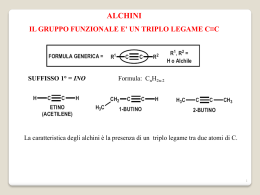
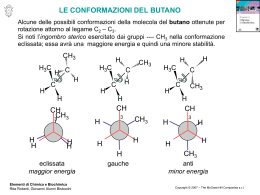
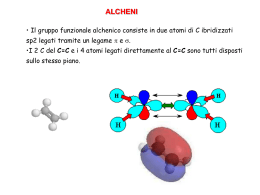
Comuni Composti Organici e Gruppi Funzionali Classi di Composti Struttura Generale Gruppo Funzionale alcani R H nessuno alogenuri alchilici R X X = F, Cl, Br, o I alcheni alchini R CH R R´ CH C R´ C Esempio CH3CH2CH2CH3 butano CH3CH2CH2Cl 1-cloropropano doppio legame carbonio–carbonio CH3CH2 triplo legame carbonio–carbonio CH3 CH CH2 C CH3 1-butene C 2-butino H composti aromatici H H anello benzenico, anche scritto come H H H benzene alcoli R OH gruppo ossidrile fenoli Ar OH gruppo ossidrile su un anello aromatico tioli R SH gruppo sulfidrilico eteri R O R´ ossigeno tra due alchili tioeteri R S R´ zolfo tra due alchili CH3CH2 OH fenolo SH CH3 metantiolo CH3CH2 O CH2CH3 CH3 S CH3 dietil etere dimetil solfuro O O epossidi C OH etanolo etere in un anello a tre termini C 1,2-epossicicloesano O O chetoni R C R´ CH3 gruppo carbonilico C acetone O aldeidi R C O H gruppo carbonilico CH3CH2 R C C propanale O acidi carbossilici CH3 H O OH CH3 gruppo carbossilico C OH acido acetico O esteri R C O O R´ gruppo carboalcossi CH3 C ammine nitrili nitroalcani R R R R C NH2 NH2 C CH2CH3 O O ammidi O acetato di etile N NO2 gruppo carbossammidico H C N(CH3)2 N,N-dimetilformammide gruppo amminico gruppo ciano gruppo nitro CH3CH2 NH2 etilammina CH3CH2 C propionitrile CH3CH2 N NO2 nitroetano Gruppi Comuni in Chimica Organica Abbreviazione Significato Ac acetile Reagenti e Solventi Comuni Struttura Abbreviazione Struttura O O CH3 allile H2C C CH R CH2 R O Boc terz-butilossicarbonile Bn benzile Bu i-Bu butile isobutile s-Bu sec-butile (CH3)3C Ph O C CH2 R R CH3 CH2 CH2 CH2 (CH3)2CH CH2 R CH3 CH2 CH R (CH3)3C terz-butile Ph benzoile benzilossicarbonile Et etile c-Es cicloesile Me metile Ph fenile Pr propile i-Pr R Sia isopropile alchile isoamile Ph C CH3 idruro di diisobutilalluminio 1,2-dimetossietano diglyme bis(2-metossietil) etere DMF C CH2 R C N C N O (CH3 CH2CH2 O R R H C N (CH3)2 DMSO dimetilsolfossido CH3 EtOH EtO− etanolo CH3CH2OH CH CH O− Et2O dietil etere LAH idruro di litio e alluminio LiAlH4 LDA diisopropilammide di litio [(CH3)2CH]2N− Li+ ione etossido S 3 CH3CH2 CH3 2 O O (i-Pr)3Si Ts para-toluensolfonile, “tosile” CH3 R MCPBA acido-metacloroperbenzoico MeOH MeO− metanolo ione metossido MVK metil vinil chetone C O CH3OH O− CH3 CH3 C CH CH2 O O R NBS N-bromosuccinimmide PCC clorocromato di piridinio Py o Pyr piridina t-BuOH alcol terz-butilico t-BuOK terz-butossido di potassio THF tetraidrofurano TMS tetrametilsilano N H R Non tutte queste abbreviazioni sono usate in questo testo, ma possono comunque essere utili come riferimento. Br O O C O Cl R S H 2C CH2CH3 O tetraidropiranile vinile O O CH3 CH2 CH2 R (CH3)2CH R generica (CH3)2CH C H R triisopropilsilil CH3 CH2CH2)2O R TIPS O AcO OAc I OAc O periodinano di Dess–Martin dimetilformammide CH3 [(CH3)2CHCH2]2AlH CH3 C H3 THP O O R CH3 DIBAL-H DME, “glyme” O C O R O CH2 dicicloesil carbodiimmide CH3 R O Cbz (o Z) DCC DMP O Bz anidride acetica R C H3 t-Bu Ac2O pyr CrO3 HCl N •• (CH3)3C (CH3)3C OH O− K + O (CH3)4Si H FONDAMENTI DI CHIMICA ORGANICA L.G. WADE, JR. W H I T M A N C O L L E G E I edizione italiana ridotta e rielaborata della VIII americana a cura di Orazio Taglialatela Scafati Professore Associato di Chimica Organica Dipartimento di Farmacia Università degli Studi di Napoli Federico II Authorized translation from the English language edition, entitled ORGANIC CHEMISTRY, 8th Edition by LEROY G. WADE, JR., published by Pearson Education, Inc., publishing as Prentice Hall, Copyright © 2013 All rights reserved No part of this book may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording or by any information storage retrieval system, without permission from Pearson Education, Inc. Traduzione autorizzata dall’Edizione in lingua inglese, intitolata ORGANIC CHEMISTRY, 8th Edition di LEROY G. WADE, JR., pubblicata da Pearson Education, Inc., per i tipi della Prentice Hall, Copyright © 2013 Tutti i diritti sono riservati È VIETATA PER LEGGE LA RIPRODUZIONE IN FOTOCOPIA E IN QUALSIASI ALTRA FORMA È vietato riprodurre, archiviare in un sistema di riproduzione o trasmettere sotto qualsiasi forma o con qualsiasi mezzo elettronico, meccanico, per fotocopia, registrazione o altro, qualsiasi parte di questa pubblicazione senza autorizzazione scritta dell’Editore. Ogni violazione sarà perseguita secondo le leggi civili e penali. ISBN 978-88-299-2300-7 Stampato in Italia Edizione italiana © 2014, by Piccin Nuova Libraria S.p.A., Padova www.piccin.it Traduzione italiana a cura di: Carmela Dell’Aversano Ricercatore di Chimica Organica Dipartimento di Farmacia Università degli Studi di Napoli Federico II Martino Forino Ricercatore di Chimica Organica Dipartimento di Farmacia Università degli Studi di Napoli Federico II Orazio Taglialatela Scafati Professore Associato di Chimica Organica Dipartimento di Farmacia Università degli Studi di Napoli Federico II Luciana Tartaglione Ricercatore di Chimica Organica Dipartimento di Farmacia Università degli Studi di Napoli Federico II V Prefazione all’Edizione Italiana Questo testo costituisce la prima edizione italiana tratta dal libro “Organic Chemistry” di L.G. Wade, tanto apprezzato negli Stati Uniti da essere giunto all’ottava edizione. Insieme alla casa editrice Piccin, abbiamo concordato un progetto editoriale piuttosto ambizioso, che non si limitasse alla traduzione del testo americano, ma ne costituisse una rielaborazione e soprattutto uno snellimento, da cui il titolo di “Fondamenti di Chimica Organica”. La riduzione delle pagine, circa 1300 nella versione americana e 700 nella nostra edizione, ha richiesto un lavoro attento e scrupoloso. Sono stati eliminati i tre capitoli di spettroscopia/spettrometria di massa e gli esercizi su questi argomenti presenti anche negli altri capitoli: benché molto interessanti, essi non rientrano quasi mai nei programmi dei corsi di Chimica Organica. Sono stati inoltre accorpati alcuni capitoli, eliminati alcuni esercizi troppo elaborati, raggruppati e riposizionati alcuni concetti. È stata invece mantenuta l’impostazione generale, incluso l’accurato riepilogo dei concetti di chimica generale trattati nei primi tre capitoli, imprescindibili per una comprensione corretta dei meccanismi di reazione, ai quali viene dedicato poi un intero capitolo tematico (Cap. 4). È stato inoltre mantenuto l’approccio meccanicistico per le varie reazioni e lo sforzo fatto dall’Autore di razionalizzare i diversi meccanismi, evidenziando sempre chiaramente il ruolo di nucleofili ed elettrofili. Questo impegnativo lavoro ha avuto l’obiettivo di produrre un testo più agile, ma che mantenesse i diversi pregi dell’edizione americana. I docenti e gli studenti apprezzeranno la chiarezza e la precisione dell’esposizione, completata da numerosi esempi ed illustrazioni. Un esempio del rigore del testo di Wade si può evidenziare nell’uso dei descrittori stereochimici: lo studente troverà uno stile che è meno frequentemente adoperato negli altri testi, ma che è visivamente più immediato e razionale. Un altro esempio riguarda la nomenclatura dei composti organici. Il testo segue infatti in maniera rigorosa le raccomandazioni IUPAC del 1993, secondo le quali il numero riferito alla posizione del gruppo funzionale va inserito appena prima del suffisso che lo indica (es. pentan-2-olo). In quest’ultimo caso abbiamo preferito adoperare in maniera relativamente più frequente, rispetto all’edizione americana, la nomenclatura IUPAC più tradizionale, secondo la quale il numero si può scrivere all’inizio del nome del composto (es. 2-pentanolo). L’organizzazione di questo testo lo rende ideale per i corsi di studio (CdS) in cui la Chimica Organica viene insegnata in un singolo modulo, come ad esempio i CdS triennali, o anche CdS quinquennali come quelli in Farmacia, Agraria e diversi CdS ad indirizzo bio-medico. Gli studenti di tali corsi troveranno particolarmente interessanti gli esempi e le applicazioni (“In pratica”) presi dal mondo della medicina, della biologia e della tossicologia. Per tali studenti saranno anche molto utili gli ultimi due capitoli (Capp. 20 e 21), in cui vengono illustrate le strutture e le proprietà dei composti bio-organici: carboidrati, acidi nucleici, amminoacidi, peptidi e lipidi. Ringrazio la casa editrice Piccin, che ha assistito con competenza e professionalità la preparazione di questa versione, ed il gruppo di colleghi che mi ha coadiuvato. Spero vivamente che questo testo accompagni e faciliti lo studio della Chimica Organica per tanti studenti e che dalla sua lettura risulti chiaro che esso è stato preparato per l’edizione americana, ed adattato per l’edizione italiana, da chimici organici innamorati di questa disciplina. VI Orazio Taglialatela Scafati Ai miei studenti e colleghi del Whitman College Notizie sull’Autore Leroy G. “Skip” Wade decise di diventare un chimico durante il secondo anno alla Rice University, mentre seguiva il corso di chimica organica tenuto dal Prof. Ronald M. Magid. Dopo la laurea alla Rice, conseguita nel 1969, Wade si spostò all’Università di Harvard, dove si appassionò alla ricerca sotto la guida del Prof. James D. White. Ad Harvard si occupò anche dei corsi di laboratorio di