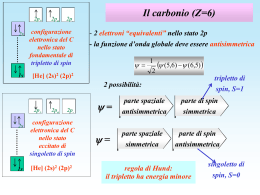
Corso di Chimica Fisica II 2013 Marina Brustolon 14. Molecole 1° parte Molecola di H2: stati eccitati, di singoletto e di tripletto. Molecole omonucleari degli elementi della seconda riga. Molecole eteronucleari: il metodo variazionale per la costruzione degli OM. Molecole con legami coniugati Il metodo di Hartree-Fock Riassunto delle approssimazioni per gli orbitali molecolari 1. Abbiamo assunto che valga l’approssimazione di Born-Oppenheimer. 2. Abbiamo usato la combinazione lineare di OA per costruire gli OM. 3. Abbiamo trascurato l’interazione elettroneelettrone e quindi assunto che la funzione d’onda di due elettroni sia data dal prodotto delle funzioni d’onda dei singoli elettroni (approssimazione monoelettronica). Come descrivere con una funzione d’onda i due elettroni della molecola di H2? 1. Usiamo gli orbitali molecolari costruiti come combinazioni lineari degli orbitali atomici 1s: 1σ e 2σ. 2. Costruiamo la configurazione elettronica: per lo stato fondamentale sarà 1σ2, per gli stati eccitati può essere 1σ1 2σ1(stato monoeccitato), o 2σ2 (stato bieccitato). 1σ2 1σ1 2σ1 2σ2 3. Scriviamo la funzione d’onda in modo che sia rispettato il principio di Pauli (funzione che cambia di segno allo scambio dei due elettroni). 1 (1,2) 1 (1)1 (2) ( (1) (2) (1) (2)) 2 funzione spaziale S funzione di spin x A = non cambia di segno cambia di segno scambiando 1 e 2 scambiando 1 e 2 Lo stato di spin è di singoletto. A Singoletto S 2 ( S1 S 2 ) 2 S z S z1 S z 2 Operatori di momento di spin totale 1 S 0 2 1 spin 1 Sz 0 2 spin 2 Gli spin dei due elettroni sono correlati e sempre antiparalleli Stato di singoletto singoletto S=0 fondamentale 2 Singoletto eccitato 1 ' (1,2) ( 1 (1)2 (2) 2 (1)1 (2)) ( (1) (2) (1) (2)) 2 funzione spaziale S 1 2 funzione di spin (singoletto) A ( (1) (2) (1) (2)) è ancora una funzione di singoletto. singoletto eccitato Tripletto eccitato 1 ' ' (1,2) ( 1 (1)2 (2) 2 (1)1 (2)) ( (1) (2) (1) (2)) 2 1 ' ' (1,2) ( 1 (1)2 (2) 2 (1)1 (2)) ( (1) (2)) 2 1 ' ' (1,2) ( 1 (1)2 (2) 2 (1)1 (2)) ( (1) (2)) 2 tripletto eccitato funzione spaziale funzione di spin (tripletto) A S Tripletto Operatori di momento di spin totale S 2 ( S1 S 2 ) 2 S z S z1 S z 2 S 2 S ( S 1) 2 2 2 S S ( S 1) 2 2 2 2 S 1 1 2 1 ( ) S ( ) 2 2 2 2 S z S z S z 0 tripletto eccitato Esercizio Usando le proprietà degli orbitali molecolari valutate la stabilità della molecola di He2. He2 ha 4 elettroni. OM Bilancio dell’energia: E 2 1 1 ( ) 2 ( ) 4 0 1 S 1 S E2 1 ( ) 1 S E1 1 ( ) 1 S La molecola non è stabile Molecola di H2: stati eccitati, di singoletto e di tripletto. Molecole omonucleari degli elementi della seconda riga. Molecole eteronucleari: il metodo variazionale per la costruzione degli OM. Molecole con legami coniugati Il metodo di Hartree-Fock Le altre molecole biatomiche omonucleari la molecola di He2 non si può formare, perché si riempiono gli OM sia di legame che di antilegame: questo dipende dal fatto che l’atomo di He ha uno shell completo. Gli orbitali degli shell completi non contribuiscono al legame. Con il Li si inizia un nuovo shell. Gli orbitali degli shell non completi contribuiscono al legame, e si chiamano quindi shell di valenza. Gli OM formati dagli OA per molecole biatomiche omonucleari della seconda riga Gli orbitali atomici che entrano in gioco sono il 2s, e i tre orbitali 2p. Legami 2px 2px X direzione del legame Legami 2py 2py 2pz 2pz Orbitali + - + - Gli orbitali sigma non cambiano di segno per una rotazione di 180°. Orbitali - + + - + + - Gli orbitali pigreco cambiano di segno per una rotazione di 180°. 