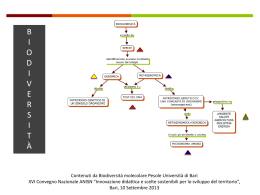
Antitumorali Neoplasia – termine con il quali si indica la abnorme e nuova crescita di un tessuto. Esse possono essere: Neoplasie benigne – hanno contorni ben definiti, si possono asportare facilmente chirurgicamente ottenendo anche la scomparsa delle sintomatologie ad esse connesse ma, soprattutto, non generano metastasi. Neoplasie maligne – invadono i tessuti circostanti e la loro rimozione completa diventa difficile se non praticamente impossibile. Inoltre diffondono in tessuti anche lontani generando le metastasi. Sono classificate in vari modi ma si preferisce chiamarli: con il nome dello scopritore (tumore di Wilm, sarcoma di Hodking, ecc…); Con il nome del tessuto dal quali derivano (Classificazione istologica). Antitumorali 2 Oma – suffisso che solitamente indica un tumore benigno (eccezione Melanoma e linfoma che sono maligni). Tra questi ricordiamo come esempio l’Adenoma (epitelio ghiandolare), osteoma (osso) e condroma (cartilagine). Lesioni quali il granuloma non sono affatto dei tumori. Blastoma – serve ad indicare neoplasie maligne. I tumori indifferenziati possono essere originati da tessuti epiteliali detti carcinomi o da tessuti connettivi detti sarcomi. Leucemia – tumore selle cellule ematiche con sovrapproduzione di globuli bianchi (fino a 1000 volte): Anche in questo caso se ne conoscono due tipi: Leucemia acuta – i precursori delle cellule bianche non arrivano a maturazione ed interferiscono con la normale produzione di globuli bianchi normali. Leucemia cronica – produce cellule ematiche mature anomale non in gradi di svolgere il loro ruolo di prevenzione e resistenza alle infezioni. Antitumorali 3 Metastasi – sono causate da cellule del tumore maligno che si staccano dalla sede primaria e vengono distribuite dal sistema vascolare o linfatico. Durante questa fase di trasporto la sopravvivenza delle cellule è minima. Quelle che sopravvivono devono riuscire ad aderire esse stesse nelle membrana basale e ivi penetrare. la formazione del tumore secondario avviene solo se la cellula metastatica viene a trovarsi in un ambiente favorevole. Principi base Le due principali fasi della vita di una cellula sono: la sintesi del DNA e la mitosi con produzione di nuove cellule; La differenziazione cellulare in grado di produrre cellule specializzate. La crescita cellulare è regolata da fattori prodotti dalla stessa cellula. Antitumorali 4 Spesso le cellule hanno un sistema di controllo (feedback) negativo in grado di controbilanciare l’effetto del fattore della crescita. Sia i fattori di crescita che quelli di inibizione esplicano la loro azione legandosi a recettori sulla superficie cellulare. Le cellule tumorali sono in grado di sovraprodurre i fattori di crescita e di esprimere meno quelli di inibizione. Il risultato è la perdita del controllo della proliferazione cellulare che così diventa anomala ed esagerata. Regolazione del ciclo cellulare G1 – momento in cui nasce una cellula nuova. La durata della fase dipende dal tessuto; S – fase di sintesi nella quale si portano le cellule che devono proliferare. Si ha replicazione del DNA. G2 –rappresenta la fase di preparazione alla mitosi o fase M. Antitumorali 5 Vi sono due momenti di controllo e precisamente all’interfacia G1-S e G2 – M e precisamente quando la cellula decide rispettivamente di replicarsi e di dividersi. È a livello dell’inizio della farse replicativa che si parla di tumore e di sua cura. Nella fase G2 la cellula cessa la sintesi del DNA ma continua la sintesi di RNA e proteine che precede la mitosi (M). Si formano così due cellule figlie che possono iniziare il ciclo o entrare in una fase di quiescenza chiamata G0 dove possono rimanere per lungo periodo ed essere successivamente immesse nel cirocolo. La cellula può anche andare incontro al processo di morte cellulare e così morire. Nei tumori la percentuale di cellule in fase proliferativa cresce notevolmente a scapito di quelle quiescenti e che muoiono. Ciò produce una crescita abnorme. È stato osservato che nei tumori le nuove cellule si formano in zone ipossiche vicine al centro del tumore e poco vascolarizzate. Antitumorali 6 Ne consegue che esse vengano a trovarsi in uno stato di quiescenza. Ciò complica la durata della cura. Infatti, gli antitumorali agiscono distruggendo le cellule in fase replicativa ma non agiscono sulle cellule quiescenti. Per questo motivo sono necessari cicli di terapia al fine di consentire alle cellule in fase quiescente di entrare in fase S ed essere così distrutte dal ciclo di cura successivo. Vi sono due tipi di mutazioni: Della linea germinale; Somatiche (causano il cancro). Il processo di carcinogenesi abbisogna di una mutazione (che non venga riparata) che rappresenta l’iniziazione ed una fase di sviluppo nella quale le cellule proliferano. A questo livello si forma un tumore benigno che consente la nascita del tumore maligno primario e successivamente si formano le metastasi o tumori secondari lontani. Antitumorali 7 Iniziatori – sono sostanze che non producono cancro, ma solo mutazioni che possono essere anche riparate. Promotori – di per sé non sono genotossici ne tanto meno mutageni. Prodotti chimici – XVII secolo scoperta della elevata incidenza di tumori negli spazzacamini londinesi (benzopirene). Si ipotizza che i prodotti chimici possano interagire con le basi del DNA generando un danno o mutazione in un singolo punto. Questo tipo di danno può essere riparato o può rimanere e pertanto essere trascritto e generare una mutazione permanente. Antitumorali 8 Queste sostanze possono dare direttamente degli addotti con il DNA o possono indurli (radiazioni). Virus oncogeni – essi si inseriscono nel genoma della cellula,ospite. Virus a DNA – portano alla produzione di proteine che possono interagire con quelle della crescita o con i geni che sopprimono i tumori. Virus a RNA – possono causare i tumori (soprattutto i retrovirus). Grazie alla trascrittasi inversa essi producono del DNA a doppia catena a partire dal RNA. Successivamente una integrasi inserisce questo DNA provirale nel genoma della cellula ospite. Si possono generare alterazioni nell’espressione genica che determinano poi le fromazioni tumorali. Alterazione genica – esistono dei protooncogeni che controllano la crescita cellulare. Se essi vengono espressi in quantità abnorme e possono alterare l’equilibrio tra geni oncogeni e anti-oncogeni. Si può avere pertanto: Una aumentata espressione degli oncogeni; Diminuita espressione dei geni antitumore (anti-oncogeni). Antitumorali 9 Si conoscono numerosi oncogeni raggruppabili in: Oncogeni che determinano una continuità nella produzione dei fattori di crescita (gene hst e int); Oncogeni che includono i geni associati ai recettori dei fattori della crescita. Oncogeni che legano i nucleotidi guaninici. Esistono anche geni soppressori dei tumori quale il gene p53 (che attraverso l’interazione le cicline rallenta la proliferazione cellulare) e la p21. Terapia del Cancro Fondamentalmente il tumore può essere curato con: Chirurgia; Terapia con radiazioni; Terapia immmunologica; Chemioterapia. Antitumorali 10 Lo scopo della chemioterapia è quello di utilizzare farmaci dotati di tossicità selettiva sull’esempio degli antibatterici. Le differenze esistenti tra cellule sane e tumorali sono minime è ciò rende difficile il raggiungimento di questo obiettivo. Perciò la maggior parte degli antitumorali agisce interferendo nel processo di divisione cellulare. Ne consegue che le neoplasie più sensibili sono quelle con un elevato indice di accrescimento (es tumori giovani con elevata % di cellule in fase replicativa). La terapia deve quindi essere iniziata il più presto possibile ed eventualmente associarla alla terapia radiante e a quella chirurgica. La logica conseguenza è che i farmaci antitumorali tendono a colpire anche le cellule sane in rapida attività. Le manifestazioni tossiche più frequenti sono: nausea, vomito, anoressia, alopecia, leucopenia ed aumentata sensibilità alle infezioni. Antitumorali 11 Classi di antitumorali Ormoni e antagonisti ormonali - tendono a variare le condizioni tissutali dove è avvenuta la crescita neoplastica. Immunostimolatori – stimolano la risposta immunitaria; Citotossici o citostatici – intervengono nei processi replicativi della cellula dove possono: Intervenire nella desossiribonucleotidi; biosintesi dei Replicazione; Processi di trascrizione e traduzione del DNA; Processo di formazione del fuso mitotico. ribo e Agenti alchilanti 1 Agenti alchilanti Rappresenta una classe di composti molto eterogenea. L’ipotesi del loro utilizzo come antitumorali nacque dall’osservazione dell’azione tossica sui tessuti proliferanti dell’Iprite, gas tossico, utilizzato per la prima volta a Ypres durante la prima guerra mondiale. Cl S Cl Nella sintesi di numerosi analoghi vennero sintetizzati anche gli isosteri azotati recanti un sostituente allo stesso atomo d’azoto. 1949 venne approvata da parte della FDA la Mecloretamina. Cl CH3 N Cl Mecloretamina Inizio dello sviluppo di nuovi farmaci ad azione antitumorale (ciclofosfamide, clorambucile, Melfasan ….). Agenti alchilanti 2 Meccanismo d’azione – agiscono tutti alchilando il DNA e in in particolare la posizione 7 della guanina O H2N H3C N O HN H2N N CH3 N N HN N Cl N DNA Cl O Cl N N DNA H3C N O + HN H2N N N+ H N NH2 N N+ N DNA N DNA L’alchilazione può interessare anche altre basi che compongono il DNA L’ossigeno della posizione 6 della Guanina; Gli atomi di azoto 1 e 3 della Adenina; L’azoto in 3 della Citosina; Può interessare anche i gruppi fosfato presenti sullo scheletro del DNA e le proteine associate al DNA. Agenti alchilanti 3 Qualora l’agente alchilante lo consenta si possono formare legami intra- o inter-filamento. La reazione che avviene procede con un meccanismo che può essere: SN1 - la reazione limitante è riferita ad una sola specie molecolare (mostarda azotata) e consiste in una ionizzazione dell’agente alchilante; lenta R+ + X- R X + R - R A + A veloci R+ + HA R A + H+ SN2 - la reazione fa riferimento a due specie (mostarda azotata e substrato) ed avviene con simultanea rottura e formazione di legami tra agente alchilante e substrato. R X + A- R X + HA lenta A R X HA R X veloci R A + X- R A + X- + H+ Agenti alchilanti 4 Questo avviene qualora l’alchilante non rechi un sostituente in grado di generare un anello a tre termini (tensionato e reattivo) caricato positivamente (ione aziridinio). In questo caso è la successiva reazione con il nucleofilo che determina l’ordine di reazione SN2 a) Mostarde alifatiche b) Mostarde aromatiche Agenti alchilanti 5 L’alchilazione del N7 stabilizza la forma enolica che a sua volta favorisce altre reazioni. CH3 N O N HN H2N Cl N + N N N CH3 N OH Cl N H2 N N H2N N N O CH3 N N N Cl H2N N N CHO NH O O O Cl