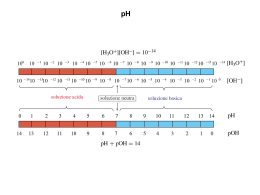
ANALISI QUANTITATIVA: valutare quanta sostanza è presente nel campione. TRATTAMENTO DEI DATI Misura con accuratezza minima = misura limite È assolutamente inutile cercare di fare misure più accurate. CIFRE SIGNIFICATIVE Numero di cifre necessario per esprimere i risultati di una misurazione consistenti con la precisione misurata. NOTA: lo zero! Cifra 0: - SIGNIFICATIVA - POSIZIONA IL PUNTO DECIMALE 92.067 → 5 cifre significative 0.92067 →“ “ “ 0.092067 → “ “ “ (lo zero prima della virgola non è significativo) Stesso numero di cifre significative. Ci sono differenti “unità” per esprimere una misura (Es. millimetri, decimetri, metri). Es. 727.0 LO ZERO È SIGNIFICATIVO? Risposta: Sì Se lo zero precede un punto decimale può sorgere ambiguità. Se cade tra due numeri interi È SEMPRE SIGNIFICATIVO. 936.600 → 1 o 2 zeri significativi? 9.3660 · 105 → 6 cifre ma 5 SIGNIFICATIVE OPERAZIONI PRODOTTO E DIVISIONE (· E :) Misure con cifra stimata incerta. Ultima cifra significativa della misura. 1 Nella · e : questa incertezza è trasmessa attraverso le operazioni matematiche. Nella risposta di una · o :, c’è un grado di incertezza relativa che c’è nell’operatore con il minimo grado di certezza, cioè quello con il minimo numero di cifre significative, numero che costituisce il limite come numero chiave. A parità di cifre significative sarà il numero con valore assoluto più piccolo a prescindere dal punto decimale. Perché? Perché la sua incertezza è più grande. SOMMA E SOTTRAZIONE (+ E -) Nelle operazioni + e – le cifre significative vengono determinate dal posizionamento del punto decimale. NON SI PUÒ DARE UN RISULTATO CON UNA PRECISIONE MAGGIORE DELLA INCERTEZZA MASSIMA ASSOCIATA AD UNO DEI DATI IN CONSIDERAZIONE PER L’ANALISI. IL RISULTATO DEVE ESSERE CONSISTENTE CON LE CIFRE SIGNIFICATIVE. Campione 20% ± 2 Errore 10% Pesata 2 g ± 0.1 Errore 5% Non è necessario pesare il campione con un’accuratezza maggiore di 0.1 g. A quale accuratezza si può eseguire una misura? Es. se si legge una misura con 3 cifre è inutile pesare il campione con più di tre cifre. ARROTONDAMENTO Numero successivo all’ultima cifra > 5 si arrotonda cifra superiore. Se < 5 si arrotonda al valore dell’ultima cifra presente. 9.47 = 9.5; 9.43 = 9.4 Se l’ultima cifra è un 5? Il numero è arrotondato alla più vicina cifra pari. 8.65 = 8.6; 8.75 = 8.8; 8.55 = 8.6 2 ERRORI DETERMINATI Possono essere evitati o corretti o che se ne possa tener conto: • Perdita solubilità precipitato • Pesi starati; vetreria starata • Errori dell’operatore • Pregiudizio nella stima delle misure • Reazioni collaterali • Reazioni incomplete LOGARITMI Log a · 10b b = caratteristica Log a = mantissa (parte decimale) Es. pH = - Log [H+] Soluzione: 2.0 · 10-3 M. HCl → pH? Risposta: pH = 2.70 (3 cifre significative) Tutti gli zero nella mantissa sono significativi Log10 N = b → N = 10b = antilog b NOTA: nel passaggio da log ad antilog e viceversa il numero su cui si deve operare e la mantissa del logaritmo hanno lo stesso numero di cifre significative. pH = - log 2.0 · 10-3 = - (-3 + 0.30) = 2.70 -3 è la caratteristica; un numero puro determinato dalla posizione del decimale. L’ACCURATEZZA è il grado di accordo tra un valore misurato ed il valore vero. La PRECISIONE è il grado di accordo tra misure ripetute della stessa quantità e non implica necessariamente l’accuratezza. 3 MODALITÀ DI ESPRESSIONE DELLA PRECISIONE Si esprime tramite s o σ S o σ = DEVIAZIONE STANDARD σ teorica; s reale Se Xi sono valori misurati valore medio N = numero di misure effettuate Se N → ∞ allora → µ = valore vero. In pratica N è un numero finito e µ viene approssimato con la media . Allora S = σ Per N misure → N deviazioni variabili riferite ad un numero di riferimento. Ma se questo numero è la ∑ deviazioni = 0, quindi le deviazioni indipendenti da sono N – 1 → 1 grado di libertà per corregge la differenza tra ad N – 1 per la precisione. In pratica N – 1 e µ. ERRORI INDETERMINATI (accidentali o casuali) Piccole differenze in misure successive condotte dallo stesso analista in condizioni virtualmente identiche e che non possono essere previsti o stimati. DISTRIBUZIONE CASUALE Si possono applicare le leggi matematiche della probabilità. Gli errori indeterminati dovrebbero seguire una distribuzione normale o curva Gaussiana. L’area totale della curva rappresenta tutti i valori della popolazione analizzata. 4 S = deviazione standard: il 68% dei valori cadono tra ± S, il 95% tra ±2S Spesso si usa il C.V. o RSD (coefficiente di variazione o deviazione standard relativa), deviazione standard espressa come percentuale della media. MODALITÀ DI ESPRESSIONE DELL’ACCURATEZZA ERRORE ASSOLUTO. Differenza tra il valore vero e il valore misurato con riguardo al segno. Es 100 ±1; 10 ± 1 ± 1 = ERRORE ASSOLUTO ERRORE MEDIO. Se il valore misurato è la media di molte misure. ERRORE RELATIVO. vero. ACCURATEZZA Errore assoluto o medio espresso come percentuale del valor Es 1/100; 1/10 RELATIVA. Valore misurato o medio espresso come percentuale del valore vero. Es. Buretta 50.0 ml Errore di lettura = 0.05 ml (errore assoluto) La precisione è diversa dall’accuratezza! 5 LIMITE DI CONFIDENZA Deviazione standard S, significato: • Il 68% delle singole deviazioni cade all’interno di 1 VOLTA (S) la deviazione standard dalla media. • Il 95% a meno di 2 VOLTE (S) ed il 99% a meno di 2.5 VOLTE (S). Ciò è vero per molte misure ed in assenza di errori determinati. Per pochi dati la media può non coincidere con µ (vero valore medio). Possiamo però stimare l’intervallo all’interno del quale il valore medio potrebbe cadere con una data probabilità definita dalla (media sperimentale) e S (deviazione standard). I limiti di questo intervallo sono chiamati limiti di confidenza (L) Dove t = fattore statistico funzione del livello di confidenza desiderato e dei gradi di libertà N considerati. 6 SCARTO DI UN RISULTATO Base pratica: - Esperienza - Senso comune Se ci si aspetta una σ di un metodo si scarteranno i punti al di fuori della media di 2σ. Ci sono molti test statistici per determinare la validità di uno scarto. È difficile determinare un intervallo dove devono cadere le osservazioni statisticamente significative. TEST Q → per numero di misure piccolo. Dati: ordine decrescente di valore X = numero sospetto Y è il numero più vicino. Questo rapporto è confrontato con valori tabulati di Q. Se il valore è ≥ di quello tabulato allora si scarta. La tabella è data con un limite di confidenza del 90 %. Quoziente di scarto, Q al limite di confidenza del 90%a N° di osservazioni Q 3 0.94 4 0.76 5 0.64 6 0.56 7 0.51 8 0.47 9 0.44 10 0.41 ∞ 0.00 a Adattato da R. B. Dean e W. J. Dixon, Anal. Chem., 23 (1951), 636. Questo test è più affidabile per più di 5 osservazioni, non si può invece usare per 3 osservazioni di cui 2 uguali. 7 MINIMI QUADRATI LINEARI CURVA DI CALIBRAZIONE Linea retta tracciata con semplice valutazione ad OCCHIO della miglior linea retta. y=mx+d Statisticamente si dimostra che la miglior retta attraverso una serie di valori sperimentali è quella per cui la somma dei quadrati delle deviazioni dei punti dalla linea stessa è MINIMA. COEFFICIENTE DI CORRELAZIONE Misura la correlazione tra due variabili. Nota. Le variabili x e y non sono direttamente dipendenti tra di loro ma correlate. Non si parla del miglior valore di y per un dato x ma di quello più probabile. Più vicini sono i valori osservati ai valori più probabili più definita è la relazione tra x e y. Si esprime con r e si calcola con la CALCOLATRICE. r = 1 → esatta correlazione; r = 0 → x e y INDIPENDENTI 8 BILANCIA ANALITICA Pesate 3 - 4 cifre significative. Dispositivo accurato e sensibile. Bilancia analitica = leva di 1° grado che confronta due MASSE! Peso di un oggetto = forza esercitata su di esso dalla attrazione gravitazionale. La massa non varia; IL PESO SÌ = L1; = L2 All’equilibrio M1L1 = M2L2 Se L1 e L2 sono il più possibile uguali allora M1 = M2 Per bilance a bracci uguali: Si usa quindi il termine PESO. NOTA: le masse note vengono chiamate PESI STANDARD Bilance: Due o singolo piatto ↓ ↓ lenta rapida Le due bilance si equivalgono in termini di precisione e accuratezza. BILANCIA TECNICA → pesa fino al decimo di grammo. BILANCE ANALITICHE → accuratezza 0.1 mg (“range” fino a 200 g) Semimicro - sensibile entro 0.01 mg. Micro - sensibile entro 0.001 mg. I limiti di carico sono ovviamente inferiori. Si usano con molta cura. NOTA: controllo dello zero per ∆T; umidità e elettrostasticità almeno ogni 30’ Errori: deriva punto zero; ∆T, pesi, controspinta. Il ∆T dipende dall’ambiente e dall’oggetto. Correnti d’aria convettive → varia lo ZERO 9 REGOLE DI PESATA • Non toccare pesi e oggetti. Usare pinze o forcipi. • Pesare a T ambiente. • Non mettere prodotti chimici direttamente sui piatti. • Usare recipienti idonei (pesafiltri). • Chiudere lo sportello della bilancia (specie se molto sensibile). • Introdurre i pesi ed i campioni con delicatezza ed in sequenze predeterminate. • Non