo perché tra gli atomi si stabilisce una forza molto piccola, dovuta alla variazione casuale
delle cariche elettrostatiche nelle singole molecole, chiamata Forza di van der Waals).
Per questo la legge dei gas perfetti non fornisce risultati accurati nel caso di gas reali, soprattutto in condizioni di
bassa temperatura e/o alta pressione, mentre diventa più precisa in caso di gas rarefatti, ad alta temperatura e a bassa
pressione, cioè quando forze intermolecolari e volume molecolare diventano trascurabili.
L'equazione dei gas reali si può ricostruire tenendo quindi conto del fatto che il volume a disposizione del gas sarà
(V - nb), dove b è il volume occupato da una mole di particelle e n è il numero di moli di gas considerate, e la
pressione sarà invece corretta di un fattore a/V2 che tiene conto delle forze di attrazione fra atomi. Dunque
l'equazione, detta anche equazione di Van der Waals, risulta:
Questa equazione non è valida in ogni caso, ma solo in particolari condizioni, ma è molto importante in quanto si
può identificare all'interno di essa un significato fisico. Un'equazione che invece ci da un'esatta visione dello stato
del gas reale è l'equazione del viriale (di cui si parla più specificamente alla parola Equazione di stato).
Note
[1] il segno "»" si legge "molto maggiore".
Voci correlate
•
•
•
•
•
•
•
•
Gas naturale
Biogas
Gas degenere
Vapore
Teoria cinetica dei gas
Legge di effusione dei gas
Cambiamento di stato
Gas criogeno
• Gas refrigeranti
Gas
157
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Gases
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/Gas
Soluzione (chimica)
In chimica una soluzione è un sistema omogeneo che può essere
decomposto per mezzo di metodi di separazione fisici.
Una soluzione si differenzia da una generica dispersione perché il
soluto è disperso nel solvente a livello di singole molecole o ioni,
ciascuno di essi circondato da molecole di solvente (si parla più
precisamente di solvatazione). Quando, in una soluzione, un soluto è
presente con atomi, ioni o molecole di dimensioni particolarmente
contenute (inferiori ad 1 nm), invisibili anche con l'ausilio del
microscopio, si parla di soluzione vera. Altrimenti, quando le
dimensioni delle particelle del soluto risultano comprese tra 1 e 1000
nm, si parla di soluzione falsa, o dispersione colloidale.
Nell'ambito delle soluzioni, si usa chiamare soluto (o fase dispersa) la
sostanza (o le sostanze) in quantità minore e solvente (o fase
disperdente o fase continua) la sostanza in quantità maggiore.[1]
Quando le sostanze sono in differenti stati di aggregazione (nelle
condizioni ambientali date) si definisce solvente la sostanza che
conserva il suo stato di aggregazione.
Nel caso di composti ionici, il meccanismo della dissoluzione è il
seguente: le molecole polari del solvente circondano i cristalli del sale,
e possono anche diffondere all'interno del reticolo cristallino; in questa
maniera vengono indebolite le forze di attrazione tra gli ioni di carica
opposta che costituiscono il cristallo, i quali quindi si trasferiranno nel
solvente sotto forma di ioni solvatati.
Soluzione salina preparata a partire da acqua e
cloruro di sodio (sale da cucina).
Nel caso di soluti polari, il fenomeno della dissoluzione avviene per attrazione reciproca tra le cariche opposte dei
dipoli delle molecole di soluto e solvente.
Soluzione (chimica)
158
Solubilità e saturazione
La quantità massima di soluto che può sciogliersi[2] in un dato solvente si chiama solubilità ed è funzione della
struttura chimica dei due composti e della temperatura.
La maggior parte dei composti liquidi e solidi ha una
solubilità proporzionale alla temperatura (si dice che il
sistema solvente-soluto è a solubilità diretta); le
solubilità dei gas hanno invece in genere un andamento
opposto (in questo caso si dice che il sistema
solvente-soluto è a solubilità inversa).
I valori delle solubilità delle sostanze nei diversi
solventi sono costanti e sono riportati in letteratura.
Una soluzione è detta satura quando contiene la
massima quantità di soluto che il solvente è in grado di
sciogliere a quella temperatura; aggiungendo ad una
soluzione satura ulteriore soluto, questo non si scioglie,
ma si separa dalla soluzione, precipitando (se è un
solido), formando una nuova fase (se è un liquido) o
gorgogliando (se è un gas).
Una soluzione è detta insatura quando contiene una
quantità di soluto inferiore a quella massima che il
solvente è in grado di sciogliere a quella temperatura;
aggiungendo ulteriore soluto, questo si scioglierà nella
soluzione.
Curve di solubilità per sistemi a solubilità diretta e inversa.
In condizioni particolari, è possibile ottenere soluzioni
soprasature, ovvero soluzioni che contengono più soluto di quanto il solvente sia normalmente in grado di sciogliere
a quella temperatura; tali soluzioni sono sistemi instabili che in seguito a perturbazioni meccaniche (agitazione,
scuotimento, aggiunta di corpi estranei) liberano l'eccesso di soluto trasformandosi in soluzioni sature. L'aggiunta di
pochi cristalli di soluto ad una soluzione soprasatura per provocare la separazione del soluto è detta semina, e viene
ad esempio sfruttata nell'ambito del processo industriale di cristallizzazione.
Solubilità e temperatura
La solubilità di una determinata sostanza dipende dalla temperatura. Esistono infatti le soluzioni endotermiche e le
soluzioni esotermiche. Le soluzioni endotermiche sono quelle in cui viene trasferita energia (sotto forma di calore)
dall'ambiente verso il sistema, ovvero si verifica un assorbimento di calore. Pertanto, in questo caso, la solubilità
aumenta proporzionalmente alla temperatura.
Una soluzione endotermica può essere descritta come:
soluto + solvente + calore → soluzione
Le soluzioni esotermiche, invece, sono quelle che cedono energia all'ambiente esterno. Pertanto, in questo secondo
caso, la solubilità diminuisce con l'aumento della temperatura.
Una soluzione esotermica può essere descritta come:
soluto + solvente → soluzione + calore
Soluzione (chimica)
Concentrazione
La misura della quantità di soluto rispetto alla quantità di solvente è detta concentrazione e viene misurata sia tramite
unità fisiche che tramite unità chimiche, e in particolare:
•
•
•
•
•
•
•
percentuale in volume: quantità di soluto in ml per 100 ml di soluzione
percentuale in peso: quantità di soluto in grammi per 100 g di soluzione
percentuale mista: quantità in grammi di soluto per 100 ml di soluzione
molarità (indicata con M): moli di soluto per litro di soluzione
molalità (indicata con m): moli di soluto per 1000 g di solvente
normalità (indicata con N): equivalenti di soluto per litro di soluzione
frazione molare (indicata con x): rapporto tra le moli di soluto ed il totale delle moli della soluzione
Soluzioni ideali: legge di Raoult
Una soluzione si dice "ideale" se soddisfa questa legge:
dove:
• i rappresenta il componente i-esimo della soluzione
•
•
è la pressione parziale del componente i-esimo
rappresenta la frazione molare del componente i-esimo in miscela liquida
•
è la pressione di vapore del componente i-esimo puro.
Inoltre, per la legge delle pressioni parziali:
essendo:
• p è la pressione della miscela
•
la frazione molare del componente i-esimo in miscela gassosa.
Una soluzione si avvicina a questo comportamento ideale quando i fenomeni di attrazione o repulsione tra le
molecole dello stesso componente sono della stessa entità dei fenomeni di attrazione o repulsione tra le molecole di
componenti differenti.[3]
Potenziale chimico di un componente in una soluzione ideale
Il concetto di potenziale chimico viene spesso utilizzato nell'ambito delle soluzioni. In particolare, il potenziale
chimico del componente i-esimo di una soluzione liquida che segue la legge di Raoult può essere scritto come:
in cui R è la costante dei gas, T la temperatura della soluzione e ln indica la funzione matematica chiamata logaritmo
naturale.
Questo significa che il potenziale chimico del componente i-esimo in soluzione liquida è uguale al suo potenziale
chimico all'equilibrio in fase liquida
più un termine che dipende dalla sua frazione molare e dalla temperatura
assoluta.
159
Soluzione (chimica)
160
Esempi di soluzioni
A seconda dello stato di aggregazione (solido, liquido, o gassoso) del soluto e del solvente, possiamo avere diverse
tipologie di soluzioni, riportate nella tabella seguente:
Esempi di
soluzioni
Solvente
gas
Soluto
gas
Ossigeno e altri gas in azoto
(aria).
liquido
solido
Vapore acqueo in aria.
Il naftalene sublima in aria, formando una soluzione.
liquido Anidride carbonica in acqua.
Etanolo in acqua; soluzioni di
idrocarburi (greggio).
Saccarosio in acqua; Cloruro di sodio (ovvero "sale da
cucina") in acqua; oro in amalgama con mercurio.
solido
Esano in paraffina, mercurio
in oro.
Acciaio, duralluminio, e altre leghe metalliche.
Idrogeno in dissoluzione nei
metalli (ad esempio platino).
Note
[1] La quantità di sostanza può essere definita in termini di massa, volume o numero di moli.
[2] Il termine "sciogliersi" è un termine utilizzato comunemente, che può risultare ambiguo. Nell'ambito delle soluzioni, il termine "sciogliersi"
va inteso come sinonimo di "dissolversi", e non di liquefarsi.
[3] Volendo semplificare, tanto più i due componenti della miscela "si somigliano" dal punto di vista chimico-fisico, tanto più a miscela che ne
risulta sarà ideale.
Bibliografia
• J. M. Smith; H.C.Van Ness; M. M. Abbot, Introduction to Chemical Engineering Thermodynamics , 6a ed. (in
inglese), McGraw-Hill, 2000. ISBN 0-07-240296-2
• K. G. Denbigh, I principi dell'equilibrio chimico , Milano, Casa Editrice Ambrosiana, 1971. ISBN 88-408-0099-9
Voci correlate
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Colloide
Cromatometro
Dispersione
Emulsione
Legame chimico
Miscuglio
Molecola
Molarità
Molalità
Normalità
Osmolalità
Saturazione (chimica)
Soluzione satura
Soluzione tampone
Sospensione
Soluzione (chimica)
Collegamenti esterni
• Soluzioni (http://docentiold.unina.it/docenti/web/download.php?id_prof=1825&id_insegn=9110&
id_madre=14314&id_doc=100130&action=dld)
Dispersione (chimica)
Una dispersione è un sistema (stabile o instabile) costituito da più fasi (di solito due) in cui la prevalente è detta
disperdente e le altre disperse. Caratteristica delle dispersioni è che le varie fasi sono eterogenee e che le fasi disperse
hanno dimensioni superiori alle grandezze colloidali (diametro > 1 μm).
Se la fase disperdente è liquida si possono avere: schiume quando la fase dispersa è gassosa, emulsioni quando è
liquida e sospensioni quando è solida.
Se la fase disperdente è gassosa, si parla di nebbia quando la fase dispersa è liquida e di fumo se è solida.
Voci correlate
• Colloide
•
•
•
•
Emulsione
Soluzione
Sospensione
Fumo
161
Elettrone
162
Elettrone
Elettrone
L'esperimento con il tubo di Crookes è stato il primo a dimostrare l'esistenza dell'elettrone
Composizione:
Particella elementare
Famiglia:
Fermione
Gruppo:
Leptone
Generazione:
Prima
Interazione:
gravitazionale, elettromagnetica e debole
Antiparticella:
Positrone
Teorizzata:
G. Johnstone Stoney (1874)
Scoperta:
J.J. Thomson (1897)
Simbolo:
e−, β−
Massa:
9,109 382 6(16) · 10-31 kg
[1]
5,485 899 094 5(24) · 10−4 u
1
⁄1822,888 4849(8) u
0,510 998 918(44) MeV/c2
Carica elettrica:
−1,602 176 53(14) · 10−19 C
Spin:
½
[2]
L'elettrone è una particella subatomica con carica elettrica negativa che, non essendo composta da altre particelle
conosciute, si ritiene essere una particella elementare.[3] Appartenente alla prima generazione della famiglia dei
leptoni,[4] è soggetto a interazione gravitazionale, elettromagnetica e nucleare debole.
L'elettrone possiede una massa a riposo di 9,109 382 6(16) · 10-31 kg, pari a circa 1/1836 di quella del protone. Il
momento angolare intrinseco, lo spin, è un valore semi intero in unità di ħ, che rende l'elettrone un fermione,
soggetto quindi al principio di esclusione di Pauli.[4] L'antiparticella dell'elettrone è il positrone, il quale si
differenzia solo per la carica elettrica di segno opposto. Quando queste due particelle collidono possono essere sia
diffuse che annichilite producendo fotoni, più precisamente raggi gamma.
L'idea di una quantità fondamentale di carica elettrica è stata introdotta dal filosofo Richard Laming nel 1838 per
spiegare le proprietà chimiche dell'atomo;[5] il termine elettrone è stato successivamente coniato nel 1894 dal fisico
irlandese George Johnstone Stoney, ed è stato riconosciuto come una particella da Joseph John Thomson e dal suo
gruppo di ricerca.[6][7] Successivamente il figlio George Paget Thomson ha dimostrato la duplice natura corpuscolare
e ondulatoria dell'elettrone, che è quindi descritto dalla meccanica quantistica per mezzo del dualismo
onda-particella.
Elettrone
L'elettrone, insieme a protone e neutrone, è parte della struttura degli atomi, e sebbene contribuisca per meno dello
0,06% alla massa totale dell'atomo è responsabile delle sue proprietà chimiche: la condivisione di elettroni tra due o
più atomi è la sorgente del legame chimico covalente.[8]
La maggior parte degli elettroni presenti nell'universo è stata creata durante il Big Bang, sebbene tale particella possa
essere generata tramite il decadimento beta degli isotopi radioattivi e in collisioni ad alta energia, mentre può essere
annichilata grazie alla collisione con il positrone e assorbita in un processo di nucleosintesi stellare.
In molti fenomeni fisici, in particolare nell'elettromagnetismo e nella fisica dello stato solido, l'elettrone ha un ruolo
essenziale: è responsabile della conduzione di corrente elettrica e calore, il suo moto genera il campo magnetico e la
variazione della sua energia è responsabile della produzione di fotoni. Tra le diverse applicazioni tecnologiche che
ne conseguono vi sono i circuiti elettrici, i tubi a raggi catodici, i microscopi elettronici, la radioterapia e il laser.
Storia
Il termine elettrone proviene dal termine greco ήλεκτρον (electron), il cui significato è ambra. Questo perché
storicamente l'ambra ebbe un ruolo fondamentale nella scoperta dei fenomeni elettrici. Gli antichi Greci, per
esempio, a partire dal settimo secolo a.c., erano a conoscenza del fatto che strofinando un pezzo di ambra o ebanite
con un panno di lana, questo acquisiva la capacità di attirare corpuscoli leggeri, quali granelli di polvere. Queste
evidenze vennero riprese nel 1600 da William Gilbert che individuò numerose sostanze, dal diamante allo zolfo, che
presentavano questo stesso comportamento. Egli poi diede il nome di forza elettrica alla forza che attirava i
corpuscoli e chiamò elettrizzati quei materiali che manifestavano questa proprietà.
Successivamente fu il fisico irlandese George Stoney a utilizzare per primo l'elettrone come unità fondamentale
dell'elettrochimica (nel 1874), e fu il primo a dare il nome alla particella nel 1894.
Negli ultimi anni del 1800, erano numerosi fisici a sostenere la possibilità che l'elettricità fosse costituita da unità
discrete, alle quali vennero conferiti vari nomi, ma delle quali non c'era ancora alcuna prova sperimentale
convincente. La scoperta della natura di particella subatomica dell'elettrone fu fatta nel 1897 da J. J. Thomson
all'interno del Laboratorio Cavendish dell'Università di Cambridge, mentre svolgeva esperimenti sul tubo catodico.
Nel 1860 William Crookes effettuò esperimenti con il tubo di Geissler: inserendovi due lamine metalliche e
collegandole a un generatore di corrente continua a elevato potenziale (circa 30.000 V) scoprì che si generava una
luce di colori diversi a seconda del gas utilizzato. Questa luce partiva dal catodo (polo negativo) e fluiva verso
l'anodo (polo positivo). Dopo circa trent'anni di sperimentazione questi raggi vennero chiamati raggi catodici e si
scoprirono essere formati da corpuscoli di materia capaci di muovere un mulinello posto sul loro cammino. La
velocità varia a seconda del potenziale applicato agli elettrodi, hanno scarsa penetrazione e carica negativa.
J.J. Thomson nel 1895 constatò, lavorando sui raggi catodici, che applicando un campo magnetico ed elettrico, il
rapporto tra la carica elettrica e la massa era uguale a 5,273 · 1017 e/g. Queste particelle furono chiamate elettroni.
Nel 1909 Robert Millikan calcolò la carica elettrica dell'elettrone con il famoso esperimento della goccia d'olio, che
era pari a 1,602 × 10−19 C. Fu quindi possibile calcolare la massa dell'elettrone che era di 9,109 × 10−31 kg.
163
Elettrone
Teoria atomica
Dal 1914, gli esperimenti dei fisici Ernest Rutherford, Henry Moseley,
James Franck e Gustav Hertz hanno stabilito definitivamente che
l'atomo è composto da un nucleo positivo massivo di cariche positive
circondato da una leggera massa di elettroni.[9] Nel 1913, Il fisico
danese Niels Bohr postula che gli elettroni risiedano in stati di energia
quantizzata, con l'energia determinata dal momento angolare delle
orbite degli elettroni attorno al nucleo. Gli elettroni possono muoversi
tra questi stati, o orbite, in seguito all'assorbimento o all'emissione di
un quanto di energia, un fotone di specifica frequenza. Questa teoria è
in grado di spiegare correttamente le linee di emissione spettrale
dell'idrogeno che questo forma se scaldato o attraversato da corrente
Il modello atomico di Bohr, in cui sono
elettrica. Ciò nonostante, il modello di Bohr fallisce nel predire
visualizzati gli stati energetici quantizzati. Un
l'intensità delle relative linee e nello spiegare la struttura dello spettro
elettrone che effettua una transizione tra due
di atomi più complessi.[9] I legami chimici tra gli atomi sono spiegati
orbite emette un fotone pari alla differenza di
nel 1916 da Gilbert Newton Lewis, come una interazione tra gli
energia fra i due livelli.
[10]
elettroni che li costituiscono.
Come è noto che le proprietà chimiche
degli elementi si ripetono ciclicamente in accordo con la legge periodica,[11] nel 1919 il chimico americano Irving
Langmuir suggerisce che questo può essere spiegato se gli elettroni in un atomo sono strutturati su strati. Gli elettroni
si dispongono in gruppi intorno al nucleo.[12]
Nel 1924, il fisico austriaco Wolfgang Pauli osserva che la struttura a strati di un atomo può essere spiegata da un set
di quattro parametri che definiscono univocamente lo stato quantico di un elettrone, e un singolo stato non può essere
occupato da più di un singolo elettrone (questa legge è nota come Principio di esclusione di Pauli).[13] Nonostante
ciò, sfuggiva il significato fisico del quarto parametro che può assumere solo due valori. Questo fu spiegato dai fisici
tedeschi Abraham Goudsmith e George Uhlenbeck quando suggerirono che un elettrone, oltre al momento angolare
associato alla sua orbita, possa possedere un proprio momento angolare intrinseco.[9][14] Questa proprietà è nota
come spin, e riesce a spiegare la misteriosa separazione delle linee spettrali osservate con la spettrografia ad alta
definizione.[15]
Meccanica Quantistica
Nella sua dissertazione del 1924 Recherches sur la théorie des quanta (Ricerca sulla teoria dei quanti), il fisico
francese Louis de Broglie ipotizzò che tutta la materia si comporti come un'onda in modo similare a quanto accade
per la luce e il fotone[16]. Questo significa, sotto le appropriate condizioni, che gli elettroni e il resto della materia
dovrebbero mostrare proprietà sia particellari sia in contemporanea ondulatorie. Le proprietà corpuscolari di una
particella si mostrano quando si cerca di osservarla in una precisa posizione nello spazio lungo la sua traiettoria a
qualsiasi dato istante.[17] La natura ondulatoria è osservata invece, per esempio, quando un fascio di luce passa lungo
fessure parallele creando le classiche figure di interferenza. Nel 1927, gli effetti dell'interferenza furono dimostrati
con un fascio di elettroni dal fisico inglese George Paget Thomson con una sottile pellicola di metallica e dal fisico
americano Clinton Davisson e Lester Germer usando un cristallo di nickel.[18]
Il successo delle predizioni di de Broglie favorirono la pubblicazione, di Erwin Schrödinger nel 1926, dell'equazione
di Schrödinger che descrive correttamente un'onda elettronica che si propaga.[19] Piuttosto che cercare una soluzione
che determina la posizione di un elettrone nel tempo, questa equazione può essere usata per prevedere la probabilità
di trovare un elettrone in un volume finito o infinitesimo dello spazio. Questo approccio fu chiamato
successivamente meccanica quantistica, che garantì la possibilità di ricavare teoricamente i livelli energetici di un
elettrone nell'atomo di idrogeno in buono accordo con i dati sperimentali.[20] Una volta che fu considerato lo spin e
164
Elettrone
165
l'interazione fra più elettroni, la meccanica quantistica è stata in grado di ricostruire l'andamento delle proprietà
chimiche tipiche degli elementi nella tavola periodica.[21]
Rappresentazione dell'orbitale atomico s,
caratterizzato da simmetria sferica.
L'ombreggiatura indica il valore della
distribuzione di probabilità relativa all'elettrone
nell'orbitale.
