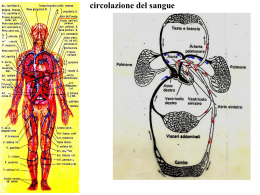
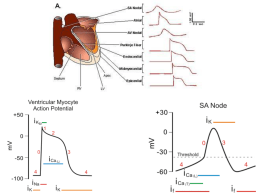
Regina di Cuori Appunti di cardiologia A cura di Antonio Romanelli ANNO ACCADEMICO 2010/2011 PROGRAMMA DI CARDIOLOGIA IV ANNO, 2° SEMESTRE prof. Federico Piscione • Fisiologia del cuore • Cardiopatia ischemica cronica e diagnostica per immagini • Sindromi coronariche acute • Infarto STEMI • Patologia valvolari ed Endocarditi • Cardiomiopatie • Aritmie • Scompenso cardiaco • ECG • Malattie cardiache congenite • Miocarditi, pericarditi e versamento pericardico TESTI CONSIGLIATI • Harrison – Principi di Medicina Interna – McGrawHill Programma ricavato su informazioni dirette del professore. CAPITOLO I: FISIOLOGIA DEL CUORE BASI CELLULARI DELLA CONTRAZIONE CARDIACA Ultrastruttura cardiaca: All’interno della struttura del cuore, possiamo riconoscere due tipi di tessuti, addetti a due funzioni differenti: - Il miocardio di lavoro; - Il miocardio di conduzione. Il cuore è un organo muscolare, formato da cellule muscolari striate, detti miociti, ma molto diverse rispetto a quelle del muscolo scheletrico. Ciascun miocita, all’ingrandimento, contiene numerosi fasci bastoncellari, con bande trasversali (miofibrille) che percorrono la cellula in tutta la sua lunghezza e che sono composte da strutture che si ripetono in serie, i sarcomeri. Il citoplasma tra le miofibrille contiene altri elementi cellulari tipici.All’ingrandimento, è possibile notare come il muscolo cardiaco o miocardio, sia costituito da fibre striate ramificate e mononucleate, ricche in mitocondri. Le singole cellule sono unite tra di loro grazie a delle specializzazioni della membrana definite dischi intercalari. Nei dischi intercalari sono presenti le giunzioni comunicanti, che permettono la comunicazione elettrica e i desmosomi, che mantengono unite le cellule tra di loro. Grazie alla presenza delle giunzioni comunicanti, che sono permeabili a ioni, le singole cellule del miocardio rispondo come un tuttuno a seguito di una stimolazione di natura elettrica, ed è per questo che il tessuto del cuore viene anche definito come un “sincizio funzionale”. Osservando al microscopio elettronico è possibile notare all’interno del citoplasma delle specializzazione dell’apparato citoscheletrico, che tende ad organizzarsi e formare sarcomeri. Il sarcomero è l’unità morfologica e funzionale della contrazione, e viene definito come l’insieme delle strutture proteiche comprese tra due linee Z. Sempre all’interno della cellula si può notare un numero enorme di mitocondri disposti a sandwich tra le miofibrille e inoltre, si può notare, la presenza dei tubuli T, che penetrano all’interno della cellula fino in prossimità delle linee Z. la distanza fra due linee Z varia in funzione del grado di contrazione, da 1,6 µm a 2,2 µm. All’interno del sarcomero, si possono notare delle aree più elettrondense e meno elletrondense. Al centro del sarcomero vi è una banda densa di lunghezza costante, 1,5 µm, la Banda A, affiancata da due linee più chiare, le bande I, di lunghezza variabile. Il sarcomero cardiaco è formato da due serie di miofilamenti intermedi: i filamenti spessi, ovvero la miosina, che attraversano la banda A, e i filamenti sottili di actina, che corrono dalla linea Z, attraversano la banda I fino alla banda A. I filamenti spessi e sottili si sovrappongono solo a livello della bandaA, mentre la banda I contiene solo filamenti sottili. Tra i filamenti sottili e spessi sono presenti le teste di miosina. Questo descritto fin qui è il miocardio di lavoro. Per quanto riguarda il miocardio di conduzione, per ora, è necessario sapere che risulta essere una specializzazione del miocardio di lavoro. Il compito di questo tessuto specializzato è quello di generare e veicolare, a tutti i miocardiociti ed in maniera ordinata, il potenziale di azione responsabile della sistole cardiaca. Il processo contrattile: Il cuore è un muscolo. Il calcio gioca un ruolo fondamentale per la contrazione, il sodio e il potassio svolgono un ruolo importante per l’attività pacemaker. Il cuore si contrae per espellere il sangue in circolo esi deve rilassare per accettare il sangue che proviene dal circolo. Le singole fibre muscolari cardiache sono in grado di proporzionare la contrazione e la forza generata tramite la concentrazione di calcio citoplasmatico. Il modello dei filamenti scorrevoli per la contrazione muscolare si basa sull’osservazione fondamentale che sia i filamenti spessi che sottili presentano un lunghezza costante durante la contrazione ed il rilassamento. Durante la contrazione la lunghezza della Banda A rimane costante, mentre la Banda I si accorcia e le linee Z si muovono l’una verso l’altra. Oltre all’actina e alla miosina, esistono altre proteine con il compito di stabilizzare la struttura del sarcomero, come la titina, e proteine regolatrici, poste sui filamenti di actina, le troponine C, I e T. Durante l’attivazione del miocita cardiaco, il calcio si lega alla troponina C, dando luogo ad un cambiamento della conformazione della proteina regolatrice tropomiosina; quest’ultima, a sua volta, espone i siti di interazione dei ponti crociati di actina. L’interazione ripetitiva fra le teste di miosina e l’actina viene denominata cross-bridge cycling. La scissione dell’ATP poi dissocia la testa di miosina e l’actina. Quando vi è l’ATP, l’interazione tra miosina ed actina sono costituite e rotte ciclicamente, a patto che sia presente una certa quota di calcio. Questi legami cessano quando la concentrazione di calcio intracitoplasmatico diventa basso e il complesso troponina-tropomiosina impedisce di nuovo le interazioni tra actina e miosina. Il calcio intracitoplasmatico è uno dei principali mediatori dello stato inotropo del cuore. Attivazione cardiaca: Il calcio gioca un ruolo molto importante nel mediare l’accoppiamento tra eccitazione e contrazione: 1) Il potenziale di azione di propaga lungo la membrana plasmatica; 2) Si verifica l’apertura dei canali del Ca2+ con ingresso dello ione all’interno della cellula; 3) L’ingresso del calcio nella cellula innesca il rilascio di altro calcio dal reticolo sarcoplasmatico e tale meccanismo viene definito come “rilascio di calcio calcio-indotto”; 4) Aumento della concentrazione intracellulare di calcio; 5) Legame del calcio alla troponina, con innesco della contrazione. La troponina impedisce l’interazione tra la testa di miosina ed i filamenti di actina; 6) Distacco del calcio dalla troponina ed inizio del rilassamento muscolare; 7) Il calcio, tramite opportune pompe, viene riportato all’interno del reticolo sarcoplasmatico; il calcio può essere trasportato o grazie ad un antiporto col sodio, oppure tramite una pompa ATP-dipendente; 8) La pompa Na+/K+ mantiene il gradiente di sodio, necessario per l’antiporto a livello del reticolo sarcoplasmatico. Se il calcio manca, se non viene rilasciato nel tempo giusto o non viene ripreso nel reticolo sarcoplasmatico o nei mitocondri al tempo giusto, salta la normofunzionalità del muscolo cardiaco. Nello stato inattivo, la cellula miocardica è polarizzata elettricamente, con un potenziale trans membrana compreso tra -80 e -100 mV. Il sarcolemma, che in condizioni di riposo è impermeabile al sodio, è dotato di una pompa per il sodio e il potassio. Tale pompa serve a mantenere i gradienti di sodio e potassio all’interno della cellula. Durante il plateau del potenziale d'azione, si osserva una corrente lenta diretta verso l'interno attraverso i canali del calcio di tipo L. La corrente depolarizzante, non solo si estende lungo la superficie della cellula, ma penetra profondamente al suo interno grazie ai tubuli T. La quantità assoluta di calcio che attraversa la membrana e i tubuli è relativamente modesta e di per sé non è in grado di determinare l'attivazione dell'apparato contrattile. Però, questa corrente di calcio innesca il rilascio di calcio dal reticolo sarcoplasmatico, un processo denominato rilascio di calcio-calcio indotto. Il calcio viene liberato dal reticolo sarcoplasmatico tramite un canale specifico, un’isoforma del recettore rianodinico (RyR2) e controlla la concentrazione del calcio e provoca le modificazioni locali della concentrazione di calcio note come calcium sparks. Vi sono numerose proteine regolatrici che inibiscono il recettore rianodinico, come la calstabina. Il calcio liberato dal reticolo sarcoplasmatico si propaga verso le miofibrille dove si combina con la troponina C. Reprimendo quest'inibizione della contrazione, il calcio attiva l'accorciamento di miofilamenti. Durante la ripolarizzazione, l'attività della pompa del calcio nelle reticolo sarcoplasmatico (SERCA), accumula ioni di calcio contro gradiente di concentrazione, e gli ioni calcio vengono immagazzinati nelle reticolo sarcoplasmatico mediante il legame con una proteina, la calsequestrina. Questo accumulo di calcio è un processo che richiede energia e riduce la concentrazione citoplasmatica di calcio fino a livelli che determinano l'inibizione dell'interazione actina-miosina. CONTROLLO DELLA PERFORMANCE E DELLA GITTATA CARDIACA L'entità del accorciamento del muscolo cardiaco, e quindi la gittata sistolica del ventricolo, dipende da tre fattori: - la lunghezza del muscolo all'esordio della contrazione, ovvero il pre-carico; - la tensione che il muscolo e chiamata a sviluppare durante la contrazione, cioè il post-carico; - la contrattilità del muscolo, ovvero l'entità e la velocità di accorciamento ad ogni pre e post-carico. Ruolo della lunghezza muscolare (pre-carico): I sarcomeri muscolari possono essere paragonati ad una molla: c’è un limite oltre il quale non si può più distendere, prima di questo limite quanto più la molla è distesa tanto con più forza si ricompatterà (legge di Starling); ci sarà un punto in cui la molla non si ricompatterà più perché la forza diminuirà. Il rapporto fra lunghezza iniziale delle fibre muscolari e forza sviluppata e di importanza cruciale per la funzione del muscolo cardiaco: questa è la base della legge di Starling, la quale stabilisce che, entro certi limiti, la forza di contrazione ventricolare è in funzione della lunghezza del muscolo cardiaco alla fine della diastole; nel cuore sano, quest'ultima è strettamente correlata al volume ventricolare telediastolico. Durante la diastole, il sarcomero presenta una lunghezza di 2,2 µm, riducendosi a 1,90 µm durante la sistole. Nell’immagine, partendo in alto a destra, il precarico è la lunghezza massima del sarcomero poco prima dell’inizio della contrazione. Durante la sistole, il volume del ventricolo diminuisce e ciò corrisponde ad una riduzione della lunghezza del sarcomero. Poi, durante la fase rapida del riempimento, la lunghezza del sarcomero aumenta da 1,90 µm a 2,15 µm. Se mettiamo su un sistema di assi cartesiani lalunghezza del sarcomero e il volume, man mano che ilsarcomero si allunga si ha un incremento del volume.Da un certo volume in poi sembra che la curva resti parallela, che non succeda niente, ma non è così, perché c’è comunque un incremento di volume, quindi significa che un minimo allungamento del sarcomero ci debba essere. Ad un certo punto, alla massima lunghezzadel sarcomero, identifichiamo il pre-load o precarico e una volta raggiunto un apparente appiattimento c’è una fase molto brusca in cui si ha una discesa rapida, in cui si abbatte la lunghezza del sarcomero e si abbatte anche la quantità di sangue. Performance cardiaca: La pressione ventricolare di riempimento o telediastolica viene, in alcuni casi utilizzata come sinonimo di volume telediastolico. La gittata sistolica è direttamente proporzionale alla lunghezza telediastolica delle fibre (pre-carico) e inversamente proporzionale alla resistenza arteriosa (postocarico), e quando il cuore divenne insufficiente la gittata sistolica e progressivamente più scarsa. La relazione fra pressione telediastolica ventricolare e lavoro sistolico del ventricolo (curva della funzione ventricolare) fornisce un’utile definizione del livello di contrattilità del cuore. Un aumento della contrattilità è accompagnato da uno spostamento verso l'alto e a sinistra della curva funzione ventricolare, mentre la depressione della contrattilità è caratterizzata da uno spostamento verso il basso e a destra. Post-carico ventricolare: L'entità di accorciamento delle fibre muscolari ventricolari a qualunque livello di pre-carico e di contrattilità miocardica è inversamente proporzionale al postcarico, ovvero al carico che si oppone all'accorciamento. Il postcarico e dipendente dalla pressione aortica, come pure dal volume e dallo spessore della cavità ventricolare. La legge di Laplace afferma che la tensione della fibra miocardica è funzione del prodotto tra la pressione ventricolare nella cavità ed il raggio del ventricolo, diviso per lo spessore della parete. Quindi, a ogni livello di pressione aortica, il postcarico supportato da un ventricolo sinistro dilatato è maggiore di quello contrapposto ad un ventricolo di dimensioni normali. Al contrario, a parità di pressione aortica e di volume diastolico ventricolare, il postcarico di un ventricolo ipertrofico è inferiore a quello di un ventricolo normale. La pressione aortica, stavolta, dipende dalle resistenze vascolari periferiche, dalle caratteristiche fisiche dell'albero arterioso e dal volume di sangue che è contenuto nel vaso all'inizio dell'edizione. LA MECCANICA DEL CUORE: DALLA DEPOLARIZZAZIONE ALLA CONTRAZIONE Le fibre cardiache muscolari sono le uniche capaci di generare stimoli elettrici e di trasmetterli.La cellula nata per fare “lavoro” in situazioni particolari diventa capace di svolgere, senza averlo mai fatto invita sua, il compito di generare e trasmettere stimoli elettrici. Da un punto di vista elettrico, distinguiamo due tipi di fibre, a risposta lenta ed a risposta rapida. Le fibre cardiache a risposta lenta sono le fibre muscolari che stanno svolgendo in quel momento la funzione di pacemaker e di trasmissione dello stimolo elettrico. Le fibre cardiache a risposta rapida sono le fibre che svolgono la funzione di lavoro, dove bisogna essere rapidi sia nel rilasciarsi sia nel contrarsi. Il potenziale di azione di queste due fibre è diverso (vedi immagine).La risposta lenta prevede correnti di sodio e potassio. Il corpo umano è un conduttore a volume ed il cuore possiede potenziale negativo nella cavità endocardica, pertanto la depolarizzazione avviene dall’endocardio all’epicardio; la ripolarizzazione avviene dall’epicardio all’endocardio. Le cellule del miocardio specifico scaricano spontaneamente; esse hanno un potenziale di membrana che, dopo ogni scarica, diminuisce per poi ritornare a livello di scarica; è definito pre-potenziale o potenziale avviatore. A riposo le mio cellule hanno un potenziale di circa -90 mV. Il meccanismo di depolarizzazione spontanea è garantito dalla presenza di particolari canali per il Calcio ed il Potassio sulla membrana delle cellule del miocardio specifico. Al primo impulso elettrico, canali IK permettono la ripolarizzazione – il potassio esce- della membrana fino a circa -60 mV,voltaggio in cui il canale I K si chiude; a questo punto si apre il canale I Ca 2+T ( T = transitori) – il calcio entra -, che portano il potenziale di membrana fino a poco meno di -40 mV; a questo punto entrano in azione i canali I Ca 2+L (L= di lunga durata) che determinano la depolarizzazione con lo spike e ricomincia il ciclo. Le cellule del miocardio specifico sono caratterizzate dall’assenza di canali per il Na+; la depolarizzazione, quindi, avviene prima. La stimolazione vagale aumenta la conduttanza per i canali I K +, perché l’acetilcolina stimola una maggiore ripolarizzazione (diminuisce la pendenza del prepotenziale);condizioni di iperpotassiemia sono le cause più frequenti di aritmia perché non si ottiene una sufficiente ripolarizzazione del miocardio specifico. Al contrario, invece, la stimolazione simpatica aumenta la conduttanza per i canali I Ca 2+T determinando una più veloce depolarizzazione (aumenta la pendenza del prepotenziale). È da ricordare, però, che il cuore ha il meccanismo di fuga dal vago, per restare funzionante nonostante una iperstimolazione vagale. Componente elettrica del cuore: La trasmissione del segnale avviene nel seguente modo: - L’attivazione cardiaca inizia dal NODO SENO-ATRIALE e si diffonde da destra verso sinistra, dall’alto verso il basso, interessando l’atrio destro, l’atrio sinistro e il NODO ATRIOVENTRICOLARE, mediante tra fasci di tessuto specifico di conduzione a destra ed un altro solo a sinistra; - Dopo l’attivazione del nodo A-V, il processo di depolarizzazione continua nel FASCIO DI HIS, che è formato, inizialmente da un primo fascio unico che si divide, poi, nella pars membranacea del setto interventricolare, in fascio destro e sinistro. La branca destra resta unica fino al raggiungimento delle fibre del Purkinje, a livello del muscolo papillare. La branca sinistra, al contrario, si divide subito dopo il suo inizio in un fascio anteriore ed uno posteriore che decorrono lungo la parte sinistra del setto interventricolare; - Attraverso i due fasci di His, lo stimolo, alla velocità di 4 m/sec raggiunge, infine, le fibre di Purkinje; - La prima parte dei ventricoli ad essere attivata - ATTIVAZIONE VENTRICOLARE – è la porzione apicale e media inferiore della superficie sinistra del setto interventricolare. Queste forze possono essere rappresentate da un vettore: il I vettore è diretto da sx a dx, in basso e in avanti. Questi risulta piccolo perché la porzione di miocardio depolarizzata (setto interventricolare) è limitata; questa fase corrisponde all’Onda R del QRS; - Successivamente l’attivazione interessa la superficie endocardica di ambedue i ventricoli dall’apice verso la base. Tuttavia le forze elettriche del ventricolo sinistro prevalgono su quelle del dx. Il processo di depolarizzazione ventricolare è rappresentato da un vettore, II vettore, 10 volte più grande di quello settale. Questo interviene dopo 0,04 sec. dall’inizio dell’attivazione del setto ed è diretto verso sinistra, posteriormente in basso; questa fase corrispondeall’Onda R del QRS; - L’ultima parte dei ventricoli ad essere attivata è la regione basale ed in particolare quella del ventricolo dx, che cade dopo 0,06 sec. dall’inizio dell’attivazione ventricolare; ha grandezza simile al primo ed è diretto verso dx, in alto e posteriormente; questa fase corrisponde all’Onda S del QRS. La depolarizzazione può essere così riassunta dai vettori: 1. Somma dei vettori dell’attivazione del setto; 2. Somma dei vettori dell’attivazione dell’apice e del corpo dei ventricoli con dominanza del sx; 3. Somma dei vettori dell’attivazione della porzione basale dei ventricoli e del setto interventricolare. In seno agli atri ed ai ventricoli esistono, però, gruppi di cellule che mostrano potenziali d’azione molto simili, nella fase diastolica, a quelli delle cellule segna passo, dette segna-passo potenziali; in condizioni normali la frequenza di scarica el’attività del nodo SA e AV ne blocca il funzionamento. Questi pace-maker latenti possono attivarsi nel caso in cui uno stimolo avviatore non percorra in tempo il suo percorso. Sono, quindi, dei meccanismi di protezione che, però, in alcuni casi, possono attivarsi anche se tutto funziona bene; questo processo è alla base della formazione delle aritmie. Eccitazione del miocardio di lavoro: Al contrario delle cellule del miocardio specifico, quelle del miocardio di lavoro sono eccitabili solo se uno stimolo adeguato è ad esse condotto. Allora queste modificano bruscamente il loro potenziale d’azione sino a raggiungere un massimo di +15 mV. La fase di salita del potenziale è rapida, molto più che nel miocardio specifico, detta fase 0. Esiste, poi, una fase di discesa rapida che costituisce la fase 1, cui segue un plateau detta anche come fase 2, fase 3, in cui il potenziale raggiunge la massima negatività diastolica; la fase 4 permette la risalita del potenziale fino al valore stabile del tessuto muscolare. Tutto parte dall’apertura del canale per il sodio voltaggiodipendente, cui segue l’attivazione di tutti gli altri canali. La Contrazione: La funzionalità del cuore è legata direttamente alla sua morfologia. L’organizzazione in sincizi, associata alla presenza della rete specifica di conduzione, è fondamentale per la contrazione del miocardio di lavoro. La stimolazione elettrica determina l’apertura dei canali Na+ voltaggio-dipendenti: inizia, così, la depolarizzazione e il raggiungimento dello spike. Successivamente inizia un lungo plateau legato all’ingresso del Ca2+attraverso i canali lenti I Ca 2+L. L’entrata di calcio determina un aumento del rilascio di calcio dal reticolo sarcoplasmatico, cui segue la contrazione muscolare. La fase di ripolarizzazione è legata, infine, all’apertura dei canali per il K+ che abbandona la cellula. Eventi meccanici durante della contrazione: Il ciclo cardiaco è formato dalla sistole e dalla diastole. La sistole si divide in contrazione isovolumetrica, periodo di espulsione rapido e lento. La diastole inizia con la fase di rilasciamento isovolumetrico, cui segue il periodo di riempimento diastolico ventricolare rapido e lento. La diastole termina con la contrazione atriale: fase telediastolica o presistole. Il ciclo cardiaco si fa iniziare col periodo telediastolico, caratterizzato dalla contrazione degli atri. Siamo qui nella fase di Telediastole. La valvola mitrale e tricuspide, la prima tra atrio e ventricolo sx, la seconda tra atrio e ventricolo dx, sono ancora aperte. Le valvole aortica e polmonare sono chiuse. La velocità del sangue che affluisce nelle cavità cardiache è sempre minore. Inizia la fase di Sistole Atriale. Durante la diastole circa il 70% della quantità di sangue che determina il riempimento ventricolare già è passato. Adesso la contrazione atriale determina la spinta del restante 30% di sangue nei ventricoli.La contrazione atriale determina anche la contrazione della muscolatura che circonda gli orifizi della vene cava e polmonare comportando, così, l’arresto del sangue, non potendo impedire un certo rigurgito. Durante la contrazione gli atri diminuiscono di volume e la pressione intra-atriale supera di alcuni mmHg la pressione telediastolica ventricolare, così da accelerare il passaggio di sangue dagli atri ai ventricoli. Alla fine della contrazione inizia il movimento di chiusura degli ampi lembi delle valvole atrioventricolari Si passa adesso alla Sistole Ventricolare. All’inizio di questa fase le valvole mitralica e tricuspide si chiudono, con ulteriore aumento della pressione intraventricolare. La pressione aortica e polmonare media sono elevate, ciò comporta che anche le valvole aortica e polmonare siano ancora chiuse. Si ha quindi una prima contrazione definita Contrazione Isovolumetrica Sistolica, che comporta un aumento netto di pressione, che dura circa 0,05 sec.. Nel momento in cui la pressione nei ventricoli supera quella delle rispettive arterie, vale a dire in media 10 mmHg per l’arteria polmonare e 80 mmHg per quella aortica, le valvole aortica e polmonare si aprono e la contrazione continua; il sangue che affluisce nelle cavità ventricolari non si ferma nel cuore prima di essere espulso, spostandosi velocemente, conservando la propria energia cinetica, dalla camera di afflusso a quella di deflusso. Qui si ha la fase di Eiezione, che dura circa 0,20 sec.. La contrazione e l’eiezione fanno sollevare nell’atrio i lembi delle valvole mitralica e tricuspide, determinando un lieve aumento della pressione atriale. La pressione ventricolare dx può raggiungere un massimo di 25mmHg, mentre quella sx può raggiungere un picco di 120 mmHg. Questa fase è divisa in eiezione rapida e lenta. Con l’apertura delle valvole aortica e polmonare l’eiezione è dapprima veloce, rallentando lentamente fino a quando la pressione ventricolare diminuisce sotto i valori delle arterie aortica e polmonare: sichiudono le valvole aortica e polmonare. Il periodo espulsivo termina immediatamente prima della chiusura delle semilunari ed il flusso del sangue si inverte nell’aorta e nella polmonare, causando l’avvicinamento dei lembi valvolari e, nell’aorta, il riempimento delle arterie coronariche. La pressione ventricolare diminuisce ancora mentre anche le valvole AV sono chiuse e si ha il periodo di Rilasciamento Ventricolare Isovolumetrico. Quando la pressione ventricolare è al di sotto di quella atriale, si aprono subito le valvole atrio-ventricolari e ricomincia il ciclo. Ci si trova, ora, nella fase di Protodiastole e si può riscontrare un periodo di rilasciamento muscolare. Questo periodo corrisponde al riempimento della cavità cardiache col sangue proveniente dalle vene cave e dalla vena polmonare. In questo momento le valvole AV, ovviamente, sono aperte. Il riempimento in questo momento è veloce. Dura circa 0,04 sec. Nella rappresentazione dell’ECG la linea isoelettrica corrisponde alla diastole. La sistole atriale comincia dopo l’onda P, la sistole ventricolare comincia dopo l’onda R del complesso QRS per terminare dopo verso la fine dell’onda T. Per pressione massima o sistolica, si intende la pressione raggiunta durante la sistole, per pressione minima o diastolica s’intende la pressione raggiunta durante la diastole, entrambe nel sistema vasale. Definizioni: VOLUME TELEDIASTOLICO (VTD o EDV, end diastolic volume) – volume massimo contenuto nei ventricoli,ovvero volume di sangue nei ventricoli alla fine della diastole (riempimento ventricolare). In media oscilla tra 75±20 ml/m2. VOLUME TELESISTOLICO (VTS o ESV, end sistolic volume) – volume minimo contenuto nei ventricoli, ovverovolume di sangue nei ventricoli alla fine della sistole (svuotamento ventricolare). Possiamo misurare lafunzione contrattile attraverso il VTD e il VTS. In media oscilla tra 25±7 ml/m2. GITTATA SISTOLICA (GS) – [VTD-VTS, 0,07 L/battito] volume di sangue che il cuore espelle a ciascuno battito.Ogni volta che il cuore si riempie e si svuota c’è una certa quota di sangue che viene espulso e una certa quota che resta nel ventricolo. Se il VTD e il VTS sono elevati significa che il cuore o non si rilascia bene o non si contrae bene. Se il cuore funziona bene vi è una quota definita di sangue che viene espulsa: 0,07L/battito. GITTATA CARDIACA (GC) – [GC = FC x GS, frequenza cardiaca x gittata sistolica, 72 battiti/min x 0,07 L/battito = 5 L/min] quantità di sangue pompata in un minuto dal ventricolo sinistro nell’aorta. Se il paziente ha una aritmia, un blocco di secondo grado, ha aritmie extrasistoliche, le GS non sono tutte uguali. Quindi rischiamo di prevedere un valore non giusto. Se noi cumuliamo le GS in una unità di tempo (1 min), alla fine avremo una media delle singole gittate. Quindi avremo in ogni minuto la portata cardiaca, che è la somma delle volte in cui il cuore si è riempito e si è contratto nell’arco di 1 minuto. Questo parametro viene indicizzato in base alla massa corporea. Il classico valore di GC (4,5 – 5,2 L/min) per alcuni può andare bene, per altri no, quindi si normalizza per l’indice cardiaco (→ rapporto gittata cardiaca/areasuperficie corporea). La GC indicizzata è un parametro reale di valore (in base alla massa corporea, capisco di quanto sangue quell’individuo ha realmente bisogno). Quindi la portata cardiaca si esprime in mL/m2 (indicizzato per la superficie del paziente). Il ciclo cardiaco: In condizioni fisiologiche, il flusso di sangue nel cuore è unidirezionale. Questo non è un concetto banale. C’è una sistole atriale, a cui segue la sistole ventricolare, a cui segue una fase di rilasciamento. La fase diastolica è la più lunga ed è in qualche modo la più delicata, perché è vero che la partita si gioca sulla quantità di sangue che viene spinta in tutto il corpo ma la parte più sensibile e che prima si può compromettere è la fase diastolica, che crea le basi della sistole successiva. Passiamo di seguito a descrivere le singole fasi del ciclo cardiaco. Nel RIEMPIMENTO VENTRICOLARE (500 ms), la diastole è già cominciata con il rilasciamento. Perché si possa avere riempimento ventricolare è necessario che la pressione atriale deve superare la pressione ventricolare. E’ il gradiente pressorio che può determinare l’apertura della valvola atrio-ventricolare. Il riempimento o filling ha due fasi: - Fase I o Filling rapido; - Fase II o Filling lento (con aumento del 10-20 % del volume di riempimento ventricolare). Nella fase I c’è un gradiente atrio-ventricolare che determina l’apertura della valvola atrio-ventricolare con“cascata”, il sangue per caduta si porta nel ventricolo. Nella fase II, poiché dopo che si è aperta la valvola, il gradiente pressorio rapidamente si annulla, il sangue continua a passare perché il ventricolo (che si è già rilassato prima dell’apertura della valvola) crea una specie di risucchio, cioè un qualcosa per cui il sangue ha ancora motivo di passare dall’atrio al ventricolo. Quando l’atrio ha fatto aprire con la sua pressione la valvola e si è svuotato di sangue, si contrae e abbiamo un ulteriore incremento ventricolare (che porta la curva a fare un bel balzo). L’atrio, all’occorrenza, assume ruolo di protagonista e anche importante perché dà un contributo significativo nel riempimento e quindi all’efficienza della sistole. Una volta che il ventricolo si è riempito, ha bisogno di “raccogliersi” per “raccogliere le proprie forze” per poi svuotarsi contraendosi (infatti si ha un marcato incremento pressorio in un tempo brevissimo). Nella fase di raccoglimento il volume non cambia perché abbiamo tutte e due le valvole chiuse.C’è un piccolo aumento della pressione ventricolare e abbiamo la cosiddetta pressione tele diastolica ventricolare che è anch’essa un marker, perché ha un range di normalità: quando sale oltre questo range (è difficile che cada al di sotto del range) è un indice di allarme. Improvvisamente la pressione sale però il volume rimane uguale, perché la pressione all’interno del ventricolo è salita ma le due valvole sono chiuse (la valvola atrio-ventricolare si è chiusa alla fine della sistole atriale, la valvola aortica si è chiusa prima ed è ancora chiusa). In questa fase la pressione aumenta e viene detta sistole ventricolare isovolumetrica (50 ms). A questo punto la pressione ventricolare sinistra ha raggiunto la pressione aortica, dopo di che l’ha superata e ha determinato l’apertura della valvola. Quindi comincia l’eiezione ventricolare (300 ms), la pressione ventricolare continua a salire e la curva del volume rapidissimamente cade giù. Nel ventricolo, una volta che ha dato il massimo, la pressione comincia a scendere. Succede che di nuovo eguaglia la pressione aortica e a questo punto scende al di sotto della pressione aortica e l’eiezione è finita. Abbiamo una fase in cui il volume rimane fisso mentre la pressione cade tanto bruscamente quanto era risalita (più o meno). Significa che in questa fase noi abbiamo avuto la massima funzione del calcio, e di tutte le proteine (actina, miosina, che partecipano al processo della contrazione) e questi stessi attori che si sono organizzati per sviluppare questo grande lavoro si riposano e riposandosi fanno in modo che la pressione scenda fino al punto da incrociare la pressione atriale sinistra e finire ad un valore più basso. Durante questa fase la caduta della pressione avviene senza modificazione di volume, e pertanto viene detta diastole isovolumetrica (80 ms). Tanto il riempimento ventricolare quanto l’eiezione ventricolare sono condizionati da un gradiente pressorio. Nel caso della diastole è tra atrio sinistro e ventricolo sinistro, nel caso della sistole (eiezione) è tra ventricolo sinistro e aorta. Ecco un semplice grafico che riassume tutto quello che ci siamo detti fin qui. Curva pressione-volume: Ecco la curva pressione volume che ci dà l’idea. Premessa - Costruire un grafico pressione/volume prima dell’eco era complicato perché significava mettere un catetere nel ventricolo sinistro che presentava un sensore di pressione in punta. Noi non possiamo misurare la pressione trasmessa attraverso la colonna liquida che sta nel catetere perché la punta del catetere sta nel ventricolo sinistro ma il trasduttore sta nel braccio, se siamo entrati nell’omerale, o all’altezza del polso, se siamo entrati nella radiale, o nell’inguine se siamo entrati nella femorale, quindi ci può stare anche qualche metro di onda di trasmissione per cui quel valore non è proprio precisissimo. Se invece il trasduttore, che misura la pressione, è sulla punta del catetere, quando entro nel ventricolo sinistro si misura la pressione esattamente in quel distretto. Il problema era il volume che si misurava tramite iniezione di mezzo di contrasto nel ventricolo, misurare la pressione durante tutto un ciclo e dopo prendere i valori di pressione e quelli di volume e riportarli su un sistema. Si poteva fare solo per raccogliere evidenze scientifiche, non per un singolo paziente. Questo si è reso possibile con dei cateteri a conduttanza che hanno un sistema di sensori che possono essere posti all’interno del ventricolo, misurano la pressione e il volume attraverso un loro software e lo restituiscono su carta stampata o dall’eco, perché dall’eco i volumi sono facilmente misurabili, basta mettere un catetere nel ventricolo. A noi non interessa misurare la curva pressione/volume, a noi interessa sapere che esiste e che attraverso di essa possiamo capire esattamente come identificare il cuore mal funzionante. Nella fase di riempimento per minimi incrementi di pressione avremo grandi incrementi di volume (cioè nella fase diastolica è il volume che varia in maniera cospicua, mentre la pressione è bassa perché il ventricolo è sceso sotto il livello atriale per riempirsi). Alla fine di questa linea (punto B) abbiamo il volume tele diastolico e la valvola mitrale si è chiusa. Dopo c’è la contrazione isovolumica fino all’apertura della valvola aortica e quindi da B a C aumenta solo la pressione, il volume rimane uguale. Dopo di che, una volta aperta la valvola (e quindi è iniziata l’eiezione), la pressione sale e il volume diminuisce (C-D). Al punto D c’è il volume tele sistolico, la chiusura della valvola aortica e il rilassamento isovolumetrico con ritorno ad A e ripresa del ciclo. Questa curva può essere più stretta, più larga, più bassa, più alta. Se la curva è spostata a destra e in alto, mi sono giocato la fase diastolica e ho aumentato il lavoro del cuore perché gli ho dato più volume da cacciare e l’ho costretto a lavorare sia in diastole che in sistole a regime pressorio più alto. Se è spostata più in basso e più a destra, mantenendo la stessa pressione interna, il cuore lavora a bassa pressione e con una normale volumetria, quindi mi dà il massimo della performance con il minimo aumento di pressione. La legge di Maestrini-Starling: Maestrini ha avuto l’intuizione, Starling ha fatto delle osservazioni di Maestrini una legge. Maestrini aveva osservato che c’è una relazione tra la lunghezza del muscolo e la tensione sviluppata dalle fibre cardiache. C’è un rapporto lineare tra entità dell’allungamento e forza contrattile esercitata. Maestrini si espresse dicendo: “Il cuore quando venga sottoposto ad una tensione maggiore del normale,mediante un peso, è capace di fornire un lavoro meccanico” (l’oscillazione della fibra che si contraeva e si allungava attraverso un sistema che prevedeva un rotolo che girava con una penna che scriveva in relazione alle variazioni del peso). Starling dice che all’allungamento della fibra muscolare c’è un aumento della contrattilità e che c’è un limite oltre il quale questo allungamento è fine a se stesso, cioè non si determina un aumento della forza e della velocità di accorciamento. Nelle fibre cardiache, il sarcomero più si allunga meglio si accorcia, cioè ci mette più forza e si accorcia ad un massimo consentito. Però man mano che ci allontaniamo, quando eccediamo un certo valore, a quel punto la legge non vale più, cioè la fibra si accorcia sempre di meno o addiritturaresta allungata. La legge di Laplace: Un’altra legge fisica molto importante nel determinare la meccanica cardiaca è quella di Laplace. Lo stress che pressione e volume esercitano sulla parete ventricolare è legato ad una serie di cose, in particolare alla quarta potenza del raggio. Ciò significa che man mano che aumentiamo le dimensioni interne della cavità ventricolare aumenta lo stress della parete. Concettualmente un incremento dello stress di parete non è qualcosa di positivo. Il wall stress diventa un determinante fondamentale dell’adattamento o meno del ventricolo al sovraccarico di volume o di pressione (come nello scompenso o nell’ipertensionearteriosa). La formula mostrata in basso deriva dalla legge di Laplace. L’aumento della pressione nel ventricolo sinistro, per via della stenosi aortica, è compensato dall’ipertrofia della parete, che diminuisce sia il raggio della cavità (radius) ed aumenta lo spessore della parete (wallthickness). In questo modo, diminusce il wall stress. MODALITA’ DI REGOLAZIONE DEL CICLO CARDIACO: IL SNA Ci sono fibre di lavoro e fibre che trasmettono lo stimolo elettrico. Il cuore non è un organo isolato e fine a se stesso, e dobbiamo considerare il sistema neurovegetativo che regola il ciclo cardiaco. Il concetto più banale è che il sistema adrenergico fa aumentare il lavoro del cuore (aumenta la frequenza, la pressione, la resistenza), il sistema parasimpatico fa il contrario (abbiamo una serie di farmaci come i beta-bloccanti, atropina, che possono enfatizzare o bloccare un determinato sistema). Il sistema neurovegetativo contribuisce anche a mettere il cuore in comunicazione con una serie di altri organi (su tutti rene e cervello ma anche la vascolatura periferica e il muscolo). Lo stimolo vagale fa diventare le cellule pacemaker ancora più lente, fa diminuire la frequenza di generazione dello stimolo e fa diminuire la velocità di conduzione. L’atropina che è il vagolitico per eccellenza, viene iniettata quando il paziente ha una sindrome vagale (quella più comune è la sindrome vasovagale, quella del prelievo di sangue, basta mezza fiala di atropina sottocute). Il parasimpatico, agisce sulle cellule pacemaker nel seguente modo. Innanzitutto la cellula si iperpolarizza in maggiore, con misura un prolungamento della fase di refrattarietà e con una diminuzione della frequenza cardiaca. Tale fenomeno è dovuto all’azione dell’acetilcolina che si lega al recettore muscarinico M 2 , che è accoppiato con una proteina G i . L’attivazione di questo recettore, tramite le subunità βγ della proteina G, determina l’apertura di un canale del potassio, che causa un’ulteriore ripolarizzazione della membrana della cellula. Tale effetto viene detto cronotropo negativo. Ma l’acetilcolina ha anche un effetto inotropo negativo, modulando la cinetica del canale del calcio di tipo L. L’attivazione del recettore blocca l’attività dell’adenilatociclasi, con una riduzione dei livelli di cAMP e ridotta fosforilazione della PKA. In questo modo la PKA non può fosforilare i canali del calcio di tipo L, che rimangono chiusi, con una riduzione delle correnti entranti di calcio e successiva diminuzione della forza di contrazione cardiaca. Per quanto riguarda l’attività del simpatico sulle cellule pace-maker, questi tende a velocizzarne la depolarizzazione, con un aumento della frequenza cardiaca. Tale effetto cronotropo positivo è mediato dai recettori β 1 , accoppiati a G s . L’attivazione del recettore determina un aumento della concentrazione intracellulare del cAMP con aumento dell’attività della PKA. La PKA fosforila i canali L del calcio, determinando un aumento delle correnti entranti di calcio, con aumento sia della frequenza cardiaca che della forza di contrazione (effetto inotropo positivo). Oltre al SNA, l’attività cardiaca viene modulata anche da tutta un’altra serie di sostanze, come la vasopressina e l’aldosterone. IL CIRCOLO CORONARICO In condizioni basali il cuore consuma circa 6,5-10 mL/min/100 gr. di tessuto. Tale dispendio serve: - 3-5 % per l’attività elettrica; - 20 % per il mantenimento dell’integrità cellulare; - 72-75 % per l’attività contrattile. Il circolo coronarico è fatto di vasi di conduttanza e di microcircolo. I vasi di conduttanza sono la coronaria sinistra, il tronco comune, la circonflessa e la discendente anteriore, la coronaria destra. L’aorta è il gasometro, le coronarie sono i tubi che arrivano sottocasa, i tubi che salgono e arrivano agli appartamenti formano il microcircolo coronarico. Quando parliamo di coronaropatie, parliamo sempre di quello che succede nei vasi di conduttanza, perché è lì che si realizza una riduzione del flusso di sangue, ma gli effetti veri si vedono a livello microcircolare. Chi è che non funziona? Non veicola l’ossigeno? Il microcircolo. E’ l’integrità del microcircolo la vera determinante del normale metabolismo miocardico. A livello miocardico per l’elevata estrazione di ossigeno (circa il 70 %), l’unico meccanismo di compenso in caso di aumentato fabbisogno di ossigeno è rappresentato da un proporzionale aumento del flusso coronarico, determinato da una vasodilatazione del distretto coronarico arteriolare (vasi di resistenza). La capacità massima di vasodilatazione secondaria a uno stimolo è soggetta ad un sistema di autoregolazione definito riserva di flusso coronarico. Che significa sistema di autoregolazione? In qualunque sistema, il flusso è in funzione della pressione di spinta. Siccome il cuore è un organo piccolo, con un letto vascolare ridotto, di tipo terminale perché i vasi di conduttanza vanno a finire in qualcosa che non vediamo (il microcircolo è concettuale, noi non lo vediamo), il problema è che il microcircolo non può essere così strettamente dipendente dalla pressione. Che significa? Significa che il flusso coronarico non può variare in maniera significativa, come in altri distretti a seconda della pressione, perché sarebbe ingestibile. Allora per un range di pressione compreso tra 80/120-130 mmHg, il flusso coronarico è costante (sistema di autoregolazione). Succede che il cuore, nel momento in cui gli si chiede un lavoro maggiore, deve avere una maggiore quantità di ossigeno. Questo è possibile aumentando il flusso tramite vasodilatazione: dobbiamo far cadere le resistenze coronariche e inevitabilmente ci sarà aumento del flusso. Il soggetto allenato riesce ad arrivare al traguardo perché il suo circolo coronarico si è adeguato perfettamente alle richieste dell’atleta. La riserva coronarica è quel quid che consente una adeguata performance nel momento in cui questa si rende necessaria. I fattori che regolano il circolo coronarico sono: - ANATOMICI (origine dei seni di Valsalva, spessore parietale ventricolo sinistro, circoli collaterali. Se ho un vaso chiuso, con un circolo collaterale chiuso, vi è una brusca riduzione della quantità di sangue che arriva); - MECCANICI (gittata cardiaca, resistenze vascolari, compressione sistolica, riflesso miogeno, viscosità ematica); - NEUROGENI (alfa-recettori, beta2-recettori, azione vagale); - METABOLICI (pO 2 , pH, K+, adenosina, prostaglandine). I vasi coronarici si dividono in: - VASI DI CONDUTTANZA (grossi rami epicardici e loro diramazioni); - VASI DI RESISTENZA (rami intramiocardici e arteriole). Il microcircolo è l’insieme del capillari in cui si realizza lo scambio di gas e metaboliti che fornisce energia almuscolo cardiaco. Le resistenze coronariche sono regolate da fattori estrinseci (azione compressiva del miocardio ventricolare) e fattori intrinseci (di natura neuro-ormonale, miogena e metabolica). L’aumentata richiesta metabolica del miocardio determina idrolisi di ATP e conseguente liberazione di adenosina nell’interstizio. L’adenosina induce vasodilatazione perché antagonizza l’ingresso di Ca2+ all’interno delle cellule muscolari lisce soprattutto a livello dei vasi di resistenza, determinando un aumento del flusso coronarico proporzionale all’aumento delle richieste metaboliche. L’adenosina non è la sola implicata nel processo (il sistema degli eicosanoidi, l’attività NO sintetasica) ma è verosimilmente la principale. L’adenosina è un potente vasodilatatore ma è di rapida azione (si fa a livello intracoronarico o intravenoso a dosi elevate finché arriva, fa il servizio e se ne va). Il flusso coronarico si attua soprattutto in diastole perché in sistole i rami intramurali sono virtualmente occlusi dalla contrazione ventricolare. Quindi la tachicardia predispone allo sviluppo di ischemia perché accorcia la diastole (per questo uno dei cardini della terapia cardio-vascolare è il beta-bloccante. E’importante avere una diastole efficace per avere il tempo di riempire il ventricolo e di perfonderlo anche). Gli strati sub endocardici sono i più esposti all’ischemia, soprattutto perché maggiormente esposti alla pressione diastolica endocavitaria. Quando il sangue tenderebbe a cadere nel ventricolo sinistro è il momento in cui si riempiono gli osti coronarici. Quando vogliamo fare una misurazione del calibro della coronaria, la facciamo in fase tele diastolica, cioè quando c’è il massimo riempimento e quindi il massimo della dilatazione; in quel momento io so esattamente qual è la dimensione del vaso: se lo prendessi in telesistole, lo sottostimerei. Per essere sicuro che non ci sia una componente vasospastica, posso fare una iniezione intracoronarica di adenosina e nitroglicerina e avrò realmente la misura del diametro di quel vaso (se misurassi il calibro coronarico in tele sistole potrei finire con il mettere un palloncino o uno stent inadeguato). Anche le arterie coronariche sono innervate dal SNA. La stimolazione del ganglio stellato (ortosimpatico) determina vasodilatazione (mediata dai recettori beta), aumento della contrattilità e della frequenza cardiaca. Il blocco recettoriale beta induce la comparsa di effetti alfa-mediati (vasocostrizione). CAPITOLO II: ESAME OBIETTIVO DEL SISTEMA CARDIOVASCOLARE Un attento esame obiettivo rappresenta un metodo economico per valutare il sistema cardiovascolare. Innanzitutto, si deve valutare l'aspetto fisico del paziente: si può osservare un atteggiamento affaticato, dovuto a cronica riduzione della portata cardiaca, oppure un aumento della frequenza respiratoria che denuncia congestione del circolo venoso polmonare; la cianosi centrale, spesso associata a dita a bacchetta di tamburo, indica uno shunt cardiaco o extra cardiaco destra-sinistra e uno stato di inadeguata ossigenazione del sangue nel circolo polmonare; la cianosi alle estremità distali, con cute fredda e aumento della sudorazione, è invece segno di vasocostrizione nei pazienti con grave insufficienza cardiaca. Alcuni segni non strettamente inerenti al sistema cardiovascolare possono essere spesso di estrema importanza. La pressione arteriosa deve sempre essere misurata in entrambi gli arti superiori, sia in decubito supino sia in posizione eretta: la frequenza cardiaca va misurata per 30 secondi. L'ipotensione ortostatica e la tachicardia possono indicare una riduzione del volume ematico, mentre il riscontro di tachicardia a riposo può rappresentare un segno di grave insufficienza cardiaca o ipovolemia. POLSO ARTERIOSO L'onda normale del polso aortico centrale è caratterizzata da una fase di ascesa relativamente rapida sino a un picco di forma arrotondata; sulla branca ascendente è osservabile l’incisura anacrota, corrispondente al momento di massimo flusso aortico, poco prima che sia raggiunta la pressione massima. La branca discendente, assai meno ripida, è interrotta da una netta deflessione verso il basso, sincrona con la chiusura della valvola aortica, detta incisura dicrota. Il polso carotideo è generalmente meglio esaminatile con il muscolo sternocleidomastoideo rilasciato e con il capo del paziente leggermente ruotato verso l'esaminatore. Per esaminare il polso arterioso brachiale, l'esaminatore può sostenere il gomito rilassato del paziente con la mano destra mentre comprime l'arteria con il pollice. La tecnica che viene normalmente usata per la palpazione di un polso arterioso prevede la compressione del vaso col pollice o con le altre dita sino a che sia percepito il massimo della pulsazione; l'esaminatore può applicare gradi variabili di pressione, concentrandosi sulle varie fasi dell'onda sfigmica. Questo metodo di palpazione, detto tripartito, è particolarmente utile per riconoscere la fase ascendente, il picco sistolico e la curva diastolica del polso arterioso. Nella maggior parte delle persone normali l'onda dicrota non è apprezzabile. Il polso piccolo e debole (pulsusparvus) è frequentemente osservabile nelle condizioni di ridotta gittata sistolica del ventricolo sinistro, bassa pressione differenziale e aumentate resistenze periferiche. Il polso ipocinetico può essere dovuto a ipovolemia, insufficienza ventricolare sinistra, a malattie pericardiche restrittive o a stenosi della valvola mitrale. Nella stenosi della valvola aortica si osserva un polso periferico ritardato (pulsustardus), risultante dall'ostacolo meccanico all'eiezione del ventricolo sinistro. Il polso ampio e scoccante o ipercinetico è in genere associato ad aumentata gittata sistolica ventricolare sinistra, ampia pressione differenziale e diminuzione delle resistenze vascolari periferiche, come avviene nel blocco atrio ventricolare completo, nell’ipercinesia circolatoria da ansia, anemia, esercizio fisico o febbre e nei pazienti con circolazione arteriosa a rapido svuotamento. Nell'insufficienza aortica il polso ad ascesa rapida, celere, è il risultato dell'aumento del volume sistolico ventricolare sinistro e del conseguente aumento della velocità di eiezione del ventricolo sinistro. Il polso bifido o bisferiens, che è caratterizzato da due picchi sistolici, è un reperto tipico del insufficienza aortica e della miocardiopatia ipertrofica. Il polso dicroto presenta due onde palpabili, una durante la sistole e una durante la diastole. È facilmente rilevabile in pazienti con volume sistolico molto ridotto, particolarmente in quelli con miocardiopatia dilatativa. Con il termine polso alternante si definisce una situazione caratterizzata da una ritmica variabilità d'ampiezza dell'onda pressoria, malgrado un ritmo regolare; è la conseguenza di un'alternanza della forza di contrazione ventricolare sinistra, e in genere e sintomo di grave deficit della funzione ventricolare sinistra ed è di frequente osservazione nei pazienti che presentano anche un terzo tono cardiaco evidente. Il polso alternante può anche essere osservato durante o dopo tachicardia parossistica, o nei primi battiti sinusali successivi a un'extrasistole anche in pazienti senza cardiopatie. Il polso bigemino è anch'esso un'alternanza regolare di ampiezza delle onde pressorie, ma è causato da un bigeminismo extra sistolico. Il polso paradosso di Kussmaul è un'accentuazione della fisiologica riduzione dell'ampiezza del polso arterioso osservabile durante l'ispirazione; nei pazienti con tamponamento cardiaco, ostruzione della via aeree od ostruzione della vena cava superiore, la diminuzione inspiratoria della pressione sistolica frequentemente supera il valore normale di 10 mmHg e il polso periferico può anche scomparire completamente. La palpazione simultanea dei polsi femorale e radiale, che normalmente sono di fatto coincidenti, è di particolare importanza per escludere la coartazione aortica, nella quale si osservano diminuzione e ritardo dei polsi agli arti inferiori. POLSO VENOSO GIUGULARE I due obiettivi principali dell'osservazione delle vene del collo nel paziente costretto a letto sono l'esame della forma dell'onda del polso venoso e la stima della pressione venosa centrale (PVC). In molti pazienti ciò si ottiene osservando la vena giugulare interna destra, che meglio si presta per entrambi gli scopi. Di solito, la massima pulsazione della vena giugulare interna è osservabile con il tronco inclinato meno di 30°. Nei pazienti con pressione venosa elevata può essere necessario elevare il tronco fino a 90°. La pulsazione della vene giugulare e osservabile con la muscolatura del collo rilassata e con una illuminazione tangente alla cute. La palpazione contemporanea dell'arteria carotide sinistra aiuta l'esaminatore a decidere quali pulsazioni siano venose e a correlare queste pulsazioni con il ciclo cardiaco. Il normale polso venoso giugulare (PVG) riflette le variazioni pressorie nell'atrio destro ed è formato da due o tre onde positive e da due onde negative. L'onda presistolica positiva a è determinata dalla distensione venosa dovuta alla contrazione atriale destra e rappresenta l'onda dominante. Onde a particolarmente ampie indicano che l'atrio destro si contrae contro un aumento delle resistenze, come in caso di stenosi della valvola tricuspide o, più comunemente, per difficoltoso riempimento ventricolare destro (ipertensione polmonare o stenosi polmonare). È possibile osservare onde a particolarmente ampie anche durante aritmie, poiché l'atrio destro si contrae contro una tricuspide chiusa dalla sistole ventricolare. Queste onde a "cannone" possono essere regolari o irregolari. L'onda a è assente nei pazienti con fibrillazione atriale, mentre vi è un aumentato ritardo del polso carotideo rispetto all'onda a in caso di blocco atrio-ventricolare di primo grado. L'onda c, spesso osservabile nel PVG, è un'onda prodotta dalla protrusione della valvola tricuspide all'interno dell'atrio durante la sistole isometrica del ventricolo destro e dall'impatto sulla vena giugulare dell'adiacente polso arterioso carotideo. La discesa x è dovuta alla combinazione del rilasciamento atriale e del movimento verso il basso della valvola tricuspide durante la sistole ventricolare. Nei pazienti con pericardite costrittiva può essere osservabile una particolare evidenza della deflessione x durante la sistole, mentre quest'onta si presenta ridotta nella dilatazione ventricolare destra e spesso è positiva nella insufficienza tricuspidale. L'onda positiva v, che compare nella fase tardiva della sistole, è dovuta al progressivo aumento del volume del sangue nelle vene cave e nell'atrio destro durante la sistole ventricolare, quando la valvola tricuspide è chiusa. Se è presente una modesta insufficienza tricuspidale, l'onda v diviene più prominente e, nel caso in cui l'insufficienza tricuspidale sia marcata, la prominenza dell'onda v e l'obliterazione della discesa x determinano una singola e ampia onda positiva sistolica (ventricolizzazione). Dopo che è stato raggiunto il picco dell'onda v, la pressione atriale destra diminuisce a causa della diminuita sporgenza dei lembi valvolari nell'atrio destro e, con il diminuire della pressione ventricolare, si apre la valvola tricuspide. Questa discesa è nota come discesa y del PVG, ed è prodotta dall'apertura della valvola tricuspide e dal conseguente rapido flusso di sangue nel ventricolo destro. In caso di una grave insufficienza della tricuspide, si osserva una rapida e profonda discesa y nella fase precoce della diastole. Il polso venoso caratterizzato da un'incisura y profonda, col rapido ritorno alla linea di base, è osservabile nei pazienti con pericardite costrittiva o con grave insufficienza del cuore destro e pressione venosa elevata. Una discesa dell'onda y lenta suggerisce un'ostruzione al riempimento ventricolare destro, che può essere presente nella stenosi tricuspidale e nel mixoma dell'atrio destro. Per ottenere una stima accurata della pressione venosa centrale, il vaso più utilizzato è la giugulare interna destra, usando l'angolo sternale come punto di riferimento, poiché nella maggior parte dei pazienti il centro dell'atrio destro si trova circa 5 cm sotto l'angolo sternale. Il paziente viene esaminato con un'elevazione del tronco ottimale per visualizzare le pulsazioni venose. La distanza verticale tra la sommità della colonna venosa oscillante e l'angolo sternale è il parametro che viene misurato e in genere e interiore agli 8 cm. La causa più comune di aumento della PVC è un'elevazione della pressione diastolica ventricolare destra. Nei pazienti con sospetto di insufficienza ventricolare destra e PVC normale a riposo può essere utile il testo delle reflusso e epato-giugulare: con la palma della mano di posta sopra l'addome del paziente si applica una pressione continua per 10 secondi o più; normalmente la pressione venoso giugulare non viene significativamente alterata, ma se viene un'alterazione nella funzione del cuore destro sarà possibile osservare un incremento della pressione venosa. Tale test è positivo se l'incremento della PVG durante una ferma compressione esercitata per 10 secondi o più al centro dell'addome, è seguita da una rapida caduta della pressione di 4 cm dopo la cessazione della compressione. La causa più comune di positività per questo test è lo scompenso cardiaco destro, secondario ad un aumento della pressione di riempimento ventricolare sinistro. Il segno di Kussmaul, ovvero un aumento invece che una diminuzione della PVC durante l'inspirazione, è causato nella maggior parte dei casi da un'insufficienza cardiaca destra; è un reperto molto comune nei pazienti con pericardite costrittiva o con infarto ventricolare destro. PALPAZIONE DEL PRECORDIO La valutazione palpatoria di sede, ampiezza, durata e direzione dell’itto cardiaco è in genere ottimale sia effettuata con i polpastrelli. Il normale itto dell'apice ventricolare sinistro è rilevabile sulla linea emiclaveare sinistra, o generalmente all'interno di questa, nel quarto o quinto spazio intercostale. L'ipertrofia del ventricolo sinistro determina un aumento di ampiezza, di durata e dimensioni del normale itto ventricolare. Questo potrà anche essere rilevabile in posizione anomala, più laterale o caudale nel sesto o settimo spazio intercostale. L'ipertrofia ventricolare destra determina un itto prolungato in regione parasternale inferiore sinistra, sincrono con l’itto apicale del ventricolo sinistro. Pulsazioni precordiali anomale possono essere rilevabili durante la sistole in pazienti con dissinergie ventricolare sinistre secondarie a cardiopatia ischemica o a miocardiopatie diffuse. Nei pazienti con grave insufficienza mitralica è possibile osservare un itto parasternale sinistro dovuto allo spostamento anteriore del ventricolo destro determinato dall'espansione dell'atrio sinistro allargato. La pulsazione dell'arteria polmonare è spesso visibile e palpabile nel secondo spazio intercostale sinistro. Questa pulsazione denota in genere ipertensione o iperflusso polmonare. I fremiti sono vibrazioni palpabili a bassa frequenza associate ai soffi cardiaci. Il soffio cupo, sistolico, dell'insufficienza mitralica può essere palpabile all'apice cardiaco. Il fremito della stenosi aortica si irradia verso la parte destra del collo, mentre quello della stenosi polmonare si irradia più frequentemente verso la parte sinistra. Il fremito dovuto a un difetto interventricolare è in genere apprezzabile nel terzo e quarto spazio intercostale, nei pressi del margine sternale sinistro. La percussione dovrebbe essere eseguita in ogni paziente col fine di identificare la posizione, normale o anomala, di cuore, stomaco e fegato. AUSCULTAZIONE CARDIACA Al fine di ottenere il massimo delle informazioni possibili dall'auscultazione cardiaca, il medico deve tenere ben presente i seguenti principi: - essa dovrebbe essere eseguita in un ambiente tranquillo; - per rilevare un tono un soffio di bassa intensità si deve focalizzare l'attenzione sulla particolare fase del ciclo cardiaco durante la quale è possibile rilevarne la presenza; - L'accurata valutazione temporale di un tono o di un soffio cardiaco deve necessariamente passare attraverso la determinazione della sua correlazione con gli altri eventi del ciclo cardiaco. TONI CARDIACI I toni cardiaci sono il risultato di vibrazioni determinate dalla brusca accelerazione o decelerazione del sangue all'interno del sistema cardiovascolare. Il primo e il secondo tono cardiaco sono prodotti principalmente dalla chiusura delle valvole atrioventricolari e semilunari. L'intensità del primo tono cardiaco (T 1 ) è influenzata da: - posizione dei lembi delle valvole all'inizio della sistole ventricolare; - velocità di ascesa dell'onda pressoria ventricolare sinistra; - presenza o assenza di alterazioni strutturali della valvola mitrale; - quantità di tessuto, aria o liquido tra il cuore e lo stetoscopio. T 1 aumenta di intensità se la diastole è accorciata, come avviene nella tachicardia, se il flusso atrioventricolare è aumentato a causa di una gittata cardiaca elevata o se è protratto a causa di stenosi della mitrale, oppure se le contrazioni atriali sono rapidamente seguite da quelle ventricolari a una distanza di tempo insolitamente breve. Un T 1 aumentato in presenza di stenosi mitralica in genere significa che la valvola è mobile e rimane aperta all'inizio della contrazione isometrica a causa dell'elevata pressione atriale sinistra. La riduzione di intensità del primo tono può essere dovuta a diminuita conduzione di suoni attraverso le strutture toraciche, al lenta ascesa dell'onda pressoria ventricolare sinistra, a un lungo intervallo PR e a un'imperfetta chiusura secondaria a diminuzione della struttura valvolare (insufficienza mitralica). Il primo tono può essere ridotto di intensità anche nella stenosi mitralica. Lo sdoppiamento del primo tono in due componenti ad alta frequenza rappresenta un fenomeno normale. La prima componente è generalmente attribuita alla chiusura della valvola mitrale, mentre la seconda a quella della valvola tricuspide. L'ampliamento della separazione delle due componenti è spesso dovuto ad un blocco di branca destro completo. Sdoppiamento del secondo tono cardiaco: lo sdoppiamento del secondo tono cardiaco (T 2 ) in due componenti distinte, aortica e polmonare, è normalmente rilevabile durante l'inspirazione, quando il maggiore ritorno venoso aumenta il volume di sangue contenuto nel ventricolo destro e di conseguenza il periodo di eiezione, ritardando la chiusura della polmonare. Uno sdoppiamento del secondo tono che persiste durante l'espirazione è in genere anomalo quando il paziente è in posizione eretta. Questo sdoppiamento può essere dovuto a numerose condizioni: ritardo di attivazione del ventricolo destro (blocco di branca destro), extrasistoli ventricolari sinistre, presenza di pacemaker nel ventricolo sinistro, prolungamento della contrazione ventricolare destra a causa di un aumento del carico pressorio ventricolare (embolia polmonare o stenosi della polmonare), ritardo della chiusura della valvola polmonare a causa di un sovraccarico del volume ventricolare destro associato a insufficienza ventricolare destra o diminuita impendenza del letto vascolare polmonare e prolungamento dell'intervallo di distensione (difetto d'effetto interatriale). Nell'ipertensione polmonare, la componente polmonare è aumentata di intensità e lo sdoppiamento può essere diminuito, normale o accentuato a seconda della causa dell'ipertensione polmonare, delle resistenze vascolari polmonari e della presenza o assenza di scompenso ventricolare destro. La chiusura precoce della valvola aortica (insufficienza mitralica, difetto del setto interventricolare) può anch'essa determinare uno sdoppiamento del secondo tono persistente anche in espirazione. Nei pazienti con ampia comunicazione interatriale, il contributo alla riempimento atriale destro determinato dallo shunt sinistro-destro e dal ritorno venoso sistemico varia in maniera reciproca durante le fasi del ciclo respiratorio, tanto che l’afflusso all'atrio rimane quasi costante. Per questa ragione, sia il volume sia la durata dell'eiezione ventricolare non aumentano in modo significativo durante l'inspirazione; si può perciò rilevare solo una modesta variazione inspiratoria dello sdoppiamento. Questo fenomeno, detto anche sdoppiamento fisso del secondo tono, possiede un valore diagnostico molto importante. I ritardi di chiusura della valvola aortica, che determinano la comparsa della componente polmonare prima di quella aortica, sono causa del cosiddetto sdoppiamento paradosso del secondo tono. In queste situazioni l'intervallo tra le due componenti è massimo durante l'espirazione e si riduce durante l'inspirazione a causa del normale ritardo nella chiusura della valvola polmonare. Le cause più comuni di sdoppiamento paradosso sono il blocco di branca sinistra e la ritardata eccitazione del ventricolo sinistro da parte di un battito ectopico ventricolare destro. Il prolungamento meccanico della sistole ventricolare sinistra, che determina l'inversione dello sdoppiamento, può essere causato da grave costruzione all’efflusso aortico, da ampio shunt aorta-arteria polmonare, da ipertensione sistolica, da cardiopatia ischemica o da miocardiopatia primitiva con insufficienza ventricolare sinistra. La componente polmonare è normalmente meno intensa di quella aortica nel secondo spazio intercostale, qualora la componente polmonare sia più intensa di quella aortica in quest'area, si deve pensare ad una ipertensione polmonare, tranne che nei pazienti con difetto del setto interatriale. Toni sistolici: il tono da eiezione è un rumore ad alta frequenza, udibile in fase protosistolica, immediatamente dopo il primo tono cardiaco. Il tono da eiezione è presente in caso di stenosi delle valvole semilunari, quando è presente lo schiocco di apertura delle valvole aortica e polmonare, nonché in condizioni di dilatazione dell'aorta o dell'arteria polmonare. I click non da iniezione o mesosistolici denotano spesso prolasso di uno o di entrambi i lembi della valvola mitrale. Questi click sono probabilmente dovuti alla diversa lunghezza funzionale delle corde tendinee di una o di entrambe le valvole atrioventricolari, e sono meglio udibili lungo il margine sternale inferiori sinistro e all'apice. I click sistolici sono in genere più tardivi del tono di eiezione. Toni diastolici:lo schiocco di apertura (SA) è un tono breve, proto diastolico, ad alta frequenza, in genere dovuto a stenosi di una valvola atrioventricolare (mitrale). È in genere meglio udibile al margine inferiore sinistro dello sterno e si irradia bene verso la base del cuore. L’intervallo temporale tra la componente aortica del secondo tono e lo SA è inversamente proporzionale al valore della pressione media nell’atrio sinistro. Lo SA della stenosi tricuspidale avviene più tardivamente in diastole di quello della stenosi mitralica ed è spesso mascherato. Il terzo tono cardiaco (T 3 ) è un rumore a bassa frequenza prodotto nel ventricolo, 0,14-0,16 secondi dopo la componente aortica del secondo tono, al termine del riempimento rapido. Questo tono è fisiologicamente presente nei bambini e nei pazienti con alta portata cardiaca. Nei pazienti con più di 40 anni, la presenza di un terzo tono deve far pensare a deficit della funzione ventricolare sinistra, scompenso cardiaco, insufficienza di una valvola AV o ad altre condizioni che aumentano la frequenza ed il volume telediastolico del ventricolo. Spesso T 3 scompare quando l’insufficienza cardiaca viene trattata. Nei pazienti con pericardite costrittiva è spesso ascoltabile un terzo tono precoce con una frequenza più alta (tono pericardico); la sua presenza è dovuta agli effetti costrittivi del pericardio adeso, che interrompe bruscamente il riempimento diastolico. Il quarto tono cardiaco (T 4 ) è un rumore presistolico, a bassa frequenza prodotto nel ventricolo durante il riempimento ventricolare associato ad una contrazione atriale efficace. Questo tono è assente nei pazienti con fibrillazione atriale. Questo tono compare quando la diminuita elasticità ventricolare aumenta la resistenza al riempimento ventricolare ed è perciò riscontrabile nei soggetti con ipertensione sistemica, stenosi aortica, miocardiopatia ipertrofica, ischemia miocardica e insufficienza mitralica acuta. Il quarto tono accompagna spesso i ritardi di conduzione AV, anche in assenza di malattia clinicamente evidente. L’incidenza del quarto tono aumenta all’aumentare dell’età. SOFFI CARDIACI La valutazione di un paziente con un soffio cardiaco può variare notevolmente. L'intensità di un soffio è in genere graduata da I a VI. Un soffio di intensità pari a I è così debole che può essere udito solo ponendo particolare attenzione, un soffio di tipo IV è solitamente accompagnato da un fremito, mentre un soffio di tipo VI è udibile con lo stetoscopio allontanato dalla parete toracica. La morfologia di un soffio può essere in crescendo, decrescendo, crescendo-decrescendo o a plateau. La determinazione temporale dell’inizio e fine di un soffio è dipendente dalla fase del ciclo cardiaco in cui compare e scompare la differenza di pressione sufficiente a generare la vibrazione. La localizzazione di un soffio sulla parete toracica e la sua irradiazione possono essere utili per identificare la struttura cardia da cui ha origine il soffio. Soffi olosistolici o pansistolici: Questi soffi sono generati dal passaggio del sangue tra due cavità che presentano un ampio gradiente pressorio durante la sistole, come i due ventricoli. Questo gradiente pressorio si instaura precocemente durante la contrazione ventricolare sinistra e viene mantenuto quasi fino alla fine della fase di rilassamento. Quindi, i soffi olosistolici iniziano prima dell’eiezione aortica, iniziando con T 1 e terminando dopo T 2 . I soffi olosistolici sono presenti nell’insufficienza mitralica, tricuspide e nei difetti del setto interventricolare. Il soffio dell’insufficienza tricuspidale associata ad ipertensione polmonare è olosistolico e frequentemente aumenta di intensità durante l’inspirazione. Comunque, non tutti i pazienti con insufficienza mitralica, tricuspidale o difetti del setto interventricolare presentano soffi olosistolici. Soffi mesosistolici: I soffi sistolici da eiezione o mesosistolici, che spesso hanno una morfologia tipo crescendo-decrescendo, sono generati da sangue che attraversa i tratti di efflusso aortico e polmonare. Il soffio ha inizio poco dopo T 1 , quando la pressione nel ventricolo aumenta fino ad aprire le valvole semilunari. Con l'aumentare della velocità di eiezione si ha un aumento del soffio, mentre con il diminuire dell'eiezione esso diminuisce. Il soffio termina prima che la pressione ventricolare diminuisca al punto da permettere la chiusura dei lembi valvolari aortici o polmonari. La maggior parte dei soffi funzionali benigni è mesosistolica e origina dal tratto di efflusso polmonare. L'ostruzione valvolare o sottovalvolare in uno dei ventricoli può causare la comparsa di un soffio mesosistolico la cui intensità è dipendente dal flusso. Il soffio secondario a stenosi aortica rappresenta il prototipo dei soffi mesosistolici sinistri. La sede e l'irradiazione di questo soffio sembrano essere influenzate dalla direzione del getto ad alta velocità contro la parete della radice aortica. Nella stenosi valvolare aortica il soffio è di regola massimo nel secondo spazio intercostale destro, con irradiazione al collo. L'età del paziente e l'area di massima intensità all'auscultazione aiutano a determinare il significato dei soffi mesosistolici. In un giovane adulto con parete toracica sottile e alta velocità del flusso ematico, un soffio mesosistolico debole o moderato, udibile presso l'area polmonare, è in genere privo di significato clinico. Mentre un soffio leggermente più forte a livello dell'area aortica può essere un segno di stenosi aortica congenita; nei pazienti anziani, al contrario, i soffi di origine polmonare sono rari, mentre sono frequenti quelli aortici, che possono essere dovuti a dilatazione del vaso, a stenosi aortica di grado importante o a deformità non stenosanti della valvola. Soffi protosistolici: Questi soffi iniziano con il primo tono cardiaco e terminano a metà della sistole. Negli ampi difetti del setto interventricolare con ipertensione polmonare, lo shunt sinistra-destra verso la fine sistole può essere assente o minimo: per questo motivo il soffio è solo protosistolico. Un soffio simile può essere presente in caso di difetti molto lievi della muscolatura del setto interventricolare, poiché lo shunt si riduce con il progredire della sistole. Un soffio protosistolico è anche caratteristico dell'insufficienza tricuspidale senza ipertensione polmonare. Soffi telesistolici: Questi soffi sono in genere ad alta frequenza, deboli o di moderata intensità, apicali. Iniziano in netto ritardo rispetto all’eiezione sistolica, senza mascherare i toni cardiaci. Sono da correlare a disfunzioni dei muscoli papillari secondarie a infarto o ischemia oppure a dilatazione ventricolare sinistra. Questi soffi possono comparire durante attacchi anginosi, ma sono molto frequenti nei pazienti con infarto del miocardio o con miocardiopatia diffusa. I soffi telesistolici udibili dopo click mesosistolici sono associati ad insufficienza mitralica tardiva causata da prolasso della valvola nell'atrio sinistro. Soffi protodiastolici: I soffi protodiastolici iniziano con il secondo tono, o immediatamente dopo, in concomitanza con la riduzione della pressione ventricolare al di sotto dei valori di pressione aortica o polmonare. I soffi ad alta frequenza dell’insufficienza aortica e dell'insufficienza polmonare sono in genere in decrescendo, poiché vi è una progressiva riduzione del rigurgito. I soffi deboli, ad alta frequenza, dell'insufficienza aortica sono difficili da udire, a patto che non vengano ricercati appositamente tramite compressione del diaframma dello stetoscopio sul tratto medio della linea margine sternale sinistra, con il paziente in posizione semiassisa. Soffi mesodiastolici: Questi soffi hanno generalmente origine dalle valvole atrioventricolari, insorgono durante la fase iniziale della riempimento ventricolare e sono dovuti ad una sproporzione tra dimensione dell'orifizio valvolare e quantità del flusso ematico. Questi soffi possono presentare un'intensità elevata. Il soffio può essere ridotto o anche assente malgrado una grave stenosi se vi è una cospicua riduzione della portata cardiaca. Se la stenosi è marcata il soffio è in genere prolungato; la gravità della stenosi è maggiormente correlata con la durata del soffio. Il soffio mesodiastolico a bassa frequenza della stenosi mitralica segue caratteristicamente lo schiocco di apertura. Frequentemente il soffio della stenosi mitralica è presente solo all'apice ventricolare sinistro e può venire aumentato di intensità da un modesto esercizio fisico in posizione supina e dall'inalazione di nitrito di amile. Nella stenosi tricuspidale il soffio mesodiastolico è in genere localizzato in un'area relativamente limitata lungo il margine sternale sinistro e può aumentare di intensità durante l'inspirazione. Soffi mesodiastolici possono essere generati dalla valvola mitrale in presenza di difetti del setto interventricolare, nella pervietà del dotto arterioso e nell'insufficienza mitralica, o possono trarre origine dalla valvola tricuspide nei difetti del setto interatriale o nell'insufficienza tricuspidale. Questi soffi sono da mettere in relazione con il flusso molto rapido che attraversa una valvola atrioventricolare. Nell'insufficienza aortica acuta la pressione diastolica ventricolare sinistra può superare quella dell'atrio sinistro, determinando quindi un soffio mesodiastolico dovuto a "insufficienza mitralica diastolica". Nella grave insufficienza aortica cronica è frequentemente presente un soffio che può essere meso o telediastolico (soffio di Austin-Flint). Sembra che esso origini dal lembo anteriore della valvola mitrale quando il sangue entra nel ventricolo sinistro simultaneamente dall'atrio sinistro e dalla radice aortica. Soffi telediastolici: I soffi telediastolici o presistolici iniziano durante il periodo di riempimento ventricolare che segue la contrazione atriale e quindi sono rilevabili solo durante il ritmo sinusale. Sono dovuti a stenosi di una valvola atrioventricolare e presentano le stesse qualità dei soffi di riempimento mesodiastolici; sono solitamente in crescendo e raggiungono l'intensità di picco in corrispondenza di un primo tono particolarmente intenso. Il soffio telediastolico corrisponde in genere al gradiente pressorio attraverso la valvola, che può essere minimo fino al momento della contrazione atriale destra o sinistra. Il soffio telediastolico è molto più caratteristico di quello mesodiastolico nella stenosi tricuspidale il ritmo sinusale. Soffi continui: Questi soffi iniziano durante la sistole, raggiungono la massima intensità prima del secondo tono e continuano durante tutta la diastole o parte di essa. Derivano da un flusso continuo dovuto a comunicazione tra aree ad alta e bassa pressione con gradiente persistente oltre la fine della sistole. La pervietà del dotto arterioso di Botallo determina un soffio continuo finché la pressione dell'arteria polmonare rimane molto al di sotto di quella dell'aorta. Questo soffio è intensificato dall'elevazione della pressione arteriosa sistemica ed è ridotto dall'inalazione di nitrito di amile. Se vi è ipertensione polmonare, la parte diastolica del soffio può scomparire lasciando il soffio solo durante la sistole. I soffi continui possono essere il risultato di fistole artero-venose sistemiche, congenite o acquisite, fistole coronariche, origine anomala della coronaria sinistra dall'arteria polmonare o comunicazione tra il seno di Valsalva e la parte destra del cuore. I soffi associati a fistole arterovenose polmonari possono essere continui, anche se più frequentemente sono limitati alla sistole. Soffi continui possono essere dovuti ad alterazioni delle modalità di flusso in arterie sistemiche o polmonari, nel caso in cui vi sia una notevole differenza pressoria tra i due estremi di un tratto stenotico. Il "soffio mammario" è un soffio innocente udibile al livello delle mammelle durante la fine della gravidanza e l'inizio del puerperio. Soffio da sfregamento pericardico: Questo soffio può presentare componenti da sfregamento sistoliche, proto- e telediastoliche e può essere confuso con un soffio o con un rumore di origine extra cardiaca se è udibile solo durante la sistole. Il soffio da sfregamento pericardico è in genere meglio udibile con il paziente in posizione eretta e piegato in avanti e può essere accentuato durante l'espirazione. CAPITOLO III: FISIOPATOLOGIA ISCHEMICA DELL’APPARATO CARDIOVASCOLARE La genesi dell’alterazione dell’omeostasi cardiaca che conduce all’insufficienza dei meccanismi autoregolatori è riconducibile a: - Una riduzione del flusso coronarico; - Un aumento del consumo miocardico di ossigeno (MVO2). Una lesione aterosclerotica (stenosi) coronarica determina a valle una caduta di pressione proporzionale alla riduzione del calibro vasale. Il gradiente pressorio che si crea stimola la dilatazione dei vasi di resistenza, allo scopo di mantenere un flusso adeguato in condizioni basali. Se la stenosi riduce la sezione del ramo epicardico di oltre il 75%, si ha una riduzione del flusso anche in condizioni basali; in questa situazione l’albero coronarico impegna gran parte della sua “riserva” per mantenere un apporto metabolico adeguato. Non è l’entità della placca a determinare l’effetto ma è sinergia tra entità della placca e entità della vasodilatazione. Se il vaso si contrae, anche una placca modesta può diventare pericolosa. Tutto questo ci riconduce all’esperimento fatto negli anni ’80 su animale: si prendeva un cane, si faceva una toracotomia, si trovava la discendente anteriore e si metteva attorno un sistema tramite il quale si può stringere o allargare la coronaria. A questo punto si determinavano delle progressive riduzioni del calibro inmaniera meccanica e si misurava il flusso. Poi le stesse misurazioni le si facevano al cane messo, su una pedana, a correre e quindi per diverse riduzioni del lume coronarico si facevano le misurazioni del flusso coronarico. In condizioni di riposo dobbiamo arrivare quasi all’80% delle riduzioni del lume perché si abbia riduzione del flusso. Quando il paziente fa uno sforzo, il flusso aumenta in maniera cospicua (la vasodilatazione aumenta, altrimenti la quantità extra di ossigeno non si può prendere). Già intorno al 20-25% di ostruzione del lume, il flusso comincia a ridursi e questo abbassamento si determina, temporalmente, un po’ prima se la stenosi è del 50-70%. Questa è la base fisiopatologica del concetto di STENOSI CORONARICA EMODINAMICAMENTE SIGNIFICATIVA o NON SIGNIFICATIVA. Cioè è una stenosi che al primo moto del paziente (nel senso di attività fisica) gli farà ridurre il flusso coronarico in funzione della stenosi. Succederà che: - se la placca è poco sviluppata, il paziente non avrà niente a riposo e neanche in caso di sforzo modesto, ma solo in caso di sforzo elevato la placca sarà clinicamente evidente; - se è più sviluppata, ciò accadrà anche a seguito di un minimo sforzo, perché la quantità di lume residuo anche in presenza di cospicua vasodilatazione non sarà in grado di fornire al miocardio la quantità di sangue necessaria. Quando parleremo di angina stabile da sforzo, angina da discrepanza, angina ingravescente, angina instabile, sindrome coronarica acuta, ritornerà il concetto di stenosi coronarica. C’è una grande differenza tra chi sta a riposo e chi fa uno sforzo. Il paziente con angina da sforzo a riposo è asintomatico, avrà un ECG normale, solo se e quando farà uno sforzo presenterà sintomi e un ECG alterato. In condizioni normali all’aumentare dell’esercizio ho un aumento della richiesta di ossigeno (tramite incremento esponenziale del flusso coronarico). La riserva coronarica è il rapporto tra il flusso in occasione dello sforzo e un flusso in condizioni normali. Il valore minimo è 2,5: io per fare una performance decente e non avere ischemia durante lo sforzo devo aumentare di due volte e mezzo il flusso coronarico rispetto a quello basale (se lo posso aumentare di più tanto di guadagnato). Quando il paziente ha una stenosi, ad un certo punto, la domanda di ossigeno aumenta ma non aumenta l’offerta, in virtù di questa discrepanza si hanno i sintomi. Più è lunga la diastole, più si perfonde il ventricolo; maggiore è l’efficacia contrattile del ventricolo, maggiore è la quantità di sangue che in diastole andrà incontro alle necessità metaboliche del miocardio. Il filling diventa cruciale per il ventricolo, le coronarie, per tutto il sistema vascolare, ovvero le periferie che aspettano questo sangue dal ventricolo sinistro. Non è che il ventricolo sinistro è un’unità sempre uguale, sempre sinergica, nel senso che tutte le fibre del ventricolo sinistro fanno la stessa cosa nello stesso momento. Questo è anche ovvio perché il ventricolo sinistro è irrorato dal discendente anteriore (parete antero-apicale e parte del setto), circonflesso (parete laterale e a volte inferiore), coronaria destra (parete settale inferiore). Quindi a seconda di dove c’è la limitazione del flusso, ci sarà una regione o più regioni che ne soffriranno. Il problema è vedere quante parti di ventricolo debbano essere coinvolte affinchè si abbia una situazione di crisi. Nel 1985 quando è iniziata l’angioplastica coronarica (che consiste nel gonfiare un palloncino all’interno di una coronaria e difatto viene interrotto il flusso per un certo periodo; in quell’arco di tempo si può misurare la reazione meccanica, elettrica, circolatoria, coronarica, come si farebbe nel cane a cui è stata legata la coronaria), è stato possibile studiare l’adattamento della funzione meccanica all’ischemia sull’uomo. L’angioplastica ha permesso di vedere quali e quante delle leggi ricavate dagli studi sugli animali erano realmente applicabili all’uomo e quindi quanto certe manovre o scelte farmacologiche che si erano rivelate efficienti nel modello animale potevano essere realmente efficienti anche nell’uomo. Se si chiude il discendente anteriore succede che, in condizioni normali, si ha una normale contrazione del ventricolo (cioè in sistole si riduce la silhouette del ventricolo ed in diastole aumenta, c’è una distanza tra le due silhouettes); quando facevamo l’occlusione del discendente anteriore succede che la parete anteriore si spancia (è aumentata) e la silhouette sistolica e diastolica non sono separate ma sono una sull’altra: significa che anche se arresto per 30 secondi il flusso all’interno del ramo discendente anteriore determino acinesia delventricolo. Quello che noi vediamo nell’infarto a distanza di ore e giorni, nella realtà accade nel giro di 30 secondi: ogni volta che c’è un paziente con il dolore in petto da 1 minuto, significa che da 30 secondi almeno una parte del suo cuore è ferma. In diastole succede che la fibra si allunga e in sistole si accorcia. Siamo sicuri che tutte le fibre si allungano e si accorciano nello stesso tempo? Perché se è vero, la famosa sinergia è rispettata e quindi abbiamo il massimo del riempimento e il massimo dello svuotamento. Se la sinergia non c’è, c’è il rischio che mentre una parte di ventricolo si rilassa, una parte si contrae: riceve meno sangue, di fatto è ferma perché non ha sangue in quel momento, però la parte che si contrae molto bene per un effetto di risucchio se la tira. Difatto ho una parete che si sta rilassando (si mette in condizione di accettare il sangue) e una che si sta contraendo (quindi costituisce un ostacolo al riempimento). Questo è il concetto di asincronia: è importante perché quello il fenomeno che si verifica è da mettere in relazione con l’entità dell’ischemia (cioè devo valutare in quel paziente quanto ventricolo ischemico c’è, perché maggiore è la quantità di ventricolo ischemico, maggiore sarà il danno possibile) ma non posso pensare che piccoli livelli di ischemia siano trascurabili. Non è così! Perché se faccio persistere quei piccoli livelli di ischemia nel tempo, non li identifico, non interrompo questo circolo vizioso, tramite le dissinergie o asincronie danneggeranno la funzione contrattile, per cui il medico e il paziente non si accorgeranno di niente ma poi il paziente dirà che ha l’affanno quando sale le scale, si gonfiano le gambe ed altri sintomi affini: in questo modo abbiamo perso la fase decisiva in cui si poteva risolvere il problema. Man mano che riduco il flusso, cosa accade per esempio al setto? Succede che in sistole lo spessore ventricolare sia del setto che della parete posteriore aumenta (deve spingere), in diastole si assottiglia (si è rilassato per accogliere il sangue). Mentre prima abbiamo visto le due silhouettes avvicinarsi, qui vediamo che dopo il decimo battito (10 battiti dopo il gonfiaggio del palloncino o dopo l’interruzione del flusso coronarico all’interno della discendente anteriore) il setto non si contrae più, cioè tra sistole e diastole non c’è differenza. Siamo scesi ancora di più. Prima stavamo a 30 secondi, ora siamo a 10 battiti. Basta un po’ di ischemia e il ventricolo va subito “a pallino”. Il concetto importante è che in 10 secondi si è fermato, non ha perso pezzi, è solo fermo. Quindi stanno venendo meno la sincronia e la sinergia, il ventricolo non funziona già più dopo 10 secondi di ischemia come dovrebbe funzionare. Il paziente è asintomatico quando i piatti della bilancia sono uguali, cioè quando sono domanda e uguali offerta di ossigeno. Abbiamo detto finora che possono bastare 10 battiti. Per fare diagnosi di ischemia ci basiamo sui sintomi del paziente e sull’ECG. Il dolore e l’ECG sono gli epifenomeni più distanti dall’evento e meno sensibili (pur essendo specifici). Perché? Perché in quei famosi 10 battiti o 30 secondi è successo subito che mancando l’ossigeno il metabolismo da aerobico è diventato anaerobico, quindi l’ATP disponibile finisce subito e dopo 10 battiti cessa di contrarsi il ventricolo ischemico. Questo significa che abbiamo subito un aumento del pH, quindi il marker più precoce dell’ischemia è (se si potesse misurare agevolmente) il pH del sangue venoso refluo dal miocardio (tra 6,9-7,1, sarà sicuramente acido); poi c’è la riduzione drammatica della funzione contrattile che si può vedere e monitoriamo con l’ecocardiogramma il paziente, dopo che si sarà determinata un’alterazione della contrattilità ci sarà un’alterazione delle correnti di sodio e potassio e quindi vedremo il segnale all’ECG e solo successivamente ci sarà il dolore. Man mano che ci allontaniamo dal primum movens, cioè dall’alterazione del metabolismo (pH) e ci avviciniamo al titolo finale più visibile (dolore) perdiamo pezzi, cioè perdiamo pazienti! Mentre il pH alterato lo troverò per definizione in tutti i pazienti con ischemia, l’eco in quasi tutti (per una questione di tempo, cioè in base a quando lo vado a fare), il dolore diventa la cosa più generica. Il dolore tipico è fortemente sospetto ma non basta per fare diagnosi se non c’è almeno l’ECG e, in caso di necrosi, il marker biochimico (si può avere ischemia in un diabetico senza dolore e senza sottoslivellamento del tratto ST, però le altre cose succedono). CAPITOLO IV: CARDIOPATIA ISCHEMICA CRONICA CARDIOPATIA ISCHEMICA Con il termine di cardiopatia ischemica s’intende uno spettro di patologie insorte in seguito ad ischemia: condizione di squilibrio tra fabbisogno di sangue ossigenato ed apporto ematico. Nel 90% dei casi, la causa risiede in una riduzione del flusso ematico coronario. La stenosi o l’occlusione di natura aterosclerotica è la principale responsabile dell’ischemia miocardica, tant’è che si parla di coronaropatia o cardiopatia coronarica. Più del 90% dei pazienti con CI hanno lesioni aterosclerotiche coronariche stenosanti in stadio avanzato, definite ostruzioni stabili; dato che il flusso è insufficiente quando l’occlusione supera il 90% del lume, la CI è caratterizzata da modificazioni repentine della placca aterosclerotica, legate a fissurazione o ulcerazione, assottigliamento della capsula e rottura; queste condizioni determinano attivazione della coagulazione che determina la formazione di un trombo che può occludere il lume della coronaria; l’ateroma, inoltre, può andare incontro a emorragie interne, con il risultato di un aumento di volume della placca. Il problema principale, è, fondamentalmente, legato alla velocità di formazione delle placche e delle ostruzioni, perché occlusioni severe, ma lente, fanno sì che si sviluppino circoli collaterali. Un altro meccanismo importante di ostacolo al flusso è legato alla vasocostrizione che può avvenire mediante diversi meccanismi: - Agonisti adrenergici stimolanti; - Fattori rilasciati dalle piastrine (trombossano); - Alterato rilascio di sostanze vasodilatatrici (NO) rispetto a sostanze vasocostrittrici (endotelina), legato all’alterazione funzionale dell’endotelio legato all’ateroma; - Mediatori rilasciati da cellule infiammatorie quali i mastociti. In generale le alterazioni principali per la genesi di una CI sono: - Aterosclerosi coronarica; - Vasospasmo; - Aggregazione piastrinica all’interno del lume; - Malattie coronariche; - Alterazioni emodinamiche. Le sindromi ischemiche sono varie e legate all’estensione del danno e alla risposta del miocardio: - Angina pectoris; - Infarto del miocardio; - Cardiopatia ischemica cronica; - Morte improvvisa cardiaca. Epidemiologia: La cardiopatia ischemica è una delle principali cause di mortalità e disabilità. Negli USA è la malattia cronica più invalidante per la vita di un individuo comune. In particolare, ne sono affette 13 milioni di persone, più di 6 milioni soffrono di angina pectoris e più di 7 milioni hanno avuto un infarto del miocardio. Una dieta ricca di grassi e calorie, il fumo e una vita sedentaria si associano alla comparsa di cardiopatia ischemica. Sia negli Stati Uniti che nell'Europa occidentale l'incidenza di tale patologia è in aumento nelle classi meno abbienti rispetto alle classi agiate. L'obesità, insulino-resistenza e il diabete mellito di tipo due stanno aumentando e rappresentano i fattori di rischio potenziali dello sviluppo di cardiopatia ischemica. Con l'urbanizzazione nei paesi in via di sviluppo, anche in queste zone si sta verificando un aumento della prevalenza dei fattori di rischio per la cardiopatia ischemica, tanto che attualmente questa patologia si sta manifestando soprattutto nei paesi a basso e medio reddito. I gruppi che sembrano essere maggiormente colpiti sono gli uomini dei paesi sud-asiatici. In ogni caso, considerata la proiezione di elevati incrementi della cardiopatia ischemica in tutto il mondo, tale patologia sembra destinata a diventare la principale causa di morte al mondo entro il 2020. Fisiopatologia: Il flusso è determinato dalla differenza di pressione diviso le resistenze a livello arteriolare. Sono quindi le resistenze intra-arteriolari quelle che condizionano il flusso intracoronarico. Che succede quando c’è una placca? Avremo due resistenze, quella a valle della placca e quella normale a livello arteriolare. In questo caso il flusso dipenderà da due resistenze. L’ischemia miocardica sostanzialmente è sempre legata ad un deficit di apporto di ossigeno. Le due diverse forme cliniche di ischemia, cioè l’ischemia acuta e l’ischemia cronica, sono caratterizzateda un modo diverso di manifestare il ridotto apporto di ossigeno: cioè possiamo avere riduzione del flusso coronarico perché si è formato un trombo, c’è stato uno spasmo della coronaria per cui praticamente si è proprio interrotto il flusso nel vaso; oppure possiamo avere un aumento del consumo miocardico di ossigeno. Il primo caso (cioè interruzione del flusso) è tipico delle sindromi coronariche acute; il secondo(aumento del consumo di ossigeno) è tipico della cardiopatia ischemica coronarica. C’è un’aumentata richiesta di ossigeno da parte del ventricolo, perché lavora, ha fatto uno sforzo, per un’emozione, e per effetto del vasospasmo o più verosimilmente di un ostacolo fisso (stenosi che riduce significativamente il flusso) nel momento in cui il flusso dovrebbe aumentare, non c’èfisicamente la possibilità che questo accada, per cui l’aumentata richiesta non viene soddisfatta dalla quantità di sangue che arriva. A livello miocardico, per l’elevata l’estrazione di O 2 (circa il 70%), l’unico meccanismo di compenso in caso di aumentato fabbisogno di O 2 è rappresentatoda un proporzionale aumento del flusso coronarico.Tale aumento è determinato dalla vasodilatazione del distretto coronarico arteriolare (vasi di resistenza). La capacità massima di vasodilatazione secondaria a uno stimolo metabolico è definita Riserva Coronarica. Autoregolazione e riserva coronarica: C’è una fascia di pressione compresa tra 80-120 mmHg in cui la pressione di perfusione non influenza (o influenza molto poco) il flusso coronarico. Quindi, quale che sia il valore di pressione, in questo intervallo il flusso coronarico che si modifica in funzione del ΔP/R si modifica poco o niente. E’ al di sotto e al di sopra di questo range che ci sono problemi. Siccome 120/80 è il range di pressione normale, verosimilmente il paziente godrà in massimo modo del fenomeno di autoregolazione del circolo coronarico. Se il flusso è regolato in funzione della pressione (secondo la linea arancione tratteggiata) non basterebbe una coronaria da 5 mm (rispetto ai soliti 2,5-3 mm) per poter reggere. In condizioni di riposo c’è bisogno che la stenosi sia molto severa per ridurre il flusso. Quando passiamo al lavoro, per valori di volume coronarico normale, abbiamo la riserva coronarica, cioè passiamo nel grafico dal valore 1 al valore 4, perché se aumenta il lavoro aumentano le richieste di ossigeno. Man mano che si riduce il lume del vaso, la riserva coronarica si riduce. Nei range di stenosi compresi tra 75 e 100 la riserva coronarica è sì e no 1, quindi il flusso che incondizioni di lavoro verrebbe quadruplicato, sì e no si raddoppia; siccome il miocardio è sempre lo stesso e le esigenze sono sempre le stesse è evidente che il flusso, solo raddoppiandosi, non riesce a venire incontro alle esigenze del miocardio. Riduzione del flusso coronarico: Una lesione aterosclerotica di un ramo epicardico determina a valle della stenosi una caduta di pressione proporzionale alla riduzione del calibro vasale. Il gradiente pressorio che si crea stimola la dilatazione dei vasi di resistenza, allo scopo di mantenere un flusso adeguato in condizioni basali. (Tale meccanismo è, ad esempio, alla base del quadro clinico dell’Angina Stabile). Se la stenosi riduce la sezione del ramo epicardico di oltre l’80%, si ha una riduzione del flusso anche in condizioni basali; in questa situazione l’albero coronarico impegna gran parte della sua “riserva” per mantenere un apporto metabolico adeguato. In caso di aumento delle richieste metaboliche, il circolo coronarico non è più in grado di far fronte alle richieste con comparsa di ischemia. L’ischemia interessa inizialmente gli strati subendocardici. Sono sufficienti moderati livelli di ischemia perché già il subendocardio ne possa soffrire; c’è bisogno di stati ischemici più consistenti perché ne abbia a soffrire l’intero spessore parietale. Quando parliamo di riserva coronarica, non parliamo semplicemente della resistenza (di come le resistenze arteriolari modificano il flusso); dobbiamo sempre tener conto del tono.Qualunque muscolo ha un tono, siccome i vasi coronarici hanno una componente muscolare è chiaro che avranno un tono. Il tono può essere particolarmente aumentato e quindi il vaso si irrigidisce e riduce il volume, così come una diminuzione del tono coronarico determina il rilassamento delle pareti e il vaso tende a dilatarsi, per cui molto più facilmente è in grado di accogliere quantità maggiori di sangue. Determinanti del consumo miocardico di ossigeno: Il cuore è un organo aerobio e, fisiologicamente, la determinazione del fabbisogno miocardico di O 2 fornisce un indice accurato del suo metabolismo complessivo. Quando parliamo del lavoro del cuore, chi ne determina l’entità? I vari fattori implicati sono: - Frequenza cardiaca; - Contrattilità; - Tensione di parete. L’aumento di ciascuna, o peggio di tutte e tre, ha sempre una valenza negativa perché aumenta il consumo di ossigeno. Che cosa determina la tensione di parete? Dipende dal post-carico, cioè da quello che c’è in termini di resistenza e di lavoro a valle del ventricolo sinistro (tutto sommato parliamo delle resistenze sistemiche: più sono alte le resistenze, più aumenta la tensione di parete della cavità cardiaca, più aumenterà l’ossigeno necessario). La Tensione di parete della cavità cardiaca (postcarico) è data da Pressione di distensione x raggio della cavità/(2xspessore parietale). La pressione sviluppata è determinata dalle resistenze all’eiezione del sangue. Il raggio medio è determinato dal riempimento della cavità, sarà tanto maggiore quanto più alto è il ritorno venoso (precarico). L’ aumento della tensione della parete di una cavità cardiaca (postcarico) determina la sua ipertrofia e quindi l’aumento della massa miocardica, a cui consegue un incremento del consumo di O 2 . In clinica è possibile rilevare alcune variabili, come la frequenza cardiaca e la pressione arteriosa sistolica. Il prodotto [Frequenza cardiaca x Pressione arteriosa sistolica x 10-3] si chiama doppio prodotto ed è l’indice di MVO2 (consumo di ossigeno) utilizzato nella pratica clinica. Ci sono dei range di normalità che variano in funzione del paziente (età, peso, altezza). Questo parametro esprime la capacità di quel miocardio ventricolare di lavorare e di ricevere per ogni livello di lavoro la quantità di ossigeno che realmente gli occorre. Quando andiamo al di sopra è perché noi siamo allenati, quando andiamo al di sotto del valore di normalità significa che il cuore non riesce a ottenere la quantità di ossigeno che gli occorre per svolgere quel tipo di lavoro (il maratoneta ha bisogno di un’energia minore rispetto ad un sollevatore di pesi ma che deve essere costante. Per 42 km deve avere una portata di una quantità di ossigeno disponibile e quindi consente di tollerare lo sforzo dei 42 km, nel corso dei quali gli consente di aumentare in caso di sforzo il consumo di ossigeno). Quando parliamo di consumo di ossigeno, di disponibilità di ossigeno, non dobbiamo pensare che sia solo una questione legata allo stato delle arterie coronariche: se le arterie coronarie sono esenti da lesioni deve andare tutto liscio, se le arterie coronarie sono sede di un processo proliferativo intraluminale allora è normale che le cose andranno male. Così non è! Ci sono una serie di fattori che determinano il consumo di ossigeno, la cui presenza può determinare una discrepanza (la cardiopatia ischemica cronica è dovuta ad una discrepanza tra richiesta di ossigeno ed effettiva possibilità di fornirlo, tutto è in relazione all’albero circolatorio, cioè a quanto sangue riesce a passare in quella coronaria o in quel ramo). E’ una discrepanza legata al fatto che l’aumento del postcarico, della contrattilità, della frequenza, del precarico, del volume, di per sé fa aumentare in maniera critica il consumo di ossigeno, per cui anche un circolo coronarico normale o para-normale non riesce a soddisfare i bisogni. Qual è la patologia presente in quasi tutti i casi? La stenosi aortica, perché determina un aumento sconvolgente della tensione di parete, del postcarico e quindi del lavoro che il ventricolo deve fare. E come fa il ventricolo a sopportare questo? Sviluppa una ipertrofia concentrica, però le coronarie non aumentano di pari passo (in studi bioptici si è visto che nel sovraccarico cronico di pressione c’è un piccolo aumento del lume coronarico ma è sempre discrepante rispetto allo spessore: la coronaria si allarga di poco, non abbastanza per veicolare la quantità di ossigeno necessaria). Altre condizioni critiche per l’instaurarsi dell’ischemia per riduzione del flusso coronarico sono: - Spasmo coronarico; - Trombi piastrinici; - Fattori emoreologici. ANGINA ED EQUIVALENTI ANGINOSI Nella figura a lato viene mostrata una classica scena cui segue l’innesco di un attacco anginoso. Il signore che esce dal ristorante, ha mangiato e bevuto, è ben coperto ma è evidente che c'è un vento freddo che determina vasocostrizione; porta un peso. Ci sono 3 componenti: sforzo legato al peso che porta, temperatura bassa, con vasocostrizione, aumento del contenuto di ossigeno e libagione abbondante, con deviazione di una quota di sangue nel distretto splancnico, per cui se c'è stenosi coronarica e aumentano le richieste di ossigeno e come se non bastasse aumentano le richieste periferiche per il freddo o c'è shift di sangue dal distretto coronarico al distretto splancnico, allora “i conti tornano”. Quali sono i sinotmi? L'angina è dolore (c'è angina di petto ma anche di gola). Gli equivalenti anginosi, che significa? Significa che la genesi di quel sintomo è la stessa dell'angina, cioè l'ischemia che è il risultato di una discrepanza tra richiesta e fornitura di sangue e quindi di ossigeno. Gli equivalenti anginosi sono l'astenia, il cardiopalmo (aumento della frequenza o da infarto o da aritmia) e la sincope. Possiamo avere angina da sforzo perché c'è stenosi dell'arteria. Ci sono condizioni che potrebbero essere considerate non collegate alla patologia di cui parliamo ma che possono esserne la causa scatenante: - anemia, perché se non c'è Hb non c'è neanche ossigeno; - ipertiroidismo, perché c'è l'equivalente dell'ipertono simpatico, quindi il metabolismo nella sua interezza aumenta (dappertutto aumenta il consumo di ossigeno); - la febbre, perché nel cuore c'è una temperatura diversa rispetto all'ambiente esterno (normalmente è 37°C, durante la febbre aumenta e aumenta anche la Frequenza Cardiaca, per la famosa regola di 8 battiti in più per ogni grado di temperatura); - la valvulopatia aortica; - la cardiopatia ipertensiva; - altre forme di ipertrofia ventricolare sinistra. La differenza tra il paziente normale e uno coronaropatico è che ad un certo punto all'incremento dello sforzo non c'è più l'incremento del flusso coronarico, per cui il flusso coronarico restava costante, ma le richieste erano aumentate. Quindi il triangolo in arancione è il deficit di flusso, cioè il flusso che manca per compensare quello sforzo per effetto dello stenosi. Il punto in cui il flusso, invece di aumentare, comincia a stabilizzarsi, viene chiamato soglia ischemica. Quando parliamo di ECG o di dolore, parliamo della fine della cascata ischemica, sono già successe un sacco di cose. L'ischemia non è in divenire, c'è già, comincia già a fare danni. La cascata corretta, quello che dobbiamo tenere di fronte al paziente con dolore è cosa è accaduto prima: c'èstata discrepanza, interruzione del flusso, mancato apporto di ossigeno che occorreva, ha determinato una eterogeneità di perfusione, perché anche se tutti e 3 i vasi coronarici principali fossero malati è difficile che siano malati tutti e 3 in ugual modo, e quindi nel momento di aumento del lavoro cardiaco ci sarà una parte di ventricolo che richiederà più flusso e una parte che ne richiederà di meno (quindi il flusso è già eterogeneo in condizioni normali, in condizioni di sforzo lo diventa ancora di più). Questa eterogeneità determina un'alterazione metabolica, perché se non c'è l'ossigeno, non c'è ciclo di Krebs e il metabolismo da aerobico diventa anaerobico, si produce acido lattico. Se noi potessimo misurare il pH del sangue refluo, vedremo che cade. Questo determina una disfunzione diastolica, a sua volta causa di dissinergia regionale, cioè viene a mancare la sinergia (c'è asincronia non solo per la parte sistolica, ma ancor più gravemente e prima nella parte diastolica, cioè il ventricolo non si riempie bene perché le sue parti non si rilassano in egual misura e nello stesso tempo). Una volta arrivato alla dissinergia, che si vede con l'ECO, c'è la modificazione dell'ECG e il dolore. Dalla eterogeneità di perfusione alla disfunzione diastolica possiamo esser certi che nell'ischemia ci saranno (se non ci sono non c'è ischemia); la dissinergia c'è sicuro però potremmo non vederla se l'eco non è fatta bene. La fase che può mancare realmente, malgrado ci sia l'ischemia, sono le modificazioni dell'ECG e il dolore (che sono le due cose su cui ci basiamo di più). Perché parliamo di angina stabile a soglia fissa e angina stabile a soglia variabile? Nell'angina stabile a soglia fissa, per un certo tipo di lavoro si creerà la crisi. C'è bisogno di raggiungere quel certo valore di doppio prodotto perché compaiano i sintomi o perché l'ECG si modifichi (Soglia fissa significa → 3 piani di scale per avere il dolore; 100 m in salita; 10 passi di corsa per avere il dolore). Quand'è che la soglia fissa diventa variabile? Posso avere dolore per sforzo e poi ho lo stesso dolore e gli stessi sintomi per un livello di sforzo estremamente minore. Non è cambiata la stenosi, il consumo di ossigeno è determinato dalla FC x PA sistolica, è cambiato il tono vascolare; il lume della coronaria si è ridotto non perché è aumentata l'entità della placca stenosante ma perché si è ridotto il lume del vaso. Se si riduce il lume del vaso, si riduce quella quantità di sangue che passava prima e che poteva essere sufficiente per quel lavoro, quindi si ha ischemia per una soglia di lavoro più bassa. IL DOLORE TORACICO Il dolore anginoso si localizza a livello pre-cordiale o retrosternale, con tipiche irradiazioni alla faccia ulnare dell'arto sinistro. Ci sono però delle variabili. Quello classico è relazionato al dermatomero T1. Le sedi di dolore toracico meno frequenti non si sovrappongono a quelle classiche. Il dolore toracico può originare da diversi organi. Il torace è piccolo, ma molto affollato. Ci sono organi estremamente sensibili. Il dolore può originare da: - Il cuore, i grossi vasi e il pericardio; - Il tratto gastrointestinale; - I polmoni e la pleura; - La parete toracica; - Il profilo psichico ansioso-depressivo. Classica è l’interferenza tra angina pectoris e dolore esofageo: - La presenza di una patologia può simulare la presenza dell’altra; - Le due patologie possono coesistere e influenzarsi: Il reflusso esofageo può ridurre la soglia per il dolore ischemico (quindi si deve avere un'ischemia maggiore per sentire il dolore); - Test stimolativi (ergonovina) per vedere se la coronaria aumenta il tono muscolare possono indurre spasmo esofageo; - Farmaci cardiologici (nitrati) possono risolvere lo spasmo esofageo e anche con un po' di fortuna coliche epatiche e renali. CLASSIFICAZIONE La Cardiopatia ischemica si divide in due grossi capitoli: - La Sindrome coronarica stabile, con angina da sforzo; - Le Sindromi coronariche acute (instabili). ANGINA PECTORIS STABILE Qualunque situazione che determini ischemia al cuore causa angina. L’angina pectoris è una sindrome clinica caratterizzata da attacchi parossistici di dolore acuto precordiale (definito come oppressivo, come una pugnalata, da compressione, soffocante). Questa è causata da una ischemia breve, della durata di15 sec. a 15 minuti, legata ad aterosclerosi coronarica e meno frequentemente alla stenosi aortica o subaortica. Esistono tre tipi di angina: angina stabile, angina variante o di Prinzmental, angina instabile o ingravescente. L'angina stabile (o angina cronica) è un'ischemia miocardica acuta transitoria, associata a sforzo fisico o emozione, che si produce in condizioni omogenee, stabili nel tempo (c'è costante e ripetibile discrepanza tra domanda di ossigeno ed offerta). L’angina stabile è la forma più frequente, denominata anche angina pectoris tipica. È legata ad aterosclerosi coronarica stenosante cronica, responsabile della riduzione del flusso, se aumenta la richiesta; è, quindi, legata a sforzi e il dolore si arresta a riposo. È legata ad una stenosi di un’arteria epicardica; per compenso le arteriole intramiocardiche si vasodilatano e rendono il flusso massimale; se esiste una maggiore richiesta e si supera il limite di flusso, si ha dolore. Nei pazienti con dolore spontaneo si ha l’angina a soglia variabile, legata ad una componente vasocostrittiva. Il paziente è, in genere, maschio nell’80%, con età tra i 50 e 60. Il dolore aumenta progressivamente, durante 1 fino a 15-20 minuti. Il dolore è tipicamente retrosternale, irradiato lungo il lato ulnare del braccio, indicato come senso di costrizione, di peso, di compressione o bruciore. Fattori precipitanti: L’angina può essere innescata da una serie di situazioni che determinano un aumento della richiesta di ossigeno da parte del miocardio. I principali fattori precipitanti sono: - Attacco provocato da uno sforzo, in particolare può innescarsi con un lavoro che comporta l’utilizzo delle braccia al di sopra del livello della spalla; - Ambiente freddo, camminare contro vento, camminare dopo un pasto abbondante; - Crisi ipertensiva; - Paura, rabbia, stati d’ansia, tensione emotiva; - Rapporti sessuali. I principali sintomi associati sono respiro corto, vertigini, palpitazioni e debolezza. Anamnesi: Il tipico paziente affetto da angina è un uomo di oltre cinquant'anni o una donna di oltre sessant'anni che lamenta disturbi al torace, di solito descritti come senso di pesantezza, pressione, oppressione, soffocamento e solo raramente come dolore franco. Quando al paziente si chiede di localizzare il punto in cui avverte tale sensazione, pone abitualmente la mano sullo sterno, a volte con il pugno chiuso, per indicare un disturbo sottosternale schiacciante e centrale (segno di Levine). L'angina ha di solito un andamento crescente e decrescente, dura in media dai due ai cinque minuti e si può irradiare sia alla spalla e ad entrambe le braccia (superfici ulnari dell'avambraccio e della mano). Può anche diffondersi o irradiarsi fino al dorso, alla regione interscapolare, alla base del collo, alla mascella, ai denti e all’epigastrio. L'angina è raramente localizzata sotto l'ombelico e sopra la mandibola. Nell'esaminare il paziente con disturbi al torace è rilevante ricordare che il disturbo ischemico miocardico non si irradia ai muscoli trapezoidali, e che tale modello di espansione è più tipico della pericardite. Gli episodi di angina sono tipicamente causati dallo sforzo o dalle emozioni, e cessano con il riposo, ma possono verificarsi in questa fase e mentre si è in posizione supina (angina da decubito). Il paziente può essere risvegliato di notte dal tipico dolore al torace e dalla dispnea. L'angina notturna può essere dovuta a tachicardia episodica, diminuita ossigenazione a causa dei cambiamenti nella respirazione durante il sonno o da un aumento del volume di sangue intratoracico che si verifica a riposo. La soglia di comparsa può variare nelle ore della giornata e in rapporto allo stato emotivo del paziente. Molti soggetti presentano una soglia fissa per l'angina. In questi pazienti, la stenosi coronarica e l'apporto di ossigeno al miocardio sono fissi, e l'ischemia viene scatenata da un aumento della richiesta di ossigeno da parte del miocardio; in questo caso si parla di angina da sforzo stabile. In altri pazienti, la soglia per l'angina può variare considerevolmente nell'ambito della stessa giornata e da un giorno all'altro. In questi casi le variazioni dell'apporto di ossigeno al miocardio, dovute soprattutto ai cambiamenti del tono vascolare coronarico, possono giocare un ruolo importante per la definizione del tipo di angina. L'angina da sforzo migliora diminuendo o interrompendo le attività in uno-cinque minuti, e anche più rapidamente a riposo e dopo assunzione di nitroglicerina sublinguale. Un dolore al torace tagliente e repentino o un dolore prolungato e lieve, localizzato nella regione sottomammaria sinistra sono raramente dovuti a ischemia miocardica. Comunque, soprattutto nelle donne e nei diabetici, l'angina pectoris può essere localizzata in aree atipiche e non risultare strettamente correlata ai fattori scatenanti. Inoltre, il sintomo può peggiorare e diminuire nel corso di giorni, settimane o mesi. La sua ricorrenza può essere stagionale, più frequente durante l'inverno nei paesi con clima temperato. Gli equivalenti anginosi sono sintomi di ischemia miocardica diversi dall'angina. Questi includono dispnea, nausea, affaticamento e debolezza, e sono più frequenti negli anziani e nei pazienti diabetici. L'interrogazione sistematica del paziente in cui si sospetta la cardiopatia ischemica è importante al fine di scoprire le caratteristiche di una sindrome instabile associata a un aumento del rischio. È anche importante evidenziare una anamnesi familiare di cardiopatia ischemica prematura e la presenza di diabete mellito, iperlipidemia, ipertensione, fumo di sigaretta e altri fattori di rischio per l'aterosclerosi coronarica. L'anamnesi dell'angina pectoris tipica suggerisce la diagnosi di cardiopatia ischemica fino a che non è provato diversamente. Esame Obbiettivo: Spesso il paziente è negativo. Se visitato durante l’angor, è riscontrabile un IV tono per ridotta compliance ventricolare, o un III tono per distensione rapida di un ventricolo con ridotta funzione sistolica. Si può riscontrare anche un soffio sistolico puntale da insufficienza transitoria mitralica. Spesso si può riscontrare un aumento di PA. All’ECG in angina da sforzo, l’alterazione tipica è rappresentata da sottoslivellamento ST e T negativa.Nell’angina spontanea si può riscontrare un sopraslivellamento ST. Le principali caratteristiche dell'Angina da Sforzo sono: - dolore tipico durante lo sforzo; - la breve durata (qual è la differenza tra angina da sforzo e sindrome coronarica acuta? La durata del dolore. Si può sospettare una sindrome coronarica acuta o un infarto del miocardio quando il dolore dura più di 20 minuti. Il suo meccanismo di insorgenza è lo stesso); - il dolore regredisce con il riposo, in genere dopo 5 minuti anche senza l'ausilio dello spray edell'NO; - il dolore è sensibile ai nitroderivati (il paziente che ha angina da sforzo se prende il carbasin gli passa; il paziente con sindrome coronarica acuta non è sensibile ai nitroderivati, perché nel primo caso c'è discrepanza tra richiesta e apporto, quindi se vasodilato il circolo coronarico, l'apporto aumenta, nel secondo caso c'è stata un'interruzione brusca del flusso, quindi la vasodilatazione è inutile perché è a monte che si è interrotto il flusso: non basterà la vasodilatazione per far vincere al flusso quello che è un ostacolo meccanico, tipicamente un trombo). Classificazione dell’angina da sforzo: Come si classifica l'angina da sforzo? Tale classificazione è quella fornita dalla società di cardiologi canadese: - CLASSE I: la normale attività fisica non induce angina (o il paziente non ha niente o ha una