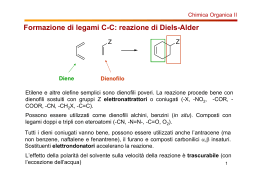
LA REAZIONE DI DIELS-ALDER E' una reazione periciclica di cicloaddizione concertata, permessa dalla teoria della simmetria degli orbitali (regole di Woodward-Hoffmann). La reazione può essere di due tipi: Il tipo di reazione mostrato a sinistra è quello HOMO del diene LUMO del diene più comune (reazione di Diels-Alder "normale"). E' favorita da dieni elettronricchi e dienofili elettronpoveri. Il tipo di reazione mostrato a destra è molto meno comune e viene chiamato "inverse demand Diels-Alder". Ha luogo quando si hanno dienofili elettronricchi (ad es. vinil eteri ed enammine) e dienofili elettronpoveri (coniugati con gruppi elettronattrattori). LUMO del dienofilo HOMO del dienofilo In entrambi i casi la simmetria degli orbitali permette una reazione termica (senza bisogno di fotocatalisi). Caratteristiche dei dienofili in reazioni di Diels-Alder normali La reazione con semplici alcheni o alchini richiederebbe condizioni molto drastiche e non è pertanto quasi mai impiegata sinteticamente. I dienofili effettivamente usati hanno sempre uno o due gruppi elettronattrattori legati al doppio legame oppure sono sistemi molto tensionati (come il benzino) esempi di dienofili CO2R acrilati R O vinil chetoni acroleina RO2C O CO2R fumarati O O anidride maleica O O R O N solfoni NO2 nitroderivati R S R O chinoni acrilammidi R O Sintecspec_5_1 Caratteristiche dei dieni in reazioni di Diels-Alder normali A differenza di quello che avviene con i dienofili, anche dieni non funzionalizzati sono sufficientemente reattivi nella Diels-Alder. Due fattori incidono sulla reattività: La presenza di sostituenti elettrondonatori fa aumentare la reattività Una maggiore percentuale di conformero s-cis fa aumentare la reattività s-cis s-trans Esempi di dieni più reattivi del comune OCH3 O Me3SiO diene di Danishefsky (reattivo grazie ai gruppi elettrondonatori + R) ciclopentadiene (reattivo perché bloccato nella conf. s-cis) furano (molto reattivo perché bloccato nella conf. s-cis e per la presenza dell'ossigeno elettrondonatore) calore orto-chinondimetano (reattivo perchè la Diels-Alder ripristina l'aromaticità e perché bloccato nella conf. s-cis) O isobenzofurano (estremamente reattivo perché combina le proprietà del furano con quelle dell' orto-chinondimetano) Sintecspec_5_2 ASPETTI REGIO E STEREOCHIMICI In principio, una reazione di Diels-Alder potrebbe portare fino a 32 composti diversi (16 coppie di enantiomeri) R1 R1 R3 * + * R4 R1 R3 * * * + * R4 R2 R2 R4 * * R3 R2 In realtà molto spesso il controllo regio e stereochimico è tale da portare a soli 2 prodotti (una coppia di enantiomeri) o addirittura ad un solo prodotto (nel caso di Diels-Alder asimmetriche con catalizzatori o ausiliari chirali). La reazione di Diels-Alder è quasi sempre un processo irreversibile e quindi il controllo è cinetico (fanno eccezione le reazioni con dieni aromatici quali il furano o l'antracene, che tendono ad essere reversibili) A REGIOSELETTIVITA' Il problema si pone quando sia il diene che il dienofilo non sono simmetrici. In questi casi il fattore dominante è quello elettronico. Si uniranno i carboni che hanno i più grandi coefficienti degli orbitali di frontiera interessati. coefficienti del' HOMO coefficienti del LUMO coefficienti del' HOMO min. magg. min. magg. ERG min. EWG magg. ERG ERG EWG EWG ERG ERG ERG Sintecspec_5_3 Quindi: EWG EWG + + EWG EWG ERG ERG ERG ERG prodotto "tipo para" prodotto "tipo orto" Gruppi alchilici piccoli sul diene hanno effetto elettrondonatore (+I) e quindi aumentano la velocità e indirizzano la reazione secondo