Nel 1928, basandosi sul lavoro di Wolfgang Pauli, Paul Dirac formulò
un modello dell'elettrone - l'equazione di Dirac, coerente con la teoria
della relatività ristretta, applicando considerazioni relativistiche e di
simmetria alla formulazione Hamiltoniana della meccanica quantistica
per un elettrone in un campo elettro-magnetico.[22] In modo da
risolvere i problemi della sua equazione relativistica (in primo luogo
l'esistenza di soluzioni a energia negativa), nel 1930 lo stesso Dirac
sviluppò un modello del vuoto come un mare infinito di particelle con
energia negativa, che fu poi chiamato mare di Dirac. Questo permise di
prevedere l'esistenza di positroni, la controparte dell'antimateria
dell'elettrone.[23] Questa particella fu scoperta sperimentalmente nel
1932 da Carl D. Anderson, che propose di chiamare gli elettroni
negatroni e di usare il termine elettroni per indicare genericamente una
delle varianti della particella sia a carica positiva che negativa. Questo
uso del termine negatroni è qualche volta occasionalmente utilizzato
ancora oggi, anche nella sua forma abbreviata 'negatone'.[24][25]
Nel 1947 Willis Lamb, lavorando in collaborazione con lo studente
Robert Retherford, trovò che certi stati quantistici dell'elettrone
nell'atomo di idrogeno, che avrebbero dovuto avere la stessa energia,
erano shiftate in relazione l'una dell'altra e la differenza fu chiamata Lamb shift. Circa nello stesso periodo, Polykarp
Kusch, lavorando con Henry M. Foley, scoprì che il momento magnetico dell'elettrone è di poco più grande di
quanto previsto dell'equazione di Dirac. Questa piccola differenza fu successivamente chiamata momento magnetico
di dipolo anomalo dell'elettrone. Per risolvere questo e altri problemi, una teoria migliore chiamata elettrodinamica
quantistica fu sviluppata da Sin-Itiro Tomonaga, Julian Schwinger e Richard P. Feynman alla fine degli anni
quaranta.[26]
Acceleratori di particelle
Con lo sviluppo degli acceleratori di particelle nella prima metà del XX secolo, i fisici iniziarono a sondare in
profondità nelle proprietà delle particelle subatomiche.[27] Il primo tentativo riuscito di accelerare elettroni usando
l'induzione magnetica fu fatto nel 1942 da Donald Kerst: il suo primo betatrone raggiunse energie di 2,3 MeV,
mentre i betatroni successivi raggiunsero i 300 MeV.[28] Nel 1947 fu scoperta la radiazione di sincrotrone con un
sincrotrone di 70 MeV della General Electric. Questa radiazione era causata dall'accelerazione di elettroni, che
raggiungono velocità prossime a quelle della luce, in un campo magnetico.[29]
Con un fascio di particelle di energia pari a 1,5 GeV, il primo collider ad alte energie è stato ADONE, che iniziò a
essere operativo a partire dal 1968:[30] questa struttura accelereva elettroni e positroni in direzioni opposte,
raddoppiando l'energia effettiva a disposizione rispetto a collisioni degli elettroni con un bersaglio statico.[31] Il
Large Electron-Positron Collider (LEP) al CERN, che operò dal 1989 al 2000, raggiunse energie di collisione pari a
209 GeV e fece importanti misure in merito al Modello Standard.[32][33]
L'LHC, l'ultimo acceleratore del CERN, sostituirà l'uso di elettroni con l'uso di adroni perché questi sono meno
soggetti alla perdita di energia per radiazione di sincrotrone e quindi è maggiore il rapporto fra energia acquisita
dalla particella e l'energia spesa per ottenerla.[34]
Elettrone
166
Proprietà fondamentali
La massa a riposo di un elettrone è di approssimativamente 9,109 · 10-31 kg o 5,485 · 10−4 u che, in base al principio
di equivalenza massa ed energia, corrisponde a un'energia a riposo di 0,511 MeV, con un rapporto rispetto alla massa
del protone di circa 1836. Misure astronomiche hanno mostrato che il rapporto fra le masse del protone e
dell'elettrone è rimasto costante per almeno metà dell'età dell'universo, come è previsto nel modello standard.[35]
L'elettrone ha una carica elettrica di −1,602 · 10−19 C, che è usata come unità standard per la carica delle particelle
subatomiche. Entro i limiti dell'errore sperimentale, il valore della carica dell'elettrone è uguale a quella del protone,
ma con il segno opposto.[36] Poiché il simbolo e è usato per indicare la carica elementare, il simbolo comune
dell'elettrone è e-, dove il segno meno indica la carica negativa, mentre per il positrone, che ha la stessa massa
dell'elettrone e la carica di segno opposto, è utilizzato come simbolo e+.[37]
L'elettrone ha un momento angolare intrinseco definito dal numero quantico di spin, pari a 1/2 in unità di ħ,[37] e
l'autovalore dell'operatore di spin è √3⁄2 ħ.[38] Il risultato di una misura della proiezione dello spin su ognuno degli
assi di riferimento può inoltre valere soltanto ±ħ⁄2.[39] Oltre allo spin, l'elettrone ha un momento magnetico
intrinseco, allineato al suo spin, che ha un valore approssimativamente simile al magnetone di Bohr,[40][41] che è una
costante fisica che vale 9,27400949(80) · 10−24 J/T. La proiezione del vettore di spin lungo la direzione della
quantità di moto definisce la proprietà delle particelle elementari conosciuta come elicità.[42]
L'elettrone non ha sotto strutture conosciute[3][43] e viene descritto come un punto materiale,[4] dal momento che
esperimenti effettuati con la trappola di Penning hanno mostrato che il limite superiore per il raggio della particella è
di 10−22 metri.[44] Esiste inoltre una costante fisica, il raggio classico dell'elettrone, con una valore di
2,8179 · 10−15 m; questa costante deriva tuttavia da un calcolo che trascura gli effetti quantistici presenti.[45][46]
Si ritiene che l'elettrone sia stabile poiché, dal momento che la particella possiede carica unitaria, il suo decadimento
violerebbe la legge di conservazione della carica elettrica.[47] Il limite inferiore sperimentale per la vita media
dell'elettrone è di 4,6 · 1026 anni, con un intervallo di confidenza al 90%.[48]
Proprietà quantistiche
In meccanica quantistica l'elettrone può essere trattato sia come
onda che come particella, in accordo col dualismo
onda-particella.[49] In base al principio di indeterminazione di
Heisenberg, inoltre, non è possibile conoscere simultaneamente la
sua posizione e la sua quantità di moto, e questo è alla base della
descrizione quantistica del'elettrone.
Le proprietà ondulatorie di una particella possono essere descritte
matematicamente da una funzione di variabile complessa, la
funzione d'onda, che è comunemente indicata con la lettera greca
psi (ψ), la quale rappresenta un'ampiezza di probabilità. Il
quadrato del valore assoluto della funzione d'onda rappresenta una
densità di probabilità, la probabilità che la particella sia osservata
nell'intorno di una determinata posizione.[50][51]
Funzione d'onda antisimmetrica per uno stato quantico
di due fermioni identici in una scatola bidimensionale.
Se le particelle si scambiassero la posizione la funzione
d'onda invertirebbe il suo segno.
Gli elettroni sono trattati come particelle identiche, ovvero non possono essere distinte l'una dall'altra per le loro
proprietà fisiche intrinseche: è possibile cambiare la posizione di una coppia di elettroni interagenti senza che si
verifichi un cambiamento osservabile nello stato del sistema. La funzione d'onda dei fermioni, di cui gli elettroni
fanno parte, è antisimmetrica: il segno della funzione d'onda cambia quando la posizione dei due elettroni viene
scambiata,[52] ma il valore assoluto non varia con il cambio di segno e il valore della probabilità resta immutato.
Questo differenzia i fermioni dai bosoni, che hanno una funzione d'onda simmetrica.[50]
Elettrone
167
L'evoluzione temporale della funzione d'onda di una particella è descritta dall'equazione di Schrödinger,[53] che nel
caso di un sistema di elettroni interagenti mostra una probabilità nulla che ogni una coppia di elettroni occupi lo
stesso stato quantico: questo fatto è responsabile del principio di esclusione di Pauli, il quale afferma che due
elettroni del sistema non possono avere i medesimi numeri quantici.
Tale principio è alla base di molte proprietà degli elettroni, in particolare genera la loro configurazione all'interno
degli orbitali atomici.[50]
Classificazione
Nel modello standard della fisica delle particelle
gli elettroni appartengono al gruppo delle
particelle subatomiche chiamate leptoni, che si
ritiene siano particelle elementari, ed hanno
massa minore rispetto a ogni altra particella
carica conosciuta. L'elettrone appartiene alla
prima generazione di particelle fondamentali,[54]
mentre la seconda e la terza generazione
contengono altri leptoni carichi, il muone e il
tauone, che possiedono idetica carica e spin, ma
massa a riposo maggiore. L'elettrone e tutti i
leptoni differiscono dai quark, costituenti i
protoni e i neutroni, per il fatto che non risentono
della forza di interazione nucleare forte.
Atomi e molecole
L'elettrone è alla base delle proprietà di atomi e
molecole, che costituiscono l'oggetto di studio
della fisica dello stato solido.
Il modello standard delle particelle elementari. L'elettrone è in basso a
sinistra.
Atomi
Insieme a protoni e neutroni, gli elettroni sono i costituenti fondamentali degli atomi. Confinati nella regione in
prossimità del nucleo atomico, sono solitamente in numero pari al numero atomico, il numero di protoni posseduti
dal nucleo. Gli elettroni sono situati negli orbitali atomici, e se il numero di elettroni è differente dal numero atomico
l'atomo è detto ione, e possiede una carica elettrica.
Classicamente un elettrone che si muove di moto circolare uniforme attorno al nucleo, essendo accelerato, emette
radiazione elettromagnetica per effetto Larmor, perdendo progressivamente energia e impattando sul nucleo. Questa
previsione è stata successivamente smentita dalla meccanica quantistica: nel 1913 l'introduzione del modello atomico
di Bohr ha fornito una descrizione semiclassica del modello atomico, nella quale un elettrone può muoversi soltanto
su alcune determinate orbite non-radiative caratterizzate da precisi valori dell'energia e del momento angolare.
Successivamente la meccanica quantistica ha costruito una descrizione completa dell'atomo sostituendo alla
traiettoria classica la funzione d'onda, che fornisce la probabilità di trovare un elettrone in una data posizione nello
spazio.
Attraverso la funzione d'onda è possibile descrivere completamente gli orbitali atomici: il numero e le caratteristiche
degli orbitali atomici sono deducibili dalla soluzione dell'equazione di Schrödinger per la funzione d'onda di un
elettrone confinato nel potenziale elettrico generato dal nucleo. I numeri quantici che caratterizzano gli elettroni in
Elettrone
un orbitale, che assumono un insieme discreto di valori, sono:
• Il numero quantico principale n, che definisce il livello energetico e il numero totale di nodi, considerando come
nodo anche una superficie sferica a distanza infinita dal nucleo. Può assumere valori interi non inferiori a 1.
L'energia di un elettrone nell'atomo nei semplici modelli non relativistici dipende unicamente da questo numero.
• Il numero quantico azimutale l, o numero quantico angolare, che definisce il momento angolare orbitale. Può
assumere valori interi positivi compresi tra 0 ed n-1 e sulla base di questa osservabile è possibile determinare
informazioni circa il numero di nodi non sferici e, indirettamente, sulla simmetria dell'orbitale.
• Il numero quantico magnetico ml, che definisce la componente z del momento angolare orbitale. Può assumere
valori interi compresi tra +l e -l ed è responsabile della geometria degli orbitali.
• Il numero quantico di spin ms, associato alla componente z dello spin dell'elettrone. Può assumere solo due valori,
+1/2 o -1/2 in unità di ħ.
Questa descrizione vale esattamente per l'atomo di idrogeno, mentre per gli atomi con più elettroni è necessario
effettuare delle approssimazioni a causa dell'impossibilità di risolvere esattamente l'equazione di Schrödinger per via
analitica. Le approssimazioni più utilizzate sono il metodo di Hartree-Fock, che sfrutta la possibilità di scrivere la
funzione d'onda degli elettroni come un determinante di Slater, l'accoppiamento di Russell-Saunders e
l'accoppiamento jj, che invece riescono ad approssimare l'effetto dovuto all'interazione spin-orbita nel caso di nuclei
rispettivamente leggeri[55] e pesanti.
Per il principio di esclusione di Pauli due o più elettroni non possono trovarsi nel medesimo stato, cioè non possono
essere descritti dai medesimi numeri quantici. Questo fatto determina la distribuzione elettronica negli orbitali.
Gli orbitali sono occupati dagli elettroni in modo
crescente rispetto all'energia o equivalentemente
al crescere del numero quantico principale, a
partire dall'orbitale a energia più bassa, detto
stato fondamentale, a quello di energia
maggiore. Lo stato di momento angolare è
definito dal numero quantico azimutale l,
corrispondente all'autovalore della parte angolare
dell'hamiltoniana. Il numero quantico magnetico
può assumere valori interi compresi tra -l e +l: il
numero di tali valori è il numero delle coppie di
elettroni, con valore di spin opposto, che
possiedono il medesimo numero quantico
azimutale.
La disposizione degli elettroni è quindi dovuta al
fatto che a ogni livello energetico corrisponde un
Funzione d'onda elettronica dei primi orbitali dell'atomo di idrogeno.
numero crescente di possibili valori del numero
quantico azimutale, a ogni valore del numero quantico azimutale corrispondono 2l + 1 valori di ml, e a ogni valore di
ml corrispondono i due valori possibili di spin.
Per ogni livello energetico ogni configurazione possibile è caratterizzata da un'energia, e la disposizione degli
elettroni al crescere del numero atomico si svolge al crescere di essa.
All'interno della nuvola elettronica è possibile che un elettrone effettui una transizione da un orbitale a un altro
principalmente attraverso l'emissione o l'assorbimento fotoni, i quanti di energia,[56] ma anche in seguito alla
collisione con altre particelle o tramite l'effetto Auger.[57] Quando un elettrone acquista un'energia pari alla
differenza di energia con uno stato non occupato all'interno degli orbitali, esso effettua una transizione in tale stato.
Una delle applicazioni più importanti di tale fenomeno è l'effetto fotoelettrico, in cui l'energia fornita da un fotone è
168
Elettrone
tale da separare l'elettrone dall'atomo.[58] Dal momento che l'elettrone è carico, poi, il suo moto attorno al nucleo, che
in una descrizione semiclassica è circolare uniforme, produce un momento di dipolo magnetico proporzionale al
momento angolare orbitale. Il momento magnetico totale di un atomo è equivalente alla somma vettoriale dei
momenti di dipolo magnetici e di spin di tutti i suoi elettroni e dei costituenti del nucleo. Il momento magnetico dei
costituenti del nucleo è tuttavia trascurabile rispetto a quello degli elettroni.[59] L'interazione tra il momento di dipolo
magnetico e il momento di spin è descritto dall'interazione spin-orbita, mentre l'interazione con un campo magnetico
esterno è descritta dai limiti di Paschen-Back e Zeeman, a seconda che l'interazione spin-orbita sia rispettivamente
trascurabile o meno rispetto al campo applicato.
Molecole e composti ionici
Nelle molecole gli atomi sono uniti dal legame chimico covalente, in cui uno o più elettroni sono condivisi fra due o
più atomi.[60] In una molecola gli elettroni si muovono sotto l'influenza attrattiva dei nuclei e il loro stato è descritto
da orbitali molecolari, più grandi e complessi di quelli di un atomo isolato, che in prima approssimazione si possono
ottenere attraverso la sommatoria di più orbitali degli atomi considerati singolarmente.[61] Differenti orbitali
molecolari hanno differenti distribuzioni spaziali di densità di probabilità: nel caso di una molecola costituita da due
atomi, per esempio, gli elettroni che ne formano l'eventuale legame si troveranno con maggiore probabilità in una
ristretta regione posta fra i due nuclei.[62]
Un composto ionico può essere definito come un composto chimico formato da ioni, atomi o gruppi di atomi con
carica elettrica complessiva neutra. Alla base dei composti ionici vi è il legame ionico, di natura elettrostatica, che si
forma quando le caratteristiche chimico-fisiche dei due atomi sono nettamente differenti e vi è una notevole
differenza di elettronegatività. Per convenzione si suole riconoscere un legame ionico tra due atomi quando la
differenza di elettronegatività Δχ è maggiore di 1,9. Al diminuire di tale differenza cresce il carattere covalente del
legame.
Conduttività
Se un corpo ha più o meno elettroni di quelli richiesti a bilanciare la
carica positiva dei nuclei, allora l'oggetto ha una carica elettrica netta.
Quando c'è un eccesso di elettroni, l'oggetto è detto carico
negativamente. Quando ci sono meno elettroni che protoni nei nuclei il
corpo è detto positivamente carico. Quando il numero di elettroni e il
numero di protoni sono uguali, le loro cariche si cancellano a vicenda e
l'oggetto è detto elettricamente neutro. Un corpo macroscopico può
sviluppare una carica elettrica attraverso lo sfregamento, per via
dell'effetto triboelettrico.[66]
Gli elettroni indipendenti che si muovono nel vuoto sono detti elettroni
liberi. Anche gli elettroni nei metalli hanno un comportamento simile a
quelli liberi. In realtà le particelle che sono comunemente chiamate
Un fulmine consiste principalmente in un flusso
[63]
elettroni nei metalli o in altri solidi sono delle quasi-particelle, che
di elettroni.
Il potenziale elettrico necessario
hanno la stessa carica elettrica, spin e momento magnetico dei reali
per il lampo deve essere generato dall'effetto
[64][65]
triboelettrico.
elettroni, ma che al contrario hanno differente massa.[67] Quando gli
elettroni liberi si muovono, o nel vuoto o in un metallo, generano un
flusso di carica chiamato corrente elettrica, che genera un campo magnetico, nello stesso modo in cui un campo
magnetico può generare corrente elettrica. Questo tipo di interazioni sono descritte matematicamente dalle equazioni
di Maxwell.[68]
169
Elettrone
A una data temperatura, ciascun materiale ha una conducibilità elettrica, che determina il valore della corrente
quando è applicato un potenziale elettrico. Esempi di buoni conduttori, materiali capaci di far scorrere facilmente al
proprio interno elettricità, sono i metalli come il rame e l'oro, mentre vetro e plastica sono cattivi conduttori. In
ciascun materiale dielettrico, gli elettroni rimangono confinati ai loro rispettivi nuclei e il materiale ha quindi le
caratteristiche globali di un isolante elettrico. La gran parte dei semiconduttori ha un livello variabile di conducibilità
che si trova nell'intorno fra i valori estremi di conduzione e isolante.[69] All'opposto, i metalli hanno un struttura
elettronica a bande in cui alcune di questo sono parzialmente riempite dagli elettroni. La presenza di queste bande
permette agli elettroni nei metalli di muoversi come elettroni liberi o delocalizzati. Questi elettroni non sono
associati a uno specifico atomo e quindi, quando è applicato un campo elettrico, si muovono liberamente come un
gas (chiamato gas di Fermi),[70] lungo il materiale come gli elettroni liberi nel vuoto.
A causa delle collisioni fra elettroni e atomi, la velocità di deriva degli elettroni in un conduttore è dell'ordine di
pochi millimetri per secondo. Ciò nonostante, la velocità alla quale un cambiamento di corrente in un punto del
materiale causa cambiamenti di corrente in un altro punto del materiale, la velocità di propagazione, è tipicamente di
circa il 75% della velocità della luce.[71] Questo accade perché i segnali elettrici si propagano come onde, con una
velocità dipendente dalla costante dielettrica del materiale.[72]
I metalli sono spesso relativamente buoni conduttori di calore, principalmente per il motivo che gli elettroni
delocalizzati sono liberi di trasportare energia termica fra gli atomi. Nonostante questo, al contrario della
conducibilità elettrica, la conducibilità termica è quasi indipendente dalla temperatura. Questo è espresso
matematicamente dalla legge di Wiedemann-Franz,[70] che dice che il rapporto fra la conduttività termica e la
conduttività elettrica è proporzionale alla temperatura. Il disordine termico nel reticolo cristallino del metallo causa
un aumento della resistività del materiale, producendo quindi la dipendenza dalla temperatura per la corrente
elettrica.[73]
Quando i materiali sono raffreddati al di sotto di un punto chiamato temperatura critica, questi possono avere una
transizione di fase dopo la quale perdono tutta la resistività alla corrente elettrica, in un processo noto come
superconduttività. Nella teoria BCS, questo andamento è modellato da coppie di elettroni che entrano in uno stato
quantico noto come condensato di Bose - Einstein. Queste coppie di Cooper si accoppiano nel loro moto nella
materia per mezzo delle vibrazioni di reticolo chiamate fononi, e quindi evitano le collisioni con gli atomi che
normalmente creano la resistenza elettrica.[74] (Le coppie di Cooper hanno un raggio di circa 100 nm, quindi si
possono scavalcare a vicenda.)[75] Nonostante questo, il meccanismo per il quale si formano superconduttori ad alte
temperature rimane incerto.
Gli elettroni all'interno dei solidi conduttivi, che sono a loro volta quasi-particelle, quando sono strettamente
confinati intorno a temperature vicine alle zero assoluto, si comportano globalmente come due nuove differenti
quasi-particelle: spinoni e oloni.[76][77] Il primo trasporta spin e il momento magnetico, mentre il secondo la carica
elettrica.