chimica organica e fu fortemente influenzato dai metodi di insegnamento dei Prof.ri Leonard K. Nash e Frank H. Westheimer. Dopo aver completato il dottorato ad Harvard nel 1974, il Dr. Wade si spostò alla facoltà di chimica della Colorado State University. Nel corso di quindici anni alla Colorado State, il Dr. Wade ha insegnato chimica organica a migliaia di studenti formandoli per le loro carriere nelle aree della biologia, chimica, medicina umana e veterinaria e scienze ambientali. È stato autore di diversi lavori scientifici in chimica organica e in didattica chimica, oltre ad undici libri riportanti le ricerche più aggiornate nel campo della sintesi organica. Dal 1989, il Dr. Wade è diventato professore di chimica al Whitman College, dove insegna chimica organica e segue ricerche nel campo della sintesi organica e della chimica forense. Nel 1993, il Dr. Wade ha ricevuto il Premio A. E. Lange per l’Eccellenza nell’Insegnamento Scientifico al Whitman. Gli interessi del Dr. Wade nella scienza forense l’hanno portato a testimoniare come esperto in diversi processi riguardanti l’uso di armi o stupefacenti, ed ha anche lavorato come istruttore e consulente delle forze di polizia per questi aspetti. Inoltre, gli piace molto riparare e restaurare vecchi violini, cosa che ha fatto in maniera professionale per molti anni. VII Sommario 1 Introduzione 1 2Struttura e Proprietà delle Molecole Organiche 29 3Struttura e Conformazione degli Alcani 57 4 Lo Studio delle Reazioni Chimiche 87 5 Stereochimica 108 6Alogenuri Alchilici: Sostituzione Nucleofila ed Eliminazione 139 7 Struttura e Sintesi degli Alcheni 190 8 Reazioni degli Alcheni 215 9 Alchini 254 10 Alcoli 273 11 Eteri, Epossidi e Solfuri 329 12 La Chimica dei Dieni 353 13 Composti Aromatici 378 14 Reattività dei Composti Aromatici 404 15 Aldeidi e Chetoni 446 16 Ammine 485 17 Acidi Carbossilici 528 18 Derivati degli Acidi Carbossilici 552 19Alfa Sostituzioni e Condensazioni dei Composti Carbonilici 589 VIII 20 Carboidrati ed Acidi Nucleici 630 21 Amminoacidi, Peptidi e Lipidi 661 Risposte ad Alcuni Problemi 691 Indice Analitico 695 Indice Generale 1 Introduzione 1 1-1 Le Origini della Chimica Organica 1 1-2 Principi della Struttura Atomica 3 1-3 Formazione del Legame: la Regola dell’Ottetto 5 1-4 Strutture di Lewis 6 1-5 Legami Multipli 7 Riepilogo: Legami e Coppie Solitarie di Alcuni Atomi 8 1-6 Elettronegatività e Polarità dei Legami 8 1-7 Cariche Formali 9 Riepilogo: Legami Comunemente Presenti nei Composti Organici 10 1-8 Risonanza 11 1-9 Formule Strutturali 14 1-10 Acidi e Basi di Arrhenius 16 1-11 Acidi e Basi di Brønsted-Lowry 17 1-12 Acidi e Basi di Lewis 23 Glossario 25 Problemi di Riepilogo 26 Struttura e Proprietà delle Molecole Organiche 29 2 2-1 Proprietà Ondulatorie degli Elettroni 29 2-2 Orbitali Molecolari 30 2-3Legame p 32 2-4 Ibridazione e Forme Molecolari 33 2-5 Come Rappresentare Molecole Tridimensionali 36 2-6 Regole Generali di Ibridazione e Geometria 36 2-7 Rotazione dei Legami 39 2-8 Isomeria 40 2-9 Polarità di Legami e Molecole 42 2-10 Forze Intermolecolari 44 2-11 Idrocarburi 48 2-12 Composti Organici Contenenti Ossigeno 50 2-13 Composti Organici Contenenti Azoto 52 Glossario 53 Problemi di Riepilogo 55 Struttura e Conformazione degli Alcani 57 3 3-1 3-2 3-3 3-4 3-5 Classificazione degli Idrocarburi 57 Formule Molecolari degli Alcani 58 Nomenclatura degli Alcani 59 Riepilogo: Regole di Nomenclatura degli Alcani 63 Proprietà Fisiche degli Alcani 64 Reazioni degli Alcani 65 IX X Indice Generale 3-6 3-7 3-8 3-9 3-10 3-11 3-12 3-13 4Lo Studio delle Reazioni Chimiche 87 4-1 4-2 4-3 4-4 4-5 4-6 4-7 4-8 4-9 4-10 Introduzione 87 Clorurazione del Metano 87 La Reazione Radicalica a Catena 88 Costanti di Equilibrio ed Energia Libera 91 Entalpia ed Entropia 91 Cinetica di Reazione 93 Energia di Attivazione e Relazione tra Cinetica e Temperatura 94 La Cinetica della Reazione di Alogenazione 97 Il Postulato di Hammond 101 Intermedi Reattivi 102 Glossario 106 5 Stereochimica 108 5-1 5-2 5-3 5-4 5-5 5-6 5-7 5-8 5-9 5-10 5-11 Struttura e Conformazione degli Alcani 67 Conformazioni del Butano 70 Cicloalcani 72 Isomeria Cis-trans nei Cicloalcani 74 Stabilità dei Cicloalcani; Tensione d’Anello 74 Conformazioni del Cicloesano 77 Conformazioni dei Cicloesani Sostituiti 80 Molecole Bicicliche 82 Glossario 83 Problemi di Riepilogo 85 Introduzione 108 Chiralità 109 Nomenclatura (R) ed (S) degli Atomi di Carbonio Asimmetrici 114 Attività Ottica 117 Miscele Racemiche 120 Eccesso Enantiomerico e Purezza Ottica 121 Composti Chirali senza Atomi Asimmetrici 122 Proiezioni di Fischer 123 Riepilogo: Le Proiezioni di Fischer ed il Loro Uso 127 Diastereoisomeri 127 Riepilogo: Classi di Isomeri 129 Stereochimica di Molecole con Due o Più Carboni Asimmetrici 130 Risoluzione degli Enantiomeri 133 Glossario 135 Problemi di Riepilogo 137 Alogenuri Alchilici: Sostituzione Nucleofila ed Eliminazione 139 6 6-1 6-2 6-3 6-4 6-5 6-6 Introduzione 139 Nomenclatura degli Alogenuri Alchilici 140 Impieghi Comuni degli Alogenuri Alchilici 142 Struttura degli Alogenuri Alchilici 143 Proprietà Fisiche degli Alogenuri Alchilici 143 Preparazione degli Alogenuri Alchilici 145 Indice Generale XI Riepilogo: Metodi di Preparazione degli Alogenuri Alchilici 146 6-7 Reazioni degli Alogenuri Alchilici: Sostituzione ed Eliminazione 147 6-8 Sostituzioni Nucleofile di Secondo Ordine: le Reazioni SN2 148 Meccanismo Chiave 6-1: La reazione SN2 149 Riepilogo: Reazioni SN2 di Alogenuri Alchilici 150 6-9 Fattori che Influenzano le Reazioni SN2: Forza del Nucleofilo 152 Riepilogo: Nucleofilicità 153 6-10 Reattività dei Substrati nelle Reazioni SN2 156 6-11 Aspetti Stereochimici della Reazione SN2 159 Meccanismo 6-2: Inversione di Configurazione nella Reazione SN2 159 6-12 Sostituzione Nucleofila di Primo Ordine: La Reazione SN1 161 Meccanismo Chiave 6-3: La Reazione SN1 162 6-13 Aspetti Stereochimici della Reazione SN1 165 Meccanismo 6-4: Racemizzazione in Una Reazione SN1 167 6-14 Riarrangiamenti nelle Reazioni SN1 167 Meccanismo 6-5: Trasposizione di un Idruro in Una Reazione SN1 168 Meccanismo 6-6: Trasposizione di un Metile in una Reazione SN1 169 6-15 Confronto tra la Reazione SN1 e la Reazione SN2 170 Riepilogo: Sostituzioni Nucleofile 171 6-16 Eliminazione del Primo Ordine: La Reazione E1 171 Meccanismo Chiave 6-7: La Reazione E1 172 Meccanismo 6-8: Riarrangiamento in una Reazione E1 174 Riepilogo: Reazioni dei Carbocationi 174 6-17 Regioselettività della Reazione di Eliminazione: La Regola di Zaitsev 175 6-18 Eliminazione di Secondo Ordine: La Reazione E2 175 Meccanismo Chiave 6-9: La Reazione E2 176 6-19 Aspetti Stereochimici della Reazione E2 177 6-20 Confronto tra i Meccanismi delle Eliminazioni E1 e E2 179 Riepilogo: Reazioni di Eliminazione 180 Riepilogo: Reazioni di Alogenuri Alchilici 183 Glossario 185 Problemi di Riepilogo 188 7 Struttura e Sintesi Degli Alcheni 190 7-1 Introduzione 190 7-2 Descrizione degli Orbitali del Doppio Legame degli Alcheni 191 7-3 Nomenclatura degli Alcheni 192 7-4 Nomenclatura degli Isomeri Cis-Trans 194 Riepilogo: Regole per Assegnare la Nomenclatura agli Alcheni 196 7-5 Stabilità degli Alcheni 197 7-6 Proprietà Fisiche degli Alcheni 200 7-7 Sintesi di Alcheni per Eliminazione da Alogenuri Alchilici 201 Meccanismo 7-1: Deidroalogenazione Mediante un Meccanismo E2 202 Meccanismo 7-2: Stereochimica della Reazione E2 203 7-8 Sintesi di Alcheni per Disidratazione di Alcoli 206 Meccanismo Chiave 7-3: Disidratazione di un Alcol Acido-Catalizzata 207 Strategie per la Risoluzione dei Problemi: Proporre meccanismi di reazione 209 XII Indice Generale Riepilogo: Metodi di Sintesi degli Alcheni 212 Glossario 213 Problemi di Riepilogo 214 8Reazioni Degli Alcheni 215 8-1 Reattività del Doppio Legame Carbonio-Carbonio 215 8-2 Addizione Elettrofila agli Alcheni 216 Meccanismo Chiave 8-1: Addizione Elettrofila ad Alcheni 217 8-3 Addizione di Alogenuri di Idrogeno (Acidi Alogenidrici) ad Alcheni 218 Meccanismo 8-2: Addizione di HX ad un Alchene 219 Meccanismo 8-3: Addizione Radicalica di HBr ad Alcheni 221 8-4 Addizione di Acqua: Idratazione degli Alcheni 224 Meccanismo 8-4: Idratazione di un Alchene Acido-Catalizzata 225 8-5 Idratazione Mediante Ossimercuriazione - Demercuriazione 226 8-6 Idroborazione degli Alcheni 227 Meccanismo 8-5: Idroborazione di un Alchene 229 8-7 Addizione di Alogeni agli Alcheni 232 Meccanismo 8-6: Addizione di Alogeni agli Alcheni 232 8-8 Formazione di Aloidrine 234 Meccanismo 8-7: Formazione di Aloidrine 234 8-9 Idrogenazione Catalitica degli Alcheni 237 8-10 Epossidazione degli Alcheni 238 Meccanismo 8-8: Epossidazione degli Alcheni 238 8-11 Apertura degli Epossidi Acido-Catalizzata 239 Meccanismo 8-9: Apertura degli Epossidi Acido-Catalizzata 240 8-12 Di-ossidrilazione Sin di Alcheni 241 8-13 Scissione Ossidativa di Alcheni 242 8-14 Metatesi delle Olefine 244 Strategie per la Risoluzione dei Problemi: Sintesi Organiche 246 Riepilogo: Reazioni degli Alcheni 246 Glossario 250 Problemi di Riepilogo 252 9 Alchini 254 9-1 Introduzione 254 9-2 Nomenclatura degli Alchini 255 9-3 Proprietà Fisiche degli Alchini 256 9-4 Struttura Elettronica degli Alchini 257 9-5 Acidità degli Alchini; Formazione di Ioni Acetiluro 257 9-6 Sintesi di Alchini a partire da Acetiluri 259 9-7 Sintesi di Alchini