PERCORSO CONDIVISO TRA ASL DI
MILANO E STRUTTURE SANITARIE
DEL TERRITORIO PER L’UTILIZZO
DELLE ERITROPOIETINE IN AMBITO
NEFROLOGICO CON PARTICOLARE
RIFERIMENTO ALL’IMPIEGO DEI
BIOSIMILARI
1
PREMESSA
I medicinali biologici
I medicinali biologici presentano caratteristiche particolari dovuti all’origine delle loro materie prime, che può
essere ad esempio umana, animale o da colture cellulari. La particolare complessità dei processi di produzione
determina procedure specifiche di controllo per la caratterizzazione e per la determinazione della loro qualità
(esami fisico-chimico-biologici e indicazioni sul processo di produzione e il suo controllo).
I medicinali biologici si differenziano per molti aspetti dai medicinali tradizionali: per esempio le tecniche di
produzione sono più complesse, la dimensione e la complessità molecolare sono maggiori.
I medicinali tradizionali sono infatti normalmente prodotti tramite sintesi chimica e hanno generalmente strutture
chimiche ben definite che possono di norma essere analizzati per individuare tutti i diversi componenti.
La maggior parte dei medicinali biologici viene invece prodotta in sistemi viventi come microrganismi o cellule
animali, e purificata attraverso un complesso processo produttivo
Questo comporta una intrinseca variabilità e i medicinali biologici sono combinazioni di molte forme diverse della
stessa proteina. Un'altra fonte di variabilità in alcuni medicinali biologici è il tipo e la lunghezza del complesso di
zuccheri o carboidrati attaccato allo scheletro della proteina (glicosilazione). .
Questa variabilità intrinseca delle molecole è strettamente controllata dai produttori e dalle autorità regolatorie e
deve rimanere entro limiti concordati e predefiniti. I processi produttivi per i medicinali biologici sono infatti
processi altamente sensibili, ed è fondamentale che vengano controllati accuratamente per ottenere risultati
costanti e garantire la sicurezza e l'efficacia del prodotto finale.
I produttori e gli importatori dei medicinali biologici approvati nell'Unione Europea, sono tenuti a possedere
un'autorizzazione a produrre ed importare (MIA) e un certificato GMP (Good Manufacturing Practices) valido,
rilasciato da un'autorità nazionale competente di uno Stato Membro dell'Unione Europea. Essi sono concessi solo
se il sito di produzione/importazione è conforme alle linee guida europee al riguardo e vengono effettuate anche
regolari ispezioni da parte delle autorità di controllo, sia sui produttori UE che su quelli con sede al di fuori
dell'UE che esportano nell'UE.
Le proteine, se assunte per via orale, subiscono l’azione del sistema digerente, di conseguenza, la maggior parte
dei medicinali biologici devono essere somministrata per via iniettiva o infusionale e possono essere
personalizzati per colpire il bersaglio desiderato.
I medicinali biologici sono generalmente più costosi dei medicinali tradizionali e la gestione del loro utilizzo
assorbe molte risorse economiche in quanto il loro impatto sulla spesa sanitaria è aumentato nel corso degli anni.
Esempi di medicinali biologici sono ormoni della crescita, insuline, eritropoietine, enzimi prodotti naturalmente nel
corpo umano, o anticorpi monoclonali, ma anche emoderivati, medicinali immunologici quali sieri e vaccini,
allergeni e prodotti di tecnologie avanzate come quelli utilizzati nelle terapie cellulari e genetiche
2
I medicinali biotecnologici
La biotecnologia utilizza sistemi viventi (cellule vegetali o animali, batteri, virus e lieviti) e tecnologie moderne
(cellule geneticamente modificate) per produrre medicinali biologici per il trattamento delle malattie e dei disturbi
genetici nell'uomo.
Le biotecnologie hanno negli ultimi anni reso possibile lo sviluppo di trattamenti per molte gravi malattie. Ad
esempio soggetti diabetici, oncologici, colpiti da attacchi cardiaci, affetti da sclerosi multipla o da artrite
reumatoide e malattie autoimmuni traggono beneficio da terapie con medicinali biologici approvati.
Ogni produttore ha le proprie linee cellulari uniche, e sviluppa i propri processi produttivi esclusivi (unici).
I primi medicinali biologici prodotti con tecniche di DNA ricombinante sono stati approvati negli anni Ottanta,
quindi i primi diritti di esclusiva (brevetti e altri tipi di protezione dei dati) per tali medicinali sono già scaduti e molti
scadranno nel prossimo decennio.
Alla fine della copertura brevettuale è possibile quindi, da parte di altre Case farmaceutiche, sviluppare medicinali
biologici simili a quelli il cui brevetto è scaduto, i cosiddetti medicinali biosimilari, ("biosimilari"): il primo
biosimilare è stato approvato e commercializzato nel 2006.
I medicinali biosimilari
Nel 2012, l'EMA ha fornito una definizione di "biosimilare, ossia:
"principio attivo di un medicinale biosimilare è un principio attivo biologico conosciuto, simile a quello del
medicinale di riferimento, ossia a un medicinale che ha ricevuto un'autorizzazione all'immissione in
commercio da uno Stato Membro o dalla Commissione europea sulla base di un dossier completo, cioè
con la presentazione di dati di qualità, preclinici e clinici. Un medicinale biologico similare e il suo
medicinale di riferimento devono avere lo stesso profilo di sicurezza ed efficacia, e sono generalmente
usati per il trattamento delle stesse condizioni"
Poiché i medicinali biosimilari sono medicinali biologici, essi rientrano nelle norme comunitarie vigenti per il
medicinale biologico. Essi devono di conseguenza seguire le linee guida scientifiche relative ai medicinali
biologici, ed essere sottoposti alla stessa valutazione da parte delle autorità regolatorie competenti come tutti gli
altri medicinali biologici.
EMA ha inoltre sviluppato linee guida scientifiche per classi di biosimilari, comprendenti linee guida
sull'immunogenicità e sulla comparabilità, per fornire un solido procedimento regolatorio attraverso il quale poter
ottenere le autorizzazioni all'immissione in commercio per i medicinali biosimilari. Queste linee guida sono riviste
a scadenze regolari per riflettere l'esperienza acquisita attraverso le domande di registrazione presentate e
approvate e alla luce dell'evoluzione della scienza e della tecnologia.
L'UE è la prima regione al mondo ad aver definito un quadro normativo e un percorso regolatorio per i biosimilari.,
che ha ispirato numerosi paesi in tutto il mondo, fra cui Australia, Canada, Giappone, Stati Uniti, oltre
all'Organizzazione Mondiale della Sanità (OMS).
