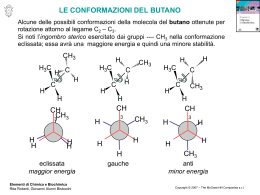
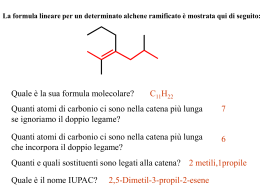
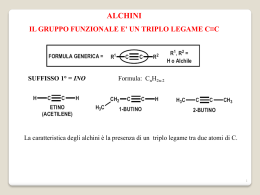
1 Prof. Renzo Ruzziconi CORSO DI CHIMICA ORGANICA SUPERIORE 2 PARTE PRIMA LA REATTIVITA’ CHIMICA 3 CAPITOLO I TEORIA DEGLI ORBITALI DI FRONTIERA 4 Perché? H+ H+ H O O lenta CH3-I I-CH3 H3C OH molto veloce lenta molto veloce O O OCH3 E+ più veloce di E+ N H N H molto lenta + O O veloce + O O O O + O ancora più veloce O OAlCl3 O O O 5 N HOO N HO C C 105 !!! più veloce di OCH3 OCH3 CHO OCH3 CHO CHO CHO N O C N C + Un'adeguata spiegazione può essere trovata nella TEORIA DEGLI ORBITALI DI FRONTIERA 6 LEGAMI OMONUCLEARI La Molecola dell'Idrogeno nodo + H H OM antilegante E * E + OM Legante H H * = c1 - c2 1 E * 2 E *> E = c1 1 + c2 2 c1 c2 +0.707 + H +0.707 H c2 = 1.00 E + H- -0.707 c2 = 1.00 H +0.707 c2 = 1.00 c2 = 1.00 7 La Molecola del Metano H H 4 nodi H H H H HH H H 2 nodi H H H HH 1 nodo H H H H H H H H H H H H H H z H H 0 nodi H H H Y H H x 8 9 10 HΨ Ψ = EΨ Ψ ∫ΨHΨ E0 = ∫Ψ2dv Applicando il principio variazionale e ponendo Ψ = c1ψ1+ c2ψ2 si ha: E= E= ∫(c1ψ1+c2ψ2)H(c1ψ1+c2ψ2)dv ∫(c1ψ1+c2ψ2)2dv c12∫ψ1Hψ1dv + c1c2∫ψ1Hψ2dv + c1c2∫ψ2Hψ1dv + c22∫ψ2Hψ2dv c12∫ψ12dv + 2c1c2∫ψ1ψ2dv + c22∫ψ22dv E= c12∫ψ1Hψ1dv + 2c1c2∫ψ1Hψ2dv + c22∫ψ2Hψ2dv c12∫ψ12dv + 2c1c2∫ψ1ψ2dv + c22∫ψ22dv ∫ψiHψidv = Hii Integrale coulombiano: energia associata ∫ψiHψjdv = Hij Integrale di risonanza: energia dell'elettrone che si ∫ψiψidv = Sii Integrale di normalizzazione: probabilità di all'elettrone localizzato sull'atomo i considerato in presenza dell'atomo j (livelli energetici degli orbitali isolati, a destra e sinistra del diagramma. muove sottol'influenza degli atomi i e j. trovare l'elettrone sull'atomo i. ∫ψiψjdv = Sij Integrale di sovrapposizione: misura della sovrapposizione dei due orbitali atomici ψiψj all'abbassamento dell'energia. E' funzione della distanza fra i nuclei. 11 12 13 LEGAMI ETERONUCLEARI σ*C-O O CC > C O Ec Ei O E0 σC-O O 2 Σc =1 CO > C C Nell'orbitale legante l'atomo più elettronegativo ha il coefficiente dell'orbitale atomico più grande Ei/Ec = % di carattere ionico del legame E0/Ec = % di carattere covalente del legame 2 E0 = Energia necessaria per rompere eteroliticamente il legame. C O C + O 2 E0 < 2 E σc-c Ec + E0 = energia necessaria per rompere omoliticamente il legame C-O 85 Kcal/mole C-C 83 Kcal/mole C O C + O !! 14 IL DOPPIO LEGAME C=O O nodo Ecc Ei Eco O E0 2 Σc =1 O Ei/Eco = % di carattere ionico del legame π C O E0/Ec = % di carattere covalente del legame π C O 2E0 = Energia minima necessaria per rompere il legame π C=O eteroliticamente (C=O C-O) E0 + E c = Energia minima necessaria per rompere il legame π C=O omoliticamente (C=O Si noti E 0 + E c > 2 E 0 Esempi: N C C-O) O OH CN N C CH2 n. r. CCl3 Cl3C CH2 Cl3C O n. r. 15 SISTEMI CONIUGATI IL SISTEMA ALLILICO E Ψ 3* LUMO nell'anione π∗ HOMO nell'anione p Ψ2 LUMO del catione π E Ψ1 HOMO nel catione 2 3 1 -.707 Σc +.500 +.500 Ψ3* 0.500φ1 - 0.707φ2 + 0.500φ3 1 0.707φ1 + 0.000φ2 - 0.707φ3 1 0.500φ1 + 0.707φ2 + 0.500φ3 1 LUMO dell'anione +.707 -.707 Ψ2 LUMO del catione HOMO dell'anione +.707 +.500 +.500 Ψ1 HOMO del catione c1 c2 c3 c1 c2 c3 Σc 2 1 1 1 2 16 IL SISTEMA ALLILICO Ψ = c1φ1 + c2φ2 + c3φ3 Equazioni secolari del sistema allilico Determinante secolare del sistema allilico c1(α α-E) + c2β = 0 c1β + c2(α α-E) + c3β = 0 c2β + c3(α α-E) = 0 (α α-E) β β (α α-E) 0 β 0 β (α α-E) =0 Confronto Allile-Etilene Allile Etilene + orb. p isolato -.707 +.500 +.500 E3= α - 1.41β β +.707 +.707 -.707 E2 = α - β -.707 E2 = α +.707 E=α +.707 +.707 +.500 +.500 E2 = α + β E1 = α + 1.41β β 2e 3e 2α α +2.82β β -(2α α + 2β β) 3α α +2.82β β -(2α α + 2β β + α) 0.82 β 0.82 β 4e 4α α +2.82β β -(2α α + 2β β + 2α α) 0.82 β 17 SISTEMI CONIUGATI - 1,3-BUTADIENE 2 E 1 4 3 Ψ4∗ π∗ π∗ Ψ3 ∗ HOMO Etilene LUMO-Diene Ψ2 HOMO-Diene E2 π π E1 LUMO Etilene Ψ1 Il butadiene è più stabile dell'etilene, ma è anche più reattivo E Σc +0.371 -0.600 +0.600 Ψ4 -0.371 1 +0.600 1 -0.600 1 +0.371 1 3 nodi Ψ3 +0.600 -0.371 -0.371 2 nodi Ψ2 +0.600 +0.371 -0.371 1 nodo +0.371 +0.600 +0.600 Ψ1 0 nodi Σc 2 1 1 1 1 2 18 L' 1,3-BUTADIENE Ψ = c1φ1 + c2φ2 + c3φ3 + c4φ4 Equazioni secolari dell'1,3-butadiene Determinante secolare dell'1,3-butadiene c1(α α-E) + c2β α-E) + c3β c1β + c2(α c2β + c3(α α-E) + c4β c3β + c4(α α-E) (α α-E) β 0 0 β (α α-E) β 0 0 α-E) β β (α 0 0 β (α α-E) =0 Confronto 1,3-butadiene - etilene -.600 +.600 -.371 +.371 E4 = α - 1.61β β +.707 -.707 E2 = α - β +.600 +.600 -.371 -.371 E3 = α - 0.628β β -.600 +.600 +.371 -.371 +.707 E2 = α + 0.628β β +.707 -0.73β β +.600 +.600 +.371 +.371 +1.24β β E1 = α + 1.61β β Etot = 4α α + 4.5β β ∆EB-E = 4α α + 4.5β β - 4α α - 4β β = +0.5β β E1 - Eet = 2(α α + 1.62β β) -2α α-2β β = +1.24β β E2 - Eet = 2(α α + 0.628β β) - 2α α - 2β β = -0.73β β E1 = α + β Etot = 2α α + 2β β 19 20 21 Da ricordare !! La coniugazione di legami π con orbitali p o altri legami π rende una molecola termodinamicamente più stabile rispetto ad una analoga molecola non coniugata. Ciò non significa, tuttavia che la prima sia meno reattiva. In realtà, molto spesso questi sistemi sono più reattivi, cioè "cineticamente meno stabili". La distinzione fra stabilità termodinamica e stabilità cinetica deve essere sempre tenuta presente. Fra i molti fattori che determinano la reattività di un diene vi è il valore dell'energia dell'HOMO e L'HOMO del butadiene (Ψ Ψ2) possiede un'energia maggiore rispetto all'HOMO dell' etilene (π π). Nel butadiene la stabilità termodinamica è associata alle energie di Ψ1 e Ψ2, mentre la stabilità cinetica è largamente (ma non solo) determinata dall'energia di Ψ2 E Eatt etilene Eatt diene Ψ2 diene (HOMO) ∆Eatt e-b πetilene (HOMO) ∆Ge-b energia del butadiene Ψ1 diene cdr 22 INTERAZIONI HOMO-HOMO e HOMO-LUMO A + B Prodotti A B LUMO A LUMO B E2 HOMO A HOMO B E1 E2 > E 1 E2-E1 è il maggior responsabile dell'Energia di Attivazione A B LUMO A LUMO B HOMO A E1 EH-L E2 HOMO B E3 E4 EH-L >> E 3 > E 4 > E 2 > E 1 23 L'EQUAZIONE DI KLOPMAN E SALEM (Stima della reattività chimica) L'energia messa in gioco nella formazione di legami da parte di due specie A e B interagenti può essere quantificata utilizzando l'equazione di Klopman e Salem. ∆E = - ∑ (qa + qb)β βabSab + occ. v occ. v +∑ r atomo k ∑-∑∑ s Qk orbitale a sull'atomo k dell'orbitale molecolare r s Ql a k ∑ εRkl + 2(∑ ∑cra csbβab)2 Er - Es r atomo l b Rkl QkQl l orbitale b sull'atomo l dellorbitale molecolare s qa e qb = Popolazione elettronica (densità elettroniche) negli orbitali atomici a e b. β e S = Integrali di risonanza e di sovrapposizione. Qk e Ql = Cariche totali sugli atomi k e l ε = Costante dielettrica totale Rk,l = Distanza fra gli atomi k e l cra = coefficienti dell'orbitale atomico a nell'orbitale molecolare r di una molecola e coefficiente dell'orbitale atomico b nell'orbitale moilecolare s dell'altra molecola. Er,s = Energia dell'orbitale molecolare r di una molecola e dell'orbitale molecolare s dell'altra molecola. 24 ANALISI DELL'EQUAZIONE DI KLOPMAN E SALEM I° Termine - Σ (qa+qb)β βabSab ab E' un termine di repulsione e deriva dall'interazione degli orbitali " pieni " della molecola A con gli orbitali "pieni " della molecola B. L'effetto totale è un effetto di antilegame. Il contributo di questo termine è, in genere, molto più grande rispetto a quello degli altri due termini. Esso è il maggior responsabile dell' entalpia di attivazione. Maggiore è il numero di legami coinvolti nella reazione più grande è il contributo di questo termine. Se una reazione ha la possibilità di procedere attraverso due diversi cammini, ad esempio, una molecola che può subire l'attacco in due siti diversi, il I° termine dell'equazione di Klopman e Salem è generalmente molto simile per ognuno dei due diversi cammini . Tale termine può dunque essere trascurato nella reattività differenziale. 25 Il 2° termine dell'equazione di Klopman e Salem + Σ k<l QkQl εRkl E' un termine che fornisce un contributo positivo alla formazione del legame. Esso rappresenta l' attrazione (o repulsione) di tipo coulombiano fra gli atomi di A e B interagenti. Questo termine diventa particolarmente importante nelle reazioni che coinvolgono specie molto polari o, ancor più, specie ioniche. Il 3° termine dell'equazione di Klopman e Salem occ. +Σ r v occ. v Σs - Σs Σr 2 (Σ cra csbβab) 2 Er - Es Anche questo termine contribuisce positivamente alla formazione del legame. Esso rappresenta la sommatoria di tutti i contributi alla formazione del legame derivanti dalle possibili interazioni di legame fra gli orbitali " pieni " della specie A e gli orbitali " vuoti " della specie B aventi la corretta simmetria. E' un termine di perturbazione di 2° ordine e vale quando Er = Es (quando Er = Es, il termine diventa: +Σ Σab 2 craCsb βab Si può notare che l'interazione che offre il maggior contributo alla formazione del legame è sempre l'interazione HOMO-LUMO. Questo termine diventa particolarmente importante nelle reazioni fra specie neutre 26 Un esempio + Trattandosi di specie cariche, il maggior contributo alla formazione del legame è dovuto al 2° termine dell'equazione di Klopman e Salem. + QkQl εRkl Σ k<l Tuttavia in questo ipotetico caso diventa particolarmente rilevante anche il contributo del 3° termine. Ciò è dovuto alla forte interazione HOMO-LUMO (E r = Es). LUMO anione -.707 +.500 +.500 HOMO anione +.707 LUMO catione -.707 E1 +.500 +.707 +.500 E2 HOMO catione (+) (-) L'interazione più efficace degli orbitali è sempre HOMO nucleofilo - LUMO elettrofilo 27 Determinazione della Popolazione Elettronica e della Distribuzione di Carica Popolazione elettronica 1. Elevare al quadrato il valore del coefficiente dell'orbitale atomico considerato 2. Moltiplicare C2 per il numero di elettroni nell'orbitale molecolare di cuo l'orbitale atomico fa parte 3. Sommare i valori di ciascun atomo su tutti gli orbitali molecolari Esempio: c2 -.707 +.500 +.500 c1 c3 HOMO anione 2 2 2 +.707 -.707 P. el. su C1 = (0.5) x2 + (0.707) x2 + (0.5) x0 = 1.5 2 2 2 su C 2 = (0.707) x2 + (0.0) x2 + (0.707) x0 = 1.0 2 2 2 su C 3 = (0.5) x2 + (0.707) x2 + (0.5) x0 = 1.5 +.500 +.707 +.500 c2 c1 c3 -.707 +.500 2 2 2 P. el. su C1 = (0.5) x2 + (0.707) x0 + (0.5) x0 = 0.5 2 +.500 LUMO catione +.707 -.707 2 2 su C 2 = (0.707) x2 + (0.0) x0 + (0.707) x0 = 1.0 2 2 2 su C 3 = (0.5) x2 + (0.707) x0 + (0.5) x0 = 0.5 +.500 +.707 +.500 28 Determinazione della Distribuzione di Carica Cambiare di segno il valore della popolazione elettronica su ciascun atomo e sommare 1. Per il sistema allilico si ottiene: popolazione elettronica 1.0 1.5 1.5 Carbanione allilico 0 -0,5 -0.5 1.0 Carbocatione allilico popolazione elettronica 0.5 0.5 + 0,5 distribuzione di carica negativa 0 + 0.5 distribuzione di carica positiva + + o 29 Acidi e Basi Acidi Hard: Specie ad alta + Σ k<l + + +Σ r ++ Sn +4 QkQl εRkl + energia LUMO, reagiscono preferibilmente con basi soft formando legami ad elevato carattere covalente domina + RC=O , BF3, Acidi Soft: Specie a bassa 2 (Σ cra csbβab) occ. v occ. v + H , Li , ..... K , Mg , +3 +3 Al , Cr , AlCl3, SO3 energia LUMO, reagiscono preferibilmente con basi hard formando legami con elevato carattere ionico domina Hard e Soft Σs - Σ Σ s 2 + ++ ++ Cu , Ag , Pd , Cd , ++ + Hg , I , C6H3(NO2)3, - + p O=C6H4=O, Br , M°, :CH2, I2, Br2 Er - E s r Soft-Soft Elettrofili Nucleofili Hard-Hard - - RSH, RS , I , SCN , CO, C 2H4, - C6H6, H , R - Basi Soft : Specie ad alta energia HOMO, reagiscono preferibilmente con acidi soft, con cui formano legami ad alto carattere covalente domina - H2O, OH , - - - RO , ClO4 , NO3 -3 PO4 - F , Cl , - occ. v +Σ r occ. Σs - Σ Σ s 2 (Σ cra csbβab) Er - Es r Basi Hard: Specie a bassa energia HOMO, reagiscono preferibilmente con acidi hard con cui formano legami ad alto carattere ionico domina + Σ k<l QkQl εRkl 30 Nucleofili ed Elettrofili Hard e Soft Nucleofili Hard hanno una bassa energia HOMO e generalmente possiedono una caraica negativa (es. OH-). Nucleofili Soft hanno un'alta energia HOMO. Essi non necessariamente possiedono una carica negativa (es. CH2=CH2). Elettrofili Hard hanno un'alta energia LUMO e generalmente possiedono una carica positiva (es. H+, BF3). Elettrofili soft hanno una bassa energia LUMO e non hanno necessariamente una carica positiva (es. Br2). Una reazione Hard-Hard è veloce principalmente a causa di una forte attrazione colombiana. Una reazione Soft-Soft è veloce a causa principalmente di una grande interazione HOMO-LUMO. 31 Valori dell’energia HOMO (Nucleofili) e dell’energia LUMO (Elettrofili) di alcuni ioni inorganici (Valori calcolati da Klopman dai potenziali di ionizzazione e dalle affinità elettroniche corretti per l’effetto di solvatazione) Nucleofili r HIHSCNBrClOHH2O F- 2.08 2.16 1.95 1.81 1.36 EHOMO (eV) -7.37 -8.31 -8.59 -8.78 -9.22 -9.94 -10.45 -10.73 -12.18 Elettrofili Al+3 La+3 Ti+4 Be+2 Mg+2 Ca+2 Fe+3 Sr+2 Ce+3 Ba+2 Ga+3 Cr+2 Fe+2 Li+ H+ Ni+2 Na+ Cu+2 Tl+ Cd+2 Cu+ Ag+ Tl+3 Au+ Hg+2 r 0.50 1.15 0.68 0.31 0.65 0.99 0.64 1.13 1.11 1.35 0.62 0.76 0.60 0.00 0.72 0.95 0.69 1.40 0.97 0.96 1.26 0.95 1.37 1.10 ELUMO (eV) 6.01 4.51 4.35 3.75 2.42 2.33 2.22 2.21 2.06 1.89 1.45 0.91 0.69 0.49 0.42 0.29 0.00 -0.55 -1.88 -2.04 -2.30 -2.82 -3.37 -4.35 -4.64 All’interno di uno stesso gruppo esiste una grossolana correlazione fra l’energia dell’HOMO o del LUMO e il raggio ionico. Maggiore è il raggio ionico, più alto è il valore dell’HOMO del nucleofilo e più basso è il valore del LUMO dell’elettrofilo. 