mediante Reazioni di Eliminazione 261 Riepilogo: Sintesi di Alchini 262 9-8 Reazioni di Addizione ad Alchini 262 Meccanismo 9-1: Riduzione con Metallo-Ammoniaca di un Alchino 265 Meccanismo 9-2: Tautomeria Cheto-Enolica Acido-Catalizzata 267 Meccanismo 9-3: Tautomeria Cheto-Enolica Base-Catalizzata 268 Riepilogo: Reazioni degli Alchini 269 Glossario 271 Problemi di Riepilogo 271 Indice Generale XIII 10 Alcoli 273 10-1 Introduzione 273 10-2 Struttura e Classificazione degli Alcoli 273 10-3 Nomenclatura di Alcoli e Fenoli 275 10-4 Proprietà Fisiche degli Alcoli 278 10-5 Acidità degli Alcoli e dei Fenoli 280 10-6 Sintesi degli Alcoli: Introduzione e Riepilogo 283 Riepilogo: Sintesi degli Alcoli Già Esaminate 283 10-7 Reattivi Organometallici nella Sintesi degli Alcoli 285 10-8 Addizione di Reattivi Organometallici a Composti Carbonilici 287 Meccanismo Chiave 10-1: Reazioni di Grignard 288 Riepilogo: Reazioni di Grignard 292 10-9 Riduzione del Gruppo Carbonilico: Sintesi di Alcoli Primari e Secondari 293 Meccanismo 10-2: Riduzione Mediante Idruri di un Gruppo Carbonilico 293 Riepilogo: Reazioni di LiAlH4 e NaBH4 295 Riepilogo: Sintesi degli Alcoli Mediante Addizione Nucleofila a Gruppi Carbonilici 296 10-10 Tioli (Mercaptani) 297 10-11 Reazioni degli Alcoli 299 10-12 Stati di Ossidazione degli Alcoli e Gruppi Funzionali ad Essi Correlati 299 10-13 Ossidazione degli Alcoli 301 10-14 Ossidazione Biologica degli Alcoli 304 10-15 Alcoli come Nucleofili e Elettrofili: Formazione dei Tosilati 304 Riepilogo: Reazioni SN2 degli Esteri Tosilati 306 10-16 Reazioni degli Alcoli con gli Acidi Alogenidrici 307 Meccanismo 10-3: Reazione di un Alcol Terziario con HBr (SN1) 308 Meccanismo 10-4: Reazione di un Alcol Primario con HBr (SN2) 308 10-17 Reazioni degli Alcoli con Alogenuri del Fosforo 310 Meccanismo 10-5: Reazione di Alcoli con PBr3 311 10-18 Reazioni degli Alcoli con Cloruro di Tionile 311 10-19 Reazioni di Disidratazione degli Alcoli 312 Meccanismo 10-6: Disidratazione Acido-Catalizzata di un Alcol 313 10-20 Reazioni Tipiche dei Dioli 316 Meccanismo 10-7: Il Riarrangiamento Pinacolico 317 10-21 Esterificazione degli Alcoli 318 10-22 Reazioni degli Alcossidi 319 Meccanismo Chiave 10-8: La Sintesi degli Eteri di Williamson 319 Strategie per la Risoluzione dei Problemi: Sintesi Multistadio 320 Riepilogo: Reazioni degli Alcoli 322 Glossario 324 Problemi di Riepilogo 326 11 Eteri, Epossidi e Solfuri 329 11-1 11-2 11-3 11-4 11-5 Introduzione 329 Proprietà Fisiche degli Eteri 330 Nomenclatura degli Eteri 332 La Sintesi degli Eteri di Williamson 335 Sintesi degli Eteri per Alcossimercurazione-Demercurazione 336 XIV Indice Generale 11-6 S intesi Industriale degli Eteri: Condensazione Bimolecolare degli Alcoli 337 Riepilogo: Sintesi degli Eteri 337 11-7 Scissione degli Eteri con HBr e HI 338 11-8 Tioeteri (Solfuri) 339 11-9 Sintesi degli Epossidi 341 Riepilogo: Sintesi degli Epossidi 342 11-10 Apertura Acido-Catalizzata degli Epossidi 343 Meccanismo 11-1: Apertura Acido-Catalizzata degli Epossidi in Acqua 343 Meccanismo 11-2: Apertura Acido-Catalizzata di un Epossido in soluzione alcolica 344 11-11 Apertura Base-Catalizzata degli Epossidi 346 Meccanismo 11-3: Apertura Base-Catalizzata degli Epossidi 347 11-12 Orientazione dell’Apertura d’Anello degli Epossidi 348 11-13Reazioni degli Epossidi con Reattivi di Grignard e di Organolitio 349 Riepilogo: Reazioni degli Epossidi 350 Glossario 351 Problemi di Riepilogo 352 12La Chimica dei Dieni 353 12-1 Introduzione 353 12-2 Stabilità dei Dieni 353 12-3 Orbitali Molecolari di un Sistema Coniugato 355 12-4 Cationi Allilici 359 12-5 Addizioni 1,2 e 1,4 ai Dieni Coniugati 359 Meccanismo 12-1: Addizione 1,2 e 1,4 ad un Diene Coniugato 360 12-6 Controllo Cinetico e Termodinamico nell’Addizione di HBr all’1,3-butadiene 361 12-7 Radicali Allilici 362 Meccanismo 12-2: Bromurazione Radicalica Allilica 362 12-8 La Reazione di Diels-Alder 364 Meccanismo Chiave 12-2: La Reazione di Diels-Alder 365 12-9 La Diels-Alder come Esempio di Reazione Periciclica 371 Glossario 374 Problemi di Riepilogo 376 13Composti Aromatici 378 13-1 13-2 13-3 13-4 13-5 13-6 13-7 13-8 13-9 13-10 13-11 Introduzione: La Scoperta del Benzene 378 La Struttura e le Proprietà del Benzene 378 Gli Orbitali Molecolari del Benzene 382 Il Modello degli Orbitali Molecolari del Ciclobutadiene 385 Composti Aromatici, Antiaromatici, e Non Aromatici 386 La Regola di Hückel 387 Ioni Aromatici 389 Composti Eterociclici Aromatici 391 Idrocarburi Policiclici Aromatici 394 Composti Eterociclici Fusi 397 Nomenclatura dei Derivati del Benzene 397 Indice Generale XV 13-12 Proprietà Fisiche di Benzene e Derivati 399 Glossario 400 Problemi di Riepilogo 401 14Reattività dei Composti Aromatici 404 14-1 Sostituzione Elettrofila Aromatica 404 Meccanismo Chiave 14-1: Sostituzione Elettrofila Aromatica 405 14-2 Alogenazione del Benzene 405 Meccanismo 14-2: Bromurazione del Benzene 405 14-3 Nitrazione del Benzene 407 Meccanismo 14-3: Nitrazione del Benzene 408 14-4 Solfonazione del Benzene 409 Meccanismo 14-4: Solfonazione del Benzene 409 14-5 Nitrazione del Toluene: L’effetto dei sostituenti alchilici 410 14-6 Sostituenti Attivanti, Orto, Para-Orientanti 412 Riepilogo: Attivanti, Orto, Para-Orientanti 415 14-7 Sostituenti Disattivanti, Meta-Orientanti 415 Riepilogo: Disattivati, Meta-Orientanti 418 14-8 Alogeni: Disattivanti, ma Orto, Para-Orientanti 419 Riepilogo: Effetti Orientanti dei Sostituenti 420 14-9 Effetto di Sostituenti Multipli sulla Sostituzione Elettrofila Aromatica 420 14-10 Alchilazione di Friedel-Crafts 422 Meccanismo 14-5: Alchilazione di Friedel-Crafts 423 14-11 Acilazione di Friedel-Crafts 426 Meccanismo 14-6: Acilazione di Friedel-Crafts 427 Riepilogo: Confronto tra Alchilazione e Acilazione di Friedel-Crafts 428 14-12 Sostituzione Nucleofila Aromatica 429 Meccanismo 14-7: Sostituzione Nucleofila Aromatica (Addizione-Eliminazione) 430 Meccanismo 14-8: Sostituzione Nucleofila Aromatica (Meccanismo via Benzino) 432 14-13 Sostituzioni Aromatiche Mediate da Reattivi Organometallici 433 14-14 Reazioni in Catena Laterale dei Derivati del Benzene 435 14-15 Reazioni dei Fenoli 438 Riepilogo: Reazioni dei Composti Aromatici 440 Glossario 442 Problemi di Riepilogo 443 15 Aldeidi e Chetoni 446 15-1 15-2 15-3 15-4 15-5 15-6 15-7 15-8 Composti Carbonilici 446 Struttura del Gruppo Carbonilico 447 Nomenclatura di Aldeidi e Chetoni 448 Proprietà Fisiche di Aldeidi e Chetoni 450 Riepilogo dei Metodi di Sintesi di Aldeidi e Chetoni 452 Sintesi di Aldeidi e Chetoni dai Nitrili 455 Sintesi di Aldeidi e Chetoni dai Cloruri Acilici e dagli Esteri 456 Riepilogo: Sintesi di Aldeidi e Chetoni 457 Reazioni di Aldeidi e Chetoni: Introduzione all’Addizione Nucleofila 460 XVI Indice Generale Meccanismo Chiave 15-1: Addizioni Nucleofile al Gruppo Carbonilico 461 15-9 La Reazione di Wittig 462 Meccanismo 15-2: La Reazione di Wittig 463 15-10 Idratazione di Aldeidi e Chetoni 465 Meccanismo 15-3: Idratazione di Aldeidi e Chetoni 465 15-11 Formazione delle Cianidrine 466 Meccanismo 15-4: Formazione di Cianidrine 467 15-12 Formazione delle Immine 468 Meccanismo Chiave 15-5: Formazione di Immine 468 15-13 Condensazione con Idrossilammina e Idrazine 470 Riepilogo: Condensazione delle Ammine con Aldeidi e Chetoni 471 15-14 Formazione degli Acetali 471 Meccanismo Chiave 15-6: Formazione degli Acetali 473 15-15 Ossidazione delle Aldeidi 476 15-16 Riduzione di Aldeidi e Chetoni 476 Meccanismo 15-7: Riduzione di Wolff-Kishner 478 Riepilogo: Reazioni di Aldeidi e Chetoni 479 Glossario 481 Problemi di Riepilogo 483 16 Ammine 485 16-1 Introduzione 485 16-2 Nomenclatura delle Ammine 486 16-3 Struttura delle Ammine 488 16-4 Proprietà Fisiche delle Ammine 490 16-5 Basicità delle Ammine 491 16-6 Effetti sulla Basicità delle Ammine 492 16-7 Sali di Ammonio 495 16-8 Sostituzione Aromatica di Arilammine e Piridina 497 Meccanismo 16-1: Sostituzione Elettrofila Aromatica della Piridina 498 Meccanismo 16-2: Sostituzione Nucleofila Aromatica della Piridina 499 16-9 Alchilazione di Ammine con Alogenuri Alchilici 500 16-10 Acilazione di Ammine Mediante Cloruri Acilici 501 Meccanismo 16-3: Acilazione di un’Ammina ad Opera di un Cloruro Acilico 501 16-11 Ammine come Gruppi Uscenti: L’Eliminazione di Hofmann 502 Meccanismo 16-4: Eliminazione di Hofmann 503 16-12 Ossidazione delle Ammine; L’Eliminazione di Cope 505 Meccanismo 16-5: Eliminazione di Cope di un Ammino Ossido 506 16-13 Reazioni delle Ammine con Acido Nitroso 507 Meccanismo 16-6: Diazotazione di un’ammina 508 16-14 Reazioni dei Sali di Arendiazonio 509 Riepilogo: Reazioni delle Ammine 512 16-15 Sintesi delle Ammine Mediante Amminazione Riduttiva 515 16-16 Sintesi delle Ammine attraverso Acilazione-Riduzione 517 16-17 Sintesi Limitate alle Ammine Primarie 518 Riepilogo: Sintesi di Ammine 521 Glossario 523 Problemi di Riepilogo 525 Indice Generale XVII 17 Acidi Carbossilici 528 Introduzione 528 Nomenclatura degli Acidi Carbossilici 529 Struttura e Proprietà Fisiche degli Acidi Dicarbossilici 531 Acidità degli Acidi Carbossilici 532 Sali degli Acidi Carbossilici 536 Sintesi degli Acidi Carbossilici 537 Riepilogo: Sintesi di Acidi Carbossili 539 Reazioni di Acidi Carbossilici e Derivati; Sostituzione Nucleofila Acilica 540 Meccanismo 17-1: Sostituzione