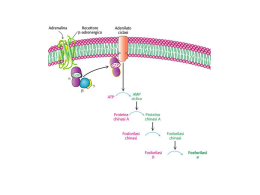
ALTERAZIONI DEL CITOPLASMA: ACCUMULI INTRACELLULARI Acqua ed elettroliti (per alterazioni di membrana o dell’osmolarità) Lipidi Trigliceridi (Steatosi) Esteri del colesterolo Carboidrati Glicogeno Glucosio Aminoacidi e derivati Pigmenti Altri Antracosi Siderosi Emosiderosi Lipofuscina (Intravescicolare) Rame Rigonfiamento idropico di epatociti Rigonfiamento di cisterne del reticolo endoplasmico Rigonfiamento di mitocondri ACCUMULI INTRACELLULARI DI ACQUA H2 O Aumento osmolarità intracell. K+ H2 O Inibizione Na+/K+ ATPasi Iponatremie Na+ H2 O FATTORI FISIOPATOLOGICI DETERMINANTI IPONATREMIE I. Deplezione del volume circolante effettivo II. Inappropriata secrezione di ADH (SIADH) Aumentata produzione di ADH III. Polidipsia IV. Cause endocrine (insufficienza surrenalica, ipotiroidismo) SIADH: Sindrome da Inappropriata secrezione di ADH I. Aumentata produzione ipotalamica: * Alterazioni neuropsichiche (infezioni meniningee ed encefaliche, accidenti vascolari (emorragie e trombosi), neoplasie, psicosi. * Farmaci * Infezioni e traumi II. Produzione ectopica: tumori III. Potenziamento effetti periferici: farmaci Dieta K+ plasmatico K+ intracell. sudore URINE feci Na+ + R-insulina K+ + b2 adr. K+ ACIDOSI H+ K+ H+ ALCALOSI Altri esempi di accumuli intracellulari Figure 1-35 Mechanisms of intracellular accumulations: (1) abnormal metabolism, as in fatty change in the liver; (2) mutations causing alterations in protein folding and transport, as in alpha1-antitrypsin deficiency; (3) deficiency of critical enzymes that prevent breakdown of substrates that accumulate in lysosomes, as in lysosomal storage diseases; and (4) inability to degrade phagocytosed particles, as in hemosiderosis and carbon pigment accumulation. Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 15 September 2005 04:26 PM) © 2005 Elsevier Ramificazione/Deramificazione Glicogeno Glicogeno sintasi I Glicogeno Fosforilasi b (defosforilata). Meno attiva Glicogeno sintasi D (fosforilata) Meno attiva Glicogeno Fosforilasi a Glucosio-1-P Fosfoglucomutasi Glucosio-6-P Glucosio-6-Pasi Glucosio GLICOGENOSI A) EPATICHE I GRUPPO: Caratterizzate da EPATOMEGALIA E IPOGLICEMIA Deficienza di glucosio-6-fosfatasi (tipo I, malattia di Von Gierke) Deficienza di enzimi deramificanti (tipo III) Deficienza di fosforilasi epatica (tipo VI, malattia di Hers) Deficienza di fosforilasi chinasi (tipo VIa) II GRUPPO: Caratterizzate da EPATOMEGALIA E CIRROSI Deficienza di enzimi ramificanti (tipo IV, malattia di Andersen) Deficienza di enzimi deramificanti (tipo III) B) MUSCOLARI I GRUPPO: Caratterizzate da DOLORI MUSCOLARI, INTOLLERANZA ALLO SFORZO, MIOGLOBINURIA. Deficienza di Fosforilasi muscolare (malattia di McArdle, tipo V) Deficienza di fosfofruttochinasi (tipo VII, malattia di Tarui) Altre deficienze II GRUPPO: Caratterizzate da DEBOLEZZA MUSCOLARE SCHELETRICA PROGRESSIVA CON ATROFIA E/O CARDIOMIOPATIA. Deficienza di enzima deramificante muscolare (tipo IIIa) Deficienza di fosforilasi chinasi cardiaca Deficienza di -glucosidasi acida lisosomiale (tipo II, malattia di Pompe) MTFR:metilen tetraidrofolato reduttasi METIL-TETRAIDROFOLATO B12 (COBALAMINA)/HMMT* metionina TETRAIDROFOLATO METILEN-TETRAIDROFOLATO S-adenosylmetionina metile S-adenosylomocisteina omocisteina VITAMINA B6 (PIRIDOSSAL 5-P) CISTATIONINA b SINTETASI cistationina cisteina Accumulo di metiltetraidrofolato ferro nel fegato (colorazione con blu di Prussia) *Omocisteina metil transferasi Ematossilina-eosina Blu di prussia Emocromatosi Transferrina* *Saturazione: 16% - 30% - 45 % Carenza di Fe2+ normale Eccesso di Fe2+ ferrireduttasi Divalent Metal Transporter LUME INTESTINALE Ossidasi Cu-dipendenti Ferroossidasi a forma multimerica costituita da una catena H e una L Che può contenere 4500 atomi di Fe3+ Nel topo inattivazione efestina determina anemia sideropenica (X-linked) EPATOCITI UBIQUITARIO, CELLS ERITROIDI Il complesso TfR-alotransferrina Ricicla sulla membr pl. ferroreduttasi CELLS TUBULI RENALI TUTTE LE CELLULE UBIQUITARIO MACROFAGI Ferro-ossidasi HEPCIDIN (un membro della famiglia delle defensine) regola l’omeostasi del ferro modulando negativamente l’espressione di ferroportina e, conseguentemente, il trasporto di Fe nel plasma Hepcidin – ferroportin – deficienza di ferro Hepcidin – ferroportin – accumulo di ferro “Bone Morphogenetic