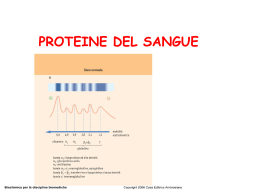
www.slidetube SIEROPROTEINE ELETTROFORESI Le proteine del siero ammontano in totale a 6-8 g/dl; nel neonato 5 g/dl. Aumento delle proteine totali: - disidratazione - cambiamenti posturali - epatopatie (accumulo di γglobuline) - tumore delle plasmacellule - infezione cronica (aumento Ig) - anemia (aumento transferrina) Diminuzione delle proteine totali: - iperidratazione (ad es. nella gravidanza) - malnutrizione - dieta non equilibrata - malattie renali - epatopatie - enteropatie protido-disperdenti - malassorbimento - ustioni - eccessivo catabolismo L’elettroforesi viene eseguito sul siero (ciò che rimane del plasma dopo l’evento coagulativo); non sul plasma il quale è ricco di fibrinogeno che si annida tra le γglobuline e C3 (posizione occupata di solito dalla componente monoclonale) A pH 7,4 prevale la dissociazione tra gli amminoacidi (gruppi COO¯). Le cariche si spostano verso il polo positivo (anodo) di elettroforesi. 1 www.slidetube L’elettroforesi permette di evidenziare 5 bande. 1) Banda A corrispondente all’albumina (ALB) 2) Banda α1 corrispondente ad α1antitripsina (AAT) ; α1antichimotripsina (AAC); α1glicoproteina acida (AAG); α-fetoproteina 3) Banda α2 corrispondente ad aptoglobina (HPT); α2macroglobulina (AMG); ceruloplasmina (CER) 4) Banda β corrispondente a transferrina (TRF) (nella parte anodica) e C3 (nella parte catodica) 5) Banda γ corrispondente alla maggior parte delle immunoglobuline 1) - Prealbumina - La prealbumina è un ottimo indicatore di nutrizione perché ha una emivita t1/2=48 h - La prealbumina trasporta gli ormoni tiroidei e partecipa all’equilibrio tra frazione libera e frazione legata; trasporta RBP (retinol binding protein), indicatore nutrizionale e nelle urine di sofferenza renale - PROTEINE DI FASE ACUTA La diminuzione di prealbumina (proteina che occupa la parte più anodica della banda A) può indicare uno squilibrio nutrizionale o la presenza di una malattia infettiva o un’infiammazione acuta. Bisogna fare gli indicatori di fase acuta (FASE RATTIVA ACUTA: risposta aspecifica con aumento della sintesi di alcune siero proteine). - proteina C reattiva (CRP) - velocità di eritrosedimentazione (VES) - In fase acuta ↑VES riflette ↑[FIB] - In fase cronica ↑VES riflette ↑[FIB]; ↑[Ig](indicano il passaggio a infiammazione cronica); ↑anemia (se non c’è microcitosi) - La VES aumenta alla fine della prima settimana di malattia e nelle gammopatie monoclonali da IgM, IgA, IgG. Intervalli di riferimento VES Età <60 anni Età >60 anni MASCHI 2-13 mm/h 2-24 mm/h FEMMMINE Età <40 anni 2-16 mm/h FEGATO (sintesi di proteine di fase acuta) IL-1 Età >40 anni 2-35 mm/h CERVELLO (febbre, sonno) IL-1 IL-6 IL-6 FAGOCITI TESSUTI (stimolazione di proteolisi 2 www.slidetube muscolare, fibtoblasti, calore) - CRP è la più precoce e sensibile proteina di fase acuta - A volte è associato ↑SAA (infiammazione acuta) - Cascata delle proteine di fase acuta - PRECOCI: ……………….CRP e AAC - INTERMEDIE: …………..HPT, AAG, AAT, C4, FIB - TARDIVE: ……………….C3, CER RISPOSTA di FASE ACUTA 120 100 80 CRP 60 PFAN INTERMEDIE 40 TARDIVE 20 0 0 1 2 3 4 5 Figura 1: PCR = proteina C reattiva (precoce); PFAN = proteine di fase acuta negativa Nell’epatopatico e parzialmente nel nefropatico la reazione di fase acuta è soppressa. 3 www.slidetube - ALB (albumina) Figura 2: rappresentazione di una molecola di albumina con siti di legame per molecole e ioni trasportati -Proteina di 580 aa; sintetizzata dal fegato (ne basta il 25%) -Livello ematico normale: 3,5-5,0 g/dl -Indicatore di stato nutrizionale (ma t1/2=15-19gg) -Carrier di anioni, soprattutto bilirubina (che nei neonati può ledere i nuclei della base); acidi grassi liberi; ormoni steroidei; cationi (Ca2+; Cu+/2+) -Sorgente di aa per i tessuti periferici -Agisce come tampone -Proteina di fase acuta negativa (diminuisce nella flogosi) Acidi grassi liberi si formano nel digiuno. l’albumina li porta al fegato che sintetizza nuovi trigliceridi e li lega alle VLDL (utili per il miocardio). Ci sono analbuminemie congenite senza edema (la sintesi di altre globuline plasmatiche mantiene la pressione oncotica) ma con alterazione del metabolismo dei grassi. Ca2+: 10 mg/% o 5 mEq/l legati per metà all’albumina. Questa metà è in effettiva; importante è la componente libera. Il rapporto tra Ca2+ libero e legato dipende dal pH. Alterazioni dell’equilibrio acido-base possono far associare Ca2+ e albumina portando ipocalcemia. Cu+: 80 mg/l legato per il 10% all’albumina e per il 90% alla cerulo plasmina. Si lega a residui di istidina (Hys). Dalla velocità di legame e dall’affinità dipende la biodisponibilità. Iperalbuminemia, rara, dovuta a disidratazione Ipoalbuminemia: (<3,2 g/dl) -epatopatie croniche -malnutrizione -edema (<2,5g/dl), ascite -perdite intestinali -malattie renali -malattie cutanee secernenti -ustioni Bisalbuminemia: banda dell’albumina sdoppiata. -Il paziente può averla parchè il fegato produce il 50% dell’albumina con 1aa diverso (alloalbuminemia) 4 www.slidetube -Può essere indotta da farmaci in caso di superdosaggio o da patologie quali ipotiroidismo e pseudo cisti pancreatica. 2) zona elettroforicamente poco rilevante - AAT (α1antitripsina) - Responsabile del picco delle α1. - E’ un inibitore delle proteasi seriche: - collagenasi ed elastasi nei processi infiammatori del polmone (si può arrivare anche all’enfisema); funzione primaria - fattori della coagulazione; funzione secondaria - Se il picco delle α1 è basso si richiede l’esame per sapere se c’è una mancanza congenita di AAT (omozigosi per allele Z); α1antitripsina inferiore a 15% del normale. - Livello ematico normale: 0,8-2 g/l - ↑ in situazioni di flogosi causate da infezioni e necrosi dei tessuti. - AAG (α1glicoproteina acida) - Costituita per il 50% da glucidi e per il 50% da aa; particolarissima - Non si conosce la sua esatta funzione. - E’ un indicatore di fase acuta: - ↑ nei processi infiammatori, pare a causa della proliferazione cellulare - ↑ anche in gravidanza e nei tumori, non è un indice molto sensibile - è ↑ dai glucocorticoidi, nelle neoplasie, nelle flogosi, nello stress chirurgico - ↓ nelle epatopatie, nelle malattie renali, estrogeni - AAC (α1antichimotripsina) - Proteina che aumenta precocemente nelle reazioni di fase acuta - α-fetoproteina - Presente nel feto, diminuisce notevolmente dopo la nascita - ↑Aumenta - in gravidanza, nell’epatocarcinoma e nel teratoma testicolare - nel liquido amniotico e nel siero materno in caso di anencefalia e spina bifida -↓Diminuisce - nel siero della madre se c’è trisomia 21 (Down) 5 www.slidetube 3) - HPT (aptoglobina) - E’ una proteina di fase acuta - ↑ nelle infiammazioni acute e croniche, nelle neoplasie e nelle nefrosi (per aumentata sintesi del genotipo di HPT z-z che non passa il filtro renale) - La sua principale funzione è però legare l’emoglobina libera a seguito di emolisi intravasale (processo patologico) formando un complesso che non viene filtrato a livello glomerulare (in corrispondenza del tubulo contorto distale e dotto collettore dove il pH si abbassa precipiterebbe formando dei cilindri che possono danneggiare il rene). Il complesso HPT-Hb viene captato dal fegato. se l’emolisi è eccessiva intervengono nell’ordine l’emopessina (si lega all’eme) e l’albumina (metalbumina). - ↓ in crisi emolitica, anemia, avvelenamento, valvole cardiache che lisano i globuli rossi. - AMG (α2macroglobulina) - E’ una proteina ad alto peso molecolare (fino a 1000000 Da) - E’ un inibitore delle proteasi e controlla il C’ - Importante ad es. nella gestosi gravidica = ipertensione arteriosa in gravidanza. Renina- proteasi: se non c’è più un inibitore la pressione arteriosa sale. - ↑ nella sindrome nefrosica - ↓ nella risposta di fase acuta, nelle pancreatiti, nel carcinoma della prostata. - CER (ceruloplasmina) - Proteina azzurra, se purificata, perché trasportatore di rame e responsabile della cupremia. Una molecola lega 8 atomi di Cu (4Cu2+ o 4 Cu+). - Ossida il ferro ferroso Fe2+ a ferro ferrico Fe3+ e altre molecole come ad es. neurotrasmettitori (amine biologiche); è uno “scavenger” dell’O2¯. - ↑ in gravidanza (bersaglio di estrogeni); nelle infiammazioni; neoplasie e leucemie come linfoma di Hodgkin - ↓ nel morbo di Wilson (alterazione del metabolismo del Cu con suo accumulo in rene, fegato, cervello e degenerazione epatolenticolare) e in cirrosi giovanile. - Se la cupremia è 30 mg/l (e CER è quindi nei limiti della norma) richiedere la cupruria (ricerca di Cu nelle urine) 6 www.slidetube 4) - TRF (transferrina) - METABOLISMO DEL FERRO: Il ferro è introdotto con la dieta sotto forma di Fe alimentare. Si lega a una FERRITINA intestinale (1 apoferritina lega 8 atomi di Fe) che lo trasporta nell’eritrocita. Sulla membrana basolaterale dell’eritrocita si trova la TRANSFERRINA, una proteina abbondante (240 – 250 mg/dl). Il Fe3+ lega la transferrina saturandola; essa lo trasporta nel torrente ematico. In questi passaggi il ferro cambia valenza da Fe2+ a Fe3+. - La ceruloplasmina ossida il ferro e permette che venga legato alla transferrina. TRF + 2 atomi di Fe3+ → TRF diferrica (1/3 delle molecole) TRF + 1 atomo di Fe3+ → TRF monoferrica (2/3 delle molecole) TRF diferrica ha maggior attività nei confronti dei recettori periferici. Tramite la circolazione sanguigna il ferro giunge al fegato. - Nel fegato c’è lo scambio di Fe tra transferrina e ferritina epatica (di deposito); essa è responsabile del mantenimento del 30% di saturazione della TRF, perché cede o acquista Fe a seconda che il livello di saturazione sia minore o maggiore del 30%. - Il midollo osseo è il principale bersaglio del ferro - Tutte le cellule necessitano ferro per la sintesi di DNA e RNA (enzima ributile reduttasi riduce il ribosio a desossiribosio) - I recettori della TRF, presenti su tutte le cellule, captano il Fe ed entrano nella cellula dove liberano il Fe per poi tornare al loro posto sulla membrana. - Esiste a questo livello un meccanismo a feedback: - Se nella cellula c’è poco ferro viene sintetizzato più recettore; i componenti del recettore vanno in circolo e con gli esami si dimostra carenza di ferro - se c’è troppo ferro il recettore è più lento nel trasportarlo all’interno della cellula. - Sideremia normale: 50-100μg/dl - Eritrociti anomali, diminuzione della sideremia, aumento della transferrina (con saturazione <30%); verificare la ferritina circolante che rispecchia il livello di ferritina di deposito - La ferritina si alza quando c’è un’infezione (come proteina di fase acuta); perciò si procede al dosaggio dei recettori solubili della transferrina. - Nelle malattie infettive la transferrina tende a diminuire ANEMIA: condizione che si verifica quando: - ↓ sideremia → < 30 – 40 μg/dl - ↓ [Hb] di un globulo rosso → piccolo con volume < 80 MCV (volume corpuscolare medio) - ↓ [Hb] totale del sangue La situazione opposta è l’emosiderosi e si verifica se la sideremia è > 150 μg/dl. La principale causa di anemia è la carenza di ferro; in questo caso si ha: - ↓ sideremia - ↑ TRF - ↓ saturazione TRF % Non è però l’unica causa, anche infezioni croniche e neoplasia possono dare anemia, senza che ci sia carenza di ferro: - ↓ sideremia - ↓ TRF - ↓ saturazione di TRF % 7 www.slidetube Per verificare se ci sia un’anemia ci sono 3 livelli di indagine: -1- Valutare TRF - TFR aumenta in stati ferrocarenziali, in gravidanza, nella terapia estroprogestinica, anemia metaemorragica - TFR diminuisce nelle sindromi protido-disperdenti, nelle anemie delle malattie croniche (processi infiammatori e neoplastici), nelle epatopatie (per diminuita sintesi epatica), malnutrizione, atransferrinemia congenita -2- Valutare la [C] della ferritina circolante che rispecchia la [C] della ferritina di deposito. Se è diminuita c’è anemia. Tuttavia essendo una proteina di fase acuta essa sarà aumentata se c’è un’infiammazione, anche questa è accompagnata da anemia -3- Ricercare i recettori solubili della TRF. Essi consentono di valutare il deficit di ferro a livello cellulare. Riflettono il numero dei recettori cellulari. E’ un indice precoce dello sviluppo di deficit funzionale di ferro rispetto ad altri indici quali la ferritina. A differenza di quest’ultima si mantiene normale (3 -9 μg/l) nei pazienti con infiammazioni, neoplasie e epatopatie ed è quindi un buon indice di discriminazione tra anemia sideropenia e anemia delle condizioni croniche. ANEMIA EMOLITICA: abnorme distruzione dei globuli rossi con: - ↑ sideremia, - ↓ TFR, - ↑ % saturazione di TRF (tutta la TRF si satura) C’è un sovraccarico di ferro e emosiderosi SATURAZIONE TRF Saturazione % = (ferro μg/dl / max[Fe] trasportabile dalle proteine) x 100 - 1 mg di transferrina lega 1,26 μg di ferro (total iron binding capacity) - 300 mg TFR lega 375 μg di Fe (bisogna vedere la percentuale di saturazione) - Valori di riferimento: 2-3,5 g/l - Hb: definisce il grado di anemia e esprime a posteriori una misura quantitativa della gravità della sideropenia. da solo è un dato poco specifico - Sideremia: rappresenta il Fe3+ legato alla TRF. Indice sensibile negli stati di lieve sideropenia 8deplezione totale delle riserve senza però ancora anemia). Ha però ritmo circadiano e alta variabilità individuale (maschi: 70 – 200 μg/dl; femmine: 50 - 170 μg/dl) ↑ sideremia: condizioni iperemolitiche, epatopatie con citolisi, anemia sideroblastica, siderocromatosi ereditaria ↓ sideremia: aumentata perdita (es. mestruazioni), diminuito apporto o assorbimento. 8 www.slidetube - C’ (complemento) - Il complemento è un sistema di diverse proteine di cui 9 sono le principali - La frazione più abbondante è C3 - Il complemento si attiva per 2 vie: - classica: Ag+Ab+C1→C4+C2→C3(amplificazione) ↓ C5C6C7(MAC) ↓ C9(lisi cellulare) - alternativa: parte dal C3b (con partecipazione di fattori B e D) - Esami: - diminuzione di C3 e diminuzione di C4 → via classica - diminuzione di C3 ma C4 normale → via alternativa - Per attivare il C’ ci vogliono 2 IgG o 1 IgM; CASCATA del COMPLEMENTO: C1 si lega al complesso immune e si attiva (C1s) ad esterasi; scinde C4 in C4a (inattivo) e C4b che lega C2 facendolo diventare substrato di C1s che lo scinde in C2b (inattivo) e C2a. Il complesso C4b2a è l’enzima C3 convertasi (esterasi) che scinde C3 in C3a (talvolta dosato come espressione di scissione di C3) e C3b che unito alle precedenti frazioni dà vita all’enzima C5- convertasi della via classica (C4b2a3b). La C5-convertasi scinde C5 in C5a (inattiva) e C5b. Il complesso C4b2a3b5b per via non enzimatica lega in successione C6, C7 con passaggio del complesso da stato idrofilo a idrofobo rendendolo adatto a inserirsi nel doppio strato lipidico della membrana cellulare. L’ingresso di C8 e C9 permette la formazione del poro sulla membrana e successiva lisi cellulare. - La concentrazione del terzo componente del complemento (C3), determinata nel siero, è considerata un indicatore di rischio dell’infarto del miocardio; infatti, esso si correla positivamente con il colesterolo-LDL ed è prodotto dai macrofagi probabilmente durante la captazione di lipoproteine LDL ossidate. - C3 aumenta per blocco del suo catabolismo (blocco C3 e C3b); nella cirrosi biliare primitiva e nelle colestasi gravi; in reazione di fase acuta - C3 e C4 diminuisce (per attivazione del C’ per via classica) in lupus eritematoso sistemico; poliartrite cronica primaria; anemie emolitiche autoimmuni; gastrite atrofica; glomerulonefrite membranosa - C3 diminuisce e C4 normale (C’ attivato per via alternativa) indice di malattie infettive - Valori di riferimento: C3: 0,90-1,80g/l C4: 0,10-0,40g/l - Nell’elettroforesi su plasma tra β e γ si forma la banda del fibrinogeno; nell’elettroforesi su siero no. - FIB (fibrinogeno) - molecola complessa costituita da 2 coppie di 3 catene α, β, γ. 9 www.slidetube - implicata nell’emostasi 5) - Ig (immunoglobuline) - Le immunoglobuline sono un gruppo di proteine eterogenee, che migrano principalmente nella zona γ, ma anche nella zona β e più raramente nella zona α; costituiscono gli anticorpi umorali, prodotti dalle plasmacellule che derivano dai linfociti B attivati. - Esistono 5 classi di immunoglobuline (IgG, IgA, IgM, IgE, IgD) con struttura molecolare simile: 2 catene pesanti (γ,α,μ,ε,δ) e 2 catene leggere (κ,λ). - Le IgG sono quelle a maggiore concentrazione nel siero - IgG: 0,6 – 1,8 g/dl - IgA: 0,08 – 1,9 g/dl - IgM: 0,09 – 0,4 g/dl - Se sono aumentate le IgM l’infezione è in atto o recente - Se sono aumentate le IgG l’infezione è pregressa (cicatrice sierologica) - Le IgM sono pentameri; le IgA dimeri (uniche Ig secretorie, presenti nel latte, nella saliva, nel muco, nelle secrezioni intestinali); le IgG sono le uniche Ig che passano dalla madre al feto attraverso la placenta (il bambino nasce con le IgG materne e assume le IgA del latte); le IgE sono usate per quantizzare le allergie di I tipo (ipersensibilità di tipo I): il frammento Fc delle IgE si lega alle mastcellule piene di granuli di istamina. Esistono 3 principali tipi di allergia: - allergie alimentari - allergie da contatto (polline, via cutanea) - allergie ai farmaci (es. alla lidocaina - anestetico – non prevedibile) - Test che identificano prima le IgE generali, poi le IgE specifiche. - Dalla ventesima settimana c’è un brusco aumento delle IgG materne che passano la barriera placentare. Comincia la sintesi da parte del feto delle IgM. Al momento del parto le IgG materne diminuiscono e scompaiono all’ottavo mese. Aumentano le IgM, IgG e IgA del bambino, ma fondamentali sono le IgA materne trasmesse con il latte. Se c’è infezione intrauterina le IgM alla nascita sono elevate. 10 www.slidetube 90 80 70 60 IgG materne 50 IgM fetali 40 IgG fetali 30 IgAs meterne 20 10 0 -40 -30 -20 -10 0 10 20 30 Figura 3: sviluppo sistema immunitario dal concepimento a 6 mesi di vita - In condizioni normali la proporzione nella produzione di catena pesante e leggera è 1:1. - Nel mieloma la plasmacellula sintetizza più catene leggere di quelle pesanti; catene leggere libere in forma dimerica→proteinuria dio Bence-Jones (catene leggere-140aa-di IgG precipita a 60°-80°) - γ-patie policlonali: l’aumento delle proteine della zona γ si estende a tutto il tratto della zona - epatopatie - malattie del collageno - infezioni - γ-patie monoclonali: si osserva un picco ristretto nella zona γ QUADRI CLINICI TIPICI: - LES (Lupus Eritematoso Sistemico), malattia autoimmune -↓C3,C4; ↑PCR; γ-patia policlonale - IMMUNODEFICIENZA -alta la banda α1 e α2 basse o in tracce IgG, IgA, IgM - SINDROME NEFROSICA -alta la α-macroglobulina La terapia sostitutiva con estrogeni (sia terapeutica che anticoncezionale) simula una reazione di fase acuta: -↑TFR, HPT, α1, α2, AAT, CER. -MIELOMA MULTIPLO -es. ↑IgG e soppressione di IgM e IgA γ-patia monoclonale - Il siero del neonato ha elevati livelli di PCR, α-macroglobulina, α-antitripsina; se sono elevate anche IgM e IgA è in corso un’infezione. 