N N O O OH HO + N N O O O N NH2 CH3 N N + N O O N N H2N O O OH NH N O CH3 N N N Cl O O OH O O O NH2 Agenti alchilanti 6 L’anello imidazolico verrebbe così indebolito con possibile formazione di derivati aperti e di composti nei quali si è scisso il legame con la catena del DNA. In particolare l’enolizzazione impedirebbe alla Guanina di instaurare legami idrogeno con la Citosina. Anzi, l’acidità del tautomero favorirebbe una analoga interazione con la Timina. Come già detto prima si possono formare anche legami inter- e intra-filamento. In particolare i legami interfilamento dati dalla Mecloretamina generano citotossicità. Come sostanze possono alchilare anche le proteine e l’RNA H H O N HN N O DNA H N H N N N H Citosina e Guanina N H HO N N R N + N N N O DNA H N H Normale legame tra le due basi Citosina e Guanina N DNA HO O DNA NH N O DNA N R N + N N H N H DNA Timina e Guanina Agenti alchilanti 7 Gli agenti alchilanti si rivelano più attivi nelle fasi G1 e S. La cellula è in grado di riparare il danno generato da queste molecole in quanto una endonucleasi taglia il tratto danneggiato mentre una esonucleasi lo allontana. Si genera una regione vuota che viene riempita in modo corretto. Le cellule in rapida replicazione non hanno sufficiente tempo per riparare il danno cosa che invece avviene nelle cellule in fase G0. Per questo motivo essi agiscono prevalentemente sulle cellule in rapida proliferazione (tumorali). Mostarde azotate Sono derivati di 2-Cloroetilamine spesso bifunzionalizzate. Mecloretamina (Clormetina) – utilizzata come cloridrato per iniezioni ev. È una sostanza molto instabile la cui soluzione acquosa (pH 3,5) deve essere utilizzata subito. Presenta una emivita di 15 minuti e soprattutto la forma aziridinica è presente nel sangue già dopo pochi minuti dalla somministrazione. CH CH CH 3 Cl Cl - N+ H 3 3 Cl Cl N Cl N+ Cl Agenti alchilanti 8 Per aumentare la selettività (verso determinate cellule maligne) e allo stesso tempo ridurne la tossicità sono stati studiati numerosi derivati. Essi consistono nell’avere il sistema dicloroetilaminico legato tramite azoto ad una catena che può essere: COOH COOH Un amminoacido; Cl Un anello aromatico, N Una base purinica; N Cl Cl N Cl Cl Uno zucchero; Novembichina Cl Cl Una fosfamide ciclica; Clorambucile Metabolita sostanze varie quali nuclei steroidici etc… Primo approccio fu l’uso della Novembichina. Clorambucile – è uno dei composti più vecchi e nasce con l’intento di ridurre il distacco dell’atomo di cloro grazie a gruppi elettron atrattori. Lo scopo è stato raggiunto con all’introduzione di un anello aromatico che diminuisce la basicità dell’atomo di N. Viene metabolizzato rapidamente a dare il derivato fenilacetico pure esso attivo. Agenti alchilanti 9 Visto che è un derivato a carattere acido si lega fortemente alle proteine plasmatiche. È indicato nel trattamento della leucemia linfocitica cronica. Sono stati fatti anche degli esteri con il Prednisolone (prednimustina) e con l’Estradiolo (Busramustina) mirato per il cancro al seno. Melfalan (L-PAM o L-Sarcolisina) – sfrutta lo stesso effetto dell’anello aromatico con l’aggiunta che l’amino acido potrebbe rivelarsi critico nel metabolismo della cellula tumorale. Sembra esservi un suo assorbimento preferenziale rispetto alla forma D per il quale sono richieste dosi maggiori. Anche lui si lega bene (30%) alle proteine plasmatiche e viene utilizzato nel mieloma multiplo. SAR - non sembra esserci differenza di attività tra isomeri ottici e i vari isomeri di posizione anche se l’isomero L-meta ha minor azione inibitrice sul sistema immunitario ed una aumentata attività antiproliferativa. H N Cl 2 COOH N Cl Melfalan L-Sarcolisina R1 Cl N Cl Agenti alchilanti 10 H L-NitroArg-L-norVal L-Ser-p-fluoro-L-Phe OH L-Prolina O R2 Me N Cl P-Fluoro-L-Phe N R2 R1 NH p-Fluoro-L-Phe L-Arg M,L-SL-L-Arg-L-Lys L-His P-Fluoro-L-Phe-Gly L-norVal COOH N Cl Bendamustina Sono entrati in uso clinico anche derivati multi alchilpeptidici o Polimelfalan (mix di prodotti) per il trattamento di malattie emolinfo proliferative. Bendamustina Treanda® – somministrata cloridrato per aumentarne la stabilità per endovena come Approvata nel 2008 dall’FDA per il trattamento del linfoma nonHodgkin durante il trattamento con Rituximab (anticorpo monoclonale). Sostituisce la ciclofosfamide. Agenti alchilanti 11 Studi condotti su mostarde azotate aromatiche ha evidenziato una stretta relazione esistente tra basicità e velocità di reazione. σ -4.02 LogKH = -1.84σ Negative slope LogK = -1.92σ σ- -1.17 Log(1/IC50) = -2.46σ σ +0.53 Mannomustina – rappresenta un esempio di mostarda azotata legata ad una porzione zuccherina. Usata nelle leucemie linfatiche per ev. Dopan e Uramustina sono derivati della Timina e dell’Uracile studiati come antitumorali senza tuttavia evidenziare l’effettiva efficacia. Cl Mannomustina H2C HO HO H H H2C N O Cl HN H H OH OH O O CH3 N H Cl DOPAN Cl Cl N Cl N HN O N H Cl N Cl Uramustina Agenti alchilanti 12 Ciclofosfamide – rappresenta un composto nel quale si è riusciti a: Ridurre la reattività dell’azoto che non è più basico (amide); Facilitarne il trasporto (profarmaco). Essa infatti risulta meno attiva nel formare lo ione aziridinio rispetto a tutti gli altri composti e risulta decisamente più stabile. La scelta del radicale ciclofosfamidico è ideale per l’effetto a livello dei tessuti tumorali che sono particolarmente ricchi di fosfatasi e fosfamidasi. Ciò rende biodisponibile il farmaco al sito d’azione con una selettiva tossicità. Richiede un’attivazione metabolica mediata dal CYP450 con ossidazione dell’anello a dare il derivato carbinolamminico che decompone facilmente a dare acroleina ed una mostarda fosforammidica. O O Cl P N La mostarda fosforamidica che si forma ed il HN derivato aziridinico sono in grado di dare Cl legami crociati nel DNA e sono citotossici Ciclofosfamide Agenti alchilanti 13 attiva attiva Agenti alchilanti 14 Cl Cl POCl3 + Cl O P N Cl HN Cl bis-(2-cloroetil)amina Cl HO NH2 NH P N O O 3-aminopropanolo Cl Cl N,N-bis-(2-cloroetil) fosfamide dicloruro Ifosfamide ed Trofosfamide – sono degli analoghi della ciclofosfamide. Ed anche loro devono essere attivati metabolicamente. 4-Idrossiperossiciclofosfamide (4-HPCy) – rappresenta un metabolita che si forma anche in vivo dalla ciclpfosfamide stessa e presenta il vantaggio di non richiedere l’attivazione metabolica da parte del CYP450 dato che quest’ultimo in alcuni tumori presenta un’attività ridotta. Cl O O P N N H O O P N N Cl Cl Cl Ifosfamide Cl Trofosfamide O O P N HN Cl Cl O HO 4-HPCy ONa P O ONa Cl Agenti alchilanti 15 Estramustina fosfato - in questo composto la parte aptofora è data dall’estradiolo mentre la O catena cloroetilaminica rappresenta il farmacoforo. N O È una mostarda modificata in quanto l’azoto è Cl legato ad un acile. L’attività sembra sia dovuta ad entrambe le componenti della molecola Principali mostarde azotate e loro indicazioni terapeutiche Agenti alchilanti 16 Alchil Solfonati Busulfan – estere del 1,4-butandiolo con l’acido metansolfonico che rappresenta il gruppo uscente. È una molecola neutra con scarsa solubilità in acqua. Questo farmaco non viene ritrovato tal quale nelle urine ad indicare che subisce reazioni di idrolisi o di alchilazione. o Come gli altri agenti alchilanti predilige reagire con l’azoto in 7 della Guanina dando luogo anche a dimeri. o È specifico nel trattamento della leucemia mielogena cronica, per la quale è il farmaco più efficace di cui si dispone. o Viene utilizzato anche per la leucemia granulocitica cronica e la mielofibrosi. Busilvex® - soluzione contenente 6 mg/mL e viene somministrato per infusione. Myleran® - compresse da 2 mg. H3C O2 S O Busulfan O S O2 CH3 Agenti alchilanti 17 o Treosulfam o (L-treo-diidrossibusulfan) che viene metabolizzato ad epossido ed è usato nel carcinoma ovarico. o Ricordiamo anche il Ritrosulfan simile alle mostarde azotate dotato di proprietà citostatiche e immunosoppressivo. o Mannosulfan – derivato del mannosio tetrametansolfonato. o Mitobronitol – corrisponde al 1,6-dibromo-1,6-dideossi-Dmannitolo. Sembra agire alchilando il DNA. Treosulfan Mannosulfan H3C O2 S OH O O OH O2 S OH O S O2 O OH CH3 SO2 O O O2S OH S O2 H3C O S O2 N H OH H N O O2 S CH3 Ritrosulfan Agenti alchilanti 18 Derivati Aziridinici Tiotepa – Thioplex® (trietilentiofosfaramide)– fa parte degli alchilanti aziridinici che rappresentano il passo successivo compiuto dopo la scoperta del meccanismo d’azione della Clormetina. È il composto più utilizzato anche perché più stabile della Tepa (Trietilenfosforamide) un composto atossico molto usato a livello industriale e come sterilizzante per insetti. A pH acidi forma lo ione aziridinio che agisce da alchilante. Triaziquone – indicato per il tumore alle ovaie. Carboquone – indicato per il trattamento del tumore ai testicoli, alle ovaie e il tumore avanzato alla prostata. Meccanismo d’azione classico con alchilazione del DNA e formazione di legami crociati che impediscono che il DNA venga separato per la sintesi e la trascrizione. Usato in associazione con Cis-platino. S N P N H S N+ P N O N P N N N N O N N TioTEPA TEPA N Triaziquone NH2 O N O O O N O O Carboquone Nitrosouree Agenti alchilanti 19 Composti attivi come antitumorali in quanto generano prodotti di degradazione in grado di alchilare il DNA e di carbamoilare le proteine. Le nitrosouree sono agenti alchilanti monofunzionali ma se recano un gruppo 2-cloroetilico il componente alchilato può a sua volta agire da alchilante con formazione di legami crociati intra- e intercatena. Inoltre, la capacità di carbamoilare il gruppo ε-amminico della Lys sembra impedire la riparazione del DNA. O Cl N N H NO R isocianato Cl O N N N O Agenti alchilanti 20 Di questa classe di sostanze fanno parte: Nimustina – usata soprattutto nella leucemia nell’encefaloma e nei tumori dell’apparato digerente. cronica, Carmustina – la prima ad essere sperimentata su larga scala. Sostanza lipofila che penetra bene nel SNC consentendo alte concentrazioni nei fluidi cerebrospinali. sebbene si pensi ad un meccanismo di alchilazione del DN e del RNA analogo alle mostarde azotate essa non presenta fenomeni di resistenza crociata. Sembra agire con un meccanismo diverso basato sulla inibizione enzimatica ottenuta tramite carbamoilazione delle proteine. Lomustina – migliore della precedente ed in particolare nel trattamento del morbo di Hodgkin. Agenti alchilanti 21 Streptozocina - isolata da brodi di fermentazione dello Steptomyces achromogenes e successivamente sintetizzata. Attiva contro tumori maligni delle isolette del pancreas. La sua scarsa tossicità a livello del midollo osseo ha suggerito la sintesi di analoghi più attivi. La molecola di zucchero che può esistere in entrambe le forme anomeriche conferisce una buona solubilità