Nucleofila Acilica di un Estere in Catalisi Basica 540 17-8 Condensazione di Acidi Carbossilici con Alcol: Esterificazione di Fischer 541 Meccanismo Chiave 17-2: Esterificazione di Fischer 541 17-9 Esterificazione con Diazometano 543 Meccanismo 17-3: Esterificazione con Diazometano 544 17-10 Condensazioni di Acidi Carbossilici con Ammine: Sintesi Diretta di Ammidi 544 17-11 Riduzione di Acidi Carbossilici 544 17-12 Sintesi ed Utilizzo dei Cloruri Acilici 546 Riepilogo: Reazioni degli Acidi Carbossilici 548 Glossario 549 Problemi di Riepilogo 550 17-1 17-2 17-3 17-4 17-5 17-6 17-7 18Derivati degli Acidi Carbossilici 552 18-1 Introduzione 552 18-2 Struttura e Nomenclatura dei Derivati degli Acidi 553 18-3Punti di Ebollizione e Punti di Fusione dei Derivati degli Acidi 558 18-4 Interconversione dei Derivati degli Acidi per Sostituzione Nucleofila Acilica 559 Meccanismo Chiave 18-1: Meccanismo di AddizioneEliminazione della Sostituzione Nucleofila Acilica 560 Meccanismo 18-2: Trasformazione di un Cloruro Acilico in un’Anidride 562 Meccanismo 18-3: Trasformazione di un Cloruro Acilico in un Estere 562 Meccanismo 18-4: Trasformazione di un Cloruro Acilico in Ammide 563 Meccanismo 18-5: Trasformazione di un Estere in un’Ammide (Ammoniolisi di un Estere) 563 MECCANSIMO 18-6: Transesterificazione 565 18-5 Idrolisi dei Derivati degli Acidi Carbossilici 566 Meccanismo 18-7: Saponificazione di un Estere 566 Meccanismo 18-8: Idrolisi Acida di un’Ammide 568 Meccanismo 18-9: Idrolisi Base-Catalizzata di un Nitrile 569 18-6 Riduzione dei Derivati degli Acidi 569 Meccanismo 18-10: Riduzione di un Estere Mediata da Idruro 570 Meccanismo 18-11: Riduzione di un’Ammide ad Ammina 571 18-7 Reazioni dei Derivati degli Acidi con Reagenti Organometallici 572 XVIII Indice Generale Meccanismo 18-12: Reazione di un Estere con due Equivalenti di un Reattivo di Grignard 572 18-8 Riepilogo della Chimica dei Cloruri Acilici 573 18-9 Riepilogo della Chimica delle Anidridi 575 18-10 Riepilogo della Chimica degli Esteri 576 18-11 Riepilogo della Chimica delle Ammidi 579 18-12 Riepilogo della Chimica dei Nitrili 581 18-13 Tioesteri 583 18-14 Esteri ed Ammidi dell’Acido Carbonico 584 Glossario 585 Problemi di Riepilogo 586 Alfa Sostituzioni e Condensazioni dei Composti Carbonilici 589 19 19-1 Introduzione 589 Meccanismo 19-1: Alfa Sostituzione 589 Meccanismo 19-2: Addizione di un Enolato ad Aldeidi e Chetoni (Reazione di Condensazione) 590 Meccanismo 19-3: Sostituzione di un Enolato su un Estere (Condensazione) 590 19-2 Enoli e Ioni Enolato 590 Meccanismo 19-4: Tautomeria Cheto-Enolica Base-Catalizzata 591 Meccanismo 19-5: Tautomeria Cheto-Enolica Acido-Catalizzata 591 19-3 Alchilazione degli Ioni Enolato 594 19-4 Formazione e Alchilazione delle Enammine 595 19-5 Alfa Alogenazione di Chetoni e Acidi Carbossilici 597 Meccanismo 19-6: Alogenazione Base-Promossa 597 Meccanismo 19-7: Alfa Alogenazione Acido-Catalizzata 598 19-6 La Condensazione Aldolica di Aldeidi e Chetoni 599 Meccanismo Chiave 19-8: Condensazione Aldolica Base-Catalizzata 599 Meccanismo 19-9: Condensazione Aldolica Acido-Catalizzata 602 19-7 Disidratazione degli Aldoli 602 Meccanismo Chiave 19-10: Disidratazione Base-Catalizzata di un Aldolo 603 19-8 Condensazioni Aldoliche Incrociate 603 Strategie per la Risoluzione dei Problemi: Proporre un Meccanismo di Reazione 604 19-9 Pianificare una Sintesi Mediante Condensazione Aldolica 606 19-10 La Condensazione di Claisen 607 Meccanismo Chiave 19-11: La Condensazione di Claisen 607 19-11 La condensazione di Dieckmann: Una Ciclizzazione di Claisen 610 19-12 Condensazione di Claisen Incrociata 611 19-13 Reazioni dei Composti b-Dicarbonilici 614 19-14 La Sintesi Malonica 615 19-15 La Sintesi Acetoacetica 618 19-16 Addizioni Coniugate: La Reazione di Michael 620 Meccanismo 19-12: Addizione-1,2 e Addizione-1,4 (Addizione Coniugata) 621 19-17 L’Anellazione di Robinson 623 Indice Generale XIX Riepilogo: Addizione di Enolati e Condensazioni 625 Glossario 626 Problemi di Riepilogo 627 20Carboidrati ed Acidi Nucleici 630 20-1 Introduzione 630 20-2 Classificazione dei Carboidrati 631 20-3 Monosaccaridi 632 20-4 Diastereoisomeri Eritro e Treo 635 20-5 Epimeri 636 20-6 Strutture Cicliche dei Monosaccaridi 636 Meccanismo 20-1: Formazione di un Emiacetale Ciclico 637 20-7 Anomeri dei Monosaccaridi; la Mutarotazione 640 20-8 Riduzione dei Monosaccaridi 642 20-9 Ossidazione dei Monosaccaridi; Zuccheri Riducenti 643 20-10 Zuccheri Non Riducenti: Formazione di Glicosidi 644 20-11 I Disaccaridi 646 20-12 I Polisaccaridi 649 20-13 Gli Acidi Nucleici: Introduzione 651 20-14 Ribonucleosidi e Ribonucleotidi 652 20-15 Le Strutture dell’RNA e del DNA 654 Glossario 658 21 Amminoacidi, Peptidi e Lipidi 661 21-1 21-2 21-3 21-4 21-5 21-6 21-7 21-8 21-9 21-10 21-11 21-12 21-13 21-14 Amminoacidi, Peptidi e Proteine 661 Struttura e Stereochimica degli a-Amminoacidi 662 Proprietà Acido-Base degli Amminoacidi 666 Punti Isoelettrici ed Elettroforesi 668 Struttura e Nomenclatura di Peptidi e Proteine 669 Classificazione delle Proteine 673 Livelli di Organizzazione Strutturale delle Proteine 674 I Lipidi 676 Le cere 677 Trigliceridi 678 Saponificazione dei Grassi e degli Oli; Saponi e Detergenti 681 Fosfolipidi 684 Steroidi 684 Terpeni 687 Glossario 689 Risposte ad Alcuni Problemi 691 Indice Analitico 695 XX Indice Generale sezioni dei meccanismi chiave CAPITOLO 6 La reazione SN2 149 La Reazione SN1 162 La Reazione E1 172 La Reazione E2 176 CAPITOLO 7 Disidratazione di un Alcol Acido-Catalizzata 207 CAPITOLO 8 Addizione Elettrofila ad Alcheni 217 CAPITOLO 10 Reazioni di Grignard 288 La Sintesi degli Eteri di Williamson 319 CAPITOLO 12 La Reazione di Diels-Alder 365 CAPITOLO 14 Sostituzione Elettrofila Aromatica 405 CAPITOLO 15 Addizioni Nucleofile al Gruppo Carbonilico 461 Formazione di Immine 468 Formazione degli Acetali 473 CAPITOLO 17 Esterificazione di Fischer 541 CAPITOLO 18Meccanismo di Addizione-Eliminazione della Sostituzione Nucleofila Acilica 560 CAPITOLO 19 Condensazione Aldolica Base-Catalizzata 599 Disidratazione Base-Catalizzata di un Aldolo 603 La Condensazione di Claisen 607 sezioni dei meccanismi CAPITOLO 6 Inversione di Configurazione nella Reazione SN2 159 Racemizzazione in Una Reazione SN1 167 Trasposizione di un Idruro in Una Reazione SN1 168 Trasposizione di un Metile in una Reazione SN1 169 Riarrangiamento in una Reazione E1 174 CAPITOLO 7 Deidroalogenazione Mediante un Meccanismo E2 202 Stereochimica della Reazione E2 203 CAPITOLO 8 Addizione di HX ad un Alchene 219 Addizione Radicalica di HBr ad Alcheni 221 Idratazione di un Alchene Acido-Catalizzata 225 Idroborazione di un Alchene 229 Addizione di Alogeni agli Alcheni 232 Formazione di Aloidrine 234 Epossidazione degli Alcheni 238 Apertura degli Epossidi Acido-Catalizzata 240 CAPITOLO 9 Riduzione con Metallo-Ammoniaca di un Alchino 265 Tautomeria Cheto-Enolica Acido-Catalizzata 267 Tautomeria Cheto-Enolica Base-Catalizzata 268 CAPITOLO 10 Riduzione Mediante Idruri di un Gruppo Carbonilico 293 Reazione di un Alcol Terziario con HBr (SN1) 308 Reazione di un Alcol Primario con HBr (SN2) 308 Reazione di Alcoli con PBr3 311 Disidratazione Acido-Catalizzata di un Alcol 313 Il Riarrangiamento Pinacolico 317 CAPITOLO 11 Apertura Acido-Catalizzata degli Epossidi in Acqua 343 Apertura Acido-Catalizzata di un Epossido in soluzione alcolica 344 Apertura Base-Catalizzata degli Epossidi 347 Indice Generale XXI sezioni dei meccanismi (continua) CAPITOLO 12 Addizione 1,2 e 1,4 ad un Diene Coniugato 360 Bromurazione Radicalica Allilica 362 CAPITOLO 14 Bromurazione del Benzene 405 Nitrazione del Benzene 408 Solfonazione del Benzene 409 Alchilazione di Friedel-Crafts 423 Acilazione di Friedel-Crafts 427 Sostituzione Nucleofila Aromatica (AddizioneEliminazione) 430 Sostituzione Nucleofila Aromatica (Meccanismo via Benzino) 432 CAPITOLO 15 La Reazione di Wittig 463 Idratazione di Aldeidi e Chetoni 465 Formazione di Cianidrine 467 Riduzione di Wolff-Kishner 478 CAPITOLO 16 Sostituzione Elettrofila Aromatica della Piridina 498 Sostituzione Nucleofila Aromatica della Piridina 499 Acilazione di un’Ammina ad Opera di un Cloruro Acilico 501 Eliminazione di Hofmann 503 Eliminazione di Cope di un Ammino Ossido 506 Diazotazione di un’ammina 508 CAPITOLO 17Sostituzione Nucleofila Acilica di un Estere in Catalisi Basica 540 Esterificazione con Diazometano 544 CAPITOLO 18 Trasformazione di un Cloruro Acilico in un’Anidride 562 Trasformazione di un Cloruro Acilico in un Estere 562 Trasformazione di un Cloruro Acilico in Ammide 563 Trasformazione di un Estere in un’Ammide (Ammoniolisi di un Estere) 563 Transesterificazione 565 Saponificazione di un Estere 566 Idrolisi Acida di un’Ammide 568 Idrolisi Base-Catalizzata di un Nitrile 569 Riduzione di un Estere Mediata da Idruro 570 Riduzione di un’Ammide ad Ammina 571 Reazione di un Estere con due Equivalenti di un Reattivo di Grignard 572 CAPITOLO 19 Alfa Sostituzione 589 Addizione di un Enolato ad Aldeidi e Chetoni (Reazione di Condensazione) 590 Sostituzione di un Enolato su un Estere (Condensazione) 590 Tautomeria Cheto-Enolica Base-Catalizzata 591 Tautomeria Cheto-Enolica Acido-Catalizzata 591 Alogenazione Base-Promossa 597 Alfa Alogenazione Acido-Catalizzata 598 Condensazione Aldolica Acido-Catalizzata 602 