11 www.slidetube INDICAZIONI allo STUDIO delle PLASMAPROTEINE 123456- Presenza componenti monoclonali Presenza e severità di un processo flogistico (se ↑ PCR) Aumento turnover eritrocitario (vedere TRF, saturazione, HPT- anemia emolitica) Attivazione del complemento (dosare C3 e C4 → malattie autoimmuni) Perché la VES è aumentata (associata a ↑ FIB o ↑ proteine di fase acuta) Stato nutrizionale (se l’albumina scende c’è un grave carente stato nutrizionale, doso ALB, pre-ALB, RBP, TRF)) 7- Patologie da deficit di proteine specifiche (AAT → enfisema, CER → morbo di Wilson) 8- Follow-up e controllo dell’efficacia della terapia Capitolo 1.1 COMPONENTE MONOCLONALE 12 www.slidetube COMPONENTE MONOCLONALE Picco γ causato da: - iperproliferazione di un clone di plasmacellule - sintesi di Ig omogenee per struttura e specificità - deficit a carico delle altre Ig Più del 95% dei soggetti ha una componente monoclonale <15 g/l Il 92% dei soggetti ha una componente monoclonale <10 g/l Il rischio di progressione è strettamente associato con la componente monoclonale. CONDIZIONI ANOMALE ASSOCIAT E alle Ig - MGUS: gammopatia monoclonale di incerto significato Benigno. Si ha nel 99% dei casi (IgM, IgG, IgA o IgD) - MIELOMA MULTIPLO: riguarda IgA e IgG. Nel MM il rapporto tra catene H e L è >1:1. Ci sono catene L singole o dimeriche libere nel sangue. Nelle urine si ritrovano le proteine di Bence-Jones. Il clone invade il midollo osseo provocando anemia, leucopenia e difficoltà alla coagulazione. - MACROGLOBULINEMIA di WALDENSTRÖM: linfoma che riguarda le IgM MALATTIE CAUSATE da Ig - CRIOGLOBULINEMIA I e II: sono Ig che precipitano ad una temperatura compresa tra 4 e 37 °C. Possono essere costituite da Ig singole monoclonali, Ig monoclonali con attività anticorpale verso Ig policlonali, Ig policlonali miste. causa della precipitabilità: forse contenuto ridotto in carboidrati. - AMILOIDOSI: deposizione IMMUNODEFICIENZE - AIDS è la più comune 13 www.slidetube STIMOLAZIONE CRONICA - Malattie autoimmuni (LES, artrite reumatoide) - Infezioni croniche TRANSIENTI - Infezioni pediatriche - Passaggi neonatali dalla madre - Infezioni virali - A seguito di un trapianto di midollo - Ipersensibilità ai farmaci (soprattutto sulfamidici) - MGUS è l’unica condizione benigna; come differenziarla dalle altre e specialmente dal MM (mieloma multiplo)? 3 parametri: - % di plasmacellule nel midollo - proteine circolanti nel siero - manifestazioni cliniche MGUS MIELOMA MACROGLOB. di AMILOIDOSI MULTIPLO PRIMARIA WALDENSTRÖM % plasmacellule ≥10 ≥10 <10 <10 % proteine ≥3 ≥3 <3 <3 sintomatico NO SI SI SI Secondo uno studio americano i soggetti con MGUS sono a rischio per lo sviluppo del mieloma. Qual è il rischio reale? L’unico fattore di rischio accertato per la progressione di MGUS a MM è la [C] di CM: ↑ CM → ↑ rischio Sospetto di MGUS; TEST RACCOMANDATI: - Visita - Emocromo - Siero, calcio, creatinina - Elettroforesi del siero → proteine totali - Elettroforesi delle urine → secrezione proteine nelle urine delle 24h - Immunofissazione del siero e delle urine → per discriminare CM - Catene L (κ e λ); non è un esame standard - Esame del midollo osseo Non si eseguono se CM<1,5 g/dl perché troppo invasivi - Biopsia dello scheletro Se la componente monoclonale è >15 g/l si proceda con: → biopsia ossea → radiografia dello scheletro PATOLOGIA MGUS (> o = 50 anni) Mieloma multiplo AL amiloidosi Waldenström (maschi) Waldenström (femmine) INCIDENZA 3,2% 40 /(milione di persone per anno) 8,9 /(milione di persone per anno) 3,4 /(milione di persone per anno) 1,7 /(milione di persone per anno) Per diagnosticare mieloma: - CM in siero e/o urine + plasmacellule nel midollo 14 www.slidetube e uno dei seguenti: - Calcium elevation - Renal insufficiency - Anemia - Bone desease - MIELOMA MULTIPLO (MM) Massa clonale - anemia - citopenia - plasmocitoma - immunodeficienza Citochine - anemia - distruzione osso - reazione di fase acuta - immunodeficienza Componente M - rene da mieloma - iperviscosità - immunodeficienza Il soggetto con mieloma deve essere trattato solo se sintomatico AMILOIDOSI - Clone molto piccolo che produce una proteina molto tossica. - Amiloidosi: deposito extracellulare di proteine autologhe che si riuniscono in fibrille con struttura a foglietto β causando un danno funzionale e strutturale dell’organo coinvolto. Le proteine tendono ad aggregare perché nella struttura β sono termodinamicamente meno stabili rispetto alle proteine normali. Stimoli principali: T°C, pH, ioni metallici, ossidazione. - Amiloidosi più diffusa è l’Alzheimer - L’amiloidosi deriva dall’azione proteolitica su un precursore che nel caso delle gammopatie sono le catene L prodotte dalle plasmacellule CAUSA BASILARE: - La amiloidosi si basa sul fatto che una proteina può assumere 2 conformazioni diverse: una ad α elica (non tossica) e una a β foglietto ripiegato (tossica); la quale provoca una malattia simile a quella dei prioni (PrP) per eziologia. - Birifrangenza verde mela con luce polarizzata dopo colorazione rosso congo; fibre non ramificate con diametro di 10 nm - Alcune forme di amiloidosi sono localizzate, altre sistemiche. si classificano sulla base della proteina coinvolta: - Proteine amiloidi: - β → Alzheimer - PrP → encefalopatie spongiformi - L (catene leggere immunoglobuline monoclonali) → sistemica - Nell’80% dei casi catene libere leggere λ che si depositano nei tessuti tranne il cervello - Macroglossia (12% dei pazienti con amiloidosi) - Porpora periorbitaria (6%); aspetto tipo procione (lesione periorbitaria, rottura di piccoli vasi) - Pseudoipertrofia muscolare (<1%) - Lesioni alle unghie (2-3%) - Shoulder pad 15 www.slidetube DIAGNOSI - Aspirato in regione periombelicale con ago sottile e colorazione. - Identificare e caratterizzare CM con elettroforesi ad alta definizione su agarosio o capillare associata a immunofissazione. Nel 56% dei pazienti con AL non si vede niente sul tracciato elettroforetico. L’immunofissazione aumenta di 10 volte la sensibilità dell’elettroforesi PROTEINA di BENCE-JONES (BJ) Proteina costituita da catene leggere libere monoclonali secrete da cellule B. E’ stato il primo marcatore di neoplasia scoperto. Il clone può essere sia maligno (solitamente associato a MM) che benigno (associato a MGUS) Cenni storici. La proteina di BJ fu osservata per la prima volta da un medico inglese (Watson) in urine riscaldate. Il calore denatura le proteine le quali precipitano. L’ALB forma un precipitato che non si scioglie mai; la proteina di BJ ha, invece, un comportamento particolare: verso la temperatura di 60°C sii forma il precipitato, che però si scioglie se continuiamo a riscaldare (T≈90°C) per poi riformarsi nuovamente durante il raffreddamento. Il dottore parlò di questa sua scoperta al dr Bence-Jones che se ne attribuì il merito in una pubblicazione. Le catene leggere possono essere di tipo κ o λ. si ritrovano entrambe in queste proporzioni 2/3 di catene κ e 1/3 di catene λ. Le catene λ formano dei dimeri che filtrano meno facilmente dei cloni κ nelle urine. Come risultato si avrà - una % maggiore di cloni λ nel siero - una % maggiore di cloni κ nelle urine Tutti gli individui producono catene leggere in eccesso. Si ha quindi una perdita giornaliera fisiologica di catene libere policlonali (PELC) di circa 5-10 mg/die. Esse formano piccole bande oligoclonali sul tracciato elettroforetico delle urine. Si parla anche di tracciato a scala. MALATTIE ASSOCIATE a BJ-P - Comuni: - mieloma multiplo - amiloidosi AL - morbo di Waldenström - malattia da deposito - Rare - linfoma - leucemia linfatica cronica 16 www.slidetube - idiopatie (benigne o di incerto significato) Le proteine di BJ vanno ricercate nelle seconde urine del mattino tramite un’elettroforesi seguita da immunofissazione. Gli antisieri utilizzati sono anti-κ, anti-λ totali con l’aggiunta dell’antisiero antiCH della CM. Da 3 anni esiste un test, chiamato FREELITE, che consente di valutare il rapporto κ/λ delle catene leggere libere nel siero. E’ un test di routine per pazienti con gammopatie monoclonali. TEST da SCORAGGIARE (inadatti per la ricerca di proteine di BJ) - proteine totali nelle urine (precipitazione, legame col colorante [dye binding]; test poco sensibile) - stick delle urine (sostanza che cambia colore in presenza di proteine acide come l’ALB e non “vede” le proteine di BJ) - test al calore (heat test); poco sensibile e poco specifico. Sensibilità dei diversi metodi - IFE: 1 proteina su 102- 103 ← STANDARD - citogenetica e southern blot: 1 su 102 - FISH in interfase: 1-5 su 103 - citofluorimetria: 1-5 su 103 - 104 - PCR: 1 su 105- 106 Capitolo 1.2 17 www.slidetube TECNICHE ELETTROFORETICHE ELETTROFORESI L’elettroforesi consiste nella migrazione di particelle elettricamente cariche in un campo elettrico E. Nel plasma a pH 7,4 le proteine che hanno punto isoelettrico minore del pH fisiologico si dissociano come anioni. Poste in E migrano verso il polo + con velocità che dipende: - dalla carica elettrica direttamente proporzionali alla v di - dalla forza di E migrazione - dimensione e forma della molecola - proprietà del mezzo di supporto inversamente proporzionali alla v di migrazione TIPI di SUPPORTO: sono vari; i principali sono agarosio, gel di poliacrilamide o soluzioni. Permettono la separazione in base a carica e dimensione. Spesso si aggiunge un detergente carico negativamente (sodio dodecilsolfato) che eguaglia le cariche di superficie e consente la separazione solo sulla base della dimensione molecolare. Dopo la separazione le proteine vengono evidenziate con opportune colorazioni. Un particolare tipo di elettroforesi è l’ELETTROFORESI CAPILLARE. In essa la separazione elettroforetica avviene in un tubo di silice fusa lungo 30-50 cm e diametro compreso tra 10 e 75 μm. 18 www.slidetube Il tubo è riempito con una soluzione tampone. Le due estremità del capillare sono immerse in due provette contenenti lo stesso tampone. la separazione avviene in seguito all’applicazione di una differenza di potenziale tra le due estremità. La v delle particelle è in funzione del rapporto carica/massa. Le proteine cariche negativamente sono attratte dall’anodo (+). Tuttavia all’interno del capillare si sviluppa anche un flusso del tampone in direzione del catodo (‒ ). Tale fenomeno è chiamato ELETTROENDOSMOSI. Esso è dovuto alla presenza di gruppi carichi negativamente sulla superficie del capillare (neutralizzati da una “nuvola” di cariche positive. Quando si applica una d.d.p. ioni positivi sono attratti dal catodo e, migrando, trascinano con sé la soluzione in cui sono immersi. - il capillare è in silice e ha cariche negative che vengono neutralizzate da ioni positivi - applicando una differenza di potenziale (V) gli ioni vanno verso il catodo (flusso osmotico, trasporto di acqua) e le proteine verso l’anodo - Il flusso endosmotico vince sulla forza elettroforetica perciò tutte le proteine vanno verso il catodo (negativo) - Ig → carica + migrano verso il catodo - ALB → carica ‒ trascinata dal flusso endosmotico verso il catodo VANTAGGI: è un metodo automatizzabile; limite CM>0,5 g/l; utile nel monitoraggio dopo una terapia. SVANTAGGI: non consente l’immunofissazione; la CM può scomparire perché interagisce col capillare. E’ un’eventualità rara, ma possibile, che può portare a una diagnosi totalmente errata. Ricordare che CM può migrare fino a zona α2 (IgA) Al posto dell’immunofissazione, dopo elettroforesi capillare è possibile applicare il metodo dell’immunosottrazione (IF-ES) che permette di valutare il contributo di ciascuna Ig al picco γ: classi e tipi di Ig sono rimossi dal campione usando Ab anti-Ig specifici per IgG, IgA, IgM, λ e κ. Dopo l’aggiunta di ogni Ab si ripete l’elettroforesi per valutare come si è modificato il tracciato. 6 5 4 IgG 3 IgM 2 IgA 1 0 0 1 2 3 4 Campione da testare → aggiungo Ab-anti-IgG Figura 4: immunosottrazione (IF-ES) 19 www.slidetube 1,5 1 IgM 0,5 IgA 0 0 1 2 3 Campione senza IgG + precipitato IgG-anti-IgG 4 IMMUNOFISSAZIONE (IFE): rende 10 volte più sensibile l’elettroforesi (perché aumenta la quantità di proteina presente). tecnica usata per caratterizzare e quantificare una proteina. Procedimento: - elettroforesi - aggiunta di un Ab specifico per un CM, incubazione - eseguire ripetuti lavaggi delle proteine che non hanno reagito - evidenziare la banda di immunoprecipitazione con opportuna colorazione La quantificazione della banda è eseguita con tecniche di densiometria Capitolo 2 20 www.slidetube LIPIDI SIERICI LIPIDI SIERICI E RISCHIO ATEROSCLEROSI Attualmente la principale causa di mortalità nei paesi sviluppati sono le malattie cardiovascolari. Esse sono un disordine generalizzato delle arterie caratterizzato dalla formazione e progressivo accrescimento di placche ateromasiche culminanti con l’occlusione parziale o totale del lume vasale. A seguito dell’occlusione si forma un danno ischemico la cui gravità è proporzionale alla natura dell’arteria coinvolta. I distretti più frequentemente coinvolti sono: - le coronarie - le carotidi e il circolo cerebrale - arterie degli arti inferiori L’aterosclerosi interessa l’intima e la media delle arterie e consiste in accumulo vascolare di lipidi, danno e attivazione endoteliale, attivazione e adesione piastrinica, infiltrazione infiammatoria, proliferazione di cellule muscolari lisce e loro degenerazione in “cellule schiumose” (cappello fibroso + core lipido-necrotico) e sovrapposizione di complicanze trombotico-occlusive. C’è una 21 www.slidetube correlazione tra il fenomeno infiammatorio (quantificato dosando la CRP) e l’infarto. La placca da sola è asintomatica. I lipidi sierici - aumentano nelle patologie degenerative (neoplasie e aterosclerosi) - sono diminuiti ma stanno riaumentando nelle patologie infettive ATEROSCLEROSI: FATTORI di RISCHIO: - MAGGIORI- iperlipidemia - fumo di sigaretta - ipertensione - diabete -età avanzata - gotta e iperuricemia - angina pectoris - morte cardiaca acuta - infarto del miocardio -PREDISPONENTI- sovrappeso - inattività fisica - storia familiare -CONDIZIONALI- ipertrigliceridemia - elevati livelli di Lp(a) - elevati livelli di omocisteina - elevati livelli di fibrinogeno - marcatori infiammatori LIPIDI di INTERESSE CLINICO --COLESTEROLO TOTALE ≤200 mg/dl ; se colesterolo >240 mg/dl → rischio elevato se colesterolo <150 mg/dl è basso; > 350 mg/dl è aterogeno per valori compresi tra 150 e 350 il rischio è determinato dalla quantità di HDL disponibile (importante per sequestrare il colesterolo trasportato dalle LDL) --HDL La [HDL] plasmatica è inversamente correlata all’insorgenza di malattie cardiovascolari. HDL ha un ruolo nel trasporto retrogrado del colesterolo, forse ha proprietà antiossidanti e antiinfiammatorie. VALORI di HDL maschi basso <35 medio 35-55 alto >55 22 www.slidetube femmine <45 45-65 >65 Nelle donne la stimolazione da parte degli estrogeni promuove un aumento delle lipoproteine HDL; effetto analogo è dato dall’assunzione di livelli moderati di vino rosso (paradosso francese) --LDL Elevate concentrazioni di colesterolo LDL producono un aggravamento del rischio cardiovascolare. - LDL > 130 mg/dl → valore di ALLARME - LDL < 100 mg/dl → se ci sono già stati problemi coronarici Valori molto elevati di LDL si riscontrano nelle varie forme di iperlipidemia, ipercolesterolemia familiare, ipotiroidismo, sindrome nefrosica, ostruzione biliare, gravidanza. Esistono due differenti forme di LDL che hanno potenzialità aterogene diverse. - Fenotipo A: lipoproteine grandi e poco dense - Fenotipo B: lipoproteine piccole e dense Il fenotipo B ha un rischio aterogeno 3 volte superiore al fenotipo A. la predominanza del fenotipo B è ereditaria ed è una condizione denominata PROFILO LIPOPROTEICO ATEROGENO (PLA); è un quadro caratterizzato da: - LDL a fenotipo B che si accumulano più facilmente delle A nell’intima delle arterie - LDL più ricche in fosfolipasi AL e più suscettibili all’ossidazione - ridotta concentrazione di HDL soprattutto fenotipo 2b, che sono le più efficienti nel trasporto inverso del colesterolo - ridotto potenziale fibrinolitico - maggiore incidenza di diabete di tipo 2 - ipertrigliceridemia Questo quadro è presente almeno nel 50% dei soggetti con malattie cardiovascolari. La valutazione del fenotipo LDL non è un esame esteso a tutta la popolazione e non è eseguibile in tutti i laboratori, ma solo in centri altamente specializzati. --TRIGLICERIDI Tra ipertrigliceridemia e rischio di malattie cardiovascolari esiste un’associazione; tuttavia non è chiaro se essa è imputabile ai trigliceridi stessi e alle lipoproteine che li trasportano o ai disordini metabolici associati all’ipertrigliceridemia. - trigliceridi > 150 mg/dl → soglia di rischio (spia di altre alterazioni metaboliche potenzialmente aterogene come ↑ LDL - trigliceridi > 200 mg/dl → rischio indipendente Esiste un dimorfismo sessuale nella concentrazione dei trigliceridi: - fino ai 20 anni uguale nei 2 sessi - nella vita adulta è superiore nell’uomo - dopo la menopausa uomo e donna tornano ad avere gli stessi valori L’ipertrigliceridemia è presente più facilmente in obesi, sedentari, iperlipidemie familiari, pancreatiti, diabete di tipo 2, insufficienza renale cronica, abuso di alcool, fumo di sigaretta, gravidanza. 23 www.slidetube Capitolo 2.1 24 www.slidetube LIPOPROTEINE e DISLIPIDEMIE METABOLISMO e STRUTTURA delle LIPOPROTEINE I lipidi sono sostanze apolari, non solubili nel plasma. Vengono trasportati dalle lipoproteine, molecole sferiche che presentano un involucro esterno composto da fosfolipidi (bipolari), apoproteine e colesterolo libero (dà stabilità alla struttura). La struttura delle lipoproteine ricorda quella della membrana cellulare. Il core della molecola è costituito da colesterolo esterificato e trigliceridi. Esistono differenti classi di lipoproteine suddivise in base alla composizione chemica e alla densità. 