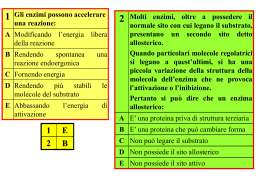
La via per accelerare i processi biologici ENZIMI Sono delle proteine altamente specializzare con attività catalitica, accelerano le reazioni chimiche rimanendo inalterati al termine della reazione stessa. La loro struttura, come quella delle proteine, è molto complessa e caratterizzata da una ben precisa configurazione tridimensionale con ripiegamenti, rientranze e sporgenze. Il sito attivo dell’enzima è un regione (estremamente piccola della molecola) a forma di fessura o di tasca che istaura legami con il substrato. E’ costituito da pochi residui di amminoacidi con una configurazione spaziale ben definita, complementare a quella del substrato con cui interagisce. Per l’interazione enzima substrato è stato suggerito il modello chiave serratura: La maggior parte degli enzimi portano nella loro molecola una parte non proteica. In questo caso l’enzima intero prende il nome di oloenzima, la componente proteica di apoenzima, la non proteica di gruppo prostetico Se il gruppo prostetico è facilmente dissociabile dall’apoenzima esso prende il nome di coenzima. La nomenclatura e la classificazione degli enzimi si basa sul tipo di reazione catalizzata, si hanno così 6 classi di enzimi: All’interno delle classi gli enzimi vengono generalmente classificati in base al nome del substrato specifico DIFFERENZE TRA ENZIMI E CATALIZZATORI INORGANICI Gli enzimi si distinguono dai comuni catalizzatori inorganici per le seguenti significative caratteristiche: Peso molecolare Specificità Potere catalitico A differenza dei catalizzatori della chimica inorganica che sono composti molto semplici spesso anche semplici atomi, gli enzimi sono molecole proteiche e, pertanto, espressione dei geni. Sono caratterizzati da un peso molecolare molto alto e da una configurazione tridimensionale piuttosto complessa, articolata nelle quattro strutture proprie delle proteine. Diversamente dai catalizzatori inorganici che molto spesso si comportano da pas-partout e catalizzano numerose reazioni anche molto diverse tra loro, gli enzimi sono altamente specifici sia verso il substrato sul quale agiscono, sia verso il tipo di reazione che catalizzano. La maggior parte di enzimi agisce su di un unico substrato o su un numero molto limitato di composti. Gli enzimi accelerano le reazioni almeno di un milione di volte. ENERGIA DI ATTIVAZIONE Perché una reazione chimica possa avvenire, si devono rompere i legami preesistenti e si devono eventualmente formare nuovi legami. Consideriamo una popolazione di molecole R-X che devono reagire per formare il prodotto R+ X-. Affinché una reazione chimica avvenga è necessario: • le molecole si urtino • urto efficace, nel senso che le molecole che si urtano devono avere un contenuto energetico tale da permettere loro di formare il complesso attivato (Rδ+--- X δ-) ad alto contenuto energetico e bassa stabilità. Il livello energetico a cui è localizzato il complesso attivato si chiama stato di transizione. La differenza di energia tra i reagenti e lo stato di transizione viene detta energia di attivazione (Ea). Ea rappresenta quindi una barriera energetica che i reagenti devono superare per trasformarsi nei prodotti. Il tutto può essere simpaticamente rappresentato come un sasso (reagente) che per arrivare a valle (prodotto) deve superare la montagna (Ea) Maggiore è il numero di molecole R-X capaci di formare, nell’unità di tempo, il complesso attivato maggiore sarà la velocità di reazione. La velocità di reazione è definita come la quantità di sostanza consumata o prodotta nell’unità di tempo: v = dc/dT Una reazione chimica può essere accelerata aumentando la temperatura. Infatti aumentando la T aumenta l’energia cinetica delle particelle aumenta il numero degli urti efficaci