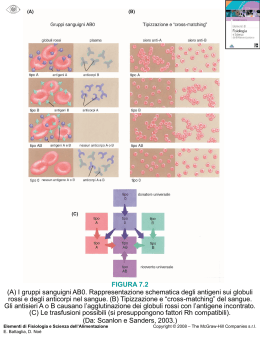
La funzione dei globuli rossi è di trasportare ossigeno ai tessuti periferici. Una riduzione in tale capacità del sangue è solitamente causata da un difetto nei globuli rosi, o anemia, definita come: riduzione sotto i limiti normali della massa totale dei globuli rossi circolanti. Comunque, dato che non è facile la misurazione di tale valore, nella pratica routinaria, l’anemia è definita come una riduzione rispetto al normale del volume di globuli rossi sedimentari, come misurato dall’ematocrito, o una riduzione della concentrazione di emoglobina nel sangue. I valori di riferimento dei globuli rossi negli adulti sono i seguenti: MISURAZIONE UOMINI DONNE Emoglobina (g/dl) Ematocrito Conta dei globuli rossi (106/µl) Conta dei reticolocita (%) Volume corpuscolare medio (µm3) Emoglobina corpuscolare media (pg) Concentrazione corpuscolare media di emoglobina (g/dl) Ampiezza di distribuzione della dimensione dei globuli rossi 13,6 - 17,2 39 - 49 4,3 - 5,9 12,0 - 15,0 33 - 43 3,5 - 5,0 0,5 - 1,5 82- 96 27 - 33 33 - 37 11,5 - 14,5 In clinica di afferma l’insorgenza di anemia quando nella femmina i livelli di Hb scendono sotto i 12, mentre nel maschio sotto i 13 g. È possibile fare una distinzione per quanto riguarda la gravità dell’anemia, e quindi individuare: 1. anemia lieve Hb < 10 g/dl 2. anemia moderata Hb 8-10 g/dl 3. anemia grave Hb < 8g/dl La classificazione maggiormente accettata delle anemie, viene effettuata in base ai meccanismi patogenetici. Distinguiamo quindi tre cause principali che portano le rispettive tipologie di anemia: 1. AUMENTATA DISTRUZIONE (ANEMIE EMOLITICHE): anomalie intrinseche dei globuli rossi: • anomalie ereditarie • anomalie della membrana del globulo rosso ♦ anomalie del citoscheletro: sferocitosi ♦ disordini della sintesi dei lipidi: aumento selettivo della lecitina di membrana • deficit enzimatici dei globuli rossi ♦ enzimi glicolitici: piruvatochinasi o esochinasi ♦ enzimi dello shunt dell’esosomonofosfato: G6PD, glutatione sintetasi • anomalie della sintesi dell’emoglobina ♦ deficit di sintesi dell’emoglobina: sindromi talassemiche ♦ emoglobinopatie: anemia falciforme, malattie da emoglobine instabili. • anemie acquisite ♦ lesione acquisita di membrana anomalie estrinseche • mediata da anticorpi ♦ isoemoagglutinine: eritroblastosi fetale 1 autoanticorpi: idiomatica, da farmaci, LES, neoplasie, infezioni da micoplasma trauma meccanico dei globuli rossi ♦ anemie emolitiche microangiopatiche: porpora trombotica, trombocitopenia, CID ♦ anemia emolitica da valvole cardiache meccaniche ♦ • infezioni • malaria, anchilostoma danno chimico • avvelenamento da piombo sequestro nelle cellule del sistema monociti macrofagico • ipersplenismo 2. ALTERATA PRODUZIONE DI GLOBULI ROSSI: anomalie proliferative e differenziative delle cellule staminali • aplasia midollare, aplasia pura della linea rossa, anemia in corso di insufficienza renale disturbi della proliferazione e della maturazione degli eritroblasti • difettosa sintesi del DNA; deficit di vitamina B12 e dell’acido folico • difettosa sintesi di Hb: ♦ ridotta sintesi dell’eme: deficit del ferro ♦ deficit di sintesi di globina: talassemia ♦ meccanismi sconosciuti: anemia sideroblastica, anemia da infezioni croniche, anemia mieloftisica da infiltrazioni del midollo osseo 3. PERDITA DI SANGUE: acuta: traumi cronica: lesioni del tratto gastrointestinale, patologie ginecologiche Un secondo utile approccio classifica le anemie in base alle alterazioni morfologiche dei globuli rossi, che spesso correla con la causa della carenza di globuli rossi. Caratteristiche morfologiche che forniscono indizi eziologici comprendono: 1. RBC = concentrazione di globuli rossi per unità di volume 2. MCV = volume corpuscolare medio 3. MCH = emoglobina corpuscolare media (27-31 pg) 4. MCHC = concentrazione corpuscolare media di emoglobina 5. conta reticolocitaria = importante per valutare il grado i produzione eritrocitaria midollare. Qualunque sia la causa, l’anemia quando è sufficientemente grave porta ad alcune caratteristiche cliniche: 1. APPARATO CARDIOCIRCOLATORIO: dispnea da sforzo, tachicardia, palpitazioni, angina, claudicatio, dispnea a riposo 2. SISTEMA NERVOSO: affaticamento, difficoltà di concentrazione, irritabilità, cefalea, vertigini, lipotimia, parestesie 3. SISTEMA GASTROENTERICO: anoressia, nausea, flatulenza, stipsi/diarrea 4. APPARATO GENITALE: irregolarità mestruali, della potenza/libido 5. CUTE, MUCOSE e ANNESSI: pallore, glossiti, disfagia, coilonichia, caduta dei capelli 2 PERDITA EMATICA ACUTA Gli effetti delle perdite ematiche acute sono principalmente dovuti alla diminuzione del volume intravascolare, che può portare a collasso cardiovascolare, shock e morte. Se il paziente sopravvive, la perdita di liquidi viene contrastata dallo spostamento dell’acqua dagli spazi interstiziali. Questa acqua va a diluire l’ematocrito. ⇓ O2 ai tessuti periferici, comprese le cellule juxtaglomerulari del rene ⇑ la produzione di eritropoietina da parte delle cellule juxtaglomerulari ⇑ proliferazione delle cellule staminali eritroidi committed (CFU-E) Dopo 5 giorni si ha la differenziazione completa in reticolocita, e quindi in eritrociti. Se la perdita ematica avviene all’interno, come nella cavità peritoneale, il ferro delle eme viene riciclato e riutilizzato per la produzione di nuova emoglobina, altrimenti, se l’emorragia è esterna si ha una perdita di ferro ed eventualmente una carenza di ferro, che può compromettere il ripristino della normale conta dei globuli rossi. PERDITA EMATICA CRONICA Una perdita ematica cronica induce anemia solo quando 1. l’entità della perdita eccede la capacità rigenerativa del midollo 2. le riserve di ferro vengono esaurite Le anemie emolitiche presentano le seguenti caratteristiche: 1. vita più breve dei globuli rossi 2. livelli elevati di eritropoietina e aumento dell’eritropoiesi 3. accumulo dei prodotti del catabolismo dell’emoglobina. L’emolisi intravascolare dei globuli rossi è causata da danno meccanico, fissazione del complemento, infezione da parassiti intracellulari o fattori esogeni e, qualunque sia la causa si manifesta attraverso: • emoglobinuria • emoglobinemia • ittero • emosiderinuria L’emoglobina libera nel plasma è prontamente legata dall’aptoglobina, formando un complesso che viene eliminato dal sistema dei fagociti mononucleati, prevenendone così l’eliminazione attraverso le urine. La diminuzione dell’aptoglobina è caratteristica dell’emolisi intravascolare, e, quando è carente questa proteina, l’emoglobina libera viene ossidata in metaemoglobina, che è di colore marrone. Le cellule renali assorbono e catabolizzato molta dell’emoglobina e della metaemoglobina filtrata, tuttavia una parte viene comunque escreta con le urine, conferendo a queste la tipica colorazione rosso-mattone. Inoltre il ferro rilasciato dal catabolismo dell’emoglobina può accumularsi nelle cellule tubulari, dando origine a emosiderosi renale. Allo stesso tempo una parte dell’emoglobina in circolo viene captata dal sistema monocito-macrofagico nel fegato e metabolizzata a bilirubina, con la comparsa di ittero. La bilirubina delle anemie emolitiche non è coniugata. Nelle anemie emolitiche extravascolari, l’emolisi ha luogo ogni qualvolta i globuli rossi sono considerati estranei o diventano meno plastici. La perdita dell’elasticità dei globuli rossi, rende 3 difficoltoso il loro passaggio attraverso i sinusoidi splenici, e porta al loro sequestro nei cordoni splenici seguito da fagocitosi. Questo meccanismo è molto importante, dato che nell’emolisi extravascolare non si verificano emoglobinemia ed emoglobinuria, e le principali manifestazioni sono anemia e ittero. Inoltre l’iperattività del sistema dei fagociti mononucleati spesso porta a splenomegalia. L’anemia falciforme è un’importante emoglobinopatie ereditaria, caratterizzata dalla produzione di una emoglobina difettosa. Le emoglobinopatie clinicamente significative derivano da mutazioni nel gene delle β-globina. L’anemia falciforme è causata da una mutazione puntiforme in posizione sei della catena della βglobina che comporta la sostituzione di un residuo di valina con uno di glutammato. Si forma così la tipologia di emoglobina indicata dalla sigla HbS. PATOGENESI Se deossigenate le molecole di HbS vanno incontro ad aggregazione e polimerizzazione. Negli stadi iniziali, l’ossigenazione riporta la conformazione del globulo rosso a quella naturale, ma le continue ossigenazioni e deossigenazioni comportano lesioni nella membrana delle cellule, che diventano col tempo irreversibilmente falciforme, mantenendo quindi la forma falcizzata anche quando ossigenate. Molti fattori influenzano l’entità e il grado di falcizzazione. ⇒ INTERAZIONE CON LE ALTRE CATENE DI EMOGLOBINA NELLA CELLULA. Infatti nei soggetti che presentano la mutazione ma sono eterozigoti, la percentuale di HbS è del 40 %. Il resto rimane HbA, che interagisce solo debolmente con l’HbS. Quindi questi soggetti saranno portatori sani asintomatici. Altra condizione di asintomaticità è costituita dai neonati, che presentano HbF che ha la capacità di impedire la polimerizzazione dell’HbS. Quindi i neonati saranno asintomatici fino a 5-6 mesi di età. L’HbC, emoglobina che ha una mutazione puntiforme della catena della β-globina portando una lisina al posto del glutammato in posizione 6, manifesta la tendenza ad aggregarsi con l’HbS, portando una sintomatologia più lieve dell’anemia falciforme. ⇒ CONCENTRAZIONE CELLULARE DI EMOGLOBINA (MCHC). Elevate quantità intracellulari di HbS aumentano la probabilità che l’emoglobina deossigenata polimerizza. Quindi tutte le condizioni che comportano disidratazione intracellulare, facilitano la falcizzazione. ⇒ DIMINUZIONE DEL PH. La diminuzione del pH riduce l’affinità dell’emoglobina per l’ossigeno, aumentando la frazione di HbS deossigenata. ⇒ INTERVALLO DI TEMPO IN CUI I GLOBULI ROSSI SONO ESPOSTI A UNA BASSA PRESSIONE DI OSSIGENO. I normali tempi di transito dei globuli rossi attraverso i capillari non sono sufficienti perché si verifiche una significativa aggregazione dell’HbS deossigenata. Quindi la falcizzazione si verificherà nei tessuti in cui il circolo è rallentato, soprattutto la milza e il midollo osseo. Inoltre un fattore che gioca un ruolo patogenetico particolarmente importante è l’infiammazione che provoca un rallentamento del flusso causato da due motivi: • adesione dei leucociti • adesione dei globuli rossi all’endotelio attivato Le manifestazioni cliniche dell’anemia falciforme sono dominate dall’emolisi cronica e dal danno ischemico del tessuto che risulta dall’occlusione dei piccoli vasi ematici. La patogenesi delle occlusioni microvascolari, una componente clinicamente importante dell’anemie falciforme, è meno chiara, comunque si è stabilito che sono principalmente responsabili le cellule reversibilmente falcizzata, che esprimono livelli di molecole di adesione più alti del normale e appaiono anormalmente adesive in diversi saggi. 4 In particolare queste cellule tendono ad aggregarsi in condizioni di flusso lento, e quindi è maggiormente probabile l’aggregazione in caso di infiammazione. Inoltre è probabile che gli stessi eritrociti falcizzata provochino la comparsa di fattori di adesione sull’endotelio, innescando una reazione a catena. EVOLUZIONE CLINICA È evidente che i pazienti sono colpiti da problemi derivanti da: 1. grave anemia 2. complicanze vaso-occlusive 3. iperbilirubinemia cronica Una complicanza molto frequente nei bambini con anemia falciforme è la maggior suscettibilità all’infezione da organismi capsulato. Questo è dovuto: 1. minor funzionalità splenica, dovuta alla congestione e allo scarso flusso sanguigno 2. difetti nella via alternativa del complemento. La scarsa funzionalità del complemento compromette l’opsonizzazione di batteri capsulati, come lo pneumococco e l’Haemophilus influenzae. Il decorso prolungato è frequentemente esacerbato da diverse crisi, classificate come: • CRISI VASO OCCLUSIVE: rappresentano episodi di danno da ipossia e infarto associato a grave dolore nelle parti affette. I distretti maggiormente soggetti a queste crisi sono polmoni, ossa, fegato, cervello, milza e pene. Nei bambini è molto frequente la crisi dolorosa dell’osso, difficilmente distinguibile dall’osteomielite acuta. Particolarmente importanti sono le crisi caso occlusive del polmone, che si presentano tipicamente con febbre, tosse, dolore toracico. • CRISI DA SEQUESTRO: si verificano nei bambini con milza intatta. Il continuo sequestro di globuli rossi comporta splenomegalia, a cui segue ipovolemia e qualche volta a shock. I frequenti infarti nella milza possono portare anche ad autosplenectomia funzionale. • CRISI APLASTICHE: in esse vie è una transitoria interruzione dell’eritropoiesi del midollo, dovuta a un’infezione acuta dei progenitori delle cellule eritroidi da parte del parvovirus B19. Oltre a queste crisi, anche l’ipossia cronica è responsabile di un’alterazione generalizzata della crescita e dello sviluppo così come del danno a organi quali milza, cuore, reni e polmoni. Il danno alla midollare del rene comporta la perdita della capacità di concentrare le urine (ipostenuria) che causa un’aumentata predisposizione alla disidratazione con tutti i rischi che ne conseguono. Altre complicanze sono: • calcolosi colecistica: a causa dell’iperbilirubinemia indiretta • megaloblastosi: a causa dell’aumento dell’eritropoiesi compensatoria. • • • • DIAGNOSI positività al sickling test: viene effettuato con metabisolfito, reagente che consuma ossigeno, comportando la falcizzazione dei globuli rossi in presenza di HbS; diagnosi prenatale mediante analisi del DNA tramite amniocentesi; Hb ↓ 6-9 g/dl elettroforesi dell’Hb: HbS 80-90 % TERAPIA È molto importante controllare le infezioni e se possibile effettuare vaccinazioni contro Pneumococco, Haemophilus e Neisseria. Un miglioramento nella terapia è stato effettuato con l’introduzione di un farmaco utilizzato per la terapia del cancro: l’idrossurea. Questo farmaco aumenta l’espressione dell’HbF, che contrasta l’aggregazione dell’HbS, è un farmaco antinfiammatorio, e quindi diminuisce il rallentamento del 5 flusso nelle zone infiammate, aumenta il volume medio dei globuli rossi e quindi diminuisce la concentrazione di HbS, e può essere ossidata per produrre NO. Le sindromi talassemiche sono un gruppo eterogeneo di patologie ereditarie causate da danni genetici che portano a una diminuzione nella sintesi delle catene dell’α- o della β-globina dell’HbA (α2β2). La β-talassemia è causata da difetti della sintesi della catena β, mentre l’α-talassemia è causata da difetti della sintesi della catena α. Le conseguenze ematologiche della minor sintesi di una catena della globina non sono soltanto provocate dalle basse concentrazioni di emoglobina intracellulare (ipocromia), ma anche dal relativo eccesso di catene difettiva. Per esempio nella β-talassemia, le catene α in eccesso si aggregano a formare inclusioni insolubili nei globuli rossi e nei precursori, portando a • una prematura distruzione degli eritroblasti che maturano nel midollo osseo (eritropoiesi inefficace) • lisi dei globuli rossi maturi nella milza (emolisi). α-TALASSEMIA Sono caratterizzate da una ridotta o assente sintesi di catene di α-globina. Ci sono quattro geni per l’α-globina, e la gravità della sindrome talassemia dipende dal numero dei geni mutati. Gli effetti sono mediati sia dalla carenza di emoglobina normale, che dall’eccesso di catene non-α. Nell’α-talassemia si assiste alla comparsa di diversi tipo di emoglobina, in base all’espressione dei geni della globina. Quindi avremo: • nel neonato: eccesso di γ-emoglobina che forma un tetrametro γ4 noto come emoglobina di Barts • negli adulti: eccesso di β-emoglobina che si aggrega formando tetrametri β4 (HbH). Poiché questi tetrametri sono più solubili rispetto alle catene α, l’eritropoiesi inefficace e l’emolisi sono meno marcate rispetto alla β-talassemia. MANIFESTAZIONI CLINICHE Le sindromi cliniche dipendono dal numero e dalla posizione dei geni meleti dell’α-globina. Questi geni sono quattro, presenti in duplice copia sul cromosoma 16. In genere ognuno di essi contribuisce per il 25% alla catena della globina risultante, ed è per questo che il numero di geni alterati influisce strettamente la struttura della globina. Quindi distinguiamo delle manifestazioni cliniche in base al numero di geni mutati, ed avremo le seguenti situazioni: 1. condizione di portatore silente: accade se viene mutato un solo gene. L’alterazione è insufficiente a causare anemia, e questi soggetti si presentano totalmente asintomatici; 2. tratto α-talassemico: è causato dalla delezione di due geni della globina α. La mutazione può interessare sia due geni dello stesso cromosoma (α/α -/-), che lo stesso gene deleto su entrambi i cromosomi (α/- α/-). Entrambe le situazioni produrranno manifestazioni cliniche molto simili (microcitosi), tuttavia i soggetti con genotipo α/α -/- sono a rischio per la generazione di una progenie con α-talassemia grave (sindrome HbH o idrope fetale); 3. malattia dell’emoglobina H: causata dalla delezione di tre geni dell’α-globina. Con un solo gene codificante per l’α-globina, la β-globina è nettamente in eccesso e si aggrega a formare i tetrametri β4. l’HbH ha un’affinità elevata per l’ossigeno, il che vuol dire che a livello tissutale non entra nel meccanismo di scambio gassoso, provocando ipossia tissutale. Inoltre 6 la HbH è soggetta ad ossidazione, diventando molto instabile. Questo porta un’anemia moderatamente grave. 