32 Tecniche per la Determinazione dell'energia HOMO e la Distribuzione Elettronica in una Molecola Insatura La Spettroscopia Fotoelettronica (PES) La spettroscopia fotoelettronica misura le energie di ionizzazione della molecole. La tecnica si basa su un principio analogo a quello dell'effetto fotoelettronico, in cui l'energia del fotone incidente è uguale alla somma dell'energia di ionizzazione più l'energia cinetica dell'elettrone espulso. Sorgente Lampada He hν = 21.22eV 1/2mv2 X+. Campione hν Ii X Analizzatore elettrostatico E = hν = Ii + 1/2mv2 UPS (ν ν ~ 58.43 nm ≡ 21.22 eV) XPS (per elettroni di orbitali più bassi) E Detector + - 33 Tecniche per la Determinazione dell'energia HOMO e la Distribuzione Elettronica in una Molecola Insatura La Risonanza di Spin Elettronico (ESR) La Risonanza di Spin Elettronico (ESR) detta anche Risonanaza Paramagnetica elettronica (EPR) fornisce importantissime informazioni di carattere elettronico, strutturale e cinetico su una grande quantità di sistemi organici, inorganici e biologici. Condizione necessaria per poter effettuare un esperimento ESR è che il sistema che si sta studiando possegga almeno un elettrone spaiato, giacchè la tecnica si basa sulla natura magnetica dell'elettrone. B S, momento angolare S = 1/2, numero quantico di spin +1/2 ∆E = geµBB -1/2 µ, momento magnetico µ = -geµBS Condizioni di risonanza: hν ν0 = geµBB In realtà la risonanza di un elettrone spaiato di un radicale può presentarsi in forma di multipletto a causa dell'accoppiamento dello spin elettronico con i diversi nuclei magneticamente attivi (I > 0). Così, ad esempio, l'assorbimento di un radicale fenile può presentarsi come doppietto, tripletto, quartetto, ecc. a seconda del numero di idrogeni legati all'anello. Le costanti di accoppiamento aH che compaiono nel multipletto sono proporzionali alla densità elettronica sll'atomo di carbonio legato ad idrogeno. aH1 aH2 aH = QHC-H ρc H H QHC-H = 24 G 34 ∂ 35 36 Esempi di Reazioni fra acidi (Elettrofili) e Basi (Nucleofili) Hard e Soft H HO H + O velocissima HOBr + Br molto meno veloce hard H hard + Br HO 2 H2O hard Br soft H2C=CH2 soft + Br-Br soft BrH2C-CH2Br velocissima H H2C=CH2 + H H3C-CH2OH O molto lenta H hard soft HgOAc RCH=CH2 soft + RCH=CH2 soft + Hg(OAc) 2 soft LiOAc hard H3CS- veloce R H H H nessuna reazione -OCH3 H3CS H C H centro hard centro soft C Br H3COH 37 LA SOSTITUZIONE NUCLEOFILA S N2 Interazione di legame X Nu + X Nu LUMO HOMO Inversione di configurazione Due orbitali possono interagire per formare un legame solo se hanno la stessa simmetria (stesso segno di φ) X Interazione di antilegame Nu NUCLEOFILI BIDENTATI Lo ione CNcentro hard centro soft : N C centro soft C N centro hard C N: CH3 C H I H CH3-CH2-CN Elettrofilo soft CH3 δ+ C H H CH3-CH2-N C I Elettrofilo hard Ag + 38 NUCLEOFILI BIDENTATI Il Sistema Enolato Ione enolato Carbanione allilico OE Ψ3 O Ψ3 Ψ2 HOMO O Ψ2 Ψ1 O Ψ1 centro hard O- + H3O+ OH + Σ k<l QkQl εRkl elettrofilo hard centro soft H2C H3C O + H3C-I elettrofilo soft C occ. v CH2 O +Σ r occ. v Σs - Σ Σ s r 2 (Σ cra csbβab) C-alchilazione I LUMO di CH 3I Er - E s 2 39 Enolati Cinetici e Enolati Termodinamici Idrogeno più acido fattore statistico favorevole Idrogeno meno acido, fattore statistico meno favorevole R R3N, DMF O H Processo irreversibile H H N R R ClTMS R H Processo reversibile LDA THF, -78°C OTMS O - OLi OTMS R ClTMS R O H N R ClTMS enolato termodinamicamente meno stabile, ma cineticamente favorito. enolato termodinamicamente più stabile, favorito in condizioni di equilibrio. Ep O R R' R R ∆G° OR R' R O- OTMS R' R cdr 40 Effetto del Solvente sulla Velocità di Alchilazione degli Enolati Solventi dipolari aprotici O δ− S-O H3C S+ O CH3 - DMSO ε = 47 M Enolato molto reattivo domina il 2° termine dell'equazione di K.-S. δ− O-S O-Alchilazione δ−O-S O O CH3 C N H N CH 3 CH3 NMP ε = 32 DMF ε = 37 O+ P N N N ε = 30 HMPA Solventi protici R O H H R O O H Hδ− M δ−H O R O − O R δO R R H Enolato poco reattivo domina il 3° termine dell'equazione di K.-S. C-Alchilazione 41 Effetto del Solvente ed Additivi sulla Velocità di Alchilazione degli Enolati Solventi chelanti O δ− M δ− S-O − O-S δ O-S O Enolato molto reattivo domina il 2° termine dell'equazione di K.-S. O-Alchilazione δ− O δ− M δ− O O M O δ− O δ−O DME (DiMetossiEtano) Diglyme Agenti chelanti O O K O O O O O 18-C-6 O O Li O O O 15-C-5 O O N Li N TMEDA 42 Regioselettività nelle Reazioni di Formazione e Alchilazione degli Enolati OR C R C O- O C R' R B H H C C C R' R H H H ka R B kb C C R H Enolato Termodinamico C R' H Enolato cinetico Ep O R R' ∆G° R OR OR R' R R' R cdr Et3N, ClTMS DMF, Riflusso OTMS O LDA, ClTMS, THF, -78°C OTMS 43 44 Dienolati O B α C8H17 C8H17 AcOH H2O - O O γ O C8H17 + O O Maggioritario CO2Et LiNR2 H2O CO2Et 87% + CO2Et 13% 45 ELETTROFILI BIDENTATI SOSTITUZIONI NUCLEOFILE ALLILICHE x centro elettrofilo soft δ+ centro elettrofilo hard LUMO Alogenuro Allilico Nucleofili soft Nucleofili hard PhS (CH3)2CuLi Nu Nu Nu X δδ+ X Nu SN2' (CH3)2CuLi SN2 D + Cl C H3C D D D D C D Cl + CH3Li D CH3 D δ- 46 ELETTROFILI BIDENTATI Il Sistema Carbonilico α,β-Insaturo α,β O Ψ4 E Ψ4 Ψ3 ∆E LUMO Ψ3 Ψ2 Ψ2 HOMO Ψ1 Ψ1 centro elettrofilo soft O δ+ Nu hard O δ+ centro elettrofilo hard 1. CH3MgBr 2. H+/H2O + OH CH3 Nu soft O 1. (CH3)2CuLi 2. H+/H2O + O 47 L'EFFETTO α Vi sono delle specie che sono estremamente più nucleofile di quanto ci si possa aspettare sulla base dei loro valori di pKa. Fra questi si annoverano: HO-O Cl-O HO-NH2 H2N-NH2 , RS-SR Tutte queste specie hanno una caratteristica comune: in tutte il l'atomo nucleofilo è legato ad un altro atomo, di uguale o diversa natura, avente almeno una coppia solitaria. HO-O HOMO ∆E HO- -O Si ricordi sempre che 1 eV = 24 Kcal/mole !! Reattività relative di HOO- e HO Elettrofilo C 6H5-C=N Softness p-O2N C6H4-COOCH3 hardness C 6H5-CH2-Br + molto hard H 3O kHOO-/kHO105 103 50 10-4 48 Effetti Stereoelectronici Gli effetti stereoelettronici sono semplicemente la conseguenza chimica e cinetica della sovrapposizione degli orbitali come, ad esempio, nella enolizzazione della norcanfora. La velocità della reazione endo è più elevata a causa della migliore sovrapposizione degli orbitali p con il sistema π del carbonile. Pertanto lo stato di transizione è stabilizzato rispetto a quello relativo alla rimozione del protone eso. Da quando furono proposti per la prima volta nel 1950, gli effetti stereoelettronici sono stati dimostrati in molti sistemi organici e la loro grandezza determinata sia sperimentalmente che computazionalmente. O O B H endo H B H H3C OCH3 O O cis Ph OCH3 O Ph cis kHcis/kHtrans = 8 O H3C O trans O H3C O + OCH3 OCH3 hν H H3C H3C OCH3 H3C O + C6H5 C6H5CH3 49 CAPITOLO II REAZIONI PERICICLICHE 50 51 REAZIONI PERICICLICHE (TERMICHE) Le reazioni pericicliche termiche sono quelle reazioni in cui un certo numero di legami vengono rotti ed altri vengono formati in maniera concertata portando alla formazione o all'apertura di un anello. La maggior parte delle reazioni pericicliche, coinvolgendo molecole neutre e spesso apolari, sono poco influenzate da interazioni coulombiane (scarso contributo del 2° termine dell'equazione di Klopman e Salem) mentre diventa importante il 3° termine (interazione degli orbitali di frontiera). TIPI DI REAZIONI PERICICLICHE (2π π 2σ σ) A) CILOADDIZIONI B) REAZIONI CHELOTROPICHE C) RIARRANGIAMENTI SIGMATROPICI D) REAZIONI ELETTROCICLICHE (π π (2π π 2σ σ) (σ σ + 2π π σ+ π) 2π π + σ) 52 LE CICLOADDIZIONI Sono reazioni pericicliche in cui due specie si combinano per formare un ciclo attraverso la formazione di due legami σ a scapito di due legami π, il tutto in maniera concertata. Un esempio è costituito dalla Reazione di Diels-Alder. O O O O O O Nelle reazioni pericicliche il segno e la grandezza dei coefficienti degli orbitali atomici negli orbitali molecolari interagenti sono di estrema importanza. I "lobi" interagenti negli orbitali di frontiera devono possedere lo stesso segno affinchè si verifichi un'interazione di legame. Da questo punto di vista è indifferente se l'interazione è del tipo HOMO di A -LUMO di B oppure LUMO di A -HOMO di B. HOMO diene LUMO dienofilo LUMO diene HOMO dienofilo La reazione è del tipo [4+2] ed è detta permessa dalla simmetria degli orbitali 53 Reazioni [2+2] O + O Nessuna reazione O LUMO etilene HOMO etilene interazione di legame Processo suprafacciale s interazione di antilegame a Una reazione [2+2] attraverso un processo del tipo supra-supra non è possibilea causa di una interazione di antilegame. Processo antarafacciale La simmetria degli orbitali potrebbe permettere una reazione del tipo supra-antara, ma in una reazione [2+2] ciò non è possibile per ovvie ragioni geometriche. [π π2a+π π2s] reazione permessa dalla simmetria degli orbitali, ma proibita dalla geometria. R R [π π2s+π π2s] reazione permessa dalla geometria , ma proibita dalla simmetria degli orbitali. R R R s s R R R NB: Le reazioni di cicloaddizione [2+2] termiche sono molto rare quelle che si conoscono hanno spesso un meccanismo radicalico (formazione di un diradicale intermedio) e non un meccanismo concerato. 54 Alcuni esempi: CO2Me + N CO2Me N N N CO2Me CO2Me [π π8s + π2s] O + O [π π6s + π4s] HOMO a 8 e (4n) s s [π π4s + π4s] proibita dalla simmetria [π π4a + π4s] permessa dalla simmetria LUMO HOMO a 10 e (4n+2) s s [π π6s + π4s] permessa dalla simmetria [π π6a + π4s] proibita dalla simmetria LUMO HOMO [π π8s + π2s] permessa dalla simmetria a 10 e (4n+2) s LUMO [π π8a + π2s] proibita dalla simmetria s 55 Cicloaddizioni 1,3-Dipolari Le cicloaddizioni non sono ristrette a molecole neutre, ma possono coinvolgere carbocationi e carbanioni. [π π2s + π2s] LUMO catione s 4e (4n) [π π2s + π4s] HOMO etilene s s Proibita dalla simmetria (infatti, mai osservata) LUMO catione Permessa dalla simmetria HOMO diene 6e (4n+2) + I [π π4s + π2s] HOMO anione s s 6e (4n+2) LUMO etilene Ph Ph Ph Ph permessa dalla simmetria H+ Ph + Ph Ph H Ph Ph Ph 56 Una sottoclasse delle reazioni di cicloaddizione sono è costituita dalle cicloaddizioni 1,3-dipolari in cui sono coinvolti eteroatomi (atomi diversi dal carbonio) O O O O O + O Si noti come la molecola dell'ozono è strutturalmente simile al carbanione allilico. La struttura degli orbitali è dunque molto simile. [π π4s+π π2s] O 6 e (4n+2) O O HOMO (ozono) s s Permessa dalla simmetria degli orbitali LUMO (etilene) H2C=N=N H2C-N=N + N 6 e (4n+2) s N CO2Me CO2Me [π π4s+π π2s] N N HOMO (diazometano) s LUMO (acrilato) permessa dalla simmetria 57 LA REGOLA DI WOODWARD - HOFFMANN Nella sua versione più semplice la regola di Woodward-Hoffmann stabilisce che: 4n+2 Le reazioni che coinvolgono un numero di elettroni pari a sono permesse dalla simmetria se il processo di approccio degli orbitali è del tipo supra-supra , mentre sono proibite se il processo è del tipo supra-antara. Le reazioni che coinvolgono un numero di elettroni pari a 4n sono permesse dalla simmetria se il processo di approccio degli orbitali è del tipo supra-antara , mentre sono proibite se il processo è del tipo supra-supra. CN NC H CN NC [π π14a+π π2s] 14 e HOMO a +2e 16 e (4n) s LUMO H 58 REAZIONI CHELOTROPICHE Le reazioni chelotropiche costituiscono una sottoclasse delle cicloaddizioni; esse si caratterizzano per il fatto che gli orbitali atomici nell'orbitale molecolare fornito da uno dei due partner di reazione appartengono allo stesso atomo. Addizione di carbeni al doppio legame C=C + CH2 (S) E' una reazione del tipo [2+2] e quindi, a prima vista, dovrebbe essere proibita dalla simmetria secondo la regola di di WoodwardHoffmann. La reazione invece è molto comune Approccio lineare LUMO HOMO HOMO LUMO Approccio non lineare HOMO LUMO LUMO HOMO 59 REAZIONI CHELOTROPICHE Reazioni di estrusione + SO2 SO2 + SO2 SO2 cis trans E' una reazione [π π4s+2s] dunque permessa dalla simmetria. Questa volta l'approccio lineare è senza problemi. HOMO s S O s s LUMO O LUMO s S O HOMO O HOMO s S O S s O LUMO O O La reazione è reversibile per semplice riscaldamento ed anche la reazione inversa può essere trattata in termini di interazioni HOMO-LUMO. 60 REAZIONI DI ESTRUSIONE LUMO CH3 - SO2 S H3C HOMO O O movimento disrotatorio LUMO - SO2 S H3 C CH3 OHOMO O movimento disrotatorio 61 Altro esempio SO2 -SO2 [π π6s+2s] SO2 s S O O s -SO2 SO2 Approccio non lineare A prima vista sembrerebbe logico il meccanismo sopra riportato,ma un tale approccio è proibito dalla dalla regola di Woodward-Hoffmann. La reazione, infatti, è del tipo [ π6 + σ2] ed è perciò una reazione 4n, per cui è permessa dalla simmetria solo se l'approccio è del tipo supra-antara e non supra-supra come sopra riportato. Infatti la reazione non è mai stata osservata. Il prodotto di estrusione del solfone cis è il trans-cis-cis triene e non il