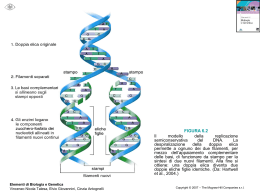
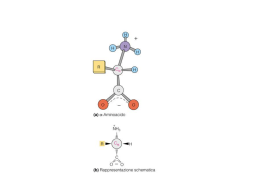
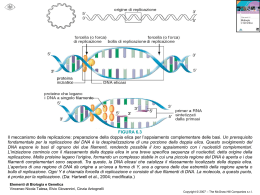
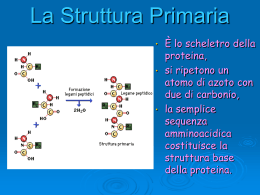
La funzione delle proteine dipende dalla loro struttura tridimensionale • La struttura dipende dal ripiegamento di particolari sequenze aminoacidiche • La sequenza aminoacidica della catena polipeptidica è la struttura primaria • Regioni diverse della sequenza formano strutture secondarie regolari (alfa eliche e filamenti beta) • Gli elementi di struttura secondaria si dispongono secondo semplici motivi strutturali. La struttura terziaria risulta dalla associazione di più motivi strutturali in una o più unità dette “domini”. • La proteina può essere formata da più catene polipeptidiche disposte in una struttura quaternaria Elementi strutturali delle proteine • L’interno delle molecole proteiche contiene soprattutto catene laterali idrofobe disposte in strutture secondarie che permettono di neutralizzare la polarità dei loro gruppi polari mediante la formazione di legami idrogeno. • Esistono 2 tipi principali di struttura secondaria: αeliche e foglietti-β (che costituisocno le regioni del core), e sono collegate da regioni loop presenti alla superficie Gli elementi della costruzione α-elica - Presenti nelle proteine quando una stringa di residui consecutivi presentano tutti coppie di angoli diedri Φ e ψ con valori compresi tra -60° e -50°. -Presenta 3,6 residui per spira -Stabilizzata da legami idrogeno che si formano tra il gruppo C=O di un residuo n ed il gruppo NH del residuo n+4. In questo modo tutti i gruppi NH e CO della catena principale sono uniti da legami idrogeno. -L’elica è sempre destrorsa Gli elementi della costruzione foglietto-β Rossiossigeno Bluazoto Biancoidrogeno NeroCα cat princ Violaposizione cat lat -Costituiti da filamenti β che possono essere tutti antiparalleli oppure paralleli tra loro. -Nel foglietto β i filamenti si trovano allineati giacendo uno accanto all’altro in modo da formare legami idrogeno tra i gruppi C=O di un filamento β ed i gruppi NH di un filamento adiacente I filamenti beta antiparalleli sono tra gli elementi a più alta stabilità foglietto β antiparallelo (direzioni opposte NC CN) foglietto β parallelo (medesima direzione biochimica NC) Gli elementi della costruzione loop -tratti di catena apparentemente disorganizzati, di lunghezza variabile -fanno da collegamento fra α-eliche o filamenti β -partecipano spesso alla formazione di siti di legame (come i loop degli anticorpi) o del sito attivo degli enzimi -si trovano alla superficie della molecola I diagrammi di topologia Topologia come gli elementi di struttura secondaria sono connesi tra loro utili per rappresentare le connessioni tra elementi di struttura secondaria aspartato carbamiltransferasi flavodossina plastocianina Gli elementi di struttura secondaria sono collegati a formare semplici motivi strutturali • MOTIVO STRUTTURALE semplice combinazione di pochi elementi fondamentali di struttura secondaria • Motivi strutturali semplici possono combinarsi a formare motivi complessi • Combinazioni di piccoli motivi strutturali costituiscono il core dei domini (unità fondamentali della struttura terziaria) I 4 motivi strutturali più ricorrenti Elica-loop-elica Forcina β Chiave greca β-α-β La combinazione dei motivi strutturali semplici dà luogo a strutture complesse Motivo Elica-loop-elica motivo strutturale specifico per il legame del DNA motivo strutturale specifico per il legame del calcio (parvalbuina,calmodulina, troponinaC) Il motivo “EF hand” -Comprende 2 alfa-eliche:E ed F, che affiancano il loop: 5 residui del loop legano il calcio quindi la loro cat lat deve contenere un atomo di ossigeno Elica E direzione che parte dalla base dell’indice Dito medio regione loop di 12 residui che lega il calcio Elica F diretta all’estremità