tra le molecole un maggior numero di particelle, nell’unità di tempo, formerà il complesso attivato. CATALISI ENZIMATICA Un altro modo per accelerare una reazione è quello di aggiungere un enzima (E). E si lega alle molecole R-X per formare il complesso attivato (E-Rδ+---Xδ-) il cui stato di transizione è più basso rispetto a quello della reazione non catalizzata. Così alcune molecole di R-X che prima non erano in grado di reagire, adesso legandosi a E entrano nello stato di transizione (più basso) per formare i prodotti. Gli Enzimi catalizzano tutte le reazioni che avvengono nell’ambiente cellulare dove le condizioni di temperatura e concentrazione esistenti comporterebbero tempi di reazione molto lunghi. Come si misura la velocità di una reazione catalizzata La velocità di reazione è definita come la quantità di sostanza consumata o prodotta nell’unità di tempo. Un altro modo per esprimere la velocità di una reazione catalizzata è il numero di turnover. Esso indica il numero di molecole di substrato che reagiscono con una sola molecola di enzima nell’unità di tempo. La velocità di reazione cambia da enzima ad enzima ed è caratteristica di ognuno di Concentrazione essi. [P] [S] vo Andamento nel tempo di una reazione enzimatica misurata come comparsa del prodotto (P) o scomparsa del substrato (S). La linea tratteggiata tangente alla curva rappresenta la velocità iniziale v0 Tempo All’inizio quando è praticamente presente tutto il substrato la velocità segue un andamento rettilineo (v0), a mano a mano che il substrato viene consumato la velocità rallenta fino a raggiungere un plateau quando tutto il substrato è stato convertito in prodotto. La velocità di una reazione catalizzata è influenzata dai seguenti fattori Concentrazione del substrato Concentrazione dell’enzima Temperatura pH Inibitori CONCENTRAZIONE DEL SUBSTRATO A parità di altre condizioni (temperatura, concentrazione di enzima, pH, ecc.) riportando in grafico la concentrazione di substrato in funzione della v0 si ottiene una curva iperbolica. Si osserva che, nel primo tratto della curva (rettilineo) la v0 è direttamente proporzionale alla concentrazione di substrato (aumentando la concentrazione di S aumenta proporzionalmente il numero di molecole che formano il complesso ES). Una volta raggiunta una certa concentrazione di S la v0 cresce più lentamente fino a raggiungere un valore massimo quando tutto l’enzima è saturato dal substrato e pur continuando ad aumentare la concentrazione di S la v0 rimane costante (Vmax). E’ possibile risalire alla velocità di una reazione enzimatica dall’equazione v0= Vmax [S] / [S] + Km proposta dai due studiosi Michaelis e Menten. Nell’equazione, v è la velocità di reazione, Vmax la velocità massima, [S] la concentrazione del substrato e Km è la costante di Michaelis-Menten. Km corrisponde alla concentrazione di S alla quale la velocità è = Vmax/2 Equazione di Michaelis-Menten Vmax [S] v0 = Km + [S] A basse concentrazioni di substrato, quando [S] è molto più piccola di Km e quindi trascurabile, si ha v0 = Vmax [S] / Km cioè, la velocità è direttamente proporzionale alla concentrazione del substrato. Ad alte concentrazioni di substrato, quando [S] è molto più grande di Km, Km diventa trascurabile e si ha v0 = Vmax, cioè, la velocità è la massima, indipendentemente dalla concentrazione del substrato. I valori di Km variano moltissimo da enzima ad enzima ed esprimono l’affinità che l’enzima ha per il substrato. Osservando la posizione di Km sul grafico velocità contro concentrazione di substrato, si può notare che se Km è bassa, in ogni istante è necessaria una bassa concentrazione di substrato per saturare metà delle molecole di enzima e questo è segno di alta affinità dell’enzima per il substrato, mentre se Km