trans-cis-trans. HOMO triene SO2 a [π π6s+2a] 4n s S O O Approccio lineare LUMO In questo caso la reazione è permessa dalla simmetria e segue la regola di Woodward-Hoffmann, essendo l'approccio del tipo supra-antara 62 HOMO O S s s O S LUMO LUMO O S s s S O HOMO HOMO O a s S O S LUMO LUMO O a S s O S HOMO 63 RIARRANGIAMENTI SIGMATROPICI I riarrangiamenti sigmatropici sono quelle reazioni in cui un legame σ (o meglio un sostituente) si sposta, attraverso un sistema coniugato, su un nuovo sito. Trasposizione 1,5 sigmatropica H H Questa reazione può essere trattata come una reazione di cicloaddizione dell'orbitale molecolare σ (HOMO) del legame C-H sull'orbitale π (LUMO) del sistema dienico. H Me LUMO D 250°C H Et Me Me D Et Me s s E' una reazione del tipo [π π4s + σ2s] (4n+2) perciò permessa dalla regola di W.H. H HOMO s Un caso esemplare H H 1. LDA 2. CH3I CH3 H3C H CH3 H H H H 64 LUMO π s H HOMO σ a I riarrangiamenti 1,3-sigmatropici sarebbero possibili, secondo la regola di W.H., solo attraverso un processo del tipo supra - antara , essendo una reazione [ π2a + σ2s]. Tuttavia, come risulta anche dallo schema sopra riportato sono geometricamente impossibili. Infatti, questi riarrangiamenti non sono mai stati osservati in reazioni termiche. In un sistema coniugato più esteso sono geometricamente possibili, magari a temperature più elevate, riarrangiamenti 1,7-sigmatropici, cioè del tipo [ π6a + σ2s] del resto previsti dalla regola di W.H. C 4H9 H C 4H9 H LUMO a H HOMO s 65 DIMOSTRAZIONE DELLA VALIDITA' DELLA REGOLA DI WOODWARD-HOFFMANN Riarrangiamento 1,7-sigmatropico C4H9 H C4H9 H LUMO [π π6a + σ2s] 8e (4n) a s H HOMO In un sistema ciclico il riarrangiamento 1,7-sigmatropico diventa geometricamente impossibile attraverso un processo del tipo supra-antara come previsto dalla regola di W.H. In realtà a temperature elevate un riarrangiamento ha luogo, ma è un riarrangiamento 1,5. H H H 150°C CH3 [1,5] 3 4 5 1 6 7 H CH3 esige condizioni più drastiche CH3 CH3 H 2 H 3 4 2 5 H 1 6 7 CH3 geometricamente impossibile 66 IL CICLO DELLA VITAMINA D R R = CH3 ergosterolo (veg.) R = H deidrocolesterolo (an) HO hν ν R' HO R' HO enzimi R' OH HO 67 Trasposizioni Sigmatropiche che Coinvolgono Legami Carbonio-Carbonio e Carbonio-Eteroatomo Le reazioni sigmatropiche non sono ristrette al solo shift di idrogeno, ma il processo può coinvolgere altri tipi di atomi quali il carbonio e l'ossigeno, e non solo legami π, ma anche legamiσ carbonio-carbonio. [3,3] H HOMO H s LUMO Si tratta praticamente di una trasposizione sigmatropica [1,5] dove però uno dei due legami π è sostituito da un legame σ carbonio-carbonio (si ricordi che il legame σ carbonio-carbonio del ciclopropano ha molte caratteristiche in comune con il doppio legameπ di un alchene) 68 Il Riarrangiamento di Cope Il riarrangiamento di Cope è un riarrangiamento sigmatropico in cui nello shift sono coinvolti legami carbonio-carbonio. [3,3] [3,3] Il riarrangiamento di Cope può essere assimilabile ad una reazione del tipo [ 4+2] che, secondo la regola di W.-H. deve avvenire con un processo di sovrapposizione degli orbitali del tipo supra-supra . In questo caso, un legame σ sirompe per formarne dueπ e due legami π formano un legame σ il tutto in maniera concertata. s HOMO "diene" s LUMO "dienofilo" LUMO "dienofilo" s HOMO "diene" s 69 Riarrangiamenti che coinvolgono un numero di elettroni pari a 4n sono molto meno comuni, infatti uno di componenti deve subire un processo antarafacciale per obbedire alla regola di W.-H. e ciò comporta restrizioni geometriche molto severe. Il riarrangiamento 1,3-sigmatropico di Berson H H OAc AcO 300°C H H H H H OAc H H LUMO OAc HOMO A prima vista questo tipo di riarrangiamento, realmento osservato, sembrerebbe contraddire la regola di W.H.. In realtà, storicamente tale argomento fu usato per mettere in crisi la teoria degli orbitali di frontiera, esso invece costituì una delle prove più schiaccianti a favore di tale teoria. s a Permessa dalla regola di W.- H. ma geometricamente impossibile AcO H H Configurazione invertita !! s AcO H AcO a H D H D D H AcO Un meccanismo di questo tipo presuppone la presenza di un orbitale p migrante; ecco perchè lo shift 1,3 di idrogeno non è mai stato osservato. H 70 IL RIARRANGIAMENTO DI CLAISEN Un caso particolare del riarrangiamento di Cope è rappresentato dal riarrangiamento di Claisen in cui nella formazione e rottura dei legami viene coinvolto un eteroatomo, più frequentemente l'ossigeno. O * O O Aldeide γ,δ-insatura γ,δ HOMO "diene" O LUMO "dienofilo" s EtO EtO + Hg(OAc) 2 EtO HgOAc HgOAc OH H O EtO AcHg H O EtO AcHg O 71 Esempi di sintesi attraverso il riarrangiamento di Claisen OEt OH O O + un feromone - O O O Br + H DMF CF3CH2OH OH centro hard + - O + - + - Na - + + O + O-alchilazione DMF solvente dipolare aprotico ROH HOR ROH O HOR centro ROH soft C-alchilazione CF3CH2OH solvente protico - O H OH 72 REAZIONI ELETTROCICLICHE Le reazioni elettrocicliche sono quella specie di reazioni pericicliche in cui un anello si forma (o si apre) intramolecolarmente trasformando un legame π in uno σ (o un legame σ in uno π). 200°C Queste reazioni possono essere trattate come cicloaddizioni ed è più semplice condiderarle nella direzione di un'apertura dell'anello. a A s B C conrotatorio B A C D D La reazione è del tipo [2 + 2] e, per la regola di W.-H. è permessa solo se le interazioni sono del tipo supra-antara ([π π2s+σ σ2a]) i movimenti di A e C avvengono entrambi in senso orario cioè conrotatorio. H3C CH3 CH3 conrotatorio H3C conrotatorio Nel caso in cui la reazione coinvolge 6e (es. [4+2]), per la regola di W.-H. essa è possibile attraverso un processo del tipo supra-supra, di conseguenza A e C devono ruotare in senso opposto uno all'altro cioè i movimento è disrotatorio . LUMO C A B HOMO disrotatorio D A B D C 73 Esempi Disrotatorio CH3 H CH3 CH3 H H H CH3 6e (4n+2) [π π4s+σ σ2s] Conrotatorio LUMO CH3 a CH3 s HOMO 8e (4n) [π π6s+σ σ2a] CH3 H3C s Cl Disrot. 2e (4n+2) Cl LUMO s H3C CH3 HOMO In questo caso particolare, la rotazione dei legami, e quindi il movimento dei gruppi, avviene verso l'interno dell'anello. In effetti una rotazione verso l'esterno provocherebbe un'interazione di antilegame fra il LUMO del legame C-Cl e gli incipienti orbitali p sui carboni 2 e 3. LUMO conrot. 4e (4n) HOMO 74 EFFETTI STEREOCHIMICI SECONDARI O veloce H H O endo + O O H H O O O O lenta Termodinamicamente meno stabile O eso Termodinamicamente meno stabile Ep Eeso Eendo ∆G° endo eso cdr Il metodo degli orbitali di frontiera è l'unico che può dare una spiegazione a questo fenomeno. Benchè le interazioni secondarie non portino alla formazione di nuovi legami, esse abbassano l'energia dello stato di transizione della reazione che porta al prodotto O endo che quindi può essere ottenuto in condizioni di controllo cinetico. Per riscaldamento l'endo si trasforma irreversibilmente nel prodotto eso termodinamicamente più stabile. HOMO O O LUMO 75 LA REAZIONE DI DIELS-ALDER 1. REATTIVITA’ La regola di Woodward-Hoffmann stabilisce che una reazione [4+2] è termicamente possibile se la sovrapposizione degli orbitali di frontiera (HOMO-LUMO) avviene con un processo del tipo supra-supra. Tuttavia non dice nulla circa l’effetto dei sostituenti, sia nel dienofilo che nel diene, sulla velocità del processo. La velocità di una reazione chimica è funzione inversa dell’energia di attivazione e, come abbiamo visto, quest’ultima, specialmente quando a reagire sono specie neutre, è condizionata dalla differenza di energia HOMO-LUMO come si evince dal 3° termine dell’equazione di Klopman e Salem. o v o v …. + ΣΣ -ΣΣ 2(Σabcracsbβ ab)2/Er-Es r s s r Più vicini energeticamente sono gli orbitali HOMO e LUMO più bassa è l’energia di attivazione e di conseguenza più alta è la velocità della reazione. Effetti dei sostituenti sia sul diene che sul di enofilo • Sostituenti come il vinile o il fenile (sostituenti C) che possono coniugare con il doppio legame fanno aumentare l’energia dell’HOMO mentre fanno diminuire l’energia del LUMO. • Sostituenti elettron attrattori (sotituenti Z) abbassano l’energia sia dell’HOMO che del LUMO. • Sostituenti elettron donatori (sostituenti X) innalzano sia l’energia dell’HOMO che quella del LUMO. 76 E 3.0 1.5 1.0 LUMO 0 -9.0 -9.1 -10.5 X C -10.9 HOMO Z E 2.5 1.0 LUMO 0.5 -0.5 -8.2 -9.1 -8.5 C X -9.5 Z HOMO 77 E 2.3 1.0 0.7 LUMO -0.3 -8.5 -8.7 -9.1 C -9.3 X HOMO Z C= Z = -CHO, X = RO-, Si ricordi che: -C=N, R2N-, -NO 2 -COR, RHN- -R, -COOH, CF 3, ecc... ecc... 1 eV = 23 Kcal/mole = 96.5 KJ/mole 78 Nella maggior parte delle reazioni di Diels-Alder è l' HOMO del diene ad interagire con il LUMO del dienifilo . Tali reazioni vengono dette reazioni di Diels-Alder a domanda elettronica normale. Vi sono, tuttavia, delle reazioni in cui è l' HOMO del dienofilo ad interagire con il LUMO del diene . Ciò accade quando un dienofilo a cui sono legati gruppi fortemente elettron donatori viene fatto reagire con dieni sostituiti con gruppi fortemente elettron attrattori. Queste reazioni vengono dette reazioni di Diels-Alder a domanda elettronica inversa. In ogni caso, è la differenza di energia HOMO-LUMO a determinare il tipo di interazione 3.0 1.0 LUMO dienofilo LUMO diene 0 LUMO dienofilo ∆E = 11.9 eV -0.5 LUMO ∆E = 12.5 eV diene ∆E = 9.1 eV ∆E = 8.5 eV -9.0 -9.1 HOMO diene -10.9 HOMO dienofilo -9.5 HOMO diene X HOMO dienofilo Z Z Reazione di D.-A. a Domanda Elettronica Normale Reazione di D.-A.a Domanda Elettronica Inversa 79 Esempio di reazione di D.-A. a domanda elettronica normale R R R R + O R= 100% in 24 h a 20°C O O 78% in 17 h a 165°C e 900 Atm R=H Esempio di reazione di D.-A. a domanda elettronica inversa R' R R N R' + N EtO R = R' = OEt 75% in 4 min a 25°C R = CH2OH, R' = H 75% in 275 min a 100°C R = CN, R' =H 75% in 1080 min a 100°C EtO CH2OH CN 80 Modulazione degli effetti nella reazione di Diels Alder O Ph Ph Ph Ph O Ph + Y Ph Ph Y Ph Y= p-N(CH3)2 p-OCH3 H p-Cl m-NO2 k2 (x106) 338 102 73 78 79 88 k2 rel. 4.6 1.4 1 1.1 1.1 1.2 d.e. inversa E p-NO2 d.e. normale d. e. inversa N Ph OCH3 Ph d. e. normale O Ph Ph LUMO Cl NO2 NO2 HOMO 81 Esiste una buona correlazione fra il log della costante cinetica delle reazioni di D.-A. e la differenza di energia ∆EH-L . L'eccezionale reattività del ciclopentadiene con un determinato dienofilo pùò essere spiegata considerando che nel ciclopentadiene la conformazione cisoide a più alta energia è bloccata dal ponte metilenico, mentre il butadiene si trova nella conformazione termodinamicamente più stabile e non reattiva. In altre parole, per poter reagire il butadiene deve prima spendere una certa quantità di energia per trasformarsi nella conformazione reattiva, cosa di cui non ha bisogno il ciclopentadiene. NC CN OCH3 NC + log k NC CN CN + NC CN O + O O O + O O ELUMO-EHOMO E Eb Ecy Prodotti cdr 82 IL RUOLO DEI COEFFICIENTI NEGLI ORBITALI DI FRONTIERA Fino ad ora nella nostra trattazione abbiamo posto l'accento solo sul significato del denominatore dell'equazione di Klopman e Salem, cioè della differenza di energia fra gli orbitali di frontiera interagenti. o v .......... + o v ΣΣ -ΣΣ r s s r 2(Σ Σabcracsbβab)2 Er-Es Tali coefficienti ( cra e csb ) influenzano in modo particolare la regioselettività, la sitoselettività e la periselettività di una cicloaddizione di Diels-Alder. La regioselettività si riferisce all' orientamento di una cicloaddizione. OCH3 OCH3 OCH3 CHO CHO + CHO ψ-meta ψ-orto La sitoselettività è la predilezione di un reagente per un sito di una molecola polifunzionale in presenza di più siti reattivi dello stesso tipo. O O CN + CN + CN O O CN O CN 62% CN O 16% 83 La periseletività è uno speciale caso di sitoselettività in base alla quale, nella reazione con un "dienofilo", un poliene può coinvolgere l'intero sistema coniugato, gran parte di esso o, addirittura, solo una piccola parte di esso (ad esempio, una sola coppia di elettroni π). Esempio: + O O O Solo un doppio legame del tropone è impegnato. E' una reazione [ π4s+π2s] Nella reazione viene coinvolto tutto il sistema p del tropone. E' una reazione [ π4s+π6s] 84 LA COMBINAZIONE DEGLI ORBITALI DI FRONTIERA La combinazione preferita nella formazione di legamiσ in una reazione periciclica è quella che prevede la sovrapposizione dell'orbitale p a coefficiente maggiore (nell'orbitale molecolare di frontiera) di una specie con l'orbitale p a coefficiente maggiore nell'orbitale di frontiera del partner di reazione. In altre parole vale la regola: Orbitale p grande di A Orbitale p grande di B Orbitale p piccolo di A Orbitale p piccolo di B come può essere dimostrato anche algebricamente. Si ricordi, infatti, che il contributo del 3° termine dell'equazione di Klopman all'abbassamento dell'energia di attivazione è funzione del prodotto dei coefficienti degli orbitali atomici interagenti facenti parte degli orbitali molecolari di frontiera dei due partner di reazione. occ. v occ. v +Σ s r sΣ - Σ x x+n y y+m xy + (x+n)(y+m) Σr 2 (Σ cra csb βab) EL - EH x x+n 2 y+m y x(y +m) + y(x+n) ∆ = xy + (x+n)(y+m)- x(y +m) + y(x+n) = nm 85 VALUTAZIONE EMPIRICA DEI COEFFICIENTI DEGLI ORBITALI ATOMICI NEGLI ORBITALI MOLECOLARI DI FRONTIERA DELLE SPECIE REAGENTI IN UNA REAZIONE DI DIELS-ALDER Olefine C - sostituite (C = vinile, fenile) I coefficienti sono in pratica quelli che si osservano su uno dei doppi legami del butadiene nell'HOMO e nel LUMO. C HOMO C 0.371 - 0.371 0.600 0.600 LUMO Olefine Z-sostituite (Z = CN, COOH, COOR, CF 3, NO2 COR, CHO..) I coefficienti sono grosso modo quelli derivati dalla mediazione fra un diene e di un carbocatione allilico. O O + 0.707 0.371 Z + -0.371 Z + 0.600 0.600 0.500 0.707 HOMO LUMO Olefine X-sostituite (R2N, OR, R, SR, ...) I coefficienti sono grossomodo quelli che si ricavano da una mediazione fra quelli dell'etilene e quelli del carbanione allilico. X X + X 0.707 -0.707 + 0.707 X -0.707 + 0.707 HOMO 0.707 0.500 LUMO 86 87 88 Dieni Sostituiti a C-1 -0.418 0.418 HOMO 0.521 0.521 LUMO Dieni Z-sostituiti Z -0.500 -0.418 Z 0.418 0 + 0.521 + 0.500 0.521 HOMO LUMO Dieni X-sostituiti -0.500 -0.600 X X 0.500 0.600 * 0 + 0.600 + 0.576 HOMO 0.600 0.500 LUMO *In questo caso la determinazione empirica non dà un risultato soddisfacente (il valore in parentesi è derivato dal calcolo teorico) 89 Semplice regola empirica per stimare l'orientamento e la regioselettività nelle reazioni di Diels-Alder 1. Stimare le energie dell'HOMO e del LUMO di entrambi i partner di reazione. 