Addizione-1,2 e Addizione-1,4 (Addizione Coniugata) 621 CAPITOLO 20 Formazione di un Emiacetale Ciclico 637 Prefazione all’Edizione Americana Per lo studente All’inizio dello studio della chimica organica, ci si può sentire sopraffatti dal numero di composti, nomi, reazioni e meccanismi con i quali ci si deve confrontare. Ci si può anche chiedere se si sarà mai capaci di imparare tutto questo materiale in un anno, o addirittura in un semestre. La più importante funzione di un libro di testo è quella di organizzare il materiale e mostrare che molto della chimica organica è costituito da pochi principi di base e molte estensioni ed applicazioni di questi principi. Se si afferrano i concetti principali e si sviluppa la capacità di utilizzarli in maniera flessibile, allora è richiesto un limitato lavoro di memorizzazione. Francamente, non ho una buona memoria ed odio memorizzare elenchi di informazioni. Anch’io non ricordo tutti i dettagli di molte delle reazioni e dei meccanismi di questo libro, ma posso arrivarci ricordando pochi principi base, come ad esempio “la disidratazione degli alcoli procede con meccanismi di tipo E1”. Nonostante ciò, nel corso dello studio sarà necessario imparare alcuni fatti e principi fondamentali che serviranno come “dizionario” di lavoro per ciascun capitolo. Da studente, mi sono scontrato con questa necessità quando ho preso una D (mediocre) al mio secondo esame di chimica organica. Pensavo che la chimica organica potesse essere come la chimica generale, dove si poteva memorizzare un paio di equazioni e cavarsela negli esami. Per esempio, nel capitolo dei gas ideali, memorizzando PV = nRT , sarei stato pronto. Quando ho provato ad adottare lo stesso approccio con la chimica organica, ho preso una D. Noi tutti dobbiamo imparare dai nostri errori, ed io ho imparato tanto in chimica organica. Nello scrivere questo libro, ho provato ad evidenziare un piccolo numero di fatti e principi importanti che bisogna imparare per risolvere i problemi. Per esempio, delle centinaia di meccanismi di reazione mostrati in questo libro, solo una ventina sono gli stadi meccanicistici fondamentali che si combinano poi in più lunghi e complicati meccanismi. Nel testo, questi meccanismi fondamentali sono evidenziati come Meccanismi Chiave per attirare l’attenzione sulla loro importanza. Quindi, non bisogna provare a memorizzare tutto di questo corso. Non funzionerebbe; bisogna capire quello che si studia e solo così lo si può applicare. Ma, allo stesso tempo, non bisogna pensare (come feci io) che si può studiare questa materia senza memorizzare niente. Bisogna leggere i capitoli, seguire con attenzione le lezioni e lavorare ai problemi. I problemi evidenzieranno bene il grado di conoscenza della materia e saranno un buon indicatore in vista dell’esame. Questa è una serie di suggerimenti che fornisco ai miei studenti all’inizio del corso: 1. Leggere il materiale del libro prima della lezione. Sapere cosa ci si aspetta e cosa c’è nel libro può aiutare a prendere meno appunti ed utilizzare il tempo per ascoltare e capire la lezione. 2.Dopo la lezione, riguardare gli appunti ed il libro e svolgere i problemi presenti nel testo. Dopo, leggere il materiale per la lezione successiva. 3.Se ci si rende conto di non aver capito qualcosa, rivolgersi immediatamente al docente. Infatti, poiché i concetti sono strettamente concatenati, non è possibile andare avanti senza aver completamente chiari gli argomenti precedenti. XXII 4. Per preparare un esame, cominciare con il rivedere ciascun capitolo insieme agli appunti e poi provare a risolvere i problemi di fine capitolo. Può anche essere utile disporre dei testi di vecchi esami scritti. Infine, molti studenti trovano utile lavorare in gruppi di studio. Prefazione all’Edizione Americana XXIII Ricordate sempre le due “regole d’oro” della chimica organica. 1. Mai rimanere indietro! Il corso procede velocemente ed è difficile raggiungerlo. 2. Risolvere molti problemi. Tutti hanno bisogno di fare pratica, ed i problemi evidenziano dove è necessario altro lavoro. Sono sempre interessato a conoscere l’opinione degli studenti sul libro. Se avete suggerimenti su come migliorare il testo, o avete trovato un errore, per favore fatemelo sapere (L.G. Wade, Whitman College, Walla Walla, WA 99362: E-mail: [email protected]). Io prendo i suggerimenti degli studenti con molta serietà e molti di questi sono presenti nel testo che state per studiare. Buona fortuna con lo studio della chimica organica. Sono certo che questo corso vi piacerà, specialmente se susciterà il vostro interesse su come i composti organici influenzano le nostre vite. Il mio obiettivo nello scrivere questo libro è stato quello di rendere questo processo un po’ più facile: costruire i concetti in maniera logica uno sull’altro, in maniera tale che essi possano fluire in maniera naturale. Anche se la vostra memoria è peggiore della mia (altamente improbabile), dovreste essere in grado di cavarvela bene in chimica organica! Per il docente Nello scrivere la prima edizione di questo testo il mio obiettivo è stato quello di produrre un testo moderno, leggibile che usasse le tecniche più efficaci per presentare i vari argomenti. Volevo un testo che presentasse la chimica organica al livello richiesto per studi superiori di chimica e biochimica, ma che allo stesso tempo porgesse il materiale in maniera tale da essere utile per tutti gli altri tipi di studenti che si avvicinano alla chimica organica. Le edizioni successive hanno esteso e raffinato questi obiettivi, con una vasta riorganizzazione e con l’aggiunta di molti nuovi aspetti. Questa ottava edizione incorpora tutti questi miglioramenti e, rispetto alla settima, presenta ulteriori revisioni nell’organizzazione, nella scrittura e nei grafici. Per esempio, per aiutare lo studente a studiare in maniera più efficace, sono stati aggiunti, all’inizio di ciascun capitolo, gli Obiettivi del capitolo. Questi riflettono gli argomenti principali e gli scopi del contenuto del capitolo. Una serie di applicazioni dei vari concetti, indicate con In pratica, riportano vari esempi di tossicologia, chimica verde, biochimica, e medicina in cui i concetti espressi trovano applicazione. I loro titoli descrittivi aiutano lo studente a capire l’importanza dell’esempio e la relazione con quanto stanno imparando nel testo. Allo stesso tempo, il contenuto del testo è stato aggiornato per garantire che questa edizione costituisca una fonte di informazioni il più aggiornata possibile. Tra i contenuti caratteristici del testo, in aggiunta alle classiche reazioni, ci sono molti argomenti che solo recentemente sono diventati di uso comune tra i chimici. La teoria degli orbitali molecolari viene spiegata abbastanza dettagliatamente ed utilizzata per razionalizzare gli effetti elettronici nei sistemi coniugati e aromatici e le reazioni pericicliche. Molte delle nuove tecniche sintetiche, come la metatesi delle olefine, sono state incluse nel testo. I meccanismi di reazione sono importanti in tutte le aree della chimica organica, ma essi presentano difficoltà per molti studenti. Spesso gli studenti cadono nella trappola di memorizzare un meccanismo senza razionalizzare perché esso proceda proprio in quel modo. Questo libro sottolinea spesso i principi usati per prevedere i meccanismi e le tecniche da utilizzare per affrontare problemi sui meccanismi, cercando di vincere la tendenza alla memorizzazione. Queste tecniche prevedono la discriminazione tra una reazione acida, basica o radicalica e l’utilizzo corretto della simbologia delle frecce ricurve per illustrare i singoli stadi. I meccanismi più importanti sono evidenziati nelle sezioni Meccanismi o Meccanismi Chiave. I vantaggi e gli svantaggi di usare l’alogenazione radicalica degli alcani per introdurre i meccanismi di reazione sono stati dibattuti per molti anni. La principale obiezione contro l’alogenazione radicalica è che non si tratta di una reazione sinteticamente utile. Ma le reazioni utili, come la sostituzione nucleofila e le addizioni elettrofile agli alcheni sono complicate dalla partecipazione del solvente e da altri effetti. L’alogenazione radicalica in fase gassosa consente una trattazione più chiara di cinetica e termodinamica, posto che i suoi svantaggi e limitazioni come reazione sintetica vengano chiaramente illustrati agli studenti. XXIV Prefazione all’Edizione Americana La sintesi organica è trattata con grande attenzione in questo testo, con discussioni di difficoltà progressivamente maggiore dei processi coinvolti nello sviluppo di una sintesi. L’analisi retrosintetica è specialmente enfatizzata e lo studente impara come muoversi all’indietro dal composto da ottenere al materiale di partenza. Per molte reazioni sintetiche vengono fornite le rese di reazione, anche se gli studenti non dovrebbero utilizzare male questi numeri. Troppo spesso gli studenti considerano la resa di una reazione come una caratteristica definita, alla stregua del punto di fusione di un composto. Nella pratica, sono molteplici i fattori che influenzano le rese dei prodotti, ed i valori presenti in letteratura per reazioni apparentemente simili spesso differiscono di E1: un fattore 2 o anche di più. Le rese Hriportate in questo libro sono valori tipici, che un bravo CH CH H CH