25 www.slidetube - CHILOMICRONI: lipoproteine sintetizzate nella mucosa intestinale costituite da una piccola frazione proteica di apoB48, apoAI, apoAIV, apoCI, apoCII, apoCIII. trasporta trigliceridi alimentari e vitamine liposolubili. la proteina ApoB (proteina principale del metabolismo lipidico) consta di due isoforme: - ApoB48: di origine intestinale per trasporto di lipidi di provenienza alimentare - ApoB100: sintetizzata nel fegato, si trova nelle LDL per trasporto dei lipidi endogeni Derivano dalla trascrizione di un singolo gene, per modificazione post-trascrizionale si differenziano. dal lume intestinale i chilomicroni passano nel sistema linfatico e da qui nella circolazione sanguigna tramite il dotto toracico. Così facendo assicurano la disponibilità di grassi ai tessuti periferici. in circolo subiscono due modificazioni principali: -1-IDROLISI dei trigliceridi ad acidi grassi e glicerolo (gli ac. grassi sono la forma in cui i lipidi sono ceduti ai tessuti) ad opera della lipoproteinlipasi (LPL) endoteliale. -2-SCAMBIO di alcune apolipoproteine: rilasciano apoAI e apoAIV e si arricchiscono di apoC e apoE Il complessivo impoverimento di trigliceridi dei chilomicroni si associa a una serie di trasformazioni biochimiche che portano a due prodotti di degradazione: -1-RENMANTS: molecole con core centrale di natura lipidica captati e distrutti dal fegato (recettore per le LDL legato ad apoE del chilomicrone) -2-HDL NASCENTI: precursori delle HDL plasmatiche - VLDL, IDL, LDL: le VLDL sono precursori di tutte le lipoproteine a bassa densità circolanti. Sono sintetizzate dal fegato e costituite principalmente da trigliceridi e colesterolo endogeno. il dominio proteico è quasi esclusivamente rappresentato da ApoB100. Se si squilibra il processo di idrolisi dei chilomicroni e sintesi delle VLDL si ha la steatosi. le VLDL sono secrete nel torrente ematico dove subiscono due modifiche: -1-azione dell’enzima CEPT (proteina di trasporto specifica per il colesterolo esterificato) che catalizza il trasferimento dei trigliceridi alle HDL in scambio con colesterolo esterificato. VLDL si trasformano in particelle più piccole e dense. -2-scambio di apolipoproteine: ApoE e ApoC sono trasferite alle VLDL dalle HDL. ApoCII attiva la LPL che idrolizza i trigliceridi ancora presenti trasformando le VLDL in molecole più dense, le IDL (intermediate density lipoproteins) Le IDL sono molecole molto instabili; per questo motivo devono essere presenti in quantità molto bassa e rapidamente convertite in LDL. Il processo di conversione in LDL coinvolge CEPT o in 26 www.slidetube alternativa l’enzima LIPASI EPATCA, che eliminano i trigliceridi rimasti e la cessione di ApoE e ApoC alle HDL2. Le LDL sono quindi lipoproteine contenenti colesterolo esterificato e con un dominio proteico costituito prevalentemente da ApoB100. Le LDL hanno un recettore periferico che al legame con LDL la interna lizza e prende il colesterolo in essa contenuto (che viene esterificato; contemporaneamente si blocca la sintesi di colesterolo endogeno e dei recettori per le LDL). Se le LDL sono troppe esse si legano anche a recettori non specifici (scavenger) e nella cellula si accumula colesterolo (cellula schiumosa) - LPL: la sua attività è dipendente da ApoCII (fegato, endotelio, muscolo, tessuto adiposo) - LIPASI EPATICA: la sua attività è indipendente da ApoCII (fegato, surrene, ovaie) - HDL: complesso costituito da un eterogeneo gruppo di particelle adibite al trasporto di colesterolo libero, esteri del colesterolo e trigliceridi. Presentano apolipoproteine interscambiabili (tipi A, C, D, E). Ci sono due classi principali suddivise sulla base della componente apolipoproteica: - HDL2: contengono ApoAI e ApoAII - HDL3: contengono ApoAI Sono sintetizzate prevalentemente nel fegato e nell’intestino. La loro funzione fondamentale è opposta a quella delle LDL: trasportano lipidi dalla periferia al fegato. Esse si legano alla superficie cellulare tramite un recettore scavenger (non specifico) che interagisce con ApoAI e assumono il colesterolo in eccesso nella cellula. Ci sono due enzimi fondamentali nel metabolismo delle HDL: - LCAT (lecitina-colesterolo-acil-transferasi): trasferisce un gruppo acilico (ac.grasso) dalla lecitina sierica al colesterolo con formazione di lisolecitina e esteri del colesterolo - CEPT: trasferisce esteri del colesterolo da HDL2 a VLDL in cambio di trigliceridi, con contemporanea cessione di ApoC e ApoE alle VLDL L’azione di questi due enzimi è responsabile di un metabolismo delle HDL che assume una forma ciclica: pre-β-HDL (proteina nascente) CEPT HDL2 LCAT HDL3 27 www.slidetube LCAT L’HDL nascente è sottoforma di pre-β-HDL di forma discoidale; LCAT, favorendo l’ingresso di colesterolo esterificato nella molecola, ne permette l’accrescimento (abbandono della struttura discoidale per quella sferica) con trasformazione in HDL3 e HDL2. CEPT, invece, rimuovendo colesterolo fa assumere a HDL2 una struttura più simile a quella delle pre-β-HDL. Il fegato capta il colesterolo delle HDL in tre modi: - capta le VLDL; è il colesterolo che esse hanno assunto dalle HDL2 - capta le HDL tramite recettori specifici - capta le HDL che contengono ApoE per la stessa via dei chilomicroni E’ importante studiare il rapporto HDL/LDL e in particolare ApoAI/ApoB100 per quantizzare il flusso di colesterolo in ingresso e in uscita dalla cellula -ApoAI = 20 mg persona normale -ApoB100 = 80 mg persona a rischio Tabella riassuntiva PROTEINA ApoAI ApoAII ApoB48 ApoB100 ApoCI ApoCII ApoCIII ApoD ApoE DOVE SI TROVA HDL HDL Chilomicroni VLDL, IDL, LDL Chilomicroni, VLDL, HDL Chilomicroni, VLDL, HDL Chilomicroni, VLDL HDL, VLDL Chilomicroni, VLDL, LDL, HDL FUNZIONE Attiva LCAT Attiva LPL Attiva LPL Attiva LPL - Lipoproteina (a) [Lp(a)]: variante genetica delle LDL da cui differisce per la presenza di una molecola denomina apolipoproteina(a) (apo-a) legata mediante un ponte disolfuro al dominio proteico della ApoB100. E’ prodotta dal fegato e immessa nel circolo ematico. A causa di apo-a la sua affinità per il recettore delle LDL è pari al 25% e quindi rimane in circolo più a lungo rispetto alle LDL con maggiore probabilità di: - essere ossidata - essere captata da recettori scavenger (i più pericolosi nella patogenesi dell’aterosclerosi perché non regolati) La Lp(a) può indurre danno endoteliale, favorisce l’accumulo di colesterolo, promuove complicanze trombotiche nella placca poichè ha un’azione anti-fibrinolitica in quanto è un analogo strutturale del plasminogeno e compete con quast’ultimo per i recettori sulla fibrina. -Lp(a) = 25-30 mg/dl -Lp(a) > 30 mg/dl soggetto normale soggetto a rischio 28 www.slidetube Pare che la [Lp(a)] sia geneticamente determinata e poco modificabile con interventi dietetici e farmacologici. Non consigliato lo screening di massa, ma il dosaggio è raccomandato per i pazienti ad alto rischio, in modo tale da impostare una terapia più aggressiva nei confronti di altri fattori di rischio modificabili con un trattamento. OSSIDAZIONE LDL Le LDL in circolo possono subire una reazione di ossidazione da parte di MIELOOSSIDASI circolanti o CERULOPLASMINA. La LDL ossidata ha un elevatissimo potenziale aterosclerotico in quanto si lega unicamente ai recettori scavenger (che non hanno il meccanismo a feedback di regolazione). E’ un meccanismo privo di controllo, che non può essere arginato dalle HDL. Il laboratorio ancora non dosa le LDL ossidate. - OMOCISTEINA: è un amminoacido contenente zolfo derivante dal metabolismo della metionina. Elevati livelli di omocisteina nel sangue sono correlati positivamente con un’incidenza maggiore di eventi cardiovascolari precoci. Essa infatti è in grado di promuovere danno endoteliale, proliferazione della componente cellulare del vaso, innalzamento del potere aterogeno delle lipoproteine (specialmente Lp(a)). I principali determinanti della [omocisteina] nel sangue sono la VITAMINA B e i FOLATI, enzimi indispensabili nel suo metabolismo. Particolare attenzione meritano quindi gli stati di carenza. -[omocisteina] < 10 μmol/l soggetto normale -[omocisteina] tra 16 e 30 μmol/l moderata iperomocisteinemia -[omocisteina] tra 31 e 100 μmol/l intermedia iperomocisteinemia -[omocisteina] > 100 μmol/l severa iperomocisteinemia - FIBRIONGENO: Elevati livelli di fibrinogeno nel sangue sono correlati positivamente con un aumento di rischio di malattie cardiovascolari. Molteplici le cause: maggiore è il fibrinogeno presente, maggiori le dimensioni del trombo; il fibrinogeno inoltre promuove la proliferazione della componente cellulare dei vasi, l’accumulo di lipoproteine nella sede del danno endoteliale, l’aggregazione piastrinica. Ci sono perplessità nell’uso del fibrinogeno quale indicatore di danno cardiovascolare. -fibrinogeno compreso tra 150 e 400 mg/dl normale DISLIPIDEMIE Le dislipidemie possono essere di 5 tipi: 29 www.slidetube - I tipo: caratterizzato da ↑ trigliceridi, colesterolo normale perché i chilomicroni non sono idrolizzati. I pazienti si questo tipo non sono ad alto rischio. - II tipo: dislipoproteinemia - tipo 2a: ↑↑colesterolo; trigliceridi normali - tipo 2b: ↑↑colesterolo; ↑ trigliceridi Sono pazienti ad alto rischio - III tipo: ↑colesterolo; ↑trigliceridi; mancanza o malfunzionamento dei recettori per le ApoE. Parziale demolizione delle VLDL con formazione delle IDL. Pazienti ad alto rischio - IV tipo: colesterolo normale; ↑↑trigliceridi; accumulo di trigliceridi dovuto alle VLDL. Sono pazienti che solitamente diventano diabetici. Nella terapia togliere i carboidrati Importante distinguere tra tipo I e tipo IV -V tipo: ↑ o normalità colesterolo; ↑↑↑trigliceridi; somma dei difetti I e IV = accumulo di chilomicroni e VLDL. La dieta deve coinvolgere sia i lipidi che i carboidrati. ELETTROFORESI delle LIPOPROTEINE E’ possibile avere un tracciato elettroforetico delle lipoproteine che permette di distinguere le dislipidemie di tipo I, IV e V. Capitolo 3 30 www.slidetube ENZIMI nella DIAGNOSTICA ENZIMI NELLA DIAGNOSTICA Alcuni enzimi sono importanti segnali di malattia. Essi hanno, in condizioni normali, una precisa ubicazione all’interno della cellula: - enzimi mitocondriali (es. GLDH) - enzimi citoplasmatici (es. GPT, LDH) - enzimi ubiquitari (es. GOT, MDH) La perdita di enzimi dalla cellula e il loro riversamento nel torrente circolatorio possono essere causate da due situazioni: - lisi cellulare conseguente a necrosi (ad es. in epatiti e pancreatiti e infarti del miocardio) - epatite: ↑transaminasi - pancreatite: ↑amilasi - infarto: ↑creatina fosfochinasi 31 www.slidetube - forme lievi e reversibili di danno cellulare (ipossia, alterazione del pH) che alterano la permeabilità della membrana. Ogni enzima ha una propria vita media e un certo andamento in circolo. Solitamente più è severo il processo patologico, più i livelli enzimatici sono elevati. Ciò però non vale per tutti gli enzimi: - enzimi tipo A: sono gli enzimi della coagulazione, la pseudo colinesterasi e altri; essi diminuiscono la loro attività in caso di patologia. Li troveremo quindi ↓ che nella norma. - enzimi tipo B: sono lipasi, amilasi e gli altri enzimi del tratto digestivo. Essi aumentano in caso di patologia. Altre situazioni importanti da verificare sono: - escrezione urinaria di enzimi: è un processo che riguarda solo alcuni enzimi, come l’amilasi, che passano attraverso la barriera renale per le loro caratteristiche molecolari - carenze congenite di enzimi. ASPETTO ENZIMATICO DEI VARI TESSUTI - GPT = glutammico-piruvico-transaminasi o ALT = alanina-transaminasi ORGANO fegato muscolo ENZIMA (unita/grammo d’organo) 35 3,5 Enzima specifico per il fegato. E’ il miglior indice di citolisi epatica. E’ più alto nell’uomo e varia con l’età. ↑GPT: epatite virale acuta, necrosi epatica su base tossica, epatite autoimmune 32 www.slidetube ↑GPT di minore entità: cirrosi epatica, ittero colestatico, stasi epatica da insufficienza cardiaca, mononucleosi infettiva - GOT = glutammico-ossalacetico-transaminasi o AST = aspartato-transaminasi ORGANO fegato cuore muscolo cervello rene pancreas polmone ENZIMA (U/gr. organo) 59 52 36 15 10 3 1 Enzima di citolisi aspecifico. Enzima presente in molti tessuti sia a livello citoplasmatico che mitocondriale. ↑GOT marcato: fisiologico nel neonato, epatite virale acuta, necrosi epatica su base tossica, infarto del miocardio, traumi muscolari, ipossia tissutale, epatite autoimmune ↑GOT moderato: cirrosi epatica, ittero colestatico, carcinoma epatico, insufficienza cardiaca, mononucleosi infettiva, malattie muscolari, emolisi - GLDH = glutammato-deidrogenasi ORGANO fegato rene cervello polmone ENZIMA (U/gr. organo) 38 4,4 3,2 2,5 Enzima specifico per il fegato. Enzima di citolisi specifico come il GPT. Il rapporto ALT/AST è importante per la diagnosi di alcune malattie: Danno lieve del fegato (lesa la membrana) GPT>GOT>GLDH epatiti acute e quasi tutte le patologie epatiche 33 www.slidetube Danno severo del fegato (lesi i mitocondri) GOT>GPT>GLDH epatite alcolica, sindrome di Reye Solitamente GPT>GOT perché GOT è inattivata più rapidamente (t1/2 minore) e GPT (contenuto nel citoplasma diffonde più velocemente di GOT. - LDH = lattico-deidrogenasi ORGANO muscolo scheletrico fegato cuore rene linfonodi pancreas globuli rossi polmone ENZIMA (U/gr. organo) 147 145 124 106 83 50 36 27 Enzima della glicolisi che si libera dopo necrosi cellulare. Enzima aspecifico di citolisi. ↑LDH: anemie emolitiche, epatopatie, infarto del miocardio (tardivamente), neoplasie, distrofie muscolari. L’enzima è costituito da quattro catene polipeptidi che di due tipi: - H (heart) la loro combinazione dà origine a 5 isoenzimi (LD-1→LD-5) - M (muscle) H4 M4 LD1 e LD2 → isoenzimi cardiaci → non totalmente specifici però LD4 e LD5 → isoenzimi epatici Nell’infarto LDH serve per monitorarne il decorso: se ↓LDH l’infarto sta guarendo → ciò ha oggi perso molta importanza clinica perché si usano le troponine Nelle neoplasie aumentano tutti gli isoenzimi con extrabande 34 www.slidetube 5 4,5 4 3,5 3 2,5 BANDA EXTRABANDA 2 1,5 1 0,5 0 0 5 10 15 20 25 - ALD = aldolasi ORGANO muscolo fegato cuore muscolo liscio rene globuli rossi ENZIMA (U/gr. organo) 48 5,7 4,9 2,6 1,1 1 Indice non totalmente specifico del muscolo ↑ALD: miopatie, epatite acuta, infarto del miocardio - CPK o CK = creatin-fosfochinasi 35 www.slidetube ORGANO muscolo cervello cuore muscolo liscio rene fegato ENZIMA (U/gr. organo) 2030 670 350 12 2 0,7 Enzima non specifico L’enzima è costituito da due subunità: - M (muscle) la loro combinazione dà origine a 3 isoenzimi - B (brain) MM MB (muscolo) (cuore) ↓ (5-30% dell’attività totale della CK) - BB (cervello) ↑CK: distrofia muscolare, polimiositi, infarto del miocardio, esercizio fisico intenso, ictus,… In caso di infarto si dosa l’isoenzima CK-MB in prima giornata. Ci possono essere 2 tipi di misurazione: - CK-MB/CK(%) → misura l’attività di CKMB rispetto a quella di CKtot. In condizioni normali è <6% ↑ in caso di infarto. Non è attendibile però se oltre all’infarto di sono dei danni muscolari MISURA DEI CK-MB FUNZIONANTI - CK-MB massa → misura la concentrazione di massa dell’enzima. Importante per diagnosi precoce MISURA DEL N° DI CK-MB - ALP = fosfatasi-alcalina enzima aspecifico 36 www.slidetube Viene prodotto da molte cellule, ma principalmente da: - epatociti e cellule dei dotti biliari - osteoblasti - epitelio intestinale ↑ALP fisiologico: durante la crescita, nel 3° trimestre di gravidanza, negli anziani (specialmente le donne) ↑ALP patologico: - malattie ossee (fratture, rachitismo, tumori degli osteoblasti, morbo di Paget) - malattie epatiche: il fegato ha 2 isoenzimi L1 e L2. Se ↑L1 (compare in elettroforesi) ci sono malattie epatiche maligne come sarcoidosi, tumori, metastasi, malattie granulomatose. Se ↑L2 può essere un’epatite complicata da colestasi o una colestasi cronica (colangite, colica biliare). Se l’ALP è di origine epatica solitamente è accompagnato da ↑γGT -FOSFATASI ACIDA = enzima che idrolizza un estere fosforico liberando Pi (fosfato inorganico) a pH = 4 o 5. MARKER ASPECIFICO di CARCINOMA PROSTATICO E’ prodotto dalla prostata e dall’osso (principalmente) ma anche da rene, piastrine, stomaco e fegato ↑patologico: carcinoma della prostata, ipertrofia prostatica, malattie ossee specialmente pediatriche, morbo di Paget. L’enzima è presente in 2 frazioni: 5a e 5b ↓ ↓ prostata solo nell’osso La frazione 5b è inibita dall’1-tartrato. Aggiungendolo si può isolare la frazione prostatica. Non è un test molto specifico e rivela il carcinoma solo quando è in fase già avanzata. Sono preferibili altri test. - γGT = gamma-glutamil-transpeptidasi → usa cisteinil-glicina come substrato a cui attacca il glutatione. Importante per recupero aminoacidi Mai trascurarlo se > 40 Enzima prodotto specialmente dal fegato, ma presente in quantità più modeste anche in rene e pancreas. E’ localizzato a livello dei microsomi e dei dotti biliari. ↑γGT: disturbo enterobiliare con microstasi, somministrazione di farmaci (benzodiazepine, antidepressivi e tranquillanti), abuso di alcool, pancreatiti (acuta e alcolica), tumori, colecistiti, cirrosi, ittero ostruttivo intraepatico, metastasi, insufficienza cardiaca (cuore dx →stasi epatica) E’ un indice di microstasi e di intossicazione epatica. E’ specifico ma bisogna stare attenti perché è inibito da estrogeni (gravidanza, contraccettivi orali) e dalla bilirubina. - AMY = amilasi Enzima che idrolizza amido e glicogeno rompendo i legami 1-4α-glicosidici. 37 www.slidetube enzima aspecifico E’ prodotto da pancreas, ghiandole salivari e tube di falloppio. ↑AMY: pancreatite acuta e cronica, occlusione intestinale alta, coliche biliari, ulcera, neoplasia pancreatica, appendicite e interessamento peritoneale, parotite, gravidanza extrauterina, insufficienza renale, macroamilasemia (nella macroamilasemia l’amilasi si lega a una IgG formando un complesso ad alto peso molecolare che no filtra nelle urine (↑amilasemia e ↓amilasuria)) Se è ↑amilasuria significa che c’è anche un’amilasemia patologica. Importante quindi un suo dosaggio ↑AMY: può essere indice di un’insufficienza renale in stadio molto avanzato. Importante di stinguere tra gli isoenzimi P3 e S ↓ ↓ pancreas ghiandole salivari e tube - LIPASI, TRIPSINA, CHIMOTRIPSINA La lipasi pare essere un segno più specifico dell’amilasi per la diagnosi di pancreatite acuta. La chimotripsina misura la funzione pancreatica esogena ed è importante per la diagnosi di pancreatite cronica. - CHE = colinesterasi (4 isoforme) o pseudocolinesterasi (11 isoforme) Enzima che idrolizza la colina e i suoi esteri tra cui la succinilcolina che è responsabile della trasmissione sinaptica. E’ in circolo in quantità abbastanza elevate in quanto indice dell’attività di sintesi proteica del fegato. ↓CHE: epatopatie di secrezione (epatite, neoplasia), condizioni in cui l’ALB è bassa, avvelenamento da esteri fosforici. ↑CHE: tireotossicosi, sindrome nefrosica. Esiste 2 forme genetiche di CHE: - normale - atipica (omozigote recessiva) Gli individui con CHE atipica non sono in grado di idrolizzare alcune anestetici (come la succinilcolina) che bloccano la respirazione. Perciò si svilupperà un’apnea prolungata. Inoltre la CHE atipica non è inibita dalla dibucaina perché il legame ha un’affinità minore del 50100% rispetto a CHE normale. 