3
La comparabilità tra il medicinale di riferimento e il medicinale biosimilare è il principio fondamentale dello sviluppo
di un biosimilare. La comparabilità era richiesta anche in caso di
modifiche al processo di produzione dei
medicinali biologici originatori, modifiche introdotte frequentemente durante tutto il ciclo di vita di un prodotto (per
es. per migliorare la qualità o aumentare il rendimento del prodotto). I principi scientifici di comparabilità necessari
per le modifiche al processo di produzione di un determinato medicinale biologico e quelli richiesti per lo sviluppo
di un medicinale biosimilare sono gli stessi, ma per i biosimilari sono sempre anche richiesti studi clinici perché, a
causa dei processi di produzione completamente indipendenti, è possibile che ci siano alcune differenze tra il
biosimilare e il prodotto di riferimento, e il potenziale impatto di queste differenze sulla sicurezza e sull'efficacia
non può essere previsto esclusivamente su valutazione analitica
Un biosimilare deve quindi presentare diverse tipologie di studi di comparabilità
1. comparabilità della qualità (comparabilità fisico-chimica e biologica determinata rispetto alla struttura
molecolare oltre che rispetto alla funzionalità); deve essere dimostrata con un'esauriente
Caratterizzazione analitica e con studi e test biologici sui legami dei recettori coinvolti, in modo
strettamente comparativo sul biosimilare e sul medicinale di riferimento
2. comparabilità non clinica (studi non clinici comparativi)
3. comparabilità clinica (studi clinici comparativi)
La comparabilità clinica e non clinica garantiscono la sicurezza ed efficacia del biosimilare rispetto al medicinale
di riferimento.
È possibile, sulla base di un’evidente comparabilità generale e di un’adeguata giustificazione scientifica,
estrapolare dei dati di efficacia e sicurezza clinica riferiti ad altre indicazioni del medicinale di riferimento non
valutati specificamente durante lo sviluppo clinico del biosimilare. Vi deve però essere almeno uno studio clinico
sulla popolazione di pazienti più sensibili, che misuri l'endpoint o gli endpoint clinici più sensibili, cioé quelli che
hanno la maggiore probabilità di evidenziare le differenze, se esistenti, tra il biosimilare e il medicinale di
riferimento. Nel caso invece in cui per le ulteriori indicazioni richieste siano coinvolti meccanismi di azione
differenti, il produttore del biosimilare dovrà fornire per tutte le indicazioni cliniche richieste, ulteriori dati rilevanti a
supporto dell'estrapolazione, nonché un’analisi dei dati disponibili in letteratura che includano il/i recettore/i degli
antigeni coinvolto/i e il/i meccanismo/i di azione.
La valutazione sulla similarità del biosimilare al medicinale di riferimento è dimostrata da parte del produttore in
coerenza con specifiche linee guida scientifiche sui biosimilari, sugli attributi di qualità del biosimilare e la sua
comparabilità con il medicinale di riferimento.
Il Comitato per i medicinali per uso umano dell’EMA valuta quindi la similarità ai fini della autorizzazione in
commercio di un farmaco biosimilare.
Come per tutti gli altri farmaci, anche le ditte produttrici di biosimilari devono predisporre un sistema di
farmacovigilanza, per monitorare la sicurezza dei propri medicinali autorizzati e individuare qualunque modifica
che ne possa compromettere il rapporto beneficio-rischio.
Il piano di gestione del rischio (EU-RMP) va presentato insieme alla richiesta di autorizzazione all'immissione in
commercio e descrive dettagliatamente il sistema di gestione dei rischi che l'azienda attuerà per il medicinale in
questione dopo la sua immissione in commercio, il profilo di sicurezza del medicinale e le misure che il richiedente
intende introdurre per prevenire o minimizzare qualsiasi potenziale rischio durante l'utilizzo del medicinale, inclusa
la misurazione della sua efficacia nella pratica clinica. Ad esempio l'immunogenicità è un problema di sicurezza
fondamentale di qualsiasi medicinale biologico e deve essere trattato nell'EU-RMP.
Inoltre, come per tutti gli altri farmaci, l'autorizzazione all'immissione in commercio di un biosimilare richiede la
presentazione di studi di sicurezza post-autorizzativi (PASS) e/o studi di efficacia post-autorizzativi (PAES) che
4
facciano parte del citato piano di gestione del rischio. Lo scopo del PAES è identificare, caratterizzare o
quantificare un rischio per la sicurezza, o confermare il profilo di sicurezza del medicinale o determinare l'efficacia
delle misure di gestione del rischio.
Come previsto per tutti i medicinali, i biosimilari
sono autorizzati alla immissione in commercio con una
denominazione di fantasia (commerciale) o il nome del principio attivo unitamente al nome dell'azienda/marchio
registrato. La denominazione, insieme al numero di lotto, è importante per una chiara identificazione del
medicinale a supporto della segnalazione di reazioni avverse e del monitoraggio dell’utilizzo sicuro del medicinale.
EMA pubblica sul proprio sito web i documenti ufficiali per ogni biosimilare autorizzato. La pagina web dedicata ai
medicinali biosimilari può essere consultata sul sito web EMA che contiene anche un link a un elenco di tutti i
medicinali biosimilari autorizzati con procedura centralizzata, con i relativi documenti di valutazione. (EPAR =
Foglio illustrativo Riassunto delle caratteristiche del prodotto Relazioni di valutazione riassunto della Relazione
pubblica di valutazione europea piano di gestione del rischio):
Come i medicinali originatori di riferimento, i biosimilari sono più difficili e costosi da sviluppare rispetto ai
medicinali generici. Essi rappresentano un'alternativa meno costosa ai medicinali biologici esistenti, che hanno
perso i diritti di esclusiva, anche se il differenziale di prezzo con l’originatore è meno rilevante rispetto ai farmaci
generici e al loro originatore. La disponibilità di biosimilari potrebbe quindi migliorare l'accesso ai medicinali
biologici per un maggior numero di pazienti e contribuire alla sostenibilità finanziaria dei sistemi sanitari.
L’introduzione dei biosimilari ha ridotto i costi dei trattamenti, anche se si osserva una tendenza generale al
passaggio a terapie con medicinali biologici ad azione prolungata ancora coperti da brevetto. Si nota infatti una
riduzione nel consumo dei prodotti originatori a breve durata di azione, indipendentemente dal fatto che essi siano
soggetti o meno alla diretta concorrenza da parte dei medicinali biosimilari. Peraltro anche per alcuni dei
medicinali biologici ad azione prolungata è oramai prossima la scadenza della copertura brevettuale e anch’essi
saranno oggetto della concorrenza diretta dei biosimilari.
La Legislazione Europea ha lasciato alle autorità nazionali dei diversi Stati l’autonomia decisionale e legislativa in
merito alla sostituibilità dei farmaci originatori con il rispettivo biosimilare, anche se l’EMA ha comunque
raccomandato che non vi deve essere intercambiabilità e che in ogni caso le decisioni vadano assunte da
personale sanitario qualificato.