2. Individuare la coppia HOMO -LUMO che fornisce il valore più basso di DE. 3. Utilizzando tale coppia HOMO-LUMO calcolarne approssimativamente i coefficienti sugli atomi di carbonio coinvolti nella formazione dei legami C-C. 4. Combinare fra loro gli atomi aventi nei due partner il coefficiente maggiore e quelli aventi il coefficiente minore. Z Z Z X Z X X X LUMO HOMO HOMO LUMO (-0.5 eV) (-9.0 eV) (-9.5 eV) +3.0 eV) ∆EH-L = 8.5 eV ∆EH-L = 12.5 eV non osservata!!! 90 CICLOADDIZIONI 1,3-DIPOLARI 1,3-Dipoli di tipo propargilico-allenico Valori in (c )2/15 eV ilide del nitrile H C N CH2 Ph C N CH2 -6.4 H C N CH2 Ph C N CH2 C N C LUMO -7.7 C N C HOMO 1.07 1.8 -9.0 1.50 C N N 0.66 N N CH2 CH2 0.64 0.9 0.6 diazalcano N N 0.69 0.56 C N N 1.57 LUMO HOMO 0.85 ossido del nitrile 1.18 H C N O H C N O Ph C N O Ph C N O -10 1.0 -0.5 H 0.17 C N OH LUMO -11 C N O HOMO 0.81 immina del nitrile 0.92 H C N NH H C N NH Ph C N NH Ph C N NH -7.5 0.1 -9.2 - 0.5 N N 0.90 0.37 azide 0.1 N N N N NH 0.36 C LUMO C HOMO 1.45 0.76 N N N LUMO N N N HOMO H N N N N N NPh -9.5 Ph N N N -11.5 1.55 - 0.2 0.96 ossido nitroso N N O 1.24 0.72 0.19 -1.1 N N O LUMO -12.9 N N O HOMO N N O 0.67 1.33 91 92 CICLOADDIZIONI 1,3-DIPOLARI Metodi di preparazione di 1,3-Dipoli di tipo allilico MeO2C O R N H MeO2C N R H N C CO2Me CH H C CH2O 1 R R C N N R C R O N R2 H2 C R1 R CO2Me O C N CH2 azometino imina H OH R1 C OH N R O C N OH R1 R1 O C N R R2 OH N R R O O H C azometino ilide R1 R1 H C N R2 O 2 R NHOH R R2 Nitrone R1 R1 O O3 O 1 R R R O O R R O O 1 R R + R1 R1 ossido del carbonile R N Ph PA O N Ph H R tmboxScCl2+ Ph H H C N O R immina del carbonile OSiMe3 R I SmI2 o Cl O R Cl R R R O R ilide del carbonile O R R = alchile R R1 93 94 95 96 97 98 99 CICLOADDIZIONI 1,3-DIPOLARI Regioselettività nelle reazione di cicloaddizione 1,3-dipolari X 3.0 LUMO -0.3 NH C 1.0 Ph N Z 0.0 N -1.4 CH H LUMO N 6.6 CH 5.6 Ph Ph -5.6 N -9 Ph N -8 C CH NH 6.6 9.5 7.6 N -8.6 8.6 -10.9 Z HOMO CH H N Ph 36 : 62 Ph + N N N Ph NPh N N Ph HOMO N+ NC N N- LUMO NC N N X HOMO 100 CICLOADDIZIONI 1,3-DIPOLARI Regioselettività nelle reazione di cicloaddizione 1,3-dipolari X 3.0 O LUMO 0.32 N 1.11 C 1.0 Z 0.3 CH H 0.0 LUMO 11.7 9.3 9.7 8.7 8.3 11.2 1.15 O HOMO C -8.7 N -8 -9 -10.9 CH H 1.11 O C O N Ph N C Ph H HOMO LUMO R R = Me 98:2 R = Ph 100:0 R = CO2Et 70:30 Ph N Ph Ph O+ N Ph O N + CO2Me Ph Z R O + X HOMO 101 CICLOADDIZIONI 1,3-DIPOLARI Ozonizzazione: una reazione di cicloaddizione 1,3-dipolare X 3.0 1.0 C Z 0.0 O LUMO LUMO 16.5 -2.2 O O 5.8 6.8 13.5 8.7 14.5 -8 C -9 Z -10.9 O HOMO -13.5 O O O O + O O O O O+ -O H O H CH2O + O Zn o PPh3 - HO O O O H O O+ X HOMO 102 REAZIONI PERICICLICHE FOTOCHIMICHE Il primo stadio di una reazione fotochimica è la fotoeccitazione di uno dei componenti (generalmente quello con il cromoforo più efficace nell'assorbire la radiazione). A hn LUMO LUMO HOMO HOMO A* Stato fondamentale Stato eccitato Il secondo stadio della reazione consiste in una interazione fra la specie fotochimicamente eccitata ( A*) ed il suo partner di reazione (spesso la stessa apecie) nello stato fondamentale. In questo tipo di reazioni vi sono due tipi di interazione, a livello degli orbitali di frontiera, particolarmente forti: 1. Un'interazione fra l'orbitale π* occupato da un solo elettrone (ex LUMO) ed il LUMO della specie non eccitata. "LUMO" - LUMO 2. Un'interazione fra l'orbitale π, occupato da un solo elettrone, (ex HOMO) della specie eccitata e l'orbitale HOMO del partner di reazione nel suo stato fondamentale. "HOMO" - HOMO 103 Entrambe le interazioni sono molto forti (specialmente nelle dimerizzazioni) poichè gli orbitali interagenti hanno energie molto simili (il secondo stadio è infatti molto veloce). Le perturbazioni sono del primo ordine ed il trattamento matematico di esse esige una modifica del 3° termine dell'equazione di Klopman e Salem ( Σab 2craCsb βab). E1 E3 E2 AB* Eccimero A* o Ecciplesso B A A*+ B AB* hν ν A* AB* C L'energia di AB* può essere molto bassa, rispetto a quella delle specie interagenti, tanto che, in qualche caso, è possibile identificare l'intermedio detto eccimero o ecciplesso . 104 La Regola di Woodward-Hoffmann per le Reazioni Fotochimiche Le foti interazioni "HOMO"-HOMO e "LUMO"-LUMO porta a riscrivere la regola di Woodward-Hoffmann per le reazioni fotochimiche nei seguenti termini: Reazioni pericicliche fotochimiche che coinvolgono (4n+2) elettroni sono permesse dalla simmetria degli orbitali solo se il processo di sovrapposizione è del tipo supra-antara. Reazioni pericicliche fotochimiche che coinvolgono (4n) elettroni sono permesse dalla simmetria degli orbitali solo se il processo di sovrapposizione degli orbitali è del tipo supra-supra. Reazione [4+2] (4n+2). a a "LUMO" "HOMO" antilegame s antilegame s HOMO LUMO Le reazioni di diels-Alder fotochimiche sono poco note e quelle conosciute non sono reazioni concertate. Reazione [2+2] (4n). "HOMO" s s "LUMO" s HOMO s LUMO 105 Cicloaddizioni Fotochimiche + hν ν + [π π2s+π π2s] * hν ν + * + [π π2s+π π2s] * * hν ν [π π4s+π π4s] O O hν ν [π π6s+π π6s] O 106 Reazioni Chelotropiche "LUMO" "HOMO" a hν ν SO2 a s s S S LUMO O O O O HOMO hν ν SO2 Riarrangiamenti Sigmatropici HOMO O hν ν [1,3] supraf. O H O H "HOMO" H hν ν HOMO [1,7] "HOMO" H H H H H CH3 + H CH3 + CH3 Reazioni Elettrocicliche H hν ν OH 4e disrot. hν ν OH 4e disrot. H hν ν 6e conrot. 107 CAPITOLO III REAZIONI RADICALICHE 108 109 REAZIONI RADICALICHE x z Radicale elettrofilo Radicale nucleofilo X Z 110 Caratteristiche Generali delle Reazioni a Catena tra Radicali e non-Radicali 1. Il carattere radicalico non viene distrutto durante la reazione, si può perciò lavorare con quantità catalitiche di iniziatore. 2. Molte reazioni radicaliche, benchè estremamente veloci, non sono controllate dalla diffusione (k< 109) e le selettività possono essere influenzate variando i sostituenti sia sul centro radicalico che nel partner di reazione. 3. La concentrazione della specie non-radicalica può essere facilmente controllata. 111 Requisiti per il Successo di una Reazione Radicalica a Catena 1. Le selettività dei radicali coinvolti nella catena devono essere diverse fra loro. 2. Le reazioni fra radicali e non radicali devono essere più veloci della ricombinazione dei radicali stessi. 2 .SnBu3 Bu 3Sn-SnBu3 + hν ν Bu 3Sn-H N C + ∆ AIBN N N Bu 3Sn. + N 2 (CH3)2CHCN C N Y R R Y Bu3SnH Bu3SnX Bu3Sn RX R Y 112 Requisiti di Reattività I processi a catena sono determinati dalle reazioni delle specie radicaliche. Perchè si abbia una reazione a catena, la specie radicalica deve essere sempre rigenerata, perciò, la velocità delle reazioni tra specie radicaliche e non radicaliche (vp) deve essere sempre maggiore della velocità di ricombinazione dei radicali (vt) che interrompono la catena. vp = kp [R.] [X] vt = kt [R.] [R.] dato che kt 10 9-1010 e [R .] 10 -7-10-8 se [x] = 0.1, affinchè sia v p > 10 2 vt kp deve essere maggiore di 10 2 Dunque, a scopo sintetico sono utili solo quelle reazioni i cui stadi di propagazione hanno kp > 10 2 113 Requisiti di Selettività La reattività di un radicale nei confronti di un alchene in una reazione di addizione, può essere modulata con una scelta opportuna dei sostituenti sia sul centro radicalico che sul doppio legame C=C. Ad esempio, radicali al carbonio aventi gruppi elettrondonatori ( alchili, OR, NR 2...) in posizione α (radicali "soft" ) reagiscono molto velocemente con alcheni sostituiti con gruppi elettron- attrattori (basso LUMO). Al contrario, radicali al carbonio aventi gruppi elettronattrattori (COR', CHO, CX 3, CN, NO2...) in posizione α (radicali "hard" ) reagiscono molto velocemente con alcheni sostituiti con gruppi elettron- donatori (alto HOMO). Ad esempio, il cosiddetto metodo allo stagno è applicabile solo nei casi in cui il processo di addizione all'alchene genera radicali di diversa polarità, o a polarità opposta. I + Bu3 SnH CN AIBN CN 95% RO2C Cl + RO2C OC4H9 Bu3 SnH AIBN RO2C RO2C OC4H9 60% 114 I + CN Bu3SnH AIBN CN 95% Y R k1 Y R. k2 k1 = 10 5-106 k3 = 10 - 10 2 k1/k3 = 10 4 R-H Bu3SnH R Y Y k1/k2 = 1 k3/k4 = 10 -4 k3 R Y Y R k4 Bu3SnH Y La reazione può avere successo solo se: [Y....] / [ Bu Y k5 3SnH] Bu 3Sn Y Bu3Sn. k6 R-X R. + Bu3SnX Onde evitare il più possibile la competizione dell'attacco di Bu3Sn. all'alkene ( k6 >> k5) è preferibile partire da ioduri alchilici piuttosto che i bromuri o cloruri ( k6-I /k6-Br = 103). > 10 2 115 Reazioni Elementari tra Radicali e non-Radicali negli Stadi di Propagazione I processi di propagazione delle catene sono per la maggior parte esotermici con energie di attivazione molto basse, per cui è possibile usare i valori di energia di legame dei legami che si formano e si rompono come un'indicazione grossolana della velocità di reazione (parametri termodinamici). A causa del carattere reactant-like degli stati di transizione, alle reazioni radicaliche può essere applicata la teoria FMO (parametri cinetici). Addizione R. + AB RAB. LUMO SOMO R. C4H9 kC6H11. 1.0 C6H5 84 X: Z SOMO CO2Me CHO 3000 R. HOMO 8500 C6H5 C 6H 5 N C6H5 CO2Et O k (EtO2C)2CH . 23 3.5 1.0 116 Riarrangiamento RAB. RAB. R R R R R R Elettron-transfer R- R. + Mn+ + M(n-1)+ oppure RX.- + RY RX + R. + Cu++ + O2 R-OO- + Cu+ R. + C6 H5 R CN RY.- CN R CN C6H5 Cu++ OR' R CN C6H5 R'OH R CN C6H5 117 Sostituzione (Estrazione) RA + B. R. + AB H O R O R' R. H H O R R' O .OR H O R O RO. è un radicale elettrofilo (basso SOMO) e preferisce agire su un orbitale -O-C-H ad alto HOMO. R. è un radicale nucleofilo (alto SOMO) e preferisce agire su un orbitale O=C-C-H a basso HOMO Eliminazione (frammentazione) RAB. R C O. RA + B. R. + OR RO. + O R' 118 Reazioni Radicaliche e Ioniche a Confronto Chemoselettività A causa dei requisiti di selettività precedentemente discussi, nelle reazioni radicaliche possono essere coinvolti solo quei gruppi che reagiscono con velocità relativamente elevate (k2 > 102). Ciò implica che le reazioni radicaliche sono compatibili con funzionalità del tipo OH, NH2, C=O, ..... senza preventiva protezione. Br Bu3SnH AIBN, C6H5, rif. HO HO CN CN Bu3SnH OH CN OH CN Regioselettività Poichè le interazioni degli orbitali (3° termine dell'equazione di Klopman e Salem) sono il fattore più importante nelle reazioni radicaliche, la regioselettività di queste è di gran lunga superiore a quella delle reazioni ioniche dove c'è un consistente contributo del termine coulombiano. SOMO soft Nu HOMO hard LUMO O 119 Confronto tra Reazioni Radicaliche e Reazioni Ioniche Ciclizzazioni prodotto cinetico prodotto termodinamico HOMO δ+ δ+ H SOMO H ST rad. ST rad. Determinanti i fattori sterici e di tensione determinanti i fattori elettronici H I Bu3SnH In. H H 60% HO TFA CF3COO H H H 30% 120 Confronto tra Reazioni Ioniche e Radicaliche Stereoselettività Br H H3 C CH3 H3 C H Br CH3 Br HBr Br -80°C H HBr Br -80°C H3C Br CH3 CH3 H Br Br CH3 H H A temperatura ambiente si ottiene una miscela quasi equimolare dei due stereoisomeri. In generale, il radicale a ponte di bromo è meno stabile dell'analogo ione bromonio, ciò comporta una notevole perdita di stereoselettività. Una buona stereoselettività si riscontra, al contrario, nelle reazioni radicaliche di sistemi ciclici. R R (H2C)n R (R) (S) + Y (H2C)n (R) (S) + (H2C)n (R) Y R Alchene OCH3 H2C=CHCN NHAc H2C=CClCN OCH3 H2C=CHCO2Me OCH3 t-EtO2CCH=CHCO2Et Y trans : cis n=1 n=2 77:23 65:35 >98 >95 88:12 70:30 98:02 92:08 121 Confronto tra Reazioni Radicaliche e Reazioni Ioniche Racemizzazione Poichè le reazioni radicaliche impiegate nella sintesi organica sono molto veloci, reazioni collaterali, come riarrangiamenti, β-eliminazioni, sono, in molti casi soppresse. Inoltre, le reazioni radicaliche vengono spesso condotte in assenza di acidi o basi cosicchè sono fortemente inibite reazioni ioniche collaterali