4. idrope fetale: è la più grave forma di α-talassemia, causata dalla perdita di tutti e quattro i geni. La grande quantità di HbH formata provoca la quasi totale ipossia dei tessuti. Durante lo sviluppo precoce, l’espressione dell’emoglobina ζ2γ2, emoglobina embrionale, evita l’ipossia, tuttavia dal terzo trimestre di gravidanza si manifesta la patologia. In passato il feto sarebbe morto nell’utero, mentre oggi, tramite trasfusione intrauterina tali neonati possono essere salvati. Il feto mostra pallore, edema generalizzato, epatosplenomegalia simile a quella dell’eritroblastosi fetale. β-TALASSEMIA Condizione geneticamente e clinicamente eterogenea, caratterizzata da una ridotta o assente sintesi di catene β, a causa di molte differenti mutazioni (riarrangiamenti, promoter, splicing, delezioni). PATOGENESI MOLECOLARE Le sindrome β-talassemiche vengono classificate in: 1. β0-talassemia, associata alla totale assenza di catene della β-globina nello stato omozigote 2. β+-talassemia, caratterizzata da una ridotta sintesi di β-globina in condizioni di omozigosi. Le mutazioni principali che possono provocare una β-talassemia sono le seguenti: • mutazioni nella regione del promotore. Questa mutazione riduce la frequenza di legame dell’RNA polimerasi, riducendo del 70-80% la produzione di β-globina • mutazione che determina un’interruzione prematura della catena. Ci sono due tipi di mutazione che possono causare l’interruzione della catena di globina: o si genera un nuovo codone di stop nell’esone o creazione di nuove sequenza tramite delezioni o inserzioni di geni che producono comunque un codone di stop che blocca la sintesi proteica. In entrambi i casi si avrà una β0-talassemia • mutazioni nello splicing. Sono la causa più frequenti di β-talassemia. Sono dovute a mutazioni di porzioni introniche o esoniche, che alterano lo splicing del RNA. In questo caso non si avrà la creazione della catena proteica, e quindi una β0-talassemia. Altre mutazioni avvengono in zone introniche creando nuovi siti di splicing. Dato anche i siti normali di splicing rimangono intatti, si avranno catene anomale di β-globina e catene normali, producendo una β+-talassemia. Il deficit nella sintesi dell’HbA produce globuli rossi poco emoglobinizzati, ipocromici, microcitici e con minor capacità di trasporto dell’ossigeno. Le catene α libere formano aggregati insolubili che precipitano all’interno dei normoblasti, formando inclusioni insolubili. Il danno principale che provocano queste precipitazioni sono danni alla membrana che causano la morte dei normoblasti in via di sviluppo nel midollo. I normoblasti che evitano la morte intramidollare produrranno globuli rossi che saranno soggetti a sequestro splenico e quindi a distruzione, a causa della loro poca deformabilità. Nei casi gravi di β-talassemia, la forte anemia che si viene a creare stimola la produzione di eritropoietina, che comporta la formazione di centri di emopoiesi extramidollare. L’emopoiesi extramidollare interessa principalmente fegato, milza e linfonodi. I precursori degli eritrociti sottraggono ulteriori quantità di O2 ai tessuti che già ne erano privi, producendo grave cachessia in pazienti non trattati. Inoltre un’atra complicanza consiste nell’eccessivo assorbimento di ferro alimentare. Questo si aggiunge a quello introdotto tramite le trasfusioni, provocando sovraccarico di ferro, a cui consegue danno agli organi parenchimali, in particolare il fegato. 7 MANIFESTAZIONI CLINICHE La β-talassemia si manifesta in due forma, major e minor. La β-talassemia major è causata da una situazione di omozigosi (β+/β+ o β0/β0), mentre la β-talassemia minor è causata da eterozigosi (β+/β o β0/β). È presente anche una variante intermedia, che può apparire in forma lieve o grave. Talassemia maggiore Chiamata anche morbo di Cooley, si instaura quando il genotipo corrisponde a β+/β+, β0/β0 o β0/β+. • L’anemia si manifesta da 6 a 9 mesi dopo la nascita, quando cioè l’HbF cessa di essere prodotta, e compare l’HbA • I bambini non trattati soffrono di ritardo della crescita e muoiono in età precoce. Nei soggetti che sopravvivono si verificano importanti malformazioni ossee. • Si verifica epatosplenomegalia a causa dell’eritropoiesi extramidollare (fegato) e dell’emolisi (milza) • Gli accumuli di ferro danneggiano fegato e testicoli. La diagnosi viene effettuata in base ai seguenti punti: • livelli di emoglobina che variano tra 3-6 g/dl • notevole anomalia morfologica nei globuli rossi (microcitosi, ipocromia, anisocitosi e poichilocitosi) • cellule bersaglio: globuli rossi con punteggiature basofile • conta dei reticolocita alta, ma a causa dell’inefficace eritropoiesi è più bassa rispetto a quanto atteso in base alla gravità dell’anemia • elettroforesi dell’Hb: assenza di HbA; HbF >80-90%. Talassemia minore È molto più comune della maggiore. La maggior parte dei pazienti sono portatori di una mutazione β+ o β0. Si tratta di pazienti totalmente asintomatici, e l’anemia, se presente, è molto lieve. La diagnosi viene effettuata tramite i seguenti punti: • anomalie degli eritrociti (ipocromia, microcitosi, punteggiatura basofila) • l’elettroforesi rivela un caratteristico aumento dell’HbA2 (dal 4% all’8%) • ridotti Hb, MCV e MCH L’identificazione del tratto β-talassemico è importante per due ragioni: 1. distinzione dall’anemia ipocromica microcitici da carenza di ferro 2. counseling genetico Questa patologia ereditaria è causata da un deficit intrinseco della membrana dei globuli rossi che li rende sferoidali, meno deformabili e vulnerabili al sequestro splenico e alla distruzione. PATOLOGIA MOLECOLARE L’elasticità della membrana del globulo rosso è dovuta ad una particolare proteina del citoscheletro, la spectrina, costituita da due catene, α e β, che formano eterodimeri intersecati e flessibili. Le deste del dimero di spectrina si associano formano tetrametri, mentre le code interagiscono con oligomeri di actina. Quindi si forma una struttura actina-spectrina che è collegata alla membrana tramite due interazioni: 1. tramite le proteine anchirina e banda 4.2 che legano la spectrina al trasportatore ionico di membrana 2. la proteina 4.1 unisce la code della spectrina alla proteina di membrana glicoforina A. 8 La sferocitosi ereditaria è causata da mutazioni della prima tipologia di legame, e principalmente mutazioni dell’anchirina. Quindi la diminuzione della stabilità della membrana porta alla sua frammentazione durante l’esposizione a sforzi di taglio lungo il torrente circolatorio. La perdita di membrana rispetto al citoplasma, porta la cellula ad assumere il più piccolo diametro possibile in un certo volume, e da ciò deriva la forma sferica. Nella milza i globuli rossi sferici non riescono ad uscire attraverso i cordoni di Billroth, e rimangono intrappolati nella milza, rallentando il circolo, provocando diminuzione di pH e l’accumulo di acido lattico. Quindi gli sferociti vengono fagocitati dai numerosi macrofaci splenici. EVOLUZIONE CLINICA La gravità della patologia varia molto da un paziente all’altro. In una minoranza dei casi, la SE si presenta alla nascita con ittero marcato, e richiede trasfusioni. Nel 20-30 % la malattia è asintomatica, poiché l’eritropoiesi aumenta e riesce a sopperire alla carenza di globuli rossi. Nella maggior parte dei pazienti, l’eritropoiesi non è in grado di compensare l’emolisi, quindi viene causata un’anemia emolitica generalmente stabile e di intensità moderata, o severa. Il decorso clinico è molte volte accompagnato da una crisi aplastica, innescata da un’infezione principalmente da parvovirus B19, che infetta e uccide i progenitori degli eritrociti finché non insorge la risposta immunitaria (entro 1-2 settimane). Fin quando la risposta immunitaria non si innesca, può essere necessario curare il paziente con trasfusioni. Le anemie emolitiche autoimmuni sono disordini essenzialmente caratterizzati da: • diminuita emivita eritrocitaria in vivo • presenza di anticorpi reattivi contro emazie autologhe La diagnosi di anemie immunoemolitiche richiede il riscontro di anticorpi e/o complemento sui globuli rossi del paziente. Questo viene effettuato mediante il test di Coombs diretto. In questo test i globuli rossi sono mescolati con antisiero eterologo specifico per immunoglobuline umane o complemento. Se uno dei due è presente i globuli rossi sono agglutinati da anticorpi polivalenti. Nel test di Coombs indiretto, il siero del paziente viene utilizzato per agglutinare specifici globuli rossi. Le malattie immunoemolitiche sono classificate per lo più in base alle caratteristiche dell’anticorpo responsabile. Avremo quindi le seguenti classi di anemie. ANEMIA IMMUNOEMOLITICA DA ANTICORPI CALDI È la forma più comune, ed è causata dalle IgG, e talvolta dalle IgA. Circa il 50 % dei casi è idiopatico (primitivo). Le forma secondarie possono essere dovute a linfomi e leucemie, neoplasie, disordini immunologici (LES), farmaci. Gli anticorpi sono definiti caldi in quanto sono attivi a 37 °C. La maggiorare dell’emolisi è extravascolare, ed è dovuta al legame delle IgG con i globuli rossi; i frammenti Fc dei monociti e dei macrofagi splenici si legano a questo complesso e provocano la perdita della membrana eritrocitica. I globuli rossi tendono quindi ad assumere una forma sferica, e, nella milza, vanno incontro ad emolisi. Quindi questa malattia provoca una modesta splenomegalia. Sono stati ipotizzati due meccanismi mediante i quali viene indotta l’emolisi da farmaci: 9 1. farmaci come cefalosporine e penicilline, se somministrati in quantità eccessive, si legano alle membrane dei globuli rossi, andando a costituire l’aptene che darà vita alla cascata immunologica 2. farmaci come l’α-metildopa provocano la formazione di anticorpi diretti contro gli antigeni intrinseci dei globuli rossi, principalmente quelli del gruppo Rh. I pazienti con AEA si presentano con anemia, la cui severità può variare da forme gravi pericolose per la vita a forme molto lievi. L’insorgenza può essere: • LENTA: prevalgono i sintomi correlati all’anemia, con lieve ittero. Questo può andare avanti per mesi ma occasionalmente può verificarsi un rapido peggioramento dell’anemia e dell’ittero. • ACUTA: con pallore marcato, febbre, ittero, epatosplenomegalia, polipnea, palpitazioni Gli esami di laboratorio evidenziano la presenza di: • anemia di grado variabile • sferocitosi, macrocitosi, policromasia • reticolocitosi, eritroblastemia • incremento della bilirubina indiretta • incremento dell’LDH • incremento della ferritina La terapia consiste in: • trasfusioni: possono essere necessarie per quei pazienti con AEA in pericolo di vita a causa di anemizzazione severa • steroidi: E’ la terapia più efficace (riduce o abolisce l’emolisi in 2/3 dei pazienti). Efficacia dose-dipendente, maggiore nelle malattie del connettivo; il 10% dei pazienti è “nonresponder”. Dose: 1-2 mg/Kg/die per 10-14 giorni, quindi ridurre a < 0.5 per 1-2 mesi. Ricadute sono comuni • splenectomia: La splenectomia rimuove il principale sito di distruzione delle emazie. Candidati alla procedura sono i pazienti in ricaduta, quelli che rispondono solo ad alti dosaggi di steroidi o necessitano di agenti immunosoppressivi non steroidei. Remissione completa o parziale in 2/3 dei casi. • farmaci immunosoppressivi: Pazienti non responsivi agli steroidi e/o alla splenectomia. Azatioprina o Ciclofosfamide sono utili in questi casi. ANEMIA IMMUNOEMOLITICA DA ANTICORPI FREDDI È causata dalle cosiddette agglutinine fredde, anticorpi IgM che si legano e agglutinano i globuli rossi a bassa temperatura (0-4°C). Rappresentano il 16-32 % dei casi di anemia immunoemolitica. Questi anticorpi appaiono soprattutto durante la risoluzione di alcuni disturbi infettivi come la polmonite o la mononucleosi infettiva. Altri agenti infettivi associati a questa forma di anemia comprendono il citomegalovirus, i virus influenzali e l’HIV. La forma cronica di questa anemia si manifesta soprattutto secondariamente a neoplasie. I sintomi clinici sono causati dal legame di IgM ai globuli rossi in sedi quali le parti esposte delle dita della mano, del piede e dell’orecchio dove la temperatura è al di sotto dei 30 °C. Questo legame provoca l’attivazione del complemento. L’emolisi è di varia entità. Clinicamente le forme possono essere: A. cronica, + ittero. B. acuta scatenata dal freddo. C. forme ricorrenti. D. fenomeni acrocianotici sono possibili, ma rari • dita delle mani e dei piedi, naso, orecchie • manifestationi ulcerative e necrotiche 10 I test di laboratorio manifestano: • Raramente anemia severa (Hb > 5-6 gr/dl); sferocitosi, schistocitosi, policromasia, reticolocitosi • Agglutinazione delle emazie, a volte evidente sugli strisci di sangue (bassa temperatura, 4° C). • Ridotti livelli di aptoglobina. Dopo le crisi emolitiche. • Positività per emosiderinuria (se è intervenuta emolisi intravascolare). • Crioagglutinine: IgM (raramente IgA o IgG) • Test di Coombs diretto positivo • Anticorpi: pressochè invariabilmente anti I/i EMOGLOBINURIA PAROSSISTICA NOTTURNA È una patologia molto rara ed è la sola anemia emolitica causata da un difetto intrinseco acquisito della membrana cellulare. L’EPN origina da mutazioni acquisite nel fosfatidilinositolo glican A, essenziale per la sintesi del glicosilfosfatidilinositolo (GPI), fosfolipidi specializzato nel legare alcune particolari proteine alla membrana cellulare. La mutazione somatica che causa questa alterazione avviene nelle cellule staminali pluripotenti; quindi tutta la loro discendenza clonale (globuli bianchi, rossi e piastrine) sarà priva di GPI. Molte proteine che legano il GPI hanno la funzione di inibire la cascata del complemento, e nell’EPN sono tre le proteine implicate: 1. fattore accelerante il decadimento (CD55) 2. inibitore della lisi reattiva (CD59) 3. proteina legante il CD8 L’EPN si osserva in soggetti giovani. L’esordio può essere caratterizzato da astenia, pallore, emissione saltuaria di urine scure al mattino e manifestazioni trombotiche di diversa entità. Il decorso è cronico e deriva dall’espressione di quattro eventi fisiopatologici: 1. IPEREMOLISI. Le crisi emolitiche possono essere innescate da molteplici meccanismi che potrebbero avere in comune la capacità di attivare molecole capaci di determinare la lisi dei globuli rossi (infezioni, stress fisico e psichico, trasfusioni) 2. TROMBOSI. Dovute all’attivazione delle piastrine da parte del complemento e alla liberazione di ADP da parte dei globuli rossi. 3. INFEZIONI. Provocate dalla neutropenia e da difetti enzimatici e funzionali dei granulociti neutrofili. 4. INSUFFICIENZA MIDOLLARE. L’analisi del sangue periferico evidenzia un’anemia severa con Hb<5 g/dl. Al momento non esiste una terapia specifica per l’EPN. La terapia trasfusionale di emazie concentrate va effettuata in rapporto al fabbisogno di ogni singolo paziente. Un fattore da considerare è quello trombotico. Le manifestazioni trombotiche vanno curate con farmaci classici, quali eparina e antiaggreganti. Il trapianto del midollo osseo allo genico può rappresentare un valido approccio terapeutico per i soggetti con EPN, soprattutto nei soggetti con età inferiore a 40 anni. 11 Le anemie spesso derivano da una carenza di nutrienti vitali necessari per la formazione di globuli rossi. Incluse in questo gruppo sono: • anemie da carenza di vitamina B12 e folati • anemie da carenza di ferro. Questo gruppo di anemie include due principali patologie: 1. anemia perniciosa (da carenza di vitamina B12) 2. anemia da carenza di folati Sono causate dalla carenza di sintesi di DNA, che comporta la produzione di precursori eritroidi e di globuli rossi abnormi, in seguito al deficit di maturazione e di divisione cellulare. ANEMIA DA CARENZA DI VITAMINA B12: ANEMIA PERNICIOSA L’anemia perniciosa è causata da un’importante carenza di vitamina B12. È una forma specifica di anemia megaloblastica causata da gastrite atrofica e una mancata produzione di fattore intrinseco che porta alla carenza di vitamina B12. METABOLISMO DELLA VITAMINA B12 La vitamina B12 è un composto complesso noto come cobalamina. L’uomo non è in grado di produrre la cobalamina, quindi dipende totalmente dai prodotti animali alimentari per il suo fabbisogno. Il fabbisogno giornaliero va da 2 a 3 mg, e una dieta bilanciata ne contiene quantità significativamente maggiori, quindi l’eccesso di vitamina B12 può durare anche diversi anni. L’assorbimento di vitamina B12 richiede il fattore intrinseco, secreto dalle cellule parietali della mucosa gastrica. Una volta ingerita la vitamina B12 segue il seguente percorso: • viene liberata dalle proteine che la legano nei cibi dall’azione della pepsina • una volta libera si lega alle cobalofiline salivari • il complesso cobalifilina-vitamina B12 arriva al duodeno dove, per azione di proteasi pancreatiche viene scisso • la vitamina B12 quindi si associa al fattore intrinseco, e questo nuovo complesso viene assorbito dagli enterociti dell’ileo. • all’interno degli enterociti la vitamina B12 si lega alla transcobalamina II, e viene liberata nel plasma • la transcobalamina trasporta la vitamina B12 al fegato e ad altre cellule, soprattutto nel midollo osseo e nelle mucose che rivestono il tratto gastroenterico Da questo è possibile dedurre alcuni meccanismi che possono portare una carenza di vitamina B12: 1. dieta inadeguata 2. acloridria e perdita della secrezione di pepsina: portano un minor rilascio di vitamina B12 dal cibo 3. gastrectomia e anemia perniciosa: provocano diminuzione di fattore intrinseco 4. perdita della funzione pancreatica: diminuiscono le proteasi 5. resezione dell’ileo 12 FUNZIONI BIOCHIMICHE DELLA VITAMINA B12 Sono conosciute due reazioni che implicano l’utilizzo della vitamina B12: OMOCISTEINA Vit B12 + metionina sintetasi Metilen-tetra-idrofolato tetraidrofolato METIONINA L’omocisteina viene metilata con la produzione di metionina. Il metilen-tetra-idrofolato viene convertito in tetraidrofolato, composto fondamentale per la conversione della deossiuridina monofosfato in deossitimina monofosfato, un precursore immediato del DNA. È stato postulato che la fondamentale causa di una ridotta sintesi di DNA nella carenza di vitamina B12, è la ridotta disponibilità di folati, la maggior parte del quale rimane intrappolato come metilentetra-idrofolato. La seconda reazione che dipende dalla cobalamina è la isomerizzazione del metilmalonil coenzima A in succinil coenzima A, che richiede adenosilcobalamina come gruppo protesico dell’enzima metilmalonil-coenzima A mutasi. Un deficit di vitamina B12 porta l’aumento nel plasma e nelle urine dei livelli dell’acido metilmalonico. L’interruzione della via succinilica e il conseguente accumulo di metilmalonato e propionato, potrebbe portare alla formazione e incorporazione di anomali acidi grassi nei lipidi neuronali, che può predisporre alla distruzione mielinica. PATOGENESI Si ritiene che l’anemia perniciosa derivi da una distruzione della mucosa gastrica mediata da un processo immunologico. Ne risulta una gastrite cronica atrofica, caratterizzata da • perdita delle cellule parietali • prominente infiltrato linfocitaria • modificazione megaloblastica nelle cellule mucose simili a quelle trovate nei precursori eritroidi Esistono tre tipi di anticorpi implicati nella patogenesi di questo processo immune: 1. anticorpo di tipo I: blocca il sito di legame della cobalamina con il fattore intrinseco 2. anticorpo di tipo II: previene il legame del complesso vitamina B12-fattore intrinseco al suo recettore ileale 3. esiste un anticorpo di tipo III che però non è implicato nel minor assorbimento della vitamina B12, e sembra invece essere una conseguenza delle lesioni gastriche, piuttosto che una causa. Comunque non è stato stabilito che questi anticorpi siano la causa scatenante la patologia; infatti si ritiene che una risposta mediata da cellule T autoreattive inizi le lesioni della mucosa gastrica, stimolando la produzione di autoanticorpi. Quando il numero di cellule parietali scende sotto la soglia, insorge l’anemia. EVOLUZIONE CLINICA E DIAGNOSI L’anemia perniciosa ha un esordio insidioso, così l’anemia è spesso abbastanza grave nel momento in cui il paziente si reca dal medico. Le caratteristiche