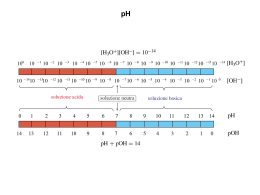
Appunti per il LABORATORIO DI CHIMICA A cura dei docenti del Laboratorio di Chimica Un particolare ringraziamento ai professori Achille Fusi Laura Maria Raimondi corso di laurea triennale in Scienze Biologiche _____________________________________________________________ Grafici e illustrazioni di Maria Antacido MISURE, OPERAZIONI FONDAMENTALI, TECNICHE DI INDAGINE DETERMINAZIONE DELLA DENSITA' La densità è una proprietà fisica di ogni sostanza solida, liquida o gassosa, a temperatura e pressione costanti. Essa esprime la massa di sostanza contenuta nell'unità di volume, viene indicata col simbolo d ed è una grandezza intensiva, cioè non dipende dalla quantità di sostanza considerata. d = m a ssa (g ) v o lu m e ( m L ) La densità di un liquido viene determinata effettuando la pesata di un volume noto del liquido. Il volume può essere misurato con accuratezza utilizzando dei recipienti tarati: pipetta a doppia tacca e matraccio (vedi più avanti). La determinazione è tanto più accurata quanto maggiore è la massa, perchè, a parità di sensibilità della bilancia, l'errore percentuale è inferiore. Per un solido la determinazione del volume viene eseguita in modo indiretto, utilizzando un matraccio ed un liquido a densità nota. I pesata massa del matraccio vuoto e tappato (tara) M II pesata massa del matraccio contenente il solido M(S) III pesata massa del matraccio con il solido e portato a volume con il liquido M(S+L) La massa del liquido si ottiene per differenza M(L) = M(S+L) - M(S) Il volume del liquido si ricava dalla sua densità V(L) = M(L) / d(L) Il volume del solido si ricava infine dal volume totale del matraccio V(S) = V(S+L) - V(L) In assenza di reazioni e/o forti interazioni, si può infatti ragionevolmente ritenere che i volumi del liquido e del solido siano additivi. PIPETTA A DOPPIA TACCA Le pipette sono tubi sottili di varia foggia e capacità predisposti per il prelievo di quantità note, fisse o variabili, di liquidi. Le pipette vengono riempite per aspirazione, utilizzando un aspirapipette, ad esempio una ampolla di gomma provvista di un'imboccatura per la pipetta e di tre valvole (per il riempimento e lo svuotamento del liquido e per l'immissione di aria) o un dispositivo equivalente. Le pipette a doppia tacca consentono l'erogazione di quantità fisse e note di un liquido; la capacità e la precisione sono stampigliate vicino all'imboccatura. Vanno riempite fino alla tacca superiore e svuotate fino a quella inferiore; infatti solo il volume di liquido compreso tra le due tacche è noto con accuratezza. Le pipette vanno utilizzate pulite ed asciutte; se sono bagnate e non è possibile asciugarle, vanno avvinate (sciacquate con alcuni mL del liquido da prelevare) prima dell'uso. MATRACCIO I matracci tarati sono contenitori a volume fisso e provvisti di un tappo ermetico; la capacità è evidenziata da una tacca sul collo del recipiente ed è stampigliata sul bulbo unitamente alla precisione. Vengono impiegati per la preparazione di soluzioni a concentrazione nota. Il matraccio viene riempito di liquido fin quasi all'inizio del collo per mezzo di un imbuto appropriato, a gambo stretto e lungo per evitare di bagnare il collo al disopra della tacca di calibrazione o, più semplicemente, utilizzando un bicchiere e asciugando poi il collo dall’imboccatura fino alla tacca. Si eliminano le eventuali bolle d’aria aderenti alle pareti inclinando e ruotando il matraccio e picchiettando leggermente. Si porta poi a volume goccia a goccia utilizzando una pipetta Pasteur finché il fondo del menisco risulti tangente superiormente alla tacca di calibrazione. Si tappa infine il matraccio e lo si capovolge alcune volte per uniformare la concentrazione della soluzione. 2 METODI VOLUMETRICI L'analisi volumetrica viene impiegata per determinare con accuratezza la concentrazione di una soluzione. Si fà avvenire una reazione, che sia completa, rapida e a stechiometria nota, tra la sostanza di cui si intende determinare la concentrazione e un'altra sostanza aggiunta in quantità nota. Le reazioni acido-base, di complessazione e di ossidoriduzione sono quelle più comunemente utilizzate. Questa tecnica di determinazione quantitativa si chiama titolazione. Si esegue aggiungendo con una buretta una soluzione a concentrazione nota (titolante) ad un volume noto della soluzione da titolare (titolando), finché la quantità del reagente sia equivalente a quella della sostanza stessa; questa procedura si dice di titolazione diretta. La fine della titolazione volumetrica, cioè il raggiungimento del punto di equivalenza, viene segnalata da un cambiamento di colore del titolando al completamento stechiometrico della reazione. Il volume erogato fino a quel punto dalla buretta viene detto volume equivalente. La colorazione può essere impartita alla soluzione da un opportuno indicatore, una sostanza aggiunta espressamente, oppure, a seconda di quale dei due sia colorato, dall’esaurimento del titolando o da un lieve eccesso di titolante. Al punto di equivalenza vale la relazione: eq1 = eq2 cioè dove eq = equivalenti V = volume N = normalità V1N1 = V2N2 1 = titolante 2 = titolando Le soluzioni a titolo noto vanno preparate con cura e il loro titolo va sempre verificato mediante una standardizzazione, cioè una titolazione con uno standard primario: questo è una sostanza disponibile con un elevato grado di purezza, non igroscopica, non volatile, stabile, con elevato peso equivalente (per minimizzare gli errori di pesata). Il procedimento di standardizzazione richiede una pesata accurata (con bilancia analitica) dello standard primario, che va poi solubilizzato con acqua deionizzata (in un bicchiere); questa soluzione serve per determinare la concentrazione della soluzione da standardizzare (nella buretta). Dalla relazione titolare si ottiene eqT; infatti e quindi, eqS = eqT = VTNT dove S = standard primario T = soluzione da eqS si ricava dalla pesata e dalla reazione di titolazione noto VT (il volume equivalente), si può risalire al titolo incognito NT. Per la standardizzazione di soluzioni di acidi forti si utilizza il carbonato di sodio (Na2CO3, purezza 99,9%; base diprotica): CO 23 + 2 H C l CO 2 + 2 C l- + H 2 O Per la standardizzazione di soluzioni di basi forti si utilizza lo ftalato acido di potassio (C8H5O4K, purezza 99,95%; acido monoprotico): DETERMINAZIONE DEL PESO EQUIVALENTE E DEL PESO MOLECOLARE Mediante una titolazione è possibile determinare una grandezza collegata alla concentrazione della soluzione: il peso equivalente della specie titolata. Dalla reazione di titolazione e conoscendo la quantità di sostanza presente nella soluzione, si applica, al punto di equivalenza, la definizione di peso equivalente: PE = g/eq Il peso molecolare si ottiene da PM = PE · n dove n indica il numero di protoni titolati (reazioni acido-base) o di elettroni scambiati (reazioni redox). 3 BURETTA La buretta è un tubo graduato, fornito di un rubinetto di erogazione e calibrato in modo da poter dispensare volumi variabili, ma determinati esattamente, di un liquido. L'inizio della graduazione è all'estremità opposta a quella del rubinetto: l'indicazione si riferisce quindi ai volumi di liquido erogati dalla buretta. La faccia interna della buretta, opposta alla scala graduata, è provvista di una linea verticale scura su fondo bianco, in modo da agevolare la lettura in corrispondenza del menisco del liquido. La capacità e la precisione sono stampigliate vicino all'imboccatura della buretta. La buretta va utilizzata pulita ed asciutta; se è bagnata e non è possibile asciugarla, va avvinata prima dell'uso. Note operative • Il punto fragile della buretta è la saldatura del rubinetto al gambo, quindi usare due mani per azionare il rubinetto: una ruota la chiavetta e l’altra tiene il corpo del rubinetto (non la ghiera di serraggio), in modo da evitare torsioni alle parti in vetro. • controllare il rubinetto: il maschio conico in teflon va serrato quanto basta per assicurare la tenuta di aria e di liquido, senza incastrarsi nella sede in vetro; • assicurarsi che il rubinetto sia chiuso (chiavetta orizzontale); • riempire la buretta col liquido utilizzando un dosatore (vedi più avanti) o un bicchiere e arrivando quasi all'orlo; • eliminare le bolle d'aria eventualmente aderenti alle pareti picchiettando leggermente sul gambo della buretta; • riempire completamente il gocciolatore fino alla estremità inferiore aprendo il