PARSERIZZAZIONE DI FILE
Un problema piuttosto comune in bioinformatica è quello di leggere un file,
tipicamente un flat-file in un qualche formato (es. FASTA) rappresentante un database
(es. un insieme di sequenze). All’interno del file si tratta di riconoscere la struttura
sintattica con cui sono organizzati i dati e quindi individuare i singoli record e
all’interno dei record i singoli campi.
Una volta “estratte” le informazioni di nostro interesse dal file è possibile sottoporle a
qualunque elaborazione, ad esempio potremo scrivere in un nuovo file i risultati
dell’elaborazione. L’elaborazione potrebbe consistere anche semplicemente nel
riscrivere i record in un nuovo formato o nello scrivere solo i record che verificano
determinate condizioni.
campo1
campo2
FILE
record1
campon
…….
…….
PROGRAMMA
PARSERIZZATORE
campo1
campo2
campon
BIOINFO3 - Lezione 35
recordn
1
FILE FASTA
Supponiamo che il file da analizzare sia in formato FASTA, ad esempio un file
di sequenze EST scaricato da GENBANK. Individuamo nei record i campi di
nostro interesse
1
2
3
4
5
1)
2)
3)
4)
5)
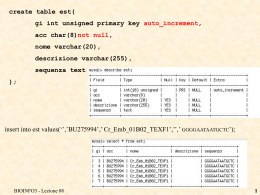
Il numero identificativo gi del record genbank
Il codice accession
Il nome della EST
La descrizione della EST
La sequenza della EST
BIOINFO3 - Lezione 35
2
ELABORAZIONE
L’elaborazione che ci interessa effettuare su ogni singolo record è quella di
inserire un record corrispondente all’interno di un database relazionale SQL
di sequenze EST. Le 5 informazioni estratte da ogni record dovranno
produrre un corrispondente comando di insert MySQL
1
2
3
4
5
insert into est (gi,accession,nome,descrizione,sequenza)
values(8777287,’AB044776’,’AB044776’,’Panax…’,‘GAAGAA…’)
BIOINFO3 - Lezione 35
3
2 ALTERNATIVE
Abbiamo visto che esistono due modi alternativi per interagire con un
database SQL e di conseguenza è possibile eseguire l’operazione di insert in
due modi alternativi
1)Dalla linea di comando del client MySQL
2)Da programma (embedding)
Nel primo caso il programma di parserizzazione creerà un file di comandi
SQL che poi daremo direttamente in pasto all’interprete MySQL.
Nel secondo caso le istruzioni di insert sono eseguite una ad una direttamente
all’interno del programma, man mano che vengono letti i singoli record.
La seconda soluzione sembrerebbe più “automatizzata”, però anche nel primo
caso, una volta scritto completamente il file, è possibile far eseguire da
programma (con una istruzione qx o system) il comando UNIX di lettura ed
esecuzione del file da parte del client mysql
BIOINFO3 - Lezione 35
4
SCHEMATIZZANDO…
campo1
campo2
1)
campon
while (riconoscimento di un record){
…
…. 1
campo
estrazione campi
campo2
scrittura del comando sul file
campon
definizione comando di insert
}
file
campo1
campo2
2)
parserizzatore
file di
comandi SQL
while (riconoscimento di un record){
campon
estrazione campi
…
…. 1
campo
SERVER MYSQL
definizione comando di insert
esecuzione del comando
campo2
campon
esecuzione
del file
DB
}
Proveremo a vedere entrambe le soluzioni in quanto la parte di programma
che cambia è minima (evidenziata in rosso)
BIOINFO3 - Lezione 35
5
LA STRATEGIA PER PARSERIZZARE IL FILE
Abbiamo detto che la parserizzazione deve essere guidata dalla struttura
sintattica del file.
In Perl una possibilità è quella di leggere l’intero file ed assegnarlo ad
un’unica stringa (che può contenere file anche molto grandi) e quindi
procedere all’individuazione dei record. I record sono chiaramente separabili
grazie al pattern “>gi|” posto all’inizio di ogni record. Si potrà pertanto
effettuare una operazione di split basata proprio su questo pattern,
ottenendo un array contenente tutti i record. Per ogni record si potrà
effettuare successivamente un pattern matching per estrarre i 5 campi.
>gi|
lettura
file
split
BIOINFO3 - Lezione 35
,
,
)
>gi|
>gi|
file
array (
stringa
pattern matching
1
2
3
4
5
6
IL PARSERIZZATORE
BIOINFO3 - Lezione 35
7
ESECUZIONE
Per evitare di “sorbirsi” tutti i cicli
di lettura fissiamo un break point!
BIOINFO3 - Lezione 35
8
ESECUZIONE
Lo split crea 22 record,
ma il primo è vuoto (perché il file
iniziava con il pattern di split).
Per questo ho aggiunto il test
if ($record)
BIOINFO3 - Lezione 35
9
ESECUZIONE
Prima di eliminare tutti gli spazi e i
new-line dalla sequenza proviamo a
vedere tutte le stringhe catturate dal
pattern matching
BIOINFO3 - Lezione 35
10
ESECUZIONE
Vediamo la sequenza $seq prima e
dopo la sostituzione del pattern
Si può anche notare che l’operazione di
pattern substitution azzera le variabili
speciali $1, $2,…
In generale ogni operazione sui pattern
cancella i risultati della precedente
BIOINFO3 - Lezione 35
11
ESECUZIONE
Si procede quindi con il riconoscimento
del secondo record e così via.
Alla fine di ogni ciclo abbiamo a
disposizione nelle variabili $gi, $ac,
$nome, $descr, $seq tutti i campi per
quel record
BIOINFO3 - Lezione 35
12
CREAZIONE DI FILE DI COMANDI SQL
Non ve l’avevo ancora detto (anche perché a fine programma i file
aperti vengono comunque chiusi automaticamente) ma è buona
norma chiudere un file quando è stato letto o scritto completamente
BIOINFO3 - Lezione 35
13
ESECUZIONE
BIOINFO3 - Lezione 35
14
ESECUZIONE DEL FILE DI COMANDI
Una volta scritti tutti i comandi SQL e chiuso il file di output
($nomefile.mysql) si può eseguire l’istruzione Perl:
qx{mysql btbm-xx <$nomefile.mysql};
Dove btbm-xx è il nome del database su cui far eseguire i comandi. Il comando
UNIX eseguito da Perl fa leggere ed eseguire al client mysql il file di comandi
appena creato.
Lo stesso comando UNIX potrà anche essere dato non da programma ma a
mano al termine dell’esecuzione del programma parserizzatore
BIOINFO3 - Lezione 35
15
COMANDI SQL DA PROGRAMMA
BIOINFO3 - Lezione 35
16
Scaricare