Chimica Fisica Industriale Modulo B
Cinetica Chimica
Saverio Santi
Esperienza n° 1
Cinetica di idrolisi di un alogenuro alchilico terziario
Effetto della Temperatura
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
2
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Meccanismo di reazione
Consideriamo l'idrolisi di un generico alogenuro alchilico. La stechiometria della reazione
è la seguente:
RX + 2H2O → ROH + H3O+ + X-
(1.1)
A temperatura e pressione costanti la velocità della reazione dipende dal meccanismo della
reazione. A sua volta il meccanismo dipende dalla natura dei reagenti e del solvente. Se R
è un gruppo alchilico terziario, ad esempio t-butile, e il solvente è una miscela acquaetanolo, il meccanismo è di tipo SN1:
Stadio 1, -1
k1
RX <=> R+ + Xk-1
(1.2)
Stadio 2
k2
R+ + H2O → ROH2+
(1.3)
Stadio 3
k3
ROH2+ + H2O → ROH + H3O+
(1.4)
Il primo è lo stadio lento, rate determining step, il secondo e il terzo sono veloci, tenendo
conto del fatto che l'acqua è anche il solvente della reazione. Possiamo scrivere le
equazioni differenziali per ogni singolo stadio:
d [ RX ]
= k1[ RX ] − k −1[ R + ][ X − ]
dt
1)
−
2)
d[R + ]
= k1[ RX ] − k −1[ R + ][ X − ] − k 2 [ R + ][ H 2 O]
dt
(1.6)
3)
d [ ROH 2 + ]
= k 2 [ R + ][ H 2 O] − k 3 [ H 2 O] [ROH2+]
dt
(1.7)
(1.5)
Poiché la velocità di formazione dell'intermedio R+ è sicuramente minore della sua velocità
di scomparsa, possiamo applicare l'ipotesi dello stato stazionario a R+:
d[R + ]
=0
dt
(1.8)
quindi dall'equazione differenziale 1.6 risulta:
k1[ RX ] − k −1[ R + ][ X − ] = k 2 [ R + ][ H 2 O]
3
(1.9)
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Ricavando l'espressione di [R+]
[R + ] =
k1[ RX ]
−
k −1[ X ] + k 2 [ H 2 O]
(1.10)
e sostituendola nell'equazione 2.5 si ottiene:
v=−
−
k −1k1[ RX ]
d [ RX ]
= k1[ RX ] −
[X − ]
−
dt
k −1[ X ] + k 2 [ H 2 O]
⎛
⎞
k −1[ X − ]
d [ RX ]
⎟
= k1[ RX ]⎜1 −
−
⎜
⎟
dt
⎝ k −1[ X ] + k 2 [ H 2 O] ⎠
(1.11)
La reazione dello stadio 2 rappresenta l'attacco nucleofilo dell'acqua sul carbocatione, in
competizione con l'anione cloruro (X- = Cl-) nella reazione di ritorno (stadio -1). L'anione
cloruro è un nucleofilo migliore dell'acqua, quindi k-1 > k2. Tuttavia l'acqua è anche il
solvente e perciò [H2O] >> [X-]. Ne risulta che k2[H2O] >> k-1[X-] e l'espressione della
velocità 1.11 diventa (e di fatto sperimentalmente si osserva):
v=−
d [ RX ]
= k1[ RX ]
dt
(1.12)
Questa espressione rappresenta l'equazione differenziale di una reazione del primo ordine
cinetico la cui integrazione definita tra [RX] = [RX]0 e [RX], e t = t0 e t:
[RX]
∫
−
[RX] 0
ln
t
d [ RX ]
= k1 ∫ dt
[ RX ]
0
[ RX ]
= − k1 (t − t 0 )
[ RX ]0
(1.13)
permette di ricavare l'andamento della concentrazione del reagente nel tempo (t0 = 0):
[ RX ] = [ RX ]0 e − k1 (t −t0 )
4
(1.14)
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Per ricavare l'andamento della concentrazione dei prodotti dobbiamo considerare il
bilancio di massa:
[RX]0 = [RX] + [X-]
(1.15)
Poiché HCl è un acido forte e quindi completamente dissociato in soluzione si ha che:
[X-] = [H3O+] = CHX
(1.16)
da cui risulta:
[X-] = [RX]0 - [RX]
sostituendo a [RX] la sua espressione nell'equazione 1.14 si ottiene:
[ X − ] = [ RX ]0 (1 − e − k1 (t − t 0 ) )
(1.17)
La variazione della concentrazione di HX può essere seguita per via conduttometrica
misurando la conduttività nel tempo χt:
χt = χ0 + χHX
(1.18)
χ0 = conduttività delle impurezze dell'acqua.