rubinetto e raccogliendo il liquido nel bicchiere predisposto (o in quello utilizzato per il riempimento); • la determinazione del volume erogato va abitualmente effettuata per differenza; all’inizio è quindi sufficiente agire sul rubinetto per portare il livello del liquido in corrispondenza di un qualsiasi punto della graduazione (non utilizzare assolutamente la pipetta Pasteur per accanirsi a raggiungere lo zero, mentre è buona norma operativa annotarsi i valori dei volumi iniziale e finale); • per una corretta lettura del volume erogato bisogna posizionare lo sguardo all'altezza del menisco; il menisco deforma la linea scura sul retro della buretta creando l'effetto di due frecce simmetriche a punte contrapposte: il livello attuale del liquido è alla congiunzione delle due frecce; • inserire la buretta nella apposita pinza di sostegno, fissata di solito all’asta di uno stativo; • erogare il liquido lentamente, per consentire al liquido di scolare lungo le pareti interne della buretta, così da evitare errori nella individuazione del volume effettivamente erogato; • terminato l'uso, la buretta va svuotata nel recipiente predisposto per il ricupero della soluzione avanzata; va quindi lavata con acqua deionizzata (usare la spruzzetta: 10-15 mL per 2-3 volte) e infine va fissata al suo sostegno col rubinetto aperto (chiavetta verticale) e rivolto verso l'alto in modo da facilitare l'asciugatura. DOSATORE E’ un apparecchio che consente di prelevare con buona accuratezza da un serbatoio volumi fissi o regolabili di un liquido. E’ sostanzialmente una siringa, provvista di tubi di pescaggio e di uscita controllati da un sistema di valvole comandato dal movimento del pistone; la corsa del pistone può prevedere un blocco a vite per selezionare il volume di liquido da aspirare ed erogare. Note operative • aspirazione – usare entrambe le mani: una tiene il corpo dell’apparecchio (non il beccuccio di erogazione) e l’altra alza il pistone fino a fondo corsa o al punto di fermo lentamente, per evitare l’errore di prelievo dovuto all’aspirazione indesiderata di aria; • erogazione: appoggiare il beccuccio sul bordo interno del recipiente di raccolta (buretta, bicchiere o matraccio) e abbassare il pistone fino a fondo corsa lentamente, per evitare spruzzi e quindi perdite della quantità erogata. 4 DETERMINAZIONE DELLA DUREZZA DELL'ACQUA Si definisce dura un'acqua con un contenuto elevato di sali di Ca2+ e Mg2+. Tali acque non formano schiuma con i saponi naturali (sali sodici di acidi carbossilici a lunga catena), perché i sali di Ca2+ e Mg2+ che si formano sono insolubili e impediscono così la funzione di tensioattivo del sapone. La durezza totale di un'acqua indica la concentrazione totale di ioni Ca 2+ e Mg2+; questa viene calcolata come CaCO3 ed espressa usualmente in gradi francesi. 1 grado francese corrisponde a 10 mg/L di CaCO3 La determinazione viene effettuata titolando con EDTA il campione di acqua in presenza di un tampone e di un indicatore appropriati. L'EDTA (acido etilendiamminotetracetico) è un acido tetraprotico (H4Y). Questa molecola possiede sei siti di coordinazione (è un legante esadentato) e forma complessi ottaedrici molto stabili (stechiometria 1:1) con i cationi bivalenti e trivalenti. Per questo motivo l'EDTA è spesso aggiunto come addolcente nei saponi e detersivi commerciali. 2- C O 2H O N H O 2C C O 2H O N N Ca N O O H O 2C Nelle analisi di complessazione si utilizza il sale bisodico biidrato (Na2H2Y·2H2O) in presenza di un tampone basico per spostare verso destra l'equilibrio: M 2+ + H 2Y 2 - MY 2 - + + 2 H L'informazione sul raggiungimento del punto finale della titolazione volumetrica viene fornita dal cambiamento di colore di un indicatore specifico per il metallo. Questi indicatori sono sostanze analoghe all'EDTA, nel senso che sono degli acidi poliprotici con la proprietà di complessare gli ioni metallici oggetto della titolazione. L'indicatore deve formare con lo ione metallico un complesso meno stabile di quello dell'EDTA e presentare colori diversi per la forma libera e per quella complessata. Il Nero Eriocromo T soddisfa queste condizioni per la coppia Ca2+, Mg2+: infatti con EDTA il complesso più stabile è quello di Ca2+, mentre con l’indicatore è quello di Mg2+. La sequenza delle reazioni e delle colorazioni è: M g 2+ + H In 2- M g In - + H C a2+ + H 2 Y C aY 2- M g In - + H 2Y 2- M gY 22- + + 2 H ro s s o v in o + + H In 2- + H + b lu Per l'indicatore è necessario operare in ambiente tamponato a pH = 10 (tampone ammoniacale) per neutralizzare l'acidità prodotta dalle reazioni di complessazione e soprattutto per favorire la formazione della specie blu HIn2- (a pH più elevati predomina la forma In3-, arancione). 5 POLARIMETRIA L'analisi polarimetrica sfrutta l'interazione tra una sostanza anisotropa (chirale non racema, ovvero otticamente attiva) ed un fascio di radiazioni polarizzate in un piano. Dopo aver attraversato la sostanza anisotropa, il piano di polarizzazione della radiazione risulta ruotato di un angolo α. La misura dell'angolo di rotazione viene effettuata con uno strumento chiamato polarimetro. L'angolo indicato dallo strumento dipende dalla lunghezza d'onda a cui si lavora, dallo spessore del campione attraversato dal fascio di radiazioni, dalla natura e dalla concentrazione del campione e dal valore del potere ottico rotatorio specifico (condizioni standard: lunghezza della cella = 1 dm; concentrazione del campione = 1 g/mL) determinato alla stessa lunghezza d'onda. In genere si opera alla lunghezza d'onda della linea D dello spettro di emissione del sodio (589 nm) e perciò si parla, in maniera semplificata, di determinazione dell'[α]D. L'angolo di rotazione dipende anche dalla temperatura e dal solvente: entrambi vanno quindi specificati. α [α]D = lc dove: α = angolo di rotazione letto allo strumento l = lunghezza della cella (dm) c = concentrazione (g/mL) [α]D = potere ottico rotatorio specifico La polarimetria è una tecnica utile per determinare la purezza enantiomerica di una sostanza chirale. Infatti l’attività ottica è una proprietà additiva e quindi l'angolo di rotazione letto dipende dalla concentrazione relativa dei due enantiomeri. Viene anche impiegata per valutare la concentrazione di una sostanza chirale enantiomericamente pura, noto il suo [α]D. 6 MISURA DEL pH, ELETTRODO A VETRO e TARATURA DEL pHMETRO Il pH di una soluzione acquosa viene misurato con un apparecchio detto pHmetro. Questo è un potenziometro, cioè misura la differenza di potenziale tra due elettrodi: uno, detto di riferimento, ha potenziale costante, mentre l'altro è detto indicatore o di misura ed il suo potenziale dipende dalla concentrazione di idrogenioni (H3O+) nella soluzione in cui viene immerso. Lo strumento è predisposto per presentare direttamente sul visore il valore del pH. Per le misure di pH si utilizza comunemente l'elettrodo a membrana di vetro. La parte attiva è un piccolo bulbo di vetro speciale e con pareti molto sottili (circa 0,1 mm), riempito con una soluzione a pH noto e stabile. Immergendo il bulbo in una soluzione a pH incognito, si genera un potenziale di membrana dovuto alla differenza di concentrazione degli ioni H 3O+ tra la soluzione interna e quella esterna al bulbo; questo potenziale varia linearmente con il pH della soluzione esterna. Note operative • L'elettrodo a vetro è molto fragile e delicato, in particolare il bulbo, che non va toccato con le dita, per evitare l'adsorbimento di unto dall'epidermide e nemmeno con tessuti o carta da filtro; • l'elettrodo in dotazione è del tipo combinato, cioè comprende anche un elettrodo di riferimento ad Ag/AgCl: è quindi necessario, per chiudere il circuito elettrico e poter effettuare la misura, che venga immerso nella soluzione almeno al disopra del minuscolo setto poroso posto poco sopra il bulbo. • prestare molta attenzione nell'immergere l'elettrodo nella soluzione, in modo da non urtare le pareti del recipiente o la barretta di agitazione magnetica (nelle titolazioni potenziometriche). • dopo l'uso (o a fine titolazione) l'elettrodo va immediatamente pulito lavando gambo e bulbo con getti di acqua deionizzata, impiegando la spruzzetta.; l’elettrodo deve poi essere subito immerso in acqua deionizzata, per evitare l'essiccamento della membrana di vetro. PROCEDIMENTO DI TARATURA Ogni elettrodo a vetro è unico (per forma e spessore delle pareti del bulbo) e inoltre si deteriora lentamente con l'uso. E' pertanto necessario