CH H CH CH CH OH CH OH studente con una buona tecnica può ottenere. H C C CH CH H C C CH CH C C + HC CH CHla In questo libro viene utilizzata anche la più recente nomenclatura IUPAC, ma CH Br CH Br − + CH O H vecchia nomenclatura e quella comune sono comunque riportate ed utilizzate. Insegnare 172 Alogenuri Alchilici: Sostituzione Nucleofila ed Eliminazione H solo la nomenclatura IUPAC più recente potrebbe essere una scelta teoricamente giusta, E2: ma questo approccio renderebbe allo studente la vita difficileCHnei suoi studi futuri e nell’uso CH O − O H della letteratura. H CH CH H CH CH 2 3 2 3 3 2 3 3 2 3 2 3 2 3 3 3 3 3 CA PITO L O 6 3 3 2 H C C CH3 Br 3 CH2CH3 C 3 C H3C CH2CH3 Br − 6–16A Meccanismo e Cinetica della Reazione E1 L’abbreviazione E1 sta per Eliminazione monomolecolare. Il meccanismo è definito monomolecolare perché lo stato di transizione limitante la velocità di reazione coinvolge una sola specie anziché la collisione tra due specie. Lo stadio lento di una reazione E1 è lo stesso della reazione SN1: la ionizzazione monomolecolare che porta alla formazione di un carbocatione. In un secondo stadio veloce, la base strappa un protone dal carbonio adiacente al C+. Gli elettroni che inizialmente formavano il legame carbonio-idrogeno sono liberi di formare un legame p tra i due atomi di carbonio. Il meccanismo generale di una reazione E1 è descritto nel Meccanismo Chiave 6–7 Sezioni dei Meccanismi Chiave I Meccanismi Chiave racchiudono i principi meccanicistici fondamentali che ricorrono in tutto il corso. Questi sono i meccanismi che compongono molti dei meccanismi più lunghi e complessi. Queste sezioni hanno lo scopo di rinforzare la comprensione dello studente con spiegazioni dei vari stadi, illustrazione di uno specifico esempio per quel meccanismo e, spesso, con un problema o una domanda finale, così lo studente può verificare la propria comprensione. 2 Na+ −OCH3 CH3OH MeCCanisMO Chiave 6–7 La Reazione E1 La reazione E1 richiede la ionizzazione che comporta la formazione di un intermedio carbocationico come nella SN1, sicché essa segue il medesimo ordine di reattività: 3°72°771°. 6-11 159 Aspetti Stereochimici della Reazione SN2 Una base (di solito debole) deprotona il carbocatione generando un alchene. difficile. gruppi alchilici lo rendono impossibile. Anche un carbocatione solo gruppo alchilico Stadio 1:Tre Ionizzazione monomolecolare con formazione di un (stadio lento). può determinare un considerevole ingombro sterico, nel caso del gruppo + terz-butile o neopentile. + −X C C C C La Figura 6–5 illustra la reazione SN2 tra uno ione idrossido e un bromuro di etile H eX (alogenuro 1°), bromuro di isopropile (2°) bromuro di terz-butileH(3°). Il nucleofilo si avvicina facilmente all’atomo di carbonio elettrofilo bromuro di etile. Nel Stadio 2: Deprotonazione mediante una base deboledel (spesso il solvente) conbromuro formazione di un alchene (stadio veloce). di isopropile l’avvicinamento è ostacolato ma comunque possibile. Invece, l’attacco + di terz-butile è reso impossibile dall’ingombro SN2 al carbonio terziario del bromuro − C C B H + C C sterico dei tre gruppi metilici. B H p R Ob l e Ma 6-10 Tra le righe ESEMPIO: Eliminazione E1 del bromocicloesano in metanolo. Non bisogna scrivere reazioni SN2 in Disporre i seguenti composti in ordine decrescente di reattività nelle reazioni SN2 con etossicui siano alogenuri alchilici Stadio 1: Ionizzazione con conseguente formazione di un carbocatione e dello ione bromuro in uncoinvolti unico stadio lento. ) in etanolo. do di sodio (Na+ -OCH2CH 3 cloruro di metile ioduro di terz-butile bromuro di neopentile H + H ioduro di metile Br cloruroCH di etile 3OH, calore bromuro di isopropile terziari. + Br − p R Ob l 2: e Ma 6-11 strappa un protone per formare il cicloesene in uno stadio veloce. Stadio Il metanolo Per ciascuna coppia di composti, individuare quale composto è un miglior substrato nelle H reazioni SN2. + H + (a) 2-metil-1-iodopropano o ioduro di terz-butile + CH3OH2 (b) bromuro di cicloesile o 1-bromo-1-metilcicloesano H CH OH 3 H (c) 2-bromobutano o bromuro di isopropile H (d) 1-cloro-2,2-dimetilbutano o 2-clorobutano (e) 1-iodobutano o 2-iodopropano PROBLEMA: Descrivere cosa accade nello stadio 2 dell’esempio (Eliminazione E1 del bromocicloesano in metanolo) se il solvente si comporta da nucleofilo invece che da base. Sezioni dei Meccanismi Le sezioni sui Meccanismi aiutano gli studenti a capire come avvengono le reazioni focalizzando l’attenzione sui singoli stadi di ciascuna reazione. Queste sezioni sono evidenziate con un colore blu in maniera tale che gli studenti possano localizzarle facilmente mentre sfogliano le pagine del capitolo. 6–11 Come abbiamo visto, la reazione SN2 richiede l’attacco del nucleofilo dalla parte posteriore di un atomo di carbonio elettrofilo (Figura 6–6). Un atomo di carbonio può avere solo quattro orbitali di legame completamente occupati da elettroni (un ottetto), sicché il gruppo uscente deve andar via man mano che il nucleofilo si lega al carbonio. Gli elettroni del nucleofilo vanno ad inserirsi nel lobo posteriore dell’orbitale del carbonio ibrido sp3 nella sua combinazione di antilegame con l’orbitale del gruppo uscente, dal momento che l’orbitale molecolare di legame risulta già pieno. Questi elettroni nell’orbitale molecolare di antilegame contribuiscono ad indebolire il legame C ¬ Br man mano che il bromuro si allontana. Nello stato di transizione il carbonio presenta legami parziali sia con il nucleofilo che con il gruppo uscente. L’attacco da retro inverte la struttura tetraedrica del carbonio esattamente come avviene per un ombrello investito da una folata di vento (Figura 6–6). Nel prodotto di reazione, il nucleofilo assume una posizione stereochimica opposta alla posizione inizialmente occupata dal gruppo uscente. Definiamo questo processo inversione di configurazione dell’atomo di carbonio. Aspetti Stereochimici della Reazione SN2 MeCCanisMO 6–2 Inversione di Configurazione nella Reazione S 2 L’attacco da retro inverte la configurazione dell’atomo di carbonio. N − + + Nuc − C Nuc X C X Nuc X C − ESEMPiO: − + + H H HO − C CH3 CH3CH2 Br (S)-2-bromobutano HO H3C C Br CH2CH3 H HO C CH3 CH2CH3 (R)-2-butanolo Br − riorità per il carconviene prose- ello in entrambe ovare un punto usare questa rminare quale lato orità sull’altra. 4 C 3 H NH2 H3C 4 1 1 (S)-alanina naturale H C H2N CH3 COOH 3 Figura 5–15 C (R)-alanina non naturale La proiezione di Fischer utilizza una croce per rappresentare un atomo di carbonio asimmetrico. Le linee orizzontali si proiettano verso l’osservatore, mentre quelle verticali si allontanano dall’osservatore. HO COOH = CH 3 Prefazione H COOH = H all’Edizione Americana XXV HO C H HO CH3 vista da questo angolo CH3 Sezioni dei Problemi Scrivere gli enantiomeri dell’1,3-dibromobutano ed assegnare la nomenclatura (R) e (S). * CH2 CH2 CH CH3 e quelli orizzontali (che puntano avanti) rimangono orizzontali. Br SOluziOne pRO bl e M a 5-8 Il terzo atomo di carbonio dell’1,3-dibromobutano è asimmetrico. L’atomo di bromo ha la priorità più alta, il gruppo ¬CH2CH2Br ha priorità 2, il gruppo metilico ha priorità 3 e l’idrogeno priorità 4. Le seguenti immagini speculari sono scritte con l’atomo di idrogeno che punta all’indietro (si trova sulla linea a strisce) e quindi sono pronte per l’assegnazione di (R) e (S). 2 2 CH2CH2Br C* H 1 Br 3 CH3 H H3C (R) Per ciascuno dei seguenti esempi indicare la relazione che esiste tra la prima struttura e ciascuna delle tre seguenti. Le possibilità sono: stesso composto, enantiometro, isomero strutturale. (a) COOH COOH H CH2CH2Br HO OH H H H3C CH3 CH3 C* 3 (S)-acido lattico proiezione di Fischer Centinaia di problemi forniscono un’immediata opportunità allo Risolvendo il Problema 5–8 si può notare come le proiezioni di Fischer che diffestudenteriscono di rafforzare ed approfondire il composto. materiale cheruotiamo hannounastuper una rotazione di 180° sono lo stesso Quando prodi Fischerprima di 180° idi legami verticali (che verticali 116 C Apuntano P I T Oindietro) L O 5 rimangono Stereochimica diato neliezione paragrafo, andare avanti. pR O bl eMa RiS Ol tO 5-2 Br (S)-acido lattico formula prospettica CH3 HO COOH OH H COOH 1 Br (b) (S ) CH2CH3 H CH3 Br Br CH3 Br CH2CH3 (R)-2-butanolo H HO Br CH2CH3 CH2CH3 CH3 OH CH2CH3 Qui sotto è mostrata la struttura di uno dei due enantiomeri del carvone. Trovare l’atomo di carbonio asimmetrico e determinare se ha configurazione (R) o (S). CH3 H H CH3 CH3 (c) pR O bl eMa RiS Ol tO 5-3 CH2CH3 H conf ha il del ¬ tilico l’idro in se H H CH2CH3 OH CH3 Una rotazione di 180° è consentita. O COOH COOH OH = H H C OH CH Strategie per la Risoluzione dei Problemi SOluziOne 246 C API TOL O 8 Reazioni Degli Alcheni L’atomo di carbonio asimmetrico è uno dei carboni dell’anello, indicato dall’asterisco nella struttura seguente. Anche se a questo carbonio sono legati due gruppi ¬CH2, si tratta di ¬CH2 differenti. Uno è un gruppo ¬CH2CO¬ e l’altro è un gruppoSt¬CH Rate2¬CH“C. G ie pe R Dopo la Riaver S O lU ZiO N e d ei p R O b leM i assegnato le priorità ai gruppi come mostrato sotto, si può determinare che si tratta dell’enanSintesi Organiche tiomero (S). (Continua) Le Strategie per la Risoluzione dei Problemi aiutano gli studenti a frazionare la moltitudine di