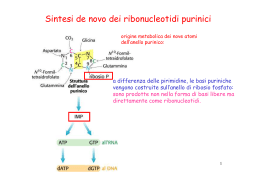
SINDROME DI LESCH-NYHAN E’ una malattia del metabolismo delle purine, dovuta alla mancanza della ipoxantina-guanina fosforibosil transferasi (HGPRT), enzima della via di riciclaggio per la sintesi dei nucleotidi Il gene dell’HGPRT è localizzato sul cromosoma X Sintomi: elevati livelli di acido urico nel sangue e nelle urine (che precipitano sotto forma di urati), calcoli renali, insufficienza renale, gotta, problemi comportamentali tra cui autolesionismo L’autolesionismo è probabilmente dovuto ad anomalie nel metabolismo della serotonina e della dopamina Non esiste cura per questa malattia Riciclaggio delle basi puriniche e pirimidiniche nelle vie di salvataggio Basi puriniche e pirimidiniche libere vengono costantemente rilasciate nella cellula durante la degradazione dei nucleotidi (turnover degli acidi nucleici endogeni e degradazione di quelli ingeriti con la dieta). Le purine libere sono in gran parte riutilizzate per la sintesi delle corrispondenti basi usando la via di salvataggio, molto più semplice di quella de novo e meno dispendiosa dal punto di vista energetico! Una reazione molto importante è quella catalizzata dalla adenosina fosforibosiltransferasi: Adenina + PRPP AMP + PPi La guanina e l’ipoxantina (prodotto di deamminazione dell’adenina) sono riciclate in maniera analoga dall’enzina ipoxantina-guanina fosforibosiltransferasi (HGPRT) Esiste una via di salvataggio del tutto simile per le basi pirimidiniche LA VIA DI SALVATAGGIO La via de novo è costituita da diversi passaggi che richiedono per sintetizzare 1 IMP: 4 aminoacidi, 1 PRPP 2 folati 3 ATP Il deficit di HPRT causa l’accumulo di guanina e ipoxantina che vengono convertire a acido urico dalla xantina ossidasi È un intermedio comune alla sintesi di diversi amminoacidi e delle purine; viene sintetizzato a partire dal ribosio 5-fosfato che si genera dalla via dei pentosi fosfati, grazie alla reazione catalizzata dalla ribosio fosfato pirofosfochinasi: Ribosio-5-fosfato + ATP 5-fosforobosil-1-pirofosfato + AMP Sintesi del 5-fosforibosil 1-pirofosfato (PRPP) Meccanismo di reazione della ipoxantina guanina fosforibosil transferasi • Converte l’ipoxantina o la guanina rispettivamente a IMP e GMP a spese del PRPP, dal quale viene rilasciato PPi • Molti parassiti sono incapaci di effettuare la via di sintesi ex novo delle basi e utilizzano unicamente la via di salvataggio La forma umana attiva è un tetramero. E’ stato dimostrato che il loop II si chiude sopra al sito catalitico dopo il legame con i substrati Nel sito catalitico 2 residui sono conservati al 100%: G69 D134 La mutazione G69E inattiva completamente l’enzima e causa la sindrome di Lesch-Nyhan La mutazione D134G lo inattiva parzialmente causando l’artrite gottosa La mutazione D193N non lega Mg2+ e inattiva completamente l’enzima • Diversi meccanismi contribuiscono alla sovrapproduzione di acido urico: 1. L’HGPRT catalizza il recupero di IMP e GMP dalle basi puriniche utilizzando il PRPP. Il deficit di HGPRT provoca un accumulo di I e G che vengono convertite ad acido urico dalla xantina ossidasi 2. L’aumentata disponibilità di PRPP per la PRPP amidotransferasi, la tappa limitante della sintesi ex-novo, aumenta la sintesi delle purine 3. Diminuisce la disponibilità di inibitori della PRPP amidotransferasi, come IMP e GMP, stimolando la sintesi ex novo TERAPIA L’allopurinolo è un analogo della xantina, inibisce la xantina ossidasi controllando i sintomi (100-600 mg/die) La colchicina allevia il dolore della gotta con una modalità non compresa Spasticità e distonia possono essere controllate con le benzodiazepine e inibitori del GABA come il baclofen Non è stato trovato alcun farmaco in grado di controllare i sintomi extrapiramidali Misure di contenimento fisico sono utili per l’autolesionismo Le immunocilline 5 fosfato, analoghi dello stato di transizione della reazione catalizzata dalla HGPRT, vengono utilizzati come farmaci antiparassitari DEFICIT DA ADENOSINA DEAMINASI (ADA) •E’ una malattia autosomica recessiva •Fa parte delle immunodeficienze combinate gravi o SCID (Severe Combined Immune Deficiency), insieme eterogeneo di malattie monogeniche caratterizzate da un difetto dei linfociti T, associato a difetti dei linfociti B e delle cellule NK •La classificazione di tali patologie è di tipo immunologico •Gli individui affetti sono privi della normali difese immunitarie •La diagnosi viene effettuata misurando l’attività enzimatica nei globuli rossi •Incidenza complessiva delle SCID: 1:50000 •L’ADA-SCID rappresenta il 15-20% di tutte le SCID
Scaricare