come la racemizzazione. C C C C + RO- OR C C C C OR COOH H3C C H OH OR CH2I H3C C H CO2 R OBn O O COOH COOH HOH2C C H NH2 CO2Bn IH2C C H NHCO2Bn CO2H CO2Bn NHCO2Bn 122 Confronto tra Reazioni Radicaliche e Reazioni Ioniche Reazioni "Umpolung" Dal punto di vista della polarità le reazioni radicaliche possono essere considerate complementari alle reazioni ioniche. Infatti, rispetto a quest'ultime, le reazioni radicaliche mostrano una polarità invertita (daltedesco "Umpolung") RO RO C Specie elettrofila RO C Specie nucleofila X CN CN Bu3SnH Br AcO AcO AcO O OAc AcO AcO AcO O OAc OSiMe3 O AcO AcO AcO ZnCl2 O OAc CO2R (RO2C)CH + CO2R RO2C CO2R CO2R (RO2C)CH + CO2R RO2C CO2R 123 Formazione del Legame Carbonio-Carbonio HX -X. R C C H + X. Y H transfer R. + R C C Y Y XZ -X. R C C Z + X. Y Atom transfer Mn+ R C C + M(n-1)+ Y electron transfer 124 Trapping con Donatori di Idrogeno Metodo dello Stagno Idruro R-X Bu3SnH + Y R Y + Bu3SnX AIBN o DBP ∆ Y R R. Y Bu3SnH Bu3Sn R R-X Y AIBN = Aza-bis-isobutirronitrile CH3 CH 3 NC C N N C CN CH3 CH 3 ∆ CH 3 2 NC C + N N CH 3 DBP = Dibenzoilperossido O O O O ∆ o hν ν O O + CO2 125 Trapping con Donatori di Idrogeno Metodo dell'Idruro di Mercurio NaBH 4 RHgX RHgH Y R Y R. RHgH Hg RHgX + NaBH4 RHg. R Y Da Alogenuri RX Mg HgX RMgX O O O 92% O + O O O O 95 O O 5 : HgCl + HC CCO2Me NaBH4 41% CO2Me H H 72 H H : O O H3 C NaBH4 HgCl + . RHgX CO2Me 28 H 126 Trapping con Donatori di Idrogeno Metodo del Di-terz-butilperossido Y R R. Y R R-H Y Da Chetoni O hν ν 2 O O. DTBP O O C 10H21 DTBP + C8 H17 O O DTBP + (CH2)CO2H (CH2)10CO2H 127 Il Metodo Ossidativo Y = Gruppo elettron-donatore Y ZC Z = Gruppo Z C. elettron-attrattore (radicale elettrofilo ) Y Radicale nucleofilo Mn+ R Y O O R' + Mn+ R R' R O R" R2 R + M (n-1)+ R' R' O Mn+ R R" O Nu O R" R R' Nu R" R R' 128 La Sostituzione Omolitica Aromatica H O + R R' . Y O . Y Mn+ O H -H + O Y + Y O 2 Ce(OAc)4 O AcOH O R OMe CAN, MeOH OMe t. a., 10 min O R OMe OMe O R Resa, % O Distribuzione isomerica orto meta para OMe 87 82 2 16 Me 59 51 21 28 Mesitilene 66 Naftalene 50 Benzotiazolo 66 Isomero α isomeri 5- e 7- 129 La Sostituzione Omolitica Aromatica Il Metodo Riduttivo OH + OH- + Fe(III) H2O2 + Fe(II) CH3SOCH3 HO CH3SO2H + CH3 O S H3 C CH3 R-I CH3-I + R H Ar R Ar-H R-I Substrato Prodotto ICH2CO2Et p-Tol N H 90 O ICH2CCN N H N H N H ICH2CO2Et O Resa CH2CO2CH3 N H p-Tol CH2CO2CH3 CH2CO2CH3 O CH2CO2CH3 S CH2CO2CH3 S O DMSO, H2O2, FeSO4 X Aux I 20-40°C X * CO-Aux ee, 80-98% Aux. = N S O2 88 65 62 ICH2CO2Et + + H+ 59 ICH2CO2Me N H O Ar-R 130 131 PARTE SECONDA COMPOSTI ORGANOMETALLICI E LORO REAZIONI 132 133 ORGANOMETALLI POLARI C M IA 2.1 H Li C ∆el. = 1.5 C.I. = 43% Na C ∆el. = 1.6 C.I. = 47% K C ∆el. = 1.7 C.I. = 51% H C ∆el. = 0.4 C.I. = 4% 1.0 2.5 C Li 0.9 1.2 Na Mg K 0.8 IV A 134 NUCLEOFILICITA' e BASICITA' degli ORGANOMETALLI POLARI Il rapporto Nu/B diminuisce con l'aumentare dell'elettrone-gatività del centro basico (in pratica, andando verso destra e salendo verso l'alto nel sistema periodico degli elementi). Perciò gli organometalli cosidetti polari, sono specie estremamente nucleofile (Preferiscono reagire con centri elettofili soft, a basso LUMO, sono determinanti i fattori cinetici). Ordine di nucleofilicità: Nu/B Eln. RC- > R2N- > RO- > F 2.5 3.0 3.5 4.0 Nondimeno gli organometalli polari sono anche basi estremamente forti come può essere facilmente dedotto dai valori di pKa degli acidi coniugati (nella reattività come basi sono determinanti i fattori termodinamici, quali la stabilità dei prodotti) Ordine di basicità: RC- > R2N- > RO- > F pKa 48 38 15.7 3.17 135 Effetto della Natura del Metallo M sulla Reattività degli Organometalli Polari C-MgX < C-Li < C-Na < C-K Si 2K Hg Si Si K Base estremamente forte Superbase Effetto della Struttura del Gruppo R sulla Reattività degli Organometalli Polari R H3C-Li < R-CH2-Li < H R pKa = 48 50 60 R Li < R R Li ? (> 60) 136 137 138 Acido Super acidi HF-SbF5 FSO3H-SbF5-SO3 FSO3H-SbF5 FSO3H RNO2H+ ArNO2H+ HClO4 HI RCNH+ RCH(=OH)+ H2SO4 HBr ArC(=OH)OR+ HCl ArCO2H2+ ArCH(=OH)+ RC(=OH)R+ ArSO3H RC(=OH)OR+ ArOH2+ RCO2H2+ ArC(=OH)R+ Ar-OH-R+ CH(CN)3 Ar3NH+ H2C=OH+ R-OH-R R3COH2+ R2CHOH2+ RCH2OH2+ H3 O+ Ar-C(=OH)NH2+ HNO3 R-C(=OH)NH2+ Ar2NH2+ HSO4HF ONOH ArNH3+ ArNR2H+ RCOOH HC(=O)CH2CHO H2CO3 H2 S ArSH CH3COCH2COCH3 HC≡N Base pKa Appr. (→ H2O) SbF6FSO3RNO2 ArNO2 ClO4IRCN RCH(=O) HSO4BrArC(=O)OR ClArCOOH ArC(=O) RC(=O)R ArSO3RC(=O)OR ArOH RCOOH ArC(=O)R Ar-O-R C(CN)3Ar3N H2C=O R-O-R R3COH R2CHOH RCH2OH H2O Ar-C(=O)NH2 NO3R-C(=O)NH2 Ar2NH SO4= FNO2ArNH2 ArNR2 RCOO[HC(=O)CHCHO]HCO3HSArS[CH3COCHCOCH3]C≡N- -12 -11 -10 -10 -10 -10 -9 -7.4 -7 -7 -7 -7 -6.5 -6.5 -6.4 -6 -6 -6 -5 -5 -4 -3.5 -2 -2 -2 -1.74 -1.5 -1.4 -0.5 1 1.99 3.17 3.29 3-5 3-5 3-5 5 6.35 7.00 6-8 9 9.2 139 Continua Acido NH4+ ArOH RCH2NO2 R3NH+ RNH3+ HCO3RSH R2NH2+ NCCH2C≡N CH3COCH2COOR CH3SO2CH2SO2CH3 EtOOCCH2COOEt CH3OH H2 O Base NH3 ArO[RCHNO2]R3 N RNH2 CO3= RSR2NH [NCCHC≡N][CH3COCHCOOR][CH3SO2CHSO2CH3][EtOOCCHCOOEt]CH3OOH- RCH2OH RCH2CHO R2CHOH R3COH RC(=O)NH2 RC(=O)CH2R RCH2O[RCHCHO]R2CHOR3CORCONHRC(=O)CHR]- pKa 9.24 8-11 10 10-11 10-11 10.33 10-11 11 11 11 12.5 13 15.2 15.74 16 16 16.5 17 17 19-20 20 Ph-C6H4-CH2SO-Ph Ph-C6H4-CH2SO2-Ph - [Ph-C6H4CHSO-Ph] [Ph-C6H4CHSO2-Ph]- 20.08 18.91 23 RCH2COOR RCH2CN HC≡CH Ph2NH EtOC(=O)CH3 PhNH2 Ar3CH Ar2CH2 H2 NH3 PhCH3 CH2=CHCH3 PhH CH2=CH2 c-C3H6 CH4 C2H6 CH3-CH2-CH3 (CH3)3CH [RCHCOOR][RCHCN]HC≡CPh2NEtOC(=O)CH2PhNHAr3CAr2CHHNH2PhCH2CH2=CHCH2PhCH2=CHc-C3H5CH3C2H5[CH3-CH-CH3](CH3)C- 24.5 25 25 24.95 25.6 30.6 31.5 33.5 35 38 40 43 43 44 46 48 50 51 140 Effetto del Solvente sulla Basicità degli Organometalli Polari Et O R O Et R Mg X Mg R X Mg X Et O Et Et O Et Et Et Li R Li Li R Li R R Li Li R R Esamero in esano O R Mg X O Li R R Li Li Li R R Tetramero in THF Reattività: R-M (esano) << R-M (DEE) << R-M (THF) 141 142 Effetto della Coordinazione sulla Basicità degli Organometalli Polari Solventi (o Co-solventi): N R H3CO OCH3 Mg R DME (DiMethoxyEthane) N + N P N O- P+ O- M -O N N P+ O N N P+ N N N N HMPA (HexaMethylPhosphorictriAmide) Crown-eteri e Criptanti: O O O O Li O O O O O K O O 18-Crown-6 (18-C-6) 15-Crown-5 (15-C-5) O N O O O N O O 143 TMEDA H3C N CH3 N Li H3C CH3 R R-CH2-Li + K-O-tBu (LICKOR) CH3 Li O C R-CH2 K CH3 CH3 Si tenga Sempre presente la forte affinità del litio per l'ossigeno !!! 144 Preparazione degli Organometalli del 1° e 2° Gruppo A) Reazione del Metallo con Alogenuri R-X + Mg DEE o THF R-Mg-X Meccanismo: R-X- R-X + Mg . R-X- + Mg+ . + Mg+ R-Mg-X Il Magnesio Superattivo Mg + BrCH2-CH2Br DEE o THF MgBr2 + K MgBr2 + CH2=CH2 Mg + 2 KBr Precipitato nero pulverulento finissimo, si incendia all'aria R-X + 2 Li (X = Cl in DEE (X = Br in THF o DEE RLi + LiX R-Li libero da sali) R-Li. LiBr) 145 B) Scambio Idrogeno - Metallo (Metallazione) R-H + R'-Li R-MgX + pKaR-H = 55 R'-C=CH R-H + R'C=C-MgX pKa = 20 C2H5-MgX + HC=CH H O R-Li + R'-H XMg-C=C-MgX + C2H6 + n-BuLi O Li Stabile fino a 40-50°C Br Li -30°C + n-BuLi O O O pKa = 33 H H CH2 H CH3 LICKOR H K CH2 H CH3 146 Alcuni Gruppi Orientanti la Metallazione più Frequentemente Usati S O N Y R N Y *** O R N R Y ** R *** O N Y NR Y *** Y ** ** N N Y O- Y O ** O OR O NR2 O ** O N Y *** Y *** O Y O C Y *** ** N OTHP Y Y ** O N Y ** * OR P OR O 147 Altri Gruppi o-Orientanti la Metallazione O OLi N R N R H O OLi R R R R HO OH O + N O H N N C O H2N OR N O O Cl O OLi O N OMe OR N 148 Effetto della TMEDA sull'Orientamento della metallazione di Aril Derivati Me Ph O Ph Me O Li Li OMe Li Li Me O Li O Ph Me Ph TMEDA OMe N N N Li Li Li Me O N H Li N N 149 150 Sintesi di Indoline e Derivati dell'Indolo Regioselettivamente Fluorurati a Rf Br b Rf Rf NH2 NHBoc NHBoc c CO2Et OH CO2Et e Rf Br d Rf N Boc Rf N Boc N Boc f PO(OEt2)2 PO(OEt2)2 Rf N Boc a. (tBuOCO)2O (Boc anidride); b. tBuLi, CBr4, THF, -75°C; c. NaH, propargil-Br, THF; d. HSnBu3/AIBN, C6H6 rifl.; e. O=C(CO2Et)2, D. f. CH2=C(POOEt)2. R R R R F R F3C N Boc F N Boc N Boc N Boc N Boc F3C F R R R R F3CO N Boc CF3 R F3C N Boc N H CO Boc 3 OCF3 F N Boc CF3 N Boc 151 C) Lo Scambio Alogeno - Metallo R H C C H t + Bu-Li R THF C -120°C Br H C H Li Quando R = alchile, la reazione è stereospecifica con ritenzione di configurazione. O + n-BuLi H THF -78°C Li O Br Li + n-BuLi THF -78°C O O Quando l'alogeno è il bromo (o lo iodio), lo scambio alogeno-metallo è più veloce dello scambio idrogenometallo. Li Br n BuLi O H THF, -78°C O H Br LIDA THF, -78°C Li N O Li = LIDA 152 δ+ H H δ- R C Br δ- δ+ HH Br C R δ- Li δ+ δ- δ+ δ- δ+ Li δ- Li + HH Br C R δ+ Br + t BuLi Br + BuLi H3CO Br + t BuLi -120°C Li -70°C -120°C Li H3CO SiMe3 + Br s BuLi SiMe3 -70°C NC NC Br + BuLi -100°C Br Li Li NO2 Br Li NO2 + BuLi -100°C Br Li 153 Sintesi di Composti Aromatici Regioselettivamente Sostituiti Br Li BuLi THF, -78°C F F CX3COCOOEt X3C F COOR X3C OH COOR DAST CH2Cl2, 0°C F F X = H, F TIPS Br TIPS a Br b Br Br c TIPS TIPS MOMO HO d O Si O a. BuLi, TIPSCl, THF, -95°C; b. BuLi, o-C6H4Br2, THF, -78°C; c. BuLi, (TMSO)2, THF, -78°C; d. NaH, ClCH2OCH3, THF, 0°C. 154 Sintesi di Schlosser del Furano di Rosa centro soft Br Br LDA O THF, -78°C H Centro hard CH3 Li O Br Li Br CH3I BuLi O O THF, -78°C O Furano di rosa D) Scambio Metallo-Metallo THP-O-CH2-C=C-H + Bu3SnH hν ν THPO H C C H SnBu3 BuLi THPO H C C H Li O = THP 155 156 Determinazione del Titolo di una Soluzione di Butillitio Impurezze presenti in una soluzione di butilitio commerciale o non correttamente usata: C4H9-Li LiOH C4H9COOLi C4H9OOLi LiH + Metodo della Doppia Titolazione 1a Titolazione C4H9-Li + LiOH C4H9COOLi C4H9OOLi LiH + H2 O LiOH Nt = VH2SO4 NH2SO4 C4H10 LiOH + C4H9COOH C4H9OOH H2 Vp 2a Titolazione C4H9-Li + BrCH2CH2Br LiOH C4H9COOLi C4H9OOLi LiH Nimp = + H2 O DEE 0°C C4H9Br + LiBr + CH2=CH2 LiOH V'H2SO4 NH2SO4 Vp NC4H9Li = LiOH C4H9COOH + C4H9OOH H2 ∆V NH2SO4 Vp 157 Il Test di Gilman Ar Ar 1. R-Li 2. H3O+ C O OH C Ar Ar R Ar = Me2NC6H4 Chetone di Michler Ar OH C Ar I2, AcOH R Ar C R Ar I N C+ R N Intensamente colorato in Verde o Blu 158 159 160 Sintesi con Reattivi di Grignard A. Preparazione di Alcooli Primari 1. CH2O 2. H+/H2O R-MgX O 1. 2. H+/H2O B. R- CH2CH2-OH Preparazione di Alcooli Secondari 1. R'CHO 2. H+/H2O R-MgX 1. HCOOEt 2. H+/H2O C. R-CH2-OH R' CH OH R R CH OH R Preparazione di Alcooli Terziari EtO R-MgX + C O EtO R R C OH R 161 162 163 164 165 La Riduzione di MeerweinPondorff-Verley Reattivi di Grignard stericamente molto ingombrati sono in grado di reagire con chetoni per dare un alchene e l'alcool prodotto di riduzione del chetone, piuttosto che il normale prodotto di addizione al carbonile. R R2 R1 H R C C + C OMgX R3 R R R R1 R2 C H C R3 C Mg O R4 X 4 R1 H R R2 C R C C R4 R3 OMgX R R' C CH3 H CH3 C O M O A B H3C CH R' 3 R H + C C OM A = Riduzione di Meerwein-Pondorff-Verley B = Ossidazione di Oppenauer O 166 COMPOSTI ORGANOMETALLICI DEL RAME: I CUPRATI centro hard H3 C C H H C C 1. CH3MgBr 2. H3O+ O CH3 1. CH3MgBr, CuI centro soft 2. H3O+ H3C H C OH C C CH3 H CH3 H H H3C C O C C CH3 CH3 Preparazione: R-Li + CuI R-Cu + LiI Cuprati di 1a generazione 2 R-Li + CuI R2CuLi + LiI Cuprati di 2a generazione 3 R-Li + CuI R3CuLi + LiI I cuprati sono basi molto deboli se paragonate ai loro precursori R-Li, ma sono ottimi nucleofili. Cl Li Cl O Cl Cl H Cu O O 167 REAZIONI DEGLI ORGANOCUPRATI A. Sostituzioni Nucleophile SN2 R' R Cu X R' R-X + R'2CuLi R-R' + R'CuX B. Sostituzioni Nucleofile al Carbonio sp2 I CH3 (CH3)2CuLi 90% Br CH3 (CH3)2CuLi 81% C. Sostituzioni Nucleofile SN2' CH3 CH3 + (CH3)2CuLi 80% OCOCH3 C C OCOCH3 C5H11 + CH3Cu-LiBr-MgBrI H C C C C H 5 11 H3C (S) 100% 168 Reazioni di Cuprati 1. tC4H9Li I 2. CuCN CuCNLi DEE, -78°C OCH3 OCH3 CuCNLi OAc C DEE, -78°C OCH3 OCH3 Spencer, T. A. et al. J. Org. Chem. 2000, 65, 1919 169 REAZIONI DEGLI ORGANOCUPRATI D. Apertura di Epossidi OH 1. (CH3)2CuLi O tipo SN2 2. NH4Cl OH 1. (CH3)2CuLi tipo SN2' 2. NH4Cl O E. Addizione Coniugata a Composti Carbonilici α,β-Insaturi O R R 1. R2CuLi R' O R 2. NH4Cl R' meccanismo proposto: O R2Cu(I) + R O t Bu t Bu R2Cu(II) R' H R2Cu(II) + H R O R 1. (CH3)2CuLi 2. NH4Cl R R' + RCu(I) O O Bu t R' O R R' O t t Bu Bu + But 170 REAZIONI DEGLI ORGANOCUPRATI F. Addizione ad Esteri Acetilenici α,β-Insaturi O O O OCH3 OCH3 (R) OCH3 Cu H CuLi 2 Per avere un'ottima stereoselettività la reazione deve essere condotta rigorosamente a -78°C !!!! La stabilità del complesso intermedio dipende marcatamente dalla temperatura e già a -60°C si può avere l'isomerizzazione del doppio legame G. Addizione al Triplo Legame C=C . C2H5Cu MgBr2 + Cu.MgBr2 H H NH4Cl ElX H El H H Stereospecifica sin 171 172 173 174 Sintesi di Arilchetoni Regioselettivamente Fluorurati Br MgBr a F b F OTMS F CuBr.LiBr COOH c O d EtO F F F O MeO OTMS Br F a MgBr F b R F CuBr .S(CH3)2 O R F F N N c MeO d O EtO F R a. Mg, THF t.a.; b. CuBr.LiBr o CuBr.S(CH3)2, 3-oxocicloesene, TMSCl; c. CAN, CH2=CHOEt, MeOH, t.a.; d. H2SO4, 80%, DDQ. 175 CAPITOLO V COMPOSTI ORGANOMETALLICI DEI METALLI DI TRANSIZIONE 176 ORGANOMETALLI DI ZINCO, CADMIO e MERCURIO II A C.I.