χHX = conducibilità di HX dissociato.
Se la concentrazione di HX nel tempo è sufficientemente bassa da poter trascurare le forze
interioniche vale la relazione ideale:
χ HX = Λ0HX [ HX ]
(1.19)
Possiamo ricavare dalle eq. 1.16, 1.18 e 1.19 l'espressione di [X-] in funzione della sua
conduttività
5
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
[X − ] =
χt − χ0
Λ0HX
(1.20)
e introdurla nell'equazione 1.17 per ricavare l'espressione (χt - χ0).
χ t − χ 0 = Λ0HX [ RX ]0 (1 − e − k1t )
(1.21)
A t∞ la concentrazione finale di HX (o di X- e H+) è uguale alla concentrazione iniziale di
RX:
CHX = [RH]0 = [X-]∞ = [H+]∞
per cui la conduttività della soluzione finale (χ∞ - χ0) è data dall'espressione
χ ∞ − χ 0 = Λ0HX [RX ]0
(1.22)
Sottraendo l'eq. 1.21 alla 1.22 si ottiene infine l'espressione di (χ∞ - χt):
χ ∞ − χ t = Λ0HX [ RX ]0 e − k1t
(1.23)
Dividendo l'eq. 1.23 per l'equazione 1.22 e passando al logaritmo naturale si ottiene un
espressione il cui termine di destra coincide con quello dell'equazione 1.13:
χ − χt
= − k1t
ln ∞
χ∞ − χ0
(1.24)
Questa espressione permette di determinare il valore della costante di velocità del 1° ordine
dalla misura della conducibiltà nel tempo o più in generale dalla misura di una grandezza
chimico-fisica che sia proporzionale alla concentrazione della specie che varia nel tempo.
6
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
5
0
(a)
χ − χt
ln ∞
-5
χ∞ − χ0
-10
(b)
-15
0
1000
2000
3000
4000
t
Figura 1.1
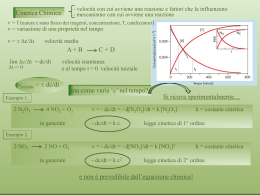
χ − χt
in funzione del tempo t. Dalla
In figura 1.1a è riportato il grafico lineare di ln ∞
χ∞ − χ0
pendenza della retta si ricava la costante del 1° ordine k1. Tuttavia la misura di χ∞ deve
essere ottenuta con un'accuratezza pari a quella della misura di χt quando la reazione è
completa. Per una reazione del 1° ordine si deve attendere 10 volte il tempo di
dimezzamento (t1/2 = ln2/k) perché la reazione sia completa al 99.9%. Tuttavia spesso
accade che il valore finale non può essere misurato con accuratezza. Tra le cause vi sono la
variazione delle condizioni sperimentali (precipitazione del prodotto), variazione delle
condizione strumentali (instabilità della linea di base, staratura) reazioni secondarie, etc.
Se si utilizza come χt l'ultimo valore misurato dopo pochi t1/2 il grafico risultante è quello
riportato in figura 1.1b (reazione al 97%).
Per ovviare a tali inconvenienti sono stati messi a punto metodi che permettono la
determinazione della costante di velocità quando il valore finale non è noto. Tra questi il
più comune è il metodo di Guggenheim.
7
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Determinazione di k1 con il metodo di Guggenheim
L'equazione 1.24 può essere riscritta nel modo seguente:
al tempo t
χ t = χ ∞ − ( χ ∞ − χ 0 )e − k1t
(1.25)
al tempo t + Δt
χ t + Δt = χ ∞ − ( χ ∞ − χ 0 )e − k1 (t + Δt )
ovvero
χ t + Δt = χ ∞ − ( χ ∞ − χ 0 )e − k1t e − k1Δt
(1.26)
con Δt incremento costante del tempo t. Dall'equazione 1.24 si ricava che
χ ∞ − χ t = ( χ ∞ − χ 0 )e − k1t
(1.27)
Sostituendo questa espressione nell'equazione 1.26 si ottiene l'equazione
χ t + Δt = χ ∞ − ( χ ∞ − χ t )e − k1Δt
(1.28)
χ t + Δt = χ ∞ − χ ∞ e − k1Δt + e − k1Δt χ t
(1.29)
che riarrangiata diventa:
La 1.29 rappresenta l'equazione di una retta y = a + bx.
x = χt
pendenza b = e − k1Δt
y = χ t + Δt
intercetta a = χ ∞ − χ ∞ e − k1Δt
I valori χ ∞ e k1 possono essere determinati mediante interpolazione lineare.