Circa la sostituibilità, l’AIFA ha escluso la sostituibilità terapeutica automatica tra farmaci biosimilari e i rispettivi
farmaci di riferimento, pronunciandosi sul fatto che essi non possono essere considerati alla stregua dei farmaci
equivalenti, essendo simili, ma non identici per le possibili differenze nelle procedure di produzione e la
conseguente variabilità biologica anche ai fini di una eventuale immunogenicità. Per AIFA i biosimilari
costituiscono un’opzione terapeutica a disposizione dei curanti, da preferire, qualora costituiscano un vantaggio
economico, in particolare per il trattamento dei soggetti naïve (che non abbiano avuto precedenti esposizioni
terapeutiche o per i quali le precedenti esposizioni in base al giudizio del clinico siano sufficientemente distanti nel
tempo.
I farmaci biosimilari non figurano pertanto nelle liste di trasparenza che consentono la sostituibilità automatica tra
farmaci equivalenti. Di conseguenza, la normativa prevede che “la decisione circa la scelta prescrittiva del
medicinale specifico da impiegare, di riferimento piuttosto che biosimilare, debba essere affidata a personale
sanitario qualificato”.
La posizione dell'AIFA è in linea con le indicazioni della Direzione Generale Salute della Lombardia di cui alla
DGR n° X/ 1185 del 20.12.2013
5
Ne discende che:
•
il prescrittore deve disporre di una informazione puntuale e aggiornata riguardo al vantaggio economico,
ovvero alla spesa effettiva sostenuta, per tutti i farmaci biologici (tutti gli originatori e tutti i biosimilari
commercializzati, Il medico che redige una prescrizione nei confronti di un paziente deve poter disporre di
fonti di informazioni complete, imparziali e obiettive sui medicinali disponibili sul mercato. Il Codice di
Deontologia Medica richiama nell'articolo 6 che il medico agisce secondo il principio di efficacia delle cure
nel rispetto dell’autonomia della persona tenendo conto dell’uso appropriato delle risorse e nell'articolo 13
che le prescrizioni e i trattamenti devono essere ispirati ad aggiornate e sperimentate acquisizioni
scientifiche tenuto conto dell’uso appropriato delle risorse, sempre perseguendo il beneficio del paziente
secondo criteri di equità.
•
ogni paziente dovrebbe essere “fidelizzato” ad uno specifico prodotto per tutta la durata della terapia,
evitando i passaggi tra diversi prodotti per quanto costituiti dalla medesima molecola biologica.
Nei
pazienti già stabilizzati in trattamento con farmaci biotecnologici, ad oggi non è raccomandato lo switch
automatico verso un altro biologico, compreso i biosimilari, a meno che non si renda necessario per
situazioni cliniche. Solo il clinico infatti è in grado di valutare sulla base della singola situazione del
paziente e delle informazioni sul farmaco, se effettuare o meno lo switch. Per questo motivo la prima
scelta terapeutica (paziente naïve cioè che non ha mai effettuato terapia con quella molecola) va condotta
privilegiando i farmaci biosimilari, a parità di indicazioni rispetto al farmaco di riferimento, perché hanno
costi inferiori per il SSN. Per il pazienti naïve si possono configurare due situazioni:
1.
il paziente non ha mai ricevuto un trattamento con il farmaco biologico
2.
il paziente è già stato trattato in precedenza con un dato farmaco biologico e si ripresenta la necessità di
ulteriore trattamento con un farmaco biologico. In questo caso, se il medico ritiene che la precedente
esposizione sia sufficientemente distante nel tempo, può usare il farmaco preferibile, cioè il farmaco
biosimilare (purché ciò comporti effettivamente una minore spesa per il sistema sanitario e per il
paziente).
Il passaggio ad altro farmaco per paziente in trattamento cronico non è esclusa, ma demandata, come sopra
ricordato, alla scelta del clinico.
Per garantire tutto ciò:
•
il prescrittore – sia lo specialista che il medico di medicina generale – deve indicare sia sulla ricetta SSN
sia sul piano terapeutico il nome commerciale del prodotto e non il nome della molecola biologicamente attiva,
così da evitare che al paziente vengano erogati, di volta in volta, prodotti commerciali diversi in farmacia della
stessa molecola biologicamente attiva
•
relativamente alla dispensazione, il farmacista è tenuto obbligatoriamente a consegnare al paziente il
prodotto commerciale indicato dal prescrittore sulla ricetta SSN e eventuali differenze di prezzo di farmaci costituiti
dalla stessa molecola biologica attiva non siano mai a carico del paziente, ma sempre sopportate dal SSN.
L'impiego (off-label) ai sensi della legge n. 648\96 (possibilità per il trattamento di una patologia di dispensare a
carico del SSN farmaci autorizzati per altra indicazione terapeutica, in assenza di una valida alternativa
terapeutica, purché disponibili dati di sicurezza ed efficacia raccolti in studi clinici almeno di fase II e previo parere
autorizzativo della Commissione consultiva Tecnico Scientifica
dell’AIFA) è ammesso anche per i farmaci
biologici; però, l'inserimento nell'elenco dei farmaci autorizzati all’impiego nelle indicazioni di cui alla Legge 648
dei corrispondenti farmaci biosimilari non è automatico e viene verificato caso per caso dalla CTS.
6
ERITROPOIETINE
L’Eritropoietina umana ricombinate è stata commercializzata fin dal 1988 per il trattamento della anemia
secondaria a malattie renali e , successivamente, per la terapia di supporto dei pazienti oncologici con anemia
secondaria a trattamento chemioterapico.
In Europa, la copertura brevettuale di Epo alfa (prima commercializzazione Janssen Cilag come Eprex) è scaduta
nel 2004.
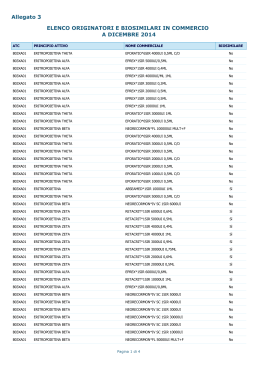
Tabella 1: principi attivi in commercio in Italia
Epoetina alfa
Epoetina beta
Epoetina zeta
Epoetina theta
Darbopoietina
Metossipolietilenglicole-epoetina beta
Attualmente sono stati prodotte due Epo alfa biosimilari, con composizione aminoacidica identica e diversa
composizione glicosidica. Ad una è stato mantenuto il nome di Epo alfa (due prodotti commercializzati : Binocrit e
Abseamed), l’altra costituisce l’Epoetina zeta (Retacrit) ; ognuna di queste Epo ha quindi
diversi dossier
registrativi. L’Epoetina theta e stata presentata all’EMA come prodotto originator e non come biosimilare.
