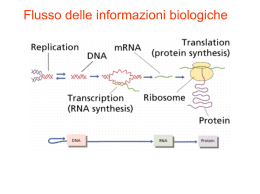
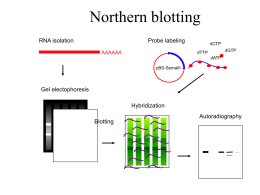
L’espressione genica e il trascrittoma Genoma Insieme delle informazioni genetiche che caratterizzano un organismo. Trascrittoma Insieme degli RNA messaggeri prodotti da una determinata popolazione cellulare. Per ogni tipo cellulare diverso sono espressi all’incirca 10000 geni diversi. Proteoma Insieme delle proteine prodotte da una determinata popolazione cellulare. Differenziamento cellulare ESPRESSIONE DEL GENOMA UMANO NELLE CELLULE DIFFERENZIATE • Tutte le cellule di un organismo hanno lo stesso corredo genomico • L’espressione genica tessuto specifica determina il fenotipo morfo-funzionale dei tipi cellulari e tissutali • In ogni cellula differenziata ed in ogni particolare momento dello sviluppo e’ attivo solo un sottoinsieme di geni In tutti gli organismi viventi le informazioni contenute nel genoma non si esprimono contemporaneamente, e sono finemente regolate Geni ad espressione costitutiva (housekeeping) Geni ad espressione condizionale (inducibili, reprimibili) Geni specializzati (tessuto-specifici, stadio-specifici, che a loro volta possono essere costitutivi o condizionali) REGOLAZIONE DELL’ESPRESSIONE GENICA • Puo’ agire su ciascuno dei livelli che caratterizzano il passare dell’informazione genica dal DNA alle proteine • Negli Eucarioti superiori la regolazione dell’espressione genica si svolge principalmente come controllo della trascrizione • Principali tipi di regolazione: Controllo epigenetico Controllo trascrizionale Controllo post-trascrizionale Attivazione/inattivazione dell’espressione genica negli eucarioti: • Decisioni cellulari durante lo sviluppo: ad es. differenziamento (geni accesi/spenti) • Regolazione del ciclo cellulare (attivazione e inattivazione ciclica) • Attivazione cellulare in risposta a mediatori esterni quali fattori di crescita, ormoni etc. (reversibile, rapida) “One-gene approach” Il gene di interesse e’ espresso in un tessuto o in un dato momento dello sviluppo ? Quanto e’ attivo dal punto di vista trascrizionale ? Real Time PCR PCR semiquantitativa Ibridazione DNA genico o cDNA con RNA totale o poly(A)+RNA (Northern blot) Ibridazione in situ “Large-scale approach” Quali geni sono espressi in un tessuto ed in un dato momento dello sviluppo ? Quanto ciascuno di essi e’ attivo dal punto di vista trascrizionale ? Profilo d’espressione del genoma (TRASCRITTOMA) Metodi per lo studio su larga scala dell’espressione genica Sequenziamento sistematico di ESTs da librerie di cDNA cDNA microarrays SAGE (Serial Analysis of Gene Expression) Preparazione librerie cDNA Clonati in batteri Sequenziamento librerie cDNA Il sequenziamento del DNA “codificante” si basa sulla purificazione dell'RNA messaggero da cellule o da campioni di tessuto e sulla sua retrotrascrizione in vitro in una sequenza di DNA complementare (cDNA). In genere i cDNA vengono frammentati e clonati in vettori batterici. Si ottengono in questo modo delle collezioni di batteri, nelle quali ogni colonia contiene un inserto corrispondente ad un frammento di sequenza di un gene espresso, dette librerie di cDNA. Conversione dell’mRNA in cDNA per trascrizione inversa AAAAA RT TTTTT RT AAAAA TTTTT Il primer oligo dT lega mRNA La trascrittasi inversa copia il primo strand di cDNA La RT digerisce e stacca mRNA e AAAAA RT copia il secondo TTTTT strand cDNA cDNA a doppio filamento Sequenziamento librerie cDNA Primers universali • Scoprire l’esistenza di nuovi geni • Associare l’espressione di geni a linee cellulari e tessuti diversi •Determinare la sequenza completa dei trascritti Cosa sono le Expressed Sequence Tags (EST)? sequencing sequencing cDNA 5’EST 