Il metodo di Guggenheim richiede che i dati ( χ t ) siano raccolti a intervalli costanti δt e il
valore di Δt scelto per l'elaborazione dei dati sia Δt >> δt.
Un esempio di applicazione del metodo Guggenheim e riportato in tabella 1.1 per T = 3600
s, δt = 60 e differenti valori di Δt = 900, 1800, 2700.
8
Laboratorio di Chimica Fisica
t
χt
Cinetica Chimica
Saverio Santi
χ t + 900
χ t +1800
χ t + 2700
0.0000
0.0000
0.0059343
0.0083470
0.0093279
60.000
0.00058235
0.0061711
0.0084433
0.0093671
120.00
0.0011308
0.0063941
0.0085339
0.0094039
180.00
0.0016473
0.0066040
0.0086193
0.0094387
240.00
0.0021337
0.0068018
0.0086997
0.0094713
300.00
0.0025918
0.0069881
0.0087754
0.0095021
360.00
0.0030232
0.0071635
0.0088467
0.0095311
420.00
0.0034295
0.0073286
0.0089139
0.0095584
480.00
0.0038122
0.0074842
0.0089772
0.0095841
540.00
0.0041725
0.0076307
0.0090367
0.0096084
600.00
0.0045119
0.0077687
0.0090928
0.0096312
660.00
0.0048315
0.0078986
0.0091457
0.0096526
720.00
0.0051325
0.0080210
0.0091954
0.0096729
780.00
0.0054159
0.0081363
0.0092423
0.0096919
840.00
0.0056829
0.0082448
0.0092864
0.0097099
900.00
0.0059343
0.0083470
0.0093279
0.0097268
960.00
0.0061711
0.0084433
0.0093671
1020.0
0.0063941
0.0085339
0.0094039
1080.0
0.0066040
0.0086193
0.0094387
1140.0
0.0068018
0.0086997
0.0094713
1200.0
0.0069881
0.0087754
0.0095021
1260.0
0.0071635
0.0088467
0.0095311
1320.0
0.0073286
0.0089139
0.0095584
1380.0
0.0074842
0.0089772
0.0095841
1440.0
0.0076307
0.0090367
0.0096084
1500.0
0.0077687
0.0090928
0.0096312
1560.0
0.0078986
0.0091457
0.0096526
1620.0
0.0080210
0.0091954
0.0096729
1680.0
0.0081363
0.0092423
0.0096919
1740.0
0.0082448
0.0092864
0.0097099
1800.0
0.0083470
0.0093279
0.0097268
1860.0
0.0084433
0.0093671
1920.0
0.0085339
0.0094039
1980.0
0.0086193
0.0094387
2040.0
0.0086997
0.0094713
2100.0
0.0087754
0.0095021
2160.0
0.0088467
0.0095311
2220.0
0.0089139
0.0095584
2280.0
0.0089772
0.0095841
2340.0
0.0090367
0.0096084
2400.0
0.0090928
0.0096312
2460.0
0.0091457
0.0096526
2520.0
0.0091954
0.0096729
2580.0
0.0092423
0.0096919
2640.0
0.0092864
0.0097099
2700.0
0.0093279
0.0097268
2760.0
0.0093671
2820.0
0.0094039
2880.0
0.0094387
2940.0
0.0094713
3000.0
0.0095021
3060.0
0.0095311
3120.0
0.0095584
3180.0
0.0095841
3240.0
0.0096084
3300.0
0.0096312
3360.0
0.0096526
3420.0
0.0096729
3480.0
0.0096919
3540.0
0.0097099
3600.0
0.0097268
Tabella 1.1
9
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Riportiamo ora nel grafico di figura 1.3 i dati riportati in tabella 1.1:
0.01
χ
t+Δt 0.008
0.006
0
0.005
χ
0.01
t
Figura 1.3
Le rette ottenute per diversi valori di Δt (900, 1800, 2700) hanno una pendenza variabile:
b = e − k1Δt
(1.30)
da cui si ricava il valore della costante di velocità:
k1 = −
ln b
Δt
(1.31)
Scelta di Δt
Supponiamo che la reazione sia stata seguita per un tempo T e ad intervalli regolari δt, con
Δt >> δt. Distinguiamo 3 casi:
1. Δt =
T
, i dati sperimentali vengono usati una sola volta (Δt = 1800 nell'esempio di
2
tabella 1.1).