Tabella 2: Eritropoietine in commercio a novembre 2014
ATC
PRINCIPIO ATTIVO
NOME COMMERCIALE
BIOSIMILARE/ORIGINATOR
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 10000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 1000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 2000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 3000UI 0,3ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 4000UI 0,4ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 5000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 6000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*1SIR 8000UI 0,8ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 10000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 1000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 2000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 3000UI 0,3ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 4000UI 0,4ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 5000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 6000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
ABSEAMED*6SIR 8000UI 0,8ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 10000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 1000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 20000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 2000UI 1ML
BIOSIMILARE
7
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 30000UI 0,75ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 30000UI 0,75ML+D
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 3000UI 0,3ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 40000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 40000UI 1ML+DISP
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 4000UI 0,4ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 5000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 6000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*1SIR 8000UI 0,8ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 10000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 1000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 2000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 3000UI 0,3ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 4000UI 0,4ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 5000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 6000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ALFA
BINOCRIT*6SIR 8000UI 0,8ML
BIOSIMILARE
B03XA01 ERITROPOIETINA THETA
EPORATIO*1SIR 10000UI 1ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*1SIR 10000UI 1ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*1SIR 20000UI 1ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*1SIR 20000UI 1ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*1SIR 30000UI 1ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*1SIR 30000UI 1ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 1000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 1000UI 0,5ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 2000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 2000UI 0,5ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 3000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 3000UI 0,5ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 4000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 4000UI 0,5ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 5000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA THETA
EPORATIO*6SIR 5000UI 0,5ML C/D
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 10000UI 1ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 1000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 2000UI 0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 3000UI 0,3ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 40000UI/ML 1ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 4000UI 0,4ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 5000UI/0,5ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 6000UI/0,6ML
ORIGINATORE
B03XA01 ERITROPOIETINA ALFA
EPREX*1SIR 8000UI/0,8ML
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*FL 100000UI MULT+F ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*FL 50000UI MULT+F
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 10000UI
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 2000UI
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 30000UI
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 3000UI
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 4000UI
ORIGINATORE
8
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 5000UI
ORIGINATORE
B03XA01 ERITROPOIETINA BETA
NEORECORMON*IV SC 1SIR 6000UI
ORIGINATORE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 10000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 1000UI 0,3ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 20000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 2000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 30000UI 0,75ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 3000UI 0,9ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 40000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 4000UI 0,4ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 5000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 6000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*1SIR 8000UI 0,8ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 10000UI 1ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 1000UI 0,3ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 2000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 3000UI 0,9ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 4000UI 0,4ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 5000UI 0,5ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 6000UI 0,6ML
BIOSIMILARE
B03XA01 ERITROPOIETINA ZETA
RETACRIT*6SIR 8000UI 0,8ML
BIOSIMILARE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 100MCG 0,5ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 150MCG 0,3ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 20MCG 0,5ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 300MCG 0,6ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 40MCG 0,4ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 500MCG 1ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 60MCG 0,3ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC 1PEN 80MCG 0,4ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 100MCG 0,5M
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 10MCG 0,4ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 150MCG 0,3M
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 20MCG 0,5ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 300MCG 0,6M
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 30MCG 0,3ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 40MCG 0,4ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 500MCG 1ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 50MCG 0,5ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 60MCG 0,3ML
ORIGINATORE
B03XA02 DARBEPOIETINA ALFA
ARANESP*SC EV 1SIR 80MCG 0,4ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 100MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 120MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 150MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 200MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 250MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 30MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 360MCG 0,6ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 50MCG 0,3ML
ORIGINATORE
B03XA03 METOSSIPOLIETILENGLICOLE-EPOETINA BETA
MIRCERA*IV SC SIR 75MCG 0,3ML
ORIGINATORE
9
Si riportano in tabella 3 un sunto delle indicazioni presenti in scheda tecnica per le Eritropoietine commercializzate
oggi in Italia.
Tabella 3: Indicazioni terapeutiche da scheda tecnica e da Legge 648
FARMACO
INDICAZIONI DA RCP
LEGGE 648/96
anemia sintomatica associata a insufficienza renale cronica (IRC): in
pazienti adulti e pediatrici di eta compresa tra 1 – 18 anni in
emodialisi e in pazienti adulti in dialisi peritoneale.
Trattamento dell’anemia
(Hgb < 10 g/dL o riduzione
dell’emoglobina > 2 g/dL
durante un qualsiasi
periodo di 4 settimane di
trattamento) nei pazienti
che ricevono ribavirina in
combinazione con
interferone standard o
peghilato per HCV e che
presentano risposta
virologica alla terapia.
anemia sintomatica associata a insufficienza renale cronica
(IRC): in pazienti adulti con insufficienza renale non ancora sottoposti
a dialisi per il trattamento dell’anemia grave di origine
renale,accompagnata da sintomi clinici nei pazienti.
in pazienti adulti in chemioterapia per tumori solidi,linfoma maligno
o mieloma multiplo e a rischio di trasfusione come indicato dallo stato
generale del paziente (situazione cardiovascolare,anemia preesistente
all’inizio della chemioterapia)
EPREX
per il trattamento dell’anemia e riduzione del fabbisogno
trasfusionale.in pazienti adulti che fanno parte di un programma di
predonazione per aumentare la quantita di sangue autologo Il
trattamento e indicato solo in pazienti con anemia di grado moderato
(concentrazione di Hb nell’intervallo compreso tra 10-13 g/dl [ 6,2 – 8,1
mmol/l], nessuna carenza di ferro) se le procedure di conservazione del
sangue non sono disponibili o sono insufficienti in caso di intervento
elettivo di chirurgia maggiore che richieda un elevato quantitativo di
sangue (4 o piu unita per le donne o 5 o piu unita per gli uomini).
in pazienti adulti, che non presentino carenze di ferro,prima di un
intervento elettivo di chirurgia ortopedica maggiore, ritenuti a elevato
rischio di complicazioni da trasfusione, per ridurre l’esposizione a
trasfusioni di sangue allogenico. L’uso deve essere limitato a pazienti
con anemia di grado moderato (concentrazione di Hb nell’intervallo
compreso tra 10-13 g/dl), per i quali non sia disponibile un programma di
predonazione di sangue autologo, e per i quali si preveda una perdita di
sangue moderata (da 900 a 1800 ml).
Trattamento dell’anemia sintomatica associata a insufficienza renale
cronica (IRC) in pazienti adulti e pediatrici:
In pazienti HIV pluritrattati
con anemia (Hgb < 8,5
g/dL) nei quali l’uso di
farmaci anemizzanti e
l’unica alternativa
terapeutica.
Sindrome mielodisplastica
(G.U. 10/03/00 n. 58)
Anemia refrattaria (AR),
con sideroblasti (RARS) e
senza sideroblasti (RA) (G.U. 10/03/00 n. 58)
Sindrome
mielodisplastica (G.U.
9/04/2014 n. 83)
- Trattamento dell’anemia associata a insufficienza renale cronica in
pazienti adulti e pediatrici emodializzati e in pazienti adulti sottoposti
a dialisi peritoneale
- Trattamento dell'anemia grave, di origine renale, accompagnata da
sintomi clinici, in pazienti adulti con insufficienza renale non ancora
dializzati
Trattamento dell’anemia e riduzione del fabbisogno trasfusionale in
pazienti adulti in trattamento chemioterapico per tumori solidi,
linfoma maligno o mieloma multiplo e a rischio di trasfusione, come
indicato dallo stato generale del paziente (situazione cardiovascolare,
anemia preesistente all’iniziodella chemioterapia).