200~500 nucleotidi 3’EST Cosa sono le Expressed Sequence Tags (EST)? Chromosome sequence Mapping back to chromosome sequence 5’EST 3’EST Cosa sono le Expressed Sequence Tags (EST)? • LeESTs sono piccoli frammenti di sequenze di DNA (200-500 nt) generati per sequenziamento di una o entrambe le estremità di un gene espresso. L’idea è sequenziare porzioni di DNA che rappresentano i geni espressi in determinate cellule, tessuti e organi da differenti organismi e usare queste “tags” per individuare un gene su una porzione di DNA cromosomico per appaiamento di basi. Identificare i geni con questo metodo può essere complicato dalla presenza di introni. Normalizzazione delle librerie di cDNA N° di copie N° di copie Al fine di trovare con la stessa probabilità sia le sequenze abbondanti che quelle rare si attua una normalizzazione delle librerie di cDNA. Per far questo si sfrutta il fatto che i cDNA più abbondanti, si appaiano o ibridizzano più rapidamente e possono essere rimossi dall’insieme di cDNA di partenza. In questo modo l’insieme rimanente si svuota delle sequenze più abbondanti ovvero si arricchisce di quelle più rare. Tipo di cDNA Tipo di cDNA Supponendo di avere il cDNA di 8 geni espressi con intensità diversa, mostriamo il grafico dell’abbondanza di copie di cDNA prima e dopo la normalizzazione della libreria. Si perdono le informazioni sul livello di espressione dei geni, si usa per scoprire nuovi geni. I microarray di cDNA Esperimenti microarray 5 fasi: • spotting del DNA sonda • preparazione cDNA target • ibridazione • lettura (SCAN) • analisi statistica e gestione dati Acquisizione immagini da microarray cDNA • Identificazione della posizione degli spot • Costruzione di un’area locale intorno ad ogni spot • Calcolo dell’intensità di ogni singolo spot (mediana dell’intensità dei pixel) • Calcolo del background locale Come si misura l’espressione dei geni? Metodo del campione di riferimento Calcolare il rapporto tra le intensità della fluorescenza, dopo adatte trasformazioni, per due campioni analizzati tramite ibridazione competitiva sullo stesso microarray. Un campione funziona come controllo, o “campione di riferimento” ed è marcato con un colorante che ha uno spettro di fluorescenza diverso dall’altro. Per convenzione una induzione (o repressione) dell’espressione genica pari a due volte il livello di espressione nel campione di riferimento indica un cambiamento significativo. Metodo del campione di riferimento = malato = sano Gene 1 Gene 2 Normalizzazione dei dati Molte variabili possono influire sui risultati è necessaria una normalizzazione dei dati per eliminare distorsioni sistematiche – efficienza diversa delle due marcature; – diverse quantità di mRNA per un canale e per l’altro (Cy3 e Cy5); – diversi parametri di scansione; – bilanciamento dei laser; – effetti di punte, effetti spaziali o di supporto. •Normalizzazione per intensita' totale •Normalizzazione con metodi di regressione •Normalizzazione con metodi di rapporto Intensità totale: assume che la quantità iniziale di mRNA sia identica nei due campioni. Le fluttuazioni sono bilanciate in modo che la quantità totale di RNA che si lega all’array per ogni campione sia la stessa. Nelle situazioni di sbilanciamento può essere calcolato un fattore di normalizzazione in grado di ricondurre alla situazione di uguale intensità totale. Metodo di regressione: Assume che usando mRNA di campioni simili, la maggior parte dei geni sia espressa allo stesso livello. In uno scatterplot i geni si raggruppano lungo una linea la cui pendenza è 1. Prevede la ricerca della migliore interpolazione con metodi di regressione Metodo del rapporto:assume che la quantità totale di