lasciare pesi sulla bilancia una volta terminata la pesata. • Riposizionare il cursore o lo zero. 10 DOSAGGIO H2O DI BaCl2 · 2 H2O (cloruro di bario bi-idrato) MATERIALE: pesafiltri, stufa elettrica termoregolata fino a 150°C. A 150°C il BaCl2 · 2 H2O perde l’H2O! Principio: DETERMINAZIONI DI UMIDITÀ Procedura • Pesare il pesafiltri • Porlo in stufa a 150°C per 15 - 20 minuti • Il pesafiltri deve essere aperto con il coperchio appoggiato sulla parte superiore del pesafiltri • Si pone il pesafiltri nell’essiccatore e si lascia raffreddare per 15 minuti • Si apre l’essiccatore dopo averlo portato vicino alla bilancia analitica • Immediatamente si chiude il pesafiltri con il coperchio • Si pone nella bilancia che è stata preventivamente azzerata • Si pesa aspettando un tempo tale che la bilancia dia un peso costante • Si pone di nuovo il pesafiltri nell’essiccatore • Si porta vicino alla stufa • Si toglie il coperchio e si mette di nuovo in stufa NOTA: non toccare il pesafiltri con le mani! SI RIPETE LA PROCEDURA SINO A COSTANZA DI PESO. Dopo aver portato a costanza di peso il pesafiltri si pesa una quantità di cloruro di bario compresa tra 0.8 – 1.0 g (distribuire uniformemente il BaCl2 · 2 H2O nel pesafiltri). Si ripete il procedimento sino a costanza di peso. CALCOLI: Peso iniziale di BaCl2 · 2 H2O = x (g) Peso dopo perdita di H2O = b (g) H2O eliminata = x – b (g) Es. Peso iniziale BaCl2 · 2 H2O = 0.9824 g 11 Peso dopo riscaldamento 150°C = 0.8378 g H2O eliminata = 0.1446 g Valore vero: P.M. BaCl2 · 2 H2O = 244.31 P.M 2 H2O = 36.03 Errore: 0.03% Esempio H2O di cristallizzazione Peso iniziale di BaCl2 · 2 H2O = g. 0.9824 Peso sostanza dopo riscaldamento = g. 0.8378 H2O eliminata = g. 0.1446 TEORICO P.M. BaCl2 · 2 H2O = 244.31 P.M 2 H2O = 36.03 12 CONCENTRAZIONE SOLUZIONI MOLARITÀ Soluzione 1 molare → 1 MOLE / LITRO Si scioglie una mole di sostanza nel solvente e si diluisce a volume finale 1 litro. Molare si abbrevia in M o . Una soluzione di AgNO3 1 M ed una soluzione di NaCl 1 M reagiranno sulla base di volumi uguali. In generale → moli = molarità · litri Nella pratica di laboratorio si lavora con i millilitri. Quindi: millimoli = molarità · millilitri NORMALITÀ Utile in analisi quantitativa. Una soluzione 1 normale → 1 1 equivalente = 1 mole · numero di unità reagenti per ciascuna molecola o atomo. Acidi, basi → unità reagenti è basato sul numero di protoni (cessione acida - reazione basica) Redox = numero di elettroni che un ossidante o riducente prenderà o fornirà. Es. H2SO4 → due unità reagenti (protoni). Ci sono 2 equivalenti di protoni per mole. Una soluzione 1 M di H2SO4 → 2 N. Il peso equivalente è la metà del Peso Molecolare. Precipitazioni. Unità reagente: carica ione metallico. Complessazioni. Peso equivalente agente precipitante o complessante = FORMALITÀ Usato per soluzioni di sali ionici che non esistono come molecole nel solido o in soluzione. Operativamente formalità = molarità. 13 MOLALITÀ Una soluzione UNO-MOLALE contiene una mole in 1000 g di solvente. Poiché le proprietà colligative (abbassamento crioscopico, innalzamento ebullioscopico, abbassamento tensione di vapore, pressione osmotica) dipendono solamente dal numero di particelle di soluto presenti in soluzione per mole di solvente, le misure chimico fisiche di queste proprietà vengono effettuate utilizzando la molalità. NOTA: le concentrazioni molali non sono dipendenti dalla temperatura. Perché? DENSITÀ: CALCOLI % in peso = peso analita espresso in grammi per 100 grammi di campione % (P/P) = g / g · 100% Acidi e basi commerciali sono forniti come % (P/P). Spesso di queste sostanze si devono preparare soluzioni MOLARI o NORMALI. Necessita quindi la alla T specificata a 20°C % in peso · densità = % volume % volume = grammi di analita per 100 ml di campione Se l’analita è un liquido sciolto in un altro liquido si può usare il VOLUME/VOLUME (ml / 100 ml campione). SITUAZIONE POCO FREQUENTE! ANALISI DI UN GAS: P / P; P / V o V / V UNITÀ ppm ppb mg % P/P (mg / kg) (µg / kg) mg / 100 g P/V mg / L µg / L mg / 100 ml UNITÀ COMUNI PER L’ESPRESSIONE DI CONCENTRAZIONE IN TRACCE NOTA: soluzioni di uguale concentrazione su una base P/P o P/V non hanno lo stesso numero di molecole, mentre lo hanno soluzioni della stessa molarità. 14 VETRERIA PER ANALISI VOLUMETRICA • Matracci • Cilindri • Pipette • Micropipette • Pipette a siringa con puntale intercambiabile matraccio cilindro pipetta tarata pipetta graduata Classe A = errore 1 ‰ TC = per contenere T.d. = per erogare BURETTE 50 ml 10 ml 2 ml (micro) 100 ml (macro) 0.1 ml (ultramicro) Essiccatori CaCl2 MATERIALI E REAGENTI • Vetro al borosilicato (PYREX; KIMAX): 200°C; T esercizio ∆T ≈ 150°C • Porcellana 1100°C – 1400°C • Pt (platino) 1500°C • Acciaio 400°C – 500°C 15 • Polietilene 115°C • Polistirene 70°C • Teflon 250°C CLASSIFICAZIONE PRODOTTI CHIMICI Tecnica; C.P. = chimicamente puro U.S.P. = United States Pharmacologia A.C.S. = Alta Purezza (American Chemical Society) Standard primario → massima purezza REAGENTE H2SO4 HCl H3PO4 C2H4O2 NH3 HNO3 PF 98.08 36.5 98.00 60.0 17.03 63.01 M 17.4 12.4 14.7 17.4 14.8 15.4 %P 94.0 38.0 85.0 99.5 28.0 69.0 D (20°C) g / cm3 1.831 1.188 1.689 1.051 0.898 1.409 Forni (500 – 1200 °C) e stufe (200 – 300 °C) Cappe (a flusso laminare = aria prefiltrata) Bottiglie di lavaggio (spruzzette), crogiuoli, filtri, tecniche di filtrazione. 16 PREPARAZIONE SOLUZIONI Campionamento ↓ Preparazione soluzione ↓ Essiccamento del campione Es. Evaporazione H2O stufa 105 – 110°C NOTA: IL CAMPIONE NON DEVE DECOMPORSI! ↓ Scioglimento del campione Distruzione totale o parziale della matrice. Es. proteine → Kjeldahl, N organico in NH4+ Acidi minerali forti sciolgono sostanze inorganiche. HCl buono in generale per metalli. HNO3 è forte ossidante buono per: METALLI, LEGHE NON FERROSE, SOLFURI HClO4 se disidratato è un forte agente ossidante, scioglie metalli comuni e distrugge le tracce di sostanze organiche. Fusione con un fondente per materiali inorganici insolubili in acidi Na2CO3 → UTILE FONDENTE BASICO (carbonati solubili negli acidi). 17 PREPARAZIONE DELLE SOLUZIONI A TITOLO NOTO Standard (si comprano); a titolo controllato. SOLUZIONI «STANDARD» Si possono preparare per: A) DISSOLUZIONE DI SOSTANZE «MADRI» B) DILUIZIONE DI SOLUZIONI PIÙ CONCENTRATE A TITOLO NOTO La sostanza madre pesata in quantità opportuna in un pesafiltri viene trasferita in matraccio tarato e portata a volume. La normalità si calcola con la formula seguente: g1 p.eq.1 g2 p.eq.2 Ricordare che: Se N1 = N2 allora Se noto il volume di diluizione, il peso della sostanza disciolta ed il relativo peso equivalente, la normalità può essere ricavata tramite la seguente formula: sono i grammi necessari per preparare un volume V in ml di una soluzione di normalità N. 18 Se N1, V1, g1 e N2, V2, g2 1000 si riferiscono alla soluzione da titolare e alla titolante Quindi V1 · N1 = V2 · N2 Tutte queste equazioni valgono quando una titolazione risulta ultimata, cioè quando si è raggiunto il punto stechiometrico della reazione o PUNTO DI EQUIVALENZA (teorico) in corrispondenza del quale si ha l’uguaglianza tra il numero di equivalenti delle sostanze reagenti. n° eq. sostanza da titolare = n° eq. sostanza titolante NOTA: la quantità di un sale poco solubile che si scioglie dipende dal SOLVENTE Ag2CrO4 → Kps = [Ag+]2 [CrO42-] Non si scioglie o dissocia a strati perché sono ELETTROLITI FORTI. Poiché il prodotto Kps è sempre valido, la precipitazione di una specie non avrà luogo a meno che il prodotto delle concentrazioni degli ioni che concorrono a formarla non superi la Kps. Se c’è un eccesso di uno ione rispetto all’altro la concentrazione di quest’ultimo viene repressa (effetto ione comune) e la solubilità del precipitato risulta diminuita. 19 DISTRUZIONE SOSTANZE ORGANICHE INCENERIMENTO Tecnica più usata. Pb; Zn; Co; Sb; Cr; Mo; Sr; Fe. RECUPERI QUANTITATIVI. CROGIUOLO PORCELLANA o Pt Si aggiungono ossidanti Mg(NO3)2 Si recupera As, Cu, Ag. INCENERIMENTO A BASSA T ≈ 100°C Una scarica di radiofrequenze produce radicali O2 che distruggono le sostanze organiche. Vantaggi: minore volatilizzazione Analisi C e H. Si esegue combustione di O2: C → CO2 H → H2O Assorbimento su tubi pre-pesati. DIGESTIONE UMIDA MISCELA HNO3, H2SO4 sino a fumi SO3 SOTTO CAPPA! HNO3 HClO4 3 : 1 \ / OSSIDANTI : H2SO4 1 CALDO Parziale distruzione sostanze di natura organica. Estrazione metalli in tracce dai terreni, estratti con NH4Cl o CH3COOH Selenio distillato come Br o Cl volatili. FILTRATI. Precipitazione di proteine CCl3 – COOH, Ba(OH2)2, ZnSO4 20 TITOLAZIONE DI ACIDI FORTI CON BASI FORTI A 25°C Kw = 1.05 · 10-14 costante di dissociazione piccola che assicura il decorso quantitativo della reazione. Poiché acidi e basi forti sono completamente dissociati l’unico equilibrio è la dissociazione di H2O, quindi è facile calcolare il pH durante la titolazione noti la quantità di acido o di base residua ed il volume della soluzione. Es. 100 ml acido forte di normalità Na si aggiunge un volume (A ml) di base della stessa normalità (Na = Nb). Si hanno le seguente condizioni: (100 – A) 100 + A Per acidi diluiti e vicino al punto di equivalenza si deve tener conto del contributo degli H+ provenienti dall’acqua. In prossimità dell’equivalenza: [H+]TOT = [H+]NON TITOLATI + [H+]H2O Ma [H+]H2O = allora [H+]2TOT - [H+]NON TITOLATI [H+]TOT – Kw = 0 Esempio: 100 ml acido forte 0.001 N + 99.99 ml base forte 0.001 N (100 – 99.99) – 0.001 100 + 99.99 Kw [H+]TOT = [H+]NON TITOLATI + [H+]H2O 21 [H+]TOT = 1.28 · 10-7 → pH = 6.89 Senza il contributo dell’acqua… [H+]TOT = 5 · 10-8 → pH = 7.3!!! Si abbiano 100 ml di una soluzione 0.1 N di HCl che debbono essere titolati con NaOH 0.1 N. All’inizio della titolazione, prima che si proceda ad aggiunta di NaOH, la soluzione è 0.1 N in HCl; il pH è quindi 1. quando si sono aggiunti 50 ml di NaOH 0.1 N, rimangono ancora 50 ml di HCl 0.1 N in un volume di 150 ml. La normalità dell’HCl nella soluzione è pertanto ml1N1 = ml2N2 50 · 0.1 = 150 · X da cui X = 0.033 N e pH = 1.5 Quando si sono aggiunti ml 99.8 di NaOH 0.1 N, la normalità della soluzione è cc NaOH aggiunti 0 50 90 98 99 99.8 99.9 100 equiv. 