è alta, occorre una più alta concentrazione di substrato per saturare metà delle molecole di enzima in ogni istante e questo vuol dire che l’enzima presenta bassa affinità per il substrato. Il valore di Km è indipendente dalla concentrazione dell'enzima e dalla concentrazione del substrato. Per un calcolo più accurato della Vmax e della Km è opportuno trasformare matematicamente l’equazione di Michealis-Menten facendo il reciproco di entrambi i lati dell’equazione Vmax [S] v0 = Km + [S] ………… 1 v0 = KM 1 Vmax [S] + 1 Vmax Si ottiene il grafico dei doppi reciproci (o grafico di Lineweaver-Burk) mediante il quale la curva iperbolica viene convertita in una retta CONCENTRAZIONE DELL’ENZIMA Km è indipendente dalla concentrazione dell’enzima v0 La velocità iniziale v0 di una reazione enzimatica è direttamente proporzionale alla concentrazione dell’enzima. 5 Unità enzimatica: quantità di enzima capace di trasformare una µmole di S in 1 minuto a 25 °C nelle condizioni ottimali del saggio. È quindi possibile misurare l’attività di un enzima (cioè la sua concentrazione espressa come unità per volume o per g di peso fresco o secco) mediante una misura della Vo ed in particolare della Vmax. Tali misure si effettuano utilizzando una concentrazione saturante di substrato (∼ 5 volte la Km quando la vo=Vmax) e monitorando la reazione nei primi minuti, quando tutto il substrato è praticamente disponibile. TEMPERATURA La velocità delle reazioni enzimatiche varia col crescere della temperatura secondo il grafico a campana riportato. Si può osservare che, inizialmente, la velocità cresce al crescere della temperatura, raggiunge un massimo in corrispondenza di una certa temperatura definita ottimale, si riduce, in seguito, per effetto della denaturazione dell’enzima. pH Come la variazione della temperatura, in modo un poco più complesso, anche la variazione del pH influenza la velocità delle reazioni enzimatiche. Anche in questo caso, la curva presenta un andamento a campana e l’attività enzimatica manifesta un massimo in corrispondenza di un valore definito pH ottimale legato alla natura del substrato. Inibitori In molti casi, molecole specifiche o ioni possono competere con le molecole di substrato nel legarsi con l’enzima ed inibire l’attività dell’enzima. L’inibizione può essere irreversibile e reversibile. IRREVERSIBILE E’ irreversibile quando l’inibitore si va a legare al sito catalitico dell’enzima con un legame molto forte, impedendo l’accesso del substrato. I+E Ki EI inattivo Un esempio di inibizione irreversibile è dato dall’azione dei gas nervini che bloccano l’azione dell’enzima acetilcolinesterasi, un enzima che ha un ruolo importantissimo nella trasmissione degli impulsi nervosi. REVERSIBILE L’inibizione reversibile può essere competitiva, quando gli inibitori sono, da un punto di vista chimico, molto simili alle molecole di substrato e si legano agli stessi siti attivi, e non competitiva, quando gli inibitori si legano a siti dell’enzima diversi da quelli che legano il substrato e, pertanto, possono legarsi sia all’enzima che al complesso ES. COMPETITIVA L’inibitore possiede una struttura molto simile a quella del substrato la similitudine porta il substrato e l’inibitore a competere per lo stesso sito attivo dell’enzima. L’esito della competizione dipende dalla concentrazione delle due molecole che si contendono il sito attivo Può essere completamente rimossa aumentando notevolmente la concentrazione di substrato NON COMPETITIVA L’inibitore si lega all’enzima in una zona diversa da quella del sito attivo dando luogo al complesso EI inattivo. Il legame dell’inibitore deforma la conformazione spaziale dell’enzima ed il suo sito catalitico pur potendosi legare al substrato risulta inattivo. E’ possibile distinguere