effettuare la taratura dello strumento, con due soluzioni tampone se si intende effettuare una misura assoluta come quella del pH. Una volta eseguita la taratura, l'apparecchio non va più spento fino al termine di tutte le misure. Su alcuni pHmetri è previsto un pulsante MEASURE/STANDBY che disattiva solamente la lettura. • • • accendere lo strumento almeno 10 minuti prima dell’utilizzo; l’elettrodo combinato a vetro è già collegato ed immerso in acqua deionizzata; impostare, dal termometro in dotazione, il valore della temperatura ambiente (nell’equazione di Nernst il termine 0.05916 è riferito a 25°C = 298.14 K); • immergere con cautela l’elettrodo combinato nel bicchierino di plastica grande contenente il tampone a pH = 7.02 , lasciar stabilizzare la misura ed eseguire la calibrazione; • immergere con cautela l’elettrodo combinato nel bicchierino di plastica piccolo contenente il tampone a pH = 4.00, lasciar stabilizzare la misura ed eseguire la calibrazione; • sciacquare l’elettrodo con la spruzzetta ed immergerlo nel bicchiere con acqua deionizzata. A seconda del tipo di pHmetro, la calibrazione comporta operazioni differenti: strumento a calibrazione automatica per ciascun tampone: premere il tasto AC strumento a calibrazione manuale tampone 7.02: ruotare la manopola SLOPE fino al 100% e poi con la manopola STANDARDIZE portare la lettura sul visore a 7.02; tampone 4.00: utilizzando solo la manopola SLOPE, portare a 4.00 il valore sul visore. 7 TITOLAZIONE POTENZIOMETRICA E' una tecnica analitica quantitativa strumentale che consente di determinare la concentrazione di una soluzione sfruttando una reazione, ad esempio di scambio protonico (acido-base) o elettronico (redox), con un appropriato reagente (soluzione titolante a concentrazione nota). E' un metodo elettrochimico che utilizza un potenziometro per misurare la variazione della differenza di potenziale tra un elettrodo indicatore ed uno di riferimento durante la titolazione; per le titolazioni acido-base l’elettrodo di misura è quello a vetro, mentre è un elettrodo di platino per quelle di ossidoriduzione. I dati sperimentali (compresi quelli iniziali), vanno raccolti in una tabella nella quale ai mL di titolante progressivamente erogati dalla buretta (non le singole aggiunte) si affiancano le corrispondenti misure di pH (per le reazioni acido-base) o di potenziale (in mV, per le reazioni redox). A differenza delle titolazioni volumetriche con indicatore, il cui viraggio (punto finale della titolazione) fornisce direttamente l’informazione del completamento della reazione (punto di equivalenza), con la tecnica potenziometrica è necessario: • riportare su carta millimetrata i dati della tabella costruendo un grafico pH/mL o mV/mL (ordinate/ascisse); • ricavare dalla curva di titolazione il valore del volume equivalente per poter applicare la relazione quantitativa della titolazione: eq1 = eq2 1 = titolante 2 = titolando V1N1 = V2N2 V = volume N = normalità La curva ha un andamento sigmoidale, crescente o decrescente, con una variazione più o meno rilevante (dipende dal valore della Ka o dei potenziali standard coinvolti) in corrispondenza del punto di equivalenza. Si riportano due esempi per le titolazioni acido forte-base forte (A) e base debole-acido forte (B): A 14 12 10 pH 8 6 4 2 0 0 5 10 15 20 25 30 35 40 25 30 35 40 mL 12 B 10 8 pH 6 4 2 0 0 5 10 15 20 mL L’esame della curva di titolazione fornisce dei suggerimenti utili per una buona esecuzione di questa tecnica. Le parti iniziale e finale sono quasi rettilinee ed orizzontali (piccola variazione di pH o di potenziale con il volume di titolante aggiunto). La parte centrale ha invece caratteristiche opposte e, per una accurata determinazione del punto finale, è importante che i due ginocchi della curva siano molto dettagliati: bisogna cioè che le misure siano molto più ravvicinate. E’ quindi buona norma eseguire aggiunte di titolante non superiori a 0.5 mL finchè non si noti una variazione più consistente di pH (o mV); a questo punto le aggiunte vanno ridotte a non più di 0.2 mL e mantenute finchè la variazione di pH (o mV) ritorni quasi costante. Si possono poi riprendere le aggiunte di 0.5 mL fino ad un volume totale di titolante non inferiore ad una volta e mezza quello erogato in corrispondenza della variazione più rilevante di pH (o mV). Con l’elettrodo a vetro non deve essere comunque superato pH = 11, perchè a pH superiori la misura non è più attendibile ed inoltre l’elettrodo si deteriora più rapidamente. 8 VALUTAZIONE GRAFICA DEL PUNTO DI EQUIVALENZA Per determinare accuratamente il punto finale della titolazione (che è una buona approssimazione del punto di equivalenza) è conveniente utilizzare il metodo grafico delle tangenti parallele. • si tracciano due rette tangenti ai ginocchi della curva e parallele tra loro (non importa la loro pendenza); • si traccia poi un segmento di retta perpendicolare alle due rette e e se ne determina il punto di mezzo; • si traccia infine una terza retta parallela alle prime due e passante per il punto di mezzo; • il punto della curva individuato da questa terza retta corrisponde al punto di equivalenza e la sua proiezione sull'asse delle ascisse è il volume equivalente Veq. 14 12 10 8 pH punto di semititolazione 6 pK a 4 2 punto di equivalenza 0 0 5 10 15 20 25 30 35 40 mL DETERMINAZIONE DEL pKa DI UN ACIDO DEBOLE MONOPROTICO Dalla reazione di scambio protonico si risale immediatamente alla espressione della costante di H A + H 2O equilibrio: + H 3O + A - K a = [H 3 O + ] [ A -] [H A ] - [A ] e passando ai logaritmi: pKa = pH – log [HA] - Ora, al punto di semititolazione, cioé quando è stato titolato metà dell'acido, si ha che [HA] = [A ] e quindi il valore del pH è uguale al pK a dell'acido (a patto che l'acido non sia troppo debole e che la soluzione non sia troppo diluita). Il pH al punto di semititolazione può essere ricavato dal grafico dalla curva di titolazione, come illustrato nella figura. 9 SISTEMI POLIPROTICI, AMMINOACIDI Un sistema poliprotico è in grado di scambiare (cedere e/o accettare) più di un protone. Due esempi già incontrati sono l' EDTA (acido etilendiamminotetracetico), un acido tetraprotico (H 4Y) -e lo ione carbonato (CO3 ), una base diprotica.. Ciascuno scambio protonico viene descritto dalla rispettiva reazione acido-base ed è governato dalla corrispondente costante di equilibrio, analogamente a quanto si verifica per i sistemi monoprotici. Le costanti di equilibrio vengono abitualmente espresse come Ka e indicate K1 , K2 , .... Kn ; esse sono ovviamente riferite alla stabilità termodinamica di ciascuna forma acida (specie protonata) e presentano valori progressivamente decrescenti,. Questo rende immediata e agevole l’individuazione del protone più acido (più forte). Dalla titolazione potenziometrica di un sistema poliprotico si ottiene una curva che è la risultante della titolazione in successione di più sistemi monoprotici. Il volume equivalente richiesto per lo scambio di ciascun protone è evidentemente costante; infatti in ciascuna reazione di titolazione le moli di titolando coincidono con quelle iniziali. Dal grafico è quindi possibile ricavare sia il volume equivalente (si osservi che Veq n = n Veq1) che, ad ogni punto di semititolazione, la corrispondente Ka. L’unica differenza rispetto ai sistemi monoprotici si verifica al completamento dello scambio di ciascuno dei primi n–1 protoni: in soluzione- è presente solo l’anfolita (vedi più avanti). Un esempio già incontrato è lo ione idrogenoftalato (C8H5O4 ) . AMMINOACIDI Gli α-amminoacidi sono composti di formula generale R-CH(NH2)-COOH, ove R è un gruppo organico diverso per ogni composto; gli α-amminoacidi naturali sono 20. Nella molecola sono presenti sia un gruppo acido (-COOH) che uno basico (-NH2); in soluzione acquosa si verifica lo scambio protonico e pertanto gli amminoacidi sono in equilibrio con il loro sale interno (betaina o zwitterione): R-CH(NH2)-COOH ◄════► R-CH(NH3+)-COO- Il pH di una soluzione acquosa del solo amminoacido viene detto punto isoionico e l'amminoacido è prevalentemente presente come zwitterione. Di maggiore interesse pratico è invece il punto isoelettrico (il cui valore differisce peraltro di circa un decimo di unità di pH dal punto isoionico), che individua la situazione di immobilità elettroforetica (nessuna migrazione); viene realizzato sciogliendo l'amminoacido in un tampone appropriato, così da avere uguali concentrazioni delle