100.1 100.2 101 102 110 HCl 1 N 0 0.5 0.13 2.0 2.3 3.0 3.3 7 10.7 11.0 11.7 12.0 12.7 pH soluzioni HCl 0.1 N HCl 0.01 N 1.0 2.0 1.5 2.5 2.3 3.3 3.0 4.0 3.3 4.3 4.0 5.0 4.3 5.3 7 7 9.7 8.7 10.0 9.0 10.7 9.7 11.0 10.0 11.7 10.7 Con calcolo analogo per un’aggiunta di ml 99.9 di NaOH 0.1 N, il pH diventa 4.3. All’equivalenza [H+] = [OH-] per cui pH = 7. Per aggiunta di 100.1 di NaOH 0.1 N si ha in soluzione un eccesso di ioni OH-; la concentrazione di ioni ossidrili risulta: 22 Valori relativi alla [H+] ed al pH di soluzioni di HCl rispettivamente 1; 0.1; 0.01 N, titolate con soluzioni di NaOH di uguali normalità: ml NaOH aggiunti 0.00 50.00 90.00 98.00 99.00 99.80 99.90 100.00 100.10 100.20 101.00 102.00 110.00 Soluzione 1 N [H+] pH 1.0 0.00 -1 3.3 · 10 0.48 -2 5.3 · 10 1.28 -2 1.0 · 10 2.00 -3 5.0 · 10 2.31 -3 1.0 · 10 3.00 -4 5.0 · 10 3.30 -7 1.0 · 10 7.00 -11 2.0 · 10 10.70 -11 1.0 · 10 11.00 -12 2.0 · 10 11.70 -12 1.0 · 10 12.00 -13 2.0 · 10 12.70 Soluzione 0.1 N [H+] pH 1.0 1.00 -1 3.3 · 10 1.50 -2 5.3 · 10 2.28 -2 1.0 · 10 3.00 -3 5.0 · 10 3.30 -3 1.0 · 10 4.00 -4 5.0 · 10 4.30 -7 1.0 · 10 7.00 -11 2.0 · 10 9.70 -11 1.0 · 10 10.00 -12 2.0 · 10 10.70 -12 1.0 · 10 11.0 -13 2.0 · 10 11.70 Soluzione 0.01 N [H+] pH 1.0 2.00 -1 3.3 · 10 2.50 -2 5.3 · 10 3.28 -2 1.0 · 10 4.00 -3 5.0 · 10 4.30 -3 1.0 · 10 5.00 -4 5.0 · 10 5.30 -7 1.0 · 10 7.00 -11 2.0 · 10 8.70 -11 1.0 · 10 9.00 -12 2.0 · 10 9.70 -12 1.0 · 10 10.00 -13 2.0 · 10 10.70 PERCENTO DI NEUTRALIZZATO CURVE DI TITOLAZIONE DI NORMALITÀ HCl 1 N, 0.1 N, 0.01 N CON NaOH DELLA STESSA **** 23 INDICATORI DI NEUTRALIZZAZIONE Sono sostanze organiche che, oltre ad essere acidi o basi deboli, hanno la proprietà di avere la forma indissociata di un colore diverso da quello della forma dissociata. Il colore della soluzione in cui è presente uno dei tali indicatori dipenderà dal suo pH. Infatti lo ione H+ condiziona il rapporto tra la forma dissociata ed indissociata. Per un indicatore acido debole, HIn, si avrà: Applicando la legge delle masse: Il rapporto Kin [H+] [In-] [HIn] è determinato dalla [H+] della soluzione. Questo rapporto ci dà il colore della soluzione. NOTA: la variazione di colore non avviene bruscamente ma gradualmente con il variare della [H+]. Se in una soluzione vi sono due specie colorate, l’occhio umano percepisce il colore di quella la cui concentrazione è almeno 10 volte superiore a quella dell’altra, quindi una variazione di colore di una soluzione (VIRAGGIO) potrà essere avvertita solo quando: In logaritmi → pH = pKIn ± 1 Quali considerazioni si possono fare? 1. viraggio indicatore confinato entro ± 1 pH (due unità) 24 2. ogni indicatore ha un determinato campo di pH in cui esso varierà sensibilmente colore 3. se pH = pKind allora l’indicatore sarà 50% [HIn] e 50% [In], la soluzione quindi avrà un colore intermedio. INDICATORI BASI DEBOLI InOH In+ + OH- Kw influisce sul campo di pH entro il quale si ha viraggio. CAMPI DI VIRAGGIO DEI PIÙ COMUNI INDICATORI A 18°C **** 25 CURVE DI TITOLAZIONE DI HCl 1 N; 0.1 N; 0.01 N CON NaOH DELLA STESSA NORMALITÀ (DA -2% A +2% DI BASE AGGIUNTA) **** Sostanza madre (standard primario) Sostanza che permette di preparare una soluzione contenente un numero di equivalenti deducibile dal PESO della sostanza Requisiti: - elevata purezza (consentita solo umidità eliminabile per essiccamento a 105110°C) - inerzia chimica - elevato peso equivalente - reazione chimica ben definita e rapida 26 Esempi: - metalli (Ag, Cu, Fe elettrolitici) - ossidi: HgO, ZnO - acidi: H2Cr2O4 · 2 H2O - sali: FeSO4, KHC8H4O4, KH(IO3)2, KIO3, K2Cr2O7, NaCl, Na2C2O4, Na2B4O7 · 10 H2O PREPARAZIONE NaOH ≈ 0.1 N - si pesano rapidamente su bilancia tecnica 4 – 5 g NaOH in pastiglie - si lava NaOH con un getto di H2O distillata, per eliminare il carbonato in superficie - si trasferisce NaOH lavato in bottiglia da 1 litro, contenente 500 ml H2O bollita per eliminare la CO2 - dopo completa solubilizzazione della soda si porta a volume con la stessa H2O bollita e si travasa in bottiglia di polietilene. STANDARDIZZAZIONE NaOH ≈ 0.1 N CON FTALATO ACIDO DI POTASSIO Reattivo: KHC8H4O4, p. eq. = 204.22 Indicatore: fenolftaleina Reazione: OH- + HC8H4O4- C8H4O42- + H2O Procedimento: - si pesano esattamente 0.6 – 0.7 g di ftalato già portato a peso costante (120°C); - si aggiungono 3 – 4 gocce di indicatore; 27 - si titola con la soda sino a viraggio da incolore a rosa. TITOLAZIONE HCl CON NaOH STANDARD Reattivo: NaOH a titolo noto! Indicatore: rosso metile, metil arancio, FENOLFTALEINA Procedimento: - dopo aver lavato 2 – 3 volte la buretta con poco acido, si riempie e si azzera!; - si preleva in beuta da 250 ml un volume esattamente noto di HCl (30 – 40 ml); - si aggiungono 2 – 3 gocce di indicatore; - si titola con NaOH sino a viraggio dell’indicatore. N.B. la buretta va lavata con H2O, H2O distillata; avvinata con NaOH, azzerata dopo averla riempita! 28 TITOLAZIONI ACIDI DEBOLI CON BASI FORTI Ci sono due equilibri di dissociazione: Il pH della soluzione è pertanto funzione dei due equilibri che hanno H+ in comune. Si potrà quindi titolare un acido debole con una base forte solo se l’acido è molto più dissociato dell’acqua (Ka >> Kw) Inoltre, la sua concentrazione deve essere tale che Ka · C ≥ 10-7 – 10-8 Condizione iniziale: in soluzione è presente solo l’acido debole ed il contributo di H+ dell’acqua è trascurabile: HA H+ + A- [H+] = [A-] e [HA] = Ca Dalla (1) [H+]2 = Ka · Ca ; pH = ½ pKa – ½ log Ca Prima del punto di equivalenza si ha [H+] < [A-] E ponendo [HA] non tit = Ca e [A-] = Cs Al punto di equivalenza l’equilibrio dominante è l’idrolisi di AA- + H2O HA + OH- Questa idrolisi è condizionata dalla dissociazione di HA e di H2O per cui si avrà: . Quindi sostituendo questo valore nel Ka… pH = ½ pKa + ½ pKw + ½ log Cs Dopo il punto di equivalenza, sarà presente il sale ed un eccesso di base, se gli OH- in eccesso reprimono l’idrolisi la base regolerà il pH. 29 VARIAZIONE DEL pH DURANTE LA TITOLAZIONE DI ACIDI DEBOLI MONOPROTICI CON DIVERSO Ka CON BASE FORTE (NaOH) DELLA STESSA NORMALITÀ Acido 0.1 N Acido 0.1 N -5 Ka = 1.8 · 10 Ka = 1 · 10-7 pH pH 0.0 2.87 4.0 10.0 3.80 6.0 50.0 4.75 7.0 90.0 5.70 8.0 99.0 6.75 9.0 99.8 7.45 9.62 99.9 7.75 9.78 PE 100.0 8.72 9.85 100.1 9.7 10.0 100.2 10.0 10.19 100.3 10.7 10.7 Il pH di equivalenza è spostato in ambiente alcalino e lo è tanto più quanto più ml NaOH 0.1 N aggiunti piccola è la Ka dell’acido e la concentrazione del sale. La titolazione di HA debole si può eseguire utilizzando un indicatore che viri in ambiente alcalino (FENOLFTALEINA) **** CURVA DI NEUTRALIZZAZIONE DI UN ACIDO DEBOLE MONOPROTICO (KA = 1.8 · 10-5) 0.1 N CON NaOH 0.1 N NOTA: il salto di pH vicino all’equivalenza è ridotto (per l’idrolisi del sale) rispetto a quello che si verifica nel caso di acidi forti di concentrazione uguale. 30 TITOLAZIONE DI ACIDI POLIPROTICI Stessa regola per questi acidi considerati come miscele di acidi deboli. 