quanto detto sopra. Quando i gruppi sono ingombrati si può però avere una diminuzione di velocità R R H CH3 tBu vel. relativa 1 4.2 < 0.05 + O O O L'effetto sterico è ancora più gravoso quando il sostituente è in cis: H R H B STEREOSPECIFICITA' H H H R H Dato che la Diels-Alder è un processo concertato, la configurazione relativa di R1 ed R2 e di R3 e R4 è decisa dalla configurazione dei 3 doppi legami. Degli 8 diastereoisomeri teoricamente possibili se ne formano solo 2. Ad esempio E R1 R1 R3 + R3 Z R2 R3 E R1 E R1 R3 R2 R3 + 3 R R R3 Z H R1 R3 2 R3 R2 R3 + + R3 E R1 R2 R3 R2 Sintecspec_5_4 C DIASTEREOSELETTIVITA' Negli esempi visti prima abbiamo visto che la stereochimica degli alcheni di partenza controlla le configurazioni relative tra R1 ed R2 e tra i due R3, ma non la configurazione relativa tra R1 ed R3 (e quindi tra R2 ed R3): si possono in principio formare due diastereoisomeri endo ed eso. R1 R1 R1 E Normalmente (sotto controllo cinetico) si ha una netta R3 R3 R3 prevalenza dell'isomero endo, anche se esso è meno Z + + stabile, quando almeno uno dei gruppi R3 è elettronattrattore ed il dienofilo è Z o terminale R3 R3 R3 E Con dienofili E (se i gruppi R3 sono diversi) la situazione è 2 2 R2 R R ovviamente più complessa. Tende comunque a stare in endo il eso endo gruppo più elettronattrattore 2 H R R3 3 R R3 R3 H 2 R3 2 R H R 2 attacco endo R 1 H R 3 R R3 H R3 H R1 R1 H H 1 R R3 R3 3 R R2 attacco eso R3 H H H R2 H 2 R 3 HR R4 H R3 R1 H D R2 1 R H R3 R3 R1 STEREOSELEZIONE π-FACCIALE Le due facce π del diene e dell'alchene possono essere omotopiche (e allora il problema non sussiste) o enantiotopiche o diastereotopiche. Per discriminare due facce enantiotopiche occorrono catalizzatori chirali Sintecspec_5_5 O ESEMPI 140°C + O 90% OAc se il diene è attivato da sost. elettrondonatori, la reazione è più facile OMe + Con dienofili monoattivati e semplici dieni la reazione richiede in genere temperature tra i 100 ed i 200°C. Ove necessario si possono usare recipienti chiusi a tenuta di pressione AcO 90°C AcO H H OAc CO2Me O 75% AcO OAc prodotto endo CO2Me Si forma un unico diastereoisomero (racemo) tra i 4 possibili grazie: a) alla configurazione relativa cis dei due acetossi, decisa dalla configurazione del diene b) alla diastereoselettività di tipo endo. I dieni ciclici sono particolarmente reattivi grazie al fatto che la conformazione s-cis è presente al 100% all'equilibrio O L'attacco è esclusivamente endo. 40°C + O 97% O O O O O T amb. O + O O O O Con il furano le reazioni sono molto veloci, ma anche reversibili. Il controllo diventa termodinamico ed il prodotto principale è l'eso. Sintecspec_5_6 I nitroalcheni sono ottimi dienofili. Grazie al fatto che il nitrogruppo risultante può subire diverse trasformazioni, questa reazione è particolarmente utile sinteticamente: 1) NaOMe, MeOH 2) TiCl3, NH4OAc, BnO BnO Et2O, 25°C OBn T amb., 16h + NO2 64% O NO2 Aspetto stereochimico: le due facce del diene sono diastereotopiche e quella meno ingombrata viene attaccata di preferenza. Le due facce del dienofilo sono invece enantiotopiche e non c'è discriminazione. Pertanto si ottiene una miscela racemica Aspetto sintetico: il nitroetilene è di fatto un equivalente sintetico del chetene, che è un pessimo dienofilo. Il meccanismo della riduzione dovrebbe essere il seguente: R1 H O R1 + MeO + 2 TiCl3 N 2 R OH R1 + H3O R O anione nitronato H R1 TiCl3 N R NH4OAc serve a tamponare HCl O R2 + 5 HCl 2 R1 N R2 + 2 TiO2 + Cl N 2 NO2 OH R1 R2 I nitroalcheni sono utili anche grazie al fatto che permettono un maggior controllo regio- e stereo-chimico MeO2C CO2Me T