Particelle virtuali
Il fisico britannico Paul Dirac fu il primo a proporre nel 1930 che lo spazio vuoto può essere visto come un insieme
infinito di elettroni che occupano gli stati a energia negativa, previsti dall'equazione da lui scoperta per descrivere gli
elettroni relativistici.[78] Questi elettroni formano il mare di Dirac e hanno lo scopo di impedire alle particelle
osservate di perdere energia senza limiti. In questo contesto, i quanti della radiazione elettromagnetica, i fotoni,
possono essere assorbiti dagli elettroni del mare, permettendo a questi ultimi di uscire fuori da esso. Come effetto
netto si generano degli elettroni a carica negativa e delle lacuna di carica positiva nel mare. Una lacuna potrà essere
rioccupata dall'elettrone che perde energia in questo modo rilasciando nuovamente un altro fotone.[79]
170
Elettrone
Attualmente la teoria dei campi quantistica reinterpreta questi
fenomeni del vuoto come le fluttuazioni quantistiche generate dai
campi delle particelle e delle forze di interazione. Secondo
l'elettrodinamica quantistica nei processi di scattering gli elettroni e
il campo elettromagnetico interagiscono fra loro puntualmente con lo
scambio fotoni e particelle cariche virtuali, di breve esistenza e non
direttamente osservabili. A causa di questo tipo di interazione e delle
fluttuazioni quantistiche, nel vuoto sono create continuamente
coppie di particelle virtuali fra le quali vi sono l'elettrone e il
positrone, che si annichilano in breve tempo senza poter essere
Rappresentazione schematica della creazione di
coppie virtuali elettrone-positrone, che compaiono
misurati effettivamente.[80] In base al principio di indeterminazione
casualmente nell'intorno di un elettrone,
di Heisenberg la variazione dell'energia necessaria a produrre la
rappresentato in basso a sinistra.
coppia di particelle e la loro vita media non si possono conoscere
contemporaneamente,[81][82] tuttavia se la vita media è estremamente
breve l'incertezza riguardo all'energia è molto ampia, e il processo e la fluttuazione possono avvenire senza violare la
conservazione dell'energia.
La presenza delle particelle virtuali, sebbene non direttamente osservabile, è responsabile tuttavia delle proprietà del
vuoto, come la sua polarizzazione e permeabilità dielettrica, superiore all'unità.[83][84] Questo tipo di polarizzazione è
stata confermata sperimentalmente nel 1997 usando l'acceleratore giapponese TRISTAN.[85] Le particelle virtuali
causano inoltre una significativa differenza sulla massa effettiva dell'elettrone,[86] e la loro interazione spiega la
piccola deviazione dal momento magnetico intrinseco dell'elettrone dal magnetone di Bohr.[40][87][88] I fotoni
virtuali, responsabili del campo elettrico, permettono infatti all'elettrone di avere un moto agitato nell'intorno della
sua traiettoria classica,[89] che genera l'effetto globale di un moto circolare con una precessione. Questo moto
produce sia lo spin che il momento magnetico dell'elettrone.[4][90] Negli atomi, poi, la creazione di fotoni virtuali
spiega lo spostamento di Lamb osservato nelle linee spettrali e il fenomeno del decadimento spontaneo di elettrone
da uno stato eccitato a uno di energia inferiore.[83]
Interazione con le forze fondamentali
L'elettrone genera un campo elettrico che esercita una forza attrattiva su particelle con una carica positiva, come il
protone, e una forza repulsiva su particelle con carica negativa e l'intensità di questa forza è determinato dalla legge
di Coulomb. Quando un elettrone è in movimento genera un campo magnetico e, tramite la legge di Ampère, questo
movimento rispetto all'osservatore viene messo in relazione al campo magnetico; è questa proprietà di induzione che
fornisce il campo magnetico che permette il funzionamento del motore elettrico.[91] Il campo elettromagnetico di una
particella carica in movimento è espresso dal potenziale di Liénard–Wiechert, anche quando la velocità della
particella è prossima a quella della luce.
171
Elettrone
172
Quando un elettrone è in moto in un campo magnetico è soggetto
alla forza di Lorentz che esercita una variazione della direzione
perpendicolare al piano definito dal campo magnetico e dalla
velocità dell'elettrone e la forza centripeta che viene generata
costringe l'elettrone a seguire una traiettoria elicoidale.
L'accelerazione che deriva da questo moto curvilineo, nel caso di
velocità relativistiche, causa una radiazione di energia da parte
dell'elettrone sotto forma di radiazione di sincrotrone.[92][93][94]
L'emissione di energia causa a sua volta un rinculo dell'elettrone,
conosciuto come forza di Abraham-Lorentz-Dirac, il quale genera
un attrito che lo rallenta; questa forza è generata da una
retro-azione del campo dell'elettrone su se stesso.[95]
Una particella con carica q (a sinistra) si muove con
velocità v in un campo magnetico B che è diretto verso
l'osservatore. Per un elettrone q è negativo, perciò
segue una traiettoria diretta verso l'alto.
In elettrodinamica quantistica, l'interazione elettromagnetica tra le
particelle è trasmessa dai fotoni: un elettrone isolato che non
subisce un'accelerazione non è in grado di emettere o di assorbire un fotone reale, poiché così facendo violerebbe le
leggi di conservazione dell'energia e della quantità di moto. Invece i fotoni virtuali possono trasferire la quantità di
moto tra due particelle cariche ed è questo scambio di fotoni virtuali che genere, per esempio, la forza di
Coulomb.[96] L'emissione di energia può avvenire quando un elettrone viene deviato da una particella carica, come
per esempio un protone; l'accelerazione dell'elettrone porta all'emissione della radiazione di bremsstrahlung, detta
anche radiazione di frenamento.[97]
Una collisione anelastica tra un fotone e un elettrone libero
produce l'effetto Compton: questo urto porta a un trasferimento
dell'energia e della quantità di moto tra le particelle, che porta alla
variazione della lunghezza d'onda del fotone incidente.[98] Il valore
massimo di questa variazione della lunghezza d'onda è h/mec, che
è noto come lunghezza d'onda Compton, che per l'elettrone vale
2,43 · 10−12 m.[99] Se la lunghezza d'onda della luce incidente è
sufficientemente lunga, come per esempio quella della luce
visibile che ha una lunghezza d'onda che va da 0,4 · 10−6 a
0,7 · 10−6 m, la variazione della lunghezza d'onda dovuta
all'effetto Compton diventa trascurabile e l'interazione tra
radiazione e particelle può essere descritta tramite lo scattering
Thomson.[100]
La radiazione di bremsstrahlung è prodotta
dall'elettrone e deviato da un campo elettrico prodotto
da un nucleo atomico. La variazione di energia E2 − E1
determina la frequenza f del fotone emesso.
La forza dell'interazione elettromagnetica tra due particelle cariche
è data dalla costante di struttura fine α che è una quantità
adimensionale formata dal rapporto di due contributi energetici:
l'energia elettrostatica di attrazione o repulsione data dalla
separazione di una lunghezza d'onda Compton e dall'energia a
riposo della carica. Il suo valore è 7,297353 · 10−3 , che è possibile
approssimare con la frazione 1/137.[101]
Quando elettroni e i positroni collidono si annichilano l'un l'altro, originando due o più fotoni dei raggi gamma. Se
invece la quantità di moto dell'elettrone e del positrone è trascurabile si può formare il positronio prima che il
processo di annichilamento porti alla formazione di due o tre fotoni dei raggi gamma con un'energia totale di 1,022
MeV.[102][103] D'altra parte i fotoni molto energetici possono trasformarsi in un elettrone e in un positrone tramite un
processo chiamato produzione di coppia, ma questo avviene solo in presenza di una particella carina nelle vicinanze,
Elettrone
173
come un nucleo atomico.[104][105]
Nella teoria dell'interazione elettrodebole la componente sinistrorsa della funzione d'onda dell'elettrone forma un
doppietto di isospin debole con il neutrino elettronico, vale a dire che a causa dell'interazione elettrodebole il
neutrino si comporta come un elettrone. Ciascuna componente di questo doppietto può subire l'interazione della
corrente debole carica tramite l'emissione o l'assorbimento di un bosone W e può essere trasformata nell'altra
componente. La carica è conservata durante questo processo poiché anche il bosone W porta una carica che annulla
ogni variazione netta durante la reazione. Le interazioni della corrente debole carica sono responsabili del
decadimento beta negli atomi radioattivi. Sia l'elettrone che il neutrino possono subire l'interazione della corrente
debole neutra tramite uno scambio di bosoni Z e questo è responsabile dello scattering elastico tra elettrone e
neutrino.[106]
Moto ed energia
In base alla relatività speciale quando la velocità di una particella si avvicina a quella della luce la massa relativistica
aumenta dal punto di vista di un osservatore esterno, di conseguenza è necessaria una forza sempre più intensa per
mantenere costante l'accelerazione. In questo modo un elettrone non può mai raggiungere la velocità della luce,
essendo richiesta un'energia infinita. Nel caso di un elettrone che si muove a una velocità molto vicina a quella della
luce c nel vuoto inserito in un mezzo dielettrico, per esempio l'acqua, essendo in tal mezzo la velocità locale della
luce significativamente minore di quella dell'elettrone, l'interazione con esso può generare un fronte d'onda di luce
compatto causato dall'effetto Čerenkov. Tale effetto è simile al boom sonico, che accade quando un oggetto superare
la velocità del suono.
L'effetto della relatività speciale è basato su una quantità nota
come fattore di Lorentz, definita da:
dove v è la velocità della particella.
L'energia cinetica Ke di un elettrone che si muove con velocità v è:
dome me è la massa a riposo dell'elettrone. Per esempio,
l'acceleratore lineare di Stanford può accelerare un elettrone a
circa 51 GeV.[107] Questo fornisce un valore per γ vicino a
100 000, dal momento che la massa a riposo dell'elettrone è circa
0,51 MeV/c2. La quantità di moto relativistica è 100 000 volte la
quantità di moto dell'elettrone che la meccanica classica
prevederebbe alla stessa velocità.[108]
Il fattore di Lorentz in funzione della velocità. Partendo
dal valore 1 raggiunge l'infinito quando v si avvicina a
c.
Dal momento che l'elettrone ha anche un comportamento
ondulatorio, a una data velocità esso ha una caratteristica
lunghezza d'onda di de Broglie. Questa è data da λe = h/p dove h è la costante di Planck e p è la quantità di moto.[16]
Per energie di 51 GeV dell'elettrone, come quelle raggiunte dall'acceleratore SLAC, la lunghezza d'onda è di circa
2,4 · 10−17 m, piccola a sufficienza per esplorare la scala infinitesima del nucleo atomico e dei protoni.[109]
Elettrone
174
Formazione
La teoria del Big Bang più comunemente accettata
dagli scienziati per spiegare gli istanti iniziali
dell'evoluzione
dell'universo:[110]
nel
primo
millisecondo dell'esistenza dell'universo noto, la
temperatura era di circa un miliardo di kelvin e i fotoni
avevano un'energia media nell'ordine del milione di
elettronvolt; questi fotoni erano sufficientemente
energetici da poter reagire l'un l'altro per formare
coppie di elettroni e positroni:
dove
è il fotone,
è il positrone e
Produzione di coppia causata dalla collisione di un fotone con un
nucleo atomico.
è l'elettrone. Contemporaneamente le coppie elettrone-positrone si
annichilivano e producevano fotoni energetici. I due processi erano in equilibrio durante la prima fase di evoluzione
dell'universo, ma dopo 15 secondi la temperatura dell'universo calò sotto la soglia di formazione delle coppie di
elettroni-positroni. La maggior parte degli elettroni e positroni rimasti si annichilirono e producendo raggi gamma
che in breve tempo irradiarono l'universo.[111]
Per ragioni non ancora ben comprese, durante il processo di leptogenesi vi era un numero maggiore di elettroni
rispetto a quello dei positroni,[112] perciò circa un elettrone ogni miliardo sopravvisse durante il processo di
annichilazione. Questo eccesso era in egual misura a quello dei protoni sugli antiprotoni, in una condizione nota
come asimmetria barionica, perciò la carica netta presente nell'universo risultava nulla.[113][114] I protoni e i neutroni
superstiti iniziarono a interagire in un processo noto come nucleosintesi, durato fino a circa 5 minuti dopo l'istante
iniziale, in cui si assistette alla formazione dei nuclei degli isotopi di idrogeno, elio e in minima parte litio.[115] I
neutroni rimasti subirono il decadimento beta, con una vita media di circa quindici minuti, con la formazione di un
protone, un elettrone e un antineutrino:
dove
è il neutrone,
è il protone e
è l'antineutrino elettronico. Per i successivi 300 000-400 000 anni gli
elettroni liberi erano troppo energetici per legarsi ai nuclei atomici;[116] seguì dunque un processo di ricombinazione,
in cui gli elettroni si legarono ai nuclei atomici per formare atomi elettricamente neutri e a causa di ciò l'universo
divenne trasparente alla radiazione elettromagnetica.[117]
Osservazioni
L'osservazione remota di elettroni richiede il rilevamento delle
loro energia irradiata. Per esempio, nell'ambiente ad alta energia
come la corona di una stella, gli elettroni liberi formano un plasma
che emette energia per gli effetti di Bremsstrahlung. Il gas
elettronico può formare delle oscillazioni di plasma le cui onde
causate dalla sincronizzazione delle variazioni in densità degli
elettroni, e queste possono produrre emissioni di energia che
possono essere rilevate usando i radiotelescopi.[119]
La frequenza di un fotone è proporzionale alla sua energia. Un
elettrone confinato a muoversi attorno a un nucleo può transire fra
i diversi livelli energetici di questo consentiti, assorbendo o
L'Aurora polare è principalmente causata dagli
[118]
elettroni energetici che precipitano nell'atmosfera.
Elettrone
175
emettendo fotoni di frequenza caratteristica. Per esempio, quando un atomo è irraggiato da una sorgente con uno
spettro continuo, appariranno delle distinte linee spettrali per la radiazione trasmessa. Ciascun elemento o molecola
esibisce un insieme caratteristico proprio di serie di linee spettrali, che lo distinguono dagli altri atomi, come per
esempio il noto caso delle serie dello spettro dell'atomo di idrogeno. La spettroscopia studia l'intensità e la lunghezza
di queste linee e le mette in correlazione con le proprietà fisico-chimiche delle sostanza in analisi.[120][121]
In condizioni di laboratorio, l'interazione di elettroni individuali possono essere osservate con l'uso di rilevatori di
particelle, che permettono misure precise di specifiche proprietà come energia, spin e carica elettrica[122]. Lo
sviluppo di focalizzatori a quadrupolo ha permesso di contenere particelle in piccole regioni dello spazio per lunghi
periodi. Questo ha permesso la misura precisa delle proprietà particellari. Per esempio in una misurazione si è riusciti
a contenere un singolo elettrone per un periodo di dieci mesi.[123] Il momento magnetico di un elettrone fu misurato
con una precisione di 11 cifre significative, che, nel 1980, è la misura migliore di una costante fisica.[124]
La prima immagine video della distribuzione di energia di un elettrone è stata catturata da un team dell'università di
Lund in Svezia, nel febbraio 2008. Gli scienziati hanno usato flash estremamente piccoli di luce, che hanno
permesso di osservare il moto di un elettrone per la prima volta.[125][126]
La distribuzione di elettroni nei materiali solidi può essere visualizzata dallo spettroscopio ARPES (Angle resolved
photoemission spectroscopy, ovvero spettroscopia fotoelettrica angolarmente risolta). Questa tecnica si basa
sull'effetto fotoelettrico per misurare il reticolo reciproco, una rappresentazione matematica della struttura periodica
di un cristallo. ARPES può essere usato per determinare la direzione, la velocità e la diffusione di elettroni nel
materiale.[127]
Applicazioni
I fasci di elettroni sono usati nella saldatura di materiali,[129]
permettendo di raggiungere densità di energia superiori ai 107
W·cm−2 nello stretto diametro focale di 0,1-1,3 mm e spesso non
richiedono un materiale di riempimento. Questa tecnica di
saldatura deve essere eseguita nel vuoto, in modo tale che gli
elettroni non interagiscano con l'aria prima di raggiungere il
bersaglio e può essere usata per unire materiali conduttori che
altrimenti sarebbero difficili da saldare.[130][131]
La litografia a fasci di elettroni (EBL) è un metodo per stampare i
semiconduttori a risoluzioni più basse del micron.[132] Questa
tecnica è limitata dagli alti costi, basse performance, dalla
necessità di operare con fascio nel vuoto e dalla tendenza degli
elettroni a essere diffusi nei solidi. L'ultimo problema limita la
risoluzione a circa 10 nm. Per questa ragione, l'EBL è
principalmente usata per la produzione di un piccolo numero di
circuiti integrati specializzati.[133]
Durante un test della NASA nella galleria del vento, un
modello dello Space Shuttle è bersagliato da un fascio
di elettroni che simulano l'effetto degli ioni degli strati
alti dell'atmosfera terrestre incontrati durante il
[128]
rientro.
La lavorazione con fasci di elettroni è usata per irradiare i materiali in modo da cambiare le loro proprietà fisiche o
per la sterilizzazione medica e la produzione di cibo.[134] Nella radioterapia, i fasci di elettroni generati da
acceleratori lineari sono usati per il trattamento di tumori superficiali: dato che un fascio di elettroni può penetrare
solamente uno spessore limitato prima di essere assorbito, tipicamente intorno a 5 cm per elettroni di energia nel
range 5–20 MeV, la radioterapia è utile per il trattamento di lesioni della cute come il basalioma. Un fascio di
elettroni può essere usato per integrare il trattamento di aree che sono state irraggiate da raggi X.[135][136]
Gli acceleratori di particelle usano campi elettrici per far raggiungere agli elettroni e alle loro antiparticelle alte
energie. Nel momento in cui queste particelle passano in una regione in cui c'è campo magnetico, questi emettono
Elettrone
radiazione di sincrotrone. L'intensità di questa radiazione dipende dallo spin e questo può permettere la
polarizzazione dei fasci di elettroni in un processo noto come effetto Sokolov–Ternov.[137] La polarizzazione di fasci
di elettroni può essere molto utile per numerosi esperimenti. La radiazione di sincrotrone può anche essere usata per
raffreddare il fascio di elettroni, in modo da ridurre la quantità di moto persa dalle particelle. Una volta che le
particelle sono state accelerate sino alla energia richiesta, i fasci separati di elettroni e positroni sono portati alla
collisione e la risultante emissione di radiazione è osservata dai rivelatori di particelle ed è studiata dalla fisica
particellare.[138]
Note
[1] Tutte le masse sono valori del CODATA accessibili tramite la pagina del NIST sulla massa dell'elettrone (http:/ / physics. nist. gov/ cgi-bin/
cuu/ Results?search_for=electron+ mass). La versione frazionaria è l'inverso del valore decimale (con un'incertezza di 4,4 × 10−10)
[2] La carica dell'elettrone è il negativo della carica elementare (che è la carica positiva del protone). Valori del CODATA accessibili tramite il
NIST alla pagina carica elementare (http:/ / physics. nist. gov/ cgi-bin/ cuu/ Value?e|search_for=electron+ charge)
[3] Eichten e Peskin, op. cit., pp. 811-814.
[4] Curtis, op. cit., p. 74.
[5] Arabatzis, op. cit., pp. 70-74.
[6] Dahl, op. cit., pp. 122-185.
[7] Wilson, op. cit., p. 138.
[8] Pauling, op. cit., pp. 4-10.
[9] Smirnov, op. cit., pp. 4-10.
[10] Gilbert N. Lewis (aprile 1916). The Atom and the Molecule. Journal of the American Chemical Society 38 (4): 762–786. DOI:
10.1021/ja02261a002 (http:/ / dx. doi. org/ 10. 1021/ ja02261a002).
[11] Eric R. Scerri, The Periodic Table, Oxford University Press US, 2007, 205–226.
[12] Irving Langmuir (1919). The Arrangement of Electrons in Atoms and Molecules (http:/ / dbhs. wvusd. k12. ca. us/ webdocs/ Chem-History/
Langmuir-1919b. html). Journal of the American Chemical Society 41 (6): 868–934.
[13] Michela Massimi, Pauli's Exclusion Principle, The Origin and Validation of a Scientific Principle, Cambridge University Press, 2005.
[14] G. E. Uhlenbeck, S. Goudsmith. Ersetzung der Hypothese vom unmechanischen Zwang durch eine Forderung bezüglich des inneren
Verhaltens jedes einzelnen Elektrons (http:/ / adsabs. harvard. edu/ abs/ 1925NW. . . . . 13. . 953E). Die Naturwissenschaften 13 (47) (in
tedesco).
[15] W. Pauli (1923). Über die Gesetzmäßigkeiten des anomalen Zeemaneffektes (http:/ / adsabs. harvard. edu/ abs/ 1923ZPhy. . . 16. . 155P).
Zeitschrift für Physik 16 (1): 155–164 (in tedesco).
[16] Louis de Broglie. Nobel Lecture: The Wave Nature of the Electron (http:/ / nobelprize. org/ nobel_prizes/ physics/ laureates/ 1929/
broglie-lecture. pdf). The Nobel Foundation, 1929. URL consultato il 30 agosto 2008.
[17] Brigitte Falkenburg, Particle Metaphysics: A Critical Account of Subatomic Reality, Springer, 2007. ISBN 3-540-33731-8
[18] Clinton Davisson. Nobel Lecture: The Discovery of Electron Waves (http:/ / nobelprize. org/ nobel_prizes/ physics/ laureates/ 1937/
davisson-lecture. pdf). The Nobel Foundation, 1937. URL consultato il 30 agosto 2008.
[19] Schrödinger, Erwin (1926). Quantisierung als Eigenwertproblem. Annalen der Physik 385 (13): 437–490. DOI: 10.1002/andp.19263851302
(http:/ / dx. doi. org/ 10. 1002/ andp. 19263851302). (DE)
[20] John S. Rigden, Hydrogen, Harvard University Press, 2003, 59–86. ISBN 0-674-01252-6
[21] Bruce Cameron Reed, Quantum Mechanics, Jones & Bartlett Publishers, 2007, 275–350. ISBN 0-7637-4451-4
[22] Dirac, Paul A. M. (1928). The Quantum Theory of the Electron. Proceedings of the Royal Society of London A 117 (778): 610–624. DOI:
10.1098/rspa.1928.0023 (http:/ / dx. doi. org/ 10. 1098/ rspa. 1928. 0023).
[23] Paul A. M. Dirac. Nobel Lecture: Theory of Electrons and Positrons (http:/ / nobelprize. org/ nobel_prizes/ physics/ laureates/ 1933/
dirac-lecture. pdf). The Nobel Foundation, 1933. URL consultato il 1 novembre 2008.