1) Si titolano solo gli H+ il cui equilibrio di dissociazione determina una costante di valore ≥ 10-7 purchè sia Ca > 10-1 M Ka · Ca > 10-7 ≈ 10-8 2) Titolare selettivamente i singoli H+ dipende dal rapporto delle costanti K1/K2 = 104 Esempio H2CO3 H2O + CO2 H2CO3 H+ + HCO3- K1 = 4 · 10-7 HCO3- H+ + CO32- K2 = 6 · 10-11 A p = 1 atm e 25°C, H2O è satura di CO2 (0.03%), allora [H2CO3] = 5 · 10-2 M K1 · Ca = 4 · 10-7 · 5 · 10-2 = 2 · 10-8 TITOLABILE K1 / K2 = 104 OK Ma solo il primo H+. Titolazione: a) soluzione di H2CO3 satura di CO2 (C.I.); b) soluzione tampone H2CO3 / HCO3-; c) soluzione HCO3- (primo punto di equivalenza: 1 equivalente di NaOH per mole di H2CO3); d) tampone dopo l’equivalenza HCO3- / CO32-; e) soluzione CO32- (secondo punto di equivalenza: 2 eq. NaOH per mole H2CO3) f) Soluzione CO32- + OH- eccesso. a) H2CO3 H+ + HCO3- [H+] + [HCO3-] pH = ½ pKa – ½ log Ca 31 K1 = 4 · 10-7 Ca = 5 · 10-2 pH = 3.85 b) aggiunta 25% NaOH della stessa normalità di H2CO3 allora [H2CO3] = [HCO3-] Per aggiunta del 45% di NaOH c) aggiunta del 50% di NaOH: H2CO3 ha reagito con la base 1 : 1 d) dopo questo punto le specie in soluzione HCO3- e CO32- debbono soddisfare l’equilibrio regolato dal K2, quindi dopo l’aggiunta di 55% NaOH si ha 45% HCO3- e 5% CO32-. Al 75% di NaOH si ha 25% HCO3- e 25% CO32-. [H+] = K2; pH = pK2 = 10.22 e) per aggiunta 95% NaOH si ha 5% HCO3- e 45% CO32-. Aggiunta 100% NaOH tutto l’acido è salificato e l’equilibrio in soluzione sarà: CO32- + H2O HCO3- + OH- [OH-] = [HCO3-] = Kw [H+]TOT K2 = [H+]TOT [CO32-] [HCO3-] [H+]TOT2 = K2 Kw Ma ora C = 2.5 · 10-2 M → pH = 11.30 f) aggiunta 101.0% NaOH 0.1 M → pH = 11.36 32 ml NaOH 0.1 N pH 0 25 45 50 55 75 95 100 101 3.85 6.40 7.35 8.31 9.25 10.22 11.17 11.30 11.36 Variazione del pH durante la titolazione di H2CO3 0.1 N con NaOH 0.1 N **** 33 TITOLAZIONE DEI CARBONATI H2CO3 – K1 = 4.2 · 10-7 – K2 = 4.8 · 10-11 pK1 = 6.38 pK2 = 10.32 Si consideri 50 ml di Na2CO3 0.1 M, titolato con HCl 0.1 M - aggiunta ml HCl = 0 pH = ½ (14 + 10.32 – 1) = 11.66 - durante la titolazione - al primo punto stechiometrico: ↓ Seconda parte della titolazione - Al secondo punto stechiometrico La soluzione H2CO3 0.033 M!! **** 34 Esempio: Reattivo: Na2CO3 p.eq. = 53 Indicatore: rosso metile (metilarancio) Reazione: CO32- + H+ HCO3- + H+ HCO3H2O + CO2 Procedimento: - si pesano esattamente circa 0.2 g di Na2CO3 - si trasferisce in beuta da 250 ml lavando accuratamente il vetrino e si aggiungono circa 100 ml di acqua distillata di recente - si aggiungono 2 – 4 gocce di rosso metile e si titola con HCl (AVVINARE!) in buretta sino al viraggio da giallo a rosa-arancio - si fa bollire per 2 minuti (si elimina CO2) - si raffredda e si aspetta che ricompare il giallo - si prosegue la titolazione (cautamente) sino a ricomparsa del colore rosaarancio. - si ripete la procedura sino a non ricomparsa del colore giallo CALCOLO DEL pH DI UN ANFOLITA (HCO3-) In generale si consideri una soluzione di molarità C di un anfolita tipo NaHA. Le concentrazioni di Na+; H+; HA-; A2-; OH- e H2A sono così collegate: BM (Na) C = [Na+] = [H2A] + [HA-] + [A2-] EN [H+] + [Na+] = [OH-] + [HA-] + 2 [A2-] BM – EN e ricavando [H+] si ha: [H+] = [OH-] + [A2-] – [H2A] [H+] + [H2A] = [OH-] + [A2-] 35 K1 [H+]2 + [H+]2 [HA-] = KwK1 + K1K2 [HA-] ESPRESSIONE RICAVATA SENZA ALCUNA APPROSSIMAZIONE!!! Approssimazioni: se si assume che le reazioni per cui da HA- si hanno A2- e H2A sono molto modeste si può approssimare HA- = C quindi Se K1 « C e C K2 » Kw allora Es. Calcolare il pH di una soluzione 0.01 M di NaHS pH = 10 – log 3.4 = 9.47 Applicando la formula più approssimata: pH = 10 – log 1.1 = 9.94 TITOLAZIONE H3PO4 K1 = 7.5 · 10-3 H3PO4 H+ + H2PO4- K2 = 6.2 · 10-8 H2PO4- H+ + HPO42- debole K3 = 5.0 · 10-13 HPO42- H+ + PO43- media forza debolissimo K1/K2 e K2/K3 > 10-4 36 Esiste la possibilità di titolare successivamente i vari H+ purché nelle immediate vicinanze dei rispettivi punti di equivalenza il sistema tampone formativi non limiti eccessivamente la variazione del pH. Punti di equivalenza 1) pH = ½ pK1 + ½ pK2 = 4.6 2) pH = ½ pK2 + ½ pK3 = 9.7 Il pH relativo al 3° punto di equivalenza si ricava dall’idrolisi di PO43-. PO43- + H2O HPO42- + OH-idr [PO43-] = C ed essendo pH = ½ pKw + ½ pK3 + ½ log C Per un acido 1 N, titolato con NaOH 1 N, pH per il terzo H+ = 12.6 Il terzo H+ non è titolabile, perché al pH di equivalenza (12.6) l’idrolisi del sale è molto spinta, per cui notevoli aggiunte di base provocano modeste variazioni del pH. 37 TITOLAZIONI DI PRECIPITAZIONE Equilibri rapidi e disponibilità di indicatori. Nelle titolazioni di precipitazione si ha la formazione di un sale poco solubile. Il Kps sostituisce il Kw dell’acqua ed il pMe o il pX sostituiscono rispettivamente il pH o il pOH. Es. Ag+ titolato con Cl- si avranno tre punti fondamentali. a) prima del punto di equivalenza: si calcola [Ag+] residua b) al punto di equivalenza si calcola la [Ag+] dal Kps (pAg = ½ p Kps) c) dopo il punto di equivalenza: Es. Calcolare pAg nella titolazione di 100 ml di soluzione 0.1 N di Ag+ con 0.1 N di Cla) Prima del punto di equivalenza Aggiunta ml Cl- = 0 [Ag+] = 10-1 g ioni/L; pAg = 1 Aggiunta ml Cl- = 50 Aggiunta ml Cl- = 90 pAg = 2.28 Aggiunta ml Cl- = 99 pAg ≈ 3.5 b) Punto di equivalenza Aggiunta ml Cl- = 100 c) Dopo il punto di equivalenza Aggiunta di Cl- = 100.04 38 Aggiunta Cl- = 101 ml pAg = 6.62 Aggiunta Cl- = 105 ml pAg = 7.30 Nota bene: nelle immediate vicinanze del punto di equivalenza si deve tener conto del contributo degli Ag+ provenienti dalla solubilità del sale. Es. aggiunta 99.99% Cl[Ag+]NON TIT = 0.01 · 0.1 199.99 = 10-3 200 = 5 · 10-6 Concentrazione di Ag+ prima del p.e. più bassa di quella al p.e (1.1 · 10-5), incompatibile! Se X = [Ag+]sale proveniente dal sale allora: [Ag+]tot = [Ag+]non tit. + [Ag+]sale Ma [Ag+]sale = [Cl-] allora [Ag+]tot · [Cl-] = Kps da cui ([Ag+]non tit. + [Ag+]sale) [Ag+]sale = Kps Sostituendo ad [Ag+]non tit. il valore trovato (5 · 10-6 + [Ag+]sale) [Ag+]sale = 1.2 · 10-10 Risolvendo per [Ag+]sale … [Ag+]sale = 8.7 · 10-6 quindi [Ag+]tot = 5 · 10-6 + 8.7 · 10-6 = 1.37 · 10-5 g ioni/L e pAg = 4.86, valore COMPATIBILE con il P.E. Ricaviamo ora il valore di [Ag+] in corrispondenza di un’aggiunta di 100.01 ml Cl[Ag+] = S condizionata dalla [Cl-]tot [Cl-]tot = [Cl-]ecc + [Cl-]sale ma [Cl-]sale = [Ag+] = S essendo [Ag+] [Cl-]tot = Kps = [Cl-]sale ([Cl-]ecc + [Cl-]sale) [Cl-]tot = [Cl-]sale + 5 · 10-6 Kps = (5 · 10-6 + [Cl-]sale) [Cl-]sale 39 2 · 10-10 = (5 · 10-6 + [Cl-]sale) [Cl-]sale Da cui [Cl-]sale = [Ag+] = 8.7 · 10-6 g ioni/L e pAg = 5.06 **** TITOLAZIONE Ag+ 0.1 N CON Cl- 0.1 N E CON I- 0.1 N Kps AgCl = 1.2 · 10-10 Kps AgI = 1.7 · 10-16 NOTA: l’andamento della titolazione è influenzato oltre che dalla concentrazione degli ioni reagenti anche dal Kps del sale che si forma! 40 METODI DI TITOLAZIONE METODO DI MOHR Metodo basato sulla precipitazione frazionata dello ione da dosare (Cl- o Br-) e di quello indicatore (CrO42-) da parte del reattivo titolante Ag+ con formazione di composti, uno bianco (o giallastro ed uno intensamente colorato, a solubilità poco diversa. Il composto colorato rosso mattone è Ag2CrO4 e si forma in una soluzione già contenente il precipitato (es. AgCl). Il precipitato Ag2CrO4 dovrebbe formarsi non appena la [Ag+] ha superato il valore dell’equivalenza, allora la [CrO42-] dovrebbe essere: Quindi una concentrazione che però impartisce una colorazione troppo intensa. Si usa allora una soluzione 3 – 5 · 10-3 mol/L per percepire più facilmente il viraggio anche se ne deriva un errore in eccesso. Che errore deriva dall’usare una [CrO42-] = 3 · 10-3? Soluzione Per questa concentrazione l’argento potrà dare inizio alla precipitazione di Ag2CrO4 quando verrà soddisfatta la reazione [Ag+]2 [CrO42-] = Kps Se si fosse usata la concentrazione teorica dell’indicatore… 2.4 · 10-5 – 1.1 · 10-5 = 1.3 · 10-5 g ioni/L = [Ag+]eccesso Quindi in presenza di [CrO42-] = 3 · 10-3 mol/L titolando 100 ml di NaCl 0.1 N con AgNO3 0.1 N la condizione di formazione di Ag2CrO4 si verificherà quando sarà presente un numero di equivalenti di Ag+ uguali a: 41 Gli equivalenti di Cl- titolati sono 10-2 allora l’errore percentuale sarà: Per poter percepire il viraggio è necessario aggiungere ½ goccia di reattivo che comporta il seguente incremento di Ag+ (½ goccia ≈ 0.025 ml) allora ERRORE GLOBALE = errore dovuto alla minore [CrO42-] + errore dovuto ad Ag+ per evidenziare il viraggio. Allora E globale = 0.026 + 0.025 = + 0.05% Inferiore a 0.1% e praticamente trascurabile! La titolazione di Ag+Cl- deve essere rapida per evitare la coagulazione di AgCl colloidale che, avendo una grande superficie, agisce da disperdente del precipitato di Ag2CrO4 favorendo la percezione cromatica del viraggio. Titolazione tra pH 6.5 – 9 Se pH > 9 precipita Ag(OH) Kps = 2.3 · 10-8 In presenza di NH4+ il pH ≤ 7.2 perché? Per evitare la formazione di NH3 che forma complessi con Ag+ solubili. In soluzione acida: 2 CrO42- + 2 H+ 2 HCrO4- Cr2O72- + H2O Poiché HCrO4- è un acido debole (Ka = 6.4 · 10-5), quindi lo ione CrO42- risente debolmente dell’acidità. La titolazione deve essere eseguita a temperatura ambiente per evitare la coagulazione di AgCl e per non aumentare la solubilità di Ag2CrO4. NOTA: la titolazione di I- e CNS- non può essere eseguita con questo metodo in quanto i precipitati di AgI e AgCNS hanno capacità adsorbenti rispetto allo ione CrO42- talmente spiccate da falsare il punto finale della titolazione. 