la inibizione competitiva da quella non competitiva 1/Vmax 1/Vmax In presenza di inibitore per ottenere la stessa velocità di reazione che in sua assenza, è necessario aumentare la concentrazione di substrato. La Vmax rimane invariata (infatti a concentrazione elevata di substrato tutta l’inibizione viene rimossa) mentre la Km aumenta 1/Km diminuisce. A qualsiasi concentrazione di substrato la velocità di reazione in presenza di inibitore è sempre minore che in sua assenza. Quindi la Vmax diminuisce 1/Vmax aumenta, mentre la Km rimane costante. STUDIO DELLE INTERAZIONI PROTEINA-LIGANDO Queste interazioni sono alla base del riconoscimento molecolare e quindi rappresentano la chiave della maggior parte dei processi biologici: Specificità degli enzimi ed allosterismo Trasporto attraverso i compartimenti cellulari Trasduzione del segnale Regolazione della trascrizione e della traduzione Riconoscimento degli antigeni da parte degli anticorpi ed altro Come si analizzano tali interazioni e come si determinano le costanti di dissociazione o di legame? Concetti basilari per lo studio del binding recettoriale I recettori sono presenti in concentrazioni molto piccole nei tessuti. Incubando il tessuto contenente i recettori R ed il ligando radiomarcato D in opportune condizioni sperimentali si formerà il complesso RD secondo l’equazione R+D RD Quando il sistema raggiunge l’equilibrio in ogni provetta avremo: Radioligando Tessuto Tampone di incubazione Incubazione Ligando radiomarcato D Equilibrio Recettore RD Recettore libero R Il recettore libero R non potrà essere misurata ma possiamo misurare la quantità di complesso ligando-recettore RD. Per poter calcolare la quantità del complesso RD è necessaria un’operazione di separazione del legato (Bound) dalla quantità di ligando radiomarcato D libero in soluzione non legato al tessuto (Free). Equilibrio Separazione Metodi di separazione del RD dal mezzo di reazione Filtrazione Il metodo più semplice e classico è quello della filtrazione su membrane in fibra di vetro Whatman GF/A, GF/B, GF/C dove varia la porosità 0.7 µm-2.7 µm Il tessuto contenente il recettore legato al radioligando aderisce al filtro mentre il radioligando libero passa attraverso la membrana. Limiti e precauzioni della filtrazione Le proteine ostruiscono il filtro Il ligando radiomarcato tende a legarsi alle fibre del filtro. Elevata velocità di filtrazione (deve essere completata in 4-5 secondi per evitare che il ligando radiomarcato si stacchi dal recettore. In genere si perde il 5% di RD) Centrifugazione Limiti e precauzioni della centrifugazione Una certa quantità di ligando radiomarcato (Free) potrebbe restare intrappolato nel pellet. Numero limitato degli alloggi nel rotore Dialisi Viene impiegata quando il recettore ha bassa affinità per il ligando radiomarcato (micromolare). Una piccola membrana simile alla cellulosa da dialisi separa due camere L L R R L L R L L L La membrana è scelta in modo tale che il ligando possa passare mentre il recettore viene trattenuto. Il ligando diffonde fino a raggiungere l’equilibrio. Nella camera azzurra [L] sarà Ligando libero Nella camera gialla [L]= ligando libero +ligando legato La differenza fra questi valori è la concentrazione di ligando legato. Quella che viene misurata è la deplezione di ligando libero Analisi di saturazione Kon Recettore-Ligando Recettore + Ligando Koff Il binding avviene quando ligando e recettore collidono mediante diffusione e quando la collisione ha un corretto orientamento e sufficiente energia. La velocità di associazione è data dal numero di eventi di binding per unità di tempo Von = [Ligando] × [Recettore] × kon Una volta avvenuto il binding, ligando e recettore restano legati per un certo periodo di tempo. La velocità di