[24] Helge Kragh, Quantum Generations: A History of Physics in the Twentieth Century, Princeton University Press, 2002. ISBN 0-691-09552-3
[25] Frank Gaynor, Concise Encyclopedia of Atomic Energy, The Philosophical Library, 1950.
[26] The Nobel Prize in Physics 1965 (http:/ / nobelprize. org/ nobel_prizes/ physics/ laureates/ 1965/ ). The Nobel Foundation. URL consultato il
4 novembre 2008.
[27] Wolfgang K.H. Panofsky. (EN) The Evolution of Particle Accelerators & Colliders (http:/ / www. slac. stanford. edu/ pubs/ beamline/ 27/ 1/
27-1-panofsky. pdf). slac.stanford.edu, 1997. URL consultato il 11 aprile 2010.
[28] Malcom W. Browne. (EN) Donald William Kerst Dies at 81; Built Particle Accelerators in 40's (http:/ / www. nytimes. com/ 1993/ 08/ 20/
obituaries/ donald-william-kerst-dies-at-81-built-particle-accelerators-in-40-s. html). nytimes.com, 20 agosto 1993. URL consultato il 11
aprile 2010.
[29] Elder, Gurewitsch, Langmuir e Pollock, op. cit., pp. 829-830.
[30] Hoddeson, Brown, Riordan e Dresden, op. cit., pp. 25-26.
[31] Bernardini, op. cit., pp. 156-183.
176
Elettrone
177
[32] (EN) Testing the Standard Model: The LEP experiments (http:/ / public. web. cern. ch/ PUBLIC/ en/ Research/ LEPExp-en. html).
public.web.cern.ch, 2008. URL consultato il 11 aprile 2010.
[33] LEP reaps a final harvest (http:/ / cerncourier. com/ cws/ article/ cern/ 28335). cerncourier.com, 1 dicembre 2000. URL consultato il 11
aprile 2010.
[34] (EN) Synchrotron Radiation (http:/ / asd. gsfc. nasa. gov/ Volker. Beckmann/ school/ download/ Longair_Radiation2. pdf).
asd.gsfc.nasa.gov, 2008
[35] Murphy, op. cit., pp. 1611-1613.
[36] Zorn, op. cit., pp. 2566-2576.
[37] Raith e Mulvey, op. cit., pp. 777-781.
[38] L'equazione agli autovalori per l'osservabile di spin al quadrato è:
da cui l'autovalore nel caso di spin 1/2:
Per approfondire si può fare riferimento a: Gupta, op. cit., p. 81.
[39] L'equazione agli autovalori per l'osservabile di spin nella direzione dell'asse z è
da cui l'autovalore nel caso di spin 1/2:
dove il segno ± indica i due stati possibili.
[40] Odom, op. cit., pp. 030801(1-4).
[41] Il magnetone di Bohr è definito come:
[42] Anastopoulos, op. cit., pp. 261-262.
[43] Gabrielse, op. cit., pp. 030802(1-4).
[44] Dehmelt, op. cit., pp. 102-110.
[45] Meschede, op. cit., p. 168.
[46] Il raggio classico dell'elettrone è ottenuto nel seguente modo: si assume la carica dell'elettrone distribuita uniformemente all'interno di una
sfera, che assume così un'energia potenziale elettrostaica. L'energia eguaglia l'energia a riposo dell'elettrone, definita dalla relatività ristretta
come E=mc2. In elettrostatica l'energia potenziale di una sfera con raggio r e carica e è data da:
dove ε0 è la costante dielettrica del vuoto. Per un elettrone con massa a riposo m0 l'energia a riposo è uguale a:
dove c è la velocità della luce nel vuoto. Uguagliando questi due termini e risolvendo l'equazione per r si ottiene il raggio classico
dell'elettrone. Per approfondire si può fare riferimento a: Haken, op. cit., p. 70.
[47] Steinberg, op. cit., pp. 2582-2586.
[48] Yao, op. cit., pp. 77-115.
[49] Tale risultato è mostrato attraverso l'importante esperimento della doppia fenditura, in cui si mostra la natura ondulatoria dell'elettrone, che
attraversa le due fenditure contemporaneamente causando una figura di interferenza.
[50] Munowitz, op. cit., pp. 162-218.
[51] La probabilità che la particella si trovi nell'intervallo
[52] Lo scambio di due elettroni comporta che la funzione d'onda
al tempo t è:
diventi
, dove le variabili
e
corrispondono rispettivamente alle posizioni del primo e del secondo elettrone.
[53] La scrittura generale dell'equazione di Schrödinger è:
dove
è la funzione d'onda,
è la costante di Planck razionalizzata, cioè divisa per
hamiltoniano.
[54] Frampton, op. cit., pp. 263-348.
[55] Per atomo leggero si intende un numero atomio minore di 30.
, ed
è l'operatore
Elettrone
178
[56] Mulliken, op. cit., pp. 13-24.
[57] Burhop, op. cit., pp. 2-3.
[58] Grupen, Claus (28 giugno – 10 luglio, 1999). "Physics of Particle Detection". AIP Conference Proceedings, Instrumentation in Elementary
Particle Physics, VIII, 3-34, Istanbul: Dordrecht, D. Reidel Publishing Company. DOI: 10.1063/1.1361756 (http:/ / dx. doi. org/ 10. 1063/ 1.
1361756)
[59] Jiles, op. cit., pp. 280-287.
[60] Löwdin, Brändas e Kryachko, op. cit., pp. 393-394.
[61] McQuarrie e Simon, op. cit., pp. 280-287.
[62] Daudel, op. cit., pp. 1310-1320.
[63] Rakov, Vladimir A.; Uman, Martin A., Lightning: Physics and Effects (http:/ / books. google. com/ books?id=TuMa5lAa3RAC& pg=PA4),
Cambridge University Press, 2007. ISBN 0-521-03541-4
[64] Freeman, Gordon R. (1999). Triboelectricity and some associated phenomena. Materials science and technology 15 (12): 1454–1458.
[65] Keith M. Forward, Daniel J. Lacks; R. Mohan Sankaran (2009). {{{titolo}}}. Journal of Electrostatics 67 (2-3): 178–183. DOI:
10.1016/j.elstat.2008.12.002 (http:/ / dx. doi. org/ 10. 1016/ j. elstat. 2008. 12. 002).
[66] Steven Weinberg, The Discovery of Subatomic Particles (http:/ / books. google. com/ books?id=tDpwhp2lOKMC& pg=PA15), Cambridge
University Press, 2003, 15–16. ISBN 0-521-82351-X
[67] Liang-fu Lou, Introduction to phonons and electrons (http:/ / books. google. com/ books?id=XMv-vfsoRF8C& pg=PA162), World
Scientific, 2003, 162,164. ISBN 978-981-238-461-4
[68] Bhag S. Guru; Hızıroğlu, Hüseyin R., Electromagnetic Field Theory (http:/ / books. google. com/ books?id=b2f8rCngSuAC& pg=PA138),
Cambridge University Press, 2004, 138, 276. ISBN 0-521-83016-8
[69] M. K. Achuthan; Bhat, K. N., Fundamentals of Semiconductor Devices (http:/ / books. google. com/ books?id=REQkwBF4cVoC&
pg=PA49), Tata McGraw-Hill, 2007, 49–67. ISBN 0-07-061220-X
[70] (http:/ / books. google. com/ books?id=UtEy63pjngsC& pg=PA260).
[71] Main, Peter (12 giugno 1993). When electrons go with the flow: Remove the obstacles that create electrical resistance, and you get ballistic
electrons and a quantum surprise (http:/ / www. newscientist. com/ article/ mg13818774.
500-when-electrons-go-with-the-flow-remove-the-obstacles-thatcreate-electrical-resistance-and-you-get-ballistic-electrons-and-a-quantumsurprise.
html). New Scientist 1887. URL consultato il 9 ottobre 2008.
[72] (http:/ / books. google. com/ books?id=D0PBG53PQlUC& pg=SA6-PA39).
[73] '.
[74] Staff. The Nobel Prize in Physics 1972 (http:/ / nobelprize. org/ nobel_prizes/ physics/ laureates/ 1972/ ). The Nobel Foundation, 2008. URL
consultato il 13 ottobre 2008.
[75] Kadin, Alan M. (2007). Spatial Structure of the Cooper Pair. Journal of Superconductivity and Novel Magnetism 20 (4): 285–292. DOI:
10.1007/s10948-006-0198-z (http:/ / dx. doi. org/ 10. 1007/ s10948-006-0198-z). arΧiv:cond-mat/0510279. URL consultato il 13 ottobre
2008.
[76] Discovery About Behavior Of Building Block Of Nature Could Lead To Computer Revolution (http:/ / www. sciencedaily. com/ releases/
2009/ 07/ 090730141607. htm) in ScienceDaily.com. 31 luglio 2009. URL consultato il 1 agosto 2009.
[77] Jompol, Yodchay (31 luglio 2009). Probing Spin-Charge Separation in a Tomonaga-Luttinger Liquid (http:/ / www. sciencemag. org/ cgi/
content/ abstract/ 325/ 5940/ 597). Science 325 (5940): 597–601. DOI: 10.1126/science.1171769 (http:/ / dx. doi. org/ 10. 1126/ science.
1171769). PMID 19644117. URL consultato il 1 agosto 2009.
[78] Huang Kerson, Fundamental forces of nature: the story of gauge fields, World Scientific, 2007, pp. 105-108. ISBN 981-270-645-3
[79] Felix Finster, The principle of the fermionic projector, American Mathematical Soc., 2006, pp. 16. ISBN 0-8218-3974-8
[80] Gordon Kane. Are virtual particles really constantly popping in and out of existence? Or are they merely a mathematical bookkeeping
device for quantum mechanics? (http:/ / www. scientificamerican. com/ article. cfm?id=are-virtual-particles-rea& topicID=13).
scientificamerican.com, 9 ottobre 2006. URL consultato il 9 aprile 2010.
[81] Nello specifico si ha che ΔE·Δt ≥ ħ
[82] Taylor, op. cit., p. 464.
[83] Genz, op. cit., pp. 241-243, 245-247.
[84] John Gribbin. More to electrons than meets the eye (http:/ / www. newscientist. com/ article/ mg15320662.
300-science--more-to-electrons-than-meets-the-eye. html). newscientist.com, 25 gennaio 1997
[85] Levine, op. cit., pp. 424-427.
[86] Murayama, Hitoshi (10–17 marzo 2006). "Supersymmetry Breaking Made Easy, Viable and Generic". Proceedings of the XLIInd
Rencontres de Moriond on Electroweak Interactions and Unified Theories. arΧiv:0709.3041
[87] Schwinger, op. cit., pp. 416-417.
[88] Huang, op. cit., pp. 123-125.
[89] Foldy, op. cit., pp. 29-36.
[90] Sidharth, op. cit., pp. 497-506.
[91] Crowell, op. cit., pp. 129-152.
[92] Munowitz, op. cit., p. 160.
[93] Mahadevan, Narayan e Yi, op. cit., pp. 327-337.
Elettrone
[94]
[95]
[96]
[97]
[98]
179
La radiazione proveniente da elettroni non relativistici è a volte chiamata radiazione di ciclotrone.
Rohrlich, op. cit., pp. 1109-1112.
Georgi, op. cit., p. 427.
Blumenthal, op. cit., pp. 237-270.
Il cambiamento della lunghezza d'onda Δλ dipende dall'angolo di rinculo θ dalla seguente relazione:
dove c è la velocità della luce nel vuoto e me la massa dell'elettrone.
[99] (EN) Compton wavelength λc>/sub> (http:/ / physics. nist. gov/ cgi-bin/ cuu/ Value?ecomwl). physics.nist.gov. URL consultato il 11 aprile 2010.
[100] Chen, Maksimchuk e Umstadter, op. cit., pp. 653-655.
[101] (EN) Fine-structure constant α (http:/ / physics. nist. gov/ cgi-bin/ cuu/ Value?alph). physics.nist.gov. URL consultato il 11 aprile 2010.
[102] Beringer e Montgomery, op. cit., pp. 222-224.
[103] Wilson e Buffa, op. cit., p. 888.
[104] Eichler, op. cit., p. 67-72.
[105] Hubbell, op. cit., p. 614-623.
[106] Quigg, Chris (4-30 giugno 2000). "The Electroweak Theory". TASI 2000: Flavor Physics for the Millennium, 80, Boulder: arXiv.
arΧiv:hep-ph/0204104v1
[107] (EN) Special Relativity (http:/ / www2. slac. stanford. edu/ vvc/ theory/ relativity. html). slac.stanford.edu. URL consultato il 5 aprile 2010.
[108] Risolvendo per la velocità dell'elettrone, e usando l'approssimazione di grandi γ, si ottiene:
[109] Adams, op. cit., p. 215.
[110] Paul F. Lurquin, The Origins of Life and the Universe, Columbia University Press, 2003. ISBN 0-231-12655-7
[111] Joseph Silk, The Big Bang: The Creation and Evolution of the Universe, 3ª, Macmillan, 2000, 110–112, 134–137. ISBN 0-8050-7256-X
[112] Christianto, Vic (2007). {{{titolo}}} (http:/ / www. ptep-online. com/ index_files/ 2007/ PP-11-16. PDF). Progress in Physics 4: 112–114.
[113] Kolb, Edward W. (7 aprile 1980). The Development of Baryon Asymmetry in the Early Universe. Physics Letters B 91 (2): 217–221. DOI:
10.1016/0370-2693(80)90435-9 (http:/ / dx. doi. org/ 10. 1016/ 0370-2693(80)90435-9).
[114] Eric Sather. The Mystery of Matter Asymmetry (http:/ / www. slac. stanford. edu/ pubs/ beamline/ 26/ 1/ 26-1-sather. pdf) (PDF).
1996. URL consultato il 12 aprile 2010.
[115] Scott Burles; Kenneth M. Nollett; Michael S. Turner. arΧiv:astro-ph/9903300 Big-Bang Nucleosynthesis: Linking Inner Space and Outer
Space]. arXiv, University of Chicago, 19 marzo 1999. URL consultato il 12 aprile 2010.
[116] Boesgaard, A.M. (1985). Big bang nucleosynthesis – Theories and observations (http:/ / adsabs. harvard. edu/ cgi-bin/
bib_query?1985ARA& A. . 23. . 319B). Annual review of astronomy and astrophysics 23 (2): 319–378. DOI:
10.1146/annurev.aa.23.090185.001535 (http:/ / dx. doi. org/ 10. 1146/ annurev. aa. 23. 090185. 001535).
[117] Barkana, Rennan (18 agosto 2006). The First Stars in the Universe and Cosmic Reionization (http:/ / www. sciencemag. org/ cgi/ content/
full/ 313/ 5789/ 931). Science 313 (5789): 931–934. DOI: 10.1126/science.1125644 (http:/ / dx. doi. org/ 10. 1126/ science. 1125644). PMID
16917052.
[118] Stuart Wolpert. (EN) Scientists solve 30-year-old aurora borealis mystery (http:/ / www. universityofcalifornia. edu/ news/ article/ 18277).
universityofcalifornia.edu, 24 luglio 2008. URL consultato il 12 aprile 2010.
[119] Gurnett, Donald A. (10 dicembre 1976). Electron Plasma Oscillations Associated with Type III Radio Bursts. Science 194 (4270):
1159–1162. DOI: 10.1126/science.194.4270.1159 (http:/ / dx. doi. org/ 10. 1126/ science. 194. 4270. 1159). PMID 17790910.
[120] W. C. Martin; Wiese, W. L.. Atomic Spectroscopy: A Compendium of Basic Ideas, Notation, Data, and Formulas (http:/ / physics. nist.
gov/ Pubs/ AtSpec/ ). National Institute of Standards and Technology, maggio 2007. URL consultato il 8 gennaio 2007.
[121] Grant R. Fowles, Introduction to Modern Optics, Courier Dover Publications, 1989, 227–233. ISBN 0-486-65957-7
[122] Murayama, Hitoshi (March 10–17, 2006). "Supersymmetry Breaking Made Easy, Viable and Generic". Proceedings of the XLIInd
Rencontres de Moriond on Electroweak Interactions and Unified Theories. La Thuile, Italy. arXiv: 0709.3041 (http:/ / arxiv. org/ abs/ 0709.
3041). —lists a 9% mass difference for an electron that is the size of the Planck distance.
[123] Staff. The Nobel Prize in Physics 1989 (http:/ / nobelprize. org/ nobel_prizes/ physics/ laureates/ 1989/ illpres/ ). The Nobel Foundation,
2008. URL consultato il 24 settembre 2008.
[124] Ekstrom, Philip (1980). The isolated Electron (http:/ / tf. nist. gov/ general/ pdf/ 166. pdf). Scientific American 243 (2): 91–101. URL
consultato il 24 settembre 2008.
[125] Johan Mauritsson. Electron filmed for the first time ever (http:/ / www. atto. fysik. lth. se/ video/ pressrelen. pdf) (PDF). Lunds
Universitet. URL consultato il 17 settembre 2008.
[126] Mauritsson, J. (2008). Coherent Electron Scattering Captured by an Attosecond Quantum Stroboscope (http:/ / www. atto. fysik. lth. se/
publications/ papers/ MauritssonPRL2008. pdf). Physical Review Letters 100. DOI: 10.1103/PhysRevLett.100.073003 (http:/ / dx. doi. org/
10. 1103/ PhysRevLett. 100. 073003).
Elettrone
[127] Damascelli, Andrea (2004). Probing the Electronic Structure of Complex Systems by ARPES. Physica Scripta T109: 61–74. DOI:
10.1238/Physica.Topical.109a00061 (http:/ / dx. doi. org/ 10. 1238/ Physica. Topical. 109a00061).
[128] (EN) Image #: L-1975-02972 (http:/ / grin. hq. nasa. gov/ ABSTRACTS/ GPN-2000-003012. html). grin.hq.nasa.gov. URL consultato il 12
aprile 2010.
[129] John Elmer. (EN) Standardizing the Art of Electron-Beam Welding (https:/ / www. llnl. gov/ str/ MarApr08/ elmer. html). llnl.gov, 3 marzo
2008. URL consultato il 12 aprile 2010.
[130] Schultz, op. cit., pp. 2-3.
[131] Benedict, op. cit., p. 273.
[132] Ozdemir, Faik S. (25-27 giugno 1979). " Electron beam lithography (http:/ / portal. acm. org/ citation. cfm?id=800292.
811744)". Proceedings of the 16th Conference on Design automation, 383-391, San Diego: IEEE Press. URL consultato il 12 aprile 2010.
[133] Madou, op. cit., pp. 53-54.
[134] Yves, Jongen; Arnold Herer (2-5 maggio 1996). " APS/AAPT Joint Meeting (http:/ / adsabs. harvard. edu/ abs/ 1996APS. . MAY.
H9902J)". {{{conferenza}}}, American Physical Society. URL consultato il 12 aprile 2010.
[135] Beddar, op. cit., p. 700.
[136] Michael J. Gazda; Lawrence R. Coia. Principles of Radiation Therapy (http:/ / www. cancernetwork. com/ cancer-management/ chapter02/
article/ 10165/ 1165822). cancernetwork.com, 1 giugno 2007. URL consultato il 12 aprile 2010.
[137] La polarizzazione di un fascio di elettroni significa che lo spin di tutti gli elettroni punta in una direzione. In altre parole, la proiezione dello
spin di tutti gli elettroni lungo il loro vettore della quantità di moto ha lo stesso segno.
[138] Chao, op. cit., pp. 155, 188.
Bibliografia
Testi generici
• (EN)Steve Adams, Frontiers: Twentieth Century Physics (http://books.google.it/books?id=yIsMaQblCisC&
printsec=frontcover&dq=Frontiers:+Twentieth+Century+Physics&cd=1#v=onepage&q=&f=false), CRC
Press, 2000. ISBN 0-7484-0840-1
• (EN)Charis Anastopoulos, Particle Or Wave: The Evolution of the Concept of Matter in Modern Physics,
Princeton University Press, 2008. ISBN 0-691-13512-6
• (EN)Theodore Arabatzis, Representing Electrons: A Biographical Approach to Theoretical Entities (http://books.
google.it/books?id=rZHT-chpLmAC&printsec=frontcover&dq=Representing+Electrons:+A+Biographical+
Approach+to+Theoretical+Entities&cd=1#v=onepage&q=&f=false), University of Chicago Press, 2006.
ISBN 0-226-02421-0 URL consultato il 1 aprile 2010.
• (EN)Gary F. Benedict, Nontraditional Manufacturing Processes (http://books.google.com/
books?id=xdmNVSio8jUC&pg=PA273), CRC Press, 1987. ISBN 0-8247-7352-7
• (EN)Eric H. S. Burhop, The Auger Effect and Other Radiationless Transitions, New York, 1952, 2–3.
• (EN)Alexander W. Chao; Maury Tigner, Handbook of Accelerator Physics and Engineering (http://books.
google.com/books?id=Z3J4SjftF1YC&pg=PA155), World Scientific Publishing Company, 1999. ISBN
981-02-3500-3
• (EN)Benjamin Crowell, Electricity and Magnetism (http://books.google.com/books?id=s9QWZNfnz1oC&
pg=PT129), Light and Matter, 2000. ISBN 0-9704670-4-4
• (EN)Lorenzo J. Curtis, Atomic Structure and Lifetimes: A Conceptual Approach (http://books.google.it/
books?id=KmwCsuvxClAC&printsec=frontcover&dq=Atomic+Structure+and+Lifetimes:+A+Conceptual+
Approach&cd=1#v=onepage&q=&f=false), Cambridge University Press, 2003. ISBN 0-521-53635-9 URL
consultato il 1 aprile 2010.
• (EN)Per F. Dahl, Flash of the Cathode Rays: A History of J J Thomson's Electron (http://books.google.it/
books?id=xUzaWGocMdMC&printsec=frontcover&dq=Flash+of+the+Cathode+Rays:+A+History+of+J+
J+Thomson's+Electron&cd=1#v=onepage&q=&f=false), CRC Press, 1997. ISBN 0-7503-0453-7 URL consultato
il 1 aprile 2010.