amb. CO2Me base + NO2 NO2 NHBoc NHBoc NHBoc NO2 Grazie all'eliminazione: eq. sint. di MeO2C CO2Me Sia la regioselettività che la diastereoselettività endo sono controllate dal nitro gruppo e non dall'estere. ma con il propiolato si otterrebe l'isomero di tipo "orto" Sintecspec_5_7 CO2Et CO2Et CO2Et 2 A : B = 83:17 NO2 A NO2 H CO2Et MeO2C B base unico prodotto CO2Et nBu3SnH, AIBN Quest'ultima trasformazione mostra come i nitroalcheni possano essere considerati equivalenti sintetici di alcheni OMe massimo coeffic. LUMO OMe 50-100°C + Me3SiO CHO CHO H2O, H3O CHO Me3SiO massimo coeffic. HOMO O Il diene di Danishefsky è un diene piuttosto reattivo. I suoi addotti possono essere facilmente trasformati in chetoni α,β β-insaturi. Si noti la regioselettività. Entrambi i sostituenti del diene innalzano il coefficiente dell'HOMO dello stesso carbonio. DIELS-ALDER CATALIZZATA DA ACIDI DI LEWIS Acidi di Lewis quali AlCl3, Et2AlCl, ZnCl2, etc. accelerano la reazione di Diels-Alder, rendendo più elettrofilo il dienofilo. Il meccanismo rimane comunque concertato e la reazione rimane stereospecifica. Si nota però un aumento di regioselettività CO2Me + + CO2Me non cat. cat. da AlCl3 CO2Me temp. 120°C 20°C rapporto 70 : 30 95 : 5 Sintecspec_5_8 DIELS-ALDER INTRAMOLECOLARE La Diels-Alder intramolecolare consente la trasformazione in un solo passaggio di un composto aciclico in un composto biciclico. Inoltre essa richiede condizioni più blande, sfruttando il vantaggio entropico e può avvenire anche con dienofili non attivati. Le IMDA procedono con elevata regioselettività. La diastereoselettività non è invece facilmente prevedibile con le regole endo-eso e dipende da una combinazione di effetti sterici ed elettronici. Vi sono due tipi di D.A. intramolecolare portano a sistemi fusi IMDA di tipo I n n n n deve essere ≥ 1 Le reazioni più frequenti hanno n = 1 o 2 e portano quindi a idrindani e decaline giunzione trans giunzione cis Per n = 1, 2 o 3 la reazione è sempre completamente regioselettiva indipendentemente dagli effetti elettronici dei sostituenti. Per esempio: IMDA di tipo II n deve essere ≥ 1 Le reazioni più frequenti hanno n = 1, 2 o 3 e sono completamente regioselettive. Anche per n = 3 il modo di attacco alternativo porterebbe ad un doppio legame anti-Bredt: portano a sistemi a ponte n n Sintecspec_5_9 Diastereoselettività nelle IMDA di tipo I L'aspetto fondamentale è la formazione di addotti con giunzione cis o trans n n giunzione cis n giunzione trans Il rapporto tra i due prodotti dipende da vari fattori • • • • La configurazione del doppio legame "interno" del diene La presenza di un gruppo attivante sul dienofilo e la sua posizione (esterna E, esterna Z, interna) Le condizioni di reazione (reazione termica o catalizzata da acidi di Lewis) L'effetto sterico di eventuali sostituenti "interni" sul diene Primo fattore: Quando la configurazione del doppio legame "interno" del diene è Z, la reazione è in generale più difficile e si possono formare solo gli addotti cis H H H H Quando la configurazione è E, in assenza di altri effetti, la reazione sembra favorire la giunzione cis, anche se la diastereoselettività non è elevata: 160°C + 76 : 24 190°C + 53 : 47 Sintecspec_5_10 La diastereoselettività dipende dal grado di formazione dei due nuovi legami nello S.d.T. (e quindi dal tipo di asincronicità della cicloaddizione): quando è più formato il legame rosso, in assenza di altri effetti, è più stabile (anche se di poco) lo stato di transizione che porta alla giunzione cis quando è più formato il legame azzurro, in assenza di altri effetti, è più stabile lo