BINOCRIT
Aumento della produzione di sangue autologo nei pazienti facent
parte di un programma di predonazione autologa. L' impiego per tale
indicazione deve essere valutato in rapporto al noto rischio di eventi
tromboembolici. Il trattamento deve essere effettuato solo in pazienti con
anemia moderata ((Hb) 10-13 g/dL (6,2-8,1 mmol/L), senza carenza di
ferro), quando le tecniche di risparmio di sangue non siano disponibili o
siano insufficienti e l’intervento programmato di chirurgia elettiva
maggiore richieda un elevato quantitativo di sangue (4 o più unità
disangue per le donne, 5 o più unità per gli uomini).
Per ridurre l’esposizione a trasfusioni di sangue allogenico in pazienti
adulti non sideropenici, ritenuti ad alto rischio di complicanze
trasfusionali, prima di un intervento elettivo di chirurgia ortopedica
maggiore. Limitare l’uso ai pazienti con anemia moderata (Hb10-13 g/dL
o 6,2-8,1 mmol/L) non facenti parte di un programma di predonazione
autologa e per i
quali si preveda una perdita ematica moderata di 900-1800 mL.
10
ABSEAMED
(da foglietto illustrativo)
Trattamento dell'anemia sintomatica associata a insufficienza renale
cronica (IRC) in pazienti adulti e pediatrici:
trattamento dell'anemia associata a insufficienza renale cronica in
pazienti adulti e pediatrici emodializzati e in pazienti adulti sottoposti
a dialisi peritoneale
trattamento dell'anemia grave, di origine renale, accompagnata da
sintomi clinici, in pazienti adulti con insufficienza renale non ancora
dializzati
Trattamento dell'anemia e riduzione del fabbisogno trasfusionale in
pazienti adulti in trattamento chemioterapico per tumori solidi,
linfoma maligno o mieloma multiplo e a rischio di trasfusione, come
indicato dallo stato generale del paziente (situazione cardiovascolare,
anemia preesistente all'inizio della chemioterapia).
Aumento della produzione di sangue autologo nei pazienti facenti
parte di un programma di predonazione autologa. L'impiego per tale
indicazione deve essere valutato in rapporto al noto rischio di eventi
tromboembolici. Il trattamento deve essere effettuato solo in pazienti con
anemia moderata (emoglobina (Hb) 10 – 13 g/dl [6,2 – 8,1 mmol/l],
sideropenia assente), quando le tecniche di risparmio di sangue non
siano disponibili o siano insufficienti e l'intervento programmato di
chirurgia elettiva maggiore richieda un elevato quantitativo di sangue (4
o più unità di sangue per le donne, 5 o più unità per gli uomini).
Per ridurre l'esposizione a trasfusioni di sangue allogenico in pazienti
adulti non sideropenici, ritenuti ad alto rischio di complicanze
trasfusionali, prima di un intervento elettivo di chirurgia ortopedica
maggiore. Limitare l'uso ai pazienti con anemia moderata (Hb 10 - 13
g/dl) non facenti parte di un programma di predonazione autologa e per i
quali si preveda una perdita ematica di 900 - 1800 ml.
EPORATIO
Trattamento dell’anemia sintomatica associata ad insufficienza renale
cronica in pazienti adulti.
- Trattamento dell’anemia sintomatica in pazienti adulti oncologici in
chemioterapia per neoplasie maligne non mieloidi.
Trattamento dell’anemia sintomatica associata a insufficienza renale
cronica (IRC) in pazienti adulti e pediatrici:
Sindromi
mielodisplastiche
• Trattamento dell’anemia associata ad insufficienza renale cronica in
pazienti adulti e pediatrici in emodialisi e in pazienti adulti in dialisi
peritoneale
• Trattamento dell'anemia grave di origine renale con sintomatologia
clinica in pazienti adulti con insufficienza renale non ancora
sottoposti a dialisi.
RETACRIT
− Trattamento dell’anemia e riduzione del fabbisogno trasfusionale in
pazienti adulti sottoposti a chemioterapia per tumori solidi, linfoma
maligno o mieloma multiplo e a rischio di emotrasfusione come
indicato dallo stato generale del paziente (situazione
cardiovascolare,anemia preesistente all’inizio della chemioterapia).
Per incrementare la quantità di sangue autologo in pazienti facenti
parte di un programma di predonazione. L’uso in questa indicazione
deve essere valutato alla luce dei rischi riferiti di eventi tromboembolici. Il
trattamento deve essere riservato solo a
pazienti con anemia di grado moderato (in assenza di sideropenia) se le
procedure di emoconservazione non sono disponibili o sono insufficienti
quando l’intervento elettivo di chirurgia maggiore previsto richiede un
notevole volume di sangue (4 o più unità di sangue perle donne, 5 o più
unità per gli uomini).
Per ridurre l’esposizione a trasfusioni di sangue allogenico in pazienti
adulti non sideropenici, ritenuti ad alto rischio di complicanze
trasfusionali, prima di un intervento elettivo di chirurgia ortopedica
maggiore. Limitare l’uso ai pazienti con anemia moderata (Hb 10 -13
g/dl) non facenti parte di un programma di predonazione autologa e per
iquali si preveda una moderata perdita ematica (da 900 a 1 800 ml).
11
trattamento dell’anemia sintomatica associata ad insufficienza renale
cronica (IRC) in pazienti adulti e pediatrici.
- prevenzione dell’anemia dei neonati prematuri con un peso alla
nascita compreso tra 750 e 1500 g e con un periodo di gestazione
inferiore a 34 settimane.
- trattamento dell’anemia sintomatica in pazienti adulti con tumore non
mieloide sottoposti a chemioterapia.
NEORECORMON
-Per incrementare la quantita di sangue autologo in pazienti facenti
parte di un programma di predonazione. Il suo uso in questa
indicazione deve essere valutato in rapporto all’aumentato
rischio di eventi tromboembolici. Il trattamento deve essere riservato solo
a pazienti con anemia di grado moderato (emoglobina 10 - 13 g/dl [6,21 8,07 mmol/l], in assenza di carenza di ferro) se le procedure di
conservazione non sono disponibili o sono insufficienti quando
l’intervento elettivo di chirurgia maggiore richiede un notevole volume di
sangue (4 o piu unita di sangueper le donne o 5 o piu unita per gli
uomini).
Trattamento dell’anemia
(Hgb < 10 g/dL o riduzione
dell’emoglobina > 2 g/dL
durante un qualsiasi
periodo di 4 settimane di
trattamento) nei pazienti
che ricevono ribavirina in
combinazione con
interferone standard o
peghilato per HCV e che
presentano risposta
virologica alla terapia.
In pazienti HIV pluritrattati
con anemia (Hgb < 8,5
g/dL) nei quali l’uso di
farmaci anemizzanti e
l’unica alternativa
terapeutica.
Trattamento della
Sindrome
mielodisplastica (G.U.