42 ARGENTOMETRIA Parte sperimentale Preparazione soluzione AgNO3 0.02 N. Si pesano sulla bilancia TECNICA circa 1.3 g del sale. Si solubilizzano in H2O distillata e si porta a volume in matraccio tarato da ½ litro. La soluzione va conservata in bottiglia scura (si evita la decomposizione fotochimica di AgNO3). STANDARDIZZAZIONE DI AgNO3 ≈ 0.02 N Si usa NaCl in stufa a 110°C. Se ne pesano tra 0.02 a 0.03 g, si trasferiscono in beuta da 250 o 300 ml. Si aggiunge H2O sino ad avere un volume di 100 – 130 ml e si procede secondo il metodo di MOHR. Alla soluzione si aggiungono 1.5 – 2 ml di K2CrO4 al 5% e si titola rapidamente al riparo dalla luce con AgNO3 sino al viraggio giallo canarino → giallo rosato. Calcolo NOTA: il nitrato di argento non può essere considerato sostanza madre in quanto presenta sempre tracce di Ag ridotto. DETERMINAZIONE DELLA PERCENTUALE DI CLORURO DI SODIO IN UN CAMPIONE Titolazione di prova Si prelevano con pipetta tarata 25.0 ml di una soluzione di NaCl ≈ 0.02 N e si trasferiscono in beuta da 250 ml. Si aggiunge H2O sino ad avere un volume di 100 – 150 ml. Si aggiungono 10 – 15 gocce di K2CrO4 al 5%, una punta di spatola di bicarbonato di sodio e si titola rapidamente al riparo della luce con AgNO3 ~ 0.02 N sino al viraggio giallo canarino → giallo rosato. 43 STANDARDIZZAZIONE DELLA SOLUZIONE DI AgNO3 ~ 0.02 N Pesare analiticamente circa 0.3 g di NaCl (precedentemente essiccato in stufa a 120°C); trasferire quantitativamente il sale in un matraccio tarato da 250 ml e portare a volume con acqua distillata; prelevare una aliquota di 25.0 ml e trasferirla in una beuta da 250 ml aggiungendo 100 – 150 ml di acqua distillata. Aggiungere 10 – 15 gocce di K2CrO4 al 5% ed una punta di spatola di bicarbonato di sodio. Titolare rapidamente con la soluzione AgNO3 ~ 0.02 N che avete a disposizione. Utilizzando la stessa procedura si determina la percentuale di NaCl in un campione incognito. Previa omogeneizzazione del campione si pesano analiticamente circa 0.3 – 0.5 g di polvere incognita contenuta nel portacampione, si trasferiscono in matraccio da 250 ml e si porta a volume con H2O distillata. Si prelevano 25.0 ml di soluzione e si trasferiscono in beuta da 250 ml. Si aggiunge H2O distillata sino ad avere un volume di 100 – 150 ml e si procede secondo il metodo di MOHR. Alla soluzione si aggiungono 10 – 15 gocce di K2CrO4 al 5%, una punta di spatola di bicarbonato di sodio e si titola RAPIDAMENTE al riparo dalla luce con AgNO3 da voi standardizzato sino al viraggio. Calcolo Dai grammi di campione inizialmente pesati e dai grammi di NaCl calcolati determinare la percentuale di NaCl nel campione. 44 METODO DI FAJANS Si sfrutta il fenomeno dell’adsorbimento e la variazione cromatica all’equivalenza è determinata dal differente tipo di ione adsorbito che determina l’adsorbimento secondario dell’indicatore. Indicatori di adsorbimento: - fluoresceina - diclorofluoresceina - eosina Gli anioni, di notevoli dimensioni, hanno legami che risentono del campo elettrico determinato dallo ione in adsorbimento primario. Quindi viene adsorbito e la deformazione dovuta al campo elettrico determinerà un riarrangiamento della sua struttura con conseguente variazione del colore. Esempio: NaBr + AgNO3 in presenza di eosinato sodico Si forma Prima dell’equivalenza Dopo l’equivalenza METODO DI VOLHARD Titolazione indiretta. L’alogenuro viene precipitato con una quantità nota ed in eccesso di Ag+. Tale eccesso viene retrotitolato in ambiente acido con una soluzione di CNS- a titolo noto, 45 usando come indicatore Fe3+ che con CNS- dà un complesso intensamente colorato. Nel caso dei cloruri si ha AgCl + CNS- AgCNS + Cl- Nella retrotitolazione è presente sia AgCl che AgCNS ed il rapporto dei Kps = 174; infatti… [Ag+] [Cl-] = Kps [Ag+] [CNS-] = K’ps Quindi AgCl costituisce una causa d’errore, infatti al punto di equivalenza il piccolo eccesso di CNS- che doveva formare il [Fe(CNS)]2+ reagirà con AgCl sino ad avere [Cl-] [CNS-] = 1.74. Allora si impedisce la reazione tra AgCl e CNS- filtrando il precipitato oppure aggiungendo un liquido immiscibile con H2O a carattere polare (nitrobenzene o etere) che rivesta AgCl con una “pellicola” protettiva. Se si titolano I- e Br- avendo un Kps < AgCNS non è necessaria la procedura precedente. NOTA: se si filtra si deve aspettare una lunga digestione del precipitato per eliminare l’adsorbimento di Ag+ su AgCl oppure si può dibattere il precipitato per 5 – 10 minuti prima della filtrazione. Si opera lontano dalla luce diretta per evitare la possibile riduzione fotochimica di Ag+. 46 COMPLESSI Molti cationi formano complessi in soluzione con sostanze che abbiano una coppia di elettroni liberi (es. atomi di N, O, S nella molecola) capaci di soddisfare il numero di coordinazione del metallo. Me = metallo accettore di un doppietto elettronico L = legante complessante donatore di un doppietto elettronico Ammoniaca L: complessa Ag+ = M Quindi ML = Ag(NH3)+ Due molecole di NH3 complessano ciascuno ione argento per cui… Kf = COSTANTE DI FORMAZIONE TOTALE Kf = Ks o Kstab Nel caso del complesso ammoniacale: NOTA: nel considerare Kd o 1/Kd = Kf si deve tener presente che l’equazione (1) non deve essere interpretata nel senso che [NH3] = 2 [Ag+] (2) La dissociazione del complesso è graduale e vi sono molti equilibri parziali che per esempio in una soluzione 0.01 mol/L di complesso Ag(NH3)2+ la [Ag+] = 1/5 [NH3] e non la metà. Quindi la Kf o Ki totale è una relazione puramente algebrica che è valida purché non si pongano relazioni come la (2). 47 EDTA Agente complessante. (HO2CH2C)2NCH2CH2N(CH2CO2H)2 ACIDO ETILENDIAMINOTETRAACETICO Un agente organico con più gruppi capaci di complessare uno ione metallico viene chiamato AGENTE CHELANTE, e il complesso si chiama chelato. L’agente complessante si chiama legante. Titolazione chelometrica: viene effettuata con un agente chelante. Nell’EDTA sono presenti 6 gruppi complessanti (4 coppie di elettroni sui gruppi carbonilici e 2 sui due atomi di azoto); l’EDTA si rappresenta come H4Y Acido tetraprotico. È il legante non protonato Y4- che forma complessi con ioni metallici. **** DISTRIBUZIONE DEI VARI COMPLESSI PER IL SISTEMA Cu2+ - NH3 La dissociazione degli ioni complessi ed i relativi equilibri possono essere alquanto complicati quindi necessitano di semplificazioni. 48 1) caso in cui il complesso si forma in presenza di notevole quantità di legante a) Me risulta complessato formando la specie con il più elevato numero di coordinazione. b) la concentrazione di Me libero e dei complessi intermedi sia molto piccola. [L] è determinato dall’eccesso di agente complessante, si trascura [L] proveniente dalla dissociazione del complesso. Poiché Y4- forma il complesso gli equilibri di complessazione saranno influenzati dal pH. NOTA: H4Y è poco solubile in H2O quindi si usa Na2H2Y · 2 H2O che si dissocia dando H2Y2- con un pH ≈ 4 – 5 Costante di formazione EDTA – Ca2+ Ca2+ + Y4- CaY2- (1) Effetto del pH Se si aumenta la concentrazione idrogenionica l’equilibrio (1) si sposta a sinistra a causa della reazione Y4- + H+ HY3- Noto quindi il pH e gli equilibri coinvolti si può usare la Kf per calcolare la [Ca2+] libero nelle varie condizioni. La [Y4-] viene calcolata a diversi valori di pH. Se [Y]tot è la concentrazione totale di tutte le forme di EDTA non complessato allora: [Y]tot = [Y4-] + [HY3-] + [H2Y2-] + [H3Y-] + [H4Y] Sostituendo questi valori nella espressione e risolvendo rispetto a H4Y; H3Y-; H2Y2-; HY3- per eliminare tutte le forme eccetto Y4- e dividendo per [Y4-] si avrà 49 α4 = frazione di EDTA totale che esiste come Y4Calcolando Y4- ad un dato pH e conoscendo [Y]tot non complessato si calcola [Ca2+] libero. dove K’ è la costante condizionale che dipende da α4 e quindi dal pH. Esempio: Ca – EDTA a pH = 10 → α4 = 0.35 Quindi Kf · α4 = 5 · 1010 · 0.35 = 1.8 · 1010 = K’ = costante condizionale Si calcola il pCa in 100 ml di una soluzione di Ca2+ 0.1 M a pH 10 dopo l’aggiunta di 0 ml; 50 ml; 100 ml; 150 ml EDTA 0.1 M. Soluzione 0 ml pCa = - log [Ca2+] = log 10-1 = 1.00 50 ml Calcoliamo le millimoli di Ca2+. 0.1 M · 100 ml = 10 m moli di Ca2+ Calcoliamo le millimoli di EDTA addizionate. 0.1 M · 50 ml = 5.0 m moli Essendo K’ grande (≈ 1010) la reazione Ca2+ + Y4- CaY2- sarà molto spostata a destra quindi si trascura la quantità di Ca2+ proveniente dalla dissociazione di CaY2-. 