in acqua rispetto alle altre nirosouree. produce sindromi diabeto-simili neglia nimali da esperimento. Clorozotocina è il risultato di queste ricerche e risulta più attiva nella leucemia sperimentale. HO HO O OH Streptozocina OH OH NO HN N CH3 O O OH OH OH NO HN N O Cl Clorozotocina Agenti alchilanti 22 Semustine – utilizzata per i tumori al cervello, i linfomi e il cancro al colon e allo stomaco. È tossica, carcinogenica (sospetta leucemia secondaria) e teratogena motivo per cui non viene più utilizzata. Fotemustine studiata per il trattamento del melanoma. Alchila la guanina generando in un legame trasversale tra N1 della guania e l’N3 della Citosina. Di conseguenza provoca l’arresto del ciclo cellulare, a seguito dell’inibizione del DNA, con apoptosi finale. L’elevata lipofilia della molecola le consente di superare la barriera ematoencefalica. H3C O O N H N NO Semustine Cl O O P N O H N NO Cl Fotemustine Agenti alchilanti 23 Altri agenti Alchilanti Dacarbazina (DTIC) – appartiene alla classe dei derivati triazinici che sono in grado di generare carbocationi alchilanti per azione enzimatica nel fegato. È un farmaco molto importante ad attività antitumorale ad ampio spettro ed in particolare attivo sul melanoma melanotico. Viene somministrata per ev, possiede una emivita di 30 minuti e viene eliminata con le urine. La sua attività è dovuta all’attivazione epatica che la demetila mediante ossidazione. Il monometil derivato si degrada a 5aminoimidocarbossiamide, azoto e catione metilico che metila la Guanina in posizione 7. Inibisce maggiormente la sintesi di RNA e proteine che quella del DNA. Possiede azione citotossica lenta e sembra non mostrare specificità per alcun momento del ciclo cellulare. O N N H NH2 N N CH3 N CH3 Dacarbazina Agenti alchilanti 24 CONH2 N CH3 N H N H O N N O NH2 N N N CH3 Cyt P450 N N N N CH3 CONH2 N CONH2 N N N H CH3 N H eliminazione spontanea N N H N N CH3 N OH CONH2 eliminazione spontanea di formaldeide H3C N NH2 NH Metildiazene CH3+ Agente alchilante Temozolomide – rappresenta un profarmaco in quanto viene convertita enzimaticamente a Dacarbazina e come tale agisce da alchilanete. Attualmente è stata approvata dalla FDA per il trattamento dei tumori al cervello. Viene somministrata oralmente e viene rapidamente assorbita Agenti alchilanti 25 Procarbazina usata nel trattamento del morbo di Hodgkin. L’ossidazione in vitro produce il metildiazene e successivamente un radicale metile. In questo modo è in grado di metilare il DNA e l’RNA. A livello del DNA agisce metilando la posizione 8 della Guanina. È un inibitore della MAO. O H3C N H H N Procarbazina O N H H3C N N N H H3 C N NH Metildiazene Agenti alchilanti 26 Etoglucide – utilizzato soprattutto per il tumore alla vescica. O O O O O O Mitobronitol – corrisponde al 1,6-dibromo-1,6-dideossi-Dmannitolo. Sembra agire alchilando il DNA. OH OH Br Br OH OH Pipobroman – derivato piperazinico in grado di alchilare il DNA, viene somministrato per os nel trattamento della polycythaemia vera. O N Br N O Br Agenti alchilanti 27 Complessi del Platino sono sostanze che agiscono in modo analogo agli alchilanti formano legami covalenti per reazione nucleofila tra le Guanine presenti nel DNA e il complesso bivalente diamminoplatino. Si srotolano e si rompono così le spire del DNA. Cisplatino - (Cis-Diclorodiamminoplatino) entrato in terapia come antitumorale nella cura del tumore ai testicoli (associazione con bleomicina) e nel carcinoma ovarico in associazione con Doxorubicina). È un complesso del Platino con due molecole di ammoniaca e due atomi di cloro in cis. Dopo somministrazione il Pt si lega alle proteine plasmatiche incluse l’albumina, la transferrrina e la gamma-globulina. - Cl NH3 Pt Cl ++ NH3 Cisplatino Agenti alchilanti 28 Carboplatino – ha legami con l’acido 1,1-ciclobutandicarbossilico e presenta un’attività minore data dalla stabilità all’idrolisi Oxaliplatino O,O)platinum) generazione. (R,R)-1,2-diaminocycloesano(etanedioatorappresenta un derivato del Platino di 3a Inibisce la replicazione del DNA per formazione di addotti con legami crociati. È stato introdotto nella terapia del cancro al colon ed al retto. Utilizzato in associazione con Rituximab, con Gemcitabina o con entrambi. In associazione con Leucovorina, Fluorouracile e radiazioni, viene utilizzato nel tumore all’esofago. O - O Carboplatino O NH3 Pt O ++ NH3 - NH2 O O Oxaplatino Pt NH2 O O Agenti alchilanti 29 Attività degli agenti alchilanti misti Antimetaboliti Sono sostanze che impediscono la formazione dei normali metaboliti cellulari ai quali assomigliano dal punto di vista strutturale. Il loro meccanismo d’azione può essere: di tipo competitivo con il substrato che mimano; Di tipo suicida in grado di bloccare l’enzima bersaglio. Alcuni di questi composti li abbiamo già incontrati come farmaci per la cura delle infezioni di tipo batterico o virale. Metotressato Questa sostanza, assieme ad i suoi analoghi, costituisce una serie di derivati pteridinici che competono con il sito attivo della diidrofolato riduttasi che già ben conosciamo. Questa classe di sostanze provocano la morte cellulare come conseguenza della mancata sintesi del DNA a causa della mancata biosintesi della Timidina e dell’acido Uridilico La sintesi della Timidina avviene grazie alla formazione del derivato N5,N10-metilentetraidrofolico che funge da agente metilante. Antimetaboliti 2 L’acido tetraidrofolico viene convertito nel corrispondente N5,N10metilentetraidrofolico ad opera di un metilene serinico in presenza di Piridossale. Il metilene viene trasferito come metile sull’anello uridinico con formazione di acido diidrofolico che torna nel ciclo biosintetico. L’inibizione della DHFR DHFR da parte del metotressato può inibire anche la biosintesi delle Serina/ Piridossale Timidilato purine. sintetasi L’acido N10- formamido tetraidrofolico (Leucovorina) che si può formare è in grado di formilare il 5-aminoimidazolo-4carbossamide nucleotide. Antitmetaboliti 3 L’acido tetraidrofolico viene convertito nel corrispondente N5,N10metilentetraidrofolico ad opera di un metilene serinico in presenza di Piridossale. Il metilene viene trasferito come metile sull’anello uridinico con formazione di acido diidrofolico che torna nel ciclo biosintetico. L’inibizione della DHFR da parte del metotressato può inibire anche la biosintesi delle purine. L’acido N10- formamido tetraidrofolico (Leucovorina) che si può formare è in grado di formilare il 5-aminoimidazolo-4carbossamide nucleotide. Antimetaboliti 4 Aminopterina è stato il primo farmaco entrato in terapia e poi sostituita per la minor tossicità del Metotrexate. Il dicloro derivato è risultato valido sugli animali, ma sull’uomo è apparso più tossico. Le differenze di tossicità sembrano imputabili a diverse velocità di metabolizzazione dei prodotti. Il Trimetrexato è un analogo chinazolinico che possiede una potente attività antifolica. La sua maggior solubilità lo rende in grado di permeare le cellule tumorali e i microorganismi (usato soprattutto comtro Pneumocystis Carinii). COOH O N H NH2 N N H2N N N N R Cl COOH N H2N R = H Aminopterina R = CH3 Metotrexate NH2 H N N N H N CH3 N H Cl Diclorometotrexate OCH3 OCH3 NH2 CH3 N H2N COOH O N H N OCH3 Trimetrexate COOH Antimetaboliti 5 La Leucovorina viene somministrata durante il trattamento antitumorale con Metotressate come terapia di salvataggio delle cellule normali in quanto sembra inibire il trasporto attivo del Metotressato all’interno cellula. COOH O N H NH2 N N H2N N N N CH3 Metotressato O COOH COOH O H N HN H2N O N N H N H COOH N H Leucovorina Il Metotrassato viene poliglutammato nel fegato e nello spazio intracellulare e successivamente viene idrolizzato enziamticamente a Metotressato. Non viene pressoché metabolizzato e lo si ritrova inalterato nelle urine. Sono stati sintetizzati alcuni suoi analoghi che non hanno dimostrato né vantaggi terapeutici né minore tossicità. Antimetaboliti 6 Sintesi del Metotressate. Antimetaboliti 7 Raltitrexed - agisce mediante l’inibizione della timidilato sintetasi impiegato dalle cellule (sia tumorali che sane) nella biosintesi delle pirimidine. È attivo nelle cellule dopo la poliglutammilazione che consente la sua persistenza nell’ambiente cellulare. Viene somministrato per via endovenosa con cicli da ripetere ogni 21 gg. Il farmaco mostra un buon profilo di tollerabilitàcon rari effetti collaterali degni di nota. Pemetrexed – usato nella neoplasia polmonare non a piccole cellule. In associazione con Cis-Platino viene utilizzato nel trattamento del mesotelioma pleurico da amianto. Raltitrexed O N N H HN S N C H3 H N CO OH H2N CO OH O COOH Pemetrexed N H N O O HN COOH Antimetaboliti 8 Analoghi dei Nucleosidi Sono strutturalmente simili ai substrati che antagonizzano ma recano piccole modifiche o nella porzione pirimidinca o in quella zuccherina. Il loro meccanismo d’azione può vederli coinvolti nella: Inibizione di una chinasi; Inibizione di un enzima coinvolto nella biosintesi delle pirimidine; Incorporazione nel DNA o RNA; Inibizione della DNA polimerasi. Derivati Pirimidinici Fluorouracile – ideato da Heidelberger nel 1957 sulla base di osservazioni sul metabolismo di alcune cellule tumorali. Egli osservò che queste cellule utilizzano l’Uracile in luogo dell’acido Orotico per la sintesi delle Pirimidine. O HN O O F N H Fluorouracile HN O O HN N H Uracile O O CH3 N H Timina COOH HN O N H Ac. Orotico Antimetaboliti 9 In particolare, la biosintesi della Timidina viene svolta da una timidilato sintetasi una proteina che presenta a livello del sito catalitico un residuo tiolico di una Cys. Si viene a formare così, a partire dall’acido desossiuridilico (dUMP), il corrispondente acido desossitimidilico (dTMP). H2N H N N HN O O HO P O OH N H N - O F HN O N O F HN N SH Timidilato sintetasi O OH N O S Timidilato sintetasi O HO P O OH O OH H2N N HN H N H N N H N O O HN O HO P O OH F N O O OH O S Timidilato sintetasi O Tutto questo è possibile perché in posizione 5 è COOH presente un H che risulta sufficientemente acido da COOH consentire di ripristinare l’enzima. L’intermedio formatosi libera l’acido diidrofolico. COOH COOH La presenza dell’atomo di fluoro rende stabile l’intermedio e non consente di ripristinare l’enzima. Antimetaboliti 10 Il composto di per sé non è attivo e deve essere trasformato nel corrispondente 5-fluorouracil riboside (5FUR) che successivamente viene convertito ad acido 5-fluoro-2’desossiuridilico (5FdUMP). Una volta trasformato in 5FdUMP, il composto si lega alla Timidilato sintetasi per dare l’intermedio stabile. Come molecola viene metabolizzato da parte di una diidropirimidina deidrogenasi a dare il 5-F-5,6-diidrouracile. Questo enzima viene a sua volta inibito dal 5-Etiniluracile e di conseguenza questo composto è cosomministrato al fine di migliorare