10
Laboratorio di Chimica Fisica
2. Δt <
Cinetica Chimica
Saverio Santi
T
, si ottengono più punti ma alcuni dati sperimentali vengono usati più di una
2
volta (Δt = 900 nell'esempio di tabella 1.1).
3. Δt >
T
, si ottengono meno punti e alcuni dati sperimentali non vengono usati (Δt =
2
2700 nell'empio di tabella 1.1)
Se la reazione viene seguita per un tempo sufficientemente lungo (T > 3t1/2) la scelta di
Δt =
T
permette di ottenere la costante di velocità k1 con il minimo errore δk1 come risulta
2
diagrammando
δk1
k1
vs. Δt (figura 1.4).
δk1
k1
T
2
Figura 1.4
Δt
Quando invece la reazione è seguita per un tempo breve (T < t1/2) il Δt che minimizza
l'errore su k1 può essere diverso da
T
.
2
Si sceglierà il valore di k1 con il minimo errore δk1 valutato sulla base dell'interpolazione
dei dati sperimentali con il metodo dei minimi quadrati.
11
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Effetto della temperatura
Per molte reazioni in fase omogenea sperimentalmente si riscontra una dipendenza della
costante di velocità dalla temperatura secondo l'equazione di Arrhenius:
k = Ae − Ea / RT
(1.32)
in cui Ea è l'energia di attivazione, ovvero l'energia termica necessaria per trasformare i
reagenti in prodotti, e A il fattore pre-esponenziale.
La teoria dello stato di transizione ha portato ad una razionalizzazione dell'effetto della
temperatura sulla costante di velocità e in particolare ha fornito un'interpretazione del
fattore pre-esponenziale A. L'espressione della costante di velocità è data dall'equazione di
Eyring:
k T
k = B K≠
h
(1.33)
kB = costante di Boltzman
h = costante di Plank
K ≠ = costante di equilibrio tra stato di transizione X≠ e reagenti.
K≠
A <=> X≠ → P
Introducendo nell'eq. 1.33 le relazioni termodinamiche
K≠ = e
−
ΔG ≠
RT
(1.34)
ΔG ≠ = ΔH ≠ − TΔS ≠
(1.35)
si ottiene la formulazione termodinamica della teoria della stato di transizione:
≠
≠
H
k BT ΔSR − ΔRT
k=
e e
h
12
(1.36)
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Per confrontare l'equazione 1.36 con equazione di Arrhenius è necessario ricavare
l'espressione dell'energia di attivazione Ea. che è definita dalla derivata secondo l'eq. 1.33
dalla derivata:
E
d ln k
= a
dT
RT 2
(1.37)
Analogamente per l'equazione di Eyring 1.33 si ottiene:
d ln k 1 d ln K ≠
= +
dT
T
dT
(1.38)
Applicando l'equazione di van'Hoff alla costante di equilibrio K ≠ si ricava:
d ln K ≠ ΔU ≠
=
dT
RT 2
(1.39)
e sostituendo il termine di destra nell'equazione 1.38 si ha:
d ln k 1 ΔU ≠
= +
dT
T RT 2
(1.40)
Eguagliando le eq. 1.37 e 1.40 è possibile ricavare l'espressione di Ea:
E
1 ΔU ≠
+
= a
T RT 2 RT 2
E a = ΔU ≠ + RT
(1.41)
E a = ΔH ≠ − PΔV ≠ + RT
(1.42)
ovvero
13
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Per una reazione unimolecolare in fase gas, la variazione di volume tra lo stato attivato e il
reagente è nullo. Infatti PΔV ≠ = Δn ≠ RT e non vi è variazione di numero di moli nella
formazione dello stato di transizione ( Δn ≠ = 0 ). Allora l'eq. 1.42 diventa:
E a = ΔH ≠ + RT
(1.43)
Introducendo l'equazione 1.43 nella 1.36 si ottiene un'espressione della costante di velocità
≠
k BT ΔSR − ERTa
e e
k =e
h
(1.44)
e dal confronto con l'equazione di Arrhenius
k = Ae − Ea / RT
(1.32)
si ottiene l'espressione del fattore pre-esponenziale:
≠
k BT ΔSR
A=e
e
h
(1.45)
Per una reazione bimolecolare Δn ≠ = −1 .