dissociazione del complesso è data dal numero di eventi per unità di tempo Voff = [ligando-recettore] × koff Dopo la dissociazione il ligando e il recettore non devono aver subito modifiche. All’equilibrio avremo: Von = Voff [Ligando] × [Recettore] × kon = [Ligando-Recettore] × koff Riarrangiando l’equazione avremo: [Ligando] [Recettore] [Ligando-Recettore] Koff = = Kon Kd Per meglio comprendere il significato della Kd poniamo [Ligando] = Kd L’equazione pertanto diviene: [Recettore] [Ligando-Recettore] da cui si evince che: = 1 [Recettore] = [Ligando- Recettore] Quando [Ligando] = Kd la quantità di recettore libero è esattamente uguale alla quantità di recettore occupato. I recettori totali sono la somma di quelli liberi più quelli legati al ligando, e quando [Ligando] = Kd avremo che metà della popolazione recettoriale sarà occupata all’equilibrio. Se il ligando ha elevata affinità per il recettore, la Kd sarà piccola perché basterà una piccola concentrazione di ligando per legare la metà dei recettori. Non bisogna confondere la Kd (costante di dissociazione all’equilibrio) con Koff (costante di dissociazione). Non sono la stessa cosa ed hanno dimensioni differenti. Kon = costante di associazione (molare-1 min-1) Koff = costante di dissociazione (min-1) Kd = costante di dissociazione all’equilibrio (molare) La legge di azione di massa permette di definire la frazione di recettore occupata all’equilibrio in funzione della concentrazione di ligando. Frazione recettoriale occupata [RL] [Rtot] = [RL] [R] + [RL] Questa equazione non è utile perché non conosciamo la concentrazione di recettore libero. Poiché [R] = [Rtot] - [RL] e sapendo che: [R] [L] Kd = [RL] [R] [L] ([Rtot] - [RL]) [L] [RL] = = Kd Kd [RL] Kd = [Rtot] [L] - [RL] [L] [RL] Kd + [RL] [L] = [Rtot] [L] [RL] (Kd + [L] ) = [Rtot] [L] [RL] = [Rtot] [L] Kd + [L] Bound (pmol/mg protein) 0.15 Binding totale Binding non specifico Binding specifico 0.10 Le tre curve corrispondenti sono riportate in figura 0.05 0.00 0.1 0.2 0.3 0.4 0.5 0.6 [3H]spiroperidolo, (nM) Visualizzando solo la curva del binding specifico possiamo individuare la Kd e la Bmax Bound (pmol/mg protein) 0.100 0.075 Bmax 0.050 0.025 0.000 0.00 Kd 0.25 0.50 0.75 [3H]spiroperidolo, (nM) 1.00 da cui la frazione recettoriale occupata [RL]/ [Rtot] sarà: [Ligando] [RL] [Rtot] [Ligando] = Ligando + Kd Frazione recettoriale occupata 0 0% 1Kd 50% 4Kd 80% 9Kd 90% 99Kd 99% Informazioni provenienti dall’analisi di saturazione KD Indica la concentrazione di radioligando che all’equilibrio occupa il 50% dei recettori presenti nel preparato biologico Bmax Indica la densità recettoriale nel tessuto studiato. Si esprime in moli/mg di proteina. Approccio sperimentale all’analisi di saturazione L’esperimento di saturazione viene effettuato determinando il Binding Totale e il Binding Non-Specifico a differenti concentrazioni di ligando radiomarcato. Binding Totale Viene definito aggiungendo al tessuto concentrazioni crescenti di radioligando Binding Non-Specifico Viene definito in presenza di ligando non marcato ad elevata concentrazione alla quale tutto il radioattivo viene spiazzato dai siti specifici di legame Esempio: Vogliamo determinare il binding specifico all’equilibrio per un determinato radioligando alla concentrazione di 0.5 nM Provetta 1 (Binding totale) Tessuto + Rx (0.5 nM) Lettura radioattività =2500 cpm Provetta 2 (Binding non specifico) Tessuto + Rx (0.5 nM) + Ligando non marcato (10 µM) Lettura radioattività= 500 cpm Alla concentrazione 0.5 nM di RX il complesso RD (Binding Specifico, Bound) sarà : 2500-500= 2000 cpm. Il valore 500 cpm rappresenta la quantità di RX che si è legato a siti non specifici di legame. Infatti l’impiego del ligando non marcato ad levata concentrazione spiazza RX solo