= 34% 1.2 Mg II B d10s2 C.I. 19% 1.6 Zn Il legame C-M(IIB) ha un carattere molto meno polare rispetto a quello del legame C-M(IIA) tali organometalli sono molto meno basici e molto più nucleofili dei reattivi di Grignard. C.I. 16% 1.7 Cd C.I. 9% 1.9 Hg Preparazione 2 RMgX + CdX2 R2Cd + 2 MgX2 2 RMgX + HgX2 R2Hg + 2 MgX2 La reazione ha la sua "drawing force" nella maggiore tendenza del metallo più elettropositivo (il magnesio) alla formazione di sali ionici spesso insolubili nel mezzo di reazione. 177 REAZIONI DEGLI ZINCOORGANICI La reazione di Reformatsky O HO O + Br-H2C OEt H3O + Zn ZnBr2 + 2 K 2 KBr + Zn Zn + HgCl2 Zn(Hg) + ZnCl2 zinco di Rieke zinco amalgama O-ZnBr+ O Br-H2C O CH2COOEt + + Zn EtO OEt 1. CH3CHBrCO2Et Zn/Hg 2. H2O/H+ HO CHCOOEt Et2O, rifl. F F H3O+ H3C CHCOOH H3C CHCOOEt DDQ F F 178 REAZIONI DEI COMPOSTI ORGANOMETALLICI DELLO ZINCO ADDIZIONE DI DIALCHILZINCO ALLE ALDEIDI R * R' OH CH3 H (R'CH2)2Zn Me (S) O (S) N Me Zn R' CH3 (S) (S) R' R' Me N Me O Zn Zn R' O (S) O R' R Zn R' Zn R' H RCHO CH3 CH3 (S) Me N Me (S) O R' Zn R' (S) N Zn Me Me R' 179 ADDIZIONE ENANTIOSELETTIVA DI DIETILZINCO ALLE ALDEIDI Aldeide = 2-Naftaldeide R (cat.) H3C H H N Zn O O Zn H3C Zn H N O O H Zn C H Fenyle 1-Naftile attacco alla faccia si attacco alla faccia re OH OH (R) (S) t-Butile i-Propile gem-Difenile Conf. cat. Resa % ee, % (config.) Rp 80 53 (S) Rp,R 98 84 (R) Rp,S 98 83 (S) Rp,R 97 88 (R) Rp,S 97 90 (S) Rp,R Rp,S 44 87 52 (R) Rp,R Rp,S 47 86 49 (R) 99 (S) Rp 100 67 (S) 99 (S) (RpS), R = tBu Aldeide Resa % ee, % (conf.) H Zn O N Zn O O ZnN 2-Naftaldeide 87 99 (S) H Zn O H3C H H3C 1-Naftaldeide 84 98 (S) Benzaldeide 84 97 (S) o-Tolualdeide 80 98 (S) m-Anisaldeide 82 98 (S) p-Anisaldeide 32 96 (S) Cicloesan-3-carbaldeide 98 > 99 (S) 3,5-bis(CF3)benzaldeide 98 99 (+) OH OH 3-piridincarbossaldeide 92 74 (S) (S) (R) Cinnamaldeide 49 84 (S) TS5 (meno favorito) attacco alla faccia si minoritario G. Ricci, R. Ruzziconi Tetrahedron: Asymmetry 2005, 16 (10), 1817. TS6 (più stabile) attacco alla faccia re maggioritario H 180 REAZIONI DEI MERCURIOORGANICI I sali di mercurio, in virtù della loro tendenza a reagire con legami π, sono impiegati molto spesso come catalizzatori nella idratazione di alcheni ad alcooli secondo Markownikoff. 1. Hg(OAc)2 2. NaBH4 OAc R H R' H + 1. HgX2 2. Nu R' Nu R Nu R' R Hg OX NaBH4 HgOX Meccanismo Nu R' R Nu R' R NaBH4 HgOX Nu R' R Nu R' R HgH Nu R' R Nu R' R Hg HgH H Hg Nu R' R 181 REAZIONI DEI MERCURIOORGANICI R ClHg Me3SiO R HgCl2 R HI H + R R O R O H +H B O O B2H6 H Hg(OAc)2 B O O R R H H Hg OAc 3B Hg(OAc)2 Br R Br2 R HgOAc REAZIONI DEI CADMIOORGANICI I composti cadmio-organici sono molto meno reattivi dei corrispondenti litio- e magnesio-derivati e sono, pertanto molto più selettivi. Essi sono utilmente impiegati in quelle reazioni dove è determinante la chemoselettività. O O O Cl + Cd(CH3)2 O CH3 182 COMPOSTI ORGANOMETALLICI DEI METALLI DI TRANSIZIONE Il Processo di Polimerizzazione Ziegler-Natta [M]-CH2-CH2R + CH3 [M]-(CH2-CH)n-CH2-CH2R H3C Il Processo Wacker (Sintesi dell'Aldeide Acetica) CH2=CH2 Pd(II) + O2 Cu(II) CH3CHO Il Processo Fischer-Tropsch [Cat.] CO + Idrocarburi Cn H2 [Cat.] = Co : ThO2 : MgO 100 5 8 L'Idrogenazione Catalitica H2 (Ph3P)3RhCl (Ph3P)3RhCl catalizzatore di Wilkinson H H 183 Effetti della Coordinazione sulla Reattività dei Composti Organici In generale la coordinazione di un composto organico con un metallo di transizione ha come effetto una marcata alterazione della struttura elettronica di tale composto da fargli subire diversi tipi di reazione altrimenti impossibili con la molecola libera. d2s2 d3s2 d4s2 d5s2 d6s2 d7s2 d8s2 4e 5e 6e 7e 8e 9e 10e 3 Ti V Cr Mn Fe Co Ni Kr, 3d104s24p6 4 Zr Nb Mo Tc Ru Rh Pd Xe, 4d105s25p6 5 Hf Ta W Re Os Ir Pt Rn, 5d106s26p6 M, dns2; M, dn+1s1; M Gas nobile dn+2s0 La Regola dei 18 elettroni Molti di questi metalli coordinano una varietà di ligandi in modo da raggiungere la configurazione stabile di 18 elettroni (configurazione elettronica del gas nobile successivo). Nel caso di Ti, Zr, Ni, Pd, Pt uno degli orbitali d ha un'energia talmente elevata per la coordinazione da rendere possibili complessi stabili a 16 elettroni. 184 TIPI DI LIGANDI Ligandi non Idrocarburici Sono classificati in base al numero di elettroni con cui partecipano al complesso. 0e Acidi di Lewis: AlX3, 1e -X (alogeno), -H, -NO, -CN 2e Basi di Lewis: :PR3, :P(OR)3, :C≡ ≡O+, R-C≡ ≡N:, R-N+≡C:-, R3N:, R2O:, R2S: 3e :NO BH3, ....... 185 TIPI DI LIGANDI Ligandi Idrocarburici Sono classificati in base al numero di carbonio legati al metallo) η1 (1 C) -R, -Ar, η2 (2 C) (numero di atomi σ n π η3 (3 C) η π η4 (4 C) η5 (5 C) η6 (6 C) η7 (7 C) η8 (8 C) Vengono considerati solo gli atomi di carbonio coinvolti nel legame con il metallo 186 Calcolo del Numero di elettroni Esterni in un Complesso N° di elettroni del metallo nel suo stato fondamentale + Somma dei valori di η dei ligandi idrocarburici + Somma degli elettroni donati da altri ligandi + Numero di cariche negative sul metallo (o del complesso) - Numero di cariche positive sul metallo 8 (o del complesso) η5 η5 Fe(8e) η5 8 + (2 x 5) = 18 e (8e) 2e Ph2P Ru Cl 1e PPh2 2e 5 + 8 + (2+2+1) = 18e η5 Mo (6e) 2e OC ON 3e η3 6 + (5+3) + (3+2) - 1 = 18e η5 Fe (8e) 2e OC OC 2e η2 8 + (5+2) + (2+2) - 1 = 18e 187 IL LEGAME DI COORDINAZIONE (LEGAME IDROCARBURO- METALLO) Il Modello di Chatt-Dewar-Duncanson per l'etilene HOMO LUMO M - - + M + - + + - - + M M LUMO HOMO - + -π La grandezza dei due effetti (donation e back-donation) dipende dalla natura del metallo per cui....... ....... un idrocarburo coordinato al metallo può risultare una specie più ricca (nucleofilo) o meno ricca (elettrofilo) di elettroni rispetto all'idrocarburo libero. 188 EFFETTI ELETTRONICI DELLA COORDINAZIONE COOH COOH OC Cr CO OC pKa = 5.68 pKa = 4.77 π COOH M COOH O2N pKa = 5.68 pKa = 4.48 COOH Cr pKa = 6.15 OC PPh3 OC π M Gli effetti elettronici di coordinazione dipendono anche dalla natura degli altri ligandi: il -C O+ è un ligando buon accettore di elettroni mentre la :PPh3 è un ligando buon donatore di elettroni. 189 EFFETTI ELETTRONICI DELLA COORDINAZIONE R R OH OH Cr OC CO OC Sostituente pKa1 pKa2 p-CH3 H 11.28 11.02 7.32 7.09 m-COOMe 10.05 6.77 m-COMe 9.99 6.82 p-COOMe 9.17 6.40 p-COMe 8.81 6.31 Ph BuOK, DMSO-d6 H Cr OC CO OC D Ce(IV) D H Cl + CH3ONa + CH3ONa Cl 65°C OC Cr OC CO nessuna reazione OCH3 Cr OC CO OC H H Cr OC CO H OC Ph t t BuOK, DMSO-d6 D D OC Cr CO H OC 190 EFFETTI ELETTRONICI DELLA COORDINAZIONE Delocalizzazione della carica C C Cr OC OC Cr CO OC OC CO AcOH CH2Cl CH2Cl OC Cr CO OC k1 CH2OAc AcOH k2 k2 = 100 k1 CH2OAc OC Cr CO OC 191 EFFETTI ELETTRONICI DELLA COORDINAZIONE La Natura del Metallo COOH COOH pKa = 7.00 pKa = 7.00 COOH COOH Fe OC CO OC OC Fe CO OC pKa = 7.50 pKa = 7.25 O CH3COCl SnCl4 OC Fe CO OC OC Fe CO OC CH3 Contrariamente al cromo tricarbonile, il ferro tricarbonile si comporta come un donatore di elettroni. Tuttavia, per effetto mesomero esso è in grado di stabilizzare anche una carica negativa. 192 EFFETTI STEREOCHIMICI DELLA COORDINAZIONE OC OC R OC R CO Fe Fe OC OC CO CO Enantiomeri CO CO Enantiomeri MeO OMe CH3 OC OC H3C Cr CO OC CO CO OMe CH3 Ligandi prochirali 1. NaH 2. CH3I O Cr CO (-) R R Cr Enantiomeri OC OC Fe R R Fe CH3 (R) OC OC O Cr decompl. H CH3 CO (-) (R) (-) O H 193 IL CICLO CATALITICO Per agire come catalizzatore il metallo deve poter cambiare il numero degli elettroni ad esso associato di 2 o 4 unità. Poichè il numero totale di elettroni non può essere superiore a 18, molte specie catalitiche possiedono 14 o 16 elettroni. Processi fondamentali coinvolti nel ciclo catalitico Coordinazione - Dissociazione coordinazione (+2e) [M] + :L [M]-L dissociazione (-2e) Addizione - Eliminazione Pericicliche addizione (+2e) [M] + Z-Y eliminazione (-2e) [M] Z Y 194 Migrazione di Ligandi Migrazione dal ligando al metallo (+2e) O [M] R Migrazione dal metallo al ligando (-2e) Migrazione dal ligando al metallo (+2e) [M] η1 H Migrazione dal metallo al ligando (-2e) (β β-elimin.) O C [M] R [M] H η2 195 IDROGENAZIONE CATALITICA L = :PPh3 L3RhCl (16e) :L L2RhCl + H-H add. peric. (14e) H H H L Rh H L Cl (16e) coord. L Rh L Cl (16e) H L Rh L Cl (16e) migr. H + H-H add. peric. H L Rh L Cl (18e) 196 IDROFORMILAZIONE HCo(CO)4 (18e) - CO + CO HCo(CO)3 (16e) coor. CO OC elim. peric. H OC OC OC H Co OC H (18e) O coor. H-H H migr. OC OC OC Co OC (16e) O O OC Co OC (16e) coor. CO migr. OC OC Co OC CO (18e) 197 NICHEL Il processo più importante che coinvolge complessi del nichel come catalizzatori è il "coupling" di alogenuri alchilici, vinilici ed arilici. La reazione procede attraverso le fasi di coordinazione o di addizione periclica e di eliminazione periciclica. A. 1 Reazioni di coupling coinvolgenti complessi η R [Ni] R-R + [Ni] R Esempi: Br 2 Ni[COD]2 2 N C Br Ni[COD]2 II N C (18e) N MeO + NiBr2 (18e) C N + NiBr2 N Ni(PPh3)4 OMe (18e) MeO OMe + NiI2 NiX2 + Zn 4 PPh3 Ni(0)(PPh3)4 + ZnX2 198 REAZIONI DI COUPLING CATALIZZATE DAL NICHEL 1. BuLi. NH2 2. H3CO H N CO2CH3 O THF OCH3 (R) HCl conc. N Br H N POBr3 O 120°C, 1.5 h NiCl2 6H2O, PPh3, Zn DMF, 50°C, 1h H3 H9s' H H N H H10s' N H9s H10s N N H3' (R,R) (R,R) NiCl2.6H2O, PPh3, Zn DMF, 50°C, 1h O NH Br POBr3 100°C, 7 h N (H3PO4)n 110°C, 30 min O O NH H2NCH2CH(OEt)2 Cl DDE, 20°C MeO OMe G. Ricci, R. Ruzziconi, E. Giorgio J. Org. Chem. 2005, 70, 1011 (R) 199 Reazioni di CROSS-COUPLING In presenza di complessi di Ni(0) si possono avere anche reazioni di cross-coupling fra aril- e vinil alogenuri e composti organometallici di Mg, Li, Zn, Zr, Al. Il primo passaggio consiste nell'addizione periciclica dell'alogenuro vinilico o arilico al metallo per dare complessi aril- o vinil- nichel. L2Ni X2 + R-MgX Ar Ar-X L2Ni (II) (0) L2Ni(II) (14e) (16e) (16e) X R-MgX Ar-R Ar L2Ni (II) (16e) Ph2P Cl + Cl Ni R PPh2 Cl MgBr 98% I+ MgBr Ni(PPh3)4 84% I + R ZnCl Ni(PPh3)4 R 87% Ni[COD]2 = Ni COD C4H9 200 Reazioni di CROSS-COUPLING Se nelle reazioni di cross-coupling catalizzate da complessi del nichel vengono impiegati reattivi di Grignard secondari, si possono ottenere prodotti di coupling riarrangiati. Ni(II) Cl + XMg Ph3P Ph3P Ni(0) Ph3P (14e) Cl Ni(II) + XMg Ph3P Cl Cl XMg Ph3P Ph3P Ni Ph3P (16e) H Ni Ph3P Cl (16e) η1 trasf. lig. Ph3P Ni H Ph3P (18e) η2 Ph3P Ni Ph3P (16e) elim. per. Ph3P + Ni Ph3P 201 B. Reazioni di coupling coinvolgenti complessi η3 del nichel La possibilità da parte del nichel di formare complessi di tipo η1 e η3 con ligandi idrocarburici permette di ottenere prodotti di eterocoupling per formazione del legame carbonio-carbonio. Così, i complessi [(η η3-allile)NiBr]2, ottenuti facilmente dalla reazione di bromuri allilici con Ni(COD)2 o Ni(CO)4, reagiscono con alogenuri alchilici per dare olefine sostituite. Ni Br Br (14e) Br 2 Ni R-X Br Ni X R (16e) Ni S Solvente d.a. (16e) el. per. R Br + Ni R-X + NiBrX + NiBrX R 2 Il processo può essere reso catalitico per aggiunta di zinco metallico. Nel sistema allilico possono essere presenti diversi gruppi , come OCH3 o COOEt. Br Br NiBrX + Zn ZnBrX + Ni(0) Ni 2 202 Reazioni dei Complessi η3 del Nichel Br MeO + Ni R-X O MeO 2 R R O MeO Br Br Il complesso acetonile MeO è l'equivalente sintetico del gruppo Ni 2 Esempi: O OCH3 Br + MeO Ni Br 2. H3O+ 2 O COOCH3 DMF O OCH3 BH4- O O O O NaH O O N H Br O Br + Ni 2 N H 203 Reazioni dei Complessi η3 del Nichel I complessi [(η η3-allil)NiX]2 reagiscono con alogenuri allilici per dare i dieni-1,5. La reazione globale consiste nel "coupling" di due alogenuri allilici. L'applicazione più importante è la sintesi di anelli medi, grandi e macrolidi attraverso il coupling intramolecolare catalizzato da Ni(CO)4 di una molecola avente due sistemi allilici terminali. Ecco alcuni esempi: Br Br Ni(CO)4 Br Ni DMSO OAc OAc 2 Ni OAc 59% n=12 Br + Ni(CO)4 Br n=14 75% n=18 80% Alcoli allilici e β-idrossichetoni possono essere sintetizzati per reazione di complessi η3- NiX2 e chetoni Br Ni + OH O 2 Br MeO Ni 2 1. R2C=O 2. H3O+ OH O R R 204 LA REAZIONE DI FELKIN + 2 CH3MgBr L2NiCl2 CH3 CH3 + OH + CH3 Meccanismo: A. B. C. L2Ni(0) + R-R + 2 MgClX L2NiCl2 + 2 R-MgX OH + R-MgX OMgX + R-H OMgX R L2Ni(0) (14e) R XMgO η2 L Ni L 2 (16e) η L (16e) R L L Ni 3 (18e) η Ni L R-MgX - MgO, MgX2 205 Aspetti Stereochimici della Reazione di FELKIN H3C H (S) OH + 2 MgBr Meccanismo probabile H3C H Ph-[Ni] L2NiX2 Inver. CH3 H CH3 H ee = 23% (-)-R Ph-[Ni] (S) OH (S) (S) CH3 XMgO H OMgX Ph-[Ni] CH3 H (R) Ph-[Ni] Ph-[Ni] CH3 H inv. CH3 H inv. Ph CH3 H (R) H3C [Ni]-Ph H Ph-[Ni] (-)-R CH3 (S) rit. CH3 H Ph-[Ni] H CH3 H Ph CH3 H Ph-[Ni] rit. Ph-[Ni] H CH3 206 Aspetti Stereochimici della Reazione di FELKIN L'impiego di ligandi chirali del nichel nella reazione di Felkin permette di ottenere olefine otticamente attive. Tuttavia gli ecessi enantiomerici non sono eccezionali. La reazione non ha un elevato valore sintetico. Esempi: C6H5 (R) Ph2P OH + R-MgX Ni (-)-DIOPNiCl2 C6H5 Ph2P Ni OMgX PPh2 R H OH + CH3-MgBr R (R) O,O'-Isopropilidene-2,3-diidrossi1,4-bis (difenilfosfino)butane ee = 15% H L2*NiCl2 H3C Ph Ph O Cl P (R) Ni (R) Cl P O Ph Ph (-)DIOPNiCl2 PPh2 R (S) L* Ni L* CH3 H ee = 15% C6H5 (R) Ph2P PPh2 Ni Cl Cl (R)-(-)-1,2-bis(difenilfosfino)1-feniletano 207 PALLADIO Nella chimica dei composti organometallici del palladio è bene tener presenti quattro aspetti fondamentali: 1. Le reazioni catalizzate dal palladio avvengono generalmente in presenza di ligandi che coordinano il palladio e possono giocare un ruolo chiave sia per quanto riguarda la reattività dell'intermedio sia per quanto riguarda la regiochimica del processo. 