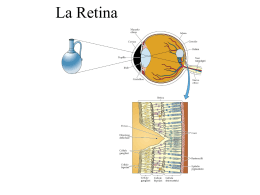
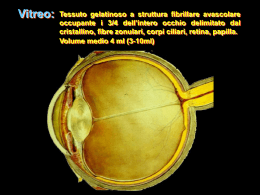
Chirurgia del distacco retinico pediatrico Giorgio Tassinari Incidenza • 3,2-5,6% del totale • 0,38-0,69/100.000 • 70% maschi (trauma; sdr. X-linked) • 30% femmine Fivgas et Al. Retina april 2001 Haiman et Al. Arch Ophthalmology 1982 Sann-ni et Al. Retina 2006 Nan-Kai et Al. Ophthalmology 2005 Incidenza • Si può ritenere che il 40% dei distacchi di retina regmatogeni in età pediatrica, contro l’11% in età adulta sia secondario ad un trauma (Dialisi giovanili post-traumatiche (23-42%) • La maggior parte dei casi risulta associato alla miopia • In minor percentuale risulta associato a sindromi o secondario ad interventi chirurgici (34% dei casi) Haiman et Al. Arch Ophthalmology 1982 Latikainen et Al Acta Ophthalmologica 1985 Il bulbo oculare presenta in età pediatrica rispetto all’adulto differenti rapporti vitreo-retina periferica e quindi differenti punti di repere anatomochirurgici. Nei primi anni di vita si verificano importanti modificazioni nelle dimensioni del bulbo, nella lunghezza assiale del globo oculare ed in modo particolare a livello del segmento posteriore. Infatti mentre il segmento anteriore è all’incirca del 20% inferiore rispetto alle dimensioni adulte, il segmento posteriore è inferiore dell’80% circa. Il segmento posteriore aumenta drammaticamente di dimensione nei primi sei mesi di vita, fino a oltre il 50% (Larsen JS, 1971). A tre anni, la lunghezza assiale è approssimativamente il 95% rispetto all’adulto (Grignolo A, Rivara A, 1968) (Sorsby A, 1989). I corpi ciliari presentano alla nascita una lunghezza media di 3,06 mm nasalmente e 3,31 mm temporalmente; a 24 mesi la lunghezza dei corpi ciliari è circa i ¾ rispetto all’occhio adulto (Aiello AL e coll., 1992). A sei anni di età, i corpi ciliari nasali raggiungono il 90% della lunghezza da adulto, e i corpi ciliari temporali l’85%. Uno sviluppo simile lo presenta la pars plana, che occupa il 75% della lunghezza totale dei corpi ciliari. Entrambe le strutture, corpi ciliari e pars plana sono presenti e distinguibili alla nascita. Questi diversi rapporti spaziali dell’occhio del bambino rispetto all’adulto condizionano le tecniche di chirurgia vitreoretinica. L’inserzione anteriore della retina (Daicker B, 1972) ( Kwitko ML, 1979) richiede la esecuzione delle sclerotomie in pars plicata (Maguire AM, Trese MT, 1992) al fine di evitare rotture di retina iatrogene. Altro problema riguarda il cristallino che nei bambini occupa una parte maggiore della camera vitrea, con maggiori difficoltà di manipolazione degli strumenti chirurgici per il rischio di danno meccanico al cristallino. (Kwitko ML 1979) (Apple DJ, Naumann GOH, 1986), Anche le correnti determinate dai fluidi di infusione in camera vitrea possono danneggiare il cristallino. In età pediatrica, il corpo vitreo presenta aderenze molto marcate con la retina e non è presente il distacco posteriore di vitreo (Seebag J, 1991). Creare un distacco posteriore di vitreo durante la vitrectomia, a questa età è molto difficile e spesso impossibile per la tenace aderenza della ialoide posteriore alla retina. Altra caratteristica da non sottovalutare è tendenza alla proliferazione vitreoretinica. E’ risaputo che la proliferazione vitreoretinica ha in età pediatrica un decorso molto più precoce e rapido rispetto agli adulti (Bodard GE, e coll,, 1978). Quando è necessario applicare un cerchiaggio episclerale bisogna ricordare che le bande in silicone larghe 2 mm o 2,5 mm, a questa età possono supportare un’ampia porzione di retina periferica, potendosi estendere dall’ora serrata fino all’equatore. Con l’età, l’ingrandimento del bulbo e il rilascio delle trazioni anteriori, può essere necessario rimuovere la banda di silicone anche per evitarne lo scivolamento posteriore. La scelta del sostituto vitreale come mezzo tamponante è condizionata dalla