del pollice -Residuo num.6 del loop è sempre glicina (poco ingombrante) La mano destra simboleggia il motivo di legame al calcio Sequenze consenso dei motivi EF in 3 proteine diverse (in arancio aa deputati al legame con il calcio; In verdeaa con cat lat idrofobe destinate a formare core idrofobo tra le alfa-eliche) Struttura della troponina C con i suoi motivi EF -E’ costituita da 4 motivi EF hand di cui 2 legano il calcio (sfere rosa) -L’atomo di calcio è legato attraverso 6 atomi di ossigeno, dei quali tre proviengono dalla cat lat di Asp (D9), Asn (N11) e Asp (D13), due sono forniti dalla cat lat di Glu (E20), ed uno dal legame peptidico del residuo 15. -Al calcio è legata anche una molecola d’acqua. Quindi questo motivo, per espletare la sua funzione, necessita sia di una specifica conform della catena principale sia della presenza di un peculiare raggruppamento di catene laterali. Il motivo elica-loop-elica fornisce l’impalcatura che ancora i ligandi degli ioni calcio in posizione corretta per poter legare e liberare questi ultimi motivo elica-loop-elica legante il DNA Motivo elica-loop-elica legante il DNA presente nella proteina Cro del fago λ…. …legato al DNA Forcine β forcina β come foglietto isolato forcina β come parte di un foglietto più complesso Motivo a greca -Quando 4 filamenti beta antiparalleli si dispongono secondo un modello simile all’unità ripetitiva di un tipico disegno ornamentale caratteristico dell’arte dell’antica Grecia, noto come “Greca”. -Si basa sull’iniziale formazione di una lunga struttura antiparallela in cui i loop sono presenti al centro di entrambi i filamenti beta. - Il motivo a greca si originerebbe in conseguenza di cambiamenti strutturali nelle regioni loop interposte tra i filamenti beta 1 e 2 e nelle regioni interposte tra i filamenti 3 e 4. Per effetto di ciò, la struttura si ripiega verso il basso ed il filamnto beta 2 va ad associarsi col filamento 1 formando con questo legami idrogeno. Ipotetica modalità di ripiegamento di struttura a forcina che porta alla formazione del motivo a greca Motivo a greca a) diagramma topologico della greca b)motivo a greca presente . nell’enzima nucleasi Staphylococcus Il motivo a greca fornisce un semplice modo di collegare filamenti beta antiparalleli che si trovino su lati opposti di una struttura a botte Motivo β-α-β a) Schema del percorso della catena principale b) Diagrammi topologici del motivo β-α-β -Permette il collegamento tra filamenti beta paralleli. -Consiste di 2 fil beta paralleli, un’alfa elica e due regioni loop. -L’alfa elica collega l’estremità carbossilica di un filamento beta con quella aminica del filamento beta successivo -Il loop che collega il terminale carbossilico del filamento beta con quello aminico dell’alfa elica spesso è implicato nella formazione del sito funzionale Motivo β-α-β a) Modalità di collegamento destrorsa b) Collegamento di tipo sinistrorso , In linea di principio questo motivo può presentare due diverse disposizioni: una in cui l’elica si trova al di sopra del piano della pagina (“connessione destrorsa” perché presenta il medesimo orientamento di una alfa elica destrorsa); ed una in cui l’elica si trova al di sotto del piano della pagina (sinistrorsa). In realtà la connessione è sempre destrorsa, con l’unica eccezione della subtilisina. I DOMINI Strutture alfa elica superavvolta fascio a 4 eliche ripiegamento della globina Strutture alfa-beta TIM barrel foglietto alfa- beta aperto ferro di cavallo Strutture beta antiparallele Beta barrel greca Up-and-down Jelly roll Strutture beta parallele Elica beta a 2 foglietti Elica beta a 3 foglietti Il dominio è • unità compatta e semi-independente (Richardson, 1981). • unità stabile di una proteina che può ripiegarsi autonomamente (Wetlaufer, ti1973). • modulo funzionale ed evoluzionistico ricorrente (Bork, 1992) icazione