stenosi coronarica che o non è severa o ha un buon circolo collaterale, non raggiunge mai la soglia ischemica, cioè non si crea mai la discrepanza tra domanda e offerta). Non significa non avere niente ma non avere i sintomi. Il paziente in classe I può anche essere normale. Teoricamente siamo tutti in classe I. Se un paziente in classe II con la terapia passa in classe I è una cosa importante perché significa che il paziente era sintomatico e per effetto della terapia è diventato asintomatico. Spesso la classificazione serve a valutare l'efficacia della terapia e per capire con che paziente abbiamo a che fare; - CLASSE II: modesta limitazione dell’attività ordinaria. L’angina insorge camminando in fretta, dopo i pasti, al freddo, al vento; - CLASSE III: forte limitazione dell’attività ordinaria. L’angina insorge camminando per 2 isolati o salendo 1 piano di scale; - CLASSE IV: impossibilità di compiere qualsiasi attività. L’angina insorge anche a riposo. Il paziente ha un vaso che si è quasi chiuso, passano gocce di sangue, non possiamo neanche parlare di flusso. Altri test: Il test da sforzo è il test diagnostico migliore, da effettuare in assenza di terapia, con una predittività del 98%, presentando diversi falsi negativi. Anche la scintigrafia perfusionale con Tallio o MIBI (metossi-isobutil-isonitrile marcato con 99Tc) è utile per riscontrare diminuita perfusione. L’ECO può essere del tutto normale.L’Holter, infine, è utile per registrare variazioni di ST-T e collegarli ad angor. Può essere effettuata la coronarografia, per lo studio dei tre rami principali. Prognosi: In questi pazienti le principali variabili prognostiche sono rappresentate dall'età, dalle condizioni funzionali del ventricolo sinistro, dalla sede e dall'entità del restringimento coronarico e dalla gravità o attività dell'ischemia miocardica. L'angina di insorgenza recente, l'angina instabile, l'angina post infarto miocardico precoce, l’angina refrattaria alla terapia medica o che si accompagna a sintomi di insufficienza cardiaca congestizia si associano a un rischio maggiore di eventi coronarici. Lo stesso vale in presenza di segni obiettivi di scompenso cardiaco. Ancora più importante per la prognosi sono i reperti di: incapacità di compiere l'esercizio richiesto per sei minuti durante la prova da sforzo; un test sotto sforzo inequivocabilmente positivo che evidenzia l'insorgenza di ischemia miocardica in seguito a bassi carichi di lavoro; sviluppo di difetti di perfusione estesi o multipli, o aumento della captazione polmonare alla scintigrafia radioisotopica di perfusione sotto sforzo; riduzione della frazione di eiezione ventricolare sinistra durante l'esercizio nel corso della ventricolografia radioisotopica o durante l'ecografia sotto sforzo. Durante il cateterismo cardiaco, l'aumento della pressione telediastolica del ventricolo sinistro e dei volumi ventricolari, nonché il calo della frazione di eiezione, rappresentano i più importanti indici diagnostici di disfunzione ventricolare sinistra e comportano una prognosi sfavorevole. I pazienti con dolore toracico, ma con funzione ventricolare sinistra normale e coronarie normali hanno una prognosi eccellente. Le lesioni ostruttive del ramo principale (superiore al 50% del diametro del lume) o di quello discendente anteriore dell'arteria coronaria sinistra, prossimali all'origine della prima arteria settale sono associate a un rischio maggiore. Le placche aterosclerotiche nelle arterie epicardiche che presentano fissurazioni o difetti di riempimento indicano un aumento del rischio. Queste lesioni vanno incontro a fasi di attività cellulare infiammatoria, degenerativa, disfunzione endoteliale, anomala vasomotricità, aggregazione piastrinica, fissurazione o emorragia. Questi fattori possono provocare un temporaneo peggioramento della stenosi e causare una anomala reattività vascolare e può esacerbare le manifestazioni dell'ischemia. La mortalità aumenta in modo significativo, indipendentemente dal grado di ostruzione dovuta alla coronaropatia, se la funzione del ventricolo sinistro è compromessa. Per ogni livello di compromissione della funzione ventricolare sinistra, la prognosi è influenzata in modo significativo dall'estensione del miocardio perfuso da vasi con lesioni critiche. Maggiore è l'estensione della necrosi miocardica, minore è la capacità del cuore di sopportare ulteriori danneggiamenti: in questo caso la prognosi risulta ancora sfavorevole. Maggiori sono il numero e la gravità dei fattori di rischio per l'arteriosclerosi coronarica peggiore è la prognosi dell'angina. TERAPIA Ciascun paziente deve essere valutato individualmente tenendo presenti le aspettative e gli obiettivi, il controllo della sintomatologia e la prevenzione di complicanze cliniche. È necessario valutare attentamente il grado di compromissione funzionale e gli stress fisici ed emotivi che possono scatenare l'angina. Il programma terapeutico dovrebbe contemplare le seguenti componenti: - spiegazione del problema e rassicurazioni circa la capacità di formulare un intervento terapeutico; - individuazione e trattamento delle condizioni patologiche aggravanti; - raccomandazioni per l'adeguamento dell'attività fisica; - trattamento dei fattori di rischio; - terapia farmacologica per l'angina; - valutazione dell'eventualità di rivascolarizzazione. Questi criteri, possono essere facilmente inquadrabili con lo schema ABCDE: - A, come, Aspirina e anti-anginosi; - B, come, beta-bloccanti e pressione arteriosa; - C, come, colesterolo e fumo (cigarettes); - D, come, dieta e diabete; - E, come, educazione e attività fisica. La riduzione del lume fa diventare il flusso da laminare a turbolento quindi la possibilità che si formi un trombo aumenta. E' vero che se la stenosi è di consistenza fibrosa, fibro-stenotica, fibro-calcifica è difficile che avrà un infarto. L'infarto lo farà chi ha una placca di qualunque entità ma con un cappuccio sottile che quando si ulcera porta all'infarto. Ciò non significa che se ho una stenosi del 90% non si può determinare per effetto del rallentamento della turbolenza del flusso una iperaggregabilità tale che il paziente con angina stabile per mesi o anni improvvisamente ha o un'angina a soglia bassa o addirittura una sindrome coronarica acuta, perché quel poco di lume residuo è stato occluso dal trombo. Gli antiaggreganti sono la terapia principe della malattia aterosclerotica: quando c'è alterazione del profilo del lume vascolare c'è indicazione alla terapia antiaggregante. Questo vale per qualunque vaso, non solo per le coronarie. I cardini della terapia aterosclerotica sono le statine e l'aspirina. Le statine non solo riducono il colesterolo ma proteggono l'endotelio dell'injury aterosclerotico e l'aspirina che impedisce l'aggregazione piastrinica. Ci sono dati in letteratura che dimostrano che le statine possono far regredire la placca. Devono essere placche in cui la componente fibrosa e calcifica deve essere modesta. La statina sull'endotelio può migliorare l'effetto dell'NO, può favorire la riendotelizzazione laddove l'injury ateromatosa ha determinato la discontinuità dell'endotelio. Uno dei motivi per cui sia in prevenzione secondaria che primaria si usano la statina è anche perché esiste la possibilità che placche non significative, ricche di lipidi (che sono tra l'altro quelle che più di altre danno luogo a sindromi coronariche acute) possano regredire. TERAPIA FARMACOLOGICA La terapia farmacologica per la cardiopatia ischemica ha lo scopo di ridurre la frequenza degli episodi di angina e diminuire l’aumento improvviso della frequenza cardiaca e della pressione arteriosa del paziente sotto sforzo, consentendogli di compiere le attività giornaliere senza avvicinarsi alla soglia ischemica. La terapia antiaggregante con acido acetilsalicilico è anche usata per ridurre gli eventi trombotici che possono verificarsi con la destabilizzazione delle placche aterosclerotiche. Nitrati: I nitrati organici sono una delle classi di farmaci più preziose nel trattamento dell'angina pectoris. Il loro principale meccanismo d'azione comprende un venodilatazione sistemica con concomitante riduzione del volume e della pressione telediastolica ventricolare sinistra, riducendo la tensione parietale miocardica e le richieste metaboliche di ossigeno, nonché dilatando i vasi coronarici epicardici e incrementando il flusso sanguigno nei vasi collaterali. I nitrati organici, se metabolizzati, rilasciano ossido nitrico che si lega alla guanilato ciclasi nelle cellule muscolari lisce vasali, causando un aumento della guanosina monofosfato ciclica, che a sua volta determina un rilassamento della muscolatura liscia nasale. L'assorbimento di questi farmaci avviene più rapidamente e completamente attraverso le membrane mucose. Per questa ragione, la nitroglicerina viene somministrata principalmente per via sublinguale, con compresse da 0,4-0,6 mg. È necessario istruire i pazienti con angina circa l'impiego di questo farmaco. Il valore dell'uso del farmaco a scopo profilattico non può essere troppo accentuato. La cefalea e le fastidiose sensazioni di pulsazione alla testa sono gli effetti collaterali più comuni della nitroglicerina, ma fortunatamente solo in rari casi questi sintomi non sono tollerabili alle dosi necessarie per prevenire o per risolvere un attacco di angina. La nitroglicerina si deteriora con l'esposizione all'aria, all'umidità e alla luce del sole; che non produce né risoluzione del dolore, né una lieve sensazione di formicolio di assorbimento e probabile che la preparazione che inattiva. Se nell'arco di due o tre minuti, con il paziente a riposo, il dolore non si attenua con la somministrazione di tre compresse di nitroglicerina, si dovrebbe consultare un medico o presentarsi immediatamente in un pronto soccorso. I nitrati aumentano la tolleranza allo sforzo nei pazienti con angina cronica e riducono l'ischemia nei pazienti con angina instabile o con angina variante di Prinzmetal. Per quanto riguarda i nitrati a lunga durata d'azione, nessuno è tanto efficace quanto la nitroglicerina sublinguale. Questi preparati di nitrato organico possono essere inghiottiti, masticati o somministrati come cerotti o pasta per via cutanea. Possono fornire livelli plasmatici efficaci fino a 24 ore, ma la risposta terapeutica è estremamente variabile. Le preparazioni più utili sono quelle a base di isosorbide dinitrato o mononitrato, unguento di nitroglicerina o i cerotti cutanei a rilascio sostenuto. L'assuefazione, con relativa perdita dell'efficacia farmacologica, si sviluppa entro 12-24 ore. Il meccanismo di sviluppo dell'assuefazione al nitrato non è completamente chiaro, ma ipotesi avanzate includono un'inadeguata deplezione dei gruppi sulfidrilici richiesti per la biotrasformazione in ossido nitrico, l'inibizione della deidrogenasi aldeide mitocondriale, l'attivazione neuroormonale di meccanismi di controregolazione con vasocostrizione e ritenzione di liquidi, è la produzione di radicali liberi dell'ossigeno che rendono inattivo l’ossido nitrico è aumentano la sensibilità della muscolatura liscia vascolare nei confronti dei vasocostrittori circolanti. Allo scopo di minimizzare gli effetti della tolleranza, sarebbe opportuno utilizzare la dose minima efficace e interrompere la somministrazione del farmaco per almeno otto ore tutti i giorni, in modo da ristabilire una risposta efficace. Bloccanti beta-adrenergici: Questi farmaci rappresentano un importante componente nel trattamento farmacologico dell’angina pectoris. Agiscono provocando una riduzione delle richieste miocardiche di ossigeno e riducendo l’incremento della frequenza cardiaca, della pressione arteriosa e della contrattilità miocardica indotte dall’attività adrenergica. I beta-bloccanti riducono queste variabili in particolare nel corso dell’esercizio fisico, mentre provocano solo piccole riduzioni nel soggetto a riposo. Quelli a lunga durata di azione o le formulazioni a rilascio sostenuto offrono il vantaggio di una sola somministrazione al giorno. Questi farmaci oltre a risolvere l’episodio anginoso o l’ischemia, sono in grado di ridurre la mortalità e la probabilità di recidiva di infarto se somministrati a pazienti recentemente colpiti da infarto miocardico; inoltre possiedono un lieve effetto antipertensivo. Controindicazioni relative al loro uso comprendono l’asma, ostruzioni delle vie aeree in pazienti con pneumopatie croniche, disturbi della conduzione AV, bradicardia grave, fenomeno di Raynaud e anamnesi clinica di depressione. Gli effetti collaterali sono astenia, ridotta tolleranza allo sforzo, incubi, impotenza, estremità fredde, claudicatio intermittens, bradicardia, alterazione della conduzione AV, insufficienza ventricolare sinistra, asma bronchiale, peggioramento della claudicatio ed intensificazione dell’ipoglicemia indotta dai farmaci ipoglicemizzanti orali e dall’insulina. I beta-bloccanti con relativa specificità per il recettore β 1 , come il metoprololo e l’atenololo, sono da preferire nei pazienti con ostruzione bronchiale moderata e nei soggetti con diabete mellito che richiedono insulina. Calcio-antagonisti: Questi farmaci sono vasodilatatori coronarici che determinano una riduzione, variabile e dose-dipendente, della domanda di ossigeno del miocardio, della contrattilità cardiaca e della pressione arteriosa. Questi effetti farmacologici combinati sono vantaggiosi e rendono questi agenti efficaci quanto i beta-bloccanti nel trattamento dell'angina pectoris. Il calcio-antagonisti vanno utilizzati qualora i betabloccanti siano controindicati, scarsamente tollerati o inefficaci. Il verapamil e il diltiazem possono provocare disturbi sintomatici della conduzione cardiaca e bradiaritmia; inoltre, possono esercitare un'azione inotropa negativa e molto probabilmente peggiorare un'insufficienza ventricolare sinistra, in particolare quando usati in pazienti che presentano una disfunzione ventricolare sinistra, che assumono contemporaneamente anche beta-bloccanti. L'angina instabile di Prinzmetal risponde particolarmente bene ai calcio-antagonisti, integrati, se necessario, ai nitrati. Il verapamil non dovrebbe, di solito, essere associato ai beta-bloccanti a causa degli effetti combinati indesiderati sulla frequenza cardiaca e sulla contrattilità miocardica. Il diltiazem può essere somministrato in associazione con i beta-bloccanti nei pazienti con funzione ventricolare normale e nessun disturbo di conduzione. L'amlodipina e i beta-bloccanti esercitano azioni complementari sull'apporto ematico coronarico e sulla richiesta di ossigeno del miocardio. Mentre la prima diminuisce la pressione arteriosa e dilata le coronarie, i beta-bloccanti rallentano la frequenza cardiaca e riducono la contrattilità miocardica. L'amlodipina e gli altri calcio-antagonisti diidropiridinici di seconda generazione sono vasodilatatori potenti e utili nel trattamento simultaneo dell'angina e dell'ipertensione. La somministrazione di diidropiridine a breve durata d'azione dovrebbe essere evitata, in quanto sussiste il rischio di accelerare l'insorgenza dell'infarto, soprattutto in assenza di beta-bloccanti. Farmaci antipiastrinici: L’acido acetilsalicilico è un inibitore irreversibile dell'attività della cicloossigenasi piastrinica e di conseguenza inibisce l'attivazione delle piastrine. Si è osservato che la somministrazione per via orale a lungo termine di 75-325 mg al giorno riduce l'insorgenza di coronaropatia nei soggetti adulti asintomatici di sesso maschile, nei pazienti affetti da angina stabile cronica e in quelli che presentano o che sono sopravvissuti ad angina instabile e infarto. Quando l'acido acetilsalicilico è usato a lungo termine, si verifica un aumento di emorragie dose-dipendente. La somministrazione di questo farmaco dovrebbe essere prese in considerazione in tutti i pazienti affetti da cardiopatia ischemica, in assenza di emorragia gastrointestinale, reazione allergica o dispepsia. Il clopidogrel è un farmaco somministrato per via orale che blocca l'aggregazione piastrinica mediata dal recettore ADP; apporta gli stessi benefici dell'acido acetilsalicilico nei pazienti con cardiopatia ischemica cronica stabile e può essere sostituito all'acido acetilsalicilico, se quest'ultimo determina l'insorgenza degli effetti collaterali. Il clopidogrel combinato con dell'acido acetilsalicilico riduce il tasso di mortalità e le manifestazioni di ischemia coronarica nei pazienti con sindrome coronarica acuta, così come riduce il rischio di formazione di trombi nei pazienti che si sottopongono all'impianto di uno stent in un'arteria coronarica. Anche se la terapia combinata di clopidogrel e acido acetilsalicilico è raccomandata per almeno un anno nei pazienti con sindrome coronarica acuta e in seguito all'impianto di uno stent a rilascio di farmaco, gli studi non hanno dimostrato alcun beneficio riguardo questa combinazione nei pazienti con cardiopatia ischemica cronica. INDAGINI DIAGNOSTICHE Per quanto riguarda le indagini diagnostiche, queste vengono suddivise in indagini non invasive ed indagini invasive. INDAGINI NON INVASIVE Le principali indagini diagnostiche non invasive sono: - ECG a riposo; - Indagini di laboratorio; - ECG da sforzo; - Ecocardiogramma; - Ecostress; - Tecniche di radiologia nucleare. Ecocardiografia: È una tecnica non invasiva che sfrutta gli ultrasuoni con frequenza 2-10 Mhz. Si basa sul principiodell'emissione di eco e della trasmissione delle onde ultrasonore. Permette di visualizzare l'anatomia del cuore e la sua funzione, in particolare è in grado di fornire informazioni sulla sua contrattilità, sulla morfologia delle valvole cardiache e sul flusso del sangue all'interno delle cavita. Nel 1985 Bonn ha messo a punto un micro-ecocardiografo, simile ad un joy-stick. La parte di sotto si metteva sul torace del paziente, la parte di sopra aveva un piccolo visore e nello schermo si vedeva l'immagine. Il cuore si può vedere sia per via transtoracica che transesofagea che intracardiaca. Dato che il cuore è racchiuso all’interno del torace, esistono delle opportune posizioni dove poggiare la sonda ecocardiografica. In questo modo è possibile ottenere varie proiezioni che mettono in evidenza determinate porzioni del tessuto miocardico: - Approccio parasternale: A. piano asse lungo: 1 radice aortica, valvola aortica, atrio sinistro, tratto di efflusso del ventricolo destro, tratto di efflusso del ventricolo sinistro; 2 ventricolo sinistro e mitrale; 3 apice del ventricolo sinistro; 4 tratto di afflusso del ventricolo; B. piano asse corto: 1 radice aortica, valvola aortica, valvola polmonare, tricuspide, tratto di efflusso del ventricolo destro, atrio sinistro, arteria polmonare, arteria coronaria; - 2 ventricolo sinistro e mitrale; 3 ventricolo sinistro e muscoli papillari; 4 ventricolo sinistro, apice; approccio apicale: A. piano quattro camere: 1 quattro camere; 2 quattro camere, con aorta; B. piano asse lungo: - 1 due camere: ventricolo sinistro e atrio sinistro; 2 due camere con aorta; approccio sub-costale: A. piano quattro camere; B. piano asse corto: - 1 ventricolo sinistro; 2 ventricolo destro; 3 vena cava inferiore; approccio soprasternale: A. piano quattro camere: arco dell’aorta, aorta discendente; B. piano asse lungo: arco dell’aorta, arteria polmonare atrio sinistro. Quando andiamo a fare la valutazione di un paziente ischemico, per effetto di quella eterogeneità di flusso è cruciale identificare i diversi segmenti del ventricolo e valutare singolarmente il comportamento. È possibile mettere in relazione il flusso coronarico alla suddivisione delle pareti del ventricolo. Cioè sappiamo ciascuno di quei segmenti da chi è irrorato. Vediamo quale segmento del ventricolo funziona male e il ramo coronarico interessato. Un tempo veniva effettuata la sola rappresentazione monodimensionale o M-mode. Questa era una sezione delle strutture cardiache secondo un asse verticale, con scarso potere di risoluzione spaziale. Il passo successivo è l’ECO 2D. L'ecocardiografia ha numerose applicazioni, con l'unico difetto di essere operatore-dipendente: - valutazione qualitativa e quantitativa delle malattie delle valvole cardiache; - valutazione della cinesi ventricolare e delle sue anomalie; - valutazione delle cardiopatie congenite; - valutazione del danno miocardico; - valutazione degli esiti di un intervento operatorio correttivo nelle cardiopatie congenite o acquisite; - valutazione delle protesi valvolari - misurazione semiquantitativa di gradienti e flussi inpresenza di valvole stenotiche o insufficienti; - diagnosi di tutte le malattie cardiache in gravidanza con possibilità anche di diagnosi intrauterina di gravi cardiopatie congenite fetali. Eco-stress: L’ecocardiografia da stress è un insieme di esami con cui si effettua un monitoraggio elettrocardiografico ed ecocardiografico bidimensionale prima, durante e dopo stress cardiovascolare. I rilievi ecocardiografici suggestivi di ischemia miocardica inducibile includono: - riduzione del movimento della parete cardiaca in almeno uno dei segmenti del ventricolo sinistro durante stress; - riduzione dello spessore di parete in almeno uno dei segmenti ventricolari durante stress (un occhio non esercitato lo vede sul setto, uno esercitato anche sulle pareti); - ipercinesi compensatoria in segmenti complementari (non ischemici). Durante lo stress fisico il paziente va in iperventilazione. Il principale nemico dell'ecografista è l'aria, che il paziente durante l'eco trans-esofageo deglutisce. L'eco da sforzo significa iperpnea. Lo stress farmacologico si fa con dipiridamolo, l'adenosina e la dobutamina. L'eco con dobutamina aumenta l'inotropismo, serve per vedere se una porzione di ventricolo apparentemente integra o acinetica ha una riserva contrattile da esprimere o se facendo aumentare il lavoro del cuore si scatena più facilmente l'evento. Il paziente che ha stenosi della succlavia (dalla succlavia nasce la vertebrale), facendo uno sforzo con il braccio dove ha la stenosi, mentre fa il lavoro va in riduzione di flusso; il problema non è la riduzione di flusso nel braccio ma la riduzione, attraverso la vertebrale, nella parte posteriore del torace. Questo significa che ad un certo punto il sangue di fronte ad una biforcazione prende una strada piuttosto che un'altra. Nel nostro caso il sangue, ad una biforcazione, andrà dove sono più basse le resistenze. Il dipiridamolo è un potentissimo vasodilatatore. Al paziente che ha la stenosi del discendente anteriore e un circonflesso normale, se iniettiamo dipiridamolo, farà vaso dilatare anche il discendente anteriore. Sappiamo però che a valle di una stenosi ostruttiva il letto è già massimamente dilatato, quindi più di tanto non può fare. Succederà che il sangue in quel paziente quando arriva nel tronco comune troverà un circonflesso molto dilatato e un discendente anteriore che più di quanto è dilatato non può essere, per cui il sangue verrà deviato dal vaso dove ce ne sarebbe bisogno al vaso in cui arrivava già in condizioni normali più che sufficiente. Tale fenomeno viene definito furto coronarico. L'adenosina è il più potente vasodilatatore che esiste ed è anche il più rapido. L'adenosina si usa un po' meno perché bisogna saperla usare, c'è il rischio che se non si coordini bene iniezione e registrazione dell'esame non si abbiano risultati soddisfacenti. L'adenosina ha uneffetto depressivo sul nodo del seno e sul nodo atrioventricolare. Per reperti suggestivi di alto rischio si intende che di fronte a quadri di questo tipo c'è un'alta possibilità che il paziente abbia una malattia ischemica: - Diverse anomalie reversibili della cinetica segmentaria (in più di un territorio di vascolarizzazione); - Gravità ed estensione delle anomalie della cinetica segmentaria (alto indice di movimento globale della parete); - Dilatazione della cavità ventricolare grave ma reversibile; - Disfunzione ventricolare sinistra a riposo. I punti di forza di tale esame sono la sensibilità e la specificità più elevate rispetto al test ergometrico perché il test ergometrico dipende molto dal paziente: il paziente può non essere abituato, può andare nel pallone e quindi il test che sarebbe più ovvio e più immediato diventa un problema. Ci sono anche pazienti che fisicamente non lo possono fare perché hanno un'artrite deformante, una patologia della testa del femore, sono poliomielitici e altro. Quindi ci sono una serie di casi in cui il test da sforzo è impossibile da far eseguire, quindi l'ecostress, che già ha una maggiore specificità e sensibilità diventa la più semplice delle possibilità diagnostiche non invasive, rispetto ad una scintigrafia costa meno ed è anche meno rischiosa. L'ecostress è il punto di incontro tra efficacia diagnostica, costi e fattibilità. Il test da sforzo in un paziente ischemico può esitare in una fibrillazione ventricolare. L'eco da stress è ancora più operatore-dipendente. I limiti sono rappresentati da: - sensibilità diminuita per l’individuazione di una malattia di un vaso o di stenosi moderate con l’imaging post-esercizio; - incapacità a visualizzare tutto il ventricolo sinistro in alcuni pazienti; - altamente operatore-dipendente per l’analisi dell’immagine; - analisi dell’immagine non quantitativa; - inadeguata finestra acustica in alcuni pazienti (per esempio, per malattia polmonare cronica ostruttiva); - zona di ischemia peri-infartuale individuata. Test ergometrico: Test provocativo di ischemia, saggia il momento di interruzione dell’equilibrio tra apporto e richiesta di O 2 . I parametri frequenza cardiaca (FC), pressione arteriosa (PA) e doppio prodotto(DP) consentono di definire una soglia di angina. La soglia di angina non sempre coincide con la soglia ischemica. In presenza di ischemia succedono determinate cose, altre non si manifestano (come l'ECG e il dolore). Quando facciamo una prova da sforzo monitoriamo l'ECG e il paziente ci dirà se c'è o meno dolore quando si arriva ad una alta soglia da sforzo. La soglia ischemica è quella che riguarda il ventricolo, cioè quando il ventricolo andrà in crisi tra domanda e apporto; la soglia di angina è quando il paziente avrà il dolore o modifiche dell'ECG, le due cose non coincidono: può esserci l'una senza l'altra, o l'una prima dell'altra. Alla soglia di ischemia, il flusso coronarico non riesce più ad incrementarsi nonostante ci sia una maggiore richiesta di ossigeno. Può accadere che la soglia di quello che vediamo succeda un po' dopo, perché probabilmente il paziente, che noi classifichiamo ad un certo livello perché ci basiamo sull'angina, dal punto di vista della comparsa dell'ischemia è messo peggio di quello che pensiamo perché nel momento in cui ci dirà di avere l'angina da sforzo probabilmente la soglia ischemica l'ha già raggiunta e superata da un po' di tempo. Nella valutazione di un paziente con la cardiopatia ischemica (cronica o acuta), la cosa che assolutamente dobbiamo fare è la STRATIFICAZIONE DEL RISCHIO DEL PAZIENTE, cioè se ho 10 pazienti, tutti e 10 riferiscono angina da sforzo, non posso considerarli uguali, devo stratificarli per capire chi è peggio combinato perché bisogna definire delle priorità. Definire la stratificazione del rischio significa avere una serie di strumenti che utilizziamo per il singolo paziente e alla fine facciamo una scala. La prova da sforzo, l'ecostress, l'anamnesi, la valutazione dei fattori di rischio servono alla stratificazione. Questo è cruciale nella sindrome coronarica acuta, in cui il tempo è muscolo, prima riperfondo più muscolo salvo. Nell'ischemia cronica, il tempo è qualità di vita. Le controindicazioni del test da sforzo si dividono in assolute e relative. Quelle assolute sono: - Infarto miocardico acuto (entro 2 giorni); - Angina instabile a elevato rischio; - Aritmie non controllate dalla terapia che causino sintomi o compromissione emodinamica; - Stenosi aortica severa sintomatica; - Scompenso cardiaco sintomatico non controllato dalla terapia; - Embolia polmonare o infarto polmonare acuto; - Miocardite o pericardite acuta; - Dissezione aortica acuta. Quelle relative sono: - Stenosi nota del tronco comune della coronaria sinistra; - Stenosi aortica moderata; - Squilibri elettrolitici; - Pressione arteriosa sistolica >200 mmHg e/o diastolica > 110 mmHg; - Tachi o bradiaritmia; - Cardiomiopatia ipertrofica o altre condizioni associate a ostruzione del tratto di efflusso ventricolare; - Incapacità fisica o mentale a compiere un adeguato esercizio fisico; - Grado elevato di blocco atrio-ventricolare. Il tempo della prova da sforzo e il suo valore ai fini della stratificazione è che il paziente deve fare lo sforzo in base alla resistenza (cioè al lavoro da compiere): in base all'impostazione del lavoro, capisco a quale carico di lavoro il paziente ha dolore o mostrerà variazioni dell'ECG (es. 75-100 W fa la terapia farmacologica e poi dopo un mese si rifà la prova; 25 W subito coronarografia). Occorre considerare anche altri fattori di rischio come l'ipertensione, il diabete mellito, l'iperlipidemia, il fumo, per la stratificazione del rischio. La durata ottimale dell’esercizio dovrebbe essere compresa tra i 6 e i 12 minuti. La tolleranza allo sforzo si valuta in equivalenti metabolici (MET) i quali vengono calcolati dall’apparecchiatura utilizzata in base al protocollo di esercizio impiegato. Un MET corrisponde al consumo d’ossigeno in condizione basale, e equivale a 3,5 ml per Kg al minuto. Il livello di affaticamento percepito dal paziente si valuta mediante la scala di Borg (0-10). Il risultato può essere: - Negativo se il paziente non ha dolore né modifiche del tracciato (vero/falso); - Positivo se il paziente ha dolore o modifiche dell'ECG (il dolore da solo può farlo entrare in un'area “grigia”. So che deve venire prima il tracciato e poi il dolore. Se viene il dolore senza il tracciato potrebbe essere un dolore toracico di altra natura che lo sforzo elicita); - Insufficiente quando il paziente non è riuscito a fare lo sforzo per arrivare ai risultati (ho dei limiti che non posso superare, ad esempio se la FC non può superare i 150, a 149 mi devo fermare! Devo rispettare parametri, come anche la pressione sanguigna, che variano in base a sesso, età ed altri fattori). Parametri rilevabili dalla prova da sforzo dotati di un ruolo prognostico indipendente e significativo nella valutazione del rischio di mortalità, si distinguono in: - ELETTROCARDIOGRAFICI: Massima depressione del tratto ST, Massima elevazione del tratto ST, Tipo di sottoslivellamento del tratto ST (discendente, orizzontale, ascendente), Numero di derivazioni che presentano alterazioni del tratto ST, Durata delle alterazioni del tratto ST durante il recupero, Aritmie ventricolari indotte dall’esercizio, Durata dell’esercizio alla soglia ischemica; - EMODINAMICI: Frequenza cardiaca massima raggiunta, Massima pressione arteriosa sistolica raggiunta, Valore di doppio prodotto (FC x PA sistolica) allo stop, Durata totale dell’esercizio, Ipotensione indotta dall’esercizio, Incompetenza cronotropa; - RELATIVI AI SINTOMI: Angina indotta dall’esercizio, Sintomi indotti dall’esercizio, Durata dell’esercizio alla soglia di angor. Indicatori di elevato rischio rilevabili alla prova da sforzo sono: - 2,0 mm o più di sottoslivellamento del tratto ST; - 1,0 mm o più di sottoslivellamento del tratto ST nello stadio 1 dell’esercizio; - Sottoslivellamento del tratto ST per una durata superiore a 5 minuti durante il periodo di recupero; - Raggiungimento di un carico di lavoro inferiore a 4 MET o della frequenza cardiaca massimale sotto uno sforzo di lieve entità; - Alterata risposta pressoria; - Tachiaritmie ventricolari. Oltre alle modifiche del tracciato, si valuta anche la comparsa di altri sintomi, come ladispnea, aritmia, abbassamento brusco della pressione. ECG da sforzo: I punti di forza di tale metodica sono: costo basso; durata breve; valutazione dello stato funzionale; elevata sensibilità per la malattia di tre vasi o del tronco comune; fornisce informazioni prognostiche utili (per esempio, ischemia a basso carico). I limiti sono rappresentati invece da: sensibilità sub-ottimale; basso tasso di identificazione di una malattia di un vaso; non diagnostico in caso di anomalie dell’ECG di base; scarsa specificità in certe popolazioni di pazienti (per esempio, nelle donne in premenopausa); necessità di raggiungere l’85% o più della frequenza cardiaca massima per una buona precisione. Scintigrafia miocardica: È il test più costoso e più complicato, con un minimo di rischio in più ma dà altre informazioni rispetto all'ECG ed è più affidabile. Dà informazioni sulla perfusione e, con particolari traccianti radioattivi, può dare anche informazioni sulla funzione ventricolare (è come se avessimo una ventricolografia). Lo studio della perfusione indica esattamente la coronaria coinvolta. Il paziente fa uno sforzo fisico o un test farmacologico, all'acme di questo si fa l'iniezione di un mezzo di contrasto radioattivo che va dove vanno i globuli rossi. Le diverse parti del ventricolo si coloreranno di più o di meno in base alla quantità di tracciante che è arrivato. Poi faremo la stessa cosa dopo un certo tempo in condizione di riposo e vedremo la differenza tra sforzo e riposo. Indicatori di elevato rischio rilevabili alla scintigrafia miocardica sono: - Diverse anomalie di perfusione (quadro totale più difetti reversibili) in più di un territorio di irrorazione; - Gravi ed estesi difetti di perfusione (indice semiquantitativo del difetto); - Aumento della captazione polmonare del 201Tallio; - Dilatazione della cavità ventricolare sinistra transitoria dopo sforzo; - Disfunzione ventricolare sinistra alla tomografia computerizzata ad emissione fotonica. Con la scintigrafia è possibile ottenere dei parametri specifici, come la wall-motion e la wall-thickening. La wall-motion è il movimento del bordo endocardico e i programmi automatici la misurano in mm (0-10 mm). La wall-thickening è l’aumento dimensionale delle pareti miocardiche è viene misurata in percentuale dello spessore telediastolico. Passiamo ora ad illustrare le principali caratteristiche dei radiofarmaci. Il tecnezio colora le cavità, come un mezzo di contrasto, così si valuta la funzione ventricolare (come in una ventricolografia), il tallio invece fa vedere la perfusione perché segue il circolo coronarico fin nelle sue più piccole diramazioni, ma non come si muove il ventricolo. Adesso ci sono nuovi traccianti che fanno vedere le due cose contemporaneamente. Il tallio appartiene alla categoria degli isotopi radioattivi che ha una lunga emivita. I principali radiofarmaci utilizzati sono: - Il Tallio-201 cloruro viene estratto rapidamente dal sangue e trasportato nelle cellule miocardiche da un meccanismo di trasporto attivo che dipende dalla integrità della pompa sodio-potassio; viene iniettato durante sforzo, la sua distribuzione miocardica dopo 5 min. riflette la perfusione al momento della iniezione; Col tempo il tallio fuoriesce dalle cellule e si instaura un equilibrio dinamico; - Traccianti tecneziati come 99mTc-sestaMIBI (attualmente più utilizzato nella diagnostica), 99mTcTetrofosmin: dopo iniezione si distribuiscono in modo proporzionale al flusso ematico; la permanenza nelle cellule miocardiche e di alcune ore; vengono eliminati dal fegato. Come facciamo la scintigrafia da sforzo o da farmaci? C'è un'iniezione all'acme dello stress (la dose di tracciante radioattivo è definita da protocolli internazionali; nei laboratori privati si usa in genere una dose inferiore per cui l'immagine è più pallida). Quando noi riteniamo che all'interno di una zona necrotica ci possano essere delle aree cellulari sopravvissute (macchia di leopardo), il primo protocollo non dà il tempo al tracciante di raggiungere le cellule vitali presenti nell'area necrotica, per cui si fa una seconda iniezione e una rilevazione tardiva che può arrivare anche a 24h (se è vero che queste cellule sono vitali ma scarsamente funzionanti perché ricevono piccole quantità di sangue, è altrettanto vero che il sangue contenente il tracciante ci metterà un tempo molto lungo per arrivare, tempo che probabilmente è superiore ai tempi del primo protocollo), per cui per valutare quanto fosse frequente il fenomeno del miocardio vitale ma disfunzionale (la cellula è viva, ha un suo metabolismo, però, siccome se volesse funzionare alla perfezione, non avendo ossigeno a disposizione, andrebbe in anaerobiosi, sta ferma. Dove ci sono cellule morte il miocardio non si muove, dove ci sono cellule vive ma disfunzionali il miocardio non si muove lo stesso). Il test fatto a 24h serve a vedere se e quanto all'interno di quell'area ci sono cellule vitali (sono vitali perché si colorano e se si fa un'eco stress con la dobutamina, le cellule iniziano a muoversi e lo vediamo perché all'eco aumenta la wall-motion regionale e abbiamo la prova che se riperfondiamo quella zona guadagniamo, a causa della contrazione, un certo numero di cellule). La regola è stabilire, secondo canoni predeterminati dalle comunità scientifiche, una volta fatto il test, quanto guadagno, cioè quante cellule riesco a “stanare” e quindi se le recuperassi ad una normale funzione di quanto migliorerei la funzione globale del ventricolo sinistro. Perché? Perché stiamo parlando di una riperfusione che se fatta con l'angioplastica ha una minima quota di rischio, ma se fatta con l'intervento chirurgico la quota di rischio aumenta. La scintigrafia con la re-iniezione o l'ecostress con dobutamina servono per capire se c'è miocardio vitale, quanto ce ne sta e in funzione della quantità si decide se vale la pena recuperarlo o no.Nella perfusione miocardica normale grosse differenze tra stress e riposo non ci sono. Le indicazioni sono l’angina da sforzo e la cardiopatia ischemica cronica. E' difficile che andiamo a fare un esame del genere con sindrome coronarica acuta perché questa è già una prova che qualcosa non va all'interno del circolo coronarico. Con la scintigrafia, nel caso dell’angina da sforzo possiamo: - Fare diagnosi di ischemia miocardica acuta in pazienti con anamnesi o modificazioni elettrocardiografiche inaffidabili; - Valutazione della funzione ventricolare; - Determinazione del territorio vascolare ischemico per l’identificazione della lesione coronarica “colpevole”; - Valutazione funzionale della coronaropatia e prognosi in pazienti efficacemente stabilizzati con la terapia medica. Nel caso di cardiopatia ischemica cronica, la scintigrafia può: - Fare diagnosi in pazienti sintomatici e in pazienti selezionati con ischemia miocardica silente; - Valutazione della funzionalità ventricolare; - Pianificazione di PTCA, identificazione delle lesioni che causano ischemia miocardica; - Stratificazione del rischio prima di interventi chirurgici non cardiaci. Anche un pregresso infarto è una cardiopatia ischemica cronica: prevenzione secondaria è quella che si fa a chi ha già avuto un evento, per cui un paziente che ha avuto un infarto, una rivascolarizzazione, un'aritmia in fase ischemica, entra in una sorta di protocollo di controllo e rivalutazione. Nel paziente che ha già avuto un infarto non trattato adeguatamente, la scintigrafia diventa uno strumento prezioso perché ci darà a distanza di tempo la situazione del paziente. Esistono forme di infarto, anche transmurale, che non vengono colte al momento perché sono paucisintomatiche o hanno una sintomatologia diversa da quella che ci si aspetta. L'ECG può essere dubbio, l'eco e la scintigrafia danno risultati certi. La scintigrafia e la TCspirale delle coronarie fanno parte del bagaglio diagnostico per la stratificazione del rischio. La prevenzione primaria e quella secondaria sono una continua stratificazione del rischio. Ogni volta che si vede un paziente e se ne valuta lo stato (abitudini di vita, farmaci, peso, test) si fa stratificazione del rischio. La scintigrafia inoltre viene utilizzata per valutare l’andamento della terapia farmacologica, valutazione di pazienti sintomatici dopo PTCA o CABG, valutazione di pazienti asintomatici dopo PTCA o CABG. La sigla di PTCA sta per “angioplastica coronarica percutanea” mentre CABG per “coronaryartery bypass graft”. Osservate la figura in alto, il riquadro A. Vedete l'immagine in basso. E' la situazione di un paziente con un infarto e verosimilmente con ischemia peri-infartuale. Durante lo stress la zona che manca è molto ampia, in condizioni di riposo un po' della zona che durante lo stress non si colorava, si colora. Quindi c'è una zona che è necrotica e una vitale, che è in grado di captare il tracciante. Il difetto è fisso ma non per tutta l'estensione di questa “ciambella”, vale la pena programmare quindi un intervento dirivascolarizzazione. Al contrario, nella figura B c'è un difetto di perfusione fisso. Faremo una re-iniezione dopo 24h. I punti di forza della scintigrafia miocardica sono: - valutazione simultanea della perfusione e della funzionalità con la gated SPECT; - sensibilità e specificità più alte di quelle del test ergometrico; - l’esame può essere eseguito in quasi tutti i pazienti; - valore prognostico addizionale significativo (la negatività del test è comunque predittiva di prognosi benigna); - precisione comparabile con sforzo farmacologico; - vitalità e ischemia valutate simultaneamente; - analisi quantitativa dell’immagine. Mentre i limiti sono: - sensibilità sub ottimale con 201Tl; - tempo di procedura lungo con agenti marcati con 99Tc; - costo superiore rispetto al test ergometrico; - esposizione a radiazioni; - immagini di scarsa qualità nei pazienti obesi, la combinazione della valutazione della perfusione e della funzione ventricolare sinistra non incrementa la capacità prognostica delle singole indagini. INDAGINI INVASIVE Tra le indagini invasive si ricordano il cateterismo, l’angiografia e la coronarografia. Coronarografia: L’angiografia coronarica o coronarografia è una metodica radiologica che, mediante l’iniezione di mezzo di contrasto, consente la visualizzazione delle arterie coronariche, al fine di studiarne la morfologia, il decorso ed eventuali alterazioni. L’esame è eseguito in una sala operatoria dedicata chiamata Sala di Emodinamica (o Laboratorio di Emodinamica), dove sono presenti una serie di strumentazioni radiologiche dedicate allo studio del cuore, le attrezzature necessarie all’interventistica cardiovascolare e alla gestione di eventuali complicanze. Viene effettuato in anestesia locale e solitamente è bene accettata dal paziente. Il catetere viene fatto avanzare nelle arterie, in aorta e fino all'imbocco delle arterie coronarie dove l'iniezione del mezzo di contrasto"colora" e opacizza i vasi rendendoli visibili ai raggi X. E' un “Gold standard” usato per confrontare la sensibilità e la specificità delle diverse tecniche non invasive per identificare la presenza di stenosi coronariche, definire le opzioni terapeutiche, e valutare la prognosi dei pazienti con sintomi o segni di CAD. La gestione di eventuali complicanze prevede farmaci ed apparecchiature rianimative. Si usano arterie periferiche (femorale, radiale o omerale). Nella coronarografia selettiva si usa un catetere che si immerge nell'ostio coronarico sinistro e destro. La coronarografia è ben accetta dal paziente perché non è molto dolorosa e se noi mettiamo insieme tutte le possibilità di complicanze arriviamo a meno del 2% in tutti i pazienti che fanno la coronarografia (compresi quelli più critici). Le controindicazioni sono indicate nella figura a lato. Il rischio più frequente è legato al sito di entrata del catetere (70%). Non bisogna trapassare l'arteria perché il buco di sopra è impegnato dal catetere, l'altro farà uno stillicidio di sangue. Si può creare uno pseudoaneurisma o un vasto ematoma (che può essere anche pulsante, cioè continuamente rifornito dall'arteria che può indurre anche la necessità di un chirurgiavascolare). intervento Un di ulteriore complicanza può essere l’occlusione acuta del vaso per trombosi o embolia. Inoltre possono aversi anche reazioni vagali, con abbassamento della pressione arteriosa e della frequenza cardiaca dovuti a riflessi scatenati dalla puntura. Quando facciamo una coronarografia dobbiamo vedere piccoli vasi mentre si muovono (perché le coronarie stanno “montate” su muscolo) e dobbiamo vederli in un ambito piccolo perché il cuore non è grandissimo. La coronaria si adatta ad una determinata patologia, è tortuosa ed ha unandamento tortuoso, è piccola, si muove, per cui se facciamo una ripresa in una sola proiezione vediamo solo alcune cose. Quindi è necessario cambiare l'asse di incidenza del fascio radiogeno. Questo si fa attraverso le diverse proiezioni. E' bene che il paziente faccia la coronarografia una sola volta, perché comunque un rischio c'è. Passiamo adesso a commentare alcune proiezioni. Questa è la visualizzazione della coronaria di destra. A - Arteria coronaria destra in una proiezione obliqua anteriore sinistra. Patologie aritmiche ipocinetiche sono molto più frequenti in pazienti che hanno una patologia infartuale della coronaria destra (perché irrora il nodo seno-atriale). Un paziente che ha un blocco atrio-ventricolare e che ha anche l'ST sopraslivellato sta avendo un infarto legato all'occlusione della coronaria destra. B - Quando cambiamo proiezione (da obliqua sinistra passiamo a obliqua destra), l'immagine cambia. Vediamo la CRUX CORDIS, cioè la parte finale della coronaria destra che si divide in due grossi rami: il ramo interventricolare posteriore (che insieme a quello anteriore irrora il setto interventricolare) e il ramo postero-laterale (che insieme al ramo circonflesso irrora la parete postero-laterale). C - Nella proiezione laterale vediamo molto bene il marginale acuto. Abbiamo essenzialmente due tipi di marginale: il marginale ottuso, che nasce dalla coronaria sinistra, e il marginale acuto, che nasce dalla coronaria destra. D - I rami del discendente posteriore vanno dal basso verso l'alto. Questa è la visualizzazione del percorso della Arteria coronaria sinistra. A - Il caudale ci fa sempre vedere molto bene l'origine del vaso che analizziamo in quel momento.La craniale ci fa ben vedere la porzione media e quella terminale che è importante per il chirurgo che per impiantare il bypass ha bisogno di un'area sana, non si può mettere il graft sia venoso che arterioso in corrispondenza di un restringimento significativo o di una placca, altrimenti il graft (che dovrebbe essere un tubo ausiliario che porta sangue) non avrà mai la possibilità di funzionare adeguatamente. Nella visione caudale anteroposteriore dell'LCA IMMAGINE vediamo bene il tronco comune, il discendente anteriore e il circonflesso. Ad un certo punto c'è una sovrapposizione dei due vasi per cui sarebbe difficilissimo vedere una patologia dell'uno o dell'altro o di tutti e due in questa zona. B - Poiché il discendente anteriore si fa tutto il solco interventricolare, i rami settali non sono esattamente paralleli tra di loro come nel discendente posteriore perché sono legati alla curva che fa il ramo. Man mano che curva il discendente anteriore, cambia l'angolo di origine dei rami settali. Nella visione craniale si vede bene la porzione mediana e terminale dei vasi, l'origine si sovrappone. È la zona dove il chirurgo fa atterrare il graft, quindi deve sapere se è in grado di accettarlo. C - Dopo l'origine del primo ramo diagonale c'è un lungo e significativo restringimento. C'è ungrosso ramo settale che origina dalla zona malata, quindi c'è una ridotta perfusione della parete antero-laterale dell'apice, ma anche della parte superiore del setto interventricolare. D - Spider view perché vediamo il tronco comune, quindi l'origine della coronaria sinistra e la biforcazione (se c'è una patologia in questa zona, tre quarti del ventricolo sinistro è a rischio). Questa visione può far propendere per un'angioplastica o una rivascolarizzazione. La dominanza dipende dal ramo interventricolare posteriore. Otto volte su dieci questo ramo origina dalla coronaria destra (circolazione destra dominante), in altri casi origina dal ramo circonflesso e quindi è sinistra-dominante. La coronaria destra dominante gode della nostra massima attenzione, cioè decidiamo se e cosa fare sulla coronaria destra a seconda se è dominante o no. Se non è dominante, la coronaria destra irrora soltanto il ventricolo destro (il ramo destro non dominante si lascia di solito così com'è anche perché di solito è piccolo e non sviluppato, quindi sarebbe anche complesso lavorarci sopra). I graft sono o in vena (in genere la safena che si isola e si capovolge, perché ci sono le valvole) o i grafts arteriosi (essenzialmente le due arterie mammarie, ma anche le radiali), che sono ideali, perché il graft venoso va incontro ad un processo di deterioramento perché essendo nato per fare la vena, è abituato quindi a lavorare a bassa pressione (18-20 mmHg), mentre qui lo si mette a funzionare a 120-140 mmHg. I grafts arteriosi sono eterni, non funzionano solo se sono stati trattati male al momento dell'impianto. I grafts venosi dopo 10 anni, solo in un terzo dei casi ancora funzionano bene. Il graft è più grande della coronaria, ma a differenza delle coronarie non ha rami. Capiamo che il graft è arrivato sulla coronaria perché c'è un tubo piuttosto grande da cui non origina niente che improvvisamente diventa un vaso un po' più piccolo da cui originano rami. Quando il tubo che non dà origine a niente, si incontra con qualcosa che dà origine a vasi, quello è il punto in cui il graft è stato anastomizzato sulla coronaria. La mammaria si isola dalla parete toracica, la si lascia in situ e la si attacca sulla coronaria. Per vedere la mammaria dobbiamo iniettare il contrasto nella succlavia. La differenza è che la mammaria è un vaso arterioso in situ, che può avere collaterali, che devono essere chiusi perché altrimenti potrebbe esserci un “furto” di sangue. Basti citare un caso clinico: Uomo che ha avuto rivascolarizzazione con mammaria che aveva angina da sforzo collegata ad attività fisiche che comportassero l'utilizzo del braccio sinistro. La scintigrafia diceva che c'era ischemia anteriore, la discendente anteriore alla coronarografia non si vedeva,quindi la mammaria doveva avere qualche problema. Il problema non era la mammaria ma la succlavia, che aveva una lesione sub-occlusiva, per cui tutte le volte che faceva un'attività che richiedeva un afflusso di sangue nell'arto superiore sinistro, questo accadeva a scapito della mammaria: il sangue veniva furtato. Per cui è bastata un'angioplastica della succlavia (la mammaria si inoscula sul discendente anteriore, che a sua volta non ha problemi). IVUS – ultrasuoni intravascolari: E' un'eco fatto all'interno di un vaso. Siccome gli ultrasuoni vengono inviati a 360°, possiamo avere un perfetto disegno del lume del vaso. Una placca calcifica si vede molto bene (il calcio si vede di bianco forte). Ci sono casi in cui l'IVUS cambia la diagnosi e cambia anche il trattamento. Ad esempio si usa prima di fare l'angioplastica di un tronco comune (l'eco fa vedere l'estensione della malattia, il tipo di placca, se c'è calcio, quanto ce ne sta, se il calcio è circonferenziale o no, perché con il calcio circonferenziale lo stent non si riesce a mettere dato che la resistenza sarà su tutta la superficie interna del vaso, invece se è un arco di cerchio lo stent potrà espandersi in maniera efficace. Virtual histology: Le immagini in bianco e nero possono venire colorate con un software che ci dice in funzione del colore cosa vediamo (verde → tessuto fibroso; giallo → tessuto fibro-adiposo; bianco → calcio; rosso → core necrotico). L'abbondanza di tessuto fibroso rende la placca stabile. Una placca con molto tessuto necrotico si può facilmente ulcerare e può dare origine più probabilmente di altre a una sindrome coronarica acuta. CENNI FINALI DI TERAPIA L’Optimal medical therapy si effettua con antiaggreganti; nitrati; beta-bloccanti; statine; terapia dei fattori di rischio, come anti ipertensivi se paziente è iperteso, ipoglicemizzanti se diabetico. Per anni ci si è chiesto se facendo rivascolarizzazione nel paziente con angina cronica cambiava il destino del paziente in termini di sopravvivenza o solo la qualità della vita. Sono stati numerosi gli studi condotti a questo proposito. Il prof. Piscione ha effettuato al riguardo un lavoro, circa 5-6 anni fa. C’è un popolazione di pazienti con un vaso cronicamente chiuso: non passa sangue, si decide di riaprirlo con angioplastica coronarica. Il fatto di riaprirlo è un vantaggio o è solo una prova muscolare (= quanto sono bravo a riaprire un vaso chiuso da 4-510 anni), una manifestazione di abilità tecnica? Quando andiamo a vedere la mortalità cardiaca per infarto o semplicemente la sopravvivenza, si vede che i pazienti che hanno avuto l’apertura a 3 anni sono quasi tutti vivi; quelli che non hanno avuto la riapertura, a partire dai 6 mesi cominciano a “perdere pezzi”: questa è una ulteriore dimostrazione che avere un vaso che funziona perfettamente, che riceve in qualunque momento della giornata e in qualunque situazione un’adeguata quantità di sangue è di gran lunga molto meglio, anche ai fini della sola sopravvivenza, che avere un vaso che zoppica o peggio che non funziona proprio. In questo studio tutti i pazienti avevano un vaso chiuso e tutti partivano dal dover essere rivascolarizzati. Siccome la rivascolarizzazione di un vaso cronicamente occluso riesce nel 60-70 % dei casi, il paradosso qual è? Se c’è un paziente con un vaso chiuso, se si riesce ad aprire, il paziente riceve un grande beneficio. Se non si riesce a riaprirlo, però comunque si è provato a farlo, il paziente starà peggio! A quel punto sta meglio un paziente con un vaso chiuso per il quale si è deciso di non fare niente. Quindi riaprire un vaso cronicamente chiuso significa o che il medico pensa di poterci realmente riuscire oppure bisogna mettere in conto che se non ci si riesce, un bypass poi si deve fare. Le metanalisi sono l’insieme di una serie di studi randomizzati. Quale è il vantaggio di una metanalisi, soprattutto se si fa partendo da dati singoli? Ho 10 metanalisi, per un totale di 20000 pazienti, mi faccio dare il database di tutti e 20000 pazienti, caso per caso e su quelli faccio l’esame statistico- Questo approccio di studio è difficile perché richiede un grosso lavoro sia per avere i dati che per elaborarli. Molto spesso la metanalisi si fa mettendo insieme le medie. Ad es: la prima metanalisi dice che ci sono un tot di morti in pazienti che fanno l’angioplastica e un tot di morti in pazienti che non la fa; poi c’è la seconda, la terza e così via. Metto insieme tutti i dati di mortalità nel “braccio” che ha fatto l’angioplastica, e tutti i dati di mortalità nel “braccio” che non l’ha fatta. Faccio lo stesso anche per l’infarto, la riospedalizzazione, per la necessità di nuove rivascolarizzazioni. Alla fine metto insieme delle medie e ne faccio uscire una media che è la media di più pazienti. Allora ho il vantaggio di vedere i risultati di molti più pazienti. Nella metanalisi del professore, la mortalità è significativamente più bassa nei pazienti che hanno avuto l’angioplastica e l’infarto non fatale è più basso, non significativamente però, ma comunque più basso, nei pazienti che hanno rivascolarizzato. E se vediamo i singoli pazienti,vediamo che tutto sommato molti di questi sembrano stare ugualmente sia dalla parte della terapia medica che dalla parte della rivascolarizzazione. Per fortuna siccome non ce n'è nessuno o quasi che sta di più dalla parte della terapia medica, bastano questi due che evidentemente hanno una discreta popolazione di pazienti che stanno da questa parte (verso la rivascolarizzazione) che fanno propendere verso una efficacia. Quindi qual è il messaggio? La metanalisi ci dà delle informazioni o meglio ci permette di tirare una informazione da una popolazione molto grande, ma può anche essere disreading (cioè una metanalisi di questo tipo dà un messaggio però bisogna considerare che quel messaggio è decisamente influenzato da alcuni studi più che da altri). Allora siamo proprio sicuri che se io piglio la mia popolazione di pazienti questa popolazione seguirà quei due studi favorevoli alla rivascolarizzazione o seguirà invece quella moltitudine per cui effettivamente io gli faccio o non gli faccio l'angioplastica non mi cambia la natura delle cose? Una cosa importante però è che noi finora abbiamo parlato di pazienti che avevano la cardiopatia ischemica. Allora introduciamo un concetto che ci servirà moltissimo per quello di cui cominceremo a parlare dopo, cioè le sindromi coronariche acute. Noi non dobbiamo fare una valutazione esclusivamente anatomica: hai tante stenosi o hai tanto di riduzione del lume del vaso per cui devi per forza essere sintomatico, devo per forza farti qualche cosa. No. Noi usando tutti glistrumenti a nostra disposizione dobbiamo valutare il potere ischemizzante di una stenosi. Quella stenosi indipendentemente da quanto è in percentuale, provoca una ischemia o no?Perché se la provoca vale tutto quello che ci siamo detti, se non la provoca che glielo faccio a farel'intervento? Gli dò solo i rischi dell'intervento e alla fine vado a fare la prova da sforzo, la scintigrafia e sarà esattamente uguale a come era prima dell'intervento. Quindi non sarà servito a niente, anzi avrà peggiorato la situazione. E quindi vedete che alla fine se noi inseriamo nella stratificazione prognostica questo semplice apparentemente banale concetto, cioè quanta ischemia fai, allora possiamo esser certi che le nostre decisioni influenzeranno o meno il destino del nostro paziente. Se facciamo l'angioplastica a un paziente che ha la lesione alla coronarografia ma che non ha l'ischemia inducibile, la percentuale di morte di questo paziente è molto più alta rispetto a quello che invece ha fatto la terapia medica. Veramente io gli vado a dare fastidio nel senso peggiore della parola. Cioè lo faccio morire di più semplicemente perché ho basato la mia decisione terapeutica solo sulla anatomia radiografica: vedo la stenosi, faccio il trattamento e i risultati sono pessimi. Invece già quando compare 1-5 % di ischemia, già la differenza c'è. Ci vuole un po' di più con l'angioplastica ma non tanto di più. Dopo di che quando arriviamo all'ischemia degna di questo nome cioè dal 10 al 20 % si abbassa, quando arriviamo ad una ischemia al di sopra del 20 % crolla, esattamente lo specchio di quello che accadeva con lo 0%. Nel caso dello 0% facevamo morti con la pala se facevamo l'angioplastica, invece nel caso del 20% facciamo morti con la pala se non gliela facciamo. Quindi a questo punto non basta dire che c'è la stenosi, bisogna dire: c'è la stenosi e c'è l'ischemia. Siccome c'è l'ischemia, questo paziente deve essere trattato, deve essere rivascolarizzato. Diventa l'ischemia il fattore predittore di eventi, non la lesione coronarica. Ecco perché la coronarografia deve venire alla fine. Altrimenti c'è il rischio veramente che il medico in presenza di una prova da sforzo dubbia, di una scintigrafia negativa, esegue una coronarografia, trova una stenosi e subito senza pensare gli fa una angioplastica. Sbagliatissimo. A parte l'eco c'è una indagine che noi possiamo fare in corso di coronarografia, una indagine invasiva che si chiama Fractional flow reserve (FFR) che mi serve per valutare sul momento la capacità ischemizzante della stenosi. Allora di fronte a una situazione del genere, col paziente che non ha una chiara storia o una chiara ischemia inducibile però con una stenosi che grida vendetta, prima di rinunciare a fare la rivascolarizzazione si procede alla FFR (che è banale: mi fa il rapporto tra la pressione a valle e a monte della lesione prima in condizioni normali, poi a seguito di una iniezione di adenosina, il vasodilatatore in assoluto più potente): se quella lesione è realmente ischemizzante a valle della lesione c'è già una vasodilatazione, quindi: - se la lesione non è ischemizzante la pressione scende in maniera uguale a monte e a valle della stenosi; - invece se il vaso è già massimamente dilatato, a valle non succede un accidente e quindi alla fine io avrò un semplice numero che è il rapporto tra le due pressioni e il valore discriminante è 0,65. Cioè ho un apparecchietto particolare con un suo algoritmo, inizialmente mi dice il rapporto tra le due pressioni in condizioni di base. Poi si fa una iniezione intracoronarica o intravenosa di Adenosina e mi rimisura il rapporto. Se esce un valore di 0,65 o meno, la lesione è sicuramente ischemizzante. C'è una serie di lavori che dimostrano che se trattiamo questi pazienti vanno bene, se non li trattiamo vanno male. Se invece è tra 0,65 e 0,80 è la zona grigia, se è superiore a 0,80 la lesione non è ischemizzante e gli stessi lavori dimostrano che questi pazienti, indipendentemente dalla stenosi, se li lasciate tranquilli, restano tranquilli per un lungo periodo di tempo. Quindi il messaggio finale di tutto quanto detto è: Se non c'è l'ischemia, sta bene così! CAPITOLO V: SINDROMI CORONARICHE ACUTE I pazienti con cardiopatia ischemica si dividono in due grandi gruppi: quelli con malattia coronarica cronica, che più comunemente si presentano con angina stabile e quelli con sindromi coronariche acute. Il secondo gruppo, a sua volta, è costituito da pazienti colpiti da infarto miocardico acuto (IMA) con evidenza di sopraslivellamento del tratto ST all’elettrocardiogramma (ST-segment elevation myocardial infarction, STEMI), e da pazienti con angina instabile e con infarto miocardico (AI/IMA) senza sopraslivellamento del tratto ST (NSTEMI). Allora,cominciamo a vedere quale è l'entità del problema. Nell'anno 2001 la principale causa di morte sono le malattie cardiovascolari comprese le vascuolopatie cerebrali seguite dalle neoplasie. Per la successiva causa di morte, i traumi accidentali, c'è un crollo; poi viene l'insufficienza respiratoria e via dicendo. Attenzione il diabete mellito è l'unico di questi per cui è previsto un incremento: cioè entro il 2050 sono previsti dei decrementi di tutte le cause di morte tranne per il diabete. Si inverte: l'ultimo diventerà il primo, e speriamo che il primo diventi l'ultimo! Vedete che vale lo stesso per maschietti e femminucce, l'infarto non guarda in faccia. Questi invece sono i decessi per patologia cardiovascolare in percentuale: riconosciamo il 54% dei decessi per malattia coronaria all'interno delle patologie cardiovascolari, più della metà dei decessi è dovuta a una malattia coronarica! Poi c'è lo scompenso, la vascolopatia cerebrale e altro. Ma come principale causa di mortalità, le forme acute di malattia coronarica la fanno da padrone. Questo sempre per darvi un'idea: corrispondono a 1,57 milioni di ricoveri ospedalieri e di questi 1,24 milioni è data dall'angina instabile soprattutto da infarto miocardio ma del senza sovraslivellamento del tratto ST (NSTEMI), mentre 0,33 milioni da miocardio infarto del con sovraslivellamento del tratto ST (STEMI). Diciamo subito una cosa: i pazienti STEMI se muoiono, muoiono subito, quindi per questi pazienti il livello di attenzione e il livello di aggressività deve essere molto alto. Invece i pazienti NSTEMI salvo casi particolari non muoiono subito, ma muoiono con la pala negli anni successivi. Quello che non deve accadere è che questo paziente che è meno delicato venga considerato figlio di un Dio minore, perché se lo consideriamo figlio di un Dio minore poi quando andiamo a tirare le somme troviamo dei gravi vuoti. Quindi questo è già un tipo diverso di atteggiamento: cioè nei confronti dello STEMI fucile puntato, riguardo lo NSTEMI fucile a palla però pronto a sparare. Perché li chiamiamo così? In passato le classificazioni erano diverse, c'era l'infarto non-Q e l'infarto Q. Adesso avendo a disposizione i marker enzimatici veloci, noi a che cosa ci riferiamo? All'ECG, perché l'ECG lo vedo immediatamente com'è, se è come a sinistra o come a destra nell'immagine. In tutti e due i casi avrò un innalzamento dei markers, perché in tutti e due i casi ho avuto perdita di tessuto, diciamo pure che nel primo caso, nel NSTEMI, questa perdita di tessuto sarà minore che nel secondo caso, salvo le dovute eccezioni. Però il messaggio fondamentale che mi dà subito l'ECG qualè? Che io nel caso del NSTEMI ho una ischemia subendocardica. La prima volta che noi abbiamo parlato della fisiopatologia a proposito di Laplace abbiamo detto che la parte di tessuto ventricolare più debole o se volete più sensibile è il subendocardio. Lì parlavamo di forza, di tensione, di spinta, di sovraccarico di pressioni e di volume. Qui diciamo la stessa cosa ma a livello elettrico e quindi metabolico. Quindi allora noi nel primo caso che cosa abbiamo? Innanzitutto, in tutti e due i casi noi abbiamo del materiale trombotico. Però vedete che nel NSTEMI insieme alla palla nera c'è anche un fondo di un bel blu notte, che è il lume pervio del vaso. Cioè c'è la palla ma non chiude completamente il lume vasale e quindi non interrompe completamente il flusso coronarico. Nello STEMI invece il materiale trombotico chiude tutto. Allora nello NSTEMI noi abbiamo una riduzione del flusso coronarico e quindi una riduzione brusca della quantità di ossigeno disponibile. Nello STEMI invece abbiamo una abolizione, interruzione del flusso coronarico e quindi della quantità del flusso disponibile. Dal punto di vista del risultato, nel caso a sinistra diciamo“no elevation” però di fatto potremmo dire “downsloping”, cioè non si alza ma si abbassa l'ST, invece nel caso di destra diciamo “elevation”, cioè l'ST si innalza almeno 3 o 4 quadratini. Questo è il primo modo di approcciare il paziente: gli facciamo l'ECG e seguiamo l'una o l'altra pista. Quindi la regola è: - STEMI, massima attenzione per la vita del paziente; - NSTEMI, un buon livello di attenzione, soprattutto per il futuro, poiché comunque è un soggetto a rischio. Questi sono una serie di studi in cui dimostrano come fino ad un certo punto la curva azzurra dello STEMI è maggiore di quello del NSTEMI, poi a 30 giorni si invertono: lo STEMI raggiunge una sorta di plateau e il NSTEMI continua a salire.Studi randomizzati su grosse popolazioni, un migliaio di pazienti seguiti nel tempo. Nell'ambio dell'ECG oltre all'ST c'è un altro segnale per noi estremamente importante in queste patologie che è il blocco di branca sinistra. Perché il blocco di branca sinistra può essere il modo di manifestarsi dell'infarto del miocardio. E vedete che è anche il modo più fetente di manifestarsi, perché quando noi andiamo a vedere la sopravvivenza a distanza a 4 anni, quelli che sopravvivono di più sono gli STEMI che sono sopravvissuti alla fase acuta, i NSTEMI vanno peggio ma peggio di tutti e due vanno i pazienti che hanno avuto il blocco di branca sinistra. Questo se voi ci riflettete è anche abbastanza logico, perché mentre con i primi due è difficile sbagliare la diagnosi, col blocco di branca sinistra uno potrebbe dire: “no ma questo potrebbe essere un fatto transitorio, quindi aspettiamo, vediamo, diciamo eccetera” quindi alla fine il paziente che si presenta con il blocco di branca sinistra probabilmente subisce un trattamento, una strategia riperfusiva più blanda, più tardiva quindi è chiaro che alla fine paga pegno perché noi nella fase in cui potevamo salvare il miocardio non l'abbiamo fatto. E' più subdolo: nella fase in cui dovevi prendere la decisione, il blocco di branca probabilmente ti ha portato fuori diagnosi. Noi abbiamo parlato di mortalità nella cardiopatia ischemica cronica, solo alla fine abbiamo detto che ci vogliono grandi numeri e molti anni di tempo per poter cogliere queste differenze. Infatti vediamo nell'immagine sotto che se mettiamo la curva dell'infarto acuto, dell'angina stabile e dell'angina instabile, l'angina stabile praticamente è una linea dritta o comunque tende molto poco verso il basso. La curva dell'infarto acuto invece parte da valori molto alti che poi si attenuano col tempo. ANGINA VARIABILE DI PRINZMETAL L’angina variabile di Prinzmental insorge a riposo ed è caratterizzata da vasospasmo; gli attacchi di angina non sono legati a sforzi e rispondono a vasodilatatori e calcio antagonisti. Il quadro clinico è sovrapponibile a quelli con le altre forme di anginainstabile. All’ECG si riscontrano sopraslivellamenti ST, segno di ischemia transmurale; questo sopraslivellamento ST ritorna normale con la cessazione del dolore. Questo riscontro può essere effettuato anche con l’Holter, utile per registrare anche tachicardie ventricolari o extrasistoli. Il test all’ergometro risulta spesso negativo. Alla coronarografia presentano una stenosi coronarica critica. Terapia: I nitrati e i calcio-antagonisti sono i principali farmaci utilizzati nei pazienti con angina variante. La nitroglicerina endovenosa o sublinguale spesso fa cessare prontamente gli episodi di angina variante e i nitrati a lungo termine sono utili per prevenire le recidive. I calcio-antagonisti sono estremamente efficaci nel prevenire lo spasmo coronarico dell'angina variante e dovrebbero essere somministrati alla massima dose tollerata. Anche la prazosina, un bloccante selettivo alfa-adrenergico, si è dimostrata utile in alcuni pazienti, mentre l'acido acetilsalicilico può aumentare la gravità degli episodi ischemici. La risposta ai beta-bloccanti è variabile. La rivascolarizzazione coronarica può essere utile nei pazienti con angina variante che presentano anche distinte lesioni ostruttive prossimali. Prognosi: Molti pazienti con angina variante attraversa una fase acuta e attiva, con frequenti episodi di angina ed eventi cardiaci nei primi sei mesi dopo la presentazione clinica. La sopravvivenza a cinque anni è eccellente (circa 90-95%). I pazienti senza evidenza di ostruzioni o con lieve stenosi coronarica tendono ad avere un decorso benigno rispetto a quelli con lesioni ostruttive gravi. Un infarto miocardico non fatale si verifica in una percentuale che giunge fino al 20% dei pazienti in cinque anni. Nei pazienti con angina variante che sviluppano un'aritmia importante durante gli episodi spontanei di dolore hanno un rischio più elevato di morte cardiaca improvvisa. Nella maggior parte dei pazienti che sopravvivono ad un infarto o al periodo iniziale di tre-sei mesi con frequenti episodi, le condizioni si stabilizzano e col tempo si registra una tendenza alla diminuzione dei sintomi e degli eventi cardiaci. ALTRI TIPI DI ANGINA Per angina intricata si intende un’angina stabile, con patologia coronarica dimostrata, associata a una patologia extracardiaca che modifica il quadro clinico sia la prognosi: patologie digestiva e scheletrica. Si presenta soprattutto di notte, irradiandosi all’epigastrio e posteriormente alle spalle. Si riconoscono due possibili cause: stimoli vasocostrittori a partenza dal tratto gastroenterico o scheletrico irritato, con sommazione algogena. L’ischemia miocardica silente si riferisce a episodi di ischemia documentata all’ECG senza sintomatologia associata. La terapia si basa principalmente sulla rivascolarizzazione. La sindrome X, infine, è caratterizzata da angina di petto tipica, anche sotto sforzo, documentata all’ECG senza segni di alterazione coronarica. La causa è attribuibile ad una ridotta soglia algogena ed una generica alterazione della muscolatura liscia micro-vascolare, associata ad alterazioni del tono vascolare. Questa sindrome prende, infatti, il nome di angina micro-vascolare. ANGINA INSTABILE La diagnosi di angina instabile è basata soprattutto sulla presentazione clinica. L'angina instabile viene definita come un'angina pectoris o un disturbo ischemico equivalente con almeno una delle seguenti tre caratteristiche: - compare a riposo o con un minimo sforzo e di solito dura oltre 10 minuti; - è intensa e di recente insorgenza, cioè entro le quattro-sei settimane precedenti; - si verifica seguendo un pattern in crescendo. La diagnosi di NSTEMI viene stabilita se un paziente con le caratteristiche cliniche di angina instabile presenta segni di necrosi miocardica evidenziati dall'aumento degli enzimi di danno miocardico. FISIOPATOLOGIA DELLA SINDROME CORONARICA ACUTA Noi qualcosa l'abbiamo già detta. Abbiamo queste possibilità per avere una sindrome coronarica acuta. Sindrome coronarica acuta significa che noi abbiamo una interruzione brusca o una riduzione brusca e severa del flusso coronarico. Quindi qua c'entra poco o c'entra molto meno l'entità della stenosi, c'entra molto di più un trombo non occlusivo su una placca preesistente. Nella stragrandissima maggioranza dei casi questa è la patogenesi di una sindrome coronarica acuta. Però possiamo avere una ostruzione dinamica. Noi abbiamo parlato di tono coronarico: se il tono coronarico aumenta e si riduce il lume vasale, questa riduzione può arrivare fino alla chiusura, cioè l'equivalente di mettere un clump sulla coronaria. Naturalmente il problema è sempre quello: se lo spasmo è momentaneo allora è facile da riconoscere e da trattare, visto e non visto. Ma in casi rari lo spasmo può essere prolungato o andare complicare una situazione come la precedente, cioè: il trombo non sarebbe stato occlusivo, se il vaso fosse rimasto nel suo lume normale, ma siccome è sopravvenuta una vasocostrizione quel trombo è divenuto occlusivo o quasi completamente occlusivo. Poipossiamo avere una ostruzione meccanica progressiva: voi sapete che quando si fa una angioplastica il risultato immediato è quasi sempre ottimo, poi però c'è il problema della restenosi, restenosi dopo pallone, dopo stent eccetera. Il problema è che noi per anni abbiamo considerato la restenosi un processo inevitabile se si faceva l'angioplastica ma tutto sommato benigno, si forma un nuovo tipo di placca all'interno di quel vaso che provoca inevitabilmente una ischemia di tipo cronico. Invece no! Noi oggi, dopo quasi 35 anni di angioplastica, abbiamo l'esperienza che ci consente di dire che circa il 20% delle restenosi si presenta sottoforma di sindrome coronarica acuta, cioè una restenosi su 5 può fare anche l'infarto! Quindi non si presenta come cardiopatia ischemica cronica ma come sindrome coronarica acuta. Poi lo abbiamo detto ci possono essere le forme secondarie che difficilmente possono dare l'infarto, però io mi ricordo di aver visto l'infarto in un paziente che si era trovato in un ambiente in cui c'era monossido di carbonio. Era sopravvissuto alla morte per asfissia ma si era fatto un infarto del miocardio, perché la sostituzione del monossido di carbonio all'ossigeno non era stata tanto lunga da farlo morire ma era stata abbastanza lunga da provocargli una significativa e abbastanza prolungata riduzione della quantità di ossigeno disponibile. Sono episodi un po' più rari ma possibili: mi è capitato anche di vedere un infarto da una profusa emorragia, ovviamente in quel caso ti devi occupare prima dell'emorragia e poi dell'infarto perché il primum movens è l'emorragia. Allora quale è il problema? Il problema è che quando parliamo della sindrome coronarica acuta come manifestazione clinica noi stiamo parlando della punta di un iceberg. Che significa? Significa che più che mai a questo fine la prevenzione primaria e la prevenzione secondaria sono cruciali. Cioè quando io avrò le manifestazioni cliniche della sindrome coronarica acuta saranno successe già una serie di cose sulla quale io avrei potuto svolgere un intervento efficace di prevenzione. Quindi attenzione, cioè vedete come noi alla stratificazione del rischio in un paziente che è già malato che è venuto da noi con dei sintomi dobbiamo aggiungere più che mai in questo caso il concetto di stratificazione del rischio in un paziente che apparentemente sta bene. Le manifestazioni, che siamo soliti riconoscere, come il dolore, le modifiche emodinamiche sono la punta dell’iceberg e sotto ci sono una serie di alterazioni, come le placche coronariche, forti induttrici dell’attività piastrinica, l’infiammazione e una serie di fattori, che sono coinvolti nella sindrome coronariche acute. Quest’immagine corso del descrive tempo la nel lesione aterosclerotica di un’arteria, una coronaria e come questa possa evolvere in due maniere, cioè la placca, che cresce e che si organizza in un trombo calcifico,con una capsula fibrosa ed è quella che abbiamo visto nella coronopatia cronica, e un’altra evoluzione possibile: si forma una capsula sottile,che si può autolesionare. Qual è la differenza? Nel primo caso c’è una capsula fibrosa spessa, con uno spesso strato di cellule muscolari liscee, un core lipidico con i macrofagi carichi di lipidi. Invece, nel secondo caso la capsula è molto sottile ed il core lipidico si trova subito al di sotto. Per cui c’è una differenza su base anatomo-patologica fra angina da sforzo, cardiopatia ischemica cronica e sindrome coronarica acuta. Per cui si tratta di un malato cronico, che si abitua alla sua malattia e noi ci abituiamo alla sua sintomatologia, mentre in quest’altro caso possiamo avere il passaggio da uno stato di benessere alla morte nel giro di qualche minuto o meno. È chiaro che se il soggetto cade a terra, il processo non è iniziato in quel momento, ma qualche attimo prima. Nel momento in cui dice: “sto male”, il trombo sta occupando una quantità di spazio enorme e cade a terra, perché l’ischemia ha innescato una serie di meccanismi, che lo hanno portato a morte repentina ed è probabile che ci sia una fibrillazione ventricolare, piuttosto che una rottura di cuore. Per cui il messaggio importante è che l’una è espressione di un fenomeno cronico, che si sviluppa nel tempo, mentre l’altra di un evento rapido, che può causare subito morte. Da studi di coronarografia dopo un evento acuto, abbiamo appreso che, a differenza di 15 anni fa, non c’è nessuna relazione tra entità della stenosi e sindrome coronarica acuta, cioè si pensava che ci volesse una stenosi piuttosto significativa, quando poi si determinava il fenomeno della trombosi, accompagnato dal fenomeno della vasocostrizione, siccome lo spazio tra stenosi e placca era modesto, bastava un nulla. Il 70% delle sindromi coronariche acute è causato da stenosi inferiori al 50%, questo significa che noi facendo una coronarografia o una angio-TC delle coronarie non possiamo dire che quella placca sicuramente darà un infarto e partiamo già con un’angioplastica. Per cui se noi facessimo un’angioplastica di tutte le lesioni inferiori al 50%, i problemi, legati alla procedura e quelli legati al fenomeno della ristenosi, farebbero sì che questi pazienti abbiano un destino peggiore rispetto al non-intervento. Questo vuol dire che in questo caso è molto più importante la prevenzione primaria e secondaria, perché su queste lesioni inferiori al 50%, che potrebbero diventare instabili, non abbiamo poteri. A qualcosa servono le statine, perché è stato dimostrato che la capacità di riduzione del processo aterosclerotico è molto più marcata per le placche inferiori al 50%. Questo perché sono placche con una storia breve, che non hanno avuto il tempo di organizzarsi, per cui sono passibili di riduzione, perché la statina agisce sulle LDL, che sono il motore di crescita delle placche. Per cui con le statine possiamo avere riduzione delle dimensioni e del numero delle placche. Per cui per avere una sindrome coronarica acuta basta una qualsivoglia placchetta. Non è la dimensione della placca, ma la sua natura dal punto di vista anatomopatologico a decidere il processo. Che cosa succede quando c’è contatto tra core lipidico e torrente circolatorio? Sono coinvolte le piastrine. In crescendo abbiamo adesione piastrinica, attivazione piastrinica e aggregazione piastrinica. L’attivazione piastrinica è il punto di svolta, perché c’è una modifica della forma, da discoide passa ad una forma a molti spilli, sulla punta dei quali ci sono recettori, che fanno legare le piastrine fra di loro tramite la fibrina. Per cui l’acquisizione di questa forma è un fattore fortemente predisponente l’attivazione piastrinica, per cui si forma un trombo bianco, che nella realtà non vedremo mai, perché ci sono i globuli rossi,che vi rimangono intrappolati,colorandolo di rosso. La differenza principale tra STEMI e NSTEMI è l’entità dell’occlusione trombotica, per cui vedete che questo sinistra è trombo localizzato, a non diffusamente occludente, per cui l’area dell’infarto è piuttosto circoscritta, confinata all’ascendente quindi del ventricolo, nella porzione sottoendocardica, per cui questa è la tipica immagine ECG. Questa in basso è la tipica immagine dell’infarto con sopraslivellamento ST, in cui vedete a sinistra il trombo che occlude in parte il vaso, a destra vedete che tutto lo spessore della parete ventricolare ha cambiato colore, è diventato bianco e questo modo di manifestarsi della sindrome coronarica acuta determina la tipica immagine del sopraslivellamento dell’infarto del miocardio. PRESENTAZIONE CLINICA DELLA SINDROME CORONARICA ACUTA Il dolore anginoso da sforzo ha una durata finita, cioè di solito recede con l’assunzione di un nitrato, ma comunque è scatenato da uno sforzo e si risolve spontaneamente nel giro di15-20 minuti, invece nel caso della sindrome coronarica acuta il dolore non recede o recede poco dopo l’assunzione del nitrato, compare a riposo per sforzi minimi e scompare in genere dopo più di 20 minuti. Un’altra caratteristica è che mentre nell’angina da sforzo il paziente sa che nel momento in cui cessa di fare lo sforzo il dolore tende a decrescere, in questo caso il dolore è ingravescente, cioè tende a crescere anche se il paziente sta fermo. Questo ci indica che il trombo sta occupando sempre più spazio, sta creando un’ischemia sempre maggiore, quindi anche la sintomatologia del paziente segue l’evolversi di questo fenomeno. L’infarto del miocardio per definizione indica la necrosi dei miocardiociti, causata da ischemia prolungata, come risultato di un’inadeguata perfusione, per squilibrio tra offerta e richiesta d’ossigeno. Questo significa che se noi avessimo un’angina da sforzo in un soggetto, che non si ferma, non curante del dolore, è chiaro che quel paziente avrà un infarto, perché lo squilibrio tra richiesta e offerta d’ossigeno, che noi siamo soliti interrompere con la fine dello stimolo ischemizzante e invece noi prolunghiamo questo stimolo, probabilmente arriveremo anche in questo caso ad una condizione, nella quale la richiesta d’ossigeno cresce in maniera esponenziale, mentre l’apporto d’ossigeno si è stabilizzato. INFARTO DEL MIOCARDIO L’infarto acuto del miocardio è la forma di CI più diffusa ed è una delle principali cause di morte. Colpisce maggiormente gli uomini (le donne, invece, sono più colpite dopo la menopausa) e il rischio aumenta con l’avanzameto dell’età. L’infarto è diviso in subendocardico e transmurale. L’infarto è, inizialmente, sempre subendocardico, perché l’apporto ematico avviene per diffusione attraverso la parete miocardica. Questi può estendersi a fronte d’onda su tutta la parete o essere direttamente transmurale, se l’occlusione dell’arteria è completa ed istantanea. Le cause principali di infarto del miocardio sono: aterosclerosi, trombosi o embolia, ipoperfusione ed arteriti. In base all’arteria colpita, si inquadrano zone specifiche di miocardio infartuato: - Coronaria Sinistra Anteriore Discendente: parete anteriore del ventricolo sx; porzione anteriore del setto interventricolare; apice; - Coronaria Destra: parete postero/inferiore del ventricolo sx; parete posteriore del setto interventricolare; parete libera del ventricolo dx; - Coronaria Cinconflessa Sinistra: parete laterale del ventricolo sx tranne l’apice. Anatomia Patologica: Le alterazioni macroscopiche dell’infarto possono essere studiate mediante colorazione con CTT (trifeniltetrazolio), perché l’area infartuata, in cui le deidrogenasi sono assenti per diffusione, perché le membrane sono rotte, appare chiara; la zona non infartuata, in cui le deidrogenasi sono presenti, appare di colorerosso-scuro. Ecco l’andamento temporale della lesione: - 12-15 ore dopo: Pallore di area infartuata; - 36 ore dopo: Emorragia attorno al pallore; - 3-4 giorni dopo: Tutto più evidente; - 1 settimana dopo: Zona che diventa necrotica e di colore giallo; - 6 settimane dopo: Tessuto fibroso infartuato sostituito da tessuto fibrosocicatriziale. L’andamento temporale delle alterazioni microscopiche è il seguente: - 3-5 ore dopo: Edema delle fibrocellule muscolari; con colorazioni speciali i danni sono quasi assenti; - 6 ore dopo: Eosinofilia del citoplasma e citoplasma granulare: segno di sofferenza cellulare e necrosi iniziale; - 12 ore dopo: Infiltrazione di PMN; presenza di fibrocellule ondulate (le cellule sane in sistole stirano le cellule morte e sembrano ondulate); - 24 ore dopo: Imponente infiltrato di PMN tra le fibre necrotiche (assenza di nuclei). Esiste una zona di tessuto subendocardico che non viene mai colpita ed ha spessore di circa 0,1 mm, legata alla diffusione ematica dall’interno delle cavità cardiache; - 48-60 ore dopo: Imponente infiltrato leucocitario e necrosi ancor più evidente (eliminazione del tessuto morto in corso); - 2-3 giorni dopo: Assenza di tessuto muscolare e visibile necrosi delle cellule infiammatorie; - 10 giorni dopo: Comparsa di fibroblasti; - 1-2 settimane dopo: Produzione di collageno; il tessuto diviene fibroso; - 6 settimane dopo: Formazione di tessuto cicatriziale. La perdita cellulare è legata solo inizialmente alla necrosi, perché prevalgono, in seguito, fenomeni apoptotici. La caratteristica fisiopatologica dell’IMA è l’alterazione segmentale della contrattilità muscolare, con sostituzione del tessuto muscolare con tessuto fibroso non contrattile. Si determina, così, aumento della rigidità della parete e riduzione della compliance, con alterato riempimento ventricolare. Se il danno è inferiore al 10% della massa del ventricolo, non si ha alterazione della funzione ventricolare. Se il danno supera il 15-20%, compaiono i segni d’insufficienza con diminuzione della FE ed aumento della pressione ventricolare sinistra.Se il danno necrotico è del 25-40% del totale, si ha marcata depressione della funzione sistolica, con edema polmonare e ipossia tissutale. L’IMA ha una mortalità del 30%. Presentazione clinica: Il dolore è il primo sintomo ed è riferito nell’85% dei casi, con le caratteristiche tipiche dell’angor: dolore profondo, viscerale, retrosternale, associato a senso d’oppressione; si irradia alla faccia ulnare del braccio sinistro o di entrambe; si irradia, spesso, dietro alle scapole, alla mandibola ed all’epigastrio. La durata del dolore è variabile, tipicamente superiore ai 20 minuti. Una buona parte dei pazienti riferisce di dolore anginoso nelle 24-48 ore prima. Per quanto riguarda la modalità di presentazionedel dolore, questi insorge con: - Sforzo fisico: 40%; non si interrompe a riposo; - Durante il sonno o a riposo: 60%; - Prime ore del mattino: 10%; - Asintomatico: 8%. Esame Obiettivo: L’EO può essere, spesso, totalmente negativo anche in fase acuta. È comune il riscontro di pallore, associato a sudorazione algida, cute fredda, aumento della temperatura corporea a 37-38 °C. La pressione e la frequenza sono variabili: - Infarto in sede inferiore: prevalenza di attività parasimpatica; bradicardia e ipotensione; - Infarto in sede superiore: prevalenza di attività simpatica; tachicardia e ipertensione. È possibile riscontrare un III tono, per elevato riempimento ventricolare, ed un IV tono per insufficienza relativa della mitrale.Si possono riscontrare rantoli, indice di stasi polmonare. Esami di laboratorio: Secondo l’OMS la diagnosi di IMA si basa su: - Anamnesi di angor di natura ischemica; - Alterazioni su ECG seriati; - Aumento e successiva diminuzione dei marcatori sierici di necrosi miocardia. I marcatori sierici più importanti sono: - Mioglobina: è il marcatore precoce più affidabile. Aumenta in circolo tra la I e laV ora dall’inizio del dolore, raggiungendo un picco in 6-12 ore, svanendo in 24-36ore; è poco specifico, aumentando anche in patologie muscolari o renali; - CK-MB: creatininchinasi, isoenzima cardiaco; aumenta in 4-6 ore dall’inizio del dolore, con un picco in 18-24 ore, normalizzandosi in 36-76 ore. Ha elevata cardiospecificità; - Troponine I e T: proteine contrattili miocardiche; compaiono dopo 5-10 ore, con un picco in 1-4 giorni e normalizzandosi in 2 settimane; hanno un’altissima specificità perché permettono, inoltre, di identificare danni miocardici di lieve entità; - AST: aspartato transaminasi, con il suo isoenzima mAST. I valori sono nella donna 35 U/l e nell’uomo 40 U/l; aumenta nelle prime 6-8 ore, con un picco nelle 24-48 ore, normalizzandosi in 4-5 giorni. L’ECG si presenta come lo strumento più utile di fronte al sospetto di IMA.L’immediata manifestazione all’ECG, soprattutto in fase acuta, è caratterizzata dalla sofferenza miocardica e si registra nelle prime ore dall’infarto ed è caratterizzata da onde T negative; è un primo reperto d’ischemia che sarà cancellato in seguito. La prima manifestazione d’ischemia è l’onda di Pardee o corrente di lesione: ampio sopraslivellamento ST, spesso coinvolgente l’onda T; compare nelle prime ore. In questa fase l’ischemia determina, a livello cellulare, perdita del potere dielettrico di membrana: - Ripolarizzazione anormalmente rapida della zona ischemica per l’accelerata apertura dei canali del potassio; - Diminuito potenziale a riposo per la perdita di ioni potassio; - Conseguente depolarizzazione lenta per scompenso elettrolitico. La fase ultima corrisponde alla necrosi, la cui manifestazione è la comparsa, nelle ore successive, di necrosi tissutale e, quindi, la presenza di zone elettricamente inerti.Ciò permette la trasmissione all’esterno dei potenziali negativi intracavitari, fenomeno della finestra elettrica, e la registrazione della classica onda Q di necrosi; per essere significativa, l’onda di necrosi deve durare almeno 0,04 sec e deve essere 2/3 dell’onda R in ampiezza. A partire dalla 12° ora si verificano ulteriori modificazioni della fase di ripolarizzazione, con ritorno graduale alla linea di base del tratto ST e successivainversione dell’onda T; questa configurazione è classica dell’infarto transmurale. Se l’infarto è subendocardico si registra sottoslivellamento di ST senza onda T; senza onde Q: IMA subendocardico o IMA non Q. L’infarto miocardico non Q è una sindrome ischemica acuta in DD con l’angina instabile. Il riscontro elevato di CK-MB, troponine T e I, dopo un episodio ischemico prolungato, distinguono l’infarto non Q. Per convenzione il livello di questi markers deve essere almeno il doppio del massimo di normalità. In base alla presentazione dell’Onda Q si distinguono gli infarti: - IMA inferiore: segni in D2 e D3 e aVF; - IMA settale: segni in V1 e V2; - IMA laterale: D1, aVL, V5 e V6; - IMA anteriore esteso: tutte le precordiali e D1 e aVL. ECO e RX: L’ECO è fondamentale per la valutazione della cinetica del miocardio sano. Si può esaminare esattamente la zona di ipocinesia o discinesia, così come si può valutare la funzionalità valvolare ed il flusso mediante la tecnica doppler. Sia l’ECO che l’RX sono fondamentali per lo studio di un’eventuale cardiomegalia che si può instaurare per insufficienza ventricolare. Trattamento dell’IMA: Il principale scopo della terapia, oltre alla riduzione del dolore con morfina e.v., è la limitazione dell’area di necrosi. Si usano, quindi, trombolitici come t-PA. I nitrati possono essere somministrati in IMA per ridurre l’area di necrosi e per alleviare il dolore; il principale è la nitroglicerina. I β-bloccanti determinano una riduzione del consumo d’ossigeno, da evitare inpresenza di bradi-aritmie. Gli ACE inibitori, inoltre, prevengono l’attivazione del sistema renina-angiotensina tipico dell’IMA, con riduzione della vasocostrizione coronarica e del pre-post-carico. La terapia antiaggregante ed eparinica è da associare al trattamento immediato. Si preferisce, quindi, l’uso di asprina 150-300 mg/die. La riperfusione, sia naturale che mediante terapia trombolitica, può determina rerestitutio ad integrum se avviene nel giro di 15-20 minuti. Gli esiti della riperfusione sono diversi a seconda del periodo in cui si effettua. Se avviene presto la lesione è reversibile, mentre, col passare del tempo, la lesione diventa irreversibile. Le manifestazioni strutturali irreversibili si manifestano dopo 20-40 minuti nel miocardio gravemente ischemico. Complicanze dell’infarto miocardico: Le conseguenze principali di un infarto sono: - Insufficienza cardiaca; - Disturbi della conduzione e del ritmo; - Pericardite fibrinosa; - Rottura di cuore; - Dissociazione elettromeccanica; - Scompenso cardiaco congestizio (tardiva); - Trombosi murale; - Aneurisma ventricolare. L’ITER DIAGNOSTICO DELLA SINDROME CORONARICA ACUTA L’iter diagnostico non può non essere quello classico, innanzitutto c’è l’esame obiettivo con l’anamnesi, in cui chiedete al paziente una serie di informazioni, poi passate alla visita (classica semeiotica). Dobbiamo,in breve, passare dall’esame obiettivo al posizionamento degli elettrodi per l’ECG e se necessario un infermiere deve posizionare una flebo per il trattamento fibrinolitico, perché se nell’angina stabile abbiamo del tempo, in questo caso il tempo non c’è, perché ogni minuto che passa, sono cardiociti che noi perdiamo. Nell’ischemia c’è un viraggio da metabolismo aerobico ad anaerobico, che si esprime sottoforma di alterazioni della contrattilità. Per cui noi possiamo non avere dolore, ECG negativo, ma se facessimo un’eco, vedremmo sicuramente le alterazioni della wall-motion. Nel caso di soggetto con febbre, positività per gli indici d’infiammazione (VES, PCR, leucocitosi), positività per gli indici di necrosi cardiaca, ECG positivo, ma eco negativa, ci deve far pensare a miocardite o pericardite. Per cui è vero che bisogna intervenire rapidamente,ma bisogna individuare la terapia più adatta. Ci sono altri elementi, che ci possono aiutare, per cui l’osservazione del malato è molto importante. Alcuni, come la tachicardia e la sudorazione, possono essere legati solo alla paura. Potremmo avere anche cute pallida e fredda per eccessivo tono simpatico, per cui il soggetto ha un’eccessiva vasocostrizione periferica, quindi ci vuole attenzione. Più cose cerchiamo, più cose troviamo, più la diagnosi è certa!! È chiaro che se compaiono terzo tono, quarto tono, il ventricolo non si sta contraendo bene, quindi c’è aumento della pressione telediastolica, c’è aumento del volume telediastolico, c’è sofferenza atriale e del circolo capillare polmonare, qualche cosa di serio sta sicuramente accadendo. Fino nel 50% degli infarti c’è sottoslivellamento transitorio del tratto ST e/o modificazione dell’onda Q, il sottoslivellamento del tratto ST è considerato significativo solo se è maggiore a 0,1 mV, cioè un quadratino. Questo si verifica nel 20-25% dei casi, un ulteriore 20% dei casi presenta sottoslivellamento minore ed è stato osservato con un grado approssimativo che è uguale a quello dei pazienti con sottoslivellamento minore, perché il sottoslivellamento in sé è foriero di eventi gravi ed il blocco di branca sinistra è ancora più foriero di eventi negativi. La prognosi peggiore è quella dei pazienti con contemporaneo sopraslivellamento del tratto ST, perché questi pazienti hanno avuto inizialmente un trombo occlusivo, che poi si è parzialmente risolto, perché non dobbiamo dimenticare che quando parliamo di fenomeni trombogenici, o quando parliamo di tono coronarico, all’interno dell’endotelio c’è un vero e proprio laboratorio, capace di produrre sostanze attive sia in senso positivo che negativo sul fenomeno della coagulazione e sul tono coronarico. Ad esempio, i primi trombolitici, come streptochinasi o urochinasi erano derivati dal veleno del cobra, allora invece di ammazzare i cobra in giro per il mondo, nel nostro organismo c’è già il t-PA, ma nemmeno da lì lo possiamo prendere, per cui siccome si tratta di un peptide biologico, lo possiamo clonare in quantità industriali. Così come ci sono gli attivatori, ci sono anche gli inibitori. In condizioni normali i due meccanismi sono in parità, nel momento in cui si forma un trombo prevarrà uno dei due (vincono i cattivi!). È vero che all’interno dell’endotelio ci sono le sostanze buone e le sostanze cattive, nel momento in cui l’endotelio è danneggiato, come nel caso dell’aterosclerosi, prevalgono i fattori negativi, come i recettori che inducono vasocostrizione, che fanno aumentare il tono vasale, prevalgono quelle sostanze, che fanno aumentare l’aggregazione piastrinica, prevalgono quelle sostanze, che tendono a mantenere attiva la cascata della coagulazione, per cui la patologia endoteliale è già di per sé, prima che si formi la placca, una condizione nella quale un minimo stimolo basta a far prevalere i cattivi sui buoni. Ad esempio, il fenomeno della trombosi può essere spontaneo, non indotto, cioè ha un andamento ciclico di adesione, aggregazione e attivazione piastrinica, tale che possiamo avere una sintomatologia più o meno rapidamente ingravescente, che poi tende a ridursi d’intensità fino a scomparire. Questo fenomeno è stato osservato principalmente in laboratorio, ma è stato osservato anche nell’uomo. Questo fenomeno è stato osservato da un lato vedendo l’andamento clinico, dall’altro facendo prelievi seriati, semmai anche prelevando il sangue refluo coronarico e vedere come si erano modificati i vari fattori, come ad esempio l’attività piastrinica e a seconda dei momenti si modificava anche la sintomatologia. Nel momento in cui i segni ECG e clinici tendevano a migliorare, anche l’attività piastrinica si riduceva. Tutto questo per dire che la prognosi peggiore si ha nei pazienti con contemporaneo sopraslivellamento, perché formatosi il trombo, che poi si è ridotto, so per certo che le capacità dell’endotelio sono limitate. Se si verificano una seconda volta i meccanismi, che portano alla formazione del trombo, sarà più difficile che il nostro organismo riesca a bloccarli. Per cui precipitatevi ad intervenire, perché le possibilità possono essere finite. Elettrocardiogramma: Dunque nel sospetto di UA/NSTEMI all’ECG bisogna ricercare: - Depressione del tratto ST; - Inversione dell’onda T. Si verifica un sottoslivellamento, o un transitorio sopraslivellamento, del tratto ST e l'inversione dell'onda T in una percentuale di pazienti che varia dal 30 al 50% a seconda della gravità della sintomatologia clinica. Nei pazienti con le caratteristiche cliniche dell'angina instabile, la presenza di una nuova alterazione del tratto ST all'elettrocardiogramma, anche se solo di 0,05 mV, rappresenta un importante fattore prognostico negativo. Le modificazioni dell'onda T sono indicatori di ischemia sensibili ma poco specifici, tranne nel caso di una profonda inversione dell'onda T, superiore a 0,3 mV, di nuova insorgenza. Enzimi di danno miocardico: Gli indici di danno cardiaco ci dicono esattamente che cosa sta succedendo e quale è l’entità, ci dicono anche se e quanto è stato efficace il nostro intervento. Se io ho un’occlusione coronarica, io avrò una necrosi dei miocardiociti a valle, cioè nel territorio di pertinenza di quel vaso occluso, la necrosi causerà il rilascio dei markers. Se però in quella zona c’è mancanza di flusso, quegli enzimi avranno difficoltà a passare in circolo, per cui ho il rischio di vederli aumentati di poco per un tempo limitato. Se io ridò sangue a quel territorio con la trombo lisi o l’angioplastica, arriverà una grossa quantità di sangue, che laverà tutti gli indici di necrosi. Per cui schizzeranno in alto, per cui potremmo pensare che la necrosi si sta allargando, quindi il discrimine sarà il prelievo successivo. Se infatti nel prelievo successivo ho un altrettanto brusca caduta degli indici, vuol dire che il loro aumento era dovuto al wash-out. Se invece vedo che gli indici tendono sistematicamente a salire e dopo24-48 ore non si sono ancora normalizzati, significa che il mio eventuale intervento, teso a risolvere il problema, non è stato efficace, perché non è arrivato abbastanza sangue da lavare tutti gli enzimi, che si erano accumulati, quindi è ancora in corso uno stimolo ischemico, per cui gli enzimi continuano a salire. Le curve ci fanno capire a quanto ci servono i markers per fare la diagnosi e per quanto tempo persistono. Per cui la curva della mioglobina, ad esempio, raggiunge il picco prima degli altri markers, ma è un picco che dura pochissimo. Invece il picco delle due troponine è un po’ più tardivo, ma si mantengono al di sopra della norma per giorni. Quindi al paziente con dolore, ST sopra o sottoslivellato, che giunge alla nostra osservazione, facciamo l’esame obiettivo, ECG e il prelievo, dove potremmo trovare la mioglobina già positiva e gli altri due indici CK-MB non ancora positivi. Se arriviamo un po’ più tardi, potremmo non trovare la mioglobina, che è già finita e non trovare ancora gli altri markers, questo è il motivo per cui in presenza di una sintomatologia sospetta e di un ECG critico, non possiamo escludere la diagnosi in presenza di un solo marker negativo. Questi markers vanno misurati ogni46 ore per 2-3 volte, cioè bisogna che passi un arco di tempo di 24 ore. Dopo di che se gli enzimi non si sono positivizzati, vuol dire che c’è stato un fenomeno ischemico, che però non è riuscito a causare necrosi, quindi infarto. In pronto soccorso se un paziente con dolore fa l’ECG o l’eco e questi sono negativi, e gli si dice che probabilmente si tratta di sindrome coronarica acuta e di ricoverarsi per ulteriori accertamenti, 9 pazienti su 10 dicono di non volersi ricoverare. Di questi 7-7,5 non hanno niente, ma i rimanenti nel migliore dei casi ritornano in ospedale con un infarto conclamato e nel peggiore muoiono. Per cui il medico di pronto soccorso va dal giudice, non per quello che ha fatto, cioè è intervenuto su un soggetto con infarto conclamato, ma per i casi sospetti. Questo perché il medico è passibile di denuncia anche nel caso in cui ha fatto firmare il paziente, ma c’era un forte sospetto da parte del medico, che quel paziente nelle ore successive poteva avere un infarto, per cui in questo caso doveva convincere il paziente e i suoi familiari a rimanere in ospedale. Quindi state attenti, prima di dire che è tutto negativo!! Prima di congedare il paziente, dobbiamo essere certi che non c’è assolutamente niente che non va. Il problema è che dobbiamo essere in grado di cogliere quelle sfumature, che non consentono di mandare il paziente a casa, perché sappiamo che svilupperà un infarto. Se il primo prelievo è negativo, si ripete dopo 4-6 ore, secondo le linee guida. Nella grande maggioranza dei casi (soggetto con dolore), il secondo prelievo sarà positivo, per cui aspettiamo se nel prossimo prelievo gli indici salgono ancora di più, ne abbiamo la certezza. STRATIFICAZIONE DEL RISCHIO La troponina da sola non ci consente di fare la diagnosi, ma ci sono una serie di fattori, che mi confermano la diagnosi. Ad esempio la stratificazione del rischio consiste nel dare un punteggio, in base al quale mi dice quale è la probabilità di un dato evento. Quella tradizionale è il TIMI RISK SCORE, che dà un punteggio anche alla positività dei markers, invece un altro score più recente, GRAIT RISK SCORE, tiene esclusivamente conto del paziente al momento della presentazione, cioè che cosa vedo. Innanzitutto vedo il sesso, l’età, FC e pressione sistolica, lo stato di compenso emodinamico, vedo se ha avuto un arresto cardiaco o un’aritmia minacciosa, ecc. Dopo di che faccio un conteggio, che mi dice come mi devo comportare con quel paziente. A questo punto posso misurare la troponina, ma se io ho già avuto informazioni su quel paziente, la troponina sarà una conferma di cosa sta succedendo, se sta avendo necrosi o no, ma non sarà il discrimine, cioè se il paziente deve rimanere in ospedale, o andare in unità coronarica, ecc…. Per cui è vero che la troponina è predittiva di eventi futuri, ma non è il discrimine. I livelli dei markers aumentano in maniera esponenziale, ma le troponine dopo 1,5 ore non si misurano più, per cui il razionale delle 4-6ore va ad essere delegittimato. Tuttavia, le troponine hanno molta importanza, perché sono i markers più sensibili e specifici, in quanto se sono negativi non sta succedendo niente, ma se sono positivi sta succedendo sicuramente qualcosa. Il TIMI RISK SCORE è più semplice da utilizzare rispetto al GRAIT RISK SCORE, in quanto è semplice da imparare a memoria le varie domande ed inoltre il punteggio per ogni domanda è dicotomico, 1 o 0.Se c’è quella cosa 1, se non c’è 0. Esso considera: - età uguale o maggiore a 65 anni; - alterazioni del tratto ST (in questo è estremamente generico, perché non considera sopra o sottoslivellamento); - 2 o più eventi anginosi nelle ultime 24 ore; - Uno o più fattori di rischio per malattia coronarica; - stenosi superiore al 50%; - uso di ASA negli ultimi 7 giorni; - innalzamento dei markers cardiaci. Il GRAIT RISK SCORE considera, invece, il paziente in quel momento, cioè è il paziente che si stratifica da solo, che come si mostra e con le informazioni che ci porta, si colloca in una data casella di rischio alto, intermedio o basso. Sempre nel TIMI RISK SCORE prendiamo in considerazione stenosi superiore al 50%, questo è in contrasto con quello che abbiamo detto prima, perché non è l’entità della stenosi, che determina l’evento, ma la presenza di una malattia coronarica, cioè se questo paziente ha almeno una lesione superiore al 50%, è molto probabile che ne abbia almeno un’altra uguale o superiore al 50%. Questo lo fa entrare nella categoria dei pazienti a maggior rischio. Considera, inoltre, l’uso di ASA negli ultimi 7 giorni, se prendeva l’aspirina evidentemente qualcun altro lo aveva già etichettato, infine innalzamento dei markers cardiaci. Quindi, questo risk score, che considera 7 parametri, considera un po’tutto, cioè il modo d’essere del paziente, la sua storia pregressa, la sua stratificazione del rischio, prima di questo evento acuto. Tuttavia, rischia di essere un po’ grossolana questa stratificazione del rischio, perché do lo stesso punteggio a tutti. Per validare un algoritmo terapeutico, lo devo provare su più persone. Con questo metodo, che è affidabile, possiamo avere da un minimo di 0 ad un massimo di 7. Dobbiamo considerare il punteggio in funzione dell’evento acuto, perché se abbiamo un punteggio di1, in quanto il soggetto ha più di 65 anni, in presenza di dolore, si tratta comunque di un malato. Per cui il fatto di essere malati, con un TIMI RISK SCORE di 3-4 è comunque associato a rischio di mortalità. Il problema è vedere come la mortalità passa da 4,7 per chi ha 0-1 a 40, 9, quindi aumenta di circa 10 volte per chi ha 6-7. Il gradiente incrementale è abbastanza costante fino a 5, poi schizza verso l’alto a 6-7. Questo significa che questi pazienti sono a basso rischio, ma questo non vuol dire che ci possiamo dimenticare di questi pazienti, per cui la terapia medica, la prevenzione secondaria vanno bene. Ai livelli 3-4 vi è un rischio intermedio, quindi grande attenzione, ma abbiamo del tempo per fare le scelte. Al livello 5 alto rischio e ai livelli 6-7 vi è un rischio molto alto. I soggetti con alto rischio non solo hanno più eventi, cioè muoiono di più, ma l’incremento è esponenziale, perché da 3 a 5-6 hanno un rischio doppio, da 6 a 7 triplo. Questo vale anche per l’infarto, per la combinazione infarto-morte e lo scompenso emodinamico, in cui passiamo da 19 a 48%. In passato era considerata solo la troponina, perché con il suo aumento, c’era un aumento delle morti. Se la troponina è espressione di necrosi ed io faccio il wash-out, il valore di troponina aumenta da 1000 a 3-4000, ma al prelievo successivo si riduceva. Se il soggetto ha raggiunto questi valori di troponina, vuol dire che la necrosi l’ha avuta, allora non è la troponina, ma l’entità della necrosi. Per cui non dobbiamo aspettare che cosa fa la troponina, ma molto prima che la troponina aumenti, dobbiamo su ampi parametri, per questo lo score ci serve, intervenire, perché il paziente può avere già avuto un danno così cospicuo, che il nostro intervento diventa inutile. L’età è un fattore di rischio a sé stante, perché si può avere un infarto anche a 100 anni, ma la probabilità di sopravvivenza è molto inferiore a quella di un soggetto, che ha un infarto a 50, 60 o 70 anni. TERAPIA DELL’UA/NSTEMI I pazienti con AI/NSTEMI devono essere messi a riposo a letto, con monitoraggio elettrocardiografico continuo del ritmo cardiaco e delle alterazioni del tratto ST. la deambulazione è permessa se il paziente non mostra alcuna recidiva di ischemia e se in un periodo di tempo che va dalle 12 alle 24 ore gli enzimi di necrosi miocardica non si positivizzano. La terapia medica comporta un trattamento anti-ischemico associato a un trattamento antitrombotico. La terapia è un insieme di cose tradizionali, sulla cui efficacia non siamo sicuri e cose più recenti, di cui conosciamo l’efficacia. Ad esempio sappiamo che l’eparina è utilizzata in tutti i casi, in cui ci sia il pur minimo sospetto di evento trombotico, ma non è mai stato fatto uno studio randomizzato sull’eparina, ma la convinzione sulla sua efficacia è talmente radicata, che nessuno pensa di fare uno studio sull’utilizzo dell’eparina. Nelle linee guida ci sono 2 modi di valutare: i livelli di raccomandazione 1, 2 e 3 e sottoprodotti A e B, poi i livelli di evidenza A ,B e C. Questosignifica: - A - ci sono una serie di studi randomizzati, che dicono che il trattamento X è più efficace del trattamento Y. Lo studio è condotto su grandi numeri; - Livello B – studio randomizzato con un numero non eccezionale di pazienti; - Livello C – non ci sono grandi studi, ma per effetto dell’esperienza quelli, che stanno scrivendo le linee guida sono tutti convinti che il farmaco A ha efficacia. Nelle linee guida dell’infarto l’eparina è classificata in 1-C, cioè non ci sono studi che dicono che l’eparina è realmente superiore ad altri farmaci, però nessuno di quelli che scrivono le linee guida si sogna di non utilizzare l’eparina in determinati casi. Anche per i nitrati vale lo stesso discorso, perché sono farmaci nati per la cardiopatia ischemica e per un certo periodo, addirittura, la gravità della sindrome coronarica acuta era valutata sulla base della sensibilità o meno ad elevati dosaggi di nitrati per periodi prolungati, cioè se il paziente continuava ad avere dolore ed alterazioni del tracciato, nonostante il trattamento con nitrati per e.v. corrispondeva al livello 7 del TIMI RISK SCORE. Quindi i nitrati fanno ancora parte del bagaglio terapeutico dei pazienti con infarto STEMI o NSTEMI, ma si tratta di farmaci di supporto. Terapia anti-ischemica: Il trattamento iniziale dovrebbe includere nitrati e β-bloccanti. I nitrati hanno un effetto vasodilatatore endotelio-indipendente. I principali effetti sono: - Aumento del flusso miocardico mediante vasodilatazione del circolo coronarico; - Vasodilatazione venosa, con riduzione del volume telediastolico per il ventricolo sx e dimunuzione della tensione di parete con diminuita richiesta di ossigeno. La principale modalità di somministrazione è per via sublinguale, 3 dosi ogni 5 minuti, ma se il dolore persiste si passa alla somministrazione endovenosa di nitroglicerina, 5-10 µg al minuto. La velocità di somministrazione può essere aumentata di 10 µg al minuto ogni 3-5 minuti, finchè non scompaiono i sintomi o la pressione arteriosa sistolica non scende sotto i 100 mmHg. Le principali controindicazioni sono ipotensione dovuta alla somministrazione contemporanea di sildenafil nelle 24 ore precedenti. Il beta-bloccante è molto importante, perché nel momento in cui riduce la richiesta ed il consumo di ossigeno, fa bene sia nella cardiopatia ischemica cronica che nella sindrome coronarica acuta, cioè fa in modo, agendo soprattutto su pressione e frequenza, che il miocardio riduca il consumo di ossigeno al minimo. Ci sono però delle controindicazioni, come tachicardia estrema, blocco AV, una storia di broncospasmo o una marcata ipotensione. È usato di solito nelle prime ore dall’infarto per via parenterale, nelle unità di terapia intensiva. I principali beta-bloccanti ad essere utilizzati sono atenololo e metoprololo. Se il dolore persiste a seguito della somministrazione di beta-bloccanti e nitrati si può procedere alla somministrazione di morfina e.v. 1-5 mg ogni 5-30 minuti. Terapia antitrombotica: Il punto di svolta della terapia della sindrome coronarica acuta è la terapia antitrombotica, con eparina, eparine a basso peso molecolare, su cui si discute molto. Poi abbiamo gli inibitori diretti della trombina, che hanno una pari efficacia anticoagulante rispetto all’eparina ed eparine a basso peso molecolare, ma con minori rischi ad esse connessi. Questo perché è chiaro che se riempio un paziente con antiaggreganti ed anticoagulanti e gli buco un’arteria, non mi devo meravigliare se poi sviluppa un ematoma. Per cui, in questi pazienti si tende sempre a tener conto del rapporto rischio/beneficio, cioè con alte dosi di antiaggreganti ed anticoagulanti sicuramente prevengono il fenomeno trombotico e le sue complicanze, ma aumenta il rischio di emorragie. Per cui se ad un soggetto ischemico faccio perdere alcuni ml di sangue, introduco un pericolo maggiore. Per quanto riguarda le modalità di somministrazione dell’eparina questa viene somministrata esclusivamente per e.v.: Bolo 60-70 UI/kg fino a max 5.000 UI, Infusione e.v. 12-15 UI/kg/h fino a max 1.000 UI. Bisogna monitorare il tempo di tromboplastina parziale attivata (aPTT): aPTT compreso tra 5075 s. Il Range di valori ottimali è 1,5-2,5 volte i valori di riferimento. L’UFH è in grado di interferire sull’attività sia della trombina che del fattore Xa. L’LMWH ha una sequenza più breve che non permette un’azione efficace sulla trombina. La maggiore attività sull’inibizione di Xa delle LMWH inibisce in maniera più efficace sulla produzione di trombina. Altri vantaggi delle LMWH sono: - Possibilità di somministrazione sottocute; - Farmacocinetica più prevedibile; - Non è necessario il monitoraggio dei parametri di anticoagulazione. Ho un risk score anche per il rischio di sanguinamento, che mi dice se e qual è il rischio di sanguinamento. Se il rischio è alto, devo usare una particolare attenzione con queste 2 categorie di farmaci, perché sono quelle che interferiscono maggiormente con la coagulazione. Tra gli altri anticoagulanti ricordiamo: - Inibitori del fattore Xa: Fondaparinux un analogo sintetico della catena penta-saccaridica dell’eparina e si lega quindi solo all’antitrombina aumentando il blocco del fattore Xa, prevenendo in maniera dosedipendente la formazione di trombina ma senza inattivarla; - Inibitori diretti della trombina: Attualmente disponibili sono irudina, argatroban, bivalirudina. Per quanto riguarda gli antipiastrinici, c’è l’aspirina; ci sono le diidropiridine, che sono ticlopidina, clopidrogel, ticagrelor, cangrelor (iniettabile). Gli inibitori della gp IIIa/IIb sono gli antiaggreganti piastrinici più specifici, perché agiscono a livello del processo finale dell’aggregazione piastrinica, tra questi c’è l’abciximab, che è una macromolecola talmente grossa, che impedisce alla piastrina di avvicinarsi ad un’altra piastrina, per cui è molto efficace e ha un’emivita lunga, ma l’insorgenza dell’azione è lenta. Poi ci sono le piccole molecole, come tiroxina, che sono meno efficaci e capaci di legare le piastrine, però hanno un‘emivita più breve, perché dopo 4 ore è come se non l’avessimo somministrato. Soprattutto quando facciamo l’eparina, dobbiamo monitorare la sua attività, cioè dobbiamo avere la certezza della coagulazione, ad esempio durante una procedura chirurgica. La misurazione deve essere fatta più volte, perché se sale significa che stiamo dando troppo anticoagulante, se invece scende ci possono essere eventi trombotici e allora dobbiamo aumentare la dose. Il valore tra 250 e 300 ci dice se la terapia anticoagulante è efficace e se ci possiamo aspettare un vantaggio. Tra tutti questi farmaci, quello che è sopravvissuto a tutti gli studi di valutazione e comparazione è la ticlopidina, perché riduce il rischio di sanguinamento, per cui è utile nei soggetti ad elevato rischio di sanguinamento, che abbiano già fatto terapie anticoagulanti o antiaggreganti per infarto miocardico acuto. Nello STEMI si abbonda con i farmaci antiaggreganti. Il fondaprinux