10/03/00 n. 58)
Anemia refrattaria (AR),
con sideroblasti (RARS) e
senza sideroblasti (RA) (G.U. 10/03/00 n. 58)
Trattamento dell'anemia sintomatica associata all'insufficienza renale
cronica (IRC) in adulti e in pazienti pediatrici
ARANESP
MIRCERA
Trattamento dell'anemia sintomatica in pazienti adulti affetti da
neoplasie non mieloidi che ricevono chemioterapia.
Trattamento dell'anemia sintomatica associata a insufficienza renale
cronica (IRC) in pazienti adulti
12
Utilizzo in ambito nefrologico
La scelta della eritropoietina con il miglior profilo costo\efficacia, in un contesto di salvaguardia dell’appropriatezza
prescrittiva e perseguimento del miglior beneficio per il paziente, in ambito nefrologico, è strettamente correlata
alle diverse tipologie di paziente che necessitano di questo trattamento.
Trattamento emodialitico \extracorporeo =
In coerenza con le disposizioni normative vigenti (D. L.vo 19\14), va favorito l’utilizzo di prodotti che posseggano i
sistemi di protezione da ferite da punta. In pazienti in trattamento assistito (ospedale, CAL) trovano convergenza
di appropriatezza, efficacia e massimo beneficio per il paziente le formulazioni con maggior frequenza di
somministrazione. Il miglior rapporto costo/efficacia può costituire motivo di preferenza per i singoli prodotti. Può
essere utilizzata sia la somministrazione e.v.(che potrebbe ridurre tra l’altro i rischi di anemia da anticorpi antieritropoietina) che la via s.c. (quando supportata dalla registrazione del prodotto), che realizza il miglior bilancio
dose/effetto
Difficilmente il paziente in dialisi è comunque un paziente naive puro, in quanto presumibilmente – nella maggior
parte dei casi – ha ricevuto eritropoietine in condizione di predialisi. Occorre considerare però che in genere
l’eritropoietina pre dialisi è somministrata sottocute; nel caso di una somministrazione ev il paziente potrebbe
essere considerato naive rispetto alla diversa via di somministrazione ( dal momento che il rischio di PRCA è stato
osservato quasi solo con l’impiego sottocute , uno shift da sottocute a endovena o da endovena a endovena
potrebbe comportare un basso rischio di PRCA ).
Comunque, come sopra ricordato, il passaggio ad altro farmaco per paziente in trattamento cronico è demandato
alla scelta del clinico.
Trattamento domiciliare della Insufficienza renale cronica pre-dialisi, dialisi peritoneale, emodialisi
domiciliare, trapianto renale =
Qualora si richieda, in base alle Linee Guida vigenti, la necessità di trattare il paziente con eritropoietina, occorre
considerare che si tratterà di un trattamento cronico, anche condotto per molti anni, in cui è fondamentale una
valutazione nella scelta del farmaco che garantisca, dato il profilo di ogni paziente, la massima compliance alla
terapia prescritta, al fine di ottenere i risultati attesi, senza inutile dispendio di risorse per scarsa compliance del
paziente alla terapia.
Una efficace valutazione della compliance attesa da un paziente non può venire condotta a priori per tipologie
prefissate di soggetti, ma deve essere fondata su una valutazione della individualità del singolo paziente, nelle
sue specifiche caratteristiche comportamentali, anagrafiche, cliniche, sociali. Inoltre la compliance è la risultante
multifattoriale di un insieme di fattori (più o meno predominanti a seconda del soggetto considerato) quali la
facilità di assunzione, l’assenza di effetti collaterali anche minori, la capacità di memorizzare le cadenze di
assunzioni.
Tutto ciò comporta che la scelta della formulazione più idonea al paziente vada condotta su base individuale e
che poi, all’interno delle formulazioni idonee per quel paziente, venga prescritto il prodotto con il costo più
favorevole per il sistema sanitario.
13
Ciò premesso, miglioramenti nella compliance attesa si possono comunemente ottenere ricorrendo a
formulazione con somministrazione sottocutanea, piuttosto che endovena, e\o formulazioni che rendano possibili
somministrazioni maggiormente dilazionate nel tempo.
La tabella 4 riporta a questo riguardo una sintesi delle vie di somministrazione e delle frequenze di
somministrazione delle diverse eritropoietine per l’indicazione anemia associata a IRC in pazienti adulti
Tabella 4 Principali schemi posologicI e vie di somministrazione eritropoietine
(fonte RCP – riassunto caratteristiche tecniche del prodotto)
Anemia associata ad IRC
ATC
B03XA01
B03XA01
B03XA01
B03XA01
B03XA01
B03XA01
B03XA01
B03XA01
B03XA01
farmaco
Eprex
Retacrit
Abseamed
Binocrit
Eprex
Binocrit
Abseamed
Retacrit
paziente
via di
somministrazi
one
adulto in emodialisi
s.c./e.v.
____________
ev
fase di correzione = 50UI/Kg 3 volte/settimana
fase di mantenimento = aggiustare il dosaggio per
mantenere Hb tra 10-12 g/dl
pediatrico in emodialisi
ev
fase di correzione = 50UI/Kg 3 volte/settimana
fase di mantenimento = aggiustare il dosaggio per
mantenere Hb tra 9,5/11 g/dl
sc
Eprex
adulto no dialisi
B03XA01
Binocrit
B03XA01
Abseamed
B03XA01
Retacrit
B03XA01
B03XA01
B03XA01
B03XA01
Eprex
Retacrit
Abseamed
Binocrit
B03XA02
Aranesp
frequenza di somministrazione e posologia
ev
____________
s.c./e.v
Sc
adulto in dialisi
peritoneale
adulto
ev
ev/sc
fase di correzione = 50UI/Kg 3 volte/settimana
incrementi di 25UI/Kg 3 voltealla settimana fino al
valore desiderato
fase di mantenimento = può essere somministrato 3
volte alla settimana e in caso di sc 1 volta alla
settimana o 1 volta ogni 2 settimane dose ed
intervalli adattati per mantenere i valori di Hb al livello
desiderato (Hb 10-12 g/dl)
fase di correzione = 50UI/Kg 3 volte/settimana
incrementi di 25UI/Kg 3 volte la settimana fino al
valore desiderato
fase di
mantenimento = aggiustare il dosaggio per
mantenere i valori di Hb al livello desiderato (Hb 1012 g/dl)
fase di correzione = 50UI/Kg 2 volte/settimana
fase di mantenimento = aggiustare il dosaggio per
mantenere i valori di Hb al livello desiderato (Hb 1012 g/dl)
fase di correzione = dose iniziale 0,45 mcg/kg sc o
ev 1 volta la settimana, in alternativa ad adulti non
dializzati dose iniziale di 0,75 mcg/kg come singola
iniezione sc 1 volta ogni 2 settimane fase di
mantenimento = iniezione singola 1 volta alla
settimana o 1 volta ogni 2 settimane.
Nei pazienti non dializzati, una volta che l’obiettivo
della concentrazione emoglobinica viene raggiunto
con una somministrazione ogni due settimane,
Aranesp può essere somministrato con iniezione
sottocutanea una volta al mese iniziando con una
dose pari al doppio di quella
precedentemente somministrata una volta ogni due
settimane.