2S 2S Orbitali atomici e molecolari degli elementi della seconda riga 2pz 2S 2S 2pz 2py L’ossigeno O2 è paramagnetico: S=1 2* 2* Questo stato di tripletto ha energia più bassa dello stato di singoletto L’ossigeno in stato di singoletto è estremamente reattivo. Molecole eteronucleari Come costruire gli OM con LCAO? i c j i j j Ora i coefficienti degli orbitali atomici non saranno più uguali. Ci vuole quindi un criterio per trovare i valori che descrivono meglio la molecola. c i j ? coefficiente dell’OA j-esimo nella combinazione lineare che dà l’i-esimo orbitale molecolare Metodo approssimato: metodo variazionale Principio variazionale H 0 E0 0 stato ad energia più bassa Principio variazionale: a qualunque altra funzione corrisponderà un valore medio dell’energia E0. p Hp pp E0 rapporto di Rayleigh Se p dipende da parametri, variandoli si può minimizzare il rapporto di R. migliorando la funzione. p c j j j c j j H c j j j j i j c c j j j H i H ij j i Sij j j c c j H i i, j c c j i j i, j j c c H c c S j i ij j i ij i, j i, j j i c c H c c S j i ij j i ij i, j i, j Per semplificare il problema, supponiamo di avere una combinazione di soli due OA Dobbiamo trovare il minimo di derivando rispetto ai ci e ponendo le derivate a zero. 2 p c j j c11 c2 2 j 1 c11 c 2 2 H c11 c 2 2 c11 c 2 2 c11 c 2 2 c12 1 H 1 c 22 2 H 2 c1c 2 1 H 2 c 2 c1 2 H 1 c12 1 1 c 22 2 2 c1c 2 1 2 c 2 c1 2 1 1 H 1 H 11 , ecc 1 1 S11 , ecc c12 H 11 c 22 H 22 c1c 2 H 12 c 2 c1 H 21 2 2 (c1 S1 c 2 S 2 c1c 2 S12 c 2 c1 S 21 ) S11 S 22 1 S12 S H 12 H 21 c12 H11 c22 H 22 2c1c2 H12 (c12 c22 2c1c2 S ) Differenziamo l’equazione rispetto a c1: Derivata del termine di sinistra: 2c1 H11 2c2 H12 Derivata del termine di destra: 2 (c1 c 22 2c1c 2 S ) (2c1 2c 2 S ) c1 Se poniamo 0 la equazione diventa: c1 2c1 H 11 2c 2 H 12 (2c1 2c 2 S ) 0 Semplificando: c1 ( H 11 ) c 2 ( H 12 S ) 0 E ripetendo rispetto a c2: c1 ( H 21 S ) c2 ( H 22 ) 0 Il sistema di equazioni lineari e omogenee c1 ( H11 ) c2 ( H12 S ) 0 c1 ( H 21 S ) c2 ( H 22 ) 0 è risolvibile solo se il determinante dato dai coefficienti delle incognite è uguale a zero. Le incognite sono i ci, quindi il determinante è: H 11 H 12 S 0 H 21 S H 22 1 2 1 2 H11 1 H 1 1 H 22 2 H 2 2 H12 1 H 2 1 S 0 S 2 determinante secolare Quindi in generale si avrà: i N k c i i 1 c (H k i i1 Si1 ) 0 i2 Si 2 ) 0 i3 Si 3 ) 0 i c (H i i c (H i N equazioni nelle N incognite ci i ................................ Il sistema è risolvibile solo se il determinante secolare (dato dai coefficienti delle incognite) è uguale a zero det H ij S ij 0 Ciascuna delle N radici della equazione di ordine N va inserita a turno nelle equazioni . Inserendo la radice k e risolvendo il sistema troveremo i coefficienti per l’orbitale molecolare k. Determinante secolare H 11 S11 H 21 S 21 H 31 S 31 H 12 S12 H 22 S 22 H 32 S 32 ... ... H 13 S13 ... H 23 S 23 ... 