stato di transizione che porta alla giunzione trans I sostituenti aggiuntivi possono quindi incidere sul decorso in due modi: stabilizzando o destabilizzando i due S.d.T. con interazioni steriche o elettroniche spostando l'asincronicità della reazione verso l'uno o l'altro dei due casi limite Secondo fattore: Quando è presente un sostituente attivante in posizione "esterna" ed E, aumenta la percentuale di isomero con giunzione trans. OHC CHO CHO 150°C + 25 CHO OHC : 75 CHO 110°C La reazione è in generale più selettiva per gli idrindani che per le decaline + 8 : 92 Sintecspec_5_11 Come è stata razionalizzata questa aumentata preferenza nei confronti della giunzione trans ? a) L'aumentata elettrofilia del dienofilo aumenta b) La regola endo favorisce la giunzione trans l'asincronicità della reazione questo legame è più formato a livello di S.d.T. H OHC il gruppo CHO è in endo H OHC Quando invece il sostituente attivante è in posizione "esterna", ma in Z, l'effetto è trascurabile NO2 NO2 O2N + 47 : 53 In questo caso i due fattori sopra citati sono in conflitto e si annullano a vicenda NO2 NO2 in eso NO2 in endo NO2 NO2 NO2 Infine quando il sostituente attivante è in posizione "interna" si forma esclusivamente la giunzione cis O O inoltre C=O è in endo O cis : trans : 97:3 questo legame è più formato a livello di S.d.T. Sintecspec_5_12 Terzo fattore: Quando è presente un sostituente attivante in posizione "esterna" ed E, l'uso della catalisi (acido di Lewis) fa aumentare notevolmente la percentuale Tale aumento non si verifica se il sostituente attivante è in Z. OMe ZnCl2, T amb. CH2Cl2 OHC di isomero con giunzione trans. CHO L'acido di Lewis aumenta l'asincronicità della reazione ed aumenta anche l'effetto endo O O unico prodotto Quarto fattore: un sostituente sul diene può destabilizzare lo S.d.T. che porta alla giunzione cis, incrementando la selettività a favore del trans nei casi problematici. R cis trans H R CO2Me HO interazione destabilizzante CO2Me HO SiMe3 > 95:5 SiMe3 Senza Me3Si la diastereoselettività è molto scarsa. Il gruppo Me3Si può poi essere rimosso e rappresenta quindi un "removable steric directing group" Sintecspec_5_13 DIELS-ALDER TRANSANNULARE Nella Diels-Alder transannulare il diene ed il dienofilo sono congiunti da catene di collegamento (tethers) su entrambi i lati. Si passa quindi da un sistema macrociclico ad un sistema triciclico. I fattori sterici ed entropici sono in questo caso decisivi nell'indirizzare la regiochimica e la stereochimica del processo. OPh O N OPh OH OH Py-BOP O N O Et3N O OMe O OMe T amb. Quando non ci sono tensioni steriche che impediscono la cicloaddizione, la Diels-Alder transannulare è fortemente agevolata entropicamente ed ha luogo in condizioni estremamente blande. La corrispondente reazione intramolecolare non avviene facilmente e a 180°C si decompone tutto. Esistono due gradi di libertà: la diastereoselezione interna (endo-eso) e la diastereoselezione relativa (rispetto al centro preesistente). Entrambe sono controllate al 100% grazie alla rigidità del sistema. conf. rel. decisa da conf. diene conf. rel. obbligatoria per la struttura del sistema OPh O * N O * H * * O conf. rel. decisa da conf. alchene conf. rel. OMe obbligatoria per la struttura del sistema Sintecspec_5_14 TRASPOSIZIONI SIGMATROPICHE Le trasposizioni sigmatropiche coinvolgono una riorganizzazione concertata di elettroni, durante la quale un legame σ migra su un sistema π adiacente con contemporaneo spostamento di elettroni π Le trasposizioni sigmatropiche principali sono di due tipi, dette [3,3] e [3,2] La classificazione delle reazioni sigmatropiche segue questa regola: si contano(sui due lati) gli atomi interposti tra il legame σ che si rompe ed il legame σ che si forma. 