due forme (acida e basica) derivate dallo zwitterione: [R-CH(NH3+)-COOH] = [R-CH(NH2)-COO-] Il comportamento acido-base degli amminoacidi è quello di sistemi poliprotici, diprotici nel caso più semplice. Le reazioni di scambio protonico di un amminoacido diprotico sono: 10 K1 e K2 hanno in genere valori ben diversi; spesso è possibile titolare separatamente i due protoni. La specie completamente protonata viene ottenuta per reazione dei gruppi basici (il solo -NH 2 nel caso più semplice) con un acido forte (di solito HCl); questo giustifica il nome d’uso adottato per individuarla (cloridrato). I cloridrati sono spesso utilizzati perchè più facilmente purificabili e molto solubili in acqua. R-CH(NH2)-COOH + HCl ────► R-CH(NH3+)-COOH + Cl- HAm·HCl In alcuni casi (acido aspartico, acido glutammico) il residuo R contiene un gruppo acido (COOH): in questo caso l'amminoacido completamente protonato è un acido triprotico. Analogamente, se il residuo R contiene un gruppo basico, la forma completamente protonata sarà un bis-cloridrato. Lo zwitterione è un anfolita, cioé una specie che può comportarsi sia da acido che da base: NH2 + H3 O R NH3 H2 O COO R NH3 H2 O COO + OH R COOH Può quindi essere titolato con un acido: R-CH(NH3+)-COO- + HCl ────► R-CH(NH3+)-COOH + Cl- o anche con una base: R-CH(NH3+)-COO- + NaOH ────► R-CH(NH2)-COO- + Na(H2O)+ Il pH di una soluzione di un anfolita è correlato alle K a adiacenti dalla relazione (per un acido diprotico): pK1 + pK2 pH = 2 Sulla curva di titolazione, questo è il pH del primo punto di equivalenza, cioé al completamento dello scambio del protone più acido. Un sistema poliprotico HnA presenterà quindi n-1 anfoliti. 11 EQUILIBRI DI OSSIDORIDUZIONE Questi processi riguardano lo scambio di uno o più elettroni tra due coppie redox: o x 1 + red red 1 + o x2 2 L'analogia con le reazioni acido-base è evidente ed agevola il richiamo dei punti essenziali. Il potere ossidante, espresso quantitativamente dal valore del potenziale redox, indica la tendenza termodinamica della forma ossidata ad accettare elettroni e consente anche di individuare la direzione spontanea della reazione; gli elettroni verranno ovviamente scambiati tra la forma ossidata di una coppia redox e la forma ridotta dell'altra coppia. E' opportuno scrivere le equazioni parziali (reazioni elettrodiche) in forma ionica e bilanciarle separatamente, per poi combinarle nel processo globale in modo da compensare esattamente gli elettroni scambiati (si assume qui che E oxa > E oxb). ox a + a e red a x b ox b + b e - red b x a La reazione globale è b ox a + a red b b red a + a ox b Dalle reazioni elettrodiche si può immediatamente passare alla valutazione del potenziale di elettrodo applicando la legge di Nernst: 0 ,0 5 9 1 6 a lo g [o x a] [re d a ] = E ° b + 0 ,0 5 9 1 6 b lo g [o x b] [re d b ] E a= E °a + E b L'analogia con i processi di scambio protonico si estende anche alle applicazioni quantitative: titolazioni volumetriche e potenziometriche. Per le prime è necessario ricorrere a un indicatore appropriato (indicatore redox) che fornisca l'informazione sul raggiungimento del punto finale sfruttando la formazione o la scomparsa di una colorazione al consumo completo del titolando o al primo eccesso di titolante. Nelle titolazioni potenziometriche ossidimetriche l’apparecchio è lo stesso già utilizzato come pHmetro, predisponendo però il visore per la lettura in mV e costruendo il sistema elettrochimico con un elettrodo di misura costituito da un filo di platino. Per le titolazioni redox non è necessaria la taratura del potenziometro, perché ad ogni aggiunta di titolante interessa solo la variazione del potenziale e non il suo valore assoluto. Bisogna comunque compilare la tabella di titolazione e poi costruire il grafico per poter risalire al volume equivalente. L'unica differenza con i processi acido-base si ha nel punto iniziale della titolazione potenziometrica. Come si può osservare dall'equazione di Nernst, all'inizio della titolazione l'argomento del logaritmo vale zero o infinito, e quindi il valore del potenziale è privo di senso; la tabella inizia quindi con i dati della prima aggiunta di titolante. La relazione quantitativa della titolazione è: Va Na = Vb Nb Tenere presente che la normalità ossidimetrica fà riferimento al numero di elettroni scambiati nella pertinente reazione elettrodica. 12 TITOLAZIONE VOLUMETRICA OSSIDIMETRICA DI Fe2+ CON MnO4Un modo semplice per valutare quantitativamente il contenuto di Fe(II) di una soluzione è di effettuare una titolazione ossidimetrica (redox) dello ione ferroso, utilizzando come ossidante il permanganato di potassio (KMnO4) in presenza di H2SO4. Nelle titolazioni con KMnO4 è importante il pH: infatti, in ambiente basico lo ione permanganato viene ridotto a biossido di manganese (MnO2), marrone e assai poco solubile, mentre in ambiente acido la riduzione procede fino a Mn2+, rosa, ma praticamente incolore se la soluzione è diluita. La soluzione di permanganato di potassio è di color viola intenso e nelle titolazioni in ambiente acido non è necessario un indicatore: infatti il punto di equivalenza viene segnalato dalla prima goccia di soluzione di permanganato in eccesso, che impartisce alla soluzione titolata una lieve colorazione rosa-violacea persistente. Va infine tenuto presente che, al termine della titolazione, il lieve eccesso di MnO 4- presente in soluzione può riossidare il Mn2+: 2 MnO4 - + 3 Mn 2+ + 2 H2O ─────► 5 MnO2 + 4 H + o anche, essendo in ambiente nettamente acido, può decomporsi: 4 MnO4 - + 4 H + ─────► 4 MnO2 + 3 O2 + 2 H2O Queste reazioni che generano MnO2 sono relativamente lente a temperatura ambiente e quindi non interferiscono con la corretta esecuzione della titolazione; è però importante sciacquare immediatamente e bene la vetreria (bicchieri e burette) utilizzata per le soluzioni di permanganato, in modo da evitare la formazione di depositi di MnO2, difficili da eliminare. Poichè il manganese è un metallo pesante, fortemente inquinante, è obbligatorio scaricare nel bidone dei reflui sia le prime acque di lavaggio della vetreria che la soluzione titolata. TITOLAZIONI IODOMETRICHE La coppia redox iodio/ioduro (I2/I-) è di uso assai conveniente in laboratorio. E' infatti disponibile un indicatore di ossidoriduzione estremamente efficace, la salda d'amido, che assume un colore blu intenso in presenza di I2, mentre è incolore quando in soluzione rimane solo I-. Il titolante specifico per lo iodio è il tiosolfato (S 2O32-), che riduce lo iodio a ioduro e si ossida a sua volta a tetrationato (S4O62-). Lo iodio è poco solubile come tale in soluzione acquosa; la presenza di un eccesso di ioni ioduro contribuisce a solubilizzarlo, perché si forma la specie ionica I 3- che viene adsorbita sull’amido. Nell'esercitazione proposta in laboratorio lo iodio viene prodotto mediante una reazione di ossidoriduzione tra una soluzione di KMnO4 a titolo noto ed un largo eccesso di KI. Gli equivalenti di iodio che si formano corrispondono ovviamente a quelli di permanganato impiegati. Lo iodio prodotto viene poi in parte utilizzato per ossidare un campione di acido ascorbico; lo iodio rimasto viene infine titolato con una soluzione di tiosolfato a molarità nota (titolazione indiretta o retrotitolazione): noti gli equivalenti totali di iodio formati e quelli titolati con tiosolfato, si può risalire agli equivalenti di iodio che hanno reagito con l’acido ascorbico e quindi alla quantità di acido ascorbico presente nel campione. O HO O O HO + I2 CH2OH HO O O O + 2I- + 2 H+ CH2OH HO 13 METODI SPETTROMETRICI La spettrometria studia le interazioni tra la radiazione elettromagnetica (una forma di energia) e la materia. Si possono osservare e studiare due tipi di fenomeni: la radiazione può essere assorbita dalla materia, oppure la materia (opportunamente eccitata) può emettere radiazione. Qui si farà riferimento solo ai fenomeni di assorbimento. La radiazione elettromagnetica può essere trattata ricorrendo a due modelli, complementari tra loro. Nel modello ondulatorio, la radiazione è un'onda elettromagnetica (descrivibile da un campo elettrico ed un campo magnetico ortogonali l'uno all'altro e oscillanti), caratterizzata da una lunghezza d'onda (λ) e da una frequenza di vibrazione (ν). Queste grandezze sono collegate alla velocità di propagazione della radiazione dall’espressione: c = λν Il secondo modello, quello corpuscolare, considera la radiazione elettromagnetica