diagnostiche comprendono: 1. moderata o severa anemia megaloblastica 2. leucopenia con granulociti ipersegmentati 3. lieve o moderata trombocitopenia 4. lieve ittero dovuto a eritropoiesi inefficace ed emolisi periferica 5. modificazioni neurologiche correlate al coinvolgimento del tratto spinale posterolaterale 6. incapacità di assorbire oralmente cobalamina (valutabile col test di Schilling, che misura la cianocobalamina eliminata con le urine) 7. acloridria anche in seguito a stimolazione con istamina 13 8. bassi livelli sierici di vitamina B12 9. elevati livelli di omocisteina e di acido metil malonico nel siero 10. marcata risposta reticolocitica e miglioramento dell’ematocrito circa 5 giorni dopo somministrazione parenterale di vitamina B12 La terapia consiste nel: • rimuovere o trattare la malattia di base • somministrare Vit. B12 e/o folati i.m. • controllare la crisi reticolocitaria e la ferritina • terapia di mantenimento ANEMIA DA CARENZA DI FOLATI La carenza di acido folico (o acido pteroilmonoglutammico) causa un’anemia avente le stesse caratteristiche di quella causata dalla carenza di vitamina B12. Tuttavia, i disturbi neurologici che si manifestano nella carenza da vitamina B12 non si osservano nella carenza di folati. L’acido folico, (FH4), agisce come un intermediario nel trasferimento di unità monocarboniose, che riceve da composti come serina e acido formiminoglutamico. I derivati del FH4 a loro volta donano i frammenti monocarboniosi in reazioni i sintesi di svariati metaboliti. Le più importanti reazioni implicate sono: 1. sintesi purinica 2. trasformazione dell’omocisteina a metionina 3. sintesi del deossimiditilato monofosfato Mentre nelle prime due reazioni, il folato viene rigenerato dai suoi derivati che trasportano unità monocarboniose, nella terza reazione viene prodotto diidrofolato che deve essere ridotto dalla diidrofolato reduttasi per rientrare nella riserva degli FH4. Questo passaggio è sensibile all’inibizione farmacologica, mediata per esempio dal timidilato. Il deossitimilidilato monofosfato è necessario per la sintesi del DNA, in quanto rappresenta un suo diretto precursore, quindi la carenza dei folati porterà una diminuzione della sintesi del DNA. EZIOLOGIA Il fabbisogno giornaliero di acido folico va da 50 mg a 200 mg. La maggior parte delle diete normali ne contiene ampie quantità, e le fonti più ricche sono i vegetali verdi come lattuga, spinaci, asparagi e broccoli. Alcuni frutti (limoni, banane, meloni) e proteine animali (fegato) ne contengono minore quantità. Nei cibi l’acido folico è contenuto sottoforma di folipoliglutammati, che sono sensibili alle alte temperature. Quindi l’ebollizione e la cottura distruggono fino al 95% di folati. Nell’intestino le coniugasi spezzano i poliglutammati in monogluammati, che una volta assorbiti, vengono modificati in 5-metiltetraidrofolato, la normale forma di trasporto di folato. Le riserve corporee di acido folico sono piuttosto modeste, e i sintomi di una carenza possono comparire dopo diversi mesi. Vi sono tre principali cause di deficit di acido folico: 1. RIDOTTA ASSUNZIONE. Può derivare da una dieta inadeguata o da alterato assorbimento intestinale. Si può riscontrare soprattutto in diete grossolane, prive di vitamine del complesso B. Tale inadeguato apporto alimentare si riscontra spesso negli alcolisti cronici, negli indigenti e nei soggetti molto anziani. Negli alcolisti cronici con cirrosi, il folato rimane intrappolato nel fegato, e oltre ad una carenza di folato si riscontra una malnutrizione generale. 2. AUMENTATA RICHIESTA. Può essere riscontrata in condizioni di maggiore richiesta, come gravidanza, infanzia, alterazioni ematologiche associate ad emopoiesi iperattiva (anemie emolitiche) 14 3. ANTAGONISTI DELL’ACIDO FOLICO. Il metotrexate inibisce la diidrofolato reduttasi e porta un deficit di tetraidrofolato, colpendo tutte le cellule in rapida crescita. Questo uguale per molti farmaci chemioterapici. DIAGNOSI L’anemia megaloblastica è identica a quella da carenza di vitamina B12 e la diagnosi può essere fatta solo dimostrando la diminuzione dei livelli di folato nel siero o nei globuli rossi. I livelli di omocisteina sono aumentati. TERAPIA La terapia consiste nel: • rimuovere o trattare la malattia di base • somministrare Vit. B12 e/o folati i.m. • controllare la crisi reticolocitaria e la ferritina • terapia di mantenimento La carenza di ferro è probabilmente il più frequente disturbo nutrizionale nel mondo. METABOLISMO DEL FERRO Ferro contenuto nel corpo di una donna: 2g. Ferro contenuto nel corpo di un uomo: 6g. Non esiste una via regolata per l’escrezione del ferro, che è limitata a 1-2 mg al giorno a causa della perdita delle cellule della mucosa e alle cellule epiteliali della cute. Con la dieta occidentale vengono assunti 10-20 mg di ferro al giorno e la maggior parte di questo si trova sottoforma di eme nei prodotti animali. Il 20 % del ferro eme è assorbibile. Nell’organismo il ferro è contenuto • nelle molecole di emoglobina (80%); • mioglobina • negli enzimi contenenti ferro come le catalasi e i citocromi Le riserve sono rappresentate dall’emosiderina e dalla ferritina che contengono circa il 15-20 % del ferro corporeo totale. La ferritina è un complesso proteina-ferro che si ritrova in tutti i tessuti, ma soprattutto nel fegato, milza, midollo osseo e muscoli scheletrici. Nel fegato la ferritina è immagazzinata nel citosol e nei lisosomi delle cellule parenchimali, e la componente lisosomiale è associata a piccole quantità di emosiderina. Quest’ultima è presente solo in minime tracce in normali condizioni di accumulo di ferro. La ferritina è presente anche nel plasma, in quantità direttamente proporzionali all’accumulo di ferro. I suoi valori plasmatici oscillano tra: • 12 µg/l quando c’è carenza di ferro • 5000 µg/l quando c’è sovraccarico di ferro. Al fegato il ferro arriva • tramite la transferrina plasmatica, dopo assorbimento intestinale • tramite le cellule del Kupffer, che catturano l’eme dei globuli rossi distrutti per l’emocateresi. 15 Nel plasma il ferro circola tramite la trasferrina, una glicoproteina che nei soggetti normali è satura per il 33% di ferro, con livelli medi di ferro pari a: • 100 µg/dl nelle donne • 120 µg/dl negli uomini Quindi la capacità del siero di legare il ferro si aggira intorno a 300-350 µg/dl. La più importante funzione delle transferrina è quella di trasportare il ferro ai tessuti, compresi il midollo osseo, in cui gli elementi eritroidi hanno bisogno del metallo per la fabbricazione delle molecole di eme. L’assorbimento del ferro avviene nel duodeno,tramite proteine transmembrana degli enterociti. Esiste una distinzione tra i trasportatori del ferro non eme e del ferro eme. Il ferro eme viene assorbito tramite trasportatori non ancora caratterizzati, mentre il ferro non eme deve subire prima una riduzione a ferro ferroso (Fe2+) da parte di un citocromo B. Quindi il Fe2+ viene trasportato all’interno dell’enterocita dalla proteina DMT-1; il passaggio attraverso la membrana basolaterale avviene tramite due proteine, ferroporina-1, che fa passare il Fe2+, e la efaestina, che riduce il Fe2+ in Fe3+, che può quindi essere legato dalla transferrina. Dopo l’assorbimento la maggior parte del ferro viene depositato con la ferritina. Tuttavia se le sedi di accumulo del ferro nell’organismo sono sufficientemente rifornite e l’attività eritropoietina è normale, gran parte del ferro assorbito viene perduto dall’intestino per sfaldamento delle cellule epiteliali. Viceversa quando le necessità di ferro sono aumentate oppure in caso di stimolo eritropoietico maggiore, una maggiore frazione del ferro assorbito viene trasferita alla transferrina plasmatica, con una concomitante diminuzione della perdita di ferro attraverso la ferritina delle cellule mucosali. Finché le perdite di ferro sono limitate, il bilancio corporeo del metallo è mantenuto regolando l’assorbimento. In condizioni di bisogno di ferro, come nel casi di eritropoiesi inefficace, per esempio durante la βtalassemia, l’assorbimento del ferro aumenta. Per arrivare a questo effetto, c’è bisogno che un segnali arrivi alle cellule mucosali, sia nel caso in cui l’assorbimento deve essere aumentato, che diminuito. Un candidato per la regolazione ormonale negativa del metabolismo del ferro è l’epcidina, un peptide che inibisce la captazione de ferro da parte delle cellule della mucosa duodenale. La concentrazione dell’epcidina diminuiscono quando le riserve di ferro si esauriscono. L’assorbimento del ferro inorganico viene aumentato da • acido ascorbico, acido citrico • aminoacidi e zuccheri EZIOLOGIA Per mantenere un normale equilibrio di ferro, è necessario ingerire almeno 1mg di ferro al giorno. Dato che ne verrà assorbito solo il 15-20 %, per l’uomo il fabbisogno giornaliero di ferro sarà di 710 mg, mentre per la donna 7-20 mg. Poiché con la dieta occidentale si ingeriscono circa 15-20 mg di ferro, la maggior parte degli uomini ingerisce una quantità più che adeguata di ferro. Una carenza di ferro più essere causata da: 1. CARENZA ALIMENTARE: raramente nei paesi industrializzati, ma frequente nei paesi in via di sviluppo. Comunque, nonostante la disponibilità di ferro nei paesi industrializzati, l’insufficienza alimentare si verifica nei seguenti gruppi: • lattanti: la dieta lattea contiene piccolissime quantità di ferro (0,3 mg/l di ferro) • bambini: nella crescita hanno un gran bisogno di ferro • poveri • anziani: solitamente hanno diete ristrette scarse di carne 2. ALTERATO ASSORBIMENTO: si riscontra nella sprue, in altre cause di steatorrea intestinale e nella diarrea cronica. 16 AUMENTATA RICHIESTA: i lattanti e i bambini in accrescimento, gli adolescenti, le donne in menopausa, donne in gravidanza, hanno un’aumentata richiesta di ferro. 4. PERDITA EMATICA CRONICA: è la causa più comune di carenza di ferro nei paesi occidentali. Se vi è sanguinamento in tessuti o cavità del corpo, l’eme viene recuperato completamente, mentre in emorragie esterne le riserve di ferro vengono depauperate. Qualunque sia la causa della perdita di ferro, questa porterà ad un’a anemia ipocromica microcitica. Gli enzimi contenenti ferro di tutto il corpo alterano la loro funzione, provocando alopecia, alterazioni atrofagiche della lingua e della mucosa gastrica e malassorbimento intestinale. La triade delle principali caratteristiche della sindrome di Plummer-Vinson è costituita da: 1. anemia microcitica ipocromica 2. glossite atrofica 3. pliche esofagee All’inizio della patologia, le riserve di ferro vengono mobilizzate, e per un certo lasso di tempo riescono a compensare la perdita. La deplezione progressiva d queste riserve dapprima causa una riduzione di ferro sierico e dei livelli di saturazione della trasferrina, senza determinare anemia. L’anemia compare sono quando i depositi di ferro sono completamente esauriti. Le manifestazioni cliniche dell’anemia sono non specifiche e sono uguali alle altre tipologie di anemie trattate: pallore, astenia, cardiopalmo, irritabilità, difficoltà di concentrazione, lipotimie, cefalea, ronzii, vertigini, distrofia cute, mucose e annessi, glossite atrofica, cheilite angolare, secchezza dei capelli, distrofia ungueale, disfagia da esofagite (S. di Plummer-Vinson). La diagnosi si basa sugli esami di laboratorio, che evidenziano • un ematocrito e l’emoglobina diminuiti • ferritina e ferro sierico bassi • capacità di legare ferro plasmatico aumentata • livello dei recettori della transferrina aumentato è importante valutare l’anemia da carenza di ferro, poiché si potrebbe diagnosticare un sanguinamento occulto o un tumore, e salvare quindi una vita. 3. TERAPIA La terapia consiste nella somministrazione di ferro per via endovenosa o orale. CON ANEMIA Marcata (Hb<9 g/100 ml) VIA ENDOVENOSA VIA ORALE 30 gg 6 mesi SENZA ANEMIA Lieve (Hb>9 g/100 ml) (Hb>12 g/100 ml) 20 gg 4 mesi 10 gg 2,5 mesi “Aplasia midollare” è un’espressione utilizzata per indicare una sindrome di insufficienza del midollo associata a pancitopenia (anemia, neutropenia e trombocitopenia). L’insufficienza del midollo deriva da soppressione o assenza delle cellule staminali multipotente mieloidi. EZIOLOGIA Le principali circostanze in cui può comparire un’aplasia midollare sono le seguenti: 1. ACQUISITA: idiopatica • difetto primitivo delle cellule staminali • immuno-mediato 17 sostanze chimiche • dose dipendente ♦ agenti alchilanti ♦ antimetaboliti ♦ benzene ♦ cloramfenicolo ♦ arsenico inorganico • idiosincratica ♦ cloramfenicolo ♦ fenilbutazone ♦ clorpromazina ♦ streptomicina ♦ insetticidi agenti fisici • irradiazione globale infezioni virali • epatite • citomegalovirus • Epstein-Barr • Herpes Zoster misto • Infrequente 2. EREDITARIA: anemia di Fanconi La maggior parte delle anemie da aplasia midollare è dovuta all’esposizione a sostanze chimiche o farmaci. Alcune sostanze mielotossiche sono en conosciute, e il loro danno è dose dipendente, e soprattutto reversibile quando si interrompe l’esposizione. Le sostanze maggiormente mielotossiche conosciute sono benzene, cloramfenicolo, agenti alchilanti e antimetaboliti. In altri casi invece la pancitopenia appare come un’apparente reazione idiosincrasica indesiderata a piccolissime dosi della sostanza. In questi casi l’aplasia midollare può essere severa. L’irradiazione totale è un fattore che può distruggere le cellule staminali emopoietiche in modo dose-dipendente. Infezioni virali (epatiti non-A, non-B, non-C e non-G) provocano aplasia midollare con meccanismi ancora non noti. L’anemia di Fanconi è una rara malattia autosomica recessiva. È causata dall’alterazione di un componente di un complesso per la riparazione del DNA. L’aplasia midollare si manifesta in età giovanile, quando il midollo diventa ipofunzionante, ed è accompagnata da altri sintomi, costituiti da altre anomalie congenite, quali ipoplasia renali e splenica, ipoplasia ossea, che spesso coinvolge i pollici e il radio. PATOGENESI È improbabile che l’aplasia midollare venga scatenata da un unico fattore. Infatti è stato ipotizzato che 1. le cellule staminali siano prima alterate antigeneticamente dall’esposizione di farmaci, agenti infettivi o altri insulti ambientali. 2. questo evoca una risposta immunitaria, nella quale le cellule T attivate producono IFNγ e TNF che prevengono la normale crescita e sviluppo delle cellule staminali. 18 Il trattamento immunosoppressivo con globuline antitimociti combinate con farmaci quali ciclosporina, provoca una risposta positiva nel 60-70 % dei pazienti, e la riuscita del trapianto osseo necessita un condizionamento con alte dosi di farmaci miolotossici o radioterapia. In entrambi i casi si suppone che queste terapie funzionano sopprimendo o uccidendo i cloni di cellule T autoreattive. Si tratta di una rara forma di insufficienza midollare caratterizzata da una marcata ipoplasia degli elementi eritroidi del midollo nel quadro di una normale granulocitopoiesi e trombocitpoiesi. Può essere: • primitiva: senza alcuna malattia associata • comparire secondariamente a neoplasie, principalmente a timomi, leucemia linfocitica a cellule grandi granulose. L’associazione a timomi fa supporre che ci sia un meccanismo immunologico alla base. Infatti l’asportazione del timo è seguita da miglioramento ematologico. Anche nella forma primitiva, molto probabilmente causata da una tipologia di autoimmunità, una terapia immunosoppressiva può essere utile. 19 Un eccessivo sanguinamento può derivare da: 1. aumentata fragilità dei vasi 2. carenza o disfunzione delle piastrine 3. alterazione della coagulazione 4. combinazione di questi disturbi I test di laboratorio utilizzati per valutare i differenti aspetti dell’emostasi sono i seguenti: 1. Tempo di sanguinamento. Misura il tempo che occorre perché un’incisione cutanea effettuata in maniera standardizzata smetta di sanguinare. I valori di riferimento dipendono dal modo impiegato e vanno da 2 a 9 minuti. Questo esame valuta un parametro altamente variabile, e quindi rappresenta un sistema non perfettamente attendibile. 2. Conta piastrinica. Ottenuta su un campione di sangue trattato con anticoagulanti. I valori di riferimento vanno da 150.000 a 300.000 /µl. 3. Tempo di protrombina. Valuta la vie estrinseca e quella comune della coagulazione. Il prolungamento del PT può essere causato da carenze o disfunzioni dei fattori V, VII, X, della protrombina o del fibrinogeno. 4. Tempo di tromboplastina parziale. Valuta la vie intrinseca e quella comune della coagulazione. Dopo aggiunta di caolino, calcio e cefalica, viene misurata in secondi. Il caolino attiva il fattore XII e la cefalica sostituisce i fosfolipidi piastrinici. Il prolungamento del PTT può essere causato da disfunzioni o carenza di fattori V, VII, IX, X, XI, dalla protrombina e dal fibrinogeno. La riduzione del numero delle piastrine costituisce un’importante causa di emorragia generalizzata. Un valore inferiore a 100.000/µl è considerato indice di trombocitopenia. Tuttavia un sanguinamento spontaneo non si manifesta se il conteggio non scendo sotto i 20.000/µl. Il sanguinamento spontaneo che si associa a trombocitopenia interessa molto spesso i piccoli vasi: le sedi tipiche sono la cute, il tratto gastrointestinali e genitourinario. Le cause che possono provocare una trombocitopenia possono essere classificate nelle seguenti quattro categorie: 1. Ridotta produzione di piastrine. Può essere associato a malattie estese del midollo osseo, come l’aplasia midollare e le leucemie, o può derivare da patologie che colpiscono i megacariociti in maniera selettiva. Nei deficit di vitamina B12 e di acido folico, la carenza di DNA porta una trombocitopenia. 2. Diminuita sopravvivenza delle piastrine. Questa è una delle più importanti cause. Può essere di origine: • Immunologica: la distruzione delle piastrine è causata da anticorpi circolanti o da immunocomplessi. Gli anticorpi possono essere rivolto contro autoantigeni delle piastrine (autoanticorpi) o contro antigeni che sono differenti fra i vari individui (alloantigeni). Tra gli antigeni sono compresi i complessi glicoproteici della membrana piastrinica IIb-IIIa e Ib-IX. • Non immunologica: può essere causata da danno meccanico, in modo analogo alla distruzione dei globuli rossi nel corso di un’anemia emolitica microangiopatica. Un’altra patologia è la coagulazione intravascolare disseminata (CID). 3. Sequestro. Una trombocitopenia si può instaurare quando si ha splenomegalia. In condizioni normali la milza sequestra il 30-40 % delle piastrine. 4. Diluizione. Può insorgere con una trasfusione massiva. 