38 www.slidetube Capitolo 3.1 INFARTO del MIOCARDIO INFARTO DEL MIOCARDIO 39 www.slidetube Nel corso degli ultimi 20 anni l’aumento dei fattori di rischio e l’allungamento della vita media hanno causato un aumento del numero di patologie cardiache legate a ischemia coronarica. La principale causa di eventi ischemici cardiaci è l’aterosclerosi coronarica, malattia che porta progressivamente all’occlusione parziale o totale delle arterie con conseguente ischemia. In questo contesto i quadri anatomopatologici dei vari pazienti sono molto diversi tra loro a seconda dell’estensione dell’infarto (transmurale o subendocardico). Analogamente le manifestazioni cliniche spaziano dall’angina da sforzo, all’angina instabile, all’infarto con o senza sottoslivellamento del tratto ST. Principali parametri da cui dipende la comparsa di danni: - n° di vasi occlusi - sede dell’occlusione - presenza di circoli collaterali - durata dell’evento occlusivo -1<20 min: generalmente la riperfusione non comporta necrosi del tessuto o danni permanenti, ma solo una modesta alterazione funzionale -2>20 min: danni da riperfusione permanenti con necrosi, edema, rigonfiamento cellulare, deposizione di Ca2+ Come si è visto le lesioni maggiori compaiono a seguito della RIPERFUSIONE del tessuto, non durante l’ischemia vera e propria. Le principali cause del danno da riperfusione sono: - l’ossigeno, che porta alla formazione di radicali liberi (ROS) che interagiscono con la membrana cellulare determinando un danno ossidativo; - l’alterazione dell’omeostasi del Ca2+ intracellulare con suo accumulo citoplasmatico (a ciò consegue la morte cellulare perché Ca2+ attiva proteasi e endonucleasi); - l’alterazione endoteliale con formazione di un essudato infiammatorio (accumulo di neutrofili, attivazione C’…). A seguito di questi eventi le cellule lesionate liberano in circolo una serie di molecole che possono assumere il ruolo di MARCATORI più o meno specifici dell’evento. In generale: quanto più il marcatore è specifico, tanto più tardivamente compare in circolo. DIAGNOSI 40 www.slidetube La diagnosi di infarto del miocardio acuto (IMA) presenta delle problematiche ancora irrisolte. IMA: necrosi di cellule miocardiche a seguito di ischemia prolungata. Nel passato la diagnosi di IMA presupponeva il riscontro contemporaneo di almeno 2 delle seguenti condizioni: - angina - alterazione ECG triade - ↑enzimi marcatori di danno miocardico Questo permette di diagnosticare solo le lesioni maggiori, non tutti gli eventi che caratterizzano la progressione della sindrome coronarica acuta. Con le moderne tecniche è possibile identificare aree di necrosi inferiori a 1g di tessuto (con ECG normale) ciò è importante perché piccoli infarti possono dare origine ad aritmie con gravi conseguenze. la diagnosi di IMA oggi si fa dosando l’aumento della [C] in circolo di marcatori specifici e sensibili di danno miocardico. Un marcatore ideale di danno miocardico deve avere le seguenti caratteristiche: - essere presente solo nel tessuto miocardico in elevata concentrazione - essere indosabile nel sangue in assenza di danno miocardico - essere rilasciato rapidamente e in quantità rilevante in presenza di danno miocardico - aumentare proporzionalmente all’entità del danno - persistere in circolo per un periodo di tempo sufficiente per consentire la diagnosi (BUONA FINESTRA DIAGNOSTICA) - essere dosato con metodi rapidi, accurati, precisi, efficienti ed economici. Secondo le più recenti linee-guida i migliori marcatori sono il complesso delle troponine e la CK-MB massa. Sono sconsigliati i dosaggi di alcuni enzimi tradizionali quali CKtot, LDH, GPT, GOT per la considerevole specificità nella localizzazione tissutale. 800 700 600 500 LDH HBDH 400 CPKtot 300 GOT GPT 200 100 0 0 1 2 3 4 5 6 7 Figura 5: Prima di 4-8h non si alza nessuno degli enzimi tradizionali. HBDH: α-butinato-deidrogenasi →esprime l’attività di LD1 e LD2 in quanto aggiungendo α-butinato come substrato LD1 lo ossida. - LDH si normalizza in 7-15gg 41 www.slidetube - HBDH si normalizza dopo 10-20gg - CPK e GOT dopo 3-6gg L’utilizzo dei marcatori di danno miocardico può portare a 2 strategie diagnostiche alternative: -1-dosaggio di 2 marcatori diversi, uno molto precoce e aspecifico e uno tardivo e altamente specifico: - enzima precoce: MIOGLOBINA (aumenta entro 2-3h) alto valore predittivo negativo - enzima tardivo: TROPONINE -2-dosaggio di un solo enzima specifico (TROPONINA). Si usa se la prevalenza di malattia coronarica è significativamente più elevata. - MIOGLOBINA <110μg/l normale Presente nel muscolo cardiaco e scheletrico. E’ un marker precoce in quanto ↑entro 1-2h dall’esordio del dolore. E’ importante per: - escludere dalle prime ore la presenza di infarto (valore predittivo negativo>95%) - valutare se la riperfusione del tessuto è stata ottenuta - valutare la comparsa di reinfarti (la troponina rimane elevata a lungo mascherando i reinfarti). E’ un marcatore aspecifico perché aumenta anche in caso di danno muscolare, traumi e insufficienza renale. -CK-MB massa <5μg/l normale Misura la concentrazione della proteina CK-MB con Ab monoclonali specifici. GRAFICO CK-MM + Ab monoclonale → si blocca CK-MB + Ab monoclonale → non si blocca Entro 3 h dall’infarto la CK-MB massa aumenta significativamente nel 50% dei pazienti. Entro 6h nell’80-100% dei pazienti. Ritorna normale in 48-72 h. Un valore normale di CK-MB dopo 6-8 h dai sintomi ha un valore predittivo negativo del 95%. In più la CK-MB è utile per: - valutare la riperfusione - valutare eventuali reinfarti E’ un marcatore non completamente specifico perché aumenta nelle malattie e nei traumi muscolari e ha scarsa sensibilità per danni miocardici minimi ( rilevati dalle troponine) La CK-MB ha 2 isoforme: - MB1: forma sierica si ottengono a partire da un precursore proteico comune che - MB2: forma tissutale subisce diverse modificazioni post-traduzionali. MB2 ha una Lys in più di MB1. MB2/MB1 = 1 MB2/MB1 ≥ 1,5 situazione normale probabile infarto in atto oggi non più molto utilizzato - TROPONINE 42 www.slidetube Il complesso delle troponine ha 3 componenti: - troponina C → lega Ca2+ non specifica - troponina I → inibisce l’attività ATPasica dell’acto-miosina - troponina T → lega la tropo miosina - TROPONINA I E’ cardiospecifica: localizzata per il 95% nelle miofibrille e la restante parte nel citosol. E’ molto importante per la diagnosi di IMA senza modificazione dell’ECG: in questi pazienti c’è una fluttuazione della [TnI] che, dosata, permette di stabilire quali pazienti sono più a rischio per eventi cardivascolari a breve termine. La TnI aumenta dopo 6 h dall’infarto (valore predittivo positivo del 95%) e resta alta per 7-10 gg. Possono quindi sfuggire eventuali reinfarti. Valori: - <0,1 μg/l soggetto normale - >0,1 e <1,5 μg/l danno cardiaco minimo (non aumenta CK-MB) 1°livello decisionale - >1,5 μg/l INFARTO 2°livello decisionale - TROPONINA T <0,01 μg/l Presente nel cuore e nel muscolo scheletrico cronicamente stressato che riesprime proteine fetali. Oggi si usano Ab specifici con selettività per la sequenza aminoacidica del TnT cardiaco, che ne fanno quindi un ottimo marcatore di infarto. Il rilascio di TnT mostra un quadro bifasico con un picco maggiore a 14 h e uno minore a 3-5 gg dall’infarto. Ciò è probabilmente dovuto alla compartimentalizzazione intracellulare della proteina. TnT resta elevato fino a 2 settimane dopo l’esordio. Aumenta fino a 100 volte rispetto al valore normale e mostra una sensibilità del 100% da 10 h a 5 gg dall’infarto. Come la TnI è importante per valutare il rischio di infarto nei pazienti con angina instabile e infarto senza sottoslivellamento del tratto ST. Esistono oggi 3 livelli decisionali a cui corrispondono considerazioni diagnostiche e terapeutiche differenti: I° livello: serve a escludere la presenza di sofferenza o danno miocardico di qualsiasi natura. E’ il valore inferiore dell’intervallo di riferimento che si ottiene integrando la [Tn] corrispondente al 99° percentile del sistema di analisi e il livello di imprecisione analitica corrispondente. Valore prossimo allo zero ↓ si esprime come coefficiente di variazione ed <10% II° livello: consente di separare il danno miocardico minimo dall’IMA vero e proprio. III° livello: consente di distinguere i danni miocardici di minore entità dai più severi 43 www.slidetube 4 3,5 3 2,5 TnI 2 TnT CK-MB massa 1,5 mioglobina 1 0,5 0 0 1 2 3 4 5 6 7 8 Figura 6: andamento markers IMA PRINCIPALI MARCATORI mioglobina CK-MB massa TnI TnT AUMENTO INIZIALE 1-4 h 2-6 h 3-12 h 3-12 h PICCO 4-12 h 12-24 h 12-24 h 12-48 h I°picco 3-5 gg II°picco RITORNO ALLA NORMA 12-24 h 72 h 7-10 gg 10-14 gg Il quadro clinico dello shock simula quello dell’infarto. ↑CPK totale, ↑GOT, ↑GPT, ↑HBDH, ↑LDH Tuttavia CK-MB non si alza e poi livelli di CPK pari a 5000 U/l sono troppo elevati per pensare a un infarto 44 www.slidetube RIASSUNTO Dolore al petto da<6h ECG positivo IMA ricovero negativo fare isoforme CK-MB positivo negativo ripetere dopo 6h positivo storia di precedenti terapia negativo nessun precedente medicina generale VALUTAZIONE DELLA FUNZIONALITA’ CARDIACA Si dosano gli ormoni natriuretici (ANP, BNP, CNP) I più noti sono: -ANP: di origine atriale → è prodotto dalle fibrocellule cardiache e immesso in circolo - BNP: di origine ventricolare Stiramento dell’atrio: ANP (tanto) e BNP (poco) Stiramento del ventricolo: ANP(poco) e BNP (tanto) - ANP: ↓P arteriosa, ↑diuresi e natriuresi; azione anti ipertrofica - BNP: ↑BNP nello scompenso cardiaco: risposta adattativa per mantenere gettata cardiaca e P venosa danno cardiaco disfunzionale→attivazione del sistema neuroendocrino→ipertrofia→↑BNP-↑ANP contrasto Anche lo stress distende il cuore. Utile il dosaggio di BNP e NT-proBNP per seguire i pazienti con scompenso cardiaco nel followup, per classificarli e screenarli e sono considerabili come indicatori prognostici (anche nei test da pronto soccorso) NT-proBNP: t1/2>60min [C] = 41 μg/ml 45 www.slidetube ENZIMI MUSCOLO SCHELETRICO Degenerazione neuromuscolare → Aldolasi più bassa che nella distrofia Test elettromiografici e aldolasi utile solo nella fase iniziale perché poi le fibre si distruggono e sono sostituite da connettivo Sarcoidosi ACE (↑P art.) Quadro radiologico come TBC o neoplasia. Se progredisce c’è dispnea gravissima ENZIMI PANCREAS -1- Pancreatite acuta Patologia che esordisce con addome acuto, ulcera perforante. La diagnosi di pancreatite acuta si fa dosando alcuni enzimi: - amilasi totale (non specifico) → sangue+amido+I →reazione - se sangue normale la soluzione è blu - se c’è amilasi il blu scompare - isoamilasi pancreatica - lipasi → lipasi+colipasi specifica - tripsina - elastasi - fosfolipasi A2 Nelle urine si trovano: -amilasi - elastasi - chimotripsina -2- Pancreatite cronica E’ di difficile diagnosi mentre è in corso di formazione. Talvolta è definita dispepsia, cattiva digestione, … Inutile il dosaggio di amilasi e lipasi per diagnosi a meno che non ci sia una fase di attivazione. Si dosa la CHIMOTRIPSINA nelle feci perché non è demolita dalle proteasi e dalla flora batterica intestinale. Le feci sono trattate con spettrofotometro a infrarossi (ne valuta la composizione). L’aspetto è untuoso con gocce di grasso e fibrocellule muscolari. L’esame è un po’ in disuso. 46 www.slidetube Capitolo 4 FUNZIONALITA’ EPATICA 47 www.slidetube FEGATO Il fegato è il principale organo metabolico del nostro organismo. Interviene in numerosi processi per cui per valutarne la perfetta funzionalità bisogna eseguire test diversi: - test di escrezione - test di sintesi e capacità metabolica - test di danno cellulare - TEST di ESCREZIONE Bilirubina → più noto test di escrezione E’ un prodotto catabolico del metabolismo dell’EME. Proviene per la maggior parte dai globuli rossi invecchiati, ma anche da mioglobina, emeproteine (citocromi) e eritropoiesi inefficace. Ogni giorno sono gestiti circa 7 g di bilirubina. globina Emoglobina apertura della proto porfirina IX EME ferrobiliverdina → biliverdina → bilirubina La bilirubina viene trasportata nel sangue legata all’albumina. E’ captata dal fegato dove subisce una reazione di glucoronazione ad opera dell’enzima UDP-glucoronil-transferasi e secreta con la bile nell’intestino. Qui è modificata dalla flora batterica (STERCOBILINA) e in parte riassorbita attraverso il circolo entero-epatico (UROBILINOGENO). La bilirubina si trova in 2 forme principali: - indiretta: non coniugata con acido glucuronico - diretta: coniugata con acido glucuronico BILIRUBINA TOTALE: 1 mg/dl è il valor normale di cui - 75% indiretta 0,1-1 mg/dl - 25% diretta <0,3 mg/dl Le due forme si mettono in evidenza con la REAZIONE PRONTA: SIERO+REATTIVO → COLORAZIONE VIOLA (diretta) SIERO+REATTIVO+SOLVENTE → COLRAZIONE VIOLA (indiretta) Se bilirubina >2 mg/dl si parla di ITTERO L’aumento di bilirubina non deve comunque superare ilo valore di 18-20 mg/dl, altrimenti passa la barriera emato-encefalica e possono svilupparsi lesioni ai nuclei della base. Importante in caso di ittero valutare sia la BILIRUBINA TOTALE che la BILIRUBINA INDIRETTA. 48 www.slidetube Esistono diverse cause di ittero: 1) ITTERO PREEPATICO→↑bilirubina indiretta da iperproduzione di bilirubina (emolisi, eritropoiesi inefficace, incompatibilità Rh) 2) ITTERO EPATICO→↑bilirubina diretta e indiretta (rigurgito di bile) - da difetto di captazione (sindrome di Gilbert = valori di bilirubina totale da 1,5 a 6 mg/dl con bilirubina indiretta fino a 5 mg/dl senza altri segni di alterazione epatica) - da difetto di coniugazione (sindrome di Crigger-Najal, ittero fisiologico del neonato, ittero da epatite o congenito grave) ↑bilirubina indiretta - da difetto di eliminazione (sindrome di Dubin-Johnson) ↑bilirubina diretta - da deficit della pompa della via biliare (ittero farmacologico dovuto al fatto che un farmaco compete con la bilirubina per un recettore; nei neonati si può verificare durante l’allattamento) 3) ITTERO POSTEPATICO→↑bilirubina diretta - da stasi biliare ( epatiti, colangiti, colangioliti, cirrosi biliare, calcolosi della via biliare, neoplasie di fegato e pancreas) INDICATORI di COLESTASI → ALP, γGT - ALP: aumenta sensibilmente in caso di ostruzione intraepatica ed extraepatica delle vie biliari ALP<455 U/l femmine da 0 a 12 anni; maschi da 0 a 15 anni ALP 34-104 U/l femmine valori normali ALP 45-130 U/l maschi valori normali - γGT: aumenta a seguito di varie patologie epatiche (microstasi, epatite, neoplasie, induzione alcolica e altre patologie non epatiche). Si considera poco specifica per malattia epatica γGT 11-97 U/l neonati γGT <33 U/l femmine γGT <50 U/l maschi -ACIDI BILIARI: prodotti dal fegato, demolendo il colesterolo, sono l’acido colico e l’acido chenodesossicolico poi coniugati con taurina e glicina. Sono secreti con la bile e riassorbiti per il 90% nel circolo entero-portale. ↑nel sangue in seguito a colestasi (valutano quindi la funzionalità escretrice del fegato). 49 www.slidetube - TEST di SINTESI a) INDICI FUNZIONALI di SINTESI PROTEICA La maggior parte delle proteine del sangue, ad eccezione delle Ig, alcuni fattori del C’ e alcune lipoproteine, è sintetizzata dal fegato. Il loro dosaggio è informativo dell’attività sintetica del fegato. Le principali proteine studiate sono: - ALB: un suo livello ridotto, escludendo altre cause come malnutrizione, malassorbimento, perdita urinaria, è indice di insufficienza epatica. Se ↓ALB il fegato è compromesso per 2/3. - pre-ALB, TRF: proteine preferite all’albumina perché hanno emivita minore, a volte si usa anche AAG (↓in caso di epatopatie, avvelenamenti, ↑se flogosi o dialisi) - fattori della coagulazione: hanno t1/2 <ALB e sono quindi un indice più fedele di insufficienza epatica. Importante il dosaggio del FATTORE VII in caso di epatite fulminante (t1/2 =2h). - tempo di protrombina (TP): misura il tempo di coagulazione del plasma. Valuta la funzione della via estrinseca (fattori X, VII, II prodotti dal fegato). Si ha un TP allungato nelle epatiti tossiche, nelle cirrosi e se il fegato produce una molecola di fibrinogeno abnorme che non polimerizza (sifibrinogenemia). - CHE: è un indice di protidi poiesi ormai no molto usato per la scarsa sensibilità di risposta b) INDICI FUNZIONALI di SINTESI LIPIDICA Colesterolo: valutazione della sua formazione ed esterificazione. Prima si calcolava il rapporto tra colesterolo libero e quello esterificato. c) INDICI FUNZIONALI di SINTESI GLUCIDICA Galattosio: nel fegato c’è l’enzima Gal1 fosfo-uridil-transferasi che converte il Gal in Glc. Se manca si sviluppa una malattia grave nel neonato → GALATTOSEMIA. per valutare la funzione di Gal-1-PUT si fa una prova da carico, si somministrano 40g di galattosio e lo si dosa nelle urine emesse 5 h dopo. Quantità di Gal >3g sono patologiche. - TEST di DANNO CELLULARE Valutano l’incremento di alcuni enzimi, specialmente GPT e GOT a seguito di un danno cellulare epatico GPT >GOT epatopatie di tipo citolitico GOT>GPT epatopatie alcoliche 50 www.slidetube EPATOPATIE ACUTE Le epatopatie acute sono caratterizzate da un aumento delle transaminasi con: SENSIBILITA’ SPECIFICITA’ ALT (GPT) 96 94 >300 U/l AST (GOT) 91 95 >200 U/l ↑di bilirubina, γGT, GLDH, LDH, ALP EPATITE A: si avvia verso la guarigione quando ALP e γGT si abbassano. Scompare CHE. Ittero nel 70% dei casi. 1400 1200 1000 GPT 800 GOT 600 ALP γ GT 400 200 0 0 sett 01-set 02-set 03-set 04-set 05-set 06-set 300 250 200 GPT 150 EPATITE B: complicata= con componente occlusiva. La forma più grave ha ALP e γGT alte. Scompare CHE. Ittero nel 30-50% dei casi. GOT ALP 100 γGT 50 0 0 sett 01-set 02-set 03-set 04-set 05-set 06-set 51 www.slidetube 3000 2500 2000 GPT EPATITE ALCOLICA γGT e ALP alte. GPT e GOT nono superano mai più di 10 volte i valori normali. Ittero nel 60-70% dei casi. GOT 1500 ALP γGT 1000 CHE 500 0 0sett 01-set 02-set 03-set 04-set 05-set 06-set 07-set AVVELENAMENTO da FUNGO Scompare CHE 600 500 400 GPT 300 GOT LDH 200 GLDH 100 0 0sett 01-set 02-set 03-set 04-set 05-set 06-set 52 www.slidetube AVVELENAMENTO da SOLVENTI Caratterizzato da LDH altissima e GOT > GPT 12000 10000 8000 GPT 6000 GOT LDH 4000 GLDH 2000 0 0set 01-set 02-set 03-set 04-set 05-set 06-set 500 450 400 350 300 GPT 250 GOT 200 MORBO di WEIL: leptospirosi emorragica GPT > GOT Caratterizzata da ittero e ematuria. Diversa dalle epatiti perché prima si alza la bilirubina e poi le transaminasi (nelle epatiti è il contrario). bilirubina 150 100 50 0 0set 01-set02-set03-set04-set05-set06-set07-set08-set GOT/GPT = quoziente di De Ritis In condizioni normali il quoziente di De Ritis > 1. Se > 1 (conversione del quoziente) ci può essere: epatite acuta, epatite tossica, mononucleosi, steatosi. 