l’indice terapeutico del 5-Fluorouracile. O F HN O O OH OH F HN N HO 5FUR O O HO O P O OH 5FdUMP N O OH 5FUTF 5FdUTP RNA DNA O O F HN O N H 5-Fluoro-5,6diidrouracile HN O N H 5-EtinilU Antimetaboliti 11 In alternativa al Fuorouracile si può utilizzare la Fluoxuridina che può essere considerata un profarmaco ed ha il vantaggio di essere solubile in acqua. Altra molecola è la Capecitabina usato per il trattamento di alcune forme tumorali, tra le quali il cancro del colon in stadio avanzato, della mammella e dell‘ovaio. Viene somministrata per os. O O O O F HN NH F N N HO O N HO O OH Floxuridina O OH Capecitabina Antimetaboliti 12 Carmofur – derivato del 5-fluorouracile Tegafur – convertito da enzimi microsomiali in 5-fluorouracile e rappresenta il trattamento di prima scelta del carcinoma colonretto avanzato. Somministrato in associazione con Uracile che blocca la Didropiridina deidrogenasi (DPD) che lo inattiva. Azacitidina – è un isostero del nucleo pirimidinico e l’azione di rottura delle catene del DNA sembra sia dovuta alla fragilità dell’anello triazinico. La FDA ha approvato Azacitidina (Vidaza) nel trattamento dei pazienti con sindrome mielodisplastica. Si ritiene che agisca ripristinando la normale crescita e differenziazione delle cellule del midollo osseo. F HN O N H F HN O N O NH2 O O N O N O N N HO O OH OH Carmofur Tegafur Azacitidina Antimetaboliti 13 Citarabina – antimetabolita al quale è stato modificato la porzione zuccherina da ribosio ad arabinosio che risulta essere un epimero in 2’. Anche in questo caso, il prodotto deve essere convertito nel mono- e poi nel trifosfato (ARA-CTP). questo derivato mostra diversi meccanismi d’azione: Inibisce la conversione dell’acido Citidilico in acido 2’desossicitidilico inibendo l’enzima ribonucleotide reduttasi (RR) a livello della subunità R1 – resistenza data da sovraespressione dell’enzima; Inibisce la DNA-polimerasi-Dna-dipendente Incorporata nel DNA (agisce da terminatore della catena) o nel RNA genera un errore di lettura del codice genetico. La sostanza è disponibile come polvere per un uso endovena, intratecale e sottocutaneo. Viene metabolizzata velocemente ad Arabinofuranosiluracile O NH2 HN N ARA-C O O N N HO HO O OH OH O OH OH Arabinofuranosiluracile Antimetaboliti 14 Gemcitabina – rappresenta uno dei recenti antimetaboliti pirimidinici e viene utilizzato nel cancro del polmone e del pancreas. Mostra una eccellente attività antitumorale in vivo in quanto viene incorporata nel DNA e porta a morte cellulare. Il derivato trifosfato viene incorporato nel RNA e così inibisce la sintesi sia del DNA che del RNA. Il principale metabolita nelle urine è l’analogo uracilico inattivo 2’-desossi-2,2’-difluorouridina (dFdU). NH2 NH2 N O N N HO O O F O HO O P O HO OH F Gemcitabina HN N O N HO O F OH F dF-dCTP O F OH F dFdU Antimetaboliti 15 Derivati Purinici 6-Mercaptopurina – anche in questo caso il derivato attivo è sempre il ribonucleotide corrispondente 6-Tioinosinato che si forma ad opera di una Ipoxantina-guanina-fosforibosil transferasi (HGPRT). Inibisce la sintesi de novo delle purine impedendo la conversione del 5-fosforibosil pirofosfato in 5-fosforibosilammina. S S N HN N N HN N H HO N O P O HO N O OH OH 6-Mercaptopurina 6-MPMP Antimetaboliti 16 Il 6-tioinosinato inibisce inoltre la conversione dell’acido inosinico ad acido adenilico come pure l’ossidazione dell’acido inosinico ad acido xantilico. Biosintesi delle Purine Antimetaboliti 17 Oltre a questo meccanismo è doveroso ricordare che il metabolita Tioinosinato può essere convertito sia a mono- che a trifosfato. Come tali questi derivati possono essere incorporati nel DNA o nel RNA al posto della Guanina inibendo l’allungamento della catena polinucleotidica. Inoltre, il tioinosinato può essere metilato al posto della Sadenosilmetionina e convertito al 6-metiltioinosinato. Come sostanza non mostra un buon assorbimento orale anche se viene somministrata per questa via. Viene metabolizzata completamente con formazione di metiltiopurine e, ad opera di una xantina ossidasi, di acido tiurico. Proprio questo metabolita ha suggerito l’utilizzo di allopurinolo, quale inibitore della xantina ossidasi, al fine di ridurre la dose del farmaco stesso. H3C S N HN HO N O P O HO N O OH OH 6-MPMP S S N N HN HO N O P O HO N O OH OH 6-Metiltioinosinato O OH H N O N H N H N N N N H Antimetaboliti 18 Azatioprina – molecola che agisce liberando lentamente la mercaptopurina e viene utilizzato soprattutto come immunosoppressivo. 6-Tioguanina – come molecola è strettamente correlata alla 6-MP e come tale viene metabolizzata a dare il derivato ribosilico monofosfato (6-TGMP) e poi il trifosfato (6-TGTP). Essi agiscono inibendo gli stessi enzimi visti precedentemente. Il 6-TGTP è in grado di essere incorporato nel RNA e, dopo trasformazione nel desossiderivato, essere inserito nel DNA. Come tale, non viene riconosciuto dagli enzimi addetti alla replicazione del DNA. N H3C N HN S N N N H3C S NO2 N H2N N N H N H H3C S N N H2N O HO P O OH N N O OH OH Azatioprina 6-TG 6-TGMP S N N H2N O HO P O OH 3 N N O OH OH 6-TGTP Antimetaboliti 19 Fludarabina fosfato – è strutturalmente correlato alla adenosina con variazioni che riguardano sia l’adenina (fluorurata) che il ribosio (sostituito con l’arabinosio). In pratica è un derivato della Vidarabina (ARA-A) della quale risulta più potente in quanto meno soggetta all’enzina adenosina deaminasi. Una volta convertita nel trifosfato è in grado di inibire la ribonucleotide reduttasi. Cladribina – l’analogo clorurato ma con il desossiribosio è in grado di inibire importanti enzimi in grado di riparare il DNA. Pentostatina (2’-Deoxicoformicina) Nipent® è un inibitore della adenosina deamminasi ed è attiva sui linfociti T tumorali. Usata nella Leucemia Linfoide Cronica. Si lega scarsamente alle proteine plasmatiche e viene pertanto escreta rapidamente a livello NH NH renale. HO 2 N F O HO P O OH 2 N N N O OH OH Fludarabina fosfato N N Cl O HO P O OH N NH N N N O OH Cladribina O HO P O OH N O OH Pentostatina Antimetaboliti 19 Clofarabina – utilizzata in pediatria nella leucemia linfoblastica acuta. Una volta convertita nel trifosfato è in grado di inibire la ribonucleotide reduttasi e la DNA polimerasi coinvolti nella produzione di materiale genetico impedendo alle cellule di produrre nuovo DNA e RNA. N H2 N N Cl HO N N O F OH Clofarabina Antimetaboliti 20 Dosi , vie di somministrazione, effetti collaterali e forme farmaceutiche di antimetaboliti Antibiotici antitumorali Numerosi composti isolati durante la ricerca di nuovi antibiotici si sono poi rivelati degli agenti antitumorali. Antibiotici antitumorali 2 Numerosi composti che allora vennero scartati per l’eccessiva tossicità furono successivamente rivisti, proprio per questo motivo, come possibili antitumorali. Bleomicine – sono state scoperte nel 1966 nei brodi di fermentazione dello Streptomyces verticillus. Sono antibiotici glicopeptidici ed attualmente viene utilizzata una miscela di A-2 e B-2 sotto forma di chelati rameosi. Antibiotici antitumorali 3 Possiedono una forte tendenza a formare chelati metallici. Si ritrovano, in natura, come chelati con un atomo di rame complessato tra l’imidazolo, la pirazina e i vari gruppi amidici. Si ritiene che formino un chelato con il Fe++ e così facciano variare il potenziale del ferro stesso favorendo la riduzione dell’ossigeno che degrada il DNA generando così l’effetto citotossico. Si pensa si formi sia una specie radicalica attiva sia il radicale ossidrile (●OH) con rottura della porzione zuccherina e relativo rilascio di basi puriniche e pirimidiniche. Antibiotici antitumorali 3 SAR – La porzione glicopeptidica laterale non è essenziale all’attività ma serve per facilitare l’attraversamento delle membrane cellulari. La rimozione non porta a perdita di attività. La catena dipeptidica non funge solo da linker bensì partecipa alla formazione di legami ad idrogeno che intensifica l’interazione e danno specificità di base. La porzione ditiazolica intercala nel DNA con le relative catene cationiche che rafforzano il complesso per interazioni elettrostatiche con i gruppi fosfato del backbone. Antibiotici antitumorali 4 Dactinomicine o Actinomicina D- Formano un gruppo di cromopeptidi (rosso-arancione) prodotto da varie specie di Streptomyces. • Sono molto tossiche e per questo utili come antitumorali. • Il cromoforo è l’acido 3-amino-1,8-dimetil-2-fenossazon-4,5dicarbossilico (detto actinocin) legato tramite i due carbossili con due depsipentapeptidi (chiusura del ciclo tra OH della Thr iniziale e il carbossile dell’ultimi la N-metil-D-Valina). • Le due catene laterali possono essere: • identiche e si parla di isoactinomicine; • Diverse e si parla di anisoactinomicine. Antibiotici antitumorali 4 . Inibiscono la sintesi di RNA. Il cromoforo si intercala tra le coppie di guanina-citosina di due molecole di DNA a doppia elica instaurando interazioni π-π π. Le catene laterali si legano nel solco minore e rafforzano il complesso. Interferisce quindi con la trascrizione del DNA in RNA. L’interferenza delle Dactinomicine con la RNa-polimerasi-DNAdipendnete è maggiore rispetto a quella con la DNA-polemerasi stessa. La loro azione è indipendente dal ciclo ccllulare anche se la loro attività è preminente in fase G-1. Antibiotici antitumorali 5 Mitomicine – Isolata negli anni ‘50 da streptomyces caespitosus come derivato C. Solo successivamente (’70) data la loro tossicità, vennero utilizzate come antitumorali. La Mitomicina A può essere sintetizzata dalla C in due soli passaggi. E’ in grado di alchilare il DNA a livello delle unità citosiniche e guaniniche dopo riduzione ad idrochinone in presenza di NADPH ed eliminazione del metossile angolare. • • O O O X NH2 X OCH3 NH2 NH2 OCH3 N H3C N R R H Mitomicina A H Mitomicina C CH3 Porfiromicina O O O X NH2 OCH3 N H3C O O O O N R O OH H2 X NH2 OCH3 N H3C OH N R OH CH3OH O NH2 OH + H X N H3C OH N R X CH2+ N H3C OH NH2-COOH + N H R Antibiotici antitumorali 6 • • • • • L’alchilazione può avvenire con entrambi i centri reattivi e preferibilmente con il gruppo amminico in 2 della guanina. Si stabilisce un ostacolo alla replicazione del DNA e al funzionamento della RNA-polimerasi DNA-dipendente con inibizione della sintesi di DNA e RNA. La mitomicina C viene assorbita poco per via orale, pertanto, è somministrata per ev e viene metabolizzata velocemente. Il sistema chinonico impartisce alla molecola una colorazione bluviolacea. Utilizzata soprattutto per il tumore al panreas. Antibiotici antitumorali 7 Antracicline Pigmenti colorati prodotti da Attinomiceti ed aventi struttura antrachinonica. L’interesse per queste sostanze emerse soprattutto dopo la scoperta dell’attività antitumorale della Daunorubicina. La ricerca di analoghi portò alla scoperta delle Doxorubicina dotata di spettro antitumorale più allargato e meno tossica. 