20 La porpora immune trombocitopenica (PIT) può manifestarsi associata ad una varietà di condizioni ed esposizioni come LES, HIV, (PIT secondaria) o in assenza di fattori di rischio (PIT primitiva). Esistono tue tipologie della patologia, cronica ed acuta. PIT CRONICA È causata dalla formazione di autoanticorpi diretti contro le glicoproteine della membrana piastrinica. Questi anticorpi possono esser rilevati nel plasma in circa l’80 % dei pazienti. Si tratta di IgG. Le piastrine con il legame di questi anticorpi vengono opsonizzate e sono più suscettibili alla fagocitosi da parte delle cellule del sistema dei fagociti mononucleati. Il sito di rimozione delle piastrine è la milza, infatti il 75-80 5 dei pazienti sottoposti a splenectomia migliora notevolmente. Sebbene il meccanismo principale della trombocitopenia sia la distruzione immunologica delle stesse piastrine, sembra che gli autoanticorpi agiscano danneggiando anche i megacariociti, compromettendo anche la produzione delle piastrine. Tuttavia si tratta solo di un meccanismo secondario, che causa una lieve riduzione della conta piastrinica. CARATTERISTICHE CLINICHE La PIT cronica colpisce maggiormente le donne (rapporto donne/uomini = 3/1). Ha un inizio subdolo, caratterizzato da sanguinamenti della cute e delle mucose. Le emorragie si manifestano sulla cute sottoforma di petecchie, che possono confluire formando ecchimosi. Può verificarsi melena, ematuria, eccessivo flusso mestruale. Conseguenze gravi della PIT cronica sono emorragie aracnoidali e intracerebrali. I segni clinici non sono specifici, quindi la diagnosi può essere effettuata mediante: • conta piastrinica ridotta • aumento di megacariociti nel midollo osseo • presenza di piastrine giganti (megatrombociti) • tempo di emorragia aumentato ma PT e PTT normali La diagnosi di PIT deve essere effettuata solo quando le altre cause di trombocitopenia sono state scartate. La terapia è basata su immunosoppressori a base di glucocorticoidi, ma molte volte bisogna ricorrere alla splenectomia. PIT ACUTA È una malattia dell’infanzia, che colpisce ugualmente entrambi i sessi. Nella maggior parte dei casi l’insorgenza della patologie è preceduta di 2 settimane circa da una malattia virale. Questa forma è autolimitante, e si risolve nel giro di 6 mesi. Comunque se la trombocitopenia è grave, è possibile effettuare un trattamento a base di glucocorticoidi. MICROANGIOPATIE TROMBOTICHE: PORPORA TROMBOTICA TROMBOCITOPENICA E SINDROME EMOLITICO-UREMICA La porpora trombotica trombocitopenica (TTP) è caratterizzata da cinque segni caratteristici: 1. febbre 21 2. trombocitopenia 3. anemia emolitica microangiopatica 4. deficit neurologici transitori 5. insufficienza renale La sindrome emolitico-uremica (SEU) presenta anch’essa anemia emolitica, ma si distingue dalle TTP per la mancanza di sintomi neurologici, per la maggior gravità dell’insufficienza renale e per la maggior incidenza nell’età pediatrica. La caratteristica fondamentale comune ad entrambe le condizioni, è la formazione diffusa di trombi ialini, composti principalmente di aggregati piastrinici, all’interno del microcircolo. Il consumo di piastrine provoca la trombocitopenia, e la presenza del trombo è la causa principale dell’anemia emolitica microangiopatica e dei disturbi d’organo generalizzati. I pazienti affetti da TTP sono spesso carenti di un enzima chiamato ADAMTS 13, metalloproteasi del vWF, cha ha la funzione di degradare o multimeri del fattore di von Willebrand di peso molecolare elevato. In assenza di ADAMTS questi multimeri si accumulano nel plasma e, in particolari condizioni, provocano la formazioni di microaggregati piastrinici nel microcircolo, causando i sintomi della TTP. Il deficit di ADAMST può essere ereditario o acquisito. È possibile che anche un’azione immunitaria diretta contro le metalloproteasi provochi la patologia. La plasmaferesi si può rivelare decisiva in quanto corregge la carenza enzimatica. Pe quanto riguarda la SEU, un’importante causa nei bambini e negli è rappresentata dalla gastroenterite causata da E. coli tipo 0157:H7. Questo produce una tossina simile a quella si Shigella, che anziani viene assorbita dalla mucosa gastrointestinale infiammata. La tossina si lega alle cellule endoteliali glomerulari e di altri organi danneggiandole, dando inizio all’aggregazione piastrinica. L’insorgenza della SEU negli adulti è più severa, in quanto insorge in patologie che comportano danno endoteliale, e spesso si tratta di disturbi cronici che sono essi stessi a prognosi infausta. Nei bambini si manifesta principalmente con diarrea ematica, e dopo pochi giorni manifestano il quadro della SEU. I deficit qualitativi della funzione piastrinica possono essere congeniti o acquisiti. Sono state descritte numerose malattie congenite caratterizzate da un prolungamento del tempo di emorragia e da una normale conta piastrinica. I disturbi congeniti della funzione piastrinica possono essere classificati in tre gruppi sulla base della specifica alterazione funzionale: 1. DEFICIT DELL’ADESIONE: sono rappresentati dalla sindrome di Bernard-Soulier, patologia autosomica recessiva dovuta alla carenza del complesso glicoproteici di membrana Ib-IX. Questa proteina è il recettore del fattore di von Willebrand, essenziale per l’aggregazione delle piastrine. 2. ANOMALIE DELL’AGGREGAZIONE: un esempio è la tromboastenia di Glanzmann, autosomica recessiva. Le piastrine non aggregano in risposta ai normali stimoli come l’ADP, l’adrenalina, la trombina, a causa del deficit o del malfunzionamento della glicoproteina IIbIIIa, che forma, normalmente, ponti tra le piastrine legandosi al fibrinogeno e al vWF. 3. DISTURBI DELLA SECREZIONE PIASTRINICA: sono caratterizzati da una normale risposta iniziale allo stimolo aggregante (collageno o ADP), ma le successive reazioni sono difettose. Tra i disturbi acquisiti della funzione piastrinica sono solo due quelli clinicamente rilavanti: 22 1. azione di FANS: inducono un significativo prolungamento del tempo di emorragia 2. uremia: la patogenesi della diatesi emorragica in corso di uremia è complessa e non completamente delucidata, comunque sono state osservate numerose alterazioni della funziona piastrinica in corso di uremia. Il quadro emorragico legato al deficit dei fattori della coagulazione differisce da quello in corso di anomalie piastriniche per il fatto che le petecchie o la porpora sono rari. • Il sanguinamento si manifesta con grandi ecchimosi o ematomi post-traumatici, o con prolungato sanguinamento dopo una ferita o dopo un qualsiasi intervento chirurgico. • È tipico il sanguinamento gastrointestinale e del tratto urinario • Un quadro tipico è quello di un soggetto che lamenta uno stillicidio della durata di giorni dopo l’estrazione di un dente o che sviluppa un emartro dopo uno stimolo di modesta entità dell’articolazione del ginocchio. Possiamo identificare anomalie acquisite o congenite. Le anomalie acquisite sono in genera caratterizzate da disturbi della coagulazione multipli. Una causa può essere la carenza di vitamina K, fattore essenziale per la carbossilazione e l’attivazione dei fattori della coagulazione. I deficit ereditari principali sono l’emofilia A, l’emofilia B e la malattia di von Willebrand. FISIOLOGIA DEL COMPLESSO FATTORE VIII-VWF Il complesso plasmatico fattore VIII-vWF è formato da: • proteina procoagulante fattore VIII: componente della via della coagulazione intrinseca necessario per l’attivazione del fattore X; viene sintetizzato dalle cellule endoteliali sinusoidali, dalle cellule di Kupffer del fegato e dalle cellule glomerulari ed epiteliali tubulari del rene. • multimeri del fattore di von Willebrand di grosso peso molecolare. Il vWF interagisce nel plasma con fattore VIII, e inoltre col collagene, con l’eparina e con le glicoproteine di membrana piastrinica. Il vWF viene prodotto dall’endotelio e si deposita nello strato subendoteliale. Quando c’è un interruzione dell’endotelio vasale, il vWF viene esposto, e interagisce con le glicoproteine della membrana piastrinica Ib e IIb, provocando un’aggregazione delle piastrine all’endotelio danneggiato. Le cellule endoteliali e le piastrine inoltre rilasciano altro vWF in circolo, dove, dopo aver formato multimeri, si lega col fattore VIII e innesca la cascata della via intrinseca della coagulazione, provocando il reclutamento di altre piastrine. Il legame del vWF col fattore VIII, prolunga l’emivita di quest’ultimo fino a 24 ore, ma se il vWF è carente, l’emivita del fattore VIII scende a sole 2,4 ore. Sembra essere una delle più frequenti malattie emorragiche ereditarie della specie umana. Esistono due principali tipi della patologia dovuti a: 1. RIDUZIONE DEL VWF: dovuti a ridotta penetranza e una variabile espressività dei geni. La grave carenza del vWF diminuisce anche l’emivita e la funzionalità del fattore VIII, e quindi 23 molte manifestazioni cliniche sono identiche all’emofilia. La mutazione può essere dovuta anche a delezione dei geni. 2. DEFICIT QUALITATIVO DEL VWF: viene prodotto un vWF anomalo che provoca un difettoso assemblaggio dei multimeri. Le manifestazioni cliniche sono le seguenti: • sanguinamento spontaneo delle mucose • eccessivo sanguinamento delle ferite • menorragia • normale conta piastrinica • riduzione plasmatica del vWF • riduzione plasmatica del fattore VIII. La terapia sostitutiva con fattore VII è l’unica possibile. È la più comune malattia ereditaria associata a emorragia di tipo grave. È causata dalla riduzione dei livelli della proteina VIII, necessaria in quanto cofattore del fattore IX per l’attivazione del fattore X. Viene trasmessa come tratto recessivo legato al cromosoma X, e quindi sono colpiti maschi e femmine omozigoti. Clinicamente si manifesta con un’ampia variabilità, correlata ai livelli del fattore VIII: • livelli < 1% del normale sviluppano una grave patologia; • tra il 2% e il 5 % i sintomi sono moderati • 6-50% la malattia è lieve. I deficit più severi del fattore VIII sono dovuti ad un insolita inversione a carico del cromosoma X, oppure a mutazioni puntiformi del gene. Le manifestazioni cliniche comprendono: • ecchimosi • emorragie massive dopo trauma o interventi chirurgici • tempo di emorragia e conta piastrinica normali • aumento del PTT • la diagnosi viene effettuata col dosaggio del fattore VIII L’emofilia è una patologia che interessa la via intrinseca della coagulazione. La via intrinseca dovrebbe assicurare un’emostasi efficiente, e invece si hanno le emorragie. Questo perché probabilmente in vitro non viene valutato alla perfezione quello che può succedere in vivo, ed è possibile che il fattore VIII possa interagire anche con la deposizione di fibrina e quindi prevenire la formazione del trombo in entrambe le vie della coagulazione. La terapia si basa sull’infusione di fattore VIII ricombinante. Questo fattore però potrebbe essere visto come non-self in pazienti con gravi manifestazioni dell’emofilia A, in quanto i valori troppo bassi di fattore VIII potrebbero non aver mai dato la possibilità all’organismo di riconoscere questa proteina come self. È una patologia provocata dalla carenza di fattore IX. Il disturbo è indistinguibile clinicamente dall’emofilia A, in quanto il fattore VIII e IX agiscono nella medesima azione di attivazione del fattore X. Anche l’emofilia B viene ereditata come carattere autosomica recessivo legato al cromosoma X. Analogamente all’emofilia A il PTT è prolungato e il PT e il tempo di emorragia sono normali. Unico esame che permette di fare diagnosi di malattia di Christmas è il dosaggio del fattore IX. 24 Il trattamento si basa sulla somministrazione di fattore XI ricombinante. La coagulazione intravascolare disseminata è un’alterazione tromboemorragica che si manifesta in forma acuta, subacuta e cronica, come complicanza secondaria di varie patologie. È caratterizzata dall’attivazione sistemica della coagulazione, che provoca la formazioni di microtrombi in tutto il microcircolo, con una distribuzione imprevedibile. Si assiste quindi al consumo di piastrine, fibrina, fattori della coagulazione, e in un secondo momento anche a fibrinolisi. EZIOLOGIA E PATOGENESI Il CID non è mai una patologia primitiva. Entrambe le vie della coagulazione confluiscono nella formazione di trombina, che a sua volta converte il fibrinogeno in fibrina. La trombina quindi favorisce il deposito di fibrina attivando essa stessa la vie intrinseca e inibendo la fibrinolisi. La trombina in eccesso segue il circolo, e quando è a contatto con l’endotelio intatto dei vasi, interagisce con una proteina chiamata trombomodulina. L’interazione con questa proteina conferisce alla trombina la capacità di attivare la proteina C, che inibisce i fattori V e VIII e inibendo quindi la formazione di coaguli. I meccanismi principali che scatenano una CID sono: 1. liberazione di fattore tissutale o di sostanze di tipo tromboplastinico in circolo: le sostanze di tipo tromboplastinico possono originare da varie fonti, come la placenta in caso di complicanze ostetriche, o dai granuli delle cellule leucemiche nella leucemia promielocitica acuta. Per quanto riguarda le complicanze ostetriche, le tromboplastina derivate dalla placenta, dalla ritenzione del feto morto o dal liquido amniotico possono entrare in circolazione. Anche nelle sepsi da gram - l’endotossina provoca una maggior espressione di fattore tissutale da parte dell’endotelio. Inoltre le endotossine inibiscono la proteina C sopprimendo l’espressione endoteliale di trombomodulina. 2. danno diffuso alle cellule endoteliali: in corso di shock settico, entrano in gioco i fattori dell’infiammazione. Il TNF aumenta l’espressione di molecole di adesione da parte dell’endotelio. Queste provocano l’adesione di leucociti che danneggiano le cellule endoteliali. Il danno endoteliale può essere provocato anche dalla deposizione di complessi antigene-anticorpo come nel casi del LES, da temperature estreme o da microrganismi. Le conseguenze della CID sono duplici: 1. la deposizione di fibrina e la formazione di trombi, può provocare anemia emolitica, che deriva dalla frammentazione dei globuli rossi, e ischemia degli organi a valle. 2. col progredire della malattia entrano in gioco le complicanze emorragiche. Queste sono dovute al sequestro di piastrine dai trombi e all’attivazione della via fibrinolitica. La piastrina infatti agisce, oltre che provocando fibrinolisi, anche esercitando la proteolisi dei fattori della coagulazione, che incrementano il sanguinamento. EVOLUZIONE CLINICA Il 50 % dei soggetti con CID sono pazienti ostetriche con complicanze della gravidanza. L’esordio può essere fulminante, tuttavia il disturbo tende a risolversi con il parto. Le principali manifestazioni cliniche sono anemia emolitica microangiopatica, dispnea, cianosi, insufficienza respiratoria, convulsioni e come, oliguria e insufficienza renale acuta, insufficienza circolatoria e shock improvvisi o progressivi. 25 La CID acuta, associata a complicanze ostetriche o traumi maggiore, è dominata da diatesi emorragica, mentre la CID cronica, associata a neoplasie per esempio, tende a presentarsi principalmente con complicanze trombotiche. L’unico trattamento risolutivo è quello di rimuovere o trattare la causa scatenante quando possibile. 26 Queste malattie possono essere classificate in diverse categorie: • Neoplasie linfoidi. Comprendono entità patologiche distinte, e in molti casi il fenotipo della cellula neoplastica è strettamente somigliante a quello di un normale linfocita a un particolare stadio differenziativo. • Neoplasie mieloidi. Derivano dalle cellule staminali emopoietiche da cui originano le cellule della linea mieloidi (eritroidi, granulocita e/o megacariocitaria). Le neoplasie mieloidi sono rappresentate da tre categorie di patologie: leucemie mieloidi acute: le cellule progenitrici immature si accumulano nel midollo osseo; sindromi mielodisplastiche: si associano a ematopoiesi inefficace sindromi mieloproliferative croniche: l’aumentata produzione di uno o più elementi mieloidi ben differenziati generalmente conducono a una conta elevata nel sangue periferico. Istiocitosi. Sono rare condizioni proliferative dei macrofagi e delle cellule dendritiche. • I disordini neoplastici dei globuli bianchi sono estremamente vari. Esistono diversi fattori che possono rappresentare una causa di queste patologie. TRASLOCAZIONI CROMOSOMICHE E ONCOGENI Le traslocazioni rappresentano anomalie cromosomiche del tutto casuali, che sono presenti nella maggior parte delle neoplasie dei globuli bianchi. • Nel caso di neoplasie linfoidi, molti riarrangiamenti degli oncogeni derivano da errori che si verificano durante l’espressione dei gene per i recettori per gli antigeni. Le cellule progenitrici della linea B e T-cellulari, esprimono un’attività ricombinasica che taglia il DNA in sequenze specifiche all’interno di loci delle immunoglobuline e del recettore T-cellulare, e molti riarrangiamenti patogeni osservate nelle neoplasie linfoidi, sono causati dall’inappropriato posizionamento di questi siti accanto a sequenze di proto-oncogeni. • Le traslocazioni cromosomiche si osservano frequentemente nelle neoplasie mieloidi, ma in questi tumori i meccanismi sottesi alla rottura del DNA sono sconosciuti. FATTORI GENETICI EREDITARI Individui con malattie genetiche che favoriscono l’instabilità gnomica, come la sindrome di Bloom, l’anemia di Fanconi e l’atassia teleangectasia, presentano un rischio aumentano di sviluppare una leucemia acuta. Inoltre la sindrome di Down e la neurofibromatosi di tipo I sono associate a una più alta incidenza di leucemia infantile. VIRUS Vengono considerati come agenti eziologici tra virus: 1. virus-1 della leucemia umana a cellule T 2. Epstein-Barr virus 3. Herpesvirus del sarcoma di Kaposi 27 AGENTI AMBIENTALI Le associazioni meglio definite tra agenti ambientali e predisposizione alle neoplasie linfoidi sono: • infezione da Helicobacter Pilori - linfoma gastrico a cellule B • enteropatia da intolleranza al glutine - linfoma intestinale a cellule T Inoltre i soggetti con HIV sono maggiormente predisposti al rischio di sviluppare linfomi B derivati da cellule B del centro germinativo. FATTORI IATROGENI Radioterapia e chemioterapia aumentano i rischio di sviluppare neoplasie mieloidi e linfoidi secondarie. Uno degli aspetti confondenti delle neoplasie linfoidi riguarda l’uso dei termini leucemia linfocitica e linfoma. • Leucemia linfocitica: termine che viene utilizzato per descrivere neoplasie linfoidi che si presentano spesso accompagnate da un diffuso interessamento del midollo osseo, e che sono spesso accompagnate da un elevato numero di cellule neoplastiche nel sangue periferico. • Linfoma: termine utilizzato per descrivere proliferazioni linfoidi che si presentano come masse tissutali distinte. Molti tipi di linfoma possono presentarsi con un quadro leucemico nel sangue periferico, con ampio interessamento del midollo: infatti la progressione a leucemia da un linfoma incurabile non è rara. Le neoplasie linfoidi giungono all’osservazione clinica in diversi modi. Per quanto riguarda due terzi del linfomi non Hodgkin si presentano con ingrossamento non dolente di linfonodi (spesso > 2cm). Il terzo rimanente insorge invece in sedi extranodali. Le leucemie linfatiche invece giungono all’osservazione clinica per la sintomatologia correlata alla soppressione della normale emopoiesi da parte delle cellule neoplastiche che invadono il midollo. La classificazione dei linfomi non Hodgkin e delle neoplasie linfoidi correlate ha creato nella storia molta confusione. Recentemente la World Health Organization (WHO) ha creato una classificazione che divide le neoplasie linfoidi in cinque grandi famiglie, sulla base della loro origine cellulare: 1. NEOPLASIE DEI PRECURSORI B-CELLULARE (neoplasie a cellule B immature) 2. NEOPLASIE DELLE CELLULE B PERIFERICHE (neoplasie a cellula B mature) 3. NEOPLASIE DEI PRECURSORI T CELLULARI (neoplasie a cellule T immature) 4. NEOPLASIE DELLE CELLULE T/NK PERIFERICHE (neoplasie a cellule T e natural killer mature) 5. LINFOMA DI HODGKIN (neoplasie a cellule di Reed-Sternberg e varianti) È necessario, prima di analizzare le specifiche entità della suddetta classificazione, analizzare alcuni importanti principi rilevanti per le neoplasie linfoidi. • Il sospetto di una neoplasia linfoide può fondarsi sulla base di segni clinici, ma per la diagnosi è necessario l’esame istologico dei linfonodi o degli altri tessuti coinvolti. • Nella maggior parte delle neoplasie linfoidi, il riarrangiamento dei geni per il recettore antigenico avviene prima della trasformazione neoplastica, quindi tutte le cellule derivanti dal progenitore avranno la stessa configurazione e la stessa sequenza del recettore per l’antigene e sintetizzano proteine recettoriale per l’antigene identiche. Questo fattore può essere utilizzato per distinguere le proliferazioni reattive da quelle tumorali: infatti le popolazioni normali di linfociti presentano molti diversi recettori antigenici. 