dei domini è essenziale: • I domini hanno dimensioni variabili. Circa il 50% delle proteine studiate hanno domini compresi fra 51 e 150 amminoacidi. Sono note proteine con molti domini (13) e le interazioni interdominio sono le stesse che stabilizzano le loro strutture interne. • i cuori idrofobici sono fondamentali per la stabilità dei domini (nelle sezioni proteiche i residui idrofobici sono in giallo) lisozima da Gallus gallus pdb: 1hel esochinasi da Homo sapiens pdb: 1hkc Non tutti i domini sono costituiti da tratti continui di polipeptide. In alcune proteine un dominio è interrotto da un tratto di sequenza che si ripiega in un dominio separato, dopo il quale continua il dominio originale. alanina racemasi da Bacillus stearothermophilus pdb: 1sft Le proteine multidominio si sono probabilmente evolute dalla fusione dei geni che un tempo codificavano per proteine separate. tioesterasi da E. coli (pdb: 1c8u) 2 domini fusi tioestere deidratasi da E. coli (pdb: 1mkb) omodimero con ciascun monomero simile ai domini della tioesterasi Il numero dei fold (motivi strutturali) proteici è vasto ma limitato. triptofano sintasi da Salmonella typhimurium α-galattosidasi da Homo sapiens Elica superavvolta Struttura ad elica superavvolta. Un’alfa elica isolata presenta una ridotta stabilità in soluzione. Perciò, nelle proteine, le alfa eliche sono stabilizzate dal reciproco impaccamento mediato dall’interazione delle catene idrofobe. Il modo più semplice di realizzare tale stabilizzazione è quello di associare 2 alfa-eliche, in modo da formare una struttura superavvolta. Una strutura superavvolta sinistrorsa formata da 2 alfa eliche destrorse riduce il numero di residui per giro in ognuna delle eliche costitutive da 3,6 a 3,5, in modo che le interazioni tra le catene laterali delle 2 eliche vengono a ripetersi ogni 7 residui. Ciò si riflette nelle sequenze aminoacidiche delle due catene, le quali risultano ripetitive, con un periodo di ripetitività di 7 residui. Elica superavvolta I residui aminoacidici nell’unità ripetitiva vengono indicati come “a-g” ed il residuo “d” è idrofobo (di solito è una leucina). La regione idrofoba tra le due alfa eliche è completata dai residui “a” che sono idrofobi anch’essi. Ripetitività degli aa in un’alfa elica superavvolta: un residuo di leucina si ripete ogni 7 aminoacidi Impaccamento delle catene laterali idrofobe tra 2 alfa-eliche in una struttura superavvolta Ruolo delle interazioni elettrostatiche a) Visione schematica dall’alto di una ripetizione di sette aa. b) Visione schematica laterale di una struttura superavvolta I residui “e” e “g” che orlano il core idrofobo sono carichi e le loro catene forniscono interazioni ioniche (ponti salini) tra le α eliche. Fascio a 4 eliche -Due alfa eliche impaccate insieme in una struttura superavvolta sono elementi costitutivi di un dominio ma non sono sufficienti a formare un dominio completo. -Infatti, il dominio ad alfa elica più semplice consiste in 4 alfa eliche disposte in un fascio con l’asse di ognuna parallelo a quelli delle altre. -Le alfa eliche sono disposte in modo tale che le eliche consecutive nella sequenza aminoacidica vengono ad essere adiacenti. Dispiegamento della catena polipeptidica in un dominio costituito da un fascio di 4 eliche Fascio a 4 eliche Proiezione del motivo su un piano perpendicolare all’asse del fascio Le catene laterali di ciascuna elica sono disposte in modo tale che le catene laterali idrofobe si trovano comprese nello spazio tra le eliche, mentre le catene laterali idrofile vengono a trovarsi alla superficie del fascio. In questo modo, nella parte centrale del fascio viene a formarsi un core idrofobo in cui le catene laterali si trovano ammassate così fittamente da escludere la presenza di molecole d’acqua. Cerchi grandi catena principale delle alfa–eliche Cerchi piccoli