come un flusso di particelle, i fotoni. L'energia dei fotoni è quantizzata ed è correlata alla frequenza dalla relazione: E = hν dove h è la costante di Plank. La quantizzazione dell'energia rende immediata l'applicazione della spettroscopia su scala molecolare. L'interazione tra radiazione e materia può infatti avvenire solo se la differenza di energia tra due livelli energetici della molecola corrisponde all’energia della radiazione incidente. Pertanto la materia interagisce con la radiazione in maniere diverse a seconda dell'energia della radiazione impiegata: - radiazioni di energia elevata (> 300 Kcal/mol: raggi γ, raggi X) sono in grado di estrarre un elettrone degli orbitali interni di un atomo, fino a provocarne la ionizzazione; - le radiazioni nel campo dell'ultravioletto (UV) e del visibile (10-780 nm; 300-40 Kcal/mol) hanno energia sufficiente solo per eccitare gli elettroni di valenza; - radiazioni di energia ancora inferiore provocano la vibrazione (stiramento o deformazione) dei legami nelle molecole (radiazioni infrarosse: 1-10 Kcal/mol) oppure la rotazione molecolare (microonde: <1 kcal/mol) o agiscono sullo spin nucleare (onde radio: 10-6 Kcal/mol). Quando una radiazione di energia opportuna attraversa un campione, una frazione di essa può essere assorbita; questo assorbimento è proporzionale alla concentrazione della specie che assorbe e allo spessore di soluzione attraversato dalla radiazione. La relazione quantitativa sperimentale che regola l’assorbimento è la legge di Lambert-Beer: dove: A = log I0/I = ε l c A = assorbanza (o densità ottica o estinzione): rappresenta la frazione di energia radiante assorbita I0 e I = intensità delle radiazioni, rispettivamente incidente e trasmessa; l = cammino ottico, cioè lo spessore (in cm) della soluzione attraversato dalla radiazione; c = concentrazione (mol/L) della specie assorbente; ε = coefficiente di estinzione molare: è l'assorbanza a concentrazione e cammino ottico unitari; dipende dalla lunghezza d'onda della radiazione, dalla specie assorbente e dal solvente. L'assorbanza di un campione viene misurata in funzione della lunghezza d'onda della radiazione assorbita con un apparecchio chiamato spettrometro. Il campione viene posto in una cella, o cuvetta, di materiale opportuno (ad esempio, per indagini con radiazioni nel campo UV, la cuvetta è in quarzo, mentre può essere in vetro se si impiegano solo radiazioni nel campo del Visibile). Misurando l'assorbanza ad una lunghezza d'onda fissa e variando la concentrazione del campione si dovrebbe ottenere una retta (in ordinate A e in ascisse c), il cui coefficiente angolare corrisponde a ε. In realtà non sempre, a parità di lunghezza d'onda, il valore di ε è costante per una specie chimica al variare della sua concentrazione, a causa di fattori di natura chimico-fisica o strumentale. Prima di accingersi ad analisi spettrometriche quantitative è necessario perciò verificare la validità della legge di Lambert-Beer costruendo una retta di taratura (vedi più avanti). Le letture allo spettrometro vanno inoltre corrette per tener conto dell'inevitabile assorbimento dovuto al solvente e alla cella: lo strumento va cioé azzerato. Si deve preparare una soluzione di riferimento (bianco) contenente il solvente e tutti gli altri reattivi eventualmente presenti in soluzione, escluso l'analita: questo spettro, registrato nella stessa cuvetta, viene sottratto automaticamente a quelli ottenuti per l'analisi vera e propria, in modo da avere come risultato l’assorbimento della sola specie di interesse. 14 DETERMINAZIONE SPETTROMETRICA DEL TENORE DI Fe(II) IN UNA SOLUZIONE ACQUOSA La spettrometria UV-Visibile viene largamente impiegata per analisi quantitative, perchè consente di determinare con accuratezza concentrazioni di analita estremamente basse (fino a 10-6 M). L'attendibilità dei dati sperimentali è legata alla costruzione della retta di taratura: si procede effettuando misure di assorbanza su alcune soluzioni dell'analita a concentrazione nota e di ordine di grandezza simile a quella della soluzione da analizzare; si riportano in grafico i dati ottenuti e si traccia la retta migliore, passante teoricamente per l'origine, a meno di piccoli scostamenti dovuti ad errori casuali. Misurando quindi l'assorbanza di una soluzione del campione, dalla retta si può risalire alla concentrazione incognita. Non tutte le specie assorbono in maniera soddisfacente per le applicazioni analitiche nel campo dell'UVVisibile; spesso è però possibile trasformarle quantitativamente in altre specie con assorbimento più intenso. E' questo il caso nella determinazione spettrometrica del Fe(II) in soluzione: lo ione metallico viene complessato con il legante 1,10-fenantrolina, formando un complesso di stechiometria definita e caratterizzato da un massimo di assorbimento nel campo del Visibile. 2+ N N + Fe2+ 3 N F e ( fe n )3 2 + N Fe N N N N (fe n ) La formazione quantitativa del complesso viene assicurata controllando il pH della soluzione: a pH acidi infatti la fenantrolina (una base) viene protonata e non può più fungere da legante, mentre a pH basici il Fe(II) precipita come idrossido e non può essere complessato. E' inoltre necessario impedire l'ossidazione del Fe(II) a Fe(III): questo processo è estremamente favorito (basta l'ossigeno dell'aria), ma il Fe(III) viene complessato con meno efficacia dalla fenantrolina e questo altererebbe i risultati dell'analisi. Alla soluzione di Fe(II) vengono pertanto aggiunti cloridrato di idrossilammina (si usa il cloridrato perché l'idrossilammina come tale non è stabile in soluzione a temperatura ambiente) e acetato di sodio. L'idrossilammina è una base debole e un riducente; l'acidità prodotta nella reazione redox forma un sistema tampone con l'acetato sodico. L'azione complessiva dei due reagenti è quindi quella di un tampone acidimetrico e redox. N H 2O H •H C l 2 N H 3O H + + 2 H 2O Fe 3+ + e CH 3 C O O - + H O 3 + N H 3O H N 2 + Fe 2+ + 2 e + - C l - + 4 H 3O + C H 3C O O H + H 2O 15 METODI CROMATOGRAFICI Il primo di questi è stato scoperto nel 1903 dal botanico russo Tswett (insignito per questo del premio Nobel) nella separazione dei pigmenti verdi delle foglie su carbonato di calcio; il nome deriva proprio dall'ottenimento di una successione di zone colorate. Si può proporre questa definizione generale: la cromatografia consiste nella distribuzione ripetuta di una sostanza tra le due fasi immiscibili che costituiscono il sistema cromatografico. La fase fissa o stazionaria è un solido o un liquido immobilizzato su un opportuno supporto; la fase mobile o eluente è un liquido o un gas che fluisce attraverso la fase stazionaria trasportandovi il campione. Le applicazioni analitiche della cromatografia sono immediatamente intuibili. I componenti di una miscela presenteranno ciascuno interazioni diverse con il sistema cromatografico. La fase mobile, mettendo a contatto ogni componente con una nuova porzione di fase stazionaria, esalterà progressivamente la differenziazione, consentendo la separazione. I metodi cromatografici si distinguono a seconda del tipo di interazione, cioè del meccanismo che determina la diversa mobilità delle sostanze nel sistema cromatografico: adsorbimento, ripartizione, scambio ionico, esclusione molecolare, affinità. Cromatografia di adsorbimento: la fase stazionaria è un solido in grado di trattenere (adsorbire) più o meno fortemente i componenti di una miscela mediante interazioni quali forze di Van der Waals e legami a idrogeno. La fase mobile può essere sia un liquido che un gas. Più la sostanza rimane adsorbita sulla fase stazionaria, più lento sarà il suo moto lungo di essa. Tanto maggiore è la solubilità della sostanza nella fase liquida, tanto meno la sostanza resterà adsorbita sulla fase stazionaria ed il suo moto lungo di essa sarà più veloce. Gel di silice, allumina (potere adsorbente forte); calcio carbonato (potere adsorbente medio); saccarosio, amido, talco (potere adsorbente debole) sono le fasi stazionarie più comuni. Sono solidi a granulometria controllata e diversa a seconda degli usi (più le particelle sono piccole, più la superficie di interazione è grande e maggiore è il potere separatore). Le fasi mobili sono liquidi puri o miscele, oppure un gas. Quando la fase mobile è liquida, sono fondamentali il suo potere solvente e la sua polarità (vedi più avanti). Ecco, in ordine di polarità crescente, alcuni degli eluenti liquidi più comuni: esano < cicloesano < etere etilico < cloroformio < diclorometano < acetone < acetato di etile < acetonitrile < etanolo < acido acetico. Cromatografia di ripartizione: si sfrutta la diversa solubilità delle sostanze in due liquidi immiscibili. All'equilibrio una sostanza si distribuisce tra i due liquidi A e B in accordo con il coefficiente di ripartizione: K = CB/CA dove CA e CB sono in prima approssimazione le concentrazioni all'equilibrio della sostanza in A e B. Sfruttando la diversa solubilità delle sostanze, queste si possono separare con una scelta appropriata dei due solventi. Cromatografia a scambio ionico: si basa su interazioni elettrostatiche selettive tra ioni di carica opposta in soluzione e la fase stazionaria solida e ionica (resina polimerica). Cromatografia di esclusione molecolare: si sfrutta la diversa dimensione delle molecole, impiegando come fase stazionaria un solido il cui reticolo cristallino è caratterizzato da una struttura regolare di canali e cavità. Solo le molecole abbastanza piccole da entrare nella struttura porosa vengono trattenute, da forze di Van der Waals (esempio: filtrazione su gel). Cromatografia di affinità: si basa sull'affinità specifica di una sostanza per un recettore ancorato sulla fase stazionaria (esempio: anticorpi immobilizzati). 16 INTERAZIONI CROMATOGRAFICHE E SCALA DI POLARITA’ Si esaminano ora in maggior dettaglio le forze responsabili del processo di separazione nelle cromatografie di adsorbimento e ripartizione. Si adotta il termine interazioni perchè le energie coinvolte sono di almeno un ordine di grandezza inferiori a quelle di un legame chimico. E’ opportuno richiamare brevemente i concetti di polarità e di polarizzabilità di una molecola; questo richiede di avere presente la distinzione tra i tipi di legame σ e π ed anche di disporre della formula di struttura della molecola. La polarità di una molecola deriva dalla presenza di atomi molto elettronegativi o di gruppi particolari di atomi (ad esempio NO2 , SO3H , CO , COOH , OR , NR2 , ...) che influenzano la distribuzione degli elettroni di legame, provocando una asimmetria tra i baricentri delle cariche positive (i nuclei) e di quelle negative e instaurando così un dipolo permanente. La polarizzabilità di una molecola dipende invece da quello che si può chiamare il grado di libertà degli elettroni all’interno della molecola stessa e vale sia per le specie neutre intrinsecamente apolari (nessun dipolo permanente) che per quelle complessivamente non polari (risultante nulla dei dipoli permanenti). Considerando in particolare la nuvola elettronica π, questa è facilmente deformabile dall’azione di un campo elettromagnetico. La presenza, vicino ad una molecola con questo tipo di legami, di una molecola polare, cioè di un dipolo permanente, influenzerà allora la distribuzione degli elettroni π, dando luogo alla formazione di un dipolo indotto. Le forze che agiscono tra le molecole della sostanza, della fase mobile e la fase stazionaria o l’adsorbente sono classificabili in tre tipi, in ordine crescente di intensità (da pochi decimi a 4-6 kcal/mol). Forze di dispersione. Il continuo movimento di tutti gli elettroni della molecola attorno ai nuclei può provocare una temporanea asimmetria della nuvola elettronica, con formazione di dipoli istantanei; la presenza di tali dipoli transienti a sua volta provoca la formazione, nelle molecole vicine, di dipoli indotti, altrettanto rapidamente e casualmente variabili sia in intensità che orientazione. L’interazione che risulta da queste momentanee polarizzazioni delle nuvole elettroniche può essere attrattiva, se i dipoli istantanei sono in fase tra di loro; oppure repulsiva, nel momento in cui le nuvole elettroniche si avvicinano troppo. Queste interazioni, la cui intensità è proporzionale al numero totale degli elettroni, sono presentate da tutte le molecole e sono le uniche che si possono esercitare tra molecole apolari. Forze di induzione. Sono originate, in maniera analoga a quelle di dispersione, dall’interazione tra il dipolo permanente di una molecola ed il dipolo indotto in una molecola polarizzabile. Si presentano se nella molecola ci sono elettroni π appartenenti a legami doppi o tripli tra atomi uguali; sono interazioni più marcate, perché i dipoli sono di maggiore intensità. Forze di orientazione. Sono dovute alle interazioni tra dipoli permanenti e portano a rilevanti associazioni molecolari. L’esempio più significativo e di maggiore intensità è la formazione dei legami a idrogeno. Una molecola può essere coinvolta in tutti e tre i tipi di interazione; se il campione è costituito da più sostanze, è senz’altro utile poter fare una previsione almeno qualitativa della diversa ritenzione che ciascun componente presenterà nel sistema cromatografico utilizzato (fase stazionaria + fase mobile). Il modo più semplice è quello di costruire delle scale di polarità. - Per il sistema cromatografico sono possibili solo due situazioni: la fase stazionaria è più polare della fase mobile oppure viceversa; nella terminologia in uso vengono indicate rispettivamente con sistema in fase normale e sistema in fase inversa. - Per il campione il criterio è esattamente lo stesso: si stabilisce quale sia il più polare in una coppia qualunque di componenti, poi si forma una nuova coppia con un altro componente e così via fino ad individuare la molecola a polarità più elevata; si ripete poi il procedimento ottenendo infine la successione dei componenti in ordine di polarità crescente. Per prevedere, con soddisfacente attendibilità, l’ordine di eluizione dei vari componenti è sufficiente richiamare che le interazioni più intense si verificheranno tra la fase più polare del sistema cromatografico ed il componente più polare; quest’ultimo verrà perciò più trattenuto (trascorrerrà un tempo maggiore) in questa fase che non nell’altra, meno polare. Se ad esempio il sistema è in fase normale l’eluizione dei componenti del campione avverrà allora in ordine di polarità crescente. Le tecniche cromatografiche, cioè le modalità di esecuzione, vengono classificate secondo due criteri: - lo stato fisico della fase mobile: cromatografia liquida o gassosa; - la disposizione della fase stazionaria: cromatografia piana o cromatografia su colonna. 17 La grandezza operativa che esprime il risultato delle interazioni di ciascun componente con il sistema cromatografico viene detta parametro di ritenzione e viene indicata in maniera diversa a seconda della tecnica utilizzata. CROMATOGRAFIA SU STRATO SOTTILE In laboratorio verrà utilizzata questa semplice tecnica cromatografica, indicata abitualmente con il suo acronimo inglese TLC (Thin Layer Chromatography). Essa fà parte delle tecniche di cromatografia piana, perché la fase stazionaria viene depositata su un supporto piano. Si tratta di una cromatografia di adsorbimento; come fase stazionaria si utilizzano prevalentemente silice o allumina, depositate in spessore sottile (0.1 – 0.25 mm) su una lastra di vetro o su un foglio di alluminio o di poliestere. La TLC è un'ottima tecnica di analisi qualitativa, ad esempio per individuare o confermare la presenza di un componente in una miscela, per controllare la purezza di un prodotto o anche per seguire il decorso di una reazione. E' una tecnica rapida che non necessita di apparecchiature sofisticate. Note operative • prendere una lastrina, tenendola per i bordi più lunghi, senza appoggiare le dita sulla fase stazionaria e tracciare leggermente con la matita una linea a circa 1 cm dal bordo di uno dei lati corti; • deporre sulla riga, equidistanti tra di loro e dai bordi, con l'aiuto di un capillare, alcune gocce di soluzione delle diverse sostanze (qui A e B si riferiscono alle sostanze pure, mentre A+B è la miscela delle due sostanze da separare); • si pone la lastrina in una vaschetta ben chiusa contenente l’eluente appropriato, il cui livello deve essere al disotto della linea di deposizione: in caso contrario, le sostanze si scioglierebbero nella fase mobile compromettendo l’eluizione; • la fase mobile sale per capillarità lungo la fase stazionaria e le sostanze vengono eluite in maniera differenziata; 18 quando il solvente è a circa 1 cm dal bordo superiore della lastrina, questa viene estratta dalla vaschetta e si segna immediatamente il punto di arrivo (fronte del solvente); • si analizza la lastrina: se tutte le macchie sono visibili l’analisi è immediata, altrimenti si ricorre ad una lampada