53 www.slidetube EPATOPATIE CRONICHE Alcune forme di epatite acuta come l’epatite B e la C tendono a cronicizzare. Ciò avviene quando: - γGT dopo un mese dall’esordio acuto > 150 U/l - transaminasi sono alte con GOT > GPT di circa 100 U/l - ↑bilirubina - ↓ALB Ci son altre 2 forme principali di epatite cronica: - PERSISTENTE: in cui GPT > GOT. E’ abbastanza benigna - FORMA CRONICA: con GPT ≈ GOT, ↑γGT e CHE↓ Per la diagnosi di epatite cronica bisogna verificare una delle seguenti condizioni: -1-↑ALT per più di 6 mesi dopo un’epatite acuta -2-↑ALT in più di un’occasione su un periodo di 6 mesi senza un’altra spiegazione ATTENZIONE! Non in tutti i pazienti con epatite cronica c’è ↑ALT perché le cellule del fegato distrutte non lo producono più. Ricercare i marker di infettività. Screening di epatopatia cronica va sempre condotto dosando ALT perché ilo test è più sensibile di AST. Monitoraggio nelle infezioni da HBV e HCV bisogna valutare la scomparsa dei marker virali per monitorare l’evoluzione della malattia. L’epatite cronica può evolvere verso la cirrosi o il carcinoma epatocellulare (HCV). CIRROSI Consiste nella sostituzione del tessuto epatico con fasci fibrosi e perdita della funzionalità epatica, ipertensione portale, ascite. Può essere causata da HBV, HCV, alcol - Da HBV, HCV: CHE bassa, modesto movimento delle transaminasi con GOT>GPT (non più di 23 volte la norma) e γGT lievemente mossa. - Da alcol: stesso quadro con γGT molto alta. Per valutare l’entità della fibrosi si possono eseguire alcuni test: -1-quoziente di De Rizis CHE diventa >1 (era <1 nell’epatite cronica non alcolica) -2-conta piastrinica -3-tempo di protrombina -4-ALB →marker di gravità della compromissione protidosintetica -5-AFP (alfafetoproteina): ↑col progredire della fibrosi (ha un buon valore predittivo positivo). 54 www.slidetube EPATOMA Complicanza dell’epatopatia cronica da HBV e HCV. Si raccomanda lo screening dei portatori cronici di HbsAg ogni 6 mesi con AFP (ma valore predittivo basso → 10-30%) e con ecografia e AFP nei pazienti con cirrosi e storia familiare di carcinoma. Oltre alle prove funzionali ci sono dei test che riguardano alcuni aspetti immunologici delle malattie epatiche. Si tratta della valutazione della [Ig] nel sangue. 90 80 70 60 50 IgG 40 IgA 30 IgM 20 10 0 epatite acuta epatite cronica aggressiva cirrosi biliare primaria cirrosi alcolica IgG: aumentano nell’epatite cronica aggressiva e nella cirrosi. Nella prima sono oligoclonali, nella seconda policlonali con ponte β-γ. IgA: aumentano nelle patologie da alcool perché sono associate a una patologia del primo tratto digerente dove sono prodotte. IgM: aumentano precocemente nell’epatite acuta e sono elevate nella cirrosi biliare primitiva. METASTASI EPATICHE Quadro con ↑ transaminasi con GOT>GPT, ↑γGT, ↑ALP, ↑LDH isoenzimi 1 e 2. Diagnosi eseguita con TAC ed ecografia EMIVITA dei PRINCIPALI ENZIMI di FEGATO E PANCREAS GOT GPT GLDH LDH1 LDH5 CPK 17±5h 47±10h 18±1h 113±60h 10±2h 15h ALP γGT CHE AMY lipasi 3-7gg 3-4gg 10gg 3-6h 3-6h 55 www.slidetube Capitolo 5 FUNZIONALITA’ RENALE 56 www.slidetube RENE I test di laboratorio che possono essere fatti sul rene si suddividono in varie categorie: - test che valutano la funzionalità glomerulare - test che valutano la funzionalità tubulare - test che valutano l’insufficienza renale acuta e cronica - test che valutano le glomerulo nefriti - TEST per la FUNZIONALITA’ GLOMERULARE -1-CLEARANCE della CREATININA Per valutare l’efficienza della funzionalità renale si misura VFG=velocità di filtrazione glomerulare che dipende: - dalla P di filtrazione Pf=P ‒ (π+Pc) - dalla superficie filtrante (numero glomeruli funzionanti) - dalla struttura della membrana filtrante VFG = volume di plasma (ml) filtrato in 1 minuto dai 2 reni VFG = 125ml/min normale riferita a una superficie corporea di 1,73m2. La VFG è pari alla CLEARANCE se la sostanza impiegata è completamente filtrata e non viene ne riassorbita, ne secreta dal tubulo, ma è completamente escreta con le urine. CLEARANCE = volume di plasma depurato da una sostanza X nell’unità di tempo. Ux = [x] nelle urine Px = [x] nel sangue V = flusso urinario Nell’uso clinico la sostanza che viene utilizzata è la CREATININA, un catabolita endogeno di origine muscolare derivante dalla creatina. - Cl creat. = 95-140 ml/min nei maschi - Cl creat. = 75-110 ml/min nelle femmine Diminuisce con l’età perché ↓ il numero dei glomeruli funzionanti. Fare attenzione al fatto che la quantità di creatinina dipende dalla massa muscolare. La misura della Cl è però spesso inaccurata e soggetta a errore (tra 10-30%) per imprecisioni compiute nella raccolta delle urine e nelle misure analitiche. più spesso l’efficienza della funzionalità glomerulare è valutata con la sola CREATININEMIA, più affidabile. - 1 mg/dl creatinine mia normale - 0,8-1,3 mg/dl nei maschi - 0,6-1,1 mg/dl nelle femmine - 0,3-0,7 mg/dl bambini<12 anni 57 www.slidetube La concentrazione plasmatica della creatinina è in rapporto inversamente proporzionale con la clearance (curva iperbolica). La VFG può diminuire del 50% prima che si verifichi un ↑della creatinina. Quando però questo si verifica è sicuro un difetto della funzionalità renale. creatininemia creatinina mg/dl 9 8 7 6 5 4 creatinina mg/dl 3 2 1 0 0 20 40 60 80 100 120 Cl% Una nefropatia è curabile e reversibile fino a che non supera il 50% di compromissione della funzionalità renale E’ una formula che permette un calcolo rapido della VFG (e volume di plasma filtrato) ↑creat.: riduzione della filtrazione glomerulare, malattie del parenchima renale, nefropatie vascolari, aumento della massa muscolare, acromegalia, traumi. -2-CISTATINA C E’ una proteina inibitrice di alcune cisteine-proteasi prodotte da tutte le cellule. Essa è totalmente filtrata dal glomerulo e catabolizzata dalle cellule del tubulo prossimale (non viene perciò riassorbita) 0,4-1,2 mg/l intervallo di riferimento normale (maschio, femmina, bambino) E’ un marker ideale della VFG (non risente di riassorbimento, secrezione e imprecisione nella raccolta delle urine) e inoltre è un indice più precoce dei compromissione renale rispetto alla creatinina in quanti rileva anche LIEVI ALTERAZIONI della FUNZIONALITA’ GLOMERULARE. Purtroppo ha una grande variabilità interindividuale e ↑in alcune neoplasie e nell’infezione da HIV. 58 www.slidetube -3-UREA Urea plasmatica = azotemia in italia L’urea è una molecola prodotta dal fegato, contenente NH3 derivante dal catabolismo degli aminoacidi. Ogni giorno sono eliminati con le urine fino a 30 g di urea. In media 15-20 g. L’urea viene utilizzata come indice di funzionalità glomerulare, ma la sua utilità clinica è inferiore a quella della creatinina perché viene riassorbita per il 40% nel TP ed è influenzata dall’apporto alimentare azotato, dallo stato di idratazione del paziente, dalla diuresi e dal catabolismo proteico. - adulti 20-50 mg/dl - bambini 10-50 mg/dl valori normali AZOTEMIA Iperazotemia è comunque da considerare perché può avere molteplici cause (anche non renali): - pre-renale: dieta ricca di proteine, emorragia gastrointestinale, febbre, compromissione del FER (shock, ipotensione). Di solito la creatinina è normale. Rapporto azotemia/creatininemia>20:1 - renale: malattie glomerulari, nefropatie croniche e acute. Rapporto azotemia/creatininemia<20:1 - post-renale: legata a impedito flusso urinario per calcoli, neoplasie, stenosi ureterali, ipertrofia prostatica. IPERAZOTEMIA ↓ accumulo di urea nel sangue ≠ IPERAMMONIEMIA ↓ accumulo di NH3 nel sangue NH3 Deriva dalla deaminazione degli aminoacidi ed è convertita in urea nel fegato ↑NH3: insufficienza epatica grave, encefalopatie di origine epatica, leucemia acuta, trasfusioni, trapianti, esercizio fisico intenso, emorragie intestinali, alcuni farmaci. - NH3 50-80 20-60 μg/dl valori normali - NH3 >150μg/dl coma epatico C’è una correlazione positiva tra NH3 e gravità del coma epatico. Ilo coma da insufficienza renale, invece è iperuremico, ma non iperammoniemico. NH3 è tossico per il cervello perché sottrae ac. chetoglutarico dal ciclo di Krebs per convertirlo in glutammato e glutammina. AMMINOACIDI Oltre all’urea e all’ NH3 nel sangue sono presenti altri composti azotati: gli aa (si valuta la presenza di una quantità di –NH2 pari a 5 g). Essi sono di origine alimentare o derivanti dalla demolizione 59 www.slidetube delle proteine. L’AMINOACIDEMIA non ha grandi implicazioni diagnostiche. Si calcola quando si sospettano difetti nella sintesi proteica. ↑aa insufficienza epatica quasi terminale. DIFETTI nella SINTESI PROTEICA: - Metabolismo della Phe: difetto più comune è la mancanza dell’enzima Phe idrossilasi che catalizza la conversione della Phe in Tyr. L’accumulo di Phe è tossico per il cervello e nel bambino non ne permette un completo sviluppo con conseguente ritardo mentale. E’ una condizione nota come OLIGOFRENIA FENILPIRUVICA o FENILCHOTONURIA (malattia a carattere autosomico recessivo). Fondamentale è una diagnosi precoce affinché al bambino sia prescritta una dieta priva di Phe. Si fa tramite il TEST di GUTHRIE (screening su tutti i neonati) che consiste nell’aggiungere il siero del neonato in una coltura di B.Subtilis. La crescita dei bacilli è inibita dalla presenza di β-2-tienilalanina (antagonista della Phe). Se nel siero aggiunto è presente un accumulo di Phe i batteri crescono; valutando la crescita batterica si può porre diagnosi di fenilchetonuria. Più importante è la valutazione della AMINOACIDURIA. Associata al dosaggio dei fosfati e del glucosio può essere utile per valutare la SINDROME di FANCONI. E’ una malattia autosomica recessiva caratterizzata da una progressiva pan citopenia associata a aumentata predisposizione per le malattie neoplastiche e a diverse anomalie congenite tra cui alcune a carico dello scheletro (RACHITISMO resistente a Vit.D) e altre del rene (disfunzione del TP con conseguente glicosuria, iperfosfaturia e ipofosfatemia, aminoaciduria, iperururicuria e ipouricemia, ipocaliemia). ACIDO URICO E’ il prodotto terminale del catabolismo delle purine. in condizioni normali ha questi valori nel sangue: 5-6 mg% nelle femmine 5-7 mg% nei maschi L’aumento dell’uricemia porta a una patologia che prende il nome di GOTTA. Le cause sono principalmente 3: -1- DIFETTO di ESCREZIONE RENALE In condizioni normali l’acido urico è parzialmente filtrato dal glomerulo con una clearance molto bassa ( 9 ml/min). E’ secreto nella porzione finale del TP e riassorbito per il 98-100% nel TD. Viene escreta circa il 6-12% della quota filtrata. ↑uricemia: insufficienza renale, acido lattico e chetoacidi bloccano la secrezione tubulare di acido urico (in caso di diabete, dislipidemia, intossicazione alcolica, digiuno, esercizio faticoso). anche alcuni farmaci possono inibire la secrezione di acido urico. -2- SOVRAPPRODUZIONE di ACIDO URICO - ↑produzione dell’enzima PRPP (fosfo-ribosil-pirofosfato-sintetasi) enzima essenziale per la sintesi dei nucleotidi. - Difetto enzimatico di HGPRT (ipoxantina-guanina-fosfo-ribosil-transferasi) Sindrome di Lesch-Nyhan: è una malattia ereditaria X-linked che ha come difetto più evidente l’iperuricemia associata a ritardo mentale, atassia, automutilazioni, aggressività, iperuricuria e [HGPRT] diminuita in eritrociti e fibroblasti. 60 www.slidetube -3- AUMENTATO TURNOVER degli ACIDI NUCLEICI L’aumento della sintesi e della demolizione di purine è tipica di alcune malattie, come le leucemie, caratterizzate da disordini mieloproliferativi. Durante la chemioterapia vengono distrutte molte cellule con liberazione di DNA (e quindi purine). Capitolo 5.1 ESAME URINE 61 www.slidetube ESAME DELLE URINE L’analisi delle urine comprende: -esame chimico-fisico -esame microscopico -1- ESAME CHIMICO-FISICO Si esegue con lo stick urinario che mette in evidenza la presenza di varie molecole grazie a reazioni enzimatiche. L’esame considera diversi parametri: - COLORE: può variare entro questa gamma: - giallo paglierino: urine normali - giallo arancio intenso: urina concentrata con pigmenti biliari e uroblilinogeno in eccesso - marsala: presenza di bilirubina - rosso limpido: emoglobina e mioglobina - rosso torbido: presenza di sangue - verde: presenza di biliverdina o infezioni batteriche - nerastra: alcaptonuria, melanina, derivati della Tyr - trasparente: urina molto diluita (es. nel diabete insipido) - ASPETTO: - limpido: urina normale - torbido: presenza di fosfati, carbonati, acido urico, proteine (leucociti, batteri, spermatozoi) - lattescente: piuria, lipuria (nefrosi o traumi), cellule vaginali molto abbondanti - presenza di precipitati: nelle urine mentre il principale precipitato è il fosfato amorfo, nelle acide l’urato di ammonio, nelle alcaline il fosfato triplo o, se flocculento, muco, leucociti, batteri e lieviti - PESO SPECIFICO: rapporto tra il peso in grammi di 1 ml di urina e 1 ml di acqua. E’ sempre > 1. Misura la capacità del rene di diluire o concentrare le urine - ps = 1003-1028 adulto normale - ps > 10023 = normale capacità di concentrazione Il peso specifico è calcolato con lo stick urinario (ma metodo poco accurato e poco specifico) o con un REFRATTOMETRO. Il campione viene attraversato da un raggio di luce che viene rifratto. L’angolo di rifrazione è α alla [soluti] nell’urina. 62 www.slidetube - ↑ps: disidratazione, glicosuria, proteinuria, ipersecrezione di ADH - ↓ps: diabete insipido, glomerulonefrite, pielonefrite, insufficienza renale - OSMOLALITA’ URINARIA: numero di particelle osmoticamente attive/Kg di solvente 100-1200 mOsm/Kg soggetto normale Non sempre coincide con il ps. Il valore dell’osmolalità dipende dallo stato di idratazione del paziente; non c’è quindi un “valore normale”. il principale determinante dell’osmolalità urinaria è la quantità di H2O riassorbita dal TD e DC in risposta all’ADH. E’ la misura più esatta della capacità del rene di concentrare le urine. Si misura con l’OSMOMETRO, strumento che valuta le variazioni del punto di congelamento dell’urina rispetto al solvente puro. ↑osmolarità: morbo di Addison, scompenso cardiaco, shock ↓osmolarità: iperaldosteronismo, diabete insipido, eccessivo itroito di acqua, necrosi tubulare - pH: il rene elimina giornalmente 80-100 mmol/l di ioni H+ tramite l’acidità titolabile (tamponi urinari) e l’ammoniogenesi (NH4+ eliminato: 30 mmol/24h) pH 5,5-6,5 normale pH 4,5-8 massima variabilità ↑pH: infezioni batteriche, dieta vegetariana ↓pH: durante la notte per acidosi respiratoria, dieta a base di amido, proteine, alcol e zucchero. pH importante nelle calcolosi (urine alcaline → calcoli di fosfato di Ca, CaCO3, NH4PO4; urine acide → calcoli di acido urico); nelle infezioni urinarie, nelle alterazioni dell’eq. acido-base, nell’insufficienza renale - GLUCOSIO: glc < 150 mg/l normale ↑glicosuria: iperglicemia o con glicemia normale in gravidanza, tubulopatie ereditarie (es. sindrome di Fanconi), tubulopatie da sostanze tossiche (Pb, Hg, tetracicline), da ipossia, malattie infiammatorie. La glicosuria si misura con le strisce reattive che sfruttano una reazione basata sulla GLUCOSIO-OSSIDASI Glu + O2 →glucosio ossidasi→ H2O2 + cromogeno →perossidasi→ cromogeno ossidato + H2O - CORPI CHETONICI: non sono normalmente presenti nelle urine; la loro presenza indica un aumento del metabolismo degli acidi grassi. ↑corpi chetonici: digiuno, diabete (associati a glicosuria), gravidanza, febbre, ipoglicemia, stress, esercizio fisico intenso 63 www.slidetube - PROTEINE: < 150 mg/24h proteinuria normale La proteinuria normale è costituita dal 30% di ALB, 30% mucoproteina di Tamm-Horsfall, RBP (retinol binding protein), β2microglobulina, catene L, IgA di secrezione. La proteinuria patologica riflette un danno renale. Può essere di 3 tipi: 1)PROTEINURIA GLOMERULARE Causata da un aumento di permeabilità del glomerulo (varia l’eziologia: glomerulonefriti, alterazione dell’emodinamica glomerulare, stenosi dell’arteria renale). Compaiono nelle urine: ALB, TNF, le α1 e le α2, poche IgG. La proteinuria può essere: - REVERSIBILE - IRREVERSIBILE - SELETTIVIA con l’aggravarsi dell’alterazione della barriera - NON SELETTIVA (sempre irreversibile) di filtrazione si perdono proteine con pH sempre maggiore fino alla perdita completa della selettività Per distinguere una proteinuria glomerulare selettiva da una non selettiva è utile calcolare l’INDICE di CAMERON: < 0,2 → proteinuria selettiva 2)PROTEINURIA TUBULARE Causata da un inadeguato riassorbimento tubulare di proteine a basso pH (normalmente filtrate e riassorbite). Si ritrovano nelle urine: α1 microglobulina, β2 microglobulina, RBP, α1 glicoproteinaacida, catene L. Le principali cause del danno tubulare sono: necrosi tubulare acuta, tossicità da farmaci e metalli pesanti, trapianti, sindrome di Fanconi. 