da tutti i siti specifici ma non da quelli non specifici. Realizzando differenti concentrazioni di RX e definendo per ciascuna il Binding Totale e il binding Non-Specifico si calcola il Binding Specifico. Bound (pmol/mg protein) 0.15 Binding totale Binding non specifico Binding specifico 0.10 Le tre curve corrispondenti sono riportate in figura 0.05 0.00 0.1 0.2 0.3 0.4 0.5 0.6 [3H]spiroperidolo, (nM) Visualizzando solo la curva del binding specifico possiamo individuare la Kd e la Bmax Bound (pmol/mg protein) 0.100 0.075 Bmax 0.050 0.025 0.000 0.00 Kd 0.25 0.50 0.75 [3H]spiroperidolo, (nM) 1.00 Come trasformare i cpm in concentrazione Esempio:Un campione dal volume totale di un 1 mL fornisce 5000 cpm per un Rx di attività specifica 27 Ci/mmole cpm = efficienza (%) dpm Conoscendo l’efficienza strumentale (es 55%) 5000 cpm corrispondono a 9090 dpm Sapendo che 2.22 x 106 dpm corrispondono a 1 µCi Avremo che 9090 dpm corrispondono a 4.1 x 10-3 µCi Dall’attività specifica del radioligando sappiamo che 27 Ci corrispondono ad 1 mmole. Cioè 27 x 106 µCi ad 1 mmole Avremo che 4.1 x 10-3 µCi corrispondono a 1.52 x10-10 mmoli Il campione in esame ha un volume totale di 1 mL La concentrazione sarà: 1.52x10-10 M (0.152 nM). Analisi di Scatchard Partendo dall’equazione: [Rtot] [L] [RL] = Kd + [L] [RL] ([L] + Kd ) = [L] [Rtot] [RL] [L] + [RL] Kd = [L] [Rtot] dividendo tutto per [L] Kd avremo: [RL] [L] [L] Kd + [Rtot] [L] [RL]Kd [L] Kd = [L] Kd quindi: [RL] [L] = [Rtot] Kd - [RL] Kd Se indichiamo con B la concentrazione di ligando legato [RL]; con Bmax la quantità massima di ligando legato [Rtot]; con F la quantità di ligando libero [L] avremo: B _ B + Bmax = F Kd Kd Riportando in grafico il rapporto tra la quantità di ligando legato e ligando libero (B/F) rispetto alla quantità di ligando legato (B) avremo: 1.00 B/F 0.75 0.50 Bmax 0.25 0.00 0.000 0.025 0.050 0.075 0.100 0.125 0.150 Bound Si ottiene una retta con Slope = - 1 Kd L’intercetta sull’asse delle ascisse (B/F = 0) permette di ricavare Bmax che rappresenta la densità recettoriale. Se è presente un solo tipo di recettore lo Scatchard sarà lineare. Se sono presenti invece due tipi di recettore in uguale concentrazione ma con differenze in Kd per il radioligando avremo: B Binding specifico A 1.00 0.75 B/F 0.50 0.25 [radioligando] 0.00 0.000 0.025 0.050 0.075 0.100 0.125 0.150 Bound Le curve dei singoli recettori sono riportate in rosso e azzurro mentre in nero la curva somma del binding totale. Nel grafico di Scatchard (B) la linea nera del binding totale (inteso come somma del binding specifico delle popolazioni recettoriali esistenti) evidenzia una curvatura e le due linee tratteggiate rappresentano il binding specifico di ciascun recettore. Dall’analisi di Scatchard si può determinare l’omogeneità di una popolazione recettoriale (sempre che vi siano differenze evidenti tra i valori di Kd del radioligando per ciascun recettore). In caso contrario avremo un’informazione apparente (di un’unica popolazione recettoriale). La soluzione del problema è realizzata ovviamente da radioligandi selettivi per ciascun tipo di recettore presente. Analisi di Hill Considerando l’equazione: [Rtot] [L] [RL] = Kd + [L] che può essere scritta: [RL] [L] + [RL] Kd = [Rtot] [L] [RL] Kd = [Rtot] [L] - [RL] [L] [RL] = [L] ([Rtot] - [RL]) Kd da cui: [RL] = [Rtot] - [RL] [L] Kd se il ligando L interagisce con n siti secondo l’equazione nL + R avremo: [RL] RLn = [Rtot] - [RL] B Bmax - B [L]n Kd = [L]n Kd passando ai logaritmi: B log Bmax - B dove n è il coefficiente di Hill (nH) log n log [L] - log Kd Se esiste un solo sito di interazione per recettore (n =1) si ha una retta con pendenza = 1 B Bmax - B Per calcolare la Kd bisogna porre log = B =0 Bmax - B 0 pendenza = nH log [L]
Scaricare