si è dimostrato efficace, ma uno dei motivi per cui nell’angioplastica o nella coronografia si utilizza l’eparina o un analogo è per evitare che nel catetere si formi un trombo e che quindi venga sparato nella coronaria durante la procedura. Il fodaparinux non è in grado di svolgere quest’azione, che l’eparina è in grado di svolgere brillantemente, perché se non siamo abbastanza veloci, vediamo che si forma un trombo bianco, che è sparato nella coronaria. Alla coronarografia vediamo il vaso in nero, con una porzione bianca, che indica la presenza di un trombo. La terapia anticoagulante va sempre accompagnata da un antipiastrinico. La scelta del farmaco, da utilizzare, dipende dalla strategia iniziale, nel caso di una strategia invasiva, la terapia deve essere fatta con anticoagulanti della classe 1C. Infatti, nessuno si sognerebbe di fare una terapia riperfusiva in un soggetto con ischemia cronica o infarto senza eparina. L’enoxaparina appartiene alla classe 2A/B, cioè se non tieni altro da fare, falla, ma non è il massimo o la ticlopidina, su cui è uscito un altro studio sull’infarto, che la colloca in 1A. In una situazione di non urgenza, nel caso in cui è scelta una strategia non invasiva, ma conservativa, si raccomanda il fondaparinux. Se non pensiamo di fare l’angioplastica o la coronarografia subito, possiamo scegliere di utilizzare il fondaparinux. L’enoxaparina deve essere utilizzata solo in presenza di danno cellulare da infarto, l’eparina frazionata e le altre eparine a basso peso molecolare non sono raccomandate in questo caso. Durante l’intervento di angioplastica deve essere continuata la terapia anticoagulante iniziale sia essa con eparina, enoxaparina o ticlopidina, mentre nel caso del fondaparinux deve essere aggiunta eparina non frazionata. La terapia anticoagulante può essere sospesa nell’arco delle 24 ore successive nella procedura invasiva, ma di solito si sospende prima, invece nella strategia conservativa i farmaci possono essere continuati. Gli anti gp IIIa/IIb sono i farmaci antipiastrinici più efficaci. In assenza di controindicazioni, l’aspirina viene utilizzata in tutti i pazienti. In tutti i pazienti è raccomandata una dose d’attacco di clopidogrel di 300mg/die, seguita da 75 mg/die. La somministrazione di clopidogrel deve essere continuata per 12mesi, a meno che non ci siano effetti avversi. Per cui, siamo entrati nella convinzione, che la placca è stabile e così la dobbiamo mantenere, perché se diventa instabile, possiamo avere problemi maggiori. Usiamo inizialmente una dose di carico, perché è un profarmaco, che deve essere convertito nel metabolita attivo, ma già dal secondo giorno possiamo ridurre la dose. Ad un certo punto nell’ambito dei pazienti si è scoperto che c’è una percentuale variabile dal 15 al 25% di non risposta. Allora se aumentiamo il carico, vediamo che succede, allora si è visto che se aumentiamo il carico a 900 mg, anche il paziente è non responsivo, a queste dosi risponderà. La dose di 1200 mg non mi dà nessun vantaggio rispetto a quella di 900 mg. Per quanto riguarda la dose di mantenimento, si è pensato a qual è la dose, che mi serve nella fase acuta e nei giorni immediatamente successivi all’infarto STEMI e NSTEMI. Per cui si è pensato di dare all’inizio 600 mg, per la settimana successiva 2 compresse da 75 mg/die e poi dall’ottavo giorno, quando ormai ci siamo allontanati dalla fase acuta, gli diamo 75 mg/die. Tutti i pazienti sia responder che non, che prendevano il prasugrel erano efficacemente scoagulati, per cui il prasugrel è migliore del clopidogrel per quanto riguarda la prevenzione dell’infarto, ma peggiore per quanto riguarda il rischio di sanguinamento. Se ho maggiore risposta all’effetto anticoagulante, ho maggiore rischio di sanguinamento, per cui sono importanti due risk score: il GRAIT RISK SCORE, che mi consente di calcolare il rischio ischemico del paziente ed un altro, che mi consente di calcolare il rischio di sanguinamento. A questo punto, la domanda è lecita: ma perché non ne abbiamo uno solo? La risposta è che forse ci arriveremo. Con le informazioni, che derivano dai due score, posso temperare la mia aggressività anticoagulante e la mia aggressività nel risolvere il sanguinamento. Per cui, possiamo aumentare l’uno e ridurre l’altro, o possiamo aumentare entrambi. È molto importante sapere quale deve essere l’aggressività del trattamento anticoagulante. Non posso concedermi il lusso di tenere la guardia bassa!! Esempio: in passato abbiamo basato la nostra conoscenza delle malattie, quindi la natura del nostro intervento sull’anatomia, cioè analizzavamo un cuore da cadavere e cercavamo di capire che cosa era potuto succedere e di ricostruire la sua storia. Sapete che i primi studi sul fenomeno dell’aterosclerosi sono stati fatti su un gruppo di soldati americani. Da questi studi è emerso per la prima volta che il processo aterosclerotico inizia alla nascita e progredisce nell’arco di molto tempo, per cui capiamo qual è l’importanza della prevenzione e l’importanza di una corretta alimentazione. Fino agli anni ’70 il principale metodo di studio era l’anatomia patologica, poi abbiamo iniziato a riprodurre nell’animale da esperimento quello, che credevamo accadesse nell’uomo. Per cui, non si è partiti dallo stato finale, ma sono stati studiati gli eventi nel loro evolversi, si è capita la fisiopatologia dell’evento. Ora con la biologia molecolare, con le nanotecnologie partiamo da lontano e quindi abbiamo la possibilità d’intervenire. Per cui attualmente, noi sappiamo quali sono le cause, quali sono i meccanismi fisiopatologici, quali saranno le evoluzioni del fenomeno e quindi quale deve essere l’intervento più adatto. Trattamento a lungo termine: Il momento della dimissione dall'ospedale è un momento educativo per il paziente con angina instabile ed infarto acuto del miocardio senza slivellamento di ST, durante il quale il medico può riconsiderare e ottimizzare il regime terapeutico. La modificazione dei fattori di rischio riveste un ruolo chiave e il medico dovrebbe discutere con il paziente importanza di smettere di fumare, di raggiungere il peso ottimale seguendo una dieta appropriata, di compiere dell'esercizio fisico quotidiano e di tenere sotto controllo la pressione arteriosa, la glicemia (per i pazienti diabetici) e i lipidi ematici. Vi sono evidenze a favore di un beneficio della terapia a lungo termine per cinque classi di farmaci, che hanno come bersaglio differenti componenti del processo atero-trombotico. I beta-bloccanti costituiscono una terapia anti-ischemica appropriata e possono ridurre i fattori scatenanti dell'infarto miocardico. Statine e ACE-inibitori sono raccomandati per la stabilizzazione a lungo termine delle placche aterosclerotiche. La terapia antiaggregante, con acido acetilsalicilico e clopidogrel per un periodo di almeno nove-12 mesi, mantenendo in seguito solo l'acido acetilsalicilico, previene o riduce la gravità di eventuali trombosi che potrebbero far seguito alla rottura della placca. L'approccio multifattoriale alla terapia medica a lungo termine è volto a prevenire i vari fattori coinvolti nel processo aterotrombotico. Questa terapia dovrebbe essere iniziata quanto prima possibile. CAPITOLO VI: INFARTO STEMI L'infarto miocardico acuto rappresenta una delle diagnosi più comuni nei pazienti ospedalizzati nel mondo occidentale. Sebbene il tasso di mortalità dopo il ricovero per IMA sia diminuito di circa il 30% negli ultimi due decenni, circa un paziente su 25 sopravvissute all'ospedalizzazione muore entro un anno. La mortalità è circa quattro volte più alta nei pazienti anziani, con età superiore ai 75 anni, rispetto a quelli più giovani. Quando i pazienti affetti da una prolungata sintomatologia ischemica riposo vengono visitati per la prima volta, l'ipotesi diagnostica che per prima va presa in considerazione è quella di sindrome coronarica acuta. L'elettrocardiogramma a 12 derivazioni è uno strumento diagnostico con un ruolo chiave, poiché è il fulcro del percorso decisionale per il trattamento. Le sindromi coronariche acute dovute a NSTEMI sono più frequenti dello STEMI. La mortalità ospedaliera è maggiore nei pazienti con STEMI rispetto a quelli con NSTEMI. Il follow-up a lungo termine nei pazienti sopravvissuti ha evidenziato un'incidenza di mortalità più elevata per i NSTEMI rispetto ai STEMI, con una differenza a quattro anni di due volte superiore. FISIOPATOLOGIA: RUOLO DELLA ROTTURA ACUTA DI PLACCA Di solito un IMA con sopraslivellamento ST si verifica quando il flusso di sangue coronarico si riduce bruscamente dopo occlusione trombotica di una arteria coronarica già affetta da un processo aterosclerotico. Generalmente la stenosi coronarica di grado severo che si sviluppa lentamente non provoca questo tipo di infarto a causa dello sviluppo, col tempo, di una ricca rete di vasi collaterali. Questo tipo di infarto si verifica quando un trombo arterioso coronarico si forma rapidamente al livello di una zona di danno vascolare. Tale danno è prodotto e favorito da diversi fattori, come il fumo di sigaretta, ipertensione e depositi di lipidi. Nella maggior parte dei casi, l'infarto miocardico con sopraslivellamento del tratto ST avviene quando si rompe la superficie della placca aterosclerotica e si creano le condizioni che favoriscono la trombogenesi. Un trombo murale si forma livello della sede di rottura della placca, determinando l'occlusione dell'arteria coronarica coinvolta. In rari casi, l' IMA con sopraslivellamento ST può essere dovuto a occlusione dell'arteria coronarica causata da un embolo coronarico, da anomalie congenite, da uno spasmo coronarico e da un'ampia varietà di malattie sistemiche, in particolare quelle infiammatorie. L'entità del danno miocardico causato dall’occlusione coronarica dipende: - Dal territorio irrorato dal vaso occluso; - Dal fatto che il vaso sia o non sia totalmente occluso; - Durata dell’occlusione; - Quantità di sangue fornita dal circolo collaterale; - Richiesta di ossigeno dell’area miocardica ischemica; - Da fattori congeniti che possono produrre una precoce lisi spontanea del trombo; - Dall’adeguatezza della perfusione miocardica nella zona infartuata quando viene ripristinato il flusso a livello dell’arteria coronarica epicardica occlusa. MANIFESTAZIONI CLINICHE In circa la metà dei casi l'IMA con sopraslivellamento ST è preceduto da un fattori precipitante, uno sforzo fisico intenso, uno stress emotivo o una patologia medica o chirurgica. Sebbene l' IMA con sopraslivellamento ST possa verificarsi in ogni momento del giorno o della notte, sono state osservate variazioni circadiane con una distribuzione preferenziale degli eventi il mattino, entro alcune ore dal risveglio. Il dolore è il sintomo di presentazione più comune, un dolore di tipo profondo e viscerale. Gli aggettivi più utilizzati per descriverlo sono pesante, a morsa, opprimente, anche se in alcuni casi viene descritto come una pugnalata o come un bruciore. Come caratteristiche, è simile a quello dell'angina pectoris, ma si manifesta a riposo ed è più intenso e più duraturo. Normalmente il dolore si localizza livello della parte centrale del torace o dell'epigastrio e occasionalmente si irradia le braccia, pur essendovi zone di irradiazione meno comuni come l'addome, la mandibola e il collo. Il dolore dello STEMI può irradiarsi verso l'alto fino alla regione occipitale, ma mai al di sotto dell'ombelico. Molto spesso si accompagna ad astenia, sudorazione, nausea, vomito, ansia e da un senso di morte imminente. Il dolore dello STEMI può simulare quello di una pericardite acuta, di un'embolia polmonare, di una dissezione aortica acuta, di patologie osteo-articolari e disturbi gastrointestinali. Pertanto, bisogna sempre prendere in considerazione tali condizioni morbose nella diagnosi differenziale. L'irradiazione del dolore al muscolo trapezio non è presente nei pazienti con STEMI e può essere un utile caratteristica distintiva che suggerisce la diagnosi di pericardite. Tuttavia, il dolore non è presente in modo uniforme in tutti pazienti e STEMI indolenti si hanno soprattutto nei pazienti con diabete mellito e avanti con gli anni. Nell'anziano l’IMA con sopraslivellamento ST può presentarsi con un'improvvisa dispnea, che può progredire fino all'edema polmonare. Esame obiettivo: La maggior parte dei pazienti è ansiosa e agitata e, nel vano tentativo di alleviare il dolore, si muove nel letto cambiando posizione. Si riscontra comunemente pallore, con sudorazione ed estremità fredde. La presenza di dolore toracico retrosternale per più di 30 minuti e sudorazione è fortemente indicativa di un IMA con sopraslivellamento ST. Molti pazienti presentano una frequenza cardiaca e una pressione arteriosa normale durante le prime ore, ma circa un quarto dei pazienti con infarto anteriore presenta manifestazioni di iperattività simpatica, mentre la metà di quelli con infarto inferiore a segni di iperattività parasimpatica. L'area cardiaca è di solito normale e l'impulso apicale può essere difficile da palpare. Nei pazienti con infarto della parete anteriore, entro i primi giorni, si può sviluppare una nuova pulsazioni sistolica, dovuta ad un movimento paradosso del miocardio infartuato. Altri segni obiettivi di disfunzione ventricolare sono la comparsa di un quarto e terzo tono, la diminuzione di intensità del primo tono e uno sdoppiamento paradosso del secondo. Può essere presente un transitorio soffio sistolico apicale meso o telesistolico dovuto a disfunzione dell'apparato valvolare mitralico. In molti pazienti si può dire uno sfregamento pericardico. Il polso carotideo è spesso di ampiezza ridotta, riflettendo una diminuzione della gittata sistolica. Durante la prima settimana dopo un IMA con sopraslivellamento ST si possono osservare aumenti della temperatura corporea fino a 38 °C. La pressione arteriosa e variabile, ma nella maggior parte dei casi si riduce di circa 10-15 mmHg. ESAMI DI LABORATORIO E STRUMENTALI L'infarto miocardico tradizionalmente progredisce attraverso i seguenti stadi temporali: - acuto, dalle prime ore fino al settimo giorno; - fase di guarigione dal settimo giorno all'28º giorno; - fase di cicatrizzazione, oltre il 29º giorno. Per valutare i risultati dei test diagnostici, deve essere tenute in considerazione la fase temporale del processo infartuale. I test di laboratorio che servono a confermare la diagnosi sono: - elettrocardiogramma; - markers sierologici di danno miocardico; - imaging cardiaco; - indici non specifici di necrosi tissutale e infiammatoria. Elettrocardiogramma: I tipici modi di manifestarsi degli infarti NSTEMI (sub endocardici) e degli infarti STEMI sono i seguenti: c’è il segmento ST/sopraslivellato espressione generalmente di una occlusione coronarica totale, invece un ST/sottoslivellato è espressione di una ischemia sub endocardica. Il segmento ST caratterizza l’una o l’altra cosa perché rappresenta l’inizio della ripolarizzazione ventricolare, e quindi normalmente è isoelettrico, cioè il tratto tra l’onda S, che è il tratto discendente del complesso QRS, e l’inizio dell’onda T è isoelettrico. Il segmento ST è la porzione del ciclo delle elettrocardiogramma che va dalla fine del complesso QRS all'inizio dell'onda T. Rappresenta l'inizio della depolarizzazione ventricolare. Generalmente è isoelettrico. L'inizio del segmento ST è detto punto J. In alcune condizioni patologiche, come l' IMA, vi possono essere delle deviazioni del segmento ST, sopra o sotto livellato. Il segmento ST sopraslivellato è tipico dello STEMI, espressione generalmente di un'occlusione coronarica totale. Il segmento ST sottoslivellato, tipico del NSTEMI, è espressione generalmente di ischemia subendocardica. In alcune condizioni patologiche possiamo avere delle alterazioni. Perché in un caso è sottoslivellato e nell’altro è sopraslivellato? Nel caso del sottoslivellato, il vettore è diretto verso la cavità endocardica, mentre nel caso dell’infarto STEMI dove c’è l’ischemia trans-murale il vettore va con andamento praticamente opposto a quello che ha nel NSTEMI, l’uno, quindi, può essere speculare dell’altro, ovvero, se in una parte dell’ECG vedete il sopraslivellamento che è indice del fatto che in quella zona c’è un’ischemia transmurale, in una derivazione che è perfettamente agli antipodi, è speculare, voi vedrete il sottoslivellameto; per cui vi dovete abituare a quello che si chiama ischemia o segni speculari. Voglio dire se voi trovate un ST sopraslivellato in una certa proiezione ad esempio in D2, D3, AVS e poi trovate l’ST sottoslivellato nelle proiezioni precordiali di sinistra, questo non significa che lui ha un infarto inferiore ed una ischemia laterale, significa che ha i segni dell’infarto transmurale e che l’elettrodo che esplora direttamente quella parte di miocardio si manifesta con il sopralivellameto mentre gli elettrodi che sono speculari per i motivi suddetti manifestano un sottoslivellamento. Quindi, ricapitolando, nel NSTEMI il vettore ST è diretto verso la cavità endocardica, mentre nell’infarto STEMI il vettore è diretto verso l’elettrodo precordiale. Quindi, i due modi di essere possono caratterizzare un tipo diverso di ischemia acuta, di interruzione brusca del flusso coronarico a riposo, però possono anche essere l’uno speculare dell’altro. NON avrete mai un sottoslivellamento che è speculare di un sopraslivellamento, la condizione è che ci deve essere tutto il miocardio coinvolto. Quindi possiamo avere un sopralivellamento con sottoslivellamento speculare il che significa che comunque da qualche parte c’è una ischemia che riguarda tutto lo spessore parietale del ventricolo, ma non potrete avere il contrario, ovvero un infarto subendocardico, in cui vedete il sottoslivellamento della derivazione che “esplora quella parte di ventricolo” e vedrete il sopraslivellamento della parte opposta: questo è impossibile perché per avere il sopraslivellamento e il sottoslivellamento speculare bisogna che tutta la porzione del miocardio non sia in grado di trasmettere correttamente la corrente di depolarizzazione, se c’è ischemia il segnale elettrico non può trasmettersi in maniera fisiologica!! Le prime alterazioni avvengono a livello del tratto ST e dell'onda T, generalmente in due fasi. Nella fase acuta, si osserva elevazione del tratto ST e talvolta onda T appuntita (onda T iperacuta). Successivamente, si può osservare inversione dell'onda T nelle derivazioni in cui si è verificato il sopraslivellamento del tratto ST. Nelle fasi più avanzate, può comparire l'onda Q ed il tratto ST ritorna all'isoelettrica. L’ECG nel caso degli infarti STEMI, transmurale, ha una storia naturale, cioè si modifica mano mano che va avanti il processo, e quindi noi possiamo ricostruire sia pure in forma approssimativa l’epoca del processo in base all’ECG. Possiamo dire cioè: questo è un paziente che se lo sta facendo ora l’infarto, sotto i nostri occhi, e quindi abbiamo grandi chance terapeutiche oppure questo è un paziente che se lo è fatto alcune ore fa, giorni, settimane o mesi fa. Ma dobbiamo tenere conto che quello che noi vediamo dall’ECG è lo specchio di una situazione che si è determinata all’interno del miocardio di questo poveretto! Muscolo cardiaco normale, ECG normale; se abbiamo invece una lesione sub endocardica ed una ischemia miocardica che sta in qualche modo procedendo, c’è una parte rossa ed una parte bianca (nell’immagine), la parte rossa è quella ormai ischemica, la parte bianca è quella che comincia a risentire dell’ischemia e che potrebbe essere coinvolta; per dirla in due parole noi qui siamo in una fase in cui non possiamo essere certi che è uno STEMI che resterà tale e non è invece un NSTEMI, quindi un trombo non occlusivo che nelle prossime ore diventerà un trombo occlusivo. Nell’ECG del NSTEMI se compare un sopraslivellamento temporaneo ha un valore prognostico negativo importante, questo significa che il trombo è cresciuto fino a diventare occludente e per un rimaneggiamento dovuto a quegli elementi che sono presenti all’interno dell’endotelio si è parzialmente o completamente dissolto. Se all’ECG notiamo un piccolo sopraslivellamento del tratto ST, piccolo significa che dopo la onda R, la S non arriva all’isoelettrica, interrompe un po’ prima la sua discesa e quindi si complessa con l’onda T che appare un po’ appuntita, quando poi questa onda di lesione procede fino a coinvolgere l’interezza della parete vedete che a questo punto noi abbiamo un sopralivellamento del tratto ST che è profondamente diverso dal precedente, praticamente poco dopo l’onda R, la S non riesce a scendere molto ed immediatamente da origine al nostro sopraslivellamento; perché evidentemente a causa dell’effetto dell’ischemia trans murale non c’è più un normale passaggio delle correnti di depolarizzazione. Quindi, siamo entro il primo giorno, abbiamo visto l’evoluzione dell’infarto (attenzione questa è la parte nella quale noi abbiamo le massime chance terapeutiche), possiamo impedire che l’infarto dia luogo a necrosi o possiamo far si che la necrosi sia estremamente ridotta. Quando parliamo di STEMI la frase canonica è: il tempo è muscolo!!! Più tempo passa più miocardio perdiamo che non recupereremo! Oltre il primo giorno c’è la comparsa dell’onda Q, l’onda R è molto piccola, l’onda Q è molto significativa, il sopraslivellamento del tratto ST è molto modesto, e vedete che la T che prima era inglobata nel tratto ST ed aveva una depressione positiva, qui invece ha una depressione negativa. (questo è il caso di un infarto anteriore letto da una derivazione che legge la parete anteriore, una V4). Osserviamo ora la parete ventricolare: noi siamo partiti da una parete normale, poi abbiamo visto una parete in cui vi era una lesione subendocardica con l’allargamento potenziale del fenomeno che nella terza immagine si è effettivamente aggravato, però attenzione, voi dovete fare una distinzione tra il nero e il rosso, cioè il nero è necrosi, il rosso è ischemia che può diventare necrosi ma che possiamo anche bloccare, cioè fare la trombolisi in occasione della terza immagine significa che noi il nero ce lo siamo già giocati ma abbiamo tutto il rosso ed il bianco attorno che possiamo riguadagnare!! Quindi l’obiettivo della nostra terapia è fare il modo che le porzioni (rosso/bianco) siano recuperate e che non accada quello che vediamo nelle immagini successive cioè una progressiva estensione della zona nera (necrosi totale trans murale) che mi da una modificazione del tracciato (dopo 2-3 giorni) quella piccola R che vedevamo prima non si vede più, quindi noi vediamo il complesso Q come una profonda onda negativa, l’ST è completamente normalizzato e l’onda T è francamente invertita. A questo punto è chiaro che il tessuto infartuato che noi non siamo riusciti a salvare sarà progressivamente sostituito da uno strato fibroso (ultima immagine a destra). Vediamo quindi che il tracciato si è ulteriormente modificato: l’onda Q è sempre presente, può comparire una piccola onda R; di solito l’onda R se ricompare in funzione dell’entità della sua ricomparsa sarebbe un segnale indiretto che li sotto c’è presenza di tessuto vitale. E’ ovvio che li dove c’era parete muscolare ora c’è fibrosi e questo tessuto fibroso non solo non si contrae, ma per effetto della contrazione tende ad espandersi e in più una parte di sangue non sarà espulsa e a questo punto cosa deve fare il cuore perché la maggior quantità possibile di sangue sia espulsa? Deve aumentare progressivamente la propria energia contrattile e fare in modo che il resto del ventricolo normale possa pompare una quantità di sangue adeguata, perché se non lo fa, noi siamo già entrati nello scompenso cardiaco: lo scompenso cardiaco non è altro che una incapacità da parte del ventricolo sinistro di pompare in periferia una quantità di sangue sufficiente alle esigenze tissutali e alle coronarie. C’è da dire ,ancora, che tutto quello che noi perdiamo come muscolo darà luogo quindi prima alla cardiomiopatia dilatativa e poi alla scompenso cardiaco. Abbiamo accennato prima al miocardio vitale. Noi per anni abbiamo pensato che quando c’era l’infarto fosse come un’onda dello tsunami, attraversa tutti i miocardiociti e fa a tutti lo stesso tipo di danno, ha un andamento frastagliato ma poi l’acqua arriva dappertutto finchè non esaurisce la propria forza di spinta ed inizia la fase di ritiro. Nel miocardio accade che l’onda di lesione non colpisca tutti i miocardiociti in egual modo, ovvero all’interno della parete ci sono ancora cellule attive (vive). Queste cellule sopravvivono in un “mondo” totalmente distrutto e quindi non hanno la capacità di nutrirsi e di avere il normale metabolismo perché naturalmente non gli arriva il sangue o gliene arriva una quantità infinitesimale, poiché la coronaria si è chiusa. A questo punto, queste cellule cadono in uno stato di ibernazione, ovvero riducono le loro necessità metaboliche e quindi cessano anche di contrarsi, per cui noi abbiamo un’area di distruzione in cui vi sono dei nuclei vitali. Qual è la manifestazione di tutto questo?? Sarà una manifestazione tipo quella descritta per l’ultima immagine cioè ci sarà una parte di ventricolo che non si muove o che ha un movimento paradosso. A questo punto noi dobbiamo capire se c’è miocardio vitale ibernato o se abbiamo perso tutto( e qui possiamo utilizzare scintigrafia ed eco-stress) perché se io a quelle cellule vitali riporto il sangue, quindi con una ripefusione, una angioplastica o by-pass, quelle cellule che erano ibernate avendo una quantità di sangue decente ricominciano a contrarsi. Questo significa che quel ventricolo che si stava avviando alla strada che lo avrebbe condotto alla cardiomiopatia dilatativa, allo scompenso cardiaco, alla necessità di un trapianto cardiaco, improvvisamente cambia direzione. Per cui se avevamo una frazione di eiezione del 40% che a seguito del mio intervento di rivascolarizzazione diventa del 50 % circa noi lo abbiamo letteralmente strappato ad un percorso che stava facendo e si sarebbe concluso con il trapianto o la morte; questo ha permesso di snellire le liste per i trapianti e le morti poiché questo intervento permette un forte guadagno della frazione di eiezione. C’è un punto da chiarire però, se ci sono 4 cellule vitali o se ce ne sono 40 o 4000 è sempre la stessa cosa? A questo punto la scintigrafia diventa importante, perché noi dobbiamo essere in grado di quantizzare quanto miocardio possiamo far svegliare. Quindi (eco e scintigrafia etc…) servono non solo a dirmi se c’è vitalità, ma anche quante cellule sono rimaste in vita; possiamo vedere i segmenti del ventricolo in varie porzioni con l’eco ad esempio e cercare di quantizzare sulla base dei segmenti una ripresa della contrattilità oppure con la scintigrafia ci rendiamo conto di quante cellule possiamo recuperare. Ricordiamo ancora quindi che la presenza dell’onda R è un modo grossolano che ci dice che li sotto c’è tessuto vitale. La cardiopatia ischemica è una malattia e come tutte le malattie è destinata a cambiare. Si ritiene che la cardiomiopatia ischemica possa scomparire a detta della società europea di cardiologia. Gli infarti STEMI stanno diminuendo forse perché le campagne sul cibo, il fumo, danno i loro effetti, forse perché l’educazione parte dai ragazzi, che a differenza degli adulti , risultano essere più sensibili al fenomeno. Markers cardiaci sierici: Noi abbiamo visto che ci sono dei markers che ci permettono di fare diagnosi di infarto e di necrosi, è particolarmente importante sapere quale di questi markers bisogna verificare nella fase in cui noi siamo, dato che non sono tutti uguali. La mioglobina ha un picco tra 1-2 ore, quindi di gran lunga la più precoce, però è anche quella che sparisce prima, fare diagnosi di infarto sulla mioglobina è importante perché ci indica un infarto imminente e noi abbiamo tutto il tempo e tutte le possibilità di rimediare. L’aumento della CK-MB comincia abbastanza presto, entro 2-3 ore, ma anche questo dura poco per cui voi capite che la troponina è effettivamente il marker per fare la diagnosi, tant’è vero che noi di fronte ad una negatività della troponina abbiamo l’obbligo di ripeterla due o tre volte a distanza di 4 o 6 ore (le regole dicono 6/8 ore, nella realtà prima sai meglio è). Le troponine possono aumentare però anche in altri casi come ad esempio un paziente che esce da un intervento cardochirurgico, in caso di dissezione aortica, di miocardite, vi dico questo perché se un paziente ha la troponina elevata bisogna dapprima trattalo come se fosse un infarto poi dopo si vede. 3. INDICI DI DANNO CARDIACO. Proteina Massa molecolare (kD) Inizio Durata Sensi Speci bilità ficità Mioglobina 16 1.5–2 h 8–12 ore +++ + CK-MB 83 2–3 h 1–2 giorni +++ +++ Troponin I 33 3–4 h 7–10 giorni ++++ ++++ Troponin T 38 3–4 h 7–14 giorni ++++ ++++ CK 96 4–6 h 2–3 giorni ++ ++ La troponina è importante ma noi abbiamo tanti altri segni che possiamo esaminare per giungere a diagnosi corretta. Ciascuno di questi indici presenta vantaggi e svantaggi. Per quanto riguarda i vantaggi dei singoli marcatori di danno miocardico, questi sono: - CK-MB: il suo picco è rapido ed è in grado di identificare un reinfarto precoce; - Mioglobina: possiede un’elevata sensibilità, normalmente si ritrova nel infarto miocardico precoce, può essere utilizzata per valutare la riperfusione; - troponine: i loro livelli vengono utilizzati per effettuare la stratificazione del rischio, possiedono maggiore sensibilità e specificità rispetto a tutti gli altri enzimi. Sono in grado di indicare la presenza di infarto avvenuto fino a due settimane prima, vengono utilizzate per monitorare l'andamento della terapia ed infine vengono utilizzate per valutare la riperfusione. Ma ciascuno di questi marcatori, possiede anche una serie di svantaggi: - CK-MB: bassa specificità con danno muscolare scheletrico e bassa sensibilità per l'infarto al miocardio precoce o tardivo dopo la comparsa dei sintomi e per danno miocardico minore; - Mioglobina: specificità molto bassa con danno muscolare scheletrico e rapido ritorno verso valori normali; - Troponine: bassa sensibilità nella fase precoce del infarto miocardico e scarsa affidabilità nelle rintracciare reinfarti minori. STRATIFICAZIONE DEL RISCHIO Noi, quando abbiamo parlato dei NSTEMI, abbiamo visto le categorie di farmaci utilizzate ma non abbiamo parlato del ruolo che ha la terapia medica rispetto all’angioplastica. Se io ho una ostruzione, fissa, data dal fenomeno della trombosi che mi ho ostacola il flusso ed ho a valle dell’ostruzione un miocardio che non riceve ossigeno in quantità adeguate e quindi non ha un normale metabolismo, l’angioplastica è quella tecnica che mi consente in maniera meccanica con un intervento invasivo, ma molto meno invasivo di un intervento chirurgico, la risoluzione del problema. Quanto vale fare l’angioplastica in un paziente con un infarto stemi o non stemi, rispetto ad una terapia farmacologica? il rischio dell’intervento ci consente di avere un grande guadagno o gli diamo una fregatura? Per anni si è discusso e solo ora i dati sono chiari e si sviluppano in base alla stratificazione del rischio: siccome noi siamo di fronte ad una placca instabile che può reagire bene ad una terapia medica, o meno, e siccome non conosciamo terapie mediche definitivamente stabilizzanti, quindi quelle placche che noi lasciamo li si possono ulcerare una seconda volta e creare danni, utilizzando l’angioplastica e imprigionando la placca al di sotto della retina metallica noi ci assicuriamo almeno per quella placca la possibilità di non dare fastidio nel futuro. Stabilizzando la placca con l’angioplastica noi otterremo un vantaggio per almeno altri 5 anni. Però, ritornando alla stratificazione del rischio e l’adozione delle opportune strategie terapeutiche in funzione delle esigenze del paziente, come rispondiamo ai rischi implicati in questa procedura? Fin’ora abbiamo osservato tutti i pazienti valutandoli allo stesso tempo, ma ora andiamo a distinguerli in funzione del rischio (riferendoci all’uso quindi dell’angioplastica rispetto ad un intervento di routine): basso rischio (qualunque cosa facciamo non interveniamo in maniera significativa sulla salute di quel paziente, possiamo evitare l’angioplastica), intermedia (ci da la possibilità di valutare attentamente e decidere), high (è necessario effettuare una coronarografia con possibilità di angioplastica perché a vista dell’indice rischio/beneficio, è favorito quest’ultimo). Esiste un TIMI risk score anche per lo STEMI costituito da una serie di punti da assegnare per ogni complicanza; in questo bisogna considerare fattori quali l’età, la pressione (soprattutto se inferiore a 100 mmHg, REGOLA DEL 100: un paziente che ha più di 100 di frequenza cardiaca e ha meno di 100 di pressione sistolica è un paziente che sta avendo lo shock cariogeno, nel giro di qualche ora diventa pallido, sudato e riprendere il paziente sarà difficile) c’è da considerare inoltre anche la classificazione di killip che serve a capire se il paziente sta avviandosi o è già in shock cariogeno (4 classi), poi ci sono ancora una serie di altre cose che valgono un punto quindi incidono di meno sul TIMI risk come ad esempio nell’ECG: si mettono sullo stesso piano un sopraslivellamento del tratto ST o un blocco di branca sinistra (il blocco di branca sx può di per sé essere un segnale di infarto del miocardio). Il punteggio da 0 ad 8 (i punti da assegnare in tutto sono 14 per il timi risk) implica un rischio di morte entro i primi 30 giorni perciò la velocità di diagnosi e di intervento sono fondamentali (IL TEMPO è MUSCOLO!!) . Una persona anziana con più di 75 anni, con pressione meno di 100, frequenza più di 100, classe killip 2-4 un po’ di diabete ed ST elevato accumulano i 14 punti ed il gioco è fatto!! Man mano che aumenta il tempo di ischemia aumenta la mortalità, questa cosa prima si sapeva genericamente non era esattamente basata sul tempo, ora invece si conosce minuto per minuto. TERAPIA DELLO STEMI Una terapia aggressiva può portare ad uno shift positivo se adoperata entro sei ore al massimo, basandosi anche su questa nuova stratificazione del tempo/d’ischemia. E’ importante quindi che il paziente arrivi in ospedale presto ed in condizioni di sicurezza. L’angioplastica primaria è quindi la terapia di scelta in un infarto STEMI ed è importante recarsi in un centro organizzato capace di operarla. Se ci sono i sintomi riferiti agli infarti STEMI possono essere valide tre opzioni ai fini terapeutici: - self decision: il paziente decide in quale ospedale andare e come deve andare; - Gp/cardiologist: è il medico di famiglia che può consigliare o un’ambulanza o il trasporto privato (operando una stratificazione del rischio); - EMS: la chiamata ai servizi d’emergenza e l’arrivo di un’ambulanza permette un trasporto sicuro e la presenza a bordo di medicinali e defibrillatori permette un trasporto anche ad un ospedale magari più lontano capace di operare una migliore terapia. Solitamente nei piccoli ospedali è operata solo una trombolisi mentre in centri ben organizzati è operata una angioplastica che permette la conservazione di una maggiore frazione di eiezione. Ciò significa che il paziente con infarto non deve andare al primo ospedale più vicino, ma in tempi brevi giungere al centro capace di dare al paziente la cura più adatta. Spesso il trombolitico può non funzionare, questo dovrebbe essere efficace entro 60 min, ma è bene comunque intervenire anche in seguito con l’angioplastica. Bisogna quindi rispettare i protocolli effettuando sempre la miglior terapia per il paziente. Le cose che noi facciamo in un paziente con lo STEMI (angioplastica o trombolitico) sono volte a ristabilire la perfusione coronarica; tra i farmaci indicati o capaci di dare un aiuto clinico ci sono: Si è visto in ogni caso che fare una angioplastica entro le 12 ore dopo la trombolisi non riportava ad una frazione d’eiezione importante (superiore al 50%), quindi è necessario intervenire prima delle 12 ore al fine di ottenere buoni risultati nella riperfusione. Se a 60 min dall’iniezione del trombolitico non c’è migliormaneto (abbassamento dell’ST) significa che il paziente deve essere immediatamente operato, prima delle 12 ore. Se invece la trombolisi è efficace, la riperfusione con angioplastica va valutata e può essere, se necessario, operata anche entro le 24ore. La trombolisi ha delle controindicazioni assolute e relative. Quelle assolute sono: - Precedente stroke emorragico o stroke di origine sconosciuta; - Stroke ischemico nei 6 mesi precedenti; - Trauma o neoplasia del SNC; - Trauma cranico o intervento neurochirurgico nelle 3 settimane precedenti; - Sanguinamento gastrointestinale nell’ultimo mese; - Malattie emorragiche; - Dissezione aortica; - Punture non comprimibili (biopsia epatica, puntura lombare). In caso di presenza di una di queste cose la trombolisi non va effettuata!! Controindicazioni relative sono: - TIA nei 6 mesi precedenti; - Terapia anticoagulante orale; - Gravidanza/ primo mese post-partum; - Ipertensione refrattaria (sistolica maggiore di 180 mmHg e/o diastolica superiore ai 100 mmHg); - Malattia epatica avanzata; - Endocardite infettiva; - Ulcera peptica attiva. TRATTAMENTO INIZIALE Assistenza preospedaliera: La prognosi è in buona parte legata al verificarsi di due classi generali di complicanze: - complicanze elettriche, aritmie; - complicanze meccaniche, insufficienza di punta. La maggior parte dei decessi in ambiente extra ospedaliero si verifica entro le prime ventiquattr'ore dall'esordio di sintomi per via della fibrillazione ventricolare, e di questi circa la metà si verifica nella prima ora. Pertanto, gli elementi più importanti del trattamento ospedaliero includono: - il riconoscimento di sintomi da parte del paziente e una pronta richiesta di assistenza medica; - il rapido arrivo di un team medico d'urgenza in grado di eseguire manovre di rianimazione; - il rapido trasporto del paziente all'ospedale con personale medico e paramedico esperto; - la rapida esecuzione di una terapia di riperfusione. Il ritardo maggiore si verifica di solito non nel trasporto all'ospedale, ma piuttosto tra l'insorgenza del dolore e della decisione del paziente di attivare soccorso medico. Questo ritardo può essere ridotto grazie all'educazione dei cittadini, da parte di operatori sanitari professionisti, sul significato del dolore toracico e sull'importanza di una rapida richiesta di soccorso medico. Sempre più, monitoraggio e trattamento sono eseguiti in ambulanza da personale addestrato, accorciando ulteriormente i tempi tra l'esordio del infarto e il trattamento adeguato. Trattamento in pronto soccorso: Nel pronto soccorso, gli obiettivi del trattamento includono il controllo del dolore cardiaco, una rapida identificazione dei pazienti candidati alla terapia di riperfusione d'urgenza, l'indirizzamento dei pazienti a basso rischio verso un’appropriata destinazione ospedaliera e l'evitare dimissioni inappropriate. Molte fasi del trattamento vengono iniziate in pronto soccorso e poi proseguite durante la fase di ricovero ospedaliero. L'acido acetilsalicilico è essenziale per il trattamento di questi pazienti ed efficace per tutto lo spettro delle sindromi coronariche acute. Nei pazienti in cui la saturazione d'ossigeno risulta normale, il beneficio clinico dell'apporto supplementare di ossigeno è limitato, se non nullo, e pertanto tale procedura è inutile. Tuttavia, in presenza di ipossiemia occorre somministrare ossigeno per via nasale o tramite maschera facciale per le prime sei-12 ore dopo l'infarto; i pazienti dovrebbero essere rivalutati successivamente per determinare la necessità di proseguire tale trattamento. Controllo del dolore: La nitroglicerina sublinguale può essere somministrata con sicurezza nella maggior parte dei pazienti con IMA con sopraslivellamento ST. Oltre a diminuire o abolire il dolore toracico, la nitroglicerina può ridurre le richieste di ossigeno, riducendo il precarico, e aumentare la disponibilità di ossigeno a livello miocardico dilatando i vasi coronarici. Nei pazienti in cui vi sia stata un'iniziale risposta favorevole alla somministrazione di nitroglicerina sublinguale seguita da una ricomparsa dei sintomi, bisogna considerare l'utilizzo di nitroglicerina per via endovenosa. La terapia con nitrati è controindicata nei pazienti che hanno una pressione arteriosa sistolica bassa, inferiore ai 90 mmHg, o in cui vi sia un sospetto di infarto del ventricolo destro. I nitrati non dovrebbero essere somministrata pazienti che hanno assunto, nelle 24 ore precedenti, il sildenafil. Infatti, può verificarsi una reazione idiosincrasica ai nitrati, consistente nell'improvvisa grave ipotensione, che di solito viene prontamente risolta dalla somministrazione rapida di Atropina per via endovenosa. La morfina è un analgesico molto efficaci per il dolore. Tuttavia, può indurre la vasocostrizione arteriolare e venosa simpatico-mediata e di conseguenza il risultante sequestro venoso può ridurre gittata sistolica e la pressione arteriosa. Questi disturbi emodinamici di solito rispondono prontamente al sollevamento delle gambe, mentre in alcuni pazienti è necessaria un'espansione della volemia tramite l'infusione endovenosa di soluzione fisiologica. Il paziente può presentare sudorazione e nausea, ma di solito questi sintomi sono transitori e lasciano il posto a un senso di benessere nel momento in cui il dolore si risolve. La morfina possiede anche un effetto vagotonico e può causare bradicardia o un blocco atrio-ventricolare di grado elevato, in particolare nei pazienti con infarto postero-inferiore. Questi effetti collaterali rispondono bene alla somministrazione di Atropina. Anche beta-bloccanti per via endovenosa sono utili nel controllo del dolore. Queste sostanze controllano efficacemente il dolore in alcuni pazienti, riducendo la richiesta miocardica di ossigeno e quindi ischemia. La somministrazione di beta-bloccanti per via endovenosa riduce i rischi di nuove infarto e di fibrillazione ventricolare. A differenza dei beta-bloccanti, i calcio-antagonisti sono di scarsa utilità nella condizione acuta e vi sono evidenze che le diidropiridine a breve durata d'azione possono essere associate a un momento del rischio della mortalità. Strategie di trattamento: Lo strumento primario per lo screening e le decisioni è l'elettrocardiogramma a 12 derivazioni. Quando è presente un sopraslivellamento del tratto ST di almeno 2 mm in due derivazioni precordiali contigue e di 1 mm in due derivazioni periferiche, il paziente dovrebbe essere considerato un candidato alla terapia di riperfusione. In assenza di sopraslivellamento del tratto ST, la fibrinolisi non è di aiuto ed esistono evidenze che possa essere dannosa. Riduzione dell'area infartuale: La quantità di miocardio che diviene necrotica a seguito di un'occlusione coronarica è determinata da fattori diversi. Mentre la zona centrale del infarto contiene tessuto necrotico, che è irrimediabilmente perso, il destino del tessuto ischemico limitrofo può essere migliorato dal tempestivo recupero della perfusione coronarica, dalla riduzione della richiesta miocardica di ossigeno, dalla prevenzione dell'accumulo di metaboliti tossici e dalla riduzione degli effetti dei mediatori del danno da riperfusione. Circa un terzo dei pazienti può andare incontro alle perfusione spontanea dell'arteria coronarica occlusa entro le 24 ore, con un miglior processo di guarigione del tessuto infartuato. La riperfusione accelera il recupero della coronaria occlusa responsabile del infarto in quei pazienti in cui si sarebbe verificata una fibrinolisi spontanea più tardi è aumenta notevolmente il numero dei pazienti in cui si raggiunge recupero di flusso nel arteria responsabile del infarto. Un tempestivo recupero di flusso nell’arteria epicardica colpita, associato miglioramento della perfusione della zona di miocardio a valle del infarto, determina una limitazione dell'area infartuale. I glucocorticoidi e i farmaci antinfiammatori non steroidei, ad eccezione dell'acido acetilsalicilico, devono essere evitati in corso di IMA con sopraslivellamento ST. Infatti possono alterare il processo di cicatrizzazione infartuale e aumentare il rischio di rottura del miocardio e il loro impiego può determinare una maggiore estensione della cicatrice infartuale. Inoltre, possono aumentare le resistenze vascolari coronariche, riducendo il flusso al miocardio ischemico. Intervento di rivascolarizzazione per via percutanea: L’intervento di rivascolarizzazione coronarica per via percutanea, consistente di solito nell'angioplastica o inserimento di stent senza precedente terapia fibrinolitica, denominato PCI (percutaneous coronary intervention) primario, è efficace nel ristabilire la perfusione, quando viene effettuato in situazioni d'emergenza nelle prime ore dopo l'infarto. Ha il vantaggio di essere applicabile ai pazienti che presentano controindicazioni alla terapia fibrinolitica, ma sono comunque considerati candidati alla riperfusione. Sempre essere più efficace della fibrinolisi nel rendere pervie le arterie coronariche occluse e si associa ad una migliore prognosi clinica. Tuttavia tale metodica è costoso in termini di personale e attrezzature, e la sua applicabilità è limitata alla possibilità di attuazione 24 ore su 24 soltanto in una minoranza di ospedali. Fibrinolisi: Qualora non vi siano controindicazioni, la terapia fibrinolitica dovrebbe essere idealmente avviata entro 30 minuti dall'accesso ospedaliero. L'obiettivo principale della fibrinolisi è un rapido ripristino della completa pervietà coronarica. Le sostanze fibrinolitiche utilizzate sono: attivatore del plasminogeno tissutale (tPA), streptochinasi, tenecteplasi (TNK), reteplasi (rPA). Tutti questi farmaci agiscono promuovendo la conversione del plasminogeno in plasmina, che lista i trombi di fibrina. Quando il flusso dell'arteria coronarica responsabile viene valutato con l'angiografia, è descritto da una semplice scala qualitativa, chiamata sistema di gradazione della trombolisi nel infarto miocardico o TIMI: il grado 0 indica occlusione completa dell'arteria che ha provocato l'infarto; il grado 1 indica una certa penetrazione del mezzo di contrasto oltre il punto di ostruzione, ma senza perfusione del letto coronarico distale; il grado 2 indica la perfusione dell'intero vasi infartuato verso il letto distale, ma con flusso ritardato rispetto a quello di un'arteria normale; il grado 3 indica la completa perfusione del vaso infartuato con un flusso normale. Quest'ultimo grado è l'obiettivo della terapia di riperfusione, poiché la riperfusione completa dell'arteria coronarica che ha provocato l'infarto si associa a risultati migliori in termini di riduzione dell’area infartuale, di conservazione della funzione ventricolare sinistra e di tassi di mortalità ridotti a breve e a lungo termine. La terapia fibrinolitica riduce il rischio relativo di morte intraospedaliero fino al 50% se somministrata entro le prime ore dall'esordio dei sintomi. Se usata in modo appropriato, la terapia fibrinolitica sembra poter ridurre l'area infartuata, limitare la disfunzione sistolica sinistra e diminuire l'incidenza di complicanze gravi. Poiché il miocardio può essere salvato soltanto prima di essere danneggiati irreversibilmente, la tempistica della terapia di riperfusione è di estrema importanza per ottenere il massimo beneficio. La terapia risulta efficace per molti pazienti a tre-sei ore dall'inizio del infarto e qualche beneficio sembra possibile fino a 12 ore. La fibrinolisi è generalmente la strategia di riperfusione preferita per i pazienti che si presentano nella prima ora dall'inizio dei sintomi, nel caso vi siano problemi logistici per il trasporto del paziente in un centro provvisto di emodinamica o se è previsto un ritardo di almeno un'ora tre momenti in cui la fibrinolisi potrebbe essere iniziata e l'esecuzione di un PCI. Sebbene i pazienti di età inferiore ai 75 anni ottengano una maggiore riduzione relativa del tasso di mortalità rispetto a quelli più anziani, il tasso di mortalità assoluta (15-25%) più alto di quest'ultimo gruppo dà luogo a riduzione analoghe della mortalità in entrambi i gruppi di età. Il tPA e gli altri attivatore del plasminogeno relativamente fibrino-specifici sono più efficaci della streptochinasi per ripristinare una completa riperfusione e hanno anche un piccolo vantaggio nel migliorare la sopravvivenza. TRATTAMENTO NELLA FASE OSPEDALIERA Unità di terapia intensiva coronarica: Queste unità sono normalmente equipaggiate con un sistema che permette il monitoraggio continuo del ritmo cardiaco di ogni paziente e il monitoraggio emodinamico in pazienti selezionati. Sono di solito disponibili anche defibrillatori, respiratori, pacemaker transtoracici non invasivi e apparecchiature adeguate all'introduzione dei cateteri per elettrostimolazione e il monitoraggio emodinamico. Molto importante è la presenza di un gruppo di infermieri altamente addestrati, in grado di riconoscere le aritmie, di adeguare i dosaggi dei farmaci antiaritmici, vasoattivi e anticoagulanti, nonché di praticare le manovre di rianimazione cardiopolmonare, incluso l'utilizzo del defibrillatore quando necessario. I pazienti dovrebbero essere ammessi in unità coronarica nelle fasi precoci di malattia, quando ci si aspetta che possono trarre beneficio dall'utilizzo di attrezzature e assistenza qualificata. La disponibilità al di fuori dell'unità coronarica di monitoraggio elettrocardiografico e di personale addestrato ha reso possibile il ricovero di pazienti a rischio più basso in unità di cura semi-intensive. La durata della permanenza in unità coronarica è dettata dall'evoluzione della necessità di terapia intensiva. CAPITOLO VII: LE MALATTIE DELLE VALVOLE CARDIACHE ANATOMIA VALVOLARE Le valvole cardiache sono le strutture che separano fra di loro le camere cardiache, gli atri e i ventricoli, e queste ultime dai grandi vasi, aorta ed arteria polmonare. Le valvole cardiache sono quattro: tricuspide, polmonare, mitrale e aorta, in grado di aprirsi e chiudersi in maniera coordinata con il battito cardiaco, così da lasciare passare il sangue solo in una direzione. Il compito delle valvole cardiache è quello di aprirsi e chiudersi in maniera coordinata con il battito cardiaco, così da lasciar passare il sangue in una sola direzione. La valvola mitrale è bicuspide ossia ha due lembi valvolari, mentre la tricuspide ne ha tre, invece le due valvole che mettono in comunicazione i ventricoli con i grossi vasi sono entrambe tricuspidi. Le valvole atrio-ventricolari hanno un sistema di ancoraggio costituito dalle corde tendinee e dai muscoli papillari cosa che non hanno le altre due valvole. Un'altra cosa importante è che la valvola mitrale ha un'area più piccola rispetto alla valvola tricuspide, proprio perché è costituita da soli due lembi ed offre una maggiore resistenza al flusso; a causa di ciò la pressione nell'atrio sinistro è maggiore della pressione presente nell'atrio di destra. In questa sezione è possibile notare come la valvola mitrale e la valvola aortica siano contigue tra di loro, mentre paradossalmente la valvola polmonare e la tricuspide sono letteralmente agli antipodi. DEFINIZIONE Le malattie delle valvole cardiache si definiscono valvulopatie e possono essere di due tipi: - Stenosi: incompleta apertura, il sangue passa attraverso un orifizio più piccolo della norma; - Insufficienza, incompleta chiusura, parte del sangue torna indietro attraverso la valvola che dovrebbe essere chiusa. Molto spesso, tuttavia, stenosi ed insufficienza coesistono, in diversa misura, nella stessa valvola, realizzando la cosiddetta stenoinsufficienza. Stenosi ed insufficienza si possono avere per tutte e 4 le valvole. Stenosi e insufficienza possono coesistere e possiamo dire che l’insieme di stenosi ed insufficienza è molto più frequente nelle forme secondarie ad una patologia infettiva-infiammatoria (malattia reumatica e le endocarditi). È possibile distinguere le valvulopatie in: - Congenite, ovvero già presenti alla nascita; - Acquisite, che compaiono nel corso della vita. A loro volta, quelle acquisite possono essere dovute a varie cause: - Degenerativa, più frequenti nei soggetti anziani, spesso ipertesi, dovute in sostanza a usura delle strutture valvolari; - Infettiva, dovute ad endocarditi; - Ischemica, in corso di infarto miocardico acuto; - Traumatica, molto raramente; - Secondaria a cospicua dilatazione del ventricolo e/o dei grandi vasi. In quelle ischemiche è fondamentalmente colpita la valvola mitrale, per rottura del muscolo papillare o delle corde tendinee; quelle traumatiche, a carico della tricuspide e della mitrale, sono spesso dovute alle cosiddette ‘’cadute in piedi’’ ossia dovute ad un forte impatto con il suolo che si trasmette totalmente verso l’alto e quindi alle valvole (con conseguente rottura delle corde tendinee) o all’aorta che può andare incontro a dissezione dato il suo stretto legame con strutture toraciche. DECORSO Il decorso delle valvulopatie è nella maggior parte dei casi lentamente evolutivo, con una fase anche molto lunga (anni) di completa asintomaticità. Qualora invece la valvulopatia insorga acutamente su una valvola fino a quel momento normale (in seguito a traumi, infarto miocardico, endocardite con perforazione dei lembi valvolari) la presentazione clinica può essere drammatica. Le malattie delle valvole del settore destro del cuore (tricuspide e mitrale), ove vige un regime pressorio più basso, sono rare e in genere dovute a problemi congeniti. Le malattie di mitrale e aorta sono invece molto più frequenti. La tricuspide spesso si ‘’ammala’’ come conseguenza di problemi a carico del cuore sinistro e successiva compromissione della circolazione polmonare. CAUSE Per quanto riguarda le cause responsabili di valvulopatie, bisogna differenziare se valvulopatia acquisita o cengenita. Nelle valvulopatie congenite si hanno alterazioni dello sviluppo embrionale delle strutture cardiache e spesso sono associate ad altre anomalie congenite che realizzano sindromi assai complesse. Le valvulopatie acquisite possono essere dovute a infezioni, degenerazione del tessuto valvolare, traumi, ischemia miocardica o a patologie del muscolo cardiaco o dell’aorta ascendente. Negli scorsi decenni una delle cause principali di valvulopatia era la malattia valvolare reumatica, che insorge come complicanza di una faringite o tonsillite causata da un particolare batterio. Le valvole cardiache sono colpite alcune settimane dopo l’infezione tonsillare. Esse vengono danneggiate e progressivamente si deformano. La deformazione determina o fusione dei lembi valvolari o eccessiva separazione dovuta a retrazione (quindi nel primo caso la stenosi e nel secondo caso l’insufficienza). Al giorno d’oggi, col miglioramento delle condizioni di vita, la riduzione delle infezioni e l’aumento della durata della vita, la causa più frequente di valvulopatia è quella degenerativa, dovuta cioè al progressivo danneggiamento della struttura valvolare che avviene con l’invecchiamento. INSUFFICIENZA MITRALICA L’apparato valvolare mitralico oltre ai lembi ha un sistema di corde tendinee che lo collegano al muscolo, quindi è il muscolo che in qualche modo contraendosi o rilassandosi condiziona, insieme ai regimi pressori dell’atrio e del ventricolo sx, l’apertura e la chiusura della valvola. L’insufficienza mitralica è determinata da un reflusso retrogrado sistolico, diretto dal ventricolo all’atrio sinistro. Quindi, durante la sistole una parte di sangue invece di passare in aorta e contribuire alla portata attiva, refluisce nell’atrio sx. Eziologia: L’insufficienza mitralica può dipendere da un’anormalità o da una patologia che colpisce una o più delle cinque componenti dell'apparato valvolare mitralico: lembi valvolari, annulus, corde tendinee, muscoli papillari e miocardio sottostante. L'insufficienza mitralica acuta può verificarsi nel contesto di un infarto miocardico acuto con rottura di un muscolo papillare, in seguito al trauma toracico o in corso di endocardite infettiva. Nell'insufficienza mitralica acuta è coinvolto molto più frequentemente il muscolo papillare postero-mediale di quello antero-laterale a causa della sua unica fonte di vascolarizzazione. Un'insufficienza mitralica acuta transitoria può verificarsi durante episodi di ischemia attiva e attacchi di angina pectoris. Nei pazienti con degenerazione mixomatosa dell'apparato valvolare la rottura di una corda tendina può causare il peggioramento acuto dell'insufficienza mitralica cronica. L'insufficienza mitralica cronica può essere dovuta a malattia reumatica, prolasso valvolare mitralico, estesa calcificazione dell’annulus mitralico, difetti valvolari congeniti, miocardiopatia ipertrofica ostruttiva o a miocardiopatia dilatativa. La malattia reumatica è la causa di insufficienza mitralica cronica solamente in circa un terzo dei casi ed è più frequente negli uomini. La flogosi reumatica provoca rigidità, deformità e retrazione delle cuspidi valvolari oltre fusione delle commessure e accorciamento, fusione e contrazione delle corde tendinee. La natura dell'insufficienza mitralica associata prolasso valvolare mitralico e a miocardiopatia ipertrofica ostruttiva è in genere dinamica. In questo caso, l'insufficienza è una conseguenza del dislocamento anteriore del muscolo papillare e del movimento sistolica anteriore del lembo anteriore della valvola mitrale nel tratto di efflusso ristretto del ventricolo sinistro. La calcificazione dell’annulus è frequente soprattutto nei pazienti con nefropatia avanzata ed è comunemente riscontrabile in donne anziane affette da ipertensione e diabete mellito. L'insufficienza mitralica può essere un'anomalia congenita più comunemente come difetto dei cuscinetti endocardici. L'insufficienza mitralica cronica è spesso secondaria a ischemia e può essere una conseguenza del rimodellamento ventricolare, della dislocazione di muscoli papillari o di impastamento dei lembi valvolari, ma anche di fibrosi di un muscolo papillare in pazienti con infarti miocardici guariti e cardiopatia ischemica. L’eziologia può essere: - di tipo degenerativo; - legata a malattia reumatica; - legata a prolasso della mitrale; - legata a processi infettivi (endocarditi); - dovuta a patologie rare di tipo congenito o acquisito come: la sindrome di marfan, il LES, le distrofie muscolari e i tumori. Indipendentemente dall’eziologia, l'insufficienza mitralica cronica grave tende ad essere progressiva, poiché la dilatazione atriale sinistra provoca lo sviluppo di tensioni anomale a carico del lembo posteriore mitralico che viene spinto lontano dal orifizio valvolare, provocando un ulteriore deterioramento dell'apparato valvolare. Analogamente, la dilatazione del ventricolo sinistro provoca un ulteriore incremento del rigurgito valvolare, il che a sua volta determina un ulteriore ingrandimento dell'atrio sinistro e del ventricolo sinistro, che causa la rottura di corde tendinee e un circolo vizioso, da cui l'aforisma: "l'insufficienza mitralica a grave insufficienza mitralica". Fisiopatologia: Distinguiamo due forme di insufficienza mitralica: cronica ed acuta. Nell'insufficienza mitralica cronica il reflusso si instaura progressivamente e quindi vengono evocati meccanismi di adattamento e di compenso. Importante in questo caso è la frazione di rigurgito, definita come la percentuale di sangue che torna in atrio sinistro durante la sistole e può arrivare fino al 70%. In questo caso il sovraccarico di volume determina progressiva dilatazione del ventricolo sinistro per il meccanismo dell'ipertrofia eccentrica. Nell'insufficienza mitralica acuta si ha un rigurgito abbondante che si instaura improvvisamente. Vengono a mancare rapidamente i meccanismi di compenso cardiocircolatorio in quanto non c'è tempo per lo sviluppo di ipertrofia. Queste due forme di insufficienza mitralica, riconoscono anche una diversa eziologia. Per l'insufficienza mitralica cronica, l'eziologia e dovuta a cause: - infiammatoria, come malattia reumatica e LES; - degenerativa, come sindrome di marfan e degenerazione mixomatosa; - infettiva, come nel caso dell'endocardite; - strutturale, come rottura di corde tendinee, disfunzione dei muscoli papillari, dilatazione dell’annulus, “leak” paraprotesico; - congenita, come il cleft e la mitrale a paracadute. Per quanto riguarda, invece, l'eziologia dell'insufficienza mitralica acuta, questa è dovuta soprattutto a: - endocardite infettiva; - disfunzione o rottura ischemica dei muscoli papillari; - disfunzione protesica. Nell'insufficienza mitralica le resistenze allo svuotamento del ventricolo sinistro si riducono. Come conseguenza, il ventricolo sinistro durante la fase sistolica invia sangue anche nell'atrio sinistro; la riduzione volumetrica del ventricolo durante la sistole provoca una brusca diminuzione della tensione parietale. Il meccanismo di compenso iniziale dell'insufficienza mitralica consiste in un maggiore svuotamento sistolico del ventricolo sinistro. Come l'aggravarsi dell'insufficienza si osserva un progressivo incremento del volume telediastolico del ventricolo, accompagnato da un deterioramento della funzione contrattile ventricolare. Questo aumento di volume del ventricolo sinistro è spesso accompagnato da una riduzione della portata cardiaca anterograda; la compliance del ventricolo sinistro è però spesso aumentata e di conseguenza la pressione diastolica ventricolare non aumenta fino alle fasi avanzate della malattia. L'entità del rigurgito varia direttamente in funzione della pressione ventricolare sistolica sinistra e della dimensione dell'orifizio rigurgitante. Poiché la frazione di eiezione aumenta nell'insufficienza mitralica grave in presenza di una normale funzione del ventricolo sinistro, anche una modesta riduzione di questo parametro riflette una disfunzione significativa. All'inizio della diastole, l'atrio sinistro disteso si svuota rapidamente e determina un'onda y particolarmente ripida, sempre che non vi sia una stenosi mitralica. Nei pazienti con insufficienza mitralica pura, il flusso di sangue molto rapido attraversa l'orifizio valvolare di normali dimensioni può determinare un gradiente pressorio protodiastolico di breve durata tra l'atrio e il ventricolo sinistro, genera un terzo tono, dovuta rapido riempimento ventricolare e un soffio mesodiastolico. Nell'insufficienza mitralica acuta severa il volume di sangue rigurgitato viene spinto in un atrio sinistro di normali dimensioni con una compliance normale o ridotta. Quindi con un aumento del volume atriale, le pressioni dell'atrio sinistro aumentano in modo marcato. L'onda v nel tracciato della pressione atriale sinistra è in genere prominente, le pressione atriale sinistra e venose polmonari sono marcatamente elevate e l’edema polmonare è comune. La funzione sistolica del ventricolo sinistro con un'insufficienza mitralica acuta può essere conservata, iperdinamica o ridotta a seconda del contesto clinico. I pazienti con insufficienza mitralica cronica sviluppano una dilatazione marcata dell'atrio sinistro e un aumento della compliance atriale che determina un minimo o assente aumento delle pressioni dell'atrio sinistro e venose polmonari in risposta ad aumenti del volume atriale sinistro. Questi pazienti lamentano in genere astenia severa secondaria alla ridotta portata cardiaca mentre i sintomi di congestione polmonare sono, almeno all’inizio, meno importanti. La fibrillazione atriale è presente a seguito della dilatazione. Se la frazione di rigurgito è esigua, è molto facile che il paziente sia asintomatico ed è possibile apprezzarla solo con una ecocardiogramma con doppler (sistema che ci permettere di misurare la velocità del sangue in transito) o un color-doppler che ci fa vedere se il sangue si avvicina o si allontana dalla nostra sonda e quindi ci permette di stabilire se il sangue in quel distretto sta seguendo il suo tragitto naturale o sta avendo un tragitto assolutamente patologico. È evidente che questo continuo reflusso di sangue determina un sovraccarico di volume con progressiva dilatazione del ventricolo sx per il meccanismo dell’ipertrofia eccentrica (ovviamente nei processi cronici). La dilatazione dell’annulus è in fondo una cosa abbastanza passiva nel senso che mano mano che il ventricolo si dilata, in qualche modo determina anche la dilatazione dell’anulus. L’anulus è la parte su cui sono ancorati i lembi valvolari e quando si dilata i lembi inevitabilmente tenderanno a seguire l’anulus, quindi se l’anulus si allarga i lembi si troveranno ad essere lontani tra di loro e non vi sarà più la perfetta coaptazione dei lembi con successivo reflusso di sangue dalla camera che sta a valle verso quella che sta a monte. Il ’’leak paraprotesico’’ è un fenomeno che avviene in un paziente in cui è stata impiantata una protesi (una valvola meccanica o biologica) dal chirurgo o dal cardiologo interventista (le valvole aortica e polmonare possono essere inserite per via transcutanea tramite puntura di un’arteria periferica). Quadro clinico: Quando compaiono i sintomi per un’insufficienza mitralica cronica non è la malattia in quanto tale che li determina, ma è la ridotta gittata cioè vuol dire che siamo arrivati in una fase in cui una frazione abbondante di sangue, invece di essere spinta dal ventricolo in aorta, torna nuovamente nell’atrio di sx . Nell’insufficienza mitralica cronica i sintomi insorgono gradualmente e dipendono dall’entità del rigurgito e dalla durata del processo. I primi sintomi sono legati alla ridotta gittata, e sono: - facile affaticabilità; - lieve dispnea da sforzo; - dispnea parossistica notturna; - dispnea a riposo. Nella dispnea parossistica notturna, il paziente, dopo esser stato per tanto tempo in posizione supina, ha un tale sovraccarico del circolo sistemico che non riesce a smaltire e che si propaga in modo retrogrado al circolo polmonare determinando difficoltà respiratorie tali da costringerlo a cambiare posizione (mettersi seduto o in piedi) per scaricare in maniera adeguata il circolo polmonare e consentirgli così di avere una adeguata ossigenazione. Nella forma di insufficienza mitralica acuta l’edema polmonare è sempre legato alla difficoltà del ventricolo di smaltire la giusta quantità di sangue. Altri sintomi sono dispnea ingravescente e shock cariogeno. Nello shock cardiogeno si ha un brusco calo pressorio dato che il ventricolo non riesce più a pompare la quantità giusta di sangue, tale da mantenere un’adeguata perfusione dei vari distretti corporei (circolo coronarico, circolo cerebrale, circolo renale etc…). Quando lo shock è grave si può avere decesso del paziente o se si riesce ad intervenire si può impiantare un contro-pulsatore costituito da un pallone che viene gonfiato con elio (viene posizionato nell’aorta discendente e si gonfia in diastole in modo da garantire un’adeguata perfusione del circolo coronarico e del circolo cerebrale) che da supporto alla funzione contrattile. Diagnosi: La diagnosi di insufficienza mitralica si basa su: - elettrocardiogramma, che tuttavia fornisce poche informazioni; - ecocardiogramma, che permette di fare una valutazione precisa delle camere cardiache; - RX torace, che consente di valutare la dilatazione atriale e ventricolare nelle forme croniche e l’edema polmonare nelle forme acute; - Cateterismo cardiaco, utile per una valutazione pre-operatoria. Un ecocolor-doppler è il sistema migliore per calcolare quanto sangue va avanti e quanto invece torna indietro. L’Rx del torace ci può dare delle informazioni indirette osservando lo stato dei polmoni e tenendo presente che cuore e polmoni sono strettamente connessi tra di loro. Il cateterismo cardiaco consiste nell’introduzione appunto di cateteri che penetrano nelle varie camere e possono misurare la saturazione dell’ossigeno e tramite l’utilizzo di mezzi di contrasto che seguono il flusso sanguigno ci permettono di visualizzare la presenza di stenosi oppure di rigurgito. Quando si presenta un rigurgito importante, per azione del mezzo di contrasto, prima si visualizzeranno l’atrio e il ventricolo insieme poi ad un certo punto l’atrio sarà visualizzato meglio del ventricolo (ovviamente nella fase di rigurgito). All’esame obbiettivo, si avrà la presenza di rantoli basali a livello polmonare, epatomegalia, edemi e ascite indicano la presenza di EPA (edema polmonare acuto) e quindi evidenziano una situazione di per se abbastanza grave. I segni ascoltatori sono: - riduzione del primo tono, dovuto alla chiusura delle valvole atrio-ventricolari, che ovviamente in questo caso non è ottimale; - sdoppiamento del secondo tono, dovuto alla chiusura delle semilunari, legato all’ipertensione polmonare; - presenza del terzo tono causato dall’improvvisa messa in tensione delle corde tendinee, muscoli papillari e lembi valvolari. Una caratteristica dell’insufficienza mitralica acuta è la presenza di un quarto tono cardiaco. Ma la caratteristica fondamentale dell’insufficienza mitralica è il soffio olosistolico, che copre e segue il primo tono. Quindi il primo tono quasi non si sente ed in questo caso la contrazione non è più isovolumetrica. Nei pazienti con rottura di corde tendinee il soffio può avere un carattere pigolante, molto simile al richiamo di un gabbiano. La valutazione del grado di rigurgito della mitrale può essere valutata con l’ecocardiografia. Dal punto di vista emodinamico, possiamo fare una valutazione del grading tramite iniezione del mezzo di contrasto. Il grading angiografico viene effettuato durante la ventricolo grafia sinistra e prevede i seguenti gradi: - grado +: minimo jet di rigurgito; - grado ++: il jet di rigurgito delinea i bordi della cavità dell’atrio sinistro; - grado +++: atrio sinistro opacizzato come il ventricolo sinistro; - grado ++++: atrio sinistro opacizzato più del ventricolo sinistro. Trattamento: Il trattamento dell’insufficienza mitralica dipende dal grading della situazione clinica del paziente e distinguiamo tra gradi: - Se l’insufficienza è poco più che moderata e il paziente è sintomatico; - Se l’insufficienza è più che moderata e vi è una diminuzione della frazione di eiezione del ventricolo sx o un aumento della pressione arteriosa polmonare; - Se il paziente ha un’insufficienza mitralica severa. Ovviamente la terapia può essere di tipo farmacologico e di tipo chirurgico. Per quanto riguarda la terapia chirurgica, si procede con la sostituzione valvolare in tutte e tre le situazioni cliniche. Nei casi meno gravi dipende tutto dal grado di sopportazione del paziente. Se sopporta male la situazione per la mancanza di meccanismi di compenso si procede con la sostituzione, anche se si cerca sempre prima di tentare una terapia farmacologica e casomai successivamente si procede con la sostituzione. Per quanto riguarda la terapia medica, il farfari deve essere impiegato quando insorge fibrillazione atriale, con l’obiettivo di mantenere l’INR compreso tra 2-3. La cardioversione deve essere presa in considerazione sulla base del contesto clinico e delle dimensioni dell’atrio sinistro. I pazienti con insufficienza mitralica acuta necessitano di un trattamento di urgenza e la preparazione immediata al trattamento chirurgico. STENOSI MITRALICA Eziologia: La stenosi mitralica è quasi sempre acquisita, se è acquisita non può che essere di origine reumatica ed ai giorni nostri è una cosa abbastanza rara. La malattia reumatica, a causa del processo infiammatorio, determina la cicatrizzazione e l'ispessimento delle cuspidi valvolari confusione dei lembi e ipomobilità degli stessi. Si manifesta generalmente dalla terza alla quinta decade. Raramente la stenosi mitralica è la conseguenza di altre condizioni patologiche quali il LES, l'artrite reumatoide, la sindrome da carcinoide e l’amiloidosi. Nell'anziano caratteristiche risultano essere le calcificazioni dell'anulus. La stenosi congenita esiste ma a quanto pare non esistono lavori in letteratura in grado di documentarla. Nella SM reumatica i lembi valvolari sono diffusamente ispessiti da tessuto fibroso e/o da depositi di calcio. Le commessure valvolari si fondono e si accorciano, le cuspidi valvolari divengono rigide. Queste alterazioni a loro volta determinano una riduzione del diametro dell’apice dell’imbuto formato dalla valvola (a bocca di pesce). Le alterazioni successive possono essere dovute all’alterazione del flusso di sangue indotta dalla iniziale deformità valvolare. La calcificazione della valvola stenotica immobilizza i lembi valvolari e riduce ulteriormente l’orifizio valvolare. Trombi ed embolizzazione arteriosa possono essere causate dalla valvola calcifica in sé, ma nei pazienti con fibrillazione atriale la trombosi si verifica nell’atrio sinistro dilatato, particolarmente nella sua auricola. Fisiopatologia: La superficie mitralica è più piccola della tricuspide per ovvi motivi anatomici, quando si verifica la stenosi la pressione atriale sx tende ad aumentare ed il flusso da laminare diventa vorticoso. Durante l’esercizio fisico l’aumento della frequenza cardiaca contribuisce alla riduzione del tempo di riempimento diastolico con aumento della pressione atriale, di conseguenza aumento della pressione nei capillari polmonari e quindi edema polmonare acuto con accumulo di liquidi nella cavità alveolare che determinano difficoltà respiratorie fino a quando il paziente inizia a cacciare dalla bocca una schiuma rosea e da li in poi avremo pochissimo tempo per salvarlo. Il risultato finale della costante presenza di elevate pressioni polmonari è l’ipertensione polmonare. In un adulto sano, l’orifizio valvolare ha una superficie di 4-6 cm2. In presenza di un’ostruzione significativa, l’area dell’orifizio si riduce ed è inferiore a 2 cm2. In questo contesto, il sangue può passare dall’atrio sinistro al ventricolo sinistro soltanto in presenza di un gradiente pressorio atrioventricolare elevato in modo anomalo. Questo è il segno emodinamico caratteristico della SM. Quando la superficie dell’orifizio è inferiore a 1 cm2, condizione denominata come stenosi serrata, per mantenere una normale portata cardiaca è necessaria una pressione atriale sinistra di circa 25 mmHg. Le elevate pressioni venosa e arteriosa di incuneamento riducono la compliance polmonare contribuendo allo sviluppo di una dispnea da sforzo. Il primo episodio di dispnea di solito viene precipitato da eventi clinici che provocano l’incremento di flusso trans mitralico, il che determina un aumento della pressione atriale sinistra. Per valutare emodinamicamente l’importanza dell’ostruzione è essenziale misurare sia il gradiente pressorio trans valvolare sia la velocità del flusso. Quest’ultima non dipende soltanto dalla portata, ma anche dalla frequenza: un incremento nella frequenza cardiaca riduce proporzionalmente più la durata della diastole che quella della sistole e quindi diminuisce il tempo disponibile al sangue per passare attraverso l’orifizio mitralico. Nella SM isolata, la pressione diastolica nel ventricolo sinistro e la frazione di eiezione sono normali. Nel caso di una stenosi mitralica pura in ritmo sinusale, in genere l’aumento della pressione atriale sinistra e polmonare di incuneamento, è elevato e la morfologia della curva pressoria di incuneamento mostra un’aumentata contrazione atriale (onda a) e una progressiva lenta discesa della pressione dopo l’apertura della valvola mitrale (discesa y). Nella stenosi mitralica serrata e quando le resistenze vascolari polmonari sono aumentate in maniera significativa, la pressione arteriosa polmonare è elevata anche quando il paziente è a riposo e durante l’esercizio si verificano ulteriori aumenti, che spesso causano incrementi secondari dlla pressione e del volume tele diastolici del ventricolo destro. Nel paziente con SM moderata (1,0-1,5 cm2) la portata cardiaca è normale o quasi a riposo, ma aumenta in maniera minore del normale durante l’esercizio. Nei pazienti con SM severa (inferiore a 1 cm2) e particolarmente in quelli in cui le resistenze vascolari polmonari sono marcatamente elevate, la portata cardiaca è minore del normale a riposo e può non aumentare o addirittura diminuire con lo sforzo. Il grado di impegno clinico ed emodinamico di questi pazienti è significativamente condizionato dal livello della pressione nell’arteria polmonare. L’ipertensione polmonare è determinata da: - Trasmissione retrograda passiva delle elevate pressioni nell’atrio sinistro; - Vasocostrizione delle arteriole polmonari, che probabilmente è innescata dall’ipertensione atriale sinistra e delle vene polmonari (ipertensione polmonare reattiva); - Edema interstiziale nella parete dei piccoli vasi polmonari; - Modificazioni obliterative del letto vascolare polmonare. La risultante ipertensione polmonare determina dilatazione del ventricolo destro con comparsa di insufficienza tricuspidale e insufficienza polmonare e, quindi, insufficienza cardiaca destra. L’unica terapia farmacologica che si può effettuare in un paziente con stenosi mitralica è l’utilizzo di Betabloccanti i quali riducono la frequenza, riducono la forza di contrazione e fanno si che anche quando il paziente fa uno sforzo la frequenza non aumenti di tanto e ci sia sempre un adeguato spazio temporale all’interno del quale l’atrio può svuotarsi all’interno del ventricolo. Nella stenosi mitralica la prima cosa fondamentale è l’aumento della pressione nell’atrio sinistro dopodiché abbiamo tre possibili strade: - Una trasmissione passiva retrograda; - Una costrizione Arteriolare Reattiva ‘’barrage’’, cioè le arterie polmonari per evitare che il circolo polmonare si ingorghi aumentano il loro regime tensivo cioè si vaso-costringono, ma ciò alla lunga può portare ad una obliterazione arteriolare irreversibile; - le resistenze arteriolari sono definitivamente aumentate e quindi avremo una ipertensione polmonare irreversibile. Parliamo di ipertensione polmonare quando la pressione è già pari a 35-40 mmHg e nei casi limite dove è presente anche la costrizione arteriolare (Barrage) la pressione può variare tra i 70 e i 120 mmHg cioè tra le due e le 4 volte superiore al normale regime pressorio. Quindi possiamo dire che la condizione terminale successiva alla stenosi mitralica è l’ipertensione polmonare. A differenza dell’insufficienza mitralica dove il danno è principalmente a carico del ventricolo, nella stenosi, invece, il danno è a carico principalmente dell’atrio e del circolo polmonare. Quadro clinico: Nei climi temperati, il periodo di latenza tra la cardite reumatica e lo sviluppo dei sintomi dovuti a SM è in genere di due decenni: la maggior parte dei pazienti comincia a mostrare disabilità intorno alla quarta decade di vita. Nei pazienti nei quali l’orifizio mitralico è sufficientemente grande da consentire un normale flusso di sangue con un’elevazione lieve della pressione dell’atrio sinistro, un aumento marcato di questa pressione con conseguente tosse e dispnea può essere determinato da variazioni improvvise della frequenza cardiaca. Nella stenosi mitralica abbiamo sintomi simili a quelli dell’insufficienza, ma in più compaiono maggiori problematiche legate ai polmoni (bronchiti invernali, emottisi legate a rottura dei vasi alveolo capillari, in più fibrillazioni atriali che possono dare embolie sistemiche e embolie polmonari). Le fibrillazioni atriali sono delle condizioni in cui l’atrio sta fermo, non si contrae e il sangue ha un andamento vorticoso ed è quindi più facile che si alteri uno dei fattori della triade di Vircov e si possa formare un trombo, in più aggiungiamo il fatto che l’atrio presenta l’auricola dove a causa dell’andamento vorticoso si possono formare numerosissimi trombi che vanno in circolo sottoforma di emboli. All’esame obiettivo, il soggetto può presentare la cosiddetta facies mitralica, con labbra viola (cianosi) con zigomi rossi uguali a quelli di chi ha la ‘’couperose’’e da turgore giugulare. Ancora, l’esame obiettivo rivelerà la presenza di rantoli basali polmonari ed il polso sarà piccolo, ineguale e deficiente. All’ascoltazione, la stenosi mitralica si caratterizza per: - Primo tono rinforzato al contrario dell’insufficienza, con sistole pulita; - Secondo tono normale seguito da un tono di apertura, detto anche OS (Opening Snap o schiocco di apertura), a sua volta seguito da un soffio diastolico con rinforzo presistolico. Quando parliamo di stenoinsufficienza la descrizione diventa più difficile. In caso di ipertensione polmonare cospicua è ascoltabile un forte soffio olosistolico dovuto alla insufficienza tricuspidale funzionale lungo il margine sternale sinistro. Questo soffio è tipicamente accentuato dall'inspirazione e diminuisce durante l'espirazione forzata (segno di Carvallo). Quando la portata cardiaca è marcatamente ridotta i tipici reperti auscultatori, incluso il rullio diastolico, possono essere assenti (stenosi mitralica silente), ma possono ripresentarsi con il ripristino del compenso. Il soffio di Graham Steell dell'insufficienza polmonare è un soffio diastolico ad alta frequenza in decrescendo, udibile lungo il margine sternale sinistro, che ha origine dalla dilatazione dell'anello della valvola polmonare in pazienti con patologia della mitrale ed ipertensione polmonare severa. Questo soffio può essere indistinguibile da quello più comune dato dalla insufficienza aortica, ma può aumentare di intensità durante l'inspirazione ed essere accompagnato ad una forte componente polmonare del secondo tono. Diagnosi: La diagnosi di stenosi mitralica si avvale di: - ECG, che tuttavia da poche informazioni; - Ecocardiogramma, che permette di fare una valutazione precisa delle camere cardiache e della entità della stenosi; - Rx torace, che permette di identificare l'ingrandimento isolato dell'atrio sinistro e, in caso di ipertensione polmonare, la dilatazione del ventricolo destro; - cateterismo cardiaco, utile per una valutazione pre-operatoria. Fin dagli anni ’80, l’ecocardiogramma m-mode ci ha permesso di vedere la stenosi mitralica, in era pre-eco si faceva l’esofago baritato che metteva in evidenza l’ingrandimento dell’atrio. All’elettrocardiogramma (ECG) è presente un’onda P un po’ più evidente, dovuta all’ingrandimento dell’atrio sinistro, e possiamo avere dei segni di ipertrofia ventricolare dx , ma molto spesso il segnale è dato dalla fibrillazione atriale, cioè il paziente ha un’aritmia totale da fibrillazione atriale. Più nel dettaglio, l’ECG presenta le seguenti modificazioni: - la durata dell'onda P è superiore a 0,12 secondi, indicativa di un ingrandimento dell'atrio sinistro; - l'asse elettrico del cuore, o asse QRS, presenta un'angolazione superiore agli 80°, dovuta alla marcata ipertrofia del ventricolo destro; - nella derivazione V 1 , il rapporto tra l'ampiezza dell'onda R e dell'onda S è superiore a uno; - si possono notare i segni della fibrillazione atriale. Il cateterismo cardiaco ci permette di registrare i valori della pressione atriale e ventricolare sx. La pressione atriale la possiamo registrare in due modi: - direttamente (pungendo il setto atriale da dx o andandoci per via retrogada); - misurando la pressione di incuneamento polmonare (tra capillari e atrio sx non ci sono valvole quindi la pressione dei capillari polmonari è la pressione dell’atrio sx); mettendo un catetere con palloncino nel circolo polmonare lo gonfiamo con elio o con anidride carbonica e misuriamo la pressione, misurando poi la pressione ventricolare destra in diastole, si è in grado di ottenere il gradiente pressorio vigente nell’atrio sinistro. All’ecocardiografia è possibile valutare la superficie dell’ostio valvolare della mitrale. Il valore normale dell’orifizio mitralico oscilla tra i 4-6 cm2. L’area dell’orifizio mitralico è possibile ottenerla anche grazie alla formula di Gorlin: Per PC si intende la portata cardiaca, Fc la frequenza, TRD è invece il tempo di riempimento diastolico e ΔP è il gradiente pressorio calcolato dalla differenza tra pressione nel ventricolo sinistro e atrio sinistro. Con l’eco si può effettuare una valutazione del grado di stenosi. Distinguiamo tre forme di stenosi mitralica: - lieve, con area compresa tra 1,5-2 cm2; - moderata, con area compresa tra 1,5-1 cm2; - severa, con area inferiore a 1 cm2. Se all’eco notiamo che la stenosi determina un’area valvolare di 1,5-2 cm2 (lieve), generalmente il paziente non presenta sintomi, se la stenosi è invece moderata (1-1,5 cm2) il paziente avrà quasi sicuramente dei sintomi, se la stenosi è severa ( inferiore ad 1 cm2) bisognerà prendere dei provvedimenti. Questa mostrata a fianco è l’immagine all’ecocardiografia di una mitrale stenotica. Trattamento: La profilassi con penicillina delle infezioni da streptococco Beta- emolitico di gruppo A è importante in tutti pazienti a rischio con stenosi mitralica. Nei pazienti sintomatici si ottiene in genere un miglioramento dell'apporto con sodico una e una restrizione dose di mantenimento di diuretici per via orale. Il glicosidi digitalici non danno beneficio ai pazienti con stenosi mitralica e ritmo sinusale, ma sono utili nel diminuire la frequenza della risposta ventricolare nei pazienti con fibrillazione atriale. A questo fine sono utili anche i beta-bloccanti e i calcio-antagonisti non-diidropiridinici (verapamil). Tutti i pazienti con stenosi mitralica e fibrillazione atriale o con una storia di trombo-embolia devono essere trattati con il warfarin a un dosaggio tale da mantenere l’INR compreso tra 2 e 3. Nel caso di stenosi serrata bisogna intervenire. In questo caso abbiamo 2 possibilità, dettate dalla morfologia della valvola: - una meno aggressiva attraverso la valvuloplastica percutanea, se la morfologia valvolare è favorevole. La valvuloplastica si effettua entrando Dalla vena femorale con un catetere, attraversiamo la cava ed arriviamo in atrio dx, pungiamo il setto interatriale, lo attraversiamo, attraversiamo la valvola mitrale e gonfiamo il palloncino che abbiamo inserito tramite il catetereguida e così aumentiamo la superficie della valvola mitrale; ovviamente tutto ciò si può fare se la morfologia è favorevole ossia se i lembi valvolari non presentano calcificazioni altrimenti si potrebbero creare danni determinando così un’insufficienza mitralica acuta e se il paziente non soffre di fibrillazione atriale; - una più invasiva, attraverso la sostituzione valvolare in caso di morfologia sfavorevole e di coesistente insufficienza mitralica; tale intervento si fa con circolazione extracorporea. PROLASSO DELLA VALVOLA MITRALE Il prolasso della mitrale (PVM) è una mancata coaptazione anatomica dei lembi valvolari (è una variante anatomica) caratterizzato all’auscultazione da un click mesosistolico, ma il decorso nelle forme lievi è ottimo. Questa patologia interessa circa il 6% della popolazione di razza bianca soprattutto nel sesso femminile. I lembi mitralici sono ridondanti perché più grandi. Se è presente insufficienza mitralica concomitante la sintomatologia è riconducibile a questa patologia. Il prolasso della valvola mitrale viene anche indicato come sindrome del click-soffio sistolico, sindrome di Barlow, sindrome della valvola basculante e sindrome del lembo mitralico ridondante. L’eziologia resta sconosciuta nella maggior parte dei casi, anche se il prolasso mitralico potrebbe essere dovuto ad una malattia del collagene di natura genetica. Il PVM è di frequente riscontro nei pazienti affetti da malattie ereditarie del tessuto connettivo, come la sindrome di Marfan, l’osteogenesi imperfetta e la sindrome di Ehler-Danlos. Il PVM a volte è associato a deformazioni scheletriche simili a quelle che si osservano nella sindrome di Marfan. Nella maggior parte dei pazienti la degenerazione mixomatosa interessa solo i lembi mitralici, mentre non sono presenti le altre manifestazioni cliniche o anatomo-patologiche della malattia; in genere il lembo posteriore è il più colpito e l’anulus spesso è molto dilatato; in molti pazienti un allungamento o rottura delle corde tendinee causa o contribuisce al rigurgito. Un prolasso della mitrale può comparire anche come sequela di febbre reumatica acuta, nella cardiopatia ischemica e in varie miocardiopatie; inoltre, è presente anche nel 20% dei pazienti con difetto del setto interatriale del tipo ostium secundum. Il prolasso della valvola mitrale può determinare una sollecitazione anomala dei muscoli papillari che è causa di malfunzionamento e ischemia di questi muscoli e del miocardio ventricolare circostante. Lo sviluppo di un'insufficienza mitralica può anche essere favorito dalla rottura delle corde tendinee e da una progressiva dilatazione dell'annulus che, determinando un aumento della tensione sul già alterato apparato valvolare, crea un circolo vizioso. Caratteristiche cliniche: Il PVM è più comune nelle donne e interessa con maggiore frequenza la fascia di età tre i 15 e i trent'anni. Il decorso clinico è spesso benigno. Il prolasso può anche essere osservato in soggetti con età superiore ai cinquant'anni, nei quali insufficienza mitralica è molto spesso grave e richiede un trattamento chirurgico. La condizione di prolasso della valvola mitrale è di grado variabile. La maggior parte dei pazienti è asintomatica e rimane tale per tutta la vita. La morte improvvisa è una complicanze molto rara e si verifica in genere in pazienti con insufficienza mitralica severa e funzione ventricolare sinistra depressa. Sempre che nei pazienti con flail di un lembo valvolare abbiano maggior rischio di morte improvvisa. Molti pazienti presentano dolori toracici di difficile valutazione; spesso sono sotto-sternali, prolungati, poco correlati all'esercizio e raramente ricordano il dolore anginoso. Il riscontro obiettivo più frequente è quello di un click mesosistolico (non di eiezione) che si manifesta circa 0,14 secondi o più dopo il primo tono, dovuto al rapido tendersi delle corde tendinee allungate o al lembo prolassante che raggiunge la sua massima escursione; si possono auscultare click sistolici multipli, spesso seguiti da un soffio telesistolico in crescendo-decrescendo che a volte assume caratteristiche musicali o a "grido di gabbiano" ed è meglio apprezzato all'apice. Esami strumentali: L’elettrocardiogramma è normale nella maggior parte dei casi, ma può mostrare un'onda T difasica o invertita nelle derivazioni II, III e aVF; talvolta si osserva la presenza di extrasistoli sopraventricolari o ventricolari. L'ecocardiografia transtoracica riveste particolare utilità in quanto consente di visualizzare il prolasso dei lembi mitralici. Una definizione ecocardiografica di prolasso mitralico è la seguente: dislocazione sistolica, in proiezione parasternale asse lungo, delle cuspidi valvolari della mitrale di almeno 2 mm verso l'atrio sinistro superiormente al piano passante per l'annulus mitralico. L'ispessimento dei lembi valvolari individua un sottogruppo di pazienti a rischio per endocardite infettiva e insufficienza mitralica grave. Terapia: La profilassi del endocardite infettiva è indicata solamente nei pazienti con una storia di endocardite. I beta-bloccanti possono alleviare i dolori toracici e controllare le palpitazioni. Se il paziente è sintomatico da insufficienza mitralica severa, è indicata la riparazione valvolare, raramente la sostituzione. Nei pazienti affetti da attacchi ischemici transitori vanno utilizzati farmaci antiaggreganti e va presa in considerazione una terapia anticoagulante. INSUFFICIENZA AORTICA L’insufficienza aortica è l’equivalente dell’insufficienza mitralica, solo che questa volta il rigurgito dall’aorta passerà nel ventricolo sx. Infatti, si definisce insufficienza aortica il reflusso diastolico dall'aorta in ventricolo sinistro per lesione dei lembi semilunari aortici o dell'aorta ascendente con il coinvolgimento dei lembi valvolari. L'insufficienza aortica può essere causata da una valvulopatia primitiva o da una malattia primitiva della radice aortica. Eziologia: In passato, la malattia reumatica e la sifilide erano le cause principali di insufficienza aortica. Attualmente, per il calo di questi due patologie, si ha un aumento dell'incidenza legato a: - malformazione della valvola aortica, classica è la aorta bicuspide; - connettivopatie; - ipertensione arteriosa; - endocardite. Per quanto riguarda il caso di valvulopatia primitiva, in circa i due terzi dei pazienti insufficienza è di origine reumatica, con ispessimento, deformazione e accorciamento delle cuspidi valvolare aortiche che impediscono la corretta chiusura della valvola durante la diastole e l'apertura durante la sistole. Un ‘eziologia reumatica è molto meno comune in pazienti con insufficienza aortica isolata senza un associata valvola che mitralica. Pazienti con valvola aortica bicuspide congenita possono sviluppare un'insufficienza aortica predominante. La stenosi sottovalvolare membranosa spesso porta a ispessimento e fibrosi dei lembi valvolari con una insufficienza aortica secondaria. Il prolasso di una cuspide aortica, che determina un'insufficienza aortica cronica progressiva, si presenta in circa il 15% dei pazienti con un difetto interventricolare, ma può presentarsi anche come fenomeno isolato oppure come conseguenza di una degenerazione mixomatosa. L'insufficienza aortica può essere dovuta endocardite infettiva che si sviluppa su una valvola precedentemente colpita da malattia reumatica, congenitamente deformata, oppure, raramente, su una valvola normale, e può portare alla perforazione o erosione di uno o più lembi valvolari. I lembi valvolari possono andare incontro a fibrosi e retrazione anche in corso di sifilide o spondilite anchilosante. Nonostante la rottura traumatica e l’avulsione della valvola aortica siano cause poco comuni di insufficienza aortica acuta, queste rappresentano le lesioni gravi più frequenti nei pazienti che sopravvivono a traumi cardiaci non penetranti. Nel caso, invece, di malattia primitiva della radice aortica (ectasia annulo-aortica), l'insufficienza aortica è dovuta ad una marcata dilatazione dell'aorta ascendente in assenza di un coinvolgimento delle cuspidi valvolari; l'insufficienza è provocata dall'allargamento dell'anulus aortico e dalla conseguente separazione dei lembi. La degenerazione cistica della media dell'aorta ascendente, la dilatazione idiopatica dell'aorta, l'ectasia annulo-aortica, l'osteogenesi imperfecta e ipertensione arteriosa severa possono tutte dilatare l’annulus aortico e portare a insufficienza aortica progressiva. In alcuni casi l'insufficienza aortica è dovuta a una dissezione aortica retrograda che coinvolge l'anello valvolare. Vi sono tre forme di insufficienza aortica: - dilatazione dell’aorta e dell’anulus (i lembi non combaciano); - forma reumatica (retrazione delle semilunari); - endocardite (cicatrizzazione e formazione di fori nella valvola ). Fisiopatologia: Come nel caso dell’insufficienza mitralica avremo un’ ipertrofia eccentrica del ventricolo sx. Distinguiamo due forme di insufficienza aortica, quella cronica e quella acuta. Nell'insufficienza cronica il reflusso di sangue nel ventricolo sinistro aumenta il riempimento diastolico determinando un sovraccarico di volume, con ipertrofia eccentrica ed è aumento del volume del ventricolo sinistro. Nell'insufficienza acuta si ha shock cardiogeno. L’insufficienza aortica acuta può portare a shock cardiogeno, ma a livello cardiaco determina un'ischemia miocardica diffusa. L'ischemia miocardica diffusa è dovuta a: - aumento del precarico, con aumento della tensione parietale, e di conseguenza aumento della richiesta di ossigeno; - ridotta diastole, con la riduzione del tempo di perfusione coronarica. Il volume totale di sangue del ventricolo sinistro è aumentato nei pazienti con insufficienza aortica. In quelli con insufficienza aortica da ampia apertura (pura), il volume rigurgitante può essere della stessa entità della gittata anterograda. Nell'insufficienza aortica tutto il volume di sangue espulso dal ventricolo sinistro in sistole deve essere spinto in una camera ad alta pressione, l'aorta. Il principale meccanismo di compenso del insufficienza aortica è rappresentato dall'aumento del volume telediastolico (aumento del precarico). La dilatazione e l'ipertrofia eccentrica del ventricolo sinistro consentono a questa camera di espellere un maggior volume di sangue senza che ciò richieda un aumento dell'accorciamento relativo delle singole miofibrille. Quindi, un paziente con insufficienza aortica grave può presentare una gittata sistolica effettiva normale e una normale frazione di eiezione, associate a un volume e una pressione telediastoliche del ventricolo sinistro aumentate. Per la legge di Laplace, la dilatazione del ventricolo sinistro aumenta la tensione sistolica di parete richiesta per sviluppare un determinato valore di pressione sistolica. Nell'insufficienza aortica cronica sono quindi aumentati sia il precarico che il postcarico. Tuttavia, questi meccanismi di compenso perdono la loro efficacia. Quando la funzione ventricolare sinistra si deteriora, il volume telediastolico, la frazione di eiezione e la gittata anterograda diminuiscono. Nell'insufficienza aortica cronica spesso si osserva un ispessimento considerevole della parete ventricolare sinistra con un aumento del peso del cuore. Durante la diastole il gradiente pressorio tra aorta e ventricolo sinistro, si riduce progressivamente e ciò spiega il caratteristico soffio in decrescendo. Nei pazienti con insufficienza aortica acuta severa il ventricolo sinistro non è in grado di sopportare il sovraccarico di volume rigurgitato. La compliance del ventricolo sinistro è ridotta o normale e la pressione diastolica del ventricolo sinistro aumenta rapidamente. Verso la fine della diastole la pressione del ventricolo sinistro può eccedere quella dell'atrio sinistro e questo gradiente pressorio inverso determina la chiusura anticipata della valvola mitrale. Manifestazioni cliniche: Circa i tre quarti dei pazienti con insufficienza aortica pura o predominante sono maschi; nelle donne è invece più frequente un'insufficienza valvolare primaria associata a patologia mitralica. Nei pazienti con grave insufficienza aortica acuta, il ventricolo sinistro esaurisce rapidamente i suoi meccanismi di compenso, con il risultato di un rapido incremento della pressione telediastolica ventricolare e quindi con l'incremento delle pressioni nell'atrio sinistro e nei capillari polmonari. Possono svilupparsi rapidamente condizioni patologiche come lei edema polmonare e/o lo shock cardiogeno. Le principali manifestazioni dell'insufficienza aortica acuta sono: - tachicardia; - ipotensione; - edema polmonare acuto. Gli individui affetti da grave insufficienza aortica cronica possono rimanere asintomatici per periodi di 10-15 anni. Tuttavia, un sintomo precoce è dato da una spiacevole percezione del battito cardiaco, soprattutto a riposo in posizione supina. La tachicardia sinusale provocata dall'esercizio fisico o da emozioni e i battiti ectopici ventricolari possono determinare fastidiose palpitazioni e cefalea pulsante. Successivamente si ha dispnea da sforzo che è il primo sintomo connesso alla riduzione della riserva cardiaca. Compaiono poi ortopnea, dispnea parossistica notturna e sudorazione profusa. Anche nei pazienti giovani sono di frequente riscontro precordialgie non necessariamente dovute a coronaropatia. Gli episodi anginosi possono essere prolungati e spesso non rispondono alla nitroglicerina sublinguale. Le fasi più avanzate della malattia sono caratterizzate dalla tendenza alla ritenzione idrica e all'epatomegalia con edema della caviglia e ascite. Nell'insufficienza aortica cronica, quindi, le principali manifestazioni sono: - asintomatica per molti anni; - astenia e dispnea da sforzo; - dispnea notturna, a riposo e edema polmonare. Il segno patognomonico dell’insufficienza aortica è una pressione di 160 su 60/50 mmHg (nell’anziano possiamo trovare un’ipertensione sistolica ossia 160 su 80 mmHg ma una pressione di 160 su 50-60 o anche meno è tipica dell’insufficienza aortica). Il paziente con insufficienza aortica grave presenta chiari segni della malattia, consistenti in pulsazioni in tutto il corpo, movimento pulsatorio della testa che accompagna ogni sistole e arterie pulsanti. Sono caratteristici dell'insufficienza aortica pura un rapido incremento del polso arterioso, detto polso celere o collassante, che collassa rapidamente quando la pressione arteriosa scende rapidamente in telesistole e proto diastole (polso di Corringan), nonché un tipico alternarsi di iperemia e pallore del letto ungueale quando viene applicata una pressione alle estremità dell'unghia (polso capillare di Quincke). Sulle arterie femorali si può ascoltare un tono detto a colpo di pistola (segno di Traube); inoltre, sempre a livello della femorale, esercitando una lieve compressione del vaso con lo stetoscopio si può apprezzare un soffio denominato "va e vieni" (segno di Duroziez). La pressione differenziale è aumentata e la misurazione della diastolica con lo sfigmomanometro può essere resa imprecisa dal fatto che spesso i toni sono percepibili anche con la cuffia dello sfigmomanometro completamente sgonfia. Comunque, il livello di pressione registrato in fase IV corrisponde abbastanza bene alla vera pressione diastolica. Con il progredire della malattia e l'aumento della pressione telediastolica del ventricolo sinistro può aumentare anche la pressione arteriosa diastolica dato che la pressione aortica diastolica non può scendere al di sotto della pressione telediastolica del ventricolo sinistro. Per lo stesso motivo l'insufficienza aortica acuta severa può essere associata ad un aumento solo leggero della pressione arteriosa differenziale. Questi pazienti sono inoltre tachicardici, poiché la frequenza cardiaca aumenta nel tentativo di preservare la portata cardiaca. Nell'insufficienza aortica grave il tono di chiusura della componente aortica del secondo tono è di solito assente. È comune il riscontro di un terzo e a volte anche di un quarto tono. All’auscultazione la caratteristica è rappresentata dal soffio diastolico diverso da quello della stenosi mitralica in quanto copre e segue il secondo tono, tendendo a scomparire (protomesodiastolico) e ciò è dovuto al rilasciamento che non è isovolumetrico (perché la valvola non è completamente chiusa). Il soffio di Austin-Flint è un soffio dovuto ad una apparente stenosi mitralica, dovuto al fatto che nel momento in cui l’atrio si contrae aumenta la quantità di sangue che entra nel ventricolo, e questa quantità di sangue ostacola la corrente di sangue che rigurgita dall’aorta e quindi si crea un rumore che è simile a quello della stenosi mitralica. Nell'insufficienza aortica acuta grave l'aumento della pressione telediastolica ventricolare sinistra può determinare una chiusura precoce della valvola mitrale e in questi casi si apprezzano un tono mesodiastolico, un primo tono lieve o addirittura assente, una pressione differenziale non particolarmente aumentata e un breve soffio diastolico. Diagnosi: Per quanto riguarda la diagnosi di insufficienza aortica, che si avvale di: - elettrocardiogramma; - ecocardiogramma; - quadro radiologico; - cateterismo cardiaco ed è esame angiografico. Nei pazienti con insufficienza aortica cronica grave l'elettrocardiogramma mostra evidenti segni di ipertrofia ventricolare sinistra. Questi pazienti presentano frequentemente una depressione del tratto ST ed un'inversione dell'onda T nelle derivazioni I, aVL, V 5 e V 6 , indice di sovraccarico ventricolare sinistro. La comparsa di una deviazione assiale sinistra e di un allargamento del complesso QRS denota una miocardiopatia diffusa, che in genere è associata a fibrosi disseminata ed è un segno prognostico sfavorevole. Con l’ecocardiogramma si è in grado di definire l’entità del rigurgito e permette anche di studiare l’apparato valvolare, valutando la presenza di vegetazioni o anomalie delle cuspidi. Il grading, invece, viene effettuato con cateterismo cardiaco utilizzando un mezzo di contrasto, e si otterrà la seguente scala: - grado +: minimo jet di rigurgito; - grado ++: jet di rigurgito che delinea i bordi della cavità del ventricolo sinistro; - grado +++: ventricolo sinistro opacizzato come l’aorta; - grado ++++: ventricolo sinistro opacizzato più dell’aorta. Trattamento: I pazienti con insufficienza aortica acuta severa possono rispondere ai diuretici ed ai vasodilatatori per via endovenosa (nitro prussiato di sodio), ma la loro stabilità clinica è di breve durata ed è necessario un trattamento chirurgico urgente. La contro pulsazione aortica è controindicata. Vanno inoltre evitati i beta-bloccanti al fine di non ridurre ulteriormente la portata e la frequenza cardiaca: la riduzione di quest’ultima prolunga la diastole e quindi la durata del rigurgito. La terapia di scelta è quindi unica: sostituzione chirurgica della valvola. Nei pazienti sintomatici bisogna sostituire la valvola per forza. Nei pazienti asintomatici bisogna dapprima valutare la frazione di eiezione. La sostituzione è indicata nei pazienti asintomatici che hanno una frazione di eiezione inferiore al 50 %. Per tutti i pazienti asintomatici che presentano una frazione di eiezione superiore al 50%, all’ecografia si valuterà il diametro telediastolico e telesistolico del ventricolo, per poter valutare l’opzione di sostituzione valvolare. Il diametro telediastolico non deve aver superato i 70 mm e quello telesistolico i 55 mm, questo perché, oltre questi valori una sostituzione valvolare non avrebbe alcun effetto dato che il ventricolo sarà iperdilatato e non recuperebbe una decente funzione contrattile. STENOSI AORTICA Questa valvulopatia interessa il 25 % di tutti i pazienti con lesioni valvolari. Circa l’80 % dei pazienti adulti con stenosi aortica valvolare sintomatica è di sesso maschile. La stenosi aortica è una malattia delle valvole semilunari caratterizzata dal restringimento valvolare e ostruzione al flusso ventricolare sinistro con sviluppo di gradiente pressorio ed ipertrofia concentrica del ventricolo sinistro. La stenosi aortica è una delle patologie più frequentemente osservate nelle persone ultra settantenni. Eziologia: La stenosi aortica può essere causata da calcificazioni degenerative delle cuspidi aortiche; può essere congenita o secondaria a malattia reumatica. Le principali cause di stenosi aortica sono: - degenerativa senile, la più frequente; - congenita; - reumatica; - forme rare. La stenosi aortica calcifica degenerativa rappresenta la più comune causa di stenosi aortica. La stenosi aortica viene definita ecocardiograficamente come un ispessimento o calcificazione delle cuspidi valvolari con un picco di velocità del flusso transitorio all’eco-Doppler inferiore o uguale a 2,5 m/s. La sclerosi aortica sembra essere un marcatore di rischio di eventi coronarici. La stenosi aortica congenita può aggravarsi con lo sviluppo di fibrosi e calcificazioni. In altri pazienti, la valvola è congenitamente alterata, generalmente bicuspide, ma senza stenosi significativa fino all’adolescenza. La sua morfologia anomala la rende però particolarmente suscettibile agli stress emodinamici, con successivo sviluppo di calcificazioni, di ispessimento e di rigidità dei lembi e, infine, riduzione dell’area valvolare. La malattia reumatica dei lembi valvolari aortici determina una fusione commissurale che provoca a volte la formazione di una valvola bicuspide. Questa conformazione la rende ancora più suscettibile agli stress emodinamici e può provocarne la fibrosi, la calcificazione e l’ulteriore restringimento. Quando l’ostruzione del tratto di efflusso ventricolare provoca sintomi importanti, di solito la valvola si è trasformata in una struttura calcifica rigida nella quale è difficile, o persino impossibile, riconoscere la valvulopatia primitiva. La stenosi aortica reumatica è suggerita dalla presenza in anamnesi di febbri reumatiche; è quasi sempre associata ad un coinvolgimento reumatico anche della valvola mitrale e, spesso, a insufficienza aortica. Oltre alla stenosi aortica valvolare, vi sono almeno tre tipi di lesioni responsabili di ostruzione del tratto di efflusso del ventricolo sinistro: - la miocardiopatia ipertrofica-ostruttiva; - la stenosi aortica sotto-valvolare congenita; - la stenosi aortica sopra-valvolare. Il valore limite dell’orifizio aortico è di 2 cm2. Le eziologie della stenosi aortica sono per la quasi totalità degenerativa (52%) e Bicuspide (36%). La bicuspide, data la sua conformazione, può facilmente nel tempo andare incontro ad una degenerazione di tipo calcifico e quindi andare incontro ad una stenosi. Una classificazione delle stenosi aortiche, le divide in tre categorie: - stenosi sopravalvolare; - stenosi valvolare; - stenosi sottovalvolare. Non dobbiamo dimenticare che oltre alla stenosi valvolare vi è la possibilità molto rara di avere una stenosi sopravalvolare cioè subito al di sopra del bulbo (dovuta alla sindrome congenita di Williams) o una stenosi sottovalvolare a diaframma o ad anello, che è possibile rinvenire all’interno del ventricolo sx prima della valvola stessa ed è come se si formassero due camere ventricolari. Fisiopatologia: L’orifizio aortico ha un area di circa 2-3 cm2. Una riduzione dell’area superiore al 30% determina lo sviluppo di un gradiente significativo trans valvolare. Per contrastare il gradiente il ventricolo sinistro va incontro allo sviluppo di ipertrofia concentrica attraverso la replicazione in parallelo delle miofibrille. L’ipertrofia determina una maggiore rigidità delle pareti del ventricolo sinistro che quindi si oppone alla distensione passiva durante la diastole determinando quindi una disfunzione diastolica. La caratteristica emodinamica principale è costituita dall’ostruzione della via di efflusso del ventricolo sinistro, che determina un gradiente pressorio tra ventricolo sinistro e aorta. In alcuni pazienti l’ostruzione è già presente alla nascita e/o aumenta gradualmente nel corso del tempo, consentendo il mantenimento di una normale portata cardiaca attraverso lo sviluppo di un meccanismo compensatorio, rappresentata dall’ipertrofia ventricolare sinistra concentrica, che tende a ridurre a valori normali lo stress sistolico sviluppato dal miocardio. Inizialmente questo è un meccanismo adattativo che ha il fine di ridurre lo stress sistolico sviluppato dal miocardio secondo la legge di Laplace. Per anni può essere presente un elevato gradiente pressorio trans valvolare aortico, senza che la portata cardiaca sia ridotta o che il ventricolo sinistro sia dilatato; a lungo termine però l’ipertrofia eccessiva diviene mal adattativa e si assiste ad un declino della funzione ventricolare sinistra. Un gradiente sistolico medio superiore ai 40 mmHg con una portata cardiaca normale o un’area effetiva dell’orifizio aortico inferiore a 1,0 cm2 è generalmente espressione di un’ostruzione severa all’efflusso del ventricolo sinistro. L’aumento della pressione tele diastolica ventricolare indica la presenza di una ridotta compliance della parete ventricolare ipertrofica. Mentre nella maggior parte dei pazienti con stenosi aortica grave la portata cardiaca a riposo è entro i limiti normali, durante l’esercizio fisico non riesce ad aumentare come accade fisiologicamente. La perdita di una funzione di pompa atriale sincrona e vigorosa che si verifica in caso di fibrillazione atriale o di dissociazione atrioventricolare può provocare una rapida progressione dei sintomi. Nelle fasi terminali della malattia la portata ed il gradiente sistolico tra ventricolo sinistro e aorta diminuiscono, mentre aumentano la pressione atriale media sinistra, la pressione arteriosa polmonare e la pressione ventricolare destra. L’aumento della massa muscolare miocardica dovuto all’ipertrofia del ventricolo sinistro fa si che aumentino le richieste miocardiche di ossigeno. Inoltre, anche in assenza di una coronaropatia, vi può essere un’interferenza con il flusso coronarico in quanto la pressione che comprime le arterie coronariche supera la pressione di perfusione coronarica e ciò causa spesso ischemia, specie a livello sub endocardico. Il paziente con ipertrofia ventricolare potrà andare incontro ad un fenomeno definito ‘’angor da discrepanza’’, ossia, in condizioni di sforzo, avrà un’angina non per una malattia coronarica ma perché in condizioni di sforzo la coronaria non riuscirà a veicolare la quantità di sangue e di ossigeno necessario a nutrire il miocardio notevolmente ispessito. Alcuni studi hanno dimostrato che una sostituzione valvolare fatta per tempo può far si che il ventricolo ritorni quasi alla normalità. Presentazione clinica: La stenosi aortica raramente assume rilevanza clinica fino a quando l’area valvolare non si riduce ad almeno 1,0 cm2. Nella maggior parte dei pazienti con stenosi aortica pura o prevalente si può avere un progressivo aumento dell'ostruzione, senza comparsa di sintomi, fino al sesto o ottavo decennio di vita. I tre sintomi principali sono: dispnea da sforzo, angina pectoris, sincope. Spesso l'anamnesi è caratterizzata da sintomi di difficile inquadramento, come astenia ingravescente e dispnea nonostante una graduale riduzione dell'attività fisica. La dispnea è dovuta all'incremento della pressione capillare polmonare provocato dall'aumento della pressione telediastolica del ventricolo sinistro, secondaria a sua volta alla sua ridotta compliance. L'angina pectoris compare più tardivamente e riflette la discrepanza tra le aumentate richieste miocardiche di ossigeno e la ridotta perfusione; l'aumento delle richieste di ossigeno è dovuto all'aumento della massa miocardica e della pressione intraventricolare, mentre la diminuita disponibilità di ossigeno è dovuta alla frequente presenza di coronaropatia e alla compressione dei vasi coronarici da parte del miocardio ipertrofico. La sincope da sforzo è provocata dall'ipotensione arteriosa dovuta alla vasodilatazione che si verifica dei muscoli che stanno sostenendo l'esercizio e alla contemporanea inadeguata vasocostrizione dei muscoli a riposo, da una situazione di portata cardiaca non aumentabile, oppure dalla rapida riduzione della portata provocata da un'aritmia. Poiché la portata cardiaca a riposo è in genere normale fino agli stadi più avanzati, di solito l'astenia, la cianosi periferica, la cachessia e le altre manifestazioni di bassa portata sono caratteristiche solo delle fasi terminali della malattia. Anche i sintomi di insufficienza ventricolare sinistra come l'ortopnea, la dispnea parossistica notturna e l'edema polmonare sono caratteristici delle fasi più avanzate della malattia, mentre il riscontro di un'ipertensione polmonare grave, di insufficienza ventricolare destra e di ipertensione venosa sistemica con epatomegalia, fibrillazione atriale ed insufficienza tricuspidale sono rilievi caratteristici delle fasi preterminali. In caso di coesistenza di stenosi aortica e stenosi mitralica, la diminuzione della portata cardiaca indotta dalla stenosi mitralica determina una riduzione del gradiente pressorio Transvalvolare aortico che può mascherare molti dei segni clinici caratteristici della stenosi aortica. L’esame obiettivo mostrerà un polso piccolo e tardo, nonché alternate. All’auscultazione abbiamo il cosiddetto soffio mesosistolico a ‘’ diamante’’ o crescente-decrescente (cresce durante l’apertura valvolare, decresce dopo che la valvola si è aperta). Il primo tono risulta essere normale o diminuito, mentre si verifica lo sdoppiamento paradosso del secondo tono, in cui la componente polmonare segue quella aortica. In alcuni casi è possibile riscontrare anche un quarto tono. La triade sintomatologica della stenosi aortica è data da: - angina; - dispnea; - sincope. La comparsa di questi sintomi predice la morte del paziente nell’arco di 2-4 anni se non gli cambiamo la valvola. Un paziente con stenosi aortica e fibrillazione atriale muore entro due anni dalla comparsa della fibrillazione, questo perché il ventricolo si riempie male e si svuota peggio. L'angina è il risultato di diversi fattori: - squilibrio tra massa muscolare ipertrofica del letto vascolare capillare; - riduzione della riserva coronarica subendocardica; - eccessiva aumento da sforzo della domanda di ossigeno; - aumento della pressione diastolica ventricolare; - compressione diretta sui vasi intramurali. Osservando l'immagine, il Vaso visibile in diastole si interrompe