catene laterali: verdi (catene laterali idrofobe ammassate); rosse (cat lat idrofile che si trovano esposte alla superficie del fascio) Fasci a 4 eliche come singolo dominio di proteine monomeriche, o come motivo di dimerizzazione Il Ripiegamento della globina Fascio di 8 eliche Collegate da brevi loop Formazione di una tasca idrofobica in cui si trova il sito attivo Strutture a dominio alfa-beta (foglietto β centrale di tipo parallelo o misto circondato da α eliche) Modalità di connessione β-α-β Le tre strutture alfa-beta sono tutte costituite da motivi β-α-β collegati tra loro in modo che i filamenti β vengono ad essere paralleli. 2 motivi β-α-β si possono collegare in 2 diversi modi: a) le alfa eliche si trovano tutte sul medesimo piano del foglietto (TIM barrel e ferro di cavallo); b) le alfa eliche si trovano sul piano opposto (foglietti beta aperti) TIM Barrel Gli 8 filamenti beta racchiudono un core idrofobo impaccato costituito dalle catene laterali dei residui presenti in filamenti beta alternati. Il core è disposto a formare tre strati, dove ogni strato contiene 4 catene laterali provenienti dai residui presenti in filamenti beta alternati. Struttura formata da 8 filamenti beta paralleli Filamento 8 forma legami idrogeno col filamento1 Per formarla sono necessari almeno 200 aa Presente in molti enzimi Filamenti beta ed alfa eliche impalcatua strutturale Regioni loop siti attivi – Rappresentazione vista dall’altodella strutturaa botte del sito attivo dell’enzima RuBisCo. Il sito di legame per il substrato (in rosso) è generato da numerose catene laterali cariche (in blu) presenti su differenti loop. Nel TIM barrel il sito attivo si trova in una tasca formata dalle regioni loop che collegano le estremità carbossiliche dei filamenti β con le adiacenti α-eliche. La Piruvato chinasi contiene diversi domini, uno dei quali con struttura TIM barrel • La struttura di questo enzima illustra perfettamente in che modo una lunga catena polipeptidica può ripiegarsi a formare domini di tipo diverso dal punto di vista strutturale. • La funzione enzimatica risulta sempre associata con il dominio che presenta struttura a botte. Il foglietto alfa-beta aperto Nei domini con struttura α/β aperta, il sito attivo si trova in una fessura localizzata esternamente all’estremità carbossilica dei filamenti β. Questa fessura è formata da 2 regioni loop adiacenti che collegano i due filamenti con α eliche presenti su facce opposte del foglietto β. Ciò è illustrato dalle dita curvate delle due mani, in cui la metà superiore delle dita rappresenta le regioni loop mentre la metà inferiore rappresenta i filamenti β. Il cilindro rappresenta un ligando posizionato nella fessura di legame. Esempi di tipi diversi di strutture α/β aperta flavodossina e adenilato chinasi esochinasi e fosfoglicerato mutasi Ripiegamento a Ferro di cavallo Struttura dell’inibitore della ribonucleasi, Ruolo dei residui conservati di leucina dei costituita da motivi β-loop-α ripetuti, motivi ricchi di leucina nella ricorda un ferro di cavallo formato stabilizzazione del modulo strutturale all’interno da un foglietto β parallelo a β-loop- α 17 filamenti e all’esterno da 16 α eliche. Il beta Barrel • Comprende enzimi, proteine di trasporto, anticorpi, proteine virali di rivestimento. • Core: costituito da filamenti β (da 4-5 a più di 10), disposti in modo antiparallelo a formare 2 foglietti β collegati ed impaccati l’uno con l’altro. • Le strutture β antiparallele presentano un core di catene laterali idrofobe all’interno di una struttura a botte, formato dalle catene laterali dei residui presenti nei filamenti β , mentre la superficie è costituita da residui provenienti sia dai filamenti β che da regioni loop β La maggior parte delle strutture rientra in pochi gruppi con topologia uguale o simile motivi a greca up and down Ogni filamento