RNA prodotto sia circa la stessa per geni essenziali come gli housekeeping. E’ possibile sviluppare una funzione di probabilità approssimata per il rapporto tra i due canali, utilizzata sia per normalizzare i dati sia per identificare geni espressi differenzialmente. Matrice di dati righe = espressione del singolo gene in diverse condizioni colonne = rappresentano le condizioni analizzate Ad ogni cella si assegna il valore relativo di espressione: rapporto tra l’intensità di un gene a una data condizione rispetto alla condizione standard (i dati sono trasformati come log in base2) Identificazione di geni differenzialmente espressi Un gene viene considerato differenzialmente espresso se la sua espressione genica si discosta dalla situazione di uguale espressione nei due canali in modo significativo. • metodo del valore soglia: valori > valore soglia positivo valori < valore soglia negativo sovraespressi sottoespressi il valore soglia ottimale dipende dalla qualità dei dati: usare controlli di qualità interni per determinare la soglia di confidenza. Analisi statistiche più rigorose Metodo dell’ANOVA Metodo dell’ANOVA (analisi della varianza) Sviluppata per verificare la significatività delle differenze tra le medie aritmetiche di vari gruppi. Confronto simultaneo tra due o più medie. I dati vengono trasformati in log2 e i canali normalizzati, quindi viene utilizzato il metodo dell’ANOVA: Livello di espressione standardizzato • sono necessarie numerose repliche per ogni esperimento • non c’è bisogno di un campione di riferimento 1 0 -1 1 2 3 4 Ripetizione dell’esperimento Microarray a oligonucleotidi GeneChip Affymetrix Ibridizzazione della sonda marcata Scansione del GeneChip con scanner laser Elaborazione dei dati Microarray a cDNA e a oligo: 2 tecniche a confronto Microarray a cDNA: • applicabili a qualunque organismo • più economici = più repliche • più flessibili per progettazione sperimentale • l’ibridazione è su migliaia di basi ( non decine) Microarray a oligo: • si possono analizzare un n > di geni • variabilità minore da chip a chip • non sono necessari macchinari, si possono acquistare • possono essere confrontati dati di diversi gruppi di ricerca Estrazione di dati da microarray Qual è il senso biologico dei dati?: individuare geni con profili di espressione simili e riunirli in gruppi. Il raggruppamento implica la co-regolazione, quindi i geni sono coinvolti in processi biologici simili. Oltre a descrivere la risposta dei geni ai diversi trattamenti, l’analisi dei microarray descrive i livelli di regolazione coordinata dell’espressione genica su scala genomica. Può portare a formulare ipotesi di funzione per geni sconosciuti. Estrazione di dati da microarray Metodi di clustering: sono metodi di statistica multivariata che raggruppano unità statistiche sulla base di misure di similarità/ dissimilarità. Simili rispetto a cosa ? Definizione di distanza I geni sono punti nello spazio: punti vicini nello spazio sono raggruppati insieme Distanze Distanza Euclidea: raggruppa geni che hanno andamenti simili a livelli di espressione simili. Correlazione di Pearson: raggruppa geni che hanno andamenti simili indipendentemente dal livello di espressione che hanno. Algoritmi di clustering Gli algoritmi di clustering si basano sulla misura di vicinanza scelta. Ogni algoritmo è caratterizzato dal metodo utilizzato per identificare i gruppi omogenei di elementi Gerarchici Non Gerarchici Algoritmi per il Clustering Divisivi Aggregativi Gerarchici: non necessitano di informazioni a priori (botton-up) Non-gerarchici: cercano di raggruppare gli elementi in un numero predefinito k di gruppi (top-down) Divisivi: da un unico cluster con