3)PROTEINURIA DA IPERAFFLUSSO Causata dall’eccessiva produzione di proteine a basso pH che filtrano nel glomerulo e non sono totalmente riassorbite per l’elevata quantità. Provocano gravi danni renali. Le principali sono: BJ (MM), lisozima (leucemia monocitica), amilasi (pancreatite), mioglobulina, Hb (emolisi intravascolare). Nelle urine si ritrovano anche numerosi enzimi il cui dosaggio non può però essere standardizzato dal momento che alcuni si bloccano nell’ambiente urinario. Ciò non vale ad esempio per: - NAG→N-acetilβ D-glucosamminidasi - α1 microglobulina 64 www.slidetube Esistono anche proteinurie benigne intermittenti come: - ALBUMINURIA ORTOSTATICA: persistente, ma presente solo nella posizione eretta. - ALBUMINURIA da SFORZO - PROTEINURIA da FEBBRE Per evidenziare la proteinuria si usano le strisce reattive. Reazione semiquantitativa specifica, ma poco sensibile. Si basa sul seguente principio: ALB a pH fisiologico sono dissociate come anioni. Sulla striscia c’è un tampone che contiene un indicatore specifico per l’ALB, giallo a pH = 3 e blu se pH > 4,2 (per farlo virare bastano 15mg/dl di ALB). La [ALB] è valutata sulla base dell’intensità di colore. - EMOGLOBINA: La rilevazione di Hb si basa sulla seguente reazione eme ↓ H2O2 + cromogeno → cromogeno ossidato + H2O Hb ha una attività perossidasica che porta al viraggio del colore del cromogeno. - UROBILINOGENO: Presente normalmente in tracce fino a 0,5-4 mg/24h. L’urobilinogeno è prodotto dalla flora intestinale a partire dalla bilirubina ed è in parte escreto con le feci, in parte riassorbito dal fegato e in parte passa nel circolo sistemico ed è escreto per via renale. La quota di urobilinogeno urinario dipende dalla quantità di bilirubina immessa nell’intestino e dalla quota riassorbita dal fegato. ↑urobilinogeno: emolisi, stipsi (↑tempo di transito nell’intestino), ↑crescita della flora batterica, pH urinario alcalino (si trova in forma ionizzata a cui l’epitelio è impermeabile). ↓urobilinogeno: ostruzione biliare, antibiotici, diminuito transito intestinale, pH urinario molto acido, ridotta funzionalità renale. - BILIRUBINA: è normalmente assente nelle urine; se presente è sempre in forma coniugata. La sua presenza indica un ostacolo intra o extraepatico al passaggio della bile. Per misurarla si dosa la AZOBILIRUBINA composto che si forma per la reazione della bilirubina con l’ac. sulfanilico-diazotato. - NITRITI: si formano dai nitrati nel metabolismo batterico. - LEUCOCITI: è possibile rilevarne la presenza con lo stick. 65 www.slidetube -2- ESAME MICROSCOPICO E’ possibile rilevare la presenza di globuli rossi, leucociti, cilindri, cristalli, occasionalmente batteri, cellule epiteliali di sfaldamento, calcoli. - EMATURIA: può essere: - macroscopica: visibile a occhio nudo - microscopica: presenza di più di 2 emazie per campo (400x). E’ possibile distinguere al sedimento la microematuria di origine glomerulare da quella avente altre cause. Infatti nel primo caso si osservano emazie piccole, dismorfiche, o tipicamente deformate con attaccate piccole spicole (ACANTOCITI). Nel secondo caso le emazie sono uniformi a disco biconcavo. - Cause ematuria: - microematuria glomerulare: nefropatia da IgA, ispessimento della membrana basale, glomerulonefriti - microematuria non glomerulare: pielonefrite, nefrolitiasi, infezioni, neoplasie vescicali o della prostata. - LEUCOCITURIA: costituita prevalentemente da polimorfonucleati. E’ espressione di un processo infiammatorio acuto in atto. - Cause: cistiti, prostatiti, uretriti, neoplasie vescicali, calcolosi, tubercolosi renale, pielonefriti. - CILINDRI: derivano da proteine (tipo Tamm-Horsfall) che prendono la forma del tubulo, precipitando a causa del pH urinario acido. All’interno dei cilindri possono rimanere intrappolati cellule tubulari desquamate, leucociti, emazie per cui si distinguono cilindri IALINI, GRANULOSI, LEUCOCITARI, EMATICI. Pochi cilindri ialini isolati non sono un reperto patologico. Cilindri ialini + granulosi→indice di sofferenza renale - CELLULE EPITELIALI: Provengono dallo sfaldamento della mucosa del tratto urinario. - CRISTALLI: La loro presenza è significativa solo se si trovano in quantità consistente. La precipitazione e il tipo di cristallo dipendono dal volume e dal pH urinari: - pH alcalino→sali di CaPO4 - pH acido→cristalli di acido urico. I cristalli di fosfato di NH4+ indicano la presenza di GRAM‒ produttori di ureasi. CONTA di ADDIS Vengono eliminati con le urine nelle 24h - fino a 1000000 di GR 66 www.slidetube - fino a 2000000 di globuli bianchi - fino a 10000 cilindri Capitolo 6 EMOSTASI 67 www.slidetube EMOSTASI EMOSTASI: processo che arresta l’emorragia - reazione infiammatoria locale - reazione vascolare: vasocostrizione per emostasi primaria fattori locali trombossano A2, serotonina, Epi, NA dalle piastrine attivate - formazione tappo emostatico: legame con endotelio danneggiato 1) Attivazione piastrinica -1-adesione delle piastrine -2-aggregazione delle piastrine legame con altre piastrine ADESIONE: interazione tra un recettore di membrana della piastrina e un ligando espresso dall’endotelio danneggito (fattore di von Willebrand) LIGANDI RECETTORI fattore di von Willebrand complesso glicoproteico Ib-IX collageno complesso glicoproteico Ia-IIa ADP trombina trombossano A2 adrenalina fattore di attivazione piastrinico il LEGAME PORTA all’EMISSIONE di PSEUDOPODI UTILI per AGGREGAZIONE AGGREGAZIONE: interazione tra glicoproteine IIb e IIIa e fibrinogeno e fibronectina, altra glicoproteina, e trombospondina e fibrinogeno Ia, IIa, IIIa, Ib, IIb → INTEGRINE 2) Reazione di rilascio Degranulazione piastrine con rilascio di serotonina, ADP, istamina, ATP, Ca2+, fattore piastrinico-4, fattore di crescita derivato dalle piastrine, proteine della coagulazione, proteine di adesione (fibrinogeno, trombospondina, fattore di von Willebrand). 68 www.slidetube 3) Coagulazione [3) e 4) → emostasi secondaria] Cascata di reazioni che portano alla conversione del fibrinogeno in fibrina 2 modalità di attivazione: INTRINSECA → contatto con superfici e precallicreina FATTORE TISSUTALE ESTRINSECA → danno tissutale o processo infiammatorio promuove coagulazione 4) Retrazione del coagulo Stabilizzazione del coagulo tramite contrazione (stimolazione trombina) degli pseudopodi di actina e miosina delle piastrine 5) Fibrinolisi → emostasi terziaria Dissoluzione del coagulo per ripristinare il flusso sanguigno. Promossa da PLASMINA Degrada fibrina → FRAMMENTI di FIBRINOGENO → azione anticoagulante Degrada alcuni fattori della coagulazione 1.blocca fattore tissutale 2.ostacola aggregazione piastrinica 3.inibisce formazione fibrina PLASMINOGENO tipo t e tipo u attivato dall’endotelio, fattore XII fattore di Hageman inibito da endotelio PAI1), fibroblasti (PAI1), monociti, placenta (PAI2), epatociti (PAI3) Collageno e LPS Fattore XII chinine C’ attivano fibrinolisi coagulazione C’ induce danno tissutale e quindi attiva coagulazione e fibrinolisi più fagociti 69 www.slidetube produce IL-1, TNFα, ROS e fattore tissutale MECCANISMI CHE BLOCCANO L’EMOSTASI 1 2 plasmina EPARANSOLFATO-ANTITROMBINA III - inattiva fattori serinproteasici della coagulazione - lega trombina all’endotelio → trombina diventa anticoagulante dopo legame con antitrombina L’eparina accelera la reazione di 2000 volte. 3 MECCANISMO PROTEINA C- PROTEINA S Si attiva se la quantità di trombina supera la capacità inibitoria dell’antitrombina III → interviene TROMBOMODULINA. La trombina si lega alla trombomodulina → attivazione PROTEINA C Proteina C si lega alla proteina S, a sua volta legata all’endotelio Complesso proteinaC-proteinaS → DEGRADAZIONE FATTORI SERINPROTEASICI Va e VIIa 4 WARFARINA Inibisce vitamina K → no sintesi fattori coagulazione. 3) Coagulazione: la cascata delle proteasi attiva la trombina che a sua volta attiva le piastrine legandosi a recettori specifici della loro membrana. Dopo il legame le piastrine espongono fosfolipidi a carica negativa e recettori come l’integrina αIIb, β3 per il legame con il fibrinogeno. TF lega VII e VIIa (forma attivata) TF-VII TF-VIIa ← attivato per danno tissutale attivano Xa, IXa, VIIa X, IX retro attivazione con amplificazione 70 www.slidetube MOLECOLA DEL FIBRINOGENO Molecola simmetrica costituita da 3 catene α, β, γ avvolte in una struttura ad α-elica, un dominio centrale E e da 2domini laterali D γ α α γ D E D β β La trombina agisce sul dominio E e stacca 2 peptidi: FIBRINOPEPTIDI A e B. Tale distacco porta alla perdita di equilibrio nella molecola e a un successivo riarrangiamento che coinvolge più molecole di fibrinogeno che si aggregano tra loro secondo una geometria ben precisa: D D E E D D D D E E D D Come ultima tappa nella stabilizzazione del complesso venutosi a formare, intervengono fattore XIII e Ca2+ che formano legami crociati tra le subunità D D D E E D D D D E E D D RIASSUNTO: molecola fibrinogeno proteolisi polimerizzazione (legami semplici tra le subunità) D D solubilizzazione (fattore XIII fa legami crociati tra le subunità) D D 71 www.slidetube TEST CHE VALUTANO IL RISCHIO EMORRAGICO 1- CONTA PIASTRINICA 2- TEMPO di EMORRAGIA E’ un indicatore dell’emostasi primaria (vasocostrizione e formazione del tappo piastrinico). E’ un test che ha un’utilità clinica limitata perché è a bassa sensibilità, specificità e riproducibilità. Esistono diversi metodi per riprodurlo: -bucare il lobo dell’orecchio e asciugare le gocce di sangue, cronometrando in quanto tempo si blocca l’emorragia -disinfettare la cute dell’avambraccio con etere, attendendo la vasocostrizione. Applicare lo sfigmomanometro al braccio e mantenerlo alla pressione di 40 mmHg. Incidere la cute e cronometrare il tempo che intercorre fino all’arresto del sanguinamento. Intervalli di riferimento: 1-7 minuti normale >10 minuti patologico → sospetta piastrinopenia 8-10 minuti da chiarire (fare prova di tolleranza all’aspirina) 3- TEMPO di QUICK o TEMPO di PROTROMBINA (PT) Test che valuta la via estrinseca: è sensibile all’attività dei fattori V, VII, X, II, ma non il IX. Il test si basa sul cronometraggio del tempo di coagulazione del plasma dopo l’aggiunta di TROMBOPLASTINA CALCICA. - TROMBOPLASTINA: miscela di proteine e fosfolipidi estratta da vari tessuti (cervello, polmone, placenta) che contiene molto fattore tissutale e fosfolipidi. - CALCICA: presenza di ioni Ca2+ che si legano al citrato, facendolo precipitare come citrato di Ca2+ e riattivando la coagulazione. La velocità di formazione della fibrina in queste condizioni è direttamente proporzionale (α) alla [VII] e dei fattori della via comune. Il PT è utile per 2 scopi: -1-monitoraggio della terapia anticoagulante orale TAO -2-diagnosi di anomalie della coagulazione o epatopatie. Metodica per eseguire il test: - Prelievo: prelevare il sangue usando citrato di Na+ come anticoagulante in un rapporto citrato/sangue = 1:10. Solitamente 4,5 ml di sangue e 0,5 ml di citrato (provette già calibrate). Agitare al provetta e centrifugare. I test di coagulazione si eseguono sul plasma. 72 www.slidetube - Test: aggiungere la tromboplastina calcica. Inclinare continuamente la provetta fino alla formazione del coagulo di fibrina e registrare il tempo. ≈ 13 sec. TEMPO NORMALE Il test deve essere standardizzato affinché sia possibile un confronto tra i vari risultati ottenuti. Il problema è che ogni laboratorio ha delle tromboplastine con sensibilità diversa. Oggi esiste una tromboplastina con PREPARAZIONE INTERNAZIONALE di RIFERIMENTO (IRP) da utilizzare per calibrare le altre tromboplastine in modo tale che: ottenuto con una tromboplastina qualsiasi possa essere convertito in una scala comune 1) eseguo il test con la tromboplastina del mio laboratorio 2) rapporto i sec del mio pz con i secondi del valore medio normale (IRP) 3) ottengo ISI = indice di sensibilità internazionale 4) converto il rapporto ottenuto con la mia tromboplastina nel RAPPORTO INTERNAZIONALE NORMALIZZATO (INR) = rapporto che si sarebbe ottenuto se il PT fosse stato eseguito con la tromboplastina IRP - INR = 1 VALORE NORMALE - INR = 2-3 PZ in TAO 4- TEMPO di TROMBOPLASTINA PARZIALE ATTIVATO (APTT) Test che valuta la via intrinseca. Si basa sul cronometraggio del tempo di coagulazione del plasma, con le stesse modalità del PT, dopo l’aggiunta di un attivatore (CaCl2) che attiva i fattori della fase di contatto e la tromboplastina parziale. - TROMBOPLASTINA PARZIALE = non contiene TF Limite del test: impossibile la standardizzazione. Si esegue principalmente per 3 ragioni: -1-screening dei disordini della coagulazione (es. emofilia) -2-monitoraggio della terapia anticoagulante con eparina -3-individuazione di inibitori dei fattori della coagulazione 34-36 sec TEMPO NORMALE ANOMALIE di TP e APTT - Allungamento APTT: deficit di un fattore della via intrinseca: precallicreina, chininogeno, fattore XII, XI, IX e VIII - Allungamento TP: deficit del fattore VII - Allungamento di entrambi: deficit di un fattore della via comune: fattore X, V, II o fibrinogeno. In caso di allungamento di APTT eseguire il TEST della MISCELA per valutare se la causa è il deficit di un fattore della coagulazione o la presenza di un inibitore di qualche fattore. Nel primo caso eseguire: DOSAGGIO dei FATTORI Fattori II, V, X, VII sono dosati determinando l’abilità del plasma del pz di correggere il PT di un plasma carente del fattore che deve essere dosato. I fattori XII, XI, IX e VIII usano invece APTT. ITER DIAGNOSTICO - test globali → tempo di emorragia ↓ - test settoriali → TP, APTT ↓ - test fattoriali → dosaggio dei fattori 73 www.slidetube TROMBOSI La trombosi ha un’incidenza > dell’emorragia. Si manifesta prevalentemente come trombosi venosa profonda che può portare a embolia polmonare. tendenza all’emorragia equilibrio tendenza alla trombosi emostatico CAUSE di EMORRAGIA trombocitopenia trombocitopatia carenza di fattori della coagulazione carenza di α2-antiplasmina (iperfibrinolisi) formazione di inibitori o Ab CAUSE di TROMBOSI carenza di inibitori della coagulazione AT-III, prot. C, prot. S attivazione della coagulazione (lesione tissutale, superfici estranee, turbolenza, tossine, chinine, stasi, neoplasie) aumento della viscosità del sangue Fattori di rischio trombotico: - diabete - placche aterosclerotiche - malattie virali - protesi artificiali - tumori - traumi e ingessature - fattori attivanti la coagulazione (danno vasale – contatto – adesione – aggregazione reversibile – aggregazione irreversibile – modificazioni morfologiche - release – trombo bianco RELEASE: ADP e TXA2 → aggreganti e vasocostrittori 74 www.slidetube TEST che VALUTANO il RISCHIO TROMBOTICO 1- ANTIROMBINA Esiste un difetto congenito di antitrombina-III che può presentarsi in 2 forme: - tipo I: riduzione [AT] - tipo II: [AT] normale e attività ridotta Per il dosaggio di AT si utilizza il metodo cromogenico: prendere plasma pz, aggiungere trombina ed eparina; la trombina non inibita, misurata con un substrato cromo genico sensibile è proporzionale all’AT presente nel plasma. 2- PROTEINA C- PROTEINA S Esistono dei difetti congeniti delle proteine C ed S. Altra causa di deficit sono le epatopatie. i metodi dosaggio sono coagulativi. 3- FATTORE V di LEIDEN E’ possibile il verificarsi una resistenza alla proteina C attivata a causa di una mutazione puntiforme del gene del fattore V geneticamente trasmissibile. Ne consegue uno stato di ipercoagulabilità. L’APC-R è evidenziata con il rapporto tra: La conferma è data dalla biologia molecolare. 4- DOSAGGIO del DIMERO-D L’azione della plasmina sulla fibrina porta alla formazione di 2 prodotti terminali: i monomeri E e i dimeri D (non può scindere i legami crociati). Il dosaggio del dimero-D può escludere una TEV recente se sono basse le concentrazioni. Il dosaggio avviene tramite l’utilizzo di anticorpi monoclonali e tecniche immunoenzimatiche (ELISA). D D dimero-D TROMBINA PLASMINA proteolisi 2 peptidi A 2 peptidi B ↓ monomeri di fibrina ↓ fibrina polimerizzata ↓ ← XIII coagulo 2 peptidi A, 2 peptidi B, 2 peptidi C ↓ FDPX ↓ FDPY + FDPD ↓ FDPE + FDPD 75 www.slidetube FDP = prodotti di degradazione del fibrinogeno 5- FRAMMENTO di PROTROMBINA e FIBRINOPEPTIDE A Al fine di valutare il rischio trombotico individuale è importante dosare nel plasma del pz anche i marcatori di attivazione. I più importanti sono il frammento di protrombina (valuta la quantità di trombina formata) e il fibrinopeptide A. ALTRE RARE CAUSE di TROMBOFILIA - anticoagulante lupico (LA): Ab che agisce contro le proteine plasmatiche con alta affinità per i fosfolipidi (protrombina, chininogeni, proteina C, proteina S) - iperomocisteinemia: causata da una mutazione al gene MTHFR e correlata positivamente col rischio di trombosi - eccessivi livelli plasmatici di F-VIII - antagonisti della vitamina K: anticoagulanti orali - inibitori della trombina EMOFILIA - inibitori del fattore Xa COAGULAZIONE INTRAVASALE DISSEMINATA (CID) Sindrome acquisita, caratterizzata da un’attivazione incontrollata della coagulazione nel microcircolo che, da una parte, porta alla formazione di trombi, e dall’altra ha manifestazioni emorragiche causate dal consumo dei fattori della coagulazione e dal calo delle piastrine. La CID è sempre una manifestazione clinica secondaria; le principali cause scatenanti sono infezioni batteriche, traumi e neoplasie. Indipendentemente dalla causa, il fattore scatenante della coagulazione è l’esposizione del TF, accompagnato dall’inibizione della fibrinolisi. In caso di CID il laboratorio documenta: - Allungamento di PT e APTT - Trombocitopenia - Diminuzione degli inibitori della coagulazione AT-III, prot. C e prot. S - Aumento del D-dimero FIBRINOGENO Il dosaggio del fibrinogeno è richiesto in numerose situazioni (è proteina di fase acuta, è correlato con le cardiopatie ischemiche e le emorragie). Esistono vari sistemi per il dosaggio. I principali sono 2: - METODO VON CLAUSS: aggiungere al plasma un eccesso di trombina, il tempo necessario per la formazione del coagulo è inversamente proporzionale al contenuto in fibrinogeno. - DOSAGGIO FIB PT-DERIVATO: disegno la curva di coagulazione di un campione di plasma. Essa riflette la differenza tra il campione prima e dopo la trasformazione del fibrinogeno in fibrina → correlato con [FIB] 76 www.slidetube 300-400 mg/dl NORMALE Capitolo 7 SCREENING e MONITORAGGIO 77 www.slidetube TEST di SCREENING per NEONATI 1- TEST per FENILCHETONURIA (PKU) 2- TEST per IPOTIROIDISMO CONGENITO (CH) 3- TEST per IPERPLASIA SURRENALE CONGENITA Test di screening per PKU e CH sono diffusi in tutti i paesi occidentali. Analisi costo-beneficio hanno dimostrato che portano a un risparmio di 215000 euro per paziente. 