in sistole perché la forza di contrazione e la massa che si è contratta tendono a schiacciarlo (ovviamente in questo caso di tratta di un vaso secondario. Il discendente anteriore è visibile in entrambi i casi). La sincope è una mancata perfusione cerebrale con caduta e perdita dei sensi. La sincope è causata da un inadeguato flusso ematico attraverso la valvola stenotica con conseguente ridotta perfusione cerebrale e coronarica. Anche la comparsa di aritmie ipercinetiche ventricolari è stata presa in considerazione come causa della sincope. La sopravvivenza media è di circa tre anni da un episodio di sincope. La comparsa di dispnea è un fattore prognostico negativo, poiché la sopravvivenza media è di circa due anni dalla comparsa della scompenso cardiaco. Infatti l'insorgenza di dispnea, astenia, tosse, dispnea parossistica notturna ed edema polmonare acuto sono chiari segni di scompenso cardiaco. Le principali complicanze di stenosi aortica sono: - embolie sistemiche, abbastanza rare; - endocardite infettiva; - aritmie ventricolari e morte improvvisa; - fibrillazione atriale; - dissezione aortica; - disturbo di conduzione. Diagnosi: La diagnosi differenziale deve essere posta nei confronti di altre patologie ostruttive della via di efflusso del ventricolo sinistro. La diagnosi si avvale delle seguenti indagini: - elettrocardiogramma; - ecocardiogramma; - quadro radiologico; - cateterismo. Nella maggior parte dei pazienti affetti da stenosi aortica grave il principale riscontro è dato da ipertrofia ventricolare sinistra. Nelle fasi più avanzate della malattia, nelle derivazioni I e aVL e nelle precordiali sinistre si osservano una depressione del tratto ST e l’inversione dell’onda T (sovraccarico ventricolare sinistro). Nel caso della stenosi aortica, l’ecocardiogramma è di grande utilità perché ci permette di valutare la stenosi e successivamente l’efficacia di un eventuale intervento. In funzione della valutazione della superfice dell’ostio valvolare, si classifica la stenosi mitralica in tre gradi: - stenosi lieve, con area valvolare superiore ad 1,5 cm2; - stenosi moderata, con area valvolare compresa tra 0,8 e 1,5 cm2; - stenosi severa, con area valvolare inferiore a 0,8 cm2. Una valutazione del grado di stenosi può essere effettuata anche valutando il gradiente pressorio transvalvolare: - stenosi lieve, con gradiente pressorio medio inferiore ai 25 mmHg; - stenosi moderata, con gradiente pressorio medio compreso tra i 25 e i 50 mmHg; - stenosi severa, con gradiente pressorio medio superiore ai 50 mmHg. Con il cateterismo cardiaco siamo in grado di effettuare lo studio coronarografico pre-operatorio e lo studio dei gradienti transvalvolari emodinamici. Tramite il cateterismo cardiaco possiamo valutare la pressione, in particolar modo possiamo calcolare il gradiente tra ventricolo e aorta, per lo stesso discorso che si è fatto per la stenosi mitralica. Anche qui è possibile calcolare l'area dell'orifizio valvolare utilizzando la formula di Gorlin. Per quanto riguarda l’area di stenosi, possiamo valutare l'entità della stenosi in: - lieve, se la superficie valvolare è superiore ad 1 cm²; - moderata, se la superficie valvolare è compresa tra 0,7 e 1 cm²; - severa, se la superficie valvolare è inferiore a 0,7 cm². Prima di passare oltre, bisogna fare un breve accenno alle forme di ipertrofia ventricolare. Esistono due forme di ipertrofia ventricolare: - ipertrofia ventricolare concentrica, dovuta ad un aumento del carico pressorio; - ipertrofia ventricolare eccentrica, dovuta ad un aumento del carico di volume, ad infarto e miopatia. In entrambi i casi, col passare del tempo, il muscolo cardiaco ipertrofico a divenire parzialmente ischemico, questo perché ad un aumento del volume non si è verificato un corrispettivo aumento del letto vascolare coronarico. Inoltre il continuo stress sul miocardio determina apoptosi delle cellule muscolari e la loro sostituzione con tessuto fibroso, evolvendo in una ipertrofia non compensata. Quindi, ricapitoliamo i punti salienti dell'evoluzione della stenosi aortica: - innanzitutto si ha un ostacolo all'eiezione del ventricolo sinistro; - segue un'ipertrofia concentrica, compensatoria, del ventricolo sinistro; - col passare del tempo, si avrà anche un'alterazione nella funzione diastolica e quindi anche una riduzione del tempo di ossigenazione del miocardio; - ciò determina un aumento inadeguato del rapporto massa miocardica e il volume di sangue che lo rifornisce di ossigeno; - tutto ciò innesca una disfunzione contrattile, con successiva dilatazione del ventricolo sinistro e dilatazione dell'anulus mitralico, innescando anche un'insufficienza mitralica. Trattamento: Nei pazienti con stenosi aortica severa l'attività fisica strenua deve essere evitata, anche durante la fase asintomatica. È necessaria attenzione al fine di evitare la disidratazione e l’ipovolemia e proteggere il paziente da una significativa riduzione della portata cardiaca. I farmaci utilizzati nel trattamento dell'ipertensione arteriosa e della coronaropatia, inclusi beta-bloccanti e ACE-inibitori, sono generalmente sicuri nei pazienti asintomatici con una funzione sistolica del ventricolo sinistro conservata. La nitroglicerina è utile nel far recedere l'angina pectoris. Il tratto chirurgico è invece indicato nelle seguenti condizioni: - area valvolare superiore o uguale a 0,8 cm²; - gradiente di picco pressorio superiore ai 50 mmHg; - in presenza di sintomi. PRINCIPI GENERALI DEL TRATTAMENTO CHIRURGICO DELLE VALVULOPATIE La terapia risolutiva delle valvulopatie è data dalla terapia chirurgica, terapia che può essere effettuata tramite due opzioni: - chirurgica propriamente detta; - percutanea. In corso di sostituzione delle valvole, vengono utilizzate protesi che possono essere: - protesi artificiali, come valvole meccaniche e valvole biologiche; - protesi “umane”, intese come trapianto homograft ed autograft. Per quanto riguarda le protesi meccaniche, queste sono vere e proprie valvole e se ne riconoscono di diversi tipi, come indicato dall’immagine soprastante. Molto importante per questo tipo di protesi è prendere in considerazione le alterazioni del flusso ematico, che da laminare tende ad assumere un andamento turbolento. È per questo che, la sostituzione con valvola meccanica è seguita da terapia anti-coagulante a vita, questo perché la superficie protesica è una superficie che determina l’attivazione piastrinica, e quindi aumenta il rischio di formazione di trombi ed emboli. Le principali indicazioni all’impianto di protesi meccanica sono: - pazienti con una lunga aspettativa di vita; - presenza di protesi meccanica in altra posizione; - pazienti con insufficienza renale, dialisi ed ipercalcemia; - pazienti già in terapia con anti-coagulanti orali per rischio trombo-embolico; - età inferiore ai 65 anni se si tratta di stenosi della valvola aortica; - età inferiore ai 70 anni se si tratta di stenosi della valvola mitrale. La prima valvola meccanica venne impiantata dal chirurgo Starr Edwards su di un paziente, Philip Admunson, affetto da stenosi mitralica. L’intervento venne effettuato il 2 settembre del 1960. Il paziente sopravvisse per 15 anni e morì per una caduta accidentale mentre verniciava casa. Le protesi valvolari biologiche sono di origine porcina, bovina od equina, e si dividono in: - protesi biologiche con supporto (stented); - protesi biologiche senza supporto (stentless). Le principali indicazioni all’impianto di protesi biologica sono: - pazienti in cui non è attuabile la terapia con anticoagulanti orali; - controindicazioni o mancata compliance alla terapia anticoagulante orale; - età superiore a 65 anni se trattasi di stenosi valvolare aortica in assenza di fenomeni trombo embolici; - età superiore ai 70 anni se trattasi di stenosi della valvola mitralica, in assenza di fenomeni trombo embolici. Ma le protesi valvolari cardiache possono essere anche di origine umana, suddividendole in homograft ed autograft (operazione di Ross). Per quanto riguarda la terapia percutanea, questa scelta può essere effettuata per risolvere: - stenosi aortica con valvuloplastica aortica; - stenosi mitralica, con valvuloplastica mitralica; - insufficienza mitralica funzionale, con il MITRACLIP; - stenosi aortica severa, tramite l’impianto di valvola aortica percutanea. La valvuloplastica aortica consiste nella dilatazione della valvola stenotica mediante gonfi aggio di catetere a palloncino. Questa modalità di trattamento viene utilizzata per: - forma calcifica dell’adulto, con una modesta riduzione della stenosi, miglioramento labile della sintomatologia, ma alto rischio di complicanze; - forma congenita, con una riduzione della stenosi, miglioramento della sintomatologia ed un basso rischio di complicanze. Per quanto riguarda la stenosi mitralica, nell’operazione per via percutanea si utilizza un particolare catetere, il catetere di Inoue, che permette, tramite una procedura rapida e sicura l’esecuzione della commissurotomia mitralica trans venosa percutanea. Nell’impianto della valvola mitralica per via transcutanea, i palloncini hanno due dimensioni a seconda del paziente. La mitral clip non è altro che una molletta che si mette al centro dei due lembi valvolari mitralici, per cui quando in sistole i due lembi tenderebbero ad estroflettersi nell’atrio e a far refluire il sangue in atrio sx, essendo legati da questa molletta i lembi si fermano e quindi il reflusso sarà minimo. Per poter effettuare questo intervento, esistono dei criteri di eleggibilità e criteri di esclusione. Tra i criteri di eleggibilità figurano: - età inferiore ai 18 anni; - insufficienza mitralica moderata o severa; - mal coaptazione dei lembi mitralici. Invece, tra i criteri di esclusione vi sono: - una frazione di eiezione inferiore al 25 % o LVESD (left ventricular end-systolic diameters) superiore ai 55 mm; - insufficienza renale; - endocardite. Per quanto riguarda le Valvole Aortiche percutanee sono essenzialmente due, auto espandibile, la quale si apre sfilando il cateterino che la circonda ed il suo vantaggio è che se la posiziono male nel caso non abbia sfilato tutto il catetere di rivestimento posso ancora riposizionarla; l’altra si espande gonfiando il pallone, quindi una volta espansa non la posso togliere più. I lati negativi sono: per quanto riguarda la prima, quando si espande può premere sul fascio di His e quindi in un quarto dei pazienti che mettono questa valvola devo impiantare un pacemaker; la seconda può essere impiantata per via transcutanea oppure effettuando una toracotomia ed entrando dall’apice del cuore (via trans-apicale) quando le vie d’accesso come la femorale sono ostruite, il difetto della seconda è che tende a spostare eventuali placche che possono ostruire le coronarie. Anche qui, vi sono criteri di eleggibilità e criteri di esclusione. Tra i primi rientrano: - stenosi valvolare aortica severa; - età superiore ai 75 anni; - logistic euroscore superiore al 15%. Tra i criteri di esclusione vi sono: - endocardite; - aneurisma e calcificazione aortica; - aterosclerosi, calcificazione e tortuosità degli accessi arteriosi. CAPITOLO VII: LE ENDOCARDITI Il termine endocardite indica un’infiammazione dell’endocardio che può interessare soprattutto, ma non esclusivamente, i lembi valvolari. Le cause di questa infiammazione possono essere infettive, immunologiche, o direttamente rappresentate da traumatismi esercitati dalla corrente sanguigna sull’endocardio. ENDOCARDITE INFETTIVA Il termine endocardite infettiva è più appropriato di quello, pure impiegato frequentemente, di endocardite batterica. Infatti, pur essendo i batteri gli agenti eziologici più comunemente implicati in questa malattia, il processo morboso può essere determinato anche da miceti. L’endocardite infettiva è caratterizzata dalla localizzazione all’endocardio, e per lo più sui lembi valvolari, di microrganismi, che anche se di per sé poco patogeni, i poteri di difesa dell’organismo non sono in grado di eliminare. Per questo motivo si verificano un danno delle strutture valvolari e la persistente immissione in circolo di microrganismi, con prolungata stimolazione del sistema immunitario. Questa stimolazione è responsabile di molti dei sintomi e dei segni propri della malattia. Se non adeguatamente trattata, l’endocardite infettiva è mortale. La lesione anatomo-patologica tipica dell’endocardite è la vegetazione. Anche se i soggetti potenzialmente suscettibili all’endocardite infettiva sono molti, la malattia è relativamente rara. CLASSIFICAZIONE Una classificazione tradizionale distingue due forme di endocardite infettiva: acuta e subacuta. Secondo questa distinzione, l’endocardite infettiva acuta è provocata da microrganismi virulenti, invasivi, che si impiantano anche su valvole cardiache sane, ove esercitano notevoli processi distruttivi. La immissione in circolo di questi microrganismi provoca frequentemente focolai infettivi metastatici in varie sedi. La durata della malattia, senza terapia, è di giorni o settimane. Anche con terapia appropriata la mortalità resta superiore al 50%. L’endocardite infettiva subacuta è invece provocata da microrganismi poco virulenti e non invasivi, che praticamente si fissano solo su valvole cardiache già lese da precedenti processi patologici. A causa della scarsa virulenza dei microrganismi, la loro immissione in circolo difficilmente dà luogo a focolai infettivi metastatici, mentre prevalgono gli effetti della prolungata stimolazione del sistema immunitario. La durata della malattia, senza terapia, va da alcuni mesi fino a 1-2 anni. Con terapia appropriata si ha una frequenza elevata di guarigione. Questa distinzione non è più corrispondente alla realtà clinica comune. L’impiego precoce di antibiotici, se pure non a dosi sufficienti per una terapia efficace dell’endocardite infettiva, fa sì che, anche quando sono in causa microrganismi virulenti e invasivi, il quadro della malattia sia spostato verso quello proprio della forma subacuta. Perciò al giorno d’oggi, quest’ultima è la variante di endocardite di gran lunga più frequente, anche se esistono casi nei quali la malattia ha un’espressione che è a metà tra i due quadri tipici. Una classificazione più appropriata dell’endocardite infettiva è perciò basata sull’agente eziologico che l’ha prodotta. EZIOLOGIA Numerosi microrganismi possono provocare l’endocardite infettiva. Essi sono per lo più derivati dal cavo orale e dalle alte vie aeree, dalla cute, dal colon, dall’apparato urogenitale. Il loro passaggio in circolazione è favorito da manovre, quali interventi odontoiatrici anche modesti, traumatismi cutanei anche banali (come nel caso di drogati da aghi per iniezione usati con scarsi accorgimenti igienici, o nella pratica medica da cateteri venosi mantenuti troppo a lungo in sede), indagini endoscopiche a livello del retto o del colon, cateterismi delle vie urinarie, raschiamenti uterini, inserimento di contraccettivi del tipo degli IUD (Intra Uterine Devices). Tuttavia, nella maggioranza dei casi di endocardite infettiva non si riesce ad individuare un fattore esogeno determinante la batteriemia (la stessa masticazione può provocare batteriemia in condizioni difettose di igiene del cavo orale). Nel 60-80% dei casi sono implicati degli streptococchi poco patogeni. In era preantibiotica gli agenti eziologici erano, nella stragrande maggioranza dei casi, rappresentati da streptococchi del tipo viridans (ossia α-emolitici), di solito provenienti dal cavo orale o dal faringe. Oggi la prevalenza di queste infezioni è diminuita e viene stimata inferiore al 50% di tutti i casi di endocardite infettiva, mentre sono aumentate le endocarditi da altri streptococchi, tra i quali particolarmente importanti lo S. faecalis (enterococco) e lo S. bovis, provenienti più spesso dall’intestino, dalle vie urinarie e dalle vie genitali femminili. Questi microrganismi hanno attitudine ad infettare valvole cardiache già precedentemente lese. Gli stafilococchi vengono al secondo posto come causa di endocardite infettiva e la loro provenienza è solitamente dalla cute (perciò sono una comune causa di endocardite nei drogati). Lo Staphylococcus aureus, che è notevolmente patogeno, può infettare anche valvole assolutamente integre (allora il quadro della malattia somiglia di più a quello descritto per la variante acuta), mentre lo Staphylococcus epidermidis colpisce più facilmente portatori di protesi valvolari. Rare sono invece le endocarditi da tutti gli altri microrganismi. Esse possono verificarsi come complicanza della malattia fondamentale (angina da streptococco (β-emolitico di gruppo A, polmonite da Streptococcus pneumoniae, gonorrea, brucellosi, febbre Q, psittacosi), ma gli agenti eziologici possono penetrare in circolo nella maniera già descritta, indipendentemente da altra patologia, per lo più attraverso la cute o le mucose. Tra questi ultimi, gli enterici Gram – e i funghi microscopici prevalgono tra i drogati e i portatori di protesi valvolari (a somiglianza dello Staphylococcus epidermidis). Molti di questi microrganismi possono insediarsi su valvole precedentemente sane. FATTORI DI LOCALIZZAZIONE Perché l’endocardite infettiva possa iniziarsi, occorre che i microrganismi si attacchino all’endocardio. Solo i microrganismi più virulenti possono farlo direttamente su cellule endoteliali integre, mentre quelli meno virulenti possono farlo solo se trovano delle circostanze favorevoli alla loro fissazione. Queste si verificano quando le cellule endoteliali vengono danneggiate e, in conseguenza di questo danneggiamento, si ha la deposizione sull’endocardio di piastrine con o senza fibrina. La condizione più propizia perché si verifichi questo processo e si abbia la fissazione di microrganismi sull’endocardio è rappresentata da un getto ematico spinto da una pressione elevata che, attraverso un orifizio ristretto, si riversi in una camera cardiaca a pressione bassa. Una condizione di questo tipo si può verificare, per esempio, in certi vizi valvolari. Nell’insufficienza mitralica, il getto è rappresentato dal sangue che rigurgita in sistole nell’atrio sinistro; nell’insufficienza aortica, dal sangue che rigurgita nel ventricolo sinistro in diastole; nella pervietà interventricolare, dal sangue che passa in sistole dal ventricolo sinistro (a pressione elevata) in quello destro (a pressione bassa). In tutti questi casi, a causa del flusso vorticoso, si ha un danno endoteliale, e una conseguente adesione dei microrganismi, immediatamente a valle dell’orifizio ristretto attraverso cui passa il getto ematico e, subordinatamente (localizzazione satellite), sulle strutture che sono investite direttamente dal getto ematico. Perciò, per restare agli esempi precedenti, nell’insufficienza mitralica la localizzazione delle lesioni avverrà preferibilmente sulla faccia atriale dei lembi valvolari e (localizzazione satellite) sulla parete dell’atrio sinistro; nell’insufficienza aortica avverrà sulla faccia ventricolare delle semilunari aortiche e (localizzazione satellite) sulle corde tendinee del ventricolo sinistro; nella pervietà interventricolare avverrà sul lato destro del setto interventricolare e (localizzazione satellite) sulla valvola tricuspidale e sulla parete opposta al setto del ventricolo destro. È importante tenere presente che, in assenza di batteriemia, in queste sedi di traumatismo endocardico possono episodicamente aversi piccoli depositi di piastrine, che però sono destinati a dissolversi rapidamente senza conseguenze. È solo quando alla deposizione di piastrine segue la fissazione di microrganismi che si innesca un processo irreversibile che conduce alla lesione tipica dell’endocardite infettiva. Il deposito di piastrine sull’endocardio è indispensabile per fornire ai microrganismi un punto di aggancio. Ma, una volta che i microrganismi si sono fissati, essi danneggiano ulteriormente l’endocardio amplificando la formazione del trombo e favorendo così, nel suo contesto, un ambiente adatto alla colonizzazione microbica. Da quanto fin qui detto risulta evidente che esistono fattori predisponenti allo sviluppo di un’endocardite infettiva. Essi sono infatti rappresentati da quelle anomalie cardiache acquisite o congenite (tra le quali i vizi valvolari) che creano le condizioni adatte alla fissazione dei microrganismi all’endocardio. I soggetti che presentano queste anomalie sono a rischio, mentre coloro che hanno un cuore integro lo sono molto meno e solamente nei riguardi di microrganismi molto virulenti. Nel passato circa il 90% dei casi di endocardite infettiva si verificava in soggetti con valvulopatie reumatiche e più spesso con malattie della valvola mitrale. Oggi, grazie alla diminuita prevalenza della malattia reumatica, una valvulopatia acquisita di questo tipo si trova in meno della metà dei casi, mentre si riconoscono sempre più spesso endocarditi infettive che si sovrappongono a lesioni valvolari degenerative negli anziani, di solito a carico delle semilunari aortiche (più facili in caso di valvola bicuspide), ma anche possibili a livello dell’anulus della valvola mitrale. Anche le cardiopatie congenite possono predisporre all’endocardite infettiva. Tra queste ultime, il massimo di rischio si ha nella pervietà interventricolare e il minimo nella pervietà interatriale (anzi, in questa il rischio sembra nullo se non coesiste un’alterazione della valvola mitrale). Attualmente la più comune diagnosi di alterazione cardiovascolare riconosciuta come fattore predisponente all’endocardite infettiva è il prolasso della mitrale. Ciò dipende dall’elevata frequenza di questa condizione nella popolazione generale. Un rischio significativo sarebbe collegato anche con la persistenza del dotto di Botallo (e in questo caso la malattia viene definita “endocardite” in un senso lievemente improprio), se questa anomalia non venisse di solito corretta chirurgicamente piuttosto precocemente. Un altro fattore predisponente importante è la presenza, in contiguità con l’endocardio, di materiale estraneo, come avviene nei portatori di protesi valvolari. Un problema particolare riguarda i tossicomani da eroina i quali, oltre ad essere a rischio di endocardite per i ripetuti traumatismi cutanei da iniezione, possono anche sviluppare lesioni predisponenti all’endocardite a causa delle sostanze adulteranti che vengono mescolate alla droga. L’iniezione endovenosa di questi contaminanti può ledere l’endocardio delle sezioni destre del cuore (le prime che vengono raggiunte dalla sede di iniezione) e creare le condizioni predisponenti alla fissazione di microrganismi. Per questo motivo l’endocardite infettiva nei drogati colpisce più facilmente la valvola tricuspide. Accanto a questi fattori predisponenti di natura meccanica, ne esistono altri di natura più propriamente biologica che è però difficile quantificare. Questi ultimi sono connessi con la risposta immunitaria dell’organismo che, anziché proteggere dalla malattia, sembra in questo caso favorirla, probabilmente perché i microrganismi parzialmente agglutinati dagli anticorpi si fissano più facilmente sull’endocardio. Si spiegherebbe in tal modo perché gli agenti più comuni di endocardite infettiva siano per lo più microrganismi largamente diffusi nell’ambiente, che hanno occasione di stimolare il sistema immunitario praticamente di tutti gli individui. MECCANISMI PATOGENETICI La lesione elementare dell’endocardite infettiva è rappresentata da una vegetazione costituita da piastrine e fibrina, adesa da un lato all’endocardio e per il resto sporgente in una cavità cardiaca. Il volume della vegetazione è per lo più modesto, ma in certi casi, particolarmente quando l’endocardite è provocata da miceti, può essere tanto grande da interferire con il passaggio del sangue attraverso la valvola colpita. Questa vegetazione costituisce un ottimo habitat per i microrganismi, i quali vi si possono moltiplicare in una situazione relativamente protetta rispetto all’azione delle cellule fagocitiche e degli anticorpi. Infatti, gli strati subendocardici, e particolarmente i lembi valvolari, non sono forniti di capillari e perciò le cellule fagocitiche (neutrofili, monociti) non possono concentrarvisi come fanno solitamente in altre sedi anatomiche quando esiste una localizzazione infettiva. D’altro canto, le stratificazioni di piastrine e di fibrina sulle vegetazioni coprono i microrganismi e li proteggono in qualche modo dall’azione degli anticorpi circolanti. È per questo motivo che, in assenza di terapia antibiotica, l’endocardite infettiva non guarisce spontaneamente. D’altro canto, la presenza delle vegetazioni cariche di microrganismi determina alcuni importanti effetti che così possiamo schematizzare: - azioni lesive locali, importanti soprattutto sui lembi valvolari; - il distacco di frammenti dalle vegetazioni che, trasportati dalla corrente sanguigna, possono provocare embolizzazione di vasi periferici; - la formazione di focolai infettivi metastatici da parte dei microrganismi veicolati dagli emboli infetti; - la persistente immissione in circolo di complessi antigene-anticorpo, nei quali l’antigene è rappresentato da prodotti microbici; - la cronica stimolazione del sistema immunitario. Azioni lesive locali: Sono rappresentate inizialmente dalla necrosi delle cellule endoteliali in corrispondenza della vegetazione e, successivamente, da un approfondimento dei fenomeni necrotici nel connettivo immediatamente sottostante. Si formano perciò delle lesioni cosiddette “ulcerative” che, in casi particolari, possono approfondirsi tanto da bucare da parte a parte un lembo valvolare, o da determinare il distacco di una corda tendinea, che resta perciò fluttuante nella cavità ventricolare. L’entità di queste lesioni ulcerative è dipendente dalla patogenicità del microrganismo: è perciò massima nelle più rare forme della variante “acuta”, mentre è più limitata nelle forme più comuni della variante “subacuta”, provocata da microrganismi poco patogeni. Alle volte, alla periferia delle vegetazioni possono formarsi dei veri e propri microascessi, i quali, se raggiungono l’anulus fibrosus di una valvola, possono farsi strada nella cavità pericardica determinando una pericardite. Questa evoluzione è, tuttavia, rara e si può verificare solo quando i microrganismi infettanti sono molto patogeni. Quando il processo endocarditico è localizzato alle valvole semilunari aortiche, la sua propagazione può anche danneggiare il sistema di conduzione cardiaco e determinare vari tipi di blocchi. Embolizzazione periferica: È più spesso determinata da frammenti molto piccoli delle vegetazioni. Questa microembolizzazione è sicuramente molto più comune di quanto sia clinicamente rilevabile e può provocare lesioni cutanee e mucose, retiniche e soprattutto renali sotto forma di una glomerulonefrite focale. Più rara, ma possibile, è anche la macroembolizzazione, con la conseguente comparsa di fenomeni ischemici in distretti dipendenti da vasi arteriosi periferici che risultano occlusi. Gli emboli di grosso volume sono provenienti più spesso da vegetazioni endocarditiche determinate da miceti (le quali sono particolarmente grandi), ma sono possibili in qualsiasi tipo di endocardite. La loro localizzazione più comune è nella grande circolazione (dato che l’endocardite infettiva è più spesso localizzata nelle cavità sinistre del cuore), ma è possibile l’embolia polmonare quando l’endocardite è nelle cavità destre (come avviene quando il fattore favorente è la pervietà interventricolare, o nelle localizzazioni alla valvola tricuspide). Formazione di focolai infettivi metastatici: È una forma rara. Più spesso il fatto che i microemboli siano infetti conferisce una caratteristica infiammatoria alle lesioni periferiche determinate da questi. Solo con microrganismi molto patogeni (per esempio Staphilococcus aureus) è possibile la formazione di veri ascessi metastatici (per esempio in sede cerebrale o renale). Meno rara e più insidiosa è l’evenienza che si verifichi la lesione delle pareti di vasi arteriosi di medio calibro, o per localizzazioni metastatiche endoarteriche o per microembolizzazione infetta dei vasa vasorum. Questo determina la formazione di piccoli aneurismi (detti micotici) particolarmente importanti nella circolazione cerebrale, ove la loro rottura può portare ad emorragie subaracnoidee mortali. Persistente immissione in circolo di complessi antigene-anticorpo: È la caratteristica patogenetica più importante dell’endocardite infettiva nella forma più comunemente osservata al giorno d’oggi. In realtà, se anche i microrganismi non esercitano un’azione lesiva diretta particolarmente grave (sia perché scarsamente patogeni sia perché antagonizzati dalla terapia antibiotica), è inevitabile che essi riversino in circolo dei prodotti di loro derivazione e sollecitino la formazione dei corrispondenti anticorpi. In eccesso di antigene si possono formare complessi antigene-anticorpo solubili che sono trasportati con la circolazione e si depositano sulle pareti dei vasi sanguigni, determinando localmente dei fenomeni infiammatori per l’attivazione della sequenza complementare. In questo processo si possono avere manifestazioni del tipo della “malattia da siero”, tra le quali meritano particolarmente di essere ricordate le artralgie (o addirittura l’artrite) e le infiammazioni delle sierose, inclusa l’evenienza che si sviluppi una pericardite. L’effetto più importante della circolazione di com plessi antigene-anticorpo nell’endocardite infettiva è però la possibilità di una glomerulonefrite acuta diffusa. Un problema controverso è se alcune o tutte le lesioni mucocutanee e retiniche dell’endocardite infettiva, tradizionalmente attribuite a microembolizzazione infetta, non siano in realtà di natura “vasculitica” e determinate perciò da complessi antigene-anticorpo circolanti. In realtà i due meccanismi patogenetici non sono mutuamente esclusivi (i microemboli potrebbero veicolare microrganismi legati ai corrispondenti anticorpi). Stimolazione cronica del sistema immunitario: È dovuta non solamente alla risposta specifica nei confronti degli antigeni propri dei microrganismi infettanti, ma anche a sollecitazioni aspecifiche. Infatti, molti prodotti batterici stimolano la proliferazione dei linfociti B, indipendentemente dalla specificità antigenica contro cui questi sono diretti. La conseguenza è un’attivazione cosiddetta “policlonale” (perché interessa simultaneamente parecchi cloni di linfociti) della produzione anticorpale, che comporta lo sviluppo di un’ipergammaglobulinemia e la comparsa a titolo significativo di vari tipi di anticorpi non direttamente correlati con l’agente infettante: fattore reumatoide (talora con le caratteristiche di crioglobuline), anticorpi anticardiolipina (con falsa positività per le reazioni sierologiche per la sifilide), anticorpi anticuore. La formazione di questi ultimi è tuttavia favorita dal danneggiamento del miocardio dovuto all’estensione del processo endocarditico. CLINICA La forma di presentazione comune dell’endocardite infettiva è quella di un processo febbrile, di entità più o meno elevata, che risulta persistente e poco influenzato (o influenzato solo transitoriamente) da eventuali terapie antibiotiche condotte “alla cieca” in dosaggi convenzionali. Il sintomo che accompagna più frequentemente la febbre è rappresentato dalle artralgie, che possono spesso interessare uno o poche delle grandi articolazioni e talora assumere l’aspetto dell’artrite migrante. I pazienti più giovani depongono spesso di avere sofferto di malattia reumatica nell’infanzia o di aver presentato molte tonsilliti febbrili (e magari di essere stati tonsillectomizzati per questo), o di essere stati nel passato riconosciuti portatori di un “soffio” cardiaco. In altri casi il paziente è consapevole di essere portatore di un vizio congenito di cuore o di una protesi valvolare o di prolasso della mitrale. Nei pazienti più anziani questi riferimenti anamnestici spesso mancano, dato che, come si è visto, il fattore favorente l’endocardite è per lo più rappresentato da lesioni valvolari degenerative, spesso non rilevate o sottovalutate. In una minoranza dei casi, nell’anamnesi recente è deposto qualche intervento odontoiatrico o qualche altra pratica strumentale favorente la batteriemia. L’esame obiettivo dimostra una tachicardia (spesso in eccesso rispetto a quanto dovuto alla febbre) e un reperto cardiaco rappresentato da soffi di vario tipo. Nei giovani questi soffi coincidono con il reperto dei vizi valvolari deposti nell’anamnesi, negli anziani più spesso viene percepito un soffio sistolico di eiezione al focolaio aortico. La diffusione dell’impiego diagnostico della ecocardiografia ha chiarito che possono darsi casi nei quali una vegetazione endocarditica è presente e un soffio non è percepibile. Questo avviene generalmente quando l’endocardite infettiva si sviluppa su valvole sane ed è in fase iniziale. Occorre precisare che, nella endocardite infettiva, i soffi percepiti inizialmente (e che sono dovuti ai fattori cardiaci predisponenti alla endocardite) possono modificarsi per il sopravvenire delle lesioni valvolari destruenti indotte dal processo patologico e per l’aggravamento dei vizi valvolari preesistenti o, in caso di rottura dei lembi valvolari, per l’instaurazione di vizi valvolari nuovi. In questo caso i soffi hanno spesso un particolare timbro musicale o sono percepiti come un “pigolio” (rumore che è stato attribuito ad una corda tendinea fluttuante libera in una cavità ventricolare). È importante ricordare che la rilevazione di un soffio cardiaco non è un reperto obbligatorio nella endocardite infettiva. Infatti nelle endocarditi del cuore destro (per es. nei drogati) e, più raramente, quando l’endocardite è localizzata in un trombo murale infetto, il reperto di un soffio può totalmente mancare. Solo raramente possono essere percepiti al cuore degli sfregamenti pericardici dovuti, come si è visto, a una pericardite indotta, o da propagazione di un focolaio infettivo o da sierosite nell’ambito di una reazione tipo “malattia da siero”. Piuttosto comunemente, nelle forme di endocardite infettiva della variante subacuta, viene constatata una splenomegalia. Se l’endocardite infettiva dura sufficientemente a lungo, si può avere una deformazione delle unghie “a vetrino d’orologio” (dita ippocratiche). Al giorno d’oggi è raro che l’esame obiettivo in una endocardite infettiva mostri molto di più di quanto precedentemente descritto. Tuttavia, il quadro classico della malattia comprende una serie di alterazioni mucocutanee e retiniche che oggi sono divenute relativamente rare, ma che meritano di essere ricordate. Le alterazioni meno infrequenti sono rappresentate da petecchie (piccoli punti rosso scuro delle dimensioni della capocchia di uno spillo che non scompaiono con la pressione) a localizzazione cutanea (ovunque sulla cute, ma soprattutto sulla porzione superiore della faccia anteriore del torace) e mucosa (palato, faringe, congiuntive). Le petecchie sottoungueali hanno una forma lineare e sono definite “a scheggia”, quelle mucose hanno spesso un centro chiaro. Più rare sono altre manifestazioni, come i noduli di Osler, le lesioni di Janeway e le chiazze di Roth. A lato viene mostrata l’immagine di una petecchia. I noduli di Osler sono a localizzazione sottocutanea, del diametro di quasi un centimetro, arrossati e dolenti e si trovano più comunemente ai polpastrelli delle dita delle mani e dei piedi. Le lesioni di Janeway sono alterazioni maculari rilevate sul palmo delle mani. Le chiazze di Roth vengono rilevate all’osservazione con l’oftalmoscopio del fondo dell’occhio e consistono in emorragie retiniche con area centrale chiara. Il quadro clinico dell’endocardite infettiva può essere notevolmente aggravato dal sopravvenire di macroembolizzazione. Possono perciò verificarsi repentinamente perdita completa del visus in un occhio (occlusione embolica di un’arteria retinica), emiplegia (embolizzazione di un’arteria cerebrale), dolore, pallore e ipotermia in un arto (embolizzazione in un’arteria periferica), dolore in sede splenica (infarto splenico), dolore in sede renale seguito da ematuria macroscopica (infarto renale), dolore addominale con quadro di addome acuto (infarto mesenterico), infarto del miocardio (occlusione embolica di una coronaria). Il sopravvenire di un’emorragia subaracnoidea (rottura di un aneurisma) provoca un quadro drammatico, caratterizzato da cefalea con rigidità nucale e gravi alterazioni neurologiche, con evoluzione più spesso mortale. ESAMI DI LABORATORIO E STRUMENTALI I cosiddetti segni “di fase acuta” sono positivi e perciò si ha un aumento della velocità di eritrosedimentazione e positività per la proteina C reattiva nel siero. L’esame emocromocitometrico dimostra una leucocitosi con neutrofilia e, se il processo infettivo dura a lungo, un’anemia normocitica che è quella delle malattie croniche (infatti, si accompagna a sideremia ridotta con transferrinemia bassa). Un reperto caratteristico nell’endocardite infettiva è la presenza di macrofagi osservabili nello striscio di sangue, specialmente se eseguito con la prima goccia di sangue ricavata da puntura del lobo dell’orecchio. È molto comune la presenza di alterazioni del reperto urinario, che riflettono la frequente implicazione del rene nel processo morboso. Queste sono rappresentate più spesso da modesta proteinuria e microematuria. Tra gli altri esami di laboratorio, meritano di essere ricordati quelli che sono significativi di una prolungata stimolazione del sistema immunitario: ipergammaglobulinemia all’elettroforesi delle proteine del siero (frequente), positività del reuma-test in circa la metà dei casi, ma non della reazione di Waaler-Rose, talora falsa positività della VDRL (reazione sierologica per la sifilide) o presenza di crioglobuline. In laboratori specializzati, possono essere messi in evidenza nel siero anticorpi anticuore in circa il 60% dei casi, complessi antigene-anticorpo circolanti nel 60-90% dei casi, e frequentemente una diminuzione dell’attività emolitica del complemento e della concentrazione della frazione C3 nel siero (segno di consumo del complemento da parte dei complessi antigene-anticorpo circolanti). La dimostrazione di batteriemia mediante emocoltura è frequentemente (ma non costantemente) positiva quando il sangue è prelevato in assenza di concomitante terapia antibiotica. Soltanto il 5-7% dei pazienti nei quali è stata formulata la diagnosi di endocardite infettiva con rigorosi criteri clinici, e nei quali non sono stati somministrati antibiotici, si presenta con emocolture sterili. Quando, in pazienti sospetti, le emocolture rimangono sterili dopo 48-72 ore, il clinico deve avvisare il laboratorio della necessità di effettuare indagini più approfondite, come un’incubazione più prolungata e subculture su mezzi più arricchiti. La tecnica della “polymerase chain reaction” è stata impiegata per dimostrare microrganismi non coltivabili su vegetazioni escisse chirurgicamente, ed è una promettente possibilità per il futuro per la ricerca di agenti infettanti nel sangue nel caso di endocarditi infettive con emocolture negative. Un’indagine strumentale molto utile è rappresentata dall’ecocardiografia, che non solo è in grado di mettere in evidenza vizi valvolari preesistenti, ma anche di visualizzare direttamente le vegetazioni endocardiche. Queste ultime possono, tuttavia, sfuggire a questo tipo di indagine se hanno un diametro inferiore a 2 mm. Esistono casi nei quali una vegetazione endocarditica è rilevabile solo con l’ecocardiogramma transesofageo (generatore di ultrasuoni posto con una sonda in esofago) e non con la tecnica classica transtoracica. Infatti, un ecocardiogramma transtoracico può essere inadeguato in circa il 20% dei pazienti adulti, a causa di obesità, broncopneumopatia cronica ostruttiva o deformità toraciche. Nel complesso, tenendo conto della possibilità di falsi negativi (che sono più comuni nelle endocarditi su valvole protesiche), la sensibilità di un ecocardiogramma transtoracico nel rivelare vegetazioni endocarditiche può essere meno del 60-70%. L’ecocardiogramma transesofageo è più costoso e più invasivo, ma ha una sensibilità tra il 75 e il 95%. Come regola generale, è stato suggerito che la decisione sul tipo di ecocardiogramma da impiegare come prima scelta dipenda dalla probabilità a priori della diagnosi di endocardite infettiva. Questa probabilità è soggettiva e dipende dal giudizio clinico, e perciò attribuirle dei numeri può apparire molto approssimativo. Tuttavia, è stato proposto che, se la probabilità a priori di questa diagnosi è inferiore al 4% (cioè piuttosto bassa) un ecocardiogramma transtoracico è efficace in relazione al costo e clinicamente soddisfacente per escludere un’endocardite infettiva. Se la probabilià a priori è superiore al 60% (cioè alta) un ecocardiogramma transtoracico come prima scelta è pure conveniente, dato che è molto probabile che risulti dimostrativo e che siano pochi i casi nei quali bisogna eseguire successivamente anche un ecocardiogramma transesofageo. Se la probabilità a priori è intermedia, conviene adottare come prima scelta l’ecocardiogramma transesofageo, dato che non sarebbero pochi i casi nei quali un ecocardiogramma transtoracico negativo dovrebbe essere comunque seguito da un ecocardiogramma transesofageo. DIAGNOSI L’endocardite infettiva dev’essere sospettata in presenza di ogni febbre di natura indeterminata che sia prolungata ed accompagnata da artralgie. Nel complesso il quadro clinico è piuttosto simile a quello di altre malattie a patogenesi immunopatologica, quali la malattia reumatica, la malattia da siero e il lupus eritematoso sistemico. Il medico che pretendesse di trovare tutti i segni classici mucocutanei e retinici per riconoscere l’endocardite infettiva rischierebbe seriamente di mancare la diagnosi (con conseguenze disastrose). Infatti, come si è già detto, questi segni sono attualmente divenuti rari. Un altro indice di sospetto può essere conferito al quadro clinico dalla rilevazione di qualche soffio cardiaco. Tuttavia, questo reperto non è patognomonico perché può essere causato da altre condizioni (per es., recidiva di malattia reumatica: in questo caso il vizio valvolare si è instaurato nel primo episodio), oppure può essere dovuto alla coincidenza di un vizio valvolare pregresso con una malattia del tutto indipendente. Per contro, come si è detto, in presenza di endocardite del cuore destro può mancare qualsiasi soffio. Molto significativa è invece una variazione delle caratteristiche del soffio che, però, deve essere clamorosa, perché variazioni lievi possono essere dovute a cause banali (per es., cambiamenti nella frequenza cardiaca). Gli elementi più utili per la diagnosi sono le emocolture (che debbono essere prelevate più volte) e l’ecocardiografia bidimensionale. Si è visto che le emocolture sono spesso, ma non sempre, positive. Una negatività può essere dovuta al fatto che il paziente è stato trattato alla cieca con antibiotici prima del prelievo (e in questo caso possono passare vari giorni prima che le emocolture divengano positive) o al fatto che l’agente eziologico è un microrganismo insolito, oppure (per es., nel caso che sia uno streptococco anaerobio) che richieda terreni colturali particolari. Si è visto che anche l’ecocardiografia non è costantemente positiva (non oltre l’80% dei casi). Tuttavia la negatività contemporanea di emocolture ed ecocardiografia è piuttosto improbabile. Per la diagnosi di endocardite vengono utilizzati i criteri di Duke. Questi costituiscono uno schema diagnostico dotato di elevata sensibilità e specificità nella diagnosi dell’endocardite batterica. Tale schema si basa sulla combinazione di dati clinici, di laboratorio ed ecocardiografici. Per porre diagnosi di endocardite batterica è necessario documentare la presenza di: - due criteri diagnostici maggiori; - un criterio maggiore e tre minori; - cinque criteri minori. Tra i criteri maggiori figurano l’emocoltura positiva e segni di interessamento endocardico. Per quanto riguarda l’emocoltura, i microrganismi che causano tipicamente endocarditi devono essere isolati in almeno 2 emocolture separate. Per quanto riguarda invece i microorganismi inusuali isolati persistentemente, questi: - devono essere isolati in due emocolture effettuate a 12 ore l’una dall’altra; - devono essere stati isolati in tre o nella maggior parte di quattro o più emocolture; - per Coxiella burneti è sufficiente una singola positività. I segni di interessamento endocardico sono refertati tramite ecocardiografia, che mostrerà: - massa intracardiaca oscillante sulla valvola o sulle strutture di supporto nell’ambito di un flusso di rigurgito; - massa adesa ad una struttura cardiaca impiantata; - visualizzazione di un ascesso; - riscontro di rigurgito valvolare di nuova comparsa; - deficienza della valvola protesica di nuova comparsa. Tra i criteri minori figurano: - presenza di condizioni predisponenti, come patologie cardiache, tossicodipendenza, immunodeficienza, ecc…; - febbre uguale o maggiore ai 38°; - fenomeni vascolari periferici, come embolo arterioso maggiore, infarto polmonare settico, aneurisma micotico, ictus o emorragia cerebrale, emorragie congiuntiovali, lesioni di Janeway; - fenomeni immunologici, come glomerulonefrite, noduli di Osler, macchie di Roth, fattore reumatoide; - emocoltura positiva ma che non soddisfa i criteri diagnostici sopra riportati. TERAPIA Se non si isola nessun microrganismo o in attesa dei risultati delle emocolture, la terapia è: - per la forma acuta gentamicina, più ampicillina, più naficillina; - se si sospetta meticillino resistenza sostituire la naficillina con vancomicina; - per la forma subacuta, ampicillina con gentamicina. Il fondamento della terapia dell’endocardite infettiva consiste nella somministrazione quotidiana, per infusione venosa continua o con molte dosi refratte per via parenterale, di elevate quantità di antibiotici battericidi per un periodo prolungato di tempo (4 settimane o più). Dato che gli streptococchi, che sono gli agenti eziologici più comuni di questa malattia, sono di solito molto sensibili alla penicillina G, questo è il farmaco che viene adoperato più comunemente, di solito alla dose di 20.000.000 U o più, quotidianamente e per via venosa. Nel caso di endocardite da S. faecalis (enterococco) è consigliata la simultanea somministrazione di streptomicina, che è sinergica con la penicillina, o di un altro aminoglicoside. Altri antibiotici possono essere scelti in base alla sensibilità del microrganismo infettante isolato nelle emocolture, purché siano battericidi e siano impiegati a dosi elevate per via parenterale. In assenza di un’indicazione clinica per una causa sospetta, nelle endocarditi con emocolture negative che insorgono su valvole native il trattamento è individualizzato e basato sull’impiego di antibiotici come la penicillina, l’ampicillina, il ceftriaxone o la vancomicina, spesso in combinazione con un aminoglicoside. Nel caso di endocardite su valvola protesica che insorga 12 mesi o più dopo l’intervento cardiochirurgico, è importante che il trattamento includa il ceftriaxone o il cefotaxime, per colpire anche alcuni organismi che raramente sono trovati sulle valvole native (cosiddetti organismi HACEK, acronimo che indica una pluralità di specie tra le quali alcune di Haemophilus, diverse da quello influenzae). Un caso particolarmente delicato è la terapia dell’endocardite della febbre Q (che, per fortuna, è assente in Italia da molti decenni). Per questa malattia, che ha una fortissima tendenza a recidivare, è indicata una terapia con doxiciclina e un fluorochinolonico per alcuni anni. L’associazione di doxiciclina con idrossiclorochina sembra dare buoni risultati anche per una durata di trattamento di “soli” 26 mesi. La terapia anticoagulante non è risultata efficace nel prevenire gli episodi tromboembolici e può facilitare le emorragie da aneurismi intracranici. Non è perciò raccomandata. La terapia chirurgica, con sostituzione della valvola ammalata, è utile in vari casi. Prima di tutto in pazienti che sviluppano uno scompenso cardiaco, ascessi perivalvolari o un’infezione che non si riesce a controllare nonostante una terapia antimicrobica molto energica. Secondariamente, in pazienti con endocardite provocata da microrganismi per i quali la terapia medica ha elevate probabilità di non essere coronata da successo, come nel caso di endocardite da Pseudomonas aeruginosa, da brucelle, da Coxiella burnetii (il già citato agente della febbre Q), da candida e altri funghi e probabilmente anche da enterococchi per i quali le indagini microbiologiche non dimostrano un regime battericida sinergistico. Le caratteristiche delle vegetazioni endocarditiche raramente giustificano un intervento chirurgico. Non sono state individuate caratteristiche “soglia”, di sede o dimensioni delle vegetazioni, che indichino una particolare predisposizione alle embolizzazioni, in modo che possa essere calcolato se i benefici dell’intervento chirurgico compensino i suoi rischi. Per di più, la persistenza delle vegetazioni, determinate ecograficamente, è comune dopo una terapia antibiotica coronata da successo e non, è necessariamente associata con complicanze tardive. Un punto delicato è se esiste un’indicazione chirurgica per prevenire ulteriori embolizzazioni, dopo che una di queste si è verificata. È dimostrato che la frequenza degli emboli decresce rapidamente con un’efficace terapia antimicrobica e perciò una singola embolia non è un’indicazione per l’intervento se è in corso una terapia medica efficace. Al contrario, l’intervento chirurgico può comportare rischi di deterioramento neurologico e di morte, e perciò una recente complicanza neurologica dell’endocardite infettiva è stata considerata una relativa controindicazione a un intervento di protesi valvolare. Un approccio prudente è di ritardare la sostituzione valvolare, se fattibile, di 2-3 settimane dopo un infarto embolico del sistema nervoso centrale e di almeno un mese dopo un’emorragia cerebrale. Diverso è il caso se l’embolizzazione si ripete. DECORSO E PROGNOSI In assenza di terapia antibiotica, l’endocardite infettiva è invariabilmente mortale. La morte consegue ad emorragia subaracnoidea, incidenti embolici o scompenso cardiaco per gravi lesioni valvolari. Al giorno d’oggi è molto improbabile che un’endocardite infettiva, se pure non diagnosticata, venga lasciata priva di ogni terapia. Più facile è che terapie antibiotiche vengano date alla cieca secondo schemi convenzionali. Questa pratica è molto dannosa perché non è in grado di guarire l’endocardite, al massimo di attenuarne l’espressione clinica e di prolungarne il decorso. Inoltre può rendere negative le emocolture rendendo difficile la diagnosi clinica ed eziologica. Può perciò capitare che pazienti non diagnosticati ed inappropriatamente trattati, vadano avanti per mesi o anni con febbri recidivanti che restano inspiegate. Questi pazienti, prima o poi, sviluppano le complicanze proprie dell’endocardite infettiva e la loro prognosi è infausta. Se il processo morboso è sufficientemente prolungato, la compromissione renale può anche esitare in insufficienza renale cronica. Quando appropriatamente trattata, l’endocardite infettiva guarisce nella maggioranza dei casi se i microrganismi sono poco virulenti e in quasi la metà dei casi se i microrganismi sono molto virulenti. La mortalità complessiva resta tra il 20 e il 25% tanto per le endocarditi su valvole native, che per quella su valvole protesiche. Dal punto di vista anatomico, la guarigione è caratterizzata dalla sostituzione delle vegetazioni e delle lesioni valvolari con tessuto cicatriziale che si retrae. Possono perciò aggravarsi vizi valvolari preesistenti o crearsene di nuovi (insufficienza mitralica o aortica). Questi danni non sono, in genere, molto gravi se la malattia è diagnosticata e trattata precocemente. Fattori prognostici positivi sono: - giovane età; - sensibilità alla penicillina; - diagnosi e trattamento precoci; - endocardite su valvola mitrale prolassata. Il coinvolgimento del SNC è il fattore prognostico negativo più significativo. Per quanto riguarda la profilassi, in persone suscettibili (portatori di protesi valvolari, pregressa endocardite, PVM, ecc…) si somministra un trattamento antibiotico profilattico prima di procedure che possono causare una batteriemia importante. CAPITOLO IX: CARDIOMIOPATIE Le miocardiopatie o cardiomiopatie sono un gruppo di malattie che colpiscono primariamente il muscolo cardiaco e non rappresentano la conseguenza di anomalie congenite o acquisite valvolari, ipertensive, coronariche o pericardiche. Si distinguono due forme fondamentali di miocardiopatia: - Un tipo primitivo, che consiste in una malattia del muscolo cardiaco, coinvolgente prevalentemente il miocardio e/o di origine sconosciuta; - Un tipo secondario, che consiste in una patologia miocardica di eziologia nota o associata ad una malattia sistemica. In molti casi non è possibile giungere ad una diagnosi eziologica specifica, e quindi è spesso preferibile classificare le miocardiopatie nell’ambito ti tre tipi morfologici. Vi sono 3 tipi fondamentali di danno funzionale: - la cardiomiopatia dilatativa (MCD); - la cardiomiopatia ipertrofica (MCI); - la cardiomiopatia restrittiva (MCR). MIOCARDIOPATIE: QUADRI MORFOLOGICI La cardiomiopatia dilatativa (MCD) è la forma più comune, ed è responsabile della maggior parte delle miocardiopatie ed è caratterizzata da dilatazione ventricolare, disfunzione sistolica e, spesso, dai sintomi dell’insufficienza cardiaca congestizia. Questo è il classico quadro del paziente che venendo da una patologia congenita, una patologia valvolare, una patologia ischemica, transita per questa fase per poi giungere allo sezione c’è scompenso. ma questa non solo un cospicuo ingrandimento sinistra, In dell’area è anche ventricolare evidente un ingrandimento dell’atrio e quindi è una patologia camere. che interessa entrambe le La cardiomiopatia ipertrofica (MDI) è caratterizzata da una ipertrofia ventricolare sinistra inappropriata, spesso con coinvolgimento asimmetrico del setto interventricolare, con preservata o aumentata funzione contrattile fino agli stati avanzati di malattia. Qui, nell’immagine a lato, lo spessore del ventricolo è più o meno uniformemente aumentato. In pazienti di questo tipo l’aumentata sporgenza del profilo settale andrà a creare un ostacolo nella fase di eiezione all’interno del ventricolo. La cardiomiopatia restrittiva è la forma meno comune caratterizzata da alterato riempimento diastolico ed, in alcuni casi, da fibrosi endocardica. Si è ridotto lo spazio per il sangue proveniente dall’atrio sinistro e si è alterata la struttura della parete, che è prevalentemente da caratterizzata fibrosi, che aumentandone la rigidità fa sì che nella fase di rilasciamento si rilasci molto poco. Accanto alle forme principali, vi sono 2 forme meno comuni, che sono meno frequenti ed anche di più recente descrizione. Una delle 2 forme è la miocardiopatia aritmogena del ventricolo destro, caratterizzata da una progressiva sostituzione fibro-adiposa del ventricolo destro e, per certi gradi, del sinistro. Vi sono 2 modi per trattare tale forma di cardiomiopatia: - Fare la terapia antiaritmica per prevenire le aritmie che possono anche risultare minacciose; - Trapianto cardiaco. Non è possibile fare un intervento di ablazione perché il rischio di perforazione del ventricolo è cospicuo. Vi sono poi delle forme cosiddette non classificate, quali la fibroelastosi, la disfunzione sistolica con minima dilatazione, la patologia mitocondriale, ecc… Le cardiomiopatie vengono classificate secondo la classificazione effettuata dalla Task Force della WHOISFC nel 1995 in: - Cardiomiopatia dilatativa; - Cardiomiopatia ipertrofica; - Cardiomiopatia restrittiva; - Cardiomiopatia aritmogena del ventricolo destro; - Cardiomiopatie specifiche. Molto importante è analizzare anche l’architettura microscopica che si ha nelle miocardiopatie. Nel riquadro A, viene mostrata l’architettura di un miocardio normale con perfetto allineamento dei miocardiociti che sono paralleli tra loro e lo spazio fibrotico tra un miocardiocita e l’altro è ridotto. In B vi è l’immagine tipica della cardiomiopatia ipertrofica: i miocardiociti, ipertrofici, sono disposti in maniera disordinata e la fibrosi interstiziale (in blu) è notevole. In C vi è l’immagine di una cardiomiopatia dilatativa: i miociti ipertrofici e fibrosi interstiziale aumentata. MIOCARDIOPATIA DILATATIVA La miocardiopatia dilatativa è caratterizzata dall’ingrandimento dela cuore e dalla disfunzione sistolica di uno o di entrambi i ventricoli. All’esame macroscopico vi è dilatazione di tutte e quattro le camere cardiache, esservi trombi possono intracavitari, specie in regioni apicali. Le arterie coronarie sono di solito normali. All’esame istologico si evidenziano estese zone di fibrosi interstiziale e perivascolare. Circa 1 caso su 4 di insufficienza cardiaca è dovuta a MCD. Nel caso però in cui la forma dilatativa è secondaria ad una malattia ischemica a più infarti, le coronarie non saranno di certo normali. Nell’immagine a lato, viene mostrata la differenza tra un cuore normale e un cuore con un ventricolo sinistro dilatato. E’ in un qualche modo mantenuto un discreto spessore della parete però quello che è aumentato è il volume. Questo è un caso di ipertrofia eccentrica per sovraccarico di volume. Considerazioni genetiche: Da un quinto ad un terzo dei pazienti è affetto da forme familiari di MCD. Sono state descritte mutazioni in oltre 20 geni, a trasmissione autosomica dominante. Le più comuni sono mutazioni dei geni che codificano proteine sarcomeri che. Si ritiene che le proteine anomale causino disfunzione contrattile compromettendo la produzione e/o trasmissione della forza. I pazienti affetti da MCD genetica possono anche presentare miopatie scheletriche, in particolare la distrofia muscolare di Duchenne e di Emery-Dreyfluss. Anche le mutazioni del gene della proteina della membrana nucleare laminina A/C vengono trasmesse in maniera autosomico dominante; esse sono responsabili dello sviluppo di MCD associata a disturbo della conduzione atrioventricolare e ad altre turbe elettrofisiologiche che possono causare morte cardiaca improvvisa. Una patologia autosomica recessiva X-linked causata dal gene della distrofina si verifica nei giovani maschi ed è caratterizzata da un decorso rapidamente ingravescente, in alcune forme di MCD sono anche state segnalate mutazioni dei geni mitocondriali. Eziologia: Più del 50% dei casi di MCD non presenta un’eziologia identificabile evidente. Tale sottocategoria di soggetti si considerano affetti da MCD idiopatica. È probabile che tale condizione rappresenti l’espressione comune del danno prodotto da una grande varietà di agenti patogeni non ancora del tutto identificati. Attualmente sono 3 i meccanismi che vengono considerati possibili: - Fattori ereditari e genetici; - Miocardite virale ed altri insulti citotossici; - Alterazioni immunologiche, come in alcuni pazienti la MCD può essere l’evoluzione di una miocardite virale acuta con il possibile intervento di fattori immunitari. La MCD interessa tutte le fasce d’età, ma più frequentemente interessa i maschi adulti e la prevalenza sembra essere in aumento. Una forma reversibile può essere associata ad abuso di alcolici, gravidanza, deficit di selenio, ipofosfatemia, ipocalcemia e tachicardia cronica non controllabile. Il 20% dei pazienti presenta una forma familiare, caratterizzata da mutazioni genetiche per la decodificazione di proteine strutturale miocardiche, come pure di fattori di trascrizione. Da un punto di vista genetico, la MCD è una malattia eterogena in quanto sono state descritte forme autosomiche dominanti, recessive ed X-linked. Quadro clinico: Le principali manifestazioni cliniche sono i sintomi dell’insufficienza cardiaca congestizia (ICC) destra e sinistra, con comparsa graduale di dispnea da sforzo, astenia, ortopnea, dispnea parossistica notturna, edemi periferici e palpitazioni. Possono comparire in alcuni pazienti precordi algie aspecifiche. A causa della presenza di trombosi cavitaria si può verificare embolia sistemica con ictus. Nell’immagine a lato, vi è un esempio di trombosi murale: essendo aumentato il volume della cavità il sangue tende ad assumere un andamento non laminare ma vorticoso e quindi è più facile che si formino i trombi. All’esame obiettivo osserverà si dilatazione cardiaca e i segni tipici ICC. Frequente è il riscontro di un terzo tono e di soffi da insufficienza mitralica e tricuspidale. Negli stadi avanzati vi è un aumento della pressione venosa centrale e una riduzione della pressione differenziale. L’ECG mostra tachicardia sinusale, tachiaritmie atriali e/o ventricolari, scarsa progressione dell’onda R nelle derivazioni precordiali, anomali aspecifiche della ripolarizzazione e blocco di branca sinistra. Si avrà tachicardia perché il primo meccanismo di compenso di un cuore che non riesce a pompare un’adeguata quantità di sangue è quello di aumentare la frequenza e ciò permette di mantenere nell’unità di tempo una quantità adeguata di sangue che viene immesso in circolo. L’Rx del torace mostrerà cardiomegalia, venosa ipertensione polmonare, interstiziale edema o alveolare. L’ingrandimento dell’ombra cardiaca è così importante che questo reperto viene anche definito come scarpa”. L’Rx al Torace è considerato “cuore il a primo strumento esplorativo. Ancora oggi non si può fare un intervento chirurgico senza aver fatto un Rx torace. Il quadro mostrato dall’RX torace però non è esclusivo in quanto lo si può trovare in tutti i casi in cui il ventricolo funziona male e ha già dato conseguenze retrograde a livello del circolo polmonare. L’ecocardiografia è importante, innanzitutto perché ci fa vedere le dimensione cavitarie. Il ventricolo è notevolmente aumentato di volume. La dimensione telediastolica e tele sistolica dei ventricolo sono aumentate, ma la loro differenza è minima, quindi ciò sottolinea quanto sia basso il grado di contrazione del ventricolo. Per quanto riguarda la geometria ventricolare, l’ecocardiografia mostra eventuali imperfezioni nella coaptazione dei lembi valvolari a seguito dell’aumento di volume ventricolare. L’ecocardiografia inoltre permette di valutare: - La frazione di eiezione; - Cinesi globale e segmentaria; - Insufficienze valvolari; - Formazioni trombotiche; - Stima delle pressioni diastoliche. Nelle metodiche di indagine l’aspetto caratteristico di un cuore affetto da CMD viene definito anche a “pallone di rugby”. Con il cateterismo cardiaco si è in grado di escludere l’eziologia ischemica, facendo così diagnosi di CMD a coronarie indenni. Una serie di dati epidemiologici, però, dicono che in una percentuale che varia dal 10 al 50% delle cardiomiopatie dilatative vi è una compromissione del circolo coronarico. Nell’immagine a lato viene mostrata la sezione del cuore in un paziente con cardiomiopatia aritmogena del ventricolo destro e involuzione di entrambi i ventricoli. Nello spessore della parete del ventricolo destro sono presenti infiltrazioni di tessuto adiposo, e a livello dell’apice ventricolo sinistro è presente un aneurisma. In alto a destra, viene mostrata una sezione istologica a livello del pavimento del ventricolo destro. Anche qui si nota l’infiltrato di tessuto adiposo, compatibile con la displasia del ventricolo destro. In basso è invece mostrata la sezione istologica del ventricolo sinistro che mostra anche qui un ricco infiltrato di tessuto adiposo l'atrofia delle cellule muscolari, compatibile con una involuzione del ventricolo sinistro. Vi sono una serie di fattori associati con un outcome avverso nei pazienti con CMD, suddivisibili in clinici, tramite l'utilizzo di metodiche non invasive e di metodiche invasive. I principali fattori clinici sono: - l'appartenenza ad una classe NYHA tipo III/IV, caratterizzata da una bassa frazione di eiezione e da alte pressioni di riempimento del ventricolo sinistro; - Età, che già di per sé comporta un aumento delle dimensioni del ventricolo; - Cuore non allenato, con raggiungimento del picco di consumo di ossigeno a basso grado di esercizio; - anomalie marcate nella conduzione intraventricolare; - presenza di aritmie ventricolari; - presenza di alterazioni all'elettrocardiogramma; - evidenza di un'eccessiva stimolazione simpatica; - presenza di galoppo protodiastolico, ovvero presenza di un terzo tono. I fattori che vengono messi in evidenza con metodiche non invasive sono: - una bassa frazione di eiezione del ventricolo sinistro; - una marcata dilatazione del ventricolo sinistro; - una bassa massa del ventricolo sinistro; - la presenza di un moderato rigurgito mitralico; - anomalie della funzione diastolica; - anomalie nella riserva contrattile; - dilatazione o disfunzione del ventricolo destro. Il fattore che viene analizzato tramite metodiche invasive è l'elevata pressione di riempimento del ventricolo sinistro. Terapia: Per quanto riguarda la terapia, questa non è specifica, ma e quella dell'insufficienza cardiaca. Per quanto riguarda le anomalie del ritmo, queste possono essere risolte tramite l'utilizzo di un defibrillatore impiantabile (ICD) oppure tramite la terapia di resincrononizzazione cardiaca (CRT). Solo il trapianto cardiaco e la terapia farmacologica a base di ACE-inibitori, beta-bloccanti, spirinolattone, idralazina con nitrati sono in grado di prolungare la sopravvivenza. Nell'immagine in alto viene mostrato lo schema terapeutico riguardo al grado di insufficienza cardiaca dovuto alla cardiomiopatia dilatativa. Tale schema terapeutico è lo stesso dell'insufficienza cardiaca. Vi sono una serie di fattori prognostici sfavorevoli, che prendono a rendere la prognosi infausta, come l'appartenenza alla classe NYHA IV, presenza di fibrillazione atriale, frazione di eiezione inferiore al 30%, diminuito apporto di ossigeno, attivazione neuro-ormonale e indice cardiatoracico superiore a 0,55. Prognosi: Circa il 5% dei pazienti muore entro i due anni dalla diagnosi. Nel 25% circa si ha una sopravvivenza superiore ai cinque anni. L'estrema gravità della prognosi è alla base della frequente indicazione al trapianto cardiaco. MIOCARDIOPATIA IPERTROFICA E’ una malattia genetica, con una frequenza di uno su 500 nella popolazione generale. L'aspetto saliente è l’inappropriata ipertrofia miocardica non correlata al carico emodinamico, prevalentemente a carico del setto interventricolare (SIV), nell'ambito di un ventricolo sinistro non dilatato ed ipercontrattile. Tale alterazione determina una disfunzione diastolica, con aumento delle pressioni di riempimento. Dal punto di vista anatomo-patologico, ma anche funzionale e sintomatico, ciò che caratterizza questi pazienti è l’alterazione diastolica. In passato tale patologia si chiamava stenosi subaortica ipertrofica. Quando si scopre che un paziente è affetto da miocardiopatia ipertrofica, bisogna caratterizzare tutti i familiari perché la probabilità che ci sia un familiare portatore o affetto è molto alta. In almeno il 50% dei casi l'eredità è autosomica dominante. Vi sono anche forme sporadiche da mutazioni spontanee. Il 40% dei casi è associato a mutazioni del gene che codifica per la catena pesante della beta-miosina cardiaca. L'espressività di tali mutazioni è alquanto variabile. Espressività variabile vuol dire che noi possiamo avere 2 genitori di cui uno portatore di questo gene e su 6 figli, 3 hanno ereditato tale gene ma soltanto 1 è affetto da miocardiopatia ipertrofica. In circa il 90% dei pazienti l'ipertrofia coinvolge il setto interventricolare dando luogo ad un quadro di ipertrofia settale asimmetrica. La classificazione di Maron è appunto una classificazione basata sull'aumento delle dimensioni delle pareti cardiache nelle cardiopatie ipertrofiche e, in funzione dell'area interessata dall'ipertrofia, distingue quattro tipi: - Tipo I – 10% dei casi: caratterizzato da ispessimento del setto interventricolare (SIV) anteriore; - Tipo II – 20% dei casi: ispessimento del SIV anteriore e posteriore; - Tipo III – 52% dei casi: ispessimento del SIV e della parete antero-laterale del ventricolo sinistro; - Tipo IV – 18% dei casi: ispessimento della parete laterale o del SIV posteriore o delle regioni apicali. Da un punto di vista anatomopatologico, la cardiomiopatia ipertrofica e caratterizzata da: - non allineamento delle fibre miocardiche; - marcata fibrosi miocardica; - alterazioni ed ispessimento delle piccole arterie coronarie intramurali. Considerazioni genetiche: Circa la metà dei pazienti affetti da MCI ha una anamnesi familiare positiva, compatibile con trasmissione autosomica dominante. Sono state identificate oltre 400 mutazioni di 11 geni differenti che codificano proteine di sarcomeri, responsabili di circa il 60% dei casi. Le più comuni sono le mutazioni del gene della catena pesante della beta-miosina sul cromosoma 14. Alcune mutazioni comportano una prognosi più sfavorevole. Molti casi sporadici sono dovuti probabilmente a mutazioni spontanee. Non è ancora disponibile un test genetico come esame di routine, tuttavia permette una conferma diagnostica definitiva di MCI ad eziologia genetica, identificando una mutazione in un gene che codifica una proteina del sarcomero. Il test genetico può identificare i familiari che rischiano di andare incontro a tale patologia e che, quindi, devono essere sottoposti a screening ecocardiografico e follow-up. È anche in grado di escludere la presenza della malattia nei familiari. Fisiopatologia: Tale patologia è caratterizzata da una disfunzione diastolica. Se la parete ventricolare è rigida, il rilasciamento sarà ridotto e ciò determina che il riempimento (filling) è abnormemente alterato. Sono tre i meccanismi di base coinvolti nella produzione ed intensificazione dell'ostruzione intraventricolare dinamica: - aumento della contrattilità del ventricolo sinistro; - ridotto precarico ventricolare; - diminuzione dell'indipendenza e della pressione aortica (postcarico). Gli interventi che aumentano la contrattilità miocardica e quelli che riducono il precarico ventricolare, riducono il volume telediastolico del ventricolo sinistro e, di conseguenza, possono causare un aumento del gradiente e il soffio. Al contrario, l'innalzamento della pressione in posizione accovacciata, mediante stretta di mano prolungata, l'aumento del ritorno venoso alzando passivamente le gambe e l'espansione del volume ematico, sono tutti fattori che incrementano il volume ventricolare e migliorano il gradiente e il soffio. In un cuore normale le coronarie sono epicardiche, cioè scorrono appoggiate al ventricolo. A volte il discendente anteriore nella sua parte media può approfondirsi all’interno del miocardio ventricolare, ciò significa che quando il miocardio si contrae si ha un occlusione transitoria del vaso in quel tratto. In un cuore disordinato, come nella cardiomiopatia ipertrofica, vi sono fenomeni multipli che riguardano più segmenti e su più pareti e quindi più ponti miocardici che al momento della contrazione vanno a ridurre il flusso coronarico in più regioni provocando ischemia. Questo è un altro motivo per cui un paziente affetto da cardiomiopatia ipertrofica deve fare la coronarografia. Quindi, le principali cause di ischemia miocardica sono: - ridotta riserva coronarica; - aumento della richiesta di ossigeno; - presenza di ponti miocardici sulle strutture vascolari coronariche. Nel 25% dei pazienti è presente un gradiente dinamico attraverso il tratto di efflusso del ventricolo sinistro, determinato dal movimento sistolica anteriore (SAM) dei lembi valvolari mitralici, spesso allungati, contro il setto ipertrofico. Sulle cause ed il significato di tale ostruzione persistono notevoli controversie. Il gradiente viene definito dinamico perché aumenta se: - vi è un aumento della contrattilità del ventricolo sinistro; - vi è una diminuzione del precarico, quindi la diminuzione del volume ventricolare telediastolico; - diminuzione del postcarico, ovvero una diminuzione della pressione sistolica e dell'indipendenza aortica. Nel momento in cui questi 3 fenomeni sono del tutto assenti non vi è più il gradiente. La gradazione del rischio del paziente con cardiomiopatia ipertrofica la si fa sulla base del gradiente: - Gradiente assente anche in presenza di stimolo (il classico stimolo è la manovra di Valsalva che serve a creare queste 3 condizioni). Il calcolo del gradiente si fa con l’ecocardiografia; - Gradiente presente a seguito dello stimolo ma temporaneo; - Gradiente fisso (nel momento della manovra questo paziente ha una sincope). Quadro clinico: All’anamnesi, spesso il paziente è asintomatico, e viene identificato nel corso di screening dei familiari. Sintomi correlati alla miocardiopatia ipertrofica sono la dispnea, l'angina pectoris, l'astenia, la presincope e sincope. Lo sforzo tende a peggiorare i sintomi e la prima manifestazione può essere la morte improvvisa. L'esame obiettivo può essere normale nei pazienti asintomatici senza gradiente o con ipertrofia moderata. I pazienti con ostruzione presentano un soffio sistolico rude, a diamante, meglio apprezzato sul margine sternale sinistro e all'apice, e non irradiato alle carotidi (diagnosi differenziale con soffio da stenosi aortica). Il soffio e più rude sulla parasternale (gradiente di efflusso) e dolce all'apice (dell'insufficienza mitralica associata). Diagnosi: In basso viene mostrato un elettrocardiogramma tipico di un soggetto affetto da miocardiopatia ipertrofica. All'ECG saranno evidenti i segni di ipertrofia ventricolare sinistra, con onde Q ampie e profonde e tachiaritmie atriali e ventricolari. Con l’ecocardiografia è possibile vedere il gradiente dinamico e lo si può vedere ancor meglio se si effettua un’ecocardiografia durante una stimolazione che può essere farmacologica oppure con la manovra di Valsalva. E precisamente, con questa metodica di indagine, sarà possibile studiare: - lo spessore telediastolico del setto; - il rapporto tra il setto e la parete posteriore, superiore a 1,3; - la localizzazione dell'ipertrofia; - la presenza del SAM o movimento sistolico anteriore; E’ chiaro che se c’è il SAM, cioè questo movimento eccessivo, questo prolasso eccessivo del lembo anteriore della mitrale, è molto probabile che il gradiente se non è fisso, quindi se non è sempre presente, comparirà sicuramente nel momento in cui noi andremo a fare lo stimolo provocativo. Questo è quello che si osserva classicamente al cateterismo cardiaco sinistro. Qui si osserva 2 volte la morfologia ventricolare; si ha una prima fase un cui la morfologia ventricolare ha un aspetto che è molto diverso e che soprattutto crea un gradiente tra ventricolo sinistro e aorta poi man mano che si tira la punta del catetere dal ventricolo verso l’aorta si arriva in un punto, cioè nella camera di afflusso del ventricolo sx, dove vi è un gradiente pressorio tra la parte del ventricolo dove arriva il sangue dall’atrio sx e l’aorta. Ritirardo ancora il catetere sempre nel tratto di afflusso ma al di sopra della “protuberanza maggiore” del setto interventricolare ispessito si nota che la pressione ventricolare eguaglia la pressione aortica. Ciò significa che all’interno del ventricolo ci sono due diversi regimi pressori. Un’immagine del genere la si può vedere o nella cardiomiopatia ipertrofica con gradiente o nella stenosi aortica sottovalvolare per presenza di membrana o anello. Essendo quest’ultima più rara, venendosi a trovare di fronte un’immagine del genere bisogna pensare ad una cardiomiopatia ipertrofica con gradiente, che è l’ipotesi più probabile. Nella tabella a lato, vengono elencati i principali fattori che aiutano nella stratificazione del rischio di un paziente con cardiomiopatia ipertrofica, soprattutto vengono indicati i fattori associati ad un out-come avverso. Terapia: Sono sconsigliati gli sforzi eccessivi e l’attività agonistica, perché l’attività agonistica, e l’ipertono simpatico ad esso connesso, sarebbe un potentissimo stimolo per far sì che il gradiente, anche quando non è presente, si realizzi proprio all’acme dello sforzo. Nel migliore dei casi il paziente ha una sincope, nel peggiore ha una morte aritmica. I farmaci da utilizzare sono essenzialmente β-bloccanti e Calcio-antagonisti perché sono quelli che diminuiscono la capacità di contrazione del cuore contribuendo, soprattutto i Calcioantagonisti a migliorare la fase di rilasciamento. Tra i calcio antagonisti viene utilizzato soprattutto il verapamil. L’evento più temibile è l’aritmia maligna che può determinare morte improvvisa e quindi come terapia si effettua l’impianto di defibrillatori impiantabili, che però non è una terapia in sé perché non migliora la funzione del ventricolo ma è una terapia preventiva della peggiore delle complicanze possibili che questa malattia può dare. Esistono due possibilità di terapia meccanica. La meno invasiva è quella dell’ablazione settale con alcol: si identifica, dalla coronarografia sinistra, il primo ramo settale o il ramo settale più ricurvato. Se provochiamo in quest’area (parte di setto che sicuramente andrà insieme al lembo anteriore mitrale a dare il gradiente) un infarto succederà che questa parte non si contrarrà più e quindi gli facciamo venire un piccolo infarto che ha piccole probabilità di essere pro- aritmogeno. Quindi purché ci sia un grosso ramo settale identificabile prossimale, che deve essere il primo o massimo il secondo ramo settale, si va dentro a questo ramo con un catetere a palloncino, si gonfia il palloncino e ci si assicura, iniettando del mezzo di contrasto, che non ci sia passaggio retrogrado cioè che il palloncino effettivamente abbia bloccato il flusso. Una volta assicuratosi di questo, a valle si iniettia dell’etanolo (etanolo puro diluito semplicemente per fare l’iniezione), il paziente avrà dolore, all’eco si vede immediatamente come si comporta quella parte di setto interventricolare, cioè se quella parte di setto interventricolare non si contrae più. E’ importante che non vi sia flusso retrogrado perché in caso contrario, cioè se l’etanolo andasse dietro, si porterebbe nel discendete anteriore. Questa terapia si fa nei pazienti che hanno gradiente fisso o che hanno un gradiente abnorme in corso di stimolo e che non hanno nessun vantaggio dalla terapia farmacologica. Uno che ha il gradiente di base o che ha facilità ad avere la comparsa del gradiente è anche un paziente fortemente sintomatico, quindi alla fin fine ci si basa sul gradiente e sull’eco ma anche su ciò che ci riferisce il paziente. L’altra possibilità è un intervento chirurgico di miotomia cioè tagliando e rimuovendo alcune fette dal setto se ne riduce lo spessore. In questo caso, però, l’intervento è a cuore aperto, l’insulto è notevole e quindi ci vuole anche un bel cuore in condizioni battenti. Questi due interventi sono riservati ai peggiori pazienti dello spettro della patologia. Prognosi: La storia naturale della miocardiopatia ipertrofica è variabile. La fibrillazione atriale è comune nelle fasi avanzate della malattia; spesso, il suo esordio causa la comparsa o l'aggravamento dei sintomi. In meno del 10% dei pazienti insorge endocardite infettiva; tuttavia, la profilassi è attualmente raccomandata sono nei soggetti che hanno già avuto un episodio precedente di endocardite infettiva. La progressione verso la dilatazione e la disfunzione ventricolare, con assottigliamento della parete è scomparsa di un gradiente pressorio in efflusso preesistente (cosiddetta miocardiopatia ipertrofica burnt out), si verifica nel 5-10% dei pazienti e può essere associata ad insufficienza cardiaca congestizia refrattaria, con necessità di trapianto cardiaco. La principale causa di mortalità è la morte cardiaca improvvisa, che può intervenire nei pazienti asintomatici o interrompere nei soggetti sintomatici un decorso peraltro stabile. I pazienti che corrono un rischio più elevato di morte cardiache improvvisa sono quelli con anamnesi positiva per rianimazione, sincope ricorrente, tachicardia ventricolare evidenziata dal monitoraggio ambulatoriale o dallo studio elettrofisiologico, marcata ipertrofia ventricolare, mancato innalzamento della pressione arteriosa durante esercizio fisico, anamnesi familiare positiva per morte cardiache improvvisa e presenza di alcune mutazioni genetiche. MIOCARDIOPATIA RESTRITTIVA La miocardiopatia restrittiva è la meno frequente. E’ caratterizzata anch’essa da un’anomala funzione diastolica; la parete ventricolare è rigida e tale rigidità può essere primitiva o secondaria all’accumulo di sostanze estranee. Nelle fasi iniziali della malattia la funzione sistolica non è compromessa. Per quanto riguarda la classificazione, esistono le idiopatica, forme non familiare, infiltrative: ipertrofica, scleroderma, diabetica, che alla fine è la più frequente fra tutte. Poi ci sono le forme infiltrative, le malattie da accumulo e infine ci sono le forme endomiocardiche. Fisiopatologia: Questi pazienti hanno una normale funzione sistolica, compliance ventricolare ridotta, aumentata pressione diastolica, modesto volume sistolico, pattern restrittivo di riempimento ventricolare, curva di riempimento ventricolare a radice quadrata. Ciò significa che, siccome in questo caso il ventricolo è incapace di rilassarsi, si rilassa al massimo nel momento in cui c’è la spinta da dietro, cioè è aumentata la pressione nell’atrio, fa aprire la mitrale, per cui questa grossa quantità di sangue scende nel ventricolo, quindi il ventricolo gioco forza deve essere in grado di accogliere questa prima fase del filling, dopodiché poiché è rigido, è incapace di rilassarsi, la pressione immediatamente sale e il filling anziché essere costituito da 3 parti (filling rapido, diastasi e il riempimento telediastolico da contrazione atriale) è composto solo dal filling rapido perché il ventricolo ha bruscamente aumentato la propria rigidità, per cui quello che accade per caduta, il riempimento per caduta, il riempimento per contrazione dell’atrio sx non trova spazio o ne trova molto poco per potersi realizzare. Quadro clinico: L’impossibilità di riempimento dei ventricoli limita la gittata cardiaca e fa innalzare la pressione di riempimento; pertanto, sono in genere dominanti l'intolleranza allo sforzo e la dispnea. In seguito alla persistenza di un'elevata pressione venosa sistemica, questi pazienti presentano comunemente edemi declivi, ascite e fegato ingrossato, dolente e spesso pulsante. La pressione venosa giugulare è alta e non scende in maniera normale durante l'inspirazione (segno di Kussmaul). I toni cardiaci possono essere distanti, ed è frequente la presenza di un terzo e quarto tono. Diversamente dalla pericardite costrittiva, che per molti aspetti è simile, l'impulso apicale è di solito facilmente palpabile ed è più spesso presente insufficienza mitralica. Diagnosi: Anche qui l’ECG non ci dice cose particolari, diciamo che non ci da nessun aiuto. Sarà possibile notare un ingrandimento atriale, con possibile fibrillazione atriale, anomalie della ripolarizzazione e blocco di branca sinistro. L’ecografia permette di visualizzare: - un ingrandimento atriale, visto che l’atrio non riesce a svuotarsi completamente nel ventricolo. Ingrandendosi però questo vuol dire anche che siccome l’atrio ha una scarsa componente muscolare, a furia di dilatarsi, si dilaterà in maniera abnorme e quindi questi pazienti andranno facilmente incontro a fibrillazione atriale; - visualizzare il pattern di flusso transmitralico di tipo restrittivo; - aumento della pressione venosa centrale con la riduzione durante le escursioni respiratorie; - ipertensione polmonare. All’ecodoppler cardiaco è possibile visualizzare il pattern di riempimento ventricolare, che è di tipo restrittivo perché all’ecodoppler vediamo che il flusso si realizza apparentemente bene subito dopo che si è aperta la valvola mitrale, però subito dopo il flusso diminuisce perché c’è l’ostacolo della rigidità della parete. Tra gli altri esami strumentali utili nella diagnosi vi sono: - Rx torace, che sottolinea un ingrandimento atriale e segni di congestione polmonare; - cateterismo, che mette in evidenza l'aumento della pressione ventricolare tele diastolica (PTD); - la biopsia endomiocardica, che è una procedura che viene effettuata meno frequentemente rispetto al passato. Ad oggi l’unica indicazione della biopsia è il trapianto al fine di monitorare la terapia antirigetto. Con la biopsia endomiocardica sarà possibile mettere in evidenza la fibrosi endomiocardica con disarrangiamento dei miociti ed eventuale presenza di sostanza amiloide. Non bisogna dimenticare che la miocardiopatia restrittiva va in diagnosi differenziale con la pericardite costrittiva. La pericardite costrittiva da un quadro simile alla cardiomiopatia restrittiva. La pericardite costrittiva ha essenzialmente un’unica genesi, quella tubercolare, quindi è anche abbastanza rara. Nel caso della cardiomiopatia restrittiva è la parete che è rigida, nel caso della pericardite costrittiva è l’astuccio pericardico che ha perso elasticità e quindi impedisce al ventricolo di rilassarsi e quindi di riempirsi. Ulteriori criteri di diagnosi differenziale vengono riportati nella tabella seguente. Prognosi e terapia: Queste forme evolvono rapidamente verso lo scompenso cardiaco, si possono avere facilmente embolie sistemiche e polmonari, a seconda che sia l’atro di dx o sx che si ingrandisce e da luogo alla formazione dei trombi. La terapia prevede l'utilizzo di ACE- inibitori, diuretici, antiaggreganti, anticoagulanti. L’unica soluzione definitiva è il trapianto cardiaco. CARDIOMIOPATIA ARITMOGENA DEL VENTRICOLO DESTRO La cardiomiopatia aritmogena del ventricolo destro è un disordine caratterizzato da progressiva sostituzione fibroadiposa del tessuto muscolare del ventricolo destro, inizialmente localizzata al livello sottotricuspidale, apice e via di efflusso del ventricolo destro. Successivamente risulta essere diffusa a tutto il ventricolo destro e in alcune forme più gravi anche a sinistro (viene in genere risparmiato il setto). La principale conseguenza di tale sostituzione è l'insorgenza di aritmie ventricolari con aspetto tipico del blocco di branca sinistro, dovute perlopiù a fenomeni di rientro. Questa patologia è una malattia familiare con trasmissione autosomica dominante, a penetranza incompleta e importante risulta essere lo screening dei familiari. È stato ipotizzato che la presenza di anomalie che desmosomiche provochi distacco dei miociti, con conseguente apoptosi e sostituzione fibroadiposa. La mutazione più comune fra i geni delle proteine desmosomiche si trova nel gene della placofilina-2 (PKP-2). Sono state descritte anche mutazioni nel gene del recettore della rianodina cardiaca (RyR2) e in altri geni. Quadro clinico,diagnosi e terapia: Molto spesso i pazienti sono asintomatici o hanno direttamente un evento aritmico maggiore che se risolto può permettere di fare la diagnosi, altrimenti la diagnosi è autoptica. Ulteriori sintomi sono dati da palpitazioni, sincope. All’ECG possiamo trovare soprattutto in un paziente giovane onde T negative in V 1 e V 2 , spesso estese a tutte le precordiali e una alterazione morfologica della parte terminale del cuore. Inoltre si evidenzia anche una impastatura terminale del complesso QRS. Invece, l’ecocardiogramma è molto più espressivo perché non solo ci mostra l’ipocinesia localizzata o diffusa del ventricolo dx ma ci da anche una ecoriflettenza con ispessimento della banda moderatrice e della struttura trabecolare. La risonanza magnetica è migliore dell’eco perché fa vedere tutto e non dipende dall’operatore, non dipende dalla struttura fisica del paziente. Il cateterismo cardiaco permette di analizzare la distribuzione orizzontale delle trabecolare a pila di piatti e di osservare le alterazioni della cinetica regionale e mammellonature parietali. La biopsia endomiocardica è sconsigliata. La terapia prevede l'utilizzo di beta-bloccanti, nonché l'impianto di un defibrillatore (ICD), in quanto il paziente può morire di aritmie ventricolari. Accanto a questa, infine, dobbiamo citare altre patologie in grado di determinare disturbi del ritmo cardiaco. Queste sono: - sindrome del QT lungo o breve: malattia dei canali ionici del sodio o del potassio; - sindrome di Brugada: malattia del canale del sodio; - tachicardia ventricolare polimorfa: anomalia del recettore rianodinico che regola il rilascio di calcio dal reticolo sarcoplasmatico e l'accoppiamento elettromeccanico. CARDIOMIOPATIA DI TAKO-TSUBO Questa cardiomiopatia è una disfunzione acuta e reversibile del ventricolo sinistro, descritta per la prima volta in Giappone. È più frequente nel sesso femminile ed è indotta dallo stress. Tako-Tsubo è un vaso a collo stretto ed ha la stessa silhouettes del ventricolo sinistro in fase telediastolica, specialmente a livello apicale. Questa cardiomiopatia sembra essere dovuta ad una sorta di tossicità miocardica dovuta ad un eccesso di increzione di catecolamine, e quindi un eccesso di ipertono simpatico, indotta dallo stress. Si manifesta come un infarto acuto massivo esteso senza però causare danni. Il paziente presenta dolore precordiale tipico, ECG con sopraslivellamento del tratto ST, all’eco acinesia della parete antero- picale, che può far nascere il sospetto di un infarto acuto anteriore. Tuttavia, gli enzimi sono poco o per niente aumentati, il paziente sopravvive e l’eco dopo un paio di giorni è perfettamente normale. Il cateterismo cardiaco rivela coronarie normali ed apical balooning. Nell’apical balooning, il ventricolo sx si muove normalmente visto in proiezione obliqua destra nella parte anteriore e inferiore, invece la parte apicale non si contrae. Se è vera l’ipotesi dell’iperincrezione di catecolamine, non si deve fare la trombolisi, neanche la coronarografia e nemmeno l’angioplastica perché le coronarie sono normali. Bisogna, invece, prendere i βbloccanti e calcio-anatgonisti per antagonizzare questo eccesso di attività simpatica. NON COMPATTAZIONE DEL VENTRICOLO SINISTRO Questo è un disordine embriogenetico per cui le pareti ventricolari non si compattano e il ventricolo ha un aspetto spongioso con persistenza di profondi recessi (sinusoidi ematici) tra le trabecolare miocardiche in comunicazione con la cavità ventricolare. La sede più frequente di tale alterazione è all'apice. Può essere isolata o associarsi a cardiopatie congenite. Può avere carattere familiare. La diagnosi si fa con ecocardiogramma, risonanza magnetica nucleare, angiografia. Le principali conseguenze di tale patologia sono: aritmie gravi, disfunzione sistolica, evoluzione verso insufficienza cardiaca e possibili fenomeni tromboembolici. AMILOIDOSI CARDIACA L'amiloidosi cardiaca è la più frequente tra le cardiomiopatie infiltrative. Il primum movens è dato dai depositi di sostanze amiloide nello spazio extracellulare. La deposizione di amiloide al livello del miocardio determina una restrizione funzionale. Infatti i miociti sono strutturalmente normali, mantenendo la capacità contrattile. Quando i depositi sono cospicui pendono lo spazio extracellulare per accogliere il loro rilasciamento diastolico. Col tempo, i cordoni di fibrille di amiloide isolano il miocita e lo privano dei rapporti strutturali con gli altri miociti. La patologia viene innescata dalla deposizione della sostanza amiloide, una sostanza di natura proteica, con una configurazione fibrillare (fibrille beta-elicoidali, di lunghezza indeterminata, con un diametro variabile tra i sette e i 10 nm, non ramificate ed identificate su campione di tessuto attraverso la colorazione rosso congo). Sono state fatte varie classificazioni dell’amiloidosi e quella biochimica raggruppa le varie amiloidosi in funzione della proteina amiloidogeniche. Oggi come oggi sono state identificate circa 21 proteine in grado di dare amiloidosi. Il meccanismo patogenetico risulta essere lo stesso in tutti i casi: - vi è danno nell'architettura del tessuto dove si depositano; - danno ischemico per coinvolgimento dei vasi epicardici ed intramurali; - danno citotossico. Qui a lato vengono mostrate le immagini di un cuore affetto da amiloidosi. Innanzitutto, si verifica anche un aumento della consistenza del tessuto miocardico, che diventa duro-elastico, assumendo un aspetto molto simile a quello della cera nei casi avanzati. Le pareti dei ventricoli e degli atri sono rigide ed ispessite. Gli atri appaiono dilatati. In questa patologia sono interessati anche gli apparati valvolari. Presentazione clinica: La presentazione clinica è molto simile nelle diverse forme di amiloidosi e dipende dall'estensione dei depositi di sostanza amiloide. Inizialmente vi è una disfunzione diastolica e nelle fasi avanzate viene intaccata anche la funzione sistolica. In alcuni casi la amiloidosi cardiaca assume un andamento ingravescente e rapidamente progressivo, fino alla "cachessia cardiaca". Tale stadio è caratterizzato da: - angina, dovuta alla compressione dei piccoli vasi e positività per il marker sierico di ischemia, la troponina; - morte cardiaca improvvisa per dissociazione elettromeccanica; - sincope, dovuta alla neuroparia autonomica; - aritmie del ventricolo sinistro e fibrillazione atriale dovuta alla dilatazione dell’atrio. Diagnosi: La diagnostica strumentale si avvale dell’utilizzo di: - ECG, che mostrerà un tracciato caratterizzato da bassi voltaggi, blocchi di branca e atrioventricolari, aritmie, pattern pseudoinfartuali; - Ecocardiogramma, che mostrerà l’ipertrofia, con dilatazione atriale, ispessimento valvolare, disfunzione diastolica-sistolica, ecogenicità (granular sparkling). L’eco-doppler tissutale evidenzierà eventuali disfunzioni in fase precoce; - Cateterismo cardiaco, tramite il monitoraggio delle pressioni; - Biopsia del grasso periombelicale, che è positiva nel 70% dei casi di amiloidosi; - Biopsia endomiocardica (BEM); - Immunoelettroforesi e immunofissazione su siero ed urine, per la ricerca delle catene leggere λ e κ; - RMN cardiaca. Terapia: Per quanto riguarda la terapia, nelle forme di amiloidosi di tipo AL il solo trapianto cardiaco è poco indicato, perché la presenza di un clone amiloidogenetico attivo, continua a produrre amiloide che si deposita sul cuore trapiantato. Si potrebbe effettuare anche un trapianto cardiaco con un autotrapianto di cellule staminali. Un’altra opzione è quella della chemioterapia, avente lo scopo di ridurre e talvolta eradicare il clone amiloidogenetico. Si può così ricorrere al trapianto cardiaco, seguito da chemioterapia con autotrapianto di cellule staminali. Diversa risulta essere la terapia per l’amiloidosi da transtiretina. La TTR è responsabile della SSA o senile sistemi amyloidosis. Questa è la forma senile poco aggressiva, tipica dell’età avanzata (età superiore 80 anni). È una forma autosomica dominante, causata dalla mutazione del gene per la transtiretina. Sino ad ora sono note circa 70 mutazioni. La transtiretina è una proteina prodotta dal fegato. Questa forma di amiloidosi, detta anche ATTR, determina un importante interessamento cardiaco e del sistema nervoso periferico ed autonomo, dando luogo ad una neuropatia progressiva. Le opzioni terapeutiche consistono in: - Trapianto di fegato, che è l’opzione curativa, ma presenta un rischio elevato se vi è importante compromissione cardiaca. Nel post trapianto nel cuore interessato dalla malattia si continua a depositare TTR normale; - Trapianto di cuore. EMOCROMATOSI L’emocromatosi è una patologia caratterizzata dall’eccessivo deposito di ferro a livello tissutale. Esistono due forme di emocromatosi: - Emocromatosi ereditaria o idiopatica o primitiva: patologia autosomica recessiva; - Emocromatosi secondaria o emosiderosi: dovuta a regime trasfusionale cronico, eritropoiesi inefficace, eccessivo supporto di ferro, porfiria cutanea tarda, atransferrinemia congenita, epatopatia alcolica. L’eccesso di ferro tende a depositarsi negli organi parenchimatosi quali, fegato (cirrosi), pancreas (diabete bronzino), cuore nel 15 % dei casi, articolazioni ed ipofisi. Il danno d’organo è dovuto sostanzialmente all’emosiderina (ovvero aggregati di ferritina) e al ferro libero, con la genesi di radicali dell’ossigeno. Nell’emocromatosi cardiaca si noterà: - Ispessimento delle pareti cardiache; - Disfunzione contrattile; - Aritmie ventricolari e sopraventricolari; - Disturbi della conduzione atrioventricolare. La terapia prevede: - Salassi periodici; - Sostanze chelanti il ferro, quali la desferrioxamina. SARCOIDOSI La sarcoidosi è una malattia sistemica restrittivo-infiltrativa con una incidenza di 11-60 casi su 100000 abitanti. La caratteristica anatomopatologica è la formazione di granulomi non necrotizzanti. I principali organi interessati sono polmoni, linfonodi, milza e cuore, con un interessamento di quest’organo nel 20-27% della popolazione e 58% nella popolazione giapponese affetta da sarcoidosi. La sarcoidosi cardiaca decorre asintomatica nella maggior parte dei pazienti e il 5% presenta una malattia cardiaca conclamata. In base alla sede ed all’estensione dei granulomi in seno al miocardio avremo: - Difetti di conduzione; - Aritmie ventricolari dovuti a fenomeni di rientro, responsabili della morte improvvisa cardiaca; - Aritmie sottoventricolari, più raramente; - Vizi valvolari, come rottura di corde tendinee; - Disfunzione sistolica e diastolica, dovuta all’infiltrazione granulomatosa del ventricolo sinistro; - Disfunzione del ventricolo destro per interessamento polmonare. La diagnosi viene posta in base al riscontro dei caratteristici granulomi non necrotizzanti. Di particolare ausilio sono le tecniche di imaging come la RMN. Molto utile è anche la scintigrafia miocardica con Tallio201 e gallio-67. Alla scintigrafia potrà notarsi: - Ipocaptazione del Tl-201 che regredisce o scompare a seguito dello sforzo; - Ipercaptazione del gallio-67, che non vede i granulomi “spenti”. La terapia prevede: - Somministrazione di corticosteroidi; - Utilizzo di farmaci immunosoppressori; - Impianto di elettrocatetere ventricolare sinistro (PM-ICD); - Trapianto cardiaco con recidiva sul graft. CAPITOLO X: LE ARITMIE Abbiamo già parlato delle aritmie e abbiamo detto che le cellule sono sempre le stesse alla nascita ma assumono un diverso compito nel corso dello sviluppo, nel corso della vita, fermo restando che, essendo figlie degli stessi genitori, hanno lo stesso corredo genetico e può accadere che una svolga la funzione dell’altra e viceversa. IL SISTEMA DI CONDUZIONE L’impulso elettrico è generato nel nodo seno atriale che si trova nell’atrio destro e attraverso gli atri è trasmesso al nodo AV dove viene filtrato e ritardato. Quindi sappiamo per certo che anche se la frequenza generata a livello degli atri è particolarmente alta, quando questo numero enorme di impulsi arriva al nodo AV, questi finiscono con l’essere in qualche modo filtrati. L’impulso segue poi una fase di rapida conduzione attraverso il fascio di His, entra nelle branche sinistra e destra e finisce quindi col determinare l’accoppiamento elettromeccanico a livello dei cardiomiociti ventricolari, responsabile della contrazione cardiaca. Questo permette al cuore, un certo numero di volte al minuto, di contrarsi e dilatarsi. Il cuore è l’unico organo in perenne attività. L’intera sequenza viene monitorizzata attraverso l’ECG di superficie. L’impulso cardiaco normale viene generato dalle cellule pacemaker del nodo seno-atriale, situato alla giunzione fra atrio destro e sbocco della vena cava superiore. Questo impulso viene trasmesso lentamente lungo il tessuto nodale fino alle complesse strutture anatomiche atriali e più rapidamente al nodo AV, generando l’onda P sull’ECG. Vi è modesto ritardo percettibile nella conduzione attraverso il nodo AV. Il tempo necessario per l’attivazione degli atri e il ritardo del nodo AV sono rappresentati dall’intervallo PR sull’ECG. Nel cuore normale, il nodo AV è l’unica connessione elettrica fra gli atri e i ventricoli. L’impulso elettrico emerge dal nodo AV e viene trasmesso al sistema di His-Purkinje, in particolare al fascio comune di His, quindi alle branche destra e sinistra e successivamente alla rete di Purkinje. In condizioni normali, i ventricoli sono rapidamente attivati in modo ben definito, modalità determinata dal decorso della rete di Purkinje e che traccia sull’ECG il complesso QRS. Il recupero dell’eccitabilità elettrica si verifica più lentamente ed è regolato dal tempo di attivazione e dalla durata dei potenziali di azione locali. La relativa brevità dei potenziali di azione epicardici, a livello del ventricolo, dà come risultato la ripolarizzazione, che avviene prima sulla superficie epicardica e procede quindi verso l’endocardio, generando un’onda T che, in condizioni normali, ha la stessa polarità del complesso QRS. La durata dell’attivazione e del recupero è determinata dalla durata del potenziale di azione e rappresentata, sull’ECG rilevato sulla superficie corporea, dall’intervallo QT. Le caratteristiche anatomiche macroscopiche del cuore creano barriere macro e microanatomiche cruciali sia per l’elettrofisiologia cardiaca normale sia per le aritmie clinicamente rilevanti. I CANALI IONICI Sappiamo anche che ci sono degli ioni che sono cruciali per lo svolgimento delle funzioni elettriche e alcuni sono coinvolti anche nelle funzioni meccaniche. Questi ioni sono, particolarmente, il potassio e il calcio. I canali ionici sono glicoproteine transmemnbrana costituite da subunità multiple connesse, che si aprono e chiudono in risposta a una serie di stimoli biologici. Altre proteine transmembrana di tipo ionico, come scambiatori e trasportatori, contribuiscono in maniera rilevante all'eccitabilità cellulare cardiaca. Le pompe ioniche stabilisco e mantengono attraverso la membrana cellulare i gradienti ionici, che fungono da forza pilota per il flusso della corrente attraverso i canali ionici. I trasportatori o scambiatori che non spostano ioni in maniera elettricamente neutra sono definiti elettrogeni. La superfamiglia più abbondante di canali ionici espressi nel cuore e quella voltafaccia-dipendente. Diversi temi strutturali sono comuni a tutti i canali ionici voltaggio-dipendenti. Innanzitutto, l'architettura modulare, consistendo di diverse subunità. Le proteine si avvolgono intorno ad un polo centrale delimitato da aminoacidi che mostrano una straordinaria conservazione della selettività. La strategia generale per l'attivazione del gating è altamente conservata: il quarto segmento transmembrana si trova sulla superficie della membrana e si muove risposta alla depolarizzazione, aprendo il canale. Infine, la maggior parte dei complessi di canali ionici comprende non solo le proteine che formano il poro, ma anche subunità ausiliarie e modificano la funzione del canale. ECG E RITMO SINUSALE Questo a lato, è l’elettrocardiogramma o meglio sono le componenti che in un ECG debbono essere presenti perché si possa definire normale e perchè il ritmo possa dirsi sinusale, ovvero originatosi dal nodo del seno. C’è un’onda P della depolarizzazione atriale, un complesso rapido cosiddetto QRS che è espressione della depolarizzazione ventricolare, composto da una piccola onda Q, quando presente, all’inizio, un’onda R alta, sia in positivo che in negativo (questo dipende da quale prospettiva esploriamo il cuore e quale elettrodo legge l’attività elettrica), e un’onda S piuttosto profonda, quindi un tratto ST e l’onda T che sono espressione della ripolarizzazione. Quindi possiamo dire che tutta le sequenza elettromeccanica di cui abbiamo parlato è rappresentata all’interno di questo disegnino. Normalmente, la durata dell'intervallo PR è compresa tra 0,12 secondi e 0,20 secondi, mentre il complesso QRS ha una durata di circa 0,08 secondi. Questo è l’ECG normale di un paziente con un normale ritmo sinusale, con una normale frequenza cardiaca, con una normale morfologia di tutte le parti dell’ECG che abbiamo visto. La frequenza deve essere compresa tra 60 e 100. Questo vale in linea generale dal momento che in un atleta allenato può essere di 38-40 senza per questo essere patologica. Se una frequenza di 38-40 è presente in un vecchio di 80 anni vuol dire che vi sono dei problemi. L’onda P deve essere positiva in D2 e negativa in aVR. Se così non è così, o c’è una patologia o si è sbagliato a mettere gli elettrodi. Il ritmo viene definito sinusale in quanto la sequenza degli impulsi ha origine dal nodo senoatriale. Le principali caratteristiche di un elettrocardiogramma normale sono: - un ritmo normale dei complessi; - una frequenza compresa tra i 60 e i 100 battiti per minuto; - ogni complesso QRS è preceduto da un'onda P; - l'onda P è positiva in D2 e negativa nella deviazione aVR. MECCANISMI DELLE ARITMIE CARDIACHE Le aritmie cardiache sono il risultato di anomalie della generazione dell'impulso, della conduzione o di entrambi. È molto difficile stabilire con certezza un meccanismo patogenetico comune per molte aritmie cliniche. Ma come si può determinare un’aritmia? Sono stati proposti una serie di meccanismi in grado di determinare aritmie, e questi sono: - automatismo; - inappropriato funzionamento del pacemaker sinusale; - presenza di pacemaker ectopico; - attività triggerata. Automatismo: Per automatismo si intende la capacità di una fibra di iniziare un impulso spontaneamente, senza necessità di stimolo precedente (non necessaria quiescenza elettrica). Ciò può accadere a carico di una cellula di conduzione ma può anche accadere ad un cardiomiocita di lavoro. La depolarizzazione diastolica spontanea è alla base della proprietà dell'automatismo (pacemaking) caratteristica delle cellule del nodo senoatriale e atrio-ventricolare, del sistema di His e Purkinje, del seno coronarico e delle vene polmonari. La depolarizzazione di fase 4 è il risultato dell'azione coordinata di una serie di correnti ioniche, comprese le correnti del potassio rettificanti in entrata (I KI ) e rettificanti ritardate (componente rapida I Kr e componente lenta I Ks ), le correnti del calcio, la pompa sodio-potassio, lo scambiatore sodio-calcio e la corrente cosiddetta funny o pacemaker (I f ). Sia la frequenza della depolarizzazione di fase 4 che la frequenza di attivazione delle cellule pacemaker sono regolate dinamicamente. Uno dei fattori più importanti nella modulazione della fase 4 è il tono del sistema nervoso autonomo. L'effetto cronotropo negativo del parasimpatico è il risultato del rilascio di acetilcolina che si lega ai recettori muscarinici, liberando subunità βγ della proteina G che attivano una corrente di potassio (I KAch ) nelle cellule atriali e nodali. Il conseguente aumento della conduttanza del potassio si oppone alla depolarizzazione della membrana, rallentando la velocità di crescita della fase 4 del potenziale d'azione. Al contrario, l'aumento del tono del sistema nervoso simpatico incrementa la concentrazione di catecolamine miocardiche, con attivazione sia dei recettori α sia di quelli β. L'effetto della stimolazione β 1 -adrenergica predomina nelle cellule pacemaker, determinando un aumento sia della corrente del calcio di tipo L sia di quella funny, aumentando quindi la tendenza della fase 4. L'aumentata attività del simpatico è in grado di incrementare notevolmente la frequenza di attivazione delle cellule del nodo senoatriale. L'aumentata frequenza di attivazione delle cellule di Purkinje ha maggiori limiti, inducendo raramente tachiaritmie ventricolari superiori a 120 battiti al minuto. Il normale automatismo può essere influenzato da numerosi altri fattori associati cardiopatia. Ipokaliemia e ischemia possono ridurre l'attività della pompa sodio potassio, diminuendo la corrente ripolarizzante e incrementato la depolarizzazione diastolica di fase 4. Il risultato finale è un aumento dell'attivazione spontanea delle cellule pacemaker. Modesti incrementi di potassio extracellulare possono rendere maggiormente positivo il potenziale diastolico massimo, aumentando di conseguenza anche la frequenza di attivazione delle cellule pacemaker. Un innalzamento più significativo della concentrazione di potassio extracellulare render cuore non eccitabile depolarizzando il potenziale di membrana. Un normale o aumentato automatismo dei pacemaker latenti sussidiari provoca ritmi di “scappamento”. La soppressione di una cellula pacemaker da parte di un ritmo più veloce determina un aumento del sodio intracellulare, e l’estrusione del sodio dalla cellula da parte della pompa sodio-potassio provoca un aumento della corrente ripolarizzante che rallenta la depolarizzazione diastolica di fase 4. A frequenze inferiori, la concentrazione del sodio intracellulare è ridotta, come pure l’attività della pompo sodio-potassio, con conseguente progressivo incremento della velocità di depolarizzazione diastolica e accelerazione della tachicardia. La soppressione da overdrive e l’accelerazione sono caratteristiche di tutte le tachicardie automatiche, ma possono anche non essere presenti. La conduzione anomala in un tessuto con aumentato automatismo può rallentare o eliminare i fenomeni della soppressione da overdrive e dell’aumento dell’attività di scarica del tessuto automatico. L’automatismo anomalo può essere alla base di forme di tachicardia atriale, ritmi idioventricolari accelerati e tachicardia ventricolare, in particolare in caso di ischemia e riperfusione. Inappropriato funzionamento del pacemaker sinusale: L’inappropriato funzionamento del pacemaker sinusale determina una frequenza sinusale troppo elevata o troppo bassa per le necessità fisiologiche del paziente. Presenza di pacemaker ectopico: Pacemaker ectopici possono ritrovarsi negli atri, seno coronarico, vene polmonari, valvole AV, giunzione AV o fibre del sistema His-Purkinje. Questi pacemaker latenti possono attivarsi con meccanismo di “scappamento” in presenza di un blocco tra il nodo SA ed il pacemaker ectopico oppure per aumento della velocità di un pacemaker ectopico che sottrae il controllo del ritmo al nodo SA. Ogni cellula del sistema di conduzione, ma anche al di fuori di tale sistema, può, ad un certo punto, prendere in mano la questione. Questo può essere positivo in certi casi, una tragedia in altri. Attività triggerata: Per attività triggerata si intende l’attività di pacemaker che risulta da un impulso o una serie di impulsi precedenti senza quiescenza elettrica, ovvero senza post-depolarizzazione. Distinguiamo due forme di attività triggerata: - precoce: prima che sia completata la ripolarizzazione; - tardiva: a recupero completato. Il termine automatismo o attività friggere si riferisce all’avvio di un impulso dipendente da postdepolarizzazioni. Le postdepolarizzazioni sono oscillazioni del voltaggio di membrana che si verificano durante (postdepolarizzazioni precoci o PDP) o dopo (postdepolarizzazione tardive o PDT) un potenziale di azione. La caratteristica comune all’induzione del PDT è la presenza di un aumento del carico di calcio nel citosol e nel reticolo sarcoplasmatico. L’inibizione della pompa sodio-potassio (digitale) favorisce, ma non è necessaria per determinare, il sovraccarico di calcio che predispone alle PDT. Le catecolamine e l’ischemia aumentano sufficientemente il carico di calcio da indurre PDT. Le PDP si osservano durante il potenziale di azione e interrompono la regolare ripolarizzazione del miocita. Si è sempre ritenuto che, diversamente dalle PDT, le PDP non dipendessero da un aumento del calcio intracellulare e che, invece, il prolungamento del potenziale d’azione e la riattivazione delle correnti depolarizzanti fossero fondamentali per la loro formazione. Il calcio citosolico può aumentare quando il potenziale di azione è prolungato; questo fenomeno, a sua volta, sembra aumentare la corrente del calcio di tipo L, prolungando ulteriormente la durata del potenziale di azione e fornendo la corrente invar che alimenta la PDP. Il sovraccarico di calcio intracellulare causato dal prolungamento del potenziale di azione può anche aumentare le probabilità di formazione di PDT. La correlazione fra la concentrazione di calcio intracellulare, PDP e PDT può spiegare la propensione del cuore sovraccarico di calcio allo sviluppo di aritmie. Le aritmie da attività triggered PDP-dipendenti mostrano di essere frequenza-dipendenti. L’ampiezza di una PDP è incrementata a basse frequenze quando i potenziali di azione sono più lunghi. Un pacing rapido abbrevia il potenziale di azione e riduce l’ampiezza della PDP, probabilmente a causa di un aumento delle correnti rettificatrici del potassio e, forse, dell’accelerazione dell’inattivazione calcio-indotta delle correnti del calcio di tipo L. Analogamente, le catecolamine incrementano la frequenza cardiaca e riducono la durata del potenziale di azione e l’ampiezza delle PDP, nonostante il ben noto effetto della stimolazione betaadrenergica sull'aumento della corrente del calcio di tipo L. Una condizione fondamentale alla base dello sviluppo di PDP è rappresentata dal prolungamento del potenziale d'azione e del tratto QT. Ipokaliemia, ipomagnesiemia, bradicardia e alcuni farmaci possono predisporre alla formazione di PDP, invariabilmente nel contesto di un prolungamento del potenziale d'azione. Gli antiaritmici dotati d'azione di classe Ia e III producono un prolungamento del potenziale d'azione e del QT con intenti terapeutici, ma frequentemente causano aritmie. Una riduzione della concentrazione extracellulare del potassio può paradossalmente ridurre la corrente di membrana del potassio a livello del miocita ventricolare, spiegando il motivo per cui l'ipokaliemia induce un prolungamento del potenziale d'azione e l'insorgenza di PDP. L'infusione di potassio accorcia l'intervallo QT in pazienti affetti dalla sindrome congenita del QT lungo e in soggetti con prolungamento acquisito farmaco-indotto del QT. L'attività triggered PDP-mediata è probabilmente alla base dell'avvio della caratteristica tachicardia ventricolare polimorfica, detta torsione di punta, osservata in pazienti affetti da forme congenite e acquisite di sindrome del QT lungo. Anche una cardiopatia strutturale può ritardare la ripolarizzazione ventricolare e favorire le aritmie correlate ad anomalie della ripolarizzazione. Aritmie da farmaci: Un’aritmia può essere associata all’uso di farmaci e tra questi, per eccellenza, gli antiaritmici. Ad esempio un eccesso nel dosaggio della digitale può indurre aritmie ipercinetiche, come una tachicardia ventricolare o un flutter ventricolare, che è l’esatto contrario di quello che ci si sarebbe aspettato in quanto si era usato il farmaco perche “bradicardizzasse”. Aritmie da blocco: Come ci possono essere accelerazioni e rallentamenti, ci possono essere delle interruzioni. Questo è il caso tipico delle aritmie da blocco, che possono essere associate al fenomeno del rientro o meno. Quelle senza rientro, a loro volta possono essere bidirezionali o unidirezionali, mentre i blocchi con rientro sono unidirezionali. Un’aritmia minacciosa è un’aritmia che può mettere a rischio la vita del paziente. Aritmie da rientro: L'attività elettrica ha inizio nel nodo senoatriale e continua fino a che tutto il cuore non è stato attivato. L'impulso cardiaco termina quando tutte le fibre sono state scaricate e sono completamente refrattarie. L'attività elettrica, quindi, riparte al successivo impulso sinusale. Se un gruppo di fibre, non attivate durante l'onda di depolarizzazione iniziale, recupera un'eccitabilità prima che l'impulso termini, questo costituisce uno stimolo per rieccitare le aree già scaricate e che hanno recuperato dalla depolarizzazione iniziale. Si costituisce così il rientro. Il rientro non è altro che un impulso, il quale percorre una struttura cardiaca in una certa direzione, torna indietro a riattivare il tessuto da cui proveniva. L’opinione corrente è che tutte le aritmie ipercinetiche abbiano alla base il fenomeno del rientro. Questo è importante dal momento che, perché ci possa essere un rientro, è necessario un circuito che grazie allo sviluppo dell’elettrofisiologia o aritmologia interventistica, può essere mappato, ricostruito e quindi interrotto. Lo interrompiamo con una folgorazione, con un eccesso di temperatura in salita o in discesa (lo possiamo cuocere o congelare) o