4 1 8 5 10 Daunorubicina Doxorubicina Iododoxorubicina Carubicina Idarubicina Epirubicina Esorubicina Antibiotici antitumorali 8 O Relazioni struttura attività Sono ben evidenti quattro regioni: a. Anello aromatico D b. Due nuclei idrochinonici B e C c. La catena idrossichetonica in 8 d. Il residuo zuccherino in 10 OH O R D C B A O OH O OH O H3C R NH2 R1 Sull’anello D sono compatibili varie modifiche tra le quali ricordiamo la demetilazione del sostituente in 1. La 1-demetossidaunomicina (Idarubicina) ha un buon assorbimento orale, minor cardiotossicità e miglior penetrabilità nelle cellule coltivate. Il nucleo farmacoforico della molecola è costituito dai due sistemi idrochinonici che devono possedere almeno un ossidrile libero radicale semichinonico. Antibiotici antitumorali 9 Il chelato triferrico della adriamicina, che si è rivelato meno cardiotossico, viene assorbito per via orale e non ha effetto immunosoppressivo. Il residuo zuccherino in 10 ha notevole importanza come aptoforo – l’aglicone delle dauno e doxorubicina è sprovvisto di attività. Composti con agliconi simili ma che contengono la Rodosamina in luogo della daunosamina hanno attività modesta e sono più tossici. O R O OH O H3C O OH NH2 O R O OH O H3C O OH N CH3 CH3 Antibiotici antitumorali 10 • La struttura β-glucosidica appare esiziale per l’attività. • L’epimero in 4’ (Epirubicina) unisce all’attività antitumorale della Dauno e Doxorubicina una minor cardiotossicità, così pure la 4’ desossidoxorubicina. • La 4’- desossi-4’-iodo (iododoxorubicina) ha una maggiore lipofila, una minore basicità, è molto efficace nei tumori al colon ed ai polmoni, è attiva per via orale e non è cardiotossica. • Le modifiche alla catena laterale in 8 tendono a ridurre l’attività. L’optimun è rappresentato dal ragguppamento chetolico (chetodiolico) della Doxorubicina. Si può accorciare ma non allungare od introdurre in essa un gruppo basico. • Vanno ricordati anche i profarmaci (esteri in posizione 14 come la Valrubicina) che rigenerano in situ il farmaco. O OH O O OH OMe O OH OR H3C O HN OH F3C O C4H9 O Antibiotici antitumorali 11 • L’ossidrile in posizione 8α α è importante non può essere eliminato o sostituito con un metile. • Nella Dauno e nella Doxorubicina l’anello A idroaromatico è in confromazione a semisedia che è cratterizzato dalla formazione di un legame idrogeno intramolecolare (8-10) che determina la disposizione spaziale dell’anello piranosico rispetto al nucleo antrachinonico. D-daunosamina 10 8 semisedia barca Metabolismo della Doxorubicina Antibiotici antitumorali 12 Riduttasi NADPH 1) Idrolasi NADPHo 2) Daunomicinolo riduttasi O-Metil Idrolasi Solfotransferasi opp. Gluconiltransferasi Doxurubicinolo D = daunosamina Antibiotici antitumorali 13 Meccanismo d’azione • L’aglicone, porzione planare della molecola, si intercala nel DNA orientandosi ad angolo retto nella scanalatura maggiore della doppia elica. Tale complesso è rafforzato da legami idrofobici e da legami idrogeno. • La porzione zuccherina interagisce elettrostaticamente con lo scheletro zucchero-fosfato del DNA. • Inibisce così la DNA- e RNA-polimerasi DNA dipendente e provoca: • Inibizione dell’incorporazione di di precursori degli acidi nucleici; • Alterazioni nucleolari nelle cellule del cuore e del fegato; • Carcinogenicità; • Frammentazione del DNA di cellule tumorali; • Anomalie cromosomiche. Antibiotici antitumorali 14 • Il principale recettore delle antracicline è il DNA del nucleo (dove si ritrova la maggior quantità di antibiotico). • La natura ossido-riduttiva delle antracicline e responsabile dell’interferenza con sistemi ox-red dell’organismoanimale (Q10, Succinicossidasi, NADPH-ossidasi, la respirazione dei mitocondriisolati, ecc…). • Questa interferenza porterebbe alla formazione di specie radicaliche (soprattutto ●OH) responsabili della frammentazione del DNA e dei danni cellulari. • La loro cardiotossicità deriva polinsaturi della membrana. dalla perossidazione di lipidi Antibiotici antitumorali 15 Sintesi • Ottenibili a partire dal sistema tetralinico di Wong. • La risoluzione degli antipodi ottici di questo intermedio, tramite Base di Schiff, fornisce l’isomero R. • La glucosidazione avviene per mezzo della 1desossi-1-cloro-3,4bistrifluoroacetildaunosamina in presenza di trifluometansolfonato di Ag. Antibiotici antitumorali 17 Meccanismo di formazione di radicali liberi a partire da antrchinoni. Antibiotici antitumorali 18 Rottura ossidativa delle catene del DNA da parte di radicali liberi genarati da antrachinoni. Antibiotici antitumorali 19 Antibiotici Cromomicinonici • Hanno in comune la struttura agliconica triciclica del Cromomicinone legato a diverse molecole di zuccheri particolari. • Isolate da Streptomyces griseus. Cromomicina A Mitromicina Antibiotici antitumorali 20 • Si legano in modo reversibile alle coppie di basi G-C all’interno del solco minore del DNA. • In particolare si lega ad almeno tre sequenze consecutive di GC inibendo così la sintesi del RNA sia nei batteri che nelle cellule animali. • Formano complessi con il DNA in cui è necessaria le presenza di ioni Mg2+, Mn2+, Co2+ e Zn2+). • L’alchilazione può avvenire con entrambi i centri reattivi. • Si stabilisce un ostacolo alla replicazione del DNA e al funzionamento della RNApolimerasi DNA-dipendente con inibizione della sintesi di DNA e RNA. • La porzione zuccherina è fondamentale per il binding della molecola con il DNA. Antibiotici antitumorali 21 Streptazocina (Zanosar®) e Clorozocina • Sono nitrosouree naturali isolate da brodi di fermentazione di Streptomyces achromogenes. • Sono antitumorali che abbinano un’attività antibatterica ad ampio spettro. • L’azione citotossica di questi composti è liberazione di gruppi alchilanti (CH3+, carbamoilanti (NH2-CO+). correlata alla ClCH2CH2+) o • Clorozocina provoca legami trasversali nel DNA e tra DNA e proteine. • La Clorozocina è mutagena e meno mielotossica rispetto ad altre nitrosouree. CH2OH O OH OH OH O HN R Streptozocina Clorozocina N NO R = CH3 R = CH2CH2Cl Antitumorali Idrossiurea Molecola piccola ben assorbita per os (80-100%) ed escreta inalterata nelle urine. Essa blocca la conversione di ribonucleotidi a desossiribonucleotidi che è uno stadio cruciale nella biosintesi del DNA. Inibisce specificatamente la fase S e provoca l’arresto cellulare nellinterfase G1-S. Questo dato è assai importante per la terapia radiante in quanto le cellule in fase G1 sono quelle maggiormente colpite dalle radiazioni. O H2N N H OH Inibitori delle topoisomerasi Diversi composti sia sintetici che naturali avevano evidenziato la capacità di intercalare il DNA. Verso la fine degli anni ’80 è stato dimostrato che l’azione citotossica è dovuta alla formazione di un complesso ternario stabile tra la sostanza, il DNA e la topoisomerasi II. La topoisomerasi II si lega al DNA in modo reversibile e successivamente esegue tagli e ricuciture dei filamenti per ridurre lo stress torsionale durante le fasi replicative. È una proteina cromosomico. omodimerica associata ad uno Scaffold Esistono due isoforme: – Topo IIα α codificata dal cromosoma 17 bersaglio della maggior parte dei farmaci che intercalano il DNA. – Topo IIβ β codificata dal cromosoma 3 e domina nelle cellule resistenti agli inibitori della topo II. Inibitori delle topoisomerasi 2 Tra i prodotti sintetici troviamo due classi di composti. Derivati 9-anilinoacridinici Studi condotti da Cain e coll (1979) portarono inizialmente al derivato metansolfonamidico dotato di maggiore solubilità, stabilità metabilica e attività biologica e successivamente all’Amsacrina primo composto sintetico in grado di intercalare il DNA con efficacia clinica nella leucemia linfoblastica acuta; è in grado di bloccare entrambe le isoforme dell’enzima. Nel complesso ternario che si forma l’acridina va ad intercalarsi nel DNA mentre la catena laterale anilinica instaura una specifica interazione con l’enzima. R HN H N SO2CH3 Metansolfonamide R = H Amsacrina N R = OCH3 Inibitori delle topoisomerasi 3 Composti antracenedionici - derivano da studi su coloranti industriali e tra essi troviamo il Mitoxantrone un derivato dialchilaminoantrachinonico in grado di intercalare il DNA, soprattutto in zone ricche di GC, e formando un complesso ternario stabile con la topoisomerasi II. Tale complesso è rafforzato dalla presenza degli ossidrili in posizione 5 e 8 (Ametantrone si dissocia molto più rapidamente). L’aspetto interessante che gli aminozuccheri sono sostituiti da catene che posseggono comunque la possibilità di formare legame idrogeno (-OH) e di essere protonate (NH). La molecola è in grado di generare radicali ossidrilici e superossidi che possono provocare danni irreparabili al DNA. Questi stessi radicali sono la causa della cardiotossicità (come nelle antracicline). Pixantrone – aza-analogo utilizzato nel linfoma non-Hodgkin R O HN H N O HN OH NH2 Mitoxantrone R = OH N Ametantrone R = H R O HN N H OH O HN NH2 Inibitori delle topoisomerasi 4 Epipodofilline Estratte da Podophyllul peltatum le podofillotossine rappresentano un vecchio rimedio contro la gotta. Esse sembrano agire nella fase G2 dove causano legami proteinaDNA e soprattutto rotture del DNA. Sebbene si leghino alle tubuline non agiscono attraverso questa via. Formano complessi ternari stabili con la topoisomerasi II ed il suo substrato (DNA). La topoisomerasi opera il taglio della catena del DNA ma le molecole impediscono il passaggio e la ricucitura che normalmente seguono. Così facendo l’enzima rimane legato ad una delle estremità di taglio del DNA e si vengono ad accumulare frammenti dello steso con morte della cellula. DNA Topoisomerasi II Isopodofillotossin glicoside Duplex DNA Complesso ternario stabile Inibitori delle topoisomerasi 5 Durante un ciclo catalitico della idrolizzate due molecole di ATP. topoisomerasi II vengono Sia l’Etoposide che il Teniposide impediscono il rilascio della molecola di ADP risultante dall’idrolisi della prima molecola di ATP. L’attività ATPasica dell’enzima viene inibita e così si impedisce il ricongiungimento dei filamenti del DNA. La loro azione prevale nella tardiva fase S e si protrae nella iniziale fase G-2 impedendo alle cellule di entrare in fase M. La Podofillotossina (isomero Cis) sostanza di partenza è un antitumorale con tossicità sistemica troppo elevata e di conseguenza si utilizzano gli isomeri semisintetici o isopodofillotossine (isomeri trans). Questi si ottengono per catalisi acida sulla molecola protetta alla porzione zuccherina. OH O O O R H3CO O OCH3 OCH3 Podofillotossina R = βH Picropodofillotossina R = αH Inibitori delle topoisomerasi 6 Etoposide – somministrato per i.v. nel il trattamento del tumore refrattario ai testicoli, tumore al polmone a piccole cellule e altri tumori