β è collegato al successivo attraverso una breve regione loop Quando in una struttura a β barrel a 8 filamenti, il numero n è collegato al filamento n+3 jelly roll La catena polipeptidica si avviluppa attorno ad un’ideale struttura a botte centrale come un dolce arrotolato coperto di gelatina Un esempio di struttura a β-barrel greca: la SOD 8 filamenti β antiparalleli disposti attorno alla superficie di una botte ideale. La struttura, a " β -barrel" , evidenzia un motivo topologico a “greca”, comune a molti sistemi biologici di grande interesse, tra cui, ad esempio, le immunoglobuline. RBP lega il retinolo all’interno di una struttura a botte up and down 8 filamenti β antiparalleli ruotati e ripiegati in modo che la struttura può anche essere vista come 2 foglietti β (verde e blu) impaccati l’uno contro l’altro. Alcuni dei filamenti β ruoati (in rosso) partecipano alla formazione di entrambi i foglietti β. La vitamina A si trova legata all’interno della struttura a botte tra i 2 foglietti β, in modo da esporre alla superficie della proteina l’unica porzione idrofila della molecola (coda contenente un gruppo OH) La neuraminidasi si ripiega a formare foglietti beta up and down Presente nel virus dell’influenza Catalizza idrolisi dell’acido sialico Omotetramero costituito da 4 catene polipeptidiche identiche, ognuna di 470 aa Ogni monomero è ripiegato a formare una superstruttura a botte costituita da 24 filamenti β disposti a formare 6 motivi strutturali simili, ognuno dei quali contiene 4 filamenti β che rappresentano le pale della struttura a forma di elica Nella neuraminidasi i motivi strutturali generati col ripiegamento formano una superstruttura a forma di elica Tetramero: 4 domini ognuno con struttura ad elica a 6 pale Monomero: (Dominio:elica a 6 pale) Topologia del monomero Motivo: (foglietto β:pala dell’elica) Ognuna delle 4 subunità del tetramero è ripiegata a formare un singolo dominio costituito da 6 motivi strutturali simili strettamente impaccati. Tale motivo strutturale è un foglietto β a 4 filamenti antiparalleli up-and-down. Questi 6 foglietti β sono disposti in modo da formare le sei lame di un’elica a 6 pale La regione strutturale e la regione del sito attivo risultano ben separate. In conseguenza della simmetria senaria dei 6 motivi strutturali a foglietti β, le 12 regioni loop derivate dai foglietti si trovano dalla stessa parte della molecola Le regioni loop che collegano i motivi strutturali formano un’ampia tasca ad imbuto contenente il sito attivo I filamenti beta rappresentano lo scheletro strutturale sul quale è incernierato il sito attivo costituito da loop di connessione tra un elemento e un altro elemento L’emoagglutinina si ripiega a formare un motivo strutturale a jelly roll Formata da 3 subunità, ognuna delle quali è ancorata alla membrana pericapsidica del virus dell’influenza. Le teste globulari contengono i siti per i recettori che legano i residui di acido sialico presenti alla superficie delle cellule eucariotiche Singola subunità dell’emoagglutinina del virus dell’influenza Il sito di legame per il recettore è formato dal dominio jelly roll La testa globulare di ogni subunità dell’emoagglutinina ha una struttura a botte jelly roll distorta. Il sito di legame è localizzato all’apice della subunità, all’interno della struttura a botte jelly roll. Domini ad eliche β parallele • Scoperta nel 1993 in California • Presente in molte proteasi batteriche extracellulari e nella proteina della coda del batteriofago P22 • La catena polipeptidica risulta ripiegata a formare un ampio superavvolgimento ad elica formato da filamenti β separati da regioni loop • I filamenti β si allineano a formare 2 o 3 foglietti β paralleli che racchiudono un core riempito dagli atomi delle catene laterali. Eliche beta parallele a 2 foglietti Elica β a 2 foglietti dove sono