difficoltà nell’utilizzo dei gas, relativa alla gestione postoperatoria, sia per il posizionamento che per il monitoraggio della pressione intraoculare, a favore dell’utilizzo del silicone (Rodriguez F, Lewis H, 1991). Eziologia • • • • Nelle anomalie congenite Nelle m. ereditarie DRR secondari a colobomi DRR nelle dermatiti atopiche (3%-tarda adolescenza • DDR post traumatico Capone et Al. Pediatric Retina, 2005 Eziologia • Miopia patologica o degenerativa (>4D) • Miopia associata a malattie non ereditarie: Prematurità Da deprivazione sensoriale (ptosi congenita, cataratta, opacità corneali) • Miopia patologica associata a sindromi Sindrome di Stickler Sindrome di Marfan Sindrome di Wagner • • • • Leucocoria Strabismo Bilateralità non infrequente Frequente coinvolgimento maculare (80% versus 56% negli adulti ) • Frequente presenza di PVR (50%) Fivgas et Al. Retina april 2001 Winslow et Al. Opthalmology 1978 Hilton et Al. Mod Probl Ophthalmol. 1969 • Bassa età postmestruale (PMA) • Zona I Sono fattori di rischio per ORGANIZZAZIONE FIBROVASCOLARE nel vitreo nelle due settimane successive al trattamento Laser (D.R. trazionale ROP st 4) Degenerazioni vitreoretiniche familiari Il distacco di retina in età giovanile insorge nella maggior parte dei casi come conseguenza di : - liquefazione del gel vitreale - distacco posteriore di vitreo - formazione di rotture retiniche a livello delle aderenze vitreo retiniche Molte condizioni determinate geneticamente si possono associare a queste alterazioni vitreo retiniche DEGENERAZIONE VITREO RETINICA EREDITARIA DI WAGNER- JANSEN- STICKLER Descritta da Wagner nel 1938 nello studio di una famiglia svizzera (18 persone 5 - 42 aa) Si caratterizza per 3 peculiarità: - cavità vitreale otticamente vuota - membrane e strands vitreali - degenerazione retinica a lattice Alterazioni pigmentarie periferiche, vasi retinici ristretti e fibrotici, atrofia ottica, cataratta. DEGENERAZIONE VITREO RETINICA EREDITARIA DI WAGNER- JANSEN- STICKLER Jansen nel1962 dimostrò una maggiore incidenza di distacco di retina ( 50- 75%) Sticker descrisse nel 1965 la presenza di anomalie facciali (naso piatto,glossoptosi), del palato (palatoschisi) e scheletriche (condrodisplasia), correlate alle alterazioni oculari. RETINOSCHISI GIOVANILE X- LINKED Descritta da Haas e Stabsarzt nel 1898. Trasmissione recessiva legata ai cromosomi sessuali maschi. Bilaterale. Regione maculare caratterizzata da spazi cistici con aspetto radiale a stella che tendono ad organizzarsi in un ampia cavità centrale. Retinoschisi periferica che interessa più frequentemente il quadrante infero temporale. RETINOSCHISI GIOVANILE X- LINKED Fori retinici dello strato interno nel 75% dei casi. Aree di demarcazione pigmentate sul bordo posteriore della schisi. Vasi retinici a ponte tra lo strato retinico interno ed esterno. Sineresi del gel vitreale, membrane vitreali con trazione sui vasi retinici a ponte. Possibili emorragie vitreali o interne alla schisi. Il distacco di retina può insorgere per la presenza di rotture retiniche su entrambi i foglietti della schisi. RETINOSCHISI GIOVANILE X- LINKED RETINOSCHISI GIOVANILE X- LINKED DEGENERAZIONE VITREORETINICA DI GOLDMANN- FAVRE Caratteristiche in comune con la retinoschisi giovanile e la Wagner. Tramissione autosomica recessiva, in genere bilaterale e simmetrica. Cavità vitreale otticamente vuota eccetto che per la presenza di filamenti vitreali. Modificazioni pigmentarie periferiche (come nella retinite pigmentosa). Cecità notturna, ERG ridotto o assente. Aree di retinoschisi centrale e periferica VITREORETINOPATIA ESSUDATIVA FAMILIARE Descritta da Schepens nel 1969, trasmissione ereditaria autosomica dominante. Bilaterale, colpisce i giovani. Classificazione in tre stadi con decadimento visivo progressivo nel tempo VITREORETINOPATIA ESSUDATIVA FAMILIARE - Stadio I variazioni dell’interfaccia vitreo retinica (distacco posteriore di vitreo, membrane vitreali, eterotopia trazionale maculare, degenerazione retinica cistoide e “ white withwithout pressure”) - Stadio II alterazioni vascolari (dilatazione e tortuosità dei vasi retinici periferici con essudati sottoretinici e distacco di retina localizzato) - Stadio III essudazione sottoretinica massiva con distacco di retina totale, cheratopatia a bandelletta, cataratta, glaucoma. DEGENERAZIONE VITREORETINICA SNOWFLAKE Descritta da Hirose nel 1974, caratterizzata da aree di degenerazione “white with pressure” associata a numerosi spot gialli, cambiamenti pigmentari e ad obliterazioni vascolari. Trasmissione autosomica dominante. Aumentata incidenza di cataratta e distacco di retina. DEGENERAZIONE VITREORETINICA SNOWFLAKE SINDROME DI MARFAN Trasmissione autosomica dominnante. Miopia assile, di almeno 7 diottrie nel 21% dei casi Ectopia lentis , presente nel 50- 80%, in genere bilaterale, simmetrica, supero temporale Distacco di retina (12%) legato all’entità di miopia, sublussazione lenticolare, afachia chirurgica. Presenza di alterazioni degenerative vitreali (distacco del vitreo anteriore) e degenerazione retinica a lattice. SINDROME DI MARFAN OMOCISTINURIA Errore metabolico congenito con mancanza dell’enzima cistein sintetasi che converte l’omocisteina in cistina. Trasmissione autosomica recessiva. Manifestazioni oculari e sistemiche simili alla Marfan. Ectopia lentis nel 90% dei casi. Bilaterale, sublussazione della lente inferiore. In 1/3 dei casi lussazione in C.A o C.V Degenerazione retinica cistica, distacco di retina nel 19% dei casi OMOCISTINURIA SINDROME DI EHLERS- DANLOS Anomalia del tessuto connettivo per incapacità delle fibrille collagene ad organizzarsi in strutture complesse. Cheratocono, miopia, sclera blu, distacco posteriore di vitreo, distacco di retina regmatogeno. Anomalie dello sviluppo PIT DEL NERVO OTTICO Anomalia di sviluppo della vescicola ottica. Localizzato in genere a livello della rima temporale del N.O di dimensioni variabili dai 0,1 ai 0,7 diametri papillari. Unilaterale nell’85% dei casi. Nell’80% distacco di retina sieroso al polo posteriore per passaggio del liquido vitreale attraverso il pit. Retinoschisi per rottura di ampie cisti intraretiniche maculari. PIT DEL NERVO OTTICO Anomalie dello sviluppo MORNING GLORY SYNDROME Caratteristiche in comune con l’optic pit. Papilla ingrandita ed escavata, con core centrale di tessuto biancastro ed anello peripapillare con modificazioni pigmentarie. Distacco di retina al polo posteriore nel 38% dei casi , presenza di rottura retinica peripapillare per effetto trazionale del tessuto epipapillare. MORNING GLORY SYNDROME Anomalie dello sviluppo COLOBOMA DEL NERVO OTTICO Si può associare a distacco di retina (66%). L’area del difetto comprende un optic pit con interessamento della retina e della coroide. Escavazione parziale o totale del canale sclerale. Papilla ottica ingrandita (50% in più del normale) Bilateralità > Optic pit Possibile associazione con coloboma della retina e della coroide. COLOBOMA DEL NERVO OTTICO Anomalie dello sviluppo PERSISTENZA DEL VITREO IPERPLASTICO PRIMITIVO Residui della guaina fibrovascolare che avvolge il cristallino fetale. Forma anteriore: membrana fibrovascolare con aspetto di massa retrolenticolare, stiramento dei processi ciliari Forma posteriore: coartazione del tessuto retrolentale con trazione sulla retina peiferica emovitreo recidivante, D.D.R trazionale In conclusione •Il DR in età pediatrica è un evento raro più frequentemente associato alla alta miopia. •Va trattato con chirurgia episclerale se non si associa a PVR permettendo un recupero nell’ 80% dei casi. •Nelle forme sindromiche (Marfan, Stickler…) va considerato il trattamento dell’ occhio adelfo con retinopessia laser su 360° e non focale per la possibilità di distacco da trazione circolare su retina sana.
Scaricare