UV, oppure si spruzza la lastrina con un reattivo appropriato che colori le macchie delle sostanze, ma non la fase stazionaria. • • si calcola il parametro di ritenzione Rf di ciascuna macchia come rapporto tra il percorso della sostanza e il percorso dell’eluente; è quindi evidente che i valori limite di R f sono 0 e 1 (qui si ha che Rf (A) < Rf (B) e infatti B viene eluito più velocemente di A). GASCROMATOGRAFIA La gascromatografia è una tecnica strumentale di grande importanza e diffusione per l'analisi qualitativa e quantitativa di miscele gassose o volatilizzabili, con limiti di rivelabilità dell'ordine delle ppm. La separazione cromatografica si attua in una colonna (generalmente di vetro o di acciaio) riempita con un’opportuna fase stazionaria: questa può essere un adsorbente solido (cromatografia di adsorbimento) o un liquido poco volatile depositato su supporto solido inerte (cromatografia di ripartizione). La fase mobile è sempre un gas (azoto, elio, argon, idrogeno), la cui scelta è dettata dalle caratteristiche del rivelatore; svolge solo un'azione di trascinamento (gas di trasporto) lungo la colonna delle sostanze da separare e pertanto le sue interazioni con i componenti del campione sono trascurabili. Schema di un gascromatografo 19 La miscela da analizzare viene sciolta in un solvente e iniettata in minime quantità (tipicamente, 1-10 µL) nella camera di vaporizzazione, dove tutti i componenti vengono trasformati allo stato gassoso e quindi trasportati dalla fase mobile attraverso la colonna contenente la fase stazionaria. La colonna è alloggiata in una camera termostatica che viene mantenuta alla temperatura opportuna. Le sostanze in uscita dalla colonna passano nel rivelatore, generando un segnale elettrico, che viene poi amplificato e inviato ad un registratore. Si ottiene così il cromatogramma: un grafico che riporta in ordinate l'intensità del segnale e in ascisse il tempo trascorso dall'introduzione del campione. Analisi gascromatografica di una benzina Ad ogni sostanza che esce dalla colonna corrisponde un picco nel cromatogramma, individuato dal tempo di ritenzione tR , cioè il tempo trascorso tra l'introduzione del campione e il raggiungimento del massimo del picco. Il parametro di ritenzione tR dipende dalla sostanza, dalla natura della fase stazionaria e dalle condizioni di esercizio dell’analisi (temperatura della colonna, flusso del gas); l’assegnazione dei picchi di un cromatogramma perciò viene effettuata esclusivamente per confronto con il tempo di ritenzione dei singoli componenti puri, mantenendo costanti le condizioni operative. E’ di fondamentale importanza che non si verifichi sovrapposizione tra i picchi del cromatogramma, perchè la quantità di sostanza è direttamente proporzionale all’area del picco, che deve essere quindi valutata in maniera non ambigua. Per ottenere una separazione ottimale si agisce sulle condizioni di esercizio dell’analisi, variando la pressione del gas di trasporto o la temperatura cui è termostatata la colonna. Il segnale prodotto dal rivelatore è però specifico per ogni sostanza, nel senso che a quantità uguali di sostanze diverse possono corrispondere aree diverse: è perciò necessaria un’operazione di taratura dell’analisi. Il procedimento più semplice ed affidabile richiede di individuare una opportuna sostanza di riferimento (standard), per stabilire la relazione esistente per ogni componente tra la sua quantità e l’area del picco corrispondente. Si riporta il procedimento per il metodo dello standard esterno (la sostanza di riferimento viene aggiunta al campione, ma non deve essere conteggiata nel calcolo della composizione): • si preparano alcune miscele di taratura a composizione nota, si aggiunge a ciascuna una quantità nota dello standard e si effettua l’analisi; dal cromatogramma si desumono le aree dei vari picchi, di norma calolate automaticamente dal dispositivo di registrazione del cromatogramma, che per questo motivo viene chiamato integratore; 20 • si applica ad ogni coppia componente/standard la seguente equazione, nella quale l’unica incognita è il termine fi, che può essere agevolmente ricavato per ogni componente della miscela : Ai ⋅ fi As ⋅ fs = qi qs dove A = area dei picchi dello standard (s) e del componente (i) q = quantità di standard e di componente (espressa in moli o in peso) f = fattore di risposta (assunto ovviamente unitario per lo standard) • si analizza il campione incognito cui è stata aggiunta una quantità nota di standard e si applica ancora la stessa relazione ad ogni picco del cromatogramma: ora la sola incognita è qi e si può così risalire alla composizione della miscela incognita. CENNI AD ALTRE TECNICHE CROMATOGRAFICHE Una tecnica molto usata è la cromatografia su colonna. In questo caso la fase stazionaria è contenuta in un tubo di vetro; la miscela da separare viene caricata in testa alla colonna e la fase mobile viene percolata per gravità attraverso la fase stazionaria, eluendo i componenti la miscela con velocità diverse. La fase mobile viene raccolta all’uscita della colonna in appositi recipienti e le diverse frazioni possono essere ulteriormente analizzate. La cromatografia su colonna è un'ottima tecnica di separazione e purificazione di prodotti da miscele di reazione. fase mobile Tra le tecniche di cromatografia piana, da segnalare la cromatografia su carta, una tipica tecnica di ripartizione: una fase liquida polare (in genere acqua) impregna un foglio di carta speciale (le fibre di cellulosa presentano tutte la stessa orientazione, parallela alla direzione di eluizione). Dopo aver deposto sulla carta impregnata le sostanze da analizzare, si eluisce con un solvente scarsamente miscibile con acqua (ad esempio fenolo, butanolo) Anche l'elettroforesi su carta, molto usata in chimica e biochimica, è una tecnica di cromatografia piana; essa sfrutta la diversa mobilità degli ioni sotto l'azione di un campo elettrico. Su una striscia di carta imbevuta di un tampone opportuno viene depositata la miscela delle sostanze ioniche da separare, poi si applica alle due estremità opposte della carta, attraverso due elettrodi, una differenza di potenziale. I vari ioni migrano con velocità diverse. HPLC (High Performance Liquid Chromatography) è una tecnica strumentale di grande importanza sia per l'analisi di miscele che per la loro separazione. La fase mobile liquida viene spinta a pressione elevata (fino a 400 atm) attraverso la colonna riempita con una fase stazionaria a granulometria estremamente fine. La procedura è analoga a quella descritta per la gascromatografia: si inietta il campione in testa alla colonna; si alimenta la fase liquida e si eluisce la miscela. Le sostanze in uscita vengono analizzate da un rivelatore che ne misura ad esempio l'assorbimento UV o l’indice di rifrazione. Il cromatogramma che si ottiene è analogo a quello delle analisi gascromatografiche. All’uscita della colonna si possono raccogliere frazioni della fase mobile e recuperare da queste i componenti separati della miscela. 21 CROMATOGRAFIA A SCAMBIO IONICO I composti ionici sono spesso separati ed analizzati nel modo migliore mediante cromatografia a scambio ionico con l’aiuto di scambiatori di ioni come fase stazionaria. Questi consistono di materiali a cui sono legati attraverso legami covalenti gruppi carichi (positivamente o negativamente). La carica è bilanciata da una un numero equivalente stechiometrico di ioni del segno opposto, i controioni. I controioni possono essere scambiati con le molecole del campione recanti carica simile. La presenza nel soluto di uno ione con la stessa carica del controione instaura un equilibrio. Per esempio, se il controione è Na+ per lo scambio cationico e Cl- in quello anionico, le equazioni di equilibrio sono le seguenti: K = [X+]resina [Na+] / [Na+]resina [X+] K = [X-]resina [Cl-] / [Cl-]resina [X-] dove [specie]resina è la concentrazione della specie in fase resina Maggiore è il valore di K, il coefficiente di ripartizione, più forte è l’interazione del soluto ionico con lo scambiatore. Il fattore di ripartizione è funzione di molti parametri, quali: pH, carica dello ione, raggio dello ione, porosità della resina, forza ionica della soluzione, solvente, temperatura, etc. A B Nella Figura è riportato uno schema pittorico di: A) interazione della fase fissa carica positivamente con un soluto carico negativamente; quanto maggiore è la carica coinvolta, tanto maggiore è la stabilità del complesso che si forma (dall’alto in basso: 1, 2, 3 cariche) B) scambio fra ioni presenti nella soluzione aventi carica simile al composto coordinato alla resina e il composto precedentemente legato; maggiore è l’interazione preesistente, maggiore è il numero di interazioni alternative che si devono instaurare per “soppiantare” il legame. 