14
NB = La dose settimanale iniziale di Aranesp (µg/settimana) può essere calcolata dividendo per 200 la dose settimanale totale di rHuEPO (UI/settimana). Ad es.4000 UI = 20 microgrammi. La dose iniziale di Aranesp da somministrare ogni due settimane (µg per
due settimane) può essere calcolata dividendo per 200 la dose totale di r-HuEPO somministrata nel corso di un periodo di due
settimane.
B03XA02
Aranesp
pediatrico
(non vi sono dati per
pz pediatrici < 1 anno
e non vi sono linee
guida per pazienti da
1 a 11 anni ).
ev/sc
Per pz con età >= 11
fase di correzione = dose iniziale 0,45 mcg/kg sc o ev
1 volta la settimana, in alternativa ai pz non dializzati
dose iniziale di 0,75 mcg/kg come singola iniezione sc
1 volta ogni 2 settimane
fase di
mantenimento = iniezione singola 1 volta alla
settimana o 1 volta ogni 2 settimene.
NB = La dose settimanale iniziale di Aranesp (µg/settimana) può essere calcolata dividendo per 240 la dose settimanale totale di rHuEPO (UI/settimana). La dose iniziale di Aranesp da somministrare ogni due settimane (µg per due settimane) può essere
calcolata dividendo per 240 la dose totale di r-HuEPO somministrata nel corso di un periodo di due settimane.
B03XA01
B03XA01
B03XA03
Neorec
ormon
Neorec
ormon
Mircera
adulto/pediatrico non in
emodialisi
adulto/pediatrico in
emodialisi
adulto non emodializzato
sc
s.c./ev
sc
fase di correzione = il dosaggio iniziale è di
3*20UI/Kg alla settimana Il dosaggio settimanale può
essere ripartito in somministrazioni giornaliere
fase di mantenimento = per mantenere il livello
dell’Hb entro un range compreso tra 10 e 12 g/dl, la
dose è inizialmente ridotta alla metà di quella
precedentemente somministrata.
Successivamente, la dose viene adattata su base
individuale per paziente (dose di mantenimento) ad
intervalli di una o due settimane.La dose totale
settimanale può essere somministrata con un'unica
iniezione settimanale o può essere divisa in tre o
sette dosi settimanali. Pazienti stabili con un regime di
singola somministrazione settimanale possono
passare ad una somministrazione ogni due
settimane. In tal caso potrebbe essere necessario
incrementare la dose.
fase di correzione = il dosaggio iniziale è di
3*40UI/Kg alla settimana Il dosaggio può essere
aumentato, dopo 4 settimane, a 80 UI/kg - tre volte
alla settimana - e con ulteriori incrementi di 20 UI/kg,
se necessario, tre volte alla settimana, ad intervalli
mensili.
fase di mantenimento = per mantenere il livello
dell’Hb entro un range compreso tra 10 e 12 g/dl, la
dose è inizialmente ridotta alla metà di quella
precedentemente somministrata. Successivamente,
la dose viene adattata su base individuale per
paziente (dose di mantenimento) ad intervalli di una o
due settimane.
Pazienti che non stanno ricevendo la terapia con un
agente stimolante l'eritropoiesi (ESA)
dose iniziale 1,2 mcg/kg sc 1 volta al mese
somministrati una volta ogni mese come singola
iniezione sottocutanea.In alternativa 0,6 mcg/kg 1
volta ogni 2 settimanein per via ev o sc
Pazienti che stanno ricevendo la terapia con un ESA
1 singola iniezione mensile sc o ev. la dose iniziale è
stabilita in base alla dose settimanale
precedentemente calcolata di eritropoietina al
momento della sostituzione della terapia
15
NB
B03XA03
B03XA01
Mircera
Eporatio
adulto in emodialisi
ev/s.c.
adulto
ev/ sc
Pazienti che non stanno ricevendo la terapia con un
agente stimolante l'eritropoiesi (ESA)
dose iniziale 0,6 mcg/kg 1 volta ogni 2 settimane per
via ev o sc Pazienti che stanno ricevendo la terapia
con un ESA =1 singola iniezione mensile sc o ev. La
dose iniziale è stabilita in base alla dose settimanale
precedentemente calcolata di eritropoietina al
momento della sostituzione della terapia
Fase di correzione =
sottocutanea: =posologia iniziale è di 20 UI/kg di peso
corporeo 3 volte / setti.
endovenosa= posologia iniziale è di 40 UI/kg di peso
corporeo 3 volte /sett
Fase di mantenimento = La dose adattata su base
individuale per mantenere il livello target di HB tra 10
g/dl e 12 g/dl.
sottocutanea: La dose settimanale può essere
somministrata in un’unica iniezione settimanale o
suddivisa in 3 somm. alla settimana.
endovenosa: I pazienti che stabili con un regime
posologico suddiviso in 3 somm. settimanali,
possono passare ad una somministrazione
bisettimanale.
Oltre alla problematica della compliance, occorre inoltre considerare l’opportunità di utilizzare formulazioni dotate
di sistemi di sicurezza contro il rischio di punture accidentali,
Altro elemento da considerare, soprattutto per i prodotti short-acting (Epo alfa, beta, zeta e theta) è la “potenza”
(cioè la quantità di prodotto “dichiarata” necessaria per mantenere una certa risposta in termini di Hb sul paziente,
convenzionalmente il rapporto dose/g di Hb) dei singoli prodotti: questa può variare in rapporto alla via di
somministrazione (più favorevole per via s.c. che e.v.), la frequenza di somministrazione (somministrazioni
dilazionate, es. 1 volta/sett, richiedono dosaggi maggiori che con somministrazioni ravvicinate, es. 3 volte/sett), e
verosimilmente caratteristiche di prodotto o di confezionamento dei singoli prodotti. La potenza diversa di singoli
prodotti viene verificata “in proprio” nella pratica clinica. Tutte queste informazioni, quando disponibili al clinico in
termini quantitativi, contribuiscono a meglio valutare “l’economicità” reale dei singoli prodotti indicando un
coefficiente correttivo rispetto al prezzo di vendita.
Questi criteri di riferimento condivisi possono quindi orientare il clinico nella identificazione del farmaco con il
miglior profilo costo\efficacia, in particolare nel paziente naive.
Perché ciò accada è però indispensabile che i Medici prescrittori siano tempestivamente a conoscenza dei costi in
Servizio Sanitario Nazionale delle diverse eritropoietine. La definizione di questi costi non è oramai identificabile
nel prezzo al pubblico del farmaco, ma è dipendente dalla via di distribuzione del farmaco in atto nella ASL, in
16
base alle indicazioni regionali (vedi paragrafo successivo “Modalità erogative”). Per questo la ASL è tenuta a
mettere al corrente i Medici prescrittori specialisti dei reali prezzi sostenuti in SSN per questi farmaci, aggiornando
periodicamente il dato quando necessario. Solo in questa condizione, il prescrittore potrà essere infatti posto nella
condizione di attuare scelte che soddisfino in contemporanea le esigenze cliniche del paziente e le necessità
economiche del sistema SSN.