mostrati 3 superavvolgimenti completi dell’elica. - Ogni unità strutturale è costituita da 18 residui che formano una struttura β-loop-β. - Ogni regione loop contiene sei residui di sequenza Gly-Gly-X-Gly-X-Asp-H-U-X dove U è un aminoacido con catena ingombrante e idrofobica, spesso una leucina. - Ioni calcio sono legati ad entrambe le regioni loop, stabilizzandole. Eliche beta parallele a 3 foglietti - 2 dei foglietti β sono paralleli l’un l’altro e perpendicolari al terzo. - Ogni giro dell’elica contiene 3 brevi filamenti β ognuno di 3-5 residui collegati da 3 regioni loop. - Uno dei loop è sempre costiuito solo da 2 residui, mentre gli altri 2 sono più lunghi e variano in dimensioni. -Quindi l’elica β comprende comprende 3 foglietti β paralleli grossolanamente disposti come le tre facce di un prisma Struttura della pectato liasi C http://www.biochem.ucl.ac.uk/bsm/cath • Banca Dati secondaria (viene fatta prima un’analisi, una selezione ed infine viene eseguito l’immagazzinamento dei dati) • Classificazione delle proteine in base a similarità di sequenza e strutturali • La funzione non è presa in considerazione in questa banca dati Livelli di classificazione • Class (α, β, mixed α-β, low 2ndry structure content) • Architecture: stesso arrangiamento spaziale tra le strutture secondarie, ma connessioni diverse • Topology: stesso fold • Homologous superfamily: proteine con probabile ancestore comune • Sequence Family: proteine con identità di sequenza (id >= 35%) e funzionalità CLASS (C-level) • Assegnata in modo automatico • Class : secondary structure composition and contacts – Class1 : Mainly Alpha – Class2 : Mainly Beta – Class3 : Mixed Alpha- Beta – Class4 : Few Secondary Structures Class1 Class2 Class3 Class4 Architecture (A-level) -forma generale del dominio -descrizione dell’arrangiamento della struttura secondaria indipendentemente dalle connessioni - Effettuata manualmente Topology (T-level) Proteine che hanno la stessa topologia hanno fold simile e core abbastanza conservato, e quindi hanno strutture che sono simili ma con funzioni diverse. prende in considerazione le connessioni tra elementi di struttura secondaria, ovvero le connessioni tra motivi strutturali Topology Il panorama non è identicamente rappresentato Metodi di classificazione -avviene in maniera gerarchica, così che ogni proteina è riconosciuta attraverso un numero. -es: 1.10.490.20 vuol dire che la proteina appartiene alla Classe 1, Architettura 10, Topologia 490 e Homologia 20. -ad ogni numero corrisponde una ed una sola proteina Criteri di classificazione Step 1 : si selezionano serie di strutture su PDB (risolte per diffrazione o NMR a risoluzione di almeno 3.0 Å) Step 2: Comparazione di sequenze: proteine con identità di sequenza >35% vengono messe a livello S Step 3: dividere proteine in domini per poi analizzarle singolarmente. L’assegnamento della classe è automatico perché utilizza una procedura che esamina la composizione della struttura secondaria analizzando il valore degli angoli Φ e ψ . Step 4: Comparazione di struttura per definire H e T in maniera automatica utilizzando il programma SSAP SSAP (Sequence Structure Alignement Program) • Programma di comparazione strutturale • Compara distanze tra residui in modo sequenziale. • Il parametro utilizzato per la classificazione è il numero S che è proporzionale all’ ’inverso della sommatoria di queste differenze. Tanto è più piccola questa differenza tanto più saranno simili le strutture e tanto più S sarà grande. Se S è uguale a 100 le strutture sono completamente identiche. • La soglia è S=70 per il livello T, e di S= 80 per il livello H. Quindi tra 70 e 80 la proteina viene classificata nel livello T o da 80 in su viene classificata nel livello H. • Codice PDB • Codice CATH • Parole chiave • Accesso tramite link da altri database (es. PDB)
Scaricare