K≠
A + B <=> X≠ → P
L'eq. 1.42 in questo caso diventa:
E a = ΔH ≠ + 2 RT
(1.46)
e si ottiene
≠
k BT ΔSR − ERTa
k =e
e e
h
2
(1.47)
Sperimentalmente si trova che le equazioni 1.44 e 1.47 vanno bene anche in soluzione.
14
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
I parametri di attivazione A, E a , ΔH ≠ e ΔS ≠ sono ricavabili sperimentalmente misurando
le costanti
di velocità a differenti temperature e diagrammando lnk
Arrhenius) o ln
(plot di
k
(plot di Eyring) in funzione di 1/T per ottenere rispettivamente A e Ea, e
T
ΔH ≠ e ΔS ≠ :
⎛ k ⎞ ΔS
ln⎜ B ⎟ +
R
⎝ h ⎠
v
lnA
lnk
k
ln
T
E
− a
R
≠
ΔH ≠
−
R
1/T
1/T
Plot di Arrhenius
Plot di Eyring
Figura 2.5
Significato dei parametri termodinamici di attivazione
La determinazione dell'entalpia e dell'entropia di attivazione di una reazione fornisce
importanti informazioni sul meccanismo di reazione. Consideriamo la reazione di idrolisi:
RX + 2H2O → ROH + H3O+ + X-
(1.1)
Se la reazione segue un meccanismo di tipo SN1 lo stadio lento della reazione è la rottura
eterolitica di RX che porta alla formazione di R+ e X-. I parametri di attivazione
ΔH ≠ e ΔS ≠ ricavati sperimentalmente si riferiscono generalmente allo stadio lento della
R-X
δ+
R
δX
15
R+ + X-
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
reazione. Nello stato di transizione sarà presente una parziale rottura del legame C-X e una
parziale separazione di carica:
Il valore di ΔH ≠ dipende dal grado di rottura del legame R-X (l'energia di legame C-Cl è
ΔH ° = 81 kcal mol-1) nello stato di transizione, mentre il termine entropico ΔS ≠ risentirà
dell'aumento di disordine dovuto alla parziale dissociazione.
Tuttavia, in un solvente polare lo stato di transizione è solvatato:
+
-
+
-
+
+
-
+
-
δ+
+
δ-
X
-
+
+
-
+
+
-
-
+
R
+
+
-
+
+
-
-
-
+ -
+
-
+
+
+
-
-
+
+
+
-
+
-
-
-
-
-
-
+
R X
-
+
+
-
+
-
+
+
+
+
+
+
-
-
-
-
-
+
-
+
+
-
-
+
Di conseguenza il ΔH ≠ è ridotto dal contributo dell'energia di solvatazione e il ΔS ≠
diminuisce a causa dell'aumento di ordine nello stato di transizione.
Per la reazione di idrolisi del cloruro di t-butile in una miscela al 30% di acqua/etanolo si
trovano i seguenti valori, ΔH ≠ = 17 kcal mol -1 , ΔS ≠ = −20 cal K -1mol -1 .
Per una reazione che segue un meccanismo di tipo SN2 lo stadio lento della reazione è
OH-, al reagente in uno stadio bimolecolare
l'addizione del nucleofilo, ad esempio
(meccanismo associativo):
δ−
HO
C
R-X + OH
-
+
+
+
+
+
+
-
+
-
++
-
-
-
-
-
+
-
+
-
δ−
HO
-
-
+
+
-
-
+
-
+
+
+
-
16
δ−
X
C
+
+
-
+
+
+
-
-
+
+
+
-
+
+
ROH + X-
+
+
-
+
+
OH+
+
-
-
-
-
-
+
-
+
-
+ +
+
+
-
+
-
-
+
+
+
+
-
-
+
-
R X
-
+
+
-
-
-
+
+
+
-
-
+
-
-
-
+
-
δ−
X
-
-
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Nello stato di transizione l'energia di parziale rottura del legame C-X è in parte compensata
dall'energia di formazione del legame C-O (l'energia di legame C-O è ΔH ° = 85.5 kcal
mol-1) e il valore di ΔH ≠ risulterà minore di quello relativo ad un meccanismo
dissociativo. Inoltre la carica relativa è maggiormente dispersa nello stato di transizione
che è quindi meno solvatato dei reagenti e il ΔS ≠ risulta positivo a meno del contributo
negativo dovuto alla diminuzione di disordine dovuto alla diminuzione del numero di
molecole nello stato attivato.