• (EN)Henning Genz, Nothingness: The Science of Empty Space, Da Capo Press, 2001. ISBN 0-7382-0610-5
180
Elettrone
• (EN)Howard Georgi, The New Physics (http://books.google.com/books?id=akb2FpZSGnMC&pg=PA427),
Cambridge University Press, 1989. ISBN 0-521-43831-4
• (EN)M. C. Gupta, Atomic and Molecular Spectroscopy, New Age Publishers, 2001. ISBN 81-224-1300-5
• (EN)Hermann Haken; Hans Christoph Wolf, W. D. Brewer, The Physics of Atoms and Quanta: Introduction to
Experiments and Theory, Springer, 2005. ISBN 3-540-20807-0
• (EN)Lillian Hoddeson; Laurie Brown, Michael Riordan, Max Dresden, The Rise of the Standard Model: Particle
Physics in the 1960s and 1970s (http://books.google.com/books?id=klLUs2XUmOkC&pg=PA25),
Cambridge University Press, 1997. ISBN 0-521-57816-7
• (EN)Kerson Huang, Fundamental Forces of Nature: The Story of Gauge Fields (http://books.google.com/
books?id=q-CIFHpHxfEC&pg=PA123), World Scientific, 2007. ISBN 981-270-645-3
• (EN)David Jiles, Introduction to Magnetism and Magnetic Materials, CRC Press, 1998, 280–287. ISBN
0-412-79860-3
• (EN)Per Olov Löwdin, Erkki Erkki Brändas, Eugene S. Kryachko, Fundamental World of Quantum Chemistry: A
Tribute to the Memory of Per-Olov Löwdin, Springer, 2003. ISBN 1-4020-1290-X
• (EN)Marc J. Madou, Fundamentals of Microfabrication: the Science of Miniaturization (http://books.google.
com/books?id=9bk3gJeQKBYC&pg=PA53), 2ª, 2002.
• (EN)Donald Allan McQuarrie, John Douglas Simon, Physical Chemistry: A Molecular Approach, University
Science Books, 1997. ISBN 0-935702-99-7
• (EN)Dieter Meschede, Optics, light and lasers: The Practical Approach to Modern Aspects of Photonics and
Laser Physics (http://books.google.com/books?id=PLISLfBLcmgC&pg=PA168), Wiley-VCH, 2004. ISBN
3-527-40364-7
• (EN)Michael Munowitz, Knowing, The Nature of Physical Law, Oxford University Press, 2005. ISBN
0-19-516737-6
• (EN)Linus C. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: an
introduction to modern structural chemistry (http://books.google.co.uk/books?id=L-1K9HmKmUUC), 3ª,
Cornell University Press, 1960. ISBN 0-8014-0333-2 URL consultato il 29 marzo 2010.
• (EN)Wilhelm Raith; Thomas Mulvey, Constituents of Matter: Atoms, Molecules, Nuclei and Particles, CRC
Press, 2001. ISBN 0-8493-1202-7
• (EN)Helmut Schultz, Electron Beam Welding (http://books.google.com/books?id=I0xMo28DwcIC&pg=PA2),
Woodhead Publishing, 1993. ISBN 1-85573-050-2
• (EN)John Taylor, The New Physics (http://books.google.com/books?id=akb2FpZSGnMC&pg=PA464),
Cambridge University Press. ISBN 0-521-43831-4
• (EN)Jerry Wilson; Anthony Buffa, College Physics, 4ª, Prentice Hall, 2000. ISBN 0-13-082444-5
• (EN)Robert Wilson, Astronomy Through the Ages: The Story of the Human Attempt to Understand the Universe
(http://books.google.it/books?id=AoiJ3hA8bQ8C&printsec=frontcover&dq=Astronomy+Through+the+
Ages:+The+Story+of+the+Human+Attempt+to+Understand+the+Universe&cd=1#v=onepage&q=&
f=false), CRC Press, 1997. ISBN 0-7484-0748-0 URL consultato il 1-4-2010.
• (EN)Boris M. Smirnov, Physics of Atoms and Ions (http://books.google.it/books?id=I1O8WYOcUscC&
printsec=frontcover&hl=it#v=onepage&q&f=false), Springer, 2003. ISBN 0-387-95550-X URL consultato il
23-1-2012.
181
Elettrone
Pubblicazioni scientifiche in inglese
• Beddar, A.S. (2001). Mobile linear accelerators for intraoperative radiation therapy (http://findarticles.com/p/
articles/mi_m0FSLis_/ai_81161386). AORN Journal 74. DOI: 10.1016/S0001-2092(06)61769-9 (http://dx.doi.
org/10.1016/S0001-2092(06)61769-9).
• Beringer, Robert (1942). The Angular Distribution of Positron Annihilation Radiation. Physical Review 61 (5-6).
DOI: 10.1103/PhysRev.61.222 (http://dx.doi.org/10.1103/PhysRev.61.222).
• Bernardini, Carlo (2004). AdA: The First Electron–Positron Collider. Physics in Perspective 6 (2). DOI:
10.1007/s00016-003-0202-y (http://dx.doi.org/10.1007/s00016-003-0202-y).
• Blumenthal, George J. (1970). Bremsstrahlung, Synchrotron Radiation, and Compton Scattering of High-Energy
Electrons Traversing Dilute Gases. Reviews of Modern Physics 42. DOI: 10.1103/RevModPhys.42.237 (http://
dx.doi.org/10.1103/RevModPhys.42.237).
• Chen, Szu-yuan, Anatoly Maksimchuk, Donald Umstadter (1998). Experimental observation of relativistic
nonlinear Thomson scattering. Nature 396. DOI: 10.1038/25303 (http://dx.doi.org/10.1038/25303).
• R. Daudel, R.F.W. Bader, M.E. Stephens, D.S. Borrett (11 ottobre 1973). The Electron Pair in Chemistry (http://
article.pubs.nrc-cnrc.gc.ca/ppv/RPViewDoc?issn=1480-3291&volume=52&issue=8&startPage=1310).
Canadian Journal of Chemistry 52. DOI: 10.1139/v74-201 (http://dx.doi.org/10.1139/v74-201). URL consultato
il 7 aprile 2010.
• Eichler, Jörg (14 novembre 2005). Electron–positron pair production in relativistic ion–atom collisions. Physics
Letters A 347 (1-3). DOI: 10.1016/j.physleta.2005.06.105 (http://dx.doi.org/10.1016/j.physleta.2005.06.
105).
• Elder, F.R., A.M. Gurewitsch, R.V. Langmuir, H.C. Pollock (1947). Radiation from Electrons in a Synchrotron.
Physical Review 71 (11). DOI: 10.1103/PhysRev.71.829.5 (http://dx.doi.org/10.1103/PhysRev.71.829.5).
• Estia J. Eichten, Michael E. Peskin (1983). New Tests for Quark and Lepton Substructure. Physical Review
Letters 50 (11): 811–814. DOI: 10.1103/PhysRevLett.50.811 (http://dx.doi.org/10.1103/PhysRevLett.50.
811).
• Paul H. Frampton (giugno 2000). Quarks and Leptons Beyond the Third Generation. Physics Reports 330. DOI:
10.1016/S0370-1573(99)00095-2 (http://dx.doi.org/10.1016/S0370-1573(99)00095-2).
• Leslie L. Foldy (1950). On the Dirac Theory of Spin 1/2 Particles and Its Non-Relativistic Limit. Physical Review
78. DOI: 10.1103/PhysRev.78.29 (http://dx.doi.org/10.1103/PhysRev.78.29).
• G. Gabrielse, D. Hanneke, T. Kinoshita, M. Nio, B. Odom (2006). New Determination of the Fine Structure
Constant from the Electron g Value and QED. Physical Review Letters 97. DOI: 10.1103/PhysRevLett.97.030802
(http://dx.doi.org/10.1103/PhysRevLett.97.030802).
• Hans Dehmelt (1988). A Single Atomic Particle Forever Floating at Rest in Free Space: New Value for Electron
Radius. Physica Scripta T22. DOI: 10.1088/0031-8949/1988/T22/016 (http://dx.doi.org/10.1088/0031-8949/
1988/T22/016).
• Hubbell, J.H. (2006). Electron positron pair production by photons: A historical overview. Radiation Physics and
Chemistry 75 (6). DOI: 10.1016/j.radphyschem.2005.10.008 (http://dx.doi.org/10.1016/j.radphyschem.2005.
10.008).
• D. Koltick, B. Howell, E. Shibata, J. Fujimoto, K. Tauchi, K. Abe, T. Abe, I. Adachi (1997). Measurement of the
Electromagnetic Coupling at Large Momentum Transfer. Physical Review Letters 78. DOI:
10.1103/PhysRevLett.78.424 (http://dx.doi.org/10.1103/PhysRevLett.78.424).
• Mahadevan, Rohan, Ramesh Narayan, Insu Yi (1996). Harmony in Electrons: Cyclotron and Synchrotron
Emission by Thermal Electrons in a Magnetic Field. Astrophysical Journal 465. DOI: 10.1086/177422 (http://dx.
doi.org/10.1086/177422). arΧiv:astro-ph/9601073v1.
182
Elettrone
• Robert S. Mulliken (1967). Spectroscopy, Molecular Orbitals, and Chemical Bonding. Science 157 (3784). DOI:
10.1126/science.157.3784.13 (http://dx.doi.org/10.1126/science.157.3784.13). PMID 5338306.
• Michael T. Murphy, V.V. Flambaum, S. Muller, C. Henkel (20 giugno 2008). Strong Limit on a Variable
Proton-to-Electron Mass Ratio from Molecules in the Distant Universe (http://www.sciencemag.org/cgi/
content/abstract/320/5883/1611). Science 320 (5883). DOI: 10.1126/science.1156352 (http://dx.doi.org/10.
1126/science.1156352). PMID 18566280. URL consultato il 5 aprile 2010.
• B. Odom, D. Hanneke (2006). New Measurement of the Electron Magnetic Moment Using a One-Electron
Quantum Cyclotron. Physical Review Letters 97. DOI: 10.1103/PhysRevLett.97.030801 (http://dx.doi.org/10.
1103/PhysRevLett.97.030801).
• Rohrlich, Fritz (1999). The self-force and radiation reaction. American Journal of Physics 68 (12). DOI:
10.1119/1.1286430 (http://dx.doi.org/10.1119/1.1286430).
• Julian Schwinger (1948). On Quantum-Electrodynamics and the Magnetic Moment of the Electron. Physical
Review 73 (4). DOI: 10.1103/PhysRev.73.416 (http://dx.doi.org/10.1103/PhysRev.73.416).
• Burra G. Sidharth (2008). Revisiting Zitterbewegung. International Journal of Theoretical Physics 48. DOI:
10.1007/s10773-008-9825-8 (http://dx.doi.org/10.1007/s10773-008-9825-8). arΧiv:0806.0985.
• R.I. Steinberg, K. Kwiatkowski, W. Maenhaut, N.S. Wall (1999). Experimental test of charge conservation and
the stability of the electron. Physical Review D 61 (2). DOI: 10.1103/PhysRevD.12.2582 (http://dx.doi.org/10.
1103/PhysRevD.12.2582).
• W.M. Yao (luglio 2006). Review of Particle Physics. Journal of Physics G: Nuclear and Particle Physics 33 (1).
DOI: 10.1088/0954-3899/33/1/001 (http://dx.doi.org/10.1088/0954-3899/33/1/001).
• Jens C. Zorn, George E. Chamberlain, Vernon W. Hughes (1963). Experimental Limits for the Electron-Proton
Charge Difference and for the Charge of the Neutron. Physical Review 129 (6). DOI: 10.1103/PhysRev.129.2566
(http://dx.doi.org/10.1103/PhysRev.129.2566).
Voci correlate
•
•
•
•
•
•
•
•
Elettrone solvatato
Fermione
Elettromagnetismo
Elettrodinamica quantistica
Protone
Scattering di elettroni
Lista delle particelle
Elettrone spaiato
183
Elettrone
184
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Electrons
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/elettrone
•
Wikiquote contiene citazioni: http://it.wikiquote.org/wiki/Elettrone
Collegamenti esterni
• Particle Data Group (http://pdg.lbl.gov/)
• Raggio di elettroni in campo elettrico e magnetico incrociato (http://www.bigs.de/Bstore/index.
php?option=com_content&view=category&layout=blog&id=46&Itemid=167)
Isotopo
Un isotopo (letteralmente nello stesso luogo) è un atomo di uno stesso elemento chimico, e quindi con lo stesso
numero atomico Z, ma con differente numero di massa A, e quindi differente massa atomica M.[1] La differenza dei
numeri di massa è dovuta ad un diverso numero di neutroni presenti nel nucleo dell'atomo a parità di numero
atomico. Stessi isotopi che differiscono solamente per lo stato eccitato vengono definiti isomeri.
Se due nuclei contengono lo stesso numero di protoni, ma un numero differente di neutroni, i due nuclei avranno lo
stesso comportamento chimico (con delle minime differenze nei tempi di reazione e nell'energia di legame,
denominate collettivamente effetti isotopici), ma avranno comportamenti fisici differenti, essendo uno più pesante
dell'altro.
Gli isotopi sono suddivisi in isotopi stabili (circa 340) e non stabili o isotopi radioattivi (circa 3000 conosciuti ed altri
6000 ipotizzati da calcoli teorici). Il concetto di stabilità non è netto, infatti esistono isotopi "quasi stabili". La loro
stabilità è dovuta al fatto che, pur essendo radioattivi, hanno un tempo di dimezzamento estremamente lungo anche
se confrontato con l'età della Terra di 4.5 Ga. Secondo teorie cosmologiche recenti nessun isotopo è da ritenersi
propriamente stabile. Ci sono 21 elementi (ad esempio berillio-9, fluoro-19, sodio-23, scandio-45, rodio-103,
iodio-127, oro-197 o torio-232, quasi-stabile) che possiedono in natura un solo isotopo stabile anche se nella
maggior parte dei casi gli elementi chimici sono costituiti da più di un isotopo con una miscela isotopica naturale,
che in molti casi è variabile in conseguenza di fenomeni idro-geologici (es: idrogeno ed ossigeno), decadimenti
radioattivi (es: piombo) e manipolazioni dovute all'uomo (es: idrogeno/deuterio/trizio e isotopi dell'uranio). Pertanto
la IUPAC aggiorna continuamente i valori delle masse atomiche medie raccomandate per i vari elementi chimici
tenendo conto di tale variabilità. Essa è ampiamente condizionata dal sito geologico di provenienza (acquifero,
terrestre, atmosferico), nonché dalla provenienza extraterrestre o molto raramente extrasolare (meteoriti). Poiché la
massa atomica media degli elementi poliisotopici è talvolta variabile, il suo valore deve essere dotato di cifre
significative in numero appropriato (ad esempio 58,933 195(5) u per il 59Co che è monoisotopico, 58,6934(2) u per
il Ni, 207,2(1) u per il Pb che è il prodotto dal decadimento delle catene radioattive naturali di 235U, 238U e 232Th).
Isotopi stabili
Tra gli isotopi stabili più studiati ci sono: l'idrogeno, il boro, il carbonio, l'azoto, l'ossigeno e lo zolfo, chiamati anche
isotopi leggeri. Di solito gli isotopi dello stesso elemento sono presenti in natura in diverse concentrazioni: uno in
alta concentrazione e l'altro, normalmente, in tracce. Per esempio in natura il carbonio si presenta come una miscela
di tre isotopi con numero di massa pari a 12, 13 e 14: 12C, 13C e 14C (questo ultimo è radioattivo ed è di origine
cosmogenica). Le loro abbondanze rispetto alla quantità globale di carbonio sono rispettivamente: 98,89%, 1,11%,
tracce (1 atomo di 14C ogni ~ 1012 atomi di 12C)[2].
Isotopo
185
Il rapporto isotopico tra due isotopi viene calcolato mettendo l'isotopo pesante al numeratore (es. R = D/H o
18 16
O/ O).A causa delle difficoltà nel gestire rapporti R con un numero così elevato di decimali (ad esempio D/H =
0,000160025) è stato deciso dal principio di evitare i valori assoluti e di usare il valore relativo del rapporto del
materiale da analizzare contro un "materiale di riferimento". Questo nuovo valore viene indicato come δ e viene
calcolato in base alla seguente formula:
La scelta di esprimere il valore moltiplicato per 1000, fa sì che si eliminino i decimali e si semplifichi così il valore
finale.
Gli standard di riferimento sono:
Elemento
Standard
Ossigeno
V-SMOW (Vienna-Standard Mean Ocean Water)
Idrogeno
V-SMOW (Vienna-Standard Mean Ocean Water)
Carbonio
PDB-1 (Pee-Dee Belemnitella)
Azoto
N2 atmosferico
Zolfo
CDT Canyon Diablo Troilite
Abbondanza isotopica relativa
Note
[1] Rolla, op. cit., p. 35
[2] WANG et al., 1998
Bibliografia
• Wang Y., Huntington T. G., Osher L. J., Wassenaar L. I., Trumbore S. E., Amundson R. G., Harden J. W., Mc
Knight D. M., Schiff S. L., Aiken G. R., Lyons W. B., Aravena R. O., Baron J. S. (1998), Carbon Cycling in
Terrestrial Environments.
• C.Kendall and J.J.McDonnell (Ed.), Isotope tracers in catchments hydrology. Elsevier, 519-576
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori, 29a ed., Dante Alighieri, 1987.
Voci correlate
•
•
•
•
•
•
•
•
•
Abbondanza isotopica
Isotono
Isobaro
Isodiafero
Numero di massa
Numero atomico
Numero neutronico
Tabella degli isotopi
Spettrometria di massa
Idrolisi
186
Idrolisi
Rientrano sotto il generico nome di idrolisi (leggasi "idròlisi" o anche "idrolìsi"[1][2], dal greco ydor",acqua, e lyo,
sciogliere) diverse reazioni chimiche in cui le molecole vengono scisse in due o più parti per effetto dell'acqua e può
talvolta essere considerata come la reazione inversa della reazione di condensazione.[3] Non è da confondersi con
l'idratazione, in cui ad una molecola viene addizionata una molecola di acqua (come nel caso dell'idratazione degli
alcheni ad alcoli).
Caratteristiche
Le reazioni di idrolisi sono tra le più varie; alcune sono spontanee, altre hanno bisogno di un catalizzatore, spesso un
acido o una base. Nei sistemi viventi le idrolisi sono spesso catalizzate da specifici enzimi della famiglia delle
idrolasi.
Un esempio di idrolisi sono le idrolisi degli esteri a dare acidi carbossilici ed alcoli; industrialmente, l'idrolisi
alcalina dei grassi animali e vegetali (saponificazione) è usata per produrre glicerina e sali di acidi carbossilici a
lunga catena, impiegati come detergenti.
CH2-O-CO-R
|
(NaOH)
CH-O-CO-R + 3 H2O ------>
|
CH2-O-CO-R
CH2-OH
|
CH-OH +
|
CH2-OH
3 R-COO-
Variazioni di pH
Con il termine idrolisi si indica anche la reazione che subiscono gli ioni derivanti da acido o base debole: questi
reagiscono con l'acqua originando in parte l'elettrolita debole di partenza e provocando una variazione del pH.
Possiamo considerare l'esempio della variazione di pH causata dall'aggiunta di un sale derivante da un acido debole e
una base forte:
CN- + H2O
HCN + OH-
che porta alla formazione di acido cianidrico (acido coniugato della base cianuro) e ioni OH-. Tale tipo di reazione è
un esempio di idrolisi basica
Consideriamo adesso la reazione di idrolisi del cloruro di ammonio, NH4Cl:
NH4+ + H2O
NH4OH + H+
In questa reazione lo ione NH4+, acido debole, reagisce ripristinando parzialmente la sua base coniugata (l'idrossido
di ammonio, NH4OH) dando idrolisi acida.
Consideriamo infine il caso di un sale binario derivato da acido e base entrambi deboli, il cianuro di ammonio,
NH4CN: in questo caso entrambi gli ioni subiscono idrolisi ma essendo la base coniugata di NH4+ più forte dell'acido
cianidrico, come si può evincere dalle relative costanti di equilibrio, prevarrà l'idrolisi basica e il pH sarà alcalino.
Idrolisi
187
Note
[1] Dizionario italiano (http:/ / www. dizionario-italiano. it/ definizione-lemma. php?definizione=idrolisi& lemma=I009DC00)
[2] Dizionario Sapere.it (http:/ / www. sapere. it/ sapere/ dizionari/ dizionari/ Italiano/ I/ ID/ idrolisi. html?q_search=idrolisi)
[3] Nella reazione di condensazione infatti due molecole reagiscono tra loro espellendo una molecola a basso peso molecolare (chiamata
"condensato"). Quando il condensato è una molecola di acqua la reazione di condensazione può dirsi inversa a quella di idrolisi.
Voci correlate
•
•
•
•
•
•
•
•
•
Reazione di neutralizzazione
Condensazione
Esteri
Ammidi
Acidi carbossilici
Alcoli
Equilibrio chimico
Teoria acido-base di Brønsted-Lowry
Idratazione (chimica)
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Hydrolysis
reactions
Collegamenti esterni
• Idrolisi salina (http://www.itchiavari.org/chimica/lab/idrolisi.html)
Attinoidi
188
Attinoidi
Numero atomico
Nome
Simbolo
89
Attinio
Ac
90
Torio
Th
91
Protoattinio Pa
92
Uranio
U
93
Nettunio
Np
94
Plutonio
Pu
95
Americio
Am
96
Curio
Cm
97
Berkelio
Bk
98
Californio
Cf
99
Einsteinio
Es
100
Fermio
Fm
101
Mendelevio Md
102
Nobelio
No
103
Laurenzio
Lr
La serie degli attinoidi (in passato attinidi) comprende i 15 elementi chimici compresi fra l'attinio e il laurenzio sulla
tavola periodica, con numeri atomici fra l'89 e il 103 inclusi. [1] [2] [3] Sono molto simili, per caratteristiche chimiche,
agli elementi della serie dei lantanoidi: gli attinoidi con il numero atomico più alto non sono reperibili in natura per
via della loro emivita molto breve: quasi tutti gli attinoidi sono fortemente radioattivi. Il plutonio, inoltre, è anche
estremamente velenoso.