50 Le millimoli di Ca2+ libero sono essenzialmente uguali al numero delle millimoli non reagite: m moli Ca2+ = 10.0 – 5.0 = 5.0 m moli pCa = - log 3 · 10-2 = 2 – 0.48 = 1.52 100 ml Punto di equivalenza. Tutto il Ca2+ si è convertito in CaY2-; allora per calcolare la [Ca2+] all’equilibrio si deve usare la: Al punto di equivalenza le millimoli di CaY2- formatesi sono uguali alla concentrazione iniziale di Ca2+, quindi Ma CaY2- Ca2+ + Y4- → Y’ e [Ca2+] = [Y’] = X All’equilibrio [CaY2-] = 0.05 – X ≈ 0.05 150 ml La concentrazione Y’ è uguale all’eccesso di EDTA aggiunto. Bisogna però trascurare la dissociazione di CaY2- che viene ulteriormente retrocessa. Quindi le millimoli di CaY2- saranno a maggior ragione uguali a 10, allora Le millimoli Y’ in eccesso saranno Y’ = 0.1 · 150 – 0.1 · 100 = 5 m moli 51 [Ca2+] = 1.1 · 10-10 mol/L pCa = 10 – log 1.1 = 10 – 0.04 = 9.96 Titolazione Si aggiunge EDTA al campione (Ca2+) e la reazione è Ca2+ + Y4- CaY2- Prima dell’equivalenza la concentrazione del Ca2+ è praticamente uguale al calcio non reagito. Infatti la dissociazione del complesso è scarsa (Kf = 5 · 10-10 e a pH = 10 K’ = 1.8 · 1010). Al punto di equivalenza il pCa è determinato dalla dissociazione del chelante ad un dato pH! 52 INDICATORI PER TITOLAZIONI CHELOMETRICHE Nero Eriocromo T Si rappresenta come H3In. È un agente chelante che ha la proprietà di formare complessi chelati con i cationi in esame meno stabili dei corrispondenti complessi Me – EDTA. Il complesso Me – In ha un colore diverso da quello dell’indicatore libero e la variazione cromatica della soluzione permetterà di individuare il punto in cui EDTA ha complessato quantitativamente il metallo sottraendolo al sistema Me – In (PUNTO DI VIRAGGIO). Es. Titolazione Mg2+ con EDTA. Si aggiunge NET in piccola quantità, si forma un complesso rosso Mg – NET. L’indicatore non complessato è NET (BLU). All’equivalenza si usa la KI COSTANTE CONDIZIONALE. Tanto più elevata è la Kf tanto più Ca2+ + Y4- CaY2- sarà spostato verso destra, ma la diminuzione di pH sposta l’equilibrio verso sinistra secondo la competizione Conclusione: più stabile è il chelato più l’equilibrio sarà spostato verso destra, più grande sarà il salto al punto di equivalenza, più basso sarà il pH a cui si può condurre la titolazione. 53 **** Quando tutti gli ioni Mg2+ sono titolati l’EDTA sposta l’indicatore dal magnesio con variazione di colore rosso - blu MgIn- + H2Y2- → MgY2- + HIn2- + H+ Il complesso Me – In deve essere 10 – 100 volte meno stabile del complesso Me – EDTA. NOTA: le Kf di EDTA – Ca ed EDTA – Mg sono troppo vicine per poterle differenziare in una titolazione (anche regolando il pH), quindi vengono titolati entrambi con Nero eriocromo T. Questa titolazione si usa per determinare la durezza di un’acqua. Tampone NH3/NH4+ pH = 10 Se il pH > 10 precipita Mg(OH)2 quindi si può titolare il Ca2+ precipitando il Mg a pH = 12. 54 **** EFFETTO DELLA COMPLESSAZIONE SULLA SOLUBILITÀ Precipitato MA si dissocia secondo MA M+ + A- Se è presente un legante L allora: Il calcolo della solubilità avviene in maniera analoga a quello dell’effetto acido sulla solubilità. Es. AgBr in presenza di NH3 Si scrivono gli equilibri AgBr Ag+ + Br55 La concentrazione totale di argento all’equilibrio [Ag’] sarà = [Br-] = solubilità di AgBr Ma [Ag’] = [Ag+] + [Ag(NH3)+] + [Ag(NH3)2+] (1) Dai valori delle Kf1 e Kf2 si ottiene [Ag(NH3)+] = Kf1 [Ag+] [NH3] [Ag(NH3)2+] = Kf2 [Ag(NH3)+] [NH3] = Kf1Kf2 [Ag+] [NH3] Allora sostituendo nella (1) [Ag’] = Kf1Kf2 [Ag+] [NH3]2 + Kf1 [Ag+] [NH3] + [Ag+] Allora Kps = [Ag+] [Br-] sarà Kps = [Ag’] β0 [Br-] da cui Es. calcolare S di AgBr in 0.1 M di NH3 Kps = 4 · 10-13 β0 per NH3 0.1 M = 4 · 10-6 Quindi in NH3 0.1 M AgBr è circa 500 volte più solubile. 56 DUREZZA TOTALE DELL’ACQUA La durezza dell’acqua è dovuta alla presenza del calcio e del magnesio, normalmente sotto forma di idrogenocarbonati. La quantità totale di calcio e magnesio viene normalmente espressa indicando la quantità equivalente di carbonato di calcio in ppm (mg/litro). Quindi, dato che il peso molecolare del CaCO3 è quasi esattamente 100 (p.m. = 100.089 g/mol), una durezza di 1000 ppm corrisponde ad una concentrazione totale di calcio e magnesio di 0.01 M. - prelevare 100 ml di acqua di rubinetto e trasferirli in beuta da 250 ml; - aggiungete 2 ml tampone ammoniacale a pH 10 e 5 - 6 gocce di NET (Nero Eriocromo T); - titolare con EDTA 0.01 M fino al viraggio dal rosso al blu. Calcoli: Durezza (°F) = ml EDTA0.01 M ogni 100 ml di acqua in esame NOTA: la durezza espressa in gradi francesi (°F) corrisponde ad un contenuto in calcio e magnesio equivalente a 10 mg/litro di CaCO3, ovvero a 10 ppm di CaCO3 Avvertenza: se la soluzione in esame fosse acida, si aggiungono 1 – 2 gocce di metilarancio e si neutralizza con NaOH diluito sino al viraggio al giallo; in tal modo, titolando con EDTA (indicatore NET), al punto finale la soluzione vira da rossoarancio a verde. 57 DETERMINAZIONE DEL CARBONATO DI CALCIO IN UN CAMPIONE INCOGNITO Si pesano esattamente circa 0.25 g. di CaCO3 seccato a 150-200 °C e si trasferiscono in beuta da 250mL. - si aggiungono 20-30 mL di acqua distillata e HCl 2N goccia a goccia fino a completa dissoluzione del campione. - Si fa bollire la soluzione per eliminare la CO2, si raffredda e si diluisce con acqua distillata a 80-100 mL. - si aggiungono 2 ml di una soluzione Mg – EDTA + 5 mL tampone ammoniacale + 3 - 4 gocce di NET; - si titola fino al viraggio dal rosso al blu. Calcolo: 58 OSSIDIMETRIA Titolazioni di ossidoriduzione. Principi: OSSIDAZIONE = perdita di elettroni RIDUZIONE = acquisto di elettroni Reazione “redox” in generale Oss1 + Rid2 Rid1 + Oss2 Agente ossidante è la specie chimica che acquistando elettroni passa ad uno stato di ossidazione inferiore: Es. Ce+4 + e- Ce+3 Agente riducente è la specie chimica che cedendo elettroni passa ad uno stato di ossidazione superiore. Es. Fe2+ Fe3+ + e- NOTA: i due processi sono concomitanti: l’uno non può avvenire in assenza dell’altro. Ossidante + riducente = coppia coniugata Es. La tendenza ossidante o riducente delle sostanze si ricava studiando le celle elettrochimiche ed i relativi potenziali elettronici CELLE ELETTROCHIMICHE Galvaniche → reazione chimica spontanea che produce energia elettrica (batterie) Elettrolitiche → si utilizza l’energia elettrica per far avvenire una reazione chimica di per sé non spontanea (elettrolisi H2O) La natura elettrica di questi processi si evidenzia facendo sì che il trasferimento di elettroni avvenga attraverso un circuito esterno realizzato con due elettrodi di platino 59 immersi rispettivamente nella soluzione dell’ossidante e del riducente separate da un setto poroso. Non è possibile misurare il potenziale di ogni singola semireazione, però si può misurare la differenza tra due potenziali elettrodici ed al potenziale della semireazione. 2H+ + 2 e- H2 è stato assegnato il valore zero (0.000 V) L’elettrodo corrispondente a questa reazione è detto: Elettrodo Normale a Idrogeno o Elettrodo Standard a H. Quindi si può misurare una ddp tra questa semireazione e le altre dovute ad altri elementi. I potenziali sono dipendenti dalle concentrazioni e tutti i potenziali che si definiscono “standard” si riferiscono a condizioni di attività unitaria o nel caso di gas alla pressione parziale di 1 atmosfera. Per convenzione i potenziali standard vengono scritti nel senso della riduzione ed il potenziale aumenta all’aumentare della tendenza del composto a ridursi. NOTA: valori di potenziale positivi alti = forte tendenza del composto a ridursi. valori di potenziale negativi alti = forte tendenza del composto a ossidarsi. Es. Due metalli Ferro e Zinco Fe3+ + eZn2+ + 2 e- Fe2+ E0 = 0.77 Zn E0 = - 0.77 Quale sarà la reazione? 2 Fe3+ + Zn 2 Fe2+ + Zn2+ Il voltaggio di cella? E = 1.54 volts, questo valore è sempre positivo Collegando i due elettrodi ad un galvanometro si osserva un passaggio di corrente 60 **** Ogni elettrodo sarà caratterizzato da un potenziale elettrico determinato dalla tendenza degli ioni a cedere o ad acquistare elettroni. Quindi tra i due elettrodi si genererà una differenza di potenziale che sarà tanto maggiore quanto più alta è la tendenza degli ioni a cedere o acquistare e-. Es. la ddp tra Fe2+ e Ce+4 può essere utilizzata per compiere lavoro elettrico. Una cella per convenzione si scrive nel seguente modo: anodo / soluzione / catodo F.E.M. = Ecatodo – Eanodo Ossidazione → anodo Riduzione → catodo Ne consegue che l’agente riducente più forte viene posto a sinistra e l’agente ossidante più forte sulla destra. Quindi il potenziale di cella sarà: Ecella = Edestra – Esinistra = Ecatodico – Eanodico Per le semireazioni: Fe3+ + eSn4+ + 2 e- Fe2+ E0 = 0.77 Sn2+ E0 = 0.15 Ecella = E0 Fe3+/ Fe2+ – E0 Sn4+/ Sn2+ = 0.77 – 0.15 = 0.62 61 **** 62 EQUAZIONE DI NERNST Prendiamo in esame la reazione Fe / Zn Fe3+ + eZn2+ + 2 e- Fe2+ E0 = 0.77 Ecatodo Zn E0 = - 0.77 Eanodo La pila sarà così schematizzata Zn/[ Zn2+] // [Fe2+] · [Fe3+] / Pt La reazione globale sarà 2 Fe3+ + Zn → 2 Fe2+ + Zn2+ Se si opera in condizioni di reversibilità allora il lavoro elettrico dell’elemento galvanico schematizzato sarà L=nFE dove L = lavoro elettrico n = numero moli di e trasferiti E = F.E.M. F = Faraday (96500 Coulomb) Il lavoro elettrico = lavoro utile = VARIAZIONE DI ENERGIA LIBERA ∆G ∆G = -L = - n F E ed in condizioni standard ∆G0 = - n F E0 Nel caso della reazione Fe / Zn la variazione di energia libera di una reazione redox sarà E per una generica reazione aA + bB aC c aD d aA a cC + dD sarà aB b Sostituendo le espressioni su descritte Dividendo per n F e cambiando segno si ha Per una reazione a Oss + n e- b Rid 63 ed E è il potenziale di riduzione ad una specifica concentrazione. Il potenziale E dipende da T, da [ ] e da altri fattori come ad es. il pH Cr2O72- + 14 H+ + 6 e- 2 Cr3+ + 7 H2O Se [Cr2O72-] = 10-3 M [Cr3+] = 10-2 M E0 = 1.33 Costante di equilibrio: sia la reazione a Oss1 + b Rid2 a Rid1 + b Oss2 Si calcoli il valore della costante di equilibrio. All’equilibrio il potenziale E del