28 • • • • La maggior parte delle neoplasie linfoidi (80-85 %) origina dalle cellule , mentre la gran parte dei restanti casi è costituito da tumori a cellule T. Le neoplasie linfoidi destabilizzano il sistema immunitario, provocando l’insorgenza di processi infettivi e di fenomeni autoimmuni. Le cellule neoplastiche B e T tendono ad imitare la loro controparte normale. Cosi i linfomi B cellulari tenderanno a crescere nelle aree B-cellulari dei linfonodi, producendo un quadro di accrescimento follicolare o nodulare, mentre i linfomi T crescono tipicamente nelle zone paracorticali. Il linfoma di Hodgkin si estende in maniera ordinata, e lo stadio finale è fondamentale per decidere la terapia. Al contrario i LNH si diffonde in maniera imprevedibile, e alla diagnosi il paziente presenta giù la malattia sistemica. La leucemia linfoblastica acuta comprende un gruppo di neoplasie costituite da precursori linfocitari immaturi B (85% dei casi). Il 15 % comprende invece quadri derivanti dal coinvolgimento dei precursori della filiera T. Circa 2500 nuovi casi di ALL sono diagnosticati ogni anno negli USA, e la maggior parte in soggetti di età inferiore a 15 anni. CLASSIFICAZIONE LA classificazione Franco-Americano-Britannica (FAB) ha distinto tre principali gruppi di ALL, in base a caratteristiche nucleocitoplasmatiche degli elementi leucemici: 1. L1: blasti uniformemente piccoli, con scarso citoplasma. Forma prevalente nei giovani sotto i 15 anni. 2. L2: elementi blastici più grandi, di dimensioni variabili, con contorno irregolare dei nuclei contenenti evidenti nucleoli 3. L3: blasti grandi, omogenei, con citoplasma basofilo vacuolizzato. A differenza delle forme di leucemia mieloide acuta ove la diagnosi morfocitochimica è sufficientemente indicativa nella discriminazione dei sottotipi, nelle caratteristiche delle forme di ALL è necessario ricorre all’applicazione di tecniche immunologiche per una completa definizione diagnostica. Particolare importanza a questo riguardo è rivestita dalla determinazione della TdT (desossiribonucleotidil-transferasi terminale), dell’antigene common CD10 di Graves, dei CD19, CD79a, CD3, CD7. della presenza Ig di superficie e citoplasmatiche, di riarrangiamenti di TCR. CITOGENETICA Circa il 90 % di pazienti con ALL hanno alterazioni numeriche o strutturali dei cromosomi nelle cellule leucemiche. Le alterazioni più comuni sono: • Iperploidia (> 50 cromosomi) • Poliploidia • Traslocazioni t(12;21), t(9;22) (cromosoma Philadelphia) e t(4;11) • Aberrazioni: interessano la funzionalità dei fattori di trascrizioni necessari per il normale sviluppo cellulare emopoietico. Le ALL pre-B e pre-T sono associate a particolari traslocazioni, cosa che suggerisce che alla base della loro patogenesi ci siano differenti meccanismi molecolari. 29 CARATTERISTICHE CLINICHE Va sottolineato che sebbene le ALL e le AML siano immunofenotipicamente e geneticamente distinte, si presentano solitamente con caratteristiche cliniche molto simili. In entrambe le patologie l’accumulo di blasti neoplastici nel midollo osseo sopprime la normale emopoiesi per occupazione dello spazio midollare, per competizione per i fattori di crescita e per altri motivi poco compresi. Ne risultano così: • Anemia di grado variabile • Neutropenia • Trombocitopenia (< 25000/mm3) • Conta leucocitaria elevata (GB > 100000) Il midollo osseo si presenta quindi all’osservazione infiltrato in maniera cospicua da parte di blasti, mentre in una piccola frazione di pazienti, l’ago aspirato si presenta vuota (punctio secca). L’evoluzione clinica si articola nelle seguenti fasi: 1. ESORDIO IMPROVVISO E IRRUENTE: i pazienti mostrano in pochi giorni o in poche settimane l’insorgenza dei sintomi. 2. SINTOMI RELATIVI ALLA SOPPRESSIONE DELLA NORMALE FUNZIONE MIDOLLARE: astenia dovuta all’anemia, febbre causata dalle infezioni, sanguinamento (petecchie, ecchimosi, epistasi, emorragie gengivali) secondario a trombocitopenia. 3. DOLORE E FRAGILITÀ OSSEA: derivati dall’espansione midollare e dall’infiltrazione del subperiostio. 4. LINFOADENOPATIA GENERALIZZATA, SPLENOMEGALIE ed EPATOMEGALIA: causate dall’infiltrazione neoplastica 5. MANIFESTAZIONI A CARICO DEL SNC, quali cefalea, vomito, paralisi dei nervi cranici, risultato della diffusione meningea. Sono più comuni nelle ALL che nelle AML. PROGNOSI Diversi fattori sono stati consistentemente associati ad una prognosi peggiore: 1. età inferiore a 2 anni: per via della forte associazione delle ALL infantili con le traslocazioni che interessano il gene MLL sul cromosoma 11; 2. esordio durante l’adolescenza o in età adulta 3. la conta dei blasti nel sangue periferico superiore a 100000, che può riflettere una grande massa tumorale 4. presenza di aberrazioni genetiche sfavorevoli, come la t(9;22) (cromosoma Philadelphia), presente nel 3 % delle ALL dell’infanzia e nel 25 % dei casi negli adulti. Al contrario fattori prognostici favorevoli comprendono 1. età fra 2 e 10 anni 2. bassa conta dei globuli bianchi 3. fenotipo pre-B precoce 4. iperploidia 5. t(12;21) TERAPIA La ALL è una delle emopatie maligne per le quali è stato raggiunto un significativo miglioramento terapeutico negli ultimi anni con l’uso della chemioterapia. Nei bambini è possibile acquisire una completa remissione nel 90-95 % dei casi, e probabilmente una guarigione in circa 2/3 dei casi. Negli adulti i risultati sono relativamente inferiori. In linea di massima, i pazienti vengono trattati secondo quattro fasi: 1. TERAPIA DI INDUZIONE. Si effettua con una combinazione di farmaci che include vincristina, prednisone e antracicline, e che consente di ottenere una remissione completa del 70-90 %. 2. TERAPIA DI CONSOLIDAMENTO. Serie di misure finalizzate al controllo della malattia minima residua, alla prevenzione delle ricadute e all’emergenza di cellule resistenti ai farmaci 30 citostatici utilizzati in precedenza. Si utilizza methotrexade che ha anche il vantaggio di controllare la diffusione nel SNC poiché passa la barriera ematoencefalica. 3. TRATTAMENTO DI MANTENIMENTO. Effettuato con 6-mercaptopurina e con methotrexade per 3 anni 4. TRAPIANTO DI MIDOLLO. È divenuta una procedura importante nella strategia di trattamento dei pazienti con ALL dell’adulto. Per quanto attiene al trapianto di midollo allo genico, questo può essere preso in considerazione per pazienti con ALL ad alto rischio. Pazienti con ALL a basso rischio possono utilizzare questa terapia in seconda remissione. Questa procedura assicura un risultato decisamente migliore rispetto alla sola chemioterapia con una sopravvivenza del 20-40 % a 3 anni. La leucemia linfatica cronica è un disordine linfoproliferativo cronico acquisito di natura monoclonale, caratterizzato dall’espansione di piccoli linfociti apparentemente maturi che si accumulano nel sangue periferico, nel midollo osseo e negli organi linfatici. • 95% dei casi: la neoplasia coinvolge un clone di linfociti B • 5% dei casi: la neoplasia coinvolge un clone di linfatici T La LLE è la forma di leucemia più frequente nei paesi occidentali, con un’incidenza annua di 5-15 casi ogni 100.000 abitanti. Colpisce con maggior frequenza il sesso maschile, soprattutto soggetti con più di 50 anni. CARATTERISTICHE MORFOLOGICHE L’architettura linfonodale è diffusamente alterata per una popolazione prevalente di piccoli linfociti. Queste cellule sono frammiste ad un numero variabile di cellule di maggiori dimensioni definite prolinfociti che, nella maggior parte dei casi si riuniscono a formare centri di proliferazione. Nelle LLC il sangue periferico contiene un alto numero di linfociti rotondi con scarso citoplasma. Queste cellule sono fragili e si rompono molto facilmente durante la preparazione dello striscio, dando luogo alla formazione di ombre cellulari. Le cellule tumorali infiltrano solitamente la polpa bianca e la polpa rossa della milza e l’area periportale epatica. ASPETTI IMMUNOFENOTIPICI Nel 95% dei casi la proliferazione leucemica interessa un singolo clone linfocitario B. La natura monoclonale della proliferazione nella B-LLC, è dimostrata dal carattere omogeneo delle immunoglobuline di superficie sintetizzate dai linfociti leucemici. Le catene pesanti sono prevalentemente del tipo IgM, seguite dalle IgD. Inoltre le catene leggere sono le stesse, con la stessa specificità idiotipica. Inoltre i linfociti della B-LLC esprimono le molecole antigeniche CD19, CD5, CD21, CD24, CD23. In corso di B-LLC anche i linfociti T sono numericamente aumentati, specialmente i linfociti citotossici e soppressori (CD8+), al contrario dei linfociti CD4+ che diminuiscono. ANOMALIE CROMOSOMICHE Le traslocazioni cromosomiche sono rare, con una spiccata presenza di delezioni (13q12-14, 11q, 17p), trisomia di 12q. CARATTERISTICHE CLINICHE 31 La maggior parte dei pazienti si presenta con un’età superiore a 50 anni. I pazienti con LLC sono spesso asintomatici e la diagnosi in questo casi può essere occasionale (leucocitosi isolata e modesta linfocitosi). Quando la sintomatologia compare, si manifesta generalmente con perdita di peso, anoressia, stanchezza, febbre. Nel 50% dei casi si manifesta linfoadenopatia (linfonodi più grandi del normale, di consistenza anormale e palpabili in numero aumentato), anemia (dovuta allo sviluppo di anticorpi contro i globuli rossi), epatosplenomegalia, infezioni conseguenti alla neutropenia. • • • • DIAGNOSI Diagnosi differenziale con una linfocitosi reattiva, in cui sono presenti linfociti T normali. Infatti generalmente negli stadi iniziali non è presente linfoadenopatia e organomegalia. Valore minimo di linfocitosi è 5000/mm3. Infezione ricorrente può indirizzarci verso una patologia del sistema immunitario. Anemia emolitica autoimmuni PROGNOSI In genere la sopravvivenza media è di 4-6 anni, ma in pazienti con massa tumorale minima all’esordio, può essere anche di 10 anni o più. Fattori prognostici negativo sono: • Stadio • Tempo di raddoppiamento dei linfociti inferiore a 12 mesi • Iniziale numero assoluto di linfociti superiore a 50.000/mm3 • Anormalità del cariotipo (trisomia 12) • Percentuale di cellule con aspetti prolinfocitoidi • Elevato indice proliferativo • Infiltrazione midollare diffusa La stadiazione della leucemia linfatica cronica viene effettuata secondo due classificazioni: Rai (1975) 0. 1. 2. 3. 4. Linfocitosi periferica e midollare 0 + linfoadenopatia 0 + splenomegalia + epatomegalia 0 + HB< 11 gr/dL 0 + trombocitopenia (< 100.000 mm3) Binet (1981) A. Linfocitosi con < 3 aree linfatiche coinvolte in assenza di anemia e piastrinopenia B. Linfocitosi con >3 aree linfatiche coinvolte in assenza di anemia e piastrinopenia C. Linfocitosi + anemia e/o trombocitopenia (Hb < 10 g/dl +/- Piastrine < 100.000/mmc ) indipendentemente dalle aree linfatiche coinvolte TERAPIA La terapia deve tener conto dei fattori di rischio presenti al momento della diagnosi. Quindi individuiamo tra classi di pazienti: • A basso rischio: stadio clinico 0 di Rai e A di Binet. Non devono essere trattati con farmaci citostatici, tranne colori che presentano una leucocitosi > 30-40.000/mm3, tempo di raddoppiamento linfocitario inferiore ad un anno, sintomatologia evidente (calo ponderale, febbre ecc.). Se dopo un periodo di follow-up la patologia sembra progredire, si utilizzano farmaci alchilanti, associati a prednisone (25mg/die per 1-4 settimane). • A rischio intermedio: stadio I e II di rai e B di Binet. Se non ci sono sintomi evidenti, si tiene il paziente in osservazione per 4 mesi, e se la malattie progredisce si utilizza 32 clorambucile (alchilanti) e steroidi. La chemioterapia sia nei pazienti a rischio intermedio che basso non determina grossi miglioramenti. • Ad alto rischio: stadio III e IV di Rai e C di Binet. Trattati energicamente con chemioterapia. L’aggiunta di corticosteroidi alla fludarabina (farmaco che porta ad una remissione completa nel 13 % e parziale nel 13 %) non migliora la percentuale di risposte cliniche. Il fine della terapia comunque non deve essere quello di eradicare la malattia, ma quello di contenere la linfocitosi e gli impegni degli organi linfatici. Il trapianto allo genico è preferibile in soggetti con meno di 40 anni di età e con malattia disseminata. La terapia di supporto si avvale di emotrasfusioni di sangue o piastrine, antibioticoterapia, Ig nel caso di marcata ipogammaglobulinemia associata ad infezioni recidivanti. È la forma più comune dei linfomi non Hodgkin, costituendo circa il 45 % dei linfomi negli adulti. Si presenta in età media e colpisce allo stesso modo maschi e femmine. Le cellule neoplastiche somigliano molto alle normali cellule B del centro germinativo. MORFOLOGIA Si osserva un coinvolgimento linfonodale con un pattern di crescita prevalentemente nodulare. I tipi di cellule osservate sono due: 1. piccole cellule con contorno nucleare irregolare 2. cellule grandi con cromatina nucleare dispersa, diversi nucleoli e una modesta quantità di citoplasma. Nell’85 % dei casi si ha coinvolgimento del midollo osseo, che appare come aggregati linfoidi paratrabecolari. Nel 15 % dei casi viene sviluppata una linfocitosi pari a 20.000/mm3. CITOGENETICA E GENETICA MOLECOLARE La modificazione genetica nel linfoma follicolare più frequente è la traslocazioni 14;18 che avvicina il locus delle IgH sul cromosoma 14 al locus di BCL2 sul cromosoma 12. Questo provoca un’iperespressione del gene BCL2, che è un’antagonista dell’apoptosi, promuovendo quindi la sopravvivenza delle cellule. CARATTERISTICHE CLINICHE I linfomi follicolari tendono a presentarsi con linfoadenopatia non dolorosa, con raro interessamento del tratto gastrointestinale, nervoso o del testicolo. La patologia segue un andamento clinico indolente che alterna fasi di crescita e di regressione. La sopravvivenza media va da 7 a 9 anni, e tende a migliorare con il trattamento. Raramente il linfoma follicolare si trasforma in tumore aggressivo come il linfoma di Burkitt, evento che solitamente associa all’acquisizione di una traslocazioni cromosomica che interessa il locus c-MYC. Questo può essere dovuto al fatto che in seguito alla prima mutazione possono verificarsene altre, che provocano l’aumento dell’aggressività della neoplasia. Questa categoria comprende tre tipologie di linfomi, differenti non istologicamente, ma per alcune caratteristiche cliniche: 1. linfoma di Burkitt Africano (endemico) 2. linfoma di Burkitt sporadico 33 3. linfomi aggressivi che colpiscono i soggetti HIV positivi. Morfologicamente questo linfoma è caratterizzato da infiltrato di cellule linfoidi di media dimensione nei tessuti coinvolti. È caratteristico un indice mitotico molto elevato, affiancato da numerosi fenomeni di apoptosi. I macrofagi benigni sono diffusamente distribuiti fra le cellule tumorali e presentano un abbondante citoplasma chiaro, che crea un caratteristico aspetto a cielo stellato. IMMUNOFENOTIPO Sono neoplasie a cellule mature che esprimono IgM di superficie, CD19, CD21, CD10 e BLC6. ASPETTI DI CITOGENETICA La modificazione genetica di tutte le tipologie di linfoma di Burkitt è la traslocazione del gene cMYC sul cromosoma 8. La traslocazione avviene con il locus delle IgH, o con il locus delle catene leggere delle immunoglobuline. Nel caso della forma endemica, il linfoma è correlato strettamente con un’infezione da EBV, presente nel 25% dei tumori associati ad HIV. Analisi molecolari dimostrano che l’infezione da parte di EBV precede l’insorgenza della neoplasia, e questo mette in evidenza il ruolo principale del virus nella linfomagenesi. CARATTERISTICHE CLINICHE Il 30 % dei linfomi pediatrici non Hodgkin è costituito dal linfoma di Burkitt endemico, che si presenta spesso come una massa che interessa la mandibola ed evidenzia una strana predilezione ad interessare i visceri addominali (reni, ovaie e surreni). Invece la forma sporadica del linfoma di Burkitt interessa più spesso l’intestino cieco e il peritoneo. L’interessamento del sangue periferico e del midollo osseo è molto rare, soprattutto nel casi del linfoma endemico. Il linfoma di Burkitt risponde molto bene alla chemioterapia ad alte dosi. Se insorge in età giovane, la prognosi non è negativa, mentre è peggiore negli adulti di età più avanzata. Rappresenta il 3% dei LNH negli USA. Colpisce soprattutto nei 50-60 anni di vita, principalmente nei maschi. Le cellule tumorali somigliano molto alle cellule B normali della zona mantellare che circonda i centri germinativi. Il linfoma mantellare esprime CD19. CD20, CD5. È associata a traslocazione (11;14) che interessa il locus IgH sul cromosoma 14 e il locus della ciclica D1 sul cromosoma 11. Quest’alterazione si trova nel 70% dei casi. La presentazione più frequente è una linfoadenopatia indolente, accompagnata da splenomegalia. La prognosi è di 3-4 anni. La chemioterapia ha scarsissimi effetti, e la maggior parte dei pazienti arriva all’exitus per l’infiltrazione organica della neoplasia. È una neoplasia molto rara (2% di tutte le leucemie). Interessa soprattutto i maschi Caucasici di mezza età. Il nome della leucemia deriva dalla caratteristica morfologia delle cellule leucemiche, che hanno sottili prolungamenti simili a capelli. 