Come i lantanoidi (Ln), anche gli attinoidi (An) sono spesso rappresentati al di fuori della tavola periodica, nello
stesso modo e per gli stessi motivi.
Attinoidi
189
Attinidi minori
Per attinidi minori si poi intendono
poi tutti gli attinoidi, ad esclusione di
plutonio ed uranio (denominati attinidi
maggiori), che compongono il
combustibile nucleare esaurito.
Note
[1] (EN) IUPAC, Periodic Table of the
Elements, 2003-2007 (http:/ / www. iupac.
org/ reports/ periodic_table)
Diagramma delle fasi degli attinoidi
[2] (EN) IUPAC, Periodic Table of Elements,
2007 (http:/ / www. iupac. org/ reports/
periodic_table/
IUPAC_Periodic_Table-22Jun07b. pdf)
[3] (EN) IUPAC, Red Book, 2005 (http:/ / old.
iupac. org/ publications/ books/ rbook/ Red_Book_2005. pdf)
Voci correlate
• Tavola periodica
• Metalli del blocco d
• Lantanoidi
Lantanoidi
190
Lantanoidi
Z
Nome
Simbolo
57 Lantanio
La
58 Cerio
Ce
59 Praseodimio Pr
60 Neodimio
Nd
61 Promezio
Pm
62 Samario
Sm
63 Europio
Eu
64 Gadolinio
Gd
65 Terbio
Tb
66 Disprosio
Dy
67 Olmio
Ho
68 Erbio
Er
69 Tulio
Tm
70 Itterbio
Yb
71 Lutezio
Lu
La serie dei lantanoidi (in passato lantanidi) è costituita dai 15 elementi chimici, che sulla tavola periodica si
trovano fra il lantanio ed il lutezio.[1][2] Essi hanno numero atomico compreso fra 57 e 71 (estremi inclusi).
Essi costituiscono, insieme a scandio e ittrio, le cosiddette terre rare[3].
Nei lantanoidi (spesso indicati con il simbolo Ln) gli orbitali 4f sono parzialmente o completamente riempiti, mentre
gli orbitali p e d, più esterni, restano ancora vuoti. Visto lo scarso effetto degli orbitali f sulle proprietà chimiche di
un elemento rispetto agli orbitali s, p e d, tutti i lantanoidi mostrano sostanzialmente lo stesso comportamento e le
stesse proprietà, rendendo molto difficile una loro separazione per via chimico-fisica.
D'altra parte gli orbitali f conferiscono loro una serie di proprietà magnetiche e ottiche molto interessanti: il samario
è molto usato in lega con il cobalto per fabbricare magneti permanenti, mentre l'erbio viene sfruttato come drogante
per fibre ottiche allo scopo di renderle attive, cioè capaci di amplificare il segnale luminoso che le attraversa
direttamente.
Per motivi di compattezza grafica, i lantanoidi e gli attinoidi sono in genere posti come nota sotto la tavola periodica,
in cui al loro posto compare solo un rimando.
I lantanoidi così come i metalli di transizione possono dare luogo a ioni complessi, sebbene le specie derivanti
abbiano caratteristiche completamente diverse.
Il tutto è correlabile al fatto che gli orbitali 4f risultano essere orbitali interni per cui non subiscono alterazioni in
seguito all'azione del campo dei leganti, cosa che invece accade per i complessi di metalli di transizione. Va inoltre
detto che per tutta la serie, lo stato di ossidazione caratteristico è il +3, gli altri stati (+2 e +4) sono molto più rari, la
spiegazione di ciò risiede sempre nella considerazione che gli orbitali 4f risultano essere orbitali interni per cui
difficilmente ionizzabili.
Caratteristica dei composti dei lantanidi è la luminescenza, qualità sfruttata per la costruzione di schermi televisivi
prima dell'avvento del display al plasma.
Lantanoidi
191
Note
[1] (EN) IUPAC, Periodic Table of the Elements, 2003-2007 (http:/ / www. iupac. org/ reports/ periodic_table)
[2] (EN) IUPAC, Periodic Table of the Elements, 2007 (http:/ / www. iupac. org/ reports/ periodic_table/ IUPAC_Periodic_Table-22Jun07b. pdf)
[3] (EN) IUPAC, Red Book, 2005 (http:/ / old. iupac. org/ publications/ books/ rbook/ Red_Book_2005. pdf)
Bibliografia
• N. N. Greenwood; A. Earnshaw, Chimica degli elementi, volume II, Padova, Piccin, 1992.ISBN 88-299-1121-6
Voci correlate
•
•
•
•
•
•
Tavola periodica
Metalli del blocco f
Metalli del blocco d
Terre rare
Contrazione lantanidica
Attinoidi
Stato di ossidazione
In chimica, lo stato di ossidazione (o numero di ossidazione) di un elemento chimico in un composto è definito
come il numero di elettroni ceduti o acquisiti virtualmente durante la formazione di un composto. Quando due atomi
vengono uniti da un legame, gli elettroni si considerano virtualmente acquisiti da quello a maggiore elettronegatività.
La somma algebrica dei numeri di ossidazione è 0 per una molecola neutra o coincide con la carica complessiva
totale nel caso di uno ione.
Conoscere i numeri di ossidazione degli elementi dei composti coinvolti in una reazione consente di distinguere le
reazioni di ossido-riduzione (redox) dalle normali reazioni di scambio: nelle prime i numeri di ossidazione degli
elementi cambiano, nelle seconde no.
Le reazioni di ossido-riduzione (dette anche redox) implicano un trasferimento di elettroni tra i reagenti. Tale
trasferimento può essere sfruttato per produrre una corrente elettrica continua, come nel caso della pila.
Prendendo l'esempio dell'acido solforico (H2SO4), sapendo che l'ossigeno è più elettronegativo dello zolfo e
dell'idrogeno, si ha che i numeri di ossidazione degli elementi che lo costituiscono sono
O-H
|
O=S=O
|
O-H
idrogeno: +1
zolfo: +6
ossigeno: -2
x 2 atomi: +2
x 1 atomo: +6
x 4 atomi: -8
Dall'applicazione della definizione, consegue che la somma dei numeri di ossidazione degli elementi di una molecola
neutra è zero. Nel caso di uno ione, occorre tenere conto che la somma dei numeri di ossidazione dei suoi elementi
deve essere uguale alla carica dello ione medesimo.
Stato di ossidazione
Regole empiriche per determinare il numero di ossidazione
• per gli atomi di una qualsiasi specie chimica allo stato elementare il numero di ossidazione è 0
• per gli elementi del gruppo I (metalli alcalini) nei composti il numero di ossidazione è +1
• per gli elementi del gruppo II (metalli alcalino terrosi), lo zinco (Zn) e il cadmio (Cd) nei composti, il numero di
ossidazione è +2
• nei suoi composti, l'idrogeno ha numero di ossidazione +1, negli idruri dei metalli ha numero di ossidazione -1
• nei suoi composti, l'ossigeno ha numero di ossidazione -2, (poche eccezioni: -1 nei perossidi, -1/2 nei superossidi,
nel difluoruro d'ossigeno OF2 è +2)
• il fluoro (F) ha sempre numero di ossidazione -1. Il cloro (Cl), il bromo (Br) e lo iodio (I) hanno numero di
ossidazione -1, tranne che nei composti in cui sono legati a fluoro o ossigeno, nei quali assumono numeri di
ossidazione positivi +1, +3, +5, +7
• per qualsiasi elemento allo stato di ione monoatomico il numero di ossidazione è uguale alla carica dello ione
• la somma dei numeri di ossidazione degli elementi presenti in una molecola neutra è uguale a zero;[1] in uno ione
poliatomico la somma dei numeri di ossidazione coincide con la carica dello ione.
Note
[1] Rolla, op. cit., p. 84
Bibliografia
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori, 29a ed., Dante Alighieri, 1987.
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996, pp. 98-102. ISBN 88-408-0998-8
Voci correlate
•
•
•
•
•
•
Lista degli stati di ossidazione degli elementi chimici
Legame chimico
Legame covalente
Legame ionico
Ossidoriduzione
Valenza
192
Equazione chimica
Equazione chimica
Un'equazione chimica descrive una reazione ponendo i reagenti con la loro formula molecolare a sinistra e i prodotti
a destra,[1] secondo lo schema:
aAsf + bBsf → cCsf + dDsf,
dove:
a,b,c e d sono i coefficienti stechiometrici di ciascun composto, ossia indicano il numero di molecole (o moli)
di ciascun composto, che partecipa alla reazione;
A,B,C e D sono i composti, scritti con la loro formula molecolare;
sf indica lo stato fisico del composto, che nelle equazioni chimiche sono quattro: solido (s), gassoso (g),
liquido (l) e acquoso (aq)
Leggere un'equazione chimica
Prendiamo ad esempio l'equazione:
CH4 (g) + 2 O2 (g) → CO2 (g) + 2 H2O(g),
L'equazione indica una reazione di combustione, in cui una mole di metano reagisce con due moli di ossigeno per
produrre una mole di anidride carbonica e due moli di acqua. La stechiometria gioca un ruolo estremamente
importante nelle equazioni chimiche, infatti grazie ad essa è possibile determinare la quantità di sostanza (in grammi)
prodotta a partire da una certa quantità di reagenti.
Osservando l'equazione chimica precedente si può dedurre che se si impiegano 200gr di metano, che sono circa 12,5
moli [dalla relazione Moli = Massa (in grammi)/ Massa (in unità di massa atomica)], si ottiene, per reazione con
25 moli di ossigeno (che sono 800gr), 12,5 moli di anidride carbonica (ossia 550gr) e 25 moli di acqua (cioè 450gr).
Bilanciare un'equazione chimica non redox
Dal postulato fondamentale di Lavoisier, nulla si crea, nulla si distrugge, tutto si trasforma, deriva necessariamente
che la somma delle masse dei reagenti è forzatamente uguale alla somma delle masse dei prodotti. Per questo
un'equazione come:
KMnO4 + Ca3(PO4)2 → K3PO4 + Ca(MnO4)2
deve essere bilanciata. Si può capire da alcune evidenti differenze tra i reagenti e i prodotti, ad esempio a sinistra il
potassio appare una volta mentre a destra tre. Bilanciare una reazione significa porre un nuovo coefficiente
stechiometrico al composto. Non è possibile cambiare in alcun modo i composti partecipanti alla reazione, ad
esempio alterando il numero di atomi di un elemento all'interno della molecola. Questo si traduce nel fatto che
mettendo un nuovo coefficiente ad un composto per bilanciare un elemento potrebbe significare dovere in seguito
bilanciare l'altro elemento del composto.
Per bilanciare l'equazione è buona norma iniziare con il bilanciare il metallo, in questo caso il potassio e il calcio:
3KMnO4 + Ca3(PO4)2 → K3PO4 + 3Ca(MnO4)2
Adesso è necessario bilanciare il nonmetallo, ossia il manganese e il fosforo ma non l'ossigeno (generalmente, a
meno che non appaia nella sua forma elementare, l'ossigeno non viene bilanciato per verificare al termine del
bilanciamento se la procedura seguita è corretta):
6KMnO4 + Ca3(PO4)2 → 2K3PO4 + 3Ca(MnO4)2
193
Equazione chimica
Da notare che bilanciando il fosforo è stato necessario cambiare di nuovo il coefficiente del potassio nel primo
composto, permettendo quindi il bilanciamento del manganese. L'ossigeno è a sua volta stato bilanciato. In definitiva
bilanciare una reazione chimica significa:
• bilanciare la carica elettrica (se la reazione è scritta in forma molecolare la carica è automaticamente bilanciata)
• bilanciare la massa in accordo con la legge di Lavoiser
• bilanciare le quantità di ogni singolo elemento che partecipa alla reazione.
Note
[1] Rolla, op. cit., p. 13
Bibliografia
• Luigi Rolla, Chimica e mineralogia. Per le Scuole superiori, 29a ed., Dante Alighieri, 1987.
Prodotto (chimica)
I prodotti di una reazione chimica sono i composti che si ottengono alla fine della reazione, dopo la ricombinazione
degli atomi dei reagenti.
Una reazione chimica è composta da due termini: nel termine di sinistra stanno i reagenti, mentre nel termine di
destra stanno i prodotti.
Per la legge di Lavoisier, la massa dei prodotti è uguale alla massa dei reagenti.
Se il prodotto della reazione viene subito dopo la sua creazione utilizzato da un'altra reazione come reagente, si parla
più propriamente di intermedio di reazione.
I prodotti dell'industria chimica vengono classificati in base al loro volume di produzione e al loro valore aggiunto a
partire dalla matrice di Kline.
Voci correlate
•
•
•
•
Velocità di reazione
Catalizzatore
Reazione chimica
Reagente
194
Fonti e autori delle voci
Fonti e autori delle voci
Chimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=46820547 Autori:: .jhc., 5Y, Albris, Alexxxx, Alpha30, Andrea.gf, AnjaManix, Antony93, Aphaia, Apollodoro, Aracuano, Archenzo,
Ares, Assinda, AttoRenato, Aushulz, Avesan, Azattoni, Baronnet, Basilicofresco, Berto, Biopresto, Blakwolf, Bouncey2k, Buggia, Capannelle, Casamadella, ChemicalBit, Cisco79, Civvì,
Contezero, Darth Kule, Demart81, Dobby, Dr Zimbu, Estel, Fiir, Flippo, FrancescoCas, Frazzone, Gabriele85, Gac, Giannib, Giuliorn71, Giuse93, Guidomac, Hellis, Henrykus, Ignlig, Jalo,
Julio.caesar, Kikka2571, Klaudio, Leonard Vertighel, Lucka997, Luisa, M7, MM, Melkor II, Micvac, Nick1915, No2, Numbo3, Osk, Ottaviano II, Paginazero, Pandit, Pantalaimon, Patafisik,
Phantomas, Ploqperq, Rael, Ramac, Restu20, Riccardo.fabris, Rob-ot, Sannita, Santuzzo, Sbisolo, Scusatesonoio, Seics, Sesekem, Shivanarayana, Simo ubuntu, Sky, Smark, Snowdog, Stan
Shebs, Suisui, SuperSecret, Svante, Taueres, Template namespace initialisation script, Ticket 2010081310004741, Twice25, Vituzzu, Von Zurich, Ylebru, Yujin, 216 Modifiche anonime
Storia della chimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=45834110 Autori:: Alfreddo, Andrea.gf, Apollodoro, Aushulz, Avesan, Biopresto, Bronaldo, Buggia, Burubuz, Cisco79,
CommonsDelinker, Dadda86, Dinwath, Doc.mari, Felyx, Gabriele85, Jacopo, Jacopo Werther, Joder, Joe123, Luisa, Marcol-it, Phantomas, Powt, Riccardov, Rob-ot, Senpai, 41 Modifiche
anonime
Legame chimico Fonte:: http://it.wikipedia.org/w/index.php?oldid=45978679 Autori:: AXRL, Alfio, Ariel, Ask21, Aushulz, Avesan, Biopresto, Blakwolf, Buggia, Bumba, Calabash,
ChemicalBit, Ciao come stai, Cisco79, Danilo, Dr Zimbu, Ernesttico, Fir, Flippo, Frazzone, Frieda, Frigotoni, Ghazi85, Giancarlodessi, Ginosal, Giovannigobbin, Jalo, Kibira, Larry Yuma,
Loroli, Lukino24, M7, Marco 27, Marcookie, Mark91, MaxDel, Metralla, Number 21, Osk, Paginazero, Pazzoide87, Phantomas, Rael, Sansa, SignorX, Tia solzago, To011, Trolley 77,
Unstoppable87, Valerio79, Ylebru, Zardos, ^musaz, 177 Modifiche anonime
Atomo Fonte:: http://it.wikipedia.org/w/index.php?oldid=46427181 Autori:: .jhc., 27182, 5Y, Aerandir09, Alec, Alexxxx, Alfio, Amarvudol, Anassagora, Andreapowoso, AnjaManix,
Aracuano, Aushulz, Bag, Barbaking, Beltd, Berto, Biopresto, Bizio, Blakwolf, Bluemask, Brownout, Buggia, Calabash, CavalloRazzo, ChemicalBit, Cisco79, Clop, Codas, Cruccone, Danilo,
DanyUP, Dark, DarkAp, Davide, Davide l'Armaro, Dome, Dr Zimbu, Elph, Elwood, FaLcON283, Felyx, Filnik, Franco3450, Franz Liszt, Frazzone, Frieda, Frigotoni, Gac, Gacio, Gggg81,
Giancarlo Rossi, Gianluigi, Gio97, Giovannigobbin, Grazianoleni, Guidomac, Hashar, Henrykus, Ignlig, Incola, Ippatsu, Istcol, Jackox, Joana, K92, Kaibou, Kal-El, Kiado, Kibira, L'alchimista,
L736E, Larry Yuma, Leonard Vertighel, Lorenz-pictures, Loshack, Lp, Lucus, M7, Maitland, MapiVanPelt, Marc Lagrange, Mark91, Matteo pregnolato, Mau db, Mauro742, Melkor II, Midnight
bird, MikyT, Nickotte, No2, Noiseplayer, Nubifer, Olando, Omino di carta, Osk, Otello Felarinnonz, Paginazero, Painlord2k, Pasquale.Carelli, Pequod76, Phantomas, Pietrodn, Pipep,
Pracchia-78, Rdocb, Remulazz, Renato Caniatti, Restu20, Retaggio, Ripepette, Rl89, Roberto Mura, Roberto1974, Rojelio, Ruthven, Salvatore Ingala, Sannita, Senet, Senpai, Shivanarayana,
Simone, Sirabder87, Snowdog, Socho-sama, Stefany, Sublime Negativo, Suisui, SuperSecret, Supernino, Svante, T137, Taueres, Template namespace initialisation script, Tenan, Ticket
2010081310004741, Tirinto, Tompase, Twice25, Vipera, Vituzzu, Webkid, Yattaman, Ylebru, 391 Modifiche anonime
Molecola Fonte:: http://it.wikipedia.org/w/index.php?oldid=46662241 Autori:: AKappa, Albris, Andrea Sivieri, Archenzo, Ask21, Aushulz, Barbaking, Baroc, Biopresto, Bramfab, Buggia,
Cesalpino, Cisco79, Danilo, Davide, Eumolpo, Franz Liszt, Gac, Globoken, Hashar, Hellis, Johnlong, Kal-El, L736E, Mark91, Massimo Macconi, Moroboshi, No2, Osk, Paginazero, Phantomas,
Retaggio, Sbisolo, Simo82, Swert, Template namespace initialisation script, Ticket 2010081310004741, Tommaso Ferrara, WK, Witesmoke, Ylebru, ^musaz, 61 Modifiche anonime
Stato della materia Fonte:: http://it.wikipedia.org/w/index.php?oldid=45517907 Autori:: ARTE, Al3xI98O, Aushulz, Berto, Biopresto, Blakwolf, Buggia, Capitanonemo, Caulfield,
ChemicalBit, Cisco79, DanGarb, Eio, ElfQrin, Filippo2192, FrAnCiS, Frigotoni, IlPisano, Kibira, Madaki, Manusha, Marcuscalabresus, Martin Mystère, Ottaviano II, Owlbuster, Phantomas,
Poldo328, Seics, Semolo75, Snowdog, Ssspera, The Guru, Ticket 2010081310004741, Truman Burbank, Ylebru, 39 Modifiche anonime
Miscela (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=46798575 Autori:: Ariel, Aushulz, Biopresto, Buggia, Bultro, ChemicalBit, Cisco79, Comune mortale, Davide, Doc.mari,
Francisco83pv, Johnlong, Lorydec, Marek96, Phantomas, Seics, Taueres, Xandi, 34 Modifiche anonime
Composto chimico Fonte:: http://it.wikipedia.org/w/index.php?oldid=44929844 Autori:: Alfio, Archenzo, Aushulz, Ayato86, Azrael555, Biopresto, Blakwolf, Buggia, Cisco79, Danilo, Fra74,
Frieda, Gabriele85, Gac, Gio97, Hashar, Incola, Johnlong, Mafejthoth, Osk, Paginazero, Pietrodn, Remo Mori, SamZane, Sandrobt, SupremoAssoluto, Svante, Taueres, Template namespace
initialisation script, Tommaso Ferrara, 13 Modifiche anonime
Reazione chimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=46548972 Autori:: 5Y, Actam, AlessandroAM, Aushulz, Avesan, Biopresto, Buggia, Cerrigno, ChemicalBit, Cisco79,
Danilo, Dr Zimbu, Eio, Felyx, Floranda, Frieda, Ghazi85, Giovannigobbin, Giuliorn71, Gori.silvio, Guidomac, Ignlig, Kaeso, Lependu, LukeWiller, Mafejthoth, Malvivent, MapiVanPelt,