22 Meccanismo della separazione cromatografica Il comportamento di uno ione durante la separazione dipende dall’equilibrio che esso stabilisce con i gruppi attivi (carichi) della resina, che prima di introdurre il campione si trovano associati ad un determinato tipo di contro ione. Legame dello ione alla resina Consideriamo una resina di tipo cationico in forma sodica (Re-Na+). Quando su questa resina passa una soluzione contenente ioni K+ , si instaura il seguente equilibrio: Re-Na+ + K+ Re-K+ + Na+ [Re-K+] [Na+] kK/Na = ——————— [Re-Na+] [K+] Questa costante (coefficiente di selettività) esprime l’affinità della resina in forma sodica per un determinato ione (K+). Eluizione Se si immette nella resina un altro catione (disciolto nella fase mobile) che ha un’affinità per la resina maggiore di K+, gli ioni K+ vengono spostati dalla resina più in basso nella colonna; lo stesso avviene quando si immette in colonna il controione Na + in elevate concentrazioni. La migrazione di K+ avviene, dunque, per effetto di fenomeni di adsorbimento-desorbimento dei gruppi attivi della resina. Fasi stazionarie A seconda della loro funzione, i riempimenti a scambio ionico sono scambiatori di cationi oppure di anioni. Gli scambiatori di cationi contengono gruppi acidi solfonici (fortemente cationici) o gruppi acidi carbossilici (debolmente cationici), mentre gli scambiatori di tipo anionico hanno gruppi ammonio quaternari. Consistono solitamente di polimeri ad alto peso molecolare o di gel di silice a cui vengono legati covalentemente dei gruppi ionici. Materiali a base silicea hanno maggior resistenza meccanica; grazie a questa, essi resistono a contropressioni ed espansioni con sbalzi di pressione e non danno luogo a rigonfiamenti o contrazioni al variare dei pH o della forza ionica come fanno i materiali a base di gel polimerici. 23 La fase mobile Vi sono due variabili della fase mobile che interessano la ritenzione di un soluto ionico in una colonna a scambio ionico: la forza ionica il controllo del pH La più importante di esse è la regolazione della forza ionica, che può essere ottenuta con l’aiuto di soluzioni tampone. La forza ionica è correlata alla concentrazione ed alla carica degli ioni disciolti nell’eluente. Il controllo della forza ionica è un elemento critico, poiché questa forza è semplicemente la misura del numero di controioni presenti nella fase mobile moltiplicata per la loro carica. Il numero di controioni presenti è critico, poiché è esso che stabilisce il delicato equilibrio sui siti attivi dello scambiatore di ioni e permette che il campione venga alternativamente attratto e spostato durante il suo movimento attraverso la colonna. Se la concentrazione di controioni nella fase mobile è troppo elevata, gli ioni del campione non troveranno alcun sito disponibile e non saranno pertanto ritardati. Al contrario, se la concentrazione di controioni è troppo bassa, gli ioni del campione non verranno eluiti dalla colonna. I campioni più complessi possono contenere componenti aventi cariche variabili da deboli a forti. Per una completa eluizione di campioni di questo tipo si richiede frequentemente l’uso di eluizioni a gradiente, nelle quali il processo di eluizione inizia con una fase mobile di bassa molarità (spesso semplicemente acqua deionizzata a pH controllato), aggiungendo gradualmente un tampone a molarità più elevata, fornendo in tal modo una sufficiente forza ionica per spostare gli ioni del campione più fortemente attratti dallo scambiatore. Nella tabella seguente sono elencati alcuni modificatori ionici (tamponi), insieme con gli intervalli utili di pH. I più frequentemente usati tra questi sono probabilmente i tamponi fosfato, l’acetato di sodio e il borato di sodio. Si noti che nella tabella non compaiono i sali dell’acido cloridrico poiché questi non hanno capacità tampone. 24 Regolazione del pH La seconda delle variabili che influenzano la ritenzione di un soluto ionico è il pH, in particolare nel caso di scambi anionici o cationici deboli. Per esempio, una diminuzione del pH (il che significa un aumento della concentrazione di ioni idrogeno) ridurrà la ionizzazione di scambiatori di cationi deboli. Allo stesso modo, un aumento di pH può diminuire la ionizzazione di deboli scambiatori di anioni. D’altro canto, con scambiatori anionici o cationici forti, o quando vengono analizzati campioni altamente ionizzati, le variazioni di pH non avranno influenza, o ne avranno molto poca, sui tempi di ritenzione dei componenti del campione. Nei casi in cui il pH influenza la separazione, l’intervallo appropriato può essere scelto tenendo conto del pKa, che misura la costante di dissociazione di un acido. Quanto più è forte l’acido, tanto maggiore è il valore di Ka e, quindi tanto minore è il valore di pH (il contrario vale per la base coniugata). Si ha che: pH = pKa + log(ionizzato/non ionizzato) perciò in caso di acidi o basi deboli il pH influenza fortemente lo scambio, perché regola la quantità di composto ionizzato, che è l’unica forma in cui il composto può effettuare lo scambio. Come regola approssimativa il pH del tampone scelto dovrebbe essere di una o due unità inferiore al pKa della base, o superiore al pKa dell’acido. Infine, in caso di sostanze con più gruppi acidi, occorre ricordare che al punto isoelettrico tali sostanze possono essere considerate neutre e, quindi, non possono essere separate tramite scambio ionico. 25 REAZIONI IN CHIMICA ORGANICA: IDROLISI DEL BENZOATO DI METILE Tutte le tecniche analitiche esaminate fino ad ora si basano ovviamente su reazioni chimiche, ma in questo caso si vuole parlare di reazioni che hanno come obiettivo la sintesi chimica ovvero la trasformazione di un composto chimico in un altro, che viene preparato allo scopo di utilizzarlo come tale o di trasformarlo ulteriormente. Come esempio di sintesi chimica, in laboratorio viene preparato l’acido benzoico, a partire da benzoato di metile, attraverso una reazione di idrolisi con NaOH. Questa reazione è facilmente realizzata per semplice mescolamento dei reagenti e breve riscaldamento della miscela. Inizialmente, il benzoato di metile è insolubile nella soluzione acquosa di NaOH ma, se l’idrolisi avviene, si forma un prodotto solubile in acqua, il sale sodico dell’acido benzoico. Quando la reazione è terminata, per ottenere l’acido benzoico bisogna aggiungere alla soluzione HCl. Infatti HCl è un acido più forte del benzoico, e quindi forma il sale Na+ Cl-, producendo l’acido benzoico che, essendo pochissimo solubile in soluzioni acquose, dà luogo ad un precipitato bianco, che è possibile separare dalla soluzione per filtrazione sotto vuoto. COOCH3 COO- Na+ + + NaOH insolubile CH3OH solubile COOH COO- Na+ + solubile + HCl NaCl insolubile FILTRAZIONE SOTTO VUOTO Serve per raccogliere solidi dai liquidi dopo precipitazione o ricristallizzazione. Si utilizza una beuta da vuoto, un imbuto di Buchner, un filtro di carta, una guarnizione di gomma ed una pompa per fare il vuoto. 26 • • • • • l’imbuto Buchner è montato su una beuta da vuoto utilizzando un adattatore si prepara un filtro di carta che copra perfettamente i fori del Buchner (la dimensione del filtro di carta deve essere tale da ricoprire tutti i buchi ma non arrivare alle pareti verticali) si bagna il filtro con lo stesso solvente della soluzione da filtrare si collega la beuta alla pompa si versa la soluzione contenente il solido nell’imbuto Al termine della filtrazione, il solido viene lavato con una piccola porzione di solvente pulito e quindi raccolto con una spatola in un contenitore adatto per essiccarlo e procedere alla sua caratterizzazione PUNTO DI FUSIONE Il punto di fusione (p.f.) è una proprietà fisica che può essere usata per il riconoscimento di un composto. Il p.f. o intervallo di fusione è definito come l’intervallo di temperatura che intercorre da quando il solido comincia a fondere (prima goccia di liquido) a quando è completamente liquido. Se una sostanza è pura il suo intervallo di fusione dovrebbe essere di circa 1 °C (es. 180-181 °C). La presenza di impurezze, spesso, fa diminuire il punto di fusione. 27
Scaricare