difficili da trattare. Data l’elevata lipofilia della molecola e l’impossibilità di trasformarla in Sali solubili, essa viene somministrata ev utilizzando per la formulazione del polisorbato 80 modificato. È in commercio l’Etoposide fosfato che rappresenta un profarmaco in quanto viene rapidamente totalmente idrolizzato ad Etoposide. Teniposide – somministrato per i.v. è indicato nel trattamento della leucemia non linfocitica acuta, Linfoma di Hodgking, sarcoma di Kaposi e neuroblastomi. OR O O Etoposide O H H3CO O OCH3 OCH3 Tenoposide O R = H3C O HO R= O O S HO O OH O OH Inibitori della topoisomerasi 7 Camptotechine Camptotechina isolata inizialmente da estratti di Camptotheca acuminata e successivamente da Mappia foetida. Rivelatasi troppo tossica è stata utilizzata come struttura base per derivati semisintetici. Irinotecan è un profarmaco nel quale l’anello chinolico ossidato è derivatizzato come carbammato. Somministrato i.v. in associazione con 5-FU nel trattamento del carcinoma metastatico al colon ed al retto. Topotecan utilizzato nel trattamento del tumore alle ovaie e di quello al polmone a piccole cellule. R R1 R2 Camptothecin H H H Topotecan H Irinotecan Et OH O H3C 16 E O 1 N 2 10 R2 N A 4 9 R1 R CH2N(CH3)2 O H OH O O N N Inibitori della topoisomerasi 8 Rubitecan viene utilizzato nella cura del carcinoma pancreatico avanzato e rappresenta un classico “farmaco orfano”. Belotecan – è attivo nel trattamento del tumore al polmone a piccole cellule. Da studi SAR è emersa l’importanza: • della funzione lattonica dell’anello E; • del gruppo carbonilico dell’anello D; • Della chiralità del C16 in quanto l’eutomero S è 10-100 volte più potente del distomero R. OH O H3C O Rubitecan R= Belutecan R = NO2 N N O R H N Inibitori della topoisomerasi 9 Meccanismo d’azione Agiscono a livello della topoisomerasi I enzima preposto a; • rompere provvisoriamente un singolo filamento del DNA; • Ruotare la molecola; • Riunire il filamento tagliato. In presenza di Camptotechine, si forma un complesso ternario stabile che impedisce di riunire i filamenti del DNA. Il DNA tagliato non serve alla cellula che rimane in fase S mentre il DNA stesso viene degradato con successiva morte della cellula. Farmaci antimitotici Interferiscono con la mitosi cellulare ed in particolare con la formazione/disgregazione del fuso mitotico. Durante la mitosi si ha polimerizzazione della tubulina la proteina che forma il fuso mitotico stesso. Questi farmaci inibiscono tale processo con: La depolarizzazione dei microtubuli; La formazione di strutture diverse dal fuso mitotico stesso. I cromosomi, in assenza del fuso mitotico, non possono separarsi correttamente e sopraggiunge la morte cellulare. Alcaloidi della Vinca – sono estratti dal fiore di pervinca del Madagascar (Vinca rosae) aventi una caratteristica struttura dimerica, non simmetrica, contente due gruppi indolici. Antimitotici 2 Si evidenziano due sottostrutture: 1. Sottostruttura vindolica; 2. Sottostruttura catarantina 1 2 Antimitotici 3 La vinblastina e la Vincristina sono importanti come antitumorali in quanto bloccano la mitosi con un arresto a livello della anafase (distribuzione del materiale genetico tra le due cellule figlie). Presentano una alta affinità e specificità per la tubulina della quale causano depolimerizzazione. Legandosi alla subunità β dei dimeri di tubulina (α α-tubuline β-tubulina) ne impediscono l’incorporazione nei microtubuli. La cellula muore per apoptosi. Nonostante la stretta somiglianza sono diverse per attività e tossicità. Antimitotici 3 Vinblastina Velbe® – somministrata come solfato per i.v. nel trattamento del tumore ai testicoli metastatizzato (in associazione con Bleomicina e Cisplatino) e per vari limfomi. Presenta minore neurotossicità ma la leucopenia ne limita la dose. Vincristina – rappresenta un componente dei cocktails antitumorali utilizzati nelle leucemie, nei linfomi, nei sarcomi e nei carcinomi. Usata sovente in associazione con corticosteroidi. L’effetto neurotossico ne limita la dose di utilizzo. Non produce serie depressioni del midollo osseo. Da uno studio SAR e emerso che fondamentale per l’attività sono sia la struttura dimerica che la stereochimica della giunzione. La riduzione del doppio legame e/o del carbonile ne diminuisce leggermente la potenza. L’attività antileucemica è fortemente legata ai gruppi funzionali presenti (es. l’idrolisi dell’acetile in C-4 della Vinblastina abolisce l’attività antileucemica). Antimitotici 4 Vindesina (Eldisine®) – corrisponde al derivato desacetilato della Vinblastinamide solfato. • Viene somministrata in dosi di 3 mg per m2 di superficie corporica e possiede un’emivita di 24 h. • Riduce la produzione di globuli bianchi predisponendo il paziente ad infezioni. Dopo 10-14 giorni dalla sospensione del trattamento si ripristina lentamente il loro normale ematico prima del successivo ciclo chemioterapico. Vinorelbine (Navelbine®) attualmente trasttamento del tumore alle ovaie. • in fase Sembra dotata di un più ampia attività antitumorale II per il Antimitotici 5 Vinflunine è un nuovo derivato fluorurato semisintetico della Vinca modificato nella porzione catarantina. • Sviluppata nel corso di ricerche per il trattamento del tumore alla vescica ed al polmone. • Negli studi in vitro ha dimostrato sinegismo con cisplatino, Mitomicina C, Doxorubicina e 5-fluorouracile. Tassoidi Antimitotici 6 Sono derivati terpenici, anche semisintetici, estratti dalle foglie del tasso del pacifico, caratterizzati da un complesso nucleo triciclopentadecanico. Questi derivati si legano alla β-tubulina e stimolano la formazione dei microtubuli e di conseguenza i microtubuli: vengono costretti a disporsi in modo parallelo; non si possono disporre nella corretta posizione del fuso mitotico. Come effetto finale la cellula non può andare in mitosi. Antimitotici 7 Attualmente vengono ottenuti per via sintetica a partire da un precursore estratto sempre dalle foglie. Il primo derivato scoperto è stato il Paclitaxel. A C B Le SAR sono state studiate a livello: • biochimico, sfruttando prove di polimerizzazione delle tubuline o di inibizione della depolimerizzazione dei microtubuli; • cellulare, dove si misura la citotossicità nei confronti delle cellule tumorali quali cellule KB, J 774, cellule della Leucemia P 388 o del melanoma B-16; • dell’organismo, valutando l’inibizione della crescita tumorale nei topi o nei tumori umani Xenografts Antimitotici 8 Area A riguardante comprendenti le posizioni 7, 9, 10 e il doppio legame 11-12: • l’acilazione in 7 e la deacetilazione in 10 non portano ad un incremento di attività • solo in posizione 7 sono tollerati sostituenti polari e voluminosi anche se questi determinano in vivo una certa diminuzione di attività: • L’ossidazione degli ossidrili presenti nelle posizioni 7 e 10 conduce ad una diminuzione di circa 1000 volte della citotossicità nei confronti delle cellule KB. A C B Antimitotici 9 Area B relativa alle posizioni 1, 2, 4 e 5; • è la regione meno studiata del Taxolo a causa degli impedimenti sterici e chimici dati dai gruppi funzionali presenti. • Nessuna modifica risulta a carico del C1 ed in C2. • è stata eseguita una sola modifica e precisamente l’idrossilazione, analoga a quella metabolica, a m-idrossibenzoato che comporta analoga attività nell’assemblaggio delle tubuline associata ad una minore citotossicità (∼ ∼39 volte minore) verso le cellule del L 1210. • L’apertura dell’anello eterociclico porta al composto 21 (figura 13) 10-5 volte meno citotossico, • la mesilazione dell’ossidrile in C1 porta al riarrangiamento dell’anello A con una citotossicità circa 100 volte minore con uguale attività sulle tubuline. Antimitotici 10 Area C - Corrisponde alla zona più studiata e più critica del taxolo in quanto è ritenuta indispensabile per l’attività. • Il Baccatin III è 50 volte meno attivo del taxolo nelle prove riguardanti le tubuline dei mammiferi così come una serie di suoi derivati privi della funzione esterea in C13. • La catena laterale deve possedere entrambi i gruppi legati al C3 e precisamente il residuo fenilico e benzamidico.. • Un’ulteriore studio su derivati fenilpropionici, variamente sostituiti, legati in catena ha determinato l’importanza della stereochimica dei centri chirali presenti e dei vari sostituenti.. • L’inversione della posizione dei sostituenti ed anche della stereochimica dei centri chirali comporta una diminuzione d’attività Baccatin III DAB Antimitotici 11 La sola inversione dei centri chirali riduce l’attività di 4-5 volte. • La rimozione della benzamide porta al derivato amminico libero molto meno attivo e la eventuale acilazione con un gruppo tosile o t-butossicarbonile non influenza ulteriormente l’attività. • L’ossidrile in posizione 2’ se acilato porta a diminuzione di attività salvo nel caso di composti appositamente progettati come profarmaci. I composti oggi in uso sono: • Paclitaxel Taxol® Anzatax®– utilizzato in combinazione con altri farmaci nel trattamento di vari tumori refrattari alle terapie (ovaie, fegato, polmoni, esofago, vescica ….). • Docetaxel Taxotere® – semisintetico preparato a partire dal 10DAB (10-deacetilBaccatin III). Ha un ruolo importante nella terapia anticancro ma essendo parecchio tossico va utilizzato con cura. Terapia ormonale Il trattamento terapico di tumori con ormoni è oggi diventato frequente basti ricordare: L’uso di antiestrogeni nella cura del cancro al seno; L’utilizzo di antiandrogeni per l’ipertrofia prostatica. Antiestrogeni Storicamente la terapia ormonale inizia nel 1896, quando Beatson praticò, per la prima volta, l’ovariectomia per il trattamento di pazienti con tumore della mammella. Dopo oltre cento anni, oggi le conoscenze scientifiche sul ruolo degli ormoni sessuali si sono notevolmente perfezionate: tuttavia, il razionale è rimasto immodificato poiché si ritiene che gli estrogeni siano coinvolti nell’insorgenza e nello sviluppo di almeno un terzo dei tumori mammari. Pertanto la efficace. loro soppressione può risultare un trattamento Terapia ormonale 2 Il meccanismo di azione dei diversi farmaci consiste, quindi, nell’interferenza con l’attività degli estrogeni. In base all’osservazione che il testosterone aveva azione antagonista sugli effetti fisiologici degli estrogeni venne introdotta la terapia androgena per la cura del tumore alla mammella. Inizialmente venne usato il testosterone proprionato che presenta collaterali effetti virilizzanti. Per ovviare a ciò si è ricorsi a derivati anabolizzante e scarsi con attività androgena. prevalentemente Il Calusterone è lo steroide più utilizzato nella terapia ormonica primaria e secondaria del cancro alla mammella. O Me O Me Me O Testosterone propionato O Me OH Me Me Calusterone Terapia ormonale 4 L’espressione del recettore per gli estrogeni nel