2. Il legame C-Pd è relativamente debole specialmente in presenza di idrogeni in posizione β. Lo stadio finale di molte reazione catalizzate dal palladio consiste nell'eliminazione di Pd(0) e H+. Questa tendenza verso l'eliminazione distingue i composti organometallici del palladio da tutti gli altri organometalli. 3. Le specie organopalladio con due sostituenti organici mostrano la stessa tendenza alla decomposizione e ricombinazione dei gruppi organici esibita dai composti organometallici del nichel. 4. I complessi organici del palladio son specie elettrofile. HOMO LUMO Pd - - + 208 A. Complessi π del Pd(II), η2 I complessi organopalladio vengono generalmente preparati "in situ"; nei processi più importanti il palladio viene usato in quantità catalitiche. Gli alcheni reagiscono con Palladio (II) per dare complessi η2 soggetti ad attacco nucleofilo. Pd+ Nu Pd++ + Pd(II) R R R η2 -H R Nu eliminazione Pd+ + -[Pd(0)] R Nu 1 η Nu [H] -[Pd(0)] R Nu riduzione 209 IL PROCESSO WACKER Un importante processo industriale che sfrutta i complessi η2 del palladio (II) è il processo Wacker per la sintesi dell'aldeide acetica per ossidazione dell'etilene. Pd+2 + Pd(II) HO H2O H Pd+2 H O HO 1/2O2 2 Cu+ Pd2+ OH- 2 Cu2+ Pd(0) HO CH3CHO Pd2+ H2O HO Pd+ H+ H H HH CH2=CH2 + 1/2 O2 CH3CHO O O PdCl2 CuCl2, H2O O 68% 210 Complessi η3 del Palladio I complessi η3 del Palladio possono essere ottenuti sia dalla reazione di acetati allilici, o altri derivati allilici con più o meno buoni gruppi uscenti, con Pd(0), sia dalla reazione di alcheni aventi idrogeni allilici con PdCl2 o Pd(OTFA)2 . (0) R' L2Pd R R R' Pd L X L X R' Pd++ R H H L L Pd++ R 2L R R' Pd+ L L R' R R' Pd+H H L L H H Pd++ R -X- Pd(0) R Pd+ X L L R - X Nu R R + CuCl2 Nu Nu L2Pd(0) +X- L Pd L X 211 Reazioni dei Complessi η3 del Palladio L OCOMe MeO2C Pd(PPh3)4 L Pd+ MeO2C CO2Et C MeO2C CO2Et C H CO2Et H CO2Et L NaH MeO2C PhO2S OCOMe NaH O Pd(PPh3)4 O O Pd(PPh3)4 + Pd+L MeO2C H PhO2S O O O O O L Pd+ PhO2S CO2Me PhO2S CO2Me L 212 SOSTITUZIONE NUCLEOFILA ALLILICA CATALIZZATO DA Pd(0) EtO OSiMe3 O CAN, MeOH, t.a. O NO2 + EtO ONO2 EtO 55:45 NaHC(CO2Me) Pd(PPh3)4 THF, t. a. O Ph3P Pd PPh3 O COOMe EtO EtO COOMe + MeOOC O COOMe 5 EtO EtO O OSiMe3 O O CAN, ONO2 + MeOH, t.a. ONO2 OEt OEt O O 60:40 N Pd(PPh3)4 THF, t. a. O O O O + OEt OEt O O 97:3 95 213 MeO2C HC(COOMe)2 [PN]cat., 5% CH2Cl2, t.a. OCOCH3 CO2Me (s) ee, 60% N P [PN]cat. P P N H N Pd H H H H H Pd H H H H (R) P H H N P Pd H H (S) N H H H H Pd H H P H H N Pd H H H (R) 214 Complessi η1 del Palladio Alogenuri alchilici, arilici o vinilici reagiscono con alcheni in presenza di quantità catalitiche di palladio(0) per dare prodotti di sostituzione dell'alogenuro con un gruppo alchenile. Questo processo prende il nome di La reazione di HECK Y Br + Y (Ph3P)2Pd(OAc)2 Base 80-90% La reazione ha inizio con l'addizione ossidativa (Add. periciclica) dell'alogenuro su una specie del Palladio (0) generata in situ da Pd(II) (in genere Pd(OAc)2. Pd(OAc)2 + 2 PPh3 + CHY=CH2 Br Pd(PPh3)2 +CHYCH2OAc + AcOH PPh3 Pd Br PPh3 PPh3 Pd Ph3 Pd(PPh3)2 Y PPh3 Ph3P Pd Y + HN+(C2H5)3 N(C2H5)3 Y H H Y 215 Complessi η1 del Palladio Esempi della reazione di HECK C4H9 Pd(0) Br + C4H9 C4H9 Br + Pd(0) Pd(OAc)2 (R)-BINAP K2CO3; Tol. H3CO H3CO (S) OTs 62%, 95% ee 216 Complessi η1 del Palladio La reazione di SONOGASHIRA La reazione di Sonogashira è concettualmente molto simile alla reazione di Heck e consiste nel "coupling" di un aril alogenuro ed un alchino terminale. La reazione è catalizzata da complessi di palladio(0) ed usa come co-catalizzatore un sale rameoso (generalmente CuI). R Br + R (I) Pd(PPh3)4/CuI R base 80-90% Il meccanismo della reazione di Sonogashira è molto simile a quello della reazione di Heck. Unica variante la presenza di un sale rameoso che forma inizialmente un alchinilcuprato che, in seguito, subisce la transmetallazione con il palladio. R H N(C2H5)3/CuI PPh3 Pd Br PPh3 Br Pd(PPh3)2 R Cu Cu PPh3 Pd Ph3 R R R 217 Complessi η1 del Palladio Esempi della reazione di SONOGASHIRA La reazione di Sonogashira è molta adatta alla sintesi di bisarilalchini non simmetrici che possono essere preparati con una reazione one-pot applicando la cosiddetta reazione silaSonogashira. R Br + R Pd(PPh3)4 R Si(CH3) Si(CH3)3 (I) 80-90% R' Br R Si(CH3) R' R Na2CO3,Pd(PPh3)4 Si(CH3)3 I Br + Si(CH3)3 Br Si(CH3)3 HO Br + B HO Pd(OAc)2 Na2CO3 CF3 OH F3C OH-/H2O CF3 OH F3C 218 Complessi η1 del Palladio La reazione di uno stannano (o composto organometallico dello stagno) con un carbonio elettrofilo catalizzata da palladio prende il nome di La reazione di STILLE R1X + R2Sn(R3)3 Pd(0)Ln R1-R2 + (R3)3SnX La reazione ha inizio con l'addizione ossidativa (Add. periciclica) dell'alogenuro su una specie del Pd (0) generata in situ da Pd(II) (in genere Pd(OAc)2 aggiungendo un leggero eccesso di stannil derivato. Pd(II) R2Sn(R3)3 Pd(0)L2 R1-X Add. Ox R1-Pd-L2-X R1-R2 R2Sn(R3)3 El. Rid. R1-Pd-L2-R2 Transmetallazione XSn(R3)3 La reazione di Stille va assumendo un'importanza crescente in chimica organica sintetica in quanto in essa possono essere usati molti e diversi tipi di substrati. 219 Complessi η1 del Palladio La tabella seguenteenumera una serie di possibili elettrofili al carbonio e di stannani che possono essere accoppiati in ogni combinazione con la reazione di STILLE. R1-X R2-SnR3 R1-X H-SnR3 Ar-CH2-X O R R2-SnR3 SnR3 (X = Cl, Br) Cl R' SnR3 X (X = Cl, Br) Ar-X (X = Br, I) SnR3 SnR3 X X (X = I, OTf) COOR (X = Br, I) SnR3 R' R'-SnR3 NB. Il trasferimento di semplicic gruppi alchilici R (-CH3, -C4H9) dallo stagno al palladio è un processo molto lento paragonato al trasferimento di R2. Esempi della reazione di STILLE Ph-Sn(CH3)3 Pd(PPh3)2Cl2 I O Br O Br THF, t.a. Ph MEMO O O O MEMO I Ph SnBu3 (S) OH O Pd(0) H3O+ O HO (S) O 220 Complessi η1 del Palladio Nel caso della reazione di Suzuki un composto organoboro, generalmente un acido boronico, viene fatto reagire con un aril (o alchenil, o alchinil) alogenuro in presenza di palladio(0) come catalizzatore. La reazione di SUZUKI R X + R'B(OH)2 (X = Br, I) Pd(0)L2 R-R' Base Come nelle reazioni di Heck e di Stille, la reazione ha inizio con l'addizione ossidativa (Add. periciclica) dell'alogenuro su una specie del Palladio (0) generata in situ da Pd(II) (in genere Pd(OAc)2. Pd(OAc)2 + 2 PPh3 + CHY=CH2 Pd(PPh3)2 +CHYCH2OAc + AcOH Pd(II) CHY=CH2 Pd(0)L2 R-R' R-X Add. Ox L R-Pd-X L El. Rid. L R-Pd-R' L OH R' B Base OH Transmetallazione Base-B(OH)2 + X- 221 Complessi η1 del Palladio Esempi della reazione di SUZUKI Br C4H9Li Li CH3 1. B(iC3H7)3 2. HCl aq. OH B OH CH3 CH3 H3C H3C OH + Cl B OH H3C CH3 OCH3 + B OCH3 Si Pd(PPh3)2 Na2CO3 acq. CH3 H3C Pd(PPh3)2 Br Na2CO3 acq. F Si F I + (HO)2B OH Pd(0), Base OH NB. La reazione di Suzuki tollara un buon numero di gruppi funzionali. Contrariamente alle reazioni di Heck e di Stille, La reazione di Suzuki non avviene in condizioni neutre. 222 Reazioni di Alogenuri Aril e Vinilici con Reattivi di Grignard Catalizzate da Pd(0) H H C6H13 I Pd(PPh3)4 + BrMg C6H13 75% Meccanismo: L Pd R L X R'-M L R - M-X Pd L R' elim. peric. R R' + L2Pd(0) Esempi: CH3 CH3 ZnCl + Br ZnCl + NO2 I Pd(PPh3)4 Pd(PPh3)4 NO2 223 Sintesi di Esteri da Alcheni Catalizzata da Palladio(II) (Carbonilazione) + CO R Pd(II) R CH3OH H3CO 2Cu+ OCH3 O R ++ Pd 2Cu++ [Pd(PPh3)2I2] Pd(0) Pd++ OMe R O R CH3OH OMe R CH3OH O + Pd OCH3 Pd+ - :C O+ I + -:C O+ R OCH3 O Pd(II) OMe MeOH Meccanismo Pd(II) I Cu++ (0) Pd + Pd-I O OMe CO MeOH Pd-C=O O Pd 224 Sintesi di Lattoni Catalizzate da Palladio(II) H3C H3C Pd(PPh3)2Cl2 I CH3 CO HO O CH3 O 99% H3C H3C PdL2Cl2 I CH3 L Pd I L HO HO CH3 :C O+ H3C L O L H3C CH3 Pd HO I L migr. OC Pd I L HO H3C (0) O O + L2Pd CH3 CH3 Cu++ L Pd(II) 2 225 RODIO COBALTO FERRO Ciascuno di questi metalli è "specializzato" in determinate reazioni: Rodio e Cobalto sono catalizzatori impiegati nelle reazioni di alcheni con H2 e CO per dare aldeidi in condizioni controllate. RODIO O Rh2O3 + CO + H2 H 100°C 60-150 Atm Il passaggio chiave di questa reazione è la migrazione del gruppo alchilico dal metallo al carbonio carbonilico. H + Rh H O H Rh C O+ Rh CO CO H Rh H O O H2 Rh 226 Reazioni di Decarbonilazione Catalizzate da Complessi del Rodio Il passaggio chiave della formilazione, cioè l'inserzione del carbonile, può essere reversibile cosicchè questa reazione viene occasionalmente impiegata nella decarbonilazione di aldeidi e acil alogenuri O R X + Ph3P Rh Cl Ph3P Ph3P Ph3P Ph3P Rh Cl Ph3P PPh3 Rh Cl O + PPh3 X R Ph3P PPh3 Rh Cl R X Ph3P + X = H, Cl R-X 227 COBALTO IDROFORMILAZIONE HCo(CO)4 (18e) - CO coor. + CO HCo(CO)3 (16e) elim. peric. CO OC Co OC H (18e) C H migr. OC OC Co OC (16e) O H OC C OC OCH O coor. H-H OC O OC Co OC (16e) coor. CO OC OC Co OC CO (18e) migr. 228 FERRO Il ferro viene spesso impiegato nella sintesi organica come dianione tetracarbonilferrato. Tale anione, altamente carbonilato, può effettuare la conversione di alogenuri e tosilati alchilici in aldeidi, chetoni e derivati degli acidi carbossilici. = R R-X + 2 Na+ Fe(CO)4= Fe(CO)3 X (18e) (18e) CO - R Fe(CO)3 + X- O H+ (16e) O2, R'OH O R R'X, PPh3 O R H O R' Fe(CO)3 PPh3 (18e) R O R' O R R' 229 CROMO Da un punto di vista sintetico, i complessi p di composti aromatici con il cromo più utili sono quelli che si ottengono con il Cr(CO)6. + Cr(CO)6 OC Cl Cr CO CO (18 e) Cl + Cr(CO)6 Cr CO CO (18 e) OC I complessi del cromo tricarbonile sono soggetti ad attacco nucleofilo (si iricordi che il cromo è un metallo "elettronattrattore"). Cl + Cr CO CO (18 e) CH3 :C CH3 CN Cr CO CO (18 e) OC OC CH3 C CH3 CN CH3 C CH3 CN 230 CROMO L'attivazione da parte del cromo tricarbonile nelle sostituzioni nucleofile aromatiche può essere così forte da permettere persino la sostituzione di un idruro in composti aromatici non alogenati. + Cr(CO)6 OC Cr CO CO Li CH3 t C CO2 Bu CH3 H3CO H OC O + Cr CO CO I2 CH3 :C CH3 CN CH3 CH3 +:C CO2tBu OC CO H OC Cr CH3 C t H3C CO2 Bu OC CO H OC Cr CH3 C H3C CN H3CO TFA H CH3 C CH3 NC H+ H3CO H CH3 C CH3 NC Tuttavia, non tutti i nucleofili hanno la capacità di addizionarsi ai complessi aromatici del cromo. Ad esempio, RLi, RMgX e enolati non reagiscono. 231 232 PARTE TERZA RIARRANGIAMENTI 233 234 235 236 237 STEREOCHIMICA DEI RIARRANGIAMENTI Vi sono molte evidenze che dimostrano una ritenzione di configurazione del gruppo migrante W in uno shift-1,2. Ciò risulta anche intuitivo, visto che la rottura del legame W-A e la formazione del nuovo legame W-B è un processo sincrono. W W * * Ritenzione di HOMO configurazione LUMO HOMO LUMO A B A B Una diversa situazione si riscontra per gli atomi A e B. In molte reazioni la questione stereochimica non si pone poiché potrebbe essere possibile una sola possibilità sterica sia per A, o per B, o per entrambi. Qualora A o B costituiscano dei centri chirali, in essi si può osservare sia inversione che racemizzazione. Ph H CH3 Ph HO Ph HNO2 H Ph HO NH2 Ph Ph CH3 H CH3 O -N2 N2 Inver sione di conf igurazione in B Il grado di racemizzazione ci da una misura grossolana della concertazione dei vari step attraverso cui passa l'intero processo. Se si osserva racemizzazione in B è molto probabile che il primo passsaggio (la formazione del carbocatione) abbia luogo prima della migrazione di R. In tal caso il carbocatione costituisce un intermedio di reazione. R R A B X A B R R A B X R Rac. 3° Step A B R Inv. A B 3° Step A B X La stereochimica all'origine della migrazione A non è quasi mai coinvolta poichè nella maggiorparte dei casi non si arriva alla tetredricità di A. Ma quando in A si verifica un'inversione di configurazione è operante un meccanismo SN2 su A A Y R R B X Y A B R R A B Y A B Y 238 RIARRANGIAMENTI: ABILITA' DEI GRUPPI A MIGRARE La trasposizione pinacolica è una reazione che si presta molto bene allo studio dell'abilità migratoria dei gruppi in un dato riarrangiamento. OH H+ H -H2O Ph Ph H Ph Ph Ph Ph HO Ph Ph OH H+ H H -H2O HO In questo caso particolare il fattore che determina la natura del gruppo migrante è la Ph stabilità del carbocatione che si forma dopo protonazione ed eliminazione di una molecola O di H2O. Ovviamente una volta eliminata la molecola di H2O, nella migrazione non può essere coilvolto che l'idrogeno con formazione della difenilacetaldeide. H O H H H Ph H H La migrazione di un gruppo rispetto ad un altro, entrambi legati allo stesso atomo di carbonio, è determinata non solo dalla capacità di migrare intrinseca del gruppo, ma anche dalla sua capacità di stabilizzare la carica che si viene a creare sul sito origine in seguito alla migrazione. I due esempi che seguono rendono meglio l'idea di quanto affermato. H3C H3C H3C H3C OTs CH3 H TsO CH C H , 80 °C H 3 6 6 Ph CH3 H H3C H3C - Ph E1 migliore assistenza anchimerica H3C H3C H H H3C H3C Ph H3C H C 3 H + H Ph CH3 H Ph H3C Ph CH3 H