Se il nucleofilo è l'acqua
δ+
H2O
C
R-X + H2O
-
+
+
-
+
+
+
+
-
+
+
+
δ−
C
-
+
-
X
-
+
+
-
+
+
+
-
+
-
+
+
+
-
-
-
+
-
δ+
H 2O
-
-
+
+
-
+
+
-
+
-
-
+
-
+
R X
-
+
+
-
-
+
+
+
-
+
+
-
-
-
-
-
+
+
+
ROH + HX
+
-
δ−
X
-
+
+
-
-
-
si ha una separazione di carica nello stato di transizione a cui corrisponde una sua
maggiore solvatazione rispetto ai reagenti e perciò un valore ridotto di ΔH ≠ e negativo di
ΔS ≠ .
17
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Esperienza 1 – Cinetica di idrolisi di un alogenuro alchilico
Obiettivi: seguire per via conduttimetrica l’idrolisi di un alogenuro alchilico terziario a
varie temperature (indicativamente 21, 27, 33, 39, 45, 50°C) e determinarne le costanti
cinetiche (di primo ordine), l’energia di attivazione Ea, il fattore pre-esponenziale A,
l’entalpia e l’entropia di attivazione.
Apparecchiatura: : conduttimetro interfacciato a PC, cella conduttimetrica, 1 matraccio
da 250 cc e 1 da 50 cc, termostato, termometro, agitatore magnetico con ancoretta, beuta
con camicia per la termostatazione, canna in gomma per raccordo con termostato.
Reagenti: acqua bidistillata, etanolo assoluto, alogenuro alchilico.
Nei matracci da 250 e 50 ml si prepara la soluzione acqua-etanolo con la percentuale
fissata di acqua (tra il 15 e il 30% in volume) mediante la seguente procedura: si pesa il
matraccio vuoto (pulito ed asciutto), lo si pesa nuovamente dopo l’aggiunta del volume
prefissato di acqua, ed infine si porta a volume con l’etanolo (riportare nella relazione il
valore della percentuale in volume di acqua).
Si termostata la soluzione nella beuta di misura per circa 20 minuti, sotto agitazione, vi si
immerge la cella conduttimetrica e si regola l’agitazione in modo da evitare la formazione
di vortici e bolle all’interno della soluzione stessa. Si aggiungono quindi, con la pipetta, 0.4
ml di alogenuro alchilico e si fa partire il campionamento. Si eseguono misure di
conducibilità ad intervalli di tempo rigorosamente costanti (ad esempio 10, 20, 30”, 45" e
1’, rispettivamente per le reazioni a 21, 27, 33, 39, 45, 50°C). Si consiglia di seguire la
cinetica, se possibile, per almeno due volte il tempo di dimezzamento dell’alogenuro.
Analizzare i dati secondo il metodo di Guggenheim-Swinbourne, calcolando la costante
cinetica di primo ordine alle temperature di lavoro; trattare prima i dati sulla base
dell’equazione (8), utilizzando come χ∞ l’ultima conducibilità misurata; ripetere alla fine
tale trattamento utilizzando il valore di χ∞ determinato con il metodo di GuggenheimSwinbourne.
Si ricorda che è importante “tarare” il conduttimetro prima di ogni determinazione,
portando il valore della costante di cella ad 1.
Si raccomanda di versare alla fine la soluzione nella bottiglia di recupero, facendo
attenzione a non perdere l’ancoretta.