2 1 3 X X X Y Y Y 1 3 2 2 1 1 [3,3] X 3 Y Z [2,3] Z 2 Le reazioni del tipo [3,3] sono le più importanti. Sono reazioni permesse dalla regola di simmetria degli orbitali. Lo stato di transizione può essere visto come l'accoppiamento di due radicali allilici: X Y X Y X orbitali di un radicale allilico Y Nell'interazione di due radicali allilici il LUMO e l'HOMO sono lo stesso orbitale. L'interazione è permessa Lo stato di transizione può essere in questo caso sia a barca che a sedia. Normalmente prevale lo S.d.T. a sedia Sintecspec_5_15 1 2 3 X Y 1 Quando X è un atomo di ossigeno e Y un carbonio, le trasposizioni [3,3] vengono chiamate trasposizioni di Claisen X Y 3 I substrati più classici delle trasposizioni di Claisen sono gli allil vinil eteri, di solito preparati in situ dagli alcoli allilici [3,3] 2 CHO 195°C + O OH O In principio le trasposizioni sigmatropiche [3,3] sono reazioni di equilibrio. Le Claisen sono però generalmente spostate verso destra (ed irreversibili) grazie all'alta energia del legame C=O Altri tipici substrati sono i fenil allil eteri O O OH L'ultima tautomeria sposta a destra l'equilibrio Un'utile variante è la trasposizione di Claisen degli ortoesteri + OH OEt − EtOH OEt OEt trietil ortoacetato In pratica si aggiunge un'unità di acetato anziché un'unità di acetaldeide O EtO O OEt OEt O OEt Sintecspec_5_16 Aspetti stereochimici Le trasposizioni [3,3] di substrati chirali sono stereospecifiche (solo 2 dei 4 possibili prodotti si formano) e stereoselettive (uno dei due prodotti prevale). (Z) HO H H (E) CO2Et (R) (S) + CO2Et H maggiore H OEt minore OEt H CO2Et O O Se il doppio legame si forma E il carbonio stereogenico deve per forza essere (R) S.d.T. favorito O O CO2Et H EtO Se il doppio legame si forma Z il carbonio stereogenico deve per forza essere (S) EtO S.d.T. sfavorito La configurazione del nuovo centro stereogenico è quindi decisa: a) dalla configurazione del vecchio stereocentro; b) dalla configurazione del doppio legame di partenza; c) da quale delle due sedie è favorita In ogni caso i prodotti che si formano hanno lo stesso e.e. del substrato. La stereospecificità della Claisen è molto utile sinteticamente. Consente infatti di trasferire la chiralità da un alcool secondario (facile da ottenere stereoselettivamente ad es. per risoluzione enzimatica, riduzione enantioselettiva etc.) ad un carbonio stereogenico non funzionalizzato (difficile da ottenere direttamente in modo enantioselettivo) Gli stessi prodotti della Claisen con ortoesteri possono essere ottenuti utilizzando enolati di esteri o silil chetene acetali. La reazione con questi ultimi (detta Ireland-Claisen) è la più usata. O R2 R2 O O R1 1 chetene acetali OEt (da ortoesteri) R silil chetene acetali OSiMe3 enolati di esteri OM Sintecspec_5_17 Quando l'enolato o il silil chetene acetale derivano da un acido diverso dall'acido acetico esistono due possibili configurazioni al doppio legame ed esse incidono sul decorso stereochimico della reazione: OSiMe3 OSiMe3 2 R R2 Me3SiO2C R2 O O 1 R1 R R1 silil chetene acetale (E) doppio legame (E) OSiMe3 OSiMe3 2 R R2 R1 O R1 O Me3SiO2C R2 R1 silil chetene acetale (Z) doppio legame (E) anti sin Lo stesso vale per gli enolati La formazione di enolati (E) o (Z) può essere controllata giocando sulle condizioni di enolizzazione: LDA THF O 2 R O 2 R O enolato (E) O R1 LDA THF-HMPA O 2 R R1 O enolato (Z) I silil chetene acetali sono quindi preparati dagli enolati per semplice