Mark91, Matgio, Miao, Numbo3, Paginazero, Phantomas, Ppalli, Restu20, Retaggio, Shivanarayana, Tia solzago, Ticket 2010081310004741, Tiziana veiga, Unriccio, ViciDig, Whiles, 84
Modifiche anonime
Processo chimico Fonte:: http://it.wikipedia.org/w/index.php?oldid=45167661 Autori:: Aerandir09, Aushulz, Buggia, Taueres, 5 Modifiche anonime
Ossido-riduzione Fonte:: http://it.wikipedia.org/w/index.php?oldid=32447474 Autori:: ARTE, Airon90, Alchimico, Aushulz, Biopresto, Buggia, ChemicalBit, Cisco79, Danilo, Eudigioia,
Ghazi85, Gionnico, Helios, Liopac, Liuksky, LoVid8, Mafejthoth, Malemar, Marcel Bergeret, Marcol-it, Mars79, Metralla, No2, Paginazero, Pavlo Chemist, Phantomas, Simo82, SkedO, Toobaz,
Trixt, Umbo86, Viames, ^musaz, 21 Modifiche anonime
Reazione acido-base Fonte:: http://it.wikipedia.org/w/index.php?oldid=45858327 Autori:: Ancar, Aushulz, Avesan, Buggia, ChemicalBit, Cisco79, Fakk, Frigotoni, Guidomac, No2,
Paginazero, Senpai, Whiles, 22 Modifiche anonime
Decomposizione (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=46438151 Autori:: Aushulz, Avesan, Buggia, Mars79, PaneBiancoLiscio
Metatesi (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=45454303 Autori:: Aushulz, Cisco79, Harlock81, IlPisano, Jacopo Werther, Mars79, Mau il chimico, 3 Modifiche anonime
Precipitazione (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=46483490 Autori:: Aushulz, Avesan, Buggia, Cisco79, Daniele Boglio, Dia^, Ithunn, Johnlong, Paginazero,
PaneBiancoLiscio, Pazzoide87, 5 Modifiche anonime
Complesso (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=41315541 Autori:: Albris, Archenzo, Aushulz, Buggia, Carlo.milanesi, Cisco79, Dello, Esculapio, Eumolpo,
Ilcontestatore, Leyo, Mars79, MaxDel, Memoria, Paginazero, Snowdog, 10 Modifiche anonime
Reazione organica Fonte:: http://it.wikipedia.org/w/index.php?oldid=42106090 Autori:: Aushulz, Cisco79, Eumolpo, Joder, KS, Mafejthoth, Paginazero, Paolo.campanella, 3 Modifiche
anonime
Reagente Fonte:: http://it.wikipedia.org/w/index.php?oldid=45250194 Autori:: Aushulz, Avesan, Blakwolf, Boombaster, Choij, Cisco79, Eio, Klaudio, Lornova, Paginazero, Snowdog, 10
Modifiche anonime
Catalizzatore Fonte:: http://it.wikipedia.org/w/index.php?oldid=40719936 Autori:: A7N8X, Alchimico, Arnade, Aushulz, Catecolamine, Cisco79, Conte sty, D r k, DaveBlack, Dello, Felyx,
Gabriele85, Giac83, Gliu, Incola, Luc106, Luciano G. Calì, Marcel Bergeret, Marsigliesenormanno, Mion, N314, No2, Paginazero, Pil56, Pinkflag, SamZane, Scexpir, Spidernik84, Suisui,
Superchilum, Tommaso Ferrara, Whiles, 33 Modifiche anonime
Solubilità Fonte:: http://it.wikipedia.org/w/index.php?oldid=46489196 Autori:: Arcade¹¹, Aushulz, Cisco79, Dedessenz, Felyx, Gnumarcoo, Guidomac, Kibira, Klaudio, Madaki, MapiVanPelt,
McGriffin, Neosg1, Nick, Paginazero, Phantomas, Ppalli, Silvano berton, Snowdog, Terrasque, Thijs!, 25 Modifiche anonime
Concentrazione Fonte:: http://it.wikipedia.org/w/index.php?oldid=45411748 Autori:: A7N8X, Andrea Gonella88, Ary29, Aushulz, Batuffolina, ChemicalBit, Cisco79, DarkAp, Donjel,
FrAnCiS, Luca.lombini, Marcoconsumi, Moongateclimber, No2, Paginazero, Pandit, Pleiade, Sbisolo, Tener, Ub, 21 Modifiche anonime
Frazione molare Fonte:: http://it.wikipedia.org/w/index.php?oldid=46783612 Autori:: Alberto da Calvairate, Archenzo, Aushulz, Blakwolf, Cisco79, Hellis, Julio.caesar, MagnInd, Paginazero,
Ripepette, Snowdog, Svante, Testadilegno, 11 Modifiche anonime
Numero di Avogadro Fonte:: http://it.wikipedia.org/w/index.php?oldid=46267573 Autori:: ATMB, Alec, Alfio, Artsakenos, Aushulz, Beta16, Blakwolf, Burubuz, Cisco79, Colom, Cruccone,
DarkBeam, EdoM, Eginardo, Elwood, F l a n k e r, Formica rufa, Fra74, Frassionsistematiche, Ginosal, Giova the star, Giuliof, Giuseppe Salustri, Guidomac, Hashar, Henrykus, Klaudio, Lm,
LoVid8, Mark91, Mars79, Paulatz, Preziusom, Restu20, Retaggio, Sassospicco, Sbisolo, Snowdog, Straw Dog, Suisui, Svante, Ylebru, 52 Modifiche anonime
Mole Fonte:: http://it.wikipedia.org/w/index.php?oldid=46736103 Autori:: Alex Pividori, Aushulz, Beta16, Burubuz, Cisco79, Codicorumus, DanGarb, Dinwath, Dome, Eio, Elwood, F l a n k e
r, Fabexplosive, Fiaschi, Fra74, Franz Liszt, Frigotoni, Gac, Gassendi, Gauss, Gianma89, Gliu, Gori.silvio, Jcs, Kibira, Klaudio, Manusha, Maquesta, Marcok, Marcus89, Mark91, Mars79,
Massic80, Mattia Luigi Nappi, N314, No2, Orzetto, Paginazero, Pietrodn, Pracchia-78, Rael, Raffamaiden, Red devil 666, Ricce, Rook, Salvatore Ingala, Sbisolo, Senza nome.txt, Sirabder87,
195
Fonti e autori delle voci
Snowdog, Soddi, Tommaso Ferrara, Trixt, Turgon, Vannyn, Vargenau, Withe, Ylebru, ^musaz, 123 Modifiche anonime
Chimica inorganica Fonte:: http://it.wikipedia.org/w/index.php?oldid=45983789 Autori:: A7N8X, Albris, Aushulz, Cisco79, Danilo, Eumolpo, Frieda, Gianfranco, Hashar, Hellis, Paginazero,
Suisui, Svante, 7 Modifiche anonime
Chimica organica Fonte:: http://it.wikipedia.org/w/index.php?oldid=45147210 Autori:: Aushulz, Avesan, Basilicofresco, Biopresto, Buggia, Cisco79, Danilo, Davide Fusco, Elwood, Giannib,
Hashar, Hellis, Hill, Jacopo, Koji, MapiVanPelt, Mark91, Nuovoastro, Paginazero, Retaggio, Rojelio, Sesekem, Stan Shebs, Suisui, Svante, Ticket 2010081310004741, Webkid, 39 Modifiche
anonime
Sale Fonte:: http://it.wikipedia.org/w/index.php?oldid=45455483 Autori:: .mau., AlessandroAM, Antoz, Aracuano, Arcade¹¹, Ary29, AsdfMovie, Augusto15, Aushulz, Avalon93, Bella
Situazione, Biopresto, Buggia, Buzz lightyear, CavalloRazzo, Cisco79, Civa61, Danilo, Davide, Dommac, Eio, Gacio, Ghazi85, Guidomac, InWind, Indoril, Lgl72, Limonadis, MapiVanPelt,
Mazz, Mela, Melefabrizio, N314, NerdEXE, NewLibertine, Nico-mix, Pacini Fabio, Paginazero, Patafisik, Phantomas, Piero, Pumpie, Rael, Raffamaiden, Rojelio, Salvo da Palermo, Sannita,
Seics, Snowdog, Svante, Taueres, Template namespace initialisation script, Ticket 2010081310004741, Trixt, Twice25, Vale maio, Whiles, 67 Modifiche anonime
Ossido Fonte:: http://it.wikipedia.org/w/index.php?oldid=46693397 Autori:: Adfc, Alfio, Angelorenzi, Augusto15, Aushulz, Blakwolf, Buggia, Cisco79, Dome, EdoM, Franco3450, Gabriele85,
Gac, Giancarlodessi, Guidomac, Helios, KeyboardSpellbounder, Limonadis, Luca Bergamasco, Luca Visentin, Lucamuna, Paginazero, Rojelio, Sky, Superchilum, Supernino, Taueres, Template
namespace initialisation script, Trikke, Trixt, Whiles, X-Dark, Zandegù, 69 Modifiche anonime
Chimica fisica Fonte:: http://it.wikipedia.org/w/index.php?oldid=46809938 Autori:: Blakwolf, Carlo.Ierna, ChemicalBit, Cisco79, Cla, Dommac, Felyx, Gori.silvio, Montinar, Paginazero,
Svante, Ylebru, 1 Modifiche anonime
Biochimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=46120867 Autori:: .anaconda, .jhc., Adoo, Annamaria.dmr, Ary29, Aushulz, Avesan, Buggia, Carlo.Ierna, Cisco79, Contezero,
Danilo, Ernesttico, Frazzone, Frieda, Gabriele85, Gaux, Ggonnell, Giac83, Ginosal, Hellis, Indoril, Joe123, LM77, Lumage, M7, Mark91, Massimo Bachetti, Nase, No2, Olando, Pinkflag,
Riccardov, Richzena, Romario81, Rupertsciamenna, Sbisolo, Sgargiulo, Skywolf, Snowdog, Ticket 2010081310004741, Towerman, 31 Modifiche anonime
Numero atomico Fonte:: http://it.wikipedia.org/w/index.php?oldid=46644597 Autori:: Alfio, Blakwolf, Cisco79, DanGarb, Danilo, Ego, Frenzul, Giannib, Gio97, Guidomac, Iron Bishop,
L-walker, Labba94, Luisa, Paginazero, Romanm, Sarcelles, Simone, Suisui, Svante, Trixt, Ylebru, 15 Modifiche anonime
Peso atomico Fonte:: http://it.wikipedia.org/w/index.php?oldid=46757114 Autori:: Alfio, Archenzo, Aushulz, Blakwolf, Buggia, Cisco79, Codicorumus, DanGarb, Dardorosso, Davide, Eio,
Eriol, Essepoint, Felyx, Frigotoni, Gianluigi, Gianpasha, Giuliof, Gliu, Hellis, MapiVanPelt, Mark91, Mela, Nucleare, Paginazero, Ritchie92, Simone, Spino, Suisui, Tomi, Twice25, Venom9,
Ylebru, 51 Modifiche anonime
Acido Fonte:: http://it.wikipedia.org/w/index.php?oldid=46775224 Autori:: Ai2007, Aidos24, Aldiaz, Alescan, Ancar, Aushulz, Bahar, Bianco.ciro, Biopresto, Buggia, Calipper, ChemicalBit,
Cisco79, Davide, Federicuccio, Franco3450, LoStrangolatore, LuigiPetrella, LukeWiller, MEGATRON-22, Mark91, Massimo Macconi, No2, Paginazero, Pequod76, Rael, Rupertsciamenna,
SamZane, Taueres, Tr6637, Waglione, Whiles, WinstonSmith, ^musaz, 28 Modifiche anonime
Base (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=45679204 Autori:: Ancar, Aushulz, Bianco.ciro, Biopresto, Buggia, ChemicalBit, Cisco79, Gac, Gce, Ggonnell, Ghazi85,
Guidomac, KeyboardSpellbounder, LukeWiller, Moloch981, No2, Paginazero, Raffaele Lovino, Waglione, 16 Modifiche anonime
Radicale libero Fonte:: http://it.wikipedia.org/w/index.php?oldid=45608005 Autori:: Albris, Aushulz, Barbaking, Carlomorino, Catellani, Cerebroleso, Cisco79, Dommac, ELEONORA
GELARDI, Fstefani, Giac83, Giancarlo Rossi, Grazianoleni, IlMonopattinoDiGiacomo, Lserni, Lunar Eclipse, Mediano88, Muthafxcka, Paginazero, Phantomas, Pierpao, Rjwilmsi, Rémih,
Semolo75, Tommaso Ferrara, Toxicologytoday, Un chien andalou, ValeBreve, Vtp saponaro, 29 Modifiche anonime
Metallo Fonte:: http://it.wikipedia.org/w/index.php?oldid=46761481 Autori:: .snoopy., A7N8X, Adfc, Alessio malan, Alexander Luthor, Aphaia, Ary29, Ask21, Aushulz, Azrael555,
Basilicofresco, Beard, Beechs, Beta16, Biopresto, Buggia, Bumba, Cisco79, DarkAp, Dj ture, Dr Zimbu, Elwood, F l a n k e r, Federicuccio, Gameover, Gianluigi, Guidomac, Henrykus,
Hotgia97, Johnlong, Kal-El, Kibira, Klaudio, Kormoran, La voce di Cassandra, Laurentius, Lenore, Lucas, Luciodem, LukeWiller, M7, MM, MapiVanPelt, Mark91, Massimiliano Lincetto, Matra
dj, MaxDel, Megalexandros, Melos, Montinar, Mpitt, Nemo bis, Numbo3, Orric, Oscaracciato, Osk, Pequod76, Phantomas, Renato Caniatti, Retaggio, Ripepette, Rojelio, Sbisolo, Shivanarayana,
Taueres, TheRedOne, Ticket 2010081310004741, Torsolo, Urli mancati, Veneziano, Vituzzu, Whiles, Ylebru, 201 Modifiche anonime
Non metallo Fonte:: http://it.wikipedia.org/w/index.php?oldid=46719638 Autori:: A7N8X, Ai2007, Alkalin, Beta16, Biopresto, Bumba, Cisco79, Cruccone, Dan 2011, Felyx, Frank.sartori,
Manusha, Maurice Carbonaro, Melos, Shivanarayana, Simòn, Supernino, Wyszinski, Ylebru, 25 Modifiche anonime
Semimetalli Fonte:: http://it.wikipedia.org/w/index.php?oldid=33338007 Autori:: A7N8X, Biopresto, Burubuz, Cesalpino, Cisco79, Frank.sartori, Hashar, Kormoran, Mac'ero, Marcok, Osk,
Paginazero, Romanm, Suisui, Svante, Ylebru, 9 Modifiche anonime
Gas nobili Fonte:: http://it.wikipedia.org/w/index.php?oldid=46795879 Autori:: Arnoux, Aushulz, Baglio, Biopresto, Cisco79, Danilo, Eio, Elwood, Frieda, Gspinoza, Guybrush Threepwood,
Hashar, Lukius, Otello Felarinnonz, Paginazero, Piper90, Rago, Romanm, Snowdog, Suisui, Svante, Taueres, Ture1989, Urban, Vituzzu, Ylebru, Θixωr89, 22 Modifiche anonime
Formula chimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=44781831 Autori:: Andrebask, Aushulz, Austro, AvdE, Cisco79, Danilo, Dariocima, F l a n k e r, Felyx, Hashar, Hellis,
Hystrix, Kal-El, Lp, Mpitt, Paginazero, Phantomas, Polveracchio, RobertoITA, Svante, 14 Modifiche anonime
Legame covalente Fonte:: http://it.wikipedia.org/w/index.php?oldid=45922887 Autori:: Aushulz, Avesan, Aytrus, BigFrank71, Biopresto, Bumba, Cisco79, DarkAp, Fabexplosive, Flippo,
Frieda, Giancarlodessi, Gianluigi, Gibao, Gnumarcoo, Gspinoza, Kibira, Kyklops, Luca.lombini, Lukino24, Marcookie, Marcus89, Nick1915, No2, Paginazero, Rael, RanZag, Rojelio, Sbisolo,
Shivanarayana, Template namespace initialisation script, Tommaso Ferrara, Twice25, Withoutwords, ^musaz, 58 Modifiche anonime
Gas Fonte:: http://it.wikipedia.org/w/index.php?oldid=46259104 Autori:: Albris, Andreabrugiony, Ary29, Aushulz, Avemundi, Basilicofresco, Biopresto, Buggia, Centurion.e75, ChemicalBit,
Cisco79, Dani4P, Davide, Dommac, DoppioM, Dr Zimbu, Elwood, Eumolpo, Felyx, Fredericks, Gabriele85, Guidomac, Jacopo Werther, Jalo, Kibira, Kormoran, Madaki, No2, Numbo3, Orric,
Paginazero, Phantomas, Poldo328, Progettualita, Retaggio, Sacchigno, Scheggia.agm, Seics, Shivanarayana, Simone, Ylebru, Yujin, 53 Modifiche anonime
Soluzione (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=45790193 Autori:: 4nT0, Adfc, AnjaManix, Ariel, Ary29, Ask21, Aushulz, Biopresto, Buggia, Bultro, ChemicalBit,
Cisco79, Danilo, DarioR, Demart81, DiZ, Fire90, Janilborntobekilled, Loutoma, Luisa, MaEr, Miao, Midnight bird, Nioger, Omegakent, Paginazero, Pazzoide87, Phantomas, Prophet 999,
Quatar, Svante, Taueres, Turgon, 35 Modifiche anonime
Dispersione (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=45563668 Autori:: Aushulz, Biopresto, Carlog3, Cisco79, Nick, SkY`, 1 Modifiche anonime
Elettrone Fonte:: http://it.wikipedia.org/w/index.php?oldid=46676943 Autori:: .anaconda, ANGELUS, Alberto da Calvairate, AlessandroAM, Alexyan, ArtAttack, AttoRenato, Aushulz,
Batmang, Blakwolf, Bobrg, Buggia, Carlog3, Cisco79, Crazyspin, CristianCantoro, Danilo, Ele piloni krati, Elph, Engineer123, Eumolpo, F l a n k e r, Felyx, Fire90, Fredericks, Freplo, Gacio,
Gazal Cotre, Giambrox, Gianluigi, Gionnico, Gspinoza, Hashar, Huntster, Intoinside, Johnlong, Koji, Lenore, Lorenzor, M&M987, Madaki, Massimiliano Lincetto, Numbo3, Olando, Paulatz,
Phantomas, Poweruser, Rago, Red Power, Remogatto, Restu20, Salli, Sassospicco, Sbisolo, Sesquipedale, Shivanarayana, Snowdog, Straw Dog, Suisui, Taueres, Template namespace
initialisation script, Trixt, WinstonSmith, Wiso, X-Dark, ^musaz, ppp-62-10-106-121.dialup.tiscali.it, 40 Modifiche anonime
Isotopo Fonte:: http://it.wikipedia.org/w/index.php?oldid=46317477 Autori:: .jhc., Aushulz, Beltd, Broc, Chemako0606, Cisco79, DanGarb, Danilo, DarkAp, Doc.mari, EdoM, Engineer123,
Eosforon, F l a n k e r, Felyx, Filippo000, Fornaeffe, Gabriele85, Giovannigobbin, Luisa, Nicchio, Nucleare, Paginazero, Patria, Remy1992, Simone, Snowdog, SolePensoso, Svante, Ylebru, 31
Modifiche anonime
Idrolisi Fonte:: http://it.wikipedia.org/w/index.php?oldid=46778934 Autori:: AttoRenato, Aushulz, Buggia, Cisco79, DanGarb, Djdmx, Fenix Felix, Helios, Jalo, Leyo, Luke94, LukeWiller,
Mark91, Mars79, Mau db, Osk, Paginazero, PaneBiancoLiscio, SoliDreamer, Ssspera, Time9, 21 Modifiche anonime
Attinoidi Fonte:: http://it.wikipedia.org/w/index.php?oldid=46828748 Autori:: Amarvudol, Broc, Cesalpino, Dwalin, Gabriele85, Kormoran, Lohe, Mac'ero, Orionist, Scriban, Superchilum, 2
Modifiche anonime
Lantanoidi Fonte:: http://it.wikipedia.org/w/index.php?oldid=46520618 Autori:: Burraco2, Cesalpino, Dinwath, Flippo, Gabriele85, Hashar, Kormoran, Lohe, Mac'ero, Paginazero, Romanm,
Suisui, Superchilum, TinyFox, 1 Modifiche anonime
Stato di ossidazione Fonte:: http://it.wikipedia.org/w/index.php?oldid=45576301 Autori:: Arriano60, Aushulz, Biopresto, Cisco79, Deko90, Garibaldino, Guido Gonzato, Iakopo, Lmercatanti,
Mars79, Mattia Luigi Nappi, Memoria, Paginazero, Physchim62, Rael, Retaggio, Snowdog, Ylebru, 20 Modifiche anonime
196
Fonti e autori delle voci
Equazione chimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=44431462 Autori:: AttoRenato, Aushulz, Avesan, Cisco79, Dommac, Felyx, Gabriele85, Johnny B. Goode, No2, Piddu,
Sl, 13 Modifiche anonime
Prodotto (chimica) Fonte:: http://it.wikipedia.org/w/index.php?oldid=45380115 Autori:: Aushulz, Boombaster, Buggia, Cisco79, Eio, 1 Modifiche anonime
197
Fonti, licenze e autori delle immagini
Fonti, licenze e autori delle immagini
File:Chemicals in flasks.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Chemicals_in_flasks.jpg Licenza: Creative Commons Attribution 2.0 Autori:: Benjah-bmm27, Fjmustak,
ITurtle, Ida Shaw, Karelj, Meisam, 1 Modifiche anonime
File:Alchemist-small.gif Fonte:: http://it.wikipedia.org/w/index.php?title=File:Alchemist-small.gif Licenza: Public Domain Autori:: Anne97432, LeastCommonAncestor, Lewenstein, Maksim,
Matthias M., Sailko, 1 Modifiche anonime
File:Fullerene-C60.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Fullerene-C60.png Licenza: GNU Free Documentation License Autori:: File:Copper sulfate.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Copper_sulfate.jpg Licenza: GNU Free Documentation License Autori:: Stephanb
File:Hydrochloric acid ammonia.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hydrochloric_acid_ammonia.jpg Licenza: Public Domain Autori:: User:Walkerma
File:Boyles Law animated.gif Fonte:: http://it.wikipedia.org/w/index.php?title=File:Boyles_Law_animated.gif Licenza: Public Domain Autori:: NASA's Glenn Research Center
File:Charles and Gay-Lussac's Law animated.gif Fonte:: http://it.wikipedia.org/w/index.php?title=File:Charles_and_Gay-Lussac's_Law_animated.gif Licenza: Public Domain Autori::
NASA's Glenn Research Center
File:Colonne distillazione.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Colonne_distillazione.jpg Licenza: Creative Commons Attribution 3.0 Autori:: User:Luigi Chiesa
File:Medikamente.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Medikamente.jpg Licenza: GNU Free Documentation License Autori:: Würfel
Immagine:Wikibooks-logo.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wikibooks-logo.svg Licenza: logo Autori:: User:Bastique, User:Ramac et al.