tutti gli elementi procede dividendolo in cluster più piccoli Aggregativi: partono con tanti cluster quanti sono i geni e procedono raggruppandoli in cluster sempre più grandi Tipico algoritmo gerarchico agglomerativo • L’algoritmo è semplice 1. Calcola la matrice di distanze a coppie 2. All’inizio, ogni punto è un singolo cluster 3. Unisci i cluster più vicini 4. Aggiorna la matrice di distanze 5. Ripetere i punti precedenti fino a quando rimane un singolo cluster • L’operazione chiave è il calcolo della vicinanza tra due cluster – Questo concetto di vicinanza costituisce la differenza principale tra algoritmi differenti Esempio di calcolo clustering gerarchico Come calcolo le nuove distanze? 3 metodi: Legame semplice Legame completo Legame intermedio Algoritmi non-gerarchici Cercano di raggruppare gli elementi in modo tale che siano il più possibile omogenei all’interno dei cluster e il più possibile disomogenei tra i vari cluster k-means Procedura iterativa: 1. Scegli un numero di classi 2. Assegna gli oggetti alle classi (a caso o in base ad un’altra classificazione) 3. Sposta gli oggetti nelle classi il cui centroide è più vicino (la varianza intra-classe diminuisce) 4. Ripeti lo step 3 finchè non c’è più nessun cambiamento nella composizione delle classi Risultati del clustering gerarchico Analisi Componenti Principali (PCA) La PCA è una tecnica per la riduzione del numero di variabili casuali che descrivono un fenomeno. L’obiettivo e’ quello di identificare un sottoinsieme di variabili casuali dalle quali dipende la maggiore varianza (‘variabilità’) del fenomeno OBIETTIVI • Ridurre la dimensionalità di un dataset, composto da p variabili tra loro correlate; • Trovare relazioni non precedentemente sospettate tra le variabili. Determinazione abbondanza assoluta La tecnica dei microarray non fornisce dati sui livelli assoluti di espressione: un metodo per determinare l’abbondanza assoluta di ciascun trascritto espresso in una data popolazione di cellule è l’analisi seriale dell’espressione genica (SAGE, serial analysis of gene expression) SAGE è un metodo sperimentale ideato per utilizzare i vantaggi del sequenziamento su larga scala per avere informazioni quantitative di espressione genica (Velculescu et al. 1995, Zhang et al, 1997) Il metodo non è influenzato da fattori come i campioni di riferimento, gli artefatti di ibridazione o la frequenza dei cloni e fornisce una misura precisa del vero numero trascritti per ogni cellula. E’ un metodo molto costoso e non consente ripetizioni di esperimenti. Sintesi DNA a doppia elica a partire dai messaggeri con primer oligo(dT) biotinilato Taglio con enzima di restrizione ed isolamento della porzione 3’ del cDNA per purificazione mediante sfere a streptavidina Separazione del cDNA in 2 aliquote, ciascuna ligata con un linker diverso, contenente un sito di taglio per un enzima di restrizione (tagging enzyme) che taglia ad una distanza definita dal sito riconociuto (20bp) Il linker con attaccato un breve tratto di cDNA (9-12 bp) viene rilasciato Ligazione tags a due a due, taglio ditags in modo da creare estremita’ coesive Analisi automatizzata dei risultati: identificazione di tutte le specie di tags, conteggio della frequenza di ciascuna, assegnazione a sequenze geniche note ed annotazione Clonaggio dei concatameri e sequenziamento Le tag possono essere unite insieme in serie, a costituire lunghe molecole di DNA, che vengono clonate e sequenziate in modo automatizzato Isolamento delle “tag” Ligazione Sequenziamento Livellodi espressione Quantificazione di ciascuna “tag” e determinazione del pattern di espressione GENE GENE Normale Normale GENE GENE Alterato Alterato Il risultato