sistema riducente è uguale a quello del sistema ossidante. Non vi sarà più la tendenza degli elettroni a passare preferenzialmente da una specie all’altra. La ddp tra le due semicoppie è zero ed il valore di E riferito all’elettrodo di idrogeno è uguale per tutti e due i sistemi. Essendo E1 = E2 si avrà: 64 Quale sistema è più adatto per ridurre Fe+3 a Fe+2? a o b? a = Sn4+ / Sn2+ b = I3 - / 3 Idai potenziali si ha E0 Fe+3/ Fe+2 = 0.77 V E0 I3- / 3 I- = 0.536 V E0 Sn4+ / Sn2+ = 0.15 V Per 2 Fe+3 + 3 I- 2 Fe+2 + I3- K = 6.3 · 107 Per 2 Fe+3 + Sn2+ 2 Fe+2 + Sn4+ K = 5 · 1020 65 CURVE DI TITOLAZIONE Variazione del potenziale di una soluzione durante la titolazione. Semicoppia riducente con E2 viene titolata con reattivo ossidante che in soluzione assumerà un potenziale E1. All’equilibrio E2 – E1 = 0 Ogni valore del potenziale della soluzione, riferito all’elettrodo ad idrogeno per ogni aggiunta di titolante è determinato dal rapporto [Oss] / [Rid] relativo alla semicoppia che risulta presente in eccesso. L’altra semicoppia, in difetto, subirà il potenziale imposto da quella in eccesso. [Oss] / [Rid] all’equilibrio per la specie in difetto si determina ricordando che E2 = E1. ALL’EQUIVALENZA = particolare punto di equilibrio in cui la quantità di titolante aggiunto è equivalente alla specie da dosare. Il potenziale della soluzione può essere dedotto indifferentemente da tutte e due le semicoppie partecipanti all’equilibrio. Calcoli per E2 o E1 - prima del punto di equivalenza per una reazione a Oss1 + b Rid2 a Rid1 + b Oss2 in soluzione si ha eccesso di riducente (specie da dosare) per cui - Al punto di equivalenza Specie da dosare = reattivo aggiunto Dopo il punto di equivalenza si ha un eccesso di reattivo ossidante aggiunto per cui Per capire queste espressioni facciamo un esempio pratico: 66 Sn2+ titolato con Fe+3 si avrà 2 Fe+3 + Sn2+ 2 Fe+2 + Sn4+ Prima del punto di equivalenza si ha aggiunta di un volume di soluzione di Fe+3 in un numero di moli minore di 2 ad una soluzione contenente 1 mole di Sn2+ Sia a il numero di moli di Fe+3 aggiunte. Si avrà per a < 2 Poiché a < 2 Al punto di equivalenza a = 2 quindi si avrà All’equivalenza si verifica che il rapporto delle concentrazioni delle specie ottenute dalla reazione è uguale a quello delle concentrazioni delle specie residue all’equilibrio. In generale il punto di equivalenza sarà indicato e soddisfatto dalla seguente equazione: Oltre il punto di equivalenza a > 2 Per ogni punto della titolazione, incluso il punto di equivalenza vi è una condizione 67 È il prodotto di potenze che hanno per base la stessa base e per esponente i valori dei coefficienti di reazione. All’equivalenza i rapporti delle concentrazioni delle specie delle singole semicoppie sono facilmente ricavabili conoscendo la K ed i coefficienti di reazione. Infatti: che sostituita nella equazione di Nernst permette di calcolare il potenziale all’equivalenza: Il valore del potenziale al punto di equivalenza può anche essere dedotto tai potenziali standard dei due sistemi redox. Scriviamo i potenziali E1 e E2 delle due semicoppie: (1) (2) Moltiplicando la (1) per «b» e la (2) per «a» si ha 68 e semplificando Es. per la coppia Fe+3/Sn4+ **** 69 **** 70 INDICATORI DI OSSIDO-RIDUZIONE Sostanze organiche (prevalentemente) che cambiano colore passando dalla forma ossidata a quella ridotta. Affinché la variazione di colore venga percepita deve essere molto intensa in un rapporto di concentrazione di 10 o di 1/10. Reazione: Indoss + n e- colore ossidato Indrid colore ridotto Applicando l’equazione di Nernst si ha Se il viraggio è condizionato da un rapporto che vale 10 o 1/10 allora si ricava: Quando l’indicatore è 50% forma ossidata e 50% forma ridotta, allora la soluzione avrà un potenziale che viene detto di transizione (Etrans) e corrisponde al valore indicato con E0 ind. Da un punto di vista pratico (scelta dell’indicatore) si sceglierà un indicatore che abbia un Etrans = Eequiv per ridurre al minimo l’errore di titolazione. Es. nella titolazione Sn2+ con Fe3+ per - 0.1% di Fe3+ il potenziale varia di - 120 mV e per + 0.1% di Fe3+ varia di + 230 mV (rispetto a Eequiv). Il salto è di 350 mV e comprende quello necessario per avere il viraggio di un indicatore se Etrans ≈ Eequiv con un errore < ± 0.1%, quale che sia il senso della titolazione. Gli indicatori redox più noti sono: 1 -7 1 - 10 INDICATORE Difenil – benzidina Dimetil – ferroina Fenantrolina sale ferroso (ferroina) Nitro – O – fenantrolina sale ferroso (nitro-ferroina) Blu Metilene Ox – Viraggio - Rid Etrans Violetto – incolore 0.76 1% H2SO4 Verde – arancio 0.88 0.025% H2SO4 Bleu – rosso 1.06 0.25% H2SO4 Bleu – rosso violetto 1.31 0.25% H2SO4 Incolore - blu 0.53 Acido 1 … 71 PERMANGANATO KMnO4 agente ossidante molto usato. - potenziale molto elevato da determinare reazioni pressoché complete con molti sistemi riducenti. - Impartisce alla soluzione un colore molto intenso - Non servono quindi indicatori. Una goccia di KMnO4 0.1 N in 300 ml di soluzione dà un colore rosa facilmente percepibile. In ambiente acido si ha: MnO4- + 8 H+ + 5 e- Mn2+ + 4 H2O In ambiente poco acido o neutro o alcalino: MnO4- + 4 H+ + 3 e- MnO2 + 2 H2O Il potenziale MnO4-/Mn2+ diminuisce all’aumentare del pH, per cui se la reazione consuma 8 H+ per mole è importante un eccesso di acido per non far variare il potenziale durante la titolazione. Si usa come acido H2SO4. Non si usa HNO3 perché NO3- + 4 H+ + 3 e- NO↑ + 2 H2O Potrebbe essere un sistema ossidante rispetto al riducente da dosare specie a bassi pH infatti Dove E0 = 0.94 Se si usa HCl si introdurrebbe il sistema riducente Cl2 + 2 e- 2Cl- Con E0 Cl2 / 2Cl- = 1.36 che, anche se minore di MnO4-/Mn2+ (E0 = 1.51), fa pensare ad un’azione riducente di Cl- quando è presente in alta concentrazione. KMnO4 non è sostanza madre perché è difficile ottenerlo esente da MnO2 4 MnO4- + 2 H2O 4 MnO2 + 3 O2 + 4 OH- 72 Reazione di ossidazione dell’H2O catalizzata da MnO2 formatosi da tracce di sostanze organiche presenti in H2O distillata o reazione catalizzata dalla luce. 2 MnO4- + 3 Mn2+ + 2 H2O 5 MnO2 + 4 H+ Reazione rapida in ambiente neutro se presente Hn3+. Standardizzazione della soluzione KMnO4 Si usa Na2C2O4 anidro Le semicoppie sono: MnO4- + 8 H+ + 5 e- Mn2+ + 4 H2O E0 = + 1.51 V 2 CO2 + 2 H+ + 2 e- H2C2O4 E0 = - 0.49 V Si pesano 0.15 – 0.23 g di Na2C2O4 anidro e si trasferiscono in un becker da 400 ml. Si diluisce a 150 – 200 ml con H2O e si aggiungono 20 ml di H2SO4 (1 : 4). Si inizia la titolazione con la soluzione di KMnO4, se ne aggiungono 1 – 2 ml, si riscalda, agitando sino a decolorazione, si raffredda e si prosegue la titolazione lentamente sino al rosa persistente per almeno 30 sec. Calcolo Che succede quando si aggiunge H2SO4? Perché bisogna scaldare e poi titolare a temperatura ambiente? H2SO4 + C2O42- → H2C2O4 + SO42Se si scalda troppo! H2C2O4 → CO2↑ + CO↑ + H2O Quindi T ≈ 60°C In presenza di Mn2+ che agiscono da catalizzatore la titolazione può essere proseguita a freddo. 73 ANALISI Fe METODO ZIMMERMAN HCl per attacco campione ematite SnCl2 al 5% per riduzione Fe3+ → Fe2+ 2 Fe3+ + Sn2+ 2 Fe2+ + Sn4+ Soluzione Z.R. consiste in MnSO4, H2SO4, H3PO4 Soluzione Hg2Cl2 Soluzione MnO4- a titolo noto Sn2+ + 2 Hg2+ + 2 Cl- Hg2Cl2 + Sn4+ Si elimina l’eccesso di Sn2+. Il precipitato bianco sericeo è dovuto alla formazione di Hg2Cl2. Se il precipitato è bianco caseoso o grigio si è avuta una riduzione spinta a formare Hg. Hg2+ + Sn2+ Sn4+ + Hg ↓ L’eliminazione dell’eccesso di Sn2+ deve essere eseguita il più rapidamente possibile ed a freddo per evitare la reazione Fe2+ + O2 + 4 H+ + 3 e- Fe3+ + 2 H2O Vediamo l’influenza di Mn2+ e H3PO4 sulla reazione MnO4- → Fe2+ in presenza di ClMnO4- + 5 e- + 8 H+ Cl2 + 2 eFe3+ + e- 2 ClFe2+ Mn2+ + 4 H2O E0 = + 1.51 V E0 = + 1.36 V E0 = + 0.77 V 74 La presenza di Mn2+ riduce il potenziale di MnO4- evitando l’ossidazione di Cl- a Cl2. H3PO4 forma COMPLESSI con il Fe3+ [FeHPO4]+ che abbassa il potenziale d’ossidazione del sistema Fe3+ / Fe2+. I sistemi da considerare sono: 1) MnO4- + 5 e- + 8 H+ 2) Fe3+ + e- Fe2+ 4) Hg2Cl2 (s) + 2 e- E0 = + 1.51 V E0 = + 0.77 V 3) 2 Hg2+ + 2 Cl- + 2 e5) Sn4+ + 2 e- Mn2+ + 4 H2O Hg2Cl2 E0 = + 0.53 V 2 Hg (l) + 2 Cl- E0 = + 0.268 V Sn2+ E0 = + 0.15 V Reazione desiderata 1 – 2 Reazione 1 – 5 ininfluente perché Sn2+ è stato tutto ossidato con l’eccesso di Hg2+. La 4 è da escludere (se si forma Hg (l) la titolazione deve essere ripetuta). Quindi la reazione deve essere 1 – 2 e NON 1 – 3 Infatti alla fine della titolazione di Fe2+ il colore deve rimanere solo per 15 secondi altrimenti si avrebbe l’ossidazione di Hg2Cl2. 