34 Il midollo osseo è sempre interessato da un diffuso infiltrato interstiziale di cellule con nuclei ovalari o reniformi. Queste cellule rimangono intrappolate in una matrice di reticolina, scleroproteina dalle fibre connettivali, che rende molto difficile l’aspirazione con un ago aspirato. CARATTERISTICHE CLINICHE Queste derivano prevalentemente dall’infiltrazione tumorale di midollo, fegato e milza. • Splenomegalia: massiva e frequente, rilevabile anche con esame obiettivo • Epatomegalia: meno frequente e meno marcata della splenomegalia • Pancitopenia: risulta dalle alterazioni del midollo osseo e dal sequestro splenico. • Circa un terzo dei pazienti manifesta infezioni soprattutto micobatteri. PROGNOSI: Sopravvivenza media 5-6 anni TRATTAMENTO: - 2-Clorodeoxiadenosina (2-CdA), Desossicoformicina (DCF) - -IFN alfa (3 MU/die x 6-12 mesi) - Splenectomia Queste categorie comprendono un gruppo eterogeneo di neoplasie che hanno fenotipo simile rispettivamente alle cellule T mature o alle cellule NK. Anche se nessuna caratteristica morfologica è patognomonica dei linfomi a cellule T periferiche, alcuni aspetti sono tipici. Le neoplasie alterano diffusamente l’architettura dei linfonodi coinvolti e sono composta da una miscela polimorfa di cellule T maligne piccole, medie o grandi. Tutti i linfomi a cellule T periferiche hanno un fenotipo a cellule T mature esprimono CD2, CD5, CD3 di superficie. La maggior parte dei pazienti presenta: • linfoadenopatia generalizzata • prurito • febbre • perdita di peso È una neoplasia delle cellule CD4+ che si osserva negli adulti infettati dal virus HTVL-1, virus che provoca leucemia a cellule T. Sono maggiormente affetti da questa malattia soggetti che vivono nelle aree dove il virus HTVL-1 è endemico, come Giappone, Africa, Carabi. L’infezione è caratterizzata da: • lesioni cutanee • linfoadenopatia • epatosplenomegalia • linfocitosi 35 Le cellule tumorali contengono il provirus clonale HTVL-1, a conferma del diretto coinvolgimento patogenetico del virus in questa patologia. La micosi fungoide e la sindrome di Sézary sono differenti manifestazioni di una neoplasia delle cellule T helper CD4+, caratterizzata da una marcata predisposizione a interessare la cute. La micosi fungoide si presenta con tre fasi: 1. fase premicotica infiammatoria 2. fase a placche 3. fase tumorale Istologicamente si possono osservare infiltrazione dell’epidermide da parte di cellule T neoplastiche, che in genere proseguono la loro diffusione a livello dei linfonodi e del midollo osseo. La sindrome di Sézary p una variante nella quale il coinvolgimento della cute si manifesta con una eritrodermia esfoliativa generalizzata. A differenza della micosi, nella sindrome di Sézary le lesioni cutanee raramente progrediscono in tumefazioni. Anche se la malattia cutanea domina il quadro clinico in queste patologia, le sensibili analisi molecolari hanno dimostrato che le cellule tumorali si riscontrano nelle fasi precoci della malattia nel sangue, nel midollo osseo e nei linfonodi. Questi tumori sono indolenti con una sopravvivenza media di 8-9 anni. 36 Il linfoma di Hodgkin rappresenta un linfoma maligno contrassegnato dalla proliferazione di cellule neoplastiche associate ad una componente polimorfa considerata di tipo reattivo. A differenza dei linfomi non Hodgkin che si presentano frequentemente in sedi extralinfonodali e si diffondono in maniera imprevedibile, il LH insorge in un singolo linfonodo o in una catena di linfonodi e diffonde prima ai linfonodi immediatamente contigui. È caratterizzato da un elemento esclusivo costituito dalle cosiddette cellule di Reed-Sternberg, che inducono l’accumulo di linfociti, mastociti e granulociti reattivi. Nonostante l’esclusività di questo reperto diagnostico, è molto difficile individuarle, dato che costituiscono solo l’1,5% dell’intera massa tumorale. Ora è chiaro che nella maggior parte dei casi, le cellule neoplastiche di Reed-Sternberg derivino da cellule B dei centri germinativi o post-centri germinativi; questo fa pensare che il linfoma di Hodgkin origini prevalentemente dalle cellule B. EPIDEMIOLOGIA Colpisce tutte le età, con maggior frequenza gli individui > 60 anni. La razza maggiormente soggetta è quella bianca. Soggetti a rischio sono quelli con HIV e HBV. MORFOLOGIA ISTOLOGICA Innanzitutto la WHO riconosce cinque sottotipi di LH: 1. sclerosi nodulare 2. cellularità mista 3. ricchi di linfociti 4. deplezione linfocitaria 5. prevalenza linfocitaria Ognuno presenta delle caratteristiche istologiche diverse. L’identificazione delle cellule di Reed-Sternberg è essenziale per l’individuazione del linfoma. Le cellule di Reed-Sternberg si presentano come cellule binucleate, e ogni nucleo presenta un grosso nucleolo. Esistono anche varianti delle cellule di Reed-Sternberg: • varianti mononucleate • cellule lacunari • varianti linfoistiocitiche Sebbene le cellule di Reed-Sternberg siano un requisito per la diagnosi, esse devono essere presenti in un appropriato background di cellule infiammatorie non neoplastiche (linfociti, plasmacellule, eosinofili). 1. Linfoma di Hodgkin, tipo sclerosi nodulare È la forma più comune di LH (50-60 %). Le caratteristiche istologiche principali sono: • variante a cellula lacunare (più nuclei multilobati) della cellula di Reed-Sternberg • bande di collagene che dividono il tessuto linfoide in noduli circoscritti. Le cellule di RS sono osservabili in un contesto di linfociti T, eosinofili e plasmacellule. L’immunofenotipo è caratteristico: CD15+, CD30+, CD45+. Interessa maschi e femmine con la stessa frequenza. Prognosi: eccellente. 2. Linfoma di Hodgkin, tipo a cellularità mista Costituisce il 20-25 % delle manifestazioni. Si presenta come un interessamento diffuso da parte di un eterogeneo infiltrato cellulari di piccoli linfociti, eosinofili, plasmacellule e macrofagi benigni, frammisto alle cellule neoplastiche. Sono abbondanti le cellule di RS mononucleate. 37 L’immunofenotipo è identico al tipo sclerosi nodulare. Questa tipologia è molto frequente in maschi con EBV, infatti le cellule di RS presentano lo stesso menoma nel 70 % dei casi. Prognosi buona. 3. Linfoma di Hodgkin, tipo ricco di linfociti È una forma di LH molto rara, nella quel il tessuto colpito si presenta infiltrato per la maggior parte da linfociti T. Si osserva però una certa modularità dovuta alla presenza di follicoli residui di cellule B. Questo linfoma ha una tipologia di immunofenotipo caratteristica: CD45-, CD20-, CD15+, CD30+. È associata a EBV nel 30 % dei casi, e la prognosi è molto buona. 4. Linfoma di Hodgkin, tipo a deplezione linfocitaria Costituisce il 5 % del LH, ed è caratterizzata da una scarsità di linfociti e un’abbondanza di cellule di RS, o di loro varianti. Il fenotipo è uguale al LH a sclerosi nodulare. Questa tipologia si osserva prevalentemente negli anziani, nei pazienti HIV+. È un linfoma molto meno favorevole rispetto agli altri sottotipi. 5.Linfoma di Hodgkin, tipo predominanza linfocitaria Rappresenta il 5 % dei LH. Caratterizzato da una grossa presenza di linfociti molto piccoli frammisti a un numero variabile di istiociti benigni. Data la massiccia componente linfocitaria, risulta difficile trovare le cellule di Reed-Sternberg. Molto frequenti sono varianti delle cellule di RS chiamate cellule linfoistiocitiche (L&H). Queste esprimono marcatori delle cellule B (CD20) e il fattore di trascrizione specifico del centro germinativo (BCL6). L’EBV non è associato a questo sottotipo. La maggior parte del linfomi a predominanza linfocitaria si trasformano in linfomi a grandi cellule B. STADIAZIONE CLINICA DEI LINFOMI DI HODGKIN E NON HODGKIN La stadiazione dei linfomi fu formulata nel 1971 da Ann Arbor, e individua 4 stadi: 1. interessamento di una singola regione linfonodale (II) o di un singolo organo o regione extralinfatica (IIE) 2. interessamento di due o più regioni linfonodali dallo stesso lato del diaframma (III) o con interessamento del singolo organo o tessuto extralinfatico (IIIE) 3. interessamento delle regioni linfonodali da entrambe le parti del diaframma (IIII). Se comprende la milza (IIIIS), e/o il solo organo o sito extralinfatico contigui (IIIIE, IIIES) 4. focolai multipli o disseminati (E E) EZIOLOGIA E PATOGENESI Studi recenti basati sulla microdissezione e sull’analisi delle cellule di Reed-Sternberg hanno dimostrato che in più casi tutte le cellule di Reed-Sternberg sono interessate da identici riarrangiamenti dei geni delle immunoglobuline con ipermutazioni somatiche, stabilendo che la cellule di origine è una cellula B di un centro germinativo o post-centro germinativo. Esiste una piccola percentuale (1-2%) in cui le cellule di Reed-Sternberg non hanno geni delle immunoglobuline nella configurazione germ line, ma hanno riarrangiamenti del recettore delle cellule T, e in questo casi le cellule di Reed-Sternberg derivano da cellule T modificate. Dato che le cellule di RS sono difficili da studiare e non esistono ancora modelli animali di LH, si conosce relativamente poco circa i fattori che contribuiscono alla loro trasformazione. Un importante indizio è la frequente presenza degli episomi del virus EB nelle cellule di ReedSternberg, nel LH a cellularità mista. Importante è che la configurazione del DN dell’EBV sia la stessa in tutte le cellule tumorali di un dato caso, cosa che indicherebbe che l’infezione precede la trasformazione tumorale. 38 Le cellule di Reed-Sternberg esprimono la proteina di membrana LMP-1, una proteina codificata dal virus EB che trasmette segnali attivanti il fattore NF-κB, fattore di trascrizione di grande importanza nell’attivazione linfocitaria. L’attivazione di NF-κB e di altri fattori, determina la mutazione della cellula che la porta ad essere maggiormente soggetta ad altre mutazioni sconosciute, che portano alla formazione delle cellule di Reed-Sternberg. Una volta formata la cellula di Reed-Sternberg inizia a produrre citochine come l’IL-5, IL-6, IL-13, TNF. Queste citochine attraggono l’infiltrato reattivo, che a sua volta supporta la crescita e la sopravvivenza delle cellule tumorali. Infatti eosinofili e cellule T esprimono rispettivamente ligandi per i recettori CD30 e CD40, ognuno dei quali crea segnali che portano all’attivazione di NF-κB. SINTOMATOLOGIA CLINICA • • • • Sintomi sistemici Febbre: può essere continua, remittente o ciclica (15 gg alternati a periodi afebbrili). Sudorazione Perdita di peso: una perdita del 10 % nei 6 mesi prima dell’esordio clinico della malattia è importante dal punto di vista prognostico, ed è riconducibile all’anoressia e al conseguente diminuito apporto di cibo Prurito: può essere molto intenso, in genere resistente a diverti trattamenti, ma sensibile alla radio-chemioterapia Linfoadenopatia superficiali I linfonodi aumentano di volume, di consistenza duro-parenchimatosa, sono indolenti e inizialmente mobili sui piani profondi. Linfoadenopatie superficiali Interessano principalmente due sedi: 1. MEDIASTINO. Il coinvolgimento di questa sede riguarda i primi due stadi sovradiaframmatici. Quando le linfoadenopatie sono di piccole dimensioni possono rimanere a lungo asintomatiche, mentre quando raggiungono le grosse dimensioni possono compromettere strutture vicine provocando tosse secca, dispnea per interessamento del nervo ricorrente, disfagia per compressione dell’esofago, edema per compressione della vena cava superiore, stenosi bronchiale, coinvolgimento pleurico con versamento. 2. LOCALIZZAZIONE SOTTODIAFRAMMATICA. L’interessamento di sedi sottodiaframmatiche è poco frequente (10% dei casi). In ordine decrescente sono interessati i linfonodi lomboaortici, paracavali e periaortici, ilo splenico, epatico, linfonodi mesenterici. Connessi con queste localizzazioni possono comparire sintomi quali nausea, vomito, ittero colestasico. Splenomegalia L’ingrandimento della milza diventa clinicamente evidente nel 35 % dei casi. Comunque questo sintomo non è un indicatore del reale interessamento splenico da parte del morbo di Hodgkin, in quanto un terzo dei pazienti con splenomegalia non ha la malattia istologicamente dimostrabile. Scheletro Nel 15 % dei casi avanzati si possono manifestare interessamenti ossei con dolore e sindrome radicolare. SNC e periferico 39 C’è la tendenza del tessuto granulomatoso a diffondersi dai linfonodi paravertebrali agli spazi epidurali provocando sindromi compressive e quindi dolore, debolezza degli arti inferiori, ipoestesia corrispondenti al metamero interessato. Rene Spesso vi è ostruzione uretrale per compressione di masse retroperitoneali. REPERTI EMATOLOGICI Nelle fasi attive della malattia è presente anemia, leucocitosi (10-20000/mm3), neutrofilia e eosinofilia, linfocitopenia, trombocitopenia. I reperti ematochimici significativi sono: • iposideremia • iperuricemia • VES elevata • iperfibrinogenemia • iper α2-globulinemia Negli ultimi anni hanno assunto importanza particolari molecole di membrana (molecole solubili) elaborate dalle cellule di Reed-Sternberg, che possono mostrare elevati livelli nel siero dei pazienti con LH, in stretto rapporto con la stadiazione clinica del morbo. La diagnosi deve essere completata da una precisa valutazione dell’estensione della malattia, non solo ai fini prognostici, ma soprattutto di una realizzazione del piano terapeutico. La prognosi di sopravvivenza per gli stadi della classificazione di Ann Arbor sono: • stadio I: 79-90 % • stadio II: 74-80 % • stadio III: 55-60 % • stadio IV: 35-40 %. TERAPIA La strategia terapeutica attuale consiste nella radioterapia con acceleratori lineari e nella combinazione polichemioterapica MOPP per il trattamento di pazienti con malattia avanzata. La radioterapia viene effettuata utilizzando campi contrapposti su diverse sedi linfonodali dopo schermature d organi vitali: I campi di irradiazione sono personalizzati per ogni singolo paziente, tuttavia fanno comunque parte di schemi di riferimento indicati con i termini convenzionali per i campi: mantelline, Y rovesciata, Sub Total Nodal Irradiation (STNI), Total Nodal Irradiation (TNI). Per quanto riguarda la chemioterapia, l’associazione di più farmaci antiblastici attivi singolarmente e con differente meccanismo d’azione ha portato all’introduzione della MOPP (mecloretamina, vincristina, procarbazina, prednisone) che ha consentito di raggiungere remissioni complete nel 45-80 % dei casi. Sono state proposte anche altre combinazioni farmacologiche, quale l’ABVD (adriamicina, bleomicina, vimblanstina, decarbazina), con gli stessi risultati della MOPP. 40 La leucemia mieloide acuta è una malattia neoplastica che origina dal coinvolgimento della cellula staminale emopoietica, caratterizzata da un’alterata proliferazione e differenziazione della stessa e delle linee cellulari derivanti mieloide e/o eritroidi e/o megacariocitaria. La leucemia mieloide acuta colpisce principalmente gli adulti, con un picco di incidenza tra i 15 e i 39 anni, però si osservano casi anche negli anziani e nei bambini FISIOPATOLOGIA La maggior parte delle LMA è associata con alterazioni genetiche acquisite, che impediscono il normale differenziamento delle cellule mieloidi. Di conseguenza il midollo sarà costituito da blasti indifferenziati che esprimono uno o più tipi delle fasi precoci della differenziazione mieloide. Le mutazioni che più spesso si ritrovano nelle cellule mutate sono riarrangiamenti cromosomici che interessano geni che codificano per fattori della trascrizione. Per esempio la traslocazione t(8;21) e l’inversione (16), interessano geni che normalmente codificano per due subunità dello stesso fattore di trascrizione eterodimerico. Queste mutazioni provocano la codifica di proteine di fusione con attività cosiddetta dominante negativa, e le cellule che deriveranno dalla cellula mutata presenteranno un blocco parziale o completo nella differenziazione. Comunque non bastano queste mutazioni per l’insorgenza della LMA, ma è necessaria una collaborazione protogenetica. Per esempio una leucemia promielocitica associata alla traslocazione (15;17) produce un gene di fusione che codifica per una porzione di un fattore di trascrizione, il recettore α dell’acido retinoico, (RARα), fusa con una porzione di un’altra proteina, PML. Normalmente RARα è un fattore che attiva la trascrizione, ma quando è legato alla proteina PML si trasforma in un fattore inibente che spegne i geni necessari per una completa differenziazione mieloide. In tutte le LMA, l’accumulo dei precursori mieloidi neoplastici proliferanti nel midollo osseo sopprime le rimanenti cellule progenitrici ematopoietiche normali attraverso una sostituzione fisica come pure attraverso altri meccanismi sconosciuti. Quindi il fallimento della normale emopoiesi determina anemia, neutropenia e trombocitopenia, che causano la maggior parte delle più rilevanti complicanze delle LMA. MORFOLOGIA La diagnosi della leucemia mieloide acuta si basa sul reperto di una percentuale di blasti mieloidi superiori al 20% delle cellule del midollo. Sono diversi i tipi di blasti mieloidi: • Mieloblasti: fine cromatina nucleare, con due o più nucleoli e citoplasma voluminoso che spesso contiene granuli azzurrofili. • Corpi di Auer: sono elementi che si colorano in rossi alla colorazione per la perossidasi. Sono presenti in molti casi e sono particolarmente numerosi nelle LMA associate alla t(15;17). • Monoblasti. Spesso hanno nuclei convoluti o lobulati e non presentano i corpi di Auer. Raramente i blasti delle LMA presentano una differenziazione eritroidi, mentre spesso presentano una differenziazione megacariocitica, che è spesso accompagnata da fibrosi midollare. La conta dei blasti nel circolo può essere più di 100.000/µl, anche se nel 10% dei pazienti è sotto i 10.000/µl. Occasionalmente gli strisci di sangue periferico potrebbero non contenere blasti (leucemia aleucemica) e per tale ragione la diagnosi deve essere fatta con l’analisi istologica del midollo. 