problemi complessi in pezzi semplici e a stabilire metodi per affrontare problemi che richiedono di proporre un meccanismo o sviluppare una sintesi in più passaggi. routare di 180° = C H COOH CH3 3 CH3 CH3 HO = HO H COOH Gli alogenuri alchilici e gli alcheni possono essere agevolmente preparati a partire da altri composti e al tempo stesso possono essere facilmente trasformati in altri gruppi funzionali. Tale versatilità rende queste due classi di composti piuttosto utili nel campo delle sintesi organiche. Gli alcheni in particolare, essendo poco costosi e disponibili in grosse quantità, trovano largo impiego nelle sintesi industriali. Per sintesi organica si intende la preparazione di un dato composto a partire da reagenti facilmente reperibili. Una sintesi può essere costituita da un’unica e semplice reazione, oppure da una sequenza più complessa di reazioni. In questo libro sono proposte diverse sintesi organiche sotto forma di problemi per gli studenti. Talvolta, la risoluzione delle sintesi proposte prevede una sola reazione; altre volte sono invece necessarie più reazioni e potrebbero esserci anche diverse soluzioni per eseguire la medesima sintesi. Nell’affrontare le cosiddette sintesi multistep (che richiedono cioè una sequenza di reazioni) è spesso utile analizzare il problema andando a ritroso. In altri termini, può essere conveniente partire dal prodotto desiderato e individuare la strategia giusta per trasformarlo via via fino ad arrivare al materiale di partenza. Tale approccio è definito analisi retrosintetica. I seguenti suggerimenti potranno essere utili nell’affrontare le sintesi organiche: 1. Non provare a tirare ad indovinare o a considerare ogni possibile reazione per trasformare il materiale di partenza nel prodotto desiderato. Prova piuttosto a partire dal prodotto desiderato e ad utilizzare un’analisi retrosintetica. 2. Nel descrivere una reazione, fai uso di equazioni semplificate, scrivendo i reagenti sopra e sotto le frecce. In tal modo, le reazioni non dovranno essere bilanciate, ma tutti i reagenti e le condizioni indispensabili al successo delle reazioni saranno descritti. A Br2, luce " B NaOH, alcol calore " C pR Scr SOl Il t rità pri die H+, H2 O " D 3. Focalizza l’attenzione sui gruppi funzionali e non utilizzare reagenti che possano interferire con i gruppi funzionali che non intendi modificare. La risoluzione di sintesi multistep a volte non è immediata, per questo è d’aiuto sia l’analisi retrosintetica che il prendere in considerazione vie sintetiche alternative. Tra le righe RiepilOGO Reazioni degli Alcheni Le sezioni denominate Tra le righe si trovano ai margini del testo ed evidenziano agli stu1. Addizioni Elettrofile denti i fatti o i concetti che possono essere più utili per risolvere i comuni tipi di problemi. Sono dei suggerimenti che normalmente ia.docenti danno ai propri studenti per aiutarli in Addizione di acidi alogenidrici vista dell’esame. C C + H X (HX = HCl, HBr, or HI) C C H X orientazione di Markovnikov (anti-Markovnikov con HBr e perossidi) Esempio Tra le righe Nell’assegnare le priorità per il carbonio di un anello conviene proseguire intorno all’anello in entrambe le direzioni fino a trovare un punto di differenza e poi usare questa differenza per determinare quale lato dell’anello ha la priorità sull’altra. pR Qu car CH3 CH3 senza perossidi CH3 C CH + HBr C CH3 Br bromuro di tert-butile (orientazione di Markovnikov) SOl L’a ziona i da una esemzatore, di idrodotto è onano essario. forma ossidata Pt forma ridotta +forma H2ossidata O N + H CH3 l’alcano prodotto è rilasciato dal catalizzatore all’Edizione XXVI Prefazione R = (CH2Americana CH C CH2)10 In pratica forma ridotta N 14–15B H + H2O N Figura 13–10 Sostituzione Elettrofila Aromatica deidi Fenoli La piridina è basica, con elettroni N + In pratica: L’annerimento della frutta non reattivi legame disponibili strappare un elettrofila aromatica L’annerimento della frutta è un tipico I fenoli sono substrati altamente verso la asostituzione piridina dell’ossigeno protonata (ione piesempio di ossidazione dei fenoli a ione piridinio, pK = 5,2 in quanto la coppia di elettroniprotone. di nonLalegame del gruppopiridina, ossidrilico pKb = 8,8 a ridinio) continua ad essere aromatica. Le sezioni denominate Instabilizza pratica ilhanno lo scopo dimostrare l’importanza della complesso sigmadiformato dall’attacco in posizione ortoChimie para. Pertanto, chinoni. Mele, pere, patate, etc. conl’ossidrile è unegruppo fortemente orto, para-orientante. I fenoli sono ca Organica per le vite degli studenti per diverse areeattivante di interesse come la Biochimica, i substra- tengono la polifenolo ossidasi (PPO), un eccellenti Verde. per le reazioni di alogenazione, nitrazione, solfonazione, e alcune delle enzima che catalizza l’ossidazione dei Farmaci, l’Ambiente, o latiChimica reazioni di Friedel-Crafts. Generalmente, a causa della loro elevata reattività, i fenoli derivati del catecolo (1,2-benzendiolo) vengono alchilati o acilati utilizzando catalizzatori deboli di Fridel-Crafts (come HF) presenti in natura mediante ossigeno In pratica: Biochimica In pratica: Antibiotici 13–8B Pirroloatmosferico. I prodotti sono degli ortoproprio per evitare la polialchilazione e la poliacilazione. Il porfobilinogeno, un pirrolo sostituito, La riduzione enzimatica di un doppio legachinoni, che sono instabili e tendono Il pirrolo è un eterociclo aromatico a cinque termini, con un atomo di az è il costituente principale me è lo stadio chiave nella formazione di OH OH OHdell’anello rapidamente a formare polimeri scuri. legami (Figura 13–11). Sebbene il pirrolo sembri possedere solamente dell’eme, una porfirina che svolge un acido grasso che viene poi incorporato OH L’annerimento può essere controlla) CH(CH 3 2azoto ha una coppia di elettroni non condivisa. L’atom ni p, l’atomo di numerose nella parete del batterio che provoca la HFfunzioni fisiologiche, come il 2to tramite l’aggiunta di agenti riducenti , e il suo orbitale p non ibrido si sovrappone con gli pirrolo è ibridato sp + CH 3 CH CH 3trasporto e il deposito dell’ossigeno. + tubercolosi. Il farmaco antitubercolare isoo di soluzioni acide che inibiscono atomi di carbonio a formare un anello continuo. La coppia solitaria de COOH CH2NH2 niazide blocca questo enzima, impedendo l’attività dell’enzima PPO. Comunel’orbitale p, e (a differenza della coppia solitaria della piridina) quest H2C la riduzione del doppio legame. Senza una mente alla frutta fresca tagliata sono ) CH(CH partecipano al sistema p di legame. Questi due elettroni, insieme ai qua 3 2 NH parete cellulare integra, il batterio muore. aggiunte soluzioni di bisolfito di sodio, dei due doppi legami, costituiscono un sestetto aromatico. Il pirrolo h O acido ascorbico (vitamina C), e succo di Lo ione fenossido, generato facilmente per trattamento del fenolo con idrossido CH2CH2COOH risonanzadidisodio, 92 kJ/mol (22 kcal/mol). è ancora più reattivo del fenolo verso la sostituzione elettrofila aromatica. In quanto cari- limone per ritardarne l’annerimento. C N so / Pt Il pirrolo (pKb = 13,6) è una base molto più debole della piridina (p porfobilinogeno NHNH2 chi negativamente, gli ioni fenossido reagiscono con elettrofili carichi positivamente sta differenza èper dovuta alla struttura del pirrolo OH protonato (Figura 13–1 isoniazide 4 dare complessi sigma neutri le cui strutture somigliano a quelle dei chinoni. derivatiè richiesto l’impiego di una coppia di elet un legame con un protone del catecolo CH2CH2COO–– H3C – – aromatico. Nel pirrolo protonato, l’atomo di azoto è legato nel sestetto O O OH O O diversi (due atomi di carbonio e R due atomi di OH idrogeno), ciò richiede excess Br Br 3 che non presenta orbitali p non ibridi. Il pirrolo protonato non è aro N Br2 –Brsp Br2 NaOH 2 H2C CH CH2CH2COO un acido con adeguata forza, protonerà O2il pirrolo PPO in posizione 2, su uno H2O N Fe – carbonio dell’anello, e non sull’atomo di azoto. OH N H Br Br H3CBr ione fenossido H N CH3 Br Br complesso sigma 13–8C Lo Studio delle Reazioni Chimiche O orto-chinone Pirimidina e Imidazolo (instabile) CH H2C attivati CH3subiscono reazione di sostituzione Gli ioni fenossido sono così fortemente che La pirimidina è un eterociclo a sei R termini conO due atomi di azoto s elettrofila aromatica con anidride carbonica (reazione di Kolbe), un debole elettrofilo. 1,3. Entrambi gli atomi di azoto sono simili all’azoto della piridina. orbitale p gruppo eme, presente in emoglobina 5–9 129 Diastereoisomeri deboleè una sintesi industriale dell’acido saliInfatti, la carbossilazione degli ioni vuoto fenossido e mioglobina coppia di elettroni di non legame nell’orbitale ibrido sp2 nello stesso p sovrapposizione cilico, il quale a sua volta viene successivamente convertito in aspirina, come mostrato H aromatico. Queste coppie non condivise non servono al sestetto aroma no di essi ha un enantiometro. Quindi esistono quattro 2-bromo-3-clorobutani stereoinelle pagine precedenti. polimeroproprio di colorecome marrone sto conferiscono basicità agli atomi, accade per la coppia sp3 somerici: due coppie di enantiomeri. Ciascun membro di una coppia di enantiomeri è + H C C 3 della piridina. sp O dell’altra coppia. un diastereoisomero di ciascun membro H H sp3 O C O O– OH O– O CH3 CH3 CH3 CH3 H – – O C H O COOH C Ocarbocatione gruppo alchilico Obiettivi del Gli del Capitolo aiutano gli studenti a H+ Obiettivi H H H Br H H Br Br H H Br H CapitOlO 4 focalizzare l’attenzione sugli argomenti che è inH2O H Cl H Cl Cl H Cl H – OH H Obiettivi del Capitolo 1 Proporre meccanismi che descrivano i diversi stadi di 2 reazioni semplici, come l’alogenazione radicalica. 