in altre maniere. Tali interventi sono detti di ablazione e spesso dopo un intervento di ablazione può essere necessario eseguirne un altro (vuol dire che qualcosa di tale circuiteria è sfuggito). I requisiti necessari affinché avvenga il rientro sono due vie diverse dal punto di vista elettrofisiologico per la propagazione di un impulso intorno a una regione non eccitabile, cosicché si verifichi un blocco unidirezionale in una delle vie e un'aria di tessuto eccitabile sul fronte dell'onda che si propaga. Una caratteristica fondamentale per classificare le aritmie da rientro è la presenza e la misura dell'intervallo eccitabile. Questo esiste quando il circuito tachicardico è più lungo della lunghezza d'onda della tachicardia, permettendo che stimoli adeguatamente temporizzati resettino la propagazione nel circuito. Aritmie rientranti possono esistere nel cuore, anche in assenza di un gap eccitabile e con una lunghezza d'onda dell’aritmia quasi della stessa lunghezza della via di propagazione. In questo caso, il fronte dell'onda si propaga attraverso un tessuto parzialmente refrattario, senza ostacoli anatomici e senza un gap pienamente eccitabile; si parla di rientro con circuito principale, una forma di rientro funzionale (rientro che dipende dalle proprietà funzionali del tessuto). Diversamente dal rientro da gap eccitabile, nelle rientro con circuito principale non vi è alcun circuito anatomico fisso e può quindi non essere possibile eliminare la tachicardia mediante la distruzione di una parte del circuito. Inoltre, nelle rientro con circuito principale, il circuito tende a essere meno stabile rispetto a quello delle aritmie rientranti da gap eccitabile, con ampie variazioni nella lunghezza del ciclo e propensione all'interruzione della propagazione. QUADRI ELETTROCARDIOGRAFICI DI ARITMIE Nella figura in basso, vengono mostrati vari quadri di elettrocardiogrammi dovuti ad aritmie. PREECCITAZIONE VENTRICOLARE E SINDROME DI WPW Oggi trovare una sindrome di WPW è raro dal momento che, quando è nata l’aritmologia interventistica, la cosa più facile da fare era identificare un circuito anatomicamente esistente. Quindi si è iniziato proprio con la WPW e tali soggetti sono stati tutti ablati con successo. C’è gente che è morta, per esempio durante una partita di pallone, dopo una serata di bagordi, etc..., in cui con la successiva analisi di pregressi ECG si è identificata una WPW misconosciuta. Un impulso che riesce ad attivare una zona miocardica distante dalla sua sede di origine, prima di quanto avverrebbe se esso venisse condotto solo attraverso il normale sistema di conduzione, determina una cosiddetta "pre-eccitazione". Un esempio di pre-eccitazione è dato dalla presenza di una via accessoria anomala, in grado di trasmettere l'impulso più rapidamente della via normale di conduzione. Questo è il caso del fascio di Kent. La presenza di una via accessoria darà un segno anche a livello dell'ECG, caratterizzato da: - tratto PR corto, dovuto all'anticipata attivazione ventricolare; - presenza di onda delta, una deflessione rallentata iniziale del complesso QRS; - QRS largo, dato dalla fusione di più complessi QRS; - alterazioni secondarie della ripolarizzazione ventricolare, con onde T negative asimmetriche. La presenza di questo fenomeno è molto importante, in quanto può dar luogo a tachicardia da rientro atrioventricolare, fibrillazione atriale e fibrillazione ventricolare. CLASSIFICAZIONE DELLE ARITMIE In basso, viene mostrata una tabella di classificazione delle aritmie. Classificazione delle aritmie DISFUNZIONE DEL NODO SENOATRIALE - Tachicardia sinusale - Bradicardia sinusale - Aritmia sinusale RITMI ECTOPICI DI SCAPPAMENTO - Arresto sinusale - Battiti o ritmi atriali di scappamento - Battiti o ritmi giunzionali di scappamento RITMI ECTOPICI PREMATURI - Di origine atriale Battiti prematuri atriali Tachicardia atriale Flutter atriale Fibrillazione atriale - Di origine giunzionali giunzionale - Di origine ventricolari ventricolare Tachicardia giunzionale - Battiti o ritmi ventricolari di scappamento DISTURBI DI CONDUZIONE - Blocco senoatriale Tipo 1; Tipo 2 Battiti prematuri- Blocco AV I grado Battiti - Blocco AV II grado tipo 2 prematuri- Blocco AV III grado (completo) - Blocco AV II grado tipo 1 Tachicardia ventricolare Fibrillazione ventricolare Le extrasistoli sono i battiti ectopici prematuri. Extrasistole è un termine più “colloquiale”. I disturbi di conduzione sono le aritmie ipocinetiche. Questa è una classica bradicardia sinusale. L’onda P ci sta, il QRS ci sta, la T ci sta, hanno una normale morfologia. Salta all’occhio l’ampio intervallo R-R. La frequenza la calcoliamo dividendo 1500 per il numero di quadratini piccoli tra due onde R-R successivi. Questo ha 50 bpm. Questa è una tachicardia sinusale perché le onde P sono sempre presenti. Probabilmente è uno che ha fatto una corsa, etc. Questa è una tachicardia atriale ectopica dal momento che l’onda P, pur presente, è negativa. Questa è una tachicardia giunzionale. Non è atriale perché manca la P e non è ventricolare perché il QRS è stretto. ARITMIE IPOCINETICHE Le aritmie ipocinetiche sono artitmie che causano bradicardia per un disturbo della formazione dell’impulso nel nodo del seno o per un disturbo della propagazione dell’impulso dal nodo del seno all’atrio destro o lungo il tessuto specifico di conduzione cardiaco (nodo atrioventricolare, fascio di HIS, branche e sistema del Purkinje). L’attivazione elettrica del cuore ha origine normalmente nel nodo seno atriale, il pacemaker dominante. L’attivazione spontanea e la contrazione del cuore sono la conseguenza della funzione pacemaker del tessuto specializzato nell’ambito di queste localizzazioni anatomiche. La bradicardia è la conseguenza di un’insufficienza nell’avvio o nella conduzione dell’impulso. Il deficit nell’avvio dell’impulso può essere causato da depressione dell’automatismo dovuta a rallentamento o mancanza della depolarizzazione diastolica di fase 4, in seguito a malattie o a esposizione a farmaci. La disfunzione del nodo SA e il blocco di conduzione AV sono le cause più comuni di bradicardia patologica. La prima può risultare difficile da distinguere dalla bradicardia sinusale fisiologica, soprattutto nei giovani. La disfunzione del nodo SA è più frequente fra la quinta e la sesta decade di vita e va considerata nella diagnosi di pazienti che presentano affaticamento, intolleranza allo sforzo o sincope e bradicardia sinusale. Il blocco AV transitorio è comune nei giovani e si verifica probabilmente a causa dell’elevato tono vagale riscontrato nel 10% circa dei giovani adulti. L’applicazione permanente di un pacemaker è l’unica terapia affidabile per la bradicardia sintomatica in assenza di eziologie estrinseche e reversibili, come aumento del tono vagale, ipossia, ipotermia e farmaci. Nell’immagine sottostante, vengono riportate le principali cause di aritmie ipocinetiche. Le cause possono essere suddivise in intrinseche ed estrinseche. INTRINSIC CAUSES Idiopathic degeneration (aging) Infarction or ischemia Infiltrative disease • Sarcoidosis • Amyloidosis • Hemochromatosis Collagen vascular disease • Systemic lupus erythematosus • Rheumatoid arthritis • Sclerodermia Myotonic muscolar dystrophy Surgical trauma • Valve replacement • Correction of congenital heart diseas • Heart trasplantation Familial disease Infectious disease • Chagas’ disease • Endocarditis EXTRINSIC CAUSES Autonomically mediated syndromes - Neurocardiac syncope - Carotid-sinus hypersensitivity - Situational disturbances (Coughing, Micturition, Defecation, Vomiting) Drugs - β-Adrenergic blockers - Calcium-channel blockers - Clonidine - Digoxin - Antiarrhythmic agents Hypothermia Hypothyroidism Neurologic disorders Electrolyte imbalances - Hypokalemia - Hyperkalemia Quando parliamo di aritmie ipocinetiche, e ciò vale in una certa misura anche per quelle ipercinetiche, dobbiamo tenere conto che ci sono cause intrinseche e cause estrinseche. Le cause intrinseche interessano fondamentalmente l’anatomia del sistema. Tra le cause estrinseche, frequenti sono le forme sincopali (che possono essere di varia natura, ad es. post-minzionale). La sincope si può avere o perché il cuore si è, seppure per poco, fermato, o perché la frequenza è scesa a valori assolutamente incompatibili con il mantenimento della stazione eretta e dello stato di coscienza. TACHIARITMIE Il termine si riferisce tipicamente a battiti prematuri isolati (depolarizzazioni) o a forme non sostenute e sostenute di tachicardia che hanno origine da foci miocardici o da circuiti di rientro. La definizione standard di tachicardia è un ritmo che produce una frequenza ventricolare superiore ai 100 battiti al minuto. Questa definizione presenta alcuni limiti, poiché le frequenze atriali possono superare i 100 battiti nonostante la frequenza ventricolare sia più lenta. Inoltre, le frequenze ventricolari possono superare la frequenza sinusale di base e rimanere al di sotto dei 100 bpm, rappresentando comunque un importante risposta tachicardica. Sintomi delle tachiaritmie: Le tachiaritmie provocano classicamente sintomi come palpitazioni o accelerazione del polso. Nel caso di battiti prematuri, il paziente può avvertire la mancanza di una pulsazione o una pausa, e perfino percepire un rallentamento della frequenza cardiaca. Nel caso di ritmi caotici e rapidi o di tachiaritmie che hanno origine nell’atrio e vengono condotte in modo variabile ai ventricoli verrà sentita un’irregolarità più intensa del polso. Nelle tachiaritmie molto rapide si può verificare una compromissione emodinamica, come pure vertigini e sincope causate da una riduzione della gittata cardiaca o affanno dovuto a un marcato aumento delle pressioni di riempimento cardiaco. Occasionalmente il paziente può presentare un malessere toracico che simula un’ischemia miocardica. Test diagnostici nella valutazione delle tachiaritmie: Nel paziente che manifesta sintomi non potenzialmente letali è essenziale la conferma elettrocardiografica. Solo per i soggetti con sintomi quotidiani va preso in considerazione un monitoraggio Holter delle 24 ore. Nei pazienti che presentano sintomi più gravi, come sincope, il monitoraggio ambulatoriale può risultare insufficiente. Nel caso in cui è presente una cardiopatia strutturale e sincope e vi è il sospetto di una tachicardia ventricolare, è necessario il ricovero ospedaliero, con esecuzione di test diagnostici elettrofisiologici e probabile impianto di un dispositivo impiantabile (ICD, implantable cardioverter/defibrillator). Il monitoraggio delle tachiaritmie asintomatiche è indicato in una serie di situazioni specifiche. Nei pazienti con sospetta miocardiopatia tachicardia-indotta, caratterizzata da dilazione cavitaria e depressione della funzione sistolica, è necessario documentare che l’aritmia è sotto controllo. Il monitoraggio dei complessi prematuri ventricolari asintomatici e della tachicardia ventricolare non sostenuta può essere utile per stratificare il rischio di morte cardiaca improvvisa nei pazienti con funzione ridotta del ventricolo sinistro in seguito ad infarto al miocardio. Infine, nei pazienti con fibrillazione atriale asintomatica, le strategie terapeutiche anticoagulanti dipendono da un’accurata valutazione della presenza di questa aritmia. Una registrazione ECG a 12 derivazioni nel corso della tachicardia può rappresentare un importante strumento diagnostico per identificare il meccanismo e l’origine di una tachicardia, non acquisiti mediante un ECG a una o due derivazioni. L’ECG a 12 derivazioni della tachiaritmia deve essere registrato e conservato, ove possibile, come parte integrante della cartella clinica. Molte tachiaritmie sopraventricolari parossistiche non si associano a un rischio significativo di cardiopatia strutturale, e non è di solito necessaria una valutazione della funzionalità cardiaca, né la ricerca di una cardiopatia ischemica, a meno che non siano presenti sintomi gravi o caratteristici. Meccanismi patogenetici delle tachiaritmie: Le tachicardie sono causate da anomalie della formazione dell’impulso e/o della sua propagazione. I principali meccanismi di genesi sono: - anomalie della formazione dell’impulso: un aumento dell’automatismo provoca, normalmente, incremento della frequenza sinusale e tachicardia sinusale. L’automatismo anomalo è dovuto a un aumento della pendenza della depolarizzazione di fase 4 o a una riduzione della soglia di depolarizzazione del potenziale d'azione in un'area miocardica diversa dal nodo del seno. Si ritiene che l'automatismo anomalo sia responsabile della maggior parte dei complessi prematuri atriali e ventricolari e di alcune tachiaritmie. Più raramente, la formazione anomala dell'impulso è causata dallo sviluppo di attività triggered. Quest'ultima è correlata a postdepolarizzazioni cellulari che si verificano alla fine del potenziale d'azione, durante la fase 3, e sono definite postdepolarizzazioni precoci, oppure avvengono dopo il potenziale d'azione, durante la fase 4, e in questo caso si indicano come postdepolarizzazioni tardive. Le postdepolarizzazioni sono ascrivibili a un aumento dell'accumulo di calcio intracellulare. Le postdepolarizzazioni precoci possono essere responsabili dei complessi prematuri ventricolari che innescano l'aritmia ventricolare polimorfica nota come torsione di punta (TDP). Si ritiene che le postdepolarizzazioni tardive siano responsabili delle tachiaritmie atriali, giunzionali e fascicolari causate dall'intossicazione da digossina; inoltre, sembra siano alla base della tachicardia ventricolare catecolamina-sensibile che si origina nel tratto di efflusso; - anomalie della propagazione dell'impulso: il rientro è dovuto a disomogeneità della conduzione miocardica o delle proprietà di recupero. La presenza di un blocco unidirezionale con conduzione lenta per permettere recupero retrogrado del miocardio bloccato consente la formazione di un circuito che può sostenere una tachicardia. Queste disomogeneità sono in qualche modo intrinseche ma ridotti al minimo in un normale ciclo di attivazione-recupero del miocardio. La difformità possono essere enfatizzate dalla presenza di vie accessorie (sindrome di WPW), oppure nelle anomalie generalizzate e geneticamente determinate dei canali ionici miocardici (sindrome del QT lungo) oppure per l'interruzione dei pattern miocardici normali di attivazione a causa dello sviluppo di fibrosi. Il rientro sembra rappresentare il meccanismo fisiopatologico alla base della maggior parte delle tachicardie sopraventricolari sostenute e delle tachicardie ventricolari. Il rientro può dipendere da un circuito fisso, dal punto di vista anatomico, costituito da barriere anatomiche naturali della conduzione come la cresta terminale, ovvero la cresta verticale sulla parete interna dell'atrio destro che separa il seno della vena cava e l'atrio posteriore, e/o fibrosi estesa creata da una patologia miocardica sottostante. Questa forma di rientro sembra essere più stabile e al luogo a una tachicardia di aspetto ripetitivo ed uniforme, spesso monomorfica. Altre forme di rientro sembrano di tipo funzionale e dipendono maggiormente da alterazioni dinamiche delle proprietà elettrofisiologiche del miocardio. Queste tachicardie tendono a essere più instabili e possono dare origine a tachicardie di aspetto polimorfico. Due esempi di rientro funzionale sono la fibrillazione ventricolare da ischemia miocardica acuta e la tachicardia ventricolare polimorfica nei pazienti con anomalia geneticamente determinata dei canali ionici. Passiamo adesso a descrivere nello specifico le aritmie più frequenti ed importanti. MALATTIA O DISFUNZIONE DEL NODO DEL SENO Il nodo SA è composto da un gruppo di piccole cellule fusiformi situete nel solco terminale sulla superficie epicardica del cuore, a livello della giunzione fra atrio destro e vena cava superiore, dove avvolgono l’arteria del nodo SA. La propagazione irregolare e lenta degli impulsi dal nodo SA può essere spiegata sulla base dell’elettrofisiologia delle cellule nodali e della struttura del nodo SA stesso. I potenziali di azione delle cellule nodali SA sono caratterizzati da un potenziale di membrana relativamente depolarizzato da -40 a -60 mV, lenta salita della fase 0 e depolarizzazione diastolica di fase 4 piuttosto rapida, rispetto ai potenziali di azione registrati nelle cellule muscolari cardiache. La disfunzione del nodo SA può avere diverse manifestazioni cliniche. La bradicardia sinusale è dovuta ad una depressa automaticità del nodo del seno. Coppi aveva una bradicardia sinusale (38 bpm). Con l’allenamento si può ottenere questo tipo di condizione ma ci si può anche nascere. Le pause o arresto sinusale sono dovuti ad un disturbo o di formazione dell’impulso nel nodo del seno o di conduzione dell’impulso fuori dalla regione nodale all’atrio circostante. Il blocco del SA di II grado è l’unico che può essere riconosciuto sull’ECG di superficie. È caratterizzato da un’intermittente mancanza dell’attivazione atriale per incapacità di conduzione dell’impulso dal nodo del seno all’atrio destro. La mancanza dell’attivazione atriale è data dalle onde P. La presenza, sia pure non costante dell’onda P, evidenzia come il fenomeno si svolga all’interno dell’atrio. Nell’immagine in alto, vengono mostrati alcuni esempi. Questo è un arresto sinusale e non AV. In tal caso vi sarebbero una serie di onde P non seguite da complesso QRS. Qui invece le onde P non ci sono. Arresto sinusale è diverso dal blocco. La linea piatta, isoelettrica, indica che non c’è alcun tipo di stimolo. La sindrome bradi-tachicardica è caratterizzata dall’assoluta impredivibilità di questo fenomeno e crea dei problemi. La terapia , dopo un tentativo con farmaci antiaritmici, è inevitabilmente il pacemaker bicamerale, cioè il pacemaker che sincronizzi l’attività atriale con l’attività ventricolare e fa in modo che sia, in condizioni normali, il nodo del seno del paziente a comandare, quando questo diventa disfunzionale (tachi o bradicardia), sopravviene il pacemaker. La sindrome bradi-tachicardica è stata la palestra in cui è stato messo a punto il pacemaker atrioventricolare (prima si usava il pacemaker ventricolare e quindi si perdeva la quota atriale dell’EDV e poi il pacemaker era fisso per cui, se la frequenza era impostata su 60, il paziente non poteva fare uno sforzo che richiedesse una frequenza maggiore perché gli veniva la sincope). Si è quindi ricostruito il circuito fisiologico atrio-ventricolo. Eziologia della malattia del nodo SA: La disfunzione del nodo SA è stata classificata come intrinseca o estrinseca. La distinzione è importante, perché la forma estrinseca è spesso risolvibile e dovrebbe generalmente essere corretta prima di considerare la terapia con pacemaker. Le cause più comuni di disfunzione estrinseca del nodo SA sono farmaci ed influssi sul sistema nervoso autonomo, che sopprimono l’automatismo e/o compromettono la conduzione. Altre cause estrinseche includono ipotiroidismo, apnea da sonno e patologie che possono insorgere in pazienti in area critica, come ipotermia, ipossia, aumento della pressione intracranica (risposta di Cushing) e suzione endotracheale, mediante attivazione del nervo vago. La disfunzione sinusale intrinseca ha una natura degenerativa e spesso si caratterizza, sotto l’aspetto anatomo-patologico, per la sostituzione fibrosa del nodo SA o delle sue connessioni all’atrio. La malattia coronarica acuta o cronica può essere associata a disfunzione del nodo SA. I processi infiammatori possono alterare la funzione del nodo SA, provocando alla fine la sostituzione con tessuto fibroso. Pericarditi, miocarditi e cardiopatie reumatiche sono state correlate a malattia del nodo SA con bradicardia sinusale, arresto sinusale e blocco d’uscita. Anche la cardite associata a lupus eritematoso sistemico, artrite reumatoide e malattie miste del tessuto connettivo può influire sulla struttura e sulla funzione del nodo SA. L’amiloidosi senile è una malattia infiltrativa che colpisce tipicamente i soggetti nella nona decade di vita; la deposizione di proteina amiloide nel miocardio atriale compromette la funzione del nodo SA. Una parte delle disfunzioni del nodo SA è iatrogena e fa seguito alla correzione chirurgica di alcune cardiopatie congenite, in particolare alla riparazione palliativa della trasposizione corretta delle grandi arterie mediante procedura di Mustard. Sono state descritte rare forme ereditarie di disfunzioni del nodo SA, e sono numerose quelle caratterizzate dal punto di vista genetico. La disfunzione del nodo SA autosomica dominante unita a tachicardia sopraventricolare, cioè la variante tachicardica-bradicardica della sindrome del seno malato (sick-sinus syndrome, SSS), è stata correlata a mutazioni del gene della sub unità della corrente pacemaker, I f , HCN4, situato sul cromosoma 15. Diverse malattie neuromuscolari hanno una predilezione per il sistema di conduzione e il nodo SA; fra queste, la sindrome di Kearns-Sayre (oftalmoplegia, degenerazione pigmentosa della retina e miocardiopatia) e la distrofia miotonica. Caratteristiche cliniche della malattia del nodo SA: La disfunzione del nodo senoatriale può essere completamente asintomatica e manifestarsi con un'anomalia dell'ECG come bradicardia sinusale, arresto sinusale e blocco d'uscita, oppure tachicardia sopraventricolare alternante e bradicardia. I sintomi associati a disfunzione del nodo senoatriale, e in particolare alla sindrome tachicardia-bradicardia, possono essere dovuti sia alla bradicardia sia alla tachicardia. In molti casi, la sintomatologia associata alla disfunzione del nodo senoatriale è il risultato di una patologia cardiovascolare concomitante. Da un terzo fino alla metà dei pazienti con disfunzione del nodo senoatriale va incontro a tachicardia sopraventricolare, di solito sottoforma di fibrillazioni o flutter atriale. L'incidenza di fibrillazione atriale cronica nei pazienti con disfunzione del nodo senoatriale aumenta con l'avanzare dell'età, ipertensione, diabete mellito, dilatazione del ventricolo sinistro, cardiopatia valvolare e pacing ventricolare. I pazienti che corrono i maggiori rischi, cioè quelli con un'età superiore ai 65 anni, o con una anamnesi positiva per ictus, cardiopatia valvolare, disfunzione del ventricolo sinistro o dilatazione atriale, devono essere trattati con anticoagulanti. Fino a un quarto dei soggetti con malattia del nodo senoatriale presenta contemporaneamente disturbi della conduzione atrioventricolare, benché solo una minoranza necessitino di una terapia specifica per un blocco atrio-ventricolare di grado elevato. La storia naturale della disfunzione del nodo senoatriale è caratterizzata da intensità sintomatologica variabile anche nei pazienti che presentano episodi sincopali. I sintomi correlati a disfunzione del nodo senoatriale possono essere significativi, ma la mortalità complessiva non è generalmente modificata in assenza di altre patologie comorbose. Le caratteristiche della storia naturale devono essere tenute in considerazione nella scelta della terapia. Elettrocardiogramma nella malattia del nodo senoatriale: Le manifestazione ECG di tale condizione comprendono bradicardia sinusale, pause sinusali, arresto e blocco sinusale in uscita, tachicardia e inadeguatezza cronotropa. È spesso difficile differenziare la bradicardia sinusale patologica da quella fisiologica. Per definizione, la bradicardia sinusale è un ritmo guidato dal nodo senoatriale con una frequenza inferiore ai 60 battiti al minuto; la bradicardia sinusale è molto frequente e tipicamente benigna. Frequenze inferiori ai 60 battiti al minuto sono comuni nei giovani sani e nei soggetti allenati. Una frequenza sinusale inferiore 40 battiti al minuto nello stato di veglia è in soggetto non allenato, è generalmente considerata anomala. Pause sinusali e arresto sinusale possono verificarsi in seguito all'assenza di stimolazione da parte del nodo senoatriale: ciò provoca una pausa con assenza di onde P sull’ECG. Un deficit intermittente della conduzione del nodo senoatriale provoca il blocco sinusale in uscita. La gravità di questo può variare in maniera simile a quella del blocco atrio-ventricolare. Il prolungamento della conduzione del nodo del seno può non essere evidente sull'ECG; il blocco senoatriale di secondo grado causa una conduzione intermittente dal nodo senoatriale e un ritmo atriale regolarmente regolare. Il blocco senoatriale di secondo grado tipo I è il risultato del prolungamento progressivo della conduzione dal nodo senoatriale e si manifesta sul ECG con un graduale allungamento dell'intervallo P-R e un accorciamento dell'intervallo R-R, seguito da una pausa. Nel blocco senoatriale di secondo grado tipo II Non vi è alcun cambiamento dell'intervallo P-R prima della pausa. Il blocco senoatriale completo, o di terzo grado, da luogo ad assenza delle onde P sul ECG. La sindrome tachicardia-bradicardia si manifesta con un'alternanza di bradicardia sinusale e tachiaritmie atriali. L'inadeguatezza cronotropa è l'incapacità di aumentare appropriatamente la frequenza cardiaca in risposta ad esercizio fisico o ad altre forme di stress. Test diagnostici: La disfunzione del nodo senoatriale viene perlopiù diagnosticati in sede clinica con un ECG. Una bradicardia sinusale o la presenza di pause sull’ECG a riposo sono raramente sufficienti per la diagnosi di malattie del nodo del seno. Sono necessarie registrazioni a lungo termine e correlazioni sintomatiche. La presenza di sintomi, in assenza di bradiaritmie sinusali, può essere sufficiente a escludere una diagnosi di disfunzione del nodo del seno. La registrazione dell'ECG ha un ruolo cruciale nella diagnosi e terapia della disfunzione del nodo senoatriale. L'incapacità di aumentare la frequenza cardiaca sotto sforzo è definita inadeguatezza cronotropa. Si parla, alternativamente, di impossibilità di raggiungere l'85% della frequenza massima prevista al picco dello sforzo, oppure dell'incapacità di ottenere una frequenza cardiaca superiore ai 100 battiti al minuto durante un esercizio fisico, o ancora di frequenza cardiaca massima sotto sforzo inferiore di almeno due deviazioni standard rispetto a quello della popolazione di controllo della stessa fascia d'età. Il test sotto sforzo può essere utile per discriminare l'inadeguatezza cronotropa dalla bradicardia riposo e può favorire l'individuazione del meccanismo patogenetico dell'intolleranza lo sforzo. L'esame del sistema nervoso autonomo è utile nella diagnosi dell'ipersensibilità del seno carotideo; pause superiori ai tre secondi sono compatibili con la diagnosi, ma possono essere presenti anche in pazienti asintomatici. I test elettrofisiologici possono avere un ruolo nella valutazione dei pazienti con presunta disfunzione del nodo senoatriale e della sincope, soprattutto nel quadro di una cardiopatia organica. In questa situazione, l'esame elettrofisiologico è utilizzato per escludere eziologie più maligne della sincope, come tachiaritmie ventricolari e blocco di conduzione atrio-ventricolare. Esistono diverse metodiche invasive per valutare la funzionalità del nodo senoatriale. La prima di queste è il tempo di recupero del nodo del seno (TRNS), definito come la pausa più lunga dopo la cessazione del pacing con overdrive dell'atrio destro in prossimità del nodo senoatriale (normale inferiore ai 1500 ms oppure, corretto per la lunghezza del ciclo sinusale, inferiore ai 550 ms). Il secondo metodo è il tempo di conduzione senoatriale (TCSA), definito come la metà della differenza tra la lunghezza del ciclo sinusale intrinseco e una pausa non compensatoria dopo uno stimolo atriale prematuro (normale inferiore ai 125 ms). La presenza contemporanea di un TRNS anomalo, un TCSA anomalo e una bassa FCI è un indice sensibile e specifico della malattia intrinseca del nodo senoatriale. Terapia: Poiché la disfunzione del nodo senoatriale non è accompagnata da un aumento della mortalità, lo scopo della terapia è di alleviare i sintomi. Una parte essenziale del trattamento consiste nell'escludere cause estrinseche di disfunzione del nodo senoatriale e correlare il ritmo cardiaco con i sintomi. L'impianto di un pacemaker è l'intervento terapeutico di prima scelta nei pazienti affetti da disfunzione sintomatica del nodo senoatriale. Numerosi farmaci sono in grado di modulare la funzione del nodo senoatriale e rappresentano cause estrinseche di disfunzione. La terapia farmacologica cronica delle bradiaritmie sinusali è limitata. Alcuni farmaci possono migliorare la funzione del nodo senoatriale. In alcune circostanze, la bradicardia sinusale non richiede alcuna terapia specifica o solo un supporto temporaneo della frequenza. La bradicardia sinusale è comune nei pazienti con infarto miocardico acuto inferiore o posteriore e può essere esacerbata dall'attivazione vagale indotta da dolore o da uso di farmaci come la morfina. Un'ischemia dell'arteria del nodo senoatriale si può verificare nelle sindromi coronariche acute, più tipicamente quando è coinvolta la coronaria destra, e anche in caso di infarto l'effetto sulla funzione del nodo senoatriale è perlopiù transitorio. La bradicardia sinusale è una caratteristica importante dell'ipersensibilità del seno carotideo e dell’ipotensione neuro-mediata associata a sincope vasovagale che risponde alla terapia con pacemaker. L'ipersensibilità carotidea con presincope o sincope ricorrente associata ad una componente cardio-inibitoria predominante risponde all'impianto di un pacemaker. TACHIARITMIE SOPRAVENTRICOLARI Inizieremo adesso a parlare delle tachiaritmie sopraventricolari, per poi passare a quelle ventricolari. COMPLESSI PREMATURI ATRIALI O CPA I complessi prematuri atriali rappresentano l'aritmia più comune. Spesso l'incidenza di CPA aumenta con l'età e con la presenza di cardiopatia organica. I CPA sono asintomatici, anche se alcuni pazienti possono manifestare palpitazioni o l'irregolarità del polso. Diagnosi: La diagnosi ECG dei CPA si basa sull'identificazione di un'onda P che appare prima del battito sinusale anticipato. Le sedi più comuni di origine dei CPA sono gli orifizi della vena cava superiore e delle vene polmonari, il seno coronarico, la cresta terminale, l'anulus delle valvole mitrale e tricuspide e l'appendice atriale destra e sinistra. Il profilo dell'onda P è diverso da quello che si osserva durante il ritmo sinusale. In risposta a un CPA, l'intervallo PR si allunga, anche se i CPA che hanno origine vicino all'area del nodo atrioventricolare possono in effetti dar luogo a un intervallo PR più breve a causa dell'accorciamento del tempo di conduzione atriale verso la giunzione. Un CPA molto precoce può non arrivare a condurre fino al ventricolo e creare un'irregolarità del polso percepibile con una pausa o un "battito perduto". Se il CPA conduce rapidamente attraverso il nodo atrioventricolare, incontrerà un sistema di conduzione in fase di parziale recupero e potrà comparire un complesso QRS con morfologia da blocco di branca destro o sinistro. I CPA hanno la caratteristica di riprogrammare il nodo del seno. La somma risultante dell'intervallo RR pre e post-CPA è inferiore a due intervalli PP sinusale. Terapia: Raramente i CPA richiedono un intervento terapeutico. Si può tentare di sopprimere i CPA con agenti farmacologici. I beta-bloccanti rappresentano la prima azione terapeutica, ma possono esacerbare i sintomi se insieme ai CPA si verifica un blocco atrio-ventricolare e di conseguenza l'irregolarità del polso diventa più intensa. L'impiego di antiaritmici di classe Ic può eliminare i CPA, ma va evitato se è presente una cardiopatia organica. COMPLESSI PREMATURI GIUNZIONALI Sono estremamente rari. Hanno origine dal nodo atrioventricolare e dalle regioni del fascio di His, e possono provocare attivazione atriale retrograda con un'onda P che distorce le porzioni iniziali o terminali del complesso QRS, a livello delle derivazioni II, III e aVF. I pazienti sintomatici possono essere trattati con beta-bloccanti oppure, in assenza di cardiopatia strutturale, con antiaritmici di classe Ic. TACHICARDIA SINUSALE La tachicardia sinusale fisiologica rappresenta una normale risposta a uno stress, ma anche alcune condizioni patologiche sono in grado di determinare tachicardia sinusale. È importante distinguere la tachicardia sinusale dalle altre tachicardie sopraventricolari. La tachicardia sinusale fa assumere all'onda P una forma che riflette la sua origine dal nodo del seno. L'onda P appare rivolta verso l'alto nelle derivazioni II, III e aVF, mentre negativa nella derivazione aVR. In V1, la morfologia dell'onda P assume un profilo bifasico, positivo-negativo. L'esordio della tachicardia sinusale è graduale e in risposta alla pressione sul seno carotideo si può verificare un certo rallentamento, modesto e transitorio, ma non un'interruzione improvvisa. La diagnosi non deve essere fatta sul intervallo PR o sulla presenza di un'onda P prima di ciascun complesso QRS. Il trattamento della tachicardia sinusale fisiologica è diretto alla patologia di base. Raramente si usano i betabloccanti. La tachicardia sinusale inappropriata è una patologia medica in frequente, nella quale la frequenza cardiaca aumenta spontaneamente o in misura sproporzionata rispetto all'entità dello stress o dello sforzo fisiologico. La tachicardia sinusale è spesso accompagnato da vertigini e anche da sincopi franche e da palpitazioni. La sindrome può essere piuttosto invalidante. Sono comuni anche sintomi come dolore toracico, cefalea e malessere gastrointestinale. In molti casi la sindrome si manifesta dopo una malattia virale e tende a risolversi spontaneamente. Anche qui i farmaci di primo utilizzo sono i beta-bloccanti. Nel caso in cui il soggetto si intollerante al betabloccante o non risponde alla terapia, può essere efficace l'ablazione transcatetere, ma dato l'alto tasso di recidive questo rimane un intervento di seconda scelta. RITMI DI RIENTRO Le principali aritmie caudate da rientro sono: - tachicardia da rientro del nodo atriventricolare (AVNRT); - tachicardia da rientro atrioventricolare (AVRT), questa suddivisibile a sua volta in ortodromica e antidromica; - flutter atriale; - fibrillazione atriale; - tachicardia ventricolare. Inizieremo ora a parlare per prima delle tachiaritmie sopraventricolari. FIBRILLAZIONE ATRIALE Una fibrillazione atriale può associarsi anche a 500-600 impulsi al minuto. Se non ci fosse il nodo AV, che rappresenta una barriera per la trasmissione degli impulsi dagli atri ai ventricoli, tale situazione si configurerebbe incompatibile con la vita. L’atrio con un numero così alto di impulsi praticamente sta fermo. Nel caso dell’arresto atriale noi dobbiamo dare l’impulso mentre nella fibrillazione atriale dobbiamo dare una scossa che blocchi e faccia ripartire il nodo sinusale (può anche accadere che dopo la defibrillazione l’impulso non parta più e in quel caso è necessario il pacemaker. Paradossalmente si è ottenuta una situazione in cui non c’è più impulso). Definiamo la fibrillazione atriale anche come aritmia totale. La caratteristica è che gli intervalli R-R sono tutti diversi tra loro e la frequenza al polso non può essere misurata ma va misurata al precordio ( per almeno 30 secondi, ma meglio se per un intero minuto). La fibrillazione atriale può derivare da multiple aree di rientro all'interno dell'atrio o da multipli foci ectopici. In basso viene mostrata un'immagine riguardo un ECG con fibrillazione atriale. I segnali elettrici sono disorganizzati e molto rapidi tra i complessi QRS (onde di fibrillazione). All'ECG l'onda P NON è individuabile e il ritmo è "irregolarmente" irregolare. La fibrillazione atriale è l'aritmia sostenuta più comune. È caratterizzata da attivazione atriale irregolare, rapida e disorganizzata; anche la risposta ventricolare all'attivazione atriale rapida è irregolare. Nel paziente non trattato, anche la frequenza ventricolare tende ad essere rapida ed è interamente dipendente dalle proprietà di conduzione della giunzione atrio-ventricolare. In alcuni pazienti può superare i 200 battiti al minuto, in altri, a causa di un aumento del tono vagale o delle proprietà intrinseche di conduzione del nodo atrio-ventricolare, la risposta ventricolare è inferiore ai 100 battiti al minuto e, occasionalmente, anche profondamente lenta. Il meccanismo di insorgenza e mantenimento della fibrillazione atriale sembra rappresentare un'interazione complessa fra gli impulsi responsabili dell'avvio e il complesso substrato anatomico che favorisce il mantenimento di piccole onde multiple di (micro) rientro. Sembra che gli impulsi abbiano origine soprattutto nella muscolatura atriale che penetra nelle vene polmonari e rappresentino un automatismo anomalo focale o una attivazione triggered modulata in qualche modo da influssi autonomici. La fibrillazione atriale è comune nella popolazione adulta, mentre è estremamente rara nei bambini, a meno che non sia presente una cardiopatia strutturale o un'altra aritmia che scatena la fibrillazione atriale. L'incidenza di fibrillazione atriale aumenta con l'età, cosicché oltre il 5% della popolazione con un'età superiore a settant'anni ha esperienza di aritmia. Poiché molti pazienti con fibrillazione atriale sono asintomatici, si ritiene che l'incidenza globale possa essere maggiore. La fibrillazione atriale acuta è particolarmente frequente durante la fase acuta o di prima convalescenza in seguito a interventi chirurgici maggiori vascolari, addominali e toracici, in cui i meccanismi autonomici o l'irritazione meccanica diretta sono in grado di potenziare l'aritmia. La fibrillazione atriale può anche essere innescata da altre tachicardie sopraventricolari, come la tachicardia rientrante del nodo atrio-ventricolare. Eziologia della fibrillazione atriale: Per quanto riguarda l'eziologia, questa è da ricercarsi in alcune condizioni patologiche che tendono ad alterare la struttura dell'atrio. Le principali cause di fibrillazione atriale sono: - alterazioni strutturali dell'atrio; - fibrosi che si interpone tra le fibre atriali; - fibrosi ed infiltrazione grassa del nodo del seno; - reazione ad un processo infiammatorio o degenerativo; - riscontro di miocardite nel 66% delle biopsia atriali di pazienti con fibrillazione atriale; - ipertrofia dilatazione dell'atrio come causa o conseguenza di fibrillazione atriale persistente. Tuttavia, nella maggior parte dei pazienti non è possibile identificare il processo anatomico sottostante responsabile del aritmia. Valutazione elettrocardiografica della fibrillazione atriale: La fibrillazione atriale è l’aritmia più diffusa. L’ECG di un soggetto affetto da fibrillazione atriale presenta una serie di modifiche patologiche: - per quanto riguarda la frequenza, quell'atriale non è valutabile e quella ventricolare è usualmente intorno ai 120-180 battiti per minuto; - il ritmo ventricolare è irregolare; - per quanto riguarda invece l'onda P, questa può essere assente, e si nota un'attività elettrica caotica o onda di fibrillazione; - l'intervallo QRS può essere normale a meno di conduzione ventricolare aberrante e l'ampiezza dell'onda R varia irregolarmente. Presentazione clinica: L’importanza clinica della fibrillazione atriale è correlata a: - perdita della contrattilità atriale; - risposta ventricolare rapida inappropriata; - perdita di contrattilità e svuotamento dell'appendice atriale con rischio di formazione di coaguli e conseguenti eventi tromboembolici. I sintomi della fibrillazione atriale sono estremamente variabili. Molti pazienti sono asintomatici e non presentano conseguenze emodinamiche evidenti. Altri soggetti hanno solo minime palpitazioni o percepiscono irregolarità del polso; molti altri, invece, hanno palpitazioni intense. L'effetto emodinamico può essere drammatico, in base alle esigenze di contrattilità atriale normale e di risposta ventricolare. In alcuni pazienti l'ipotensione, la congestione polmonare e i sintomi anginosi possono essere gravi. Nei pazienti che presentano disfunzione diastolica del ventricolo sinistro che si verifica in caso di ipertensione, miocardiopatia ipertrofica e valvulopatia aortica ostruttiva, i sintomi possono essere ancora più intensi, soprattutto se la frequenza ventricolare non permette un adeguato riempimento del ventricolo. L'intolleranza allo sforzo e la facilità di affaticamento sono caratteristiche di uno scarso controllo della frequenza durante l'esercizio fisico. Occasionalmente, l'unica manifestazione della fibrillazione atriale è rappresentata da intense vertigini o sincope associate alla pausa che intercorre fra il termine della fibrillazione atriale e la ripresa del ritmo sinusale. Clinicamente, si distinguono tre forme di fibrillazione atriale: - parossistica, caratterizzata da risoluzione spontanea; - persistente, che necessita di cardioversione per la sua interruzione; - permanente, che resiste alla cardioversione farmacologica e/o elettrica, in questi casi può essere presa la decisione di non cardiovertire. Le forme permanenti sono in genere collegate ad alterazioni strutturali: dilatazione atriale. Oggi abbiamo diverse possibilità per il trattamento (antiaritmici, ablazione) della fibrillazione atriale. Tali trattamenti sono tanto più efficaci quanto meno organica è l’eziologia. Con un atrio di 6,5 (v.n. 4 e qualcosa) gli potete fare che cavolo volete voi, quello fibrillante era e fibrillante rimane. Potete solo combattere gli effetti collaterali. Questo è un ECG di fibrillazione atriale. Tutto dipende dall’azione del filtro del nodo atrioventricolare. Gli stimoli arrivano a flotte al nodo AV ma possono passare in varia maniera (tantissimi o molto pochi). Talora la frequenza ventricolare è molto alta potendosi alternare a momenti in cui la frequenza è bassa (intervalli RR maggiormente distanziati). Ricordatevi che l’atrio sta fermo e viene a mancare il contributo atriale del riempimento ventricolare (1/4 circa). Ricordatevi inoltre che si associa a modificazione del flusso e quindi possibilità di formazione di trombi che più probabilmente andranno a localizzarsi a livello delle auricole. Terapia: Il trattamento della fibrillazione atriale deve tener conto della situazione clinica in cui si manifesta l'aritmia, la cronicità della fibrillazione atriale, lo stato della terapia anticoagulante, i fattori di rischio per ictus, la sintomatologia, l'impatto emodinamico da fibrillazione atriale e la frequenza ventricolare. In assenza di una compromissione emodinamica che richieda una cardioversione urgente, gli obiettivi iniziali della terapia consistono in: - stabilire un controllo della frequenza ventricolare; - affrontare il problema della anticoagulazione. Nella fibrillazione atriale, la frequenza ventricolare viene tenuta sotto controllo efficacemente da betabloccanti, i calcio-antagonisti come il verapamil o il diltiazem. La terapia anticoagulante è particolarmente importante nei pazienti con fattori di rischio per ictus associati a fibrillazione atriale. I fattori di rischio più importanti sono anamnesi positiva per ictus, attacco ischemico transitorio o embolia sistemica, oppure presenza di stenosi mitralica reumatica. Altri fattori di rischio ben identificati sono l'età superiore a 65 anni, anamnesi positiva per insufficienza cardiaca congestizia, diabete mellito, ipertensione, disfunzione del ventricolo sinistro e segni evidenti di dilatazione atriale sinistra (superiore ai 5 cm). La terapia anticoagulante cronica con warfarin è raccomandata nei pazienti con fibrillazione atriale parossistica persistente o frequente e di lunga durata e fattori di rischio. Può essere necessario interrompere immediatamente la fibrillazione atriale, sulla base dei parametri clinici e dello Stato emodinamico. La cardioversione transtoracica diretta in corso di anestesia di breve durata è un metodo affidabile per porre fine alla fibrillazione atriale. Una terapia farmacologica per mantenere il ritmo sinusale può essere istituita una volta ristabilito il ritmo, oppure prima della cardioversione. Un singolo episodio di fibrillazione atriale può non richiede alcun intervento, o solo un breve ciclo di terapia con beta-bloccante. Per prevenire la fibrillazione atriale ricorrente che non risponde beta-bloccanti può essere necessaria una terapia antiaritmica, soprattutto se la fibrillazione atriale è associata a frequenza rapide o a sintomi significativi. La scelta degli antiaritmici deve essere diretta soprattutto dalla presenza o assenza di malattia coronarica, depressione della funzione del ventricolo sinistro non imputabile a una miocardiopatia indotta da tachicardia reversibile, e di grave ipertensione con segni di marcata ipertrofia del ventricolo sinistro. La presenza di una cardiopatia organica significativa restringe il campo terapeutico all'uso di sotalolo, amiodarone o dofetilide. Una funzione gravemente depressa del ventricolo sinistro può precludere la terapia con sotalolo, o imporre bassi dosaggi. Nei pazienti senza segni di cardiopatia strutturale o ipertensiva senza segni di grave ipertrofia, sembrano ben tollerati i antiaritmici di classe Ic come flecainide o propafenone, che non presentano rischi proaritmici significativi. Nessun farmaco è uniformemente efficace, e durante il follow-up a lungo termine è prevedibile la recidiva di aritmie in oltre la metà dei pazienti. È inoltre importante osservare che, benché il mantenimento del ritmo sinusale migliori la sopravvivenza a lungo termine, la sopravvivenza dei soggetti randomizzati al mantenimento farmacologico del ritmo sinusale non era superiore a quella dei pazienti trattati con controllo della frequenza e terapia anticoagulante (trial AFFIRM e RACE). Molti dei farmaci usati per il controllo del ritmo favoriscono il rallentamento della conduzione del nodo atrio-ventricolare. L'assenza di sintomi induce spesso a sospendere la terapia anticoagulante, e la fibrillazione atriale asintomatica in assenza di scoagulazione aumenta il rischio di ictus. L'intenzione di sospendere gli anticoagulanti deve sempre, perciò, essere accompagnato da un periodo prolungato di monitoraggio all'ECG. Il controllo della frequente è spesso difficile da ottenere nei soggetti con fibrillazione atriale parossistica. Nei pazienti affetti da forme più persistenti di fibrillazione atriale, è possibile tenere sotto controllo la frequenza con associazioni di farmaci, onde evitare alcuni degli effetti collaterali più comuni osservati in caso di monoterapia ad alte dosi. Nei pazienti con sintomi derivanti da un controllo inadeguato della frequenza con la terapia farmacologica o peggioramento della funzione del ventricolo sinistro dovuto alla tachicardia persistente, è possibile eseguire un'ablazione del fascio di His/giunzione atrio-ventricolare. L'ablazione va accompagnata dall'impianto di un pacemaker. Oltre il trattamento farmacologico, la fibrillazione atriale può essere trattata con della terapia ablativa transcatetere. La maggior parte delle procedure chirurgiche include tecniche che i sono i lembi muscolari atriali che penetrano nelle vene polmonari; questi lembi sono stati identificati come la fonte della maggioranza degli stimoli responsabili dell'avvio della fibrillazione atriale. La terapia ablativa è attualmente un'alternativa alla terapia farmacologica nei pazienti con fibrillazione atriale sintomatica ricorrente. La terapia ablativa transcatetere è promettente anche per i pazienti con forme più persistenti di fibrillazione atriale e per quelli che presentano grave dilatazione atriale. L’ablazione chirurgica della fibrillazione atriale viene eseguita tipicamente nel corso di intervento sulle valvole cardiache o sulle coronarie e più raramente come procedura a sé. La procedura di Cox-Maze è stata concepita per interrompere tutti i circuiti di macrorientro che potrebbero in teoria svilupparsi negli atri, così da precludere la possibilità di fibrillare. Nel tentativo di semplificare l’operazione, le incisioni multiple della procedura tradizionale di Cox-Maze sono state sostituite da tratti lineari di ablazione e isolamento delle vene polmonari con l’impiego di varie fonti di energia. FLUTTER ATRIALE Le aritmie da macrorientro che coinvolgono il miocardio atriale vengono definite collettivamente con il termine di flutter atriale. Il circuito di flutter tipico, il più comune, ruota in senso orario o antiorario nell’atrio destro, intorno all’anulus della tricuspide. Il flutter atriale destro antiorario rappresenta l’80% circa di tutti i flutter atriali con attivazione diretta in senso superiore del setto interatriale, che produce l’aspetto a sega (flutter comune o tipico) dell’onda P nelle derivazioni all’ECG II, III e aVF. La rotazione in senso orario dello stesso circuito atriale destro provoca onde P prevalentemente positive nelle derivazioni II, III e aVF. Il flutter atriale destro classico presenta una frequenza atriale di 260-300 bpm, con una risposta ventricolare che tende ad essere 2:1 o, tipicamente, 130-150 bpm. Nel quadro di una patologia grave della conduzione atriale e con una terapia farmacologica antiaritmica, la frequenza atriale può rallentare sotto i 200 bpm. Molto spesso, il flutter atriale è associato a valvulopatia mitralica o tricuspidale, a cuore polmonare cronico o acuto ed a coronaropatia. Questo è il flutter che ha morfologia a dente di sega. Le onde hanno una loro frequenza,un loro ordine. Il flutter atriale è frequentemente associato a patologie. A differenza di una fibrillazione atriale che si può avere anche perché uno ha fatto una mangiata eccezionale o perche ha fatto sforzi, il flutter è quasi sempre legato ad una patologia. A differenza della fibrillazione atriale possiamo, in questo caso, stabilire la frequenza di contrazione atriale. Caratteristiche all’ECG: Per quanto riguarda il quadro elettrocardiografico, questo è caratterizzato da: - una frequenza atriale compresa tra 220-350 battiti al minuto, mentre la frequenza ventricolare è generalmente intorno a 110-170; - il ritmo atriale è regolare, mentre il ritmo ventricolare è regolare se vi è un blocco atrio-ventricolare costante, diventa irregolare se il blocco atrio-ventricolare è variabile; - è possibile notare onde di flutter, ovvero l'onda P, nelle derivazioni II, III e aVF. Se la conduzione è 2:1 o 1:1 è difficile individuare le onde di flutter, che possono essere messi in evidenza tramite il massaggio del seno carotideo o la somministrazione di adenosina endovena; - l'intervallo PR è normale; - l'intervallo QRS è normale a meno di conduzione ventricolare aberrante. Terapia: A causa della frequenza ventricolare rapida anticipata, associata con il flutter atriale, e della mancata risposta alla terapia farmacologica volta al rallentamento della frequenza ventricolare, i pazienti vengono spesso sottoposti a cardioversione a corrente continua. I soggetti asintomatici con flutter atriale possono andare incontro a sintomi di insufficienza cardiaca, con grave disfunzione del ventricolo sinistro tachicardia-indotta. In tutti i pazienti bisogna sforzarsi di tenere sotto controllo la frequenza ventricolare tramite farmaci o di ripristinare il ritmo sinusale. Può essere difficile normalizzare la frequenza con calcioantagonisti, beta-bloccanti e digossina. Anche un rallentamento ventricolare di grado più elevato può essere transitorio e viene spesso superato con l'attività o uno stress emotivo. A causa della frequenza ventricolare tipicamente più rapida il flutter atriale tende ad essere meno tollerato rispetto la fibrillazione atriale. In pazienti selezionati ad alto rischio per anestesia, è appropriato un tentativo di cardioversione farmacologica con procainamide, amiodarone o ibutilide. La terapia farmacologica antiaritmica può anche aumentare l'efficacia della cardioversione elettrica. I pazienti che presentano flutter atriale ricorrente sembrano rispondere bene alla terapia ablativa transcatetere. Qui in basso viene raffigurato l'algoritmo per il trattamento delle tachiaritmie a complessi stretti. TPVS: TACHICARDIA PAROSSISTICA SOPRAVENTRICOLARE La TPSV è una sindrome clinica, caratterizzata da ripetuti episodi di tachicardia con insorgenza improvvisa e durata da alcuni secondi a molte ore. Qui di seguito viene riportato un tracciato ECG di TPVS con le caratteristiche. ALTRE TACHICARDIE SOPRAVENTRICOLARI Tra le altre tachicardie sopraventricolari, si ricordano: - la tachicardia da rientro nodale; - la tachicardia da rientro AV; - la tachicardia automatica giunzionale. TACHICARDIA RIENTRANTE DEL NODO AV (TRNAV) La tachicardia rientrante del nodo atrioventricolare è la tachicardia sopraventricolare regolare parossistica più comune. Si osserva spesso nelle donne e si manifesta tipicamente nel secondo-quarto decennio di vita. Poiché la TRNAV tende verificarsi in assenza di cardiopatia strutturale, è di solito ben tollerata. In presenza di ipertensione o di altre forme di cardiopatia organica e limitano il riempimento ventricolare, può presentarsi ipotensione o sincope. La TRNAV si sviluppa per la presenza di due vie elettrofisiologiche distinte per la conduzione nel complesso sinciziale di fibre muscolari e compone il nodo atrioventricolare. La via rapida localizzato nella parte più alta del nodo ha un periodo refrattario più lungo, mentre la via più bassa, nella regione del nodo atrioventricolare, conduce più lentamente ma ha un periodo refrattario più breve. In seguito alle disomogeneità di conduzione e refrattarietà, si può sviluppare un circuito rientrante. Benché la conduzione si propaga lungo entrambe le vie nel corso del ritmo sinusale, si manifesta solo la conduzione lungo la via rapida e, di conseguenza, l'intervallo di PR è normale. I CPA che si verificano in un intervallo critico d'accoppiamento vengono bloccati nella via rapida, a causa del periodo refrattario più lungo, e sono condotti lentamente lungo la via lenta. Quando si presenta un rallentamento sufficiente della conduzione, la via rapida bloccata può recuperare l'eccitabilità con conseguente attivazione atriale, così da completare il circuito. L'attivazione ripetitiva verso il basso (via lenta) e verso l'alto (via rapida) dà luogo alla tipica tachicardia rientrante del nodo atrioventricolare. Reperti dell'ECG: Il CPA che dà il via alla TRNAV è caratteristicamente seguito da un lungo intervallo PR, coerente con la conduzione lungo la via lenta. La TRNAV si manifesta tipicamente con una tachicardia con complessi QRS stretti e frequenze fra i 120 e i 250 battiti al minuto. Il pattern QRS-P associato alla TRNAV tipica è peculiare, con attivazione simultanea degli atri e dei ventricoli dal circuito di rientro del nodo atrioventricolare. L'onda P è frequentemente inclusa nel complesso QRS e non è visibile o distorce la porzione iniziale o terminale dello stesso complesso QRS. Poiché l'attivazione atriale ha origine nella regione del nodo atrioventricolare, la depolarizzazione atriale retrograda creerà una deflessione negativa. Trattamento: Il trattamento è volto ad alterare la conduzione nell'ambito del nodo atrioventricolare. La stimolazione vagale può rallentare la conduzione del nodo atrioventricolare in maniera sufficiente a porre fine alla TRNAV. Nei pazienti in cui le manovre fisiche non funzionano, l'obiettivo è spesso raggiunto somministrando adenosina. Come seconda opzione va considerata la terapia endovenosa con beta-bloccanti o calcio-antagonisti. Se è presente compromissione emodinamica una cardioversione può risolvere la tachiaritmia. TACHICARDIE GIUNZIONALI AV Possono verificarsi nel quadro di un aumento del normale automatismo, di un automatismo anomalo o di attività triggered. Queste tachicardie possono o meno essere associate a conduzione retrograda agli atri, e le onde P possono apparire dissociate o provocare conduzione intermittente e attivazione precoce della giunzione. Possono rappresentare un aumento del tono adrenergico o un effetto farmacologico in pazienti con disfunzione del nodo del seno o in seguito ad ablazione chirurgica o transcatetere. L'aritmia può anche costituire un segno di intossicazione da digossina, ma in questo caso non vi è conduzione retrograda. L'attività sinusale può apparire dissociata o dar luogo a battiti intermittenti con un lungo intervallo PR. Se la frequenza e compresa tra i 50 e i 100 battiti al minuto, sia il termine di ritmo giunzionale accelerato. Terapia: Il trattamento è volto a ridurre la stimolazione adrenergica e a contrastare l'intossicazione da digossina, se è presente. La terapia con digossina va sospesa, nel caso di intossicazione bisogna somministrare frammenti di anticorpi digossina-specifici. La tachicardia giunzionale dovuta ad automatismo anomalo può essere trattata farmacologicamente con beta-bloccanti ed è possibile una terapia con farmaci antiaritmici di classe Ia o Ic. È anche possibile eseguire un'ablazione transcatetere, associata però a un aumento del rischio di blocco atrio-ventricolare. TACHICARDIE ASSOCIATE A VIE ACCESSORIE Le tachicardie che coinvolgono vie accessorie fra atri e ventricoli si manifestano comunemente con un complesso QRS normale e con un intervallo RP breve o lungo. La maggior parte delle tachicardie associate a vie accessorie implica la presenza di un ampio circuito macrorientrante e comprende i ventricoli. Le vie accessorie sono tipicamente in grado di condurre rapidamente sia in direzione anterograda che retrograda. Normalmente, l'impulso sinusale attiva i ventricoli tramite il nodo atrioventricolare e il sistema di His-Purkinje, dando luogo a un intervallo PR compreso tra 120 e 200 ms. Quando è presente una via accessoria anterograda, l'impulso scavalca il nodo atrioventricolare e attiva direttamente i ventricoli, dando luogo a pre-eccitazione ventricolare; l'intervallo PR, in questo caso, è più breve. Inoltre, poiché l'attivazione ventricolare iniziale è dovuta a conduzione da muscolo a muscolo, la porzione iniziale del complesso QRS è imperfetta e crea la caratteristica "onda delta". La rimanente parte del complesso, nel ritmo sinusale, è formata da una funzione del fronte d'onda dell'attivazione ventricolare che origina dalla rete di Purkinje e dalla propagazione continua di attivazione dalla sede di insorgenza della via accessoria. La pre-eccitazione ventricolare si manifesta, nel ritmo sinusale, con un intervallo PR breve e l'onda delta. La via accessoria più comune collega l'atrio sinistro al ventricolo sinistro; al secondo posto c'è la via accessoria del setto posteriore, seguita da quelle della parete libera destra e del setto anteriore. Le vie accessorie si inseriscono tipicamente dall’atrio nel miocardio ventricolare adiacente, tuttavia, alcuni vie possono presentare un'inserzione ventricolare in una sede distante dal nodo atrioventricolare, nei fascicoli. Queste vie sono molto lunghe e conducono più lentamente e pertanto vengono definite vie accessorie atriofascicolari. Possono esistere anche altri vie accessorie dal nodo atrio-ventricolare ai fascicoli e vengono definite fibre di Mahaim. I pazienti con pre-eccitazione manifesta e sindrome di WPW sono tipicamente soggetti sia a tachicardie macrorientranti, sia a una risposta rapida alla fibrillazione atriale. La tachicardia macrorientrante più comunemente associata alla sindrome di WPW è definita rientro atrio-ventricolare ortodromico. L'attivazione ventricolare si verifica attraverso il nodo atrioventricolare e il fascio comune. Quindi, la conduzione torna o rientra agli atri lungo la via accessoria. Il circuito rientrante si sviluppa a causa della disomogeneità di conduzione e della refrattarietà a livello della via accessoria e del nodo atrioventricolare normale. La via accessoria presenta una conduzione più rapida ma un periodo refrattario più lungo. Un CPA può causare un blocco nella via accessoria e condurre in maniera sufficientemente lenta o calante attraverso il nodo atrioventricolare per permettere un recupero retrogrado dell'attivazione della via accessoria e, in seguito, degli atri. Questa attivazione retrograda degli atri attraverso la via accessoria è definita "battito eco". Se il pattern si ripete, si sviluppa una tachicardia. Raramente, il circuito rientrante può essere invertito e in questo caso si parla di rientro atrio-ventricolare antidromico e/o macrorientro pre-eccitato, in cui l'intera attivazione del ventricolo ha origine dalla sede di inserzione della via accessoria. Meno frequenti sono le tachicardie sopraventricolari antidromiche. La seconda aritmia più comune, e più grave, associata alla sindrome di WPW, è la fibrillazione atriale a conduzione rapida. Nei soggetti che presentano conduzione anterograda rapida dagli altri ai ventricoli lungo la via accessoria, questa può condurre rapidamente in risposta alla fibrillazione atriale, dando origine a una frequenza ventricolare più rapida di quella che si avrebbe normalmente attraverso il nodo atrioventricolare. Le frequenze ventricolari rapide possono determinare problemi emodinamici e perfino scatenare fibrillazione ventricolare. Nei pazienti con pre-eccitazione manifesta, il complesso QRS durante la fibrillazione atriale può apparire bizzarro e cambiare di battiti in battito, a causa della variabilità del grado di fusione dell'attivazione del nodo atrioventricolare. Vie accessorie occulte: Nel 50% circa dei pazienti con vie accessorie, non vi è conduzione anterograda lungo di esse; tuttavia, la conduzione retrograda è conservata. Quindi, la via accessoria non è palese nel ritmo sinusale e lo è solo durante la tachicardia sostenuta. La presenza di una via accessoria occulta è suggerita dalla timing e dal pattern dell'attivazione atriale durante la tachicardia: l'onda P fa seguito all'attivazione ventricolare dopo un breve intervallo RP. Poiché molte vie accessorie connettono il ventricolo sinistro all'atrio sinistro, il pattern di attivazione atriale nel corso della tachicardia spesso produce onde P negative nelle derivazioni I e aVL. Occasionalmente, le vie accessorie conducono in maniera estremamente lenta in direzione retrograda, dando luogo a conduzione retrograda più lunga e a sviluppo di un lungo intervallo RP nel corso della tachicardia (tachicardia con RP lungo). Per la presenza di questa conduzione drammaticamente rallentata, non è necessario un ulteriore rallentamento della conduzione causato da complessi prematuri atriali affinché si presenti la tachicardia. Questi pazienti sono più soggetti ad episodi frequenti di tachicardia e possono manifestare tachicardie incessanti e miocardiopatia del ventricolo sinistro tachicardia-indotta. La diagnosi corretta può essere suggerita dal pattern di inizio e dalla morfologia dell'onda P. Spesso, comunque, per confermare la diagnosi è necessaria un'analisi elettrofisiologica. Terapia: Il trattamento acuto delle tachicardie ortodromiche macrorientranti mediate da vie accessorie è simile a quello delle rientro nodale atrio-ventricolare ed è volto ad alterare la conduzione del nodo atrioventricolare. La stimolazione vagale con la manovra di Valsalva e la pressione sul seno carotideo possono indurre un rallentamento nodale atrio-ventricolare sufficiente a porre fine alla tachicardia da rientro atrio-ventricolare. La terapia farmacologica di prima scelta consiste nella somministrazione endovenosa di adenosina, ma possono risultare efficaci anche i calcio-antagonisti o i beta-bloccanti, sempre per via endovenosa. Nei pazienti con pre-eccitazione e fibrillazione atriale, la terapia dovrebbe tendere alla prevenzione di una risposta rapida ventricolare. In questo caso può essere utilizzata la cardioversione. La somministrazione cronica per via orale di beta-bloccanti e calcio-antagonisti può essere utilizzata per prevenire le tachicardie rientranti sopraventricolari ricorrenti associate a vie accessorie. I pazienti con anamnesi positiva per episodi sintomatici ricorrenti di tachicardia sopraventricolare, e frequenza superiore ai 200 battiti al minuto, devono essere candidati all’ablazione transcatetere, come pure i soggetti che presentano conduzione anterograda rapida. L'ablazione delle vie accessorie para-hissiane è accompagnata dal rischio di blocco cardiaco, mentre l'ablazione dell'atrio sinistro è associata a un rischio basso ma ben definito di fenomeni tromboembolici. I soggetti che mostrano segni di pre-eccittazione ventricolare in assenza di qualsiasi indizio precedente di aritmia meritano un'attenzione particolare e dovrebbero essere informati sui rischi che corrono e sulle opzioni terapeutiche, in previsione di un evento aritmico. TACHIARITMIE VENTRICOLARI Passeremo adesso a trattare delle principali tachiaritmie ventricolari. COMPLESSI VENTRICOLARI PREMATURI O CVP Un complesso ventricolare prematuro è una depolarizzazione precoce che origina da uno dei ventricoli, precedendo il battito sinusale atteso. I due ventricoli non vengono attivati contemporaneamente. L'origine dei battiti prematuri ventricolari in sedi lontane dalle fibre di Purkinje provoca una attivazione ventricolare lenta e un complesso QRS largo, tipicamente della durata su di periore ai 140 ms. I CVP sono comuni e aumentano con l'età e con la presenza di cardiopatia strutturale; possono verificarsi con una certa periodicità. I CVP possono presentare pattern bigemini, in cui ogni battito sinusale è seguito da un CVP, o trigemini, in cui due battiti sinusali sono seguiti da un CVP. Possono avere morfologie differenti e sono quindi definiti multiformi. Due CVP successivi sono denominati paia o coppie. Tre o più CVP consecutivi vengono definiti tachicardia ventricolare se la frequenza è superiore ai 100 battiti al minuto. Se i CVP ripetitivi terminano spontaneamente e durano più di tre battiti, l'aritmia è denominata tachicardia ventricolare non sostenuta. Per quanto riguarda l'ECG, tipicamente si verifica un allungamento del QRS, che assumono una forma bizzarra e anche la sequenza di ripolarizzazione è alterata, con il segmento ST e onda P con direzione opposta al complesso QRS. I CVP sono associati a una pausa pienamente compensatoria; in pratica, il periodo fra l'ultimo QRS prima del CVP e il complesso QRS successivo è il doppio della frequenza sinusale. Nella pagina precedente, viene rappresentato un classico ECG dovuto a complessi ventricolari prematuri. Se un CVP cade sull'onda T può determinare la comparsa di tachicardia ventricolare o fibrillazione ventricolare. Alle volte, anche i CVP e cadono dopo l'onda T possono determinare tachicardia ventricolare. Una pausa compensatoria è la pausa successiva al battito precoce. Come facciamo a stabilire che l’extrasistole è sicuramente ventricolare? Attraverso la pausa compensatoria, che può essere decalante o realmente compensatoria. Si misura la distanza tra la R sinusale che precede il complesso prematuro e quella successiva. Se questo intervallo di tempo è esattamente il doppio dell’intervallo R-R dell’ECG di quel paziente, quella è un’extrasistole, è ventricolare (indipendentemente da come appare). Se è inferiore al doppio dell’intervallo R-R allora l’extrasistole è giunzionale o atriale. Adesso, siccome la morfologia delle extrasistoli ventricolari è la stessa della tachicardia atriale, come facciamo a distinguerle? Allorquando siano presenti 6 o più complessi ventricolari prematuri in successione si parla di tachicardia ventricolare. Tra tachicardia ventricolare e tachicardia ventricolare sostenuta c’è una differenza. TV sostenuta significa che il ventricolo si contrae male, si contrae un numero eccessivo di volte, la diastole è più corta e la perfusione coronarica, che come sapete si realizza fondamentalmente in diastole, è ridotta. Per cui se sentite parlare di TV sostenuta sappiate che non è una cosa buona. Prima, tale condizione, si interrompe e meglio è. Se un complesso ventricolare cade sull’onda T, siccome si è realizzato mentre ancora sta finendo il fenomeno precedente, quella singola extrasistole può essere l’innesto di una tachicardia ventricolare, o peggio, di una fibrillazione ventricolare. Mentre la tachicardia ventricolare sono tante extrasistoli l’una appresso all’altra, la fibrillazione ventricolare si caratterizza con onde irregolari che possono assumere l’aspetto di una fisarmonica. Possiamo distinguere i complessi ventricolari prematuri in monomorfi e polimorfi. Una extrasistole monomorfa più difficilmente potrà trasformarsi in TV, TV sostenuta o fibrillazione ventricolare, perché è molto più facile che sia espressione di una sofferenza transitoria e di tipo funzionale. Quelle monomorfe sono dovute alla rientro dallo stesso focus e l'intervallo tra il precedente battito sinusale e la PVC (intervallo di accoppiamento) è costante. Extrasistoli ventricolari frequenti e polimorfe correlano più spesso con un substrato organico (ischemia, infiammazione, infiltrazione, etc…) e più spesso si complicano. Gli intervalli di accoppiamento e la morfologia del complesso QRS sono variabili, originando da siti diversi, oppure originano dallo stesso sito con conduzione ventricolare variabile. A seconda della periodicità tra battiti sinusale e complessi prematuri ventricolari si potranno osservare fenomeni di bigeminismo, trigeminismo e quadrigeminismo. Di seguito verranno mostrati vari ECG riguardo a tale fenomeno. Bigeminismo: dopo ogni battito regolare è presente una PVC. Trigeminismo: dopo ogni due battiti regolari è presente una PVC. Quadrigeminismo: dopo tre battiti regolari, il quarto è una PVC. Criteri elettrocardiografici: All’ECG saranno presenti una serie di alterazioni caratteristiche, che facilitano il riconoscimento di tale anomalia. Le principali alterazioni consistono in: - il complesso QRS non ha una normale morfologia e la durata è superiore a 0,12 secondi; - per quanto riguarda l'onda P, questa usualmente è nascosta dal complesso QRS, dal segmento ST o dall'onda T della PVC. Terapia: La soglia per il trattamento dei complessi prematuri ventricolari è elevata, e il trattamento è principalmente volto a eliminare i sintomi gravi associati a palpitazioni. CPV di frequenza sufficiente possono causare una miocardiopatia reversibile. Nei pazienti con cardiopatia strutturale, frequenti CPV e salve di tachicardia ventricolare non sostenuta hanno significato prognostico e possono comportare un aumento del rischio di morte cardiaca improvvisa. Le terapie che rallentano la conduzione miocardica o aumentano la dispersione o la refrattarietà possono aumentare il rischio di aritmie potenzialmente letali, nonostante la loro efficacia nell'eliminare i CPV. RITMO IDIOVENTRICOLARE ACCELERATO Il termine ritmo idioventricolare accelerato (RIVA) si riferisce a un ritmo caratterizzato da tre o più complessi a frequenza compresa tra 40-120 battiti al minuto. Si ritiene che il meccanismo aritmico causa di ciò sia un automatismo anomalo. Siccome il RIVA tende a essere benigno, con implicazioni terapeutiche differenti, è opportuno tentare di distinguerlo dalla tachicardia ventricolare lenta. Il RIVA presenta caratteristicamente esordio ed esaurimento graduali, e maggiore variabilità di lunghezza del ciclo. È tipicamente un'aritmia breve e autolimitante. Si può osservare in assenza di una cardiopatia strutturale, ma è spesso presente nei quadri clinici dell’infarto miocardico acuto, dell'intossicazione da cocaina, della miocardica acuta, dell'intossicazione da digossina e nel periodo successivo a un intervento cardiochirurgico. Nel RIVA sostenuto può verificarsi una compromissione emodinamica a causa della perdita di sincronia atrio-ventricolare. TACHICARDIA VENTRICOALRE La tachicardia ventricolare ha origine al di sotto del fascio di His, alla frequenza superiore ai 100 battiti al minuto. A causa della sovrapposizione delle frequenze con il RIVA, le caratteristiche all’ECG dell’aritmia e le circostanze cliniche possono talvolta essere utilizzate per distinguere le due forme di tachicardia. La TV sostenuta lenta ha minori probabilità di evidenziare un marcato riscaldamento della frequenza e le notevoli oscillazioni della lunghezza del ciclo che si osservano nel RIVA, e si verifica più facilmente in un quadro d’infarto cronico o di miocardiopatia, mentre è più rara nell’infarto acuto o nella miocardite. Nelle forme tipiche, la TV lenta si avvia con una stimolazione programmata e rappresenta un ampio circuito macrorientrante in un miocardio con patologia cronica, in grado di sostenere una conduzione marcatamente lenta. Durante la TV, il complesso QRS può essere uniforme (monomorfico) o può variare da battito a battito (polimorfico). La TV polimorfica nei pazienti che mostrano un intervallo QT lungo nel ritmo basale è tipicamente definita TDP (torsades des pointes). Le conseguenze emodinamiche dipendono dalla presenza o meno di disfunzione miocardica e dalla frequenza della TV. La TV monomorfica suggerisce la presenza di un focolaio tachicardico stabile in assenza di cardiopatia strutturale, o un substrato anatomico fisso che può fornire la base per un circuito TV rientrante stabile quando è presente una malattia strutturale. La TV monomorfica tende a rappresentare un fenomeno riproducibile e ricorrente e può essere indotta dal pacing e dalla stimolazione ventricolare programmata. La TV polimorfica suggerisce un processo più dinamico e/o instabile e, per la sua stessa natura, è meno riproducibile. La TV polimorfica può essere causata da ischemia acuta, miocardite o alterazioni dinamiche dell’intervallo QT e aumento della dispersione della refrattarietà ventricolare. Una durata di 30 secondi è spesso considerata la soglia per distinguere la TV sostenuta da quella non sostenuta. Anche una TV emodinamicamente instabile, che richiede di essere interrotta prima di 30 secondi o una TV che viene interrotta mediante terapia con defibrillatore impiantabile è classificata come sostenuta. Il flutter ventricolare appare come un’assenza di onde sull’ECG e presenta una frequenza superiore ai 250 battiti al minuto. La fibrillazione ventricolare è caratterizzata da attivazione ventricolare completamente disorganizzata visibile sull’ECG di superficie. Aritmie ventricolari polimorfiche, flutter ventricolare e fibrillazione ventricolare provocano sempre collasso emodinamico, se lasciate proseguire. La stabilità emodinamica di una TV monomorfica dipende dalla presenza e gravità della cardiopatia strutturale di base, dalla localizzazione della sede di origine dell’aritmia e dalla frequenza cardiaca. Il tracciato ECG a 12 derivazioni sinusale o di base fornisce importanti informazioni che aiutano a formulare la diagnosi corretta di una tachicardia con complessi larghi. La presenza di complessi QRS aberranti, che coincidono esattamente con quelli del ritmo a complessi larghi avvalora la diagnosi di TSV. Un pattern QRS da blocco di branca destro o sinistro che non coincide con il QRS e/o che è di durata più lunga rispetto al QRS nel corso della tachicardia a complessi larghi conferma la diagnosi di TV. La maggior parte dei pazienti con TV è affetta da cardiopatia strutturale e mostra segni di infarto miocardico pregresso con onda Q durante il ritmo sinusale. La presenza di un pattern QRS pre-eccitato sull’ECG a 12 derivazioni durante il ritmo sinusale indica che il ritmo a complessi larghi rappresenta un’aritmia atriale, con flutter atriale o tachicardia atriale focale, con conduzione rapida lungo una via accessoria o tachicardia macrorientrante antidromica. Se l’aritmia è irregolare, con variabilità dei complessi QRS, va considerata l’ipotesi diagnostica di una FA con pre-eccitazione ventricolare. La maggior parte delle TV non risponde alla stimolazione vagale provocata da massaggio del seno carotideo, manovra di Valsalva o somministrazione di adenosina. La somministrazione endovenosa di verapamil e/o adenosina non è consigliata. Il verapamil è causa di collasso emodinamico. I pazienti con TV mostrano spesso disaccoppiamento AV. All’esame obiettivo, il riscontro di onde a giganti intermittenti e variabilità del primo tono cardiaco è compatibile con la dissociazione AV, caratteristicamente indicata dalla presenza di cattura sinusale o battiti di fusione. Una conduzione ventricolo-atriale 1:1 non esclude una diagnosi di TV. Criteri ECG: All’ECG la TV presenta le seguenti caratteristiche: - frequenza superiore a 100 bpm e inferiore a 200 bpm; - ritmo generalmente regolare; - onda P, nella TV rapida non visibile, mentre in quella lenta le onde P possono essere visibili, ma l’attività elettrica atriale e quella ventricolare sono completamente separate; - L’ampiezza del QRS è superiore a 0,12 secondi, con morfologia bizzarra con incisura; - Segmento ST e onda T con polarità opposta al QRS. Quando è multifocale l’intervallo di accoppiamento e la morfologia del QRS variano. In basso viene mostrato un classico ECG, tipico della TV. Trattamento: Cosa dobbiamo fare in un paziente con tachicardia ventricolare senza polso. La prima cosa è il pugno precordiale, che risulta essere una sorta di defibrillazione meccanica. Se non c’è un monitoraggio, il pugno precordiale e il massaggio cardiaco servono nelle manovre di rianimazione, in attesa dell’attivazione del defibrillatore. Solo il defibrillatore potrà fare la differenza. Ormai i defibrillatori sono dappertutto. Si defibrilla fino a tre volte aumentando il carico in Joule della scarica che daremo. La sigla RCP (rianimazione cardio-polmonare) consiste nella respirazione assista con massaggio cardiaco. L’accesso venoso ci serve per somministrare adrenalina che comunque rappresenta uno stimolo alla ripresa contrattile. Se al termine di tutti i passaggi, definiti dall’algoritmo di intervento, la fibrillazione non si è risolta, è probabile che siamo di fronte ad una situazione irrisolvibile (occlusione tronco comune in paziente che aveva la destra già malata, rottura di cuore, infarto in paziente poli-infartuato, etc…). Allora quando fermarsi? Sappiamo che il paziente resuscitato, perché parliamo di questo, “resuscitation”, ha un destino molto peggiore di uno che non ha mai avuto bisogno di resuscitazione. Un infarto resuscitato “va peggio” di uno non resuscitato. Quindi un soggetto del genere rappresenta sempre un paziente critico. Il paziente resuscitato è un cerino che in qualunque momento si può riaccendere e bruciare. Questo è invece lo schema di trattamento della tachicardia ventricolare a complessi larghi. FIBRILLAZIONE VENTRICOLARE La fibrillazione ventricolare è l’anarchia totale ed è il ritmo più importante che l’operatore dell’emergenza deve riconoscere. Il più comune meccanismo di arresto cardiaco dopo l’infarto o l’ischemia miocardica è rappresentato appunto dalla fibrillazione ventricolare. In corso di fibrillazione ventricolare vi sono multiple aree del ventricolo che presentano notevoli variazioni di depolarizzazione e ripolarizzazione. Per questo motivo la gittata cardiaca è pressoché nulla e il polso non è rilevabile. Si distinguono due tipi di fibrillazione ventricolare: - FV a onde ampie, di recente insorgenza e risolvibile con defibrillazione precoce; - FV a onde lenti, di insorgenza non recente la cui risoluzione con la cardioversione è meno probabile. ECG: In corso di FV, l’ECG presenta le seguenti caratteristiche: - Assenza di QRS normali; - La frequenza è troppo rapida per poter essere calcolata; - Il ritmo è irregolare, i complessi elettrici variano per forma e dimensione. Non ci sono complessi QRS, tratto ST, onda P e T sono assenti. In basso viene mostrato un caratteristico ECG di tale situazione. “Torsione di punta” vuol dire che il complesso ventricolare che era molto alto prima si riduce poi dopo si inverte per poi crescere ma non con deflessione positiva bensì con deflessione negativa. La fibrillazione ventricolare che si manifesta con torsione di punta è ancora peggio della sola fibrillazione ventricolare da un punto di vista prognostico. ASISTOLIA L’asistolia è la totale assenza di attività elettrica ventricolare, quindi non vi è contrazione ventricolare. Può verificarsi come primo evento in un arresto cardiaco o può seguire una FV. La diagnosi differenziale va posta tra FV ad onde fini e asistolia senza battiti di scappamento. All’ECG non sarà rinvenibile attività ventricolare, vi saranno onde P occasionali e QRS di scappamento ventricolare (battiti agonici). In basso viene mostrato un ECG. RITMI DEI BLOCCHI DI CONDUZIONE Struttura e fisiologia del nodo AV: L’asse di conduzione AV è strutturalmente complesso. Il nodo AV è una struttura sub endocardica che ha origine nella zona di transizione, composta da aggregati di cellule situati nella parte postero-inferiore dell’atrio destro. I fasci atrio nodali transazionali superiore, mediale e posteriore convergono sul nodo AV compatto. Questi ha dimensioni di 1 x 3 x 5 mm, è situato all’apice del triangolo di Koch, che è limitato posteriormente dall’ostio del seno coronarico. Anteriormente dall’anulus della tricuspide e superiormente dal tendine del Todaro. Il nodo AV compatto prosegue come fascio AV penetrante dove questo attraversa immediatamente il corpo fibroso centrale e si trova in stretta prossimità con gli anulus delle valvole aortica, mitrale e tricuspide. Pertanto è soggetto a lesioni in caso di cardiopatia valvolare o della sua correzione chirurgica. Il fascio AV penetrante continua attraverso l’anulus fibroso ed emerge nel setto interventricolare in una zona adiacente al setto membranoso, come fascio di His. La branca destra emerge dal fascio AV distale formando una banda che attraversa il ventricolo destro (banda moderatrice). Al contrario, la branca sinistra è costituita da un’ampia lamina sub endocardica di tessuto sul lato settale del ventricolo sinistro. La rete delle fibre di Purkinje emerge dalle branche destra e sinistra e si ramifica estesamente sulle superfici endocardiche rispettivamente dei ventricoli destro e sinistro. L’apporto ematico al fascio AV penetrante è fornito dall’arteria del nodo AV e dal primo ramo settale perforante della coronaria discendente anteriore sinistra. Il nodo AV è abbondantemente innervato da nervi parasimpatici e simpatici post-gangliari. Il fascio di His e il sistema di conduzione distale sono influenzati dal tono autonomino solo in minima misura. Le cellule che compongono il complesso del nodo AV sono eterogenee presentando variazioni nelle caratteristiche dei potenziali di azione. I miociti che compongono il nodo compatto sono depolarizzati, presentano potenziali di azione con scarsa ampiezza, lenta salita della fase 0 e della depolarizzazione diastolica di fase 4, resistenza a impulsi elevati e relativa insensibilità alla concentrazione di potassio extracellulare. Classificazione dei blocchi: I blocchi AV sono più comuni rispetto a quelli del seno-atriale. I blocchi AV vengono a loro volta suddivisi in categorie, che vengono riassunte nell’immagine successiva. Allora, i blocchi AV vengono suddivisi in: - il blocco AV di primo grado è caratterizzato solo dall’aumento dell’intervallo PR. Normalmente tale intervallo dovrebbe essere 0.2 s; - Blocco AV di secondo grado: l’intervallo PR aumenta progressivamente fino a che compare una P non seguita da un complesso rapido ventricolare. Questo si chiama blocco AV di secondo grado Mobitz tipo I con periodismo di Luciani-Wenckeback. Voi capite bene che se la P ci mette dieci battiti per bloccarsi è un conto, se ci mette cinque battiti è un altro conto, se ce ne mette tre è un altro conto ancora. Questo (guarda immagine) invece è il Mobitz di tipo II. L’intervallo PQ si è allungato ma è sempre uguale dopo di che compare un’onda P non seguita da un complesso QRS. Anche questo è un blocco AV di secondo grado, ma in questo caso ci sono due P che non sono seguite da un complesso QRS.; - Blocco totale o AV di terzo grado, le onde P se ne vanno per fatti loro ma ci sono e i complessi QRS, ovviamente dissociati dall’onda P, perché lo stimolo non è mai passato dall’atrio al ventricolo e quindi si è dovuto creare un centro ectopico ventricolare per cui necessariamente deve avere la morfologia del blocco di branca. Qui tutto dipende dal ventricolo e in particolare da dove il centro ectopico è localizzato. Mano mano che vi allontanate dal fascio di His diminuisce la frequenza per cui, se il centro ectopico è localizzato a livello delle fibre del Purkinje la frequenza sarà di 20 bpm. Quindi per blocco AV intenderemo il ritardo o l’interruzione della conduzione dell’impulso dall’atrio ai ventricoli. Eziologia della patologia della conduzione AV: Un blocco della conduzione dall’atrio al ventricolo può verificarsi per diverse ragioni e il blocco di conduzione AV può essere classificata in vari modi. Le eziologie possono essere funzionali od organiche, in parte simili alle cause intrinseche ed estrinseche della disfunzione del nodo senoatriale. Il blocco può essere classificato, sulla base della gravità, come blocco di primo, secondo, terzo grado o blocco atrioventricolare completo, oppure, sulla base della localizzazione, nell'ambito del sistema di conduzione atrio-ventricolare. Un innalzamento del tono vagale durante il sonno o in soggetti ben allenati può essere accompagnato da tutti i gradi del blocco. L'ipersensibilità del seno carotideo, la sincope vaso-vagale e la sindrome da tosse e minzione possono essere associate a rallentamento del nodo atrioventricolare e blocco della conduzione. Anche un certo numero di alterazioni transitorie metaboliche ed endocrine può provocare blocco di conduzione atrio-ventricolare reversibile. Numerose malattie infettive presenta inoltre un'affinità per il sistema di conduzione (malattia di Lyme e di Chagas). Alcune malattie autoimmuni ed infiltrative possono provocare blocchi atrioventricolari. La fibrosi progressiva idiopatica del sistema di conduzione è una delle cause più comuni e degenerative del blocco di conduzione atrio-ventricolare. L'invecchiamento è accompagnato da alterazioni degenerative dell'estremità superiore del setto interventricolare, del corpo fibroso centrale e degli anulus aortico e mitralico, descritte come "sclerosi dello scheletro cardiaco sinistro". Il blocco di conduzione atrio-ventricolare è anche correlato ad alcune malattie neuromuscolari ereditarie. Un blocco atrio-ventricolare congenito può essere osservato in caso di anomalie cardiache congenite complesse. Un blocco atrio-ventricolare iatrogeno può verificarsi in corso di interventi chirurgici sulla valvola mitrale o aortica, raramente in caso di irradiazione toracica e come conseguenza di ablazione transcatetere. Il blocco atrio-ventricolare è una complicanza decisamente rara della correzione chirurgica di difetti del setto interatriale o interventricolare, ma può complicare gli interventi di Fontan o Mustard per la correzione della trasposizione dei grandi vasi. La malattia coronarica può provocare blocco atrio-ventricolare transitorio o persistente. Il blocco atrioventricolare di secondo grado o di grado più elevato tende a verificarsi più spesso nel infarto miocardico inferiore che in quello anteriore; tuttavia, nell’infarto miocardico inferiore il blocco è di solito a livello del nodo atrioventricolare con ritmi di scappamento più stabili e limitati. L'infarto miocardico anteriore acuto è accompagnato da blocco a livello del complesso nodale atrioventricolare distale, del fascio di His o delle branche, e da luogo a ritmi di scappamento instabili, ampi e complessi; la prognosi è più sfavorevole e la mortalità è alta. Elettrocardiografia ed elettrofisiologia del blocco di conduzione atrio-ventricolare: Il blocco di conduzione atrio-ventricolare viene tipicamente diagnosticato tramite l'ECG. Ed è appunto in base all'ECG che si riesce a differenziare tra i vari blocchi. Il blocco atrio-ventricolare di primo grado è caratterizzato dal ritardo di passaggio dell'impulso dall'atrio ai ventricoli. In basso viene riportato un ECG di BAV di primo grado. Si può notare, come l'intervallo PR è superiore ai 200 ms. Nelle forme tipiche la sede del ritardo si trova nel nodo atrioventricolare, ma può essere situata anche negli atri, nel fascio di His o nel sistema His-Purkinje; un complesso QRS ampio favorisce la diagnosi di alterata conduzione distale, mentre un complesso QRS stretto ritarda a livello del nodo o, più raramente, nel fascio di His. Nel blocco atrio-ventricolare di secondo grado si osserva una assenza intermittente della conduzione dell'impulso elettrico dall'atrio al ventricolo. Il blocco atrio-ventricolare di secondo grado è ulteriormente suddiviso in due sottocategorie: - Mobitz tipo I o Wenckebach; - Mobitz tipo II. Nel blocco atrio-ventricolare di secondo grado, tipo uno, vi è il ritardo di passaggio dell'impulso dal ventricoli con un certo periodismo. Alcuni impulsi sono condotti, altri invece sono bloccati. Da un punto di vista elettrocardiografico questo blocco è caratterizzato da un progressivo allungamento dell'intervallo PR, finché un impulso resta completamente bloccato. Inoltre si verifica una deviazione dell'intervallo RR e una pausa inferiore al doppio dell'intervallo RR immediatamente precedente. L'ECG dopo la pausa evidenzia un intervallo più breve di quello immediatamente precedente. Tale aspetto dell'ECG insorge soprattutto a causa della conduzione difettosa degli impulsi elettrici a livello del nodo atrioventricolare. Di seguito viene mostrato un ECG tipico di tale situazione. È molto importante distinguere il blocco atrio-ventricolare di secondo grado tipo uno da quello di tipo due, poiché quest'ultimo ha implicazioni prognostiche più serie. Nel blocco atrio-ventricolare di secondo grado tipo due, viene un ritardo di passaggio dell'impulso dall'atrio e ventricoli. Di conseguenza alcuni impulsi sono condotti ed altri vengono bloccati. Di seguito viene mostrato l'ECG. Questo tipo di blocco è caratterizzato da assenza intermittente della conduzione dell'onda P senza alterazioni degli intervalli PR o RR precedenti. Questo tipo di blocco si verifica soprattutto nei sistema di conduzione distale ed è spesso associato a ritardi della conduzione intraventricolare e ha maggiori probabilità di progredire verso blocchi atrioventricolari di grado più elevato. In particolare il tipo due può essere associato a una serie di onde P non condotte, definite blocco atrio-ventricolare parossistico, e denota una patologia significativa del sistema di conduzione. Nel blocco atrio-ventricolare di terzo grado nessun impulso e condotto ai ventricoli. Questo tipo di blocco è quasi sempre distale rispetto al nodo atrioventricolare e la durata del complesso QRS può dare indicazioni utili per determinare il livello del blocco stesso. Di seguito viene mostrato l'ECG di tale situazione. Molto importante è il concetto che le anomalie del complesso QRS possono essere associate alla sede dove viene blocco dell'impulso. Se la sede del blocco dell'impulso è prossimale (intra-nodale), i complessi QRS saranno stretti, il massaggio del seno carotideo porterà ad un peggioramento della conduzione mentre la somministrazione di Atropina tenderà a migliorarla. Se la sede del blocco è localizzata in una porzione distale, all'interno del fascio di His, i complessi QRS saranno stretti, il massaggio del seno carotideo può determinare nessun effetto o miglioramento della conduzione, mentre la somministrazione di Atropina può determinare nessun effetto o peggioramento della conduzione. Per i blocchi che si trovano in sede distale (sottohissiano) valgono gli stessi principi esposti per i blocchi del fascio di His, con l'eccezione che in questi casi si avranno complessi QRS larghi. Trattamento: Dopo aver escluso la presenza di eventuali cause organiche, i blocchi vengono trattati con impianto di un pacemaker. Attenzione ragazzi, qual è l’indicazione al pacemaker? L’indicazione al pacemaker non si ha per i primi due tipi di blocco e ciò vale particolarmente per il blocco di tipo II, il periodismo di L-W, che è in genere un fenomeno transeunte talora secondario all’uso di farmaci (antiaritmici) utilizzati per contrastare un’altra situazione. Negli altri casi, invece, pur potendosi fare la osservazione o l’elettrostimolazione temporanea con pacemaker esterno e vedere “come va”, in 9 pazienti su 10 deve essere applicato un pacemaker. La maggior parte degli stimolatori oggi in uso sono a domanda: si attivano quando la frequenza del paziente scende al di sotto di un limite Quindi, prefissato. un tracciato normale può presentare sia battiti sinusale, sia battiti da pacemaker, come mostrato nell'immagine. BASI PER IDENTIFICARE UN’ARITMIA SCONOSCIUTA Molto spesso il paziente può presentare la rapida insorgenza di un’aritmia, oppure averla già ma essere del tutto asintomatica. In questi casi spetta al medico cercare di identificare il tipo di aritmia e valutarne la gravità. Innanzitutto, la prima osservazione da fare riguarda la frequenza cardiaca, se è bassa (meno di 60 bpm) oppure alta (maggiore di 100 bpm): - Una frequenza bassa suggerisce bradicardia sinusale, arresto sinusale o blocco di conduzione; - Una frequenza alta suggerisce un’aumentata/anormale automaticità o un rientro. Successivamente si valuta la presenza o meno di un ritmo irregolare. Un ritmo irregolare suggerisce la presenza di fibrillazione atriale, blocco AV di II grado, tachicardia multifocale atriale, o flutter atriale con blocco variabile AV. Molto importante è l’analisi dei complessi QRS, se questi sono stretti o larghi: - Complessi QRS stretti, il ritmo potrebbe originare dal nodo AV o oltre; - Larghi, il ritmo potrebbe originare dappertutto. L’onda P permette di aggiungere ulteriori informazioni. Se questa è assente suggerisce la presenza di fibrillazione atriale, tachicardia ventricolare o di un ritmo che origina dal nodo AV. L’analisi della relazione tra onde P e complesso QRS permette di approfondire la conoscenza sul fenomeno: - Se vi sono più onde P rispetto ai complessi QRS, ciò suggerisce un blocco AV di II o III grado; - Se vi sono più complessi QRS rispetto alle onde P ciò suggerisce un ritmo giunzionale accelerato o un ritmo ventricolare. Infine si passa a vede se l’inizio/terminazione del ritmo si verifica all’improvviso oppure è graduale: - Interruzione del ritmo improvvisa, suggerisce la presenza di ritmi di rientro; - Interruzione del ritmo graduale, suggerisce un’automaticità alterata. Qui di seguito verranno riportati gli algoritmi diagnostici. Algoritmo Diagnostico delle Tachiaritmie a QRS Stretto Intervalli RR Regolari? Fibrillazione atriale No Flutter atriale T. atriale focale T. atriale multifocale Attività atriale ? Algoritmo Diagnostico delle Tachiaritmie a QRS Stretto Intervalli RR Regolari? Fibrillazione atriale T. sinusale Si No Flutter atriale T. atriale focale T. atriale focale T. atriale multifocale TRNAV TRAV Attività atriale ? T. giunzionale Si Polimorfe Flutter atriale Onde P Morfologia Monomorfe Onde F No Algoritmo Diagnostico delle Tachiaritmie a QRS Stretto Intervalli RR Regolari? Fibrillazione atriale T. sinusale No Flutter atriale Si T. atriale focale Flutter atriale T. atriale focale T. atriale multifocale TRNAV TRAV Onde P Visibili? T. giunzionale P > QRS ? Si RP < PR RP < 70 msec RP / PR RP > 70 msec No Si RP > PR TRNAV T. giunzionale T. atriale focale Algoritmo Diagnostico delle Tachiaritmie a QRS Stretto Intervalli RR Regolari? T. sinusale Si Flutter atriale T. atriale focale TRNAV No Flutter atriale T. atriale focale T. atriale multifocale TRAV Onde P Visibili? No Fibrillazione atriale T. giunzionale Manovre vagali / Adenosina Interruzione Rallentamento della conduzione AV Migliore valutazione dell’attività atriale Algoritmo Diagnostico delle Tachiaritmie a QRS Largo Fibrillazione atriale Dissociazione A-V Flutter atriale T. atriale focale T. atriale multifocale Manovre di stimolazione vagale* Si TRAV No TRNAV T.V. T.V. TSV preeccitata Inefficaci QRS-nadir S > 100msec Morfologia nelle precordiali BBDx Interruzione Aspetto concordante nelle precordiali No No Slatentizzazione Onde P ectopiche o F Si Si TV BBSx Per ovviare ad errori nella trattazione, vi è in allegato il capitolo X BIS, preso dal Rugarli. C A P I T O L O 6 ARITMIE G. FERRARIO Per aritmia si intende ogni condizione nella quale viene a mancare la normale frequenza o la regolarità del battito cardiaco, ovvero è alterata la fisiologica sequenza dell’attivazione atrioventricolare. Anatomia del sistema di conduzione cardiaco L’attivazione normale del cuore inizia nel nodo del seno. Il nodo del seno è una struttura cilindrica lunga 15 × 5 mm, situata a livello della giunzione fra vena cava superiore e atrio destro. Esso è costituito da almeno tre tipi di cellule, di cui quelle funzionalmente più importanti sono le cellule P (pacemaker o segnapassi). Queste cellule hanno la proprietà di depolarizzarsi spontaneamente a intervalli regolari, con una frequenza che è condizionata principalmente da due fattori: dall’attività del sistema nervoso autonomo e dalle richieste emodinamiche dell’organismo. L’impulso sorto a livello del nodo del seno si diffonde negli atri e raggiunge il nodo atrioventricolare. È stata affermata, ma non dimostrata in modo definitivo, l’esistenza a livello atriale di alcuni fasci costituiti da cellule specializzate (distinte da quelle contrattili); lungo queste “vie internodali”, che connetterebbero il nodo del seno con il nodo atrioventricolare, l’impulso procederebbe più velocemente che nel miocardio comune. Sicuramente specializzate, ma sede di un rallentamento della velocità di conduzione, sono le cellule che costituiscono il nodo atrioventricolare (A-V) attraverso il quale l’impulso proveniente dagli atri può giungere ai ventricoli. Il nodo A-V, unica struttura muscolare che unisce fisiologicamente atri e ventricoli, è sito alla base del setto interatriale, a ridosso dell’anulus tricuspidale, 1-2 cm avanti al seno coronarico. Superato il nodo A-V, l’impulso prosegue attraverso il fascio di His, le due branche ed il sistema di Purkinje. Fascio di His, branche e sistema di Purkinje sono costituiti da tessuto di conduzione specializzato e in particolare da cellule tipo Purkinje (Fig. 6.1). Conviene aggiungere che l’insieme Nodo del seno Atri Nodo A-V Fascio comune Branche Fibre di Purkinje Ventricoli Figura 6.1 - Rappresentazione delle vie di conduzione cardiaca. formato dal nodo atrioventricolare e dal tronco comune del fascio di His viene spesso denominato “giunzione atrioventricolare”. Il fascio di His è lungo circa 15 mm: nasce dal bordo antero-inferiore del nodo A-V e, correndo lungo la parte membranosa del setto interventricolare, raggiunge il setto interventricolare muscolare. Qui originano le branche. La branca destra è un fascio sottile, ben individualizzato, che scende lungo il lato destro della porzione muscolare del setto interventricolare. La branca sinistra è rappresentata invece da una serie di fibre che si sfilacciano dal fascio di His correndo lungo il lato sinistro del setto interventricolare muscolare. L’anatomia della branca sinistra mostra una spiccata variabilità individuale. Anche se ai fini clinici è utile considerare la presenza di due principali fascicoli (rispettivamente il 122 MALATTIE DEL SISTEMA CIRCOLATORIO fascicolo anteriore e il fascicolo posteriore), è per lo più impossibile individuare nell’ambito della branca sinistra suddivisioni costanti e ben definite da un punto di vista anatomico. In prossimità dell’apice dei due ventricoli, branca destra e branca sinistra si suddividono in molti fascetti, che danno origine a una fitta rete di fibre, la quale si distribuisce sulla superficie endocardica dei ventricoli: è il sistema di Purkinje. Il sistema di Purkinje è in stretta connessione con le cellule miocardiche contrattili: a loro trasmette finalmente l’impulso dal quale prenderà avvio l’attività meccanica dei ventricoli. La funzione emodinamica del cuore si realizza in maniera ottimale solo quando la contrazione atriale e la contrazione ventricolare si susseguono con la sequenza che risulta dall’ordinata trasmissione dell’impulso dal nodo del seno ai ventricoli attraverso il tessuto atriale, il nodo A-V, il fascio di His, le branche e la rete di Purkinje. Il nodo del seno viene irrorato da un’arteria centrale, che prende origine in circa la metà dei casi dalla coronaria destra, nell’altra metà dall’arteria circonflessa della coronaria sinistra. Il nodo A-V è irrorato da un’arteria che origina dalla coronaria destra. Il fascio di His è irrorato per lo più da vari rami della coronaria sinistra. Elettrofisiologia Il normale ciclo cardiaco prende origine dalla spontanea insorgenza dell’eccitazione nel nodo del seno; da qui l’eccitazione si propaga a tutto il miocardio. Sia la formazione degli impulsi sia la propagazione dello stato di eccitazione derivano da caratteristiche funzionali delle cellule, che possono essere descritte analizzando il potenziale d’azione di esse. Per potenziale d’azione, come è noto, si intende la curva che descrive le variazioni che subisce il potenziale elettrico esistente all’interno di ogni singola cellula in relazione alle fasi del ciclo cardiaco. A riposo, durante la diastole elettrica, l’interno delle cellule miocardiche ha un potenziale elettrico negativo, cioè inferiore a quello che esiste sulla superficie esterna della membrana cellulare (e nei liquidi circostanti): se in questi il potenziale è 0, all’interno di esso è di circa –90 mV. Tale potenziale di transmembrana a riposo dipende dal fatto che in questa fase del ciclo cardiaco la cellula è dotata di un patrimonio di ioni negativi che è leggermente maggiore del patrimonio di ioni positivi. Il contrario si verifica nel liquido extracellulare che circonda le cellule: esso contiene, cioè, un leggero eccesso di cariche positive. Per attrazione elettrostatica, il plus di cariche negative endocellulari si dispone in gran parte a ridosso della faccia interna della membrana cellulare, mentre il plus di cariche positive esocellulari si dispone a ridosso della faccia esterna della stessa membrana. È il “doppio strato” di cariche, che caratterizza la membrana “polarizzata”. Se, per influenze esterne, cariche positive esterne si allontanano, anche le cariche negative interne, non subendo più l’attrazione elettrostatica, si allontanano dalla membrana (che perde così la sua polarizzazione). L’eccesso di cariche negative all’interno della cellula è conseguenza della struttura della membrana cellulare e del fatto che fisiologicamente le cellule hanno una distribuzione di ioni diversa da quella dei liquidi interstiziali. In particolare, dentro alle cellule vi è una concentrazione di ioni potassio (K) 30 volte superiore a quella che vi è all’esterno delle cellule: la concentrazione degli ioni sodio (Na) è 15 volte inferiore, quella degli ioni calcio (Ca) è 10.000 volte inferiore. L’ineguale distribuzione di questi ioni è controllata dall’attività di pompe scambiatrici di ioni che, usando energia, espellono ioni non voluti dalla cellula, scambiandoli con altri. Occorre energia perché questo scambio avviene contro la tendenza degli ioni stessi, che tenderebbero a parificare le rispettive concentrazioni ai due lati della membrana cellulare. Un eventuale deficit metabolico cellulare altera necessariamente i rapporti ottimali degli ioni in questione fra esterno ed interno delle cellule. L’attività delle pompe non sarebbe certo sufficiente a mantenere lo squilibrio descritto, se la membrana cellulare fosse liberamente permeabile agli ioni. Una descrizione semplificata ma sufficiente dei fenomeni permette di dire che, in effetti, la membrana è impermeabile agli ioni, salvo che in corrispondenza di particolari strutture proteiche, dette “canali”: questi possono essere chiusi, oppure più o meno aperti, e in questo caso il passaggio degli ioni attraverso la membrana è possibile. Si conoscono tre tipi di canali fisiologicamente molto importanti: per il Na, per il Ca, per il K. Dato che gli ioni sono dotati di carica elettrica, si può esprimere la chiusura o l’apertura di canali in termini di conduttanza della membrana rispetto ai singoli ioni; per esempio: conduttanza al Na nulla, scarsa, elevata. Come si dirà meglio in seguito, vi sono due tipi fondamentali di cellule miocardiche: le cellule del miocardio comune o “da lavoro” (atriali e ventricolari) e le cellule pacemaker. Entrambi i tipi di cellula sono caratterizzati da tre proprietà: l’eccitabilità, la conducibilità e la refrattarietà. Solo le cellule pacemaker hanno una quarta proprietà, che è l’automatismo. Per eccitabilità si intende l’attitudine di una cellula miocardica a sviluppare il potenziale d’azione (si veda in seguito). L’eccitazione subentra criticamente alla fase di riposo quando il potenziale di transmembrana viene ridotto (da eventi che saranno in seguito specificati) dal valore (indicativo) di –90 mV a un valore (indicativo) di –60 mV: a questo livello di potenziale, che si chiama potenziale-soglia, si scatena la serie di variazioni che determina il potenziale d’azione e che si traduce in perdita completa della polarizzazione caratteristica delle cellule a riposo. Per conducibilità si intende la velocità con la quale l’eccitazione di una cellula del miocardio si trasmette alle cellule vicine. Questa trasmissione avviene perché la cellula eccitata è completamente depolarizzata, cioè priva di cariche elettriche positive in superficie; questo “vuoto” di cariche positive attrae le cariche elettriche 6 - ARITMIE positive che tappezzano la faccia esterna della membrana delle cellule vicine. In conseguenza, il potenziale di transmembrana di tali cellule diventa minore di –90 mV, e quando raggiunge –60 mV le cellule si eccitano a loro volta. È evidente che la velocità con cui l’eccitazione si propaga da un punto all’altro del miocardio è funzione della velocità con cui si depolarizzano le singole cellule. Per refrattarietà si intende un fenomeno ben noto ai fisiologi: se una cellula opportunamente stimolata entra in stato d’eccitazione, occorre del tempo perché essa possa essere nuovamente eccitata. In un primo periodo nessuno stimolo potrà rieccitarla (periodo refrattario assoluto o effettivo); in un secondo periodo la rieccitazione è possibile, ma solo a opera di stimoli molto forti (periodo refrattario relativo). Si può dire che durante il primo periodo la cellula, essendo depolarizzata, non può ovviamente eccitarsi; durante il secondo periodo la cellula, che ha recuperato solo in parte la polarizzazione di riposo, si eccita lentamente e fornisce un potenziale d’azione imperfetto. Tutto ciò può essere trasferito in termini di conduzione nell’ambito del cuore: se un tratto di miocardio M (per es., i ventricoli) è stato eccitato da uno stimolo 1, esso può rieccitarsi ad opera di uno stimolo 2 solo se è trascorso un certo tempo. In particolare, se lo stimolo 2 segue lo stimolo 1 dopo un intervallo di tempo molto breve, il miocardio M, tuttora in periodo refrattario effettivo, non risponde affatto; se lo stimolo 2 segue lo stimolo 1 un po’ più tardi, quando M è in periodo refrattario relativo, M si eccita, ma lentamente; se lo stimolo 2 segue lo stimolo 1 quando M ha recuperato completamente, M si eccita subito. Nel primo caso la velocità di conduzione dell’eccitamento nell’ambito del miocardio M è zero, nel secondo caso è rallentata rispetto alla norma, nel terzo caso è normale. Per automatismo (caratteristica, come si è detto, esclusiva delle cellule pacemaker) si intende la capacità che queste cellule hanno di autoeccitarsi, mentre tutte le altre cellule miocardiche si eccitano solo se ricevono uno stimolo dall’esterno. Le cellule non pacemaker in stato di riposo, infatti, tendono a mantenere questo stato indefinitamente; le cellule pacemaker, invece, non hanno un potenziale di transmembrana a riposo stabile: questo potenziale tende a ridursi spontaneamente col tempo e, quando raggiunge il valore-soglia, la cellula si eccita. Nel cuore umano ci sono cellule con proprietà pacemaker nel nodo del seno, qua e là negli atri (specie vicino al nodo atrioventricolare, lungo le presunte vie internodali), nel nodo atrioventricolare e ve ne sono nel fascio di His con relative diramazioni, fino al sistema di Purkinje. In più, vi sono cellule appartenenti al sistema di conduzione (specie a livello fascio di His e distalmente a questo) che non sono pacemaker in condizioni normali, ma che possono acquisire automatismo in condizioni patologiche (per es., ischemia). Nell’ambito delle cellule pacemaker, esiste una fisiologica differenza fra i vari gruppi di cellule sopra citati: la rapidità con cui avviene la depolarizzazione spontanea durante la fase di riposo è maggiore nelle cellule del nodo del seno, è intermedia nelle cellule del nodo atrioventri- 123 colare, è minore negli elementi pacemaker dei rami periferici del tessuto di conduzione. In relazione a ciò, la frequenza base di autoeccitazione è 70/min a livello del nodo del seno, 60 a livello della giunzione A-V, 30 a livello della rete di Purkinje. In condizioni normali la proprietà dell’automatismo si esprime solo a livello del nodo del seno. Le cellule degli altri pacemaker, infatti, vengono eccitate dall’impulso che origina dal nodo del seno prima di potersi autoeccitare. Questi pacemaker vengono definiti “sussidiari” o “latenti”. Lo svolgimento fisiologico del ciclo cardiaco trae origine, come già si è detto, dalla ritmica autoeccitazione di cellule pacemaker nel nodo del seno. Ogni volta, la depolarizzazione di queste cellule sottrae cariche elettriche positive dalla superficie esterna delle cellule circostanti; questo fatto riduce il potenziale di transmembrana di tali cellule fino al potenziale-soglia. A questo punto le cellule in questione si eccitano depolarizzandosi e ciò sottrae cariche positive dalla superficie delle cellule successive e così via: così si realizza la conduzione, che finisce per coinvolgere nell’eccitamento tutto il cuore. Per comprendere bene lo svolgimento dei fenomeni fisiologici e le possibili alterazioni patologiche, conviene ora descrivere il potenziale d’azione, che è già stato definito come una successione di eventi critici. Questi sono un po’ diversi nelle cellule pacemaker e in quelle che non lo sono; conviene cominciare con queste ultime. Le cellule in questione (tutto il miocardio comune atriale e ventricolare e buona parte delle cellule costituenti il tessuto di conduzione) hanno, come ripetutamente spiegato, un potenziale di transmembrana a riposo stabile di circa –90 mV (Fig. 6.2). Quando l’eccitazione delle cellule vicine sottrae cariche dalla superficie cellulare, il potenziale si riduce passivamente fino verso i –60 mV (potenziale-soglia). A questo punto si scatena una serie di eventi rapidi e importantissimi. Il primo di essi è costituito dall’improvvisa apertura dei canali per il Na della membrana cellulare. Attraverso questi “canali veloci”, nel giro di pochissimi millisecondi una certa quantità di ioni Na entra nelle cellule annullando il preesistente deficit di cariche positive intracellulari, fino a un leggero eccesso di cariche positive. Il potenziale di transmembrana passa perciò rapidissimamente (fase 0 del potenziale d’azione) da –60 mV a +20 mV. A un certo punto i canali del Na si chiudono e il Na non può più entrare. Intanto, però (per un comando potenziale-dipendente), si sono aperti i canali “lenti” del Ca, che permettono l’ingresso (più lento ma che dura più a lungo) di altre cariche positive nelle cellule. Quest’ulteriore squilibrio è peraltro controbilanciato da una quasi contemporanea apertura dei canali per il K, che, in relazione al rapporto di concentrazione fra interno ed esterno della membrana di questo ione, permette l’uscita di ioni K. In termini di potenziale di transmembrana questi fenomeni portano a: • una breve fase 1 del potenziale d’azione in cui prevale l’uscita di K+ sull’ingresso dei Ca++, sicché il potenziale scende attorno a zero; 124 MALATTIE DEL SISTEMA CIRCOLATORIO 0 1 +20 20 0 2 60 0 3 4 –90 Na+ Ca++ Na+ K+ K+ Cl – Figura 6.2 - Rappresentazione schematica del potenziale d’azione di una fibrocellula cardiaca con i principali flussi ionici nelle diverse fasi. • una fase 2 del potenziale in cui l’afflusso di Ca++ e l’efflusso di K+ si bilanciano, per cui il potenziale resta più o meno sullo zero (fase di plateau); • una fase 3 durante la quale cessa l’afflusso di Ca++ mentre prosegue l’efflusso di K+: al termine di questa fase (detta di ripolarizzazione) il potenziale di transmembrana a riposo si è ripristinato; durante questa fase 3 si ripristina pure l’eccitabilità cellulare, che passa dal periodo di refrattarietà effettiva a quello di refrattarietà relativa, verso la normalizzazione; • finita la fase 3, inizia la fase 4, che dura finché la cellula non venga eccitata di nuovo. Il potenziale è stabile intorno a –90 mV, ma all’inizio con oscillazioni, che stanno alla base del fatto che, finita la fase di refrattarietà relativa, l’eccitabilità può essere superiore alla norma (fase supernormale). Questa fase 4 non vede la cellula inoperosa, perché essa, con l’aiuto delle pompe scambiatrici di ioni (e con impiego di energia), provvede a ripristinare la composizione qualitativa del patrimonio ionico intracellulare: in particolare espelle ioni Na e Ca, scambiandoli con ioni K. Nelle cellule pacemaker le vicende sono un po’ diverse, perché in queste cellule non vi sono i canali veloci per il Na. Al momento in cui viene raggiunto il potenzia- Figura 6.3 - Potenziale d’azione delle cellule del nodo del seno. I caratteri distintivi principali sono rappresentati da una fase 4 instabile (automatismo) e da una fase 0 relativamente lenta. le-soglia, si aprono i canali lenti del Ca, con ingresso di Ca++: essendo però questo ingresso relativamente lento, la fase 0 del potenziale d’azione non è quasi verticale, ma obliqua (Fig. 6.3); la variazione di potenziale è alquanto graduale e non oltrepassa il valore 0 (per cui mancano la breve fase di positività intracellulare e successiva fase 1 del potenziale d’azione). Anche la fase 2 è breve, e la 3 è più graduale e prolungata. La 4, poi, è caratterizzata dal fatto (già descritto) che il potenziale di transmembrana non si mantiene: da un valore massimo iniziale, il potenziale si riduce lentamente, fino ad arrivare al potenziale-soglia. Come già detto, nel nodo del seno l’obliquità della fase 4 porta il potenziale endocellulare al valore soglia in circa un secondo, nelle cellule pacemaker intraventricolari l’obliquità è minore e per arrivare al valore-soglia occorrono circa 2 sec. In precedenza si è anche accennato a cellule che normalmente non sono pacemaker e che possono diventare tali in condizioni patologiche: la variazione intervenuta in questo caso sembra essere soprattutto l’inattivazione dei canali per il Na. Da quanto si è detto risulta anche comprensibile come diverse parti del cuore possano avere una diversa velocità di conduzione: nel nodo A-V, dove buona parte delle cellule non ha canali per il Na, la velocità di conduzione è di circa 200 mm/sec; nel fascio di His, branche e rete di Purkinje, dove le cellule tipo Purkinje hanno numerosi canali del Na, la conduzione è 4000 mm/sec; nei ventricoli è intermedia: 400 mm/sec. Tutto ciò perché, se la fase 0 di una cellula è ripida, essa sottrae rapidamente cariche elettriche positive dalla superficie delle cellule vicine e quindi le fa eccitare in poco tempo; se la fase 0 è obliqua, la propagazione dello stimolo sarà rallentata. Ultima considerazione generale: vi sono vari sottotipi di cellula cardiaca e in ognuno i passaggi sopra descritti sono concatenati fra loro; è evidente che se un qualunque fattore (stimolazione nervosa, stimolazione ormonale, farmaco, processo patologico) altera uno qualunque dei processi connessi con l’eccitazione cellulare, ciò può determinare varie e talora impreviste ripercussioni. Per esempio, la stimolazione del sistema nervoso simpatico aumenta la frequenza di autoeccitazione del nodo del seno, ma aumenta anche la velocità di conduzione, diminuisce la durata dei periodi refrattari e aumenta (facendo aprire più canali per il Ca) la contrat- 125 6 - ARITMIE tilità. Però la stimolazione simpatica non agisce allo stesso modo su tutte le cellule cardiache: alcune sono molto influenzate da questa stimolazione, altre molto di meno (per es., le cellule pacemaker intraventricolari non aumentano la frequenza di autoeccitazione). La generalizzazione di questo concetto permette di capire come gli effetti di un singolo evento morboso o di un farmaco possano essere molteplici e spesso variabili da caso a caso. Meccanismi di formazione delle aritmie Non è possibile parlare dei meccanismi di formazione delle aritmie senza averne preliminarmente distinto le più importanti varianti. Secondo la nomenclatura attualmente in uso in Italia, le aritmie possono essere distinte in aritmie ipercinetiche ed aritmie ipocinetiche. In linea generale le aritmie ipercinetiche sono quelle in cui si tende ad avere un numero di impulsi maggiore del normale a livello degli atri (aritmie ipercinetiche atriali) o dei ventricoli (aritmie ipercinetiche ventricolari). Spesso accade, ma non necessariamente, che l’aumentato numero di impulsi atriali determini un aumentato numero di impulsi ai ventricoli. Non è detto pertanto che, per interferenze di vario tipo, il fenomeno iniziale “aumentato numero di impulsi sopraventricolari o ventricolari” si traduca in un aumentato numero di battiti nelle unità di tempo. Le aritmie ipocinetiche, atriali o ventricolari, sono viceversa quelle che tendono a produrre un minor numero di impulsi a livello atriale e/o ventricolare. Anche in questo caso, non è detto che fenomeni secondari non interferiscano con l’effettiva estrinsecazione di un numero di battiti che non è diminuito. Nonostante queste limitazioni, la nomenclatura risulta clinicamente utile. I meccanismi di formazione delle aritmie ipercinetiche sono quattro: • aumento della frequenza propria del nodo del seno; • acquisizione da parte di un pacemaker (latente o patologico) di una frequenza superiore a quella del nodo del seno; • fenomeno del rientro; • la cosiddetta “triggered activity”. 