CH3 CH3 Ph + H3C H3C H CH3 Migliore stabilizzazione del carbocatione Ph H3C CH3 + CH3 H3C H3C CH3 Ph 239 240 TRASPOSIZIONI AL CARBONIO ELETTRON-DEFICIENTE La trasposizione Pinacolinica Quando un diolo vicinale (glicole) viene trattato con un acido, esso riarrangia per dare un'aldeide o un chetone, benchè esistane esempi di reazioni di eliminazione senza riarrangiamenti. Il gruppo migrante può essere un alchile, un arile, un idrogeno e persino un gruppo metossicarbonile (-COOCH3). Nella maggior parte dei casi ciascun carbonio ossidrilico tiene legato almeno un gruppo alchilico o arilico e molto spesso è tetrasostituito. Con glicoli non simmetricamente sostituiti, si possono ottenere più prodotti a secondo del gruppo che migra. R1 R R2 HO 4 R H+ R3 R4 R2 HO OH O R2 HO R4 R3 OH2 R1 R4 R2 R1 R1 3 OH -H+ OH 1 R 4 R R2 R3 R2 R1 R4 R3 R3 Esistono diverse varianti di questa trasposizione che differiscono essenzialmente per il modo con cui si ottiene il carbocatione intermedio. O R1 R2 R3 L.A. 4 R M O M O R1 R2 R3 R1 R2 4 R H 2O R3 4 R R1 O R2 R4 R3 M = H+, BF3-Et2O, MgBr2 Et2O, Cu+2, Fe+3ecc. T rasposizione "Semipinacolinica" R1 R2 HO R3 HNO 2 R4 + H NH2 R1 R1 R2 HO R3 R4 N2 -N2 O R2 HO R4 R3 R2 R1 R4 R3 241 TRASPOSIZIONI AL CARBONIO ELETTRON-DEFICIENTE Il riarrangiamento di Demyanov (contrazione o espansione d'anello) Quando una carica positiva viene generata su un carbonio di un cicloalcano, si può osservare la migrazione di un gruppo alchilico con formazione di un anello avente un numero di atomi di carbonio inferiore di un'unità (contrazione d'anello). OH2 -H2O N2 NH2 HNO2 OH -N2 H3O+ OH O -H+ O O- R OH R R1 Br NH2 e on C t on e on i z ra i ns pa Es H3O+ HNO2 NH2 OH -H2O i PrMgBr R Br R1 R1 MgBr2 O R R1 O O CH2Br O CH2 Bu3SnH COOEt COOEt COOEt O O Bu3SnH COOEt CH2 COOEt 242 TRASPOSIZIONI AL CARBONIO ELETTRON-DEFICIENTE Riarrangiamenti di Aldeidi e Chetoni Acido-Catalizzati. Riarrangiamenti di questo tipo, in cui un gruppo in posizione a ad un carbonile scambia con un groppo legato direttamente al carbonio carbolico, avvengono quando c'è una buona attitudine di un gruppo alla migrazione. Aldeidi possono essere convertite in chetoni e chetoni in altri chetoni, ma non è mai stata osservata la conversione da chetone ad aldeide. 1 4 R2 R H R4 3 R R2 R + R1 3 R O O R2, R3, R4 = alchile o idrogeno. Meccanismi 1 R2 R A) H+ 1 R4 R3 O R2 R 3 R R2 R4 R 3 OH H+ B) R4 -H+ R1 R O R1 R 4 R OH R3 HO R1 R2 -H+ R4 R1 3 OH 2 1R R3 2 R4 R R2 O R3 R4 Secondo il primo meccanismo, si ha la migrazione dei soli gruppi alchilici, mentre il secondo meccanismo provede la migrazione anche dell'ossigeno. Il meccanismo può essere individuato attraverso il marcamento del carbonio carbonilico con 14C. 243 244 IL RIARRANGIAMENTO DI FAVORSKII Quando un -alochetone (X = Cl, Br, I) viene trattato con un base alcossido o idrossido, si osserva un riarrangiamento con formazione di un estere. Tale reazione vva sotto il nme di riarrangiamento di Favorskii. O 3 R O X R1 R O - R3 R R1 O R2 -epossichetoni fornisce -idrossiesteri. La reazione con O Ph C H2 R2 O OR1 Cl O 1 OR 1 Ph OR Ph CH3 Cl 3 * 4 2 5 Cl 1 O 6 * = * O RO14 C + * O OR OR 50 : 50 Il meccanismo prevede la formazione di un ciclopropanone intermedio che viene aperto dall'alcossido per dare l'estere. ovviamente, l'apertura del ciclopropanone avviene preferenzialmente in modo da formare il carbanione intermedio più stabile. Questo spiegherebbe l'esito della prima reazione, la formazione dello stesso prodotto da due diversi alogeno chetoni. R3OO O R3O- X R1 RHC R R1 R3O- R3 O 1 R + R3 O R 2 R2 R2 O R O R R1 R2 In assenza di idrogeni a la reazione può aver luogo ugualmente. In tal caso si ha una migrazione vera e propria di un gruppo alchilico o arilico. R3O- O O X R R1 R2 R1 3 R Inversione!!! O R R2 245 LA SINTESI DI ARNDT-EISTERT Nella sintesi di Arndt-Eister un alogenuro acilico viene convertito in un acido carbossilico avente un atomo di carbonio in più. Il primo step consiste nella formazione di un -diazochetone che, successivamente riarrangia in un chetene il quale, per trattamento con H2O e AgO (o benzoato di argento + Et3N o altri catalizzatori propri della Sandmeyer), si trasforma in acido carbossilico (riarrangiamento di Wolff). Usando un alcool al posto dell'acqua si ottiene un estere, mentre il trattamento con ammoniaca fornisce la corrispondente ammide. O O R R Cl O H 2O CH2N2 CH2N2 R Ag2O OH Meccanismo O O O H2C N N R R Cl C H N N R C H N O N -N2 R CH Nu Nu = -OH, -NH2, -OR Nu O CH2 R H2O, ROH, NH3 O C C H R La reazione può avvenire anche in assenza di catalizzatori, per semplice riscaldamento. Il processo ha una vasta applicabilità, in pratica, con qualsiasi R (alchile, arile, vinile, ecc. ). Spesso la reazione viene effettuata anche con altri diazoalcani ed è stata utilizzata per la contrazione di anello a partire da diazocicloalcanoni. O N C N O Nu C Nu O O Nu = -OH, -NH2, -OR meccanismo alternativo 14 C O 246 OMOLOGAZIONE DI ALDEIDI E CHETONI Un processo analogo alla sintesi di Arndt-Eisterche passa attraverso il riarrangiamento di Wolff è l'omologazione di aldeidi e chetoni. O 1 R O CH2N2 R R O R1 R O CH2N2 R H CH3 Meccanismo H2C N N O H2C N N R 1 N N H C 2 O R1 R R Con le aldeidi l'epossido diventa il prodotto collaterale più impor tante, f ino a diventar e il prodotto principale quando su R vi sono gruppi elettr on attrattor i che inibiscono la migr azione di R1 O - R C H2 betaina, in qualche caso isolabile O R R1 CH2 R1 Chetoni ciclici a tre termini e superiori reagiscono particolarmente espandendo l'anello. N O H2C N N O N -N2 O 247 MIGRAZIONE AL CARBONIO DA PARTE DI UN ETEROATOMO Idrossi-debromo-cine-sostituzione Quando una sostituzione nucleofila viene effettuata su un substrato avente un gruppo nucleofilo sul carbonio adiacente al sito di sostituzione, si può generare un intermedio ciclico che può essere aperto dal nucleofilo sul lato opposto. Il risultato è una migrazione del gruppo nucleofilo sul sito originale di sostituzione. Ad esempio, nel caso di b-amino-a-alochetoni, la reazione ha luogo attraverso la formazione di un aziridinio intermedio che viene aperto per attacco nucleofilo dell'H2O. H NR2 O Ph H2O Ph H Br H NR2 Ph O Ph H2O H Br- Ph -HBr H O Ph HO H NR2 a-Alo- o a-acilossi epossidi hanno tendenza a riarrangiarsi e spesso lo fanno spontaneamento senza catalizzatori, benchè nella maggior parte dei casi necessitano di catalisi acida. O R O R1 R X R1 Cl In presenza di un catalizzatore di rame, vinil epossidiriarrangiano a 2,5- diidrofurani. Come pure vinil tiirani sono trasformati in 2,5diidrotiofeni. O S O S Un importante riarrangiamento sfruttato sinteticamente è il riarrangiamento di Meyer-Schuster, secondo il quale un alcool propargilico riarrangia in catalisi acida per dare un composto carbonilico a,b-insaturo. OH H+ -H2O H2O H+ -H+ H O OH 248 249 250 251 252 253 254 MIGRAZIONI CARBONIO AZOTO LA REAZIONE DI SCHMIDT Reazione_di_alcoli e alcheni_con_HN3 Gli alcoli e gli alcheni rreagiscono con HN3 dando alchilazidi (R-N3), che durante la reazione riarrangiano per dare immine. La conversione di alcoli nelle corrispondenti azidi può essere effettuata attravesro la reazione di Mitsunobu anche utilizzando un reagente alternativo (PhO)2PON3. :PPh3 O N N EtO OEt HN3 PPh3 O N H EtO O N N3OEt O O EtO R-OH R-N3 N3- N H N (PhO)2PON3 DBU R2 R3 1 4 R DBU N3- + R-OPO(OPh)2 R HN3 TiCl4 cat. o AlCl3 N3 R2 R1 N R3 H R4 Esempio N N HN3 H2SO4 N CH3 N H3C OEt O R-O PPh3 oppure R-OH H N + N2 255 MIGRAZIONI CARBONIO AZOTO IL RIARRANGIAMENTO DI STIEGLITZ E REAZIONI RELATIVE Certe N-aloammine, quando solvolizzate in presenza di AgNO3, subiscono un riarrangiamento. AgNO3 N Cl N CH3OH OCH3 La reazione è simile al riarrangiamento di Wagner-Meerwein ed inizia con la rimozione di uno ione cloruro ad opera dell'Ag+. Reazioni simili sono state impiegate per espansioni e contrazioni di anelli. HO Cl N Ag+ R HO N R HO N R R O - H+ N - AgCl Il nome di riarrangiamento di Stiegklitz si applica, in genere, al riarrangiamento delle tritil N-aloammine e idrossilammine e sono simili al riarrangiamento delle delle alchil azidi. Ar Ar Ar NHX Base Ar Ar Ar Ar N Ar Ar NHOH PCl5 Ar N Ar Ar Una reazione simile è quella delle tritilammine con Pb(OAc)4. - Ar Ar Ph NH2 Pb(OAc)4 C 6H 6 Ar Ar Ar Ar H N Pb(OAc)2 OAc N H OAc Ar N Ar Ar Pb(OAc)2 + AcOH 256 MIGRAZIONI CARBONIO OSSIGENO IL RIARRANGIAMENTO DI BAEYER-WILLIGER Il trattamento di un chetone con peracidi (m-CPBA, PACA) in condizioni di catalisi acida porta alla formazione di esteri per migrazione di un gruppo alchilico. Dal perossiacido, per riduzione, si forma l'acido carbossilico corrispondente. Oltre ai peracidi citati è stato usato anche l'oxone KHSO4KHSO5-K2SO4. O R O R 1 O [ H+] 2 + R R O OH O O R1 + R2 OH La reazione è spesso usata per ottenere lattoni da chetoni ciclici O HO O R 2 O O O O OH O R2 O [H+] Con chetoni non simmetrici l'ordine di migrazione dei gruppi R è approssimativamente: terziario > secondario >primario > metile. L'abilità di migrazione del fenile è aumentata dalla presenza di gruppi elettron-donatori. -dicheoni (enolizzabili) non reagiscono, mentre da -dichetoni si ottengono anidridi. Aldeidi danno acidi, e questo può spiegare il meccanismo di ossidazione delle aldeidi ad acidi carbossilici. R2 R2 Meccanismo O O R OH H+ R1 R2 O OH R R1 O O HO O R R1 La scarsa capacità di migrare del metile fa si che questa reazione sia adatta alla formazione di fenoli a partire da metil aril chetoni dopo idrolisi dell'acetato arile risultante. O O HO O R1 R - R2CO2H O R O R1 257 258 259 MIGRAZIONI AZOTO CARBONIO IL RIARRANGIAMENTO DI STEVENS R3 R2 N C H H R1 Z R3 R2 N C H R1 Z Base Esempi Ph Ph N+ CH3 N+ CH3 CH3 Ph N NaNH2 -NH3 Ph Ph t H3C N+ Ph BuOK DMSO Ph H3C N+ Ph H3C N Ph Diverse reazioni possono competere con il riarrangiamento di Stevens. Ad esempio, se Z = arile, si può osservare il riarrangiamento di Sommelet-Hauser, mentre in presenza di idrogeni in b all'azoto può essere possibile l'eliminazione di Hofmann. Riarrangiamento di Sommelet-Hauser R N+ R NaNH2 CH3 - NH3 CH3 C H2 R R N+ R N+ R CH3 CH2 R R R N R H N R C H2 N R + CH2 260 261 262 263 INDICE PARTE PRIMA LAREATTIVITA’ CHIMICA CAPITOLO I – Orbitali di Frontiera e Reattività Chimica Legami omonucleari Legami etero nucleari Il sistema allilico Dieni coniugati, l’1,3-butadiene Interazioni HOMO-LUMO e HOMO-HOMO L’equazione di Klopman e Salem Nucleofili ed elettrofili hard e soft Nucleofili bidentati Enolati Elettrofili bidentati L’effetto a Effetti stereo elettronici 6 14 16 18 23 24 31 39 40 46 48 49 CAPITOLO II – Reazioni Pericicliche Termiche e Fotochimiche Cicloaddizioni Reazioni [2+2] Ciclo addizioni 1,3-dipolari La regola di Woodward Hoffmann Reazioni chelotropiche Riarrangiamenti sigma tropici Il riarrangiamento di Cope Il riarrangiamento di claisen Reazioni elettrocicliche Effetti stereo elettronici secondari La reazione di Diels-Alder Cicli addizioni 1,3-dipolari Reazioni fotochimiche Regole di Woodward-Hoffmann “fotochimiche” 52 53 55 57 58 63 66 70 72 74 75 90 101 103 CAPITOLO III –-Reazioni Radicaliche Caratteristiche generali Confronto tra reazioni radicali che e ioniche Formazione del legame C-C radicalica Il metodo ossidativo La sostituzione omolitica aromatica 108 117 121 125 126 264 PARTE SECONDA COMPOSTI ORGANOMETALLICI E LORO REAZIONI CAPITOLO IV –-Organometalli Polari Classificazione Nucleofilicità e basicità degli organometalli polari Effetto del solvente sulla reattività degli organometalli polari Effetto di additivi coordinanti sulla reattività degli organometalli polari Preparazione degli organometalli polari Lo scambio idrogeno-metallo Lo scambio alogeno-metallo Lo scambio metallo-metallo Determinazione del titolo di organometalli polari in soluzione Sintesi con reattivi di Grignard La riduzione di Meerwein-Pondorff-Verley Composti organometallici del rame Reazioni degli organocuprati 129 130 136 138 140 141 147 150 152 156 161 162 163 CAPITOLO V –- Organometalli di Metalli di Transizione Organometalli dello zinco, del cadmio e del mercurio Composti organometallici dei metalli di transizione Tipo di ligandi dei metalli di transizione Il modello di Chatt-Dewar-Ducanson Effetti elettronici della coordinazione Effetti stereochimici della coordinazione Il ciclo catalitico Organometalli del Nichel e loro reazioni La reazione di Felkin Organometalli del palladio Complessi η3 del palladio eloro reazioni Complessi η1 del palladio e loro reazioni Complessi organometallici del rodio, del cobalto e del ferro Complessi organometallici del cromo 172 176 180 182 184 188 189 193 200 203 206 210 221 225 PARTE TERZA RIARRANGIAMENTI CAPITOLO VI –- Riarrangiamenti Classificazione Riarrangiamenti nucleofili 229 230 265 Stereochimica dei riarrangiamenti Trasposizioni al carbonio elettron deficiente Migrazione di un eteroatomo al carbonio elettron deficiente Migrazione dal carbonio all’azoto elettron deficiente Migrazione dal carbonio all’ossigeno elettron deficiente Migrazione dall’azoto al carbonio elettron deficiente Migrazione dall’ossigeno al carbonio elettron deficiente 231 233 241 243 250 252 255
Scaricare