1) IPERFENILALANINEMIE Derivano dalla mancata o ridotta conversione di Phe in Tyr. La forma più comune è la PKU caratterizzata dalla ridotta attività della Phe-idrossilasi. Sono note più di 200 mutazioni nel gene di questo enzima, diverse nei vari gruppi etnici. La mutazione R408W è associata a un severo quadro clinico e richiede un’attenta restrizione dietetica della Phe, mentre la mutazione I65T è associata a un quadro più lieve. E’ autosomica recessiva in 1/10000 nati (1:5400 in Irlanda, 1:16000 in Svizzera) Clinica: progressivo deficit mentale, microcefalia, iperattività, convulsioni, ritardo mentale. La restrizione dietetica deve avvenire entro le prime 3 settimane. DIAGNOSI - screening di tutti i neonati con il test di Guthrie: si basa sull’impiego di un antagonista della Phe, la β-L-tienilalanina che aggiunta a un terreno di coltura, impedisce la crescita di Bacillus Subtilis. Tale inibizione è rimossa dalla Phe ([C] di almeno 4 mg/dl) e la crescita del batterio è tanto più rigogliosa quanto più è alta la [C] di Phe - altri metodi di dosaggio: fluorimetrico, GLC - diagnosi di portatore: carico di 100 mg/kg di Phe 9 mg/dl a 1 h NORMALE 5 mg/dl a 4 h 19 mg/dl a 1 h PORTATORE 78 www.slidetube 2) TIROSINEMIA e CONDIZIONI CORRELATE - albinismo oculo-cutaneo: dovuto a una ridotta sintesi della melanina, nella maggior parte di questi casi l’attività della tirosinasi risulta assente o ridotta. - tirosinemia-I (tirosinosi): ridotta attività della fumarilacetoacetato-idrolasi e della acido idrossifenil-piruvico ossidasi con aumentata [C] plasmatica della tiroxina e dei suoi metaboliti (DOPA) e della Met. Risulta in un danno epatico (epatite acuta infantile, cirrosi nell’adulto), danno tubulare renale (sindrome di Fanconi). - tirosinemia-II: deficit dell’enzima epatico tirosina-amino-transferasi. Risulta in danni oculari (erosioni corneali) e lesioni alla cute del palmo delle mani e dei piedi (formazione di cristalli di Tyr intracellulari), a volte ritardo mentale. - tirosinemia transitoria del neonato: dovuta a immaturità epatica. La Tyr accumulata inibisce la Phe-idrossilasi (false diagnosi di PKU). - alcaptonuria: dovuta a deficit di acido-omogentisico (HGA)-ossidasi. HGA si accumula e lega al collagene. Risulta in artrite e pigmentazione (acronosi). AMINOACIDURIA Danno TP. SE associata a rachitismo: sindrome di Fanconi (autosomica recessiva con depositi intracellulari di Cys). Il difetto è un ostacolato efflusso di Cys dai lisosomi. Nel bambino la causa più comune è la cistinosi. Sindrome di Fanconi: disfunzione generalizzata del TP - iperfosfaturia e ipofosfatemia - glicosuria - aminoaciduria - ipourirecemia - ipocaliemia - rachitismo resistente a Vit.D 79 www.slidetube MONITORAGGIO dei FARMACI BIODISPONIBILITA’: rapporto tra sostanza somministrata/dose assorbita [CONC] STAZIONARIA: si ottiene quando la velocità di eliminazione è uguale alla velocità di assorbimento. Il monitoraggio dei farmaci è particolarmente importante per i farmaci con indice terapeutico ristretto ([C] ottimale terapeutica e [C] tossica molto vicine). FARMACI SOTTOPOSTI a MONITORAGGIO anticonvulsivanti (fenobarbital) immunosoppressori (ciclosporina) antibiotici (vancomicina) antitumorali (metotrexate) cardioattivi (amiodarone) antipsicotici (litio) antiretrovirali (ritonavir) antivirali (ganciclovir) FARMACOGENETICA e FARMACOGENOMICA FARMACOGENETICA: studio della relazione tra le varianti di un singolo gene e gli effetti di un farmaco. FARMACOGENOMICA: studio della relazione tra le varianti di vari geni, fino all’intero genoma, e gli effetti di un farmaco. geneticamente predisposti seri effetti collaterali risposta inadeguata pazienti in trattamento All’interno di ogni popolazione esiste un sottogruppo di individui geneticamente predisposti a soffrire per effetti collaterali di un farmaco o a rispondere in modo inadeguato. Ciò rimarrà celato fino a che essi non sono esposti al farmaco. Quindi solo l’esposizione al farmaco o test eseguiti a priori riveleranno la predisposizione genetica. 80 www.slidetube La FAMIGLIA del CITOCROMO P-450 Famiglia di citocromi che assorbe la luce a 450 nm con aggiunta di CO. Ci sono 10 famiglie di CYP che comprendono più di 100 geni. Sono enzimi localizzati nel reticolo endoplasmatico dell’epatocita. Importante per la detossificazione degli xenobiotici (es. idrossilazione del fenobarbitale ne facilita l’escrezione). Una delle funzioni più rilevanti del citP450 è il suo ruolo nel metabolismo dei farmaci. gene deleto MUTAZIONE PUNTIFORME no enzima enzima instabile enzima normale alterata specificità no metabolismo ridotto metabolismo normale metabolismo possibilità di nuovo metabolismo duplicazione del gene alti livelli di enzima aumentato metabolismo Test in clinica: CYP2D6 → per antidepressivi, antipsicotici, antiaritmici, β-bloccanti, antiipertensivi. ↓ si richiede la ricerca di mutazioni (frequenza 30% nella popolazione) per evitare episodi di tossicità. Esempio: ISONIAZIDE (anti TBC) può dare epatite fulminante se è mutato l’N-acetil-transferasi-II. Iniziare con una dose bassa e monitorare l’attività epatica. 2 modalità di diagnosi DOSAGGIO ATTIVITA’ TEST GENETICO ENZIMATICA potenzialmente evidenzia tutti i facile e veloce VANTAGGI difetti (sensibilità 100%) consumo di tempo, non è sensibilità limitata (≈75%), SVANTAGGI automatizzato, richiede costoso e richiede tempo esperienza, falsi negativi dopo trsfusione SNP: ogni individuo può avere un nucleotide o una base diversa dagli altri in una data locazione o su un cromosoma. Esistono delle mappe degli SNP che possono essere usate per predire la risposta a un trattamento: si classificano i genotipi verso cui il farmaco è efficace e quelli verso cui non lo è. ANALISI FARMACOLOGICA - FARMACOLOGIA - studi di farmacocinetica - studi clinici 81 www.slidetube - BIOCHIMICA - attività enzimatica, funzione recettoriale, funzione dei trasportatori - GENETICA - analisi SNP - sequenziamento genico TEST GENETICO malattie genetiche rare mendeliane (geni causali) farmaco genetica comuni (suscettibilità genica) geni del metabolismo dei farmaci profili SNP per metabolismo dei farmaci OTTIMA RISPOSTA RISCHI: implicazioni etiche, legali e sociali. ORIENTAMENTO della TECNOLOGIA nella CLINICA di LABORATORIO -TEST GENETICI: con il mappaggio del genoma umano, il campo della diagnostica molecolare, che include i test genetici, crescerà molto rapidamente. -FARMACOGENOMICA: studio delle basi genetiche della differenza nella risposta ai farmaci. -PROTEOMICA: analisi sistematica delle proteine espresse da una cellula o un tessuto usando una sensibile, veloce e accurata tecnica di separazione delle proteine e metodi quantitativi e strumenti di identificazione e caratterizzazione. - MICRODEVICES BIOMEDICI, NANOTECNOLOGIA, e BIOSENSORI: i micro e nano devices, con componenti da centinai di micron a pochi nanometri, hanno un potenziale uso nella diagnosi, prevenzione e trattamento delle malattie. MEDICINA PERSONALIZZATA: UNA SFIDA PER IL LABORATORIO L’uso di marker nella diagnosi e terapie bersaglio derivati da un profilo molecolare individuale rivoluzionerà la cura del paziente. Principi e conoscenze scientifiche richiesti: - accuratezza dei test e comparabilità dei risultati tra i laboratori 82 www.slidetube - attenta selezione, in accordo con i colleghi, di test che contribuiscono alla decisione clinica - perdita di fiducia nei confronti di test sostitutivi - markers di superficie cellulare e sistemi di trasduzione - genomica del paziente, genomica della patologia, proteo mica e farmaco genomica - sviluppo della bioinformatica e di sistemi di informazione medica CITOMETRIA A FLUSSO Capacità di misurare le proprietà delle cellule in un flusso. SORTING o FLUORESCENCE-ACTIVATED CELL SORTING (FACS) Capacità di separare le cellule sulla base delle loro proprietà misurate in un flusso. ANALISI CELLULARE CLASSICA MICROSCOPIA - 100 o 1000 cellule/saggio - + di 5 min/campione - risultati soggettivi - risultati positivi-negativi - bassa riproducibilità - lavoro intensivo - non c’è controllo di qualità - Parte fluidica - Parte ottica - Parte elettronica CITOMETRIA a FLUSSO - 10000 o 1000000 di cellule - ‒ di 1 min/campione - risultati oggettivi - risultati numerici - alta riproducibilità - pienamente automatizzato - controllo di qualità BASI della CITOMETRIA A FLUSSO cellule in sospensione fluiscono in singole file attraverso un volume illuminato dove loro spargono la luce e emettono una fluorescenza che è raccolta, filtrata e convertita i valori digitali che sono fissati su un computer Le cellule sono iniettate in una camera in cui è stato creato un flusso laminare che le spinge in un capillare una alla volta. Qui sono colpite da un laser. Se la cellula è marcata a fluorescenza i raggi che emette sono catturati da sensori. la luce è emessa principalmente in 2 direzioni: FORWARD ANGLE LIGHT SCATTER (FSC) → in avanti laser 0 sensore 83 www.slidetube 90° LIGHT SCATTER (SSC): → angolo di deviazione laser sensore 0 sensore La luce catturata a FSC è proporzionale alla dimensione delle cellule. L’entità di SSC è proporzionale alla rugosità cellulare e alle proprietà antigeniche. Con SSC e FSC nel sangue distinguiamo: LINFOCITI MONOCITI GRANULOCITI piccoli e lisci medi e media rugosità grandi e rugosi Essi sono rappresentati con un istogramma o in DOT PLOT. Le cellule possono essere marcate con vari fluorocromi. La fluorescenza emessa da ciascuno è solitamente rilevata in un unico CANALE di FLUORESCENZA. La specificità della rilevazione è controllata dalla selettività della lunghezza d’onda (λ) dei filtri ottici e degli specchi. Ogni marcatore rileva una caratteristica diversa florocromo 2 POSITIVO FL2 POSITIVO FL1 e FL2 NEGATIVO FL1 e FL2 POSITIVO FL1 PROCEDURA -prendere campione biologico -Ab monoclonali -metterlo in soluzione -analizzarlo per sangue, urine, CSF, lavaggio bronco-alveolare, liquido sinoviale. Bastano 200 μl di materiale. SORTING: allarga il campo della citometria a flusso con la capacità di dividere e raccogliere le cellule che esibiscono un set di caratteristiche identificabile sia meccanicamente che tramite strumenti elettrici. Le caratteristiche da trovare le programma l’operatore. Es: le cellule sono caricate con un determinato Ag. Sotto ci sono 2 piastre (+/‒ ). Le cellule + entrano nella provetta sotto la piastra ‒ e viceversa. Si possono isolare cellule presenti in circolo per l’1% con una purezza del 95%. APPLICAZIONI della CITOMETRIA A FLUSSO 84 www.slidetube - determinazione eterogeneità cellulare - analisi multiparametrica - dimensione cellulare e struttura interna - Ag di superficie cellulare - studio dell’attività cellulare - attività enzimi e localizzazione - citochine intracellulari - attività dei fagociti - metabolismo ossidativo - contenuto totale DNA e RNA - composizione del DNA - apoptosi - sorting CICLO CELLULARE Il sorting permette di valutare quante cellule sono in duplicazione. Per rilevare in quale fase del ciclo sono si utilizza il DNA: - cellule G0-G1 → euploidi – diploidi - cellule G2-M → tetraploidi - cellule ≠ G0 e G1 → aneuploidi n° cellule G0-G1 G2-M S contenuto DNA Il sorting può essere utilizzato anche per ricerca di cellule non frequenti (es. eritroblasti fetali dopo un mese) o per valutare come un soggetto risponde a terapia (leucemie). 85 www.slidetube EMOCROMO Può essere determinato con tecniche manuali (microscopio) o automatizzate (10 volte più sensibili) CELLULE CONTATE ERRORE TECNICHE MANUALI eritrociti globuli bianchi piastrine reticolociti ±11% ±16% ±22% ±33,9% TECNICHE AUTOMATIZZATE ±1% ±1,5% ±2% ±5% Valori di riferimento M 4,6-6,2 13,5-18 4,3-10,8 130-400 0,42-0,5 80-98 F 4,2-5,4 12-16 4,3-10,8 130-400 0,35-0,45→sempre<50% 81-99 Emocromo 12 n° degli eritrociti (10 /litro) Hb(cianmetaemoglobina)(g/dl) leucociti (109/litro) piastrine (109/litro) Hct (ematocrito) MCV (femtolitri) HCT(%)/RBC(x108/μL)x10 MASSA ERITROCITARIA ← MHC (Hb corpuscolare media) pg (10‒ 12 g) valori: 26-32 MCHC (concentrazione Hb corpuscolare) g/dl valori: 32-36 N.B. Importante perché le 2 anemie più diffuse sono la sideropenia e la talassemica INDICE di ANISOCITOSI ← RDW (ampiezza di distribuzione del volume dei globuli rossi) valori: 11,5-14,5 Sapere anche il valore degli elettroliti Neutrofili 0,5x109/litro TALASSEMIA ETEROZIGOTE: RDW normale; ↓MCV SIDEROPENIA: ↑RDW; MCV basso-normale 86 www.slidetube Falso ↑MCV: agglutinine fredde, iperglicemia (già attiva nella cellula la soluzione del flusso durante l’esame) Falso ↑MCH: iperlipidemia. Per la conta delle cellule a mano si usa il reticolo di Burker METODI AUTOMATICI PRELIEVO: minimizzare l’impiego del laccio per minimizzare l’emoconcentrazione; mescolare bene con l’anticoagulante. DILUIZIONE: il tipo di fluido e l’entità della diluizione dipendono dal tipo di cellula da valutare. CONTEGGIO: sistema ad impedenza, sistema ottico, sistema misto IMPEDENZA = resistenza elettrodi flusso→ → → Se non ci sono cellule la corrente passa tranquillamente. Se ci sono si calcola la resistenza. apertura Oggi un potente strumento per l’analisi delle cellule del sangue (specialmente globuli bianchi e reticolociti) è COULTER 3D-VCS tecnology che mostra elevata sensibilità, specificità ed efficienza 87 www.slidetube APOPTOSI Programma di morte cellulare energia-dipendente. APOPTOSI processo attivo shrinkage cellulare condensazione nucleare membrana intatta non flogosi NECROSI processo passivo rigonfiamento cellulare rottura membrana reazione flogistica L’apoptosi gioca un ruolo attivo nella regolazione della risposta immune e probabilmente nella patogenesi di alcune malattie autoimmuni (molti autoAg sono substrati per le caspasi). Caratteristica principale è l’autodigestione controllata della cellula per mezzo dell’attivazione di proteasi endogene (caspasi). Queste causano la distruzione del citoscheletro (BLEBBING) con esposizione sulla superficie cellulare esterna della FOSFATIDILSERINA, segnale per i macrofagi. L’attivazione delle endonucleasi provoca la frammentazione del DNA con condensazione del nucleo. La perdita della funzione mitocondriale è un evento iniziale e irreversibile. CASPASI (almeno 10 note) Proteasi cisteiniche aspartato-specifiche intracellulari. I proenzimi sono attivati dalla rimozione proteolitica di un frammento N-terminale e dalla formazione di 2 subunità. Substrati: - proteine coinvolte nella riparazione cellulare - proteine coinvolte nel controllo del ciclo cellulare - proteine coinvolte nella trasduzione del segnale - proteine coinvolte nell’integrità strutturale della cellula PROTEINE DELLA FAMIGLIA BCL-2 Importanti regolatrici della sopravvivenza cellulare. La famiglia comprende sia proteine anti-apoptotiche (BCL-2) sia pro-apoptotiche (BID). Modificano la soglia di suscettibilità della cellula all’apoptosi. Possiedono una regione C-terminale idrofobca che permette l’ancoraggio alla membrana del RE, del mitocondrio e del nucleo ( di cui regolano la permeabilità). → impediscono il rigonfiamento del mitocondrio con fuoriuscita del cytC Legano inoltre APAF-1, proteina coinvolta nell’attivazione delle caspasi. CITOCROMO C 88 www.slidetube Normalmente localizzato nello spazio intermembrana mitocondriale. Nelle fasi precoci dell’apoptosi esce nel citosol. Si lega a APAF attivando la caspasi 9. PROTEINA FAS TNF RECETTORE-RELATA→ SEGNALE ESTERNO Il ligando FAS è una proteina trimerica, che legando al suo recettore lo trimerizza (è omotrimero), attivandolo. Ciò porta al reclutamento della proteina FADD che contiene un dominio della morte in grado di attivare le caspasi 8, per clivaggio autocatalitico. La caspasi 8 attivata determina l’attivazione a cascara delle altre caspasi. Questo processo può essere modulato da proteine della famiglia BCL-2. METODI di STUDIO dell’APOPTOSI 1- Metodo più chiaro e indiscusso: microscopia elettronica 2- Valutazione della frammentazione della cromatina nucleare mediante: -elettroforesi del DNA estratto produce una scala caratteristica di frammenti di oligonucleosomi -metodi di citometria a flusso che combinano la frammentazione specifica del DNA e l’integrità della membrana cellulare per distinguere l’apoptosi dalla necrosi TUNEL: tecnica che rileva le rotture del DNA marcando l’estremità 3’OH con nucleotidi coniugati a fluorocromi o a biotina per mezzo di una reazione catalizzata da DNA-polimerasi. Può dare però risultati falsamente positivi in cellule che sono andate incontro a rotture del DNA non apoptotiche. Metodo complesso e difficilmente automatizzabile. 3- Annexina V (metodo semplice e robusto, probabilmente il migliore). Durante le fasi precoci dell’apoptosi la fosfatidilserina (PS) è esposta sulla membrana cellulare esterna. L’annessina V è una proteina legante i fosfolipidi in presenza di Ca2+ con alta affinità per la PS (è unasonda sensibile per l’apoptosi). Si usa citometria a flusso con doppia colorazione e 2 markers: - FITC-annessina V - ioduro di propidio che colora le cellule necrotiche (valuta integrità cellulare) ioduro di propidio NESSUNA CELLULE CELLULA NECROTICHE CELLULE SANE CELLULE APOPTOTICHE annessina V 4- Metodo dell’Apo2.7 L’Ab apo2.7 riconosce una proteina di membrana mitocondriale che è esposta nelle cellule nelle prime fasi dell’apoptosi. E’ possibile valutare questo Ag in citofluorimetria. 5- Determinazione dell’attività delle caspasi 89 www.slidetube Utilizzo di un substrato caspasi-sensibile e misurazione fluorimetrica Capitolo 8 EQUILIBRIO ACIDO-BASE 90 www.slidetube EQUILIBRIO ACIDO-BASE ACIDO: sostanza in grado di cedere protoni BASE: sostanza in grado di accettare protoni Il pH del sangue arterioso è determinato dal rapporto tra la [HCO3‒ ] e la pCO2 Il rapporto intercorrente tra queste variabili è espresso dall’equazione di Henderson-Hasselbach HCO3‒ ]/[H2CO3] Ricordando l’equilibrio tra queste reazioni: CO2 + H2O H2CO3 H+ + HCO3‒ L’equazione può essere espresso in questo modo: pH = pK + Log [HCO3‒ ]/(pCO2⋅0,03) - [HCO3‒ ] = 24 mMoli/L - pCO2 = 40 mmHg -pK = 6,1 per il sistema tampone H2CO3/ HCO3‒ Da qui si ricava che pH(sangue) = 6,1 + Log 20 = 7,4 pH 7,35-7,45 → normale 6,8-7,8 → compatibile con la vita [HCO3‒ ]/pCO2 = 20 pCO2: componente respiratoria dell’equilibrio acido-base. Il suo valore dipende dalla funzione polmonare. HCO3‒ : componente metabolico dell’equilibrio acido-base. Il suo valore dipende dalla funzionalità renale e da quella epatica ( la produzione di urea avviene a partire da NH4+ e HCO3‒ ). Il controllo della ventilazione è effettuato dal centro respiratorio bulbare che è attivato dall’↑ di pCO2. Il rene rigenera HCO3‒ con 3 meccanismi principali: - riassorbimento di HCO3‒ nel TD e DC con il sodio 91 www.slidetube - scambio di HCO3‒ con H+ (acido titolabile) - formazione di HCO3‒ ex novo a partire dalla glutammina. Le cause che modificano il pH sono: METABOLICHE → compenso respiratorio (rapido 3-6 h) RESPIRATORIE → compenso metabolico (lento, anche giorni). 