della SAGE e’ di tipo digitale: una lista di tags e la frequenza di ciascuna di esse La fase in cui si stabilisce la corrispondenza tra tag e gene e’ cruciale per una corretta stima del livello di espressione del gene La corrispondenza tag-gene non e’ sempre biunivoca,come ci si aspetterebbe Gli errori di sequenziamento hanno effetti molto pesanti sui dati SAGE (1% 10% che ci sia almeno 1 errore su 10 bp) Le assegnazioni tag/EST sono affette da un errore maggiore La tecnica consiste nel sequenziamento da messaggeri cellulari di brevi oligonucleotidi, che fungono da etichette di sequenza (TAG). Il numero di volte in cui una singola “tag” viene osservata permette di quantificare l’abbondanza del messaggero identificato nella popolazione dei messaggeri e, indirettamente il livello di espressione del gene corrispondente Tecnologia basata su MICROSFERE Metodo MegaCloneTM Permette un clonaggio parallelo in un sistema acellulare di centinaia di migliaia di cloni genomici o do DNA. • una tag (etichetta) è legata all’estremità 3’ di ciascun frammento di DNA (cDNA) e la sequenza è amplificata mediante PCR • gli amplificati vengono legati su microsfere mediante ibridazione con la sequenza complementare (anti-tag) legata con legami covalenti alla microsfera • le sequenze vengono separate per citometria di flusso e clonate e sequenziate oppure sequenziate con il metodo del sequenziamento su larga scala con contrassegni in parallelo (MPSS) • negli studi comparativi è possibile separare le microsfere in base all’ abbondanza dei trascritti Clonaggio in parallelo con l’uso di TAGs I. Costruzione della library Brenner et al., PNAS 97:1665-70. cDNA TTTTTTT TTTTTTTT AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA TTTTTTT TTTTTTTT AAAAAAA AAAAAAA AAAAAAA TTTTTTTT AAAAAAA AAAAAAA TTTTTTTTT AAAAAAA TTTTTTTT GATC GATC TTTTTTTT AAAAAAA AAAAAAA TTTTTTTTTT AAAAAAA AAAAAAA AAAAAAA AAAAAAA Ogni microsfera contiene il prodotto derivato dal terminale 3’di un singolo trascritto 1) Marcatura con Tag 2) Amplificazione tramite PCR AAAAA TTTTT 3) Ibridazione con microsfera III. Sequenziamento diretto II. Separazione per citometria a flusso + NNNN 4321 Brenner et al., Nat. Biotech. 18:630-4. NNNX RS CODEX1 NNXN RS CODEX2 NXNN RS CODEX3 XNNN RS CODEX4 2) Sequence by hybridization 16 cycles for 4 bp Caso studio sull’utilizzo dei microarray Oltre a costruire atlanti di espressione genica, molti studi del trascrittoma hanno iniziato a identificare le differenze di espressione genica nelle cellule tumorali e quelle associate ad altre malattie umane. Gli scopi di questi studi sono: • ottenere una migliore classificazione dei tipi di tumori e identificare i tipi cellulari da cui i tumori provengono • caratterizzare i profili di espressione che possono aiutare a prevedere la risposta terapeutica • raggruppare i geni per formulare ipotesi riguardanti il loro meccanismo di azione nella cancerogenesi • identificare nuovi bersagli genici per la chemioterapia Ross et al. 2000 : tipi di tumori simili tra loro tendono a condividere profili di espressione genica, in parte correlati alle differenze caratteristiche del loro tessuto di origine. Hanno inoltre identificato marcatori che possono rivelarsi utili nella diagnosi clinica e suggeriscono funzioni per geni non ancora caratterizzati. Scherf et al. 2000: l’esposizione di linee cellulari tumorali a oltre 70000 composti ha permesso di ottenere una classificazione in parallelo dei tipi di tumore in base all’attività di inibizione da parte del farmaco. Profili di espressione in risposta ai farmaci 3 analisi di clustering di tipo gerarchico: • 1 cluster per valutare il livello di espressione genica • 1 cluster per valutare