3 Costruire un diagramma della coordinata 3di reazione CH CH ed utilizzarlo per identificare i fattori che controllano (2S,3R) (2R,3S) termodinamica e cinetica di una reazione. enantiomeri 3 Usare meccanismo, termodinamica e cinetica di rea- zione per prevedere quale tra i suoi possibili prodotti è quello principale. 3 3 CH CH 4 Identificare gli(2S,3S) intermedi reattivi e spiegare le loro (2R,3R) proprietà. enantiomeri dispensabile comprendere durante lo studio di ciaacido salicilico N H H N H scun capitolo. H Riepilogo Uno degli aspetti più utili ed interessanti della chimica organica è lo studio delle reazioni. È impossibile ricordare migliaia di specifiche diastereoisomeri reazioni organiche mentre è pos- Introduzione sibile organizzare queste reazioni in gruppi basati sul modo in cui esse hanno luogo e quali intermedi coinvolgono. Cominciamo questo studio considerando l’alogenazione degli alcani, una reazione relativamente prevede che la sostituzione un Abbiamo ora visto tutti isemplice tipi diche isomeri bisognadi conoscere e possiamo riportaidrogeno con un alogeno in fase gassosa, senza solventi che complichino lo studio re in un diagramma loro e riassumere definizioni. della reazione. È conveniente le partire da relazioni questa reazione perché abbiamo le giàloro studiato struttura e proprietà degli alcani. Tuttavia, va ricordato che, in pratica, gli alcani sono così poco reattivi da essereClassi usati raramente come materiali di partenza per le sintesi di Isomeri organiche di laboratorio. Scrivere l’equazione complessiva di una reazione, con i reagenti sulla sinistra ed gli isomeri i prodotti sulla destra è solo il primo passo nello studio di una reazione.tutti Se veramente si vuole capire una reazione bisogna conoscerne il meccanismo, cioè il percorso che porta passo dopo passo dai reagenti ai prodotti. Inoltre, per capire la tendenza dei reagenti a trasformarsi in prodotti, la posizione dell’equilibrio e l’energia coinvolta bisogna studiare la termodinamica della reazione.strutturali isomeri stereoisomeri Anche se l’equilibrio dovesse favorire la formazione di un prodotto, la reazione potrebbe non avvenire ad una velocità accettabile. Per utilizzare una reazione in un tempo ragionevole (o per evitare che diventi esplosiva) ne va studiata la cinetica, cioè l’andamento della velocità della reazione al variare di diverse condizioni, tra cui le concentrazioni dei reagenti. La comprensione della cinetica aiuta anche a proporre diastereoisomeri enantiomeri meccanismi di reazione che siano in accordo con il comportamento osservato. RiepilOGO H H 4–1 pirrolo struttura orbitalica del pirrolo (sei elettroni p, aromatico) Figura 13–11 I Riepiloghi, sistemati in posizioni strategiche all’interno dei capitoli, sottolineano le inStruttura di legame p del pirrolo. L’atomo di azoto del pirrolo è ibridato sp2, c formazioni importanti usando diagrammi e grafici, quando possibile. pia solitaria di elettroni nell’orbitale p. Questo orbitale p si sovrappone con gl isomeri cis-trans (isomeri geometrici) 4–2 restanti atomi di carbonio in modo da formare una nube circolare continua. Co quattro elettroni dei doppi legami e i due elettroni nell’orbitale p dell’azoto, il in totale sei elettroni p. altri diastereoisomeri (due o più centri di chiralità) La clorurazione del metano è un’importante reazione industriale, con un meccanismo relativamente semplice e che può essere utilizzato per illustrare molti dei principi im- Clorurazione portanti di una reazione. La reazionedifferenti del metanocon con illacloro produce una miscela di del Metano Isomeri sono composti stessa formula molecolare. prodotti clorurati la cui composizione dipende dalla quantità di cloro aggiunto e dalle Isomeri strutturali sono isomeri che differiscono per l’ordine condizioni di reazione. La luce o il calore sono necessari per far avvenire la reazione con il quale gli atomi sono legati tra di ad una velocità accettabile. Quando si aggiunge cloro al metano, la prima reazione è Stereoisomeri sono isomeri che differiscono solo per l’orientazione degli atomi nello spazio. 87 loro. Enantiomeri sono isomeri immagini speculari l’uno dell’altro. Diastereoisomeri sono stereoisomeri che non sono immagini speculari l’uno dell’altro. Isomeri cis-trans (isomeri geometrici) sono diastereoisomeri che differiscono per la disposizione cis-trans su un anello o su un doppio legame. Ringraziamenti È un piacere ringraziare tutte le persone che mi hanno aiutato nella revisione di questo testo. Più degli altri, Jan William Simek mi ha dato una serie di eccellenti consigli e valutazioni fondate per tutte le diverse edizioni di questo libro. In questa edizione, Jan mi ha dato dei suggerimenti sulle revisioni dei vari capitoli e mi ha aiutato a scrivere e correggere tutti i nuovi paragrafi. Ha anche preparato molti dei nuovi problemi e tutte le Risposte ai Problemi a fine libro. Particolari ringraziamenti sono anche dovuti a John Murdzek, Developmental Editor, che mi ha dato migliaia di utili suggerimenti durante il processo di scrittura e revisione, che ha contribuito a definire la struttura di questa nuova edizione. Vorrei ringraziare i revisori per i preziosi commenti e suggerimenti. Anche se non li ho adottato tutti, i loro suggerimenti sono stati comunque utili ed hanno contribuito alla qualità del prodotto finale. Revisori dell’Ottava Edizione David A. Boyajian Hasan Palandoken Susan Schelbe Alline Somlai Jon Antilla Eric Brown Timothy B Clark James Fletcher Keith Osbourne Pascoe Anthony J Pearson Owen Priest K.C. Russell Solomon Weldegirma Palomar College California Polytechnic State University Metro State College of Denver Delta State University University of South Florida Loyola University Lake Shore Western Washington University Creighton University Georgia State University Case Western Reserve Northwestern University Northern Kentucky University University of South Florida Revisori delle Edizioni Precedenti Jung-Mo Ahn David Alonso Merritt B. Andrus Arthur J. Ashe Bill Baker Dan Becker John Berger Bob Bly Mary Boyd Hindy Bronstein David Brown Philip Brown Christine Brzezowski Patrick Buick David Cantillo Dee Ann Casteel Amber Charlebois Cai Chengzhi Barry Coddens Jamie Lee Cohen Barbara Colonna Richard Conley Robert Crow Maria de Graca Vicente Chris Gorman Geneive Henry William Jenks Przemyslaw Maslak Kristen Meisenheimer Stephen A. Miller Guillermo Moyna Rabi Musah Anthony J. Pearson Allan Pinhas University of Texas at Dallas Andrews University Brigham Young University University of Michigan University of South Florida Loyola University Montclair State University University of South Carolina Loyola University, Chicago Fordham College at Lincoln Center St. John’s University North Carolina State University University of Alberta Florida Atlantic University Hillsborough Community College Bucknell University William Paterson University University of Houston Northwestern University Pace University University of Miami Middlesex County College St. Louis College of Pharmacy Louisiana State University North Carolina State University Susquehanna University Iowa State University Pennsylvania State University Cal Polytechnic at San Luis Obispo University of Florida University of the Sciences in Philadelphia University at Albany Case Western Reserve University University of Cincinnati XXVII XXVIII Ringraziamenti Stanley Raucher Suzanne Ruder David Son Joseph B. Wachter University of Washington Virginia Commonwealth University Southern Methodist University Michigan State University Infine, voglio ringraziare le persone alla Pearson, la cui dedizione e flessibilità hanno contribuito al completamento di questo progetto. Jeanne Zalesky, Executive Editor, e Jennifer Hart, Senior Project Editor, hanno garantito che il progetto andasse avanti, assicurando le risorse necessarie ed hanno proposto molti utili commenti e suggerimenti. Marisa Taylor, Kate Thomas e Shari Toron, Project Managers, hanno organizzato il processo di produzione con la giusta tempistica. È stato un piacere lavorare con tutte queste persone professionali e competenti. Mi è piaciuto molto lavorare a questa nuova edizione e spero che venga percepita come un utile miglioramento rispetto alla settima edizione. Ho provato a rendere questo libro il più possibile privo di errori, ma sono sicuro che alcuni errori sono comunque sfuggiti. Se trovate errori o avete suggerimenti su come migliorare questo libro, per favore fatemelo sapere (L.G. Wade, Whitman College, Walla Walla, WA 99362; e-mail: [email protected]). Ho già preparato un file con possibili variazioni e miglioramenti per la prossima edizione e spero che molti di quelli che useranno questo testo possano contribuire a questo file. Spero che questo libro renda il vostro lavoro più facile, sia che voi siate studenti che docenti. Questa è la ragione più importante per la quale l’ho scritto. L. G. Wade, Jr. Walla Walla, Washington
Scaricare