Immagine:Commons-logo.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Commons-logo.svg Licenza: logo Autori:: SVG version was created by User:Grunt and cleaned up by
3247, based on the earlier PNG version, created by Reidab.
Immagine:Wikinews-logo.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wikinews-logo.svg Licenza: logo Autori:: Vectorized by Simon 01:05, 2 August 2006 (UTC) Updated by
Time3000 17 April 2007 to use official Wikinews colours and appear correctly on dark backgrounds. Originally uploaded by Simon.
Immagine:Wiktionary-ico-de.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wiktionary-ico-de.png Licenza: logo Autori:: Bobit, F l a n k e r, Melancholie, Mxn, Nodulation,
Rocket000, Saibo
Immagine:Wikiquote-logo.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wikiquote-logo.svg Licenza: Public Domain Autori:: -xfi-, Dbc334, Doodledoo, Elian, Guillom, Jeffq,
Krinkle, Maderibeyza, Majorly, Nishkid64, RedCoat, Rei-artur, Rocket000, 11 Modifiche anonime
Immagine:JosephWright-Alchemist.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:JosephWright-Alchemist.jpg Licenza: Public Domain Autori:: Goldfritha,
LeastCommonAncestor, Luigi Chiesa, Mattes, PKM, Plindenbaum, Ragesoss, Shakko, Svajcr, Warburg
Immagine:Robert Boyle 0001.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Robert_Boyle_0001.jpg Licenza: Public Domain Autori:: Johann Kerseboom
Immagine:David - Portrait of Monsieur Lavoisier and His Wife.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:David_-_Portrait_of_Monsieur_Lavoisier_and_His_Wife.jpg
Licenza: Public Domain Autori:: AnRo0002, Badzil, Bohème, Churchh, Cybershot800i, Didactohedron, Diligent, Ecummenic, Elian, FxJ, Jastrow, Kilom691, Kirtap, Mattes, Mutter Erde, Natl1,
QWerk, Serge Lachinov, Shakko, Sir Gawain, Slomox, Svencb, Urban, Zolo, 2 Modifiche anonime
Immagine:Lavoisier Nomenclature01.gif Fonte:: http://it.wikipedia.org/w/index.php?title=File:Lavoisier_Nomenclature01.gif Licenza: Public Domain Autori:: User:Walkerma
File:Urea Synthesis Woehler.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Urea_Synthesis_Woehler.png Licenza: Public Domain Autori:: Jü
Immagine:Friedrich Witte Fabrik.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Friedrich_Witte_Fabrik.jpg Licenza: Public Domain Autori:: AnRo0002, Ingolfson, Schiwago
Immagine:Mendeleev Table 5th II.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Mendeleev_Table_5th_II.jpg Licenza: Public Domain Autori:: Ragesoss, 4 Modifiche anonime
Immagine:Heinrich von Angeli - Friedrich August Kekulé von Stradonitz.jpg Fonte::
http://it.wikipedia.org/w/index.php?title=File:Heinrich_von_Angeli_-_Friedrich_August_Kekulé_von_Stradonitz.jpg Licenza: Public Domain Autori:: Heinrich von Angeli (1840-1925)
Immagine:Plum pudding atom.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Plum_pudding_atom.svg Licenza: Public Domain Autori:: User:Fastfission
Immagine:Atome de Rutherford.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Atome_de_Rutherford.png Licenza: GNU Free Documentation License Autori:: User:Jean-Jacques
MILAN
Immagine:Bohr-atom-PAR.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Bohr-atom-PAR.svg Licenza: sconosciuto Autori:: Original uploader was JabberWok at en.wikipedia
Immagine:Erwin Schrödinger.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Erwin_Schrödinger.jpg Licenza: Public Domain Autori:: Harp, JdH, Mdd, NielsF, Orgullomoore,
Threedots, UV, Yerpo
Immagine:ADN animation.gif Fonte:: http://it.wikipedia.org/w/index.php?title=File:ADN_animation.gif Licenza: Public Domain Autori:: brian0918
Immagine:Giulio Natta.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Giulio_Natta.png Licenza: Public Domain Autori:: Giac83
Immagine:Supramolecular Assembly Lehn.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Supramolecular_Assembly_Lehn.jpg Licenza: GNU Free Documentation License
Autori:: Original uploader was M stone at en.wikipedia
Immagine:BZGraphScheme.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:BZGraphScheme.png Licenza: Creative Commons Attribution-ShareAlike 3.0 Unported Autori::
User:RedAndr
File:Legame_chimico_covalente_puro.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Legame_chimico_covalente_puro.PNG Licenza: Public Domain Autori:: Disegnato da
--Paginazero 11:44, Ago 31, 2004 (UTC). Original uploader was Diablo at it.wikibooks
File:Legame_chimico_covalente_polare.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Legame_chimico_covalente_polare.PNG Licenza: Public Domain Autori:: Disegnato da
--Paginazero 11:46, Ago 31, 2004 (UTC). Original uploader was Diablo at it.wikibooks
File:Helium atom QM.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Helium_atom_QM.png Licenza: Public domain Autori:: en:User:FrankH
File:String theory.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:String_theory.svg Licenza: Creative Commons Attribution 3.0 Autori:: MissMJ
File:A New System of Chemical Philosophy fp.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:A_New_System_of_Chemical_Philosophy_fp.jpg Licenza: Public Domain Autori::
haade
File:Esperimento Rutherford.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Esperimento_Rutherford.PNG Licenza: GNU Free Documentation License Autori:: Original uploader
was Diablo at it.wikibooks
File:Atom.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Atom.svg Licenza: GNU Free Documentation License Autori:: Svdmolen/Jeanot (converted by King of Hearts)
File:Molecola saccarosio modello.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Molecola_saccarosio_modello.png Licenza: Public domain Autori:: Utente:Paginazero
Immagine:potenziale intermolecolare.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Potenziale_intermolecolare.PNG Licenza: sconosciuto Autori:: Utente:Gugliottavincenzo
File:Wave functions binding.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wave_functions_binding.svg Licenza: Public Domain Autori:: Kreuvf
File:Wave functions anti-binding.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wave_functions_anti-binding.svg Licenza: Public Domain Autori:: Kreuvf
Image:Dihydrogen-HOMO-phase-3D-balls.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Dihydrogen-HOMO-phase-3D-balls.svg Licenza: Public Domain Autori:: derivative
work: Pbroks13 (talk) Dihydrogen-HOMO-phase-3D-balls.png: Benjah-bmm27
Image:Dihydrogen-LUMO-phase-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Dihydrogen-LUMO-phase-3D-balls.png Licenza: Public Domain Autori::
Benjah-bmm27, Mion, Pieter Kuiper
File:H1F1.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:H1F1.png Licenza: Public Domain Autori:: Draabaar. Original uploader was Draabaar at de.wikipedia
File:H2o.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:H2o.png Licenza: GNU Free Documentation License Autori:: Luigi Chiesa, Maksim, Wst
File:Ch4-hybridisation.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ch4-hybridisation.png Licenza: GNU Free Documentation License Autori:: by Sinuhe
File:Methaan.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Methaan.png Licenza: Creative Commons Attribution 2.5 Autori:: Effeietsanders
File:He2 antibonding orbital.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:He2_antibonding_orbital.png Licenza: Creative Commons Attribution-Share Alike Autori:: Helvet
File:O2 orbitali molecolari.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:O2_orbitali_molecolari.jpg Licenza: Creative Commons Attribution-Sharealike 3.0 Autori:: Laghi.l
File:MOdiagramCO.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:MOdiagramCO.png Licenza: Creative Commons Attribution-Sharealike 3.0 Autori:: Jcwf
File:Molecular energy levels en.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Molecular_energy_levels_en.svg Licenza: Creative Commons Attribution-Sharealike 3.0 Autori::
Lvzon
198
Fonti, licenze e autori delle immagini
File:Morse-potential.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Morse-potential.png Licenza: Creative Commons Attribution-ShareAlike 3.0 Unported Autori:: Kri, Pieter
Kuiper, Somoza
Image:Vibrationrotationenergy.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Vibrationrotationenergy.svg Licenza: Public Domain Autori:: David-i98 (talk) 20:52, 3 January 2008
(UTC) Original uploader was David-i98 at en.wikipedia
File:Ir hcl rot-vib mrtz.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ir_hcl_rot-vib_mrtz.svg Licenza: Creative Commons Attribution-Sharealike 2.5 Autori:: mrtz
Image:Franck-Condon-diagram.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Franck-Condon-diagram.png Licenza: Creative Commons Attribution-ShareAlike 3.0 Unported
Autori:: Mark M. Somoza
Image:Fisica materia passaggi stato 1 it.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Fisica_materia_passaggi_stato_1_it.svg Licenza: Creative Commons Attribution-Sharealike
3.0 Autori:: ElfQrin
File:Chemicals-HP.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Chemicals-HP.jpg Licenza: Creative Commons Attribution 2.0 Autori:: FlickrLickr, FlickreviewR, Nilfanion
Immagine:P&ID.JPG Fonte:: http://it.wikipedia.org/w/index.php?title=File:P&ID.JPG Licenza: Creative Commons Attribution-Sharealike 3.0 Autori:: Ub
File:Arrhenius2.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Arrhenius2.jpg Licenza: Public Domain Autori:: ChVA, Duesentrieb, HenkvD, JdH, LX, Lukius, Väsk, Yelm
File:Potenziale di decomposizione - Daneell.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Potenziale_di_decomposizione_-_Daneell.png Licenza: Public Domain Autori::
Heinrich Daneell
Immagine:Silberchlorid.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Silberchlorid.jpg Licenza: Public Domain Autori:: Siegert
File:Tris(bipyridine)ruthenium(II) chloride.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Tris(bipyridine)ruthenium(II)_chloride.png Licenza: Public Domain Autori:: Edgar181
File:Linear-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Linear-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Trigonal-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Trigonal-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Tetrahedral-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Tetrahedral-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Square-planar-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Square-planar-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27, Zzyzx11
File:Trigonal-bipyramidal-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Trigonal-bipyramidal-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Square-pyramidal-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Square-pyramidal-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Octahedral-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Octahedral-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Pentagonal-bipyramidal-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Pentagonal-bipyramidal-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
Image:Cis-dichlorotetraamminecobalt(III).png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Cis-dichlorotetraamminecobalt(III).png Licenza: Public Domain Autori:: Benjah-bmm27
Image:Trans-dichlorotetraamminecobalt(III).png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Trans-dichlorotetraamminecobalt(III).png Licenza: Public Domain Autori::
Benjah-bmm27
Image:Fac-trichlorotriamminecobalt(III).png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Fac-trichlorotriamminecobalt(III).png Licenza: Public Domain Autori:: Benjah-bmm27
Image:Mer-trichlorotriamminecobalt(III).png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Mer-trichlorotriamminecobalt(III).png Licenza: Public Domain Autori:: Benjah-bmm27
Image:Delta-tris(oxalato)ferrate(III)-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Delta-tris(oxalato)ferrate(III)-3D-balls.png Licenza: Public Domain Autori::
Benjah-bmm27, Ephemeronium, Tpa2067, Wickey-nl
Image:Lambda-tris(oxalato)ferrate(III)-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Lambda-tris(oxalato)ferrate(III)-3D-balls.png Licenza: Public Domain Autori::
Benjah-bmm27, Ephemeronium, Wickey-nl
Image:Delta-cis-dichlorobis(ethylenediamine)cobalt(III).png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Delta-cis-dichlorobis(ethylenediamine)cobalt(III).png Licenza: Public
Domain Autori:: Benjah-bmm27
Image:Lambda-cis-dichlorobis(ethylenediamine)cobalt(III).png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Lambda-cis-dichlorobis(ethylenediamine)cobalt(III).png Licenza:
Public Domain Autori:: Benjah-bmm27
File:LinkageIsomers.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:LinkageIsomers.png Licenza: Public Domain Autori:: Smokefoot
File:Diagramma attivazione.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Diagramma_attivazione.svg Licenza: GNU Free Documentation License Autori:: Originally uploaded by
Jerry Crimson Mann, vectorized by Tutmosis, corrected by Fvasconcellos, translated by Gia.cossa
File:Catalizzatore al Rodio.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Catalizzatore_al_Rodio.png Licenza: Creative Commons Attribution-Sharealike 3.0,2.5,2.0,1.0 Autori::
Aushulz
File:Hydrogenation on catalyst.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hydrogenation_on_catalyst.png Licenza: Creative Commons Attribution 1.0 Generic Autori::
Michael Schmid
File:Solubilità diretta e inversa.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Solubilità_diretta_e_inversa.svg Licenza: Creative Commons Attribution-Sharealike 3.0 Autori::
Aushulz
File:Dilution-concentration simple example.it.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Dilution-concentration_simple_example.it.jpg Licenza: Creative Commons Attribution
2.5 Autori:: Original uploader was AMarkov at bg.wikipedia
File:Elettrolisi per numero di Avogadro.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Elettrolisi_per_numero_di_Avogadro.png Licenza: sconosciuto Autori:: Utente:Paulatz
File:Ampolla_da_elettrolisi_per_numero_di_Avogadro.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ampolla_da_elettrolisi_per_numero_di_Avogadro.png Licenza: Public
domain Autori:: Utente:Paulatz
Immagine:Inorganic-montage.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Inorganic-montage.png Licenza: Public Domain Autori:: Benjah-bmm27, Karelj
Immagine:Potassium-oxide-3D-vdW.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Potassium-oxide-3D-vdW.png Licenza: Public Domain Autori:: Benjah-bmm27,
Ephemeronium, Luigi Chiesa
Immagine:CoEDTA-anion-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:CoEDTA-anion-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
Immagine:Tetrasulfur-tetranitride-3D-vdW.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Tetrasulfur-tetranitride-3D-vdW.png Licenza: Public Domain Autori:: Benjah-bmm27
Immagine: Cisplatin-3D-vdW.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Cisplatin-3D-vdW.png Licenza: Public Domain Autori:: Benjah-bmm27
Image:N-butyllithium-tetramer-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:N-butyllithium-tetramer-3D-balls.png Licenza: Public Domain Autori:: Aushulz,
Benjah-bmm27
Immagine:Decaborane-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Decaborane-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
Immagine:Fe4S4-3D-vdW.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Fe4S4-3D-vdW.png Licenza: Public Domain Autori:: Benjah-bmm27, Rhadamante
Immagine:Vitamin-B12-Co-centre-3D-balls.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Vitamin-B12-Co-centre-3D-balls.png Licenza: Public Domain Autori:: Benjah-bmm27
Immagine:YBa2Cu3O7.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:YBa2Cu3O7.png Licenza: Creative Commons Attribution-Sharealike 3.0 Autori:: Gadolinist
Immagine:Ferricyanide-3D.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ferricyanide-3D.png Licenza: Public Domain Autori:: Benjah-bmm27, Luigi Chiesa, Pieter Kuiper
Immagine:Nitrogen-dioxide-3D-vdW.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Nitrogen-dioxide-3D-vdW.png Licenza: Public Domain Autori:: Benjah-bmm27
File:Jöns Jacob Berzelius from Familj-Journalen1873.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Jöns_Jacob_Berzelius_from_Familj-Journalen1873.png Licenza: Public
Domain Autori:: Celsius, Den fjättrade ankan, Thuresson
File:Friedrich woehler.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Friedrich_woehler.jpg Licenza: Public Domain Autori:: Kresspahl, PDH, Rex, Stefi, Väsk, 1 Modifiche
anonime
File:NaCl-zoutkristallen op Schott Duran 100 ml.JPG Fonte:: http://it.wikipedia.org/w/index.php?title=File:NaCl-zoutkristallen_op_Schott_Duran_100_ml.JPG Licenza: GNU Free
Documentation License Autori:: ARTE, Bios
File:Rareearthoxides.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Rareearthoxides.jpg Licenza: Public Domain Autori:: Peggy Greb, US department of agriculture. Original
uploader was Materialscientist at en.wikipedia
File:Rust screw.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Rust_screw.jpg Licenza: Creative Commons Attribution 2.0 Autori:: User:Paulnasca. Original uploader was
Paulnasca at en.wikipedia
File:Saccharose.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Saccharose.svg Licenza: Public Domain Autori:: Booyabazooka, Calvero, Edgar181, 1 Modifiche anonime
199
Fonti, licenze e autori delle immagini
File:Hemoglobin.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hemoglobin.jpg Licenza: GNU Free Documentation License Autori:: Grafite, Habj, Lennert B, Noca2plus, Shizhao
File:Amino acids 1.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Amino_acids_1.png Licenza: GNU Free Documentation License Autori:: Arria Belli, CommonsDelinker,
CommonsDelinkerHelper, Edgar181, Karelj, Matanya (usurped), OsamaK, 1 Modifiche anonime
File:Biologia molecolare - Biochimica - Genetica.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Biologia_molecolare_-_Biochimica_-_Genetica.jpg Licenza: GNU Free
Documentation License Autori:: Ggonnell
File:Sulfuric acid burning tissue paper.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Sulfuric_acid_burning_tissue_paper.jpg Licenza: Public Domain Autori:: Rifleman 82
Immagine:Radicale allilico formule.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Radicale_allilico_formule.PNG Licenza: Public domain Autori:: Utente:Paginazero
Immagine:Radicale benzilico formule.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Radicale_benzilico_formule.PNG Licenza: Public domain Autori:: Utente:Paginazero
File:Iron electrolytic and 1cm3 cube.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Iron_electrolytic_and_1cm3_cube.jpg Licenza: Free Art License Autori:: Alchemist-hp (talk) (
www.pse-mendelejew.de)
File:Gallium1 640x480.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Gallium1_640x480.jpg Licenza: sconosciuto Autori:: File:Fe,26.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Fe,26.jpg Licenza: GNU Free Documentation License Autori:: User:RTC
File:Hot metalwork.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hot_metalwork.jpg Licenza: GNU Free Documentation License Autori:: Contributor, Fir0002, Jahobr, Wst, 1
Modifiche anonime
Immagine:Cis-2-butene.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Cis-2-butene.PNG Licenza: Public Domain Autori:: User:H Padleckas
Immagine:Trans-2-butene.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Trans-2-butene.PNG Licenza: Public Domain Autori:: User:H Padleckas
Immagine:Formula_chimica_esempi.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Formula_chimica_esempi.PNG Licenza: Public domain Autori:: Utente:Paginazero
File:Kinetic theory of gases.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Kinetic_theory_of_gases.svg Licenza: Creative Commons Attribution-ShareAlike 3.0 Unported Autori::
Sharayanan
File:SaltInWaterSolutionLiquid.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:SaltInWaterSolutionLiquid.jpg Licenza: GNU Free Documentation License Autori:: User:Chris 73
Image:Crookes_tube-in_use-lateral_view-standing_cross_prPNr°11.jpg Fonte::
http://it.wikipedia.org/w/index.php?title=File:Crookes_tube-in_use-lateral_view-standing_cross_prPNr°11.jpg Licenza: sconosciuto Autori:: D-Kuru, GianniG46, RJHall
File:Modello atomico di Bohr.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Modello_atomico_di_Bohr.svg Licenza: Creative Commons Attribution-Sharealike 3.0 Autori::
derivative work: Angelus (talk)
File:Orbitale atomico S.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Orbitale_atomico_S.png Licenza: Creative Commons Attribution-Sharealike 3.0 Autori:: RJHall
File:Asymmetricwave2.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Asymmetricwave2.png Licenza: Creative Commons Attribution 3.0 Autori:: TimothyRias
File:Standard Model of Elementary Particles it.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Standard_Model_of_Elementary_Particles_it.svg Licenza: Creative Commons
Attribution 3.0 Autori:: Standard_Model_of_Elementary_Particles.svg: MissMJ derivative work: Restu20 (talk)
File:Hydrogen Density Plots.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hydrogen_Density_Plots.png Licenza: Public domain Autori:: PoorLeno (talk)
File:Lightning over Oradea Romania cropped.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Lightning_over_Oradea_Romania_cropped.jpg Licenza: Public Domain Autori::
Mircea Madau (crop by Lucas)
File:Virtual pairs near electron.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Virtual_pairs_near_electron.png Licenza: Creative Commons Attribution-Sharealike 3.0 Autori::
RJHall
File:Lorentz force.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Lorentz_force.svg Licenza: GNU Free Documentation License Autori:: User:Jaro.p
File:Bremsstrahlung.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Bremsstrahlung.svg Licenza: Public Domain Autori:: Journey234, Pieter Kuiper, RJHall, Trex2001
File:Lorentz factor.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Lorentz_factor.svg Licenza: Public Domain Autori:: egg, Graph created with KmPlot, edited with Inkscape
Trassiorf (talk) 21:54, 2 March 2010 (UTC)
File:Produzione di coppia.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Produzione_di_coppia.svg Licenza: Creative Commons Attribution-Sharealike 3.0 Autori:: derivative
work: Angelus (talk)
File:Aurore australe - Aurora australis.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Aurore_australe_-_Aurora_australis.jpg Licenza: sconosciuto Autori:: Diti, Ehquionest
File:Nasa Shuttle Test Using Electron Beam full.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Nasa_Shuttle_Test_Using_Electron_Beam_full.jpg Licenza: Public Domain
Autori:: NASA
File:actinide phases.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Actinide_phases.svg Licenza: Public Domain Autori:: James L. Smith, LANL.
200
Licenza
Licenza
Creative Commons Attribution-Share Alike 3.0 Unported
//creativecommons.org/licenses/by-sa/3.0/
201
Scaricare