tumore è un importante criterio per la scelta della terapia ormonale: infatti, è stato ampiamente dimostrato che, più elevato è il contenuto di recettori nel tumore (definiti ER-positivi) , maggiori sono le probabilità di un effetto terapeutico. Tra i farmaci SERM (Selective Estrogen Receptor Modulator) troviamo: Il Tamoxifene (Kessar®, Ledertam®, Nolvadex®, Nomafen®, Tamoxene®, Virtamox ®) , farmaco di riferimento per la terapia ormonale del tumore della mammella da oltre 20 anni, è oggi l’antiestrogeno di prima scelta e viene assunto giornalmente da milioni di donne. È stato ampiamente utilizzato sia nella malattia avanzata sia come trattamento adiuvante (dopo l’intervento chirurgico). È stato rilevato che, nelle pazienti trattate per 5 anni, diminuisce significativamente l’incidenza di tumore mammario controlaterale. STEROIDI 62 Antagonisti “Impeded” - Tra questi composti dobbiamo ricordare lo Estratriolo che qualora venga utilizzato in concetrazioni elevate è in grado di bloccare il recettore ed impedire che l’Estradiolo esplichi la sua attività. Inibitori dell’aromatasi - Sono raggruppabili in due classi: 1. Derivati dell’Androstendione – servono a bloccare la conversione degli androgeni in estrogeni. Vengono utilizzati nel trattamento della funzione riproduttiva e nella cura di forme tumorali estrogeno-dipendenti. Essi competono con l’Androstendione per il legame al sito attivo dell’enzima aromatasi. L’analisi struttura attività su questi derivati indica che essi devono essere analoghi al substrato e differire da esso per piccole variazioni strutturali O O all’anello A e in 19. O O O O O O OH O 4-idrossiandrostendione O O S O 6-metilenandrostendione 1-metilandrostandiendione S 10β β-propinilandrostendione NH2 NH2 STEROIDI 63 2. Derivati triazolici – essi inibiscono selettivamente in modo competitivo la trasformazione del testosterone a estrogeni in tutti i tessuti. riducono la concentrazione di Estradiolo, Estrone e dell’Estrone solfato circolanti senza intaccare la sintesi di ormoni corticosteroidi, dell’Aldosterone e degli ormoni tiroidei. Dato che l’estrogeno agisce da fattore di crescita per il tumore al seno, la riduzione delle concentrazioni plasmatiche inibisce lo sviluppo della malattia. Essi si legano con il triazolo al gruppo eme del complesso enzimatico del CYP19. N N N N N N N N OH N N N NC CN Anastrazolo N Cl Vorozolo CN NC Letrozolo NC C N Metabolita Terapia ormonale 3 Nel momento in cui le cellule tumorali richiedono gli estrogeni per la propria proliferazione, l’approccio più semplice per impedire loro di svilupparsi è quello di privarle di tali sostanze. I meccanismi fondamentali utilizzabili sono due: impedire alla cellula tumorale di utilizzare gli estrogeni prodotti (antiestrogeni) inibire la produzione dell’aromatasi). degli estrogeni (inibitori Il meccanismo di azione di entrambe le classi è perciò molto semplice. • Gli antiestrogeni impediscono che gli estrogeni, entrati nelle cellule tumorali, possano essere utilizzati. • Gli inibitori dell’aromatasi, invece, impediscono la formazione degli estrogeni a partire dagli androgeni nei tessuti periferici e, pertanto, la cellula tumorale non è rifornita di queste sostanze. Terapia ormonale 5 Il suo meccanismo d’azione è prevalentemente di tipo tumoristatico in quanto blocca a livello di fase G1 il ciclo cellulare delle cellule tumorali (viene antagonizzato dall’uso di estradiolo). È stato dimostrato che, in campioni di cellule tumorali, riattiva un gene oncosoppressore che nei tumori funziona poco o per niente e così impedisce la moltiplicazione cellulare Per la presenza del doppio legame vi è isomeria cis-trans: – l’isomero cis (Z) risulta attivo come antiestrogeno ed è impiegato nella cura del tumore mammario; – Il derivato trans (E) ha attività estrogenica e non viene utilizzato. Suo analogo è il Toremifene (Fareston®) che si lega al ER impedendo agli estrogeni di svolgere la loro azione. N O N O Tamoxifene Toremifene Cl Terapia ormonale 6 Altri composti SERM sono: Roloxifene, Arzoxifene e Basedoxifene indicati nel tumore della mammella dell’osteoporosi. sono composti simili e nella prevenzione Lasofoxifene è un derivato dell’antiestrogeno Nefoxidine, testato negli anni ‘70 per il tumore al seno e scartato per fenomeni di sensibilizzazione. Il composto attivo è l’isomero L che è 20 volte più attivo dell’isomero D verso gli ER (estrogen receptor) ed ha una biodisponibilità doppia rispetto a D. Si pensa che tale differenza sia imputabile ad una glucuronazione enantioselettiva dell’isomero L. O O N N O Raloxifene O Arzoxifene OH HO OMe S HO S O O Lasofoxifene R=H Nefoxidine R= Me N O Basedoxifene N OH HO R O N Terapia ormonale 7 L’aromatasi è l’enzima che converte l’Androstendione in Estrone e di conseguenza bloccare questo enzima vuol dire ridurre la concentrazione di estradiolo circolante. Questo enzima è CYP450 dipendente e essi si legano proprio alla subunità CYP450 dell’enzima ed agiscono in modo competitivo. Tale inibizione può avvenire con analoghi steroidei del substrato naturale o con molecole non steroidee strutturalmente correlate agli antifungini azoici. Me O Me O NADPH Me HO NADPH O O OH O O O Fe3+ Me O Me O Me O H O2 O O -H 2O O2 O2 O HO OH NADPH Me O Me O O HO Terapia ormonale 8 • Inibitore di prima generazione è la aminoglutetimide – Orimeten® che ha un uso limitato in quanto inibitore non specifico dell’aromatasi e capace di inibire diverse attività enzimatiche della steroidogenesi. • Tra gli inibitore di seconda generazione ricordiamo il Formestane molecola dotata di buona efficacia clinica, il cui limite è rappresentato principalmente dalla via di somministrazione (iniezione intramuscolare). • Inibitori di terza generazione introdotti in terapia sono: • Anastrozolo-Arimidex® è stato il primo inibitore dell’aromatasi a mostrare un significativo vantaggio in termini di sopravvivenza rispetto al Megestrolo. Me O H2N O Me O Amiglutetimide Me Me CN N N NH O N OH Formestane Me CN Me Anastrozolo Terapia ormonale 9 Fadrozolo –1995 utilizzato come racemo. Letrozolo – Femara® - 1996 dotato di: – attività farmacologica 200 volte superiore 10000 volte rispetto all’aminoglutetimide; all’Anastrozolo – Maggiore selettività verso l’enzima e non altera la sintesi di altre importanti sostanze. Inibisce l’aromatasi periferica per il 98% e sopprime i livelli ematici ed urinari di estrogeni per il 95% dopo 2 settimane di trattamento. Exemestrano – sviluppato dalla Carlo Erba ed introdotto in terapia da Pharmacia nel 2000 per il trattamento di tumori estrogeno dipendenti e del cancro della mammella. Me O N N N CN N N Me HCl O NC Letrazolo CN CH2 Fadrozolo Exemestano Terapia ormonale 10 I trattamenti ormonali di riferimento in prima linea in caso di carcinoma mammario sono: – il tamoxifene nelle donne in pre-menopausa; – il tamoxifene o un inibitore dell’aromatasi in quelle in postmenopausa. Questa terapia viene utilizzata: – sia come terapia adiuvante dopo la chirurgia per prevenire le recidive in forme localmente avanzate quando i recettori ormonali sono positivi; – sia come trattamento delle forme metastatiche. In seconda linea il trattamento di riferimento per il carcinoma metastatico è basato: – nelle donne in premenopausa che abbiano impiegato il tamoxifene in adiuvante, sull’utilizzo degli inibitori dell’aromatasi per via orale (anastrozolo, letrozolo, exemestane); – nelle donne in post-menopausa che abbiano utilizzato in adiuvante gli inibitori dell'aromatasi, sull’utilizzo di tamoxifene; – nelle donne in post-menopausa che abbiano utilizzato il tamoxifene in adiuvante, sull’utilizzo degli inibitori dell'aromatasi. Terapia ormonale 11 Agonisti del GnRH ed antiandrogeni Il trattamento ormonale nel caso del tumore alla prostata è ormai di uso comune. Si cerca di ridurre gli effetti stimolanti degli ormoni maschili (testosterone e diidrotestosterone) sulle cellule tumorali prostatiche. La terapia farmacologica presenta diversi approcci uno dei quali consiste nell’utilizzare agonisti del GnRH (ormone che rilascia l’ormone luteinizzante LHRH). Questi composti, dopo un iniziale incremento di testosterone, provocano, tramite retroregolazione, una drastica diminuzione dello stesso fino a livelli di castrazione. È un processo che richiede circa 4 settimane e siccome inizialmente tendono ad alzare i livelli di testosterone e quindi ad incrementare la prostata (vampata), vanno somministrati in associazione con antiandrogeni. I due agonisti più usati sono la Goserelina e la Leuprotide. Terapia ormonale 12 Come nascono questi composti? Con la scoperta del GnRH (1971) è stata confermata l’ipotesi neuroendocrinologica della connessione tra ipotalamo e ipofisi. inoltre sono state aperte nuove strade al trattamento clinico delle disfunzioni riproduttive e non solo. • Questo ormone prodotto dall’ipotalamo esercita la sua azione sull’ipofisi stimolando la secrezione sia dell’ormone follicolostimolante sia dell’ormone luteinizzante. Regola indirettamente la secrezione degli ormoni delle ovaie e dei testicoli come pure quelli steroidei. • La sequenza aminoacidica è pressoché identica per mammiferi, uccelli e pesci Terapia ormonale 13 • • • Il GnRH è un decapeptide che presenta il gruppo N-terminale sottoforma ciclica di piroglutammato e la C-terminale sotto forma di glicinammide. La molecola possiede notevole flessibilità ma la forma biologicamente attiva possiede una struttura β nell’interno della sequenza Tyr-Gly-Leu-Arg avvicinando così le due estremità. La Prepro-GnRH è una proteina di 92 residui. I primi 23 rappresentano la sequenza segnale. Terapia ormonale 14 La scissione a livello dei residui 23-24 libera la glutammina terminale che subito ciclizza ad acido piroglutammico. Enzimi tripsino- e carbossipeptidasi simili agiscono poi a livello della sequenza Gly-Lys-Arg formando un endecapeptide avente una Gly in più. La Gly presente in eccesso serve per trasformare la Gly seguente in Glicinammide. • Una volta liberato possiede une emivita di 5-7 minuti e la sua degradazione inizia a livello del legame pGlu-His ad opera di una piroglutammico amminopeptidasi. • Un enzima post-Pro spezza contemporaneamente il legame ProGly. Terapia ormonale 15 Il recettore del GnRH è una catena polipeptidica di 327 residui amminoacidici con 7 domini trans membranali. La sequenza Cterminale all’interno del citoplasma è pressoché inesistente in quanto comprende solo Ser e Leu. Relazione struttura attività Sono stati preparati numerosi analoghi al fine di studiare le relazioni struttura attività e di ottenere composti più attivi. Se eliminiamo la Glicina C terminale si ottiene un nonapeptide avente solo il 10% dell’attività. Ciò succede anche eliminando il pGlu (<0.01 La sequenza N-terminale pGlu-His-Trp è importante per l’attività e qualsiasi variante ad essa apportata