18
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
Trattamento dei dati
Considerato che la reazione di idrolisi dell’alogenuro terziario RX segue un meccanismo
per il quale la cinetica di decadimento risulta di primo ordine rispetto alla sua
concentrazione, cioè
dCRX/dt = −k CRX
(1)
dove k rappresenta la costante specifica di velocità della reazione, integrando si ottiene:
CRX = C°RX exp[−k(t − to)]
(2)
Data la stechiometria della reazione, ad ogni istante t si avrà inoltre che:
CRX + CHX = C°RX
(3)
per cui, sostituendo:
CHX = C°RX {1 − exp[−k(t − to)]}
(4)
La cinetica di tale reazione viene seguita per via conduttimetrica, misurando la
conducibilità della soluzione nel tempo. Se si suppone, infatti, che la conducibilità
specifica della soluzione, ad ogni istante t della reazione, sia la somma di un contributo
dovuto alle specie presenti al tempo zero, χ0, e da un contributo dovuto all’acido HX (che
risulta completamente dissociato) che si genera per effetto della reazione, si ha che:
χt = χ0 + χHX
(5)
Il contributo χHX viene supposto proporzionale alla concentrazione dell’acido CHX, tramite
la relazione:
χHX = ΛHX CHX
(6)
19
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
dove ΛHX rappresenta la conducibilità molare dell’acido nel sistema in esame, che si
suppone costante al variare della concentrazione dell’acido stesso (dato che la soluzione
risulta sufficientemente diluita nell’acido), e quindi pari alla conducibilità molare a
diluizione infinita, Λ°HX.
Mediante opportune sostituzioni si ottiene quindi che:
χt − χ0 = Λ°HX C°RX {1 − exp[−k(t − to)]}
(7)
Indicando con χ∞ la conducibilità specifica della soluzione a reazione ultimata, cioè
per t → ∞, con opportuni passaggi e sostituzioni, essendo, dalle relazioni precedenti, χ∞ −
χ0 = Λ°HX C°RX, si ottiene che:
ln[(χ∞ − χt)/ (χ∞ − χ0)] = −k(t − to)
(8)
da cui si potrebbe ricavare facilmente il valore di k note χ∞ e χ0.
Poiché la prima quantità non è facilmente determinabile, è necessario effettuare un
diverso trattamento dei dati sperimentali, noto come metodo di Guggenheim-Swinbourne.
Se si considera la conducibilità al tempo t ed al tempo Δt, dalle relazioni viste sopra si
può ottenere che:
χt = Λ°HX C°RX + χ0 − Λ°HX C°RX {exp[−k(t − to)]} = χ∞ − (χ∞ − χ0) {exp[−k(t − to)]}
(9)
da cui:
χt+Δt = χ∞ −(χ∞ − χ0){exp[−k(t + Δt − to)]} = χ∞ −(χ∞ − χ0)[exp(−k Δt)]{exp[−k(t − to)]}
(10)
che collegata alla relazione precedente fornisce la:
χt+Δt = χ∞ − (χ∞ − χt) [exp(−k Δt)]
(11)
che può essere anche espressa come:
20
Laboratorio di Chimica Fisica
Cinetica Chimica
Saverio Santi
χt+Δt = χ∞ − χ∞ [exp(−k Δt)] + χt [exp(−k Δt)]
(12)
la quale, considerando di esprimere χt+Δt in funzione di χt per un dato valore di Δt,
rappresenta l’equazione di una retta di pendenza exp(−k Δt) e di intercetta χ∞ − χ∞ [exp(−k
Δt)].
Sulla base di tale equazione, fissato un valore di Δt si può quindi ottenere un valore della
costante cinetica k mediante regressione lineare dei dati. Tale operazione può essere inoltre
ripetuta, utilizzando sempre lo stesso set di dati sperimentali, variando il valore di Δt; il
valore di k migliore tra quelli ottenuti sarà quello fornito dalla migliore regressione lineare.
Una volta ricavato il valore di k per un certo Δt, dal valore dell’intercetta si può ricavare
quello di χ∞ ed analizzare l’andamento rappresentato dall’equazione (8).
Osservando le equazioni (12) e (8) si può infine notare che non è necessario trattare i
dati utilizzando la conducibilità specifica χ, ma che può essere analogamente utilizzata la
conducibilità C direttamente misurata e ad essa proporzionale, senza dover determinare
quindi il valore della costante di cella.
Con i valori di k ottenuti ad ogni temperatura calcolare A ed Ea utilizzando l’equazione di
Arrhenius (13) e ΔH≠ e ΔS≠ dall’equazione di Eyring (14):
k = A e-Ea/RT
(13)
k = kBT/h e ΔS≠/R e-ΔH≠/RT
(14)
Discutere il meccanismo della reazione sulla base dei valori dei parametri di attivazione
ottenuti.
21
Scaricare