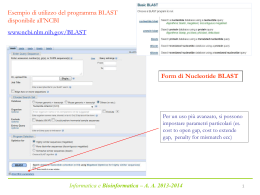
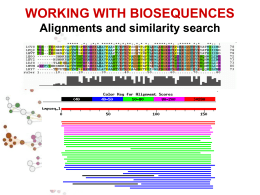
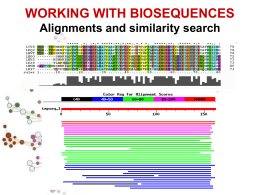
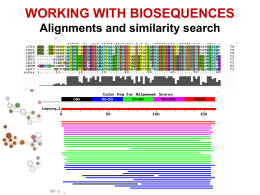
Allineamenti di sequenze biologiche Allineamento di sequenze Scopi Date due o più sequenze biologiche (DNA, RNA o proteine) può essere utile: Misurare quanto sono simili; Sapere quali sono le parti simili; Fare ipotesi sulla funzione di una proteina; Inferire relazioni evolutive. Per fare ciò è necessario definire in modo quantitativo la misura di similarità tra sequenze. Ovvero dobbiamo assegnare per ogni corrispondenza residuo-residuo delle sequenze in esame dei valori numerici e poi, usando queste misure di corrispondenza puntiforme, costruire in qualche modo l'intero allineamento tra le sequenze. Si definisce allineamento tra sequenze l'identificazione delle corrispondenze residuo-residuo che preserva l'ordine dei residui all'interno delle sequenze stesse. Possono essere introdotte gaps (lacune). Esistono dunque molti allineamenti possibili tra sequenze. Ad esempio date le due sequenze generiche: Seq1: a b c d e Seq2: a c d e f allineamento1: a b c d e | a c d e f allineamento2: a b c d e | | | - a c d e f allineamento3: a b c d e | | | | a - c d e f Quale dei tre è un allineamento migliore? E' necessario stabilire dei criteri che permettano di selezionare l'allineamento migliore. Bisogna cioè trovare un modo per valutare tutti i possibili allineamenti sistematicamente assegnando ad ognuno di essi uno score. Gli allineamenti si possono dividere in allineamenti a coppie e allineamenti multipli. Un modo per visualizzare in modo rapido se due sequenze sono in qualche modo correlate è l’uso dei dot-plot Il limite dei dotplots è che non evidenziano relazioni tra sequenze distanti dal punto di vista evolutivo. Possono essere un primo passo per l’analisi di sequenza. Per misurare la similarità di due sequenze si possono usare principalmente due misure di distanze: 1. 2. La distanza di Hamming; numero di posizioni con caratteri diversi in due sequenze lunghe uguali La distanza di Levenshtein; numero minimo di operazioni elementari di editing richieste per trasformare una stringa nell’altra. RAZZO PAZZO * d. Hamming=1 R--AZZO PALAZZO *** d. Levensthein=3 RA--ZZO PALAZZO * ** d. Levensthein=3 In biologia però bisogna essere più fini e bisogna riconoscere che certi cambiamenti sono più frequenti di altri e che quindi è necessario introdurre schemi di scoring più raffinati. IDENTITY A C T A 1 C 0 1 T 0 0 1 G 0 0 0 G 1 Date le seguenti sequenze di DNA allinearle usando una matrice di scoring binaria e con gap=-1 AGATA GATTA Significatività di un allineamento Date due sequenze allineate attgcctcgtg agtccttccag * * * ** * Come posso distinguere se l’allineamento è casuale o le due sequenze sono veramente omologhe? Significatività di un allineamento Si applica il test delle ipotesi modellando opportunamente il problema (modelli parametrici e non). Si identifica l’ipotesi H0 (ipotesi nulla) con la casualità dell’allineamento Si identifica l’ipotesi H1 con la non casualità dell’allineamento Si calcola la probabilità che l’allineamento sia casuale Si accetta o si rigetta l’ipotesi H0 Significatività di un allineamento Ricerche in DataBase di sequenze BLAST Per fare ricerche di similarità in database che contengono milioni o miliardi di sequenze bisogna far uso di algoritmi euristici che non garantiscono di trovare l’allineamento ottimo. Blast è un programma che fa l’uso di un euristica opportuna per allineare velocemente sequenze nucleotidiche e proteiche. Si basa sull’indicizzazione di brevi “parole” contenute nella sequenza query BLAST Viene creata una tabella di parole lunghe W (sottosequenze) appartenenti alla sequenza query Vengono cercate tutte queste parole in tutto il database e viene costruita una tabella con tutte le sequenze del database che contengono queste sottosequenze. Solo le sequenze contenute nella tabella precedente vengono allineate completamente Alla fine si ottiene un allineamento ottimale approssimato.
Scaricare