41 CLASSIFICAZIONE Nel sistema attualmente più usato, la classificazione FAB rivista, le LMA sono suddivise in otto categorie (M0 - M7). Questo schema prende in considerazione due caratteristiche: 1. grado di maturazione (M0 - M3) 2. derivazione dei blasti leucemici (M4 - M7) CLASSE M0 LMA Minimamente differenziate M1 Leucemia mieloide acuta senza maturazione M2 Leucemia mieloide acuta con maturazione M3 Leucemia acuta promielocitica M4 Leucemia mielomonocitica acuta M5 Leucemia monolitica acuta M6 Eritroleucemia M7 Leucemia megacarioblastica acuta INCIDENZA (%) MORFOLOGIA DEL MIDOLLO OSSEO 2-3 % Blasti privi di marker citologici e citochimica completi, ma esprimono gli antigeni della linea mieloide 20 % Molto immaturo, pochi granuli e corpi di Auer 30-40 % Serie completa di maturazione mieloide attraverso i granulociti; corpi di Auer presenti 5-10 % Le cellule sono per la maggior parte promielociti ipergranulari, con molti bastoncini di Auer per cellula. (traslocazione 15;17) 15-20 % Evidente differenziazione mielocitica e monocitica 10 % Due sottotipi: M5a) predominano monoblasti i promotori; M5b) predominano monociti maturi nel sangue periferico 5% Predominano i promotori della linea eritroidi. Le cellule non eritroidi sono per lo più mieloblasti 1% Predominano i blasti della linea megacariocitica. CARATTERISTICHE CLINICHE La maggior parte dei pazienti presenta una serie di sintomi correlati con l’anemia, la neutropenia e la trombocitopenia, in particolare: • astenia • febbre • emorragia spontanea cutanea e/o mucosa • comunemente insorgono petecchie cutanee ed ecchimosi, emorragie delle sierose che rivestono le cavità del corpo e i visceri, emorragie delle gengive e nel tratto urinario • infezioni frequenti, soprattutto nel cavo orale, cute, polmoni, tratto urinario e colon, da parte di batteri opportunisti come Pseudomonas. PROGNOSI La leucemia mieloide acuta è difficile da trattare. Il 60% dei pazienti trattati con chemioterapia ottiene una remissione completa, però solo il 15-30% rimano libero dalla malattia per 5 anni. Prognosi abbastanza favorevole viene formulata per LMA associate a t(8;21) e inv(16), mentre la prognosi è infausta nei pazienti con LMA con precedente sindrome mielodisplastica. Nuovi approcci basati su una migliore comprensione delle basi patogenetiche delle specifiche forme di LMA promettono di migliorare questa situazione. L’esempio attualmente più calzante è il caso della LMA associata a t(15;17) e dalla presenza della proteina di fusione PML-RARα. L’azione della proteina di fusione, che inibisce la differenziazione cellulare, è superato da dosi farmacologiche di acido all-trans-retinoico, che provoca la differenziazione delle cellule neoplastiche in neutrofili. Questi neutrofili derivati da cellule neoplastiche hanno una emivita breve è vengono eliminati. Quindi non si permette alla patologia di alterare la funzionalità midollare. 42 TERAPIA La terapia della LMA di basa soprattutto sulla chemioterapia citotossici e sul possibile trapianto di midollo osseo dopo chemioterapia. Lo scopo della terapia citotossici è quello di danneggiare irreparabilmente la capacità replicativi delle cellule mediante l’uso razionalmente combinato dei farmaci. Gli effetti collaterali della chemioterapia sono: • ipercaliemia • iperuricemia → nefropatia da urati • aplasia midollare (i farmaci citotossici non sono selettivi per le cellule neoplastiche). Questo a sua volta può provocare infezioni, emorragie e altre patologi correlate alla carenza di cellule prodotte dal midollo. Per evitare i primi due effetti collaterali, prima dell’inizio della terapia si somministra al paziente allopurinolo associato a idratazione del paziente con alcalinizzazione delle urine. L’obiettivo della terapia è quello di raggiungere la remissione completa dopo che il paziente ha superato il periodo di aplasia midollare e ricostituito il midollo normale. La terapia può essere schematizzata in: 1. induzione: ha lo scopo di provocare un effetto citocida su quasi tutte le cellule leucemiche 2. consolidamento: serve ad eliminare le cellule leucemiche residue (soprattutto quelle che dalla fase G0 ritornano in ciclo) 3. mantenimento: mediante l’uso continuo di farmaci citostatici si impedisce l’insorgenza e/o la crescita di cellule neoplastiche 4. intensificazione: impiego di farmaci a dosi più elevate per eradicare qualsiasi eventuale clone leucemico sviluppatosi e non ancora rilevabile con le comuni indagini Tutti i pazienti con i citotipi FAB M0-M7, eccetto M3 sono trattati con daunomicina + citosina arabinoside per l’induzione dell’effetto citotossici. La terapia della M3 è stata descritta nel paragrafo della prognosi. Il trapianto del midollo allo genico nelle LMA viene effettuato in pazienti al di sotto dei 45 anni per permettere la ricostruzione del sistema emopoietico, dopo che il paziente è stato sottoposto a chemioterapia e irradiazione del corpo intero. 43 Il termine sindromi mielodisplastiche (SMD) si riferisce ad un gruppo di malattie clonali delle cellule staminali, caratterizzate da difetti nella maturazione associati a ematopoiesi inefficace e da un aumentato rischio di trasformazione in leucemia mieloide acuta. Le SMD si presentano in due modi distinti: • SMD idiopatica o primaria: si verifica principalmente in soggetti con più di 50 anni • SMD terapia-correlata: è una complicanza di terapie genotossiche o radioterapia che si manifesta 2-8 anni dopo il trattamento. Tutte le forma di SMD possono trasformarsi in LMA, ma la trasformazione si verifica più rapidamente e con maggiore frequenza nelle t-SMD (terapia-correlata). CLASSIFICAZIONE DELLE SINDROMI MIELODISPLASTICHE La classificazione oggi in uso per le SMD è la classificazione FAB, che in base agli elementi morfologici dei blasti, individua cinque principali sottotipi delle sindromi mielodisplastiche: 1. anemia refrattaria 2. anemia sideroblastica idiopatica acquisita 3. anemia refrattaria con eccesso di blasti 4. anemia refrattaria con eccesso di blasti in trasformazione leucemica 5. leucemia mieloide cronica. ASPETTI ANATOMOPATOLOGICI Il reperto più caratteristico è costituito dalla differenziazione disordinata, che colpisce tutte le linee non linfoidi (eritroidi, granulocitica, monocitica e megacariocitica). Nell’ambito della serie eritroide, le alterazioni più frequenti comprendono: • sideroblasti ad anello: eritroblasti con mitocondri carichi di ferro • maturazione megaloblastica: simile a quella osservata nei deficit di vitamina B12 e di folati • anomalie nucleari di gemmazione: nuclei deformi e polipoidi I neutrofili appaiono con pochi granuli secondari. Le cellule pseudo-Pelger-Huet sono neutrofili con solo due lobi nucleari, e si osservano molto frequentemente. I megacariociti si osservano con lobi nucleari singoli o multipli che separano i nuclei (megacariociti pawn ball). I mieloblasti possono essere aumentati. PATOGENESI La patogenesi delle SMD è sconosciuta. Anche se il midollo osseo è solitamente ipercellulato al momento della diagnosi, può anche essere normocellulato, o ipocellulato. I progenitori mielodisplastica del midollo osseo vanno incontro ad apoptosi con maggior frequenza, e quindi è difficile comprendere come questi progenitori possano rimpiazzare del tutto i progenitori midollari normali rimanenti. Questo può suggerire che le SMD si sviluppano in un background di cellule staminali deplete o danneggiate. Le anomalie cromosomiche clonali più frequenti per lo sviluppo delle SMD sono: • sindrome da 5q-. La delezione delle braccia lunghe del cromosoma 5 si ritrova soprattutto nelle SMD secondarie a terapia. In circa il 70 % dei pazienti che presentano l’aberrazione 5q-, l quadro mielodisplastica si configura come una anemia refrattaria o una anemia sideroblastica idiopatica acquisita. Nel caso dell’anemia refrattaria, l’aberrazione 5q è associata ad una buona prognosi • monosemia 7 e 7q-. Sono state ritrovate informa di SMD sia primitive che secondarie e sono generalmente associate a decorso rapido e fatale per il paziente. 44 ASPETTI CLINICI Gli aspetti clinici delle SMD sono l’espressione del blocco differenziativo delle tre filiere midollari, con conseguente estrinsecazione di un quadro citopenico periferico. 1. ANEMIA. Quasi tutti i pazienti si presentano con i valori dell’emoglobina inferiori alla norma. L’anemia si presenta macrocitica, e solo poche volte normocitica, con anisopoichilocitosi. 2. NEUTROPENIA. Il quadro clinico è caratterizzato da numerose infezioni che nel 21 % dei casi portano a morte (escherichia,pseudomonas, staphylococcus aureus, klebsiella). Accanto alla riduzione quantitativa dei neutrofili, c’è anche un’alterazione nella funzionalità di questi, soprattutto nei riguardi della capacità chemiotattica, fagocitica e del killing. 3. TROMBOCITOPENIA. Generalmente la diminuzione della quantità piastrinica non è grave. Spesso comunque la trombocitopenia è associata anche ad alterazione morfologica delle piastrine (giganti o agranulari) che portano a disturbi funzionali quali tempo di sanguinamento allungato anche in presenza di valori piastrinici normali, ridotta aggregazione all’adrenalina e al collagene). QUADRI CLINICI SPECIFICI Anemia refrattaria (AS) L’anemia è il sintomo principale di questa condizione. Sangue periferico: • anisopoichilocitosi • macrocitosi lieve delle emazie • reticolocitopenia Il midollo presenta le seguenti caratteristiche: • iperplasia della filiera eritroide • le cellule blastiche non eccedono il 5%, mentre le periferiche risultano inferiori all’1% • midollo normocellulato o ipercellulato • le alterazioni non riguardano generalmente la serie megacariocitica e granulocitica. Anemia sideroblastica idiopatica acquisita (ASIA) A livello midollare si riscontrano cellule che prendono il nome di sideroblasti, e che sono in percentuale superiore al 15% della cellularità midollare totale. Questi sideroblasti sono praticamente eritroblasti in cui si accumulano grosse quantità di ferro che formano granuli siderotici evidenziabili con la colorazione al blu di Prussica. Questo ferro può accumularsi attorno alla membrana nucleare formando anelli (sideroblasti ad anello). Nel sangue periferico è presente un dimorfismo eritrocitaria: 1. eritrociti normali 2. eritrociti alterati da un abnorme emoglobinizzazione. Anemia refrattaria con eccesso di blasti (AREB) Questa forma può rappresentare: 1. quadro di esordio di una SMD 2. evoluzione di una AR o ASIA Le alterazioni morfologiche riguardano non più la sola serie eritroide, ma anche megacariocitica e granulocitica, con un eccesso di cellule blastiche > 5% a livello midollare. Anemia refrattaria con eccesso di blasti in trasformazione leucemica (AREB-T) 45 È caratterizzata da una quota blastica compresa tra il 20 e il 30%, valore oltre il quale si ricade nella LMA. Rappresenta quindi uno stadio intermedio tra una AREB e la LMA, infatti le cellule nel sangue periferico possono presentare anche corpi di Auer. Leucemia mieloide cronica Vedi sindromi mieloproliferative croniche DIAGNOSI DIFFERENZIALE La diagnosi delle malattie mielodisplastiche viene fatta mediante osservazione del quadro morfologico del sangue. Tuttavia questo non è patognomonico, e quindi bisogna escludere: • anemia aplastica • sindromi mieloproliferative croniche • disordini alimentari • complicazioni dell’alcolismo • intossicazione da metalli pesanti • LMA PROGNOSI La prognosi delle sindromi mielodisplastiche segue la loro classificazione. Infatti la sopravvivenza media del paziente tende a decrescere da 60 mesi nelle AR e ASIE a 12-6 mesi nelle forma AREB e AREB-T. Nell’ambito prognostico hanno acquisito rilevanza i parametri più strettamente biologici, quali i dati dell’analisi citologica. È nato quindi un indice prognostico internazionale, che affianca alla percentuale di blasti e al grado della citopenia anche le alterazioni del cariotipo. A seconda dei punteggi quindi possono essere distinti quattro gruppi a rischio, ulteriormente stratificabili per età. TERAPIA Le SMD sono caratterizzate da una grossa varietà di aspetti clonico-ematologici, e questo non ha consentito di indirizzarci verso una terapia en precisa. Comunque si utilizzano schemi terapeutici raggruppati nelle seguenti tipologie: • TERAPIA DI SUPPORTO. Riguarda le forme croniche, in cui la terapia viene fatta utilizzando opportune trasfusioni di globuli rossi • TERAPIA ORMONALE. In passato venivano usati steroidi, ma l’effetto sull’eritropoiesi viene considerato aleatorio e di breve durata, a fronte di numerosi effetti collaterali provocati da queste sostanze. • CHEMIOTERAPIA. Nel trattamento AREB e AREB e -T la chemioterapia è associata a notevoli effetti collaterali. In linea generali le remissioni complete ottenute sono di breve durata e le recidive rispondono assai poco al trattamento riduttivo. Ultimamente la chemioterapia si effettua utilizzando citosina arabinoside che sembra agisca incrementando la maturazione degli elementi mieloidi . Tuttavia molti pazienti vanno incontro a ipoplasia midollare. • FATTORI INDUCENTI LA DIFFERENZIAZIONE. I risultati di questa terapia sono tuttora controversi, e per esempio l’utilizzo dell’acido retinoico si è dimostrato insoddisfacente. Anche con IFN-γ e α solo pochi pazienti hanno acquisito una risposta significativa per quanto attiene a riduzione del fabbisogno trasfusionale. • FATTORI DI CRESCITA. • TRAPIANTO DI MIDOLLO OSSEO. Appare attualmente come il solo trattamento in grado di operare la sostituzione del clone displastico e di indurre guarigione nel 30-50% dei pazienti. 46 Il fattore limitante è l’età, in quanto il trapianto si può effettuare in soggetti con età inferiore ai 40 anni. Nella maggior parte delle malattie di questo gruppo le cellule neoplastiche sono progenitori multipotente, in grado di dar vita a eritrociti, granulociti, piastrine, monociti e linfociti. Le quattro più comuni malattie mieloproliferative croniche sono: 1. leucemia mieloide cronica 2. policitemia vera 3. trombocitemia essenziale 4. mielofibrosi primitiva. A differenza delle neoplasie linfoidi e delle LMA, le caratteristiche patologiche delle malattie mieloproliferative croniche non sono specifiche, avendo un considerevole grado di sovrapposizione l’una con l’altra. È una malattia principalmente degli adulti di età compresa tra i 25 anni e i 60 anni. La LMC si distingue dalle altre malattie sindromi mieloproliferative croniche per una caratteristica alterazione molecolare, vale a dire • traslocazione che interessa il gene BCR sul cromosoma 9 e il gene ABL sul cromosoma 22. Ne risulta un gene di fusione BCR-ABL che codifica per una proteina di fusione con attività tirosin-chinasica. • Nel 90% dei casi il cariotipo rivela il cromosoma Philadelphia (traslocazione (9;22)). L’accoppiamento BCR-ABL provoca l’attivazione di meccanismi a valle che hanno come risultato l’inibizione dell’apoptosi e la divisione cellulare. Il midollo osseo nella LMC si presenta al 100% cellularizzato con precursori granulocitici in maturazione che costituiscono la maggior parte dell’aumentata cellularità. Spesso l’aumento della cellularità riguarda anche la linea megacariocitica, mentre quella eritrocitica di solito non viene interessata. L’analisi del sangue periferico manifesta una aumentata leucocitosi (>100.000/mm3). Inoltre è presenta splenomegalia dovuta a ematopoiesi extramidollare. La storia naturale della LMC ha inizio di solito con una sensazione di fastidio all’addome causata dalla splenomegalia, o dal dolore acuto dovuto dall’infarto splenico. La progressione della malattia è lenta e la sopravvivenza media si aggira intorno ai tre anni. Dopo infatti tre anni il 50% dei pazienti entra in una fase accelerata in cui l’anemia peggiora insieme alla trombocitopenia. Dopo 1 anno la fase accelerata si conclude con un quadro simile alla leucemia acuta (crisi blastica). Nell’altro 50% dei casi si verifica una crisi blastica senza fase accelerata intermedia, in cui il 70% dei blasti hanno le caratteristiche di mieloblasti. La conoscenza della patogenesi molecolare della LMC ha portato all’introduzione di farmaci che inibiscono la chinasi BCR-ABL, e che inducono remissione completa nel 90 % dei casi. Tuttavia gli inibitori di BCR-ABL sopprimono ma non eliminano il clone LMC e quindi non possono prevenire la progressione in una crisi blastica. Per queste ragioni il trapianto di midollo osseo è la terapia preferita in pazienti giovani, col 75% di guarigioni. 47 La policitemia vera è una neoplasia che ha origine da una cellula staminale mieloide multipotente, caratterizzata soprattutto dall’aumento della massa eritrocitaria, accompagnato da modesta trombocitosi e leucocitosi. PATOGENESI I meccanismi patogenetici invocati nella policitemia vera ipotizzano: 1. fattore responsabile di una proliferazione non controllata delle cellule midollari 2. abnorme sensibilità delle cellule proliferanti ai fattori di crescita (come l’Epo) Nel midollo osseo di un paziente con policitemia vera coesistono sia il clone neoplastico, ipersensibile all’Epo, che la linea cellulare normale, che però sembra essere inattivata, con meccanismi ignoti, dal clone neoplastico. Poiché i meccanismi di produzione dell’Epo sono inalterati, è ipotizzabile che il difetto della PV risieda in un’alterata risposta del clone neoplastico ai fattori di regolazione dell’eritropoiesi. CLINICA Colpisce soggetti con più di 60 anni. Spesso la diagnosi è casuale, ma l’esordio della sintomatologia è caratterizzato da sintomi minori dovuti all’aumento della viscosità ematica: • cefalea • vertigini • disturbi visivi • claudicatio • tromboflebiti • fenomeno di Raynaud • prurito: può essere dovuto alla liberazione di istamina dagli elementi granulocitari. Tuttavia nel 15-20% dei pazienti l’esordio può avere forma grave, come processi trombotica o emorragie, che possono manifestarsi anche all’encefalo, nel polmone o a livello cardiaco. Queste manifestazioni emorragiche rappresentano la causa di morte nel 40% dei pazienti con PV. ESAME DEL MIDOLLO OSSEO Il midollo osseo appare ipercellulato con diminuzione netta della componente adiposa. La biopsia osteomidollare può rilevare presenza di fibrosi midollare che può essere lieve o raggiungere quadri conclamati. L’esame del midollo osseo è utile nella diagnosi differenziale tra PV e: eritrocitosi secondaria: si osserva solo iperplasia eritroide e non panmielosi • • trombocitemia esenziale: evidente soprattutto iperplasia megacariocitica • LMC: vie è netta predominanza di granulociti e riduzione della serie eritroide • mielofibrosi idiopatica: notevole quantità di fibre reticolari. CRITERI DIAGNOSTICI Esistono due tipologie di criteri per la diagnosi della policitemia vera: 1. criteri maggiori • incremento della massa eritrocitaria (maschi > 36ml/kg, donne > 32ml/kg) • normale saturazione O2 > 92% • splenomegalia 2. criteri minori • trombocitosi (>400.000/µl) • leucocitosi (>12.000/µl) • fosfatasi alcalina leucocitaria elevata 48 • aumento della vitB12 o della capacità legante la B12 libera La diagnosi di PV può essere effettuata solo se sono presenti tutti e tre i criteri maggiori oppure due maggiori e due minori. DECORSO CLINICO Il decorso della PV è cronico, e le complicanze più severe sono quelle vascolare, spesso causa di exitus. Durante la sua evoluzione la malattia si modifica passando dalla fase puramente policitemica, alla fase di metaplasia mieloide (fase spenta), nella quale si osservano: 1. splenomegalia aumentata 2. riduzione della massa eritrocitaria 3. incremento della fibrosi midollare L’evoluzione a metaplasia mieloide si osserva dopo 10 anni nel 25% dei pazienti e presenta anche deficit di ferro, folati e/o vitamina B12. TERAPIA La terapia viene effettuata in due modi: 1. terapia con deplezione eritrocitaria: effettuata con salassi periodici. 