provoca perdita di attività. La Glicinamide terminale può essere sostituita vantaggiosamente con gruppi alchilici. Il derivato con C-terminale Pro-NHEt è 3 volte più attivo mentre il derivato aza glicina (Pro-NH-NH-CONH2) ha un’attività doppia. Terapia ormonale 16 Introducendo in posi zione 6 dei residui Daminoacidici, si otten gono analoghi da 1,8 a 13 volte più potenti. Infatti la presenza di un amminoacido a configurazione D stabi lizza la conformazione b biologicamente atti va e riduce la degrada zione enzimatica. Ulteriore aumento di attività si ha introdu cendo in 6 ammino acidi protetti o idrofo bici non naturali. Terapia ormonale 17 • Questi agonisti oltre ad avere una maggiore attività posseggono anche un’emivita maggiori rispetto al GnRH. • Gli agonisti Fertirelina, Leuprolide (leuprorelina), Triptorelina, Buserelina, Goserelina e Nafarelina sono stati oggetto di numerosi studi. • LA Goserelina Zoladex® è impiegata nel trattamento ormonale del carcinoma prostatico in quanto, agendo sull’asse ipotalamoipofisi-gonadi, abbassa la produzione di testosterone sino a livelli di castrazione (anche Buserelina e Triptorelina). • La buserelina, la Goserelina e il Leuprolide sono impiegati nella terapia dei tumori mammari ormono-dipendenti in quanto sopprimono la funzione ovarica e riducono gli estrogeni circolanti. • L’acetato del Leuprolide Eligard® Enantone® è somministrato giornalmente per iniezioni oppure mensilmente come iniezione di deposito oppure ogni 3-4 mesi come trattamento palliativo del carcinoma dlla prostata. • La triptorelina Decapeptyl® Gonapeptyl Depot® ha dato buoni risultati nella cura della pubertà precoce sia maschile che femminile. Terapia ormonale 18 Antiandrogeni Trovano impiego nel tarttamento dell’acne giovanile, delle sindromi virilizzanti nella donna e contro il carcinoma della prostata. Prostata ingrossamento o Iperplasia Prostatica Benigna (BPH) frequente dopo i 50-55 anni risolvibile chirurgicamente. tumore della prostata – diffuso tra gli anziani e rappresenta il secondo tumore più diffuso e il terzo come causa di morte. Approcci: Uso di antagonisti del recettore degli androgeni a struttura steroidica (Ciproterone acetato, Zanosterone, Oxendolone e RU38,882) e non (Flutamide, Bilucamide, Nilutamide etc.) O OH OH OAc O2S N N H3C O Cl Ciproterone acetato H Zanosterone O OAc O Oxendolone RU-38,882 Terapia ormonale 19 Inibitori della 5a-reduttasi Quest’ultimo è un enzima intracellulare, NADPH dipendente, necessario per la conversione del Testosterone in DHT ed esiste in due isoforme: quella di tipo 1 prevale nei tessuti normali (ruolo catabolico) mentre quella di tipo 2 abbonda nei tessuti bersaglio degli androgeni con effetto anabolico (prostata). Il primo farmaco è la Finasteride Prostide® Proscar® Genaprost®, in commercio dal 1992, in grado di inibire in modo selettivo la isoforma 2 dell’enzima. È un 4-aza-steroide che agisce come inibitore competitivo del testosterone del quale innalza i livelli ematici. Suoi analoghi sono Turosteride e L-654,4066. L’Epristeride non è un aza-steroide bensì un derivato di un acido bicarbossilico ed agisce come inibitore non competitivo. O H N O O N O H N H N O O N H H Finasteride O N H H Turosteride O N H H L-654,4066 HOOC Epristeride Terapia ormonale 20 Preparazione della Finasteride O H N O H N N aIO 4 N H3 KM n O 4 H OOC O H2 Pt O A m m id e d ell' acid o 3 -c h et o -∆ ∆ 4-e tian ico O ch e to ac id o O H N D DQ Cl O N H Cl O O CN CN O lat ta m e DDQ = 2,3-dicloro-5,6-dicianobenzochinone N H F in as ter id e H N Terapia ormonale 21 Antiandrogeni anche in questo caso l’uso di questi farmaci per la cura del cancro alla prostata è diventato ormai una pratica comune. Sono stati ormai approvati dalla FDA tre farmaci: Flutamide Flutamide® Fluprost® Eulexin®, Bicalutamide Casodex® e Nilutamide. Sono degli antiandrogeni non steroidei che impediscono la traslocazione dei recettori degli androgeni nel nucleo a livello dei tessuti bersaglio (ipotalamo e prostata).. Flutamide Bicalutamide Nilutamide. Ne consegue un blocco dell’azione di O HN NO2 F O CH3 HN CH3 OH CF3 CF3 NO2 S O2 CH3 H3C NH O N O CF3 NO2 Terapia ormonale 22 La Flutamide viene metabolizzata a dare l’isrossi derivato che mantiene inalterata l’attività. La Nilutamide viene usata come singolo farmaco in aggiunta alla alla castrazione chirurgica per bloccare l’azione di del Diidro- e del Testosterone. Per la cura della prostata è allo studio anche l’uso di antagonisti del GnRH (Abarelix) che blocca i recettori del GnRH a livello della ghiandola pituitaria. Questo blocco sopprime il rilascio degli ormoni luetinizzante (LH)e follicolo stimolante (FSH) riducendo di conseguenza la secrezione del testosterone. O HN O CH3 CH3 CH3 OH CH3 CF3 CF3 NO2 Flutamide HN NO2 metabolita Terapia ormonale 23 Per concludere va ricordato che nella terapia antitumorale un trattamento efficace lo si ottiene utilizzando combinazioni di farmaci. A questo fine andrebbero utilizzate dosi usuali di più farmaci aventi però meccanismi d’azione diversi. Anche se ciò è vantaggioso, non sempre è possibile applicare questa strategia in virtù della tossicità dei farmaci. Infatti non è detto che farmaci con differente meccanismo d’azione non presentino lo stesso tipo di effetto collaterale. Resistenza agli agenti antitumorali La chemioterapia rappresenta trattamento dei tumori. la principale approccio per il La progettazione di un protocollo terapeutico razionale richiede l’utilizzo di composti: – ai quali il tumore sia sensibile; – che abbiano differenti meccanismi d’azione; – non superino la dose tossica limite; – che colpiscano le cellule nei differenti cicli cellulari. Tuttavia le cellule tumorali possono acquisire, durante il trattamento chemioterapico, dei cambiamenti pleitropici che possono rendere il tumore refrattario sia ai farmaci utilizzati sia a quelli non ancora assunti. Resistenza agli antitumorali 2 La maggior parte dei fenotipi di cellule tumorali hanno la capacità di: – Non accumulare il farmaco nel proprio interno; – Di non consentire una duratura interazione del farmaco con la proteina target; – Modificare la proteina target al fine di ridurre l’interazione con il farmaco. 1. Modifica dei targets La cellula tumorale può: – Apportare modifiche alle proteine bersaglio dei farmaci ottenendo come risultato o la diminuzione o l’incremento dell’attività. – Aumentare il numero dei geni codificanti la proteina stessa al fine di sopraffare l’effetto citotossico del farmaco. Resistenza agli antitumorali 3 Esempi: Metotrexate - Cellule che aumentano la concentrazione intracellulare di DHFR dopo esposizione al farmaco. aumentare la dose di farmaco 5-FU – il suo metabolita attivo 5-Fd-UMP inibisce la Thymidylate Synthase (TS) che blocca la sintesi de novo delle pirimidine. Esiste un polimorfismo della regione enhancer/promoter della TS caratterizzato da un Variable Number of Tandem Repeats (VNTRs) che comporta una variabilità di risposta al farmaco stesso. cellule contenenti 3 repliche possiedono una elevata attività della TS e risultano meno sensibili al trattamento con 5-FU Cellule contenenti 2 repliche possiedono una bassa attività della TS e risultano più sensibili al trattamento con 5-FU Resistenza agli antitumorali 4 Epipodofillotossine e Antracicline – inibitori della topoisomerasi II. Sono stati evidenziati cellule tumorali aventi varie mutazioni di questo enzima. Queste mutazioni hanno evidenziato in vivo una resistenza al farmaco dovuta a: – una ridotta attività dell’enzima; – Una variazione strutturale della topoisomerasi impedisce l’interazione con il farmaco. II che 2. Alterazione della ritenzione del farmaco all’interno della cellula Le cellule possono: a) ridurre la velocità di ingresso del farmaco nel citoplasma; b) aumentare la velocità di efflusso dello stesso. Resistenza agli antitumorali 5 a) Diminuzione delle velocità di ingresso Metotraxate – viene trasportato all’interno della cellula da uno specifico carrier per i folati ridotti (RFC). Cellule tumorali, sia umane che murine, resistenti al farmaco presentano modifiche a questo carrier che sono state aevidenziate da una diminuita concentrazione endocellulare del Metotraxate. Alcune cellule leucemiche umane hanno evidenziato anche una ridotta espressione di questo RFC. b) Aumento della velocità di efflusso del farmaco dalla cellula. Le cellule presentano la superfamiglia dei trasportatori definiti ATP Binding Cassette (ABC). È la più grande e vasta superfamiglia nota di proteine espresse. Resistenza agli antitumorali 6 La maggior parte delle ABC è responsabile del trasporto attivo, attraverso la membrana cellulare, di una notevole varietà di composti quali fosfolipidi, ioni, peptidi, steroidi, polisaccaridi, amminoacidi, ioni organici, acidi biliari, farmaci e xenobioti. Nell’uomo sono stati descritti ben 49 geni per gli ABC e molti di questi geni sono coinvolti in un disordine genetico ben definito e ciò ha portato condotto ad una suddivisione in sottofamiglie. Le proteine trasportatrici multifarmaco ABC sono glicoproteine plasmatiche di membrana coinvolte in funzioni protettive contro sostanze tossiche in diverse cellule e tessuti È stato dimostrato che qualora le cellule sovraesprimano queste proteine, esse acquistano resistenza multipla a farmaci anche non correlati strutturalmente. Esse includono le proteine MultiDrug Resistance (MDR) e le Multidrug Resistance-related Protein (MRP). Resistenza agli antitumorali 7 3. Alterazione della ritenzione del farmaco all’interno della cellula. Incrementando il metabolismo di fase II le cellule tumorali possono eliminare rapidamente le sostanze citotossiche. Alterare la via metabolica di coniugazione con Glutatione può comportare una differente sensibilità delle cellule ai farmaci antitumorali. Es: alcune cellule cancerogene resistenti sia a Vinblastina che ad Adriamicina, hanno evidenziato concentrazioni intracellulari di Glutatione due o tre volte superiori. Per contro altre cellule tumorali hanno livelli di glutatione intracellulari ridotti a causa delle MRPs che lo utilizzano come cotrasportatore nell’espellere il farmaco dalla cellula. Resistenza agli antitumorali 8 4. Aumentata capacità di riparare il DNA La O6-alchilguanina alchil transferasi (OGAT) svolge l’azione di riparare il DNA rimuovendo i gruppi alchilici dell’ossigeni in 6 della guanina. cellule che possiedono elevate concentrazioni di OGAT risultano resistenti ad agenti alchilenti quali le nitrouree e i derivati triazineci (es procarbazina). 5. Apoptosi anomala Nelle cellule normali il gene p53 viene attivato in seguito a danno al DNA e stimola o l’arresto della crescito o l’apoptosi in funzione dello stadio del ciclo cellulare. È stato dimostrato che più del 50 5 delle cellule tumorali evidenzia una inattivazione funzionale del p53.
Scaricare