la sensibilità ad un gruppo di farmaci • 1 cluster per valutare la correlazione tra il livello di espressione genica e la sensibilità ai farmaci Questo tipo di analisi consente di identificare i geni candidati coinvolti nella risposta ai farmaci. Marcatori di prognosi Con i microarray è possibile prevedere la mortalità o la risposta terapeutica delle leucemie. Alizadeh et al 2000: identificazione dei profili di espressione che raggruppano le leucemie in gruppi correlati con la prognosi a lungo termine. Variabilità di espressione di circa 8000 geni unici tra 60 linee cellulari provenienti dal National Cancer Institute Analisi del pattern di espressione genica e la loro relazione con le proprietà fenotipiche di 60 linee cellulari METODI • 9703 cDNA umani che includono 8000 geni differenti • campione di riferimento mRNA da 12 linee cellulari • la variazione in espressione si ottiene normalizzando il rapporto Cy5/Cy3 • algoritmo di clustering gerarchico e matrice di visualizzazione • l’obiettivo è raggruppare linee cellulari con repertori simili di geni espressi e raggruppare quei geni i cui livelli di espressione variano in modo simile tra le 60 linee cellulari • campioni in triplicato per valutare la varianza delle analisi • analisi di clustering effettuata due volte usando sotto-gruppi di geni per valutare la robustezza dell’analisi Pattern di espressione relativo al tessuto di origine • 1161 cDNA che variavano di almeno 7 volte rispetto al riferimento • nella matrice le righe rappresentano i livelli di espressione aggiustati sulla media, le colonne le linee cellulari • linee cellulari che hanno origine dallo stesso tessuto raggruppano insieme • le linee cellulari del carcinoma del polmone e del tumore al seno presentano patterns più eterogenei Pattern di espressione relativo ad altri fenotipi cellulari • 6831 con le misurazioni più attendibili nel set di riferimento • i tre cluster d, e, f sono arricchiti con geni con variazione dei livelli di espressione correlata con il tasso di proliferazione della linea cellulare • i geni ridondanti clusterizzano insieme confermando la riproducibilità e consistenza delle misurazioni • l’elevata espressione di geni coinvolti nel metabolismo dei farmaci può riflettere una selezione per la resistenza ai chemioterapici Clusters genici relativi alle caratteristiche del tessuto nelle linee cellulari a geni altamente espressi nelle linee cellulari derivate da leucemia b cluster di geni espressi nel colon e seno, moderatamente espressi ovaio e polmone c cluster di geni espressi nelle linee del melanoma d geni altamente espressi in tutti i glioblastoma: la > parte derivano dal carcinoma renale Confronto espressione genica campioni clinici di tumore al seno e colture di linee cellulari (tumore al seno e leucemia) Confronto del pattern di espressione di due biopsie di cancro al seno con con un campione di tessuto normale e le linee cellulari derivate da tumore al seno e leucemia. Il tumore al seno ha una complessa organizzazione istologica. L’analisi ha permesso di individuare il contributo di ogni tipo cellulare che costituisce la struttura della ghiandola. Caratteristiche istologiche delle biopsie le linee cellulari hanno espressione > di geni del cluster di proliferazione dovuto alla coltivazione in vitro CONCLUSIONI • Microarray a cDNA - costruzione delle librerie Analisi di clustering - normalizzazione • Microaray a oligonucleotidi - creazione dei genechip • metodi per valutare l’abbondanza assoluta - metodo SAGE - tecnica delle microsfere
Scaricare

![Alimentazione, squilibrio emozionale e s[...]](http://s2.diazilla.com/store/data/000199131_1-4df9f30b101216c63cae2f89e397f485-260x520.png)