trattamento con Me3SiCl Me3SiCl Me3SiCl OSiMe3 R2 O OSiMe3 2 R1 R1 R O R1 Sintecspec_5_18 Esempi di applicazioni sintetiche: O Br Li Br O MeO OMe 1) KN(SiMe3)2 −78°C 2) Me3SiCl O 1) OSiMe3 O O Br O O 2) (EtCO)2O MeO MeO OMe OMe enolato (E) 1 solo diastereoisomero HO2C * (S) i trimetilsilil esteri si idrolizzano facilmente nel work-up CO2H (S) (E) sono possibili in teoria 8 diversi stereoisomeri si forma solo questo Br O * Br O anti MeO OMe MeO OMe Br O MeO Br OMe CO2H OSiMe3 (R) (R) MeO OSiMe3 MeO favorito O O anti (Z) Br O sfavorito O MeO OMe Sintecspec_5_19 La reazione di Claisen dei silil chetene acetali può servire a costruire stereoselettivamente un substrato per una RCM: O 1) KN(SiMe3)2 −78°C HO2C OH DCC 2) Me3SiCl BnO O + O O DMAP BnO OBn OBn a Cat. Grubbs 1 gen. CO2Me CH2Cl2, riflusso 1) 0°C CO2Me 2) CH2N2 87% 79% O Me3SiO O O BnO O sin enolato (Z) La presenza dell'ossigeno favorisce l'enolato (Z) per chelazione del potassio OSiMe3 OSiMe3 BnO O R R O BnO O OBn CO2H O OR Si noti che i due frammenti sono stati uniti mediante un legame C− −O, che è poi divenuto un legame C− −C in base alla trasposizione Una trasposizione simile alla Claisen è il passaggio chiave della sintesi degli indoli di Fischer H N O NH2 + H+ R R N H H N NH R Sintecspec_5_20 Trasposizione di Cope La trasposizione di Cope è la [3,3] in cui tutti gli atomi sono carboni. A differenza della Claisen è in genere una reazione di equilibrio e richiede condizioni più drastiche. Nel caso mostrato qui sotto l'equilibrio è spostato verso destra dalla coniugazione del doppio legame con il fenile Ph Ph Ph (E) (R) + (S) (Z) maggiore Ph minore Ph Ph favorito Ph Ph Ph sfavorito La reazione può essere catalizzata (e procedere quindi in condizioni più blande) con palladio (II): (R) Ph (E) Pd(CH3CN)2Cl2 T amb (Z) Ph senza catalisi ci vogliono 240°C Ph Ph Pd(II) + (S) (E) (R) 70 Ph Pd(II) Ph : 30 + Pd(II) la presenza del carbocatione rende quest'ultimo passaggio favorito rispetto alla β-eliminazione di idruro. Quindi Pd(II) non viene ridotto Sintecspec_5_21 Trasposizione di ossi-Cope Forse più utile della Cope è la ossi-Cope che ha il vantaggio di avere, come la Claisen, un equilibrio spostato verso destra HO OHC HO OH la formazione del carbonile sposta a destra l'equilibrio La ossi-Cope avviene molto più velocemente se l'alcool è convertito nel corrispondente alcolato (ossi-Cope anionica) H KH, THF riflusso OH H O O H H O H O H Sintecspec_5_22 TRASPOSIZIONI SIGMATROPICHE [2,3] Sono sempre promosse da un termine anionico, che può essere un anione vero e proprio o un ilide 2 1 X Y 1 2 1 3 Z 2 X X Y Y Z 1 X 3 Y Z 2 trasposizione [2,3] neutra Z trasposizione [2,3] anionica La trasposizione dei solfossidi allilici appartiene al primo tipo: O O O S OH S (MeO)3P La reazione viene condotta in presenza di (MeO)3P che sposta l'equilibrio rompendo il legame S− −O del solfinato. La reazione è stereospecifica. Al secondo tipo appartiene la trasposizione di Wittig: Z Z O O base Z R R O Z OH R R Z deve essere un gruppo che faciliti la formazione dell'anione (un gruppo elettronattrattore o un eteroatomo come S, Si) Sintecspec_5_23 OH O SiMe3 nBuLi RO RO E' simile alla trasposizione degli allil solfossidi, ma in questo caso comporta la formazione di un nuovo legame C− −C. A differenza della Claisen (via ortoesteri o silil chetene acetali) viene aggiunta una unità di un C anziché un'unità di due C. La trasposizione di Wittig è stata spesso utilizzata per sintesi di macrocicli mediante contrazione di anello: Williamson nBuLi Cl O OH OH O Sintecspec_5_24
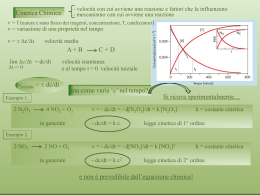
Scaricare