1. Come agisce il primo meccanismo, è di per sé evidente. Deve essere aggiunto che questo è il meccanismo meno importante per le aritmie ipercinetiche, non già perché si verifichi raramente, ma perché di significato patologico non primario e spesso indicativo di patologie extracardiache. 2. Considerazioni più interessanti si possono fare sul secondo meccanismo. Si è già visto che l’automatismo (cioè la capacità della cellula di depolarizzarsi spontaneamente) è una proprietà presente in tutte le cellule del tessuto di conduzione specializzato, ma che in condizioni normali si esprime solo a livello del nodo del seno. In alcune condizioni patologiche, un gruppo di cellule pacemaker latenti aumenta la rapidità della pro- pria depolarizzazione spontanea, che diventa maggiore di quella delle cellule del nodo del seno. In conseguenza di ciò l’eccitazione che si origina nel “focus ectopico ipereccitabile” si diffonde ai tessuti vicini prima che questi siano raggiunti dall’onda di eccitazione che proviene dal nodo del seno. Si dirà allora che il focus ectopico ha preso il sopravvento sull’attività sinusale ed è esso a generare l’impulso “guida” che diffonde al resto del cuore. Con una similitudine efficace, l’assunzione del governo del ritmo cardiaco da parte di un focus ectopico è stata paragonata all’ammutinamento da parte dell’equipaggio di una nave: il capitano (il nodo del seno) viene esautorato da un suo subordinato (un pacemaker). In questo caso il ritmo ectopico è perciò detto “ritmo attivo”. Deve essere aggiunto che l’attività del focus ectopico può essere ripetitiva e durare nel tempo, ma può anche trovare il modo di esplicarsi solo saltuariamente con singoli battiti che si sovrappongono, disturbandolo, al ritmo sinusale di fondo. 3. Sono invece disturbi della propagazione dell’impulso (vale a dire della velocità di conduzione dell’eccitamento) quelli che costituiscono la base del cosiddetto meccanismo di “rientro” (terzo) che è responsabile di numerose forme di aritmia. Il meccanismo si spiega con lo schema che segue. In condizioni di normalità (Fig. 6.4, ➀) l’impulso che proviene dall’alto, quando incontra uno sdoppiamento segmentario del tragitto attraverso il quale viene condotto, si propaga parallelamente lungo le fibre di miocardio (atriale o ventricolare) che costituiscono la via sdoppiata (A e B nella figura) riunendosi nuovamente in un solo percorso quando le fibre sdoppiate si ricongiungono (C nella figura). Non esiste alcuna possibilità che l’impulso che si è propagato lungo il percorso A possa risalire in un senso retrogrado lungo il percorso B perché quest’ultimo, attraversato parallelamente ad A dall’impulso che proviene dall’alto, si troverà in periodo refrattario. Questa situazione può essere alterata da condizioni patologiche capaci di ridurre la conduttività delle due vie A e B. È possibile che l’impulso proveniente dalla via prossimale comune (Fig. 6.4, ➁) venga bloccato (per condizioni di refrattarietà assoluta) a livello della via B e rallentato (per condizione di refrattarietà relativa) a livello della via A. Quando l’eccitazione arriva attraverso A alla fibra distale comune C (Fig. 6.4, ➂), esso si diffonde regolarmente in C ma contemporaneamente si diffonde anche per via retrograda lungo B. Quando l’impulso arriva al tratto prossimale di B, al punto cioè in cui si era precedentemente bloccato, può diffondersi liberamente perché il rallentamento presente lungo la via A ha dato modo al tratto B di recuperare una condizione di normale eccitabilità (di superare cioè il periodo refrattario). Nel frattempo ha recuperato anche A, sicché l’impulso che ha percorso in via retrograda B può rieccitare A (“rientrare” lungo A) e trasmettere alla via distale comune C un secondo impulso: questo può dare origine a un terzo impulso e così via. Perché il rientro possa avere luogo, sono necessarie per- 126 MALATTIE DEL SISTEMA CIRCOLATORIO POSTDEPOLARIZZAZIONE PRECOCE Via prossimale comune A B A. C Via distale comune POSTDEPOLARIZZAZIONE TARDIVA Via prossimale comune B A Ritardo di conduzione Blocco unidirezionale B. C Via distale comune Via prossimale comune Figura 6.5 - Postdepolarizzazioni e attività “triggered”. A. Postdepolarizzazione precoce. La ripolarizzazione viene interrotta da due depolarizzazioni secondarie, che possono attivare fibre adiacenti e indurre aritmie. B. Postdepolarizzazione tardiva. Si può manifestare quando il potenziale d’azione è tornato al valore di riposo. Se viene raggiunto il potenziale di soglia, si può indurre un secondo potenziale d’azione anomalo. B A grande (come per esempio nelle tachicardie da sindrome di Wolff-Parkinson-White), di “macrorientro”. C Via distale comune Figura 6.4 - Schema di rientro. (Vedi testo per la spiegazione). tanto tre condizioni fondamentali: deve esistere un circuito anatomico o funzionale, piccolo o grande, caratterizzato da due vie comuni (distale e prossimale) e due vie che costituiscono uno sdoppiamento segmentario; deve instaurarsi un blocco unidirezionale (ovverossia l’impulso deve essere bloccato nella diffusione anterograda ma deve poter essere trasmesso per via retrograda) in una delle due vie sdoppiate (B nell’esempio della figura); la conduzione lungo la via non bloccata deve essere però rallentata, per consentire al tessuto in B prossimale al blocco di recuperare una condizione di eccitabilità. Se il circuito interessato al rientro è piccolo, il fenomeno viene chiamato di “microrientro”; se è 4. Il quarto meccanismo di formazione delle aritmie ipercinetiche è stato descritto recentemente ed è comunemente definito con il termine inglese di “triggered activity” (letteralmente: attività messa in moto premendo un grilletto). Si tratta di potenziali d’azione aggiuntivi (singoli o ripetuti) che si sovrappongono alla fase terminale di un potenziale d’azione normale o lo seguono di poco. La prima evenienza viene chiamata “postdepolarizzazione precoce” e la seconda “postdepolarizzazione tardiva” (rispettivamente “early” e “delayed afterdepolarization”). Nella postdepolarizzazione precoce cause patologiche fanno sì che in una particolare fibra miocardica la fase conclusiva di un potenziale d’azione normale non esiti nel ritorno alla ripolarizzazione (e al ristabilimento del normale potenziale di membrana a riposo), ma sia interrotta da uno o più cicli di nuova depolarizzazione (Fig. 6.5A). Il fenomeno può sorgere a livello delle fibre di Purkinje quando queste vengono stirate, per esempio quando si instaura acutamente una dilatazione ventricolare. 127 6 - ARITMIE La postdepolarizzazione tardiva non è altro che un’accentuazione di quella instabilità del potenziale di membrana a riposo che si verifica subito dopo la fine del potenziale di azione (e alla quale si è già fatto cenno). In condizioni patologiche questo potenziale di membrana a riposo tende a oscillare e può capitare che l’oscillazione sia di entità tale da raggiungere il potenziale di soglia che è in grado di scatenare un nuovo potenziale d’azione (Fig. 6.5B). Questo potenziale d’azione può essere isolato o seguito da altri, anch’essi indotti con meccanismo analogo. Il fenomeno si verifica a livello del fascio di His e delle fibre di Purkinje in corso di intossicazione digitalica. Sia nella sua forma di postdepolarizzazione precoce, sia in quella di postdepolarizzazione tardiva, la “triggered activity” è un’imperfezione del completamento di un potenziale di azione normale. Come si vede, tanto l’aumentato automatismo di un pacemaker latente quanto il fenomeno del rientro e la “triggered activity” fanno originare il ritmo cardiaco in una sede diversa dal nodo del seno e perciò detta ectopica. A questo riguardo ha un certo rilievo clinico se il ritmo cardiaco è generato negli atri o nella giunzione atrioventricolare (aritmie ipercinetiche sopraventricolari) o a livello dei ventricoli (aritmie ipercinetiche ventricolari). I meccanismi di formazione delle aritmie ipocinetiche sono tre: • diminuzione della frequenza del nodo del seno, che conserva il governo del ritmo cardiaco; • assunzione, per carenza di attività del nodo del seno, del governo del ritmo da parte di un pacemaker latente che agisce mantenendo invariato il proprio automatismo; • disturbi della conduzione A-V. 1. Il primo meccanismo può agire semplicemente rallentando la frequenza di fondo propria dell’automatismo del nodo del seno. Si realizza anche, però, quando la frequenza di fondo del nodo del seno non è ridotta, ma alcuni dei battiti che ne dovrebbero derivare vanno perduti. Quest’ultimo fenomeno dipende da episodici arresti dell’attività del nodo del seno (arresto sinusale) o dall’episodica incapacità dell’impulso ritmicamente generato dal nodo del seno di essere condotto alle cellule miocardiche contigue (blocco senoatriale). 2. Il secondo meccanismo si verifica quando i fenomeni descritti a proposito del primo meccanismo riducono talmente la frequenza degli impulsi efficaci generati dal nodo del seno, da renderla inferiore a quella di un pacemaker latente sottostante con una frequenza propria più elevata. In questo caso è per carenza di attività di pacemaker fisiologico che un altro centro pacemaker riesce ad esprimere la propria attività automatica, dando luogo a quelli che vengono chiamati ritmi di “sfuggita”. Per ripetere la similitudine precedente, è come se il capitano di una nave diventasse incapace al comando perché colto da malore e venisse sostituito dal suo secondo. In questo caso il ritmo ectopico è perciò detto “ritmo passivo” (o anche ritmo di sfuggita o di scappamento). Quando difetta l’attività del nodo del seno, il centro ectopico che assume il governo dell’attività cardiaca è più spesso la regione giunzionale, la cui frequenza propria è più bassa di quella del nodo del seno ma più alta di quella di tutti gli altri pacemaker latenti. 3. Il terzo meccanismo si verifica quando esistono delle anomalie nella propagazione dell’eccitazione lungo il sistema di conduzione e in particolare nel suo passaggio dagli atri ai ventricoli. Perché si manifesti un’aritmia, occorre che si abbia una di queste due possibilità: episodicamente (in maniera periodica o meno) un impulso non passa dagli atri ai ventricoli; tutti gli impulsi non passano dagli atri ai ventricoli e questi si contraggono per l’attività (ritmo passivo) di un pacemaker latente collocato nel loro contesto. Nel primo caso si ha l’episodica mancanza di un battito ventricolare; nel secondo caso la frequenza cardiaca è molto bassa perché il centro ectopico che la determina ha una frequenza propria molto inferiore a quella del nodo del seno. Deve essere aggiunto che non tutti i disturbi di conduzione procurano aritmie. Alcuni determinano solo delle sindromi elettrocardiografiche che, per ragioni di convenienza, considereremo in appendice alle aritmie ipocinetiche. Eziologia In base a quanto esposto precedentemente, è chiaro che qualsiasi fattore in grado di modificare l’automatismo, l’eccitabilità e la conduttività delle cellule miocardiche può essere causa di aritmia. In primo luogo fattori fisiologici: la stimolazione dei recettori -adrenergici (o una scarica catecolaminica da parte dei surreni) accentua l’automatismo delle cellule pacemaker per aumentata pendenza della fase 4 e l’eccitabilità e conduttività di tutte le cellule per aumentata rapidità della fase 0 e accorciamento delle fasi 2 e 3. La stimolazione parasimpatica agisce in modo grossolanamente opposto. Questi effetti, dovuti al sistema neurovegetativo, agiscono evidentemente sulla frequenza propria del nodo del seno aumentandola o diminuendola. Inoltre, la stimolazione dei recettori -adrenergici può esercitarsi in modo elettivo in zone diverse dal nodo del seno, rendendo possibile ritmi ectopici attivi. Non si deve pensare che quest’ultimo tipo di aritmie ipercinetiche dovute a iperattività simpatica siano un caso limite eccezionale. Al contrario costituiscono un’evenienza che può verificarsi nella vita di tutti i giorni favorita dagli sforzi, dai processi digestivi, dall’assunzione e dall’abuso di sostanze eccitanti: caffè, tè, tabacco. Ne consegue che le aritmie sono possibili (ed anche frequenti) nel cuore sano, ed in tal caso sono dette aritmie funzionali (Tab. 6.1). Più frequentemente le aritmie si manifestano nel cuore malato, sia nel caso che il cuore abbia una malattia primitiva, sia che esso sia coinvolto secondariamente a malattia di altri organi. Squilibri elettrolitici ed attività indesiderate di farmaci sono altre cause comuni di aritmie. 128 MALATTIE DEL SISTEMA CIRCOLATORIO Tabella 6.1 - Cause principali di aritmie. Funzionali • Sforzi • Processi digestivi • Assunzione/abuso di sostanze eccitanti Organiche • Cardiache primitive: – Cardiopatie ischemiche – Cardiopatie valvolari – Cardiomiopatie primitive – Miocarditi • Cardiache secondarie a: – Tireotossicosi – Feocromocitoma – Squilibri elettrolitici – Farmaci Le malattie primitive del cuore che più spesso inducono la formazione di aritmie sono: la cardiopatia ischemica, le cardiopatie valvolari, le cardiomiopatie primitive, le miocarditi. Si può affermare, in generale, che tutte le malattie del cuore possono manifestare ogni forma di aritmia. Esempi di malattie extracardiache che possono indurre aritmie sono la tireotossicosi ed il feocromocitoma (Tab. 6.1). Lo squilibrio elettrolitico che è più frequentemente responsabile dei disturbi del ritmo è l’ipopotassiemia. Tra i farmaci va ricordata la tossicità digitalica. Parlando dei meccanismi patogenetici delle aritmie ipercinetiche, si è detto che essi richiedono l’aumento dell’automatismo cellulare in focolai ectopici e la formazione dei circuiti di rientro. Entrambi i meccanismi possono essere indotti da condizioni che sono variamente presenti nelle situazioni sopra elencate. Fra le condizioni più spesso presenti si possono elencare la distensione delle fibre miocardiche, l’ipossia delle cellule, l’alterazione degli elettroliti, l’azione di ormoni, di sostanze eccitanti o di farmaci. Non esistono criteri sicuri per distinguere le aritmie che si manifestano per un esaltato automatismo, per rientro, o per “triggered activity”. Spesso tutti i meccanismi sono variamente in gioco nella genesi dell’aritmia; altre volte un meccanismo innesca l’aritmia e gli altri la perpetuano. Fisiopatologia Le aritmie interferiscono con la corretta funzione del cuore attraverso numerosi meccanismi. Gli effetti di questi meccanismi possono essere così sintetizzati: • effetti legati alla modificazione della frequenza cardiaca; • effetti legati alla perdita della contrazione atriale (che può verificarsi in alcune aritmie); • effetti legati al consumo di ossigeno da parte del miocardio e alla riduzione del flusso coronarico; • effetti sul sincronismo della contrazione ventricolare. A. Effetti secondari alla variazione della frequenza cardiaca. In condizioni di normalità il cuore sa mantenere costante la portata cardiaca (quantità che il sangue espelle in un minuto) anche con ampie variazioni della frequenza (da circa 40 a circa 160 battiti/min). Ciò è possibile perché il cuore normale varia adeguatamente la quantità di sangue espulsa ad ogni singola sistole (gettata sistolica): l’aumenta in caso di bradicardia e la riduce in caso di tachicardia. Quando il miocardio è malato, il cuore non riesce ad incrementare adeguatamente la gettata sistolica in corso di bradicardia e, nella situazione opposta, può ridurre eccessivamente la gettata sistolica in corso di tachicardia. Nell’un caso e nell’altro diminuisce la portata cardiaca e la prestazione di pompa del cuore è compromessa. B. Effetti secondari alla perdita della contrazione atriale. Gli atri, normalmente, completano il riempimento diastolico dei ventricoli. In alcune aritmie l’attivazione atriale è assente o incoordinata, in maniera tale che non si ha una contrazione efficace delle pareti atriali (per es., la fibrillazione atriale); in altre la sistole atriale si verifica, ma senza effetto emodinamicamente utile perché fuori tempo. Ciò avviene in certi blocchi atrioventricolari, nei ritmi giunzionali, nelle tachicardie ventricolari e, in molti casi, nelle tachicardie parossistiche e nei flutter atriali. Nel cuore normale la perdita della contrazione atriale comporta, in condizioni di riposo, solo un modesto effetto emodinamico. Durante l’esercizio fisico o in presenza di un’alterata funzione contrattile, la perdita della normale sistole atriale comporta una significativa riduzione della portata cardiaca. La sistole atriale è particolarmente importante ai fini di un riempimento ventricolare diastolico corretto nelle condizioni caratterizzate da ipertrofia del ventricolo. Quando il ventricolo è ipertrofico, la compliance (distensibilità) delle fibre miocardiche è compromessa: la riduzione della compliance ostacola il riempimento protodiastolico ventricolare. Ne deriva che il riempimento ventricolare dipende, in buona parte, dalla validità della sistole atriale. Condizioni patologiche caratterizzate da ipertrofia ventricolare sono: la stenosi aortica, l’ipertensione arteriosa e le cardiomiopatie ipertrofiche primitive. C. Effetti legati al consumo di ossigeno miocardico e al flusso coronarico. La frequenza cardiaca è uno dei determinanti principali del consumo di ossigeno miocardico. L’aumento della frequenza cardiaca aumenta il consumo di ossigeno; la riduzione della frequenza lo riduce. La frequenza cardiaca, inoltre, condiziona l’entità del riempimento coronarico. Le coronarie si riempiono durante la diastole e si svuotano durante la sistole. La diastole è più lunga in corso di bradicardia ed è più breve in corso di tachicardia. La conseguenza è che in presenza di coronaropatia il flusso coronarico può essere normale se la frequenza cardiaca è bassa, ma può ridursi criticamente se insorge una tachicardia. D. Effetto sul sincronismo della contrazione ventricolare. La funzione ottimale del ventricolo dipende in buona parte dalla contrazione sincrona, quasi simultanea, delle sue fibre. 129 6 - ARITMIE Alcune aritmie comportano un asincronismo della contrazione del ventricolo. Il caso estremo è rappresentato dalla fibrillazione ventricolare, in cui la contrazione del ventricolo è del tutto caotica e inefficace. Esistono gradi minori di asincronismo legati soprattutto a una diffusione irregolare dello stimolo a livello dei ventricoli: nella tachicardia ventricolare, nella extrasistolia ventricolare e nella tachicardia sopraventricolare con conduzione aberrante. L’effetto negativo di una contrazione asincrona è particolarmente importante quando si instaura in soggetti con gravi malattie a carico del miocardio. Sintomi In generale non è possibile abbinare i vari disturbi del ritmo con sindromi e segni specifici. La formazione di essi è determinata dall’aritmia presente e da altre numerose variabili: condizioni contrattili del miocardio, stato del circolo coronarico, stato del circolo arterioso periferico, durata dell’aritmia, ecc. Queste variabili tendono a fare del sintomo un parametro molto personale. Ciononostante, è possibile costruire dei criteri di massima che, opportunamente corretti a seconda delle variabili presenti, possono costituire una traccia razionale per connettere fra loro i disturbi del ritmo con i sintomi. In linea generale si può dire che i sintomi determinati dalla presenza di un’aritmia possono derivare da tre meccanismi differenti: • interferenza da parte dell’aritmia con la funzione di pompa del cuore; • interferenza col riempimento coronarico; • percezione del battito cardiaco irregolare. Nelle aritmie ipercinetiche sopraventricolari il sintomo dominante è in genere legato alla percezione del cuore che batte velocemente (cardiopalmo). Il cardiopalmo può essere “ritmico” o “aritmico” a seconda dell’aritmia presente. Se il circolo coronarico è compromesso, possono comparire sintomi legati all’ipoperfusione coronarica (angina) perché si riduce il tempo di riempimento diastolico delle coronarie. Solitamente l’aritmia ipercinetica sopraventricolare consente una buona funzione dei ventricoli; quando però l’aritmia si manifesta in cuori con funzione contrattile ridotta, l’aumento della frequenza può ridurre significativamente la portata cardiaca. La riduzione della portata cardiaca si accompagna a segni di scompenso retrogrado (dispnea) e anterogrado (pallore, confusione mentale, sudorazione). Nelle aritmie ipercinetiche ventricolari dominano solitamente i sintomi legati alla riduzione della portata cardiaca. La portata cardiaca è variamente compromessa a seconda del tipo di aritmia: in genere poco o niente nei battiti prematuri (extrasistoli) ventricolari sporadici; gravemente nella tachicardia ventricolare e nella torsione di punta; nella fibrillazione ventricolare la portata cardiaca si riduce a zero. Nelle aritmie ipocinetiche la gittata sistolica aumenta per compensare la bradicardia. Se questo meccanismo di compenso non è sufficiente, la perfusione degli orga- ni periferici è inadeguata. I sintomi maggiori derivano dall’ipoperfusione del cervello e sono: vertigini, lipotimie e sincopi. La durata e la gravità della sintomatologia dipendono dalla gravità dell’ischemia cerebrale; questa, a sua volta, dipende dalla durata e dalla gravità dell’aritmia ipocinetica. Diagnosi Questo argomento sarà trattato in parte nella descrizione delle singole aritmie, in parte alla fine del paragrafo Problemi diagnostici particolari nelle aritmie. È opportuno però anticipare la descrizione di una manovra semplice e diagnosticamente importante: il massaggio del seno carotideo (MSC). La manovra è utile nella diagnosi differenziale di alcune aritmie ipercinetiche. Il principio su cui si basa è di indurre uno stimolo vagale che agisce soprattutto inibendo la conduzione a livello del nodo A-V. Il MSC si effettua a paziente supino con il capo esteso e ruotato dalla parte opposta a quella dove si intende applicare il massaggio. Il massaggio dev’essere praticato a livello della biforcazione della carotide, immediatamente al di sotto dell’angolo della mandibola. È necessario che durante il massaggio si registri l’ECG. Il massaggio non deve essere prolungato (non più di 5-6 sec), dev’essere inizialmente praticato con cautela (possibilità di riflessi eccessivi) e dev’essere evitato in presenza di un fremito carotideo. La presenza di un fremito, infatti, segnala un certo grado di ostruzione a carico del circolo carotideo; il massaggio del glomo potrebbe, pertanto, far precipitare una condizione di ischemia cerebrale. Terapia A. Principi generali. Non sempre l’aritmia necessita di un trattamento farmacologico specifico. L’indicazione alla terapia si pone per tre ordini di motivi: 1) quando la presenza dell’aritmia compromette la funzione di pompa del cuore, alterandone l’emodinamica in maniera significativa; 2) quando l’aritmia anticipa potenzialmente aritmie più gravi (in particolare la fibrillazione ventricolare); 3) quando l’aritmia determina sintomi molesti. Queste condizioni possono essere presenti singolarmente o associate una all’altra. Si crea così una vasta gamma di circostanze nelle quali l’indicazione al trattamento è più o meno imperativa e urgente. Da quanto detto, risulta evidente che la stessa aritmia può richiedere o meno una terapia, secondo il contesto clinico nel quale si manifesta. Il concetto può essere illustrato meglio da alcuni esempi. I battiti prematuri ventricolari, che sono sporadici, insorgono in cuori sani, non vengono avvertiti dal paziente, non alterano la funzione cardiaca in modo significativo e non necessitano di un trattamento farmacologico antiaritmico. I battiti prematuri ventricolari devono invece essere trattati nella fase acuta dell’infarto (quando possono dare il 130 MALATTIE DEL SISTEMA CIRCOLATORIO via ad aritmie più gravi), in presenza di anomalie contrattili del miocardio (dove possono precipitare una situazione di compenso labile) o quando sono avvertiti dal paziente in modo molesto. L’osservazione dei tre parametri ricordati (sintomi, potenzialità di aritmie gravi, conseguenze emodinamiche) guida anche il criterio di tempestività con la quale la terapia va instaurata. Altro esempio: la terapia di una tachicardia atriale parossistica oligosintomatica, che insorge in un soggetto giovane e privo di cardiopatia apparente, può essere dilazionata. La stessa aritmia che si presenti in un anziano con compromissione del sistema cardiovascolare e che induca sintomi (di ischemia cerebrale o miocardica, di shock o di scompenso cardiaco) richiede un trattamento d’urgenza. B. Farmaci antiaritmici. Come già visto, i meccanismi che inducono le aritmie ipercinetiche sono principalmente due: l’alterato automatismo e il rientro. La terapia farmacologica delle aritmie ipercinetiche deve quindi mirare a intervenire variamente su questi due meccanismi: modificando la pendenza della fase 4 del potenziale d’azione delle cellule automatiche e variando opportunamente la velocità di conduzione delle cellule che costituiscono un circuito di rientro. In alcuni sporadici casi il meccanismo patogenetico dell’aritmia è palese (per es., nelle aritmie da rientro della sindrome di Wolff-Parkinson-White). In questi casi la scelta del farmaco deve poggiare su basi razionali. In altri casi il meccanismo patogenetico è ipotetico e la scelta dei farmaci è più empirica. I farmaci antiaritmici modificano le proprietà bioritmiche delle cellule miocardiche, intervenendo variamente sul potenziale d’azione. Attualmente i farmaci antiaritmici impiegati nel trattamento delle aritmie specifiche vengono suddivisi in quattro classi, a seconda delle modalità con cui intervengono sul potenziale d’azione (Tab. 6.2). Alla I classe appartengono farmaci cosiddetti stabilizzatori di membrana, perché deprimono principalmente il canale rapido del sodio durante la fase 0 del potenziale d’azione. La classe I viene a sua volta suddivisa in tre gruppi (A, B e C) a seconda dell’effetto procurato sulla fase 4 delle cellule automatiche, sulla velocità di conduzione e sulla durata del periodo refrattario. La II classe dei farmaci antiaritmici raggruppa i cosiddetti -bloccanti. I -bloccanti esercitano la loro azione antiaritmica antagonizzando l’azione aritmogena delle catecolamine (adrenalina e noradrenalina). La III classe è costituita da farmaci che allungano la fase 3 del potenziale d’azione, allungano la conduzione e il periodo refrattario delle cellule. La IV classe, infine, è costituita da alcuni dei cosiddetti farmaci calcio-antagonisti. Questo gruppo di antiaritmici inibisce la corrente lenta del calcio nella fase 2 del potenziale d’azione, rallenta la fase 4 delle cellule automatiche, riduce la velocità di conduzione e allunga il periodo refrattario. Il trattamento delle aritmie ipocinetiche dipende da due fattori principali: a) se l’aritmia è sintomatica; b) se esistono segni che fanno ragionevolmente ritenere che l’aritmia evolverà verso forme più gravi, pericolose per la vita del paziente. La terapia dell’aritmia ipocinetica può essere farmacologica o elettrica. La terapia farmacologica viene impiegata solo in condizioni di emergenza per via parenterale e per lo più quando si è nell’impossibilità pratica di ricorrere alla terapia elettrica. I farmaci impiegati sono: l’atropina endovena, che svolge un’azione vagolitica e i farmaci simpaticomimetici -stimolanti (isoproterenolo). Quando l’aritmia è sintomatica o minaccia una condizione più grave, è indicato il trattamento elettrico. Il trattamento elettrico consiste nella posa di un catetere stimolatore, che può essere temporaneo o definitivo a seconda che la causa dell’aritmia sia reversibile o meno. Tabella 6.2 - Farmaci antiaritmici. • I Classe A) Chinidina Procamide Diisopiramide Ajmalina Propafenone B) Lidocaina Difenilidantoina Mexiletina C) Flecainide Encainide FASE 4 (PENDENZA) VELOCITÀ DI CONDUZIONE PERIODO REFRATTARIO ↓↓ ↓ ↑↑ ↓ ↓↑ ↓ ↓↓↓ ↓↓↓ ↑↓ • II Classe -bloccanti ↓ ↓ ↑ • III Classe Amiodarone ↓ ↓ ↑↑ • IV Classe Verapamil Diltiazem ↓ ↓ ↑ Riduzione: ↓ lieve Aumento: ↑ lieve ↓↓ moderata ↑↑ moderato ↓↓↓ marcata ↑↑↑ marcato 6 - ARITMIE L’elettrostimolazione consiste nell’inserire un catetere attraverso una vena periferica. Il catetere viene spinto fino alle sezioni destre del cuore. In particolare la punta del catetere dev’essere a contatto con l’endocardio del ventricolo destro. All’estremità opposta il catetere viene collegato con un pacemaker artificiale, che invia impulsi ritmici della frequenza desiderata. Se la stimolazione è temporanea, il catetere viene per lo più inserito attraverso la vena femorale. Il pacemaker viene collegato col catetere esternamente al paziente e ancorato a un supporto rigido (per es., il letto). Nella stimolazione definitiva il pacemaker è di piccole dimensioni. Viene applicato nel sottocute del paziente in un’apposita “tasca” precedentemente preparata. La tasca sottocutanea solitamente è situata sopra il muscolo pettorale. Nel caso della stimolazione definitiva, si introducono, a volte, due cateteri: l’estremità del primo viene posta a contatto con la parete dell’atrio, l’estremità del secondo con la parete del ventricolo destro. Il pacemaker artificiale al quale vengono collegati è in grado di emettere due impulsi separati fra loro da un piccolo intervallo: il primo impulso stimola l’atrio (e determina quindi la contrazione atriale), il secondo il ventricolo (contrazione ventricolare). L’intervallo fra due stimoli può essere variato dall’operatore. La stimolazione bicamerale consente di riprodurre una condizione molto simile a quella fisiologica. In particolare, viene conservato il vantaggio emodinamico fornito dalla sistole atriale. La stimolazione artificiale effettuata attraverso il catetere (o i cateteri nel caso della stimolazione bicamerale) dev’essere programmata secondo il tipo di aritmia ipocinetica che si vuole curare. Vale, di norma, il principio secondo il quale è opportuno preservare quel che rimane dell’attività elettrica spontanea del cuore. Per esempio, nel blocco atrioventricolare di III grado associato a una normale attività del nodo del seno (si veda il paragrafo sulle Aritmie ipocinetiche) è necessario garantire un’efficace stimolazione ventricolare artificiale che dev’essere sincronizzata in modo opportuno con quella, fisiologica, degli atri. Per ottenere questo risultato occorre inserire (come già detto) due cateteri: uno nell’atrio destro e uno nel ventricolo destro. Il catetere atriale, però, in questo caso non serve per stimolare gli atri (gli atri sono già attivati dall’onda di depolarizzazione che origina come di norma dal nodo del seno); ha solo la funzione di “sentire” l’onda di depolarizzazione spontanea e di trasmetterla al pacemaker artificiale. Quando il pacemaker artificiale riceve l’impulso atriale emette (dopo un intervallo ottimale stabilito dall’operatore) l’impulso di stimolazione ventricolare. Il vantaggio è evidente: viene preservata la normale attività sinusale e viene contemporaneamente garantito un rapporto temporale ottimale fra la contrazione degli atri e quella dei ventricoli. Diversa è la condizione del paziente che presenta una riduzione critica dell’attività sinusale. In questo caso è necessario che il pacemaker stimoli sia gli atri sia i ventricoli. Anche l’elettrodo atriale, quindi, dev’essere in grado di stimolare. 131 Come già detto, la stimolazione attraverso l’elettrodo ventricolare avviene dopo un intervallo che può essere modificato dall’operatore. Quelli descritti sono solo due dei possibili, numerosi esempi che si configurano nelle diverse forme di aritmia ipocinetica. Per cercare di descrivere in modo semplice e immediato le caratteristiche dei vari pacemaker artificiali è entrato nell’uso corrente un codice a più lettere. Quello correntemente utilizzato è a tre lettere. La prima lettera stabilisce quale camera viene stimolata: V vuol dire ventricolo, A atrio e D sta per “Dual Chamber” ovvero: sia atrio sia ventricolo vengono stimolati attraverso due cateteri. La seconda lettera definisce la camera dove il catetere ha la cosiddetta funzione di “sensing” (dove, cioè, “sente” l’eventuale impulso spontaneo). Oltre alle ricordate lettere V, A e D in questo caso si aggiunge la O che sta ad indicare l’assenza di una funzione di sensing. La terza lettera definisce il tipo di stimolazione effettuata dal pacemaker in risposta al segnale di sensing. La lettera O stabilisce che il pacemaker impiegato non prevede alcun tipo di risposta ad un eventuale segnale di sensing; la lettera I indica che il pacemaker inibisce l’elettrodo stimolatore in risposta al segnale di sensing; la lettera T sta per “triggering”. Ovvero: quando il segnale di sensing raggiunge il pacemaker questo emette (dopo un intervallo programmato) una stimolazione che raggiunge la camera stimolata (definita dalla prima lettera). La lettera D, infine, indica una risposta duplice: a) l’attività spontanea atriale e ventricolare inibisce sia la stimolazione atriale sia quella ventricolare; b) l’attività atriale non seguita da quella ventricolare determina (secondo la modalità di “triggering”) la stimolazione ventricolare. La scelta del tipo di pacemaker da impiegare dipende dalla situazione clinica e dal tipo di aritmia ipocinetica da correggere. Il pacemaker più impiegati sono del tipo VVI e DDD. Il pacemaker DDD consente una stimolazione bicamerale; ha una funzione di sensing sia in atrio sia in ventricolo e una risposta al segnale di sensing che è di volta in volta di inibizione bicamerale o di stimolazione ventricolare sequenziale all’attività atriale spontanea. Il pacemaker VVI viene applicato soprattutto nei casi di fibrillazione atriale cronica a bassa frequenza ventricolare. Ha un singolo catetere che stimola in ventricolo (prima lettera V); “sente” in ventricolo (seconda lettera V) e risponde allo stimolo di sensing con un’inibizione (terza lettera I) della stimolazione. Il tilting test è positivo in circa il 40% delle sincopi di incerta natura: quelle, cioè, nelle quali l’eziologia è ignota nonostante l’esecuzione di tutti gli altri test diagnostici. In alcuni casi prevale la componente cardiodepressiva (si ha una spiccata bradicardia); in altri prevale la componente vasodepressiva (spiccata ipotensione); in altri, infine, si ha una risposta mista (bradicardia più ipotensione marcate). Il tipo di risposta ottenuto col tilting test fornisce alcune indicazioni sul tipo di provvedimento terapeutico da adottare; per esempio, l’impianto di un pacemaker stimolatore nei casi in cui la risposta cardiodepressiva è particolarmente marcata. 132 MALATTIE DEL SISTEMA CIRCOLATORIO A tutt’oggi mancano dati definitivi sulla reale efficacia di queste procedure a lungo termine. C. Cardioversione elettrica. È una metodica universalmente accettata per la risoluzione di numerosi disturbi del ritmo. Essa consiste nell’applicazione sul torace di una grossa scarica o shock elettrico a corrente continua. Lo shock ha lo scopo di provocare una depolarizzazione simultanea di tutto il miocardio, con conseguente egualizzazione rapida e omogenea dei potenziali transmembrana delle fibre miocardiche: si spengono così i foci ectopici e i rientri, dando modo ai centri fisiologici dell’automatismo di riprendere il comando della depolarizzazione del cuore. Le aritmie per le quali è indicata la cardioversione mediante shock elettrico sono le seguenti: fibrillazione atriale, tachicardia atriale parossistica, tachicardia ventricolare e fibrillazione ventricolare. Lo shock elettrico si effettua mediante un apparecchio chiamato defibrillatore. Il defibrillatore è in grado di erogare una corrente continua in quantità variabile attraverso due elettrodi. Gli elettrodi del defibrillatore vengono posti sul torace del paziente. La scarica elettrica viene erogata attraverso un comando manuale. Se lo shock elettrico è programmato e il paziente è cosciente, è necessario effettuare una breve anestesia perché lo shock è molto doloroso. Il paziente in fibrillazione ventricolare è incosciente. In questo caso lo shock deve essere erogato immediatamente. Se il primo shock non ha successo, nella cardioversione della fibrillazione ventricolare le scariche devono essere ripetute. D. Ablazione con radiofrequenza e procedimenti chirurgici. La moderna aritmologia ha sviluppato tecniche di “ablazione” di zone specifiche del miocardio, sottoponendole all’azione mirata di radiofrequenze veicolate da cateteri. Sono anche stati elaborati procedimenti chirurgici volti a interrompere circuiti rientranti. Di alcuni di questi interventi si parlerà a proposito della fibrillazione atriale. Qui vale la pena di sottolineare l’enorme importanza di questi sviluppi terapeutici che hanno portato alla nascita di una sottospecializzazione nell’ambito della specializzazione più vasta della cardiologia. ARITMIE IPERCINETICHE Come si è anticipato, viene definita ipercinetica un’aritmia nella quale a livello sopraventricolare o a livello ventricolare, o a entrambi i livelli, singoli battiti o sequenze di battiti sono “guadagnati” rispetto al ritmo sinusale. La classificazione delle aritmie ipercinetiche si basa su due parametri: sede d’origine dell’aritmia (Tab. 6.3), tipo del disturbo del ritmo (Fig. 6.6A, B e C). L’aritmia può originare dal nodo del seno o, come più spesso avviene, da un focus ectopico (da aumentato automatismo o da rientro). In quest’ultimo caso, come si è già detto, ha rilevanza se l’origine dell’aritmia è negli Tabella 6.3 - Classificazione delle aritmie ipercinetiche. • Extrasistoli (o battiti ectopici o battiti prematuri) – Atriali – Giunzionali – Ventricolari • Tachicardie sopraventricolari – Sinusale – Tachicardia atriale ectopica (con blocco) – Ritmo giunzionale accelerato – Sindromi da preeccitazione – Tachicardia parossistica sopraventricolare • Tachiaritmie sopraventricolari – Flutter atriale – Fibrillazione atriale • Tachicardie ventricolari – Ritmo ventricolare accelerato – Tachicardia parossistica ventricolare – Torsione di punta – Fibrillazione ventricolare atri o nella regione giunzionale (aritmie ipercinetiche sopraventricolari) o a livello dei ventricoli (aritmie ipercinetiche ventricolari). Per quanto riguarda il tipo del disturbo del ritmo, esso può essere sporadico (per es., battiti prematuri), può determinare aumento ordinato della frequenza (tachicardia), oppure determinare aumento disordinato della frequenza (tachiaritmia). Extrasistoli (o battiti ectopici o battiti prematuri) Si dicono extrasistoli impulsi singoli che episodicamente nascono in sedi diverse dal nodo del seno. Essi si sovrappongono al ritmo di base e ne interrompono la normale sequenza ritmica. Secondo la sede di origine, si distinguono in atriali, giunzionali (nodo atrioventricolare e fascio di His) e ventricolari. A. Extrasistoli atriali. Le extrasistoli possono insorgere in cuori sani o in cuori malati. Nel sano possono essere favorite dallo stress (psichico e fisico) e dall’abuso di sostanze eccitanti (tè, caffè, tabacco). Nel malato esse, solitamente, si manifestano nelle condizioni caratterizzate da una distensione della parete degli atri (cardiopatia mitralica e scompenso congestizio) e nelle cardiopatie ischemiche. A volte le extrasistoli atriali precedono aritmie più gravi, quali la fibrillazione atriale o la tachicardia atriale ectopica. Le extrasistoli atriali sono impulsi che nascono a livello atriale in foci distinti dal nodo del seno. All’ECG sono caratterizzate dalla presenza di onde P che hanno morfologia diversa da quella dell’onda P determinata dal normale ritmo sinusale. Solitamente l’onda P ectopica è separata dall’onda P sinusale del complesso precedente da un intervallo inferiore all’intervallo compreso fra due P sinusali. È per questo motivo che tali battiti vengono chiamati prematuri. 133 6 - ARITMIE A. ARITMIE IPERCINETICHE Extrasistole ventricolare (2A = B + B’) Extrasistole sopraventricolare (2A > B + B’) Pre-eccitazione ventricolare (onda delta) Tachicardia sinusale Tachicardia atriale Tachicardia sopraventricolare Flutter atriale (con blocco variabile) Fibrillazione atriale Ritmo ventricolare accelerato Tachicardia ventricolare Torsione di punta Fibrillazione ventricolare Figura 6.6 - A. Rappresentazione schematica dei principali disturbi del ritmo: aritmie ipercinetiche. (Segue) L’impulso che nasce dal focus ectopico atriale diffonde al nodo atrioventricolare e al fascio di His. Tale diffusione avviene per lo più in maniera del tutto normale e in questi casi la morfologia del complesso QRS del battito prematuro atriale è identica a quella dei battiti del ritmo sinusale di fondo. Tuttavia, se l’impulso ectopico è molto prematuro, diffonde al fascio di His mentre questo è ancora in condizioni di refrattarietà assoluta o relativa. Nel primo caso (refrattarietà assoluta) l’impulso ectopico viene “bloccato” e non raggiunge i ventricoli (all’onda P ectopica non segue il QRS): nel secondo caso (refrattarietà relativa) la conduzione a livello del nodo atrioventricolare e del fascio di His viene rallentata con conseguente allungamento del tempo di conduzione atrioventricolare (P-R dell’ECG). Il recupero dal periodo refrattario a livello della parte più periferica del sistema di conduzione non è uniforme: in particolare, il periodo refrattario del ventricolo destro è più lungo di quello del ventricolo sinistro. L’onda di depolarizzazione di un battito ectopico atriale particolarmente precoce può quindi giungere alle branche mentre la branca destra è ancora in condizioni di refrattarietà e cioè funzionalmente bloccata. In questo caso l’unica via percorribile è la branca sinistra. Il QRS che segue la P ectopica è “aberrante” e mostra abitualmente una morfologia tipo blocco di branca destra (Fig. 6.7). 134 MALATTIE DEL SISTEMA CIRCOLATORIO B. ARITMIE IPOCINETICHE Bradicardia sinusale Battito sfuggita giunzionale Ritmo giunzionale Blocco A-V di I grado PQ > 0,20 sec Blocco A-V di II grado, Mobitz 1 Blocco A-V di II grado, Mobitz 2 Blocco A-V di III grado C. BLOCCHI DI BRANCA Blocco di branca sinistra completo Blocco di branca destra completo Figura 6.6 - B. Aritmie ipocinetiche. C. Blocchi di branca. V1 Figura 6.7 - Extrasistolia sopraventricolare bigemina con conduzione aberrante tipo BBD. 135 6 - ARITMIE L’impulso ectopico atriale può penetrare nel nodo del seno e depolarizzare le cellule del segnapassi sinusale. Questa depolarizzazione precede quella del ritmo di base e “scarica” precocemente le cellule sinusali mentre queste sono avviate a raggiungere il potenziale di soglia nella fase 4 del potenziale d’azione. Il risultato è che anche il recupero delle cellule segnapassi e la successiva depolarizzazione spontanea vengono anticipati. L’esito finale è: a) la sequenza salta un battito (quello interrotto dalla depolarizzazione ectopica); b) il battito sinusale che segue il battito prematuro avviene prima del tempo che si avrebbe per la regolare cadenza del ritmo sinusale, se il battito interrotto dalla depolarizzazione ectopica non fosse stato saltato. Tuttavia, l’intervallo che segue il battito prematuro atriale è più lungo dell’intervallo tra due battiti normali, essendo costituito dalla somma del tempo impiegato dall’impulso prematuro a raggiungere e depolarizzare il nodo del seno più il tempo che il nodo del seno successivamente impiega per raggiungere nuovamente il potenziale di soglia, grazie al suo normale automatismo (è solo questa seconda Figura 6.8 - Lo schema serve a spiegare perché solitamente la pausa che segue un’extrasistole sopraventricolare è “parzialmente compensatoria”, mentre quella che segue un’extrasistole ventricolare è “compensatoria”. In questo tipo di rappresentazione l’asse orizzontale rappresenta il tempo, mentre quello verticale le diverse zone anatomiche del cuore: dall’alto al basso, il nodo senoatriale (S), gli atri (A), il nodo atrioventricolare (A-V) e i ventricoli (V) (questi ultimi sono indicati solo nella sezione B della figura). A. Sono rappresentati due battiti sinusali normali seguiti da un’extrasistole atriale (EX): questa si propaga in basso, verso il nodo atrioventricolare e i ventricoli, e in alto verso il nodo del seno che depolarizza. In questo caso, la terza depolarizzazione del nodo del seno (3) è rappresentata tra parentesi perché non è localmente generata, ma propagata dall’extrasistole. Dopo la depolarizzazione (3), il nodo del seno riprende il suo automatismo e l’intervallo tra (3) e 4 è uguale all’intervallo tra 1 e 2. A livello del nodo atrioventricolare (e dei ventricoli) l’intervallo tra l’extrasistole e il battito numero 4 è più lungo di un intervallo normale (come tra 1 e 2) perché è determinato, a livello del nodo del seno, dalla somma dell’intervallo normale (3)-4 più il tempo che l’impulso generato dall’extrasistole impiega a raggiungere il nodo del seno. Tuttavia la somma dei due intervalli, quello precedente e quello seguente l’extrasistole, è minore della somma di due intervalli normali (ossia la pausa non è compensatoria) perché in (3) il nodo del seno è stato depolarizzato in anticipo. B. Sono rappresentati due battiti sinusali normali, seguiti da un’extrasistole ventricolare (EX): questa si propaga in basso, verso le zone ventricolari non ancora eccitate; e in alto, verso il nodo atrioventricolare e gli atri. La propagazione verso gli atri è rallentata nel nodo atrioventricolare, cosicché l’impulso non fa in tempo a raggiungere il nodo del seno prima che questo si scarichi secondo il suo ritmo normale (impulso 3). Tuttavia, l’impulso 3 generato nel nodo del seno trova il tessuto miocardico in stato di refrattarietà per effetto dell’extrasistole ed arresta la sua propagazione. Solo l’impulso successivo, 4, che deriva dal nodo del seno, riesce a propagarsi. Il battito numero 4, a livello ventricolare, è separato dall’extrasistole da un intervallo più lungo rispetto a quello normale. La somma dei due intervalli, quello precedente e quello seguente l’extrasistole, è uguale alla somma di due intervalli normali (ossia la pausa è compensatoria) perché gli impulsi 2 e 4 sono stati generati normalmente nel nodo del seno, mentre l’impulso sinusale 3, pure generato normalmente, manca ed è sostituito dall’extrasistole. componente quella che determina l’intervallo tra due sistoli normali). Se si sommano gli intervalli che separano il battito prematuro da quelli rispettivamente precedente e seguente, si ottiene un valore inferiore al doppio dell’intervallo tra due battiti normali: questo dipende dal già citato anticipo del battito sinusale che segue quello prematuro. Si può dire perciò che la pausa (allungata) che segue il battito prematuro non compensa l’accorciamento dell’intervallo che lo precede (pausa non compensatoria) (Fig. 6.8). Le extrasistoli atriali, solitamente, non modificano l’attività di pompa del cuore in modo significativo. Esse interferiscono con la gettata sistolica, essendo ridotte la durata della diastole ventricolare comandata dal battito prematuro e l’efficienza della contrazione atriale, ma queste alterazioni riguardano solo i battiti prematuri e sono di limitata importanza se le extrasistoli non sono numerosissime. Le extrasistoli atriali possono essere avvertite dal paziente oppure essere asintomatiche. Se vengono avvertite, vengono descritte come sensazione di cuore che si ferma o di sfarfallio nel petto. 1 2 (3) 4 S A EX A-V 1 2 EX 4 A. 1 2 3 4 5 S A A-V V B. EX 1 2 EX 4 136 Il medico che visita il paziente apprezza un polso nel quale il ritmo regolare è interrotto più o meno frequentemente dall’apparente salto di un battito o da un battito anticipato di scarsa ampiezza (la diminuita gettata sistolica del battito prematuro fa sì che l’onda sfigmica corrispondente sia ridotta e sia mal percepita al polso radiale). All’ascoltazione del cuore si avvertono, di tanto in tanto, dei toni anticipati. All’esame obiettivo è pressoché impossibile distinguere le extrasistoli atriali da quelle che originano in altre sedi (giunzione e ventricoli). Solitamente le extrasistoli atriali sono aritmie benigne che non richiedono uno specifico trattamento antiaritmico. Può essere utile suggerire l’abolizione degli eventuali fattori favorenti l’aritmia. Nei casi caratterizzati da distensione della parete atriale, si deve cercare di migliorare il compenso emodinamico. B. Extrasistoli giunzionali. Le extrasistoli giunzionali sono meno comuni di quelle atriali e ventricolari. Hanno origine nella giunzione (nodo atrioventricolare e fascio di His) e possono diffondere sia verso gli atri che verso i ventricoli. L’ordine con cui l’impulso ectopico diffonde verso atri e ventricoli dipende dalle condizioni in cui questi si trovano quando l’impulso ha origine (condizione di normale eccitabilità o di refrattarietà assoluta o relativa). Gli atri possono pertanto essere attivati prima, dopo o contemporaneamente ai ventricoli. All’ECG l’onda P ha morfologia chiaramente anomala ed è posta prima, dopo il QRS, oppure è sovrapposta ad esso (in questo caso non si vede). Solitamente le extrasistoli giunzionali diffondono ai ventricoli in modo regolare determinando la formazione di QRS con morfologia analoga a quella dei complessi di base. Se il battito prematuro giunzionale è molto precoce, la diffusione dello stimolo nei ventricoli può essere aberrante. MALATTIE DEL SISTEMA CIRCOLATORIO Cause, fisiopatologia, sintomi e segni clinici delle extrasistoli giunzionali sono analoghi a quelle dei battiti prematuri atriali. Anche le extrasistoli giunzionali hanno solitamente andamento benigno e non richiedono un trattamento specifico. Per le somiglianze elencate ed essendo talora difficile distinguere all’ECG le extrasistoli atriali da quelle giunzionali, i due tipi di extrasistoli vengono spesso raggruppati sotto il termine di extrasistoli sopraventricolari. C. Extrasistoli ventricolari (battiti ectopici, battiti prematuri ventricolari). Le extrasistoli ventricolari costituiscono la forma di aritmia più frequente. Si possono manifestare sia in presenza che in assenza di cardiopatia. In assenza di cardiopatia, riconoscono gli stessi fattori favorenti descritti per le extrasistoli atriali. In presenza di cardiopatia, si manifestano molto frequentemente in corso di infarto miocardico, di scompenso ventricolare o di sovradosaggio digitalico. All’ECG le extrasistoli ventricolari sono caratterizzate da complessi QRS più larghi di quelli normalmente condotti (di solito la durata è superiore a 0,14 sec); non sono precedute da onde P; sono separate dal complesso QRS che li precede da un intervallo più breve di quello che separa due complessi QRS del ritmo di base (Fig. 6.9). Nella maggior parte dei casi l’impulso ectopico ventricolare non penetra nel nodo del seno e non interferisce con la formazione ritmica dell’impulso sinusale. Tuttavia il battito del ritmo sinusale successivo a quello che ha preceduto il battito prematuro è mancante, perché quando l’impulso che dovrebbe dargli origine parte dal nodo del seno, trova il miocardio in periodo refrattario a causa del battito prematuro di origine ectopica che si è verificato immediatamente prima (Fig. 6.8). La pausa che separa l’extrasistole dal successivo battito sinusale normalmente condotto (primo battito post- Figura 6.9 - Ritmo sinusale in un soggetto con infarto acuto del miocardio interrotto da due coppie di extrasistoli ventricolari. La prima extrasistole di ciascuna coppia è precoce secondo le caratteristiche del fenomeno R/T. 6 - ARITMIE extrasistolico) è pertanto completamente compensatoria: l’intervallo fra il complesso normale che precede e quello che segue il battito prematuro è uguale a due cicli sinusali. Quando la frequenza del nodo del seno è sufficientemente bassa, è possibile che il battito del ritmo sinusale successivo al battito prematuro giunga ai ventricoli quando questi hanno già recuperato una normale eccitabilità. In questo caso l’extrasistole ventricolare si pone fra due battiti del ritmo sinusale senza interferire con la loro naturale cadenza. Se si verifica questa circostanza, si parla di “extrasistole interpolata”. Alcuni battiti prematuri ventricolari possono diffondere per via retrograda agli atri (battiti “retrocondotti”): la retroconduzione determina la formazione di un’onda P chiaramente anomala che si inscrive sul tratto ST del complesso ectopico. A volte un battito prematuro insorto molto precocemente può dare luogo a quello che viene chiamato fenomeno “R su T” (R/T): il QRS ectopico si inscrive sull’onda T del complesso che lo precede. Questa inscrizione coincide con il periodo cosiddetto vulnerabile dei ventricoli, e quindi il fenomeno R/T può dare il via ad aritmie ventricolari gravi (tachicardie e fibrillazione ventricolare). Se ogni complesso normale viene seguito regolarmente da un’extrasistole, si parla di bigeminismo extrasistolico; se il battito ectopico compare ogni due complessi normali, si parla di trigeminismo e così via. Il bigeminismo è particolarmente frequente in corso di intossicazione digitalica. Le extrasistoli ventricolari possono o meno essere pericolose in base a due parametri: il quadro clinico nel quale si manifestano e i caratteri elettrocardiografici. Il contesto clinico di maggior pericolo è l’infarto miocardico acuto, dove l’extrasistole ventricolare può innescare aritmie mortali. I caratteri elettrocardiografici potenzialmente più pericolosi sono: a) la precocità (associata a fenomeno R/T); b) la ripetitività (battiti prematuri in sequenze di due o tre); c) il polimorfismo (battiti prematuri che originano da foci ventricolari differenti); d) la frequenza (numero in un’ora). In sintesi, si può dire che l’extrasistolia ventricolare sporadica del sano è un’aritmia benigna che non va trattata. L’extrasistolia frequente con eventuali fenomeni R/T, coppie (due battiti prematuri in sequenza) o triplette (tre battiti prematuri in sequenza) che insorge nella fase acuta dell’infarto è una grave emergenza medica. I battiti prematuri interrompono la successione del ritmo cardiaco sinusale e compromettono la sequenza temporale fisiologica di riempimento e svuotamento delle camere cardiache. In particolare, il battito prematuro ventricolare interrompe il riempimento protodiastolico rapido del ventricolo e priva il riempimento ventricolare della quota dovuta alla sistole atriale. Questa interferenza è tanto maggiore quanto più il battito ectopico è precoce. La sistole indotta dal battito prematuro svuota un ventricolo che ha potuto riempirsi solo parzialmente e genera pertanto una gettata sistolica ridotta. Il battito sinusale che segue il battito prematuro ha un tempo di riempimento diastolico più lungo (pausa com- 137 pensatoria). Ne consegue che la quantità di sangue presente al termine della diastole ventricolare del battito sinusale postextrasistolico è maggiore di quella presente al termine della diastole di base (sequenza sinusale regolare) e la gittata sistolica è maggiore. La sistole postextrasistolica può essere avvertita dal paziente come “colpo nel petto”. L’interferenza dei battiti extrasistolici ventricolari sull’emodinamica cardiaca è insignificante quando questi sono isolati, ma può essere grave se i battiti prematuri sono frequenti e il miocardio è malato. I battiti prematuri ventricolari possono essere sintomatici oppure no. Se non sono sintomatici, di solito sono rari. Se sono sintomatici, possono determinare due tipi di disturbo. Il primo è legato alla percezione dell’aritmia: il malato avverte l’irregolarità del battito cardiaco e lo descrive come “cuore che si ferma, perde colpi, dà colpi più forti”, ecc. Il secondo è determinato dai segni dello scompenso (dispnea e segni di bassa portata periferica). I disturbi legati allo scompenso si presentano per lo più quando il cuore ha già gravi deficit contrattili di base. Il medico che visita un paziente con battiti prematuri ventricolari registra l’irregolarità del ritmo del cuore, avvertendo, alla palpazione del polso periferico, il battito extrasistolico come una pulsazione anticipata di scarsa ampiezza, oppure come assenza di un battito. Il primo battito postextrasistolico è invece particolarmente ampio. I battiti extrasistolici precoci, infatti, inducono contrazione dei ventricoli ancora vuoti di sangue o quasi, cosicché la gettata pulsatoria è scarsa o nulla. In questo caso l’ascoltazione del cuore fa percepire, in coincidenza con l’extrasistole, un primo tono isolato, vale a dire non seguito da secondo tono (questo manca perché le valvole semilunari non hanno fatto in tempo ad aprirsi). Questa è la ragione per cui l’ascoltazione del cuore deve sempre accompagnare la palpazione del polso radiale. La sola palpazione, infatti, può essere fonte di errore in presenza di un’extrasistolia ventricolare bigemina molto precoce: in questo caso, dato che al polso si sentono solo i battiti sinusali e non quelli extrasistolici, si avrebbe l’impressione di una bradicardia sinusale ritmica (per es., 50 battiti/min) mentre l’ascoltazione permette di capire che la frequenza cardiaca è di 100 (50 battiti efficaci e 50 extrasistoli inefficaci). La terapia delle extrasistoli ventricolari deve essere principalmente rivolta alla rimozione della condizione che ha favorito il manifestarsi dell’aritmia: in particolare, va interrotta l’eventuale assunzione di sostanze eccitanti, va interrotta la digitale in caso di sovradosaggio, va corretto uno stato di scompenso cardiaco o un’eventuale ipopotassiemia. L’indicazione a un trattamento antiaritmico farmacologico è legata alla frequenza con cui l’aritmia si manifesta (numero di extrasistoli all’ora), ai sintomi che induce e alla condizione patologica alla quale è associata. Durante infarto miocardico acuto, in particolare, i battiti prematuri ventricolari richiedono un trattamento farmacologico. I farmaci principalmente impiegati sono quelli della I classe e l’amiodarone. Nell’infarto miocardico si usa la lidocaina per somministrazione endovenosa. 138 MALATTIE DEL SISTEMA CIRCOLATORIO Tachicardie sopraventricolari A. Tachicardia sinusale. La tachicardia sinusale dell’adulto è caratterizzata da una sequenza ritmica di battiti con frequenza superiore a 100/min che partono, come di norma, dal nodo del seno e danno luogo a una regolare successione dell’onda di attivazione del cuore. La frequenza di battiti è variabile da un momento all’altro, ma solitamente è compresa fra 100 e 180. La tachicardia sinusale si presenta all’ECG con una successione regolare di onde P di morfologia normale seguite regolarmente dal QRS. L’intervallo PQ è uguale o lievemente accorciato rispetto a quello di base. Il massaggio del glomo carotideo procura solo una lieve riduzione della frequenza nel momento in cui viene effettuato. Al termine del massaggio la frequenza risale rapidamente ai valori precedenti. La tachicardia sinusale, solitamente, insorge e termina senza quelle variazioni repentine della frequenza cardiaca che sono proprie della tachicardie ectopiche, come effetto riflesso di circostanze fisiologiche (esercizio fisico, emozioni) e patologiche (febbre, ipertiroidismo, scompenso cardiaco, ipotensione). All’ascoltazione il medico avverte toni frequenti, leggermente variabili con gli atti del respiro: spesso l’intensità dei toni è ridotta quando la tachicardia è secondaria a scompenso cardiaco. In questo caso sono presenti anche gli altri segni dello scompenso. Il polso periferico è piccolo e frequente. B. Tachicardia atriale ectopica con blocco. Si tratta di una forma non molto frequente ma importante dal punto di vista clinico; si manifesta praticamente solo in soggetti malati. La tachicardia atriale ectopica esprime, nella maggior parte dei casi, un quadro di intossicazione digitalica, che si manifesta specie in presenza di ipopotassiemia. A volte l’aritmia complica il decorso clinico di una pneumopatia ostruttiva cronica. Nella tachicardia atriale ectopica con blocco l’attivazione nasce a livello atriale, in una sede diversa da quella del nodo del seno. Il focus ectopico stimola gli atri con una frequenza compresa fra i 150 e i 220 impulsi/min, ma una parte degli impulsi non raggiunge i ventricoli perché viene bloccata a livello della giunzione. Il blocco è dovuto al fatto che il tessuto che costituisce le strutture della giunzione non riesce a recuperare, fra uno stimolo e l’altro, una condizione di normale eccitabilità. La frequenza con cui i ventricoli vengono stimolati è dunque più bassa di quella di stimolazione atriale: tanto più bassa quanto maggiore è il grado di blocco giunzionale. All’ECG si osservano onde P di aspetto anomalo in successione rapida, a volte seguite e a volte no da un QRS che solitamente ha una morfologia analoga a quella del complesso normale. Quando non precedono un complesso QRS, le onde P sono seguite da un tratto isoelettrico (Fig. 6.10). La frequenza del QRS deriva ovviamente dal grado di blocco atrioventricola- Figura 6.10 - Tachicardia atriale con conduzione 1:1. Durante la registrazione di base l’onda di attivazione atriale può essere confusa con l’onda T del complesso che precede. Con la stimolazione del seno carotideo si ottiene un transitorio arresto dell’attività atriale (↓); segue (*) una ripresa dell’attività atriale, che conduce per una volta ai ventricoli. Dopo altre due onde atriali non condotte (↓↓), riprende la tachicardia atriale ectopica con conduzione 1:1. 139 6 - ARITMIE re. Il blocco può essere fisso (il QRS segue regolarmente ogni seconda, terza, quarta, ecc., P ectopica) e variabile. L’effetto del massaggio del glomo carotideo è quello di aumentare temporaneamente il grado di blocco con una conseguente riduzione della frequenza del QRS. I pazienti con tachicardia atriale ectopica avvertono cardiopalmo che di solito inizia all’improvviso. La tachicardia prolungata e con una frequenza ventricolare elevata (basso grado di blocco atrioventricolare) può fare precipitare un quadro di scompenso cardiaco o di angina. Il medico, all’ascoltazione e alla palpazione del polso, avverte una frequenza cardiaca elevata per lo più (quando il blocco è fisso) rigorosamente ritmica; mancano le variazioni respiratorie della tachicardia sinusale. Si può registrare un calo pressorio e possono essere presenti segni di una bassa portata periferica. La terapia dev’essere tempestiva se sostiene un quadro di scompenso o di angina. In questi casi può essere utile ricorrere senz’altro alla cardioversione elettrica. Se l’aritmia è tollerata bene, la cardioversione può essere dilazionata e deve essere preceduta dalla correzione di eventuali squilibri elettrolitici oltre che dalla sospensione della digitale. C. Ritmo giunzionale accelerato. Il ritmo giunzionale accelerato (detto anche tachicardia nodale non parossistica) è caratterizzato da un esaltato automatismo di pacemaker siti intorno al nodo atrioventricolare. In condizioni normali la frequenza di attivazione intrinseca di questi pacemaker è di circa 40-60 battiti/min. In condizioni normali la frequenza aumenta e il ritmo ectopico prende il sopravvento sull’attivazione fisiologica del nodo del seno. L’aritmia è frequente in condizioni di tossicità digitalica, di febbre reumatica o di infarto miocardico diaframmatico. Da un punto di vista elettrocardiografico, il ritmo giunzionale accelerato si esprime come la successione regolare di complessi QRS di morfologia normale con una frequenza compresa fra i 70 e 140 battiti/min. La frequenza di attivazione aumenta con l’esercizio fisico e non risente del massaggio del glomo carotideo. Gli atri possono essere attivati per via retrograda (a questo proposito, si confronti quanto già detto a proposito delle extrasistoli giunzionali). In altri casi l’attivazione atriale può derivare come di norma dal nodo del seno; si hanno così onde P di aspetto normale, ma dissociate dall’attività ventricolare. In altre parole atri e ventricoli sono sottoposti a due ritmi-guida differenti, che nascono da due pacemaker distinti e che non hanno alcun rapporto temporale fra di loro. Il ritmo giunzionale accelerato di solito non dà sintomi e raramente richiede una terapia specifica; è indicata solo la terapia delle condizioni che ne hanno favorito l’insorgenza. La diagnosi coi soli segni fisici è pressoché impossibile: occorre l’ECG. D. Sindromi di pre-eccitazione ventricolare. Col termine di sindrome di pre-eccitazione ventricolare si intende un gruppo di condizioni patologiche nelle quali una parte del miocardio ventricolare riceve precocemente l’impulso di eccitazione che proviene dagli atri. In altre parole l’attivazione che proviene dagli atri stimola alcuni settori dei ventricoli prima di quanto avverrebbe se l’impulso seguisse solo la normale via di conduzione rappresentata dalla giunzione atrioventricolare. Si è già visto che atri e ventricoli, in condizioni normali, sono elettricamente isolati e che lo stimolo proveniente dagli atri può raggiungere i ventricoli solo attraverso le strutture nel nodo A-V. Le sindromi di pre-eccitazione ventricolare sono possibili per la presenza di vie accessorie anomale, che connettono atri e ventricoli in aggiunta alle strutture normali del nodo A-V. Tali vie accessorie, a loro volta, sono caratterizzate da strutture muscolari lungo le quali lo stimolo viene condotto più velocemente che lungo il nodo A-V. Le vie accessorie possono essere congenite o acquisite. Esse possono connettere (Figg. 6.11 e 6.12): • la muscolatura atriale con la muscolatura ventricolare (fascio di Kent); • la muscolatura atriale con la parte distale delle strutture del nodo A-V (fibre di James); • il nodo A-V con la muscolatura ventricolare (fibre di Mahaim). La forma più frequente di pre-eccitazione ventricolare è quella dovuta alla presenza del fascio di Kent responsabile della sindrome di Wolff-Parkinson-White (WPW). Fascio di Kent Fibre di James Fibre di Mahaim Figura 6.11 - Nel disegno sono rappresentate le principali vie anomale di connessione atrioventricolari. Il fascio di Kent connette la muscolatura atriale con quella ventricolare; il fascio di James la muscolatura atriale con le vie di conduzione atrioventricolari; le fibre di Mahaim mettono in connessione le vie di conduzione atrioventricolari con la muscolatura ventricolare. 140 MALATTIE DEL SISTEMA CIRCOLATORIO FASCIO DI KENT DEVIAZIONE DI JAMES PR corto Onda ⌬ QRS ampio PR corto QRS normale FIBRE DI MAHAIM JAMES + MAHAIM PR normale Onda ⌬ QRS ampio PR corto Onda ⌬ QRS ampio Figura 6.12 - Nello schema sono riassunti i principali aspetti elettrocardiografici che caratterizzano la presenza di vie anomale. Il fascio di Kent è un fascio muscolare anomalo, formato per lo più da fibre contrattili, che perfora lo scheletro fibroso del cuore e connette direttamente la muscolatura dell’atrio destro o sinistro con quella del ventricolo corrispondente a livello del setto e della parete libera. La sindrome di WPW è caratterizzata dalla presenza di: • normali onde P; • intervallo P-R più corto del normale (inferiore o uguale a 0,11 sec.); • presenza di un impastamento iniziale nel QRS (onda delta); • durata del QRS aumentata e complessi QRS di morfologia anomala; • elevata tendenza all’insorgenza di aritmie ipercinetiche sopraventricolari (in particolare tachicardie parossistiche). L’onda P è normale perché l’eccitazione atriale avviene come di norma. Il P-R è più corto del normale perché lo stimolo raggiunge i ventricoli attraverso la “scorciatoia” rappresentata dal fascio anomalo. L’onda ⌬ è dovuta all’eccitazione di quel settore di muscolo ventricolare che riceve l’impulso precocemente attraverso la via anomala. Il QRS è allargato perché l’inizio è precoce per la presenza dell’onda ⌬. La morfologia del QRS è alterata perché il QRS rappresenta la “fusione” fra lo stimolo che scende precocemente lungo la via anomala e quello che raggiunge i ventricoli attraverso la via normale. La presenza della via anomala rende ragione dell’elevata incidenza di aritmie ipercinetiche sopraventricolari presenti nei pazienti con WPW. Le aritmie dei pazienti con WPW sono dovute al fenomeno del rientro. Il rientro si attua utilizzando la via anomala e la normale via di conduzione A-V. Solitamente, il periodo refrattario della via anomala è maggiore di quello del nodo A-V. Un battito prematuro sopraventricolare può trovare la via anomala in condizioni di refrattarietà e il nodo A-V normalmente eccitabile. In conseguenza di ciò, l’impulso segue la via normale per via anterograda iniziando la depolarizzazione ventricolare. Quando il miocardio ventricolare viene eccitato, la via anomala può avere recuperato una normale eccitabilità: l’impulso può, pertanto, essere condotto per via retrograda lungo la via anomala. Dopo il rientro, l’impulso può nuovamente ripercorrere per via anterograda la via normale e così via. Ha inizio, pertanto, un’aritmia ipercinetica sopraventricolare che può essere interrotta da un nuovo battito sopraventricolare. Il battito prematuro può infatti modificare ancora le caratteristiche di refrattarietà delle due vie e interrompere il rientro. Classicamente vengono distinti due tipi di sindromi di WPW in relazione alla morfologia dell’onda delta e del QRS. La diversa morfologia dell’onda delta è espressione di una diversa collocazione del fascio di Kent. Il punto in cui il fascio di Kent “abbocca” il ventricolo stabilisce quale parte di miocardio viene eccitata per prima. La successione dell’eccitazione ventricolare rende ragione sia dell’aspetto dell’onda delta, sia di quello del QRS. Semplicisticamente si può distinguere un WPW di tipo A e uno di tipo B. 141 6 - ARITMIE Nel tipo A l’attivazione precoce è a carico del ventricolo sinistro; nel tipo B è a carico del ventricolo destro. Nel tipo A l’onda ⌬ è diretta in avanti e i complessi QRS sono per lo più positivi nelle precordiali destre; nel tipo B l’onda ⌬ è diretta posteriormente e a sinistra e i complessi QRS sono per lo più negativi nelle precordiali destre. E. Tachicardia atriale parossistica o tachicardia parossistica sopraventricolare. La tachicardia atriale parossistica deriva da un ritmo ectopico che insorge nella parte bassa degli atri o meglio nella giunzione atrioventricolare. È indicata con vari nomi (tachicardia giunzionale, tachicardia parossistica sopraventricolare reciprocante, tachicardia nodale parossistica), di cui i più usati sono quello di tachicardia parossistica atriale (è il termine più tradizionale) o di tachicardia parossistica sopraventricolare (questo termine sottolinea che la tachicardia origina sopra i ventricoli, senza compromettersi sull’esatta localizzazione anatomica dell’origine, che difficilmente può essere definita con certezza, e ciò, del resto, non ha molta importanza). Il termine di “parossistica” sottolinea che la tachicardia è generalmente accessionale, con inizio improvviso (percepito dal paziente) e fine improvvisa (non sempre percepita subito). La tachicardia atriale parossistica è la forma più comune di tachicardia parossistica nel bambino e nel giovane. Può manifestarsi per la prima volta a qualunque età e solitamente è innescata da un’extrasistole atriale, giunzionale o ventricolare che dà il via al rientro. Il meccanismo patogenetico ritenuto più frequente nella genesi di quest’aritmia è quello del rientro. Il rientro può avvenire o interamente nelle strutture del nodo atrioventricolare oppure utilizzando il nodo atrioventricolare e una via precostituita nell’ambito della giunzione atrioventricolare. Spesso si tratta di vie accessorie che connettono atri e ventricoli in modo anomalo (si veda la sindrome di Wolff-Parkinson-White). Nella tachicardia atriale parossistica gli atri vengono attivati per via retrograda. All’ECG la tachicardia atriale parossistica è caratterizzata dalla sequenza ritmica di complessi compresa fra i 150 e 230 e che nella maggior parte dei casi hanno morfologia normale. L’onda P ha morfologia chiaramente anomala e ha un rapporto fisso con il QRS. L’aritmia di solito esordisce e cessa in modo improvviso. Il massaggio del glomo carotideo eseguito in corso di tachicardia o non fa nulla o risolve l’aritmia ripristinando il ritmo sinusale. La sintomatologia e l’obiettività della tachicardia atriale parossistica sono simili a quelle già descritte per la tachicardia atriale ectopica. La terapia della tachicardia atriale parossistica può essere distinta a seconda che si debba trattare l’evento acuto o se ne voglia impedire l’insorgenza. Nel primo caso si devono applicare le comuni manovre vagali (massaggio del glomo carotideo, manovra di Valsalva) eventualmente seguite dalla somministrazione di verapamil per via venosa. Qualora manovre vagali e verapamil fossero inefficaci, si può ricorrere alla cardioversione elettrica. La cardioversione elettrica è indicata specie se l’aritmia è prolungata e male tollerata dal paziente. Il trattamento cronico consiste nel cercare di prevenire le extrasistoli responsabili dell’avvio del rientro. Allo scopo si impiegano i farmaci antiaritmici della I classe o l’amiodarone. È possibile anche cercare di intervenire sul meccanismo di rientro impiegando farmaci che modificano la conduzione a livello del nodo atrioventricolare: digitale, propranololo, ecc. Tachiaritmie sopraventricolari A. Flutter atriale. Il flutter atriale è caratterizzato da un’attivazione atriale anomala molto rapida (compresa solitamente tra 220 e 360 impulsi/min) e regolare. L’impulso che proviene dagli atri viene vanamente “bloccato” a livello della giunzione atrioventricolare. Dall’entità di questo blocco dipende la frequenza di stimolazione ventricolare. Il flutter atriale solitamente insorge in pazienti affetti da: a) cardiopatia caratterizzata da distensione delle pareti degli atri (scompenso congestizio, valvulopatie mitralica e tricuspidale); b) processi infiammatori a carico degli atri; c) processi infiltrativi atriali. Nel flutter atriale all’elettrocardiogramma l’attivazione atriale è rappresentata dalla successione regolare di onde d’aspetto costante (cosiddette F) ben visibili nelle derivazioni D2 D3, aVF e V (Fig. 6.13), specie quando il blocco atrioventricolare è più marcato (quando cioè più di due onde F in successione non trasmettono l’impulso al ventricolo). Le onde F non sono separate da tratti di linea isoelettrica, ma si succedono una dopo l’altra con un aspetto complessivo che è stato definito “a denti di sega”. Il complesso QRS segue l’attivazione atriale in modo per lo più irregolare (blocco variabile). La morfologia del complesso ventricolare è solitamente analoga a quella normale. Il massaggio del seno carotideo aumenta il grado di blocco giunzionale e riduce temporaneamente la frequenza dei QRS. Il flutter atriale determina un’alterazione dell’emodinamica del cuore caratterizzata dalla perdita di una sistole atriale efficace e dalla riduzione del tempo diastolico di riempimento ventricolare e coronarico. La riduzione del tempo diastolico di riempimento ventricolare e coronarico è tanto maggiore quanto è maggiore la frequenza ventricolare. Il paziente avverte cardiopalmo in misura proporzionale alla frequenza cardiaca. Il cardiopalmo può essere “ritmico” e “aritmico”, a seconda che il blocco sia regolare o variabile. Possono essere presenti angina e i sintomi dello scompenso. L’obiettività è caratterizzata dall’ascoltazione di toni frequenti (ritmici o aritmici) accompagnati eventualmente da una riduzione della pressione arteriosa e da segni di bassa portata. Il trattamento del flutter atriale deve 142 MALATTIE DEL SISTEMA CIRCOLATORIO V1 F F F V2 V3 Figura 6.13 - Flutter atriale con blocco 2:1. La frequenza delle onde F è di 280/min; quella dei QRS di 140. essere tempestivo se c’è angina o scompenso. Il farmaco di prima scelta è la digitale. Se la digitale non è efficace, è opportuno ricorrere alla cardioversione elettrica. B. Fibrillazione atriale. Nella fibrillazione atriale gli atri vengono eccitati in maniera caotica, disorganizzata, con una frequenza di attivazione variabile da 400 a 650 impulsi/min. Per lo più questo deriva dalla formazione di multipli, piccoli circuiti rientranti originati negli atri che collidono, si estinguono e si riformano. Un secondo meccanismo è stato riconosciuto recentemente ed è dovuto alla presenza di un focus che si scarica rapidamente e che è di solito collocato vicino allo sbocco delle vene polmonari. Dal punto di vista elettrocardiografico questo focus può simulare la comparsa di fibrillazione atriale. In entrambi i casi l’impulso viene variamente bloccato a livello del nodo atrioventricolare: il grado di questo blocco determina la frequenza di attivazione dei ventricoli. L’attivazione dei ventricoli avviene in modo del tutto irregolare con una frequenza solitamente elevata: 140-160 impulsi/min (Fig. 6.14). La fibrillazione atriale può insorgere in soggetti normali (specie se anziani) o malati. Nel giovane, la fibrillazione atriale si associa spesso alla cardiopatia reuma- Figura 6.14 - Fibrillazione atriale. tica, ischemica, alla tireotossicosi e alla cardiopatia ipertensiva. Esistono, tuttavia, anche casi di persone che senza cause organiche apparenti presentano crisi di fibrillazione atriale. In questa forma, il disturbo del ritmo viene chiamato “fibrillazione atriale solitaria”. Da un punto di vista elettrocardiografico, la fibrillazione atriale è caratterizzata dall’assenza di onde P. Le onde P sono sostituite da deflessioni di piccola ampiezza assolutamente irregolari; a queste seguono complessi QRS che si succedono a intervalli sempre diversi tra loro. L’irregolarità della risposta ventricolare rende variabile il tempo di recupero della normale eccitabilità. Questa variabilità spiega la frequente formazione di complessi QRS con morfologia aberrante. Come si è visto, il tempo di conduzione del tessuto del ventricolo destro è più lungo di quello del ventricolo sinistro: ne consegue che il complesso QRS condotto con aberranza ha, di solito, una morfologia tipo blocco di branca destra. La fibrillazione atriale può verificarsi in crisi episodiche o può essere persistente. Esperimenti compiuti su animali nei quali erano indotti ripetuti episodi di fibrillazione atriale hanno dimostrato che alla fine l’aritmia diveniva persistente. L’effetto elettrofisiologico era un 6 - ARITMIE marcato accorciamento del periodo refrattario degli atri e la perdita del normale allungamento della refrattarietà atriale quando la crisi di fibrillazione era superata e la frequenza cardiaca rallentava. Questo fenomeno, che può essere reversibile se il ritmo sinusale è mantenuto, viene chiamato rimodellamento elettrico atriale. La persistente tachicardia ha anche effetti sul miocardio, che presenta alterazioni ultrastrutturali cui corrispondono disfunzioni ventricolari. Questa cardiomiopatia che è mediata dalla tachicardia è spesso reversibile dopo ritorno al ritmo sinusale o se, pur persistendo la fibrillazione, la frequenza ventricolare viene mantenuta bassa con la terapia. Il paziente con fibrillazione atriale può essere sintomatico oppure no. La mancanza di sintomi è abituale se la somministrazione di digitale ha reso la frequenza ventricolare normale. I sintomi possono essere legati alla percezione dell’aritmia e alle sue conseguenze emodinamiche. La percezione dell’aritmia consiste nel sentire battiti frequenti e irregolari. Le conseguenze emodinamiche sono secondarie sia alla variabilità della durata del riempimento ventricolare e coronarico, sia alla perdita completa della contrazione atriale. Se la frequenza è molto elevata, la contrattilità è compromessa e il circolo coronarico è insufficiente, la fibrillazione atriale può fare precipitare una condizione di scompenso o angina. Il medico può diagnosticare facilmente una fibrillazione atriale con l’ascoltazione del cuore: i toni sono completamente aritmici, con pause sempre diverse tra un tono e il corrispondente successivo; anche l’intensità del primo tono varia continuamente. In presenza di fibrillazione atriale la frequenza cardiaca va valutata con l’ascoltazione. Il polso periferico, infatti, consente di registrare solo le sistoli emodinamicamente efficaci; non possono essere percepite quelle che insorgono molto vicino alla sistole che le precede quando il riempimento ventricolare è insufficiente (si veda quanto già detto a proposito delle conseguenze emodinamiche delle extrasistoli ventricolari precoci). A parte alcuni casi di fibrillazione atriale solitaria, la prognosi di questo disturbo del ritmo deve esser considerata sfavorevole. È certamente vero che molte persone con questo disordine possono condurre a lungo una vita soddisfacente, ma è anche vero che questa condizione facilita l’insorgenza di scompenso cardiaco (per la perdita di quella quota del precarico dovuta alla contrazione atriale, oltre che per la cardiomiopatia dovuta alla tachicardia), il peggioramento della cardiopatia ischemica (per l’aumento del consumo di ossigeno dovuto alla tachicardia) e, soprattutto, espone al rischio di tromboembolie. Per ragioni anatomiche, queste si localizzano prevalentemente nel distretto encefalico e sono un’importante causa di attacchi ischemici e ictus cerebrali. Qui di seguito riportiamo le linee generali di terapia della fibrillazione atriale, precisando che sono ricavate da una sola fonte (Falk, 2001), che i dettagli vanno ricercati sui testi di terapia e che molti procedimenti terapeutici è bene che siano lasciati nelle mani di specialisti. 143 1. Fibrillazione atriale di recente insorgenza. La cardioversione elettrica si rende necessaria in presenza di edema polmonare o angina instabile, negli altri casi è sufficiente un trattamento farmacologico. Occorre distinguere i procedimenti volti a ridurre la frequenza dei ventricoli e a dar sollievo all’ammalato e quelli diretti a reintrodurre il ritmo sinusale. La digossina è piuttosto efficace nel rallentare la frequenza cardiaca, ma la sua massima azione è conseguita soltanto dopo varie ore. Un controllo più rapido della frequenza cardiaca è ottenuto con la somministrazione endovenosa di farmaci -bloccanti o di calcio-antagonisti, come il diltiazem e il verapamil. La conversione spontanea a ritmo sinusale entro 24 ore dall’inizio della fibrillazione atriale è comune e si verifica in circa i due terzi dei casi. Dopo ritorno al ritmo normale, una terapia antiaritmica di fondo e un trattamento anticoagulante sono indicati solamente in pazienti che hanno avuto o rischiano un episodio tromboembolico. Naturalmente, non è possibile sapere in anticipo se la regressione della fibrillazione atriale avverrà spontaneamente e una terapia antiaritmica precoce può essere considerata allo scopo di ottenere una cardioversione farmacologica, ma solo in pazienti nei quali l’aritmia è durata meno di 48 ore, altrimenti esiste il rischio che si siano formati coaguli negli atri fibrillanti e che si possano verificare embolie. Se non c’è questa controindicazione si possono impiegare vari farmaci per via endovenosa (per es., amiodarone o procainamide) o anche orale (per es., flecainide, propafenone, chinidina o disopiramide). In molti pazienti non è possibile stabilire da quanto tempo duri effettivamente la fibrillazione. In questo caso è consigliabile instaurare una terapia anticoagulante e tentare la cardioversione farmacologica solo dopo tre settimane. Un metodo per accorciare i tempi è di controllare preventivamente l’esistenza di trombi negli atri mediante l’ecocardiografia transesofagea. Se in seguito a questo esame si può escludere la presenza di trombi negli atri, si può eseguire il trattamento antiaritmico anche dopo un periodo più breve di terapia anticoagulante. 2. Crisi parossistiche ricorrenti di fibrillazione atriale. Questi pazienti presentano un significativo rischio di episodi tromboembolici e perciò debbono essere posti in terapia anticoagulante. Questa indicazione non vale per i ripetuti episodi di fibrillazione solitaria, riconoscibili perché si verificano in soggetti di età inferiore ai 65 anni, normotesi ed esenti da qualsiasi cardiopatia rilevabile. I farmaci antiaritmici impiegati in questi casi includono il propafenone, la flecainide e il sotalolo. 3. Fibrillazione atriale persistente. Si definisce così quest’alterazione del ritmo quando dura più di sette giorni. In questo caso la decisione sull’opportunità di cercare di riportare al ritmo sinusale il paziente è delicata. In una fibrillazione atriale persistente la probabilità di successo della cardioversione farmacologica è compresa tra il 10 e il 30%. Comunque, nel caso di un recupero del ritmo sinusale, sia con cadioversione far- 144 macologica che con quella elettrica, è opportuna una terapia continua per evitare la recidiva della fibrillazione. Non è detto che, se uno specifico farmaco antiaritmico ha fallito nel riportare la fibrillazione a ritmo sinusale, quello stesso farmaco non sia efficace nel mantenere il ritmo sinusale dopo cardioversione elettrica. Vi sono pochi studi che comparano a questo riguardo i vari farmaci antiaritmici, ma è stato dimostrato che l’amiodarone è più efficace del sotalolo e del propafenone. Ovviamente, in casi di fibrillazione persistente è molto raccomandata una terapia anticoagulante a lungo termine, per esempio per via orale con dicumarolici. Si è discusso se un trattamento con aspirina possa essere ugualmente efficace, ma non esistono dimostrazioni sicure in questo senso. In genere, il trattamento anticoagulante con dicumarolici viene considerato indicato in presenza dei seguenti fattori di rischio per episodi tromboembolici: pregressi attacchi ischemici o veri e propri episodi di ictus, scompenso cardiaco o comunque disfunzione del ventricolo sinistro, età superiore ai 75 anni. Questa indicazione deve essere pesata tenendo conto della compliance del paziente nei riguardi di un buon controllo della terapia anticoagulante e del fatto che le persone anziane, con questi trattamenti, sono più facilmente soggette a emorragie. Inoltre, uno scompenso che determini una stasi cronica a livello epatico rende molto sensibile un paziente all’azione dei dicumarolici. Perciò, nel giudizio sulla terapia anticoagulante si devono valutare vantaggi e pericoli. 4. Fibrillazione resistente ai trattamenti farmacologici. La combinazione di una fibrillazione atriale persistente e una disfunzione sistolica è un problema significativo quando la digossina non riesce a controllare la frequenza ventricolare. In questi casi sono necessari trattamenti più interventistici, che elenchiamo qui di seguito. • Ablazione del nodo atrio-ventricolare con radiofrequenze e impianto di un pacemaker. In questo modo si determina un blocco atrio-ventricolare completo e il ritmo cardiaco è comandato dal pacemaker. • Ablazione del focus a rapida scarica che, se collocato nei pressi dello sbocco delle vene polmonari, può essere causa di alcuni casi di fibrillazione atriale. • Intervento del “labirinto”. È un intervento cardiochirurgico e il suo impiego è stato suggerito quando esistono anche altre indicazioni per la cardiochirurgia. Consiste nell’esecuzione di incisioni nella parete degli atri, in modo da creare una via stretta e tortuosa per la propagazione dello stimolo dal nodo seno-atriale a quello atrio-ventricolare. Le incisioni sono collocate in modo tale che nessuna area sia larga abbastanza da consentire multipli circuiti di rientro. • Terapia con pacemaker. Questa serve a prevenire le crisi di fibrillazione ed è basata sul principio che, se gli atri sono stimolati da un pacemaker, si evita la conduzione interatriale o intra-atriale inomogenea o ritardata che predispone allo sviluppo della fibrillazione atriale. MALATTIE DEL SISTEMA CIRCOLATORIO • Defibrillatori atriali impiantabili. Anche questi sono indicati nella fibrillazione atriale ricorrente e sono programmati in maniera da terminare la fibrillazione con uno shock elettrico ogni volta che questa interviene. Tachicardie ventricolari A. Ritmo idioventricolare accelerato (RIVA). Il RIVA si manifesta per esaltato automatismo di un pacemaker situato nel tessuto di conduzione distalmente al fascio di His. I pacemaker ventricolari hanno una frequenza di eccitazione intrinseca che è di circa 30-40/min. In condizioni normali la loro attività è quindi completamente mascherata dall’attività automatica del nodo del seno. In condizioni patologiche l’automatismo idioventricolare può aumentare fino a 60120/min e costituire il ritmo-guida del cuore. Il RIVA compare soprattutto in presenza di infarto diaframmatico acuto, di tossicità digitalica, di ipopotassiemia e di blocco atrioventricolare completo. All’ECG l’aritmia si presenta con una frequenza ritmica di complessi QRS di aspetto chiaramente anomalo e con una durata superiore ai 0,14 sec. A volte sono visibili delle onde P sinusali, dissociate dai QRS; altre volte possono essere presenti dei “battiti di fusione”. I battiti di fusione sono complessi QRS con morfologia intermedia fra il complesso anomalo e quello normale: essi sono dovuti alla contemporanea diffusione nei ventricoli dello stimolo condotto dall’onda P sinusale attraverso il nodo atrioventricolare e quello originato dal pacemaker ventricolare ectopico. Solitamente il RIVA non dà sintomi: ha un’evoluzione favorevole e non richiede trattamento. Può essere necessaria una terapia medica quando la dissociazione fra attività atriale e ventricolare comporta una compromissione emodinamica. In questi sporadici casi sono solitamente sufficienti piccole dosi di atropina per via venosa per fare aumentare la frequenza sinusale e sopprimere l’attività del centro ectopico. La diagnosi di RIVA senza l’ausilio dell’ECG è spesso impossibile. B. Tachicardia ventricolare. La tachicardia ventricolare è un’aritmia ipercinetica caratterizzata dalla sequenza di un minimo di 3 (spesso sono molti) battiti ectopici a origine ventricolare in immediata successione, aventi una frequenza superiore ai 120/min. La tachicardia ventricolare può essere non sostenuta o sostenuta. È non sostenuta se dura meno di 30 sec; è sostenuta se dura più di 30 sec. La frequenza, contrariamente a quello che si osserva nelle tachicardie parossistiche sopraventricolari, non è rigidamente fissa, ma varia leggermente (più o meno di 5 battiti/min, per es.) da un minuto all’altro. La tachicardia ventricolare insorge quasi sempre in soggetti portatori di gravi cardiopatie: ischemica, reumatica, primitiva. In rapporto a ciò, gli accessi di tachicardia ventricolare tendono a ripetersi, più o meno prolungati, finché la condizione che li ha favoriti non sia migliorata. 6 - ARITMIE 145 Figura 6.15 - Tachicardia ventricolare. All’ECG la tachicardia ventricolare si presenta con una successione ritmica, rapida di complessi QRS di durata superiore ai 0,14 sec. La morfologia dei complessi è diversa a seconda della sede da cui origina l’aritmia (ventricolo destro o ventricolo sinistro; Fig. 6.15). La tachicardia ventricolare deve essere distinta dalla tachicardia sopraventricolare che viene condotta ai ventricoli con aberranza. La diagnosi differenziale spesso è possibile col solo ECG di superficie, osservando i seguenti tre criteri: a) nella tachicardia atriale il complesso QRS è spesso preceduto dall’onda P in rapporto costante; b) nella tachicardia atriale condotta con aberranza, il complesso QRS ha spesso morfologia tipo blocco di branca destra; c) nella tachicardia ventricolare possono essere visibili onde P dissociate dai complessi ventricolari. La tachicardia ventricolare interferisce con la normale funzione del cuore attraverso tre meccanismi: a) priva il riempimento diastolico ventricolare della componente dovuta alla sistole atriale; b) l’elevata frequenza cardiaca riduce il tempo di riempimento diastolico del ventricolo e delle coronarie; c) la contrazione ventricolare indotta dall’impulso ectopico non è coordinata in maniera fisiologica e pertanto non è completamente efficace. Tutto ciò in cardiopatici. La tachicardia ventricolare è un’emergenza medica da trattare con urgenza perché: riduce la portata cardiaca, riduce la perfusione coronarica e può anticipare l’arresto cardiaco per fibrillazione ventricolare. Il paziente con tachicardia ventricolare avverte principalmente i sintomi legati alla bassa portata e alla ridotta perfusione coronarica: scompenso e angina. Il medico riscontra i segni obiettivi della bassa portata: ipotensione, ipoperfusione cutanea, dispnea, sudorazione, oliguria. All’ascoltazione del cuore i toni sono di scarsa intensità, frequenti, ritmici. La dissociazione fra attività atriale e ventricolare determina un caratteristico fenomeno che si nota ascoltando il cuore: di tanto in tanto le sistoli atriali e ventricolari si sovrappongono dando origine a un tono molto intenso (colpo di cannone). Il polso è piccolo e frequente. La pressione arteriosa è ridotta. La terapia dev’essere instaurata con urgenza. Vanno distinte due possibilità in base al quadro clinico. Se il paziente è in shock, va effettuata subito la cardioversione elettrica in anestesia. Se la pressione sistolica si mantiene su valori superiori a 85 mmHg, la cardioversione elettrica può essere dilazionata di 10 min. In questo caso è possibile procedere a un tentativo di trattamento medico con lidocaina endovenosa. Se il tentativo non è efficace, bisogna effettuare subito la cardioversione elettrica. La terapia dell’aritmia deve sempre essere accompagnata dall’individuazione e dalla correzione di eventuali fattori che ne hanno favorito l’insorgenza. In particolare vanno corretti eventuali squilibri elettrolitici. C. Torsione di punta (“torsade de pointe”). La torsione di punta è un’aritmia ipercinetica ventricolare con caratteri morfologici elettrocardiografici prossimi alla tachicardia ventricolare; da questa, però, va tenuta distinta per alcune peculiarità terapeutiche. Da un punto di vista elettrocardiografico la torsione di punta è costituita da tratti di tachicardia ventricolare nei quali il QRS cambia progressivamente la propria morfologia, come se ruotasse gradualmente intorno a un’immaginaria linea isoelettrica di base. La torsione di punta insorge in soggetti in ritmo sinusale che presentano un intervallo QT notevolmente allungato. Questo fatto indica che vi sono zone del miocardio ventricolare che si ripolarizzano con molto ritardo. Ciò costituisce una condizione favorevolissima per l’instaurazione di rientri. L’allungamento del QT può essere congenito, secondario a squilibri elettrolitici (ipopotassiemia), secondario all’uso di alcuni farmaci (chinidina, disopiramide, alcune fenotiazine, antidepressivi triciclici). L’aritmia viene per lo più iniziata da un’extrasistole ventricolare tardiva. Può risolversi spontaneamente, recidivare o degenerare in fibrillazione ventricolare. La terapia della torsione di punta deve mirare a: a) rimuovere la causa che ha favorito l’allungamento dell’intervallo QT (correzione degli squilibri elettrolitici e sospensione di eventuali farmaci); b) aumentare la frequenza cardiaca. La frequenza può essere aumentata con la stimolazione endocavitaria mediante catetere stimolatore. Se non è rapidamente disponibile un catetere stimolatore, è opportuno tentare di indurre un aumento della frequenza del cuore con isoproterenolo. D. Fibrillazione ventricolare. La fibrillazione ventricolare definisce una condizione di attività elettrica e meccanica caotica e disorganizzata. I ventricoli sono percorsi da onde di eccitazione multiple e incoordinate che si modificano in continuazione, in rapporto al fatto che ognuna di esse si trova davanti cellule che hanno recuperato o che sono ancora in fase refrattaria (Fig. 6.16). Una volta instaurata, la fibrillazione non tende a cessare e quindi l’esito abituale è la morte. Durante la fibrillazione i ventricoli non si contraggono in maniera efficace, per cui il circolo è interrotto. La fibrillazione ventricolare, in assenza di cardioversione 146 elettrica immediata, è una condizione terminale. Quest’aritmia è frequente causa di morte improvvisa nei pazienti con infarto acuto del miocardio. Unica terapia della fibrillazione ventricolare è la cardioversione elettrica. Non potendo praticare subito la cardioversione, è indispensabile eseguire il massaggio cardiaco esterno e la ventilazione assistita. L’interruzione del circolo per un tempo superiore a 2-3 min comporta, infatti, lesioni cerebrali irreversibili. E. Sindrome del Q-T lungo. Si tratta di una condizione geneticamente determinata riconoscibile per un allungamento, nell’elettrocardiogramma, dell’intervallo Q-T corretto per la frequenza cardiaca. Quest’alterazione è dipendente da una tra sei possibili mutazioni genetiche (le varianti genetiche sono designate con una sigla che va da LQT1 a LQT6) che producono anomalie del funzionamento nel cuore dei canali del sodio o del potassio. La penetranza di questi geni è però ridotta e l’espressione fenotipica è variabile. I soggetti con la sindrome del Q-T lungo possono stare benissimo, ma hanno una predisposizione a sviluppare episodi di fibrillazione ventricolare o torsione di punta. Possono perciò andare incontro a ripetuti episodi di sincope, ma anche a morte improvvisa. Esistono argomenti per ritenere che almeno una parte delle cosiddette “morti in culla” siano dovute a quest’alterazione genetica. La “morte in culla”, o SIDS (Sudden Infant Death Sindrome), è una condizione ben nota che si verifica repentinamente in neonati apparentemente sani nel primo anno di vita. In passato, per evitare questo drammatico incidente ci si limitava a raccomandare di evitare di mettere a dormire il neonato in posizione prona. Attualmente il Q-T lungo può essere dimostrato all’elettrocardiogramma nei primi giorni di vita e un trattamento efficace può essere istituito con mexiletina (un bloccante dei canali del sodio che accorcia in questi pazienti l’intervallo Q-T) o con propranololo. In ogni caso una sindrome del Q-T lungo dev’essere sopettata in tutti i casi di persone giovani che presentano ripetuti episodi di sincope, soprattutto se esiste familiarità per questo disturbo, dato che un trattamento appropriato può salvare la vita. Altre forme predisponenti a queste stesse aritmie e geneticamente determinate sono la cardiomiopatia ipertrofica (Cap. 8) e altre anomalie ancora più rare, che qui ricordiamo a titolo di esempio, come la sindrome di Brugada (associata con elevazione del segmento S-T in V1, V2 e V3) e la displasia aritmogenica del ventricolo destro (spesso caratterizzata da inversione dell’onda T in V1, V2 e V3). Trovare in una persona giovane un’ano- Figura 6.16 - Fibrillazione ventricolare. MALATTIE DEL SISTEMA CIRCOLATORIO malia elettrocardiografia non spiegabile dovrebbe spingere ad approfondire il caso consultando la letteratura. ARITMIE IPOCINETICHE Il termine di aritmia ipocinetica definisce una condizione nella quale la frequenza atriale o ventricolare si riduce al di sotto di 60 impulsi/min: ciò per bradicardia ritmica, o perché molti impulsi vanno perduti. Questa condizione, come si è visto, si può manifestare come anomalia della formazione dell’impulso, oppure come anomalia della sua propagazione. Disfunzioni del nodo del seno A. Bradicardia sinusale. Nella bradicardia sinusale l’impulso nasce come di norma dal nodo del seno, ma con una frequenza inferiore ai 60 battiti/min. La bradicardia sinusale è una condizione fisiologica nei soggetti giovani e negli atleti; è una condizione patologica (si veda la malattia del nodo del seno) quando si presenta in circostanze che di norma si associano a un aumento della frequenza cardiaca: scompenso di cuore, esercizio fisico, emozioni, ecc. B. Malattia del nodo del seno. Il termine di malattia del nodo del seno viene riferito a una serie di anomalie elettrocardiografiche eterogenee che hanno come carattere comune una disfunzione del nodo del seno. Solitamente la malattia del nodo del seno comprende una o più delle seguenti condizioni: • bradicardia sinusale persistente e inopportuna (si veda sopra); • blocco seno-atriale; • arresto seno-atriale senza comparsa di ritmi di scappamento; • sindrome bradicardia-tachicardia. Nel blocco seno-atriale l’impulso nasce a livello del nodo del seno, ma viene bloccato prima che possa diffondere agli atri. Da un punto di vista elettrocardiografico, il blocco seno-atriale è rappresentato dall’assenza di uno o più complessi P-QRS che interrompe una loro successione regolare. L’intervallo fra l’onda P che precede e quella che segue l’impulso bloccato (quando è uno solo) è uguale a due volte l’intervallo P-P normale. L’arresto seno-atriale (cioè la mancata formazione dell’impulso a livello del nodo del seno) è, sull’ECG di 147 6 - ARITMIE superficie, simile al blocco seno-atriale. Nel primo caso, infatti, l’impulso non si forma; nel secondo si forma, ma non diffonde agli atri. In entrambi i casi gli atri non vengono attivati e non danno luogo alla formazione dell’onda P e del seguente QRS. La mancata formazione degli impulsi a livello del nodo del seno dovrebbe consentire ai pacemaker latenti più distali (atriali e giunzionali) di emergere e di dare vita a quelli che vengono chiamati “ritmi di sfuggita o scappamento” (Fig. 6.17). Nella malattia del nodo del seno il blocco o l’arresto sinusale non si accompagna a ritmi di sfuggita efficaci e può determinare periodi di asistolia prolungati. La sindrome bradicardia-tachicardia si verifica in conseguenza del mancato intervento di ritmi di sfuggita efficaci (per es., un ritmo giunzionale regolare, avente frequenza intorno a 50/min) in caso di inattivazione prolungata del nodo del seno. Dopo una breve asistolia completa, si scatena qualche ritmo atriale patologico, solitamente una tachicardia atriale parossistica o una fibrillazione atriale. Pertanto la sindrome è caratterizzata dall’alternanza di periodi di aritmia ipercinetica atriale con tratti di bradicardia sinusale di grado marcato. Caratteristicamente, durante le fasi di bradicardia mancano i ritmi di sfuggita dei pacemaker latenti. La malattia del nodo del seno è solitamente determinata da una lesione a carico del tessuto di conduzione. La lesione può essere circoscritta al nodo del seno o più estesa; in genere è determinata da un processo ischemico, infiammatorio o degenerativo. I sintomi della malattia del nodo del seno sono per lo più secondari a ipoperfusione del cervello e si manifestano quando la frequenza cardiaca è molto bassa. Il paziente accusa vertigini, lipotimie o sincopi. Le manifestazioni di ischemia cerebrale, che seguono al deficit acuto della funzione cardiaca per la presenza di pause più o meno prolungate della attività del nodo del seno, sono raggruppate nella sindrome di Morgagni-Adam-Stokes (MAS). La sindrome di MAS può essere così riassunta: se l’ischemia cerebrale dura dai 2 ai 5 sec il paziente avverte una vertigine; se dura 510 sec perde conoscenza e cade; se l’ischemia persiste per più di 15 sec, il paziente perde feci e urine e compaiono convulsioni, cianosi e respiro stertoroso. Se l’ischemia si protrae oltre i 30 sec il paziente è apparentemente morto. La mancata perfusione del cervello che non si risolve entro 3-4 min porta a lesioni ischemiche irreversibili delle cellule del sistema nervoso centrale. Nella fase tachicardica della sindrome bradicardia-tachicardia i sintomi e segni sono quelli già descritti nelle aritmie ipercinetiche. Se il medico visita il paziente nel momento dei sintomi, riscontra una frequenza cardiaca molto bassa con toni ritmici o aritmici a seconda dell’aritmia presente. La pressione arteriosa sistolica è in genere aumentata mentre la diastolica è ridotta. Ciò è dovuto all’aumento della gettata sistolica, che aumenta per compensare la riduzione della frequenza cardiaca. Se il paziente viene visitato lontano dalla crisi, l’obiettività può essere assolutamente normale. C. Ritmo giunzionale. Il ritmo giunzionale nasce dai pacemaker della giunzione atrioventricolare; esso si esprime quando viene a mancare l’impulso “guida” a origine più prossimale (nodo del seno). Da un punto di vista elettrocardiografico, l’aritmia è caratterizzata dalla successione ritmica di complessi QRS di aspetto normale con frequenza che solitamente varia da 40 a 60/min. A volte si può osservare un’attivazione atriale per via retrograda: tale attivazione retrograda si esprime con la formazione di onde P di aspetto chiaramente anomalo, che hanno peraltro un rapporto costante col QRS. Il ritmo giunzionale è spesso espressione di un sovradosaggio digitalico. D1 D2 D3 Figura 6.17 - Arresto seno-atriale con ritmo di sfuggita giunzionale. 148 MALATTIE DEL SISTEMA CIRCOLATORIO Solitamente l’aritmia è asintomatica. Può generare sintomi e segni di scompenso quando l’aumento della frequenza e la componente sistolica atriale del riempimento ventricolare sono necessari per mantenere una portata cardiaca normale. In assenza di scompenso l’esame obiettivo può essere normale: il medico avverte solo la bradicardia. Blocco atrioventricolare Il blocco atrioventricolare è una condizione caratterizzata da un’anomala diffusione dell’impulso dagli atri ai ventricoli. Classicamente il blocco atrioventricolare viene suddiviso in tre gradi a seconda della gravità. • Nel blocco atrioventricolare di I grado tutti gli impulsi che originano a livello atriale vengono condotti ai ventricoli con una velocità ridotta rispetto al normale. Ciò si esprime all’ECG con un allungamento dell’intervallo PQ che supera il limite normale massimo che è di 0,20 sec. Il complesso QRS che segue il blocco di I grado è normale. • Nel blocco atrioventricolare di II grado alcuni impulsi atriali raggiungono i ventricoli, altri no. Il blocco di II grado viene ulteriormente suddiviso in due varietà: il tipo I (Mobitz 1) e il tipo II (Mobitz 2). (Mobitz è l’Autore che propose questa classificazione). Nel tipo I la conduzione a livello del nodo atrioventricolare si allunga progressivamente fino al blocco completo. L’ECG mostra una serie di complessi P-QRS in cui, mentre le P sono ritmiche, cioè con intervalli P-P sempre uguali, l’intervallo PQ è progressivamente più lungo, fino alla comparsa di un’onda P bloccata, vale a dire non seguita dal QRS. Il complesso P-QRS che segue l’onda P bloccata solitamente riprende con un intervallo PQ normale; esso inizia una nuova serie, che finirà con la P bloccata (Fig. 6.18). Il progressivo allungamento del tempo di conduzione atrioventricolare (segno di conduzione progressivamente “affaticata”) è denominato “fenomeno di LucianiWenckebach”. Si può precisare che nel blocco atrioventricolare di II grado tipo Mobitz 1 la lunghezza dei cicli RR (intervallo fra onda R di un QRS e onda R del QRS che segue) tende a ridursi progressivamente. Questa riduzione si ha perché l’entita degli incrementi percentuali dell’intervallo PR tende progressivamente a ridursi. Ne deriva che l’intervallo RR più breve è quello immediatamente precedente l’onda P bloccata. Nel blocco atrioventricolare di II grado tipo Mobitz 2 vige, per quanto riguarda la conduzione atrioventrico- lare, la legge del tutto o nulla: alcune volte l’impulso atriale raggiunge i ventricoli; altre volte no. Quando l’impulso raggiunge i ventricoli il tempo di conduzione atrioventricolare è costante. All’ECG si sono osservate onde P seguite regolarmente dal QRS dopo un intervallo PR costante. A queste si alternano onde P bloccate, prive cioè del complesso QRS seguente (Fig. 6.19). Il blocco atrioventricolare può insorgere o meno secondo una cadenza fissa. Se il blocco ha cadenza regolare, si parla di blocco 2:1, 3:1, 4:1, ecc. a seconda che il blocco insorga regolarmente dopo una, due, tre, quattro ecc. onde P normalmente condotte ai ventricoli. • Nel blocco atrioventricolare di III grado nessun impulso che origina negli atri può essere condotto ai ventricoli. All’ECG si osserva una sequenza di onde P prive del QRS corrispondente. I ventricoli vengono attivati da un pacemaker posto distalmente al nodo atrioventricolare: nella parte distale della giunzione o nei ventricoli. Nel primo caso, i complessi QRS si succedono con una frequenza di 40-60/min e hanno una morfologia analoga a quella dei QRS normalmente condotti (QRS stretti: essi durano 0,08-0,10 sec) (Fig. 6.20). Nel secondo caso, la frequenza è più bassa (inferiore a 40 battiti/min) e il complesso QRS è largo (più di 0,10 sec e, di solito, più di 0,12 sec) (Fig. 6.21). Nel blocco atrioventricolare di III grado i complessi QRS si succedono indipendentemente dalle onde P: si dice che il ritmo è “dissociato” perché i rapporti temporali fra complesso atriale e ventricolare sono del tutto casuali e dipendono dalla frequenza indipendente di ciascuno dei due. Da un punto di vista pratico, è importante diagnosticare la sede dei blocchi atrioventricolari, differenziando i blocchi situati sopra il fascio di His da quelli situati sotto. È importante, cioè, stabilire in quale punto l’impulso che proviene dagli atri viene interrotto. La distinzione (blocchi sopra e sottohissiani) porta con sé conseguenze prognostiche e terapeutiche determinanti. Nei blocchi posti sopra al fascio di His, infatti, l’attività automatica del cuore e l’eccitazione ventricolare sono in genere garantite dalla funzione dei pacemaker posti a livello del nodo atrioventricolare e del fascio di His. La frequenza intrinseca di questi pacemaker (40-60/min) garantisce una buona funzione cardiaca e raramente è responsabile di riduzioni critiche della frequenza cardiaca. Figura 6.18 - Blocco A-V di II grado tipo Mobitz 1 (fenomeno di Luciani-Wenckebach). L’intervallo PQ si allunga progressivamente fino a quando una P non conduce. Nel primo tratto il blocco della P si ha dopo 4 attivazioni atriali; nel secondo dopo 3. 149 6 - ARITMIE D1 D2 D3 aVR aVL aVF Figura 6.19 - Blocco atrioventricolare di II grado tipo Mobitz 2. Il blocco è di tipo 2:1. Si ha infatti un’onda P regolarmente condotta ai ventricoli e pertanto seguita dal QRS-T; ad essa segue un’altra bloccata e pertanto priva del complesso ventricolare che dovrebbe seguire. P P P P P Figura 6.20 - Blocco atrioventricolare completo. Il ritmo delle onde P è dissociato da quello dei QRS. La frequenza delle P è di 100/min; quella dei QRS di 36. Quando l’onda P cade casualmente durante il QRS-T, può essere nascosta o deformata (si vedano la terza e la quinta di quelle evidenziate). Figura 6.21 - Blocco atrioventricolare completo. Il ritmo della P è di circa 4 volte superiore a quello del QRS. A un primo esame può sembrare che il rapporto fra l’onda P e il QRS sia costante e che pertanto si configuri il blocco atrioventricolare di I grado Mobitz 2, 4:1. In realtà l’intervallo PQ varia: si tratta di un blocco di III grado, con una dissociazione completa. Il ritmo dei QRS è di circa 20/min, quello delle P è di 82. 