2. terapia con antiblastici: viene utilizzata maggiormente l’idrossiurea per la sua efficacia e la buona tollerabilità. La trombocitopenia essenziale è un disordine mieloproliferative cronico caratterizzato da trombocitosi (>600.000/µl), con iperplasia megacariocitaria nel midollo osseo e a volte da leucocitosi modesta. EZIOPATOGENESI La TE deriva dalla proliferazione clonale della cellula staminale totipotente a prevalente differenziazione megacariocitaria. Comunque nella patologia sono aumentati tutti i precursori staminali mieloidi (CDF-GM, CFUMeg e BFU-E). Inoltre le CFU-Meg rispondono in modo eterogeneo agli stimoli della trombocitopoietina, dimostrandosi alcune estremamente ipersensibili. CLINICA La malattia interessa soggetti adulti con più di 50 anni. Può esordire con manifestazioni trombotiche e/o emorragiche. Queste ultime sono le più frequenti, soprattutto a carico dell’apparato gastrointestinale (melena e ematemesi). Una splenomegalia di modesta entità è presente nel 60% dei casi, mentre nel 40% si verifica anche una epatomegalia. DIAGNOSI La diagnosi di TE può essere fatta basandosi sui seguenti criteri: • conta piastrinica > 600.000/µl • midollo ipercellulare con iperplasia megacariocitica • fosfatasi alcalina leucocitaria normale o aumentata • no cromosoma Philadelphia • eritrociti normali 49 • fibrosi midollare di modesta entità. Ci sono diverse condizioni in cui potrebbe verificarsi piastinosi: • splenectomia • neoplasie • interventi chirurgici • malattie renali croniche • osteoporosi • sideropenia. Tutte queste situazioni sono caratterizzate da una piastinosi transitoria, e questo ci permette di fare diagnosi differenziale. TERAPIA 1. Farmaci antiaggreganti. Utilizzati soprattutto nei soggetti che hanno gia presentato episodi trombotici. 2. Farmaci antiblastici. Questa terapia ha l’obiettivo di riportare la conta piastrinica al di sotto di 500.000/µl. 3. Idrossiurea. Dosi tra 15-30 mg/kg controllano la trombocitosi e inducono remissione completa parziale della stessa. 4. IFN-α, IFN-β, IFN-γ. Inibiscono la crescita delle colonie di CFU-Meg. La sospensione della terapia con interferone induce una riespansione della massa piastrinica, e quindi è necessario che una terapia di soppressione venga seguita da una terapia di mantenimento. La peculiarità della mielofibrosi primitiva è il rapido sviluppo della fibrosi midollare degenerativa. La mielofibrosi sopprime l’ematopoiesi del midollo osseo, portando citopenia del sangue periferico e una estesa emopoiesi extramidollare neoplastica nella milza, nel fegato e nei linfonodi. FISIOPATOLOGIA La mielofibrosi ha inizio da un iperilascio di collagene da parte dei fibroblasti nel midollo. Questa fibrosi va a sostituirsi agli elementi ematopoietici del midollo, comprese le cellule staminali, causando l’insorgenza di una emopoiesi extramidollare che interessa principalmente fegato, milza e linfonodi. Il deposito di collagene da parte dei fibroblasti non neoplastici è un effetto causato dalla notevole produzione di fattori fibrogenici da parte di megacariociti neoplastici. Di questi fattori sono stati identificati: • TGF-β. Promuove la fibrosi e causa angiogenesi • PDGF Sono entrambi fitogeni dei fibroblasti. Man mano che procede la mielofibrosi, le cellule staminali ematopoietiche che sono in circolo si localizzano negli organi emopoietici extramidollari, cioè milza, fegato e linfonodi, che diventano quindi le maggiori sedi di emopoiesi. EVOLUZIONE CLINICA La mielofibrosi primitiva è rara nei soggetti al di sotto dei 60 anni. Solitamente giunge all’osservazione clinica per: • Anemia • Splenomegalia Il paziente può sentire una sensazione di riempimento nel quadrante superiore sinistro. 50 I riscontri di laboratorio comprendono: • anemia normocromica normocitica da moderata a grave • leucoeritroblastosi • conta piastrinica normale o elevata, ma con il progredire della malattia sopravviene la trombocitopenia Per la diagnosi è necessaria comunque una biopsia del midollo osseo che appare come segue: • midollo ipercellulato negli stadi precoci e ipocellulato negli stadi avanzati • degenerazione mielofibrotica dello spazio midollare • megacariociti atipici PROGNOSI Il decorso clinico è molto variabile, e questa variabilità dipende soprattutto dall’eterogeneità delle casistiche valutate, soprattutto dal limitato numero di pazienti e dalla difficoltà di stabilire criteri diagnostici assoluti. In genere la sopravvivenza è compresa in un range ristretto tra 5 e 15 anni. Le cause più frequenti di exitus sono individuate nello scompenso cardiaco congestizia, nelle complicanze emorragiche, specie intestinali, nell’evoluzione in leucemia acuta. TERAPIA La maggior parte dei pazienti asintomatici non viene sottoposta ad alcun trattamento, e il paziente viene sottoposto solo a controlli periodici. Il trattamento viene instaurato solo quando comincia a manifestarsi l’anemia (che all’inizio può essere trattata con terapia trasfusionale). • terapia con androgeni:nel tentativo di indurre l’eritropoiesi • terapia con idrossiurea • radioterapia splenica (per ridurre il volume della milza) • splenectomia. Viene presa in considerazione solo: dolore intenso e persistente anemia emolitica refrattaria ipersplenismo con piastrinopenia severa. 51 La caratteristica di questo gruppo di malattie è la proliferazione di un clone di cellule B che sintetizza e secerne un unico e omogeneo tipo di immunoglobulina o suoi frammenti. Tutte insieme le gammapatie monoclonali rappresentano il 15 % di tutti i decessi per neoplasie dei globuli bianchi. L’immunoglobulina prodotta viene indicata come “componente M”. Essa ha un P.M. ≥ 160.000, quindi sono confinate nel plasma e nei fluidi extracellulari, e sono escluse dalle urine in assenza di lesioni dei glomeruli. Le plasmacellule neoplastiche secernono, oltre che il clone di immunoglobulina, anche frammenti di Ig, che nella maggior parte dei casi sono catene pesanti L. Queste catene sono molto piccole, e vengono escrete con le urine. Quindi saranno riscontrabili nel sangue sono se c’è danno renale, o la loro produzione è smisuratamente grande. Il MM è una malattia tumorale a elettiva localizzazione nel midollo osseo, caratterizzata da interessamento dello scheletro in sedi multiple. La malattia ossea è prevalente, ma può interessare anche linfonodi e sedi extralinfonodali, come la cute. Il MM determina l’1 % delle morti per neoplasia nei paesi occidentali. È verosimile che la cellule clonogenica nel MM sia una cellula della fase differenziativa antigenedipendente (plasmoblasto o cellula B della memoria), cioè elementi che hanno gia passato la selezione antigenica nel centro germinativo. EZIOLOGIA La proliferazione e la sopravvivenza delle cellule del mielosa sono dipendenti da diverse citochine, soprattutto IL-6. L’interleuchina-6 è prodotta dalle stesse plasmacellule neoplastiche, oltre che dalle cellule stromali del midollo. Nella malattia in atto, i livelli sierici di IL-6 aumentano notevolmente, e maggiore è il livello, peggiore sarà la prognosi. Inoltre le cellule neoplastiche producono fattori che favoriscono la distruzione ossea, quali i fattori MIPα e NF-κB che attivano gli osteoclasti e offrono l’indicazione per eventuali interventi terapeutici. Le alterazioni cromosomiche che si riscontrano nel MM sono: • delezione di 13q • traslocazione del locus per le catene pesanti delle Ig con: cromosoma 4p16: codifica per un recettore tirosin-chinasico implicato nella proliferazione cellulare geni della ciclica D1 o D3 che controllano il ciclo cellulare gene per il fattore di trascrizione cMAF MORFOLOGIA Il MM si presenta molto spesso in tutto il sistema scheletrico sotto forma di lesioni tumorali multifocali osteolitiche e composte da plasmacellule. Sono colpite maggiormente le ossa dello scheletro assiale (colonna 66%, coste 44%, cranio 41%, pelvi 28%, femore 24%, clavicola 10%, scapola 10%). La lesione origina nella cavità midollare, erode l’osso trasecolare e distruggono progressivamente la corticale. Quindi si capisce il perché la frattura ossea è una delle manifestazioni principali di questa neoplasia, oltre il dolore osseo. 52 Le lesioni ossee appaiono come lesioni a margini netti, costituite da masse tumorali molli e rosse. Al microscopio il midollo presenta numerose plasmacellule che possono presentarsi sia normali, che anormali, per esempio con più di un nucleo, con citoplasma rosso acceso (cellule a fiamma), con numerosi granuli citoplasmatici (cellule di Mott). L’elevato livello sierico della componente M porta all’impilamento dei globuli rossi negli strisci di sangue periferico, reperto noto col nome di formazione di rouleaux. Non si tratta di un reperto specifico, poiché si può ritrovare anche nel LES o nell’HIV. Se la malattia avanza, le plasmacellule possono invadere anche il sangue periferico, provocando una leucemia plasmacellulare. SINTOMATOLOGIA E DIAGNOSI I sintomi sono correlati all’instaurarsi di due eventi: 1. infiltrazione di organi e tessuti ad opera delle plasmacellule mielomatose 2. eccessiva produzione di immunoglobuline monoclonali I segni della malattia in atto sono i seguenti: 1. DOLORI OSSEI. Sono dovuti ad una lesione ossea di tipo litico correlata alla proliferazione delle plasmacellule mielomatose nella cavità midollare. Le plasmacellule sembra che producano un fattore chiamato OAF in grado di esaltare l’attività osteoclastica. Ne consegue ipercalcemia, che rappresenta un vento di frequente osservazione in questi pazienti. Sono quindi utili ai fini diagnostici anche radiografie, che mostrano il caratteristico aspetto delle lesioni litiche delle ossa, oltre alla scintigrafia. 2. INFEZIONI. L’infezione caratterizza il 20 % di questi pazienti, e può provocare anche la morte del soggetto. L’insorgenza è causata da: carenza di Ig efficienti, dato che la maggior parte di esse sono monoclonali carenza della funzione monocitaria e granulocitaria immunodeficienza causata dalla chemioterapia neutropenia da carenza produttiva midollare 3. INSUFFICIENZA RENALE. In circa il 30 % dei pazienti. I livelli di creatinina e di azoto ematico si trovano aumentati in misura diretta al grado di insufficienza renale. I fattori che intervengono nel determinare l’insufficienza renale sono: ipercalcemia iperuricemia deposizione di crioglobuline infezioni renali disidratazione 4. SINDROME DA IPERVISCOSITÀ. Si osserva raramente, soprattutto nel mielosa IgA e nei sottotipi IgG1 e IgG3. è caratterizzata da cefalea, vertigini, parestesie, epistassi, emorragie, insufficienza cardiaca congestizia. 5. ALTERAZIONI DELL’EMOSTASI. È dovuta alla piastrinopenia causata dall’inefficienza midollare 6. MANIFESTAZIONI NEUROLOGICHE. 7. AMILOIDOSI. Nel 5 % dei pazienti con MM. ESAMI DI LABORATORIO L’esame dell’elettroforesi delle proteine sieriche pone in evidenza la presenza di un picco a banda stretta nel 90 % dei casi che migra in regione γ o meno frequentemente in zona β o α2. La componente monoclonale è nel 60 % dei casi di tipo IgG, nel 20 % IgA e nell’1% IgD. Tramite la tecnica di immunofissazione è possibile evidenziare una componente monoclonale nel siero e nelle urine nel 90 % dei casi, anche se la tecnica che meglio può approfondire l’indagine è l’immunoelettroforesi. Dal punto di vista ematochimico si evidenziano: 53 • • • • • • • • • • ipercalcemia iperuricemia aumento della viscosità plasmatica aumento della β2-microglobulina proteinuria di Bence-Jones: produce lesioni molto spesso irreversibili a livello renale. Infatti questa proteina viene filtrata dai glomeruli renali, ma a livello dei tubuli distali precipita provocandone l’occlusione. VARIANTI CLINICHE Mieloma non secernente. Il clone plasmacellulare produce, ma non secerne immunoglobuline Mieloma micromolecolare. Il clone plasmacellulare produce sono la catena leggera delle immunoglobuline. Non si osserva il picco a livello γ nell’elettroforesi, ma è presente la proteinuria di Bence-Jones. Mieloma non secernente e non producente. Non produce né secerne Ig. La diagnosi viene formulata solo osservando l’infiltrazione midollare. Plasmocitomi solitari dell’osso o plasmocitomi extramidollari. La proliferazione delle plasmacellule non invade altri tessuti, e si concentra solo su un segmento osseo o un ornano extramidollare. La diagnosi viene effettuata mediante evidenzia radiologica (nel caso dell’osso), o TAC (nel caso di sedi extramidollari). Leucemia plasmacellulare. Le plasmacellule sono rilevabili nel sangue periferico. Esordisce soprattutto in età giovanile e determina lesioni osteolitiche e picchi monoclonali all’elettroforesi molto evidenti. PROGNOSI È piuttosto variabile ed oscilla intorno a 20-60 mesi. Durie e Salmon hanno suddiviso l’evoluzione del MM in tre stadi, in base ai quali è possibile ipotizzare una prognosi. Il primo stadio prevede una prognosi di 60 mesi, mentre lo stadio III di 14 mesi. TERAPIA Varia a seconda dello stadio clinico della malattia. Gli stadi II e III vengono trattati con chemioterapia. Per lo stadio I, in cui la prognosi è buona, la terapia è costituita da phalan e prednisone, a cicli che vanno ripetuti ogni 6 settimane. La sopravvivenza a 2 anni è del 55-60%. Pazienti trattati con IFN-α e steroidi hanno mostrato una riduzione della componente monoclonale, anche se senza miglioramento della sopravvivenza. GAMMAPATIE MONOCLONALI DI INCERTO SIGNIFICATO La proteina M può essere identificata nel siero dell’1% di individui asintomatici con età superiore a 50 anni. Questa disproteinemia non associata a malattia è chiamata gammapatie monoclonale di incerto significato (MGUS). Circa l’1 dei pazienti con MGUS progredisce fino allo sviluppo del MM; infatti le modificazioni che scatenano la MGUS, sono le stesse che si osservano nel MM, e questo fa pensare che la MGUS sia una manifestazione precoce del mieloma multiplo. I livelli di proteine monoclonali nel MGUS è 3 g/dl, e non presentano proteina di Bence-Jones. Non è possibile valutare se un paziente con MGUS avrà un decorso benigno della malattia, quindi necessita la valutazione periodica dei livelli sierici di componenti M e della proteinuria di BenceJones. 54 È una patologia nota anche col nome di magroglobinuria essenziale o primitiva, o porpora macroglobulinemica. Si caratterizza per la produzione di una componente monoclonale del tipo IgM prodotta da elementi linfoidi o linfoplasmociti che proliferano in maniera disordinata infiltrando il midollo osseo. Questa IgM è rilevabile soprattutto mediante elettroforesi delle proteine plasmatiche, oltre che a tecniche di immunofissazione e immunoelettroforetiche. Il morbo di Waldenström è annoverato tra i linfomi maligni, e tende a manifestarsi nei maschi in età avanzata. SINTOMATOLOGIA I sintomi principalmente rilevabili sono: • astenia, dispnea da sforzo, emorragie e porpora cutanea • alterazioni oculari: interessano soprattutto la retina con edema, emorragia e congestione venosa • anemia, adenopatie, epatomegalia: derivano dall’espansione del clone linfocitario. • a differenza del MM non si verifica insufficienza renale • possono verificarsi fenomeni di autoemolisi dovuta alle immunoglobuline prodotte • le IgM hanno una grande facilità nell’aggregarsi tra di esse e con le emazie, quindi sono riscontrabili complessi di emazie tipici, a pila. PROGNOSI La sopravvivenza media è di 5 anni, e la morte sopraggiunge per l’infiltrazione del clone cellulare degli organi, o per accidenti vascolari. • • • • • LABORATORIO picchi dell’elettroforesi nella fascia γ anemia normocromatica test di Coombs positivo nel 30 dei casi viscosità plasmatica aumentata cellule linfoidi midollari reagiscono con antisieri anti-IgM. TERAPIA Clorambucile e prednisone. 55 La mononucleosi infettiva è un’adenopatia benigna causata dal virus di Epstein-Barr (EBV). Questo virus è associato allo sviluppo di leucoplachia villosa e di varia neoplasie, soprattutto particolari linfomi. La mononucleosi infettiva è caratterizzata da: • febbre • linfoadenopatia • splenomegalia • faringodinia • comparsa nel sangue di linfociti T atipici • meningite, polmonite ed epatite (solo in rari casi) Nelle nazioni più sviluppate, la malattia si manifesta principalmente nella tarda adolescenza, o nei giovani adulti. PATOGENESI L’EBV si trasmette attraverso contatti interumani, frequentemente tramite la saliva con il bacio (infatti viene definita malattia del bacio). Una volta ingerito il virus, l’infezioni virale inizia nel tessuto linfoide della rinofarige e orofaringe, soprattutto nelle tonsille. L’EBV penetra nel tessuto linfoide sottomucoso dopo aver infettato le cellule epiteliali. Quindi interagisce con il recettore CD21 delle cellule B e a questo punto l’infezione può assumere due forme: 1. infezione produttiva: le cellule B vengono infettate e si assiste a lisi cellulare con liberazione di virioni; 2. infezione latente I geni implicati nell’infezione da EVB sono almeno 11. Per esempio: • EBNA1 che ha importanza nella replicazione del DNA • EBNA2 e LMP-1, che hanno importanza nell’attivazione delle cellule B. Le cellule B così attivate si disseminano in circolo e producono anticorpi specifici diversi, tra cui gli anticorpi eterofili anti-emazie di montone, utilizzati per la diagnosi della mononucleosi infettiva. Infatti soggetti con mononucleosi infettiva producono immunoglobuline in grado di agglutinare le emazie di montone. I sintomi della mononucleosi infettiva compaiono all’inizio della risposta immunitaria dell’ospite. La risposta immunitaria è di tipo cellulare, mediata dalle cellule T citotossiche CD8+ e dalle cellule NK. Infatti i linfociti atipici osservati nel sangue nel corso della malattia sono proprio linfociti CD8+ e NK. La proliferazione di questi linfociti è localizzata a livello dei tessuti linfoidi, e questo spiega la linfoadenopatia e la splenomegalia. All’inizio della malattia vengono prodotto IgM che reagiscono contro gli antigeni virali, e in seguito si producono IgG che rimangono per tutta la vita. I sintomi principali sono: • Febbre • Faringo-tonsillite (angina monocitica) • Adenopatie Cervicali (90%) • Splenomegalia (40%) • Epatomegalia (10%) • Rush Maculo-papulare (10%) • Epatite (< 5%) • Emorragie cutaneo-mucose (< 5%) 56 • • Complicanze gravi (< 1%) (encefalite, meningite, miocardite, aplasia midollare) Astenia marcata DIAGNOSI La diagnosi viene posta sui seguenti reperti • linfocitosi con caratteristici linfociti atipici nel sangue periferico • reazione positiva agli anticorpi eterofili (monotest) • presenza di anticorpi specifici per gli antigeni dell’EBV Negli esami di laboratorio avremo i seguenti risultati: • Leucocitosi con monocitosi assoluta, cellule mononucleate con l’aspetto di: - cellule linfoidi - cellule monocitoidi - cellule plasmocitoidi • Trombocitopenia e anemia (rare) • Aumento delle transaminasi (80% dei casi) • Sierologia: anticorpi anti-EBV, inizialmente IgM poi IgG (> 90% dei casi), anti-CMV (<10%) • Sierologia della infezione EBV • Fase Acuta anti-VCA IgM→IgG anti-EA IgG • Convalescenza anti-EBNA • Test di Coombs positivo (5%) 57
Scaricare