Una volta poi effettuata la scelta della formulazione più costo\efficace per un determinato paziente, occorre
evitare, per le motivazioni in premessa illustrate, che avvengano shift tra specialità diverse, anche all’interno del
medesimo principio attivo; gli shift devono infatti essere attuati quando sono resi inevitabili per modificazioni dello
stato clinico o operativo (ad esempio: passaggio di metodica di trattamento). Per evitare quindi shift impropri, è
bene che sul piano terapeutico sia identificato con chiarezza il prodotto prescritto.
Poiché la prima prescrizione di ESA in pazienti naive non ancora in dialisi avviene non infrequentemente in corso
di un ricovero ospedaliero, è da prevedere una prassi che permetta ai singoli ospedali di avere disponibilità, in
aggiunta ad eventuali prodotti alternativi in uso, anche del prodotto che verrà individuato come a miglior rapporto
costo/efficacia a livello territoriale.
17
Modalità erogative
La tabella 6 riassume le possibili modalità erogative delle eritropoietine:
Tabella 6: modalità erogative
FASCIA A \PHT
= dispensabile al
pubblico con ricetta
medica da rinnovare
di volta in volta
rilasciata da centri
ospedalieri o da
specialisti
in
nefrologia,
ematologia,
medicina
interna,
chirurgia, anestesia,
pediatria,
emotrasfusionista,
oncologia
Ricetta SSN
redatta dal Medico
di Medicina
generale, presa
visione del piano
terapeutico
Piano
terapeutico
redatto da
specialista di
branca
Ritiro del
farmaco presso
le farmacie della
Lombardia
Oppure
Ricetta SSN
redatta specialista
di branca
Distribuzione
diretta da parte
dell’ospedale che
ha in carico il
paziente
La ASL acquista il farmaco e lo fa
distribuire alle farmacie,
riconoscendo loro il costo della
distribuzione (distribuzione per
conto DPC)
Le farmacie acquistano il farmaco e
chiedono il rimborso alla ASL
Rendicontazione
da parte
dell’ospedale in
tipologia 6 del
File F (legge 648
solo tramite
ospedale)
L’Asl rimborsa in toto l’ospedale,
ribaltando i costi dei cittadini non
milanesi sulle altre ASL in regime
di compensazioni
La distribuzione diretta da parte dell’ospedale e la distribuzione per conto attuata dalla ASL (testo evidenziato in
grigio) consentono di realizzare dei risparmi rispetto al prezzo con cui sono stati registrati i farmaci al momento
della loro immissione in commercio (prezzo al pubblico), in quanto le ditte produttrici sono obbligate a fornire
farmaci alle ASL e agli Ospedali applicando una scontistica ulteriore rispetto al prezzo al pubblico. Inoltre i
meccanismi di acquisizione dei farmaci, in Ospedale e in ASL, tramite procedure di gara centralizzata, possono
assicurare scontistiche ancora superiori. Ovviamente va comunque garantita al paziente NON naive, anche al
momento di scadenza di gara, la continuità terapeutica con il prodotto precedentemente in suo.
Pertanto, la razionalizzazione dei costi di terapia deve prevedere due distinti e correlati interventi decisionali: la
scelta del farmaco con il profilo costo\efficacia più favorevole, effettuata dal clinico in base alla tipologia del
paziente, e l’applicazione di una strategia distributiva, da parte di ASL e Regione, che consenta una diminuzione
dei costi della terapia.
Occorre considerare che la recente DGR 2566 del 31\10\2014 ha unificato in tutte le Asl della Regione le
modalità distributive dei farmaci classificati in fascia A/PHT, ponendo l’obbligo per tutte le ASL lombarde di
distribuire farmaci a base di in regime di distribuzione per conto .
Pertanto l’analisi costo \efficacia va condotta sulla base di prezzi di cessione che le Ditte produttrici faranno per
questi farmaci nell’ambito della gara a valenza regionale tutt’ora ancora in corso e della quale non sono disponibili
al momento della stesura del presente protocollo i prezzi di aggiudicazione. E’ quindi fondamentale che la ASL
comunichi, non appena possibile, questi prezzi, comprensivi dell’onere di distribuzione da riconoscere alle
farmacie territoriali per la prestazione, ai Medici prescrittori specialisti, così che essi possano compiutamente
valutare ogni formulazione sotto il profilo costo/efficacia, attuando quindi la scelta migliore per la compatibilità
economica del Servizio Sanitario Nazionale e la salute di ogni paziente.
Il monitoraggio dell’utilizzo dei biosimilari può essere efficacemente osservato analizzando la percentuale di
biosimilare sui consumi di Eritropoietine
forniti alle nefrologie/dialisi da parte delle farmacie ospedaliere ,
specificando nel dettaglio il dato relativo all’impiego della Eritropoietina Theta. , non strettamente “biosimilare” ma
con profili sovrapponibili
18
FARMACOVIGILANZA
Come ricordato in premessa, la nuova normativa europea di farmacovigilanza (Dir. 2012\84\EC Regolamenti
1235\01 e 520\12) prevede che l’autorizzazione in commercio di un farmaco sia subordinata alla esecuzione di
studi post autorizzazione di sicurezza e alla presenza di un piano di gestione del rischio (RMP).
E’ quindi fondamentale da parte del clinico prescrittore
la segnalazione spontanea e tempestiva di reazioni
avverse; essa indica la comparsa di una reazione avversa durante la somministrazione di un determinato farmaco
e non deve essere quindi una valutazione certa di correlazione tra le reazione avversa e il farmaco somministrato,
ma serve per implementare un sistema di monitoraggio centralizzato (banca dati europea Eudravigilance) che,
da analisi statistiche sulle segnalazioni sovrapponibili, trae le eventuali conseguenze per ulteriori approfondimenti
post marketing o
alert a carattere generale che possono arrivare anche al ritiro di un prodotto dalla
commercializzazione.
Gli RMPs delle eritropoietine biosimilari sono rivolti a monitorare il rischio di immunotossicità, in particolare le
reazioni avverse per formazione di anticorpi anti-Epo , alla luce di passati casi di aplasie midollari conseguenti alla
somministrazione di Epo sottocute . Viene inoltre monitorato il rischio di tromboembolismo venoso, embolie
polmonari e altre reazioni avverse.
Pertanto la segnalazione di sospette reazioni avverse è uno strumento fondamentale per la valutazione dei
rapporti rischio \beneficio dei farmaci, nelle reali condizioni di impiego territoriale.
La segnalazione va trasmessa al Responsabile di Farmacovigilanza della struttura sanitaria di appartenenza o,
qualora provenga da clinici operanti presso strutture sanitarie private, al Responsabile di Farmacovigilanza della
ASL competente per territorio.
19
Scaricare