1. ACIDOSI METABOLICA Causa dell’acidosi è l’aumento degli ioni H+, che vebgono tamponati da HCO3-, la cui [C]↓ nel plasma. La base HCO3- è sostituita da Cl- (acidosi IPERCLOREMICHE) o con anioni patologici quali ac. lattico, corpi chetonici (es. cheto acidosi diabetica) (acidosi IPOCLOREMICHE con ALTO GAP ANIONICO). Compenso respiratorio (↑VP con ↓pCO2) e infine compenso renale con escrezione di ioni H+ - CARATTERISTICHE PRINCIPALI - ↓pH - ↑H+ - ↓pCO2 - ↓HCO3- ↑Cl o normale 2. ALCALOSI METABOLICA Causa dell’alcalosi è l’↑ [HCO3-] nel plasma per vomito prolungato, somministrazione di mezzi di contrasto,… Il compenso è respiratorio (↓VP con ↑pCO2); in seguito si attiva il compenso renale che ↑ l’escrezione di HCO3-. - CARATTERISTICHE PRINCIPALI - ↑pH - ↓H+ - ↑HCO3- ↑pCO2 3. ACIDOSI RESPIRATORIA Causata da ↑pCO2 per difficoltà di ventilazione, insufficienza respiratoria, asma, grave broncopatia ostruttiva che impedisce l’eliminazione della CO2. Il compenso è renale con ↑HCO3- CARATTERISTICHE PRINCIPALI - ↓pH - ↑H+ - ↑pCO2 - ↑HCO392 www.slidetube 4. ALCALOSI RESPIRATORIA Causata da ↓pCO2 legata ad aumento dell’attività respiratoria (febbre, anemia, stati d’ansia). Compenso renale che trattiene H+ ed elimina HCO3- CARATTERISTICHE PRINCIPALI - ↑pH - ↓H+ - ↓pCO2 - ↓HCO3- GAP ANIONICO E’ una valutazione di ioni plasmatici non misurati che si ottiene sottraendo dalla somma dei cationi (Na+ e K+) la somma degli anioni (HCO3- e Cl-) G.A. = Na+ + K+ ‒ Cl‒ ‒ HCO3‒ G.A. = 8 ‒ 12 μmol/L VALORI MEDI G.A. > 17 μmol/L PATOLOGICO Il GA è utile nella diagnosi differenziale di acidosi metabolica, dal momento che molte sono caratterizzate dal tipo di anione che aumenta (ac. lattico, corpi chetonici,…) MIDALITA’ di PRELIEVO EMOGASANALISI Il prelievo arterioso si esegue sull’arteria radiale. Sono indispensabili stretta anaerobiosi, conservazione in ghiaccio e processazione rapida. 93 www.slidetube Capitolo 9 MARCATORI TUMORALI 94 www.slidetube MARCATORI TUMORALI La diagnosi definitiva di tumore si compone di 3 fasi: - 1 – diagnostica per immagini - 2 – indagini sierologiche - 3 – indagini anatomo-patologiche Approccio del laboratorio di analisi alla malattia tumorale: analisi del prelievo ematico per giungere a una diagnosi legata al fatto che il tumore rilascia in circolo molecole che lo rappresentano. La trasformazione neoplastica è infatti associata all’espressione di proteine anomale che poi vanno in circolo (situazione ideale). In realtà: 1) solo una piccola parte delle proteine è implicata nella trasformazione neoplastica; 2) sono rilasciate in piccole quantità Per questo la biopsia è più sicura per una diagnosi. Breve storia dei marcatori tumorali. 1846: proteina di BJ 1928: sindrome ormone ectopico 1930: hCG (tumori placentari) 1932: ACTH 1949: delezione Ag gruppi sanguigni 1959: isoenzimi 1963: AFP (α-feto-proteina) 1965: CEA (Ag carcino-embrionale) 1969: oncogeni 1975: Ab monoclonali (utili come reagenti per evidenziare markers) 1980: 1985: geni oncosoppressori Potenziale uso dei markers tumoral. - screening della popolazione - diagnosi differenziali su pz. 95 www.slidetube - valutazione stadio clinico del tumore - stima del volume del tumore - indicatore prognostico di progressione - radioimmunolocalizzazione della massa tumorale - valutazione successo del trattamento - valutazione eventuale recidiva - monitoraggio risposta alla terapia - determinazione direzione di immunoterapia Nella diagnostica più comune l’uso del marker è valutare il successo della terapia e le eventuali ricadute. ENZIMI COME MARKERS ALT: LDH: fegato fegato ALP: osso, fegato (isoenzima L1), leucemia, sarcoma AFP: Ag oncofetale (presente nella vita fetale, è riespresso in caso di tumore). Utile nella diagnostica del carcinoma epatocellulare (ATTENZIONE! non tutti lo secernono). E’ come ALB col 4% di glicosilazione. CEA: 160 aa, differentemente glicosilato. Presenta grande eterogeneità e quindi è di difficile standardizzazione. Associato a tumore del pancreas, colon, stomaco, polmone, mammella. PROTEINE COME MARKERS FERRITINA: aumenta in caso di neoplasia del fegato Ag CELLULO-SQUAMOSO: cervice, pelle, polmone, faringe Ag TISSUTALI: riconducibili a frammenti di citocheratine 8- 18- 19 (sono elementi del citoscheletro). Il più famoso è SARIFA (da citocheratina 19). Associati a tumore della mammella, ovaio, colecisti, colon, vescica. MUCINE COME MARKERS Oligosaccaridi complessi legati a proteina transmembranaria. Composti da 50-80% carboidrati e acido sialico. Danno viscosità alla soluzione in cui sono identificate. Le mucine compaiono a seguito di un’alterazione della glucosiltransferasi che altera a sua volta la struttura cellulare. La cellula perde così la sua polarità e le mucine vanno in circolo. Esse vengono indicate con la sigla CA (Carboidrato Ag) + numero corrispondente all’Ab monoclonale che mette in evidenza l’Ag. 96 www.slidetube CA 15.3: MCA: proteina MUC-1 con dominio centrale e un numero variabile di tandem repeat (VNTR). Tumore della mammella tumore della mammella (non si usa più) CA 125: oligosaccaride, aumenta nel tumore dell’ovaio DU-PAN Z: per il pancreas, ma non si usa più CA 19.9: costituito da un epitopo glucidico sialilato denominato SIALIL…….. Pancreas, colon, retto ALTRI MARKERS RECETTORI per ESTROGENI e PROGESTERONE: mammella METABOLITI CATECOLAMINE: nelle urine. Neuroblastoma (si forma nel cranio e nei gangli addominali) e feocromocitoma IDROSSIPROLINA: per metastasi ossee ONCOGENI TROVATI in TUMORI UMANI mutazione proteina RAS FUNZIONE trasduzione del segnale traslocazione C. myc regolazione della trascrizione amplificazione C. erb B2 recettore per EGF PRODOTTI TUMORE GTP/GDP leucemia, binding protein neuroblastoma, linfoma lega DNA linfoma cellule B e T, carcinoma polmone a piccole cellule tirosina-chinasi mammella, ovaio, intestino Non sono specifici dei tessuti GENI ONCOSOPPRESSORI mutazione p53: mammella, colon, vescica, polmone, rene, sarcoma, colecisti mutazione APC: colon, retto 97 www.slidetube Obiettivo clinico - diagnosi differenziale (distinguere tumore benigno da maligno) - bilancio di base - risposta al trattamento primario - riconoscimento della progressione della malattia - monitoraggio della malattia avanzata ATTENZIONE!!! Molte condizioni benigne ↑ i markers CONDIZIONE - abitudini voluttuarie -fumo -alcol MARCATORI CEA, TPA (antigene polipeptidico tissutale) CEA, TPA - condizioni fisiologiche -allattamento -ciclo mestruale -gravidanza MCA CA 125 AFP, βhCG, TPA, CEA, CA 125, MCA - patologie non neoplastiche -ittero -pancreatite -nefropatia cronica -ipertrofia prostatica -endometriosi -epatite cronica -polmonite -malattie reumatiche CA 125, CA 19.9, TPA CA 125, CA 19.9 CEA, TPA, β2microglobulina PSA, PAP CA 125 CEA, TPA, AFP, CA 19.9, CA 125, CA 15.3 CEA, TPA CA 19.9 98 www.slidetube MARCATORI di EVIDENTE UTILITA’ CLINICA AFP CA 15.3 epatocarcinoma, tumore germinale di testicolo e ovaio mammella CA 125 ovaio CA 19.9 pancreas, stomaco, colon CEA colon-retto, fasi avanzate di tutti i tumori HER 2 (rec. EGF) mammella NSE (enolasi neuro-specifica) PSA (Ag prostatico specifico) carcinoma micro cellulare del polmone e tutti quelli che derivano dalla cresta neuronale prostata fPSA (frazione libera) prostata hCT calcitonina carcinoma midollare della tiroide hTC tireoglobulina carcinoma differenziato della tiroide catecolamine carcinoide, tumore neuroendocrino Ag cellulo-squamoso collo dell’utero 99 www.slidetube hCG vescica, corio-carcinoma, ovaio, testicolo CM-BJ Mieloma Multiplo CgA (cromogranina A) tumori neuroendocrini CARATTERISTICHE dei PRINCIPALI MARKERS - CEA - t1/2 = 6-8 gg ↓CEA: intervento chirurgico, successo terapia ↑CEA: patologia benigna del tratto gastroenterico, fegato, polmone, insufficienza renale cronica Se[CEA] è costante per lungo tempo probabilmente la patologia è maligna - AFP - t1/2 = 5-6 gg ↑ gravidanza, epatopatia cronica - CA 15.3 e CYFRA 21.1 - t1/2 non noto ↑epatopatie - NSE ↑ emolisi campione - CT - t1/2 = 2-15 minuti → utile fare più prelievi per valutare l’operazione già in sede intraoperatoria ↑ insufficienza renale cronica, iperparatiroidismo, morbo di Paget - TPS ↑ patologia benigna del tratto gastrointestinale - PSA marker tumorale callicreina. Membro delle proteasi a callicreina, è prodotto dalle cellule epiteliali secretorie della prostata. Si trova nel liquido seminale (mg/dl) in cui ha la funzione di proteasi (semelogelina e fibronectina) per aumentare la mobilità degli spermatozoi. Nel siero la [C] è nell’ordine dei ng/ml. ↑ a seconda del numero delle cellule tumorali colon-retto I^ scelta: CEA (bilancio di base) II^ scelta: CA 19.9, TPA 100 www.slidetube stomaco CA 19.9, CA 72.4, CEA fegato AFP, ferritina ovaio AFP (germinale), hCG prostata PSA totale e lbero polmone NSE (microcitoma), CYFRA (spino cellulare), CEA (adenocarcinoma=origine epiteliale) Capitolo 10 MALATTIE AUTOIMMUNI 101 www.slidetube MALATTIE AUTOIMMUNI Causa: errore del sistema immunitario che produce anticorpi diretti contro Ag-self Si distinguono in - malattie sistemiche: LES, artrite reumatoide - malattie d’organo: tiroidite di Hashimoto, malattie renali, epatiti, cirrosi biliare, colite ulcerosa, gastrite atrofica, piastrinopenie, anemia emolitica, malattie emorragiche (Ab contro fattori della coagulazione) MALATTIE SISTEMICHE Nelle malattie autoimmuni si ricercano gli Ab autoimmuni con tecniche immunoenzimatiche, immunofluorescenti e radioimmunologiche. Gli Ab possono avere vari bersagli: - Ab anti-nucleo (ANA o FAN) Si utilizzano vetrini con cellule a cui viene aggiunto il siero del pz. Se ci sono Ab antinucleo questi si legano alle cellule. Si aggiunge un Ab anti-Ig marcato con fluoro cromo. Osservare con microscopio a fluorescenza. ENA: sottoclasse degli Ab anti-nucleo. Ab diretti contro strutture nucleari estraibili con fisiologica (ribinucleoprotine)→ pattern centromerico. Per identificare gli ENA si usa CRITHIDIA LUCILIAE, un parassita dotato di un nucleo e di un flagello con DNA a doppia elica e presenza di DNA a singola elica nel nucleo. - Ab contro nucleoli (RNA, non DNA) - Ab anti topo-isomerasi - Ab anti-mitocondriali Dosare poi il titolo anticorpale 102 www.slidetube - LES: - ANA anti ds-DNA (double strand), tranne nel caso di LES farmacologico → (Ab anti-istone) - ENA - SCLEROSI SITEMICA: - ANA - ENA - Ab anti-topoisomerasi I - ARTRITE REUMATOIDE: - Ab antinucleo negativi - IgM anti-IgG = fattore reumatoide (FR) NON SPECIFICO - Ab anti-peptide citrulli nato - SCLERODERMIA CUTANEA: - Ab anti SCL-70 - Ab anti-actina - Ab anti-mitocondriali MALATTIE d’ORGANO - TIROIDITE: - Ab anti-tireoglobulina - Ab anti-perossidasi (da richiedere per diagnosi e monitoraggio) - IPERTIROIDISMO AUTOIMMUNE (morbo di Graves): - Ab anti recettore del TSH (si legano al recettore e lo attivano) - MORBO CELIACO: - IgA anti-glutine → NON SPECIFICI Per diagnosi ricercare: - Ab anti-transglutaminasi - Ab anti-endomisio - DIABETE TIPO 1: - Ab anti-isole - Ab anti-recettore insulina 103 www.slidetube Capitolo 11 EMOGLOBINOPATIE 104 www.slidetube EMOGLOBINOPATIE Le emoglobinopatie si distinguono in qualitative e quantitative. - QUANTITATIVE: riduzione dell’espressione di una o più catene globiniche → TALASSEMIA - QUALITATIVA: derivano dall’alterazione strutturale della molecola che causa falcizzazione (HbS), alterazione dell’affinità per O2, ecc… Struttura dell’Hb L’Hb è una proteina tetramerica contenente 4 gruppi eme, uno per ogni subunità. HbA1 (normale, dell’adulto) contiene 2 tipi di globina: 2 catene α e 2 catene β. L’eme, costituito dalla proto porfirina IX e da Fe2+, è situato in una tasca costituita a sua volta da Leucina, isoleucina e altri amminoacidi idrofobi che impediscono l’ingresso di H2O e l’ossidazione del ferro. Il gruppo eme è legato alla globina da 2 residui di istidina distale e prossimale. Nell’adulto sono presenti, in condizioni normali 3 tipi di Hb: - 97% HbA1 costituita da 2 catene α e 2 catene β - 2,5-3,2% HbA2 costituita da 2 catene α e 2 catene δ - HbF in una minima quota costituita da 2 catene α e 2 catene γ Il 5-6% di HbA1 è glicata (HbA1c). La catena β è più soggetta a mutazioni, rispetto alla catena α e ciò trova spiegazione a livello evoluzionistico: l’Hb nei primi organismi viventi si è differenziata dalla mioglobina e si è specializzata nel trasporto di O2 nella forma tetramerica α2β2. Da questa forma si è costituita HbF (α2γ2) che ha la caratteristica di avere un’affinità > per O2 e infine, per ulteriore evoluzione si è costituita la catena δ. La α è rimasta invariata e quindi è più facile riscontrare una patologia a carico della β. PRINCIPALI VARIANTI dell’Hb - HbS: dovuta alla sostituzione nella catena β di un Glu con Val. Ciò comporta l’assunzione della caratteristica forma a falce dell’eritrocita a basse concentrazioni di O2 e polimerizzazione in lunghe file 105 www.slidetube - HbE: dovuta alla sostituzione nella catena β di Glu con Lys. HbE è sintetizzata in quantità ridotta perché la sostituzione crea un sito di splicing alternativo in un esone. Pertanto l’individuo con HbE mostra caratteristiche della talassemia β - HbC: dovuta alla sostituzione nella catena β di un Glu con una Lys. I soggetti eterozigoti sono asintomatici, gli individui omozigoti presentano emolisi associata a splenomegalia, litiasi biliare - HbM: divuta alla sostituzione di Ist con Tyr. L’Hb non è più in grado di legarsi all’O2 e si stabilizza in metaemoglobina (Fe3+). Porta a cianosi TALASSEMIA E’ causata da una mutazione genetica delle catene globiniche dell’Hb. I difetti possono essere molti e a vari livelli della molecola. Principalmente comunque le talassemie sono suddivise in: - β-talassemia o anemia mediterranea. è la più frequente. L’individuo eterozigote è asintomatico, ma possiede globuli rossi in numero maggiore, più piccoli (MICROCITEMIA) e poveri di Hb. Gli omozigoti hanno microcitemia, anemia e povertà di Hb più marcate. Devono subire trasfusioni ogni 15-20 gg che portano però ad un accumulo di Fe in organi quali cuore, gh. endocrine, fegato compromettendone la funzione. Necessario somministrare Fe-chelanti. La malattia può avere diversi gradi di gravità: la sintesi della catena β può essere annullata o rallentata. In entrambi i casi per compenso ↑ la produzione di HbA2 e HbF. Nella forma più lieve aumenta solo HbA2 (da 3 a 5%), nelle forme più gravi anche HbF. - α-talassemia. causata dalla sintesi rallentata o annullata della catena α. Per compenso si ha nell’adulto un accumulo di catene β e nel feto di catene γ. Ciò porta alla formazione di emoglobine patologiche: - HbH → β4 nell’adulto - Hb di BART → γ4 nel bambino Se γ4 e β4 > 20% INCOMPATIBILI con la VITA METODI di STUDIO Punto di partenza dell’indagine di laboratorio: ANEMIA riscontrata con un emocromo. L’indagine comincia con l’ELETTROFORESI dell’Hb. Viene eseguita su emolisati ottenuti lavando i GR con fisiologica e emolizzandoli. Vengono poi sottoposti a centrifuga per allontanare le membrane. Per visualizzare le emoglobinopatie correttamente è utile eseguire 2 elettroforesi una a pH basico e una a pH acido. ……………………………… ……………………………….. 106 www.slidetube ……………………………….. ……………………………. ………………………………. - CROMATOGRAFIA HPCL (cromatografia liquida ad alta selezione) Esame che permette di valutare le differenze aa delle varianti di Hb in aggiunta a elettroforesi e FINGER PRINTING. E’ un sistema che permette il confronto di frammenti di Hb tagliati a livello di Arg (emolisato + tripsina) ANEMIE EMOLITICHE Sono causate da Ab diretti contro gli Ag eritrocitari (malattia autoimmune). Varie le cause (es. incompatibilità Rh tra medre e feto, farmaci …). Per la diagnosi esistono test diretti e indiretti per evidenziare Ab sul globulo rosso. ENZIMOPATIE ERITROCITARIE Gli eritrociti non possiedono mitocondri; perciò le principali vie metaboliche sono: - glicolisi anaerobia 90% - via del pentoso-fosfato 10% - produzione del 2,3-BPG E’ possibile dosare l’attività enzimatica degli enzimi coinvolti nel metabolismo eritrocitario. I principali sono: - G6PD = glucoso-6-fosfato-deidrogenasi: enzima eritrocitario chiave della via dei pentoso-fosfati. L’enzima riduce un NADP a NADPH, che la cellula usa per ridurre Fe3+ a Fe2+ (enzima METAHb REDUTTASI). Se non funzione G6PD il soggetto mostra particolare sensibilità a sostanze e farmaci che hanno potenziale ossidativo (es. FAVISMO → anemia emolitica dopo l’ingestione di legumi; malattia X-linked). L’attività della G6PD è valutata basandosi sulla riduzione del NADP in un emolisato con aggiunta di G6PD - PK = piruvato chinasi (ENZIMA GLICOLITICO) Catalizza la reazione: fosfoenolpiruvato → piruvato + ATP La cellula non riesce a produrre sufficiente ATP e muore PORFIRIE PORFIRIA: malattia di origine genetica che interessa il metabolismo delle porfirine PORFIRINURIA: alterazione biometabolica associata con un’escrezione anomala di sostanze tetrapirroliche, ma indotta da altre condizioni (es. intossicazione da Pb) METABOLISMO dell’EME SuccinilCoA + glicina → ac. δ-aminolevulanico (ALA) → porfobilinogeno (PBG) → →protoporfirina IX e copro porfirina 107 www.slidetube Le porfirie possono essere di tipo eritropoietico o epatico, a seconda che l’alterazione metabolica risieda nel midollo osseo o nel fegato. Sono dovute a difetti parziali di attività enzimatiche che comportano l’accumulo di uno o più metaboliti (ALA e porfobilinogeno) nei tessuti e la loro escrezione con le urine. La porfiria più diffusa è di tipo epatico (PORFIRIA ACURA INTERMITTENTE) caratterizzata da fotosensibilità (che porta a fotodermatosi), anemia, coliche addominali intervallate da periodi di benessere. L’alterazione biochimica è il deficit della uroporfirinogeno sintasi che induce l’ALA sintetasi con accumulo di ALA e porfobilinogeno. Dosare le porfirine urinarie. Avvolgere il contenitore in un panno perché le porfirine sono sensibili alla luce. - 5-10 mg/die valori normali PORFIRINURIE Sono le malattie in cui un’aumentata escrezione di porfirine nelle urine non costituisce la manifestazione più saliente della patologia. In questi casi il dosaggio delle porfirine urinarie è utile per la diagnosi. - Avvelenamento da Pb: inibisce alcuni enzimi del metabolismo dell’eme. www.slidetube.it 108
Scaricare