150 Nei blocchi più distali (sottohissiani) l’eccitazione dei ventricoli è affidata ai pacemaker ventricolari: questi pacemaker hanno solitamente una frequenza ventricolare troppo bassa (25-35) per garantire un compenso emodinamico duraturo. Spesso, inoltre, frequenze così basse anticipano aritmie più gravi (fibrillazione ventricolare). La diagnosi di sede del blocco è possibile con certezza solo mediante elettrocardiogramma endocavitario. Esistono comunque alcuni dati desumibili dall’ECG di superficie che consentono di porre una diagnosi di buona probabilità. I criteri sono i seguenti: a) i blocchi di I grado e quelli di II grado tipo Mobitz 1 sono solitamente soprahissiani; b) i blocchi di III grado nei quali il QRS dissociato (originato cioè dal pacemaker secondario che dà origine al ritmo di scappamento) ha durata normale sono probabilmente soprahissiani; c) i blocchi di II grado tipo Mobitz 2 e quelli di III grado nei quali il ritmo di scappamento è sostenuto da complessi QRS allargati (di durata superiore agli 0,10-0,11 sec) sono probabilmente sottohissiani. La morfologia del QRS, infatti, deriva da due fattori: la sede di origine dell’impulso e la via seguita dall’impulso stesso per attivare i ventricoli. Se l’impulso nasce in una posizione prossimale al fascio di His, l’attivazione dei ventricoli avviene come di norma (con attivazione contemporanea delle due branche) e il QRS è normale. Se la sede di formazione dell’impulso è distale al punto in cui il fascio di His si divide nelle branche, la morfologia del QRS è alterata perché l’attivazione dei ventricoli avviene in modo anomalo. In particolare, se la sede del pacemaker sussidiario che dà il via al ritmo di scappamento è situata nel ventricolo sinistro, la morfologia del QRS è analoga a quella che si osserva nel blocco di branca destra; se la sede è nel ventricolo destro, la morfologia del complesso QRS è analoga a quella presente nel blocco di branca sinistra (si veda in seguito). L’origine dell’impulso in uno dei due ventricoli fa sì che l’onda di eccitazione raggiunga il ventricolo controlaterale attraverso il miocardio contrattile, proprio come avviene quando la branca controlaterale è bloccata. I blocchi atrioventricolari possono essere congeniti o acquisiti. I blocchi congeniti sono percentualmente rari, possono avere andamento familiare ed eventualmente associarsi ad altre cardiopatie. I blocchi acquisiti possono essere secondari a processi infettivi, possono manifestarsi per la prima volta in corso di cardite reumatica, nella cardiopatia ischemica e nelle cardiomiopatie primitive. Forme acquisite di blocchi sono pure quelli indotti da farmaci e quelli chirurgici. I blocchi chirurgici sono causati dall’involontaria lesione delle vie di conduzione da parte del chirurgo in corso di intervento sul cuore. I farmaci più frequentemente responsabili di blocco atrioventricolare sono gli antiaritmici del gruppo I A, il verapamil, l’amiodarone e i -bloccanti. La conduzione atrioventricolare è sottoposta alla regolazione vagale. Forti stimoli vagali possono aggrava- MALATTIE DEL SISTEMA CIRCOLATORIO re turbe della conduzione preesistenti; a volte uno stimolo vagale molto intenso può indurre blocchi atrioventricolari di grado elevato anche in soggetti sani. Tra i blocchi atrioventricolari elencati meritano un commento quelli che insorgono in corso di infarto miocardico acuto. Da un punto di vista pratico, è opportuno distinguere i blocchi che compaiono in corso di infarto inferiore da quelli che compaiono in corso di infarto anteriore. Nell’infarto inferiore i blocchi sono in genere soprahissiani e le vie di conduzione poste al di sotto del blocco sono indenni. Nell’infarto anteriore il blocco è più distale (sottohissiano) e si associa alla compromissione delle vie di conduzione. Nell’infarto anteriore il blocco evolve rapidamente con una tendenza elevata verso il blocco atrioventricolare totale. I sintomi dei blocchi dipendono da due fattori: a) dalla gravità del blocco e quindi dalla frequenza dei pacemaker sussidiari; b) dalla cardiopatia di base che ha determinato il blocco e da altre cardiopatie eventualmente associate. Il blocco di I grado non complicato è asintomatico e non consente una diagnosi senza ECG. Il blocco di II grado può essere sintomatico se è di grado elevato e soprattutto nel corso del passaggio a blocco di III grado: la frequenza cardiaca si riduce in maniera critica e compaiono i sintomi neurologici già descritti per la malattia del nodo del seno. In particolare può verificarsi la sindrome di Morgagni-Adam-Stokes. Il blocco di III grado è quasi sempre sintomatico; i sintomi sono legati alla scarsa irrorazione cerebrale o, eventualmente, allo scompenso cardiaco. I sintomi secondari allo scompenso compaiono soprattutto quando il blocco si manifesta in soggetti in compenso labile a causa della cardiopatia di base. Il medico che visita un paziente in blocco avverte una frequenza molto bassa sia all’ascoltazione che alla palpazione del polso. Tipicamente, nel blocco di III grado la bradicardia non si riduce per effetto dell’esercizio fisico. All’esame fisico il medico avverte che il primo tono cardiaco ha intensità variabile. Ciò dipende dal fatto che il rapporto temporale fra la sistole atriale e la sistole ventricolare varia continuamente. Quando la sistole atriale è sincrona con la sistole ventricolare, il primo tono è particolarmente intenso e viene chiamato “colpo di cannone”. APPENDICE Blocchi di branca Si definisce blocco di branca la condizione nella quale la conduzione dell’impulso trasmesso attraverso il fascio di His è alterata a livello di una delle sue suddivisioni. Si parla di blocco di branca destra se la conduzione è bloccata a livello della branca destra; di blocco di branca sinistra quando il blocco è a livello della branca sinistra. I blocchi di branca possono essere completi o incompleti. Nel blocco di branca completo la conduzione 6 - ARITMIE è completamente interrotta; nel blocco incompleto la conduzione è solo rallentata. Nei cenni di anatomia si è già visto che la branca destra è un’entità anatomica discreta mentre la branca sinistra si sfilaccia, poco dopo la suddivisione del fascio di His, in una specie di ventaglio di fibre. Sia da un punto di vista elettrocardiografico che funzionale si è soliti raggruppare queste fibre in due gruppi: il gruppo antero-superiore e il gruppo postero-inferiore. In alcune condizioni patologiche la noxa patogena interessa uno solo dei due gruppi di fibre e risparmia l’altro. Si possono pertanto avere degli “emiblocchi”: anteriore sinistro, se è leso il gruppo di fibre antero-superiore; posteriore sinistro, se è leso il gruppo postero-inferiore. Se è bloccata una branca o una delle suddivisioni della branca sinistra, lo stimolo che proviene dall’atrio può raggiungere comunque i ventricoli. L’impulso percorre la branca indenne e diffonde al ventricolo controlaterale (omolaterale rispetto alla branca lesa) attraverso il tessuto contrattile non specifico. Lo stesso avviene se si stabilisce una condizione di blocco cosiddetto bifascicolato. Nel blocco bifascicolato si ha il blocco della branca destra e di uno dei due gruppi di fibre che costituiscono la branca sinistra. In questo caso l’unica via percorribile è il gruppo indenne della branca sinistra. Se anche questo gruppo non riesce più a condurre lo stimolo, l’impulso che proviene dagli atri non raggiunge i ventricoli: si ha un blocco atrioventricolare completo (di III grado). Esiste anche una condizione definita blocco trifascicolato. Il blocco trifascicolato si ha nelle seguenti condizioni: blocco di branca destra più emiblocco sinistro (anteriore o posteriore) e allungamento della conduzione atrioventricolare (perché il fascicolo integro della branca sinistra rallenta la conduzione dello stimolo); blocco di branca destra più emiblocco anteriore sinistro (o posteriore) che si alterna con emiblocco posteriore (o anteriore); blocco di branca destra alternato a blocco di branca sinistra. Il blocco trifascicolato esprime, cioè, una condizione nella quale l’unica struttura che in un dato momento è ancora in grado di condurre l’impulso tra atri e ventricoli “zoppica”. Il blocco trifascicolato anticipa molto spesso un blocco atrioventricolare completo. La diagnosi di blocco di branca all’elettrocardiogramma è di grande importanza per i seguenti motivi: a) perché evidenzia l’interessamento del tessuto di conduzione da parte di una determinata noxa patogena; b) perché consente di seguire nel tempo l’eventuale evoluzione del danno a carico del tessuto di conduzione. Questo secondo punto riveste una grande importanza pratica. L’esempio più significativo è costituito dai blocchi di branca che si instaurano in corso di infarto acuto. Se durante infarto acuto insorge un blocco di branca o un emiblocco, vuol dire che è danneggiato il circolo coronarico deputato al nutrimento della branca o del fascicolo leso. Il blocco acuto isolato di una branca è una condizione d’allarme. La conduzione atrioventricolare però è garantita dalla branca indenne. Se al blocco di 151 branca isolato segue un blocco bi- o trifascicolato, l’ischemia coinvolge entrambe le branche. Ciò significa che il danno è grave e ha carattere evolutivo. In corso d’infarto, una condizione di blocco di branca seguita da un blocco bi- o trifascicolato anticipa spesso un blocco atrioventricolare completo e richiede senz’altro la posa di un pacemaker artificiale di “protezione”. Il carattere evolutivo del blocco delle branche in corso di malattia cronica (ischemica o degenerativa) non richiede soluzioni ugualmente urgenti. L’indicazione al pacemaker artificiale va rivalutata periodicamente di volta in volta, a seconda della rapidità con cui la malattia del tessuto di conduzione evolve e dei sintomi che produce. A. Blocco di branca destra. Nel blocco di branca destra la durata dell’attivazione ventricolare è allungata. Questo allungamento si esprime con un aumento della durata del QRS. Per convenzione, il blocco è detto completo quando determina una durata del QRS uguale o maggiore di 0,12 sec. L’aumento di durata è dovuto al fatto che l’onda di attivazione del ventricolo destro non proviene come di norma dalla branca destra. Il ventricolo destro viene attivato da un’onda che proviene dal ventricolo sinistro attraverso il miocardio contrattile del setto interventricolare. Ma la velocità di conduzione del miocardio contrattile è molto meno rapida di quella del tessuto di conduzione che costituisce la branca, pertanto il completamento dell’attivazione ventricolare è prolungato. Per semplicità, la sequenza di attivazione ventricolare in presenza di blocco di branca destra può essere arbitrariamente distinta in quattro fasi (Fig. 6.22). Prima fase. È la fase di attivazione del setto. Il setto viene attivato come di norma da sinistra a destra perché la branca sinistra è intatta. L’attivazione del setto determina l’inscrizione di un’onda R normale in V1 e di una piccola q in V6 e in D1. Seconda fase. L’attivazione del setto continua sempre in direzione sinistra-destra perché la branca destra è bloccata. Durante la seconda fase inizia anche l’attivazione del ventricolo sinistro. I potenziali determinati dall’attivazione del ventricolo sinistro sono maggiori di quelli dovuti all’attivazione settale. Ne deriva che la risultante dei vettori di attivazione del setto e del ventricolo sinistro è diretta sempre in avanti, ma un po’ a sinistra. Nella seconda fase si ha il completamento dell’onda R nelle precordiali destre e l’inizio dell’onda R nelle precordiali sinistre e in D1. Terza fase. Continua l’attivazione del setto (sempre da sinistra a destra) e si completa quella del ventricolo sinistro nelle regioni antero-laterale e basale. Anche in questa fase i potenziali del ventricolo sinistro sono dominanti. Il vettore risultante è diretto verso sinistra, inferiormente e, più o meno, posteriormente. Durante questa fase si inscrive l’onda S nelle precordiali destre e l’onda R nelle precordiali sinistre e in D1 raggiunge lo zenit. 152 MALATTIE DEL SISTEMA CIRCOLATORIO DEPOLARIZZAZIONE VENTRICOLARE NEL BLOCCO DI BRANCA DESTRA VETTORI DELLA ATTIVAZIONE VENTRICOLARE SUL PIANO ORIZZONTALE Fase 1 Iniziale attivazione del setto (0,01 sec) Fase 2 Continua l’attivazione del lato sinistro del setto e della porzione antero-apicale del ventricolo sinistro (0,04 sec) Fase 3 Si completa l’attivazione del ventricolo sinistro e continua l’attivazione del setto (da 0,06 a 0,08 sec) Fase 4 Si completa l’attivazione del setto e del ventricolo destro (0,12 sec o più) COMPLESSI QRS NELLE DERIVAZIONI PRECORDIALI 3 3 3 3 V6 1 4 V2 2 4 2 V5 2 1 V1 3 4 V3 1 V4 2 2 2 4 1 1 2 4 1 4 1 4 3 3 V1 V2 V3 V4 V5 V6 Figura 6.22 - Rappresentazione schematica della depolarizzazione ventricolare nel blocco di branca destra. Quarta fase. Si ha il completamento dell’attivazione settale, l’attivazione della parete apicale del ventricolo destro e infine della parete libera e della parte restante del ventricolo destro. Il vettore risultante della quarta fase è diretto a destra e anteriormente. Sul piano frontale può essere diretto sia superiormente che inferiormente. Durante questa fase si inscrive un’onda R nelle precordiali destre e una S nelle precordiali sinistre e in D1. Il blocco di branca destra apporta anche delle modificazioni dell’ripolarizzazione che derivano direttamente dall’alterata depolarizzazione ventricolare. Le alterazioni più significative si osservano nelle precordiali destre dove il segmento ST è slivellato verso il basso e l’onda T è invertita (Fig. 6.23). 1. Criteri riassuntivi elettrocardiografici di blocco di branca destra. • QRS di durata superiore a 0,12 sec. • Complesso RSR⬘ nelle precordiali destre (V1 e V2). L’onda R⬘ è solitamente di ampiezza maggiore di R. • Nelle precordiali sinistre (V5 e V6) complesso QRS con ampia S finale. • Ritardo di inscrizione della deflessione intrinsecoide in V1 e V2 (oltre gli 0,07 sec). (Si dice deflessione intrinsecoide la durata dell’intervallo fra l’inizio del QRS e il momento in cui comincia la branca discendente della R. È una misura che si esegue solo nelle derivazioni precordiali.) • In D1 il complesso QRS mostra un’ampia S finale. • Il segmento ST è slivellato in basso e la T è negativa nelle precordiali destre. 2. Blocco di branca destra incompleto. I criteri elettrocardiografici sono analoghi a quelli del blocco completo, con la differenza che il QRS dura in genere fra 0,10 e 0,12 sec. B. Blocco di branca sinistra. Nel blocco di branca sinistra la durata dell’attivazione ventricolare è aumentata per motivi analoghi a quelli già descritti per il blocco di branca destra. Anche nel blocco di branca sinistra la sequenza di attivazione ventricolare può essere arbitrariamente divisa in quattro fasi (Fig. 6.24). Prima fase. È la fase di attivazione del setto che inizia dal lato destro perché la branca sinistra è bloccata. L’attivazione settale si dirige da destra a sinistra 6 - ARITMIE 153 porzioni posteriore e basale del ventricolo sinistro. Il vettore risultante è diretto senz’altro a sinistra e posteriormente. In questa fase si ha il nadir della S in V1 e lo zenit della R in V6 e D1. Quarta fase. Si completa l’attivazione delle pareti anteriore e laterale del ventricolo sinistro. Il vettore della quarta fase è sempre diretto a sinistra, ma un po’ più anteriormente rispetto a quello della terza fase. Questo vettore determina l’inscrizione di una R in V6 e D1 e completa l’inscrizione della S in V1. Anche nel blocco di branca sinistra la ripolarizzazione è alterata secondariamente all’alterata sequenza della depolarizzazione ventricolare. In particolare, il segmento ST è slivellato verso il basso nelle precordiali sinistre e in D1. In queste derivazioni l’onda T è invertita. Figura 6.23 - Blocco di branca destra completo. e dall’apice verso la base. Contemporaneamente l’attivazione diffonde all’apice e alla porzione anteriore del ventricolo destro. Perciò nella prima fase abbiamo la coesistenza di vettori del setto e del ventricolo destro (quello sinistro non è ancora depolarizzato). Il vettore settale è diretto a sinistra, posteriormente e inferiormente. Il vettore del ventricolo destro è diretto a destra, in avanti e inferiormente. Il vettore risultante deriva dalla grandezza e dalla direzione dei due vettori che lo costituiscono. Se prevale il vettore settale, il vettore risultante è diretto a sinistra, inferiormente e posteriormente. In questo caso in V1 si inscrive l’inizio di un complesso QS e in V6 e in D1 l’inizio di un’onda R. Se prevalgono invece i potenziali del ventricolo destro, il vettore risultante è diretto ancora inferiormente e a sinistra ma è un po’ ruotato in avanti. In questo secondo caso, in V1 si inserisce una piccola r e in V6 e D1 si avrà sempre l’inizio di un’onda R. Seconda fase. Continua e si completa l’attivazione del setto (sempre in direzione destra-sinistra) e si completa l’attivazione del ventricolo destro. I potenziali settali sono dominanti. Ne deriva che il vettore risultante è diretto a sinistra e posteriormente. All’ECG continua l’inscrizione del QS (o rS) in V1 e della R in V6 e D1. Terza fase. Ha luogo l’attivazione del ventricolo sinistro. L’attivazione è anomala: inizia e si completa nelle 1. Criteri di diagnosi di blocco di branca sinistra (Fig. 6.25). • QRS di durata superiore a 0,12 sec nelle derivazioni periferiche. • Complesso QS o rS nelle precordiali destre. L’onda S è ampia e profonda. • Complesso R o RsR nelle precordiali sinistre. L’onda R è ampia e impastata. • La deflessione intrinsecoide in V6 è spesso ritardata (oltre 0,07 sec). • Nelle derivazioni periferiche: D1 e aVL sono simili alle precordiali sinistre; D2, D3, aVF sono simili alle precordiali destre. • Il segmento ST è slivellato in basso e la T è negativa nelle precordiali sinistre e in D1. 2. Blocco di branca sinistra incompleto. I criteri di diagnosi elettrocardiografica di blocco di branca sinistra incompleto sono analoghi a quelli descritti per il blocco completo. La differenza viene fatta sulla base del QRS, che nel blocco incompleto è più breve: 0,10-0,12 sec. C. Emiblocchi. Normalmente l’onda di eccitazione diffonde in modo uniforme lungo i due gruppi di fibre in cui schematicamente viene distinta la branca sinistra: l’antero-superiore e la postero-anteriore. Se la conduzione viene bloccata a livello di uno dei due gruppi di fibre, la sequenza di attivazione ventricolare viene alterata. Nell’emiblocco anteriore è bloccato il gruppo antero-superiore: l’onda di eccitazione percorre il gruppo postero-inferiore di fibre e diffonde solo in un secondo tempo in avanti e in alto, attraverso il miocardio contrattile. La conseguenza elettrocardiografica è che il vettore QRS terminale è diretto a sinistra e in alto; l’asse elettrico è ruotato a sinistra; il QRS può essere lievemente allungato. Se è bloccato il fascicolo postero-inferiore, l’onda di attivazione percorre il gruppo di fibre antero-superiore e poi diffonde inferiormente e posteriormente attraverso il tessuto di conduzione non specializzato. Come conseguenza si avrà che il vettore QRS terminale è diretto inferiormente e posteriormente; l’asse elettrico è ruotato inferiormente a destra; il QRS può essere lievemente allungato. 154 MALATTIE DEL SISTEMA CIRCOLATORIO DEPOLARIZZAZIONE VENTRICOLARE NEL BLOCCO DI BRANCA SINISTRA VETTORI DELLA ATTIVAZIONE VENTRICOLARE SUL PIANO ORIZZONTALE Fase 1 Attivazione del lato destro del setto e della porzione apico-anteriore del ventricolo destro (0,02 sec) Fase 2 Completamento dell’attivazione del ventricolo destro e attivazione da destra a sinistra del setto (0,06 sec) Fase 3 Attivazione della regione posterobasale del ventricolo sinistro (0,080,10 sec) Fase 4 Attivazione della regione antero-laterale del ventricolo sinistro (0,12 sec o più) COMPLESSI QRS NELLE DERIVAZIONI PRECORDIALI 3 3 4 4 2 4 3 V6 1 1 1 1 4 1 1 2 1 2 4 4 2 V5 4 2 2 2 3 3 3 V4 V1 V2 V3 3 V1 V2 V3 V4 V5 V6 Figura 6.24 - Rappresentazione schematica della depolarizzazione ventricolare nel blocco di branca sinistra. 1. Criteri di diagnosi di emiblocco. • Emiblocco anteriore sinistro – Asse elettrico del QRS deviato a sinistra oltre i –30°. – Piccole onde q in D1 e aVL. – Piccole onde r in D2, D3, aVF. • Emiblocco posteriore sinistro – Presenza di piccole onde r in D1 e aVL. – Piccole onde q in D2, D3 e aVF. – Asse elettrico ruotato verso destra oltre i 120°. • Blocchi bifascicolati. Il blocco bifascicolato è caratterizzato dal blocco della branca destra e di una delle due emibranche della branca sinistra. I criteri di diagnosi sono per lo più una somma dei criteri diagnostici dei singoli disturbi di conduzione. • Blocchi trifascicolati. Nel blocco trifascicolato l’ECG può mostrare: a) un blocco bifascicolato con un allungamento patologico (superiore a 0,20 sec) dell’intervallo PR; b) un blocco di branca destra e un emiblocco anteriore sinistro (o posteriore), alternato a un blocco di branca destra ed emiblocco posteriore sinistro (o anteriore); c) un blocco di branca destra alternato a un blocco di branca sinistra. PROBLEMI DIAGNOSTICI PARTICOLARI NELLE ARITMIE L’aritmia può essere un evento costante, frequente o raro. Se è costante, è facilmente documentabile con un elettrocardiogramma di superficie eseguibile all’atto della visita del paziente. Un esempio di aritmia costante è la fibrillazione atriale cronica. Se l’aritmia è frequente (ma non costante), può essere assente nel momento in cui il medico visita il malato. In questo caso la diagnosi può a volte avvalersi del solo elemento anamnestico (è il caso dell’extrasistolia descritta come “la sensazione di perdita di battiti, di colpi irregolari, di sfarfallio”, ecc.) specie se chi la descrive non presenta segni obiettivi di cardiopatia e non manifesta sintomi riferibili a una compromissione emodinamica coincidente con la presunta aritmia. Diverso è il caso dell’aritmia frequente ma potenzialmente pericolosa, perché chi l’avverte è un cardiopatico (portatore per esempio di una cardiopatia ischemica), o perché l’aritmia si accompagna a sintomi gravi (neurologici o di scompenso). In questo caso lo strumento diagnostico più idoneo alla diagnosi è costituito dall’elettrocardiogramma dinamico (o ECG di Holter). 155 6 - ARITMIE D1 aVR D2 aVL D3 aVF V1 V2 V3 V4 V5 V6 Figura 6.25 - Blocco di branca sinistra completo. L’ECG di Holter consiste nella registrazione continua (in genere per 24 h) su nastro magnetico di una o due derivazioni (solitamente una derivazione degli atri e una precordiale) dell’elettrocardiogramma del paziente. La registrazione avviene mediante un registratore a batteria che il paziente indossa (le dimensioni del registratore sono quelle di una piccola radio a transistor) e porta con sé durante la normale attività di tutti i giorni. L’apparecchio è costruito in modo tale che il paziente può immettere un segnale nella registrazione del proprio ECG nel momento in cui avverte, eventualmente, sintomi. Il nastro viene poi stampato (o osservato su monitor o letto in maniera completamente automatizzata a seconda del modello dell’apparecchio), dando così modo di mettere in evidenza: a) il tipo di aritmia presente; b) se l’aritmia è responsabile o meno dei sintomi segnalati dal paziente. Diverso ancora è il caso dell’aritmia rara e potenzialmente pericolosa. L’elettrocardiogramma dinamico in questo caso è poco utile perché la probabilità di cogliere l’evento aritmico è bassa. Si ricorre allora a metodiche di tipo provocativo da riservare esclusivamente allo specialista fornito degli strumenti adeguati. Il modo più semplice per cercare di indurre l’aritmia è l’esercizio fisico. Si sottopone quindi il paziente a una prova da sforzo (il più delle volte viene impiegato il cicloergometro) con incrementi progressivi del carico di lavoro e con la registrazione continua dell’elettrocardiogramma (la registrazione dev’essere mantenuta per qualche minuto anche nella cosiddetta fase di recupero dello sforzo, durante la quale frequentemente si manifestano alcune forme di aritmia). Nell’impossibilità clinica di eseguire una prova da sforzo o quando la prova da sforzo non è conclusiva, si può ricorrere allo studio elettrofisiologico. Lo studio elettrofisiologico è una metodica invasiva da riservare a un ambiente altamente specializzato, che consiste nel tentativo di creare, attraverso stimolazione elettrica dell’atrio e del ventricolo destro, le condizioni fisiopatologiche che sono alla base della formazione dell’aritmia. L’induzione programmata dell’aritmia consente di studiarne in dettaglio le caratteristiche, specie per quanto concerne la risposta ai vari farmaci antiaritmici. Lo studio elettrofisiologico viene effettuato introducendo un elettrodo stimolatore attraverso una vena periferica (solitamente la femorale) e spingendolo nel ventricolo destro sotto controllo fluoroscopico. L’elettrodo stimolatore è collegato all’esterno con un apparecchio in grado di inviare al ventricolo degli “extrastimoli”. Gli extrastimoli sono, di fatto, dei battiti prematuri indotti artificialmente, che possono essere sincronizzati con l’attività elettrica spontanea del cuore. Questa sincronizzazione consente all’operatore di fare “cadere” l’extrastimolo durante un momento prescelto della ripolarizzazione ventricolare. Questa tecnica cerca, in sostanza, di ricreare in laboratorio la condizione patologica che dà il via all’aritmia. Insieme col catetere dell’elettrodo stimolatore viene inserito (sempre per via femorale) anche un catetere re- 156 MALATTIE DEL SISTEMA CIRCOLATORIO ventricolare. La registrazione dei potenziali endocavitari consente di conoscere esattamente la sede nella quale è situato il blocco: nell’atrio se è allungato il tempo P-A; nel nodo atrioventricolare se è allungato il tempo A-H; nelle branche e nel sistema di Purkinje se è allungato il tempo H-V. La conoscenza della sede del blocco orienta verso le misure terapeutiche più idonee. In particolare, può suggerire l’impianto di un pacemaker artificiale. La conoscenza della via seguita dall’impulso è particolarmente importante nella diagnosi e nella terapia di alcune aritmie da rientro. La via seguita da un determinato impulso può essere ricostruita osservando la successione con la quale avviene l’inscrizione dei vari potenziali endocavitari. Normalmente l’attivazione è quella già descritta: P-A-H-V; in condizioni patologiche l’ordine può essere incompleto e in parte invertito. Si prenda l’esempio di un’extrasistole ventricolare che induce un rientro a livello del nodo atrioventricolare. Alla deflessione V del battito ectopico ventricolare (inizio QRS ectopico) segue un’onda H da attivazione retrograda del fascio di His. A questa segue poi un’altra V d’attivazione ventricolare (inizio di un secondo QRS ectopico) che consegue al rientro attraverso il nodo atrioventricolare. La conoscenza di questo meccanismo patogenetico consente di orientare la scelta del farmaco antiaritmico (nel caso citato, per esempio, si sceglierà un farmaco gistratore, il cui compito è quello di registrare i cosiddetti potenziali endocavitari (Fig. 6.26). L’ECG di superficie consente di registrare solo variazioni di potenziale elettrico di voltaggio relativamente elevato: l’onda P di attivazione atriale; il QRS di attivazione ventricolare; l’onda T di ripolarizzazione ventricolare. La registrazione endocavitaria fornisce utili informazioni supplementari. Mediante questa tecnica, infatti, possono essere registrati il potenziale determinato dall’attivazione del fascio di His (deflessione H) e il potenziale originato dall’attivazione della parte distale, bassa dell’atrio destro (deflessione A). La registrazione contemporanea dei potenziali endocavitari e di una derivazione elettrocardiografica consente di avere due tipi di informazione: a) scomporre il tempo di conduzione atrioventricolare (PR nell’ECG di superficie) in tre componenti: P-A; A-H; H-V (P rappresenta l’inizio della P dell’ECG; V rappresenta l’inizio del QRS sempre nell’ECG di superficie); b) conoscere in dettaglio la via seguita dall’impulso. Si è già visto che il significato prognostico di un blocco atrioventricolare è diverso a seconda della sede del blocco: soprahissiano o sottohissiano. L’ECG di superficie fornisce solo un dato complessivo e si limita a segnalare che esiste un disturbo di conduzione compreso fra il momento in cui la depolarizzazione atriale ha origine e il momento in cui inizia la depolarizzazione R P VCS D2 Q AS AD NAV S V A H H HBE A VS VD AD AS D2 H HBE NAV V VCI VCS VD VS = Potenziale di attivazione dell’atrio destro = Atrio destro = Atrio sinistro = Derivazione dell’ECG di superficie = Fascio di HIS e potenziale hissiano sull’ECG endocavitario = Elettrocardiogramma endocavitario = Nodo atrioventricolare = Complesso ventricolare = Vena cava inferiore = Vena cava superiore = Ventricolo destro = Ventricolo sinistro VCI Figura 6.26 - Elettrocardiogramma endocavitario. Si registra introducendo un catetere rilevatore attraverso una vena periferica (di solito la femorale) e posizionandolo al di sotto della commissura mediale della tricuspide in corrispondenza del fascio di His. È così possibile registrare i potenziali di attivazione dell’atrio destro e del fascio di His. Il tempo di conduzione fra l’inizio dell’attivazione atriale (inizio onda P) e quello dell’attivazione ventricolare (inizio onda Q) può così essere scomposto in alcune componenti (P-A; A-H; H-V) e mettere in evidenza con maggiore precisione la sede di eventuali disturbi di conduzione. 6 - ARITMIE che allunghi la conduzione a livello del nodo atrioventricolare per impedire il rientro nodale). Il catetere stimolatore può essere utilizzato anche a scopo terapeutico con la tecnica detta “dell’over-drive”. L’over-drive si può usare nella terapia del flutter e della tachicardia atriale e per alcuni casi di tachicardia ventricolare. La tecnica dell’over-drive consiste nello stimolare la camera (atriale o ventricolare) dalla quale prende origine l’aritmia con una frequenza superiore a quella intrinseca dell’aritmia che si vuole curare. La stimolazione viene mantenuta per 10-15 sec poi viene bruscamente interrotta. Se la pratica ha successo, dopo l’interruzione della stimolazione segue un breve periodo di asistolia, seguito a sua volta dalla ripresa del normale ritmo sinusale. L’over-drive atriale (utile quindi solo nel flutter e nella tachicardia atriale) può essere effettuato con tecnica non invasiva mediante catetere esofageo. L’esofago è adiacente alla parete posteriore degli atri. Per via orale è possibile inserire un piccolo elettrodo collegato a un filo contrassegnato da alcune tacche equidistanti. Il paziente può deglutire l’elettrodo aiutandosi con qualche sorso d’acqua. Il medico regge con una mano il filo collegato all’elettrodo e, aiutandosi con le tacche poste sul filo, calcola la distanza percorsa dall’elettrodo nell’esofago del paziente. L’elettrodo dev’essere po- 157 sto a ridosso della parete posteriore degli atri. L’elettrodo ha due funzioni: di registrazione (il filo dev’essere collegato a una derivazione dell’elettrocardiografo) e di stimolazione (filo collegato a un pacemaker). La posizione corretta dell’elettrodo viene segnalata dalla registrazione di ampie onde di depolarizzazione atriale. Collegando il filo a un pacemaker e stimolando, è possibile “catturare” gli atri ed eseguire correttamente un over-drive secondo le modalità già descritte. La registrazione dell’attività atriale mediante catetere esofageo è molto utile anche per la diagnosi differenziale fra una tachicardia ventricolare e una tachicardia sopraventricolare condotta con aberranza. Nella tachicardia ventricolare l’attività atriale è dissociata da quella ventricolare. Nella tachicardia sopraventricolare condotta con aberranza il rapporto fra onda P e onda QRS è costante. Sia nella tachicardia ventricolare sia nella tachicardia sopraventricolare condotta con aberranza il complesso QRS è allargato oltre 0,10-0,12 sec. L’elevata frequenza del complesso ventricolare e l’allungamento del QRS tendono a mascherare l’attività atriale (onda P). La registrazione mediante elettrodo esofageo amplifica l’onda P che può essere facilmente registrata. Se è dissociata dal QRS, la tachicardia è ventricolare; se è in rapporto fisso con il QRS, è sopraventricolare condotta con aberranza. CAPITOLO XI: L’ELETTROCARDIOGRAMMA SISTEMA DI CONDUZIONE INTRAMIOCARDICO Sappiamo che lo stimolo nasce a livello del nodo seno-‐atriale poi passa al nodo atrio-‐ ventricolare, da qui si trasmette al fascio di His e attraverso le due emibranche di sx e alla branca di dx arriva alle fibre di Purkinje che sono le fibre che raggiungono i cardiomiociti nello spessore del ventricolo. La velocità con cui gli impulsi elettrici sono condotti lungo il sistema di conduzione varia da punto a punto. La conduzione dell’impulso è più lenta attraverso le fibre in nodo AV e più veloce attraverso le fibre di Purkinje. Il rallentamento che si realizza a livello del nodo AV ha un significato funzionale: consente cioè il riempimento dei ventricoli prima che giunga l’onda di depolarizzazione cardiaca. Questa caratteristica ci fa ancora più comodo nella fibrillazione atriale, perché impedisce che tutti gli stimoli che si originano nell’atrio possano propagarsi ai ventricoli determinando una condizione che sarebbe incompatibile con la vita. DEPOLARIZZAZIONE E RIPOLARIZZAZIONE Il miocardio cita va incontro a cicli di depolarizzazione e ripolarizzazione. A riposo il cardiomiocita è polarizzato, cioè vi sono delle cariche negative all’interno e positive all’esterno. Uno stimolo elettrico determina una depolarizzazione e quindi le cariche positive si accumulano all’interno e quelle negative all’esterno (parliamo in questo caso di cardiomiocita attivato). Il ritorno alle condizioni di base avviene mediante ripolarizzazione Quindi possiamo affermare che così come il cardiomiocita di lavoro ha un suo ciclo (sistolediastole) così il cardiomiocita ‘’elettrificato’’ ha un suo ciclo che è sempre lo stesso (polarizzazione- depolarizzazione). Se noi ponessimo un elettrodo sulla cute saremmo in grado di percepire questa attività elettrica. Il principio banale per cui noi osservando un tracciato, parliamo di onda positiva o negativa dipende dalla posizione dell’elettrodo rispetto alla corrente, rispetto quindi alla direzione di propagazione dell’impulso. Se l’impulso, l’onda di depolarizzazione procede verso l’elettrodo positivo che registra avremo una deflessione positiva del tracciato (ossia verso l’alto), se l’impulso si allontana dall’elettrodo la deflessione sarà negativa (ossia verso il basso). Tale tracciato prende il nome di elettrocardiogramma o ECG. Adesso analizziamo i vari componenti dell’ elettrocardiogramma in base a questo concetto appena espresso. L’onda P è l’onda di depolarizzazione degli atri, cioè significa che all’interno dell’atrio i cardiomiociti sono passati da una condizione di riposo ad una condizione di attivazione (quindi le cariche positive sono passate all’interno mentre quelle negative all’esterno). Il complesso rapido QRS, che segue l’onda P, rappresenta la depolarizzazione dei ventricoli. L’onda T rappresenta infine la ripolarizzazione ventricolare. La ripolarizzazione atriale è nascosta nel complesso QRS in condizioni normali. LA REGISTRAZIONE DELL’ECG Per poter correttamente monitorare l’attività elettrica cardiaca, deve essere preposto un buon sistema scrivente atto a registrare le correnti di depolarizzazione e ripolarizzazione. Innanzitutto la carta utilizzata presenta una serie di quadrati, che corrispondono sull’asse delle ascisse all’andamento temporale e sull’asse delle ordinate alle variazioni di potenziale. Generalmente l’ECG si registra ad una velocità di 25 mm/sec (sarebbe la velocità di scorrimento della carta), quindi tutti i calcoli che noi facciamo si basano su questo concetto. Quindi primo concetto importante è che 1 secondo corrisponde a 25 mm sul tracciato. La carta che noi utilizzeremo sarà quadrettata e millimetrata; avremo in 25 mm 5 quadrati contornati da una linea più marcata e, all’interno di questi, 5 quadrati tracciati con una linea più sottile. Ciascun quadrato grande (5mm per 5 mm) corrisponde a 0,2 secondi, ogni quadratino di 1 mm corrisponde a 0,04 sec (questo ci permetterà di valutare ad esempio un eventuale QT lungo). Un quadrato grande corrisponde in verticale a 5mm oppure possiamo dire a 0,5 mV( sull’asse verticale valutiamo ad esempio un sovraslivellamento o un sottoslivellamento del tratto ST). All'interno di un ECG possiamo caratterizzare una serie di tratti. L’intervallo PR si misura dall’inizio dell’onda all’inizio del P complesso rapido QRS e rappresenta il tempo necessario affinché lo stimolo passi attraverso gli atri e raggiunga la giunzione atrio-ventricolare. Negli adulti varia tra 0,12sec (3 quadratini) e 0,20 sec (5 quadratini). Quindi, è patologico al di sotto di 0,12 cioè indica che c’è una pre-eccitazione ventricolare e al di sopra di 0,20 (0,24 potrebbe essere un blocco atrioventricolare di I grado). L’intervallo tempo QRS rappresenta necessario affinché il uno stimolo depolarizzi i ventricoli. Negli adulti l’intervallo QRS è inferiore a 0,10 sec. Se la diffusione dello stimolo attraverso i ventricoli è rallentata, come accade nei blocchi di branca,allora il QRS si allunga. Il segmento ST è la porzione del ciclo elettrocardiografico che va dalla fine del complesso QRS all’inizio Rappresenta dell’onda l’inizio T. della ripolarizzazione ventricolare. Generalmente è isoelettrico (piatto). L’inizio del segmento ST è detto punto J (è una finezza diagnostica usata spesso in ergometria). In alcune condizioni patologiche , come nell’infarto del miocardio, vi possono essere delle deviazioni del segmento ST (sopraslivellato o sottoslivellato). Un ST sopraslivellato indica un infarto di tipo STEMI che è generalmente l’espressione di un’occlusione coronarica totale che determina un’ischemia transmurale (a tutto spessore) della parete miocardica. Un ST sottoslivellato come accade nell’NSTEMI è espressione di un‘ischemia subendocardica. L’ST sopraslivellato è per definizione molto più grave dell’ST sottoslivellato. Ma come mai in un tipo e nell’altro si verificano queste variazioni nell’ECG così caratteristiche? Per una maggiore chiarezza, guarda l’immagine in altoNell’infarto NSTEMI il vettore ST è diretto verso la cavità endocardica, quindi si allontana dall’elettrodo e viene quindi descritto sul tracciato con un sottoslivellamento. Questo fenomeno è dovuto all’ischemia sub endocardica. Nell’infarto STEMI, il vettore ST è diretto verso l’elettrodo precordiale e viene quindi descritto sul tracciato con un sopraslivellamento. A seconda di dove notiamo il sopraslivellamento possiamo determinare con certezza la sede dell’evento ischemico e quindi qual è la coronaria che si è chiusa. Tale fenomeno è dovuto all’ischemia transmurale. ECG A 12 DERIVAZIONI Le 12 derivazioni ci permettono di ‘’fotografare’’ il cuore in tutte le sue parti (davanti, dietro, di lato etc). Abbiamo 6 derivazioni dagli arti e 6 derivazioni precordiali (I, II, III, aVR, aVF, aVL, V1, V2, V3, V4, V5, V6). Ciascuna derivazione è ottenuta tramite la disposizione su determinate aree del corpo di elettrodi: - La derivazione I si ottiene mettendo l'elettrodo positivo sul braccio sinistro e l'elettrodo negativo sul braccio destro; - la derivazione II si ottiene mettendo l'elettrodo negativo sul braccio destro e l'elettrodo positivo sulla gamba sinistra; - la derivazione III si ottiene mettendo l'elettrodo positivo sulla gamba sinistra e l'elettrodo negativo sul braccio sinistro. Queste tre derivazioni, proprio perché prevedono il posizionamento di due poli sulla superficie corporea, vengono definite bipolari. Le derivazioni unipolari misurano il voltaggio in un punto relativo a un elettrodo, definito centrale terminale o indifferente, caratterizzato da un potenziale praticamente nullo. Le tre derivazioni unipolari sono: - aVR, sul braccio destro; - aVL, sul braccio sinistro; - aVF, sulla gamba sinistra. La lettera "a" minuscola indica che questi potenziali unipolari vengono aumentati elettricamente del 50%. L'elettrodo posto sull'arto inferiore destro costituisce la "terra" (terminale centrale di Wilson). L’insieme di queste prime sei derivazioni permette di analizzare l'attività elettrica del cuore sul piano frontale, e vengono rappresentate sul diagramma esassiale. Le sei derivazioni toraciche sono registrazioni unipolari ottenute mediante elettrodi posti nel seguente modo: - V1, quarto spazio intercostale sulla linea marginosternale destra; - V2, quarto spazio intercostale sulla linea marginosternale sinistra; - V3, in posizione intermedia tra V2 e V4; - V4, sulla linea emiclaveare in corrispondenza del quinto spazio intercostale; - V5, sulla linea ascellare anteriore allo stesso livello di V4; - V6, sulla linea ascellare media allo stesso livello di V4 e V5. Questi elettrodi esplorano l'attività elettrica del cuore lungo un piano orizzontale. Nell'insieme, le 12 derivazioni permettono una rappresentazione tridimensionale dell'attività elettrica cardiaca. Tanto per cominciare, possiamo notare come l’elettrodo aVR è lontano da qualunque corrente di attivazione del cuore che normalmente va dall’atrio verso il ventricolo, quindi tale derivazione è la derivazione dove tutte le onde sono negative (negativa la P, negativo il QRS e negativa l’onda T). Se qualcosa è positivo in AVR vorrà dire che c’è qualcosa che non va!!! Ciascuna delle altre 5 derivazioni provenienti dagli elettrodi posti sugli arti avrà delle positività o delle negatività in funzione della morfologia del cuore (ad esempio come si è ingrandito, sviluppato o ipertrofia di un ventricolo). Le derivazioni toraciche sul piano orizzontale (precordiali) sono generalmente positive perché seguono la normale propagazione dello stimolo dall’atrio al ventricolo. Ovviamente, le derivazioni più vicine al ventricolo sinistro mostreranno le onde di maggiore ampiezza. Quindi con le prime 6 derivazioni (periferiche o degli arti) andiamo a valutare il piano frontale, mentre con le precordiali o toraciche andiamo a valutare il piano orizzontale MORFOLOGIA DEI COMPLESSI QRS NELLE 12 DERIVAZIONI La prima fase della depolarizzazione ventricolare è relativamente breve (<0.04 sec). È dovuta alla depolarizzazione del setto interventricolare, che inizia a livello della parte sinistra del depolarizzazione setto del setto stesso. La determina contemporaneamente una deflessione positiva in V1 (onda R) ed una negativa in V6 (onda Q). La seconda fase della depolarizzazione ventricolare coinvolge entrambi i ventricoli, che si depolarizzano dall’endocardio verso l’epicardio. Dal momento che nel cuore normale la massa del ventricolo sinistro è maggiore di quella del ventricolo destro il vettore del QRS è diretto a sinistra e verso l’esterno. La depolarizzazione dei ventricoli determina contemporaneamente una deflessione negativa in V1 (onda S) ed una positiva in V6 (onda R). Passando da V1 a V6 le onde R tendono ad aumentare di voltaggio, mentre le onde S diventano relativamente più piccole, come mostrato nell’immagine sottostante. Questo incremento di voltaggio dell’onda R, che raggiunge il massimo in V4V5, è detto normale progressione dell’onda R. Generalmente a livello di V3-V4 il rapporto tra onda R ed onda S diventa uguale ad 1. La derivazione nella quale l’onda R ed S tendono ad eguagliarsi prende nome di zona di transizione. Il concetto della normale progressione dell’onda R nelle precordiali è importante poiché in alcuni casi, come a seguito di un infarto anteriore, si può avere scarsa progressione dell’onda R, per la perdita dei miociti. Quindi se mi manca la progressione dell’onda R potrei sospettare una pregressa necrosi!!! COSA E’ POSSIBILE RILEVARE DALL’ECG Dall’ECG possiamo rilevare: - La frequenza; - il ritmo; - l’asse; - la presenza di ipertrofia; - l’infarto o comunque l’ischemia. Frequenza: La frequenza normale è compresa tra 60 e 100 bpm, parliamo di bradicardia quando la frequenza è inferiore ai 60 bpm e di tachicardia quando la frequenza è superiore ai 100 bpm. Nel bambino una frequenza di 100 bpm difficilmente può rappresentare un indice patologico, mentre nell’adulto e nell’anziano è più facile che lo sia. La frequenza si calcola prendendo un onda R che cade precisamente in corrispondenza dell’inizio di un quadrato grande e da qui si contano i successivi quadrati grandi fino al raggiungimento di un’altra onda R (1 quadrato = 300bpm, 2 quadrati =150bm, 3 quadrati= 75bmp etc etc). Questo ovviamente lo facciamo se non abbiamo a disposizione un righello apposito per l’ECG. Questo metodo pratico è riassunto nell’immagine seguente. Ritmo: Il ritmo sinusale è il ritmo che normalmente nasce dal nodo del seno-atriale. Come facciamo a dire che c’è un ritmo sinusale? Per poter rispondere si passa ad analizzare i seguenti parametri: - presenza dell’onda P prima di ogni QRS e tali onde P devono essere positive in DII e negative in aVR; - intervallo PR costante (0,12-0,20 secondi); - aspetto della P costante; - frequenza compresa tra i 60 e i 100 bpm; - intervallo PP costante. In linea generale si può affermare che la caratteristica fondamentale del ritmo sinusale è l’equidistanza tra onde simili. Asse: L’asse è una cosa che ha perso nel tempo la sua importanza dato l’avvento dell’ecocardiogramma. L’asse elettrico esprime l’orientamento in gradi del vettore medio di attivazione ventricolare. La sua posizione è determinata all’interno di una circonferenza disegnata idealmente sul torace del paziente. Abitualmente è orientato verso il basso e a sinistra (tra -30° e +90°). Un asse compreso tra 30° e 90° corrisponde ad una deviazione assiale sinistra. Un asse compreso tra +90° e +180° corrisponde ad una deviazione assiale destra. Per valutare approssimativamente l’asse bisogna guardare D1 e aVF. A tal proposito guarda l’immagine a lato. In funzione dell’altezza dell’onda R, sia essa positiva o negativa, noi riportiamo i valori su di un asse ideale. Se il QRS è positivo in D1 e positivo anche in aVF, il vettore QRS sarà orientato in basso ed a sinistra risultante del paziente, avrà quindi l’asse una direzione normale. Se il QRS è positivo in D1 e negativo in aVF, il vettore QRS sarà orientato nel quadrante superiore risultante sinistro, avrà quindi l’asse una deviazione assiale sinistra. Se il QRS è negativo in D1 e positivo in aVF si avrà una deviazione assiale destra. Le principali cause di deviazione assiale destra sono: - artefatto: inversione degli elettrodi braccio destro-braccio sinistro; - variante normale; - destrocardia; - sovraccarico ventricolare destro, che può essere acuto per via di un’embolia polmonare, oppure cronico dovuto a BPCO, ipertensione polmonare, stenosi polmonare; - infarto miocardico coinvolgente la parete laterale; - emiblocco posteriore sinistro. Le principali cause di deviazione assiale sinistra sono: - ipertrofia del ventricolo sinistro; - emiblocco anteriore sinistro; - enfisema; - iperpotassiemia; - atresia della tricuspide; - iniezione di contrasto nell’arteria coronaria sinistra. Ipertrofia: L’ECG ha una sensibilità minore rispetto all’ecocardiogramma nell’identificare l’ipertrofia. Mediante l’ECG è comunque possibile osservare segni ECGrafici riferibili a: - Ipertrofia ventricolare destra; - Ipertrofia ventricolare sinistra; - Ipertrofia atriale destra/ingrandimento atriale destro; - Ipertrofia atriale sinistra/ingrandimento atriale sinistro. L’ECG ha un valore diagnostico molto elevato nella cardiopatia ischemica e nelle aritmie. Il criterio abituale per la diagnosi di ingrandimento atriale destro è un’onda P alta più di 2,5 mm. Generalmente si osserva meglio nelle derivazioni inferiori II, III e aVF, dal momento che l’asse dell’onda P (per le aumentate forze elettriche) è diretto verso il basso. L’ingrandimento atriale destro è spesso causato da un sovraccarico di pressione o di volume a livello dell’atrio stesso. L’immagine mostrata riguarda proprio tale situazione. Dato che tale aspetto ECGrafico è legato così frequentemente all’ipertensione polmonare, le onde P caratteristicamente alte ed appuntite vengono denominate “P polmonari”. L’ipertrofia atriale sinistra o ingrandimento atriale sinistro si associa con alcuni possibili segni ECGrafici: - Onda P allargata, con durata superiore 0,11 sec in tutte le derivazioni; - Un’incisura o “doppia gobba” nell’onda P, con i due picchi separati da ≥0,04 sec; - Deflessione negativa nella porzione terminale dell’onda P in V1 ≥1 mm di ampiezza e profondità. Dato che tale aspetto ECGrafico è frequentemente legato ad una patologia mitralica, le onde P da ingrandimento atriale vengono denominate “P mitraliche”. L’immagine a lato mostra tale situazione. L’ipertrofia ventricolare destra si associa con alcuni possibili segni ECGrafici: - Onda R alta nella derivazione V1, con rapporto onda R: onda S≥1; - Deviazione assiale destra; - Onde S relativamente più profonde nelle precordiali sinistre rispetto a quanto si osserva comunemente. Tale situazione viene mostrata nell’immagine a lato. L’ipertrofia ventricolare sinistra è caratterizzata, in V5 e V6, da un’onda R molto positiva e in V1 abbiamo una P mitralica che sta a significare come l’atrio soffra per via del ventricolo sinistro e un un’onda S molto profonda. Per poter fare diagnosi di ipertrofia ventricolare sinistra esistono una serie di criteri, ciascuno dotata di una propria sensibilità e specificità. Questi criteri sono: - La somma dell’onda S in V1 e dell’onda R in V5 o V6 deve essere maggiore di 35 mm. Tale criterio mostra una sensibilità del 43% ed una specificità del 95%; - La somma dell’onda R precordiale più alta e dell’onda S più profonda deve essere superiore a 45 mm. Tale criterio mostra una sensibilità del 45% e una specificità del 93%; - L’utilizzo del Romhilt-Estes Point system, un algoritmo che presenta una sensibilità del 50-54% ed una specificità del 95-97%. Infarto: L’irrorazione cardiaca è assicurata da: - Arteria coronaria Dx: parte diaframmatica del cuore e ventricolo destro; - Arteria discendente anteriore: setto interventricolare e gran parte della parete libera del ventricolo sinistro; - Arteria Circonflessa: parete laterale del ventricolo sinistro. Tuttavia tale schema può presentare possibili variazioni. Le immagini precedenti riassumono le variazioni dell’ECG durante un infarto transmurale. Nell’infarto transmurale, le prime alterazioni avvengono a livello del tratto ST e dell’onda T, generalmente in due fasi. Nella fase acuta, si osserva elevazione del tratto ST e talvolta onda T appuntita (onda T iperacuta). Successivamente, si può osservare inversione dell’onda T nelle derivazioni in cui si è verificato il sopraslivellamento del tratto ST. Nelle fasi più avanzate, può comparire l’onda Q ed il tratto ST ritorna all’isoelettrica. La localizzazione dell’infarto avviene a livello: - Infarto anteriore: • Antero-settale; • Anteriore; • Antero‐laterale o antero‐apicale; - Infarto inferiore; - Infarto posteriore; - Infarto ventricolare destro. Come fa l’ECG a dirci qual è la coronaria che si è chiusa? Ce lo dice in base alle derivazioni. E’ facile dire come le derivazioni precordiali siano specifiche della parete anteriore del ventricolo così come DII, DIII e aVF sono molto più specifiche della parete inferiore, mentre DI e aVL esplorano bene la parete laterale. Con questo criterio io posso sapere quando è coinvolta la discendente anteriore, quando il circonflesso e quando la coronaria destra. Osservando l’ECG possiamo avere molte informazioni, ad esempio se facciamo una trombolisi e il tratto ST non scende o facciamo un’angioplastica e l’ST non scende qualcosa è andato storto Nel caso di infarto posteriore vero, questi non va confuso con l’ipertrofia ventricolare destra , ossia va posta una diagnosi differenziale in caso di patologia polmonare acuta. Quindi dato che il reperto elettrocardiografico potrebbe essere confuso, la presenza di una patologia polmonare acuta ci dirigerà verso un’ipertrofia ventricolare destra, altrimenti sarà quasi sicuramente un infarto posteriore causato da un problema relativo alla coronaria destra e ciò lo vedremo solo in V1. Seguiranno adesso una serie di esempi che ricapitolano praticamente tutto quello che ci siamo detti fin qui. Frequenza: 60 bpm Ritmo: sinusale Asse: normorientato Ipertrofia: IVS+Ipertrofia/dilatazione atriale sinistra Infarto: no Criteri IVS: Somma dell’onda R precordiale più alta e dell’onda S più profonda > 45 mm La somma dell’onda S in V1 and dell’onda R in V5 o V6 > 35 mm Criteri Ipertrofia/dilatazione atriale sinistra: 1. Onda P allargata, con durata >0,11 sec in tutte le derivazioni. 2. Un’incisura o “doppia gobba” nell’onda P, con i due picchi separati da ≥0,04 sec. 3. Deflessione negativa nella porzione terminale dell’onda P in V1 ≥1 mm di ampiezza e profondità • DONNA DI 58 ANNI CON STORIA DI BPCO Frequenza: 55 bpm Ritmo: sinusale Asse: normorientato Ipertrofia: Ipertrofia/dilatazione atriale DESTRA Infarto: no Criteri Ipertrofia/dilatazione atriale DESTRA: • • Il criterio abituale per la diagnosi di ingrandimento atriale destro è un’onda P alta più di 2,5 mm, Generalmente si osserva meglio nelle derivazioni inferiori II, III e aVF, dal momento che l’asse dell’onda P (per le aumentate forze elettriche) è diretto verso il basso. • Uomo di 51 anni con pregresso infarto Frequenza: 60 bpm Ritmo: sinusale Asse: alterato Ipertrofia: no Infarto: Onde Q in sede inferiore II-III-aVF Uomo di 63 anni con dolore toracico, fumatore, iperteso, dislipidemico e familiarità per CAD. INFARTO ANTERO-SETTALE 1) Onda Q in V1 e V2 (“derivazioni settali”) 2) ST sopraslivellato in V1 e V2 3) Onde T invertite da V1 a V5 Frequenza: circa 60. Ritmo: sinusale Frequenza: 60 bpm Ritmo: sinusale Asse: alterato Ipertrofia: no Infarto: Onde Q in sede inferiore II-III-aVF Uomo di 63 anni con dolore toracico, fumatore, iperteso, dislipidemico e familiarità per CAD. INFARTO ANTERO-SETTALE 1) Onda Q in V1 e V2 (“derivazioni settali”) 2) ST sopraslivellato in V1 e V2 3) Onde T invertite da V1 a V5 Frequenza: circa 60. Ritmo: sinusale Uomo di 40 anni con dolore toracico oppressivo, retrosternale, con irradiazione al giugulo. Insorto da circa 1 ora dopo assunzione di cocaina. INFARTO ANTERIORE 1) Onda Q in V2-V6 2) ST sopraslivellato da V1 a V5 Frequenza: 100 bpm Ritmo: sinusale Uomo di 74 con dolore precordiale insorto da 10 ore. Marcatori di danno cardiaco positivi (CK-MB e Troponina I). Diabete mellito in terapia insulinica, fumatore di circa 20 sigarette/die, dislipidemia. DIAGNOSI: NSTEMI Alterazione dell’onda T da V2 a V6. Ritmo sinusale Frequenza 65 bpm Ragazza di 23 anni con stenosi polmonare. Ipertrofia ventricolare destra 1. Onda R alta nella derivazione V1, con rapporto onda R: onda S≥1. 2. Deviazione assiale destra. CAPITOLO XII: CARDIOPATIE CONGENITE PREMESSA Per malattie cardiovascolari congenite (congenital heart disease, CHD) si intendono le anomalie della struttura o della funzione cardiocircolatoria presenti alla nascita. Si ritiene che circa lo 0,8% dei bambini nati vivi sia affetto da cardiopatia congenita. Complessivamente i soggetti di sesso maschile sono più affetti di quelli di sesso femminile, ma esistono notevoli differenze nel rapporto maschi/femmine tra forma e forma. Le malformazioni cardiovascolari congenite sono in genere dovute ad un alterato sviluppo embrionale di una struttura normale o l’arresto di sviluppo di questa struttura in uno stadio precoce embrionale o fetale. L’incidenza delle cardiopatie congenite è intorno a 8 casi per 1000 nati vivi, per cui si può stimare che attualmente in Italia nascono circa 4000 neonati all’anno con cardiopatia congenita. Nell’immagine soprastante viene mostrato uno schema riguardo la circolazione fetale, che risulta essere completamente diversa rispetto a quella dell’adulto. Alla nascita si verificano tutta una serie di modificazioni: - I polmoni da inespansi si espandono, in questo modo la circolazione polmonare diventa un circolo a bassa pressione; - Da una circolazione in parallelo, si passa ad una circolazione in serie; - Il dotto arterioso pervio si chiude, in questo modo non vi è più commistura di sangue proveniente dalla circolazione polmonare con quello della circolazione generale; - Chiusura del forame ovale. In questo modo la pressione nell’atrio destro aumenta e il sangue viene così spinto nel ventricolo destro e nel circolo polmonare. EZIOLOGIA Nella maggior parte dei casi la causa della cardiopatia congenita non è nota. Sono state però formulate alcune correlazioni eziologiche di carattere generale: - La rosolia contratta dalla madre durante il primo trimestre di gravidanza aumenta l’incidenza di pervietà del dotto arterioso; - L’abuso di bevande alcoliche durante la gravidanza aumenta il rischio complessivo di cardiopatie congenite. - L’uso di alcuni farmaci da parte della madre durante la gravidanza favorisce l’insorgenza di cardiopatie congenite: la talidomide, la difenilidantoina, i barbiturici, alcuni antitumorali; - I nati nella cui famiglia vi sono casi di cardiopatie congenite hanno più probabilità di avere vizi cardiaci congeniti; - Anomalie genetiche, come la sindrome di Noonan, Kartagner, di DiGeorge; - Associazione con altre malattie genetiche come trisomia 18, 21 e monosomia X0. CONSEGUENZE PATOLOGICHE DELLE CHD Le principali conseguenze delle CHD sono: - Insufficienza cardiaca congestizia; - Cianosi centrale; - Ipertensione polmonare; - Sindrome di Eisenmenger. L’insufficienza cardiaca congestizia insorge precocemente ed è già diagnosticabile in periodo pre-natale, con segni quali edema del cuoio capelluto, ascite, versamento pericardico, riduzione dei movimenti fetali. Nel neonato pre-termine la causa più comune è data dalla pervietà del dotto arterioso, invece nel neonato a termine le cause più comuni sono: coartazione aortica e cuore sinistro ipoplastico. Dopo la prima o seconda settimana di vita (quando la riduzione delle resistenze vascolari polmonari consente uno shunt sinistro-destro significativo, le cause più comuni sono: DIV, TGA, tronco arterioso, difetto del setto AV. La cianosi centrale è dovuta a desaturazione arteriosa di ossigeno conseguente ad uno shunt o mixing di sangue venoso sistemico nel circolo arterioso. L’entità dello shunt o del mixing e la quantità di flusso ematico polmonare determinano la gravità della desaturazione. CHD che causano cianosi centrale sono: TGA, Fallot, Anomalia di Ebstein, atresia della tricuspide, stenosi critica o atresia della polmonare e ritorno venoso polmonare anomalo totale. L’ipertensione polmonare è conseguente all’incremento del flusso e/o delle resistenze vascolari polmonari (RVP). I pazienti con ipossiemia, elevati livelli di ematocrito, aumentato flusso polmonare, possono sviluppare una vascolopatia polmonare la cui eziologia è legata a modificazioni anatomiche dei vasi polmonari con ispessimento della tonaca media, proliferazione intimale, ialinizzazione e fibrosi che comportano modificazioni ostruttive/occlusive del circolo polmonare e quindi aumento delle RVP. La sindrome di Eisenmenger è una vascolopatia ostruttiva del circolo polmonare che si sviluppa a seguito di un preesistente ampio shunt sinistro-destro e che eleva la pressione del circolo polmonare ai livelli di quella sistemica e rende lo shunt bidirezionale o addirittura invertito. Le principali manifestazioni cliniche sono: fibrillazione o flutter atriale, emottisi, trombo embolia polmonare, insufficienza cardiaca congestizia, angina, sincope, endocardite e cianosi. CLASSIFICAZIONE Esistono molte classificazioni di cardiopatie congenite. Quella più seguita (e che adotteremo) divide i difetti cardiaci congeniti in due gruppi: cianogeni e non cianogeni. Nel primo gruppo i pazienti hanno cianosi, nel secondo gruppo no. Ciascun gruppo viene poi ulteriormente suddiviso in due sottogruppi a seconda che il flusso di sangue nel circolo polmonare sia aumentato oppure normale (o ridotto). Le Cardiopatie non cianogene sono: • Flusso polmonare aumentato: - Pervietà del dotto arterioso di Botallo; - Difetti del setto interatriale; - Difetti del setto interventricolare; - Ritorno venoso polmonare anomalo; • Flusso polmonare normale: - Stenosi polmonare; - Stenosi aortica; - Coartazione aortica; - Malattie del miocardio e dell’endocardio; - Ventricolo sinistro ipoplastico. Le Cardiopatie cianogene sono: • Flusso polmonare ridotto: - Tetralogia di Fallot; - Atresia della tricuspide; - Anomalia di Ebstein; • Flusso polmonare aumentato: - Trasposizione dei grossi vasi; - Ventricolo destro a doppia uscita; - Fistola arterovenosa polmonare. Le cardiopatie citate rappresentano circa il 90% di tutte le anomalie cardiache congenite. La distinzione in cardiopatie cianogene e non cianogene non è sempre rigorosa. Possono esistere cardiopatie cianogene nelle quali l’ipossiemia del sangue arterioso è lieve e in questi casi il paziente non appare evidentemente cianotico. E vi sono, al contrario, casi di cardiopatie non cianogene che sviluppano cianosi per una serie di modificazioni emodinamiche. DIAGNOSI CLINICO STRUMENTALE DELLE CHD All’esame obiettivo verranno valutati: - Aspetto fisico sindromico; - Pressione arteriosa e caratteristiche del polso; - Dimensioni e pulsatilità del fegato; - Valutazione dei rumori cardiaci; - Valutazione dell’accrescimento. La diagnosi strumentale non invasiva consiste in: - Elettrocardiogramma; - Tecniche di imagin del torace; - Ecocardiografia fetale, ecocardio color doppler. La diagnosi strumentale invasiva si avvale invece del cateterismo cardiaco. Molto importante è il ruolo della cardiologia pediatrica nella diagnosi prenatale, che si avvale di metodiche diagnostiche non invasive, nonché di terapia interventistica o chirurgia precoce. Sono state messe a punto anche tecniche di chirurgia percutanea e chirurgia mini invasiva. Per ciascuna CHA esiste un periodo ottimale nel quale effettuare la correzione del difetto, ad esempio: - Il TGA viene corretto alla nascita; - L’anomali del ventricolo unico viene corretta intorno alla sesta-ottava settiamana di vita; - Il DIV ampio e il CAVC completo vengono corretti intorno al primo-secondo mese di vita; - Il ToF, viene corretto intorno al III-VI mese di vita. Il trattamento percutaneo viene effettuato nei seguenti casi: - Stenosi aortica critica neonatale; - Stenosi/atresia polmonare critica neonatale; - Stenting del dotto di Botallo; - Chiusura percutanea di DIA, DIV, dotto di Botallo, MAPCAs, fistole artero-venose. Nell’ambito delle CHD alcune sono decisamente più frequenti di altre: - DIV, 32 %; - DIA, 9 %; - Pervietà del dotto di Botallo, 8 %; - Coartazione aortica, 7,6 %; - Tetralogia di Fallot, 6 %; - La trasposizione completa dei grossi vasi, 3,1 %. CARDIOPATIE NON CIANOGENE CON FLUSSO POLMONARE AUMENTATO PERVIETA’ DEL DOTTO ARTERIOSO DI BOTALLO A lato viene mostrata un’immagien relativa al difetto trattato. Il dotto arterioso di Botallo è un vaso che, durante la vita fetale, connette l’arteria polmonare con l’aorta. Esso abbocca l’aorta in immediatamente una posizione distale all’origine dell’arteria succlavia sinistra. Durante la vita fetale il sangue passa dall’arteria polmonare in aorta. Alla nascita il dotto si stringe fino a chiudersi. Lo stimolo che induce l’obliterazione del dotto di Botallo è l’aumento della pressione di ossigeno nel sangue arterioso che segue ai primi atti respiratori. La chiusura del dotto arterioso avviene solitamente nel primo giorno di vita extrauterina; a volte è più tardiva. La pervietà del dotto dopo i tre mesi di vita è da considerare senz’altro patologica. La pervietà del dotto arterioso può essere un difetto isolato o essere associata ad altri difetti cardiaci congeniti. Le conseguenze emodinamiche del difetto isolato sono legate principalmente a due fattori: le dimensioni del dotto e l’entità delle pressioni e delle resistenze vascolari (sia del circolo polmonare sia del circolo sistemico). Il diametro del dotto può variare da pochi mm a circa 1,5 cm. Normalmente, nella vita extrauterina la pressione presente nell’aorta supera quella della polmonare. Il flusso ematico nel dotto arterioso pervio è pertanto diretto dall’aorta alla polmonare. Se il dotto è di dimensioni ampie, il flusso di sangue nel circolo polmonare aumenta di molto. L’aumentato flusso polmonare aumenta la pressione del piccolo circolo. Inizialmente tale aumento pressorio è reversibile e le pressioni ritornano a valori normali se il dotto viene chiuso chirurgicamente. A lungo andare, però, può verificarsi un processo noto come reazione di Eisenmenger: le arterie polmonari sottoposte a costante aumento pressorio vanno incontro ad alterazioni anatomiche irreversibili, con aumento persistente delle resistenze polmonari, che diventano superiori alle resistenze del circolo sistemico. Di necessità, la pressione nell’arteria polmonare diventa molto alta, maggiore di quella aortica. Quando questo avviene, la direzione del flusso del dotto arterioso si inverte: dall’arteria polmonare il sangue (non ancora ossigenato) va nell’aorta. L’inversione del flusso comporta un’immissione di sangue venoso nel circolo arterioso sistemico e il paziente diventa cianotico. A questo punto, il quadro clinico risulta completamente cambiato e si dice che il paziente ha una sindrome di Eisenmenger. Si parla di sindrome di Einsenmenger perché il fenomeno “reazione di Einsenmenger”, precedentemente descritto, può verificarsi, oltre che nella persistenza del dotto di Botallo, anche in altre cardiopatie congenite, in particolare nei difetti interventricolari e, raramente, nella comunicazione interatriale. Quando la reazione di Eisenmenger si è verificata, ne deriva un quadro clinico che è lo stesso qualunque sia stata la cardiopatia originale. Tale quadro clinico si chiama, appunto, sindrome di Einsenmenger. Sintomi: Se il dotto è piccolo, il paziente è asintomatico. Se il dotto è più ampio, il ventricolo sinistro è sottoposto a un sovraccarico di volume; la quantità di sangue che deve pompare, infatti, è costituita dalla somma di due quantità: quella che serve a nutrire i tessuti più la quantità che ricircola lungo il percorso aortapolmoni-atrio sinistro-ventricolo sinistro. In questo caso possono comparire i segni iniziali dello scompenso cardiaco: dispnea da sforzo e facile affaticabilità. Se il difetto non viene corretto, possono manifestarsi in seguito i sintomi dell’insufficienza ventricolare sinistra conclamata. Il dotto arterioso pervio può essere facilmente sede di infezione: si tratta dell’endoarterite infettiva del dotto che ha le stesse caratteristiche dell’endocardite infettiva. Nei casi che vanno incontro a reazione di Eisenmenger si stabilisce una condizione di sovraccarico di pressione a carico del ventricolo destro. Di qui, scompenso cardiaco di tipo destro. Segni: La maggior parte dei pazienti con pervietà del dotto di Botallo è di sesso femminile. Il segno di maggiore rilevanza ai fini della diagnosi è il soffio continuo tipico della pervietà del dotto. Solitamente, la pressione in aorta supera quella in arteria polmonare sia in sistole sia in diastole. Il flusso ematico attraverso il dotto è pertanto sia sistolico sia diastolico. Il passaggio di sangue attraverso il dotto determina un soffio intenso, sistodiastolico, che ha la sua massima intensità sul focolaio di ascoltazione della polmonare. Il gradiente diastolico fra aorta e polmonare è massimo quando le valvole semilunari si chiudono all’inizio della diastole. Il soffio, pertanto, è in crescendo-decrescendo con acme in prossimità del II tono. Se il soffio è particolarmente intenso è accompagnato da fremito alla palpazione. Il reperto ascoltatorio classico può modificarsi se il vizio si complica con un aumento significativo delle pressioni polmonari. Se la pressione del piccolo circolo aumenta in modo significativo, il gradiente fra aorta e polmonare si riduce. La riduzione del gradiente comporta una riduzione di intensità del soffio. L’aumento della pressione polmonare determina la comparsa dei segni ascoltatori dell’ipertensione polmonare: in particolare l’intensità della componente polmonare del II tono aumenta. Può essere presente cianosi solo se il vizio è grave e l’ipertensione polmonare inverte la direzione del flusso attraverso il dotto. Elettrocardiogramma: L’elettrocardiogramma è normale quando il dotto è piccolo. Se la pervietà del dotto è ampia, il ventricolo sinistro è sottoposto a un aumento significativo di lavoro: all’elettrocardiogramma compaiono i segni di ipertrofia del ventricolo sinistro. Se c’è reazione di Einsenmenger è il ventricolo destro a essere sottoposto a un sovraccarico di lavoro. In questo caso l’elettrocardiogramma mostra i segni dell’ipertrofia ventricolare destra (o segni di ipertrofia destra e sinistra combinati). Radiografia del torace: Sono presenti i segni radiologici di ipertrofia del ventricolo sinistro, ma soprattutto l’arteria polmonare e i rami arteriosi polmonari appaiono distesi e dilatati in relazione al grande flusso di sangue che percorre il circolo polmonare. Nella radiografia del torace in proiezione antero-posteriore l’arteria polmonare appare dilatata e la trama dei vasi arteriosi polmonari è aumentata (quadro radiologico di “iperafflusso polmonare”). La sindrome di Eisenmenger è invece caratterizzata dal persistere della dilatazione dell’arteria polmonare e dei suoi rami principali, ma le zone periferiche dei campi polmonari sono particolarmente povere di trama (perché i vasi sono ristretti). Ecocardiografia: L’ecocardiografia bidimensionale può mettere direttamente in evidenza il dotto arterioso pervio. Possono essere presenti i segni di sovraccarico mono o biventricolare. Con la tecnica Doppler è possibile identificare la direzione anomala dei flussi. Nei casi non complicati, in diastole il flusso di sangue è diretto dall’aorta alla polmonare. Ponendo il volume campione nell’arteria polmonare, è possibile registrare un flusso anomalo diastolico turbolento. Col color-Doppler è possibile visualizzare direttamente il dotto. Cateterismo cardiaco: L’arteria polmonare riceve sangue arterioso proveniente dall’aorta. La misurazione del grado di ossigenazione del sangue (ossimetria) mette pertanto in evidenza una differenza di ossigenazione fra i campioni di sangue prelevati nell’arteria polmonare e quelli prelevati nel ventricolo destro. Mediante cateterismo si possono anche misurare direttamente le pressioni e le resistenze del circolo polmonare. Le pressioni e le resistenze del piccolo circolo possono, come si è visto, essere normali o aumentate a seconda del flusso di sangue che passa attraverso il dotto. Classificazione del PDA: La classificazione si basa sulle dimensioni del dotto e sul rapporto delle portate, e si avrà: - Silente: PDA con minimo shunt; - Piccolo: soffio continuo e rapporto tra portata polmonare e portata sistolica inferiore a 1,5; - Moderato: soffio continuo e rapporto tra le due portate compreso tra 1,5 e 2,2; - Grande: con rapporto tra le portate superiore a 2,2. Caratteristiche clinico/fisiopatologiche: La presentazione clinica varia in funzione della grandezza del PDA: - PDA silente: paziente asintomatico, senza alterazioni emodinamiche; - PDA piccolo: paziente asintomatico, senza alterazioni emodinamiche, piccolo shunt e predisposizione ad endoarterite; - PDA moderato: dispnea, palpitazioni ed aritmie atriali, alterazioni emodinamiche con sovraccarico di volume dell’atrio e del ventricolo sinistro con conseguente disfunzione ventricolare sinistra e fibrillazione atriale; - PDA grande: sovraccarico di volume ventricolare sinistro, con progressivo incremento della pressione arteriosa polmonare fino a sviluppare la sindrome di Eisenmenger. Diagnosi: La diagnosi si effettua con la visualizzazione all’eco-color- doppler di un jet che indica lo shuntsinistro-destro dall’aorta discendente al ramo polmonare sinistro. Tale situazione viene mostrata nell’immagine a lato. Decorso: Il decorso naturale della pervietà del dotto arterioso dipende dalle dimensioni del dotto e dalle condizioni del circolo polmonare. Se il dotto ha dimensioni piccole, il paziente è asintomatico e le sue prospettive di vita non sono influenzate dalla cardiopatia. Se il vizio è molto ampio, il paziente può morire nei primi mesi di vita per scompenso cardiaco. Nei più frequenti casi intermedi il decorso clinico è caratterizzato da: assenza di sintomi fino all’adolescenza; dispnea da sforzo e affaticabilità precoce intorno ai vent’anni di età; scompenso cardiaco verso i quarant’anni. Il flusso ematico anomalo nel dotto pervio crea condizioni favorevoli per l’insorgenza di infezioni batteriche (endoarteriti) a carico del dotto stesso. Le endoarteriti del dotto possono manifestarsi in qualsiasi momento della vita. Terapia: La terapia è di tipo chirurgico: consiste nella chiusura del dotto. L’intervento è semplice e presenta una bassa incidenza di complicazioni. Non richiede l’impiego della circolazione extracorporea. Nei casi complicati da ipertensione polmonare il rischio operatorio è maggiore. Va praticata di regola la profilassi antibiotica per l’endoarterite batterica. Sono indicati all’intervento: - Neonati prematuri con shunt significativo ed alterazioni emodinamiche, in questi casi si somministra ibuprofene o indometacina per inibire la sintesi di PGE e favorire la chiusura del dotto. La terapia chirurgica si avvale della legatura del dotto; - Bambini e adulti con PDA moderato/grande vengono direttamente trattati con la chiusura chirurgica del dotto. La tecnica adoperata per la chiusura del dotto è quella della chiusura percutanea con device dedicato. DIFETTI DEL SETTO INTERATRIALE I difetti del setto interatriale (DIA) rappresentano la più frequente anomalia cardiaca congenita. È stato calcolato che i difetti interatriali costituiscono da soli il 15% dei difetti cardiaci congeniti nei soggetti che arrivano al primo anno di vita. La diagnosi di difetto interatriale viene frequentemente posta per la prima volta nell’età adulta. I soggetti di sesso femminile ne sono interessati in misura circa doppia rispetto ai maschi. Nella vita extrauterina il circolo venoso e il circolo arterioso sono completamente separati. In particolare a livello atriale, il setto interatriale impedisce che il sangue arterioso presente nell’atrio sinistro si mescoli col sangue venoso presente nell’atrio destro. Nei difetti interatriali questa condizione viene a cadere: l’atrio sinistro comunica tramite il difetto settale con l’atrio destro e, dato che in quest’ultimo la pressione è minore, si ha passaggio di sangue (shunt) da sinistra a destra. I difetti interatriali vengono suddivisi in tre varietà principali a seconda della sede: - il difetto tipo ostium secundum; - il difetto tipo ostium primum; - il difetto del seno venoso. Nel tipo ostium secundum il difetto interatriale è posto nella porzione media del setto a livello della regione del forame ovale. Nel tipo ostium primum il difetto è posto nella parte inferiore del setto a ridosso del piano atrioventricolare. Nel difetto del seno venoso la comunicazione interatriale avviene in prossimità dello sbocco della vena cava superiore nell’atrio destro. Più dell’80% dei DIA sono del tipo ostium secundum. Le dimensioni del difetto interatriale variano da pochi millimetri a 4-5 cm. DIFETTO INTERATRIALE TIPO OSTIUM SECUNDUM Il DIA consente una comunicazione fra atrio sinistro e atrio destro. Nell’atrio sinistro la pressione è più alta che nell’atrio destro, pertanto il flusso del sangue attraverso la comunicazione va da sinistra a destra. Conseguenze: parte del sangue arterioso ossigenato presente in atrio sinistro si mescola col sangue venoso dell’atrio destro; il flusso nel circolo polmonare aumenta di una quota corrispondente alla quantità di sangue che passa da sinistra a destra attraverso il difetto. L’entità di questa conseguenza è proporzionale a due fattori: le dimensioni del difetto e la pressione presente nel circolo polmonare. Solitamente il difetto ha dimensioni superiori ai 2 cm2 e consente un significativo passaggio di sangue nell’atrio destro: ciò comporta un aumento del flusso di sangue nel circolo polmonare. Inizialmente l’aumentato flusso polmonare non modifica significativamente le pressioni polmonari. La pressione polmonare può aumentare nell’età adulta: dapprima in maniera reversibile e talora irreversibile (si veda quanto detto per il dotto arterioso pervio: reazione di Eisenmenger). Nei rari casi in cui l’ipertensione polmonare secondaria all’iperafflusso di sangue determina un aumento della pressione nel ventricolo destro, e secondariamente nell’atrio destro, il flusso attraverso la comunicazione si inverte: sangue venoso entra nel circolo arterioso e il paziente diviene cianotico; si ha allora la sindrome di Eisenmenger. Sintomi: La sintomatologia dipende dalle dimensioni del difetto. Nei difetti di media grandezza il paziente è asintomatico fino ai 16-18 anni di età. I primi sintomi sono la dispnea da sforzo e la facile affaticabilità; questi sintomi tendono a progredire col passare degli anni. I grossi difetti interatriali possono favorire l’insorgenza di infezioni delle vie respiratorie. La distensione degli atri dovuta all’iperafflusso di sangue facilita l’insorgenza di aritmie ipercinetiche sopraventricolari. Segni: La diagnosi di DIA viene solitamente sospettata all’ascoltazione cardiaca e viene poi confermata dagli esami strumentali. Il segno più caratteristico dell’ascoltazione è costituito dallo sdoppiamento del II tono cosiddetto fisso. Il II tono è composto dalla componente aortica e dalla componente polmonare. In condizioni normali la componente aortica precede quella polmonare. L’intervallo che separa le due componenti dipende dagli atti del respiro: in inspirazione l’intervallo è maggiore; in espirazione è minore, fino ad essere praticamente nullo. L’inspirazione, infatti, riduce la pressione intratoracica e facilita il ritorno di sangue venoso nell’atrio destro. L’aumento del ritorno venoso nell’atrio destro aumenta il volume di sangue nel ventricolo destro. Questo a sua volta aumenta il tempo necessario per lo svuotamento sistolico del ventricolo destro. L’aumento della durata della sistole del ventricolo destro fa ritardare la chiusura della valvola polmonare e perciò ritarda l’inscrizione della componente polmonare del II tono. Il contrario avviene nell’espirazione. In presenza di DIA, il flusso attraverso atrio destro, ventricolo destro e arteria polmonare è aumentato e costante sia in inspirazione che in espirazione. In espirazione, infatti, la quota di sangue “persa” dal circolo refluo venoso periferico (per aumento della pressione intratoracica) viene compensata dall’aumento del flusso attraverso il difetto. L’intervallo che separa P2 (componente polmonare del II tono) da A2 (componente aortica) è pertanto ampio e non varia con le fasi del respiro. Allo sdoppiamento fisso del II tono si accompagnano altri due segni ascoltatori: il soffio sistolico eiettivo udibile sul focolaio della polmonare e il rumore mesodiastolico che si apprezza sul focolaio di ascoltazione della tricuspide. Il primo è dovuto all’aumento del flusso di sangue in sistole attraverso la valvola polmonare; il secondo al flusso diastolico torrenziale attraverso la tricuspide. Elettrocardiogramma: L’aumento di flusso nel ventricolo destro determina il quadro elettrocardiografico di sovraccarico di volume del ventricolo destro: in V1 è presente un complesso rSr' che è molto caratteristico. Nei casi in cui compare ipertensione polmonare, l’elettrocardiografia può mostrare il quadro classico dell’ipertrofia del ventricolo destro con sovraccarico sistolico: R in V1 con T negativa. Radiografia del torace: Alla radiografia del torace compaiono i segni di dilatazione dell’atrio destro, del ventricolo destro, dell’arteria polmonare e dei suoi rami. La vascolarizzazione polmonare è accentuata (quadro di iperafflusso polmonare). All’esame radioscopico i rami principali dell’arteria polmonare appaiono vistosamente pulsanti (danza ilare). Ecocardiogramma: I segni ecografici di DIA sono per lo più indiretti e sono rappresentati dal sovraccarico del ventricolo destro. In particolare l’ecocardiogramma TM mostra un ventricolo destro dilatato e un movimento paradosso del setto interventricolare. I difetti di una certa entità sono visibili direttamente con l’ecografia bidimensionale. Con la tecnica Doppler si può mettere in evidenza il flusso anomalo di sangue che, nelle condizioni non complicate, è diretto dall’atrio sinistro all’atrio destro attraverso il difetto settale. Il color-Doppler mostra direttamente il flusso di sangue che attraversa il DIA nella direzione sinistra-destra. Cateterismo: Il cateterismo cardiaco serve per misurare il contenuto di ossigeno nel sangue e per determinare le pressioni nelle camere destre del cuore e del circolo polmonare. L’ossimetria mette in evidenza un gradiente nel contenuto di ossigeno fra i campioni di sangue prelevati nell’atrio destro e quelli prelevati nella vena cava. Solitamente, le pressioni nel circolo polmonare dei DIA di media gravità sono normali. Decorso: La chiusura spontanea del DIA tipo ostium secundum è molto rara. Nella massima parte dei casi i sintomi compaiono nell’età adulta e sono progressivi in assenza di una correzione chirurgica. Il 75% dei soggetti non operati muore per insufficienza cardiaca verso i 50 anni d’età. Terapia. La terapia è chirurgica e consiste nella chiusura del difetto o per sutura diretta o per applicazione di un “patch”. DIFETTO INTERATRIALE DI TIPO OSTIUM SECUNDUM Durante lo sviluppo embrionale la parte distale del setto interatriale si completa insieme con la formazione del piano atrioventricolare e della parte prossimale membranosa del setto interventricolare. La costruzione ottimale di ciascuna di queste strutture dipende dallo sviluppo armonico delle altre. È per questo che, spesso, le anomalie di formazione del setto interatriale distale (tipo ostium primum) si accompagnano a vizi congeniti a carico del setto interventricolare membranoso e delle valvole atrioventricolari. I difetti più frequenti dell’apparato valvolare mitralico e tricuspidale che si accompagnano al DIA tipo ostium primum sono: - fissurazione (cleft) dei lembi valvolari; - corde tendinee corte; - inserzione anomala dei muscoli papillari. Queste anomalie (da sole o variamente combinate fra loro) determinano un’insufficienza delle valvole atrioventricolari. Al DIA tipo ostium primum può associarsi anche un difetto di formazione del setto interventricolare nella sua porzione membranosa: il ventricolo sinistro è in diretta comunicazione col ventricolo destro. Il complesso DIA tipo ostium primum più difetto del setto membranoso interventricolare prende anche il nome di canale atrioventricolare comune. Le caratteristiche emodinamiche dell’ostium primum dipendono dalla presenza o meno di vizi associati e dalla loro gravità. L’ostium primum isolato, privo cioè di vizi associati a carico del setto interventricolare e delle valvole atrio-ventricolari, ha caratteri emodinamici sovrapponibili a quelli descritti per l’ostium secundum. Anche l’esame obiettivo, radiologico ed ecocardiografico dell’ostium primum isolato sono simili a quelli descritti nell’ostium secundum. L’elettrocardiogramma dell’ostium primum ha (rispetto all’elettrocardiogramma dell’ostium secundum) una peculiarità. Questa peculiarità deriva dall’anomala formazione del setto interatriale distale (ed eventualmente del setto interventricolare membranoso) che spesso coinvolge il tessuto di conduzione. Il gruppo di fibre anteriori della branca sinistra è il distretto più frequentemente coinvolto: da ciò un’anormale diffusione dell’eccitamento nel ventricolo sinistro. L’elettrocardiogramma mostra (in circa il 90% dei casi di ostium primum) il quadro dell’emiblocco anteriore sinistro: deviazione assiale sinistra marcata (–30°) associata ad onda q evidente in D1 e aVL. L’evoluzione naturale dell’ostium primum è più grave di quella dell’ostium secundum: la mortalità è più precoce e complessivamente più elevata. La terapia è di tipo chirurgico. Il DIA tipo seno venoso è il più raro delle tre forme di DIA. Il quadro clinico del DIA tipo seno venoso è sovrapponibile a quello tipo ostium secundum. DIFETTI DEL SETTO INTERVENTRICOLARE Per difetto del interventricolare intende un setto (DIV) difetto si di formazione del setto che separa i due ventricoli. Il difetto mette in comunicazione diretta il ventricolo destro con il ventricolo sinistro, così come mostrato nell’immagine a lato. Fisiopatologia: In presenza di difetto interventricolare, il sangue arterioso presente nel ventricolo sinistro può passare nel ventricolo destro. direzione del flusso La è da sinistra a destra perché la pressione nel ventricolo sinistro è maggiore di quella nel ventricolo destro. In presenza di DIV parte del sangue arterioso del ventricolo sinistro si mescola col sangue venoso presente nel ventricolo destro. L’entità del flusso attraverso il difetto dipende dalle dimensioni del difetto e dal gradiente pressorio presente fra ventricolo sinistro e ventricolo destro. Se il difetto è piccolo, il flusso di sangue verso il ventricolo destro è scarso; le pressioni nel ventricolo destro e nel circolo polmonare restano normali. Si usa spesso indicare questa condizione anomala, ma clinicamente innocua, come malattia di Roger. Se il difetto è grande, il flusso di sangue da sinistra a destra aumenta significativamente: aumenta la pressione del circolo polmonare. Inizialmente l’incremento pressorio è reversibile; col tempo l’ipertensione polmonare può diventare irreversibile per la comparsa di anomalie dei vasi del piccolo circolo secondarie all’aumento del flusso (reazione di Eisenmenger). I DIV sono un vizio cardiaco congenito frequente e vengono diagnosticati per lo più in età prepuberale; è rara la diagnosi di DIV nell’età adulta. Il DIV può presentarsi come vizio isolato o associato ad altre anomalie congenite. I DIV possono essere distinti in base a tre parametri: le dimensioni, la sede e il numero. La dimensione del DIV può variare da pochi mm2 fino ad alcuni cm2. La maggior parte dei DIV è posta a livello della porzione prossimale membranosa del setto. I difetti della porzione muscolare sono chiamati difetti settali muscolari. I difetti della porzione membranosa vengono ulteriormente suddivisi a seconda della loro collocazione relativamente alla crista ventricularis. La crista ventricularis è quella cresta muscolare che attraversa la parte posteriore del canale di efflusso del ventricolo destro. Si distinguono DIV sopracristali e inferocristali, a seconda che siano posti sopra o sotto la crista ventricularis. La maggior parte dei DIV membranosi è sottocristale. I difetti sottocristali sono posti in prossimità della valvola aortica e possono accompagnarsi a difetti congeniti di questa valvola. I difetti sopracristali giacciono immediatamente al di sotto della valvola polmonare. I difetti della porzione membranosa sono solitamente singoli. I difetti della porzione muscolare del setto interventricolare possono essere multipli. L’aumento di pressione nel circolo polmonare si ripercuote sul ventricolo destro. L’aumento di pressione del ventricolo destro riduce il flusso sinistra-destra attraverso il DIV. Se la pressione in ventricolo destro diventa simile a quella sistemica, il flusso attraverso il difetto può invertirsi: sangue venoso passa nel circolo sistemico arterioso con comparsa di cianosi (a questo punto si parlerà di sindrome di Eisenmenger). Classificazione morfologica: I DIV possono essere suddivisi in funzione della localizzazione del difetto. In questo modo i DIV vengono suddivisi in: - DIV membranoso: localizzazione nella porzione superiore del setto, dove aorta, mitrale e tricuspide sono tra loro in continuità fibrosa; - DIV muscolare: localizzato in qualsiasi parte della porzione trasecolata del setto, è di variabile grandezza e prende il nome dalla posizione che occupa nel setto: apicale, muscolare anteriore o medio-muscolare. Può essere unico, ma più spesso sono multipli dando il caratteristico aspetto a Swiss cheese. Un’altra classificazione è quella fisiopatologica, e avremo quindi: - DIV piccolo o restrittivo: quando per le sue dimensioni offre resistenze al passaggio di sangue. Se il difetto è restrittivo lo shunt sinistro destro è piccolo. La pressione nel ventricolo destro sarà in questi casi normale e il QP/QS inferiore 1.5; - DIV moderato: shunt moderato con QP/QS compreso tra 1,5 -2,2; - DIV ampio o non restrittivo: Il difetto è definito ampio o non restrittivo quando non offre resistenze al flusso. Di conseguenza la pressione del ventricolo destro sarà uguale a quella del ventricolo sinistro ed il QP/QS è superiore a2,2. Il DIV restrittivo non determina alterazioni emodinamiche, è asintomatico e presenta la tendenza spontanea alla chiusura. Nel DIV moderato/ampio si ha un carico emodinamico al ventricolo sinistro con conseguente dilatazione e disfunzione atriale e ventricolare sinistra. Ciò determina dispnea, insufficienza cardiaca congestizia, deficit dell’accrescimento nel II-III mese di vita. Il progressivo aumento della pressione arteriosa polmonare può comportare aumento della RVP, con sindrome di Eisenmenger ed inversione dello shunt. Sintomi: Se il difetto è piccolo, il paziente è asintomatico. I difetti più ampi determinano un aumento significativo del flusso e, al di là di una certa portata cardiaca, della pressione polmonare. I pazienti lamentano i sintomi della congestione polmonare: facile affaticabilità, dispnea da sforzo. Più tardi si può avere edema polmonare acuto. L’aumento del flusso polmonare altera i normali equilibri di ventilazioneperfusione e facilita (come nel DIA) la comparsa di infezioni a carico dei polmoni. La cianosi è presente solo nei casi con grave ipertensione polmonare e inversione del flusso attraverso il difetto. Segni: Il principale elemento di diagnosi di DIV deriva dall’ascoltazione del cuore. Il passaggio di sangue attraverso il difetto avviene in sistole, quando esiste un gradiente significativo tra la pressione del ventricolo sinistro e quella del ventricolo destro. Il flusso di sangue dal ventricolo sinistro al destro determina un rumore olosistolico. Il rumore si apprezza meglio sulla marginosternale sinistra a livello del IV spazio intercostale. Il soffio ha solitamente un’intensità compresa fra 4 e 6 sesti; ha forma per lo più “a plateau” (infatti il gradiente ventricolo sinistro-ventricolo destro si mantiene significativo per tutta la durata della sistole) e può accompagnarsi a fremito. In presenza di ipertensione polmonare grave, il flusso ventricolo sinistro-ventricolo destro è molto ridotto: il rumore tipico del DIV può essere assente e possono essere presenti solo i segni ascoltatori dell’ipertensione polmonare. I segni classici dell’ipertensione polmonare sono: un aumento d’intensità della componente polmonare del II tono, un soffio sistolico eiettivo sul focolaio polmonare ed eventualmente un click protosistolico di apertura della valvola polmonare. Se il DIV è sufficientemente ampio, il flusso polmonare è aumentato in modo significativo. L’aumento del flusso polmonare ritarda la chiusura della valvola polmonare: la componente P2 del II tono è ritardata. Lo sdoppiamento del II tono (più ampio quindi del normale) è comunque mobile con le fasi del respiro. Esame obiettivo: Nel DIV restrittivo si può notare la presenza di un soffio olosistolico che diviene evidente quando si riducono le RVP dopo la nascita. Nel caso di DIV ampio, l’uguagliarsi delle pressioni attraverso il DIV evita la genesi del soffio. Elettrocardiogramma: Se il difetto è piccolo, l’elettrocardiogramma è normale. Se il difetto è ampio, sia il ventricolo sinistro che il ventricolo destro sono sottoposti ad un sovraccarico di lavoro. Questo aumento del lavoro determina la formazione di quadri elettrocardiografici di ipertrofia ventricolare sinistra, destra o biventricolare. Radiografia del torace: L’aumento del lavoro dei ventricoli e l’aumento del flusso polmonare determinano i segni radiologici di dilatazione ventricolare (destra e sinistra), dilatazione dell’arteria polmonare e dei suoi rami principali, aumento della trama vascolare polmonare (iperafflusso polmonare). Ecocardiogramma: L’ecocardiogramma TM evidenzia i segni indiretti del sovraccarico del cuore: soprattutto aumento di diametro delle cavità. L’esame bidimensionale può, in alcuni casi, consentire la visione diretta del difetto. Per quanto riguarda la tecnica Doppler, l’esame mette in evidenza un flusso anomalo di corrispondenza del difetto. Il color-Doppler, inoltre, mette direttamente in evidenza il flusso anomalo. L’iimmagine a lato mostra un quadro all’eco-colordoppler. Cateterismo cardiaco: Il ventricolo destro riceve sangue arterioso dal ventricolo sinistro attraverso il DIV. Il sangue prelevato dal ventricolo destro è perciò più ossigenato di quello presente nell’atrio destro. Il cateterismo consente anche la misurazione delle pressioni nell’arteria polmonare. L’ossimetria e la misurazione delle pressioni polmonari consentono una stima indiretta dell’entità del difetto, mediante valutazione comparativa della portata (gettata/min) nel circolo polmonare e nel circolo sistemico. Evoluzione: I piccoli DIV possono chiudersi spontaneamente. Il difetto interventricolare piccolo che non si chiude ma ha scarsa rilevanza emodinamica (malattia di Roger) non necessita di intervento chirurgico. I difetti ampi non si chiudono spontaneamente e vanno sempre operati. Un parametro utile per decidere se un difetto è ampio è quello della gettata polmonare, che si considera eccessivamente alta se è 1,5 volte quella sistemica. I difetti ampi non operati portano a morte il 40% dei soggetti prima dei vent’anni e l’80% prima dei quaranta. Il flusso attraverso il difetto interventricolare crea una situazione emodinamica che facilita l’impianto di batteri a ridosso del difetto. L’infezione (endocardite) può avvenire anche se il difetto è piccolo. Terapia: La terapia è chirurgica e consiste nell’applicazione di un patch di materiale sintetico. Il DIV di qualunque dimensione richiede sempre la profilassi dell’endocardite batterica. L’intervento è indicato nei seguenti casi: - DIV piccolo, asintomatico che non risponde a terapia medica; - DIV moderato/ampio, sintomatico con rapporto tra le gittate superiore a 1,5; - Pressione arteriosa sistolica polmonare superiore ai 50 mmHg. TRASPOSIZIONE DEI GROSSI VASI Questa condizione viene comunemente detta destro o D-trasposizione dei grandi vasi. L’aorta origina dal ventricolo destro,anteriormente e a destra; l’arteria polmonare invece origina dal ventricolo sinistro, posteriormente e verso sinistra. Vengono così a crearsi due circoli separati e paralleli, ma affinchè la vita extrautirana sia possibile, è necessaria una comunicazione tra i due circoli. I soggetti presentano cianosi severa e la loro sopravvivenza è appunto dovuta alla presenza di una qualche comunicazione tra i due circuiti (dotto-dipendenza). Generalmente si associa una comunicazione interatriale; nei due terzi dei casi vi è la pervietà del dotto arterioso e in un terzo circa dei casi è presente anche un DIV. La trasposizione dei grossi vasi è più comune nei maschi e rappresenta il 10% circa di tutte le cardiopatie cianogene congenite. Il decorso dipende dal grado di ipossia tissutale, dalla capacità dei ventricoli di tollerare condizioni di sovraccarico in presenza di diminuita ossigenazione del sangue coronarico, dalla natura delle malformazioni cardiovascolari associate e dalle condizioni del letto vascolare polmonare. Entro la terza decade di vita il 30% circa dei pazienti avrà sviluppato una diminuzione della funzione sistolica del ventricolo destro e un’insufficienza tricuspidalica ingravescente che può portare a scompenso cardiaco congestizio. Gravi alterazioni morfologiche a carico del letto vascolare compaiono a 1-2 anni dalla nascita nella maggior parte dei pazienti. Trattamento: La procedura più semplice per garantire un aumento della commistione intracardiaca di sangue venoso sistemico e polmonare è rappresentata dalla creazione o dall’ampliamento di una comunicazione tra i due atri del neonato. La creazione di un’anastomosi fra aorta ed arteria polmonare è indicata nei pazienti con grave ostruzione all’efflusso ventricolare sinistro e ipoperfusione polmonare. I primi interventi che si possono effettuare sono: - Somministrazione di prostaglandine per mantenere l’apertura duttale fino all’intervento chirurgico; - Settostomia secondo Rushkind per consentire il mixing. In sede intracardiaca, si può intervenire modificando il ritorno venoso (scambio interatriale secondo la tecnica di Mustard o Senning), in modo da dirigere il sangue venoso sistemico verso la valvola mitrale, quindi nel ventricolo sinistro e nell’arteria polmonare, mentre il sangue venoso polmonare è deviato attraverso la valvola tricuspide e il ventricolo destro verso l’aorta. La sopravvivenza a lungo termine dopo queste correzioni chirurgiche è buona, ma circa la metà dei pazienti, nei 30 anni che seguono alla chirurgia di switch atriale, sviluppano aritmie o difetti di conduzione. La terapia chirurgica definitiva consiste nella correzione anatomica secondo Senning (buffle margini del setto interatriale) e Mustard (buffle pericardico autologo). Lo svantaggio è che il ventricolo sistemico diventa il ventricolo destro. Un’altra tecnica è quella dello switch delle grandi arterie, ripristinando la normale anatomia del sistema e rappresentando, per questo, un grande vantaggio. Di seguito viene mostrata un’immagine della correzione secondo Sennind/Mustard. Molti chirurghi preferiscono correggere la malformazione in età infantile, impiantando le due arterie coronarie sull’aorta spostata posteriormente ed eseguendo poi la resezione di aorta e arteria polmonare, le quali vengono poi anastomizzate con i ventricoli in forma incrociata. Nei pazienti con DIV e grave ostruzione a carico del tratto di efflusso ventricolare sinistro si ricorre a un deflettore di flusso (bypass) ventricolare intracardiaco, con impianto di un condotto protesico extracardiaco che sostituisce l’arteria polmonare (intervento di Rastelli). CARDIOPATIE NON CIANOGENE CON FLUSSO POLMONARE NORMALE STENOSI POLMONARE Nella stenosi della polmonare è ostacolato lo svuotamento del ventricolo destro. L’ostruzione congenita allo svuotamento del ventricolo destro può avvenire per la presenza di malformazioni diverse. Queste malformazioni possono essere poste a livello della valvola polmonare ovvero nell’infundibolo sottovalvolare. L’ostruzione a livello valvolare è caratterizzata dall’assenza della valvola semilunare; la valvola è sostituita da un diaframma concavo che al centro ha una piccola apertura. L’ostruzione a livello dell’infundibolo può essere dovuta: - alla presenza di una struttura fibrosa, posta immediatamente sotto il piano della valvola polmonare; - a una diffusa ipertrofia muscolare dell’infundibolo. In ciascuna di queste varietà il ventricolo destro è sottoposto a un sovraccarico di lavoro, la cui entità dipende essenzialmente dalle dimensioni dell’ostruzione. Il sovraccarico di lavoro determina un’ipertrofia del ventricolo destro. L’ostruzione all’efflusso dal ventricolo destro fa aumentare la pressione all’interno del ventricolo e crea un gradiente pressorio sistolico fra ventricolo destro e arteria polmonare. Questo gradiente pressorio sistolico è necessario per mantenere il flusso attraverso l’ostruzione. L’aumento della pressione nel ventricolo destro può determinare, a monte, un aumento della pressione dell’atrio destro. Se l’aumento della pressione nell’atrio destro supera quella dell’atrio sinistro, si può avere l’apertura del forame ovale e passaggio di sangue venoso nel circolo arterioso. Il forame ovale, infatti, in molti casi si comporta come una porta incernierata su uno stipite: se viene spinta contro lo stipite resta chiusa, se viene spinta in direzione opposta si apre. In condizioni fisiologiche la struttura che chiude il forame ovale viene spinta da sinistra verso destra contro il profilo del forame (la pressione nell’atrio sinistro, infatti, supera quella del destro). Se la situazione pressoria si inverte, il forame ovale può aprirsi e consentire il passaggio di sangue da destra a sinistra. I sintomi dipendono dalla gravità dell’ostruzione. Se l’ostruzione è lieve, i sintomi sono assenti. L’ostruzione di media gravità solitamente è asintomatica fino all’età adulta. L’ostruzione grave è sintomatica anche nel bambino. I sintomi principali sono la dispnea e la facile affaticabilità. L’inversione del flusso attraverso il forame ovale può determinare la comparsa di cianosi. Segni: Il sangue che viene spinto dal ventricolo destro attraverso l’ostruzione determina la formazione del principale elemento di diagnosi: un rumore sistolico intenso, rude, udibile soprattutto sul focolaio di ascoltazione della polmonare. Il rumore della stenosi polmonare ha conformazione “a diamante” (in crescendo-decrescendo con acme centrale). L’intensità del soffio e la sua morfologia costituiscono una guida per stabilire la gravità dell’ostruzione. Quando il sangue passa attraverso l’ostruzione, infatti, il flusso cessa di essere laminare e diventa vorticoso, con rumore tanto più intenso quanto maggiore è l’ostruzione. Un secondo parametro è la morfologia del soffio. L’acme del soffio corrisponde al momento in cui è massimo il flusso di sangue attraverso l’ostruzione. Al flusso massimo corrisponde il massimo gradiente pressorio fra ventricolo destro e arteria polmonare. Se il massimo gradiente è precoce, l’ostruzione è lieve; se è tardivo, l’ostruzione è grave. Infatti, in presenza di una lieve ostruzione l’aumento di pressione endoventricolare, necessario per garantire il flusso attraverso l’ostruzione, è modesto. Tale modesto aumento viene raggiunto rapidamente. Se l’ostruzione è grave, l’incremento di pressione richiesto è più elevato e viene raggiunto solo tardivamente. L’ostruzione aumenta il tempo necessario per lo svuotamento del ventricolo destro. La valvola polmonare si chiude in ritardo e la componente polmonare del II tono è distante dalla componente aortica. Maggiore è l’intervallo, più grave è la stenosi. Elettrocardiogramma e radiografia del torace: Quando la pressione sistolica del ventricolo destro raggiunge o supera i 60 mmHg si ha sviluppo di un’ipertrofia ventricolare destra significativa che si manifesta all’ECG con onda R maggiore di 7 mm in V1 e V2, più deviazione a destra dell’asse di QRS e T negativa in V1-V2 e in D2, D3, aVF. La radiografia del torace mette in evidenza una dilatazione sovrastenotica dell’arteria polmonare e l’eventuale dilatazione del ventricolo destro. Sia la radiografia sia l’ecocardiografia sono alterate in misura proporzionale all’entità dell’ostruzione. Più l’ostruzione è grave, maggiore è il sovraccarico di lavoro cui è sottoposto il ventricolo destro. Il sovraccarico di lavoro determina un’ipertrofia del ventricolo destro che può essere apprezzata con gli esami strumentali. Ecocardiogramma: L’ecocardiogramma bidimensionale consente la visualizzazione diretta sia dell’infundibolo del ventricolo destro sia della valvola polmonare. Con la tecnica Doppler si può mettere in evidenza il flusso turbolento sistolico presente nell’arteria polmonare a valle dell’ostruzione. La velocità massima del flusso registrata dopo l’ostruzione consente di risalire al gradiente pressorio transvalvolare. Cateterismo cardiaco: Mette in evidenza il gradiente pressorio a cavallo dell’ostruzione. A gradiente maggiore corrisponde un’ostruzione più grave. Evoluzione: Dipende dal grado di ostruzione. Ostruzioni lievi hanno un decorso favorevole: il paziente è asintomatico ed ha un’aspettativa di vita normale. Le ostruzioni gravi portano a morte il paziente prima della pubertà. La causa di morte principale è lo scompenso cardiaco intrattabile. I pazienti con stenosi polmonare sono esposti al rischio di endocarditi infettive. Il rischio è indipendente dalla gravità dell’ostruzione. Terapia. È di tipo chirurgico. È indicata in tutti i casi di stenosi polmonare ad eccezione dei casi lievi. Va praticata la profilassi dell’endocardite batterica. STENOSI AORTICA Aorta bicuspide: L’aorta bicuspide è la cardiopatia congenita più frequente: può determinare stenosi oppure no. Nei casi di stenosi il quadro clinico è sovrapponibile a quello descritto per la stenosi acquisita. La bicuspidia che non genera stenosi è asintomatica. La diagnosi è per lo più occasionale. Viene posta spesso nel corso di un esame ecocardiografico eseguito per altri motivi. Coartazione dell’aorta: Per coartazione aortica si intende una costrizione congenita del vaso: l’aorta è di calibro ridotto per un tratto più o meno lungo. La sede più frequente di coartazione è a livello della parte superiore dell’aorta discendente. La coartazione aortica viene distinta in due forme principali: la forma “postduttale” o dell’adulto; la forma “preduttale” o infantile. La forma post-duttale viene anche chiamata dell’adulto perché consente la sopravvivenza fino all’età adulta. La forma infantile è più grave: difficilmente il paziente raggiunge l’età adulta. Sia la forma preduttale sia la forma postduttale ostacolano il regolare flusso di sangue dall’arco aortico all’aorta discendente. La diversità di prognosi dei due tipi di coartazione dipende dal diverso grado di ostruzione creato, il quale, a sua volta, dipende dalle diverse caratteristiche anatomiche. Nella forma dell’adulto la costrizione del vaso è solitamente circoscritta. L’ostruzione è situata in una posizione immediatamente distale al legamento arterioso (il residuo fibroso del dotto di Botallo). La forma infantile interessa l’aorta nel segmento prossimale al dotto di Botallo ed è di solito caratterizzata dall’ipoplasia di un tratto esteso dell’arco aortico. L’aumento della pressione sistolica fa aumentare il lavoro del ventricolo sinistro. A valle dell’ostruzione la pressione diminuisce: è ridotta, in particolare, nell’aorta toracica e addominale e nei suoi rami principali. Si crea perciò un gradiente di pressione fra i vasi che originano dal tratto di aorta posto sopra e quelli che originano dal tratto posto sotto la coartazione; questo gradiente facilita il flusso di sangue attraverso le arterie che mettono in comunicazione i due distretti a lato della coartazione. Si sviluppa, cioè, un circolo collaterale costituito da arterie dilatate e tortuose. Il circolo collaterale contribuisce a ridurre il gradiente di pressione e di flusso determinato dalla coartazione. Il circolo collaterale principale è rappresentato dalle arterie succlavie e dai loro rami. Questi vasi, attraverso le arterie intercostali, portano sangue alle arterie mammarie interne. Le arterie mammarie interne sono variamente connesse col circolo arterioso collegato con l’aorta nella sua porzione distale alla coartazione. Sintomi: La varietà infantile di coartazione aortica è solitamente grave. Si manifesta molto precocemente con scompenso cardiaco. I sintomi della forma post-duttale dipendono dal grado di ostruzione. Se l’ostruzione è lieve, il paziente può essere asintomatico fino all’età adulta. Nelle ostruzioni di media gravità i sintomi compaiono nell’adolescenza. La sintomatologia è quella dello scompenso ventricolare sinistro. A volte il paziente manifesta una facile affaticabilità, che viene riferita soprattutto agli arti inferiori. La forma infantile si associa spesso ad altri vizi cardiaci congeniti. I vizi più frequenti sono la valvola aortica bicuspide e la pervietà del dotto arterioso. Fisiopatologia: La coartazione aortica ostacola il regolare flusso di sangue attraverso il tratto prossimale dell’aorta discendente verso l’aorta toracica. Le conseguenze di questo ostacolo dipendono dalla gravità dell’ostruzione. La gravità dell’ostruzione, a sua volta, deriva dal calibro del vaso nel tratto coartato e dalla lunghezza della costrizione. L’ostacolo del flusso attraverso la coartazione determina due conseguenze emodinamiche principali: tutte le strutture vascolari che stanno a monte dell’ostruzione sono sottoposte ad aumento della pressione ematica; tutte quelle che stanno a valle sono sottoposte a riduzione della pressione. Aumenta perciò la pressione del sangue sia nell’arco aortico sia nei vasi che da esso originano: l’arteria succlavia sinistra, la carotide comune sinistra e l’arteria anonima. L’aumento di pressione nell’arco aortico aumenta la pressione sistolica del ventricolo sinistro. L’aumento di sede di impianto di germi: i sintomi dell’arterite possono essere la prima manifestazione del vizio. Segni: I segni fisici principali della coartazione aortica sono rappresentati: - dalla riduzione dei polsi femorali; - dalla differenza di pressione arteriosa sistolica fra gli arti superiori e gli arti inferiori. Nel bambino piccolo questa differenza è dovuta essenzialmente alla riduzione della pressione arteriosa negli arti inferiori. Nell’adolescente e nell’adulto la differenza si accentua per un incremento progressivo della pressione negli arti superiori. Solitamente, il gradiente di pressione sistolica fra arti superiori e arti inferiori è superiore ai 30 mm di mercurio. Il sangue che passa attraverso l’ostruzione determina la formazione di un rumore sistolico che si apprezza sia sulla parasternale sinistra sia sul dorso in corrispondenza della colonna vertebrale toracica. Il soffio è in crescendo con un acme telesistolico. L’elettrocardiogramma mostra un’ipertrofia del ventricolo sinistro tanto più grave quanto maggiore è l’ostruzione. La radiografia del torace è caratteristica. Sono presenti i segni dell’ipertrofia ventricolare sinistra. I margini inferiori della parte posteriore delle coste presentano delle irregolarità. Queste irregolarità sono dovute all’erosione ossea determinata dalle arterie intercostali che sono dilatate per la presenza del circolo collaterale. Le incisure costali sono un segno radiologico tardivo, che solitamente compare dopo i 12 anni di età. L’ecocardiogramma bidimensionale può fornire la visione diretta della coartazione aortica. Il Doppler evidenza il flusso turbolento sistolico presente dopo la coartazione. La velocità massima del flusso registrata dopo l’ostruzione consente di valutare il gradiente di pressione posto a cavallo della coartazione. Se la coartazione non viene corretta, il paziente vive mediamente fino a 30-35 anni. Terapia: La terapia è chirurgica. L’età elettiva di intervento è fra 8 e 12 anni. CARDIOPATIE CIANOGENE CON RIDOTTO FLUSSO POLMONARE TETRALOGIA DI FALLOT La tetralogia di Fallot, mostrata in basso, è una cardiopatia congenita costituita da quattro fattori fondamentali: - difetto del setto interventricolare; - ostruzione all’efflusso dal ventricolo destro; - destro posizione aortica; - ipertrofia ventricolare destra. La tetralogia di Fallot è la più frequente cardiopatia cianogena congenita: rappresenta il 6% di tutti i vizi cardiaci congeniti. Il difetto del setto interventricolare di solito è ampio; è per lo più localizzato a livello del setto membranoso. Nella maggior parte dei casi il difetto è posto sopra la crista ventricularis. Solitamente l’ostruzione all’efflusso dal ventricolo destro è a livello infundibolare. L’alterazione anatomica può essere rappresentata sia da una costrizione di tutto l’infundibolo sia dalla presenza di una struttura fibrosa posta nella parte più bassa dell’infundibolo stesso. In circa il 20% dei casi l’ostruzione all’efflusso è rappresentata da una stenosi della valvola polmonare. L’aorta, di solito, è posta a cavallo del setto interventricolare. Origina in parte dal ventricolo destro e in parte dal ventricolo sinistro. L’ipertrofia del ventricolo destro deriva dalle condizioni emodinamiche che si vengono a creare per la presenza dei precedenti tre difetti. Fisiopatologia: Il sangue spinto dal ventricolo destro verso l’arteria polmonare viene ostacolato dall’ostruzione dell’infundibolo e solo in parte prende la via del circolo polmonare. I vasi polmonari sono pertanto ipoperfusi. La quota di sangue che non può passare nel circolo polmonare ha due vie alternative: l’aorta (che in parte origina dal ventricolo destro) e il difetto settale. L’aorta nasce a cavaliere del difetto settale. Pertanto, anche il sangue che attraversa il setto entra nell’aorta, utilizzando la porzione del vaso che ha origine dal ventricolo sinistro. In entrambi i casi (direttamente in aorta o previo passaggio attraverso il setto) sangue venoso entra nel circolo sistemico. L’ingresso di sangue venoso nel circolo sistemico determina ipossiemia arteriosa e cianosi. Il ventricolo destro opera in condizioni di sovraccarico a causa delle elevate resistenze al flusso contro le quali deve lavorare: l’ostruzione polmonare e le pressioni sistemiche presenti sia nell’aorta sia nel ventricolo sinistro al di là del difetto settale. Il ventricolo destro, pertanto, diventa ipertrofico. L’entità del flusso relativo attraverso l’ostruzione polmonare e nel circolo periferico dipende dall’entità dell’ostruzione e dalle resistenze del circolo sia polmonare sia sistemico. Sintomi: La diagnosi avviene di solito precocemente. Il bambino piccolo è irritabile, piange con facilità e mangia poco. Durante il pianto compare o si accentua la cianosi. Ciò è dovuto a un aumento delle resistenze del circolo polmonare: l’aumento delle resistenze polmonari è secondario all’aumento della pressione intratoracica dovuta alla manovra di Valsalva effettuata durante il pianto. La stessa cosa avviene in altre condizioni caratterizzate da una manovra di Valsalva: tosse e defecazione. L’ipossigenazione arteriosa dovuta al passaggio di sangue venoso nel circolo arterioso comporta ipossigenazione del cervello. Se l’ipossigenazione cerebrale è marcata, si possono avere sincope e convulsioni. Il bambino con tetralogia di Fallot assume spesso la posizione di “squatting”. La posizione di squatting consiste in un accovacciamento a ginocchia piegate con le braccia che cingono le gambe. Questa posizione aumenta le resistenze arteriose periferiche. L’aumento delle resistenze del circolo arterioso ostacola il flusso di sangue verso l’aorta e verso il difetto settale. La manovra di squatting riduce pertanto l’ingresso di sangue venoso nel circolo sistemico e riduce la cianosi. I pazienti con tetralogia di Fallot accusano dispnea da sforzo. La dispnea è determinata dall’ipossigenazione del sangue arterioso e dall’ostacolo al circolo polmonare. L’ipoafflusso polmonare determina la cianosi il cui grado riflette la gravità dell’ostruzione. Si verifica lo shunt destro-sinistro per via del DIV. Il soggetto presenterà crisi asfittiche ad improvvisa e drammatica caduta della saturazione. Da qui l’assunzione, da parte del soggetto della posizione di squatting che aumenta le resistenze periferiche. L’intensità e la durata del soffio sistolico eiettivo è inversamente proporzionale alla gravità dell’ostruzione. Segni. Il paziente è cianotico. La cianosi si aggrava in corso di manovra di Valsalva. I segni ascoltatori sono dovuti principalmente all’ostacolo al flusso dal ventricolo destro e sono riconducibili a quanto già detto per la stenosi polmonare. L’ E C G m ette in evidenza un quadro massiccio di ipertrofia e sovraccarico del ventricolo destro. Le radiografie del torace sono presenti i segni di ipertrofia del ventricolo destro. L’aorta ascendente solitamente è dilatata. I campi polmonari sono ipoperfusi. L’ecocardiografia mostra: - i segni dell’ipertrofia del ventricolo destro; - la collocazione a cavaliere dell’aorta. Col cateterismo cardiaco si mette in evidenza: - l’aumento di pressione del ventricolo destro; - il passaggio diretto del catetere dal ventricolo destro nell’arco aortico; - la ridotta ossigenazione del sangue arterioso. Evoluzione: Senza intervento, i pazienti con tetralogia di Fallot muoiono mediamente a 12 anni. Solo il 10% dei pazienti sopravvive fino all’età adulta. Terapia. La terapia è chirurgica e consiste nella chiusura del difetto interventricolare e nella rimozione dell’ostruzione del ventricolo destro. I neonati che si presentano con flusso polmonare dotto-dipendente ricevono prostaglandine per mantenere l’apertura duttale fino all’intervento chirurgico. L’intervento iniziale potrebbe essere palliativo, come la creazione di un bypass arterioso sistemico-polmonare, ma la tendenza dei Centri di eccellenza è quella di eseguire un intervento definitivo già nel neonato. L’intervento di riparazione elettiva nei neonati asintomatici dovrebbe avvenire entro i 6 mesi di vita. ANOMALIA DI EBSTEIN L’anomalia di Ebstein è una malformazione rara. Rappresenta circa l’1% di tutti i difetti cardiaci congeniti. La caratteristica principale dell’anomalia di Ebstein consiste nello spostamento verso il basso della valvola tricuspide. Nella sezione destra del cuore si creano due camere: una posta al di sopra e una al di sotto della tricuspide. La prima è costituita dall’atrio destro più la porzione di ventricolo destro posta al di sopra della tricuspide; la seconda dalla restante parte del ventricolo destro. Molto spesso la valvola tricuspide e l’apparato sottovalvolare hanno vari difetti di formazione. L’anomalia di Ebstein si associa spesso ad altre cardiopatie congenite. La più frequente è il difetto interatriale. L’effetto emodinamico principale dell’anomalia di Ebstein è costituito dalla ridotta “performance” del ventricolo destro. Questa, a sua volta, è responsabile della riduzione di flusso di sangue nei polmoni e dell’ipertensione a monte: atrio destro e circolo venoso sistemico. Se la pressione nell’atrio destro supera quella nell’atrio sinistro, ci può essere passaggio di sangue in direzione destra-sinistra attraverso la pervietà del setto atriale. I sintomi principali sono la dispnea da sforzo e la facile affaticabilità. Nel 70-80% dei casi il paziente è cianotico per la presenza di un flusso destra-sinistra attraverso il setto interatriale. All’elettrocardiogramma si osservano P di tipo polmonare. Sono frequenti le aritmie ipercinetiche sopraventricolari dovute alla distensione dell’atrio destro. Alla radiografia del torace l’atrio destro è dilatato e la vascolarizzazione polmonare è ridotta. La diagnosi è confermata all’ecografia bidimensionale, dove è possibile avere la visione diretta dell’impianto anomalo della tricuspide. L’anomalia di Ebstein isolata ha di solito un decorso benigno. In assenza di cardiopatie associate, è sufficiente di solito la terapia medica per lo scompenso del ventricolo destro e per le aritmie. CAPITOLO XIII: LO SCOMPENSO CARDIACO DEFINIZIONE L’insufficienza cardiaca è una condizione fisiopatologica nella quale, il cuore a causa di una alterazione funzionale e/o strutturale, è incapace di pompare una quantità di sangue adeguata alle richieste metaboliche dell’organismo oppure può farlo, ma con un abnorme aumento del lavoro cardiaco, per cui stiamo parlando di un cuore che non funziona bene, ma che non dà ancora segno di sé, cioè il paziente è ancora asintomatico in questa fase, ma c’è già la possibilità di passare allo scompenso cardiaco. Che cos’è lo scompenso cardiaco? Non è una malattia, ma una sindrome clinica. La sindrome è un quadro clinico, che può avere diverse patogenesi, per cui diverse cause possono determinare quel quadro. Per cui si tratta di una sindrome clinica complessa, causata da qualsiasi alterazione funzionale e/o strutturale, che compromette la capacità del cuore di riempirsi adeguatamente di sangue e di espellerlo. Perciò, fino a questo momento non è tanto diversa dall’insufficienza cardiaca, ma vi differisce, perché le alterazioni, che ne sono alla base, causano l’insorgenza di segni e sintomi caratteristici. Questa è la definizione più recente, che abbiamo, perché tratta dalle linee guida del 2008. EPIDEMIOLOGIA L’epidemiologia è importante, perché lo scompenso cardiaco è uno degli eventi più frequenti, in quanto colpisce oltre 30 milioni di europei. Circa il 40% dei pazienti muore entro un anno dal ricovero e solo il 25% degli uomini ed il 38% delle donne sopravvive oltre 5 anni dalla diagnosi. Ogni anno in Europa sono diagnosticati 3,3 milioni di nuovi casi. Purtroppo, lo scompenso è molto più comune dei tumori. I ricoveri per scompenso sono più che raddoppiati negli ultimi 20 anni. L’aspettativa di vita è peggiore più di ogni altra malattia cronica. Entro il 2020 il numero di decessi causati dallo scompenso ogni anno raggiungerà i 9 milioni. Questo ci fa capire quanto dobbiamo essere aggressivi, attenti, dobbiamo tenere il paziente sotto controllo, dobbiamo fornirgli la migliore terapia farmacologica, perché non vogliamo assolutamente che questo paziente arrivi alla situazione dello scompenso, perché la situazione dello scompenso è molto grave. 