75 IODIMETRIA E IODOMETRIA Il sistema I2 / 2I- (E0 = + 0.535 V) può agire da ossidante o da riducente a seconda del potenziale del composto con cui reagisce. Titolazioni dirette: IODIMETRIA lo iodio ossidante Titolazioni indirette: IODOMETRIA lo ioduro riducente Composti riducenti titolati con lo iodio: S4O62- + 2 H+ + 2 eS + 2 H + + 2 eSn4+ + 2 e- 2 HS2O3- H2S E0 = + 0.2 V E0 = + 0.142 V Sn2+ E0 = + 0.15 V SO42- + 4 H+ + 2 e- H2SO3 + H2O E0 = + 0.172 Sistemi ossidanti rispetto allo ioduro H 2 O 2 + 2 H + + 2 e- 2 H2O E0 = + 1.78 V Br2O3 + 6 H+ + 5 e- ½ Br2 + 3 H2O E0 = - 1.52 V MnO4- + 8 H+ + 5 e- Mn2+ + 4 H2O E0 = + 1.51 V Cl2 + 2 e- 2 Cl- Cr2O72- + 14 H+ + 6 e- E0 = + 1.36 V 2 Cr3+ + 7 H2O E0 = + 1.33 V E I2 / 2 I- è indipendente dal pH per valori < 8; a pH maggiori: I2 + 2 OH- IO- + I- + H2O e 3 IO- IO3- + 2 I- (disproporzione) Lo iodio è scarsamente solubile in H2O (5 · 10-…) Si opera in eccesso di I- che forma un complesso solubile I3- (I2 + I- I3 - ) NOTA: i metodi iodimetrici e iodometrici sono caratterizzati dall’avere un indicatore molto sensibile a tracce di iodio (salda d’amido) oppure amido colloidale che adsorbe I- quindi fissa in superficie tracce di iodio come I3- ed impartisce al colloide un colore BLU VIOLA che rivela una concentrazione di iodio pari a 2 · 10-5 M purché sia I- > 4 · 10-4 mol/L 76 Nelle titolazioni iodometriche la salda d’amido va aggiunta in prossimità dell’equivalenza per evitare l’assorbimento troppo profondo di I3- che renderebbe il viraggio poco netto. Evitare alta temperatura (minore sensibilità dell’indicatore) e l’ambiente troppo acido (idrolisi dell’indicatore). Il metodo più diffuso è la Iodometria (I- funziona da riducente). Il metodo si dice indiretto in quanto la soluzione di ioduro è trattata a pH opportuno con l’ossidante in esame. Lo iodio liberato è titolato con adatto riducente (tiosolfato) in presenza di salda d’amido sino a scomparsa di colore. NOTA: se l’ambiente è troppo acido (es. quando è presente un sistema riducente come MnO4-, Cr2O72-) si possono avere 2 errori: 1) favorisce la O2 + 4 H+ + 4 e- 2 H2O E0 = + 1.231 Ossidazione dello ioduro da parte dell’O2 dell’aria Allora o si opera in ambiente neutro, oppure si riduce [O2] in soluzione aggiungendo NaHCO3 che libera CO2, sposta l’aria disciolta e diminuisce la pO2 nella fase gassosa. Si titola con una beuta a tappo smeriglio, avendo cura, dopo la titolazione (formazione di iodio) di sollevare poco il tappo e di lavarlo con una soluzione di KI per riportare in soluzione lo iodio che per la sua volatilità aveva aderito al tappo e alle pareti della beuta. Preparazione della salda d’amido 5 gr di amido solubile sono stemperati in un mortaio con poca acqua distillata sino ad avere una pasta piuttosto fluida. Si versa la pasta cautamente in 1 litro di H2O bollente e si prosegue l’ebollizione per circa 1 minuto. Dopo raffreddamento al 77 liquido (limpido) si aggiungono circa 0.03 g di HgI2 che impedisce la fermentazione dell’amido. (conservazione per qualche mese) Preparazione soluzione di Na2S2O3 ≈ 0.1 N Il tiosolfato è facilmente ottenibile allo stato puro ma essendo efflorescente non può essere considerata sostanza madre. Na2S2O3 è un forte riducente ed ossidandosi dà prodotti come S4O62-, S2O32-, SO42-. Il potenziale di S4O62- + 2 H+ + 2 e- 2 HS2O3- a pH = 0 è + 0.2 V, per cui se l’ossidante ha un potenziale più alto del tiosolfato quest’ultimo può essere ossidato a tetrationato e lo I2 è l’ossidante adatto (E0 = + 0.535). In ambiente molto acido lo S2O32- è instabile e reagisce con H+ (disproporzione) S2O32- + H+ HS2O3- HSO3- + S Teniamo conto delle reazioni di ossidazione che lo ione S2O32- e HSO3- danno con lo I2 . 2 S2O32- + I2 S4O62- + 2 I- HSO3- + I2 + H2O HSO4- + 2 I- + 2 H+ (1) (2) Quindi se S2O32- si decompone in HSO3- e S la sua normalità aumenta. Infatti nella reazione con I2 il peso equivalente del tiosolfato è pari al peso molecolare, mentre il peso equivalente del solfuro è la metà del peso molecolare. Es. una soluzione di S2O32- a normalità iniziale N in cui una parte X (eq / L) si trasforma in HSO3- si avrà (può avvenire per la CO2 in H2O) La normalità all’equilibrio sarà N–X+2X=N+X Si ha quindi un aumento del titolo ed un intorbidimento della soluzione dovuto ad S. In ambiente alcalino e luce diretta si ha l’ossidazione del tiosolfato da parte di O2 dell’aria: S2O32- + 2 O2 + H2O 2 SO42- + 2 H+ 78 Con conseguente diminuzione del titolo della soluzione. Preparazione di una soluzione stabile - usare H2O bollita di recente - aggiungere 2 - 3 gocce per litro di CHCl3 o 5 – 10 mg di HgI2 come batteriostatico - evitare lunghe esposizioni ad intensa luce diretta Standardizzazione della soluzione di Na2S2O3 circa 0.1 N Il titolo di S2O32- è effettuato con I2 ottenuto trattando un noto numero di equivalenti di un acido ossidante in soluzione acida per acido minerale con un eccesso di KI si può usare come sistema ossidante: KMnO4 titolato o K2Cr2O7 o KIO3 come sostanze madri. Con sostanze madri Reattivi: HCl, NaHCO3, KI, K2Cr2O7 o KIO3 puri Indicatore: salda d’amido Reazioni Cr2O72- + 6 I- + 14 H+ 3 I2 + 2 Cr3+ + 7 H2O Oppure: IO3- + 6 H+ + 5 II2 + 2 S2O32- 3 I2 + 3 H 2 O 2 I- + S4O62- Calcoli: Il viraggio è blu → verde per il bicromato Il viraggio è blu → incolore per lo iodato 79 Soluzione KMnO4 a titolo noto Reattivi: H2SO4, NaHCO3, KI, KMnO4 Indicatore: salda d’amido Reazioni: 2 MnO4- + 10 I- + 16 H+ I2 + 2 S2O32- 2 Mn2+ + 5 I2 + 8 H2O 2 I- + S4O62- Calcolo Procedimento In una beuta a tappo smeriglio da 300 ml si versano 50 ml di H2O, 15 ml H2SO4 e 0.2 – 0.3 g di NaHCO3. Terminato lo sviluppo gassoso si aggiunge un volume noto (25 – 35 ml) di soluzione standard di KMnO4. Si lavano con poca H2O le pareti della beuta, si aggiungono 5 - 6 g di KI e si chiude rapidamente il tappo smerigliato. Dopo riposo al buio per 3 - 4 minuti si sfila il tappo, si lava con KI e si titola rapidamente con tiosolfato sino a colore giallo bruno. Si aggiungono 2 ml di salda d’amido e si titola lentamente sino a viraggio da blu ad incolore. 80 Il “titolo” di H2O2 nei prodotti commerciali rappresenta il contenuto di prodotto attivo ed è espresso in: a) volumi di O2 (a 0°C e a 760 mmHg) ottenibili dalla decomposizione di H2O2 presente in un volume unitario del prodotto commerciale. b) % in volume (grammi di H2O2 in 100 ml soluzione) P.M. H2O2 = 34.02 1 equivalente di H2O2 a quanti litri di O2 a C.N. corrisponde? H2O2 → H2O + ½ O2 1 mole di O2 a C.N. = 22.4 litri ½ mole di O2 a C.N. = 11.2 litri 1 equivalente di H2O2 darà 5.6 litri di O2. H2O2 si conserva in polietilene perché la rugosità del vetro favorisce la decomposizione. Dosaggio H2O2 Reattivi: Na2S2O3 a titolo noto, (NH4)2MoO4 (sol 3%); H2SO4 2 N, NaHCO3, KI Indicatore: salda d’amido Reazioni: H 2O 2 + 2 H + + 2 I2 S2O32- + I2 I2 + 2 H 2 O 2 I- + S4O62- Calcolo: 81 TITOLAZIONE DI UN CAMPIONE DI RAME Reagente tiosolfato di sodio, Na2S2O3 0.100 N Titolazione di prova con permanganato In una beuta a tappo smeriglio da 250 mL si versano 50 mL di H2O distillata, 15 ml di H2SO4 (1 : 4) e 0.2 – 0.3 grammi di NaHCO3. Terminato lo sviluppo gassoso si aggiunge un volume esattamente noto [compreso tra 25 (dosare 25 ml con pipetta tarata) e 35 ml] di KMnO4 circa 0.1 N. Si lavano con poca acqua distillata le pareti della beuta, si introducono 5 - 6 grammi di KI e si chiude rapidamente il tappo. Dopo riposo al buio per 5 minuti si sfila il tappo lavandolo con un getto (usare una spruzzetta) di KI in soluzione (preparata di recente) al 10% e si titola rapidamente con la soluzione di tiosolfato sino a colore giallo – bruno. A questo punto si aggiungono 2 ml di salda d’amido e si titola lentamente fino a viraggio da blu – violaceo a incolore. Reazioni: 2 MnO4- + 10 I- + 16 H+ → 2 Mn2+ + 5 I2 + 8 H2O I2 + 2 S2O32- → 2 I- + S4O62DI SEGUITO SI TITOLA IL CAMPIONE INCOGNITO DI RAME Le semireazioni da considerare per la determinazione iodometrica del rame (II) sono le seguenti: Cu2+ + I- + e- → CuI (s) E0 = 0.82 V e I3 - + 2 e- → 3 I- E0 = 0.536 V La differenza dei potenziali standard (∆E0 = 0.284 V), come pure un eccesso di ioni ioduro, assicura la completezza della reazione. 82 2 Cu2+ + 5 I- → I3- + 2 CuI (s) Tale reazione, infatti ha una costante di equilibrio pari a 102 ∆E0/0.059 = 102 0.284/0.059 = 4.2 · 109 Procedimento Una quantità esattamente pesata (4.4 – 4.5 g) del campione, opportunamente omogeneizzato con la spatolina nel contenitore, viene sciolto e portato a volume con acqua distillata in un matraccio tarato da 250 ml. Si prelevano quindi esattamente 50.00 mL (pipetta tarata) e si introducono in beuta a tappo smeriglio da 250 mL. Dopo diluizione a circa 150 ml si tratta goccia a goccia con NH4OH (1.5) sino a intorbidamento persistente. Si aggiungono 5 mL di CH3COOH concentrato e 5 grammi di KI. Si chiude la beuta e dopo cauta agitazione si pone al riparo dalla luce per circa 5 minuti. Quindi, si sfila il tappo lavandolo con la soluzione di KI al 10% contenuta nella spruzzetta e si titola con il tiosolfato fino al colore giallo paglierino. Si aggiungono 2 mL di salda d’amido e si prosegue la titolazione fino a scomparsa del colore blu - violaceo e comparsa di un colore bianco-latte del CuI (s) precipitato. Si aggiungono 2 g di tiocianato di ammonio (NH4CNS), si agita e si completa la titolazione nel caso di ricomparsa della colorazione violacea. Calcolo 83
Scaricare