Protein”: citochine della famiglia del TGFb.BMP6 viene prodotta in modo direttamente proporzionale all’accumulo intracellulare di Fe Hemojuvelin solubile: inibisce. Tf-Fe3+ La proteasi che cliva HJV di membrana è regolata da Fe (negativamente) e ipossia (positivamente). Mutazioni nel gene che codifica per questa proteasi causano anemia sideropenica TFR1 HIF-1 è in grado di regolare neg. HAMP transcription Alte concentrazioni di Transferrina-Fe3+ spiazzano HFE da interazione con TFR1 e ne favoriscono interazione con TFR2. tale interazione è ulteriormente stabilizzata da legame Transferrina-Fe3+ a TFR2. Conseguenza: maggiore satura zione transferrina, maggiore Trascrizione di Hepcidin. Transcriptional regulation of hepcidin by the BMP/Smad pathway. Hepcidin transcription depends upon signaling through BMP receptors (BMP-Rs) and downstream Smads. BMPs can act as autocrine or paracrine hormones. Binding of BMP to cell surface HJV positions BMP to activate BMP receptors. Activation of BMP receptors leads to the generation of phosphorylated RSmads, which dimerize with Smad4. The RSmad/Smad4 heterodimer translocates into the nucleus and activates transcription of the HAMP gene, which encodes hepcidin. Soluble HJV binding to BMP prevents the formation of a cell surface BMP-HJV complex and blocks activation of BMP receptors. Inflammatory cytokines such as IL-6 bind to IL-6 receptors (IL-6Rs), activating Stat3, which also binds to the HAMP promoter. Stat3 activation requires the presence of Smad4, as deletion of the Smad4 gene prevents IL-6 induction of hepcidin. Smad4 is downstream of TFR2 and HFE, which suggests that the signal provided by these proteins also activates the HAMP promoter or that these membrane proteins affect BMP receptor signal transmission. In their study in this issue of the JCI, Babitt and colleagues demonstrate in vivo that soluble HJV binds BMPs produced by the liver, leading to alteration in iron homeostasis J Clin Invest. 117:1755-1758, 2007 Pathophysiology of Wilson disease. The gene has been specifically localized to chromosome 13 and has been found to code for a copper transport protein. The abnormal gene results in decreased hepatic excretion of copper into bile either because of a defect at the lysosomal membrane or because of a defect in transporting copper across the cell membrane into bile. Biopsia epatica (colorazione con rodanina) Malattia di Wilson Figure 1-31 A, Schematic representation of heterophagy (left) and autophagy (right). (Redrawn from Fawcett DW: A Textbook of Histology, 11th ed. Philadelphia, WB Saunders, 1986, p 17.) B, Electron micrograph of an autophagolysosome containing a degenerating mitochondrion and amorphous material. Accumulo di lipofuscina in lisosomi di epatociti Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 15 September 2005 04:26 PM) © 2005 Elsevier Figure 1-31 A, Schematic representation of heterophagy (left) and autophagy (right). (Redrawn from Fawcett DW: A Textbook of Histology, 11th ed. Philadelphia, WB Saunders, 1986, p 17.) B, Electron micrograph of an autophagolysosome containing a degenerating mitochondrion and amorphous material. Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 15 September 2005 04:26 PM) © 2005 Elsevier ASPETTI GENERALI MALATTIE DI ACCUMULO LISOSOMIALE Autosomiche recessive (a parte due X-linked: sindrome di Hunter, malattia di Fabry). Molto eterogenee dal punto di vista molecolare e clinico. Malattie progressive: normalità alla nascita, comparsa di sintomi, a carattere ingravescente, dopo mesi o anni. Da sospettare in tutti i casi di: - DISORDINI NEUROLOGICI PROGRESSIVI (alterazioni intellettive, convlusioni, riduzioni acuità visiva e uditiva) - EPATOSPLENOMEGALIA ( e organomegalia) - ALTERAZIONI OSSEE (fratture, limitazione movimenti articolari, dolore) Patogenesi malattie d’accumulo lisosomiale GOLGI REL 4. trasporto difettivo per alterazioni di Una molecula comune a più idrolasi 5. Difettiva glicosilazione e trasporto dal Golgi al lisosoma 3. trasporto difettivo dal REL al Golgi lisosoma substrato 1.Difetto di espressione o funzione di enzima enzima prodotto Proteina attivatrice 2. Difetto di espressione o funzione regolatore DEFICIENZA MULTIPLA DI SOLFATASI C-formylglycine generating enzyme Sulphatase modifying factor-1 STRATEGIE TERAPEUTICHE PER MALATTIE D’ACCUMULO 5:554-565, 2004
Scaricare