15 9,89,7 10 5 0 0,10,1 0,10,1 0,70,5 1,81,3 6,86,6 6,2 3,4 Maschi Femmine 20-24 25-34 35-44 45-54 55-64 65-74 75+ Se andiamo a vedere la prevalenza in funzione dell’età, mostrata in alto, vediamo che dai 50 anni la prevalenza sale in maniera ripida, perché il paziente anziano è un paziente con comorbilità ed è un paziente fragile dal punto di vista non solo fisico, ma anche cognitivo, infatti spesso si tratta di un paziente con demenza, che non si rende conto della sua malattia, per cui si tratta di una condizione molto difficile. Per cui, secondo il professore, è la malattia, fra quelle croniche, che merita la massima attenzione da parte nostra. Oggi si parla molto di telemedicina, di terapia a domicilio dello scompenso, ma si dovrebbe passare ad una fase operativa, perché l’intervento classico dello scompenso è il compenso, cioè il soggetto va in ospedale, lo rimettono a posto, poi va a casa e se vive con un coniuge che non è tanto più giovane di lui o vive con la badante, che non si impegna più di tanto, oppure è un paziente con comorbidità, nel giro di qualche settimane o mese scompensa di nuovo e va di nuovo in ospedale. La qualità di vita di questo paziente non è buona, con grandi costi per la sanità. Di solito, il paziente scompensato non è ricoverato in cardiologia, ma in medicina, dove è letteralmente abbandonato, salvo pochi casi. È noto, infatti, che pazienti con patologie cardiache curate in medicina generale, hanno dei risultati pessimi, e molto peggiori di quelli di pazienti con la stessa patologia, ma curati in cardiologia. EZIOLOGIA La cardiopatia ischemica dà luogo al 70% dei casi, di cui è una conseguenza a lungo termine. Le altre cause sono molto meno frequenti e ricordiamo le valvulopatie, le cardiomiopatie ed altre cause come l’endocardite. Di solito, parliamo di disfunzione sistolica o scompenso sistolico e di disfunzione o scompenso diastolico, questa distinzione è soprattutto didattica, perché lo scompenso nasce come sistolico o diastolico e poi tende a diventare un unicum. Le cause della disfunzione sistolica sono l’insufficienza contrattile, il sovraccarico emodinamico volumetrico e sovraccarico pressorio, invece le cause della disfunzione diastolica sono un inadeguato rilasciamento, un inadeguato riempimento diastolico e disturbi del ritmo cardiaco. MODELLI PATOGENETICI Poi ci sono i cosiddetti modelli patogenetici, che ci fanno capire come ogni tipo di scompenso si inscriva in determinate sottocategorie. Sono proposti vari modelli: - Modello cardiorenale, per cui vuol dire che in quel caso la comparsa dello scompenso e la sua sintomatologia saranno dovuti all’eccessiva ritenzione idrosalina, causata da una disfunzione renale, che è conseguente alla disfunzione contrattile; - Modello emodinamico, con deficit di pompa, con caduta di portata e vasocostrizione; - Modello neuro-ormonale, con l’attivazione di meccanismi di tipo neuro-endocrino, che determinano modificazioni funzionali, strutturali ed endocrine, che tendono a modificare l’attività cardiaca. Nella realtà questi tre modelli tendono a mischiarsi tra di loro, ma serve solo a capire quale modello più degli altri contribuisce a determinare la sintomatologia dello scompenso cardiaco. Il modello neuro-ormonale ad un certo punto diventa biomeccanico, perché le alterazioni indotte dallo scompenso diventano automantenenti, non più dipendenti dallo stimolo neuro-ormonale e non più modificabili dalla terapia. Questo è importante, perché il modello neuro-ormonale, ma soprattutto il compenso neuro-ormonale è il più efficace, ma per certi aspetti anche il più pericoloso, perché se non lo controlliamo arriviamo alla condizione nella quale, il modello di compenso è diventato di scompenso e su quelle modificazioni non abbiamo più possibilità di vincere. Per cui dobbiamo ricorrere a terapie più aggressive, come il cuore artificiale oppure il trapianto cardiaco, cioè un cuore, che non è più capace di reagire ai nostri tentativi terapeutici è un cuore perso, per cui l’unica cosa da fare è sostituirlo. PATOGENESI Partiamo dall’infarto del miocardio o da più infarti, la frazione d’eiezione tenderà ad abbassarsi, però i meccanismi di compenso tenteranno, in qualche modo, di tamponare questa situazione. Per cui in questa guerra tra danno e meccanismo di compensazione, la frazione d’eiezione decresce fino ad arrivare ad un valore critico, che sarà diverso a seconda dei casi, ma per parlare d’insufficienza questo valore deve essere necessariamente inferiore a 4550%. Questo discorso viene riassunto nell’immagine a lato. A mano a mano che il tempo passa e la frazione d’eiezione decresce, il paziente da asintomatico diventa sintomatico, questo ci fa capire che possiamo avere anche una fase lunga d’insufficienza cardiaca senza sintomi, ma se trattiamo con una terapia adeguata il deficit contrattile o di riempimento, che con l’eco o altre forme d’imaging possiamo facilmente diagnosticare, siamo ancora in tempo per interrompere l’evoluzione verso lo scompenso. Per cui se interrompo il progressivo ridursi della frazione di eiezione, ed i meccanismi di deterioramento dello scompenso, lo blocco in una situazione, in cui sicuramente non va avanti, e addirittura con una terapia adeguata potrebbe tornare indietro, cioè ad una situazione di quasi normalità della funzione contrattile. Per cui capire lo stato del paziente, soprattutto se cardiopatico, cioè che ha già avuto eventi, che possono portare allo scompenso, è fondamentale. Qualunque sistema perturbato tende a ritornare ad uno stato di equilibrio tramite l’adozione di meccanismi di compenso. Lo stesso discorso vale per l’apparato cardiovascolare. I principali meccanismi di adattamento all’alterazione della funzionalità cardiaca sono: - meccanismo di Frank- Starling; - ipertrofia cardiaca e rimodellamento cardiaco; - ridistribuzione della gittata cardiaca a favore di organi vitali, come cervello e cuore, a discapito di cute, muscolatura scheletrica e reni, per cui abbiamo vasocostrizione cutanea, vasocostrizione a livello della muscolatura scheletrica, con astenia del soggetto, a livello renale c’è riduzione della diuresi. In realtà, questo accade molto prima, quando la perfusione ematica a livello coronarico e renale è normale; - meccanismi neurormonali. Il meccanismo di Frank–Starling prevede che all’aumento del grado di stiramento del miocardio, l’allungamento delle fibre determina entro certi limiti l’aumento della forza di contrazione, per cui il cuore risponde ad un aumento del ritorno venoso con l’aumento della contrattilità, che corrisponde ad un aumento della gittata cardiaca. L’unico problema è che arrivato al valore di 2,2 micron, la relazione fra allungamento e forza di contrazione viene a mancare. La legge di Laplace afferma che la tensione, che il miocardio ventricolare deve sviluppare durante l’eiezione, dipende dalla pressione aortica e dallo stesso volume della camera ventricolare, per cui questa legge dice che lo stress di parete è direttamente proporzionale alla pressione ed al raggio, per cui per piccoli aumenti del raggio aumenta lo stress di parete, che è indirettamente proporzionale allo spessore parietale, per cui quanto è maggiore lo spessore, minore sarà lo stress. Ci sono 2 percorsi principali, mostrati nell’immagine a lato, quello del sovraccarico pressorio e quello del sovraccarico Attraverso di volume. 2 percorsi questi arriviamo a diversi stati di ipertrofia. Il sovraccarico pressorio determina ipertrofia concentrica, mentre il sovraccarico di volume determina ipertrofia eccentrica. Questi meccanismi di adattamento vengono innescati in funzione della legge di La Place, onde ridurre lo stress di parete. Tuttavia, in entrambe le situazioni, il contesto evolve verso una perdita dell’efficacia di pompa. Efficacia di pompa che viene col tempo deteriorata dell’innesco di a seguito meccanismi di rimodellamento. Il rimodellamento, che è l’evento su cui con i mezzi a nostra disposizione in ultima analisi, vogliamo intervenire, è regolato da meccanismi molecolari, anche se noi facciamo interventi grossolani, come intervenire su una valvola o un intervento di rivascolarizzazione, e non interveniamo a livello molecolare. Tale rimodellamento è caratterizzato da degenerazione e morte dei cardiociti, con apoptosi, accumulo di collagene e matrice interstiziale, attivazione del sistema renina-angiotensina, attivazione del simpatico con aumentato rilascio di catecolamine, alterazioni dell’espressione delle metalloproteinasi, alterazione del rapporto eccitazione/contrazione, quindi alterazioni dei flussi intracellulari del calcio, quindi una nuova permeabilità agli elettroliti, ai loro flussi, alle loro correnti, alterazioni strutturali delle proteine contrattili, come ad esempio dei ponti della miosina, alterazioni della fornitura energetica del miocardio, riduzione dell’espressione dei recettori beta-adrenergici, cioè down-regulation. La down-regulation è figlia dell’adattamento neuro-ormonale, cioè se nel modello neuro-ormonale faccio aumentare la secrezione delle catecolamine e faccio in modo che i recettori alfa e beta adrenergici siano in grande attività, allora il mio organismo risponderà in modo da ridurre la stimolazione, per cui ad un certo punto i recettori per sovrasaturazione andranno in sciopero. Questo ha fatto sì che i beta-bloccanti, che ora sono una componente importante della terapia, non fossero utilizzati, perché riducono la forza di contrazione, inoltre in questo caso c’è una down-regulation dei recettori. Ma proprio per il fatto che dobbiamo intervenire sull’insufficienza prima che diventi scompenso, dobbiamo ridurre la down-regulation dei recettori, perché ci servono per bloccare le catecolamine e per utilizzare i beta-bloccanti, che è vero che riducono la contrattilità, ma riducono soprattutto il consumo d’ossigeno, la frequenza cardiaca, per cui aumentando il tempo diastolico, migliora la perfusione coronarica, ma soprattutto diminuisce la pressione, per cui interviene in maniera positiva sul rimodellamento. Abbiamo detto che nello scompenso vi è un aumento delle pressioni di riempimento, per cui se riduciamo le pressioni di riempimento, riduciamo lo stress di parete, ne miglioriamo il rilasciamento, ne miglioriamo il riempimento, ne miglioriamo la contrattilità. Tutti questi meccanismi, come apoptosi, alterazione dell’espressione genica, alterazioni dello stato energetico, stress ossidativo, disequilibrio neuro-ormonale, aumento dell’espressione delle citochine, aumento dei markers dell’infiammazione ecc…, determinano oltre a pressione e volume, che esercitano i loro effetti attraverso meccanismi , che abbiamo già visto, determinano l’evoluzione verso lo scompenso. A mano a mano che peggiora il rimodellamento, aumenta il raggio, e quindi lo stress di parete, ma peggiora l’attività contrattile. Per cui si capisce perché è fondamentale l’intervento terapeutico sul rimodellamento, sia nel soggetto già scompensato, che nel soggetto, che sta per diventarlo. Perché una volta che c’è stato il rimodellamento, quel ventricolo rimane così, non ha più la possibilità di migliorare, per cui l’unica soluzione sarebbe quella di sostituirlo. Il modello di rimodellamento classico è quello dell’infarto (mostrato a lato), dove una parte della parete muore o più pareti muoiono e le rimanenti parti si contraggono e si agitano, per vicariare la parte di miocardio andata persa. Se ad un ventricolo tolgo il 20-25%, e pretendo che la rimanente parte svolga elegantemente il proprio ruolo e quello della parte persa, sono un pazzo furioso. Per cui significa nella prima fase che sia per effetto della cicatrizzazione che della iperfunzione della parte che si è salvata, c’è iperespansione della parte ipertrofica. Questo vuol dire che se il ventricolo si contrae e questa parte non si contrae, allora una parte del sangue o gran parte del sangue uscirà attraverso l’aorta, ma una parte andrà a sbattere contro la parete ventricolare, di cui una parte è assolutamente incapace di contrarsi, per cui avremo un progressivo allargamento, per cui aumenterà progressivamente lo stress di parete sulla parte che si contrae, per cui anche la parte che si contrae, si contrarrà di meno e tenderà a sfiancarsi. Tra i meccanismi neuro-ormonali dobbiamo ricordare il sistema renina-angiotensina-aldosterone. C’è un aumento dell’attività di questo sistema, a causa della riduzione della gittata cardiaca, che causa riduzione della perfusione renale, i recettori – sensori che si trovano a livello delle arteriole renali. Questo porta a vasocostrizione, ritenzione idro-salina con aumento del pre-carico e del post-carico, a livello cardiaco l’aldosterone e l’angiotensina II promuovono il rimodellamento e la fibrosi interstiziale. L’iperattivazione adrenergica e l’aumento delle catecolamine circolanti causano aumento della frequenza cardiaca, dell’inotropismo, ma anche del post-carico, con un aumento del rischio di aritmie. In passato i livelli di catecolamine erano considerati importanti nella fase iniziale, ma poi erano considerate un marker prognostico per il paziente, cioè quando si dosavano le catecolamine circolanti, e queste superavano un certo limite, il paziente era considerato finito, cioè o si faceva un trapianto o il paziente non aveva più speranze. Per cui avevamo un marker prognostico, che diceva sempre la stessa cosa: “devi morire”. Altri fattori che intervengono sono: - aumento della secrezione di vasopressina o ADH, che causa aumento della ritenzione idrosalina, quindi del pre–carico; - l’endotelina (ET-1) induce vasocostrizione, con aumento del post–carico; - Negli ultimi tempi sono comparsi altri fattori, come TNF–alfa, IL-1, NO, che determinano una sorta di stato infiammatorio cronico, inducendo un aumento dell’apoptosi e dell’ipertrofia ventricolare; - Il peptide natriuretico atriale è rilasciato in seguito all’aumento della pressione di riempimento atriale, che provoca vasodilatazione e natriuresi. L’iniezione di ANP causa aumento della diuresi, senza perdita di sodio, con riduzione della pressione di riempimento e miglioramento della frazione d’eiezione, il problema però è che questo peptide somministrato per via parenterale ha un’emivita estremamente breve, allora ci furono circa 20 anni fa una serie di tentativi, per trovare un ANP o qualcosa di simile di sintesi, che fossero resistenti ai succhi gastrici, ma per ora non abbiamo ancora niente a nostra disposizione. DALL’ADATTAMENTO AL MALADATTAMENTO Nella fase iniziale, ci sono una serie di meccanismi, descritti precedentemente, che consentono al nostro organismo di adattarsi alla ridotta capacità del ventricolo di pompare un’adeguata quantità di sangue, come aumento del pre–carico, aumento della gittata, aumento della pressione, adeguata perfusione degli organi vitali. Inizialmente sono meccanismi di compenso, ma poi peggiorano lo scompenso, per cui non bisogna pensare che tanto c’è il meccanismo di compenso, perché se non interveniamo, ci siamo giocati il paziente. Tali meccanismi di adattamento vengono riassunti qui di seguito. Quindi, a lungo andare i meccanismi di determinano compenso un circolo vizioso, con aumento del sovraccarico emodinamico, rimodellamento patologico, peggioramento funzione di della pompa, che determinano a lungo andare la comparsa dei sintomi dello scompenso. Quindi si ha il passaggio dalla fase di disfunzione cardiaca asintomatica (insufficienza cardiaca) alla fase di scompenso cardiaco conclamato. CAUSE PRECIPITANTI DI SCOMPENSO CARDIACO Lo scompenso può esacerbarsi a seguito di cause precipitanti. Quali sono le cause precipitanti lo scompenso? Le infezioni, questo ci fa capire perché nel caso degli anziani si è così aggressivi nel prevenire l’infezione dei virus influenzali , infatti ad una certa età il vaccino è obbligatorio. Proprio per questo motivo agli anziani con storia di insufficienza respiratoria si fanno dei cicli di disinfezione con la terapia antibiotica, perché una banale bronchite in un soggetto con insufficienza cardiaca può far scoppiare un scompenso cardiaco in tutta la sua drammaticità. Inoltre anemia, embolia polmonare, tireotossicosi, gravidanza sono altri fattori precipitanti. Per cui vediamo che all’inizio ci sono una serie di fattori, che non hanno niente a che vedere con la funzione cardiaca. È chiaro che se di base ho un problema cardiovascolare, come una miocardite, un’endocardite, ipertensione, un secondo infarto possono rappresentare un ottimo trigger per passare da un’insufficienza ad uno scompenso cardiaco, però bisogna considerare che anche cose che apparentemente non coinvolgono la funzione cardiaca, possono comunque precipitare uno scompenso. FORME DI SCOMPENSO CARDIACO Possiamo avere 2 forme cliniche di scompenso, quello cronico, stabile o in aggravamento, per cui il paziente che è passato allo scompenso vi resta, semmai con acuzie, ma rimarrà scompensato, poi c’è quello acuto, in cui abbiamo un improvvisa comparsa di segni e sintomi, e che richiede una terapia urgente, può essere una situazione de novo, oppure verificarsi in un soggetto, che già ha uno scompenso cardiaco cronico. Un soggetto con scompenso cardiaco cronico che ha un infarto ha elevate probabilità di avere uno shock cariogeno, un edema polmonare, tutte cose che rientrano nello scompenso cardiaco acuto. Un paziente che ha un infarto, in cui si rompe una corda tendinea o una parte del setto interventricolare, andrà incontro sicuramente a scompenso cardiaco acuto, che può portare a shock cardiogeno, che può provocare morte. Per cui lo scompenso cardiaco acuto è una condizione nella quale non c’è il tempo che entrino in funzione i meccanismi di compenso, perciò o interveniamo noi in maniera più o meno radicale, o il paziente è andato. Abbiamo un’altra classificazione, che si basa sulla presentazione clinica. Quindi abbiamo: - Scompenso di nuova insorgenza, presentazione iniziale ad insorgenza acuta o lenta; - Transitorio, recidivante o episodico; - Cronico, stabile persistente o instabile, in aggravamento Dal punto di vista clinico, possiamo avere le forme a bassa gittata, in cui il cuore non riesce a mandare in circolo una quantità di sangue adeguata alle esigenze dell’organismo. Inoltre possiamo avere lo scompenso cardiaco ad alta gittata, in cui c’è un aumento della gittata cardiaca, in risposta della riduzione delle resistenze periferiche. Raramente queste condizioni possono causare un’insufficienza cardiaca, ma possono precipitare uno scompenso, in presenza di cardiopatia sottostante, fino ad allora compensata. Le condizioni caratterizzate da aumentata gittata cardiaca sono l’anemia, tireotossicosi, fistole artero–venose, mieloma, malattia di Paget, però queste sono condizioni rare, ad eccezione della gravidanza e dell’anemia. Ci può essere uno scompenso cardiaco sistolico e uno diastolico, ma abbiamo già detto che all’inizio ci può essere il prevalere di uno dei due, ma poi i due quadri tendono a sovrapporsi. Le caratteristiche delle due forme di scompenso vengono riassunte nell’immagine sottostante. Forme di scompenso cardiaco (III) Scompenso cardiaco sistolico Scompenso cardiaco diastolico Dilatazione ventricolare Ipertrofia ventricolare concentrica, cavità ventricolare ristretta Pressione arteriosa normale o ridotta Ipertensione arteriosa sistemica Frazione di eiezione ridotta Frazione di eiezione conservata (≥ 50%) Ritmo di galoppo S3 Quarto tono Sintomi da ipoperfusione periferica Sintomi da congestione vascolare a monte del ventricolo interessato (debolezza, affaticamento, ridotta tolleranza allo sforzo, vertigini) Più frequente negli uomini Destro: edemi periferici fino all’anasarca, epatomegalia, ascite ecc) Sinistro: congestione vascolare polmonare fino all’edema polmonare Più frequente nelle donne anziane Nell’ambito dello scompenso sistolico c’è la sindrome da ipoperfusione periferica, per cui il sangue non va in circolo, nel caso del diastolico ci sono sindromi di congestione a monte del ventricolo interessato, perché il ventricolo non riesce a raccogliere tutto il sangue, che gli arriva. Il sistolico è più frequente negli uomini, invece il diastolico nelle donne anziane; nel sistolico la pressione può essere normale o ridotta, invece nel diastolico c’è ipertensione arteriosa sistemica; nel sistolico c’è riduzione della frazione d’eiezione per definizione, invece nel diastolico può essere ridotta. Un’altra modalità per classificare lo scompenso è quello di suddividerlo in destro e sinistro. Ciascuna di queste due forme presenta anche diverse manifestazioni cliniche. Il soggetto con scompenso destro è pieno d’acqua, perché non riesce ad espellere il dovuto, mentre quello con scompenso cardiaco sinistro presenterà soprattutto ingorgo nel circolo polmonare. DIAGNOSI Sir Thomas Lewis nel 1933 affermò che l’essenza profonda della medicina sta nell’individuare i primi segni di scompenso, ma fino a che non è entrata in gioco l’ecocardiografia questo era molto difficile. Abbiamo scoperto che ci sono una serie di fasi più o meno lunghe, in cui il paziente è asintomatico, che possiamo individuare solo con l’eco, per cui oggi l’aspettativa di vita per un soggetto con scompenso è molto migliorata. Quando parliamo di scarsa sopravvivenza, parliamo di pazienti, in cui questa diagnosi non è stata fatta o la terapia non è stata adeguata. Non per sminuire la gravità della condizione, ma oggi rispetto a 20 anni fa, abbiamo a disposizione mezzi più efficaci sia dal punto di vista diagnostico–terapeutico che prognostico, per cui possiamo non solo allungare la vita del paziente, ma anche migliorarne la qualità. Di fronte ad un paziente, in cui sospettiamo o sappiamo già uno scompenso, dobbiamo sapere come costruire la nostra terapia, come ci dobbiamo muovere. Anamnesi: L’anamnesi è importantissima, per cui dobbiamo passare in rassegna prima i fattori di rischio, quali ipertensione arteriosa, fumo di sigaretta, dislipidemia, storia familiare e diabete mellito. Dobbiamo sapere se è portatore di una cardiopatia acquisita o congenita, se si è già sottoposto ad interventi, che intendevano risolvere questa cardiopatia strutturale. Molto importante è il referto di una valvulopatia nota. Inoltre, vedere se c’è una vasculopatia periferica, se ha avuto una cardiopatia reumatica, se assume farmaci cardiotossici, droghe o alcool. Infatti, dovete sapere che la cardio-oncologia sta diventando un ambito di grande interesse, perché ci sono molti chemioterapici, che possono provocare gravi cardiopatie. Inoltre, se ci sono malattie sistemiche associate a cardiopatia, quali amiloidosi, connettivopatie, ecc… Quali sono i sintomi? Innanzitutto dispnea, se non c’è dispnea, non possiamo fare diagnosi. Esistono varie forme di dispnea: - Dispnea da sforzo, dapprima moderata, poi progressivamente ingravescente al compimento di uno sforzo sempre più leggero; - Ortopnea: necessità di mantenere il torace in posizione eretta, per evitare che compaia la dispnea, si verifica perché in posizione supina c’è un aumento del volume massimo di riempimento, se però le pressioni di riempimento non sono in grado di rispondere all’aumentato ritorno venoso, si verifica ipertensione polmonare e quindi dispnea, che di solito si ritrova entro 1 o 2 minuti dall’assunzione della posizione supina. L’ortopnea si verifica per esaurimento del meccanismo di Frank–Starling, per incapacità della gittata cardiaca di essere efficace in proporzione al ritorno venoso; - Poi c’è la dispnea parossistica notturna, in cui il paziente si sveglia mentre sta dormendo con sensazione di fame d’aria, che lo porta a sedersi in poltrona o portarsi alla finestra in cerca d’aria. Di solito, abbiamo bisogno di 30 minuti per risolvere la crisi, c’è depressione del centro del respiro e c’è un aumento dell’attività del vago, che si verifica durante la notte e che in aggiunta all’aumento del ritorno venoso, causa un peggioramento della sintomatologia respiratoria. - Infine, dispnea a riposo, che è sempre un marker di aumentata gravità dello scompenso e del suo avanzare. Considerando queste 4 forme di dispnea, possiamo intervenire ancora sulla prima, ma sull’ultima molto meno. L’edema è un’emergenza, che si verifica, con aumento della pressione capillare polmonare. L’edema dal punto di vista clinico è quello descritto nei libri ed è l’evento peggiore per un medico, perché quando si verifica, il paziente ha la piena consapevolezza che sta per morire, cioè il paziente rimane lucido fino ad un attimo prima di morire. Un tempo osservare un edema polmonare era più frequente, e si interveniva con un salasso. Il catetere era collegato con una sacca da prelievo di sangue e si poteva osservare che a mano a mano che il sangue veniva tolto, il soggetto cambiava colore, cioè diventava sempre meno congesto. Il paziente appare estremamente sofferente, ansioso, tachipnoico e tachicardico; obiettivamente si hanno rantoli polmonari a marea montante, estremità fredde e sudate, talora tosse con escreato roseo schiumoso. Altri sintomi sono il dolore toracico, che può essere dovuto ad una malattia coronarica pre-esistente, ma molto spesso si verifica in seguito ad ischemia miocardica da ridotto afflusso, palpitazioni,che si hanno in tutte le aritmie ipercinetiche, astenia, ridotta tolleranza allo sforzo per ridotta perfusione del muscolo scheletrico, poi ci sono altri sintomi, che compaiono più tardivamente, perché il circolo coronarico e cerebrale sono preservati fino all’ultimo. Tra i sintomi cerebrali si annoverano: - Confusione; - Cefalea; - Disturbi della memoria; - Psicosi con incubi, delirio, allucinazioni (raramente e prevalentemente nei soggetti anziani con vasculopatia cerebrale). Al contrario il soggetto con scompenso del cuore destro, avrà sintomi digestivi, quali nausea, vomito,tensione addominale, dolore addominale al fianco destro, perché il fegato è diventato congesto, in quanto con l’aumento del volume c’è stiramento della capsula, che lo avvolge e quindi dolore. Esame obbiettivo generale: L’esame obiettivo generale inizia con la valutazione del livello di attenzione, stato nutrizionale onde notare la presenza di cachessia miocardica, edemi declivi. A livello dell’apparato cardiovascolare, sarà possibile osservare: - Ingrandimento dell’aia cardiaca; - Tachicardia sinusale, cioè eventuali variazioni e loro entità; - presenza di terzo e quarto tono se lo scompenso è grave, con polso alternante; - ci può essere una riduzione delle pressione differenziale. A livello polmonare saranno presenti rumori, possibili sibili espiratori detti asma cardiaco, versamento pleurico, perché il cuore nello scompenso di destra causa un versamento pleurico, pericardico e peritoneale. A livello dell’addome vi sarà epatomegalia congestizia, ascite, reflusso epato-giugulare positivo, cioè il paziente è talmente pieno di sangue che se gli spingete il fegato verso l’alto, il sangue affluisce nella giugulare e c’è turgore, per passaggio del sangue dalla cava inferiore a quella superiore. A livello dell’apparato urinario, c’è nicturia o oliguria. Per cui lo scompenso cardiaco è una sindrome con coinvolgimento dell’intero organismo. Criteri diagnostici: Esistono dei criteri per porre la diagnosi che sono importanti dal punto di vista clinico, ma anche dal vista di vista medico-legale, per conoscere i danni. I criteri che vengono maggiormente utilizzati sono quelli di Framingham. Secondo questo schema, vi sono criteri maggiori e criteri minori. Noi facciamo la diagnosi di certezza, quando ci sono un criterio maggiore e almeno 2 criteri minori. I criteri maggiori sono abbastanza sensibili e specifici, cioè se c’è un criterio maggiore e 2 minori, la diagnosi è fatta. Tra i criteri maggiori figurano: - dispnea parossistica notturna; - rantoli polmonari; - edema polmonare; - cardiomegalia; - ritmo di galoppo sistolico; - aumento della pressione venosa; - distensione delle giugulari; - reflusso epato-giugulare positivo. Tra i criteri minori figurano: - dispnea da sforzo; - tosse notturna; - versamento pleurico; - capacità vitale ridotta; - tachicardia sinusale, superiore ai 120 bpm; - edema periferico; - epatomegalia. La dispnea parossistica è un criterio maggiore, mentre la dispnea da sforzo è un criterio minore. Indagini di laboratorio: Una serie di analisi sono raccomandate in tutti i pazienti, e sono: - l’esame emocromocitometrico completo, con test utili nel valutare l’alterata funzione renale, se sono alterati gli elettroliti, se è alterata la funzione epatica. Dobbiamo vedere se ci sono comorbidità, come il diabete, se il paziente è iperuricemico, perché la funzione renale è ulteriormente a rischio, inoltre va anche effettuata l’analisi delle urine; - dosare il peptide natriuretico atriale, in particolare il tipo B e della sua porzione terminale pro-BNP, che ci consente di fare la diagnosi, per cui se è alto c’è scompenso, se non è alto non c’è scompenso, elevati valori per tale test ha valore predittivo negativo. Alcuni markers vengono utilizzati prettamente per la ricerca, e sono: - noradrenalina; - endotelina; - renina; - aldosterone; - arginina-vasopressina. Però dò per scontato che la renina e l’aldosterone siano alti, per cui devo ricorrere ad una terapia che li antagonizzi. Un possibile algoritmo di trattamento di pazienti, non alla prima diagnosi, è esame obbiettivo del torace, elettrocardiogramma, ecocardiogramma e ANP, per confermare la certezza, per cui sulla base del valore del ANP, la diagnosi di scompenso cardiaco cronico è improbabile o dubbia o se è probabile e la sua gravità. In basso viene riportato un algoritmo per la diagnosi di scompenso cardiaco nei pazienti non trattati che presentano sintomi suggestivi. Esami strumentali: Gli esami strumentali sono raccomandati in tutti i pazienti. L’elettrocardiogramma è raramente normale, perché comunque c’è una cardiopatia a monte, il soggetto può avere delle aritmie, può avere dei segni d’ischemia miocardica, semplicemente da discrepanza, cioè non arriva una quantità di sangue sufficiente per far funzionare bene il cuore. La radiografia del torace è meno importante dal punto di vista diagnostico rispetto alle altre metodiche, ma l’elettrocardiogramma e la Rx, non si negano a nessuno, nel senso che fanno parte della prassi. Ci può solo dire che lo scompenso è in fase avanzata, ma in realtà questo ce lo dice già il quadro clinico del paziente. L’Rx del torace può mostrare cardiomegalia, congestione polmonare, versamento pleurico e/o pericardico, presenza di eventuali pneumopatie che determinano o contribuiscono alla dispnea. Tali reperti non sono specifici e vanno considerati indicatori di scompenso cardiaco solo in un contesto clinico caratteristico. A lato viene mostrato un Rx del torace di un paziente con scompenso dimostrano cardiomegalia rapporto cardio-toracico, cardiaco: con si elevato versamento pericardico, segni di congestione polmonare con concomitanti fenomeni flogistici. L’ecocardiogramma è molto diffuso, molto operatore dipendente, perché dipende da quanto l’operatore conosce quel paziente e la sua patologia. L’ecocardiogramma trans toracico permette di valutare in modo accurato e non invasivo l’anatomia cardiaca, la cinesi parietale globale e segmentaria, la funzione sistolica e diastolica e, tramite metodica Doppler, la funzione valvolare. Inoltre può dare informazioni fondamentali per comprendere l’eziologia dello scompenso. Nella tabella in basso vengono riportate le principali modificazioni visibili all’eco. L’ecocardiogramma trans esofageo viene utilizzato per i pazienti con inadeguata finestra ecocardiografica transtoracica (pazienti obesi o ventilati), con patologia valvolare complessa, nei casi di sospetta endocardite o di cardiopaita congenita, oppure nei pazienti con FA quando sia necessario escludere la presenza di trombi nell’auricola sinistra. Nel caso in cui il soggetto abbia un importante edema polmonare non possiamo fare l’eco, per cui ricorriamo all’eco trans esofageo, in cui la sonda passa esattamente dietro il cuore, l’atrio sinistro, per cui cambiano le proiezioni, cambiano i punti di vista. La risonanza magnetica cardiaca è particolarmente utile nel definire la vitalità del ventricolo, ci può ad esempio far vedere delle forme infiltrative, come amiloidosi, collagenosi, per cui in alcuni casi ci può far capire la genesi dello scompenso, non tanto la sua entità. La risonanza magnetica permette di valutare con estrema accuratezza e riproducibilità i volumi cardiaci, lo spessore e la cinesi parietale, le valvole cardiache e il pericardio; è controindicata nei pazienti con aritmie e nei portatori di dispositivi impiantabili (PM, ICD). La spirometria ci può far capire se la compromissione cardiaca è primaria e quindi si tratta di un cuore polmonare, o se è lo scompenso, che ha compromesso i polmoni. La spirometria è indicata nei casi in cui sia necessario confermare o escludere cause respiratorie della dispnea e per esaminare il potenziale contributo della patologia polmonare alla sintomatologia dispnoica. La coronarografia è utilizzata nei casi di sospetta origine ischemica, supportata da un’anamnesi positiva per angina da sforzo, alto profilo di rischio per cardiopatia ischemica. La biopsia endomiocardica serve poco, è utile solo nel caso di diagnosi di rigetto, in tutti gli altri casi non serve e può essere pericolosa, come nel caso di ventricolo non compatto o disfunzione aritmogena del ventricolo sinistro, per cui in un ventricolo fatto male può essere fonte di perforazione. Deve essere presa in considerazione in pazienti con scompenso cardiaco acuto ad eziologia sconosciuta accompagnato da aritmie ventricolari e/o blocco AV, nei pazienti non responsivi alla terapia standard e in quelli con scompenso cardiaco cronico in cui si sospetta un disordine infiltrativo o una cardiomiopatia restrittiva. Vi sono poi una serie di test funzionali utili per la valutazione oggettiva della capacità di esercizio e della relativa sintomatologia. Tra questi ricordiamo il six-minute walking test ed il test cardiopolmonare. Nelle terapie non farmacologiche la riabilitazione del paziente scompensato è cruciale, cioè una volta che abbiamo risolto o riteniamo di aver risolto la causa dello scompenso, passiamo alla riabilitazione, che comprende modifiche dello stile di vita. Possiamo valutare la riabilitazione con la capacità d’esercizio e con la sintomatologia, valutando il paziente sotto sforzo e facendo un test cardiopolmonare, che mi dà contemporaneamente informazioni sulla funzione cardiovascolare e polmonare, quindi mi dice quanto di quello che ha il soggetto dipende da un deficit di ossigeno a livello cardiaco o polmonare. CLASSIFICAZIONE CLINICA DELLO SCOMPENSO CARDIACO La principale classificazione dello scompenso è la NYHA, che forse è stata la prima classificazione in ambito cardiologico, e comprende quattro categorie, riassunte nella tabella successiva. Classe NYHA Disturbi soggettivi I Nessuna limitazione dell’attività fisica: l’esercizio fisico abituale non provoca affaticabilità, palpitazioni né dispnea. II Lieve limitazione dell’attività fisica: l’esercizio fisico intenso provoca affaticabilità, palpitazioni e/o dispnea. III Severa limitazione dell’attività fisica: il minimo esercizio fisico provoca affaticabilità, palpitazioni e/o dispnea. IV Impossibilità a svolgere qualunque attività fisica senza dolore: i sintomi sono presenti anche a riposo e peggiorano con l’esercizio fisico. Alla prima classe appartengono i soggetti con nessuna riduzione della capacità di svolgere attività fisica, cioè il soggetto ha l’insufficienza cardiaca, ma questa non è diventata ancora scompenso. Poi mano mano abbiamo lieve riduzione della capacità di svolgere attività fisica, severa riduzione della capacità di svolgere attività fisica, infine incapacità di svolgere qualsiasi attività fisica. Di solito consideriamo i pazienti di fase 1 o 2 a basso rischio, i pazienti di fase 3 e 4 ad alto rischio. Infatti, sono stati fatti vari studi randomizzati per testare l‘efficacia delle varie terapie soprattutto nei pazienti 3 e 4, che sono quelli a maggior rischio. Per cui questa classificazione dello scompenso ha un’importanza pratica. Recentemente è sorta la necessità di classificare il livello di scompenso sulla base delle alterazioni funzionali e/o strutturali, proprio in virtù della definizione di scompenso. Tale classificazione viene riportata nella tabella che segue. Stadio ACC/AHA Alterazioni strutturali e/o funzionali miocardiche A Alto rischio di sviluppare scompenso cardiaco in assenza di anomalie cardiache, strutturali o funzionali, né segni o sintomi manifesti (ipertensione arteriosa, coronaropatia, uso di farmaci potenzialmente cardiotossici, anamnesi personale positiva per febbre reumatica, anamnesi familiare positiva per cardiomiopatie), B Presenza di anomalie strutturali cardiache fortemente associate allo sviluppo di scompenso cardiaco, in assenza di segni e sintomi (pregresso infarto miocardico, ipertrofia e/o dilatazione del ventricolo sinistro, diminuzione della funzione contrattile) C Scompenso cardiaco sintomatico associato a sottostante alterazione strutturale. D Scompenso cardiaco terminale. Per cui abbiamo i livelli A, B, C e D. Al livello A corrisponde un alto rischio di sviluppare lo scompenso in assenza di anomalie strutturali e/ o funzionali e di sintomi manifesti, per cui si tratta delle forme genetiche, per cui questa classe indica il soggetto, che per il momento non ha niente, ma è ad alto rischio di sviluppare lo scompenso, poi c’è lo scompenso cardiaco sintomatico, in presenza di alterazioni strutturali e/ o funzionali, infine scompenso cardiaco terminale. Perciò si tratta di una classificazione molto grossolana, in quanto dice che o il soggetto per ora non ha niente, ma lo potrà avere o dice che già c’è qualcosa di concreto o dice che il soggetto ha già una forma grave. Nella quotidianità si continua ad usare la vecchia classificazione, che anche è più affidabile, ma potremmo fare dei paralleli tra le 2 classificazioni, come ad esempio i livelli A e B, che corrispondono esattamente alla prima categoria NYHA. PROGNOSI La prognosi è prevalentemente variabile, perché dipende dall’eziologia, dall’età, dalle comorbilità, per cui più sono numerose sono le comorbilità, maggiore è l’età del paziente, più numerosi sono gli eventi, che hanno caratterizzato la storia clinica del paziente, tanto peggiore sarà la prognosi. Senza dimenticare che anche la classificazione in base ai sintomi e alle alterazioni strutturali e/o funzionali è una prognosi, se ad esempio vedo un paziente in classe 4 o a livello C, avrà una prognosi peggiore rispetto ad un soggetto in classe 1 o 2 o a livello A o B. Le donne muoiono di più solo per cancro del polmone rispetto allo scompenso, ma muoiono di meno per tumori dell’ovaio, dell’intestino, delle mammelle e per diabete. La stessa cosa vale anche per gli uomini, cioè l’unica cosa, che li fa morire più frequentemente rispetto allo scompenso è il cancro del polmone, compresi tumori della prostata e vescica. TERAPIA DELLO SCOMPENSO CARDIACO CRONICO Qual è l’obbiettivo della terapia dello scompenso cardiaco? I principali obiettivi sono: - Trattare la causa, che ne sta alla base; - ridurre i sintomi; - rallentarne la progressione. Per cui la terapia dello scompenso è una terapia curativa. Nell’ambito della terapia la seconda classificazione ci aiuta, perché nella prima classe il soggetto non ha niente, anche se ha ottime possibilità di averlo in futuro, per cui dobbiamo intervenire riducendo questa probabilità. Poi dobbiamo trattare la dislipidemia, il diabete, utilizziamo ACE- inibitori, beta-bloccanti. Dopo di che passiamo a ACE-inibitori in tutti i pazienti, betabloccanti in pazienti selezionati, ACE-inibitori con beta-bloccanti in tutti i pazienti, restrizione di sodio nella dieta, diuretici, digitale, stimolazione cardiaca se c’è un blocco di branca, rivascolarizzazione, antagonisti dell’aldosterone, trapianto, ospizio (cioè a questo punto la frase: “devi morire”, diventa filosofia). La terapia dello scompenso cardiaco cronico risulta essere quindi una terapia a gradini, così come mostrato dalla figura in basso. Trattamento non farmacologico: Per quanto concerne il trattamento non farmacologico, questo consiste soprattutto nel: - Riduzione dell’apporto giornaliero di sodio; - Riduzione dell’apporto giornaliero di alcol (abolire in caso di cardiomiopaita alcolica, negli altri casi massimo 10-20 g/die, pari a 1-2 bicchieri di vino/die); - Riduzione del peso corporeo nei pazienti obesi (BMI > 30); - Abolizione del fumo di sigaretta; - Abolizione dei lunghi viaggi e il soggiorno a grandi altezze; - Vaccinazione annuali contro l’influenza e lo pneumococco; - Programmi di riabilitazione cardiaca nelle fasi II e III; - Restrizione dell’apporto dei liquidi solo nei pazienti con scompenso severo (soprattutto se associato a iponatriemia); - Monitoraggio del peso corporeo (un aumento di peso è spesso associato ad un aggravamento dello SC e a ritenzione idrica, mentre la perdita di peso non intenzionale [se >6% rispetto al peso abituale in assenza di ritenzione idrica in 6 mesi si parla di cachessia cardiaca] è di frequente riscontro nei pazienti con SC severo ed è un importante indice prognostico sfavorevole); - Riposo nelle fasi di instabilità emodinamica. Trattamento farmacologico: Il trattamento farmacologico risulta essere molto valido e prevede l’utilizzo di beta-bloccanti, inotropi, ACE-inibitori, diuretici. Gli ACE-inibitori sono i farmaci di prima scelta nei pazienti con funzione sistolica ridotta, sia in presenza che in assenza di sintomatologia, e determinano miglioramento dell’attività contrattile, sono controindicati solo nel caso di stenosi renale bilaterale e storia di angioedema. L’ACE-inibitore, dato per 5 anni, riduce significativamente la mortalità per scompenso, ma ancora più importante è efficace subito. Per cui se vediamo un soggetto scompensato, dobbiamo subito somministrare un ACE-inibitore, non solo perché è efficace subito, ma anche perché la sua efficacia cresce nel tempo. I beta-bloccanti carvedilolo, bisoprololo, metoprololo e nebivololo, si somministrano in tutti i pazienti scompensati, sintomatici, con frazione d’eiezione inferiore al 40%, in condizioni emodinamiche stabili. Nel caso di shock dobbiamo utilizzare altri farmaci. I beta-bloccanti determinano un miglioramento della funzionalità del ventricolo sinistro, della tollerabilità allo sforzo fisico, riduzione della classe NYHA, riducono la mortalità alla ospedalizzazione e la morte improvvisa. In basso viene riportata una tabella riguardo la posologia di somministrazione di tali composti. Beta-Bloccante Dose iniziale (mg) Incremento (mg/die) Dose finale Periodo di (mg/die) titolazione Bisoprololo 1.25 2.5-3.75-5-7.5-10 10 settimane-mesi Metoprololo 12.5-25 25.50-100-200 200 settimane-mesi 3.125 6.25-12.5-25-50 50 settimane-mesi succinato Carvedilolo Nebivololo 1.25 2.5-5-10 10 settimane-mesi I diuretici dell’ansa, come furosemide, torasemide e bumetanide, sono essenziali nel trattamento dei pazienti non solo con congestione polmonare, ma anche sistemica. Nel caso di eccessiva ritenzione dei liquidi possiamo associare un diuretico tiazidico. Gli effetti collaterali sono soprattutto gli squilibri elettrolitici, primo fra tutti l’ipokaliemia, che può causare altri sintomi. Per cui in questo caso possiamo usare i diuretici risparmiatori del potassio, che possono essere utilizzati anche da soli nelle fasi iniziali, ma che sono usati più frequentemente in associazione ai diuretici dell’ansa, in quanto ne potenziano l’effetto diuretico, ma riducono la perdita di potassio. I diuretici risparmiatori di potassio amiloride, triamterene, dovrebbero essere somministrati in caso di persistenza di ipokaliemia in corso di terapia diuretica nonostante la somministrazione di un ACE-inibitore. Poi ci sono gli antagonisti recettoriali dell’aldosterone, che sono usati soprattutto in associazione agli altri farmaci, per cui si somministrano solo nelle fasi avanzate dello scompenso. L’aldosterone stimola la ritenzione di acqua e sodio, induce fibrosi e rimodellamento cardiaci, per cui bloccando questi effetti, c’è una riduzione della portata. L’utilizzo di tali composti determina una riduzione della mortalità. Poi ci sono farmaci, che vengono utilizzati poco e che sono stati creati soprattutto per la terapia dell’ipertensione. Stiamo parlando degli antagonisti recettoriali dell’angiotensina II o sartani. Non è stato dimostrato che abbiano un’efficacia maggiore o un effetto sinergico a quello degli ACE-inibitori, per cui sono utilizzati solo nel caso di mancata risposta agli ACE-inibitori o nel caso di comparsa di effetti collaterali, di cui quello principale è la tosse, ma poiché questo è un effetto collaterale comune di questa classe di farmaci, allora non possiamo utilizzare un altro ACE-inibitore, perché la tosse comparirà comunque, ma dobbiamo cambiare categoria di farmaci. I vasodilatatori non hanno un ruolo specifico nel trattamento dello SC. I nitrati possono essere utilizzati per migliorare la dispnea o nel trattamento di un’angina concomitante. I calcio-antagonisti diidropiridinici possono essere utilizzati nei casi i ipertensione arteriosa non controllabile con altri farmaci ma, in generale, il loro uso non è raccomandato nella terapia dello SC. Per anni la digitale è stato il farmaco di scelta nel trattamento dello scompenso, perché aumenta l’inotropismo, ma contemporaneamente aumenta il consumo di ossigeno e aumenta anche il batmotropismo, con possibilità di aritmie ipercinetiche, originate da centri ectopici, ma rallenta anche la conduzione atrioventricolare con la possibilità di aritmie ipocinetiche. Per cui oggi questo farmaco si usa solo nei soggetti con scompenso, con possibilità di aritmie ipercinetiche, per il suo effetto bradicardizzante. Poi ci sono altri farmaci inotropi, rappresentati dai beta-agonisti (dopamina, dobutamina) e dagli inibitori delle fosfodiesterasi (milrinone); sono indicati nei pazienti con SC acuto ed evidenza di shock cardiogeno. Devono essere utilizzati solo negli anziani o nelle fasi avanzate o nelle fasi acute, e che sono a rischio di shock cardiogeno. Gli antitrombotici sono utilizzati nel caso in cui il paziente ha già avuto un evento trombo-embolico in precedenza o nel caso in cui sappiamo che c’è un trombo nel sistema venoso periferico o nelle cavità di sinistra del cuore, da cui è partito lo scompenso, allora nella terapia dobbiamo aggiungere anche gli antitrombotici. DISPOSITIVI MECCANICI Spesso i pazienti con scompenso presentano disturbi della conduzione interventricolare, a causa della dilatazione delle camere ventricolari. I disturbi della conduzione interventricolare determinano asincronia della contrazione tra i due ventricoli, peggiorando ulteriormente la funzione sistolica, e peggiorando la sintomatologia dello scompenso e la prognosi. La terapia di risincronizzazione cardiaca o CRT è indicata nei pazienti con FE ≤ 35% in classe funzionale NYHA III o IV nonostante terapia medica ottimale e QRS di durata > 120 msec. La CRT prevede il posizionamento di due pace -maker, che comandano il ventricolo destro e quello sinistro, in modo che tutto si contragga in modo più o meno sincrono. Non va impiantato a tutti i soggetti con scompenso, ma a quelli con frazione d’eiezione <35%, nei soggetti 3 e 4, nei soggetti con durata del QRS >120 msec, perché se il QRS è allargato vuol dire che la conduzione nel ventricolo è ritardata, per cui abbiamo un ventricolo che si contrae tardi. In realtà ci deve preoccupare di più un blocco di branca sinistra, in cui i 2 ventricoli non si contraggono contemporaneamente, rispetto ad un blocco di branca destra, ma non ci deve interessare la causa. L’intervento di risincronizzazione cardiaca, a 6 mesi, è associato ad una sopravvivenza maggiore, ma è indicato solo in presenza di determinate condizioni del paziente, che abbiamo già visto, in quanto in soggetti con una gravità minore dello scompenso non apporta benefici, ma addirittura può causare peggioramenti, perché altera un meccanismo fisiologico, che già funziona. Come si posiziona? Posizioniamo un catetere in atrio destro catetere in ed un ventricolo destro, per cui stimoliamo il ventricolo sinistro indirettamente, perché il catetere potrebbe dare fastidio nella parte sinistra del cuore. Il catetere è posizionato in un ramo della discendente o dell’interventricolare anteriore, e siccome la parete del vaso è in contatto diretto con la parete ventricolare, l’impulso è trasmesso al ventricolo immediatamente. Per cui utilizzo l’atrio destro come generatore d’impulsi e faccio in modo che l’impulso raggiunga il fascio di His, e si distribuisca in tempi adeguati. Per cui ho un atrio e due ventricoli e faccio in modo che i due ventricoli si contraggano in modo più simultaneamente possibile. Sappiamo che fisiologicamente i due ventricoli non pompano il sangue esattamente nello stesso istante, perché il circolo sistemico è a più alta resistenza di quello polmonare, ma c’è un tempo che li separa, quando questo tempo diventa più evidente, allora vuol dire che c’è un disturbo nella conduzione interventricolare. Siccome quando abbiamo parlato delle aritmie, abbiamo detto che è la fibrosi la causa principale del meccanismo di rientro e che lo scompenso cardiaco nel 70% dei casi è causato da una cardiopatia ischemica, allora anche nel paziente scompensato ci sono aree di fibrosi, con possibilità di aritmie ipercinetiche ventricolari, per cui la possibilità di avere una morte elettrica per questo paziente è alta. Per cui si è cercato di trovare farmaci anti-aritmici efficaci e sono stati anche trovati, ma ci si è chiesti se si poteva inventare un dispositivo, che prevenisse la morte cardiaca improvvisa. Da uno studio iniziale, si è cercato di capire qual è il vantaggio di impiantare un defibrillatore ad un soggetto con scompenso. Da questo studio sono emersi i criteri di Madit II, che definiscono quali sono le indicazioni all’impianto del defibrillatore o ICD. Innanzitutto, sono indicati per prevenire la morte cardiaca improvvisa, inizialmente venivano indicati nei pazienti giovani con aritmie da rientro non correggibili con l’ablazione o con cardiomiopatie croniche, per cui si tratta di pazienti con meccanismi complessi. L’impianto è indicato nei soggetti sopravvissuti ad un infarto del riduzione miocardio, della funzione sistolica, frazione d’eiezione <50%, in terapia ottimale e con aspettativa di vita superiore ad 1 anno. Il defibrillatore è anche prevenzione nella indicato primaria, in soggetti che hanno avuto un episodio ischemico acuto, entro 40 giorni dall’infarto, con frazione d’eiezione <35%, in terapia ottimale e con aspettativa di vita superiore ad 1 anno. Il contropulsatore aortico o IABP è un catetere con un palloncino, in cui è iniettato un gas nobile, perché se il pallone si rompe non deve dare embolia e viene impiantato per via femorale nell’aorta discendente fino a raggiungere il cuore ed è accompagnato dall’ECG. In diastole il pallone si gonfia e migliora la perfusione del ventricolo. È indicato nei pazienti con scompenso cardiaco, shock cardiogeno, nei soggetti in attesa di impianto di un dispositivo. Questo rappresentata a lato è un dispositivo per la circolazione assistita prolungata, i soggetti sono attaccati alla macchina cuorepolmone, ma questa soluzione non può essere eterna. circolazione indicati nei I dispositivi assistita pazienti per prolungata con la sono scompenso cardiaco severo refrattario come “ponte” verso il trapianto cardiaco. Sono costituiti da una pompa che attraverso due cannule preleva il sangue dal ventricolo sinistro e lo immette nell’aorta ascendente. La pompa può essere esterna (extracorporea) o interna (posizionata in una tasca chirurgica nella fascia peritoneale dell’addome). I problemi principali legati all’uso di questi dispositivi sono rappresentati da complicanze infettive e tromboembolismi. TRAPIANTO CARDIACO Il trapianto cardiaco è indicato nel caso di scompenso cardiaco in fase terminale, refrattario a qualsiasi tipo di trattamento. È considerato come ultima possibilità per il miglioramento della qualità di vita e dell’aspettativa di vita. I soggetti candidati devono avere meno di 70 anni, non avere insufficienza di altri organi, ipertensione polmonare o neoplasie. Infatti si sente sempre parlare di soggetti trapiantati in età giovane, perché un soggetto ad esempio di 88 anni ha un tale carico di altre morbilità, che è inutile impiantargli un cuore nuovo. TERAPIA DELLO SCOMPENSO CARDIACO CON FRAZIONE DI EIEZIONE CONSERVATA (SCOMPENSO DIASTOLICO) In tale situazione, si utilizzano soprattutto: - ACE-INIBITORI: possono migliorare il rilasciamento ventricolare sinistro mediante i loro effetti anti-ipertensivi , l’inibizione della risposta ipertrofica e della fibrosi miocardica; - DIURETICI: sono necessari in caso di ritenzione idrica ma devono essere usati con cautela per non ridurre eccessivamente il precarico e dunque il riempimento ventricolare sinistro; - BETA-BLOCCANTI: diminuendo la frequenza cardiaca allungano il periodo di rilasciamento diastolico e dunque migliorano il riempimento ventricolare sinistro; - VERAPAMIL: effetto simile al beta-bloccante. Tali raccomandazioni terapeutiche sono solo speculative in quanto per lo scompenso cardiaco diastolico non esistono dati sperimentali a favore di uno specifico trattamento. SCOMPENSO CARDIACO ACUTO Lo scompenso cardiaco acuto si intende una rapida insorgenza o modificazione di sintomi e segni di scompenso cardiaco che comporta l’avvio di una terapia urgente. Può manifestarsi de novo oppure come riacutizzazione di uno SC cronico persistente. Lo scompenso cardiaco acuto generalmente è caratterizzato da congestione polmonare, ma talvolta il quadro clinico è dominato da sintomi e segni di ridotta gittata e ipoperfusione periferica. L’insorgenza o la precipitazione di uno scompenso cardiaco acuto può essere dovuto a molteplici cause, cardiovascolari e non cardiovascolari, riassunte nella tabella a lato. La vautazione del paziente con scompenso cardiaco acuto è molto importante, e prevede: - Anamnesi se è possibile e esame obiettivo; - ECG; - Rx torace; - EGA o emogasanalisi; - Ecocardiogramma; - Esami di laboratio; - Peptidi natriuretici. Lo scompenso cardiaco acuto è caratterizzato dall’edema polmonare acuto. L’edema polmonare acuto è dovuto ad insufficienza ventricolare sinistra con aumento acuto della pressione in atrio sinistro e quindi nelle vene polmonari. Nella prima fase l’aumento del flusso linfatico riesce a mantenere costante la quantità di liquido perivascolare; nella fase successiva il liquido filtrato supera la capacità di smaltimento del sistema linfatico determinando la comparsa di edema interstiziale; aumentando ulteriormente la pressione nelle vene polmonari si rompono il liquido supera la membrana alveolo-capillare determinando la comparsa di edema alveolare. Clinicamente, i pazienti con edema polmonare alveolare mostrano insufficienza respiratoria severa, con tachipnea, ortopnea, rantoli polmonari ed escreato schiumoso. La saturazione è generalmente < 90% in aria ambiente. Gli interventi terapeutici immediati sono: - porre il paziente in posizione ortopnoica con gambe pendenti (per favorire la distribuzione extratoracica del volume ematico); - sedazione con morfina o diazepam; - somministrazione ossigeno con sonda nasale con aspirazione del secreto. Se necessario, respirazione assistita a pressione positiva; - riduzione del precarico: nitroglicerina s.l. o e.v., con cautela in caso di ipotensione, furosemide 2040 mg e.v. La terapia causale prevede la rimozione immediata della causa scatenante lo scompenso (ad es., terapia di una crisi ipertensiva, rivascolarizzazione in caso di IMA). Infatti, uno scompenso cardiaco acuto può verificarsi in circa il 15 % dei casi delle sindromi coronariche acute. Può essere dovuto a insufficienza acuta di pompa (ad esempio in caso di estesi infarti anteriori), a complicanze meccaniche in corso di IMA o all’insorgenza di aritmie. Il trattamento dello scompenso cardiaco acuto deve verificarsi nel più breve tempo possibile, onde evitare il rischio di shock. Lo shock cariogeno è una sindrome determinata da ipoperfusione tissutale a causa di una ridotta gittata sistolica. Clinicamente è presente ipotensione (PAS < 90 mmHg) e segni di ridotta perfusione periferica (cute fredda e pallida, oliguria, alterazioni dello stato mentale fino al coma, con tachicardia compensatoria) in presenza di un adeguato volume ematico. Esistono criteri emodinamici per la valutazione di shock cardiogeno: - ipotensione sostenuta (PAS < 90 mmHg per più di 30 minuti); - ridotto indice cardiaco (CI < 2.2 L/min/m2); - elevata pressione capillare polmonare ( > 15 mmHg). La principale causa di shock cariogeno è rappresentata dall’infarto miocardico acuto. Terapia: Gli obiettivi della terapia possono essere distinti in: - Obiettivi immediati: miglioramento della sintomatologia, ripristino dell’ossigenazione, miglioramento delle condizioni emodinamiche e della perfusione d’organo e limitazione dei danni cardiaci/renali; - Obiettivi intermedi (degenza ospedaliera): stabilizzazione del paziente e ottimizzazione della terapia, inizio di una corretta terapia farmacologica, utilizzo di device ausiliari (PM, ICD, CRT) se necessari, ridurre al minimo la degenza ospedaliera; - Obiettivi a lungo termine (prevenzione secondaria): pianificazione di un adeguato follow-up, istruzione del paziente sulle opportune modifiche dello stile di vita, miglioramento della qualità della vita e della sopravvivenza. Sono stati elaborati una serie di algoritmi per il trattamento dello scompenso cardiaco acuto. Ciascuno di questi algoritmi prende in considerazione determinati parametri, ed in base al loro valore viene iniziata una specifica terapia. Qui di seguito ne verranno mostrati 2. Infine la tabella sottostante riassume gli obiettivi della terapia in funzione della gravità della situazione. CAPITOLO XIV: MIOCARDITI Chiudiamo con questa ultima parte, trattante le miocarditi e le pericarditi, non perché siano figlie di un Dio minore ma sono comunque patologie più rare. MIOCARDITE E' una Patologia infiammatoria del muscolo cardiaco associata a disfunzione cardiaca e diagnosticata tramite criteri istologici, immunologici e immunoistochimici. Sono state identificate forme infettive, autoimmuni, tossiche, chimiche e fisiche. Tuttavia, ancora oggi spesso la causa non viene riconosciuta, cioè noi siamo di fronte a un quadro di infiammazione del muscolo cardiaco ma non siamo certi della genesi, della motivazione per cui quel muscolo è andato incontro a ìnfiammazione: si parla dunque di miocardite idiopatica. Eziologia: Queste, riportate anche in tabella, sono le cause infettive, ce n'è per tutti i gusti e questo è uno dei motivi per cui si dice che a volte l'influenza con una carica virale particolarmente aggressiva in un soggetto con una particolare depressione immunologica può addirittura evolvere in altre forme, con localizzazioni parenchimali e viscerali importanti tra cui la miocardite. Essa si può manifestare come una sindrome coronarica acuta, con un dolore abbastanza tipico, con la salita degli indici ematici, più difficilmente con una modificazione tipica dell'ST o meglio se c'è una modificazione dell'ST di solito non è così settoriale. Cioè noi abbiamo visto che per l'infarto nell'ECG ci sono delle zone ben precise in cui troveremo un segnale elettrocardiografico e quindi ci faremo anche un'idea della regione interessata dall'ischemia. Nel caso della miocardite questo è molto più difficile per non dire estremamente raro perché il fenomeno è molto più globale, nel senso che non è che colpisce una parte ed un'altra no, quindi l'eventuale segnale dell'ECG lo possiamo trovare su tutte le derivazioni. Poi ci sono le cause non infettive, quindi: - le forme da ipersensibilità: una reazione allergica caratterizzata come tutte le reazioni allergiche da una ipereosinofilia periferica e un infiltrato infiammatorio. Una volta si usava in maniera molto più liberale la biopsia miocardica per fare questo tipo di diagnosi, oggi invece sia l'eco ma soprattutto la RM ci sono di grande aiuto e sono ovviamente diagnostica non invasiva, mentre con la biopsia miocardica un minimo di rischio ce l'ha perché può anche perforare il cuore, causare un tamponamento. I farmaci che la possono causare sono: antibiotici, antinfiammatori, anticonvulsivanti e addirittura alcuni diuretici; - Poi ci sono ovviamente le forme tossiche a cominciare dal monossido di carbonio, passando per l'etanolo. Naturalmente la miocardite da etanolo, specie se non curata, tenderà a diventare cardiomiopatia dilatativa cronica. Altra causa sono gli agenti fisici, soprattutto le radiazioni ma anche il colpo di calore e l'ipotermia che possono avere un coinvolgimento miocardico, magari transitorio ma che dà la tipica sintomatologia dell'insufficienza ventricolare con astenia, pallore, dispnea; - Poi ovviamente se non riusciamo a identificare nessuna di queste cause, la forma diventa automaticamente idiopatica. Fisiopatologia: Ma qual è la via attraverso cui noi passiamo dal virus alla miocardite? E' ovvio che il virus una volta entrato (e le porte di accesso saranno verosimilmente le vie respiratorie e la via orale, quindi la via gastrointestinale), determinerà una situazione infiammatoria, con produzione di chemochine e citochine. Dopo di che a questo punto il virus viene ceduto all'organo bersaglio: noi non possiamo sapere il virus una volta penetrato, una volta attivato, attraverso il processo di disseminazione chi andrà a colpire: possiamo avere l'encefalite, la miocardite, una qualunque altra forma. Dopo di che una volta entrato nel miocita va incontro a un processo analogo a quello che si determina tutte le volte che il virus arriva in un organo bersaglio: replicazione, moltiplicazione e coinvolge a seguire tutto il parenchima. Una volta entrato in una cellula si trasmette alle cellule vicine e così via e il fenomeno diventa generale. Una volta determinato il processo ci sarà una reazione da parte dell'organismo e produzione di anticorpi: quindi paradossalmente potremo fare la diagnosi eziologica anticorpi più che gli antigeni. Lo schema di seguito riassume tutto ciò che abbiamo detto fin qui. andando a cercare determinati Classificazione clinico-patologica: Distinguiamo una forma fulminante, subacuta, cronica attiva e persistente. L'esordio è chiaro nel caso della fulminante: solitamente a distanza di qualche giorno dall'infezione virale, si manifesta con una disfunzione severa, con un ventricolo non dilatato e a pareti ispessite ma non per l'ipertrofia bensì per l'edema interstiziale. La biopsia se fatta è positiva, è fatale in una minoranza di pazienti entro due settimane. Nella maggioranza dei casi va incontro a guarigione completa. Dal punto di vista istologico non lascia segno di sé, è un po' come la Tako-Tsubo, cioè viene, fa il danno, simula un infarto terrificante e poi saluti e baci, tutto torna come prima. Per la forma fulminante e per la forma subacuta possiamo avere la restitutio ad integrum con la terapia. Quando invece passiamo nella cronica attiva o persistente, l'aggettivo stesso dice che per qualche motivo (o per nostra incapacità o per la particolare aggressività del virus) il fenomeno è destinato a non risolversi, a non esaurirsi, quindi è ovvio che dovrà dare sia istologicamente che clinicamente delle conseguenze. Clinicamente le conseguenze saranno: - nel caso di una cronica attiva una cardiomiopatia restrittiva con normali volumi cavitari, ma noi sappiamo che una cardiomiopatia restrittiva per quanto possa avere un normale volume cavitario non si dilata a sufficienza e quindi interferisce con la normale funzione eiettiva del cuore; - nel caso invece della persistente, noi abbiamo una infiammazione persistente che può anche per lungo tempo non compromettere la funzione ventricolare propriamente detta, ma che mantenendo attivo il processo di infiammazione potrebbe essere per esempio l'evento scatenante per una rottura o perforazione di una placca coronarica o carotidea o periferica e determinare così una sindrome coronarica acuta o un evento cerebrovascolare acuto o un evento ischemico acuto degli arti superiori. Io ricordo che fino allo sviluppo attuale dell'eco e all'avvento della RM, la stragrande maggioranza delle biopsie che ho fatto in vita mia erano per diagnosi di miocardite sospetta, però credo che pur inviando da un certo momento in poi questi frustoli di tessuto miocardico in centri altamente specializzati, Pavia per fare un nome a caso, o Trieste, molto spesso la diagnosi era ipotetica, verosimile, altamente probabile, fortemente sospetta, ma mai che mi dicessero “guarda è così!”. Quindi voglio dire può essere falsamente negativa o può essere positiva nel senso che c'è qualcosa ma non siamo in grado di stabilire con assoluta certezza quel qualcosa quale nome e cognome abbia. Quanto detto fin qui viene riportato nella tabella in basso. Classificazione clinico-patologica delle miocarditi Miocardite FULMINANTE Miocardite SUBACUTA Miocardite CRONICA ATTIVA Miocardite PERSISTENTE ESORDIO Manifesto, solitamente a distanza di giorni da un’infezione virale Spesso misconosciuto Spesso misconosciuto Spesso misconosciuto FUNZIONE VENTRICOLARE SINISTRA Disfunzione severa con ventricolo non dilatato e a pareti ispessite (per l’edema interstiziale) Disfunzione moderatosevera con lieve dilatazione Disfunzione moderata Normale BIOPSIA Positiva Positiva o border-line Positiva o border-line Positiva o border-line STORIA CLINICA Fatale in una minoranza di pazienti entro 2 settimane; negli altri casi guarigione completa Miocardiopatia dilatativa Miocardiopatia restrittiva con normali volumi cavitari Funzione ventricolare sinistra normale in presenza di infiammazione persistente EVOLUZIONE ISTOLOGICA Restitutioad integrum Restitutioad integrum Fibrosi a cellule giganti Infiammazione persistente Manifestazioni cliniche: A monte dell'esordio della miocardite c'è una infezione delle alte vie respiratorie o del tratto gastrointestinale, quindi una delle cose più banali e più frequenti di questo mondo. Dobbiamo sospettare un interessamento del miocardio quando compaiono affaticamento, dispnea, dolore toracico, palpitazione, tachicardia, riduzione del primo tono cardiaco, ritmo di galoppo, soffi da insufficienza mitralica o tricuspidale, sfregamento pericardico. Laboratorio: Il laboratorio non ci dice niente di particolare se non che ci può essere leucocitosi (abbastanza tipica delle forme batteriche ma non necessariamente presente nelle forme virali). In un terzo dei pazienti, ma probabilmente in molti di più, c'è un aumento, un discostarsi dai valori normali della troponina e ovviamente si può avere quando il danno è esteso un aumento degli altri indici quali il CK. Esami strumentali: L'elettrocardiogramma è alterato ma specifico, cioè può dare delle alterazioni che ricordano quelle dell'ischemia soprattutto subendocardica: modifiche del tratto ST (sottoslivellamento) o inversione dell'onda T; però possiamo avere sia aritmie iper che ipocinetiche e soprattutto se il processo infiammatorio colpisce un tratto di miocardio in cui scorre o in cui è presente un pezzo importante del tessuto di conduzione, possiamo avere ritardi di conduzione atrioventricolare o intraventricolare, quindi il blocco atrioventricolare o di branca di varia entità, completo o incompleto che sia. La radiografia del torace difficilmente ci è di grande aiuto nelle patologie cardiovascolari se non per segnalarci delle conseguenze sul circolo polmonare. L'ecocardiografia può essere negativa ma più spesso può mostrare i segni di ispessimento parietale che resta tale sia in sistole che in diastole, perché ovviamente l'edema o l'infiltrazione non se ne fregano un accidente della fase della rivoluzione cardiaca. Quindi un ispessimento della parete che non si modifica nelle varie fasi del ciclo cardiaco insieme alle altre cose ci dovrebbe far pensare alla miocardite. La RM ha una sensibilità del 100% e una specificità del 90-100%. Quindi la RM ci può dire con ragionevole certezza che c'è la miocardite e in alcuni casi anche quale è la causa o quantomeno (se riteniamo che sia importante la diagnosi di certezza) può indirizzare la biopsia: se tu vuoi trovare un marker, un qualcosa che ti dica di che cosa si tratta, questa è l'area in cui ti conviene andare a fare la biopsia. Una volta che la biopsia abbiamo deciso di farla o siamo stati costretti a farla perché non veniamo a capo della situazione in nessuna maniera, per tirare fuori dall'esame bioptico una diagnosi relativa alla miocardite ci sono i cosiddetti criteri di Dallas, riportati nella tabella successiva. Alla prima biopsia se la miocardite è nella fase attiva, di massima espressione, dovremo avere un infiltrato associato a necrosi delle cellule con o senza segni di fibrosi. E' chiaro che la fibrosi sarà tanto maggiore quanto da più tempo sarà stata attiva la miocardite, perché è un processo reattivo non acuto. Poi possiamo avere una forma borderline quando non c'è la necrosi miocellulare, però potremmo aver preso un frustolo in una zona dove non c'è necrosi per cui è necessario un ulteriore prelievo bioptico. Solo se abbiamo fatto più prelievi in zone diverse e tutti sono privi di necrosi allora è verosimile che siamo in una forma borderline. Nelle biopsie successive, se è una miocardite persistente, troveremo sempre lo stesso tipo di quadro microscopico; se è una miocardite in via di guarigione, potremo trovare o meno la fibrosi; lo stesso nella miocardite guarita. E' chiaro che noi ci auguriamo sempre di non trovare la fibrosi perché questo significherebbe che la miocardite è guarita completamente con restitutio ad integrum. In basso vengono mostrate immagini con la descrizione istologica di quanto detto. Nell’immagine a sinistra si vede l'infiltrato infiammatorio del miocardio con necrosi e degenerazione dei miociti adiacenti. Tutti questi puntini neri sono tutti espressione dell'infiltrato infiammatorio e ci sono delle aree bianche abbastanza evidenti all'interno del tessuto: è proprio il focolaio dove troviamo sia segni di infiammazione che segni di necrosi. Nel caso centrale invece l'infiltrato infiammatorio è un po' più scarso, più diluito e non c'è danno miocitario però c'è un certo grado di fibrosi, cioè qui abbiamo l'impressione di un certo grado di distribuzione e di organizzazione di miociti mentre a destra nell'area di massima infiammazione non si capisce un accidente, è successo il massimo di quello di cui abbiamo parlato. Nel danno miocardico irreversibile ci sono quindi foci di infiammazione interstiziale, tutti diffusi all'interno dell'interstizio, che occupano uno spazio che noi ci aspetteremmo occupato dal tessuto fibrotico. Inoltre all'interno di alcune cellule ci sono chiari segni di degenerazione come i vacuoli intracitoplasmatici, superfici irregolari ed altro. Per fare questo tipo di diagnosi oltre al frustolo giusto per locazione di prelievo e per grandezza del frustolo stesso c'è bisogno di un occhio esercitato. Siccome le miocarditi non si vedono tutti i giorni e non tutti i giorni riusciamo ad avere una biopsia chiara, leggibile di una miocardite, è la classica diagnosi che finisce per esser fatta non in un qualunque laboratorio di anatomia patologica ma che viene riferita a dei centri di eccellenza che hanno acquisito una casistica sufficientemente numerosa da poterci dare credibilità, quello che leggono è verosimilmente la verità perché a furia di leggerne tante hanno sviluppato una capacità diagnostica che altri non sono in grado di avere. Prognosi e terapia: La miocardite virale è nella maggior parte dei casi una malattia autolimitantesi senza sequele; talvolta però può recidivare e nei casi peggiori può evolvere verso la cardiomiopatia dilatativa rapidamente ingravescente e quindi l'unica soluzione in questi casi è il trapianto cardiaco. Di solito quando sentiamo parlare di trapianti cardiaci in persone giovani la prima cosa che dobbiamo pensare è una cardiomiopatia dilatativa che forse non è primitiva ma è l'evoluzione di una miocardite virale, tossica, sempre se si riesce a identificare la possibile eziopatogenesi. La cosa importante è che abbiamo una cardiomiopatia dilatativa andata talmente avanti da non essere più sensibile ad alcun tipo di terapia che non sia il trapianto. La diagnosi precoce laddove possibile è dunque molto importante per instaurare un’opportuna terapia e monitorare l’evoluzione della patologia, la migliore garanzia contro l'evoluzione verso la cardiomiopatia dilatativa. Non c'è una terapia mirata, più che curare le virosi noi cerchiamo di prevenire le possibili complicanze batteriche e altre. In tutti i pazienti è indicata una limitazione dell’attività fisica fino alla risoluzione dei sintomi e alla normalizzazione dell’ECG, per evitare che nel momento di maggiore debolezza del miocardio si possa favorire l'estensione del processo e l'evoluzione verso la cardiomiopatia dilatativa. I pazienti che sviluppano un’insufficienza cardiaca o peggio ancora uno scompenso conclamato devono ricevere le consuete misure terapeutiche (ACE-inibitori, diuretici, digitale). Se ci sono aritmie, vanno curate: nel caso di aritmie ipercinetiche, con farmaci antiaritmici che deprimano il batmotropismo, l'eccitoconduzione e quant'altro; se ipocinetiche, vanno curate o con farmaci che aumentino il dromotropismo (quindi la conduzione) o in casi estremi con l'elettrostimolazione permanente. In casi rari le forme fulminanti, nella fase di acuzie clinica, possono portare a una tale compromissione da non poter aspettare la fase di risoluzione naturale della patologia, perché sarebbe preceduta da una grave e irreversibile alterazione della funzione cardiovascolare. FORME PARTICOLARI: MALATTIA DI CHAGAS Con i flussi migratori è arrivata anche dalle nostre parti, capita di farne diagnosi anche se non tutti i giorni né in tutti gli ospedali. E' una realtà però con cui ci dobbiamo confrontare. E' provocata dal protozoo Trypanosoma Cruzi trasmesso all’uomo tramite il morso di una cimice. Rappresenta una delle cause più comuni di cardiopatia in America centrale e in Sudamerica. Solo una minoranza di pazienti sviluppa una forma acuta di miocardite, caratterizzata da insufficienza cardiaca e aritmie che nel 90% dei casi si risolve con restitutio ad integrum entro pochi mesi. Circa un terzo dei pazienti sviluppa invece una forma cronica di miocardite, che diviene evidente anni dopo l’infezione iniziale e si manifesta come una vera e propria cardiomiopatia dilatativa, determinando un danno miocardico esteso, con dilatazione delle cavità cardiache, fibrosi e assottigliamento della parete ventricolare, con formazione di aneurismi nelle zone di assottigliamento (particolarmente all’apice) e formazione di trombi murali. L'immagine della ventricolografia (immagine sopra) è molto peculiare e naturalmente questa è un'altra delle forme nelle quali il trapianto rischia di essere l'unica soluzione terapeutica. La diagnosi si può fare se noi troviamo gli anticorpi anti-Trypanosoma nel siero. Nella forma cronica della malattia di Chagas è sempre presente un’insufficienza cardiaca progressiva generalmente a prognosi infausta. L’ ECG mostra quasi sempre blocco trifascicolare (blocco di branca destro con emiblocco anteriore sinistro) con possibile progressione verso il blocco atrioventricolare completo. Sono frequenti aritmie ventricolari, soprattutto durante sforzo, che possono essere pericolose per la vita. L’ecocardiogramma mostra una particolare ipocinesia confinata alla parete posteriore del ventricolo sinistro. La diagnosi richiede la dimostrazione di anticorpi anti-Tripanosoma nel siero del paziente. Le principali cause di morte sono rappresentate da un’insufficienza cardiaca intrattabile, aritmie ventricolari o eventi embolici a partenza dai trombi murali ventricolari. La terapia è volta a controllare lo scompenso e le aritmie. Queste ultime sono generalmente ben controllate dall’Amiodarone. La presenza di disturbi della conduzione può richiedere l’impianto di un pace-maker. La somministrazione di anticoagulanti può ridurre il rischio embolico. La prognosi è solitamente infausta; la principale raccomandazione è l’uso di misure preventive (insetticidi per eliminare il vettore). CARDITE DI LYME E' veramente rara. E' causata da una spirocheta trasmessa da zecche, la Borrelia burgdoferi. Questa una volta fino a qualche tempo fa era frequente perché le condizioni igieniche erano mediamente una schifezza, l'acqua corrente era un bene che non raggiungeva le case, l'acqua calda si otteneva solo tramite riscaldamenti di legna e bollimenti vari, il concetto di igiene personale era un concetto assolutamente diverso, c'era una costante e continua convivenza con gli animali. Qualche tempo fa io sono stato a visitare i sassi di Matera, c'è la ricostruzione ed è noto che quando il presidente della Repubblica dell'epoca a metà degli anni 50 andò a Matera e visitò i sassi e vide che in questi monovani scavati nella roccia c'erano uomini e bestie, che andavano dai pulcini alle galline al maiale al gatto al bovino o al mulo o quello che fosse, sentenziò che queste cose erano intollerabili, la gente andava tolta da lì, i sassi dovevano essere chiusi e bonificati, cessare di essere una realtà abitativa di quel tipo. Infatti adesso se andiamo ai sassi troviamo ristoranti, alberghi, bed and breakfast, civili abitazioni. Però una situazione del genere nei paesi a basso tenore di vita era comunissima ed è ancora comune. Esordisce generalmente nei mesi estivi con un caratteristico rash cutaneo (eritema cronico migrante) seguito dopo settimane o mesi da segni e sintomi di coinvolgimento neurologico, articolare o cardiaco. L’interessamento cardiaco è presente in circa il 10% dei pazienti ed è generalmente rappresentato da un blocco atrioventricolare di grado variabile (che può determinare sincope), tachicardia ventricolare, disfunzione ventricolare sinistra (solitamente modesta e asintomatica). Si può associare pericardite. La terapia si basa sulla somministrazione di penicillina o ceftriaxone per via endovenosa, monitoraggio elettrocardiografico e, qualora necessario, applicazione di un pace-maker temporaneo. Una cronicizzazione della malattia è molto rara. CAPITOLO XV: LA PARICARDITE Il pericardio è una membrana sierosa che ricopre il cuore. Esistono due facce della stessa medaglia: c'è il pericardio viscerale che ricopre come una pellicola il sacco ventricolare e poi c'è il pericardio parietale, cioè quello volto verso la parete. Tra questi due foglietti c'è uno spazio virtuale che costituisce lo spazio pericardico all'interno del quale c'è una minima quantità di liquido che evita, nel corso dei movimenti di sistole e di diastole, la frizione tra foglietto viscerale e foglietto parietale. Questa quantità di solito è contenuta entro i 50 ml e chiaramente è impossibile da estrarre: ce n'è talmente poca che se noi fossimo in grado di estrarla automaticamente creeremmo un contatto, una frizione tra i due foglietti. Il pericardio non è indispensabile per la vita, difatti durante gli interventi cardiochirurgici che implicano l'apertura del cuore, la prima cosa che fa il chirurgo è aprire il pericardio, dopo di che non si dedica a richiuderlo ma lo lascia così com'è o ne asporta una parte senza nessuno scrupolo né problema. Funzioni: Delimita, cioè è una specie di confine: il cuore non può andare oltre questo confine. Dall'altro lato è un confine anche nei confronti di organi circostanti, quindi praticamente riduce gli attriti con le strutture adiacenti. “Omogenizza” la contrazione del ventricolo destro e sinistro e del cuore rispetto alla respirazione. Quando noi diciamo che gli atri si contraggono e si rilasciano sinergicamente, i ventricoli si contraggono e rilasciano sinergicamente, noi pensiamo ad ognuno di loro separatamente. Il compito del pericardio è proprio quello del tenere insieme queste quattro camere: mentre le due di sopra si contraggono e le due di sotto si dilatano e viceversa, fa in modo che non ci sia questo disequilibrio spaziale tra la parte superiore e la parte inferiore del cuore, cioè confina loro all'interno di questo spazio delimitato. Infine funge da barriera meccanica per i patogeni e questo ci spiega anche perché di tutte le patologie fatte finora la miocardite per fortuna non è certamente frequente. PERICARDITE La pericardite è una patologia infiammatoria del pericardio. Classificazioni: Di solito noi la classifichiamo in base al decorso clinico: − Pericardite acuta, che è comparsa da meno di sei settimane e si è risolta in questo limite di tempo; − Pericardite subacuta tra sei settimane e sei mesi; − La pericardite cronica oltre i sei mesi. Però questa classificazione non ci dà soddisfazione, perché a noi non importa sapere di quanto è vecchia la pericardite, per noi è importante scoprirne la causa, il responsabile, perché riteniamo che questo sia il modo migliore per raggiungere il successo terapeutico. Quindi una migliore classificazione è fatta in base all'eziologia: − infettiva; − non infettiva; − da ipersensibilità. Nell'infettiva può essere virale (abbiamo detto che il pericardio sta subito sopra il miocardio, che è la barriera contro i patogeni, quindi per contiguità se c'è una infezione del miocardio il rischio che ci sia anche a carico del pericardio non è così peregrino), da piogeni, tubercolare (molto spesso può essere l'unica manifestazione della forma tubercolare), micotica o da parassiti cosiddetti minori che minori non sono. Poi ci sono le forme non infettive: idiopatica, traumatica, legata ad infarto miocardico acuto (in cui abbiamo due forme di pericardite: la forma acuta, poi quella che fa parte della sindrome di Dressler che compare a distanza di alcune settimane dall'infarto), metabolica (uremica, quando la gotta era particolarmente diffusa, nelle classi benestanti per esempio, dovuta ad un eccesso di alimentazione di dieta carnea e quindi portava ad un alterazione del metabolismo delle purine; così come può essere da mixedema, in cui molto spesso una ridotta funzione viene a manifestarsi proprio con la comparsa della pericardite). Poi vi è la pericardite da ipersensibilità: malattia reumatica (può esserci una pancardite reumatica, cioè endocardite, miocardite e pericardite, tutte le parti che definiscono il viscere cuore coinvolte nella forma da ipersensibilità), da farmaci, secondaria a danno miocardico. PERICARDITE ACUTA La pericardite acuta è dovuta all’infiammazione acuta dei foglietti pericardici. Le cause più frequenti sono infettive (virali o batteriche), traumatiche, da ipersensibilità (collagenopatie, IMA). Può essere accompagnata da versamento pericardico E’ caratterizzata da 3 elementi fondamentali: dolore toracico, sfregamento pericardico e modificazioni elettrocardiografiche. Il Dolore toracico può essere simile a quello dell'infarto, infatti la principale diagnosi differenziale è tra pericardite acuta ed infarto. Ci può aiutare l'età del paziente: un ragazzo di 15 anni che ha una sintomatologia dolorosa protratta simile a quella dell'infarto (certo per carità può avere una anomalia di origine delle coronarie, una fistola tra coronaria e la camera cardiaca), l'esperienza mi insegna che la prima cosa a cui si pensa è la miocardite o la pericardite. La diagnosi si fa sempre per esclusione, soprattutto se in quell'ospedale c'è una emodinamica, allora una coronarografia al giovanotto o alla signorina non gliela scansa nessuno, salvo poi pensare ad un'altra diagnosi in caso di analisi normali. Molto importanti sono anche le modificazioni elettrocardiografiche. Anche queste sono tendenzialmente diffuse (cioè presenti in quasi tutte le derivazioni) e soprattutto non hanno le stesse caratteristiche dell'infarto (nel caso della necrosi noi abbiamo il sopraslivellamento nella regione che legge quella regione di cuore, ma abbiamo il sottoslivellamento nelle derivazioni che leggono l'opposto, quindi c'è questa immagine speculare. Io credo di aver fatto vedere una immagine in cui il sopraslivellamento era di entità pari al sottoslivellamento di un'altra derivazione; in questo caso questo criterio diagnostico non vale, perché il pericardio abbiamo detto avvolge l'interezza del muscolo e quindi il danno è diffuso). Le principali alterazioni dell’ECG sono: - Sopraslivellamento del tratto ST diffuso (due o tre derivazioni periferiche e precordiali da V 2 a V 6 ) - Sottoslivellamento del tratto PR; - Onda T totalmente innalzata rispetto all’isoelettrica, che si inverte solo dopo il ritorno del tratto ST all’isoelettrica; - Complesso QRS normale o con diminuzione del voltaggio in caso di versamento pericardico; - Talvolta fibrillazione atriale o battiti ectopici sopraventricolari e/o ventricolari. Questo a lato è un tipico ECG di un soggetto con pericardite acuta. Clinica: Clinicamente avremo (vedi immagine alla pagina precedente) dolore retrosternale intenso trafittivo irradiato al dorso e al muscolo trapezio, ma si può anche irradiare alle braccia e quindi anche complicare (o rendere più difficile o più necessaria) differenziale. Può la diagnosi localizzarsi all'epigastrio e quindi può simulare un addome acuto, però abbiamo dei criteri per poter fare la diagnosi differenziale con il dolore toracico coronarico. Per esempio un dolore da pericardite acuta si esacerba con la tosse e con l'ispirazione mentre è mitigato dalla posizione genupettorale, quindi basta fare queste piccole manovre e già ci si aiuta nella diagnosi differenziale. Il segno obiettivo caratteristico (se c'è) è lo sfregamento pericardico. Anche questi rumori, così come il dolore, solitamente cambiano con il variare della posizione (disteso, seduto, genuflesso). Naturalmente il modo migliore per ascoltare loro è tenere il paziente in piedi e un poco chinato verso avanti. Importante è differenziare lo sfregamento pericardico da quello pleurico. Lo sfregamento pleurico scompare se noi facciamo trattenere l'aria: se teniamo il paziente in apnea i due foglietti della pleura non hanno occasione di sfregarsi perché abbiamo conservato il polmone in una posizione fissa; se continua ad esserci sfregamento vuol dire che è il pericardio, perché il cuore ovviamente non abbiamo possibilità di fermarlo. Esami strumentali: L'ECG è importante per la diagnosi (più spesso presuntiva che di certezza), ma è importante soprattutto nella diagnostica differenziale con gli eventi coronarici acuti. Possiamo avere: - sopraslivellamento del tratto ST, ma di solito è un sopraslivellamento diffuso; - sottoslivellamento del tratto PR; - onda T totalmente innalzata rispetto all'isoelettrica; - complesso QRS normale o con diminuzione del voltaggio (cioè abbiamo un QRS basso. Allora voi capite bene che in un paziente normale, caso mai un pezzo d'uomo, se ha avuto la febbre ecc… è verosimile che abbia una pericardite essudativa, che cioè si è manifestata con una iperproduzione di liquido pericardico al punto di avere allontanato il muscolo cui si genera il vettore che leggiamo con l’ ECG dall'elettrodo che è posto sul torace, quindi è un criterio); - spesso abbiamo dei battiti ectopici, delle extrasistoli atriali o ventricolari. Questo significa che l'ECG non consente una diagnosi di certezza, però ci indirizza se noi eravamo un po' confusi perché il paziente aveva troppo dolore per essere un fatto solo coronarico, non si capiva se aveva avuto o non avuto febbre, raffreddore, gastroenterite, l'eco non si vedeva perché tutto sommato il ventricolo si contraeva bene: in questo panorama nebuloso, repertare modifiche dell'ECG che non sono comuni ma neanche chiaramente indicative di una specifica patologia, consente di poter indirizzarsi verso la pericardite, cercare degli altri segnali di certezza, indirizzare diagnosi e terapia in questa direzione, perché è verosimilmente la più probabile. Quindi per quanto ci sia di tutto di più nell'ECG, è proprio questo di tutto di più, cioè delle cose che finora non avevano motivo di essere presenti contemporaneamente, che ci inducono a pensare che questa potrebbe verosimilmente essere una forma di infiammazione del pericardio. Questo è l'elettrocardiogramma. Nella pericardite c'è il sopraslivellamento del tratto ST però è generalizzato: lo vediamo in D1, D2, D3, non lo vediamo in aVR, lo vediamo ancora in aVL, un po' in V2, V3, V4, V5, un po' di meno in V6. Se noi dovessimo chiederci “Ma questo l'infarto dove lo ha avuto? Un po' qua, un po' là!”. Possibile che abbia avuto contemporaneamente un infarto anteriore esteso e un infarto antero-laterale? E che speranze tiene di venirne fuori? Si è già candidato allo scompenso se non al trapianto addirittura. Invece con l'elettrocardiogramma a destra, ci troviamo perfettamente: nelle derivazioni precordiali vediamo un sopraslivellamento non dei più tipici ma chiaro, ma quel che più conta nelle derivazioni periferiche vediamo un sottoslivellamento, perciò questo ECG parla da solo. Quello di sinistra invece dice e non dice e proprio per questo ci deve far pensare che infarto non è, sindrome coronarica acuta non è, angina da sforzo non è. C'è di tutto e c'è di più: pensiamo alla pericardite che male non facciamo! Esami di laboratorio: E' ovvio che è una malattia infiammatoria, quindi VES, PCR, IL-6, citochine, tutto quello che è di ambito infiammatorio, sarà inequivocabilmente incrementato. Ci può essere la leucocitosi, un rialzo lieve o moderato dei marcatori di danno miocardico. In caso di sospetto di pericardite secondaria ad eziologie specifiche dobbiamo fare delle indagini specifiche: se noi pensiamo sia di origine tubercolare dobbiamo fare una reazione alla tubercolina, se noi pensiamo che sia uremica dovremo andare a vedere creatinina e azotemia e via di questo passo. Ancora bisogna valutare i livelli di ormoni tiroidei (mixedema), la presenza di ANA, ENA, ANCA e fattore reumatoide (collagenopatie) e crioglobuline (pericardite da micoplasma). Questo vi deve mettere in testa che la troponina (marker importantissimo per sindrome coronarica acuta con necrosi cellulare, STEMI e NSTEMI), non è che ad ogni innalzamento di troponina ci dobbiamo imbarcare in una diagnosi di sospetta sindrome coronarica acuta, il che avviene sistematicamente, o per ignoranza o per eccesso di medicina difensiva. La troponina sale nello scompenso, nell'insufficienza renale, nell'embolia polmonare, nella miocardite, nella pericardite. Il problema è QUANTO sale e IN QUANTO TEMPO. Per esempio se un paziente presenta troponina 0,46, troponina 7, troponina 16, troponina 40 in due giorni. Ma che dubbio c'è? Dici: ma questo c'ha il blocco di branca destra quindi il sopraslivellamento non lo possiamo vedere. Vabbè, non lo possiamo vedere, ma la troponina che in 24 ore da 0,46 arriva a 40, cioè si è centuplicata, questo lo possiamo vedere e non abbiamo più dubbi. Ma se la troponina era 0,2, poi 0,4 e poi scendeva, a quel punto può aver anche avuto una necrosi, ma non necessariamente da ischemia protratta. Anche la miocardite porta un po' di necrosi. Allora non dobbiamo prendere i singoli marker o i marker più probabili con assoluta rigidità: la troponina deve aumentare di una certa entità, con una certa rapidità, poi ovviamente se prima di avere la troponina ho altri segni non sto a perdere tempo. Quante volte ci capita che il paziente arriva, dolore sì e no, l'ECG si vede non si vede, allora aspettiamo un attimo gli enzimi (un attimo!), dopo di che si decide se portarlo in emodinamica subito o aspettare altre 6 ore e fare un altro prelievo. Ma se il paziente arriva con dolore tipico, l'ST alto, all'eco si vede che c'è una zona franca di ipoacinesia, ma che ci importa di sapere della troponina? Tu fammi il prelievo e portamelo in emodinamica, dopo che gli ho riaperto il vaso mi farà anche piacere sapere se: la troponina aveva iniziato a salire e quindi l'ho preso a tempo; era già salita di poco quindi così così; era già salita tantissimo quindi per quanto io sia intervenuto con la riperfusione ormai una parte di miocardio me la sono già giocata. Quindi attenzione: la troponina è un marker ma non la semplice presenza, ma la cinetica e l'entità della modificazione. Radiografia del torace: Se hai un fatto miocardico o cardiologico non serve a niente. I dettami della corretta didattica mi impongono di dire qual è il quadro radiografico delle varie patologie ma non ci attaccheremo certamente su questo. Può essere normale nei casi non complicati di pericardite acuta. Può dare importanti indizi diagnostici (infiltrati polmonari o versamenti pleurici- infezione virale o da Myocoplasmafocolai polmonitici- eziologia batterica- presenza di masse o ingrandimenti linfonodali- malattia neoplastica). Può rivelare un versamento pericardico e/o pleurico concomitante. Ecocardiogramma: L'ecocardiogramma invece è molto utile, perché ovviamente la prima cosa che repertiamo è l'iper-rifrangenza dei foglietti. Essi saranno inevitabilmente inspessiti per effetto dell'infiammazione, quindi invece di vedere una cosa quasi virtuale vediamo proprio una linea che segna la presenza del foglietto pericardico. Se c'è il versamento lo vediamo: se io ho il sospetto durante una angioplastica che ci sia stata una piccola lesione della parete coronarica e quindi ci sia il rischio di passaggio di sangue dal lume coronarico al pericardio, non faccio altro che chiedere che mi portino in sala l'ecocardiogramma e analizzo lo spazio pericardico per cinque-dieci minuti. In quei cinque minuti se lo spazio pericardico resta virtuale o comunque la distanza tra i due foglietti non si modifica, io sono ragionevolmente certo che la lesione di parete non c'è stata. Se invece in quei cinque-dieci minuti vedo che questo spazio tende ad aumentare, comincia a diventare qualcosa di più che un sospetto e quindi io mi comincio ad attrezzare per fronteggiare un possibile tamponamento cardiaco. Quindi l'ecocardiogramma del torace è qualcosa di estremamente importante. Terapia: In tutti i casi è indicato il riposo a letto fino alla scomparsa dei sintomi e la normalizzazione degli indici di flogosi. Nel caso di pericardite acuta ad eziologia specifica (ad es., tubercolare-battericaautoimmune), la terapia è quella della malattia di base. Nel caso di pericardite uremica, è indicato l’inizio o l’intensificazione della terapia diuretica e la somministrazione di farmaci anti-infiammatori o glucocorticoidi (anche direttamente nel sacco pericardico). Nelle più comuni forme virali o idiopatiche, il trattamento è asintomatico e consiste nella somministrazione di acido acetilsalicilico o FANS. Nei pazienti che restano sintomatici, dopo aver escluso un’eziologia batterica o tubercolare, è indicata la somministrazione di corticosteroidi (prednisone). Nella prevenzione delle recidive si è dimostrata utile la somministrazione di COLCHICINA. Tuttavia, se le recidive sono frequenti e si presentano ancora dopo 2 anni dall’episodio iniziale, è indicata l’asportazione chirurgica del pericardio. Oggi l’Ibuprofene è preferito all’indometacina in virtù della migliore tollerabilità. La colchicina sta emergendo attualmente anche come valida alternativa ai FANS nel trattamento iniziale. VERSAMENTO PERICARDICO Si parla di versamento pericardico quando il liquido pericardico eccede i fatidici 50 ml. Può essere presente in qualunque tipo di pericardite: è l'infiammazione che determina l'iperproduzione di liquido, non uno specifico agente eziologico. E’ frequente dopo interventi di cardiochirurgia e solitamente si risolve in alcune settimane. Può presentarsi in forma silente in pazienti senza segni di pericardite (versamento pericardico cronico). Può avere carattere essudatizio, trasudatizio, emorragico (di solito emorragico ci fa pensare alla forma neoplastica, qualche volta alla forma tubercolare). Può evolvere verso la risoluzione o verso il tamponamento cardiaco. Spesso capita di avere pazienti che hanno una prima diagnosi di pericardite con essudato, quindi con versamento pericardico, dopodiché si seguono nel corso dei giorni finché cominciano a comparire segni di insufficienza ventricoloare, di sbandamento del ventricolo destro: sono segni fortemente indicativi di un imminente tamponamento cardiaco, quindi a quel punto bisogna eseguire la pericardiocentesi e soprattutto osservare poi il paziente, perché se la pericardite è dovuta ad un episodio acuto o subacuto, nel momento in cui svuotiamo il liquido in eccesso di solito si risolve; se invece la pericardite è legata a una forma neoplastica o tubercolare ancora attive, noi dopo qualche giorno rischiamo di trovarci di nuovo lo stesso paziente, con la stessa sintomatologia, lo stesso eco e quindi dobbiamo un'altra volta fargli la pericardiocentesi. Clinica: Clinicamente è presente il dolore toracico tipico della pericardite, anche se talvolta può attenuarsi per la riduzione dell’attrito tra i due foglietti pericardici. I rumori da sfregamento pericardico si riducono o scompaiono. All’ EO cardiaco si ha aumento dell’aia cardiaca, toni cardiaci lontani e riduzione o scomparsa dell’itto (se il versamento è cospicuo). La base del polmone può subire una compressione da parte del liquido pericardico, dando luogo ad un’area di ipofonesi sotto l’angolo scapolare sinistro (segno di Ewart). Tutti questi segni sono indicativi per una cardiomegalia. Imaging: Di ausilio sono Rx del torace, l’ecocardiogramma e la RM cardiaca. All’Rx torace vi è la tipica immagine cardiaca “a fiasco”; aumento del rapporto cardiotoracico; a differenza della cardiomegalia, nel versamento pericardico gli angoli cardiofrenici sono concavi (freccia). L’Ecocardiogramma è la metodica diagnostica di scelta per le elevate sensibilità e specificità e la semplicità di esecuzione; dimostra uno spazio ecoprivo che separa i due foglietti pericardici; per essere classificato come versamento, questa separazione deve essere mantenuta per l’intera durata del ciclo cardiaco. La RM cardiaca è di difficile e non rapida esecuzione, può essere eseguita come esame ausiliario in pazienti stabili quando sia necessaria una più accurata localizzazione di versamenti regionali o quantificare l’inspessimento del pericardio. Qual è la quantità di liquido? 50 ml è la quantità normale, al di sopra c'è il versamento pericardico. Mediamente il versamento diventa tamponamento quando supera i 200 ml, quando cioè diventa 4 volte la quantità fisiologicamente consentita! Ciò significa che la tolleranza del pericardio è grossomodo 4 volte il suo contenuto normale. Terapia: L'unica terapia del tamponamento cardiaco ingravescente o già determinato è la pericardiocentesi. Però è una metodica salvavita. Cinque minuti in più o in meno fanno la differenza tra mancato collasso del ventricolo sinistro o il suo collasso. Infatti per le coronarie e per il ventricolo ci vuole poco, non è che passano chissà quanti minuti da cessazione di perfusione alla morte. Non ci vuole niente. Quindi la pericardiocentesi si rischia di doverla fare nelle condizioni più incredibili, per cui è molto più della toracentesi (che è una cosa che allevia la sintomatologia ma non è salvavita). Ficcandogli quest'ago all'interno del pericardio, si salva la vita delle persone. Se non ce la fate, non ci riuscite o sbagliate la diagnosi, il paziente muore. Semplicemente per la mancanza di una manovra facilissima. La pericardiocentesi può essere effettuata per via percutanea o chirurgica (pericardiotomia). Comunemente viene effettuata per via percutanea sotto guida ecografica o fluoroscopico, con il paziente in posizione semiseduta e sotto monitoraggio pressorio ed elettrocardiografico. La pericardiocentesi chirurgica è indicata in caso di emopericardio acuto traumatico. In caso di versamento che non provochi tamponamento, la pericardiocentesi è indicata se la falda di versamento misurata all’ecocardiogramma supera i 20 mm o per chiarire l’eziologia del versamento (pericariocentesi esplorativa). Unica controindicazione assoluta alla pericardiocentesi è la dissezione aortica. Controindicazioni relative sono rappresentate da coagulopatia, terapia anticoagulante orale, trombicitopenia (PLT < 50,000/mm3). Il metodo più semplice è mettere il paziente in decubito semisupino (quindi non disteso ma neanche completamente seduto) e introdurre un ago sufficientemente lungo, di solito 9 cm (quello che si usa per le punture arteriose in emodinamica). Si introduce al di sotto dell'appendice xifoidea, con andamento da destra a sinistra, verso il ventricolo destro. Si può fare in scopia o meglio ancora ecoguidato, perché mentre in scopia io vedo il profilo del cuore ma non so dove comincia il pericardio e dove comincia l'epicardio, in eco invece questa differenza la posso fare quindi è più facile, non prende raggi il paziente, non li prendo io e ho una guida molto più sicura. Nel momento in cui l'ago entra all'interno del pericardio, uscirà del liquido che sarà di solito trasparente o appena giallino che noi aspireremo con la siringa. Se vogliamo sapere se è un essudato o un trasudato c'è il vecchio sistema di raccoglierlo in una provetta, fargli cadere una goccia di acido acetico, se ci sono le proteine si fa la forma del fumo di sigaretta, se non ci sono il liquido mantiene esattamente lo stesso aspetta che aveva prima (prova di Rivalta). Il problema è che io posso entrare nel ventricolo destro perché la diagnosi ad esempio è stata sbagliata e quindi almeno nella zona in cui ho punto non c'è una grande quantità di liquido pericardico (perché casomai è tutto sulla faccia posteriore), per cui entro nel ventricolo destro e quando vado ad aspirare mi esce sangue. Però io posso anche essere entrato in sacco pericardico e trovarmi un essudato pericardico emorragico. E allora in quel caso per esempio l'eco mi aiuta molto di più della scopia perché mi dice che sono ancora ben lontano dalla parete del ventricolo destro quindi il sangue che sto tirando fuori è all'interno del sacco pericardico. Distinguiamo: - una pericardiocentesidiagnostica: naturalmente deve esserci una quantità di liquido sufficiente, perché noi entriamo nel pericardio e non nel muscolo, dopodiché da questo liquido che noi tiriamo possiamo fare una analisi chimico-fisica, batteriologica, ricerca di specifici agenti patogeni, o ricerca di cellule neoplastiche; - una pericardiocentesi terapeutica: Quando il liquido è tale da far immaginare che possa sopravvenire in tempi brevi un tamponamento cardiaco (cioè la quantità di liquido è diventata tale che non c'è spazio per il ventricolo di riempirsi). TAMPONAMENTO CARDIACO Ritorniamo a quello che ho detto prima: che cos'è il pericardio? E' il sacchetto di plastica con cui andate a fare la spesa. Allora immaginate di comprare un melone che ha esattamente le dimensioni del sacchetto, quindi lo fate entrare con un poco di sforzo dopodiché il sacchetto è perfettamente pieno. Ora provate a immaginare che il melone abbia una sua dinamica, per cui questo sacchetto dà normalmente un certo gioco e consente al melone di espandersi e contrarsi. Nel momento in cui in questo sacchetto che già lascia un po' di spazio ci mettiamo del liquido, anche semplicemente dell'acqua, l'acqua voi sapete bene che se contenuta in uno spazio ristretto esercita una resistenza bestiale. Per spostare l'acqua in questo sacchetto ci vuole una forza incredibile. Il melone in quel momento la forza non ce l'ha non riesce ad espandersi e se il melone non si espande, collassa. E se il ventricolo sinistro collassa, secondo voi che succede? Non ci sarà passaggio di sangue dal ventricolo sinistro all'aorta, non ci sarà sangue nel circolo sistemico e quindi la mancanza di ossigeno è incompatibile con la vita. Quindi rischio di tamponamento cardiaco significa rischio di morte immediata. Il versamento pericardico può evolvere verso la risoluzione, può rimanere stabile o può determinare un tamponamento cardiaco. Il tamponamento cardiaco è la condizione in cui il liquido presente all’interno del pericardio determina un’ostruzione all’afflusso di sangue ai ventricoli. Si tratta di una grave complicanza che può essere fatale se non viene trattata tempestivamente. La quantità di liquido necessaria per causare un tamponamento può essere pari a soli 200 ml se il versamento si sviluppa rapidamente, o superiore ai 2 l se il versamento si sviluppa lentamente. Clinica: L’ostacolato riempimento ventricolare determina ipotensione, aumento della pressione venosa giugulare e ipoperfusione d’organo. I pazienti con tamponamento cardiaco sono sofferenti, dispnoici o ortopnoici, e presentano segni che riflettono i diversi gradi di diminuzione della gittata cardiaca (tachipnea, diaforesi, estremità fredde, cianosi periferica, sensorio alterato) fino allo shock conclamato. I principali reperti obiettivi sono: - pressione venosa giugulare elevata; - polso paradosso (più che paradosso, polso di Kussmaul, cioè se misuriamo il polso, la pressione arteriosa tende a salire, poi tende a discendere, poi tende a salire, poi tende a discendere: questo è un segno certo di versamento pericardico importante che evolverà sicuramente in tempi molto brevi verso il tamponamento); - tachipnea; - tachicardia; - ipotensione arteriosa (PAS < 100 mmHg); - aumento aia cardiaca delimitabile con la percussione; - toni cardiaci lontani; - rumori da sfregamento pericardico. Il polso paradosso è un segno caratteristico ma non specifico di tamponamento cardiaco. Può essere presente anche in caso di pericardite costrittiva, shock ipovolemico, BPCO ed embolia polmonare. Consiste in una caduta della pressione arteriosa sistolica > 10 mmHg durante l’inspirazione. In corso di tamponamento cardiaco, l’aumento inspiratorio del volume ventricolare destro determina un’abnorme riduzione del volume ventricolare sinistro, determinando così la riduzione della gittata. Il tamponamento cardiaco che si verifica per valori di pressione intrapericardica pari a 5-10 mmHg. Si può verificare quando vi è una diminuzione del volume ematico in un quadro preesistente di versamento pericardico che non aveva in precedenza determinato alterazioni emodinamiche significative. Si manifesta tipicamente in pazienti in emodialisi durante il trattamento dialitico, nei pazienti disidratati, nei pazienti con perdite ematiche o in corso di terapia diuretica. Esami strumentali: Ci si avvale dell’ausilio di ECG, Rx del torace e esame doppler. L’ECG mostra tachicardia sinusale, diminuzione dei voltaggi e alternanza elettrica dei complessi QRS (variazione del voltaggio da battito a battito, segno specifico ma poco sensibile dovuto alla rotazione antero-posteriore del cuore ad ogni battito cardiaco). L’Rx del torace dimostra il versamento pericardico con la tipica immagine cardiaca “a fiasco” e polmoni solitamente oligoemici. L’ecocardiocolordoppler è la metodica di scelta per la diagnosi; quando il versamento pericardico determina tamponamento, sono evidenziabili 2 fondamentali segni ecografici: il collasso telediastolico del ventricolo e dell’atrio di destra ed una esagerata variazione del flusso valvolare nei distretti destro e sinistro con gli atti del respiro (aumento del flusso nelle sezioni destre e riduzione nelle sinistre durante l’inspirazione e viceversa). Terapia: La terapia è lo svuotamento. Se sappiamo che c'è una causa che alimenta quel passaggio di liquido all'interno del pericardio, dobbiamo certamente da un lato assicurarci di risolvere la causa del tamponamento ma non ci dobbiamo contentare (sappiamo che l'alimentazione è continua) di aver svuotato il pericardio. Cioè noi una volta che siamo entrati all'interno del pericardio dobbiamo connettere questa ago cannula con un sistema di aspirazione a bassa pressione, per cui mentre ci occupiamo di risolvere la causa del tamponamento (quindi la fissurazione o la rottura della coronaria), dobbiamo esser certi che quel passaggio di sangue o di liquido che ci sarà nel pericardio nei minuti successivi non sarà tale da determinare un tamponamento con esito fatale. Quindi io vi garantisco che il pericardio più di tanto non si può riempire, se si riempie poi lo svuotiamo della stessa quantità di cui si è riempito, poi a quel punto con maggiore serenità mi vado ad occupare della causa del tamponamento. Cioè, si è rotta una coronaria? Vado in quella coronaria, metto uno stent ricoperto. Si è determinata una dissezione dell'aorta? Avvio il paziente dal cardiochirurgo però glielo porto in condizioni di decente stabilità emodinamica. PERICARDITE POST-INFARTUALE È una forma di pericardite acuta che si manifesta secondariamente ad un infarto miocardico acuto. Sono descritte due forme a diverse eziologia e presentazione clinica. L’incidenza di entrambe le forme è notevolmente diminuita grazie alle terapie di riperfusione. Forma post-infartuale precoce o pericardite epitenocardica: Si manifesta entro 1 settimana (solitamente 1-3 giorni) dall’IMA ed è dovuta all’infiammazione del pericardio viscerale e parietale adiacente ad una zona di necrosi transmurale. I pazienti sono generalmente asintomatici o lamentano dolore di tipo pleurico (DD con angina post-infartuale!!!!). All’EO è presente il rumore da sfregamento pericardico. Le alterazioni elettrocardiografiche possono essere meno chiare rispetto alle altre forme di pericardite acuta (lieve sopraslivellamento ST localizzato e precoce normalizzazione dell’onda T). Nei pazienti sintomatici, il trattamento è in genere rappresentato da un aumento del dosaggio dell’acido acetilsalicilico (fino a 650 mg 3-4 volte/die per 2-5 giorni), mentre l’uso di FANS e corticosteroidi deve essere attentamente valutato in quanto questi farmaci possono interferire con la cicatrizzazione dell’area miocardica infartuata. Forma post-infartuale tardiva o sindrome di Dressler: Si manifesta da 1 settimana a pochi mesi dopo un IMA ed è dovuta ad una reazione autoimmune ad antigeni miocardici liberatisi al momento della necrosi. Il quadro clinico è caratterizzato da febbre e dolore pleuritico. All’EO sono presenti sfregamenti pleurici, pericardici o entrambi. La radiografia del torace evidenzia versamento pleurico e/o pericardico. Le alterazioni elettrocardiografiche sono quelle tipiche della pericardite acuta. Si tratta generalmente di una manifestazione autolimitantesi, tuttavia l’ospedalizzazione è indicata in caso di presenza di cospicuo versamento pericardico. Il trattamento è sintomatico e analogo a quello della pericardite acuta idiopatica. PERICARDITE CRONICA Un versamento pericardico cronico Può rappresentare un riscontro occasionale all’EO o ad una radiografia del torace eseguita per altre ragioni. Le cause più importanti sono rappresentate da: - TUBERCOLOSI; - MIXEDEMA; - COLLAGENOPATIE; - NEOPLASIE; - RADIAZIONI; - INFEZIONI FUNGINE o PIOGENE. La diagnosi può essere suggerita da altre manifestazioni della malattia di base. Negli altri casi, è indicata l’esecuzione di una pericardiocentesi diagnostica (esplorativa). La terapia di versamenti cronici voluminosi è rappresentata dalla pericardiocentesi; in caso di recidiva è necessario ricorrere all’asportazione del pericardio per evitare l’insorgenza di pericardite costrittiva. PERICARDITE COSTRITTIVA Succede che per effetto dell'infiammazione e del protrarsi della malattia i due foglietti aderiscono tra di loro e per effetto della reazione infiammatoria si ispessiscono fino a diventare anche calcifici, per cui questo astuccio che normalmente si adattava a quello che conteneva, diventa rigido. Quindi non c'è più gioco: il ventricolo destro e sinistro, nel momento in cui si espandono per riempirsi di sangue, vanno a sbattere contro un ostacolo che non è più dinamico, elastico, ma è un pezzo di calcio, una parete dura. E' un fenomeno costante, la ridotta funzione cardiaca diventa un fatto cronico e si può manifestare uno scompenso, una insufficienza. Le cause più frequenti sono la pericardite tubercolare, la pericardite uremica, l’irradiazione mediastinica e i traumi toracici. Clinica: Il quadro clinico è dominato da sintomi e segni di scompenso diastolico: - SINTOMI: dispnea, ortopnea, astenia, disturbi addominali; - SEGNI: edemi declivi, epatomegalia, ascite, turgore delle giugulari, versamento pleurico, polso paradosso (1/3 dei casi), segno di Kussmaul, toni cardiaci lontani, schiocco pericardico, retrazione sistolica dell’itto. Il segno di Kussmaul è dovuto all’aumento inspiratorio della pressione venosa sistemica (evidenziabile dalla comparsa/aumento di turgore delle giugulari durante l’inspirazione). Il terzo tono precoce (protodiastolico), è auscultabile generalmente lungo il margine sternale sinistro o all’apice cardiaco, che corrisponde al brusco arresto del riempimento ventricolare già nella prima fase della diastole. Esami strumentali: L’lettrocardiogramma non mostra reperti caratteristici. Le alterazioni più diffuse sono la riduzione dei voltaggi e anomalie diffuse della ripolarizzazione (appiattimento/inversione onda T); in circa il 30% dei pazienti si riscontra FA. L’Rx torace può mostrare calcificazioni pericardiche (soprattutto in caso di eziologia tubercolare), ingrandimento dell’atrio destro, versamento pleurico e, in caso di elevate pressioni di riempimento ventricolare sinistro, segni di congestione polmonare. L’ecocardiocolordoppler dimostra l’inspessimento del pericardio, l’ingrandimento atriale, un movimento diastolico ventricolare anomalo (brusco arresto in protodiastole), segni di aumentata pressione venosa (dilatazione vena cava inferiore e vene epatiche), funzione contrattile conservata (normale FE), alterazioni del flusso transvalvolare analoghe a quelle riscontrate in corso di tamponamento cardiaco. Nell’immagine a sinistra, possiamo notare il versamento pericardico fibrinoso, mentre in quella di destra vi è una proiezione apicale col tipico aspetto a “tela di ragno” dovuto ai fasci di fibrina organizzati che obliterano lo spazio pericardico. Il cateterismo cardiaco fornisce preziose informazioni per l’identificazione del profilo emodinamico restrittivo; utile per la diagnosi differenziale con la cardiomiopatia restrittiva. Mostra due reperti fondamentali: - Le pressioni di riempimento di entrambi i ventricoli mostrano una netta e precoce caduta diastolica seguita da una profonda onda di plateau (aspetto dip and plateau, presente anche nella cardiomiopaita restrittiva); - Le pressioni di riempimento delle camere cardiache sono pressochè equivalenti (< 3-5 mmHg, diversamente dalla cardiomiopatia restrittiva). Terapia: Riguardo la terapia quindi, qua c'è poco da svuotare, da analizzare, bisogna togliere! L'unica terapia della pericardite costrittiva è la rimozione del pericardio o pericardiectomia. Tale intervento presenta una mortalità perioperatoria compresa tra il 5 e il 15%, con tassi di sopravvivenza a 5 e 10 anni rispettivamente pari all’80 e al 60%. La mortalità perioperatoria dipende dall’estensione e dalla profondità dei depositi di calcio, dalla compromissione del miocardio e dalla presenza di eventuali comorbidità. La mortalità a lungo termine è essenzialmente connessa al grado di funzionalità miocardica e all’ entità di disfunzione epatica e/o renale secondaria. Per tali motivi, il ricorso alla chirurgia è indicato nelle fasi precoci della malattia. Nel periodo pre-operatorio o nei pazienti inoperabili, un miglioramento del quadro clinico può essere ottenuto tramite somministrazione di diuretici e restrizione di sodio con la dieta (anche se la maggior parte dei pazienti diventa refrattaria nel lungo termine). I farmaci con effetto cronotropo negativo dovrebbero essere evitati o usati con cautela in quanto la tachicardia rappresenta un fondamentale meccanismo di compenso al ridotto riempimento diastolico. La pericardiectomia è un evento raro però io mi ricordo che un paio di anni fa ho avuto un paziente di 3435 anni che aveva avuto una pericardite e nonostante che con la terapia avesse risolto l'evento acuto, c'era stata una evoluzione infiammatoria cronica e aveva sviluppato una pericardite costrittiva e cominciava ad avere i sintomi dello scompenso (dispnea, facile affaticabilità, aritmie etc...), per cui a 36 anni ha dovuto subire una sternotomia per poi incidere il pericardio che a quel punto è come il guscio dell'uovo: lo incidete, aprite, sfilate il tuorlo da dentro, lo buttate nel coso giallo dei rifiuti speciali e l'intervento è finito e gli avete risolto tutto.
Scarica