0 H 33 S 33 ... ... ... Le radici di che soddisfano questa equazione sono i valori approssimati dell’energia. Per ogni valore di si ottiene un set di equazioni che dà i coefficienti corrispondenti. Torniamo al caso della molecola biatomica: Poniamo la condizione che il determinante secolare sia eguale a zero: 1 S 0 S 2 Sviluppando troviamo l’equazione di secondo grado: (1 ) ( 2 ) ( S ) 2 0 da cui si ottengono le radici: E 1 2 2S [( 1 2 2S ) 2 4(1 S ) 2 ( 1 2 2 )]1 / 2 2(1 S ) 2 Proviamo a vedere se questo risultato è in accordo con quello trovato per la molecola omonucleare biatomica, per la quale ci eravamo basati solo sulle proprietà di simmetria: Ponendo 1 = 2 = (molecola biatomica omonucleare) otteniamo: 2 2S [( 2 2S ) 2 4(1 S ) 2 ( 2 2 )]1 / 2 E 2(1 S ) 2 che sviluppando e semplificando dà: E (1 S ) come già trovato basandosi sulle proprietà di simmetria e sulla normalizzazione. Un’importante informazione qualitativa sulla forza di un legame chimico la possiamo ottenere semplificando l’espressione: E 1 2 2S [( 1 2 2S ) 2 4(1 S ) 2 ( 1 2 2 )]1 / 2 2(1 S ) 2 ponendo formalmente S = 0 (anche se sappiamo che certamente S0 quando si forma un legame chimico). Otteniamo: E 1 2 [( 1 2 ) 2 4( 1 2 2 )]1 / 2 2 1 2 [ 12 22 2 1 2 4 1 2 4 2 )]1 / 2 2 ( 1 2 ) [( 1 2 ) 2 4 2 )]1 / 2 2 2 Dividendo e moltiplicando il secondo termine per 1 - 2: 1/ 2 ( 1 2 ) 2 4 2 ( 1 2 ) 1 ( 1 2 ) E 2 2 2 ( 1 2 ) 1/ 2 2 ( 1 2 ) 1 2 ( 1 2 )1 2 2 ( 1 2 ) Se 2 (1 2 ) possiamo approssimare 2 1 ( 2 ) 1 2 1/ 2 2 2 1 ( 1 2 ) 2 Trovando alla fine: 2 2 E 1 , E 2 1 2 1 2 (1 x) 1/ 2 1 1 x 2 Molecole eteroatomiche A-B Se 1 2 la differenza in energia tra gli orbitali atomici e gli orbitali molecolari è molto piccola. 0 2 2 1 2 1 Energie e OM Trovando le radici di (E ) che annullano il determinante secolare, siamo in grado di costruire i sistemi di equazioni che ci danno i coefficienti per i due OM di legame (che indichiamo con +) e di antilegame (che indichiamo con -). c1 ( H 11 E ) c 2 ( H 12 E S ) 0 c1 ( H 21 E S ) c 2 ( H 22 E ) 0 c j j c11 c2 2 j 1 c1 ( H 11 E ) c 2 ( H 12 E S ) 0 2 c1 ( H 21 E S ) c 2 ( H 22 E ) 0 2 c j j c11 c2 2 j 1 Per esempio per la molecola HF troviamo: Momento di dipolo • Si noti che a differenza delle molecole omonucleari le molecole eteronucleari hanno l’importante proprietà del momento di dipolo diverso da zero. + + H Cl • La direzione della polarita’ del legame e’ simboleggiata dal vettore con la convenzione (positive end) (negative end) H Cl MO The molecular orbital (MO) is an important concept in chemistry, and molecular orbital theory is employed extensively to describe chemical behavior. Not only has MO theory become a ubiquitous set of tools used to explain chemical behavior, such as reactivity and kinetics, but it also provides an indispensable conceptual construct for the description of other phenomenon involving molecular electronic structure including charge transfer processes, photoexcitation, magnetism, and molecular electronics. In fact, it is quite common to extract trends in molecular behavior based on simple MO properties. For example, molecules with large HOMO-LUMO gaps are generally stable and unreactive; while those with small gaps are generally reactive. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the two most important molecular orbitals. At the Hartree-Fock (HF) level, Koopmans’ theorem suggests that the energy of the HOMO is a good approximation to the negative experimental ionization potential (-IP). Similarly, it suggests that the electron affinity (EA) for an N-electron system is equal to the negative of the LUMO energy, assuming that the orbitals do not relax.
Scaricare